

SUMMARY
Autosomal-recessive cerebellar hypoplasia and ataxia comprise a group of heterogeneous brain disorders, caused by disruption of several fundamental cellular processes. Here, we identified 10 families showing a neurodegenerative condition involving pontocerebellar hypoplasia with microcephaly (PCHM). Patients harbored biallelic, mutations in genes encoding the spliceosome components Peptidyl-Prolyl Isomerase Like-1 (PPIL1) or Pre-RNA Processing-17 (PRP17). Mouse knockouts of either gene were lethal in early embryogenesis, whereas PPIL1 patient mutation knockin mice showed neuron-specific apoptosis. Loss of either protein impacted splicing integrity, predominantly affecting short and high GC-content introns and genes involved in brain disorders. PPIL1 and PRP17 form an active isomerase-substrate interaction, however, we found isomerase activity is not critical for function. Thus, we establish disrupted splicing integrity and ‘major spliceosome-opathies’ as a new mechanism underlying PCHM and neurodegeneration, and uncover a non-enzymatic function of a spliceosomal proline isomerase.
Keywords: Pontocerebellar hypoplasia, microcephaly, neurodegeneration, brain development, spliceosome, cyclophilin, proline isomerase, alternative splicing, PCHM, PPIL1, PRP17, NMR, recessive disease
eTOC Blurb
Chai et al. discover that loss of splicing factors PPIL1 and PRP17 lead to a neurodegenerative brain disease, and even though they are an enzyme-substrate pair, they function instead to scaffold the spliceosome.
INTRODUCTION
Pontocerebellar hypoplasia (PCH) refers to a group of severe pediatric-onset neurodegenerative disorders affecting cellular survival in the brainstem and cerebellum, resulting in impaired neurological function and early death (Cassandrini et al., 2010). Humans with PCH show near-normal early embryologic development, followed by midgestational slowing or cessation and later regression in select neuroanatomical regions (Joseph et al., 2014). Most genes implicated in PCH are involved in tRNA splicing or GTP availability, suggesting a potential effect on protein translation (Breuss et al., 2016; Budde et al., 2008; Karaca et al., 2014; Schaffer et al., 2014). While postnatal progressive microcephaly can be part of the clinical spectrum, children are mostly born with normal or only mildly reduced head circumference (van Dijk et al., 2018).
Pre-mRNA splicing, mediated by the spliceosome complex, is essential for gene expression and regulation in higher organisms (Shi, 2017; Will and Luhrmann, 2011). Increased splicing complexity results in dramatically enlarged diversity in both the transcriptome and proteome: for instance, 95% of multi-exon human genes undergo alternative splicing (AS). Not surprisingly, AS is especially prevalent in the brain, corresponding to its complex composition of cell types and functions (Raj and Blencowe, 2015; Zhang et al., 2016). Remarkably, while aberrant splicing of individual genes due to defective cis- or trans-regulation has been widely reported in human brain diseases, global splicing defects by mutations in core major spliceosome complex (MSC) components have rarely been associated (Chabot and Shkreta, 2016; Scotti and Swanson, 2016).
Incorporated within the MSC are eight cyclophilin peptidyl-prolyl isomerases (PPIase), enzymes initially found as targets of immunosuppressants, but later found to promote conformational changes of substrates by catalyzing cis-trans isomerization of Xaa-Proline peptide bond (Agafonov et al., 2011; Bessonov et al., 2010; Davis et al., 2010; Evans et al., 1987; Rajiv and Davis, 2018; Teigelkamp et al., 1998; Zhou et al., 2002). Functions and substrates of most PPIases remain unknown. Here, we report that biallelic, hypomorphic mutations in two spliceosomal genes PPIL1 and PRP17, encoding an active PPIase-substrate pair, disrupt RNA splicing integrity and cause converging neurodegenerative phenotypes in human and mouse. While both proteins are required for spicing integrity and neuronal survival, surprisingly, mutations preventing PRP17 isomerization catalyzed by PPIL1 are tolerated, thus revealing a predominant non-enzymatic function of a spliceosomal PPIase.
RESULTS
Identification of biallelic mutations in PPIL1 in PCHM families
From our cohort of 7,288 patients with recessive congenital neurological disorders, we identified rare damaging homozygous missense variants in PPIL1 among 8 index cases from 5 families (Figures 1A–1C). All patients showed both features of PCH as well as congenital microcephaly (−3SD to −6SD HC); the latter phenotype progressed postnatally for all. Patients did not show features of known syndromic PCH subtypes (Akizu et al., 2013; Namavar et al., 2011) (Table 1 and Table S1). All subjects were enrolled with IRB-approved protocols at referral institutions and provided signed consent. Through Genematcher (Sobreira et al., 2015), we identified 4 additional families with 8 affected patients in which PPIL1 mutations were independently identified as the likely cause (Table S1 and S2). As all individuals exhibited PCH with microcephaly, we defined this presentation as a unique clinical entity, which we termed PCHM (PCH+Microcephaly). Further common phenotypes included hypotonia, difficulty swallowing, failure to control the airway, seizures, and delayed motor and language development. Brain MRI showed cortical changes in most affected (Figure 1B and Table S1), notably simplified gyri pattern, which was rarely reported for other subtypes of PCH.
Figure 1. Biallelic mutations in PPIL1 lead to neurodegenerative pontocerebellar hypoplasia with microcephaly (PCHM) in human.
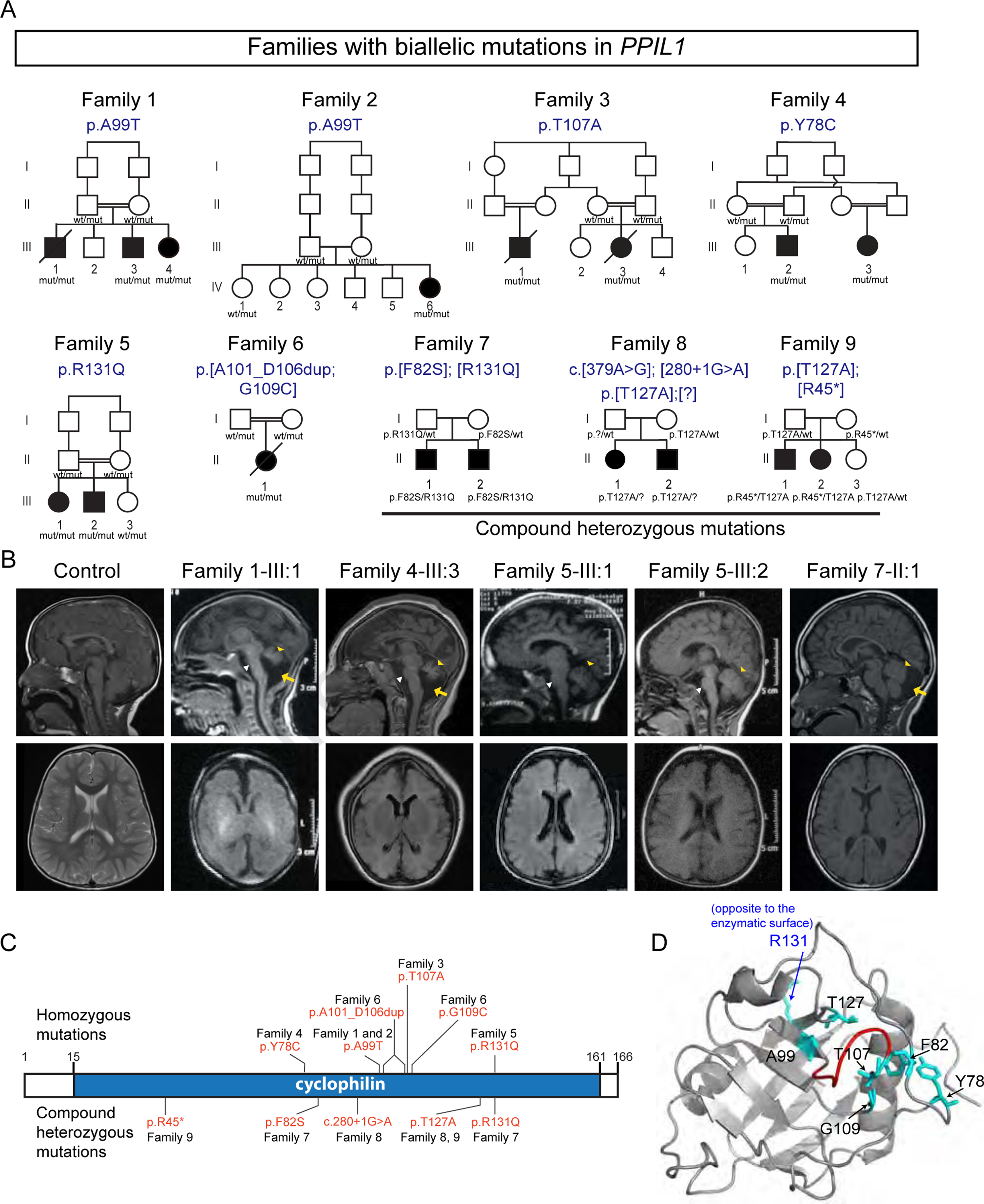
(A) The families with predicted effects of PPIL1 variants listed above pedigree. All variants are homozygous in affected individuals, except Family 7–9, which are compound heterozygous. All pathogenic variants segregated as a recessive trait. Filled symbols: affected; p.[?]: splice donor site mutation, c.280+1G>A; square: male; circle: female; double bar: consanguinity; diagonal line: deceased. wt, reference allele; mut, patient variant allele.
(B) Sagittal (top) and axial (bottom) T1-weighted brain MRIs show reduced cerebellar volume (yellow arrowhead), atrophic pons (white arrowhead) and posterior fossa fluid accumulation (yellow arrows) indicative of cerebellar atrophy in affected individuals. Simplified gyri pattern is most apparent in the affected from Family 1 and 4.
(C) Identified PPIL1 mutations. Above: homozygous variants. Below: compound heterozygous mutations.
(D) En face view of enzymatic surface with labeled variant residues in NMR-resolved PPIL1 structure (PDB: 2K7N). All except R131 (blue) localized to the enzymatic surface. Red: duplicated region in Family 6 (A101-D106).
See also Figure S1.
Table 1.
Clinical information of selected patients
| Patient | Family 1–III:1 | Family 3–III:1 | Family 4–III:3 | Family 5–III:1 | Family 6–II:1 | Family 7–II:1 | Family 8–II:1 | Family 9–II:1 | Family 10–V:3 |
|---|---|---|---|---|---|---|---|---|---|
| Mutation gRNA (hg19) | chr6:g.368237 95C>T | chr6:g.368237 71T>C | chr6:g.368244 09T >C | chr6:g.368236 98C>T | chr6:g.[3682377 2 36823789dup; 36823765C>A] | chr6:g.[36824 397A>G];[368 23698C>T] | chr6:g.[36823 711T>C];[368 24361C>T] | chr6:g.[36823 711T>C];[368 39572G>A] | PRP17 hg19:chr6:110 550122T>G |
| Mutation cDNA PPIL1 NM 016059.1, PRP17 NM_015891.2 | c.295G>A | c.319A>G | c.233A>G | c.392G>A | c.[301_318dup; 325G>T] | c.[245T>C]; [392G>A] | c.[379A>G]; [280+1G>A] | c.[379A>G];[1 33C>T] | PRP17 c.1505T>G |
| Mutation Protein PPIL1 NP 057143.1, PRP17 NP_056975.1 | p.Ala99Thr | p.Thr107Ala | p.Tyr78Cys | p.Arg131Gln | p.[Ala101_Asp10 6dup;Gly109Cys ] | p.[Phe82Ser; Arg131Gln] | p.[Thr127Ala];[ ?] | p.[Thr127Ala];[Arg45*] | PRP17 p.Phe502Cys |
| Gender | Male | Male | Female | Female | Female | Male | Female | Male | Female |
| Ethnic origin | Egyptian | Pakistani | Mexican | Egyptian | Pakistani | Chinese | European-American | European-American | Egyptian |
| Parental consanguinity Head | + | + | + | + | − | − | − | − | + |
| circumference (HC) at birth | 28cm (−6SD) | 30cm (−4SD) | 28cm (−4.5SD) | 29cm (−4SD) | 30cm (−4SD) | 30cm (−2.5SD) | 29cm (−4SD) | 32cm (−2SD) | 30cm (−3SD) |
| HC at last examination | 36cm (−8SD) at 9 mos | 39cm (−5SD) at 1 y | 36cm (−8SD) at 9 mos | 37cm (−6SD) at 9 mos | 39cm (−5SD) at 1 y | 45cm (−4SD) at 4 y | 43cm (−8SD) at age 10 y | 45.5cm (−6SD) at 13 y | 42 cm (−5SD) at 4y |
| Pontocerebellar hypoplasia | + | + | + | + | + | + | + | + | + |
| Simplified cortical gyral patterning | + | + | + | + | + | − | − | − | |
| Agenesis of corpus callosum | + | + | Partial | + | Partial | Partial | Partial | ||
| Cerebellar hypoplasia | + | + | + | + | + | + | + | + | + |
| Brainstem hypoplasia | + | + | + | + | + | + | + | + | + |
| Hydrocephalus | − | − | − | − | + | − | − | − | |
| White matter abnormalities | − | − | Delayed myelination | − | − | − | Delayed myelination | − | |
| Intellectual Disability | Severe | Severe | Severe | Severe | Severe | Severe | Severe | Severe | Severe |
| Seizure Onset | Birth | - | Infancy | Infancy | - | Infancy | Infancy | Infancy | Infancy |
| Seizure Type | Focal | - | Focal | Generalized | - | Myoclonic | Infantile spasms | Infantile spasms | Myoclonic / GTC |
| Seizure Frequency | Intractable | - | Infrequent | Infrequent | - | Monthly | Intractable | Daily, Intractable | Intractable |
| Gross motor | Absent | Absent | Absent | Delayed | Absent | Delayed | Absent | Absent | Delayed |
| Fine motor | Absent | Absent | Absent | Absent | Absent | Delayed | Absent | Absent | Absent |
| Language | Absent | Absent | Absent | Absent | Absent | Delayed | Absent | Absent | Absent |
| Social | Absent | Absent | Absent | Absent | Absent | Delayed | Absent | Absent | Absent |
| Hypertonia | Mild | - | - | Mild | - | Mild | Mild | Mild | |
| Hypotonia | - | Severe | Severe | Mild | Severe | - | Mild | Mild | - |
| Deep tendon reflexes | Brisk | Brisk | Brisk | Brisk | Brisk | - | Brisk | Brisk | Brisk |
| Spastic tetraplegia | + | + | + | + | ++ | − | + | + | + |
| Other | Died at 8mos; Inguinal hernia | Died at 2 mos; Persistent thrombocytop aenia | Dystonia; Chronic neutropenia | Died at 2 mos | Dystonia; Chronic neutropenia | Dystonia | Chronic anemia and thrombocytope nia |
The mutations in each family segregated with the phenotype according to recessive inheritance. Families 1–5 showed homozygous missense variants, and Family 6 showed two separate homozygous variants predicting a 6 aa duplication and a p.G109C substitution. Family 7–9 showed compound heterozygous variants: Families 5 and 7 and Family 8 and 9 shared an identical variant. All substituted residues were highly conserved (Figure S1A), predicted ‘damaging’ by MutationTaster (Schwarz et al., 2014), and clustered within PPIL1’s cyclophilin PPIase domain, suggesting deleterious functions (Figure 1C).
Patient mutations affect PPIL1 function
PPIL1 joins the MSC together with its interacting protein SKIP, two of the NineTeen complex-related proteins, during B complex formation, and remains until splicing is complete (Rajiv and Davis, 2018; Wang et al., 2010). We mapped human mutations onto the ordered globular PPIL1 structure (Xu et al., 2006), and found all the affected residues except p.R131 were located on the enzymatic face (Figure 1D). To test the impacts of patient variants, we expressed FLAG-tagged mutant protein in HEK293T cells and found most variants led to unstable proteins (Figures S1B and S1C). Specifically, p.Y78C, p.A99T, p.[A101_D106dup;G109C], p.F82S and to a lesser degree p.T127A were barely detectable. Likewise, reduced endogenous p.A99T PPIL1 protein was also observed in patient fibroblasts, which showed a slightly higher RNA possibly due to compensatory upregulation (Figure S1D–S1F).
Two variants, including p.T107A and p.R131Q, did not show altered protein levels in HEK293T cells. However, we found that both purified mutant proteins showed reduced thermal stability and increased aggregation propensity (Figures S1G–S1I). PPIL1 associates with SKIP prior to its incorporation into the spliceosome (Wang et al., 2010; Xu et al., 2006). The SKIP binding interface is located on the non-enzyme face (Wang et al., 2010; Xu et al., 2006), and previous studies suggested that PPIL1 p.R131 was involved in the binding to SKIP (Wang et al., 2010; Xu et al., 2006). As expected, we found that purified p.R131Q PPIL1 failed to associate with SKIP in both surface plasmon resonance and immunoprecipitation assays (Figures S1J and S1K). PPIL1 p.T107A, although localized to the enzymatic face, showed reduced SKIP interaction (Figure S1J). Thus, all assessed PPIL1 patient mutations either affect protein stability or interaction with SKIP.
Defective brain development and neuron-specific apoptosis in knockin mice
To reveal functions of PPIL1 in brain development, we studied expression during development. RNA in situ hybridization showed ubiquitous Ppil1 expression in the developing cortex (Figures 2A and 2B). Due to a lack of specific PPIL1 antibodies, we generated a CRISPR knockin mouse introducing an N-terminal HA epitope in Ppil1, which confirmed ubiquitous protein expression (Figures 2C, 2D, and S2A–S2D). We next generated a Ppil1 frameshift mouse line (fs, c.302delC, p.N102Tfs*13), but found no viable homozygous embryos any time after embryonic day (E) 12.5 (0 in 41 embryos, p < 0.00001, Binomial test), while Ppil1fs/+ pups were indistinguishable from wild-type (WT) littermates. However, at E9.5 we recovered several partially resorbed embryos, all of which were genotyped as Ppil1fs/fs (13 mutants in 46 embryos, with the expected 25%, Figures 2E and S2E). We conclude that Ppil1 is essential for embryogenesis.
Figure 2. Patient PPIL1 mutation knockin mice exhibit PCHM-like phenotype.
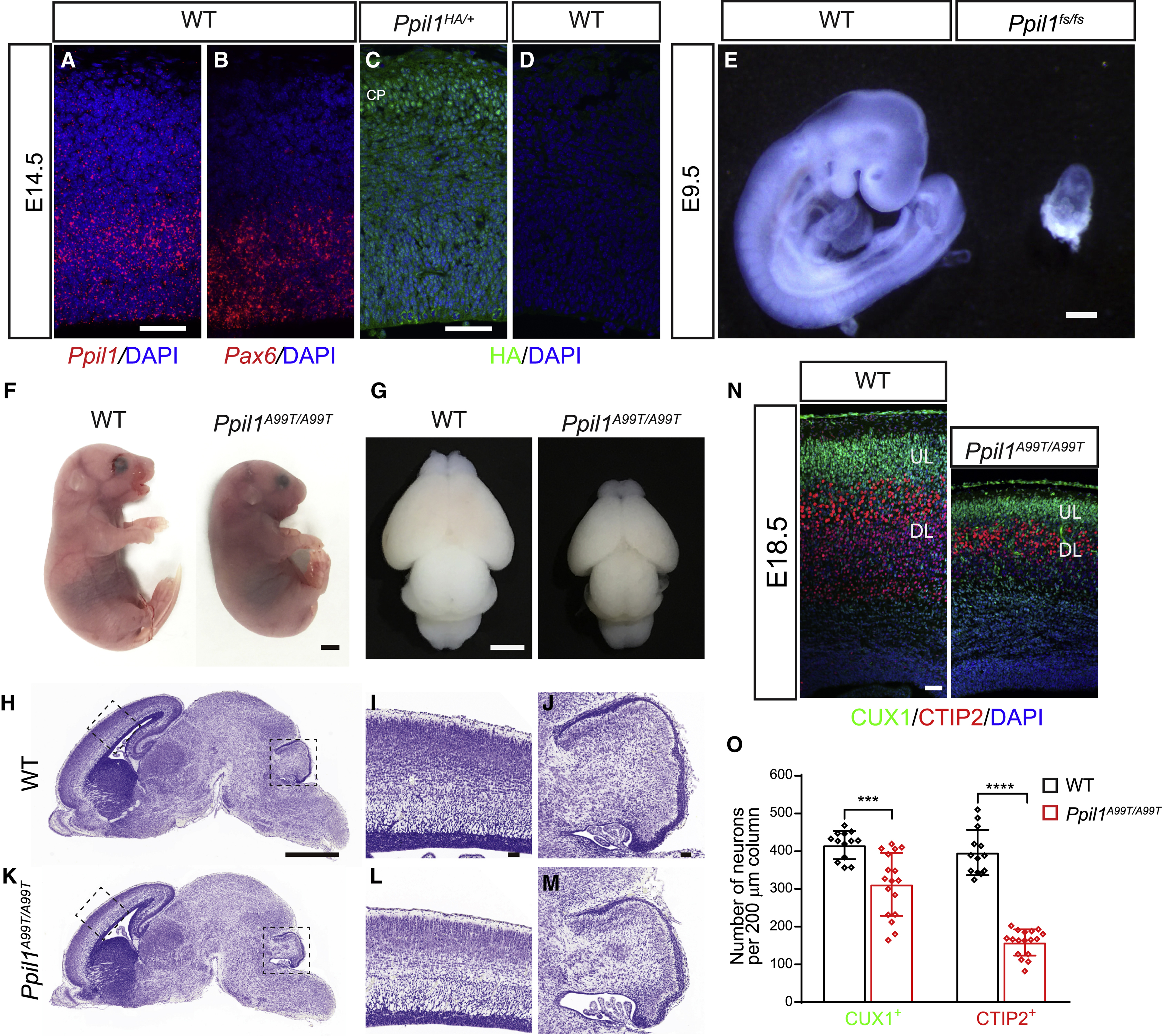
(A and B) Fluorescent in situ hybridization (FISH) on coronal sections of E14.5 brain cortex hybridized with Ppil1 (A) and Pax6 (B) probes using RNAscope. Scale bar: 50 μm.
(C and D) Coronal sections of E14.5 embryos from Ppil1HA/+ (C) and WT (D) embryos immunostained with an anti-HA antibody showing ubiquitous expression of PPIL1. CP: cortical plate. Bar: 50 μm.
(E) Ppil1 fs/fs mouse embryos showed reabsorption at E9.5. Bar: 2 mm.
(F and G) Homozygous patient variant p.A99T knockin mouse with microcephaly at E18.5.
(H-M) Nissl stained sagittal sections of E18.5 Ppil1A99T/A99T brains, magnified for dashed regions in the cerebral cortex and cerebellum.
(N) E18.5 Ppil1A99T/A99T cortex (coronal) shows reduced thickness but with intact lamination based upon immunostaining against CUX1 (upper layer neurons) and CTIP2 (lower layer neurons).
(O) Reduced density of cortical CUX1+ and CTIP2+ neurons in E18.5 Ppil1A99T/A99T cortex. n = 4 mice/genotype. Mean ± s.d.; p = 0.0003 CUX1+ cells; p < 0.0001 CTIP2+ cells; two-tailed unpaired t-test.
Scale bar: 1 mm in H and K; 50 μm in I, J, and L–M.
See also Figure S2.
We next generated a patient p.A99T knockin (KI) mouse line, chosen because it was the first allele we identified. Ppil1A99T/A99T mice were born at the expected Mendelian ratios (22 Ppil1A99T/A99T, 42 Ppil1A99T/+, and 23 WT), but died within 24h. Newborns showed smaller head size, severely reduced cerebral and cerebellar size, reduced cortical surface area and thickness (Figures 2F–2M atching human PCH While the cortex showed normal lamination, neuronal numbers were severely reduced, with CUX1+ upper layer neurons decreased by ~25% and CTIP2+ deep layer neurons by ~60% (Figures 2N and 2O). Like patient fibroblast, PPIL1 protein was also severely reduced by ~80% in Ppil1A99T/A99T bryo lysates (Figures S2F and S2G). Additionally, compound heterozygous Ppil1A99T/fs mutant embryos showed much more severe phenotypes across the body at E18.5 (Figure S2H), suggesting that PPIL1 p.A99T is a hypomorphic mutation.
Based on the severe reduction in cortical thickness and neuronal numbers, we hypothesized that this could be caused by cell death during embryonic development. We examined apoptosis by assessing cleaved caspase 3 (CC3) and p53 expression in the embryonic brains. Indeed, the apoptosis showed a striking accumulation in TUJ1+ neurons within deep layers of the lateral cortical margin in the mutant brains starting at E12.5 and dramatic at E14.5 (Figures 3A–3F), with some apoptotic cells are GAD65/ interneurons (Figures S2I and S2J). The apoptosis in the rest of the cortex appeared at E14.5 and turned to be massive at E16.5 (Figures 3A–3N). Apoptotic cells predominantly localized in the cortical plate and did not overlap with SOX2+ neural stem cells (NSCs) and TBR2+ intermediate neural progenitors (INPs) (Figures 3G–3N and S2K–S2M). ~70% of p53 positive cells were positive for CTIP2 and ~30% for CUX1, consistent with a more severe reduction of deep layer neurons. CC3 upregulation was also observed in the cerebellum and pons at E18.5 (Figure S2N and S2O). Consistent with the predominant impact of the brain in the patients and KI mutant mice, no significant upregulation of apoptosis was observed in other major organs (Figures S2P and S2Q).
Figure 3. Ppil1 knockin mice show increased neuron-specific apoptosis and depletion of neural progenitor cells.
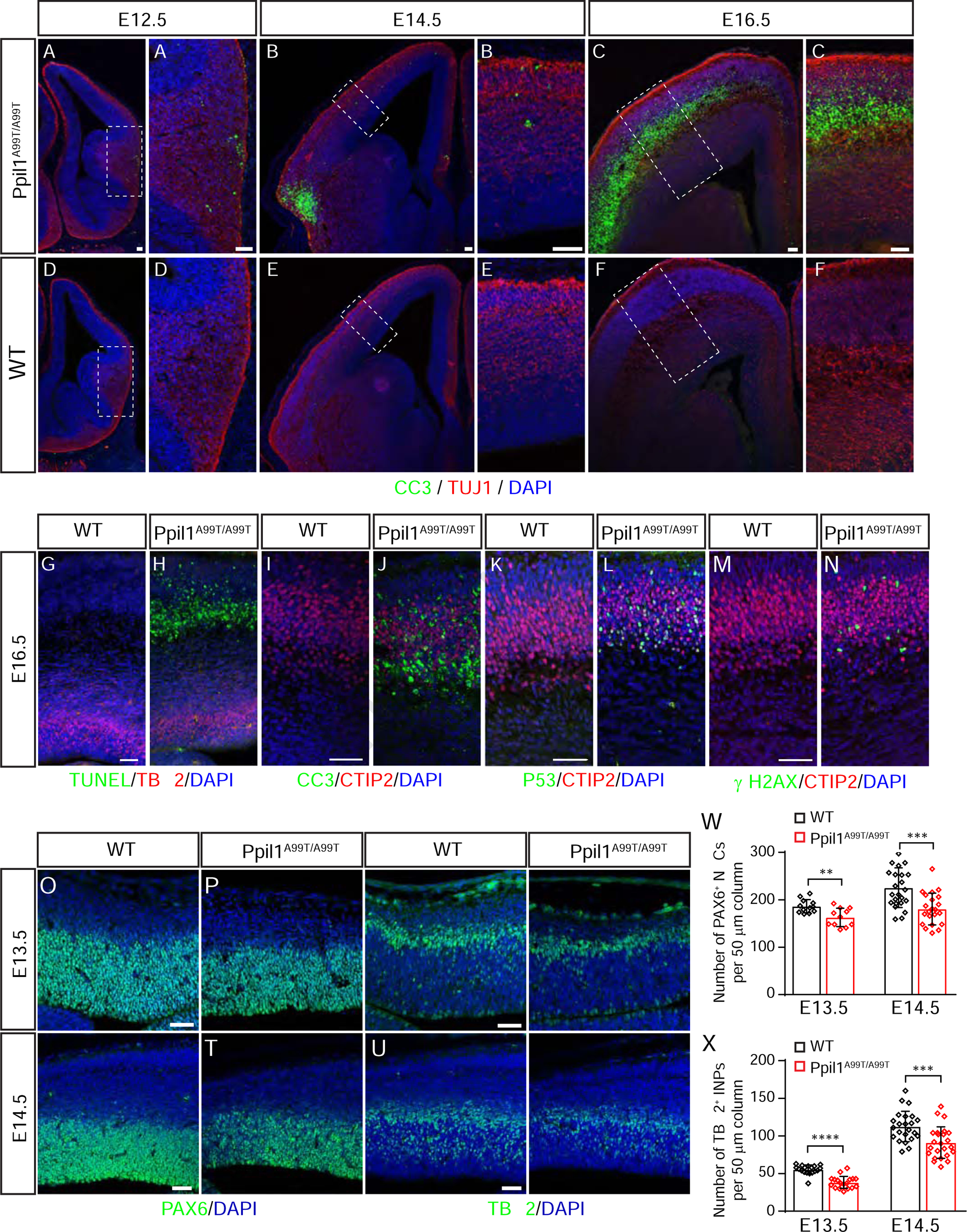
(A-F) Embryonic Ppil1A99T/A99T brains (coronal) shows increased cleaved Caspase 3 (CC3, green).
(G-N) Coronal sections of E16.5 brain cortex from WT and Ppil1A99T/A99T embryos stained for TUNEL and TBR2 (G and H), CC3 and CTIP2 (I and J), p53 and CTIP2 (K and L), γ-H2AX and CTIP2 (M and N).
(O-V) Embryonic Ppil1A99T/A99T cortex (coronal) shows reduced PAX6 (neural stem cells, NSC) and TBR2 (intermediate neural progenitor, INP) positive cells.
(W and X) Reduced density of cortical PAX6+ neural stem cells and TBR2+ intermediate progenitor cells in E13.5 and E14.5 cortex. Mean ± s.d.; p = 0.0029 E13.5 PAX6+, p = 0.0002 E14.5 PAX6+; p < 0.0001 E13.5 TBR2+; p = 0.0008 E14.5 TBR2+; two-tailed unpaired t-test.
Scale bar: 50 μm.
See also Figures S2 and S3.
Recent studies proposed MSC defects result in genome instability, due partially to accumulated R-loops, transient RNA:DNA hybrid structures that displace the non-templated strand and generate susceptibility to DNA damage (Jangi et al., 2017; Paulsen et al., 2009), evidenced by γ-H2AX and p53 accumulation (Denis et al., 2005; Sorrells et al., 2018). Like p53, we indeed observed a dramatic upregulation of γ-H2AX in the mutant brains (Figures 3M, 3N, and S2L), suggesting similar mechanisms.
In contrast to other genetic models of microcephaly (Gruber et al., 2011; Insolera et al., 2014; Silver et al., 2010), no premature neurogenesis was observed at E12.5 (Figures S3A and S3B), and apoptosis initiation through p53 was predominantly localized to postmitotic neurons but not neural progenitors; consequently, the numbers of both neural stem cells (NSCs) and intermediate progenitors (INPs) only showed a slight reduction (Figures 3O–3X). The cell cycle of the mutant progenitors seems not to undergo dramatic alterations, as we only observed a slightly increased percent of INPs at the G2/M phase (Figures S3C–S3F). Together, our results suggest genotoxic stress, neuronal apoptosis, and perturbations of progenitors lead to brain volume loss in Ppil1 KI mutants.
PPIL1 is required for alternative splicing integrity
Despite its discovery in the spliceosome many years ago (Rappsilber et al., 2002), the function of PPIL1 in RNA splicing remains mostly unknown. Since Ppil1fs/fs embryos survived until E9.5, we suspected that some cells might tolerate a complete loss of PPIL1. Thus, we generated PPIL1 knockout (KO) human HAP1 cells (Figure S4A), which were viable and subjected to RNA-seq. Five different forms of alternative splicing (AS), including skipped exons (SE), mutually exclusive exons (MXE), alternative 5’ and 3’ splice sites selection (A5SS and A3SS), and retained introns (RI), were evaluated using rMATS software (Shen et al., 2014). We benchmarked rMATS by comparing significant differential splicing events (SDSE) were identified from 231,850 total AS vents (Table S3). In contrast, 8,602 (i.e. 3.4%) SDSEs were identified comparing 3 KO vs.3 controls, with the number of SDSEs scaling with number replicates in each group (Figures S4B and S4C). We repeated the computational analysis using LeafCutter (Li et al., 2018), which identified 951 SDSEs between 3 KO vs. 3 controls, compared with 8 and 0 SDSEs for 3 controls vs. 3 controls vs. 3 KO vs. 3 KO (Table S3), respectively. All these revealed a dramatic disruption of global AS integrity upon loss of PPIL1.
We also confirmed altered AS by assessing ‘sashimi-plot pile-ups’ of RNAseq and performing RT-PCR validation on selected RI and SE events (Figures S4D–S4H). Loss of PPIL1 predominantly impacted the splicing of short and high GC-content introns, without significant bias towards splice site strength (Figures S4I–S4M). The most severe changes were present in introns ≤ 75 bp in length and with > 70% GC content (Figures S4N and S4O), where 12.6% of such introns were retained at higher levels in KO cells, compared with only 0.66% higher in controls.
Finally, we compared the list of SDSE genes in KO with OMIM disease categories based upon organ system involved and found overrepresentation for neurodevelopmental disease (p = 3.45 X 10−20, Bonferroni corrected Chi-squared test, Figure S4P), but not for cancer, cardiac or immune disease (p > 0.05). We also found that SDSE genes were significantly enriched in genes known to undergo AS in the brain (p = 1.12 X 10−287, Figure S3L). All these findings are consistent with its predominant impact on the brain.
Disrupted alternative splicing integrity in Ppil1A99T/A99T brains
To test the impact of its loss on AS integrity in the brain tissue, we performed RNAseq on 3 WT and 3 Ppil1A99T/A99T KI E14.5 brains before the accumulation of apoptotic cells. Using rMATS, we detected 3,797 SDSE among 236,870 total AS events (i.e. 1.6%, Figures 4A and S5A–S5C). Splicing alterations were also confirmed with Leafcutter, which identified 115 SDSE of 16,528 total AS events (i.e. 0.7%). A randomly selected group of significant RI and SE events were verified in semi-quantitative RT-PCR (Figures S5D and S5E). Two ‘minigene’ constructs transfected into MEFs for the SDSE introns of Atg4d and Evi5l confirmed that the splicing defects were not secondary to non-specific effects (Figures 4B and S5F). Similar to HAP1 cells, KI brains showed significant RI events for short and high GC-content introns (Figures 4C and 4D). However, unlike HAP1 KO cells, we also observed a preference for weak 5’ and 3’ splicing sites among significant events (Figures 4E–4H and S5G), which may reflect competition for reduced PPIL1 protein between strong and weak splicing sites. Profiling of misspliced genes revealed protein translation, RNA processing, and DNA damage response as the most significantly disturbed modules (Figures 4I, S5H, and S5I), whose disruptions are major causes of cerebellar ataxia (Synofzik et al., 2019). Moreover, genes involved in axon development and cell cycle were also significantly affected (Figure 4I), reinforcing the phenotypes observed in KI brains.
Figure 4. Global splicing integrity defects in Ppil1A99T/A99T developing brain.
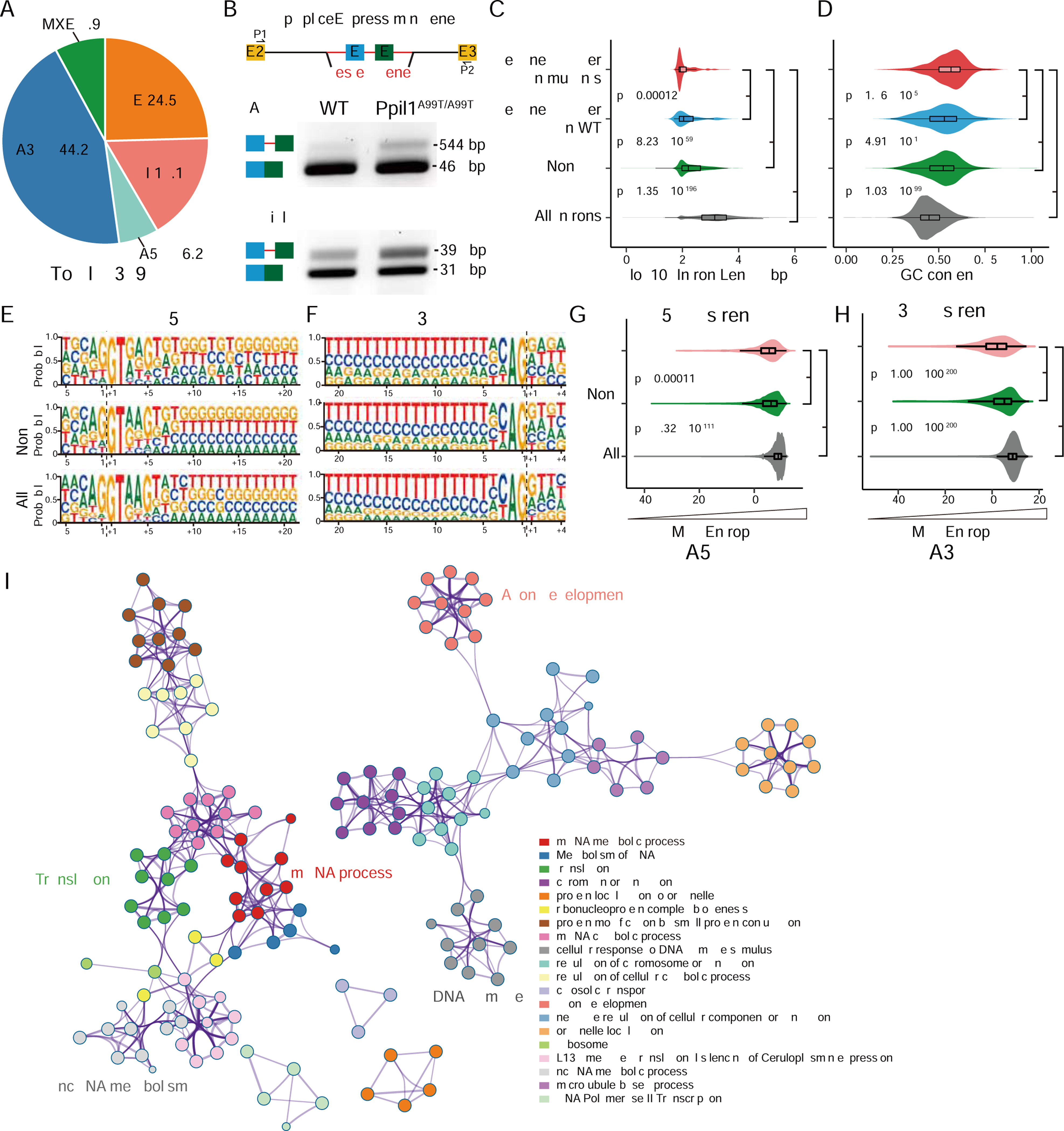
(A) Impact of p.A99T mutation on five major types of AS events detected with rMATS in E14.5 brain hemispheres (3 KI vs. 3WT). A3SS was most impacted, followed by SE, RI, MXE, and A5SS.
(B) Minigene splicing reporter assays in transfected Ppil1A99T/A99T mouse embryonic fibroblasts show higher intron retention levels in mutant cells for both Atg4d and Evi5l. (C and D) Distribution of differential splicing identified by rMATS in KI or control, based upon intron length and GC content. Introns with short length or high GC content show significantly retained higher in Ppil1 KI brains. All introns represent all identified introns from mouse reference genome. p-value: Wilcox test.
(E-H) Splice-site strength analysis of 5’SS and 3’SS in all introns (All, gray), non-significant A5SS or A3SS events (Non-sig, green), and significant A5SS or A3SS events (Sig, red) identified by rMATS. The 5’SS and 3’SS strength show lower maximum entropy for choice points that were significantly different in KI compared with control. p-value: Wilcox test.
(I) Metascape visualization of enriched networks and pathways among all misspliced genes in E14.5 Ppil1A99T/A99T brains (n = 2134 misspliced genes), showing several key modules represented including “mRNA metabolic process” among others.
See also Figures S4 and S5 and Table S3.
PPIL1 catalyzes the isomerization of PRP17 Gly94-Pro95 95 in vitro
Recent cryo-EM structures allowed for detailed analysis of proline isomerases within the MSC (Bertram et al., 2017; Fica et al., 2019; Haselbach et al., 2018; Yan et al., 2015a; Zhan et al., 2018; Zhang et al., 2017; Zhang et al., 2018; Zhang et al., 2019), six of which were evident in one or more MSC complexes (Table S4). However, only for PPIL1 was it possible to identify a Pro from an adjacent protein within the enzymatic pocket, where we identified Gly94-Pro95 of PRP17 within the Bact, C, C*, P, and ILS complexes (Fica et al., 2019; Zhan et al., 2018; Zhang et al., 2017; Zhang et al., 2018; Zhang et al., 2019), conserved from S. pombe (Yan et al., 2015a) to human (Figures 5A, 5B, and S6A–S6E). This finding suggests that PRP17 may be a substrate of PPIL1 in the MSC.
Figure 5. PPIL1 P95 is positioned in the enzymatic pocket of PRP17 in the activated spliceosome.
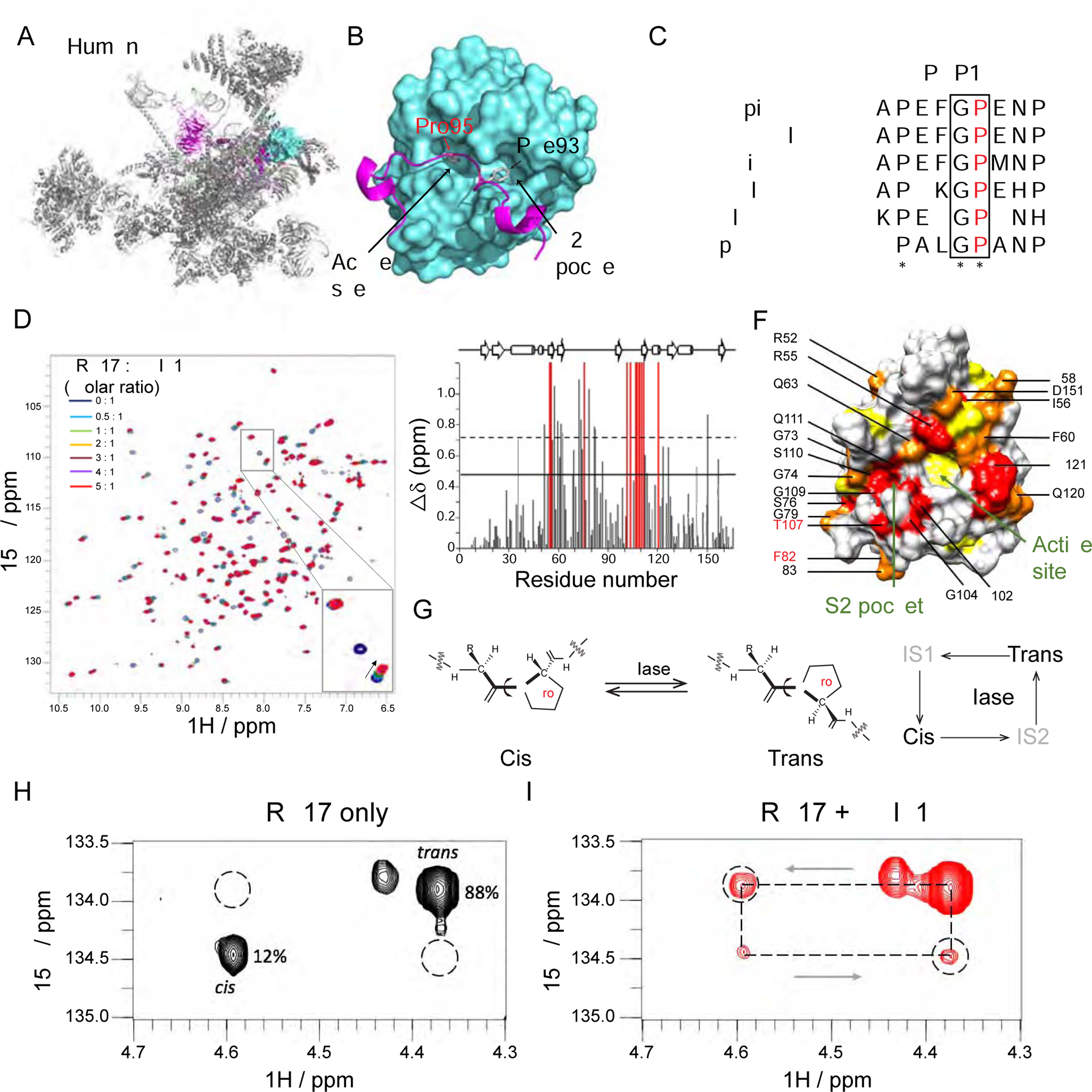
(A and B) Cryo-EM structure of human spliceosome C* complex (PDB: 5XJC) shows an N-terminal loop of PRP17 (cartoon in purple) bound to PPIL1 (teal) enzymatic surface with Pro95 buried inside the S1 enzymatic pocket.
(C) Protein sequence alignment of PRP17 from 6 species shows an evolutionarily conserved Gly-Pro (G-P) motif in PRP17. * stands for identical residues, : indicates similar residues.
(D) Overlaid 1H-15N HSQC spectra of PPIL1 with PRP17 peptide titrations (0–5 molar equivalents, aa 89–101: FAPEFG[P]ENPFRT). Specific resonance shifts or broadening beyond detection indicates specific binding of PRP17 to PPIL1. Inset: Examples of PPIL1 resonance changes that shift (arrowhead), broaden beyond detection (open arrowhead) or were unchanged (arrow) as a result of PRP17 titration.
(E) Average chemical shift perturbations of PPIL1 residues upon titration with PRP17. Gray: shifted resonances. Red: broadened beyond detection resonances. Solid or dashed lines: shifts >1 or >2 SD above mean, respectively.
(F) Space-filling model of PPIL1 (PDB: 2X7K) showed significantly perturbed residues upon PRP17 peptide binding. Red: residues broadened beyond detection, Orange: residues >2 SD, Yellow: residues >1 SD above mean. Residues affected in patients are labeled in red.
(G) Schematic of cis-trans Xaa-Pro peptide bond isomerization catalyzed by PPIase. IS: Intermediate State.
(H and I) 2D 1H, 15N-H(Cα)N ZZ exchange spectra of PRP1 peptide in the absence (H) or presence (I) of sub-stoichiometric concentrations (1% molar ratio) of PPIL1, with appearance of ‘exchange signals’ (dashed circles), i.e. significant interconversion between cis-trans states.
See also Figures S5 and S6, Table S4, and Movie S1.
Gly94-Pro95 in PRP17 occurs within an intrinsically unstructured region, between two alpha-helical domains, conserved from S. pombe to human (Figure 5C). There is an adjacent residue (human Phe93 or S. pombe Leu67) within the S2 pocket, which likely determines substrate specificity (Davis et al., 2010; Teigelkamp et al., 1998). To test their interaction, we performed heteronuclear single-quantum coherence (HSQC) spectral analysis with 15N-labeled PPIL1, and a 13-mer PRP17 peptide (aa 89–101) containing Pro95. We confirmed that PRP17 peptide interacts with PPIL1 along its enzymatic surface (Figures 5D–5F). Using isothermal titration calorimetry assay, we defined a dissociation constant (KD) of 111.9 ± 4.0 μM (Figure S6F). To investigate whether PPIL1 catalyzes PRP17 isomerization, we used a PRP17 peptide (aa 89–101) with 15N, 13C double-labeled P95, and demonstrated the Gly94-Pro95 bond was present in both cis and trans conformations (Figures S6G and S6H). Addition of catalytic concentrations of PPIL1 accelerated the rate of proline isomerization in PRP17 peptide, evidenced by the appearance of exchange peaks in 1H 15N H(Cα)N ZZ exchange spectra (Figures 5G–5I). This was also confirmed using 2D 1H-15N ZZ exchange spectra of a uniformly 15N labeled PRP17 peptide (aa 84–101) with catalytic concentrations of PPIL1 WT (Figure S6I). We conclude that PPIL1 is capable of catalyzing PRP17 isomerization in vitro.
PRP17 loss associates with pontocerebellar hypoplasia
Only a few of the >100 MSC components are associated with human disease (Lines et al., 2012; Pellagatti and Boultwood, 2017; Ruzickova and Stanek, 2017; Xu et al., 2017), so we considered genes encoding PPIL1-associated proteins as candidates for PCHM. In addition to PRP17, we found SKIP and RBM22 bound to PPIL1 (Figures S6A–S6C and Movie S1). We thus searched our unsolved pediatric brain disease cases and identified a multiplex consanguineous family with PCHM (Family 10), also with chronic anemia and thrombocytopenia, and with a homozygous PRP17 variant, predicting a damaging p.F502C protein change (Figures 6A, 6B, and Table S1 and S2). No further families were identified in Genematcher.
Figure 6. PPIL1 and PRP17 control neuronal survival independent of catalysis.
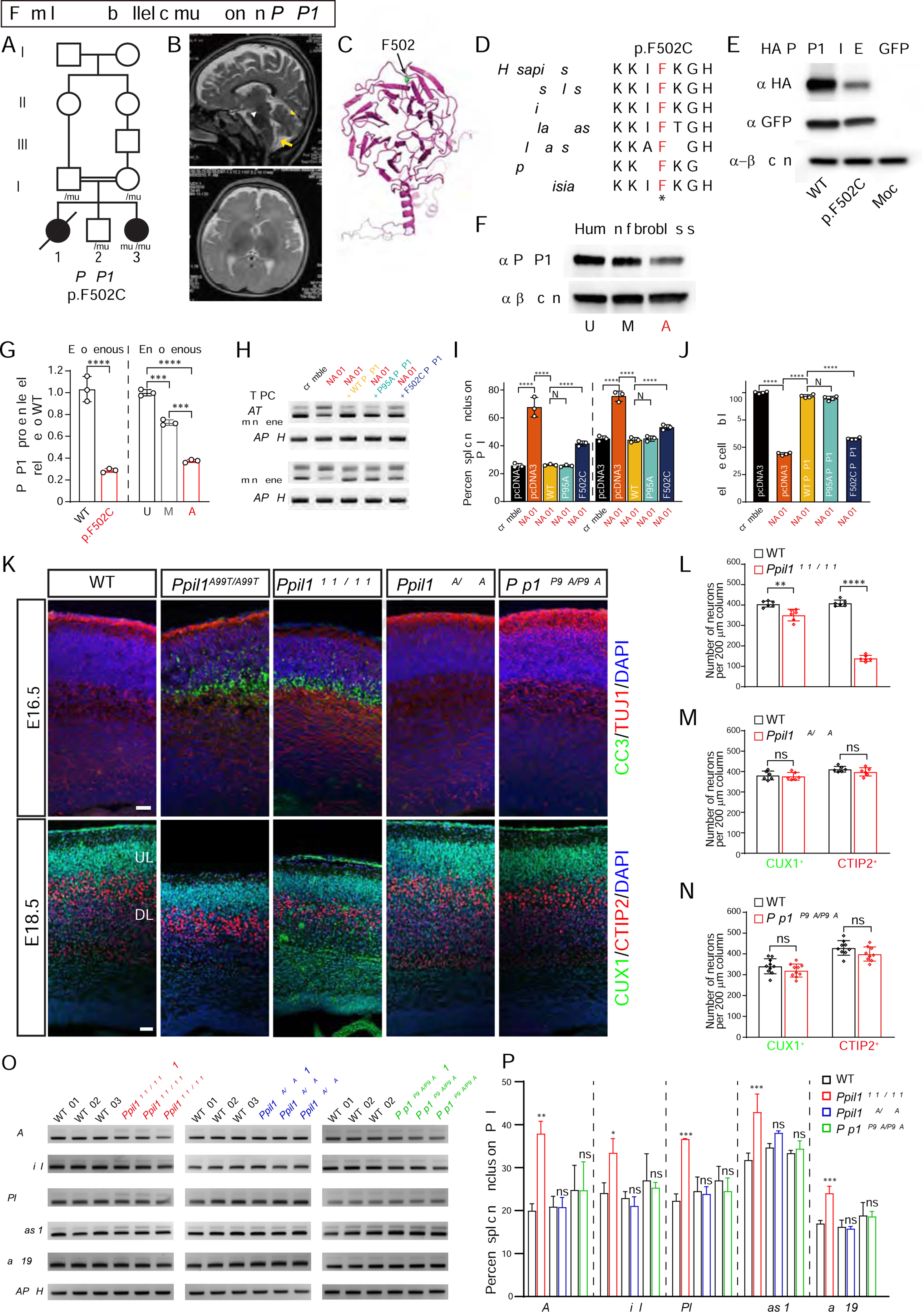
(A) Pedigrees of PCHM Family 10 with homozygous PRP17 p.F502C variant segregating as a recessive trait. Filled symbols: affected; square: male; circle: female; double bar: consanguinity; diagonal line: deceased.
(B) T2-weighted brain MRI shows reduced cerebellar volume (yellow arrowhead), atrophic pons (white arrowhead) and posterior fossa fluid accumulation (yellow arrows) indicative of cerebellar atrophy in the living affected.
(C) The structure of PRP17 resolved from the cryo-EM structure of spliceosomal C complex (PDB: 5XJC) showing mutated residue F502 within the C-terminal WD40 domain.
(D) Protein sequence alignment of PRP17 showing mutated F502 residue highly conserved across eukaryotes. *: identical.
(E) Western blot of overexpressed HA-tagged PRP17 shows that the p.F502C substitution destabilized the protein.
(F) Western blot of endogenous PRP17 from dermal fibroblasts demonstrating reduced protein levels from affected (A) compared with mother (M) and unaffected control (U).
(G) Quantification of exogenous and endogenous PRP17 protein in transfected HEK293T cells and human dermal fibroblasts, respectively. n = 3.
(H) RT-PCR based minigene splicing assay following PRP17 repression in HEK293T cells, showing full rescue by WT or p.P95A PRP17 but only partial rescue by p.F502C PRP17.
(I) Quantification of percent splicing inclusion (PSI) for minigene splicing reporters. PSI was calculated as percent of inclusion form transcripts among all transcripts (inclusion and exclusion forms). n = 3.
(J) Reduced cell viability following PRP17 repression was fully rescued by WT or p.P95A PRP17 but only partially by p.F502C PRP17. n = 4.
(K) Coronal sections of E16.5 (top) and E18.5 (bottom) mouse brains with upregulated cleaved caspase 3 (CC3) and reduced cortical thickness in Ppil1A99T/A99T and Ppil1R131Q/R131Q, but not in Ppil1R55A/R55A or Prp17P95A/P95A. CUX1 and CTIP2 label upper and deep layer cortical neurons, respectively. Scale bar: 50 μm.
(L–N) Quantification of cortical CUX1+ and CTIP2+ neurons in E18.5 cortex of Ppil1R131Q/R131Q (L), Ppil1R55A/R55A (M), Prp17P95A/P95A (N), and littermate controls. n = 3 mice/genotype.
(O) Semi-quantitative RT-PCR analysis of significant RI events in Ppil1A99T/A99T among E14.5 brains of Ppil1R131Q/R131Q (red), Ppil1R55A/R55A (blue), Prp17P95A/P95A (green), and littermate controls. GAPDH as loading control.
(P) Quantification of percent splicing inclusion (PSI) for RI events. n = 3 for each genotype.
Mean ± s.d.; p-value: ns > 0.05; * <0.05; ** <0.005; *** <0.001; **** <0.0001; one-way ANOVA test for all panels.
See also Figure S7.
PRP17 contains a C-terminal WD40 domain, where F502 resides, which is fully evolutionarily conserved (Figures 6C and 6D). In cryo-EM MSC structure, the WD40 assumes a classical ‘7-propeller’ architecture, and associates with U2 snRNA and the U2/branch point sequence (BPS) duplex, stabilizing the catalytic site (Movie S1) (Bertram et al., 2017; Haselbach et al., 2018; Zhan et al., 2018; Zhang et al., 2017). We assessed the impact of p.F502C on protein using HA-tagged cDNA expressed in HEK293T cells and found dramatic protein destabilization, which was also confirmed in patient fibroblasts (Figures 6E–6G and S7A).
More severe than Ppil1fs/fs, Prp17 homozygous frameshift mice were lethal prior to E9.5 (c.277_287del11, E9.5, 0 mutant in 47 embryos, p < 0.00001, Binomial test). We further examined RNA splicing and cell survival in HEK293T cells after CRISPRi induced repression of PRP17 followed by expression of rescue PRP17 cDNAs (Gilbert et al., 2013). Both Atg4d and Evi5l minigene reporters showed higher intron retention levels after PRP17 knockdown, rescued by WT but not p.F502C PRP17 (Figures 6H, 6I, S7B, and S7C). We also observed that cell viability was significantly decreased after PRP17 repression, which was almost fully rescued by WT but only slightly by p.F502C cDNA (Figure 6J). These results suggest that p.F502C impairs PRP17’s function within the MSC in a fashion similar to PPIL1 patient variants.
PPIL1 mediated proline isomerization of PRP17 is not required for function
Given PPIL1 and PRP17 are both essential for embryonic development, and form an active PPIase-substrate pair, we hypothesized that catalyzed isomerization of PRP17 Gly94-Pro95 by PPIL1 is required for function. First, to rule out an effect of PPIL1 outside the spliceosome in the brain, we knocked in PPIL1 p.R131Q in mouse, since our results showed that this substitution selectively prevents its binding to SKIP, which recruits PPIL1 to the spliceosome (Wang et al., 2010). Homozygous Ppil1R131Q/R131Q mice showed perinatal lethality, microcephaly, and evidence of neurodegeneration in a fashion similar to Ppil1A99T/A99T (Figures 6K–6N and S7D–S7F). Selected splicing defects identified in Ppil1A99T/A99T brains were also confirmed in Ppil1R131Q/R131Q brains (Figures 6O and 6P). Although we obsered a slightly reduced PPIL1 protein (~ 30%) in Ppil1R131Q/R131Q embyros (Figures S7D and S7E), this is not sufficient to cause PCHM since heterozygous Ppil1A99T/+ showed even a higher reduction (~ 40%) but did not show any phenotype (Figures S2F and S2G). Together, we conclude that PPIL1 mediates its effect in the brain by subserving its role in the spliceosome.
Second, we generated two additional knockin mouse lines: isomerase-inactive PPIL1 p.Arg55Ala (Davis et al., 2010; Zhang et al., 2013; Zydowsky et al., 1992), and non-isomerizable PRP17 p.Pro95Ala. We reasoned that if isomerization catalysis is crucial, these mutations should phenocopy patient mutations. Surprisingly, we observed no phenotype in either homozygous mutant: both Ppil1R55A/R55A and Prp17P95A/P95A were viable, fertile, and showed no microcephaly, cell death, or defective cortical lamination (Figures 6K–6N). Likewise, splicing defects were not observed in Ppil1R55A/R55A or Prp17P95A/P95A mice (Figures 6O and 6P). To confirm these results in vitro, we performed rescue assays in cultured cells by re-expressing either p.R55A PPIL1 or p.P95A PRP17. We found both mutants rescued proliferation and splicing defects as well as WT (Figures 6H–6J and S7G–S7J). Thus, while PPIL1 is capable of catalyzing PRP17 isomerization, and both are required for RNA splicing and mutated in human brain disease, their enzymatic interaction is not an essential part of the function of either protein. This data suggests the two proteins maintain a scaffolding rather than an enzyme-substrate interaction.
DISCUSSION
Here, we report that biallelic, partial loss-of-function mutations in PPIL1 and PRP17, encoding two core spliceosomal components, disrupt RNA splicing integrity and lead to neurodegenerative pontocerebellar hypoplasia with microcephaly in human and mouse. The reported patients showed severe congenital microcephaly and simplified cortical gyri, which are rarely observed in other reported PCH types. We described this unique form of PCH as a new clinical entity PCHM, wherein spliceosome genes are the major contributors. Thus, our study highlighted the essential role of global splicing integrity in brain development and neurodegeneration, and uncovered a new pathway and mechanism underlying this heterogeneous group of degenerative brain disorders.
In general, clinical severity correlated with degree of impairment of protein stability or function by different patient variants, although T107A only reduced SKIP binding moderately, yet the affected children were severely affected, suggesting a potential unexplored mechanism or possible environmental contributions.
PPIL1 and PRP17 form an active enzyme-substrate pair in the spliceosome, and both are required for RNA splicing and neuronal survival. Surprisingly, our results showed PPIL1-mediated isomerization of PRP17 was not critical for their functions in splicing and neuronal survival, thus revealing a predominant non-enzymatic function of a spliceosome isomerase. Previous in vitro work also suggested that the enzymatic activity of PPIL1 is not required for splicing of selective pre-mRNA substrates (Adams et al., 2015). In line with this, only trans PRP17 G94-P95 was observed in MSC structures reported to date (Figures S7K and S7L). In the spliceosome, PPIL1 interacts with PRP17, RBM22, and SKIP. RBM22 is an RNA interacting protein, grasping the 5’ intron right after the 5’SS region where U6 snRNA hybridizes (Bertram et al., 2017; Haselbach et al., 2018; Zhan et al., 2018; Zhang et al., 2017), whereas the PRP17 WD40 domain binds to the intron branching point/U2 duplex (Movie S1). Both N-terminal SKIP and PRP17 are intrinsically disordered and undergo disorder-order transition upon PPIL1 binding (Wang et al., 2010; Zhang et al., 2017; Zhang et al., 2018). We propose that PPIL1 stabilizes these structures, allowing the MSC to function as a ‘torque wrench’ to bend ‘challenged’ introns to bring the 5’SS and the branching point into proximity. Loss of PPIL1 may reduce the ability to ‘torque’ the more rigid introns, which would explain the primary impacts on short and high GC content introns.
Although mutations in MSC genes had been linked to several types of diseases such as cancer and autosomal-dominant retinitis pigmentosa (Nik and Bowman, 2019; Scotti and Swanson, 2016; Singh and Cooper, 2012), our study is, to our knowledge, the first to connect MSC gene mutations and global splicing integrity to neuronal survival and neurodegeneration.
Why do mutations in these ubiquitously expressed spliceosomal genes lead to brain-specific disease? In our mouse model, apoptosis was limited to postmitotic neurons in homozygous KI mice, strikingly different from other microcephaly models which affect mitosis and survival of neural progenitors or show premature neurogenesis (Gruber et al., 2011; Insolera et al., 2014; Silver et al., 2010). Moreover, defects were observed across the body of compound heterozygous Ppil1A99T/fs mice. These suggest a distinct mechanism in PCHM and also reveals a higher susceptibility of neurons to global splicing perturbation. Alternatively, the developing brain expresses longer genes with more AS (Lipscombe and Lopez Soto, 2019; Raj and Blencowe, 2015; Yeo and Burge, 2004), which might render neurons more susceptible. Patient mutations may affect some neural-specific splicing events critical for neuronal survival (Lin et al., 2020). Postmitotic neurons might accumulate higher levels of misspliced mRNA, impacting protein production or accumulating toxic or unfolded proteins. Loss of PPIL1 predominantly impacted AS of brain-expressed or brain-disease genes, involved in protein translation, DNA repair, and noncoding RNA processing, whose disruptions are the main causes for degeneration in cerebellar ataxia. Moreover, we further found evidence for DNA double-strand breaks with upregulated p53 and CC3 expression, consistent with accumulated transcriptional R-loops (Costantino and Koshland, 2018), although the exact mechanism remains to be explored. All these suggest a potentially shared mechanism of neurodegeneration underlying several genetic forms of PCH.
STAR+METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Joseph G. Gleeson (jogleeson@ucsd.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The raw RNA_seq data related to this manuscript, including human HAP1 cells and mouse E14.5 brain samples, has been deposited on SRA under accession number PRJNA669300 (https://www.ncbi.nlm.nih.gov/sra).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subjects
We studied patients from 10 unrelated families (Figures 1A and 6A). Information about gender, age, and health status are listed in Table S1. All work with patients was approved by the UCSD IRB protocol 140028 or local protocols, and performed according to accepted guidelines. All patients and/or parents/guardians signed a consent form for participation. All mutations were confirmed with Sanger sequencing according to the base change and inheritance within the family.
Animals
All mice used were maintained on a C57BL/6 background and bred under standard group housing laboratory conditions with 12 hours light/dark cycle and free access to food and water. Age and number of mice used for each experiment are detailed in the figure legends. Sex of embryos used was not tested. All work with mice was performed in accordance with UCSD IACUC protocol S15113. To generate mutations in mice, the mixture of gRNA (0.6 μM), Cas9 protein (0.6 μM, NEB, #M0646T), and single-strand DNA oligo (10 ng/μl, only for knockin) was injected to mouse zygotes at UCSD Transgenic Mouse Core, offspring genotyped by PCR Sanger sequencing. Mice with correct genotypes were backcrossed with C57BL/6J WT mice for at least two generations before breeding to generate homozygous mice for analyzing phenotypes.
To produce Ppil1 knock-out mice (1 bp deletion, c.302delC; NM_026845.4), gRNA targeting GTCTGGTCCTGCGTTGGCCA was transcribed and purified as described previously (Ran et al., 2013). For the generation of patient mutation knockin mice in Ppil1p.A99T, single-strand DNA oligo (TGCCCTTCATGCTCTTCTCTCTCCTTATGTCCCCAGGGGCTGGGATTCTCACGATGGCCAACGCAGGACCAGACACCAATGGCAGCCAGTTCTTTGTGACC) was co-injected with gRNA and Cas9 protein.
To generate Ppil1 N-terminal HA epitope knockin mice, synthesized crRNA (0.6 μM) targeting GATACCTTCGCTCAGCATGG was co-injected with tracrRNA (0.6 uM, IDT DNA), Cas9 protein, and single-strand repair DNA (CCGGGTTAACTCCGCCGGAAGT AGTGATTGCTAGCGGGGGGGATACCTTCGCTCAGCATGTACCCATACGATGTTCC AGATTACGCTCTCTGGCGGCGATTCCCCCAGACACCTGGCAGCCGCCCAACGTCT ACCTGGAGACTAGGTGAG).
To generate patient mutation Ppil1p.R131Q knockin mice, synthesized crRNA targeting TCCCTATACCCTGGCACACT was co-injected with tracrRNA, Cas9 protein, and single-strand repair DNA(GGTCCTGGGAGTTTGTTTCCACCATGCCCACTCGATTCACCATCCCTATACCCTGGCACACTTGTCCAAAAATAGTATGCTTGCCGTCCAGCCATTGCGTGGGGGCCAGGGTCACAAAGAAC).
To produce Prp17p.P95A knockin mice, synthesized crRNA targeting TTCCTTATAT CGTTGCAGTT was co-injected with tracrRNA, Cas9 protein, and single-strand repair DNA(GGATTAGAGTTGAAAATACATTGTAATTTCAGGATCCTTCTTTTCCTTCCTTATATCGTTGCAGTTTGGAGCAGAAAATCCCTTTCGAACACAGCAAATGGCTGCCCCTA GAAATATGCTTTCTGGGTATGCAGAGCCAGC). Besides Prp17p.P95A knockin mice, we also got a Prp17 frameshift mouse (11 bp deletion, c.277_287delTTTGGACCAGA; NM_027879.2) and was bred to homozygosity after back-crossing with WT mice.
To generate Ppil1p.R55A knockin mice, synthesized crRNA (0.6 μM) targeting TGAAGTCCTTGATGATCCTG was co-injected with tracrRNA (0.6 μM, IDTDNA), Cas9 protein, and single strand repair DNA(GTCATTGTCCTGGAGCTATACTGGAAGCATGCGCCCAAGACCTGCAAGAACTTCGCGGAGCTGGCTCGGCGGGGCTACTACAATGGCACCAAGTTTCACGCGATCATCAAGGACTTCATGATCCAAGGCGGCGACCCGACAGGCACAGGTACACTTAAGCCACCATTGGGGAGGAACTGGGTGGTAAGGCAGCCACAGCT).
Mammalian Cells
Dermal punch biopsy was obtained under UCSD IRB protocol 171094, patients underwent sterile 0.5cm biopsy, which was mechanically dissociated and then cultured in 20% FBS in DMEM and 100U/ml penicillin-streptomycin until confluent as described (Vangipuram et al., 2013). Cells at low passage were used for protein expression analysis. HEK293T were cultured in DMEM supplemented with 10% FBS, 2mM L-glutamine and 100U/ml penicillin-streptomycin. Mouse embryonic fibroblast (MEF) cells were isolated from E13.5 embryos using mouse embryonic fibroblast isolation kit (ThermoFisher, #88279) and cultured in DMEM supplemented with 10% FBS, 2mM L-glutamine and 100 units/ml penicillin-streptomycin on gelatin-coated dishes. PPIL1 knockout HAP1 cells was generated in Horizon using the CRISPR/Cas9 system, and carries a 22 bp deletion in the first exon (22 bp deletion, c.28_49del, NM_016059). HAP1 cells were grown in Iscove’s Modified Dulbecco’s Medium (IMDM) with 10% FBS and 100 units/ml Pen/Strep. All cells used were tested negative for mycoplasma.
METHOD DETAILS
Analysis of patient phenotype
Patients were recruited as part of a multiyear effort to identify pedigrees showing multiple affected children with neurodegenerative or neurodevelopmental phenotypes, in the presence of parental consanguinity, in order to identify causes of recessive pediatric brain disease. Families were recruited at several locations around the world including the US, UK, Egypt, Pakistan, and Turkey. Subjects underwent detailed phenotyping analysis including standard medical, genetic, and neurological evaluations, serial evaluations over the course of months to year to characterize the natural history of disease progression, pedigree analysis, exclusion of previously identified genetic syndromes through the use of the London Dysmorphology Database and OMIM, followed by brain MRI or CT scan to evaluate for structural defects, and candidate gene sequencing where appropriate to exclude previously reported syndromes. Phlebotomy was performed on the entire family including all genetically informative members, for segregation analysis and linkage.
Human Brain MRI
Imaging was performed on standard clinical radiology equipment (0.5–1.5T) GE instruments, using standard T1, T2, and FLAIR settings. Hard copies of brain images were available in all cases, whereas digital files were available on a minority. This precluded quantitative analysis of brain morphology but allowed for comparison of images in the sagittal, axial and coronal orientations.
DNA extraction, whole-exome, and whole-genome sequencing
Patient DNA extraction and whole-exome sequencing libraries were performed using the Agilent SureSelect Human All Exon v2.0 (44Mb baited target) and sequenced on an Illumina HiSeq 2500 with v2 chemistry (Read Length: 151). Variant calling and filtering were performed using in-house software with Annovar, Variant Effect Prediction software to define population-specific allele frequencies from 1000 Genomes, the Greater Middle East Variome, dbSNP, and gnomAD. Variants were prioritized according to allele frequency, conservation, and predicted effects on protein function.
Variant prioritization
Variant calling and filtering were performed following an established exome sequencing pipeline (Lee et al., 2019). Identified variants were filtered out if not consistent with recessive monogenic inheritance, if the minor allele frequency (MAF) of gnomAD was >1:10,000, if MAF of local cohort was > 1:1,000, if not moderate or high impact, if CADD PHRED score ≤20, or if not predicted as damaging by either SIFT, PolyPhen, or MutationTaster.
Sanger sequencing
Primers for Sanger sequencing were designed using the Primer3 program (U. Massachusetts) and tested for specificity using the Alamut Visual 2.7.1 software. PCR products were treated with Exonuclease I (Fermentas) and Shrimp Alkaline Phosphatase (USB Corp) and sequenced using the Big Dye terminator cycle sequencing kit v.3.1 (Applied Biosystems) on an ABI DNA analyzer (Applied Biosystems). Sequence data were analyzed using Snapgene software.
PPIL1 protein expression and purification
Full-length PPIL1 (GenBank NM_016059.4) open reading frame was cloned into NdeI/XhoI sites in pET22b expression vector. Plasmids were transformed into BL21(DE3) E. coli cells, grown at 37 °C in 2YT media (5 g/l NaCl, 10 g/l yeast extract, 16 g/l tryptone), or M9 minimal media supplemented with 0.05% w/v 15N NH4Cl (for 15N labelled protein), with 100 μg/ml ampicillin. Protein expression was induced using 1 mM isopropyl β-D-1thiogalactopyranoside (IPTG) at OD600 0.5–0.8 and subsequently incubated at 18 °C overnight. PPIL1 protein was purified from cell lysate using a 1 ml Ni-NTA column (GE Healthcare) and eluted using an imidazole gradient (10 mM to 500 mM imidazole, 0.5 M NaCl, 20 mM sodium phosphate). Purity and identity of purified proteins were confirmed using SDS-PAGE Western analysis with an anti-PPIL1 antibody, and mass spectrometry.
Protein aggregation assay
Protein unfolding and aggregation were measured over a temperature gradient using an Optim machine (Unchained Labs). Protein unfolding was assessed using the barycentric mean wavelength of intrinsic protein fluorescence. Fluorescence was excited using a laser at 266 nm and emission monitored from 280 nm to 450 nm. Protein aggregation was detected by measuring static light scattering at 266 nm. Assays were carried out in PBS buffer (pH 7.4).
Immunoprecipitation assay and western blotting
HEK293T cells were seeded into 6-well plates and transfected with indicated plasmids using Lipofectamine 2000. 36 h after transfection, cells were lysed in RIPA buffer with protease inhibitor cocktail (Roche Applied Science, 11836170001). Cell lysates were centrifuged at 14,000g for 15 min at 4 °C. 45 μl supernatant was mixed with 15 μl 4× SDS-loading buffer and further heated at 95 °C for 2 min as total lysates. The remaining supernatant was incubated with 10 μl prewashed anti-FLAG M2 magnetic beads (Sigma-Aldrich, M8823) for 3 hours at 4 °C. The beads were washed four times with lysis buffer and eluted in 40 μl 2× SDS-loading buffer. Total lysates and immunoprecipitates were further separated by SDS-PAGE and analyzed by immunoblotting. Primary antibodies used include mouse anti-Flag M2 (1:10,000, Sigma-Aldrich, F1804), mouse anti-SKIP (1:1,000, Santa Cruz, sc-393856), mouse anti-beta-actin (1:1,000, Santa Cruz, sc-47778), rabbit anti-CDC40/PRP17 (1:2,000, Abcam, ab175924) and rabbit anti-PPIL1 (1:2,000, Proteintech, 15144–1-AP).
SKIP protein expression and purification
SKIP 59–129 was cloned into pGEX-6P1 expression vector (linearised using SalI and NotI restriction enzymes) using In-Fusion HD cloning kit (Clontech). The vector was transformed into BL21(DE3) Escherichia coli cells and grown at 37 °C in 2YT media (100 μg/ml ampicillin). SKIP 59–129 expression was induced with 1 mM isopropyl β-D-1thiogalactopyranoside (at OD600 0.5–0.8) for three hours at 37 °C. 1 ml glutathione sepharose columns (GE Healthcare) were used to purify GST-SKIP 59–129 from cell lysates and protein was eluted with a 10 ml glutathione injection (10 mM glutathione, 50 mM Tris-HCl pH 7.4).
Surface plasmon resonance (SPR) assay
Anti-GST antibodies were immobilized on a CM5 chip (GE Healthcare) using an amine coupling reaction, in 100 mM sodium acetate buffer (pH 5.6). The chip surface was activated using 35 μl, 0.05 M N-hydroxysuccinimide/ 0.2 M N-ethyl-N’-(dimethylaminopropyl) carbodiimide injections (GE Healthcare amine coupling kit). 20 μl anti-GST antibody (GE healthcare, 27457701) was injected at 30 μg/ml and unreacted material was subsequently eluted with 40 μl 1 M NaCl. Unreacted sites were capped using 35 μl 1 M ethanolamine.HCl pH 8.5. Amine coupling was performed in 100 mM sodium acetate running buffer (pH 5.6). N-terminal GST tagged SKIP 59–129 protein was injected across the chip surface to allow immobilization on anti-GST antibodies. In order to measure binding, PPIL1 proteins were injected across the chip surface (50 μl/min, 2.5 minutes) in PBS, 0.05 % v/v IGEPAL running buffer. Experiments were carried out using Biacore 3000 system at 25oC and the data were analyzed using Biacore BiaEvaluation software.
Nissl staining
Dissected mouse brains were fixed overnight in Bouin’s solution and embedded in paraffin. Sagittal sections were collected at a thickness of 5 μm and stained with Cresyl violet after deparaffinization and rehydration. Images were taken using a Leica Aperio AT2 scanner and a Keyence BZX-700 microscope.
Immunofluorescent staining and Fluorescence in situ Hybridization
Embryos were fixed in 4% paraformaldehyde (PFA), cryoprotected in 30% sucrose and then cryosectioned for immunostaining. Primary antibodies and reagents used include: rabbit anti-CUX1 (1:100, Santa Cruz, sc-13024), rat anti-CTIP2 (1:300, Abcam, ab18465), rabbit anti–cleaved caspase3 (1:400, Cell Signaling, 9661), rabbit anti-HA (1:300, Cell Signaling, 3724), rabbit anti-P53 (1:500, Leica, P53-CM5P), rabbit anti-PAX6 (1:300, Biolegend, 901301), rabbit anti-TBR2 (1:1,000, Abcam, ab183991), Rabbit anti-γ-H2AX (1:400, Cell Signaling, 9718), Mouse anti-SATB2 (1:400, Abcam, ab51502), and Goat anti-SOX2 (1:400, R&D systems, AF2018).
Fluorescence in situ Hybridization (FISH) was performed following the manufacturer’s instructions (Advanced Cell Diagnostics) of RNAscope Multiplex Fluorescent Assays V2 kit. TUNEL staining was performed with Apoptag Fluorescein in situ apoptosis kit (Millipore, S7110) following the provided manual. EdU fluorescence staining was performed with Click-iT™ EdU Alexa Fluor™ 488 Flow Cytometry Assay Kit (ThermoFisher, C10420). Images were taken using a Zeiss LSM 880 confocal microscope and Keyence BZX-700 fluorescent microscope and analyzed with Adobe Photoshop and Illustrator.
RNA-sequencing and data analysis
Total RNA was extracted from cultured cells or dissected mouse tissues following the manual of RNeasy Plus Mini Kit (Qiagen), yield and quality of RNA assessed by NanoDrop (Thermo Fisher Scientific ) and Agilent Bioanalyzer (Agilent Technologies), respectively, enriched by poly-A capture. Paired-end libraries were prepared according to the manufacturer’s protocols (TruSeq Stranded mRNA, Illumina) and sequenced using Illumina HiSeq2500 or 4000 system (paired-end 100, Illumina). 80–100 million reads were collected for each sample.
rMATS computational analysis
FASTQ files containing RNA-seq data from WT and mutant samples were trimmed with Cutadapt(Martin, 2011), aligned to reference human hg19 or mouse (GRCm38) with GTF release M16 using STAR aligner(Dobin et al., 2013) with ENCODE standard options, and with ‘end-to-end alignment type’ checked as required by rMATS (Shen et al., 2014). Alternative splicing events were summarized by rMATS 3.2.5, with ‘novel splice site detection’ turned on. Heatmap representation of inclusion levels for each sample was plotted using R package pheatmap(Kolde and Kolde, 2015). All intron positions for mouse were downloaded as one BED file from UCSC browser. Sequences from the mouse genome file were extracted using known start and end positions with bedtools getfasta function(Quinlan and Hall, 2010). Then the length and GC-content distributions of all introns, as well as introns residing in alternative splicing events, were calculated in R ggplot2(Wickham, 2016), with significance determined by FDR <0.05 and the absolute value of inclusion level difference > 0.05. A one-sided Mann-Whitney test was performed to obtain p-value between each group. Sequences of 40bp in length (19bp extension towards each direction) around 5’ and 3’ consensus splice sites of all mouse introns as well as our splicing event introns were extracted and stacked as sequence logo plots using WebLogo(Crooks et al., 2004). Splice sites strength was calculated using maxentpy python wrapper for MaxEntScan(Yeo and Burge, 2004) for both 5’ and 3’ sites, and scores plotted in R.
Leafcutter computational analysis
LeafCutter release 0.2.8 with STAR aligned bam files were performed according to published methods (Li et al., 2018). Intron clustering was performed by leafcutter_cluster.py with default options (maximum intron length as 100,000 bp, minimum reads in a cluster as 10, and the minimum fraction of reads in a cluster that support a junction as 0.001, respectively). Differential intron excision analysis was performed by leafcutter_ds.R with default options but --min_coverage=30 to increase specificity and --min_samples_per_intron=3 as recommended. Significance was determined by adjusted p-value (FDR) < 0.05 from the final output.
Sashimi plot
Sashimi plots were generated with python script rmats2sashimiplotm, with sorted BAM files from STAR used as inputs. Event files contain selected events extracted from rMATS outputs. Plots for different splicing types were specified by -t argument.
Enrichment analysis for misspliced genes in diseases
rMATS output produced lists containing the genes of significant events (misspliced genes) and genes in non-significant events (un-misspliced genes). Brain AS genes (n=16052) derived from (Yan et al., 2015b), OMIM genes associated with neurodevelopmental disorders (n=2755), cancer (n=589), cardiac/heart disease (n=446), and immune disorders (n=386) derived were requested from NCBI.
A 2×2 contingency table was generated for each disease gene list and the number of genes was calculated for inclusion or exclusion in the misspliced and unmisspliced genes, respectively, or calculated with 95% confidence interval. p-values were generated using Fischer’s Exact test and corrected for multiple tests with the Bonferroni method.
Pathway and Process Enrichment Analysis
Enrichment of Gene Ontology (GO) Biological Processes and KEGG pathways was carried out with KOBAS 2.0 as previous described(Xie et al., 2011). The list of genes (n = 2134) with altered AS in the mutant mouse brains were created from all the significant AS events in rMATS analysis. All expressed genes (n = 15169) in E14.5 mouse brain hemisphere were obtained from RNA-seq data with RPKM>1.0 and used as a background list. Networks of enriched pathways were generated by Metascape(Zhou et al., 2019), with ontology sources: GO Biological Processes, KEGG Pathway, Reactome Gene Sets, and CORUM. Terms with a p < 0.01, a minimum count of 3, and an enrichment factor > 1.5, grouped into clusters based on membership similarities.
Semi-quantitative RT-PCR
Total RNA was extracted from cultured cells or tissue with RNeasy Plus Mini Kit (Qiagen), according to the manufacturer’s instructions. 1 μg RNA was reverse transcribed with cDNA Synthesis Kit (Maxima First Strand cDNA Synthesis Kit for RT-qPCR, with dsDNase, ThermoFisher) following the provided manual. Minus reverse transcription (RT) negative control was added for each sample to check for DNA contamination. Negative control for PCR was included for each experiment. Primers sequences used were listed in Table S5.
Splicing reporter assay
Splicing minigene constructs were generated as previously reported (Kishore et al., 2008). Briefly, DNA fragment containing 3’ intron-exon-intron-exon-5’ intron was amplified by PCR using mouse genomic DNA as a template, which was further cloned into pSpliceExpress reporter vector using gateway recombination cloning technique. For splicing analysis, cells were transfected with pSpliceExpress plasmids and cultured for additional 36–48 hours before the extraction of total RNA. RNA was then reversed transcribed and semi-quantitative RT-PCR was performed to check the splicing of the introns. Primer sequences were: CTCTCTACCTGGTGTGTGGG (forward), and AGTGCCAAGGTCTGAAGGTC (reverse). GADPH was amplified as control for each experiment.
Purification of uniformly 15N labeled PRP17 peptide
His-SUMO-PRP17 (18 mer: T84-T101, TYETMFAPEFGPENPFRT) was cloned into NcoI/XhoI sites in pET-28b expression vector, transformed into BL21(DE3) E. coli cells, grown in 5 ml LB medium at 37°C for 6 hours and 500 μl cultured LB medium was then added to 100 ml M9 minimal media supplemented with 0.05% w/v 15N NH4Cl for overnight culture at 37 °C. Cells were further transferred to 4 L 15N minimal medium. Protein expression was induced using 1 mM IPTG at OD600 0.7–0.8 and cells were subsequently incubated at 37 °C for 8 hours. The His-tagged recombinant protein was purified from cell lysates using Ni-NTA agarose (GE healthcare) and eluted protein was further dialyzed into SUMO cleavage buffer (25mM Tris-HCl, 100 mM NaCl, PH=8.0). Purified protein was cleaved by SUMO protease (MC-LAB, SP-100) at 30 °C for 6 hours. After cleavage, His-SUMO and SUMO proteases were absorbed by Ni-NTA agarose and the collected flow-through containing PRP17 peptide was loaded to reverse-phase HPLC column (WATERS, C18) with 0.1% TFA (Trifluoroacetic acid). The peptide was eluted using an increased gradient of elution buffer (90% Acetonitrile, 0.1% TFA, and 10% H2O). Purified 15N labeled PRP17 peptide was collected and further lyophilized. Purity and identity of purified peptide were confirmed using peptide SDS-PAGE with 16.5% Mini-PROTEAN Tris-Tricine Gel (BioRad, Inc), MALDI-TOFMS analysis for molecular weight, and mass spectrometry.
1H15N HSQC of PPIL1
1H 15N heteronuclear single quantum coherence (HSQC) spectra of 15N labeled PPIL1 were recorded in the presence and absence of PRP17 peptide (Bio-FAPEFGPENPFRT-NH2; purchased from Peptide Synthetics, Inc). PPIL1 concentration was 80 μM and PRP17 peptide was titrated in at 0–5 molar equivalents in 10 mM sodium phosphate, 100 mM NaCl, 5% D2O at pH 6.5. Spectra were collected on a Bruker 600 MHz NMR spectrometer equipped with a quadruple-resonance QCI-P cryo-probe (QCI-P CP). Resonances were identified using the published assignment of PPIL1 (Stegmann et al., 2010; Xu et al., 2005). Average chemical shift perturbations were calculated as described elsewhere (Hewitt et al., 2017).
1H 15N H(Cα)N ZZ exchange spectra of PRP17 peptide
1H 15N-H(Cα)N ZZ exchange spectra (based on spectra used by previous study (Dujardin et al., 2015)) were acquired of 500 M PRP17 (residues 89–101 PRP17, Ac-FAPEFGPENPFRT-NH2, 15N, and 13 labeled Pro95) in the presence and absence of catalytic concentrations of PPIL1 (5 μM). The sample buffer used was PBS buffer (pH 7.4), 5% D2O and spectra were acquired using a 950 MHz triple resonance spectrometer equipped with a TXO triple resonance cryo-probe (TXO-CP).
1H 15N ZZ exchange spectra of uniformly 15N labeled PRP17 peptide
1H 15N ZZ exchange spectra were acquired of uniformly 15N labeled PRP17 peptide (residues 84–101 PRP17; 400 concentrations of PPIL1 (4 120(×2_, 150, 200, 250(×2), 350, 450, 600 and 750 ms. The sample buffer used was: 25 mM sodium phosphate buffer, 100 mM NaCl, 1 mM dithiothreitol, 5% D2O at pH 7.0. Spectra were acquired on a Bruker 950 MHz NMR spectrometer with a TXO triple resonance cryo-probe (TXO-CP).
Isothermal Titration Calorimetry (ITC) assay
PRP17 peptide (2 mM) was titrated into PPIL1 WT protein (40 μM), in PBS buffer (pH 7.4). Experiments were carried out using Microcal ITC 200 calorimeter at 25 °C. PRP17 titrations were carried out using 2 μl, 4-second injections spaced 2 minutes apart. Results were analyzed using Microcal Origin 7 software. Binding curves were fit to a one-site interaction model using a fixed stoichiometry (n = 1), which is recommended for low-affinity interactions(Turnbull and Daranas, 2003).
Assignment of cis and trans resonances, PRP17 peptide
A 1H-13C heteronuclear single quantum coherence (HSQC) spectrum of PRP17 peptide (PRP17 89–101; 13C and 15N labeled pro95) were recorded with 500 μM peptide in PBS (pH 7.4), 5 % v/v D2O using a Bruker 750 MHz NMR spectrometer equipped with a triple-resonance TCI triple resonance cryoprobe (TCI-CP). Cis and trans PRP17 Gly94P-ro95 peptide bond assignment was based on the 13C chemical shift for Cγ and Cβ resonances, as reported (Shen and Bax, 2010).
CRISPRi knockdown assay in HEK293T cells
Empty CRISPRi plasmid (PX330-U6–2XBsmBI-gRNA-CBh-dCas9-KRAB-T2a-Puro) was generated on the modified PX330 with 2× BsmBI gRNA cloning sites. dCas9-KRAB-T2a-Puro was amplified from vector pLV-hU6-sgRNA-hUbC-dCas9-KRAB-T2aPuro (Addgene #71236) and cloned inside PX330 to replace original WT Cas9. gRNAs targeting PRP17 or scramble gRNA was further cloned between 2XBsmBI sites. CRISPRi plasmids containing PRP17 or scramble gRNAs were co-transfected into HEK293T cells with either empty pcDNA3 or PRP17 cDNAs. 24 hours after transfection, cells were treated with 5 μg/ml puromycin for 36 hours to kill untransfected cells and then cultured in medium without puromycin for additional 24–36 hours before used for RNA extraction or Resazurin cell viability assay.
Resazurin (cell viability/proliferation) assay
Cultured cells were treated with 20% Resazurin (R&D, AR002) in the medium for 2 hours at 37 °C. 100 μl resazurin medium was further transferred to a well in 96-well plate. Fluorescence was read using Ex544nm/Em590 nm by Spectramax M5 microplate reader (Molecular Devices) and the final reading was subtracted from the background control (Resazurin in medium without cells).
Splicing rescue assay in HAP1 cells
WT and PPIase-inactive (p.R55A) PPIL1 CDS with a stop-codon was cloned into Doxycycline (DOX)-inducible pINDUCER20 expression vector, using Gateway cloning system (ThermoFisher). Lentivirus was generated by co-transfecting pINDUCER20 (empty, WT PPIL1, and R55A PPIL1, respectively), pMD2.G envelope plasmid (Addgene 12259) and psPAX2 packaging plasmid (Addgene 12260) into HEK293T cells, using Lipofectamine 2000 (ThermoFisher Scientific). The viral supernatant medium was collected at 48 and 72 hours, respectively, and then pooled and concentrated 100× using Lenti-X Concentrator (Clontech, # 631232). WT and PPIL1 knockout HAP1 cells were infected with concentrated lentivirus overnight in the presence of 5 μg/ml polybrene. 72 hours after infection, cells were selected with 1 mg/ml G418 for 2 weeks. Stable HAP1 cells were further treated with different concentrations of DOX (0, 0.1, and 0.5 μg/ml, respectively) for 72 hours and then lysed in RIPA buffer. Western blot was then applied to test the expression level of DOX-induced WT and R55A PPIL1. For splicing minigene assay, stable HAP1 cells were first cultured with 0.1 ȝg/ml DOX for 5 days and then transfected with the minigene splicing plasmid using lipofectamine 3000 (ThermoFisher Scientific) in the presence of DOX. Total RNA was extracted from transfected HAP1 cells 48 hours later and then reverse transcribed followed by semiquantitative RT-PCR. For checking the splicing of endogenous genes, stable HAP1 cells were cultured in the presence of 0.1 μg/ml DOX for 7 days and then lysed for total RNA extraction, reverse transcription and semi-quantitative RT-PCR.
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless specifically stated, each experiment was performed at least twice for each condition. Due to variabilities in the absolute values obtained in each experiment, for some assays, data from one experiment is presented and noted in the figure legend. For normally distributed data, unpaired t-tests were performed using Graphpad Prism 7 and p values were labeled in the figure panels with ns (p > 0.05), *(p < 0.05), **(p < 0.01), ***(p < 0.001), and ****(p <0.0001), and the exact values were shown in either panels or figure legends. Other statistics used include one-way ANOVA test, non-parametric Mann-Whitney test, Wilcox test, and Fischer’s Exact test, and were described in the figure legend and methods.
Supplementary Material
Movie S1. Visualization of PPIL1 and its interacting proteins in the cryo-EM structure of the spliceosome C* complex, related to Figure 5 and Figure 6 Cryo-EM structure of spliceosome C* complex (PDB: 5XJC), rotating around y- (first half) and x-axis (second half). The backside of PPIL1 (surface in teal) associates with the N-terminal of SKIP (surface in slate blue), and RBM22 (surface in orange), while its enzymatic side associates with the N-terminal of PRP17 (surface in purple). U5 snRNA (light blue) associates with the 5’ exon junction, and U6 snRNA (green) forms a duplex with the 5’ splice site (5’SS). RMB22 associates with the intron downstream 5’SS, which traverses a positively charged channel within the N-terminal domain of RBM22. U2 snRNA (light orange) forms a duplex with branching point site (BPS) region, which binds to the C-terminal WD40 domain of PRP17. PPIL1 is positioned to stabilize these associations within the spliceosome.
Figure S1-S7
Table S1. Clinical information of all the affected individuals, related to Table 1
Table S2. Clinical variants table, related to Table 1
Table S3. Summary of altered RNA splicing events in PPIL1 KO HAP1 cells using rMATS and Leafcutter, related to Figure 4
Table S4. Summary of the eight cyclophilin PPIases in the spliceosomal complexes, related to Figure 5
Table S5. Primer Sequences for RT-PCR, Related to Method Details
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-Flag M2 | Sigma-Aldrich | Cat# F1804; RRID:AB_262044 |
| Goat anti-GST | GE Healthcare | Cat# 27457701; RRID:AB_771432 |
| Mouse anti-SKIP | Santa Cruz | Cat# sc-393856; RRID: N/A |
| Mouse anti-beta-actin 1 | Santa Cruz | Cat# sc-47778; RRID:AB_2714189 |
| Rabbit anti-CDC40/PRP17 | Abcam | Cat# ab175924; RRID:N/A |
| Rabbit anti-PPIL1 | Proteintec | Cat# 15144–1-AP; RRID:AB_2169603 |
| Rabbit anti-CUX1 | Santa Cruz | Cat# sc-13024; RRID:AB_2261231 |
| Rat anti-CTIP2 | Abcam | Cat# ab18465; RRID:AB_10015215 |
| Rabbit anti–cleaved caspase3 | Cell Signaling | Cat# 9661; RID:AB_2341188 |
| Rabbit anti-HA | Cell Signaling | Cat# 3724; RRID:AB_1549585 |
| Rabbit anti-p53 | Leica | Cat# P53-CM5P; RRID:AB_2744683 |
| Rabbit anti-PAX6 | Biolegend | Cat# 901301; RRID:AB_2565003 |
| Rabbit anti-TBR2 | Abcam | Cat# ab183991; RRID:AB_2721040 |
| Rabbit anti-γ-H2AX | Cell Signaling | Cat# 9718; RRID:AB_2118009 |
| Mouse anti-SATB2 | Abcam | Cat# ab51502; RRID:AB_882455 |
| Rat anti-PH3 | Abcam | Cat# ab10534; RRID:AB_2295065 |
| Mouse anti-GAD65 | Abcam | Cat# ab26113 RRID:AB_448989 |
| Mouse anti-GAD67 | Millipore | Cat# MAB5406; RRID:AB_2278725 |
| Bacterial and Virus Strains | ||
| BL21(DE3) E. coli cells | ThermoFisher | #C600003 |
| Rosetta™(DE3) Competent E. coli Cells | Millipore | #ab70945 |
| Biological Samples | ||
| Skin punch biopsy | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cyclosporin A | Cell Signaling | #9973 |
| Bio-FAPEFGPENPFRT-NH2 | Peptide Synthetics | This study |
| N-succinyl-Ala-Ala-Pro-Phe-p-nitroanilide | Sigma | #S7388 |
| SUMO protease | MC-LAB | SP-100 |
| Critical Commercial Assays | ||
| Click-iT™ EdU Alexa Fluor™ 488 Flow Cytometry Assay Kit | ThermoFisher | #C10420 |
| HPLC column | WATERS | C18 |
| Mini-PROTEAN Tris-Tricine Gel | BIO-RAD | |
| cDNA Synthesis Kit | ThermoFisher | #1671 |
| RNeasy Plus Mini Kit | QIAGEN | |
| RNAscope Multiplex Fluorescent Assays V2 kit | Advanced Cell Diagnostics | # 323100 |
| Deposited Data | ||
| RNA-seq data for HAP1 cells and mouse brains | This paper | SRA: PRJNA669300 |
| Experimental Models: Cell Lines | ||
| HEK293T cells | ATCC | N/A |
| Control HAP1 cell | Horizon | C631 |
| PPIL1 knockout HAP1 cell | Horizon | HZGHC005943c001 |
| WT and PPIL1A99T/A99T MEF cells | This paper | N/A |
| Patient-derived skin fibroblast cells | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse, C57BL/6J, Ppil1 frameshift (1bp del) | This paper | N/A |
| Mouse, C57BL/6J, Ppil1A99T knockin | This paper | N/A |
| Mouse, C57BL/6J, Ppil1 HA-tag knockin | This paper | N/A |
| Mouse, C57BL/6J, Ppil1R131Q knockin | ||
| Mouse, C57BL/6J, Ppil1R55A knockin | This paper | N/A |
| Mouse, C57BL/6J, Prp17P95A knockin | This paper | N/A |
| Mouse, C57BL/6J, Prp17 frameshift knockin | This paper | N/A |
| Oligonucleotides | ||
|
Ppil1 frameshift mice, gRNA: GTCTGGTCCTGCGTTGGCCA |
This paper | N/A |
| For the generation of patient mutation Ppil1p.A99T knockin mice, gRNA: GTCTGGTCCTGCGTTGGCCA; single-strand repair DNA oligo: TGCCCTTCATGCTCTTCTCTCTCCTTATGTCCCCAGGGGCTGGGATTCTCACGATGGCCAACGCAGGACCAGACACCAATGGCAGCCAGTTCTTTGTGACC. |
This paper | N/A |
| To generate Ppil1 N-terminal HA epitope knockin mice, gRNA: GATACCTTCGCTCAGCATGG; single-strand repair DNA: CCGGGTTAACTCCGCCGGAAGTAGTGATTGCTAGCGGGGGGGATACCTTCGCTCAGCATGTACCCATACGATGTTCCAGATTACGCGTCTCTGGCGGCGATTCCCCCAGACACCTGGCAGCCGCCCAACGTCTACCTGGAGACTAGGTGAG. |
This paper | N/A |
| For the generation of patient mutation Ppil1p.R131Q knockin mice, gRNA: TCCCTATACCCTGGCACACT; single-strand repair DNA oligo: GGTCCTGGGAGTTTGTTTCCACCATGCCCACTCGATTCACCATCCCTATACCCTGGCACACTTGTCCAAAAATAGTATGCTTGCCGTCCAGCCATTGCGTGGGGGCCAGGGTCACAAAGAAC. |
This paper | N/A |
| To produce Prp17p.P95A knockin and knockout mice, gRNA: TTCCTTATATCGTTGCAGTT; Single-strand repair DNA: GGATTAGAGTTGAAAATACATTGTAATTTCAGGATCCTTCTTTTCCTTCCTTATATCGTTGCAGTTTGGAGCAGAAAATCCCTTTCGAACACAGCAAATGGCTGCCCCTAGAAATATGCTTTCTGGGTATGCAGAGCCAGC. |
This paper | N/A |
| To generate of Ppil1p.R55A knockin mice, gRNA: TGAAGTCCTTGATGATCCTG; single-strand repair DNA oligo: GTCATTGTCCTGGAGCTATACTGGAAGCATGCGCCCAAGACCTGCAAGAACTTCGCGGAGCTGGCTCGGCGGGGCTACTACAATGGCACCAAGTTTCACGCGATCATCAAGGACTTCATGATCCAAGGCGGCGACCCGACAGGCACAGGTACACTTAAGCCACCATTGGGGAGGAACTGGGTGGTAAGGCAGCCACAGCT. |
This paper | N/A |
| CRISPRi scramble gRNA: GCACTACCAgaGCTAACTCA |
This paper | N/A |
| CRISPRi PRP17 gRNA 01: CTCACTGTCCGAGTCCGATT |
This paper | N/A |
| CRISPRi PRP17 gRNA 02: GGACCCTGAACCCGAACCAT |
This paper | N/A |
| CRISPRi PRP17 gRNA 03: GCGATTTAGTCAAGTGCATG |
This paper | N/A |
| CRISPRi PRP17 gRNA 04: GGCAGTGGACTCGGCTCCGG |
This paper | N/A |
| Recombinant DNA | ||
| pET22b-PPIL1 | This paper | N/A |
| pcDNA3.1-PPIL1-FLAG | This paper | N/A |
| pET28b-His-SUMO-PRP17(18aa) | This paper | N/A |
| pGEX-6P1-GST-SKIP(aa59–129) | This paper | N/A |
| PX330-U6–2XBsmBI-gRNA-CBh-dCas9-KRAB-T2a-Puro | This paper | N/A |
| pLV-hU6-sgRNA hUbC-dCas9-KRAB-T2a-Puro | Addgene | #71236 |
| pMD2.G envelope plasmid | Addgene | #12259 |
| psPAX2 packaging plasmid | Addgene | #12260 |
| pSPLICEEXPRESS-Atg4d | This paper | N/A |
| pSPLICEEXPRESS-Evi5l | This paper | N/A |
| pcDNA3-HA-PRP17-IRES-GFP | This paper | N/A |
| pINDUCER20-PPIL1 | This paper | N/A |
| Software and Algorithms | ||
| Dynamic NMR (DNMR) module of the TopSpin 3.2 acquisition and processing software | Bruker | N/A |
| Origin 7 | OriginLab Inc. | http://originlab.com |
| Graphpad Prism 7 | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| Adobe Illustrator | Adobe | N/A |
| Adobe Photoshop | Adobe | N/A |
| Inkscape | N/A | https://inkscape.org/ |
| Image Lab | Bio-rad | http://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| DESeq2 | Love et al., 2014 | http://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| rMATS 3.2.5 | Shen et al., 2014 | http://rnaseq-mats.sourceforge.net/rmats3.2.5/ |
| Leafcutter | Li et al., 2018 | https://davidaknowles.github.io/leafcutter/ |
| WebLogo | Crooks et al., 2004 | https://weblogo.berkeley.edu/logo.cgi |
| MaxEntScan | Yeo and Burge, 2004 | http://hollywood.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq_acc.html |
| Pymol | N/A | N/A |
| Other | ||
HIGHLIGHTS.
Loss of spliceosomal components PPIL1 and PRP17 occurs in pontocerebellar hypoplasia
Modeling shows neural apoptosis and disrupted global RNA splicing integrity
PPIL1 and PRP17 form an enzyme-substrate pair within the active spliceosome
PPIL1 and PRP17 control splicing independent of enzyme activity
ACKNOWLEDGMENTS
This work was supported by SFARI 51486313, NIH R01NS048453, R01NS09800, QNRF NPRP 6-1463-3-351 (to J.G.G.), UM1HG008900/Broad Institute Center for Mendelian Genomics U54HG006504/Yale Center for Mendelian Disorders, N01 268201700006I-0-26800029 to CIDR for genotyping. J.G.G. is an investigator with the Howard Hughes Medical Institute and received support from Rady Children’s Institute for Genomic Medicine. We thank Peter Jackson (Stanford) for mass spectrometry analysis, Xiang-Dong Fu, Susan Taylor, Joshua Mayfield, and Jack Dixon for comments, UCSD Mouse Transgenic Core, UCSD CTRI (UL1TR001442) for biostatistics support, UCSD Neuroscience Core (P30 NS047101) and UCSD IGM Genomics Core (#S10 OD026929). We are also grateful to the Astbury BioStructure Laboratory BioNMR Facility funded by the University of Leeds and the Wellcome Trust (094232).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adams BM, Coates MN, Jackson SR, Jurica MS, and Davis TL (2015). Nuclear cyclophilins affect spliceosome assembly and function in vitro. Biochem J 469, 223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agafonov DE, Deckert J, Wolf E, Odenwalder P, Bessonov S, Will CL, Urlaub H, and Luhrmann R. (2011). Semiquantitative proteomic analysis of the human spliceosome via a novel two-dimensional gel electrophoresis method. Mol Cell Biol 31, 2667–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akizu N, Cantagrel V, Schroth J, Cai N, Vaux K, McCloskey D, Naviaux RK, Van Vleet J, Fenstermaker AG, Silhavy JL, et al. (2013). AMPD2 regulates GTP synthesis and is mutated in a potentially treatable neurodegenerative brainstem disorder. Cell 154, 505–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram K, Agafonov DE, Liu WT, Dybkov O, Will CL, Hartmuth K, Urlaub H, Kastner B, Stark H, and Luhrmann R. (2017). Cryo-EM structure of a human spliceosome activated for step 2 of splicing. Nature 542, 318–323. [DOI] [PubMed] [Google Scholar]
- Bessonov S, Anokhina M, Krasauskas A, Golas MM, Sander B, Will CL, Urlaub H, Stark H, and Luhrmann R. (2010). Characterization of purified human Bact spliceosomal complexes reveals compositional and morphological changes during spliceosome activation and first step catalysis. RNA 16, 2384–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuss MW., Sultan T., James KN., Rosti RO., Scott E., Musaev D., Furia B., Reis A., Sticht H., Al-Owain M., et al. (2016). Autosomal-recessive mutations in the tRNA splicing endonuclease subunit TSEN15 cause pontocerebellar hypoplasia and progressive microcephaly. Am J Hum Genet 99, 228–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budde BS, Namavar Y, Barth PG, Poll-The BT, Nurnberg G, Becker C, van Ruissen F, Weterman MA, Fluiter K, te Beek ET, et al. (2008). tRNA splicing endonuclease mutations cause pontocerebellar hypoplasia. Nat Genet 0, 1113–1118. [DOI] [PubMed] [Google Scholar]
- Cassandrini D, Biancheri R, Tessa A, Di Rocco M, Di Capua M, Bruno C, Denora PS, Sartori S, Rossi A, Nozza P, et al. (2010). Pontocerebellar hypoplasia: clinical, pathologic, and genetic studies. Neurology 75, 1459–1464. [DOI] [PubMed] [Google Scholar]
- Chabot B, and Shkreta L. (2016). Defective control of pre-messenger RNA splicing in human disease. J Cell Biol 212, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino L, and Koshland D. (2018). Genome-wide map of R-Loop-Induced damage reveals how a subset of R-Loops contributes to genomic instability. Mol Cell 71, 487–497 e483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, and Brenner SE (2004). WebLogo: a sequence logo generator. Genome Res 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis TL, Walker JR, Campagna-Slater V, Finerty PJ Jr, Paramanathan R, Bernstein G, MacKenzie F, Tempel W, Ouyang H, and Lee WH (2010). Structural and biochemical characterization of th an cyclophilin family of peptidyl-prolyl isomerases. PLoS biology 8, e1000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denis MM, Tolley ND, Bunting M, Schwertz H, Jiang H, Lindemann S, Yost CC, Rubner FJ, Albertine KH, Swoboda KJ, et al. (2005). Escaping the nuclear confines: signal-dependent pre-mRNA splicing in anucleate platelets. Cell 122, 379–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujardin M, Madan V, Montserret R, Ahuja P, Huvent I, Launay H, Leroy A, Bartenschlager R, Penin F, Lippens G, et al. (2015). A proline-tryptophan turn in the intrinsically disordered domain 2 of NS5A protein is essential for Hepatitis C virus RNA replication. J Biol Chem 290, 19104–19120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans PA, Dobson CM, Kautz RA, Hatfull G, and Fox RO (1987). Proline isomerism in staphylococcal nuclease characterized by NMR and site-directed mutagenesis. Nature 329, 266–268. [DOI] [PubMed] [Google Scholar]
- Fica SM, Oubridge C, Wilkinson ME, Newman AJ, and Nagai K. (2019). A human postcatalytic spliceosome structure reveals essential roles of metazoan factors for exon ligation. Science 363, 710–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. (2013). CRISPR-mediated modular RNAguided regulation of transcription in eukaryotes. Cell 154, 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber R, Zhou Z, Sukchev M, Joerss T, Frappart PO, and Wang ZQ (2011). MCPH1 regulates the neuroprogenitor division mode by coupling the centrosomal cycle with mitotic entry through the Chk1-Cdc25 pathway. Nat Cell Biol 13, 1325–1334. [DOI] [PubMed] [Google Scholar]
- Haselbach D, Komarov I, Agafonov DE, Hartmuth K, Graf B, Dybkov O, Urlaub H, Kastner B, Luhrmann R, and Stark H. (2018). Structure and conformational dynamics of the human spliceosomal B(act) complex. Cell 172, 454–464 e411. [DOI] [PubMed] [Google Scholar]
- Hewitt SH, Filby MH, Hayes E, Kuhn LT, Kalverda AP, Webb ME, and Wilson AJ (2017). Protein surface mimetics: understanding how ruthenium tris(Bipyridines) interact with proteins. Chembiochem 18, 223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insolera R, Bazzi H, Shao W, Anderson KV, and Shi SH (2014). Cortical neurogenesis in the absence of centrioles. Nat Neurosci 17, 1528–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jangi M, Fleet C, Cullen P, Gupta SV, Mekhoubad S, Chiao E, Allaire N, Bennett CF, Rigo F, Krainer AR, et al. (2017). SMN deficiency in severe models of spinal muscular atrophy causes widespread intron retention and DNA damage. Proc Natl Acad Sci U S A 114, E2347–E2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph JT, Innes AM, Smith AC, Vanstone MR, Schwartzentruber JA, Bulman DE, Majewski J, Daza RA, Hevner RF, Michaud J, et al. (2014). Neuropathologic features of pontocerebellar hypoplasia type 6. J Neuropathol Exp Neurol 73, 1009–1025. [DOI] [PubMed] [Google Scholar]
- Karaca E., Weitzer S., Pehlivan D., Shiraishi H., Gogakos T., Hanada T., Jhangiani SN., Wiszniewski W., Withers M., Campbell IM., et al. (2014). Human CLP1 mutations alter tRNA biogenesis, affecting both peripheral and central nervous system function. Cell 157, 636–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishore S, Khanna A, and Stamm S. (2008). Rapid generation of splicing reporters with pSpliceExpress. Gene 427, 104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolde R, and Kolde MR (2015). Package ‘pheatmap’. R Package 1. [Google Scholar]
- Lee S, Chen DY, Zaki MS, Maroofian R, Houlden H, Di Donato N, Abdin D, Morsy H, Mirzaa GM, Dobyns WB, et al. (2019). Bi-allelic Loss of Human APC2, Encoding Adenomatous Polyposis Coli Protein 2, Leads to Lissencephaly, Subcortical Heterotopia, and Global Developmental Delay. Am J Hum Genet 105, 844–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YI, Knowles DA, Humphrey J, Barbeira AN, Dickinson SP, Im HK, and Pritchard JK (2018). Annotation-free quantification of RNA splicing using LeafCutter. Nat Genet 50, 151158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Zhang M, Stoilov P, Chen L, and Zheng S. (2020). Developmental Attenuation of Neuronal Apoptosis by Neural-Specific Splicing of Bak1 Microexon. Neuron 107, 1180–1196 e1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lines MA, Huang L, Schwartzentruber J, Douglas SL, Lynch DC, Beaulieu C, GuionAlmeida ML, Zechi-Ceide RM, Gener B, Gillessen-Kaesbach G, et al. (2012). Haploinsufficiency of a spliceosomal GTPase encoded by EFTUD2 causes mandibulofacial dysostosis with microcephaly. Am J Hum Genet 90, 369–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipscombe D, and Lopez Soto EJ (2019). Alternative splicing of neuronal genes: new mechanisms and new therapies. Curr Opin Neurobiol 57, 26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet journal 17, 10–12. [Google Scholar]
- Namavar Y, Barth PG, Kasher PR, van Ruissen F, Brockmann K, Bernert G, Writzl K, Ventura K, Cheng EY, Ferriero DM, et al. (2011). Clinical, neuroradiological and genetic findings in pontocerebellar hypoplasia. Brain 134, 143–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik S, and Bowman TV (2019). Splicing and neurodegeneration: Insights and mechanisms. Wiley Interdiscip Rev RNA 10, e1532. [DOI] [PubMed] [Google Scholar]
- Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee MC, Guan A, Hesley JA, Miller SC, Cromwell EF, Solow-Cordero DE, et al. (2009). A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol Cell 35, 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellagatti A, and Boultwood J. (2017). Splicing factor gene mutations in the myelodysplastic syndromes: impact on disease phenotype and therapeutic applications. Adv Biol Regul 63, 5970. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj B, and Blencowe BJ (2015). Alternative splicing in the mammalian nervous system: recent insights into mechanisms and functional roles. Neuron 87, 14–27. [DOI] [PubMed] [Google Scholar]
- Rajiv C, and Davis TL (2018). Structural and functional insights into human nuclear cyclophilins. Biomolecules 8, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Ryder U, Lamond AI, and Mann M. (2002). Large-scale proteomic analysis of the human spliceosome. Genome Res 12, 1231–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzickova S, and Stanek D. (2017). Mutations in spliceosomal proteins and retina degeneration. RNA Biol 14, 544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer AE, Eggens VR, Caglayan AO, Reuter MS, Scott E, Coufal NG, Silhavy JL, Xue Y, Kayserili H, Yasuno K, et al. (2014). CLP1 founder mutation links tRNA splicing and maturation to cerebellar development and neurodegeneration. Cell 157, 651–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Cooper DN, Schuelke M, and Seelow D. (2014). MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11, 361–362. [DOI] [PubMed] [Google Scholar]
- Scotti MM., and Swanson MS. (2016). RNA mis-splicing in disease. Nat Rev Genet 17, 19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, Zhou Q, and Xing Y. (2014). rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci U S A 111, E5593–5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, and Bax A. (2010). Prediction of Xaa-Pro peptide bond conformation from sequence and chemical shifts. J Biomol NMR 6, 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y. (2017). Mechanistic insights into precursor messenger RNA splicing by the spliceosome. Nat Rev Mol Cell Biol 18, 655–670. [DOI] [PubMed] [Google Scholar]
- Silver DL, Watkins-Chow DE, Schreck KC, Pierfelice TJ, Larson DM, Burnetti AJ, Liaw HJ, Myung K, Walsh CA, Gaiano N, et al. (2010). The exon junction complex component Magoh controls brain size by regulating neural stem cell division. Nat Neurosci 13, 551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RK, and Cooper TA (2012). Pre-mRNA splicing in disease and therapeutics. Trends Mol Med 18, 472–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobreira N, Schiettecatte F, Valle D, and Hamosh A. (2015). GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat 36, 928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrells S, Nik S, Casey M, Cameron RC, Truong H, Toruno C, Gulfo M, Lowe A, Jette C, Stewart RA, et al. (2018). Spliceosomal components protect embryonic neurons from R-loop-mediated DNA damage and apoptosis. Dis Model Mech 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegmann CM, Luhrmann R, and Wahl MC (2010). The crystal structure of PPIL1 bound to cyclosporine A suggests a binding mode for a linear epitope of the SKIP protein. PLoS One 5, e10013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synofzik M, Puccio H, Mochel F, and Schols L. (2019). Autosomal recessive cerebellar ataxias: paving the way toward targeted molecular therapies. Neuron 101, 560–583. [DOI] [PubMed] [Google Scholar]
- Teigelkamp S, Achsel T, Mundt C, Gothel SF, Cronshagen U, Lane WS, Marahiel M, and Luhrmann R. (1998). The 20kD protein of human [U4/U6.U5] tri-snRNPs is a novel cyclophilin that forms a complex with the U4/U6-specific 60kD and 90kD proteins. RNA 4, 127141. [PMC free article] [PubMed] [Google Scholar]
- Turnbull WB, and Daranas AH (2003). On the value of c: can low affinity systems be studied by isothermal titration calorimetry? J Am Chem Soc 125, 14859–14866. [DOI] [PubMed] [Google Scholar]
- van Dijk T, Baas F, Barth PG, and Poll-The BT (2018). What’s new in pontocerebellar hypoplasia? An update on genes and subtypes. Orphanet J Rare Dis 13, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangipuram M, Ting D, Kim S, Diaz R, and Schule B. (2013). Skin punch biopsy explant culture for derivation of primary human fibroblasts. J Vis Exp, e3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhang S, Zhang J, Huang X, Xu C, Wang W, Liu Z, Wu J, and Shi Y. (2010). A large intrinsically disordered region in SKIP and its disorder-order transition induced by PPIL1 binding revealed by NMR. J Biol Chem 285, 4951–4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. (2016). ggplot2: elegant graphics for data analysis (Springer; ). [Google Scholar]
- Will CL, and Luhrmann R. (2011). Spliceosome structure and function. Cold Spring Harb Perspect Biol 3, a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, Kong L, Gao G, Li CY, and Wei L. (2011). KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res 39, W316–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Xu Y, Tang Y, Wu J, Shi Y, Huang Q, and Zhang Q. (2005). Backbone and side chain assignments of human Peptidylprolyl Isomerase Like 1 (hPPIL1). J Biomol NMR 31, 179180. [DOI] [PubMed] [Google Scholar]
- Xu C, Zhang J, Huang X, Sun J, Xu Y, Tang Y, Wu J, Shi Y, Huang Q, and Zhang Q. (2006). Solution structure of human peptidyl prolyl isomerase-like protein 1 and insights into its interaction with SKIP. J Biol Chem 281, 15900–15908. [DOI] [PubMed] [Google Scholar]
- Xu M, Xie YA, Abouzeid H, Gordon CT, Fiorentino A, Sun Z, Lehman A, Osman IS, Dharmat R, Riveiro-Alvarez R, et al. (2017). Mutations in the Spliceosome Component CWC27 Cause Retinal Degeneration with or without Additional Developmental Anomalies. Am J Hum Genet 100, 592–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C, Hang J, Wan R, Huang M, Wong CC, and Shi Y. (2015a). Structure of a yeast spliceosome at 3.6-angstrom resolution. Science 349, 1182–1191. [DOI] [PubMed] [Google Scholar]
- Yan Q., Weyn-Vanhentenryck SM., Wu J., Sloan SA., Zhang Y., Chen K., Wu JQ., Barres BA., and Zhang C. (2015b). Systematic discovery of regulated and conserved alternative exons in the mammalian brain reveals NMD modulating chromatin regulators. Proc Natl Acad Sci U S A 112, 3445–3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, and Burge CB (2004). Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 11, 377–394. [DOI] [PubMed] [Google Scholar]
- Zhan X, Yan C, Zhang X, Lei J, and Shi Y. (2018). Structure of a human catalytic step I spliceosome. Science 359, 537–545. [DOI] [PubMed] [Google Scholar]
- Zhang X, Chen MH, Wu X, Kodani A, Fan J, Doan R, Ozawa M, Ma J, Yoshida N, Reiter JF, et al. (2016). Cell-type-specific alternative splicing governs cell fate in the developing cerebral cortex. Cell 166, 1147–1162 e1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yan C, Hang J, Finci LI, Lei J, and Shi Y. (2017). An atomic structure of the human spliceosome. Cell 169, 918–929 e914. [DOI] [PubMed] [Google Scholar]
- Zhang X, Yan C, Zhan X, Li L, Lei J, and Shi Y. (2018). Structure of the human activated spliceosome in three conformational states. Cell Res 28, 307–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhan X, Yan C, Zhang W, Liu D, Lei J, and Shi Y. (2019). Structures of the human spliceosomes before and after release of the ligated exon. Cell Res 29, 274–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XC, Wang WD, Wang JS, and Pan JC (2013). PPIase independent chaperonelike function of recombinant human Cyclophilin A during arginine kinase refolding. FEBS Lett 587, 666–672. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Licklider LJ, Gygi SP, and Reed R. (2002). Comprehensive proteomic analysis of the human spliceosome. Nature 419, 182–185. [DOI] [PubMed] [Google Scholar]
- Zydowsky LD, Etzkorn FA, Chang HY, Ferguson SB, Stolz LA, Ho SI, and Walsh CT (1992). Active site mutants of human cyclophilin A separate peptidyl-prolyl isomerase activity from cyclosporin A binding and calcineurin inhibition. Protein Sci 1, 1092–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Visualization of PPIL1 and its interacting proteins in the cryo-EM structure of the spliceosome C* complex, related to Figure 5 and Figure 6 Cryo-EM structure of spliceosome C* complex (PDB: 5XJC), rotating around y- (first half) and x-axis (second half). The backside of PPIL1 (surface in teal) associates with the N-terminal of SKIP (surface in slate blue), and RBM22 (surface in orange), while its enzymatic side associates with the N-terminal of PRP17 (surface in purple). U5 snRNA (light blue) associates with the 5’ exon junction, and U6 snRNA (green) forms a duplex with the 5’ splice site (5’SS). RMB22 associates with the intron downstream 5’SS, which traverses a positively charged channel within the N-terminal domain of RBM22. U2 snRNA (light orange) forms a duplex with branching point site (BPS) region, which binds to the C-terminal WD40 domain of PRP17. PPIL1 is positioned to stabilize these associations within the spliceosome.
Figure S1-S7
Table S1. Clinical information of all the affected individuals, related to Table 1
Table S2. Clinical variants table, related to Table 1
Table S3. Summary of altered RNA splicing events in PPIL1 KO HAP1 cells using rMATS and Leafcutter, related to Figure 4
Table S4. Summary of the eight cyclophilin PPIases in the spliceosomal complexes, related to Figure 5
Table S5. Primer Sequences for RT-PCR, Related to Method Details
Data Availability Statement
The raw RNA_seq data related to this manuscript, including human HAP1 cells and mouse E14.5 brain samples, has been deposited on SRA under accession number PRJNA669300 (https://www.ncbi.nlm.nih.gov/sra).