

Abstract
Background
SHOX enhancer CNVs, affecting one or more of the seven recognized evolutionary conserved non‐coding elements (CNEs) represent one of the most frequent cause of SHOX‐haploinsufficiency. During the diagnostic workflow deletions/duplications have been identified downstream SHOX not including any of the these CNEs.
Methods
Fine tiling aCGH and breakpoint PCR were used to characterize the critical interval and to search for novel alterations in a cohort of selected patients.
Results
Screening of 252 controls provided evidence that duplications in this area represent likely benign variants whereas none of the deletions were detected. These findings suggested that other alterations relevant for SHOX‐haploinsufficiency might be missed by the standard diagnostic methods. To identify such undisclosed elements, the aCGH was used to reanalyze 52 unresolved cases with clinical features strongly suggestive of SHOX‐haploinsufficiency. This analysis followed by the screening of 210 patients detected two partially overlapping small deletions of ~12 and ~8 kb in four unrelated individuals, approximately 15 kb downstream SHOX, that were absent in 720 normal stature individuals.
Conclusion
Our results strengthen the hypothesis that alterations of yet unidentified cis‐regulatory elements residing outside those investigated through conventional methods, might explain the phenotype in ISS/LWD patients thus enlarging the spectrum of variants contributing to SHOX‐haploinsufficiency.
Keywords: array CGH, enhancers, LWD, short stature, SHOX
The characterization of deletions identified in Short Staure patients through fine tiling aCGH narrowed down a critical interval downstream the enhancer that might contains elements relevant for SHOX expression. To identify undisclosed

1. INTRODUCTION
The short stature homeobox‐containing gene (SHOX; OMIM * 312865) is located at the tip of both sexual chromosomes within the pseudoautosomal region 1 (PAR1). Like other pseudoautosomal genes, it escapes X inactivation and two functional copies are required to provide normal function. Genetic alterations leading to the loss of one functional allele result in SHOX‐haploinsufficiency responsible for the 70%–90% of Léri–Weill dyschondrosteosis (LWD; OMIM #127300) and of a variable percentage (~5%) of idiopathic short stature (ISS; OMIM #300582) cases. The complete SHOX deficiency caused by loss of both the copies of the gene causes Langer mesomelic dysplasia (LMD; OMIM #249700), a rare syndrome characterized by severe disproportionate short stature with mesomelic and rhizomelic shortening of the upper and lower limbs.
Seven cis‐acting conserved non‐coding elements (CNEs) located either upstream (CNE‐5, CNE‐3, and CNE‐2; Durand et al., 2010) or downstream (CNE4, CNE5, ECR1, and CNE9/ECS4; Benito‐Sanz et al., 2012; Fukami et al., 2006; Sabherwal et al., 2007) the coding sequence regulate SHOX expression. Functional studies showed that they act as enhancers for SHOX promoter activity in human U2OS cell line (Benito‐Sanz et al., 2012; Fukami et al., 2006; Verdin et al., 2015), in developing chicken limb (Durand et al., 2010; Sabherwal et al., 2007) and in zebrafish embryos (Kenyon et al., 2011). Recently, a further regulatory sequence (zeugopodal enhancer downstream of SHOX, ZED) was identified about 7 kb upstream CNE9 that showed limb‐specific enhancer activity in a transgenic mouse model (Skuplik et al., 2018).
Micro‐rearrangements affecting the entire gene and/or the downstream enhancer represent the most frequent genetic defect underlying SHOX‐haploinsufficiency. These rearrangements are likely originated by events of nonallelic homologous recombination (NAHR) and non‐homologous end‐joining (NHEJ) mediated by Alu repeats of which PAR1 is enriched(Benito‐Sanz et al., 2012; Blaschke & Rappold, 2006; Fukami et al., 2006). Moreover, in PAR1 the recombination rate is nearly 17 times higher than the rest of the genome during male meiosis (Hinch et al., 2014), thus enhancing the probability of unbalanced rearrangements.
The deletions of the entire SHOX coding gene with or without the enhancers represent a well‐established cause of LWD and ISS. Moreover, deletions encompassing only the enhancer regions have been detected in these patients and in particular, a recurrent 47.5 kb deletion represent the most common SHOX alteration identified in patients, albeit with variable penetrance (Benito‐Sanz et al., 2012; Bunyan et al., 2013; Kant et al., 2013; Shima et al., 2016).
Unlike deletions, the significance of duplications remains unknown and controversial in several cases (Bunyan et al., 2016; Hirschfeldova & Solc, 2017). It has been proposed that larger duplications involving the entire gene and its enhancers may lead to SHOX overexpression, as observed in subjects carrying triple dose of PAR1 (47, XXY and 47, XXX karyotypes; Ottesen et al., 2010). On the other hand, microduplications involving only a part of the downstream or upstream regulatory regions have been observed both in short and in normal stature subjects (Benito‐Sanz et al., 2011; Bunyan et al., 2016; Fukami et al., 2015; Hirschfeldova & Solc, 2017; Monzani et al., 2019; Thomas et al., 2009) and a similar frequency between patients and controls has been reported with the exception of duplications encompassing the CNE7‐CNE9 that are significantly more frequent in patients (Hirschfeldova & Solc, 2017). Partial duplications involving the coding region and some of the CNEs seem to be more deleterious as they might reduce the gene expression by altering the distance between SHOX and its flanking regulatory elements (Fukami et al., 2015).
During the diagnostic screening for SHOX alterations performed through conventional procedures (MLPA and Sequencing) we encountered deletions and duplications in the SHOX downstream area not encompassing any of the described functionally relevant CNEs in patients with phenotypes strongly suggestive of SHOX‐haploinsufficiency, that were classified as VUS (variant of uncertain significance). To better characterize and interpret these rearrangements a fine tiling array CGH with high density probe in the PAR1 region was used. The same platform was then utilized to reanalyze a cohort of 52 short stature patients that had been previously tested negative through the SHOX standard test.
2. MATERIALS AND METHODS
2.1. Subjects
A total of 1102 patients referred for SHOX genetic analysis were recruited from multiple Italian centers from 2010 to date. Informed written consent was provided from all study participants. The patients were selected for short stature, i.e., height SDS <−2 for age, sex and population group (Cacciari et al., 2006), or stature below the genetic target according to Tanner's method (for males: [paternal height + maternal height]/2 + 6.5 cm; for females: [paternal height + maternal height]/2 − 6.5 cm). Patients with SDS >−2 were also included in presence of other clinical characteristics suggestive of SHOX deficiency as cubitus valgus, short forearm, bowing of the forearm, muscular hypertrophy, dislocation of ulna, and Madelung deformity. The Rappold Score (RS) was calculated when all the requested parameters were available (Rappold et al., 2007). A clinically proved diagnosis of LWD was present in 10% of these subjects. Individuals with abnormal karyotypes, severe neurological impairments and clear evidences of systemic, endocrine, or nutritional diseases were excluded from the study.
For every patient, height, weight, and BMI were stratified according to the Italian growth charts (Cacciari et al., 2006); measurements of standing height, sitting height (measured from the highest point of the head to the sitting surface; Bogin & Varela‐Silva, 2010), arm span (length from the fingertips of one hand to the other with the arms raised parallel to the ground; Bogin & Varela‐Silva, 2010) were performed. Growth velocity (cm/year) (difference of mean heights obtained from two consecutive visits, divided by the time between the visits) was considered as a useful parameter (Genoni et al., 2018).
2.2. SHOX genetic analysis
Genomic DNA was extracted from whole blood samples using the ReliaPrep Blood gDNA Miniprep System (Promega) following the manufacturer's instructions.
The MLPA analysis for deletions/duplications detection was performed using the commercial kit SALSA MLPA P018‐G1 SHOX probemix (MRC‐Holland) according to manufacturer's instructions. This MLPA test contains 26 probes spaced 0.2–6.7 kb in the coding region and 0.4–338 kb in the non‐coding region. The amplified fragments were analyzed by capillary electrophoresis on an ABI PRISM 3100 Genetic Analyzer with the GeneMapper software (Applied Biosystems). The data analysis was performed by MRC‐Holland Coffalyser v9.4 software.
Sequencing analysis was performed by direct sequencing of the coding exons (1‐6a/6b). Each exon was PCR amplified using GoTaq G2 Flexi DNA Polymerase (Promega; Table S1). PCR products were visualized on a 1.5% agarose gel, purified using EuroSap ‐ PCR enzymatic clean‐up kit (Euroclone), and then sequenced in the forward or reverse direction with the BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems) and analyzed on an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems).
2.3. Array CGH analysis
Array‐based comparative genomic hybridization (CGH) analysis was performed using a SurePrint G3 Custom CGH 8x60K platform (Agilent Technologies) designed using the Agilent SureDesign online tool (https://earray.chem.agilent.com/suredesign/). The custom array CGH was designed with high resolution in PAR1 (371 bp average probe spacing). A number of probes targeting short stature causative genes were also added along with a low‐resolution genome‐wide backbone (Table S2). Array CGH experiments were performed according to manufacturer's instructions using Agilent's Human DNA Male and Female as references. Test DNA samples labeled in Cy5‐dUTP and reference DNA samples labeled in Cy3‐dUTP were coupled on the basis of similar yield ([ng/µl DNA × 10 µl]: 1000 ng/µg) and specific activity (pmol/µl Dye: µg/µl DNA) for a final volume of 16 µl. Slides were scanned 24 h after hybridization through the Agilent SureScan Dx Microarray Scanner Bundle (G5761A) and the Agilent Microarray Scan Control software. The data extraction was performed using the Feature Extraction for CytoGenomics (Agilent) software and the data analysis was performed through the Agilent CytoGenomics (Edition 3.0.6.6) software. For each spot of the array, the value of log2 ratios between the Cy5‐labeled test DNA and the Cy3‐labeled reference DNA were calculated.
The possible pathogenicity of the observed variants was evaluated based on their presence in public databases: UCSC Genome Browser (https://genome.ucsc.edu/cgi‐bin/hgGateway), Database of Genomic Variants (DGV, http://dgv.tcag.ca/dgv/app/home), and Database of Genomic Variation and Phenotype in Humans using Ensembl Resources (DECIPHER, https://decipher.sanger.ac.uk/index). The assembly GRCh37/hg19 was used as reference genome.
2.4. Deletion breakpoints mapping
Based on array CGH results PCR primers were designed to amplify across the deletion breakpoints on genomic DNA (primer sequences in Table S3) identified in patients #1, #2, #7, #8, and #9. The forward and the reverse primers were designed within the undeleted regions upstream and downstream the boundaries defined by the aCGH probes: the alleles carrying the deletions are amplified whereas the wild‐type (non deleted) alleles are too large for being amplified under standard PCR conditions. To establish the genotype in a panel of patients and controls for the deletions identified in patients #7 and #8/#9, a further reverse primer was designed within the deleted sequence to amplify the wild‐type alleles (Table S3).
The PCR reactions were performed using the Phusion High‐Fidelity DNA Polymerase kit (Thermo Fisher Scientific) according to manufacturer's instructions. The cycling conditions were as follows: Patient#1 (96℃ for 5 min, 35 cycles at 96℃ for 30 s, at 60℃ for 30 s and at 72℃ for 2 min, and at 72℃ for 10 min); Patient #2 (96℃ for 5 min, 35 cycles at 96℃ for 30 s, at 60℃ for 30 s and at 72℃ for 2 min, and at 72℃ for 10 min); Patient#7 (96℃ for 5 min, 35 cycles at 96℃ for 30 s, at 64℃ for 30 s, and at 72℃ for 2 min, and at 72℃ for 10 min); Patient#8/#9 (96℃ for 5 min, 35 cycles at 96℃ for 30 s, at 60℃ for 30 s, and at 72℃ for 3 min, and at 72℃ for 10 min). PCR products were sequenced using the BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems) and analyzed on an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems).
3. RESULTS
3.1. Identification and characterization of rare deletions in ISS/LWD individuals
The diagnostic screening for SHOX alterations led to the identification of point mutations and deletions encompassing the gene and/or the well‐known regulatory regions in the 8.7% of the 1102 individuals referred to the Genetic Unit during the last 10 years (data not shown). Besides these well‐known alterations we identified deletions and duplications that did not affect any of the already described enhancers in six individuals. All the rearrangements mapped within a region of ~430 kb downstream the CNE9 (Figure 1a).
FIGURE 1.
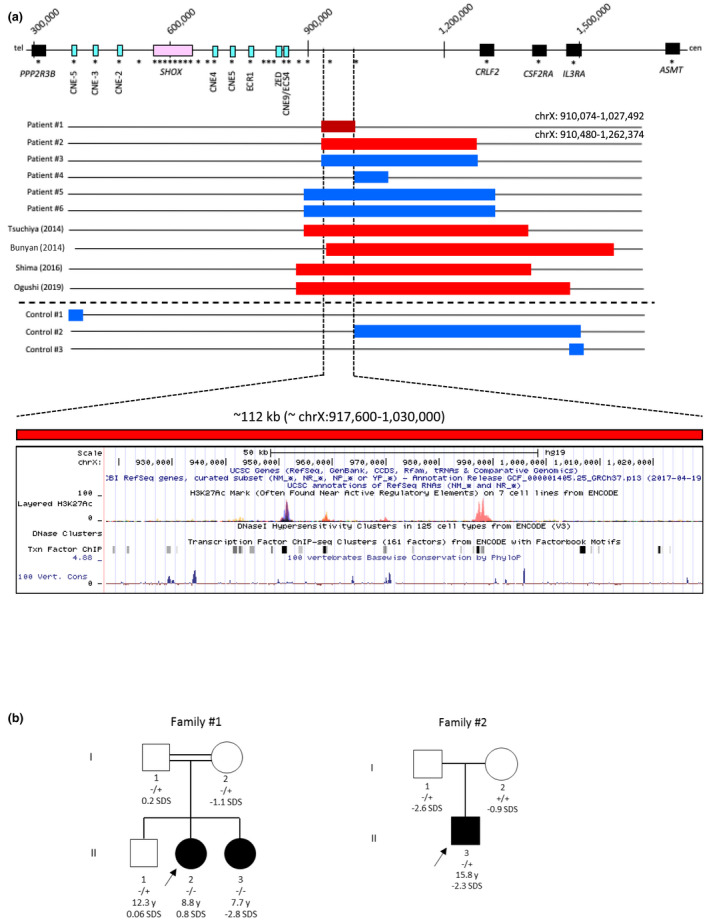
(a) Duplications/Deletions identified in patients #1 to #6 during the SHOX diagnostic routine and in a normal stature control cohort through MLPA. In the upper part of the figure is reported a schematic representation of PAR1 (assembly GRCh37/hg19). MLPA probes from the commercial P018‐G1 kit (MRC‐Holland) are indicated as black asterisks. Duplications are depicted as blue bars and deletions as red bars (the deletion at homozygous state observed in patient #1 is indicated in dark red). Deletions in the same area previously reported in literature are also indicated. In the lower panel, the interrogation of the UCSC Genome Browser for the minimum deleted region shows the presence of evolutionarily conserved sequences, H3K27Ac marks from ENCODE and transcription factors binding predictions from ChIP‐seq. (b) Pedigrees of patients #1 and #2. The proband is indicated by an arrow. The wild‐type and variant alleles are indicated as − and +, respectively
Two hundred fifty‐two normal stature controls were also screened through the same MLPA assay. Three different duplications, one upstream of SHOX and two downstream of the CNE9, were identified in three controls (Figure 1a). These findings confirmed previous observations (Hirschfeldova & Solc, 2017) that duplications downstream CNE9 may represent likely benign CNVs and for this reason they were not further considered. In contrast, no deletion was identified in any of the analyzed controls. As the deletions identified in patients #1 and #2 (Figure 1a) did not include any of the already described CNEs they were classified as VUS. However, similar rearrangements were previously reported in short stature individuals (Bunyan et al., 2014; Ogushi et al., 2019; Tsuchiya et al., 2014; Figure 1a) suggesting a possible pathogenicity for these imbalances. To better characterize these rearrangements, a custom array CGH with high probe density in PAR1 was used to analyze these patients’ DNA.
Patient#1: this 16‐year‐old female (II‐1, Figure 1b) presented at the first access at the age of 9 years with normal height (0.9 SDS), precocious puberty (Tanner 3 stages), advanced bone age (hand‐wrist x‐rays: +3.5 years), cubitus valgus, and short forearm. The karyotype analysis did not reveal any chromosomal aberration. She started treatment with a GnRH analog (triptorelin) with a partial regression of Tanner stages (Tanner 2). However, progressively the growth velocity decreased more than that expected, and at 11 years old triptorelin treatment was stopped. Furthermore, some clinical features characteristic of SHOX‐haploinsufficiency became more evident such as cubitus valgus, muscular hypertrophy, short forearm, arm span/height ratio (0.89), and sitting height/height ratio (0.56) with a RS of 16. The hypothalamic‐pituitary magnetic resonance imaging was normal, and biochemical tests revealed normal glucose levels, absence of insulin resistance, normal hepatic and renal function, normal levels of IGF‐1(463 ng/dl, SDS +0.50), and normal response to arginine test for GH secretion. The patient was thus addressed to SHOX molecular analysis that revealed a deletion at the homozygous state in the SHOX downstream area that did not encompass any known enhancer (Figure 1a). Such deletion was inherited from the two consanguineous normal stature parents (I‐1 and I‐2, Figure 1b). She started treatment with rhGH that was stopped at the age of 13 years. The patient was lost at follow‐up and came back to our attention at the age of 15 years when she underwent endoscopic removal and local chemotherapy with mitomycin C for a papillary urothelial carcinoma. Her height at the age of 16 years was 147.5 cm (−2.42 SDS), puberty was completed, and she was eumenorrheic. Her sister (II‐3, Figure 1b) with disproportionate short stature (−2.8 SDS), cubitus valgus and slight micrognathia was also homozygous for the deletion whereas the normal stature brother (II‐2, Figure 1b) was heterozygous. Neither the brother nor the parents showed clinical signs characteristic of SHOX‐haploinsufficiency.
The array CGH analysis revealed that this rearrangement had a minimum size of 117 kb (Figure S1a). A precise breakpoint mapping performed through PCR and sequencing analysis revealed that such deletion spans 119,258 bp (chrX: 909,966–1,029,224) (Figure S1a). Both upstream and downstream breakpoints are embedded within the Alu repetitive elements AluSx1/AluSp that likely provided the conditions for a nonallelic homologous recombination (NAHR) event.
Patient #2: this patient (II‐1, Figure 1b) was a short stature 15.8‐year‐old male (−2.3 SDS) presenting slight bowing of the forearm suggestive of Madelung deformity and ulnar dislocation (RS = 7). Previous investigations excluded common causes of short stature and an arginine stimulation test showed normal GH secretion (peak GH 12.10 ng/ml). Considering the age, the advanced bone age and stature within target height at time of diagnosis, this patient was not treated with rhGH. The molecular SHOX analysis showed the presence of a deletion at the heterozygous state detected by two MLPA probes (Figure 1a), that was inherited from the short stature father (−2.6 SDS; I‐1, Figure 1b).
The fine tiling aCGH analysis combined with the breakpoint mapping revealed that the patient carried two distinct deletions: a smaller deletion of 719 bp (chrX:949,397–950,116, hg38) and a larger one of 235,154 bp (chrX:950,396–1,185,550, hg38) separated by 280 bp (Figure S1b), that were both inherited from the father and thus on the same allele. All the breakpoints, except for the telomeric breakpoint of the smaller deletion, reside within repetitive elements type Alu (also present in the 280 pb region) that probably mediated a complex rearrangement. This deletion did not include any of the described CNE elements and overlapped to the deletion detected in family#1 (Figure 1a).
3.2. Search for alterations through fine tailing aCGH
The deletions identified in patients #1 and #2 suggest that pathogenic unbalances might occur in regions that are not covered by the probes included in the MLPA kit. To search for such alterations, 52 subjects with phenotype suggestive for SHOX‐haploinsufficiency that tested negative to the conventional SHOX genetic test, were screened through the custom fine tiling aCGH platform. The patients presented a stature between −3.1 and −1.2 SDS (mean −2.01 SDS ±0.89). Among these, 9.6% presented shortening of the upper and/or of the lower limbs, 48.7% were characterized by body disproportion with or without short stature, 35.4% had skeletal dysplasia and 16.1% presented characteristics of LWD. The RS was available for 32 patients: 7 (21.9%) had a RS between 4 and 7 while 15 (46.9%) had a RS >7.
None of the patients carried alterations outside PAR1 in the other short stature genes tagged by the aCGH. Conversely this analysis revealed the presence of two small deletions of ~12 and ~8 kb in three patients (patients #7, #8, and #9) not reported in the Database of Genomic Variants (DGV) at approximately 15 kb downstream the SHOX coding region (Figure 2a and Figure S2). A specific PCR assay and sequencing analysis were set up to map the breakpoints in all the three patients and to extend the analysis to the other family members (Figure 2a and Figure S2).
FIGURE 2.
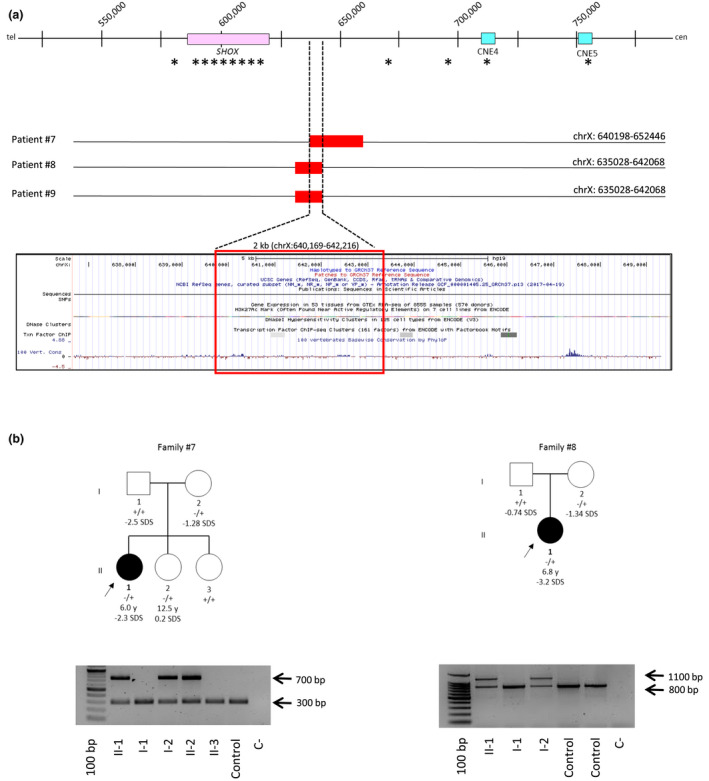
(a) Deletions identified through custom array CGH in patients #7, #8, #9. The asterisks indicate the MLPA probes. The UCSC Genome Browser shows poor evolutionary conservation within the minimal deleted region is indicated. (b) Pedigrees of patients #7 and #8. The parents of patient #9 were not available for the analysis. The proband is indicated by an arrow. The wild‐type and variant alleles are indicated as − and +, respectively. A PCR assay was specifically designed to detect each of the deletions identified through the fine tiling aCGH. The forward and the reverse primers were designed just upstream and downstream the putative 5′ and 3′ breakpoints, respectively; the PCR product is detectable only in deletion carriers (upper band). A further reverse primer was designed within the deleted sequence to amplify wild‐type alleles (lower band)
Patient# 7: this patient (II‐1, Figure 2b) was a 6‐year‐old female referred as LWD, with short stature (−2.3 SDS), short forearm, and muscular hypertrophy (RS = 12). She presented a target height SDS of −2.15 and a growth velocity SDS of −1.28 before treatment that increased to >3 SDS after GH therapy. The sequencing analysis revealed that the deletion identified through the aCGH spanned 12,837 bp (chrX:639,875–652,712) (Figure S2a). No repetitive element is present in the proximity of the telomeric breakpoint while the centromeric breakpoint resides within the repetitive element AluSx1.
The extension of the PCR analysis to the family revealed that the deletion was also present in a sister (II‐2, Figure 2b) and that it was inherited from the mother (I‐2, Figure 2b), both with normal stature.
Patient #8: the patient (II‐1, Figure 2b) was a 6.8‐year‐old female with a diagnosis of LWD, presenting severe short stature (−3.2 SDS) and muscular hypertrophy with a RS = 9. The growth velocity SDS before treatment was −2.05 that increased >3 SDS after GH therapy.
The breakpoint sequencing analysis revealed that the deletion had a size of 8,875 bp (chrX:634,213–643,088) (Figure S2b). The telomeric breakpoint is located within the repetitive element AluSx3, while the centromeric breakpoint resides within a low complexity repeat. This deletion was inherited from the normal stature mother (I‐2; Figure 2b).
Patient #9: this 12.6 years old male presented disharmonic normal height (−0.8 SDS) characterized by shortening of the lower limbs accompanied by muscular hyperthrophy (RS = 9). The array CGH analysis identified the same deletion reported in patient #8 and sequencing analysis confirmed that both deletions presented identical breakpoints (Figure S2c). The DNA of the parents was not available for segregation analysis.
3.3. Rapid screening for the deletions in other patients and controls (patient#10)
The same PCR assays were then used to test the presence of these two microdeletions in further 210 patients tested negative after routine SHOX analysis and in a cohort of 740 normal stature controls. The 12,837 kb deletion was identified in another patient (Patient #10), a 13‐year‐old female with short stature (−2.4 SDS), normal secretion levels of GH after stimulation and normal levels of IGF‐1. The hand‐wrist x‐rays revealed a 2 years advanced bone age. The parents were not available for the genetic analysis.
Both the deletions were absent in the group of normal stature controls.
4. DISCUSSION
Whereas deletions including the SHOX coding region and the CNEs can be easily interpreted as pathogenic or likely pathogenic, it is harder to draw solid conclusions and communicate information to carriers of either deletions/duplications that do not include any regulatory sequence or nucleotide variants lying outside the coding regions.
Nonetheless it is important to investigate the role of VUS that might be encountered during SHOX molecular analysis either to establish a potential causative role and address patients to an effective GH replacement therapy (Blum et al., 2007) or to exclude the presence of pathogenic variations in individuals that would not benefit from the expensive GH treatment. In this regard we recently demonstrated that non‐coding variations within the 5′UTR affect SHOX expression through different mechanisms and should be considered and referred as likely pathogenic (Babu et al., 2021).
During the routine diagnostic screening over the last 10 years besides deletions and duplications involving SHOX and/or its enhancers we identified duplications and deletions that did not include any of the already described enhancers (Figure 1a). A control cohort of 252 normal stature subjects was screened through MLPA to assess the frequency of CNVs encompassing PAR1 in the general population and three duplications were identified (Figure 1a). These results, along with previous observations (Hirschfeldova & Solc, 2017) support the hypothesis that most of the micro‐duplications downstream SHOX that do not affect the CNEs might represent rare benign CNVs. Instead, deletions downstream the last conserved regulatory element (CNE9) were absent in the controls and were previously reported in four patients with SHOX‐related phenotype (Blum et al., 2007; Bunyan et al., 2014; Ogushi et al., 2019; Tsuchiya et al., 2014; Figure 1a). It is thus conceivable that this interval might contain further elements crucial for SHOX expression. The deletion identified in patient#1 is to our knowledge the smallest deletion in this area reported in individuals referred for SHOX‐haploinsufficiency allowing to narrow down the critical interval to a region of approximately 112 kb (chrX:917,600–1,030,000) (Figure 1a). It is likely that this alteration exerts a recessive effect as both the homozygous siblings (II‐1 and II‐3, Figure 1b) showed a phenotype strongly suggestive of SHOX deficiency whereas the heterozygous member of this family (I‐1, I‐2 and II‐2) presented with normal stature and no clinical signs. Although any of the described CNE maps within the imbalance, in silico analysis performed through the UCSC Genome Browser (https://genome.ucsc.edu) revealed the presence of sequences with high evolutionary conservation within such interval approximately between chrX: 917,000 and 1,029,000 (Figure 1a), and chromatin signatures suggestive of active transcription as the acetylation of lysine 27 of the H3 histone. Moreover an expression quantitative trait locus (eQTL) of approximately 20 kb (chrX:901,465–920,235) containing several SNPs was identified from the GTEx database that correlate with SHOX expression as observed in a previously reported family (Ogushi et al., 2019). The presence of a cis‐regulatory domain within the here characterized critical interval (chrX:917,600–1,030,000, hg19) is supported by the experimental results demonstrating through 4C‐seq that the cis‐regulatory landscape of SHOX extended beyond the boundaries of the already functionally characterized CNEs (i.e., chrX:398,357–835,567, hg19) (Verdin et al., 2015). Taken together, these considerations suggest that the region between 917,600 and 1,030,000 represents a good candidate to contain further putative elements regulating SHOX expression, although this hypothesis requires experimental validation.
The use of the same custom array CGH platform allowed to identify in four ISS/LWD patients that tested negative through the standard diagnostic methods two small partially overlapping deletions of 12,837 kb (patient #7 and #10) and 8,875 kb (patient #8 and #9) just downstream the SHOX coding region (Figure 2a). The two deletions encompass a genomic region without a significant evolutionary conservation (Figure 2a). However, the lack of conservation does not exclude a functional role as in the case of the regulatory sequence (ZED) removed by the commonest recurrent 47.5 kb deletion that was probably not considered before because of the lack of evolutionary conservation among vertebrates, except for mammals (Skuplik et al., 2018). On the other hand there are several example in humans of non‐conserved and weakly conserved enhancer sequences (Blow et al., 2010; Chatterjee et al., 2011; Chen et al., 2008) suggesting that the functional conservation of genes across species is not necessarily associated with sequence conservation of their regulatory elements. Alternatively, we could speculate that these deletions might exert their pathogenic role through the alteration of the proper distance between the promoter and the downstream regulatory elements.
For deletions identified in patients #7, #8, #9, and #10, although absent in the tested controls, the significance remained uncertain as in at least the two available families they were inherited from the normal stature parents and no other similar unbalance had been previously reported in literature. They might represent low penetrance variations that exert a pathogenic effect in combination with a predisposing genetic background, that is not uncommon for micro‐rearrangements in the SHOX area (Bunyan et al., 2013).
In conclusion, this study provides further evidence for the presence of regulatory elements outside the already described regulatory regions. The array CGH provides an additional tool that supports the standard methods in the fine characterization of rare unbalances and might be useful to reanalyze patients that tested negative with standard methods but that are strongly suggestive of SHOX‐haploinsufficiency.
CONFLICT OF INTEREST
The authors have nothing to declare.
AUTHOR CONTRIBUTIONS
MG Conception and coordination of the work and drafted the article. SV, AM, AS, SB, FP: clinical assessment and management of the patient. AFa, AC, DB, WA: molecular analysis and aquisition of the data. SM, AFo interpretation of data. All authors contributed to the article and approved the submitted version.
ETHIC STATEMENT
This study was performed with a written, informed consent of the patient's parents.
Supporting information
Fig S1‐S2
Table S1‐S3
ACKNOWLEDGMENTS
The authors are grateful to all the patients and their families.
Fanelli, A. , Vannelli, S. , Babu, D. , Mellone, S. , Cucci, A. , Monzani, A. , Al Essa, W. , Secco, A. , Follenzi, A. , Bellone, S. , Prodam, F. , & Giordano, M. (2022). Copy number variations residing outside the SHOX enhancer region are involved in Short Stature and Léri‐Weill dyschondrosteosis. Molecular Genetics & Genomic Medicine, 10, e1793. 10.1002/mgg3.1793
Funding information
Fondi vari, Dipartimento di Scienze Mediche, Università del Piemonte Orientale.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available upon request from the corresponding author.
REFERENCES
- Babu, D. , Vannelli, S. , Fanelli, A. , Mellone, S. , Baffico, A. M. , Corrado, L. , Essa, W. A. , Grandone, A. , Bellone, S. , Monzani, A. , Vinci, G. , De Sanctis, L. , Stuppia, L. , Prodam, F. , & Giordano, M. (2021). Variants in the 5'UTR reduce SHOX expression and contribute to SHOX haploinsufficiency. European Journal of Human Genetics, in press. 29(1), 110–121. 10.1038/s41431-020-0676-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito‐Sanz, S. , Barroso, E. , Heine‐Suñer, D. , Hisado‐Oliva, A. , Romanelli, V. , Rosell, J. , Aragones, A. , Caimari, M. , Argente, J. , Ross, J. L. , & Zinn, A. R. (2011). Clinical and molecular evaluation of SHOX/PAR1 duplications in Leri‐Weill dyschondrosteosis (LWD) and idiopathic short stature (ISS). Journal of Clinical Endocrinology and Metabolism, 96(2), E404–E412. [DOI] [PubMed] [Google Scholar]
- Benito‐Sanz, S. , Royo, J. L. , Barroso, E. , Paumard‐Hernández, B. , Barreda‐Bonis, A. C. , Liu, P. , Gracía, R. , Lupski, J. R. , Campos‐Barros, Á. , Gómez‐Skarmeta, J. L. , & Heath, K. E. (2012). Identification of the first recurrent PAR1 deletion in Léri‐Weill dyschondrosteosis and idiopathic short stature reveals the presence of a novel SHOX enhancer. Journal of Medical Genetics, 49(7), 442–450. [DOI] [PubMed] [Google Scholar]
- Blaschke, R. J. , & Rappold, G. (2006). The pseudoautosomal regions, SHOX and disease. Current Opinion in Genetics & Development, 16(3), 233–239. 10.1016/j.gde.2006.04.004 [DOI] [PubMed] [Google Scholar]
- Blow, M. J. , McCulley, D. J. , Li, Z. , Zhang, T. , Akiyama, J. A. , Holt, A. , Plajzer‐Frick, I. , Shoukry, M. , Wright, C. , Chen, F. , Afzal, V. , Bristow, J. , Ren, B. , Black, B. L. , Rubin, E. M. , Visel, A. , & Pennacchio, L. A. (2010). ChIP‐Seq identification of weakly conserved heart enhancers. Nature Genetics, 42(9), 806–810. 10.1038/ng.650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum, W. F. , Crowe, B. J. , Quigley, C. A. , Jung, H. , Cao, D. , Ross, J. L. , Braun, L. A. , & Rappold, G. (2007). Growth hormone is effective in treatment of short stature associated with short stature homeobox‐containing gene deficiency: Two‐year results of a randomized, controlled, multicenter trial. Journal of Clinical Endocrinology and Metabolism, 92(1), 219–228. 10.1210/jc.2006-1409 [DOI] [PubMed] [Google Scholar]
- Bogin, B. , & Varela‐Silva, M. I. (2010). Leg length, body proportion, and health: a review with a note on beauty. International Journal of Environmental Research and Public Health, 7(3), 1047–1075. 10.3390/ijerph7031047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunyan, D. J. , Baffico, M. , Capone, L. , Vannelli, S. , Iughetti, L. , Schmitt, S. , Taylor, E. J. , Herridge, A. A. , Shears, D. , Forabosco, A. , & Coviello, D. A. (2016). Duplications upstream and downstream of SHOX identified as novel causes of Leri‐Weill dyschondrosteosis or idiopathic short stature. American Journal of Medical Genetics. Part A, 170A(4), 949–957. [DOI] [PubMed] [Google Scholar]
- Bunyan, D. J. , Baker, K. R. , Harvey, J. F. , & Thomas, N. S. (2013). Diagnostic screening identifies a wide range of mutations involving the SHOX gene, including a common 47.5 kb deletion 160 kb downstream with a variable phenotypic effect. American Journal of Medical Genetics. Part A, 161A(6), 1329–1338. [DOI] [PubMed] [Google Scholar]
- Bunyan, D. J. , Taylor, E. J. , Maloney, V. K. , & Blyth, M. (2014). Homozygosity for a novel deletion downstream of the SHOX gene provides evidence for an additional long range regulatory region with a mild phenotypic effect. American Journal of Medical Genetics. Part A, 164A(11), 2764–2768. [DOI] [PubMed] [Google Scholar]
- Cacciari, E. , Milani, S. , Balsamo, A. , Spada, E. , Bona, G. , Cavallo, L. , Cerutti, F. , Gargantini, L. , Greggio, N. , Tonini, G. , & Cicognani, A. (2006). Italian cross‐sectional growth charts for height, weight and BMI (2 to 20 yr). Journal of Endocrinological Investigation, 29(7), 581–593. 10.1007/BF03344156 [DOI] [PubMed] [Google Scholar]
- Chatterjee, S. , Bourque, G. , & Lufkin, T. (2011). Conserved and non‐conserved enhancers direct tissue specific transcription in ancient germ layer specific developmental control genes. BMC Developmental Biology, 11, 63. 10.1186/1471-213X-11-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H. P. , Lin, A. , Bloom, J. S. , Khan, A. H. , Park, C. C. , & Smith, D. J. (2008). Screening reveals conserved and nonconserved transcriptional regulatory elements including an E3/E4 allele‐dependent APOE coding region enhancer. Genomics, 92(5), 292–300. 10.1016/j.ygeno.2008.07.009 [DOI] [PubMed] [Google Scholar]
- Durand, C. , Bangs, F. , Signolet, J. , Decker, E. , Tickle, C. , & Rappold, G. (2010). Enhancer elements upstream of the SHOX gene are active in the developing limb. European Journal of Human Genetics, 18(5), 527–532. 10.1038/ejhg.2009.216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukami, M. , Kato, F. , Tajima, T. , Yokoya, S. , & Ogata, T. (2006). Transactivation function of an approximately 800‐bp evolutionarily conserved sequence at the SHOX 3′ region: Implication for the downstream enhancer. American Journal of Human Genetics, 78(1), 167–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukami, M. , Naiki, Y. , Muroya, K. , Hamajima, T. , Soneda, S. , Horikawa, R. , Jinno, T. , Katsumi, M. , Nakamura, A. , Asakura, Y. , Adachi, M. , Ogata, T. , & Kanzaki, S. (2015). Rare pseudoautosomal copy‐number variations involving SHOX and/or its flanking regions in individuals with and without short stature. Journal of Human Genetics, 60(9), 553–556. 10.1038/jhg.2015.53 [DOI] [PubMed] [Google Scholar]
- Genoni, G. , Monzani, A. , Castagno, M. , Ricotti, R. , Rapa, A. , Petri, A. , Babu, D. , Giordano, M. , Prodam, F. , Bona, G. , & Bellone, S. (2018). Improving clinical diagnosis in SHOX deficiency: the importance of growth velocity. Pediatric Research, 83(2), 438–444. 10.1038/pr.2017.247 [DOI] [PubMed] [Google Scholar]
- Hinch, A. G. , Altemose, N. , Noor, N. , Donnelly, P. , & Myers, S. R. (2014). Recombination in the human Pseudoautosomal region PAR1. PLoS Genetics, 10(7), e1004503. 10.1371/journal.pgen.1004503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschfeldova, K. , & Solc, R. (2017). Comparison of SHOX and associated elements duplications distribution between patients (Lėri‐Weill dyschondrosteosis/idiopathic short stature) and population sample. Gene, 627, 164–168. 10.1016/j.gene.2017.06.034 [DOI] [PubMed] [Google Scholar]
- Kant, S. G. , Broekman, S. J. , de Wit, C. C. , Bos, M. , Scheltinga, S. A. , Bakker, E. , Oostdijk, W. , van der Kamp, H. J. , van Zwet, E. W. , van der Hout, A. H. , Wit, J. M. (2013). Phenotypic characterization of patients with deletions in the 3'‐flanking SHOX region. PeerJ, 1, e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon, E. J. , McEwen, G. K. , Callaway, H. , & Elgar, G. (2011). Functional analysis of conserved non‐coding regions around the short stature hox gene (shox) in whole zebrafish embryos. PLoS One, 6(6), e21498. 10.1371/journal.pone.0021498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monzani, A. , Babu, D. , Mellone, S. , Genoni, G. , Fanelli, A. , Prodam, F. , Bellone, S. , & Giordano, M. (2019). Co‐occurrence of genomic imbalances on Xp22.1 in the SHOX region and 15q25.2 in a girl with short stature, precocious puberty, urogenital malformations and bone anomalies. BMC Medical Genomics, 12(1), 5. 10.1186/s12920-018-0445-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogushi, K. , Muroya, K. , Shima, H. , Jinno, T. , Miyado, M. , & Fukami, M. (2019). SHOX far‐downstream copy‐number variations involving cis‐regulatory nucleotide variants in two sisters with Leri‐Weill dyschondrosteosis. American Journal of Medical Genetics. Part A, 179(9), 1778–1782. [DOI] [PubMed] [Google Scholar]
- Ottesen, A. M. , Aksglaede, L. , Garn, I. , Tartaglia, N. , Tassone, F. , Gravholt, C. H. , Bojesen, A. , Sørensen, K. , Jørgensen, N. , Rajpert‐De Meyts, E. ,. , Gerdes, T. , Lind, A.‐M. , Kjaergaard, S. , & Juul, A. (2010). Increased number of sex chromosomes affects height in a nonlinear fashion: a study of 305 patients with sex chromosome aneuploidy. American Journal of Medical Genetics. Part A, 152A(5), 1206–1212. 10.1002/ajmg.a.33334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappold, G. , Blum, W. F. , Shavrikova, E. P. , Crowe, B. J. , Roeth, R. , Quigley, C. A. , Ross, J. L. , & Niesler, B. (2007). Genotypes and phenotypes in children with short stature: clinical indicators of SHOX haploinsufficiency. Journal of Medical Genetics, 44(5), 306–313. 10.1136/jmg.2006.046581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabherwal, N. , Bangs, F. , Röth, R. , Weiss, B. , Jantz, K. , Tiecke, E. , Hinkel, G. K. , Spaich, C. , Hauffa, B. P. , van der Kamp, H. , Kapeller, J. , Tickle, C. , & Rappold, G. (2007). Long‐range conserved non‐coding SHOX sequences regulate expression in developing chicken limb and are associated with short stature phenotypes in human patients. Human Molecular Genetics, 16(2), 210–222. 10.1093/hmg/ddl470 [DOI] [PubMed] [Google Scholar]
- Shima, H. , Tanaka, T. , Kamimaki, T. , Dateki, S. , Muroya, K. , Horikawa, R. , Kanno, J. , Adachi, M. , Naiki, Y. , Tanaka, H. , Mabe, H. , Yagasaki, H. , Kure, S. , Matsubara, Y. , Tajima, T. , Kashimada, K. , Ishii, T. , Asakura, Y. , Fujiwara, I. , Ogata, T. (2016). Systematic molecular analyses of SHOX in Japanese patients with idiopathic short stature and Leri‐Weill dyschondrosteosis. Journal of Human Genetics, 61(7), 585–591. 10.1038/jhg.2016.18 [DOI] [PubMed] [Google Scholar]
- Skuplik, I. , Benito‐Sanz, S. , Rosin, J. M. , Bobick, B. E. , Heath, K. E. , & Cobb, J. (2018). Identification of a limb enhancer that is removed by pathogenic deletions downstream of the SHOX gene. Scientific Reports, 8(1), 14292. 10.1038/s41598-018-32565-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, N. S. , Harvey, J. F. , Bunyan, D. J. , Rankin, J. , Grigelioniene, G. , Bruno, D. L. , Tan, T. Y. , Tomkins, S. , & Hastings, R. (2009). Clinical and molecular characterization of duplications encompassing the human SHOX gene reveal a variable effect on stature. American Journal of Medical Genetics. Part A, 149A(7), 1407–1414. [DOI] [PubMed] [Google Scholar]
- Tsuchiya, T. , Shibata, M. , Numabe, H. , Jinno, T. , Nakabayashi, K. , Nishimura, G. , Nagai, T. , Ogata, T. , & Fukami, M. (2014). Compound heterozygous deletions in pseudoautosomal region 1 in an infant with mild manifestations of langer mesomelic dysplasia. American Journal of Medical Genetics. Part A, 164A(2), 505–510. 10.1002/ajmg.a.36284 [DOI] [PubMed] [Google Scholar]
- Verdin, H. , Fernández‐Miñán, A. , Benito‐Sanz, S. , Janssens, S. , Callewaert, B. , De Waele, K. , De Schepper, J. , François, I. , Menten, B. , Heath, K,. E. , Gómez‐Skarmeta, J,. L. , & De Baere, E. (2015). Profiling of conserved non‐coding elements upstream of SHOX and functional characterisation of the SHOX cis‐regulatory landscape. Scientific Reports, 5, 17667. 10.1038/srep17667 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S2
Table S1‐S3
Data Availability Statement
The data that support the findings of this study are available upon request from the corresponding author.