

Abstract
Raman spectroscopy (RS) is used to analyze the physiochemical properties of bone because it is non-destructive and requires minimal sample preparation. With over two decades of research involving measurements of mineral-to-matrix ratio, Type-B carbonate substitution, crystallinity, and other compositional characteristics of the bone matrix by RS, there are multiple methods to acquire Raman signals from bone, to process those signals, and to determine peak ratios including sub-peak ratios as well as the full-width at half maximum of the most prominent Raman peak, which is nu1 phosphate (ν1PO4). Selecting which methods to use is not always clear. Herein, we describe the components of RS instruments and how they influence the quality of Raman spectra acquired from bone because signal-to-noise of the acquisition and the accompanying background fluorescence dictate the pre-processing of the Raman spectra. We also describe common methods and challenges in preparing acquired spectra for the determination of matrix properties of bone. This article also serves to provide guidance for the analysis of bone by RS with examples of how methods for pre-processing the Raman signals and for determining properties of bone composition affect RS sensitivity to potential differences between experimental groups. Attention is also given to deconvolution methods that are used to ascertain sub-peak ratios of the amide I band as a way to assess characteristics of collagen type I. We provide suggestions and recommendations on the application of RS to bone with the goal of improving reproducibility across studies and solidify RS as a valuable technique in the field of bone research.
Keywords: Raman spectroscopy, mineralization, crystallinity, collagen, carbonate, bone quality, fiber optics, polarization, secondary structure, collagen crosslinking
1. Introduction
The first published application of Raman spectroscopy (RS) to bone appeared in a short communication describing the acquisition of a Raman spectrum from defatted ox bone with a custom RS system1. There were only few RS studies of bone over the next two decades2–4 until commercial Raman instruments became widely available in the 1990’s. Since the publication of the first review article on the application of vibrational spectroscopy to the analysis of bone by Carden and Morris that covered RS studies published between 1994 and 19995, over 630 articles involving the use of RS to assess mineralized tissue, bone regeneration, and osteoblast differentiation appear in scientific journals and proceedings. Despite the ability of RS to provide insight into the contribution of composition to bone strength, there is not a comprehensive, systematic review of how the acquisition and processing methods of Raman spectra affect the compositional and chemical assessment of the bone extracellular matrix (ECM). Information on the analysis of bone by RS, namely Raman micro-spectroscopy (i.e., light passes through a microscope objective), is available in book chapters6,7 and in recent review articles8,9 including a comparison to Fourier transform infrared (FTIR) spectroscopy10, but such sources do not necessarily provide a practical guide on the best practices to use when assessing the physiochemical properties of bone by RS.
RS relies on a spontaneous light scattering that occurs in every ~1 out of 100 million photons6. Light interactions with a material cause chemical bonds to vibrate leading to two scattering phenomena: elastic (Rayleigh) and inelastic (Raman). Unlike the dominant elastic scattering in which there is no change in the energy of photons, the weak inelastic scattering of photons can have either higher (anti-Stokes) or lower (Stokes) energy than the energy of the incident photons (Fig. 1A). Making RS analysis of bone possible, the energy difference between the incident and Raman scattered photons is measurable as a shift in the wavelength of the light. Moreover, the amount of light (number of photons) undergoing a particular shift (change in energy) is also measurable. The shift characterizes the various chemical bonds that the light vibrates, and the number of photons characterizes the amount of the various chemical bonds within a volume of illumination. The outcome of Raman spectroscopy then is a graph of photon intensity in arbitrary units (a.u.) of multiple Raman peaks or bands as a function of the Raman shift, which is typically plotted as a wavenumber, 1/wavelength, between 300 cm−1 and 2000 cm−1 (known as the fingerprint region).
Figure 1: A schematic depiction of a Raman micro-spectroscopy instrument.
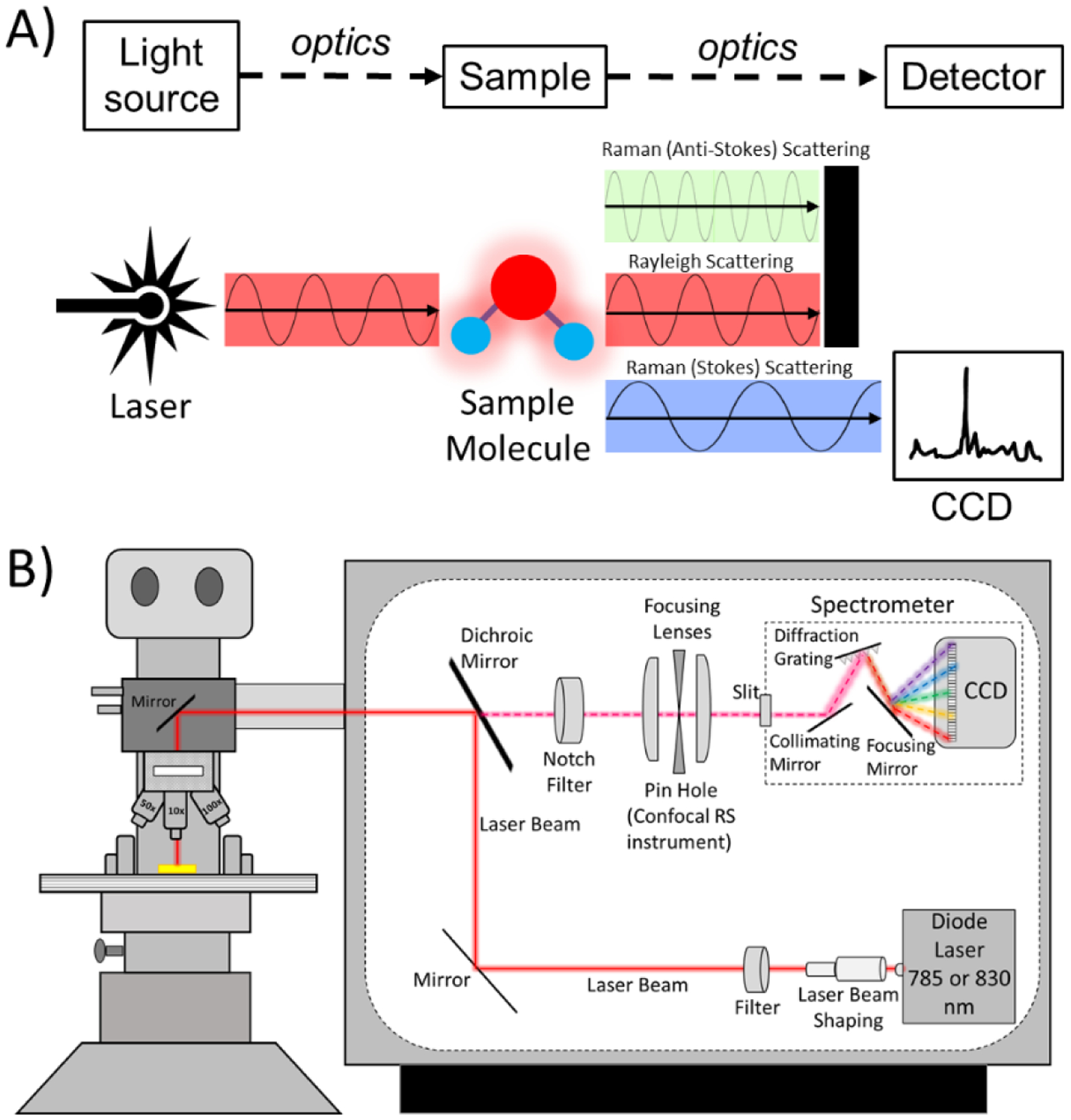
A) A Raman spectroscopy instrument has 4 primary components: a light source, a stage for the sample being analyzed, optics, and a detector. B) A commercial RS instrument includes mirrors, optical filters, focusing lenses, and objective lens to deliver the laser onto the sample and guide the collection of Raman scattered photons to the spectrometer (i.e., grating and detector, which is a charged-coupled device or CCD). The grating separates photons according to their wavelength in space so that the pixels of the CCD captures their intensity. Raman micro-spectroscopy has a confocality option which is provided by a pin hole aperture and slit. The optics of Raman micro-spectroscopy can preserve the polarization axis of the laser.
Compared to other analytical techniques that provide chemical and compositional characteristics of bone (e.g., transmission FTIR, quantitative backscattered electron microscopy, qBEI, and thermal gravimetric analysis, TGA), the advantages of RS include: minimal sample preparation, non-destructive analysis, in vivo capability11, specificity to molecular bonds that pertain to both the mineral phase and the organic phase of bone12, and sensitivity to collagen organization13 as well as water14. There are of course limitations in the ability of RS to assess the bone matrix: relative measurements without a straightforward way to obtain absolute values by incorporating appropriate reference materials in the analysis, weak photon signals necessitating long acquisition times without thermally damaging the tissue, background fluorescence that complicates the identification of the baseline of each peak, and numerous overlapping bands making it difficult to quantify unique features of the matrix (e.g., crosslinks, proteoglycans, lipids). Nonetheless, these limitations can be minimized to some extent as discussed herein.
When applied to bone, RS provides 3 primary measurements of the ECM (Table 1) as follows: 1) mineral-to-matrix ratio (MMR, often interpreted as the degree of mineralization15), 2) type B carbonate substitutions (often considered to be the amount of carbonate, , that has replaced phosphate, , in the crystal lattice causing distortion in the atomic arrangement of bone mineral16), and 3) crystallinity (an indicator of how well the bone mineral is arranged into a crystal structure, as opposed to being amorphous, and is related to crystal size17). There are however numerous peaks and overlapping bands in the Raman spectrum of bone such that other secondary characteristics of the ECM can be quantified (Table 2). Moreover, different methods for measuring the primary and secondary Raman properties of bone can be found in the literature because the quality of the acquired spectra and sensitivity of RS to differences in composition vary among RS instruments, whether from a commercial manufacturer or custom-built.
Table 1.
A list of primary Raman metrics along with their method of determination and characterization that influences how they are interpreted.
| Metric | Measurement | Method | Characterization | Ref |
|---|---|---|---|---|
| Mineral-to-matrix ratio | ν1PO4/amide I | Peak intensity ratio (PI) | MMR was significantly higher in interstitial vs. osteonal tissue, except when determined by ν1PO4/Amide I | 29 |
| ν1PO4/amide III | ||||
| ν1PO4/Proline | ||||
| ν1PO4/CH2-wag | ||||
| ν1PO4/(Pro + Hyp) | Integrated peak area ratio (IA) | Each MMR reciprocally correlated with ash fraction, R2 = 0.894 to 0.976a | 118 | |
| ν1PO4/amide I | ||||
| ν1PO4/amide III | ||||
| ν1PO4/Phe | ||||
| ν1PO4/CH2-wag | ||||
| ν2PO4/amide III | IA ratio | MMR directly correlated with the % weight of Ca from qBEI, R2=0.75 | 105 | |
| ν1PO4/Phe | PI ratio | Direct correlation between RS and FTIR, R2 = 0.8331 | 119 | |
| ν1PO4/amide I | PI & IA ratios | MMR linearly correlated with vBMD from μCT, r=0.709; Raman-PI ratio linearly correlated with IR-IA ratio | 120 | |
| ν1PO4/(Pro+Hyp)b | ||||
| ν1PO4/amide I | IA ratio | Assessed from the cortical bone of young rats, MMR increases with tissue age | 121 | |
| ν1PO4/amide I | IA ratio | Higher MMR but lower ash fraction in cortical bone from diabetic ZDSD rats vs. non-diabetic Sprague-Dawley rats | 122 | |
| ν1PO4/CH2-wag | ||||
| ν1PO4/amide I | PI ratio | Higher MMR in cortical bone from diabetic ZDSD rats vs. non-diabetic Sprague-Dawley rats but no difference in Ct.TMD between groups | 123 | |
| ν1PO4/Pro | ||||
| Carbonate substitution | ν1CO3/ν1PO4 | PI ratio | RS and FTIR did not correlate | 119 |
| ν1CO3/ν1PO4 | IA ratio | Carbonate-phosphate ratio highly positively correlated with carbonate percentage (R2 = 0.965) | 55 | |
| ν1CO3/ν1PO4 | IA ratio | Carbonate in HA of mouse cortical bone significantly increased by 6.4%, on average, from 4–5-mo. to 19-mo. | 124 | |
| ν1CO3/ν1PO4 | PI ratio | Carbonate in HA of rat cortical bone significantly increased by 1.75%, on average, from 12-mo. to 24-mo. and was higher within the outer tissue than within the inner tissue of the midshaft | 125 | |
| Crystallinity | FWHM[ν1PO4]−1 | Maximum intensityc | Crystallinity directly correlated with c-axis of the crystal as determined by X-ray diffraction of human bone, buffalo enamel, boar tusk, and HA, R2 = 0.992 | 126 |
| FWHM[ν1PO4]−1 | ν1PO4 peak = 1 | Crystallinity of mouse cortical bone significantly increased by 1.9%, on average, from 4–5-mo. to 19-mo. | 124 | |
| FWHM[ν1PO4]−1 | Peak intensity | Crystallinity increased as mouse age increased: 4-wk, 10-wk, and 24-wk; fluoridation reduced crystallinity | 127 | |
| FWHM[ν1PO4]−1 | Peak intensity | Direct correlation between RS and FTIR, R2 = 0.8589 | 119 | |
| FWHM[ν1PO4]−1 | Maximum intensityd | High correlation of ν1PO4 peak position R2 = 0.92 and ν1PO4 FWHM R2 = 0.80 with apatite crystallite size assessed by XRD | 17 | |
| FWHM[ν1PO4]−1 | Peak intensity | Crystallinity decreases in femoral cortical bones of the rats with chronic kidney disease (CKD) owing to the increased Mg/Ca ratio in the hydroxyapatite | 128 |
y = C/(100-x); range identified by local minima
958 cm−1 / [917 + 873 + 851 cm−1]
Fitted Gaussian curves to the upper 2/3rds of ν1PO4 peak and determined FWHM of the Gaussian curve at 960 cm−1
Fitted one Lorentzian curve to ν1PO4 peak (baselined 920 to 990 cm−1) and determined FWHM and band position
Table 2.
A list of secondary Raman metrics along with their method of determination.
| Metric | Measurement | Method | Characterization | Ref |
|---|---|---|---|---|
| Pentosidine | Peak at 1305 cm−1 or 1362 cm−1/amide I | Likely PI ratio | Pentosidine to matrix ratio increased in the cortical bone of rats following thyroparathyroidectomy (TPTx) and partial nephrectomy (Nx) compared to TPTx only rats | 129 |
| Band at ~1495 cm−1/CH2-wag | IA ratio | Ratio increased implying diabetic mice had greater pentosidine (p = 0.017) content on the periosteal surface as compared to non-diabetic mice; propagation toughness correlated inversely with pentosidine (diabetic: r = −0.97, p = 0.001 and non-diabetic: r = −0.94, p = 0.005)a | 130 | |
| CML | Band at ~1150 cm−1/CH2-wag | IA ratio | Diabetic mice had greater carboxymethyllysine (CML) (p = 0.003) content compared to non-diabetic mice; propagation toughness correlated inversely with CML (diabetic: r = −0.99, p < 0.001 and non-diabetic: r = −0.94, p = 0.005)a | 130 |
| Proline hydroxylation | Hyp/Pro | PI ratio | Ratio significantly increased (by ~10%) in the osteolytic samples than healthy controls | 131 |
| Lipid content | Band at ~1298 cm−1/amide III | IA ratio | Analyzing iliac crest biopsies, relative lipid content was lower in older tissue than in youngest tissue (near active osteoblasts), irrespective of menopausal status; directly correlated with osteoblast surface per bone surface (r=0.685, p=0.042) in only the premenopausal group. | 132 |
| Proteoglycan content | 1365–1390 cm−1/ν2PO4 and 1365–1390 cm−1 / amide III | IA ratio | Proteoglycan content normalized to mineral was significantly higher in both idiopathic osteoporosis and idiopathic low BMD group than in control group (p<0.01). Proteoglycan content normalized to organic matrix was not significantly different among the three study groups | 133 |
See Table 3 for correlations between other Raman properties and mechanical properties of bone.
Therefore, the goal of the present review is to describe the key components in RS that affect sensitivity and review RS methods commonly reported in the bone literature. In doing so, we provide the best available evidence for i) sample preparation, ii) selecting the signal acquisition parameters, iii) pre-processing the spectral data, iv) ensuring consistent spectra in which Raman peaks are not masked by noise, and v) calculating the Raman-based measurements of bone. In addition, we describe difficulties commonly encountered and offer suggestions on how best to inform others about the RS methods that were used to assess bone. Special emphasis is given to Raman micro-spectroscopy because most published RS studies of bone used a commercial research-grade RS instrument integrated with a microscope. Advantage of using a microscope include: providing sub-micrometer lateral spatial resolution which allows the identification of small features of bone such as lamellae to analyze and acquiring compositional maps in a specific region. In Section 6, we discuss fiber optic, probe-based RS instruments, which are more versatile than commercial instruments, as well as discuss high wavenumber RS. By describing the extrinsic factors that affect the ability of RS to provide useful information about bone tissue and explaining the methods that aid in the quality and rigor of the RS data, the unmet potential of RS in bone research may be realized.
2. Raman spectroscopy instruments
Practical applications of RS require an instrument configuration that causes Raman scattering within a material and acquires the weak Raman signals. At its most basic, a Raman instrument has a light source, optics to guide the light to the sample being analyzed, a stage to secure the specimen, and optics to guide the inelastic scattered light to a detector that generates the Raman spectrum (Fig. 1A). The present work primarily focuses on spontaneous dispersive RS which is commonly applied to the compositional assessment of bone. Commercial RS instruments ensure that optics are aligned with the laser source, provide calibration, and offer software to both acquire the Raman spectra and process it. Nonetheless, a basic understanding of the components and the calibration of the instrument helps the user identify when something is wrong with the acquisition of the Raman signal.
2.1. Components of a Raman instrument
In a typical commercial RS instrument, the light or excitation source is a diode laser; the detector is a spectrometer; the sample is placed on a microscope stage below an objective lens; and the optics include mirrors, beam expander, lenses, and filters (Fig. 1B). All these components are essential to maximize the collection of weak Raman signals, thereby maximizing the sensitivity and selectivity of the RS assessment technique.
2.1.1. Light Source:
The sensitivity of Raman spectroscopy to differences in composition depends on the power of the laser (i.e., how much light is interacting with chemical bonds) and the wavelength (λ) of the laser18,19. Diode lasers are a typical light source in commercial Raman instruments due to their ability to stably convert electrical current into light and relative inexpensiveness18,20. Other light sources, especially in previous versions of RS, include argon-ion (Ar+), krypton ion (Kr+), helium–neon (He-Ne), Titanium–sapphire (Ti:sapphire), Neodymium-doped yttrium aluminum garnet (Nd:YAG) lasers. A laser with a short wavelength has higher intensity of light scattering, hence greater Raman intensity and sensitivity than a laser with a long wavelength (1/ λ4 dependence) but also higher fluorescence background20,21. On the other hand, a laser with a long wavelength has higher penetration depth within the samples being analyzed. For RS analysis of bone, the choice of laser wavelengths typically ranges from mid-ultraviolet (mid-UV: 200 nm > λ > 300 nm) to near-infrared (NIR: ~700 nm < λ < 1400 nm), but 785 nm and 830 nm lasers are commonly applied to bone because many fluorophores are inactive when excited with NIR lasers18,19,21. These laser wavelengths minimize background fluorescence, minimize overheating the sample, and optimize the volume being analyzed18,22.
While increasing the laser power improves the quality of the Raman spectrum (Fig. S1), higher power also increases the likelihood of burning the bone sample being analyzed (Fig. S2). Therefore, practically speaking, laser power is kept below 50 mW for mid-UV lasers and below 150 mW for NIR lasers. Laser power herein is the intensity of light deposition measured at the sample surface using a meter. The power of the laser at the source is typically different than the laser power at the sample surface because laser power decreases when the light travels through various optical components such as filters, lenses, and gratings. The change in laser power between source and sample depends on the distance that the light travels and the number of optical components that the light passes through. It is important to note though that heat generation is not simply a function of the laser power but rather depends on the total laser energy density (the total amount of energy delivered per unit area, in Joules per square centimeter (J/cm2)) and the time that the specimen is exposed to the laser. The energy density of the laser depends on the illumination spot size, a function of the objective NA, if using an RS instrument with a microscope, and how the light is emitted from the diode laser (i.e., as a circular beam or as a line in which the beam is swept back and forth at a set distance). Line focus has the advantage of collecting Raman scattered photons from a larger area, thereby reducing heat deposition on the sample, and is good for mapping composition in a selected region. The disadvantage is that the signal quality is lower than it is for the circular beam or spot focus.
A laser with lower wavelength provides higher signal intensity, but as a trade-off, more background fluorescence must be removed (discussed in Section 4.2). Therefore, depending on the goals of a study (e.g., whether numerous fluorophores are present in the sample), an 830 nm laser can help minimize fluorescence23. Otherwise, 785 nm laser may be a more suitable choice as it favors higher peak intensities for nearly the same noise as observed when the 830 nm laser is the excitation source (Fig. S3). Although they generate greater signal intensity, 532 nm and 633 nm lasers should be used with caution since they may readily damage the bone specimen. Lasers with λ=1064 nm are now more widely available but costly. Thus, they are less commonly used option for when high penetration depth with low autofluorescence is desired24.
2.1.2. Optics:
The microscope in a Raman instrument provides the ability to focus the incident light at a desired site and optical magnification of the inelastically scattered light as well as direct visualization of the bone surface when using white light. Providing superior spatial resolution due to confocality (i.e., acquiring Raman signals from a focus plane), commercial RS instruments can collect single-point spectral data from a small area (typically 0.5 μm to 5 μm) as well as maps of spectra covering a defined area (typically 100 μm × 100 μm). The penetration depth of the light in biological tissue is typically 100 μm, mainly dictated by the turbidity/scattering properties of the tissue, not the axial (longitudinal) focusing & penetrating power of the objective lens. The spatial resolution of the RS instrument depends on the diffraction limit of the objective lens, which cannot resolve the physiochemical properties of two adjacent objects located closer than λ / 2 × NA, where λ is the wavelength of the laser and NA is the numerical aperture. Accordingly, the objective lenses with higher NA (higher magnification) provide higher spatial resolution along with a shorter working distance and more sensitivity to polarization bias (as discussed in the section of 2.4). Conversely, the objective lenses with lower NA (lower magnification) provide lower spatial resolution along with longer working distance and less sensitivity to polarization bias (discussed in Section 2.3).
In addition to reducing ambient light to the lowest levels possible, a key step to collecting Raman signals with high signal-to-noise ratio (SNR) is to properly focus the laser on the specimen (Fig. 2). This is a concern when a microscope is part of the instrument but not when a fiber optic probe is the conduit of light (see Section 6). With the aperture reduced to reveal a spot of white light on the bone surface, the focus knobs can be turned until a sharp border appears. Next, after opening the aperture back up, the light passing through the objective is switched from the lamp emitting white light to the laser source. Since both light sources are passing through the same magnification lens, the laser is in-focus. Focusing the laser on the bone surface does not mean however that the collected Raman spectra represent chemical composition at the bone surface only. Even when a pinhole aperture is providing confocality (see section 2.1.3), Raman spectra include signals arising several hundred microns below the bone surface.
Figure 2. Effect of focusing on peak intensity.
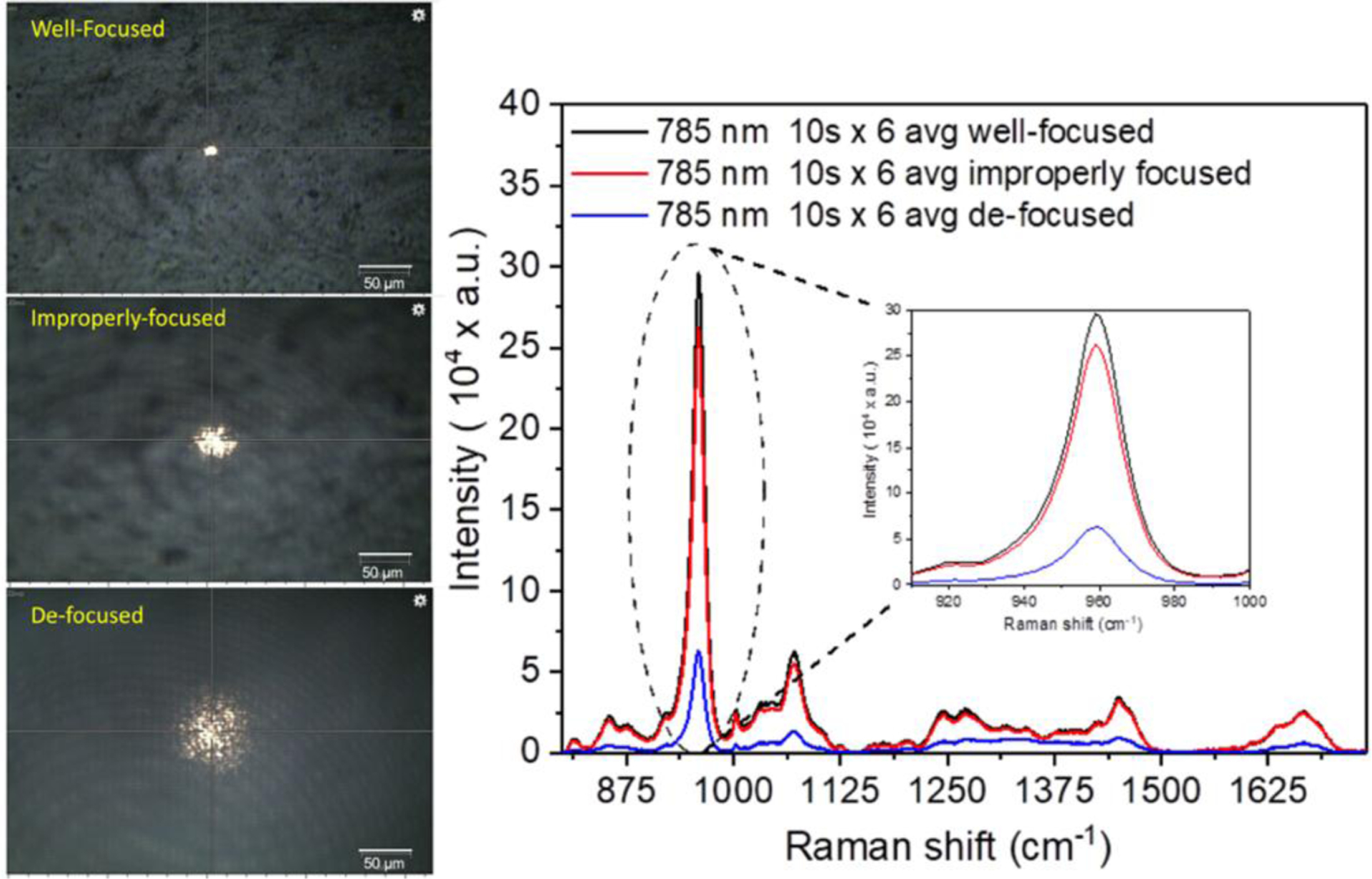
Raman spectra were acquired from a human cortical bone using a 785 nm laser source under a well-focused laser (black), improperly focused laser (red) and defocused laser (blue). When the laser is not properly focused onto the unpolished bone surface, the intensity of peaks such as ν1PO4 (inset) is considerably lower.
2.1.3. Detector (Spectrometer):
A dispersive spectrometer is an optical system in RS instruments, such as the Renishaw inVia™ and Horiba XploRA™ Raman microscopes, that split the Raman scatter (i.e., photons at different wavenumbers) in space onto the detector. The spectrometer has a high-quality diffraction grating with an entrance slit aperture and spherical mirrors that collimate the Raman scattering light onto the diffraction grating such that this light is focused onto the detector (Fig. 1B). Before the spectrometer, a pinhole aperture may be present to provide confocality. The number of grooves per length (g/mm) of a diffraction grating governs the amount of dispersion such that a higher density of grooves causes a higher dispersion of the Raman shift over a larger number of pixels of the detector25,26, hence higher spectral resolution (cm−1). The groove density is typically fixed but selecting a groove density may be an option when purchasing a commercial RS instrument or a commercial spectrometer for a custom-built RS instrument. There is a trade-off in which increasing the groove density improves spectral resolution (e.g., 1 cm−1 vs. 8 cm−1) but lowers wavenumber coverage of the acquired spectrum (e.g., 750–1750 cm−1 vs. 300–2300 cm−1).
The entrance slit aperture – as well as the preceding pinhole in confocal RS instruments – is an opening that controls how much Raman scattered light enters the spectrometer. It typically ranges from 10 μm up to 200 μm with a height of 1 mm. Commercial RS instruments include adjustable slit apertures or pinholes. This slit size (or width) is a key factor in governing the spectral resolution. As the slit narrows, spectral resolution increases unless the slit size is lower than the pixel size (width) of the detector18,25. Notably, a narrow-slit decreases Raman signal strength because fewer photons are passing through the grating. Therefore, the slit size is selected to balance between strong spectral intensity and high spectral resolution.
As the last part of the spectrometer, the detector in modern RS instruments is a charged-coupled device (CCD)20. The pixel size of the CCD also determines the spectral resolution such that the achievable spectral resolution increases as the pixel size decreases. The performance of a CCD detector in creating a high-quality spectrum (high SNR) is related to the efficiency of the chip in detecting photons and the processing speed of readout electronics18,27.
2.1.4. Sample:
Prior to turning on the laser and acquiring the Raman signals that are generated by light interactions with molecules in bone, there are several practices that can improve the quality of the Raman spectrum. One, the ex vivo bone specimen can be cleaned by wiping the surface with a Kimwipe®, sonicating the bone sample, and/or irrigating the surface with a stream of water to remove particle contamination. Two, keeping the sample holder and microscope objective lens clean also minimizes noise in the acquired signal. Three, analyzing thick bone specimens is another way to avoid ‘contamination signals’ from materials near the bone. Four, if the surface of the bone sample can be ground and polished, doing so increases the number of photons that are reflected back to the microscope objective and hence boosts signal-to-noise (SNR). Long bones such as the femur from small animals (e.g., mice) are amendable to direct RS analysis, but care must be taken to properly focus the light (Section 2.1.3) since the surface is not flat. If the study question requires Raman mapping of properties or specific locations, then small bones must be embedded in plastic and sectioned. Embedding media and ethanol to dehydrate the bone for embedding or long-term storage are factors that affect peak ratios when using RS28,29, but as discussed later (Section 5), they do not necessarily obscure differences in matrix characteristics among groups. If embedding the bone for RS is unavoidable, we recommended grinding and polishing the surface of interest to improve the signal and selecting a media whose Raman peaks do not overlap peaks of interest.
2.2. Importance of Calibration
Proper calibration is essential to acquiring comparable and consistent Raman spectra. Without it, the locations of Raman peaks could deviate from those reported in the literature, and the relative intensities of selected peaks could be incorrect. Fortunately, the calibration procedure is done at the factory prior to the delivery of a commercial RS instrument and typically checked by the service engineer upon installation at a research site. Briefly, there are two general calibration procedures: spectral (x-axis) calibration ensures all pixels on the CCD correspond to actual wavenumbers, and intensity (y-axis) calibration ensures that the spectrometer response is dependent on the intensity of the Raman scattering. In particular, such calibration procedures are vital for any custom-built RS instruments to make sure the collected spectra are correct and reliable24. For full calibration procedures of custom-built RS instruments, we refer the interested reader to the following papers20,21,30,31. Herein, we describe ways to ensure a commercial Raman instrument is properly operating.
There are 8 common chemicals that have been established for calibrating the Raman shift (x-axis) by the American Society for Testing and Materials (ASTM). They include naphthalene, sulphur (sulfur), 1.4-bis(2-methylstyryl) benzene, acetonitrile/toluene, N-acetyl-para-aminophenol (i.e., acetaminophen or paracetamol), benzonitrile, cyclohexane and polystyrene (ASTM E1840)19. Periodically, Raman spectra can be acquired from one of these standards to verify that the location of the peaks match reported wavenumbers (Fig. S4). Additionally, if the location of a prominent peak such as ν1PO4 (959–961 cm−1) for bone changes by more than 2 wavenumbers in a commercial, research-grade RS instrument, then the calibration could be off necessitating a service call to the manufacturer of the instrument.
Built-in software automatically calibrates the y-axis (intensity) of the spectrum by implementing the mathematical manipulation derived from the calibration procedure at the factory21,32. Replacing or adding optical components (e.g., objective lens, notch filter, polarizer) can affect this intensity calibration (or instrument response function), and so changes to the instrument may require a new calibration to correct for the wavelength-dependent sensitivity of all optical components in a Raman system. Commercial RS instruments often have sensors to indicate when a new calibration of intensity axis is needed.
Some commercial RS devices have integrated a neon lamp source to automatically re-calibrate wavelength axis if necessarily. A sample of silicon (often provided by the manufacturer) can be also used to quickly check the spectral calibration of the instrument if there is no automated wavelength calibration option. For a well-calibrated, research-grade RS instrument, the silicon standard has one sharp peak at 520 cm−1 (Fig. 3A). A wavenumber shift in this peak indicates a possible issue with the spectral calibration (Fig. 3B), but the software of commercial Raman instruments can be used to correct an offset in the silicon peak of several wavenumbers. The peak height (signal intensity) can also be examined for a potential y-axis calibration issue. If there is a substantial change in the intensity (i.e., spectral counts) from previously collected silicon spectra under similar conditions, then a problem with the instrument may exist such as misaligned optics, faulty laser, or improper focus. We recommend routinely collecting silicon spectra before and after RS analysis of bone so that the signal intensity at 520 cm−1 can be tracked overtime. For custom-built RS instruments with a low-grade spectrometer (i.e., a grating with a low number of groves per length), a 3 cm−1 to 4 cm−1 shift in Raman peak locations can be expected. The power of the laser at the surface of the sample for a given objective lens can also be readily measured using a meter before and after each session of data acquisition. Again, if there is a significant change in the laser power under similar conditions from a previous session, then there may be a problem with the diode laser itself, which has a finite life, or with the alignment of the laser between excitation source and the microscope lens.
Figure 3. Effect of improper calibration of the Raman shift axis on peak locations.
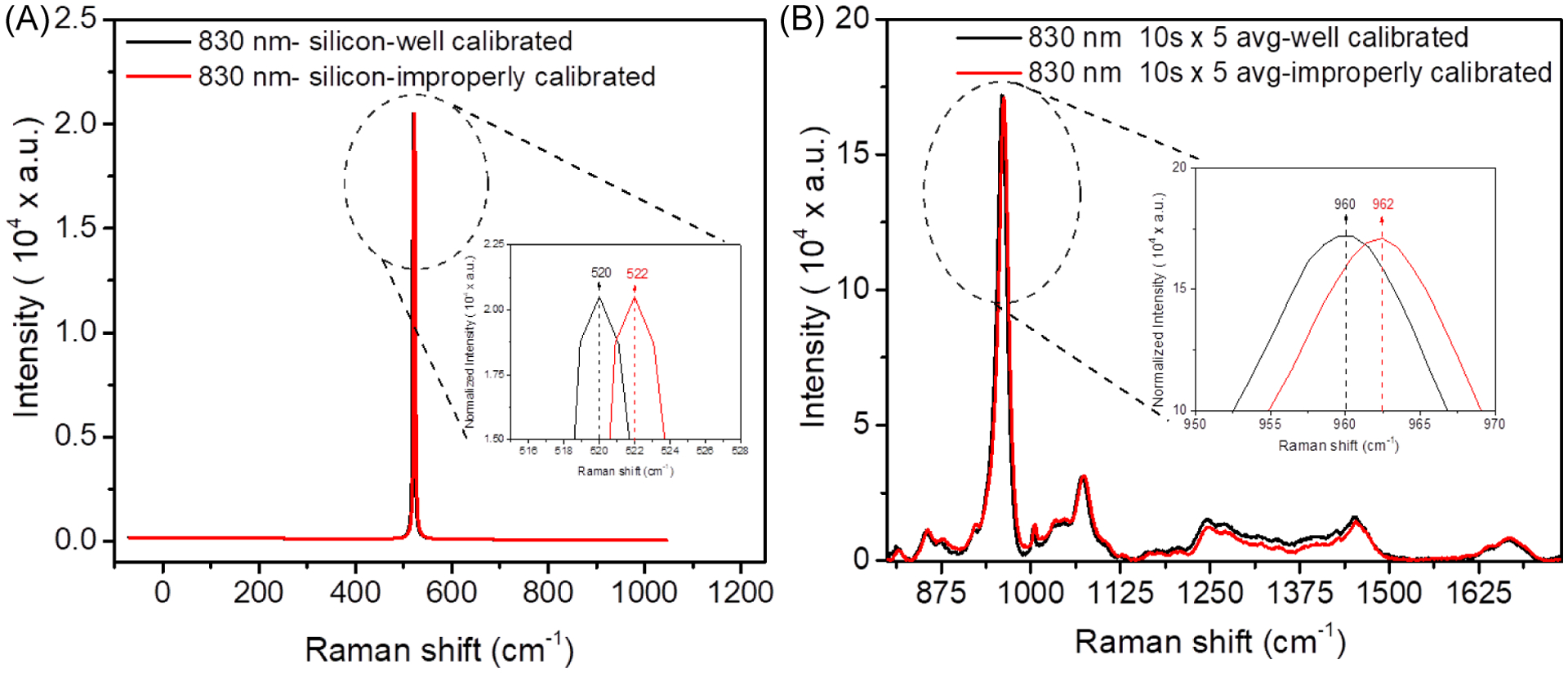
Raman spectra were acquired from silicon using an 830 nm laser source before (red) and after (black) calibration (B). When a commercial research-grade RS instrument is not properly calibrated, the wavenumber location of peaks such as ν1PO4 (inset) are shifted from their known location (B).
2.3. Polarization bias
Diode lasers are inherently polarized meaning they emit light with a preferential orientation (Fig. 4A), and this orientation is typically preserved by the optics of most commercial RS instruments. Since bone is a birefringent material, the relative height of the mineral peaks and organic peaks in Raman spectra depends not only on composition but also on the orientation of the collagen fibrils (or c-axis of the mineral crystals) relative to the polarization axis of the Raman instrument (Fig. 4B). Despite the exploitation of this phenomenon in RS analysis of bone in order to assess collagen orientation13,33–37, the dependence of peak ratios on the orientation of the bone sample relative to the polarization axis of the instrument (known as polarization bias) is typically ignored or not discussed in the vast majority of bone studies utilizing RS. As previously described in detail34,36, the Raman spectrum of bone, especially the intensities of amide III (1247–1248 cm−1) and amide I (1666–1670 cm−1) peaks, is not the same when the orientation of the human bone sample (e.g., axial direction of the osteons) is rotated 90 degrees from being parallel to being perpendicular to the polarization axis of the laser (Fig. 4B). Since polarization bias affects the intensity of each peak differently, peak ratios vary between different orientations of the bone (Fig. 4C).
Figure 4. Effect rotating bone 90° relative to the polarization axis of the laser on Raman peaks of human cortical bone.
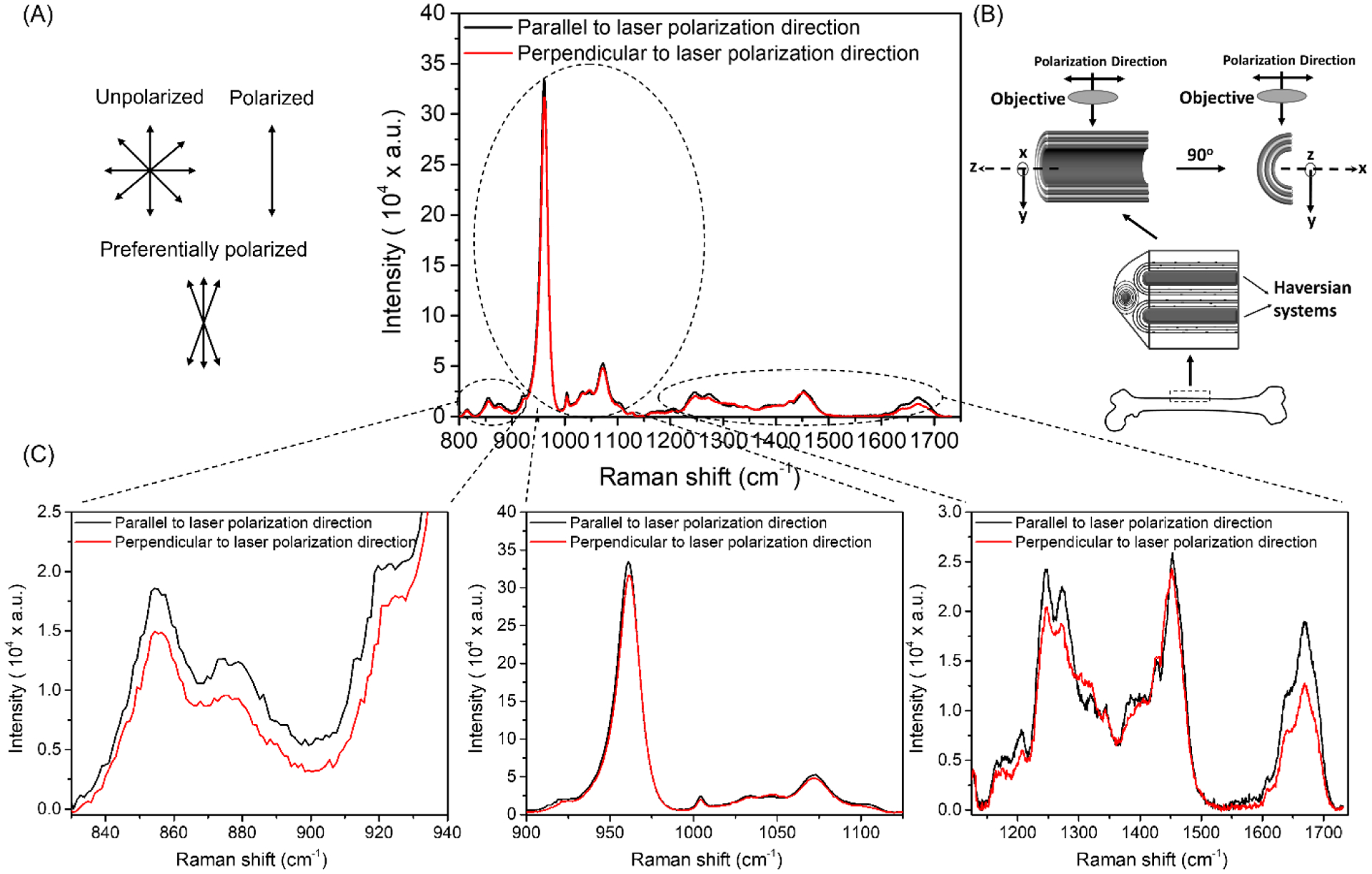
The diode laser is preferentially polarized such that the orientation of the light waves is narrowly distributed about an axis unlike unpolarized light or fully polarized light (A). By knowing the direction of the polarization axis of the laser, a bone sample can be rotated 90° such that the orientation of the osteons is parallel (black) or perpendicular (red) to the polarization direction (B). The Raman peaks of cadaveric cortical bone are higher when the osteons are parallel than when they are perpendicular to the polarization direction, but the change in height upon rotation is not the same across all peaks (C).
There are multiple ways to minimize the influence of polarization bias on Raman spectra of bone. One, by knowing the direction of the polarization axis of the RS instrument and consistently orienting the bone samples parallel to this axis, differences in peak ratios will primarily be due to differences in composition, not differences in the directionality of collagen fibrils. Two, homogenizing the tissue removes the birefringent nature of bone as the orientation of collagen fibrils becomes random (isotropic), though this precludes mechanical testing of bone after Raman analysis. Three, including optics that depolarize the light before acquisition by the spectrometer removes the instrument’s polarization bias, but not the bone orientation bias. However, there is significant cost in SNR with this approach. Four, acquiring the spectra with a low magnification objective is less sensitive to polarization bias than acquiring spectra with a high magnification objective38. Also, when acquiring Raman spectra from cross-sections of bone in which the direction of the osteons is in-line with the laser, the acquisition of Raman spectra from multiple sites can ‘average out’ the variance caused by polarization bias. Lastly, there are peak ratios that are less sensitive to polarization bias such as v2PO4/Amide III36 and v1PO4/Proline34 because the peaks are in-phase.
3. Acquisition of Raman spectra from bone
Along with laser power per area, integration time of the laser on the bone specimen can be manipulated to improve SNR. Likewise, adjusting the accumulation of spectra during an acquisition affects the quality of the Raman signal intensity. Generally speaking, the parameters for acquiring Raman spectra from bone are optimized to provide the highest SNR in a short amount of time while keeping laser power minimal. Therefore, we discuss the effects of integration time and accumulation using relatively low laser power (45 mW) on the quality of bone spectra along with providing guidance in the acquisition of the Raman signal from bone.
3.1. Acquisition parameters – accumulation vs. time
Since Raman scattering generates a weak signal, a long acquisition or scan time is necessary to enhance SNR. In setting the acquisition time, there is a balance between the quality of the acquired spectra and the time spent acquiring spectra. Two basic strategies include longer acquisition time with fewer accumulations and shorter acquisition time with greater accumulations. Selecting which strategy to use is not straightforward as both can generate high quality spectra depending on the sample being analyzed and the RS instrument being used. Practically speaking, a short acquisition time of a few seconds is a good place to start when optimizing an acquisition protocol for a given bone sample. Then, the number of accumulations can be increased until the desired quality of the Raman spectrum is achieved in the time that is perceived to be reasonable (Fig. S5). To provide an example of how spectral quality can be affected by different acquisition parameters when using a commercial, research-grade Raman spectroscopy such as the Renishaw InVia Raman microscope instrument, we varied the scan time and the number of accumulations keeping the total time of acquisition constant (Fig. 5). Given the fact that composition of bone is heterogeneous, as many spectra as is practically possible can be collected at multiple sites within region of interest to capture an overall average.
Figure 5. Effect of acquisition time and number of accumulations on Raman spectrum of bone.
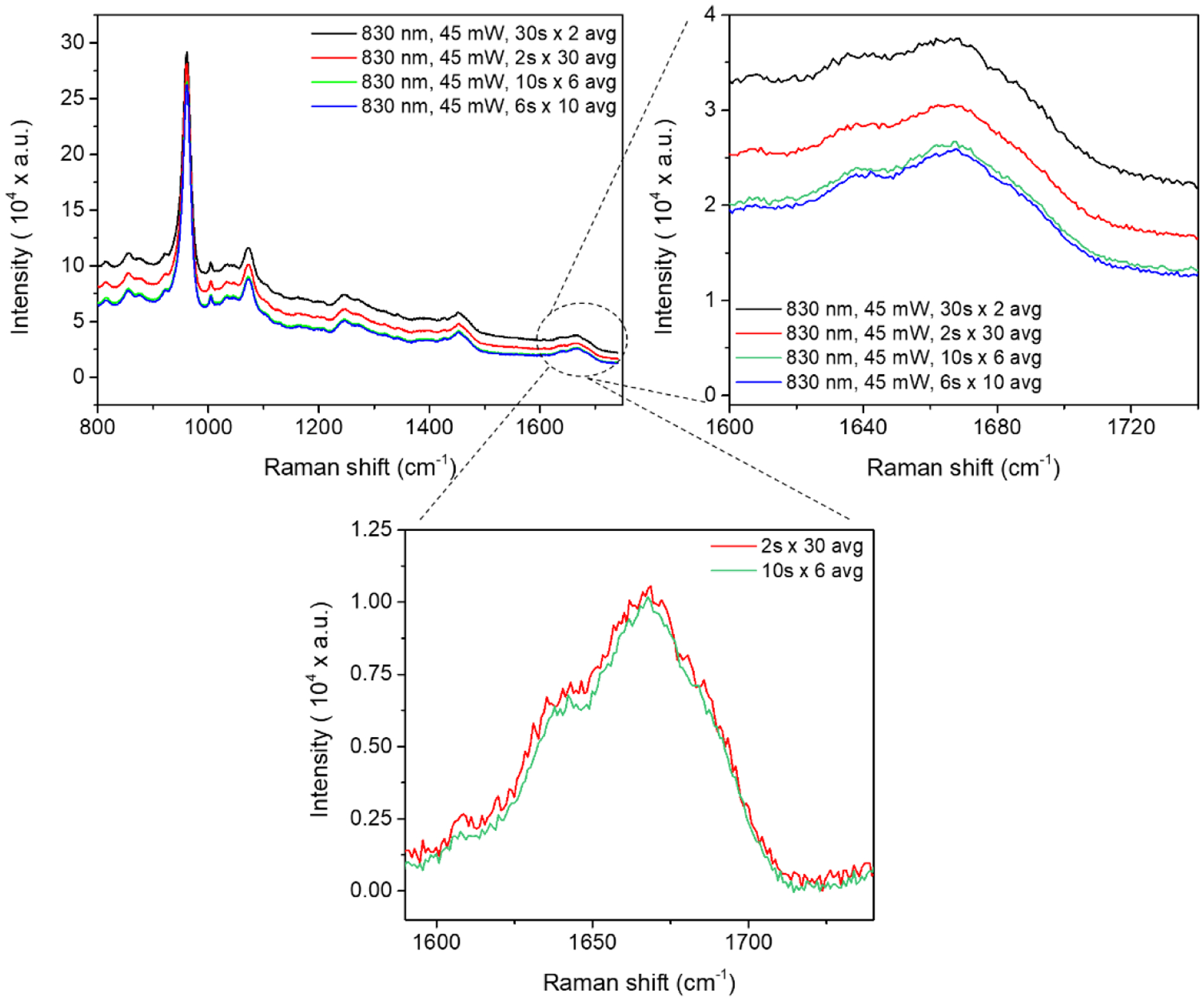
Raman spectra were acquired from the native unpolished human cortical bone obtained from a cadaveric femur using an 830 nm laser source while keeping the total acquisition time constant (e.g., scan time × number of accumulations = 60 s) but different combinations of scan time × number of accumulations. The spectral resolution of the Raman instrument was 1 cm−1.
4. Processing Raman spectra of bone
Although minimal pre-processing is the best way to characterize bone samples, the initial quality of the collected Raman spectra often does not reach sufficient SNR and negligible fluorescence background. Therefore, before calculating the physiochemical properties of bone, the raw Raman spectra are processed so that cosmic spikes are not present, the fluorescence background is negligible, and the spectral noise does not overly influence peak ratios or identification of peaks. There is not a standard method for removing the fluorescence nor is there one way to minimize noise because each RS instrument – commercial or custom-built – generates its own unique Raman spectra. Unfortunately, the selected pre-processing procedures can lead to erroneous results as demonstrated by Bocklitz et al. for 3 datasets – mixture of 4 liquids (ethanol, 2-propanol, DMSO, 1-octanol), 3 ratios of glucan (Saccharomyces cerevisiae) to chitin (crab shell), and 2 bacteria cultured for 1–10 days – using custom RS instruments39. However, by maximizing signal quality and applying consistent methods, differences in matrix characteristics among groups can be detected. When not applying advanced computer algorithms to determine processing methods, the valleys of the spectra (i.e., consecutive wavenumbers having intensity values near zero) are fit with a curve (e.g., polynomial, linear, or piecewise functions) that then serves as the baseline of each Raman peak. Upon subtracting the baseline curve from the raw spectrum, an algorithm is then applied to the intensity counts vs. Raman shift data to digitally ‘smooth out’ the spectrum. While this improves the identification of sub-components of Raman bands and the location of each Raman peak, it can mask subtle peaks arising from molecules with low density in bone. Here, we describe i) the most common methods used to process Raman spectra of bone and to calculate Raman metrics of bone and ii) the effect of processing methods on sensitivity to detecting differences in the mineral-to-matrix ratio, type B carbonate substitutions, crystallinity, and amide I sub-peak ratios. For an in-depth information on pre-processing methods (cosmic spike removal, background correction, smoothing, and normalization), the interested readers are referred to these papers30,39–41.
4.1. Subtracting background fluorescence
Bone tissue like other biological samples contains fluorophores that generate fluorescence upon exposure to a laser as previously discussed (section 2.2). This fluorescence is often several orders of magnitude more intense than the Raman signals. Thermal fluctuations within the CCD can also contribute to background signals. Thus, the background signal of a Raman spectrum is dependent on both the RS instrument used and the samples being analyzed. In most cases, removal of background signals (fluorophores + CCD) is achieved by applying mathematical functions to the raw Raman spectrum so that the Raman signals become prominent. In the event that background fluorescence overwhelms the weak Raman signals (e.g., when the bone is subjected to thermal treatment42 or to excessive accumulation of advanced glycation end-products (AGEs)23 or illuminated by a 532 nm laser43) or even saturates the ability of CCD to collect distinguishable signals (Fig. S6), preparation techniques can be used. In the past, it was reported that bleaching the surface of bone with 30% hydrogen peroxide for 2 to 4 h prior to Raman acquisition reduces background signals due to fluorescence43 but at the risk of altering the chemical composition of bone44. Therefore, this approach is not recommended. Photobleaching on the other hand takes advantage of the inherent instability in the molecular structure of fluorophores subjected to constant excitation. That is, when illuminated for an extended period of time by high-intensity light source, fluorophores stop emitting fluorescence. Therefore, by illuminating the bone surface with a laser for 5 min to 10 min, the photobleaching phenomenon reduces the background signal during the subsequent Raman acquisition45,46. Of course, there is a limit to how long the bone surface can be illuminated to cause photobleaching because the heat generated by the laser can damage the tissue (Fig. S2).
In general, polynomial curve fitting techniques47–49 are the most widely used method to remove background fluorescence from Raman spectrum of bone (Fig. S7) and other biological tissues. Commercially available software in spectroscopy implements various semi-automated techniques to eliminate the background fluorescence (e.g., Wire, LabSpec, GRAMS, Origin, and Opus software). In most cases, the user selects the polynomial order. The user may also be able to select the wavenumber range and move the locations of the valleys to which the polynomial curve is fit. A description of the rubber-band method of fitting a baseline is provided in a spectroscopy book by Wartewig50. Here, we discuss polynomial curve fitting technique as it can be easily implemented in computing software like MATLAB (MathWorks®) and is a direct, simple method to remove background fluorescence.
There are two important parameters when fitting a polynomial curve to the raw Raman spectrum of bone: the order of polynomial function and the wavenumber range to which the curve is fit. To follow description of how polynomial order affects the removal of background fluorescence, we used a Raman spectrum of bone acquired with a Renishaw InVia Raman microscope instrument. Starting with a low polynomial order avoids over-fitting the background fluorescence, but typically 1st (line) and 2nd (quadratic) degree polynomial curves are insufficient (Fig. 6). As the degree of the polynomial increases, there is a trade-off between underfitting one region and overfitting another region of the spectrum. If the extremes of the acquired spectra (e.g., 300–350 cm−1 and 1750–1800 cm−1) are not important, then higher order polynomials may provide the best fit to the valleys.
Figure 6. Effect of polynomial order on the fit of the non-linear curve to the apparent baseline of a Raman spectrum of bone.
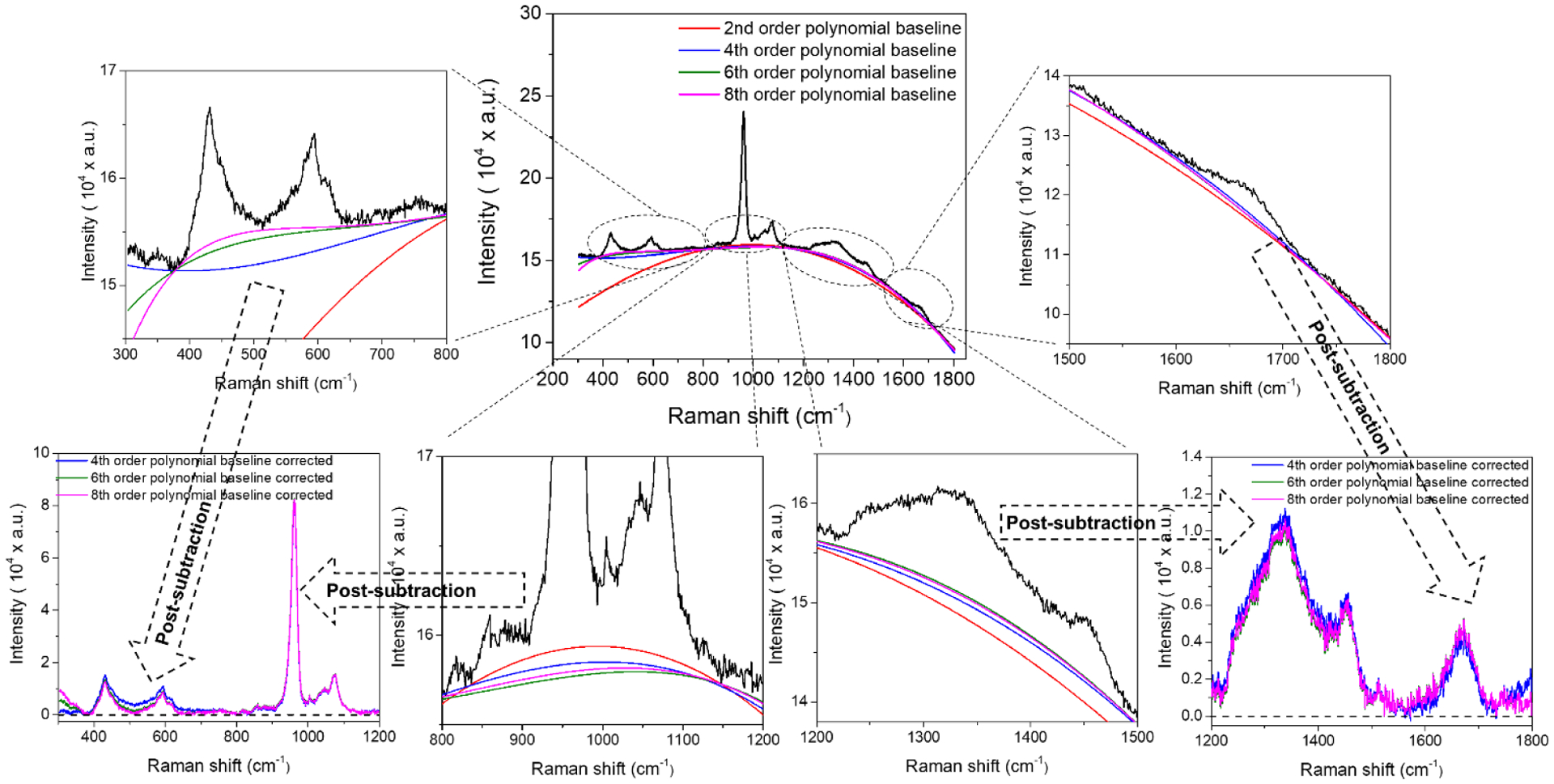
Because bone tissue contains fluorophores that auto-fluoresce upon exposure to laser light, all raw Raman spectra of bone have background fluorescence that obscures the relative of heights of various peaks. To remove or subtract the background fluorescence, a polynomial curve of some specified order (e.g., quartic) is fit to the apparent baseline of the Raman spectrum. Selecting too low of an order (e.g., a*x2 + b*x + c) or too high of an order (a*x10 + b*x9 + c*x8 + d*x7 + e*x6 + f*x4 + g*x3 + h*x2 + i*x2 + j*x + k) under-fits certain regions of the spectrum or over-fits the polynomial baseline curve. The poor fit at the extreme ends of the Raman shift can be ignored if the region does not contain important peaks (i.e., can be truncated after removing background fluorescence).
Another important part of the polynomial curve fitting method is the choice of spectral points (i.e., the valleys) where the fitted baseline passes through. As a general practice, these spectral points are selected from regions of the Raman spectrum that do not include intense Raman bands (Fig. S8). When changing the spectral points from regions of the Raman shift with low spectral intensity to either regions with high spectral intensity or at the end points of spectrum, the polynomial baseline goes from being well-fitted to over-fitted or under-fitted (Fig. S9), respectively. Under-fitting causes artificially elevated intensities while over-fitting causes negative intensities in some part of the spectrum (Fig. S10). In fact, manually selecting the spectral regions to ensure a proper fit of the polynomial baseline to each data set separately is rather time consuming and ill-advised. Fortunately, automated approaches exist in commercial software (e.g., Horiba’s LabSpec, Renishaw’s WIRE or Bruker’s OPUS) to determine the ideal spectral locations for fitting the baseline curve and the order of polynomial curve that defines the baseline47,48.
Following the subtraction of background fluorescence by fitting a polynomial curve to the apparent baseline of each spectrum, additional baseline corrections may be beneficial when residual background fluorescence is still present. For example, we recently reported that the addition of piecewise linear baselines to the Raman spectra after polynomial fitting and subtraction improved the correlations between Raman parameters and fracture toughness properties of human cortical bone51.
4.2. Minimizing spectral noise
Even when using optimized laser power and acquisition parameters, spectral noise exists in the Raman spectrum of bone. The noise obscures subtle Raman peaks with low signal intensity. One simple approach to minimize noise is to average a collection of Raman spectra from different locations in the region of interest into a single spectrum if spatial distribution of Raman parameters is not a concern (e.g., changes in Raman parameters within an osteon or between newly formed bone and older bone). As a general practice, multiple sites of Raman acquisition is distributed throughout the region of interest and cover as many unique features (e.g., thick and thin lamellae) as is practically possible. Doing so captures the overall physicochemical features of a given bone specimen. We recently reported that averaging multiple Raman spectra from osteonal and interstitial sites into a single Raman spectrum improves the initial Raman spectrum quality (improved SNR), which in turn increases the RS sensitivity to detect subtle changes in amide I sub-band ratios due to glycation treatment of human cortical bone samples23, and improves the ability of Raman properties to explain the variance in fracture toughness properties of human cortical bone51. Although averaging multiple Raman spectra improves spectral quality, filtering the spectra to digitally remove noise is often still a necessity to avoid undue influence of noise on calculations of Raman parameters.
Various smoothing or de-noising methods have been effectively used for the treatment of noisy Raman spectra52,53, but importantly, all smoothing/de-noising methods affect the outcome of the analysis39. Among others, the Savitzky–Golay (S-G) filter is the most common method applied in the RS of bone because this digital filter technique tends to preserve key spectral features such as the width and height of peaks. The S-G filter involves consecutively fitting sub-sets of adjacent data points (window) with a polynomial curve by ordinary least squares and replacing raw Raman signals with fitted Raman signals52,54. As such, there are two parameters that influence how the noise in the spectrum is minimized: 1) the order of the polynomial curve that is used to fit discrete spectral intensities and 2) the fixed wavenumber range (i.e., size of the window) that defines which spectral data is being fit by the curve. While the order of polynomial dictates how well the filtered spectral intensities match the raw spectral intensities (i.e., low root-mean squared error), the window size governs which noise frequency is preferentially eliminated or ‘smoothed out’ by the S-G filter. The window size basically dictates the number of data points being processed as the filter marches through the entire spectrum. To the best of our knowledge, there is no published method for selecting such filtering parameters for RS analysis of bone. Ultimately, the selection depends on the quality of the spectra that was acquired. In general, as SNR decreases, the window size increases to limit the identification of peaks based on noise. For the most part when analyzing spectra from commercial RS instruments, a 2nd order polynomial works as well as a 4th order polynomial for a given window size between 7 and 21 data points. While increasing the window size results in smoother spectra (Fig. S11), the downside is the increased likelihood that subtle, but important, peaks and shoulders are lost.
Recently, we examined the effects of smoothing/de-noising/filtering parameters on the sensitivity of RS to detect glycation-mediated changes in human cortical bone specimens from fresh-frozen cadaveric femurs obtained from 10 donors (5 males and 5 females, aged 46–60 years old)23. We compared S-G filters with 2nd and 4th order polynomial functions as well as window sizes of 11, 15 and 21. The comparisons also included the proprietary de-noising algorithm provided by the LabSpec software which involves a self-adapting window size to select an optimal wavenumber range for fitting the spectral data with a 2nd order polynomial. The results suggest that detecting subtle differences in sub-peaks ratios of amide I were dependent on the smoothing method for relatively noisy spectra. In the case of spectra with less noise, the smoothing method had little effect on the ability of RS to detect glycation-mediated changes to organic matrix of bone23.
4.3. Calculating the characteristics of bone from Raman spectra
Raman spectrum of bone is reported most often in the Raman shift range of 400–1800 cm−1 (Fig. 7). This spectral range is rich with the characteristic Raman bands of mineral and organic matrix (except CH and NH stretching bands which are located at wavenumber ≥ 2500 cm−1). The reported location of major peaks in Raman bone spectrum may show some variability due to the RS instrument or sometimes the sample used (e.g., due to the age, health status, differences among human subjects, and among the rodent animal models being investigated). The most intense band in a Raman spectrum of bone is a phosphate band located at ~960 cm−1 (ν1PO4). The other two phosphate bands are located at ~430 cm−1 (ν2PO4) and ~600 cm−1 (ν4PO4). Although these three Raman bands emerge from phosphate content in bone mineral, the reason why they are located at different wavenumbers is related to the vibration modes of the covalent bond between oxygen and each phosphorus atom. The nu labeling – ν1, ν2, and ν4 – in front of PO4 refers to different vibration modes: symmetric stretch, antisymmetric stretch, and antisymmetric bend, respectively. Another intense mineral band is the carbonate located at ~1070 cm−1 (ν1CO3) which is a measure of Type-B carbonate ions in the hydroxyapatite lattice. The carbonate ion occupies two different positions in the structure of bone mineral: the hydroxide position (A) and the phosphate position (B). Type B carbonate refers to carbonate ions occupied in the phosphate position55,56. Table 1 summarizes primary Raman metrics along with their correlations with other techniques.
Figure 7. Differences in Raman spectra of bone due to noise filtering and different methods to determine peak ratios.
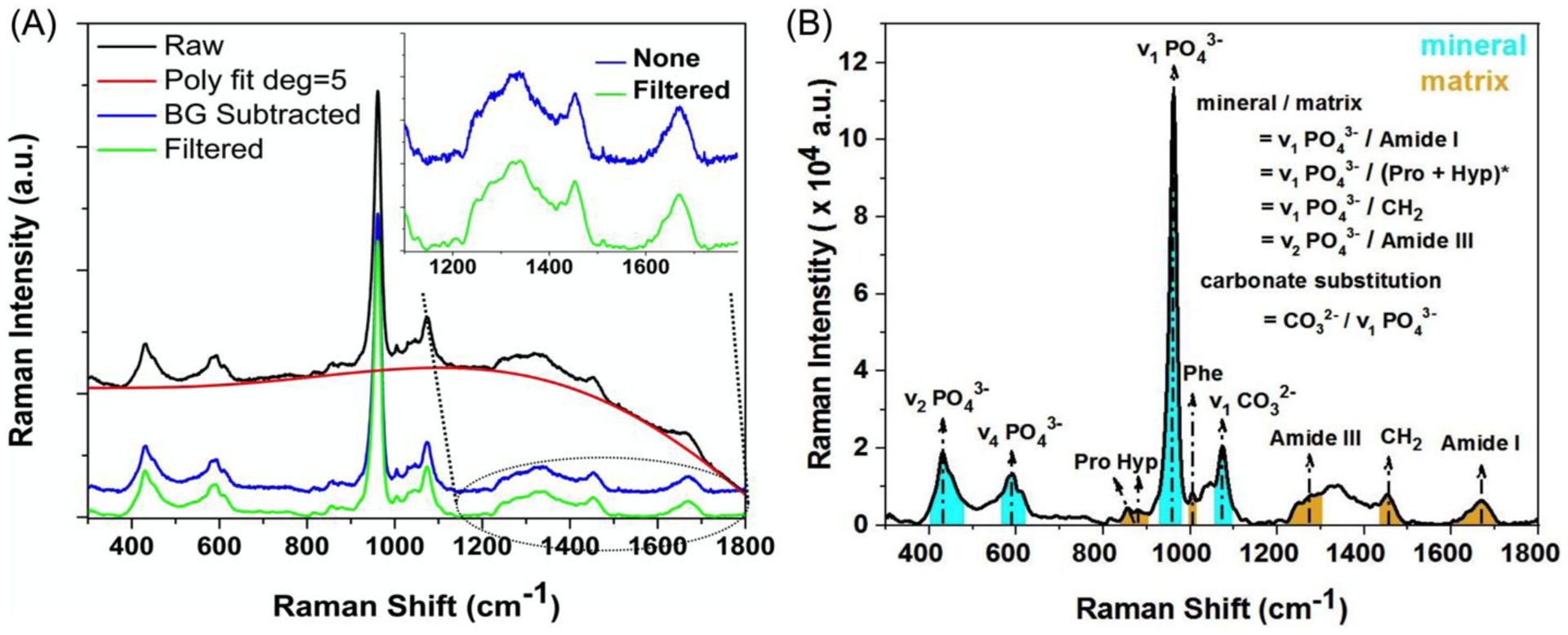
A fitted 5th order polynomial curve subtracted the background fluorescence from the raw spectrum of human cortical bone, and then a S-G filter (4th order & window of 21) was (green) or was not (blue and offset) applied (A). Two typical methods for determining peak ratios include dividing one peak intensity (PI) height by another PI height or dividing an integrated area (IA) of a band by an IA of another band (B). *When using the PI method or IA method, the mineral peak is divided by proline peak or the combination of Pro and the hydroxyproline peak, respectively.
The Raman peaks of the organic matrix are Proline (Pro, ~855 cm−1 or ~920 cm−1), Hydroxyproline (Hyp, ~875 cm−1), Phenylalanine (Phe, ~1002 cm−1), Amide III (~1243–1320 cm−1), CH2-wag (~1450 cm−1) and Amide I (~1600–1720 cm−1). The intensity of the CH2-wag Raman band depends on the amount of all organic matrix components in bone (collagen, lipids, and non-collagenous proteins), whereas the other organic Raman bands are specific to collagen type I or collagen I. Pro, Hyp and Phe Raman bands emerge from the amino acids of collagen I molecule, and amide III and amide I emerge from different side-chains and backbone of α1(I) and α2(I) peptide chains and thus are sensitive to the triple helix structure and local orientation of collagen type I (discussed in section 5). Using these Raman bands, several bone quality measurements were established or adopted from the FTIR literature (Table 1).
There is currently no consensus on the best method for determining the physicochemical properties of bone using RS. Methods include: 1) peak intensity (PI) ratios (Table 1), 2) integrated area (IA) ratios (Table 1), and 3) band area ratios in which each area is determined by a deconvolution procedure. Peak intensity is the maximum value at a specified wavenumber for the peak of interest, whereas integrated area is the sum of values within wavenumber range for the peak of interest. These values may or may not be corrected by a prescribed baseline to the peak of interest. When we calculated Raman properties (e.g., MMRs and carbonate substitution) using either PI or IA, there were significant linear correlations between MMR (or carbonate substitution) and crack initiation toughness as well as final J-integral regardless of whether PI or IA was used. However, the IA approach required linear baseline corrections to the ν1PO4 and amide I bands after background fluorescence subtraction to detect significant correlations51. Compared to IA ratios, PI ratios are not as affected by residual background fluorescence, but they are more vulnerable to poor initial SNR33. Of the Raman studies of bone reporting correlations with mechanical properties, most used the PI method of determination (Table 3).
Table 3.
Significant correlations between Raman properties and mechanical properties of bone.
| Mechanical test | Species bone | Raman property | Mechanical property | Correlation coefficient (r) | Ref |
|---|---|---|---|---|---|
| Nanoindentation | Mouse tibia | ν1PO4/Prolinea | Modulus | +0.565 | 134 |
| PY toughness | +0.770 | ||||
| Modulus | −0.592 | ||||
| Fracture stress | +0.258 | ||||
| 1/FWHM[ν1PO4] | KIce | −0.774 | |||
| CO3/ν1PO4a | ΔKIc/Δaf | −0.278 | |||
| CO3/ν1PO4a | Ultimate strain | −0.750 | |||
| CO3/ν1PO4b | ID1 | −0.653 | |||
| Nanoindentation | Rat humerus | ν1PO4/Amide Ia | Modulus | +0.894 | 140 |
| CO3/ν1PO4a | Modulus | +0.583 | |||
| Plastic index | +0.914g | ||||
| CO3/ν1PO4a | Hardness | +0.632 | |||
| Nanoindentation | Human vertebrae | ν1PO4/CH2 waga | Modulus | +0.690 | 142 |
| PY toughness | −0.735 | ||||
| I1670/I1640a | KIce | −0.569 | |||
| Nanoindentation | Human femur | 1/FWHM[ν1PO4] | Modulus | +0.514 | 143 |
Peak intensity ratio
Band area ratio
Correlation between osteogenesis imperfecta (OI) control and OI anti-TGF-β for Col1a2+/p.G610C missense model are reported
Post-yield (PY) displacement (PYD)
Crack initiation toughness was determined as critical stress intensity factor K in mode I opening (KIc)
Crack growth toughness was determined as the change in per change in crack length 90 ΔKIc/Δa
Partial correlation
To illustrate how the method of determining peak ratios can affect the findings in a Raman study of bone with S-G filtering (fourth order polynomial and window size of 21) or without filtering (Fig. 7A), we processed Raman spectra acquired from the anterior surface of intact mouse femurs in the mid-diaphysis as follows: peak intensity ratios and integrated area ratios (Fig. 7B). The femurs were from 6-mo. (n=20) and 20-mo. (n=18), male BALB/c mice57, but the extended spectra (300–1800 cm−1) were not previously reported. As indicated by three-way analysis of variance (ANOVA) in which the main effects, filter (yes or no) and ratio (PI or IA) were factors with repeated measures and age group (6-mo. or 20-mo.) was an independent factor, the values of selected peak ratios significantly depended on whether the Raman spectra were filtered and whether PI or IA method was used (Table S1). Also, the ratio method depended on whether the spectra were filtered (significant interaction term in Table S1). More importantly, the age-related difference in each peak ratio depended on the method but not on filtering (p>0.050 for Filter × Age). The one exception was the carbonate substitution (CO3/ν1PO4) in which the age-related difference depended on the interaction between filter and method (p=0.0401 for Filter × Method × Age).
To determine which method might be preferable, we compiled the standard mean difference (Cohen’s effect size d) using the mean of each group, the SD of each group, and the number of mice in each group (Table 4). Since the likelihood of detecting an age-related difference in bone composition by RS improves as the effect size increases, PI was more sensitive than IA for all peak ratios. Except for carbonate substitution, filtering the noise from the spectra (i.e., digitally smoothing the spectra) did not improve the sensitivity of the PI method or the IA method (Table 4). Regardless of the approach taken, all peak ratios were higher with age of the mouse (Fig. S12). Whether PI is always more sensitive than IA to differences in physiochemical properties of bone depends on the quality of the Raman spectra and spectral resolution.
Table 4.
Age-related difference in Raman spectroscopy properties of cortical bone as determined by peak intensity ratio (PI) and integrated area ratio (IA) with and without 2nd order Savitky-Golay filter (window size = 21). The highest standard mean difference (d) was bolded for each property.
| Property | Method | Filter | 6-mo. | 20-mo. | Effect size (d) | p-value | ||
|---|---|---|---|---|---|---|---|---|
| Mean (SD) | COV (%) | Mean (SD) | COV (%) | |||||
| ν1PO4 / Amide I | PI | S-G | 16.1 (1.4) | 8.95 | 18.5 (1.7) | 9.03 | 1.549 | <0.0001 |
| None | 15.8 (1.5) | 9.25 | 18.4 (1.7) | 9.17 | 1.627 | <0.0001 | ||
| IA | S-G | 6.06 (0.56) | 9.29 | 6.80 (0.54)a | 7.99 | 1.343 | 0.0002 | |
| None | 6.06 (0.56) | 9.28 | 6.80 (0.54)a | 7.99 | 1.343 | 0.0002 | ||
| ν1PO4 / Pro or / (Pro+Hyp) | PI | S-G | 21.7 (2.4) | 11.1 | 25.5 (2.1) | 8.03 | 1.679 | <0.0001 |
| None | 21.1 (2.1)a | 9.98 | 25.1 (2.1) | 8.22 | 1.904 | <0.0001 | ||
| IA | S-G | 12.5 (1.3)a | 9.98 | 14.2 (0.92) | 6.46 | 1.495 | <0.0001 | |
| None | 12.5 (1.2)a | 9.93 | 14.2 (0.92) | 6.48 | 1.578 | <0.0001 | ||
| ν2PO4 / Amide III | PI | S-G | 2.73 (0.32) | 11.6 | 3.11 (0.27) | 8.55 | 1.277 | 0.0003 |
| None | 2.67 (0.29) | 11.1 | 3.05 (0.26) | 8.57 | 1.375 | 0.0002 | ||
| IA | S-G | 1.60 (0.18) | 11.4 | 1.83 (0.19) | 10.3 | 1.244 | 0.0005 | |
| None | 1.60 (0.18) | 11.4 | 1.83 (0.19) | 10.3 | 1.244 | 0.0005 | ||
| ν1PO4 / CH2-wag | PI | S-G | 12.9 (1.7)a | 12.9 | 15.4 (1.5) | 9.74 | 1.554 | <0.0001 |
| None | 12.7 (1.7)a | 13.4 | 15.2 (1.5) | 9.75 | 1.554 | <0.0001 | ||
| IA | S-G | 7.95 (1.1) | 13.5 | 9.44 (0.91) | 9.61 | 1.468 | <0.0001 | |
| None | 7.95 (1.1) | 13.5 | 9.44 (0.91) | 9.64 | 1.468 | <0.0001 | ||
| CO3 / ν1PO4 | PI | S-G | 0.18 (0.005) | 2.88 | 0.19 (0.003) | 1.72 | 2.394 | <0.0001 |
| None | 0.18 (0.005) | 2.76 | 0.19 (0.005) | 2.42 | 2.000 | 0.0005 | ||
| IA | S-G | 0.22 (0.005) | 2.21 | 0.23 (0.004) | 1.61 | 2.195 | <0.0001 | |
| None | 0.22 (0.005) | 2.22 | 0.23 (0.004) | 1.61 | 2.195 | <0.0001 | ||
Data did not pass the Anderson-Darling normality test; therefore, the p-value was determined by the Mann-Whitney test. Otherwise, the comparison between 6-mo. and 20-mo. old mice was tested for significance using a two-sided t-test.
The choice among the different peak ratios that assess the degree to which the organic matrix is mineralized (MMR in Table 1) is not straightforward and depends on several factors including the range of the Raman shift with prominent signals, polarization bias, and embedding media (if used to polish the bone surface for spatial assessments). For certain commercial instruments, a narrow range is much quicker to acquire because the motorized grating does not change (i.e., it’s static) but precludes the use of both ν2PO4/amide III and ν1PO4/amide I to measure MMR. In general, selecting Raman peaks of bone that do not overlap the Raman peaks of the embedding media, if applicable, minimizes the influence of the media on the Raman properties of bone, though the background contribution does not necessarily obscure differences between groups if all bones are processed and embedded in the same way29.
When calculating carbonate substitution using either PI or IA, identifying the proper location of carbonate band (1020 cm−1 – 1100 cm−1) is important. This band overlaps several other peaks including ν3PO4 located at ~ 1045 cm−1 and ~1076 cm−1 55 and proteoglycan/lipids at ~1060 cm−1 58. Thus, the integrated region of the carbonate band (Fig. 7B) should be selected with care or judicious band fitting. For the peak intensity method, this selection is more straightforward since the carbonate peak (at 1070±3 cm−1) is the most intense peak in this region and can be easy distinguished from other peaks.
Although mineral crystallinity in RS is nearly always reported as the inverse of the full-width at half maximum (FWHM) of the ν1PO4 peak, the method for determining the height of this peak at ~960 cm−1 varies widely in the literature: a local, linear baseline between the valleys to the left and right of the ν1PO4 band59, a linear baseline between the valley to the left of proline peak (~860 cm−1) and the valley to the right of carbonate peak (~1060 cm−1)51, no baseline such that FWHM occurs at the maximum peak intensity / 229, a fit of a single Gaussian curve15, or deconvolution of the band between 901 cm−1 and 990 cm−1 with 4 sub-bands as Gaussian-Lorentzian functions60.
To investigate the effect of the method for determining the FWHM of ν1PO4, we analyzed the aforementioned Raman spectra acquired from male mouse femurs as follows: 1) with or without a local linear correction (Baseline) and 2) direct determination of FWHM or the use of a single Gaussian fit (Method). Background subtraction and digital noise filtering procedures were the same as before for all spectra prior to normalization. Defining a local linear baseline lowers the FWHM (Fig. 8A) and adjusts the Gaussian fit (Fig. 8B). Crystallinity depended on the method of determining the FWHM (direct vs. single Gaussian fit), whether or not the peak was baseline corrected, and age (Table S2). Moreover, the age-related difference in crystallinity depended on the method but not on the linear correction (LC) of the ν1PO4 peak (p=0.121 for Baseline × Age). Interestingly, the mean standardized difference in crystallinity between 6-mo. and 20-mo. old mice was higher for the single Gaussian fit than for the direct method of calculating FWHM, regardless of the baseline correction (Table 5).
Figure 8. Effect of baseline on crystallinity.
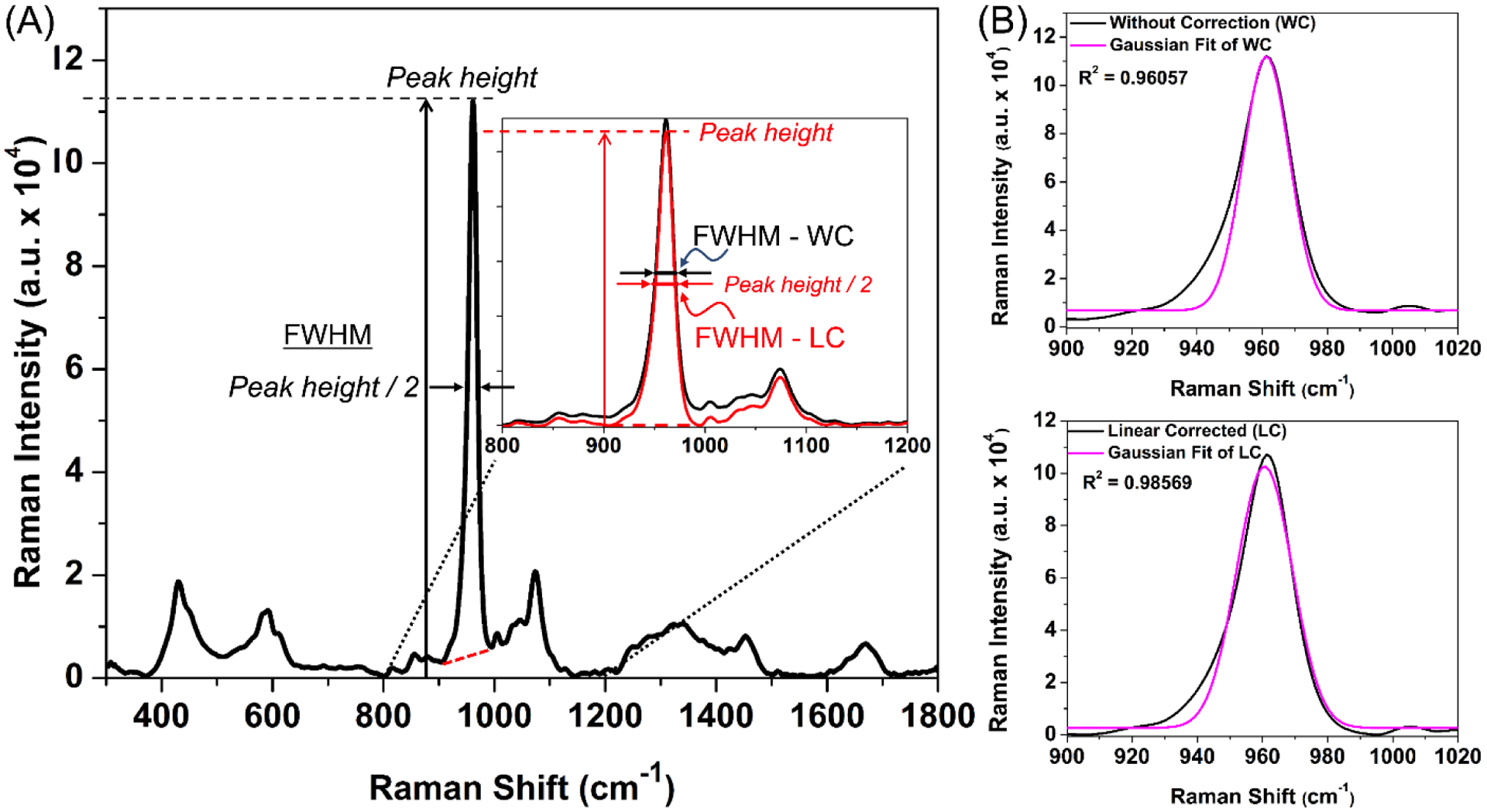
When a local linear baseline (red line) ‘corrects’ the height of the ν1 phosphate peak at ~960 cm−1, the FWHM decreases meaning crystallinity increases (A). This linear correction (LC) also increases the correlation of determination (R2) of the fit of the Gaussian curve to the peak (B) when compared to a fit without correction (WC).
Table 5.
Age-related difference in Raman spectroscopy properties of cortical bone as determined by baseline linear correction with and without single peak Gaussian fitting. The highest standard mean difference (d) was bolded for each property (filtered spectra).
| Baseline | Method | 6-mo. | 20-mo. | Effect size (d) | p-value | ||
|---|---|---|---|---|---|---|---|
| Mean (SD) | COV (%) | Mean (SD) | COV (%) | ||||
| Without correction | Direct | 0.0512 (0.0005) | 1.05 | 0.0521a (0.0011) | 2.09 | 1.065 | 0.0042 |
| Gaussian Fit | 0.0496a (0.0004) | 0.867 | 0.0505a (0.0008) | 1.63 | 1.389 | 0.0003 | |
| Linear correction | Direct | 0.0534 (0.0004) | 0.825 | 0.0542 (0.0009) | 1.80 | 1.077 | 0.0031 |
| Gaussian Fit | 0.0501 (0.0004) | 0.851 | 0.0509a (0.0008) | 1.67 | 1.208 | 0.0014 | |
Did not pass the Anderson-Darling normality test; therefore, the p-value was determined by the Mann-Whitney test. Otherwise, the comparison between 6-mo. and 20-mo. old mice was tested for significance using a two-sided t-test.
4.4. Normalizing Raman spectra
For the purposes of displaying multiple spectra from different experimental groups however, the spectra are typically normalized by 1 of 2 methods (for other methods in spectroscopy, see41): i) divide each spectral intensity (SI) by the spectral intensity of a selected peak (most often ν1PO4) or ii) divide each SI by the mean of all intensities (). The advantage of the latter method is that all potential differences are apparent (e.g., when plotting a difference spectrum) because the former method cannot reveal a potential difference at the peak being used to normalize the spectrum. Neither of these methods are suitable for multivariate analysis of Raman spectra such as principal component analysis (PCA), partial least squares regression (PLSR), types of discriminant analysis, and machine learning algorithms because certain Raman peaks (e.g., ν1PO4) are dominant over other Raman peaks (e.g., Proline) without necessarily being more important. In order to remove this dominance such that all peaks are equally weighted, spectra are normalized to their mean absolute difference61, , or by the standard normal variate method or ‘z-scoring’35, , in which each intensity is subtracted from the overall mean and divided by the standard deviation (σ) of the Raman spectrum or spectral intensities (SIi). After applying the latter technique, the mean and SD of the normalized spectrum is 0 and 1, respectively. Multivariate analysis techniques fall into 2 general categories, unsupervised (e.g., PCA) and supervised (e.g., support vector machines), as reviewed by Gautam et al.41.
5. The amide I band
The organic matrix of bone, primarily collagen type I, consists of proteins that are rich in vibrational modes arising from the amide backbone (i.e., peptide chains) as well as the α-helices and β-sheets that form the secondary structure of collagen and non-collagenous proteins. The amide I band has been widely used in the spectroscopy literature to infer differences in or changes to collagen structure (Table 6). In RS analysis of bone, the band occurs between ~1590 cm−1 and ~1720 cm−1 with a peak at ~1670 cm−1 62. It is composed of several partially resolved components or sub-peaks whose relative intensities depend on the secondary structure of proteins in the matrix and triple helical structure of collagen type I63,64. Typical assignments for the sub-peaks of the amide I band in the literature are reported as follows (±5 cm−1): ~1607 cm−1 is a stretching vibration of a carbon ring such as those found in tyrosine or phenylalanine65, ~1638 cm−1, a shoulder in the amide I band, represents ordered structures in the form of an α-helix65, ~1660–1670 cm−1 is a stretching vibration of carbonyl or C=O component and causes the strongest sub-peak in RS64,66, ~1685–1690 cm−1, another shoulder, represents disordered secondary structure with a lack of hydrogen bonds67.
Table 6.
A list of sub-peak ratios from the amide I band along with their method of determination and interpretation.
| Measurement | Methoda | Interpretation | Ref |
|---|---|---|---|
| % area at 1660 cm−1 | 2nd Deriv. / 8 to 13 sub-bands / GRAMS / 1600–1700 cm−1 or 1580–1720 cm−1 | Underlying sub-peak area at 1660 cm−1 correlated with the pyridinoline (PYD) content in collagen. (p < 0.01, r2 = 0.63). | 83 |
| 1660 cm−1 / 1690 cm−1 | 2nd Deriv. / 3 sub-bands / Matlab / 1650–1690 cm−1 | Ratio is 5% lower in the osteolytic vertebral bodies (VBs) than in the non-tumor bearing VBs suggesting metastasis but no significant difference in pyridinoline crosslinks by HPLC. | 131 |
| 1660 cm−1 / 1690 cm−1 | NSb/ 5 sub-bands / MATLAB / NS but sub-peaks at 1630, 1645, 1660, 1676, and 1690 cm−1 | No differences within intact tibiae and contralateral fracture calluses were detected between wild-type and Brtl/+ mice or with alendronate treatment, irrespective of side. | 144 |
| 1660 cm−1 / 1683 cm−1 | NS / NS / GRAMS / NS | β‐aminopropionitrile (BAPN) treatment, an inhibitor of enzymatic crosslinking, increased matrix maturity of new tissue in murine cortical bone but decreased the overall mature-to-immature crosslink ratio by HPLC. | 82 |
| 1660 cm−1 / 1683 cm−1 | NS / NS / GRAMS / NS | Four consecutive daily 5-Gy doses to the right hind limb of mice caused matrix maturity to be higher compared to non-irradiated limb. The relative difference in the ratio decreased from 1 week to 26 weeks post-irradiation. | 145 |
| 1660 cm−1 / 1690 cm−1 (~1666 & ~1686) | NS / 6 sub-bands / GRAMS / 1580–1710 cm−1 | Targeted radiotherapy (XRT) of rat bone lowered this ratio compared to non-irradiated controls, whereas in the amifostine-XRT treated group it remained largely unchanged. | 81 |
| 1656 cm−1 / 1684 cm−1 | NS / 4 sub-bands / NS / NS | Ratio significantly increased within 7-days in staphylococcus aureus infected bones suggesting an increase in the amount of the trivalent, non-reducible cross-links and a decrease in the reducible collagen crosslinks. | 80 |
| 1670 cm−1 / 1640 cm−1 1670 cm−1 / 1610 cm−1 1670 cm−1 / 1690 cm−1 | 2nd Deriv. / Intensity ratios of 3 sub-bands / NS / NS | Higher 1670/1640 cm−1and 1670/1610 cm−1 in 20-month BALB/c mice compared to 6-month mice, irrespective of gender, while the so-called matrix maturity 1670/1690 cm−1 was lower in old male mice (no significant age-related change in female mice). | 57 |
| 1660 cm−1 / 1685 cm−1 | 2nd Deriv. / 4 sub-bands, Deconv. Gauss-Lorentz func. / Peakfit / 1590–1710 cm−1 | Ratio significantly increased in ovariectomized rats compared to controls over 145 days post-surgery. | 146 |
| 1656 cm−1 / 1637 cm−1 | NS / 4 sub-bands / NS / 1575–1700 cm−1 | Ratio decreased as the MMR of the bone increased as follows: Red deer antler (1.7), Ovine femur (1.2), Fin whale bulla (0.6) | 63 |
| Peak Int. of 1660 cm−1, 1681 cm−1 | Deconv. Gauss-Lorentz func. / 4 sub-bands / Peakfit / 1590–1790 cm−1 | Relative intensity at a wavenumber (wn) = 1660 cm−1 or 1681 cm−1 decreased or increased, respectively, as the demineralization time (dt = minutes in 1.2% HCl) increased. Relative intensity at wn = 956 cm−1 decreased with demineralization time. Relative intensity at wn was intensity at wn at dt divided by the product of intensity of wn at dt = 0 and intensity of 2938 cm−1 at dt. | 147* |
| 1660 cm−1 / 1690 cm−1 | NS / 4 sub-bands / Peak Fitter / 1620–1700 cm−1 | Collagen maturity was lower in the formaldehyde fixed group at both 2 h and 6 h than in the unfixed group. | 148 * |
| 1670 cm−1 / 1640 cm−1 | 2nd Deriv./ NS / Labspec /NS | Ratio significantly increased when bovine cortical bone samples were boiled in water for 90–120 min. The ratio also significantly increased when extensive microdamage occurred at notched cortical bone samples loaded at 3 μm/s in tensile test until the emergence of damage zone at the notch tip. | 67 * |
| 1670 cm−1 / 1640 cm−1 | Gaussian func. / NS / Labspec / NS | Ratio increased at the fatigue site of cortical bone for all cyclic stress levels (25 to 55 MPa stress level with 10 increment). However, no significant difference in the ratio at the fatigue site between differing stress levels whereas the pooled-data from all non-fatigue and fatigue sites, regardless of stress level, detected ~ 4% increase in the ratio. | 99 * |
| 1668 cm−1 / 1638 cm−1, 1668 cm−1 / 1610 cm−1, 1668 cm−1 / 1690 cm−1 | 2nd Deriv., local maxima of difference spectrum (LMDS), Deconv./ 4 sub-bands / Labspec, MATLAB / 1575–1725 cm−1 | 1668 / 1638 cm−1 and 1668 / 1610 cm−1ratio decreased in both ribose and glucose treated samples having 0.1 and 0.5 M conc incubated for 4 and 16 weeks. Direct sub-peak identification method (2nd Deriv.or local maxima) was more sensitive to detective ratio changes as compared to the band fitting method (Deconv.) Glycation did not affect matrix maturity 1668 / 1690 cm−1 ratio. | 23 * |
| 1667 cm−1 / 1640 cm−1, 1667 cm−1 / 1690 cm−1 | NS / NS / NS / NS | 1667/1640 cm−1 ratio significantly increased when the cortical bone is baked to 200°C, also when boiled and pressure heated at 110°C. However, 1667/1690 cm−1 ratio decreased starting at 120°C to 200°C baking whereas increased in boiling and pressure heating starting at 110°C to 130°C | 42 * |
| 1665 cm−1 / 1685 cm−1 | NS/ NS / OriginPro / NS | Col2.3Δtk mice treated ganciclovir and pamidronate to create an adynamic bone condition had a higher collagen maturity ratio than control animals. Old adynamic mice had ~68% more collagen maturity compared to young adynamic mice (p < 0.001) and ~31% more compared to old control mice (p < 0.01). | 149 |
| 1660 cm−1 / 1690 cm−1 | Int. at specified wavenumbers? / NS / MATLAB / NS | Ratio decreased implying sequential treatment with an anti-resorptive followed by anabolic therapy followed by the anti-resorptive produced lower collagen maturity, compared to groups given any of anti-resorptive monotherapy. | 150 |
| 1670 cm−1 / 1640 cm−1 1670 cm−1 / 1610 cm−1 1670 cm−1 / 1690 cm−1 | 2nd Deriv. / i) Intensity ratios of 3 sub-bands ii) 4 sub-bands, Deconv. Gauss-Lorentz func. / MATLAB / 1590–1710 cm−1 | ~1670 / 1610 cm−1 and ~ 1670 / 1640 cm−1 negatively correlated with all three fracture toughness properties, whereas matrix maturity ratio does not correlate. Sub-peak parameters also correlated when acquired through research graded Raman system and optical probe-based Raman system. | 51 |
In sub-peak analysis of the amide I band, the method to identify the locations of sub-peaks / the number of sub-bands fitted / software used to fit the bands varies from study to study / wavenumber range of the amide I band.
Not specified
The fibril organization of collagen I within the bone matrix is maintained by the covalent crosslinks between neighboring collagen molecules and contributes to the strength and resiliency of bone68. Therefore, this covalent crosslinking is vital to making a healthy fibril network capable of withstanding the loads acting on bones during daily activities. The crosslinks of collagen can be formed through both enzymatic and non-enzymatic pathways. Enzymes facilitate the post-translational modifications that enable the crosslinking of C-terminal telopeptides to a N-terminal helix of collagen molecules as well as the maturation of divalent crosslinks to trivalent crosslinks69; whereas, non-enzymatic reactions involving sugar and oxidation form crosslinks that connect the helical tropocollagens within fibrils70 and possibly between fibrils. Both types of crosslinks affect fibril mechanics and the mechanical behavior of bone as described in multiple review articles70–72. Assessing the amide I band provides an indication of protein conformation due to the role of the amide moiety in crosslinking64,73.
Identifying the partially resolved and overlapping sub-peaks in the amide I band is not a straightforward process. The following section describes the two methods that are commonly used in the bone literature for the identification of sub-peaks comprising the amide I band envelope. Although any pre-processing procedure of spectra may affect the overall outcomes of sub-peak analysis, pre-processing the overall spectrum to minimize noise may still be necessary before doing the amide I sub-peak analysis because it is not always possible to obtain noise-free, high quality Raman spectral bands especially for less intense peaks like the amide I. That is, to achieve a ‘reasonable fit’, removing background fluorescence, smoothing or filtering noise throughout the spectrum, and defining the baseline of the band are done prior to identifying the amide I sub-peaks.
5.1. Identifying the sub-peaks
In RS and FTIR, locating the position of the sub-peaks is based on the minima of the second derivative spectrum (i.e., d2f(I)/dw2 where f(I) is the signal intensity as a function of wavenumber or w). The second derivative of course amplifies noise, hence the need to filter the spectrum to minimize small spikes imposed on top of the Raman signal. The chosen wavenumber range of the amide I band can also affect the sub-peak ratios. An alternative technique to identify sub-peaks is the local maxima of the difference spectrum23,67. It involves a subtraction of each intensity value from the neighboring intensity values such that the local maxima of the difference spectrum highlight the locations of peaks.
5.2. Comparison in amide I sub-peak ratios between RS and FTIR
The use of amide I sub-peak ratios to assess characteristics of bone was originally developed for FTIR spectroscopy74,75 and then later adapted to RS without any of the original validation experiments (i.e., comparing amide I sub-peak ratios from RS to ratios of collagen crosslinks from gold standard, biochemical techniques). FTIR yields spectra with fairly high SNR on the order of 1000:1 compared with RS in which spectrometer efficiency is on the order of 30–80:176. Moreover, vibrational modes that cause a strong signal in FTIR tend to cause a weak signal in RS and vice versa. For example, the intensity of the amide I band (1580–1720 cm−1) is much higher in FTIR than in RS, whereas the peak intensity of ν1PO4 (960 cm−1) is much higher in RS than in FTIR. The overall shape of the amide I band also differs between the 2 spectroscopic techniques77. Thus, the interpretation of amide I sub-peak ratios does not necessarily cross from one spectroscopic technique to the other.
The most reported amide I sub-peak ratio is calculated from peaks at ~1660 cm−1 and at ~1690 cm−1. In FTIR spectroscopy, resolved sub-peaks at 1660 cm−1 and 1690 cm−1 were related to the amount of non-reducible enzymatic collagen crosslinks (mature) and reducible enzymatic collagen crosslinks (immature)74 leading to the use of this sub-peak ratio in the assessment of bone by FTIR as an indicator of mature-to-immature crosslink ratio78 (often denoted as XLR79). In published bone studies using RS (Table 6), this ratio was also interpreted to indicate the amount of mature enzymatic collagen crosslinking (trivalent) relative to the amount of immature enzymatic collagen crosslinking (divalent). XLR or matrix maturity ratio has also been determined using slightly different wavenumbers such as the area of the sub-band at 1656 cm−1 (B1656) per sub-band area at 1684 cm−1 (B1684)80, B1666/B168681, and B1660/B168382 (Table 6) owing to the location of the amide I band and the method used to fit the sub-bands across studies.
Whether RS-derived XLR is a true indicator of the maturity of enzymatic collagen crosslinking came into question when McNerny et al. reported that treating young mice with an inhibitor of lysl oxidase (β-aminopropionitrile) for 3 weeks reduced mature enzymatic collagen crosslinking (pyridinoline, PYD & deoxy-pyridinoline, DPD) without affecting immature enzymatic crosslinking (di-/hydroxylysinorleucine or DHLNL & HLNL) in the femur but increased XLR when measured by RS at sites of newly formed tissue82. Gamsjaeger et al. subsequently reported that the percent area of the sub-band at 1660 cm−1 (one out of 8 to 13 sub-bands fitted to the amide I as dictated by the second derivative spectrum) directly correlated with moles of pyridinoline per dry weight from 12 fractionated peptides extracted from demineralized human bone (R2 = 0.63) and that the sub-peak at 1660 cm−1 did not appear in samples without trivalent crosslinks83. The study did not include biochemical measurements of divalent crosslinks and so could not determine whether a sub-band at 1690 cm−1 is a marker of DHLNL or HLNL. Regardless, sub-peaks of the amide I band are sensitive to the structure of the collagen I molecule67, which is a helix of two α1(1) chains and one α2(1) chain as indicated by multiple studies involving manipulations of the matrix (references with a * in Table 6).
5.3. Sub-band fitting (deconvolution)
Sub-band fitting requires the placement of overlapping non-linear curves that fit a continuous curve to the shape of the amide I band when summed (Fig. 9). Such fitting is an ill-posed problem since each non-linear curve can have any shape and can be centered at any position within the band. To provide constraints, the non-linear curves are either a Gaussian function or a mixture of Gaussian and Lorentzian functions; and as previously mentioned, the position and number of sub-bands are based on the local minima of the second derivative spectrum (Fig. 9A). The initial position may be relaxed such that the center of each sub-band can slide to a lower or higher wavenumber (e.g., ± 3 cm−1). Most commercial software for the processing of Raman spectra (e.g., GRAMS, OriginPro, LabSpec, OPUS) is proprietary and so the method for fitting the sub-bands may or may not be known, though spectroscopy software programs typically offer several options for the fitting parameters (e.g., type of non-linear curve, fixed or unfixed sub-peak positions, number of sub-bands, offset of the base of the sub-bands, etc.). The general approach of the deconvolution algorithm is to vary the width and height of the Gaussian function or the blending fractions of Gaussian and Lorentzian functions, which are centered at the aforementioned minima, until the maximum coefficient of determination (R2) from least squares regression is achieved. Next, the sub-peak ratios can then be calculated as the ratio of the area or height of any 2 sub-peaks within the amide I band.
Figure 9. Deconvolution procedure to fit sub-peaks to the amide I band.
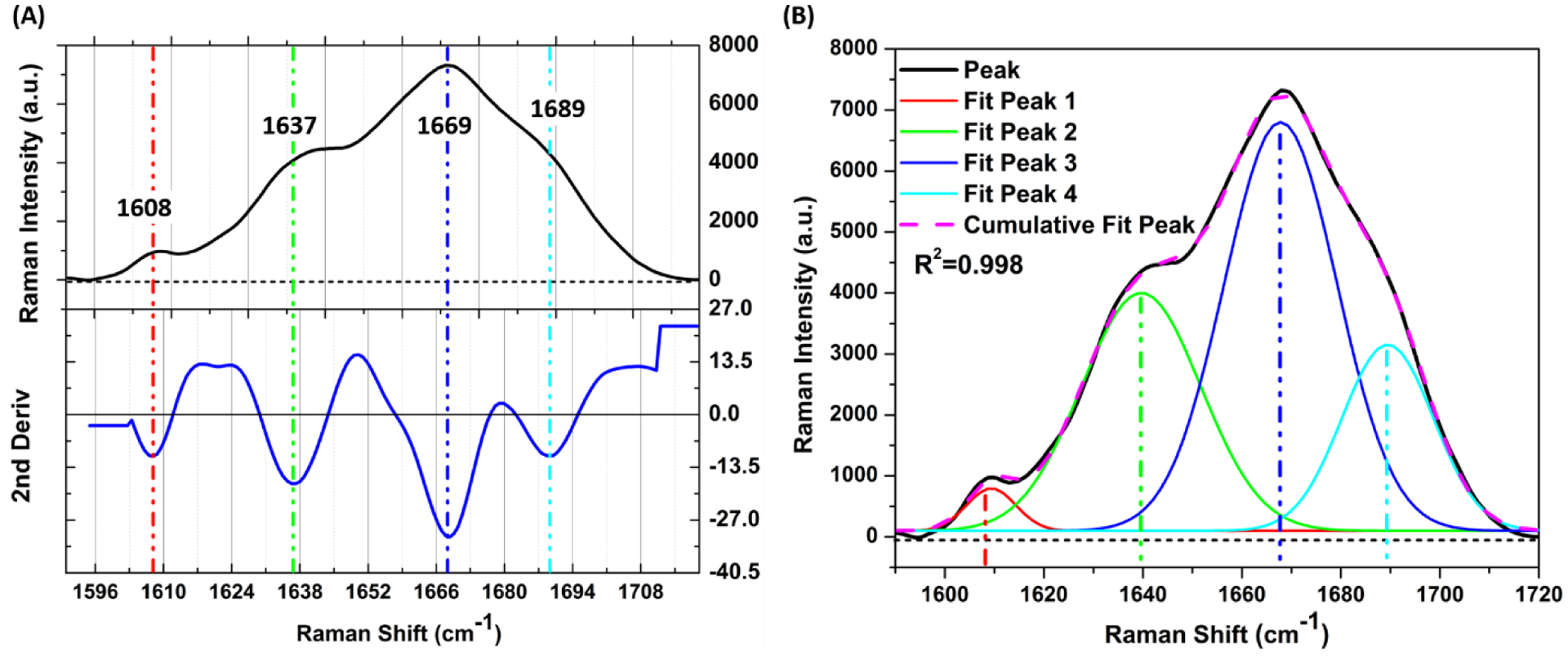
Initial peak positions under the amide I envelope are based on the minima of the 2nd derivative spectrum below zero (A). To deconvolve the sub-peaks, Gaussian functions are fit to the amide I band such that their location is close to initial location identified by the 2nd derivative spectrum and their sum achieves a high coefficient of determination (R2 > 0.990) (B). The spectrum in this example was baseline corrected using 5th order polynomial fit, and a linear baseline was applied to the amide I band before the curve fitting. Origin Pro 8.5 was used for sub-peak finding and band fitting amide I.
To demonstrate how the deconvolution method affects sub-peak ratios of the amide I band, we compared 100% Gaussian (Gauss) to a blend of Gaussian and Lorentzian functions (Gauss/Lorent) and calculated sub-peak ratios using either the area or the height of each sub-band. In these comparisons, positions of each sub-band were either allowed to move away from the initial position (unfixed) or kept at the initial position (fixed) as determined by the local minima of the 2nd derivative spectrum. The details of the RS acquisition parameters, the donor age and sex, and preparation of the human cortical bone samples can be found in our previous publication42. The fit of 4 sub-bands was excellent regardless of the type of deconvolution (Fig. S13). The unfixed option was slightly better than the fixed option going from R2 > 0.997 to R2 > 0.995, respectively; and on average, fits using Gauss/Lorent were slightly better than the fits with just Gauss. As indicated by Friedman tests, amide I sub-peak ratios depended on the deconvolution method whether based on the area (Fig. S14A) or height ratios (Fig. S14B). The sub-peak ratios were lower for unfixed than for fixed positions with the height of the sub-band at 1670 cm−1 per height of the sub-band at 1610 cm−1 being the one exception.
These sub-peak ratios did not necessarily correlate between 100% Gaussian functions and the blend of Gaussian and Lorentzian functions (Gauss/Lorent). None of the selected sub-peak ratios correlated when the position of each sub-band was allowed to move in order to achieve the best possible fit (Fig. 10A). On the other hand, the sub-peak ratios correlated between Gauss and Gauss/Lorent when the position of each sub-band was fixed, although the correlation was poor for B1670/B1610, irrespective of area ratio and height ratio (Fig. 10B). For the Gauss/Lorent functions, sub-peak ratios at unfixed positions are not good surrogates for sub-peak ratios at fixed positions (Fig. S15). Thus, deconvolution method does affect the findings from sub-band analysis of the amide I band. Additional issues with deconvolution of the amide I can be found in the supplemental information (Fig. S16–S19).
Figure 10. Sub-peak ratio correlations between 2 deconvolution methods.
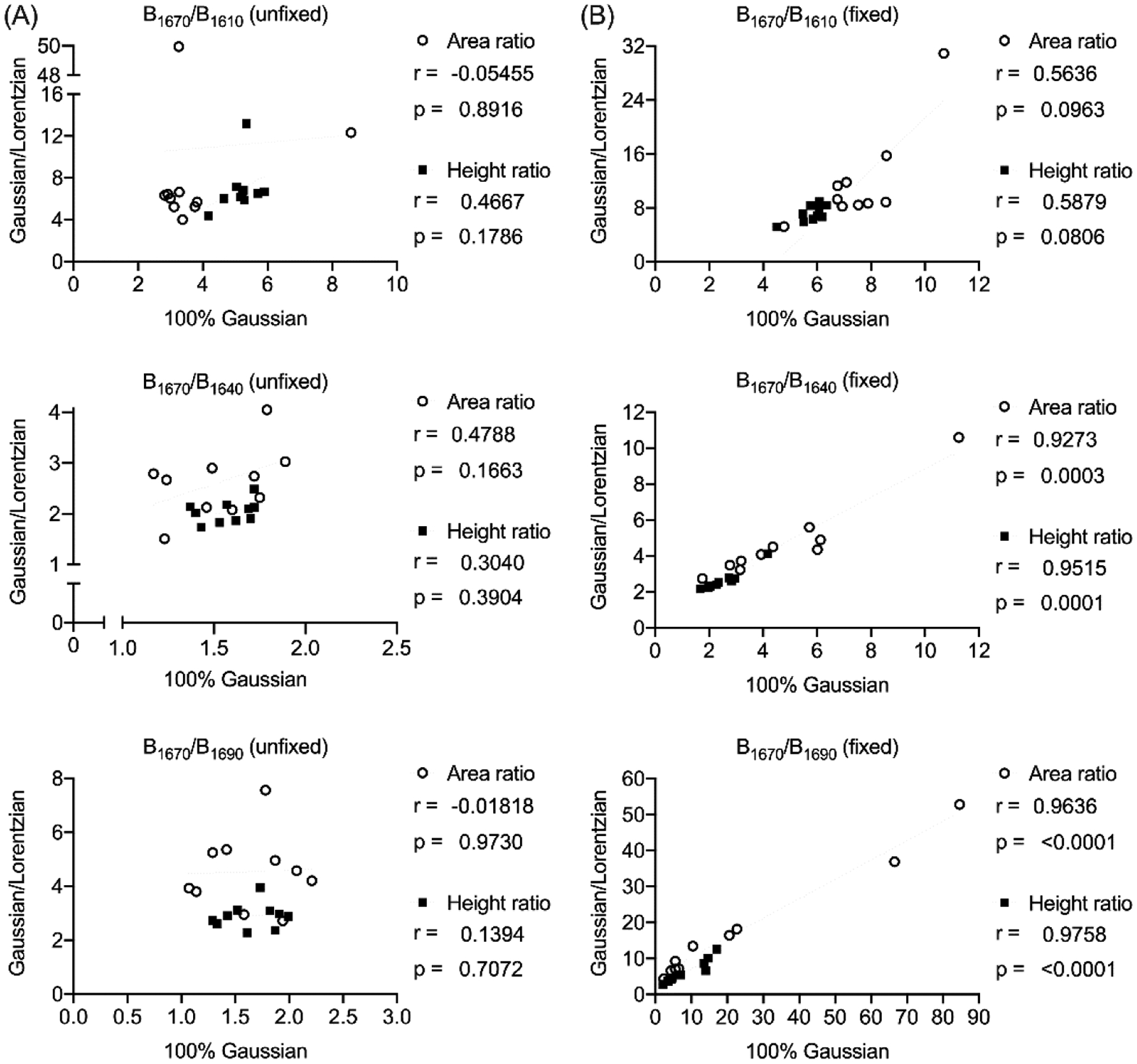
When the sub-bands are allowed to move during the fitting process, 100% Gaussian curves do not produce sub-peak ratios that correlate with their corresponding sub-peak ratio as determined by a blend of Gaussian and Lorentzian functions (A). On the other hand, when fixing the position of the sub-bands to the local minima of the 2nd derivative spectrum, the 2 different deconvolution methods determine sub-peak ratios that correlate, at least for 2 of the 3 ratios that were selected (B). This is not a justification for fixing the positions during the curve fits, but rather the choice of selecting Gaussian vs. Guassian/Lorentzian functions is less problematic when the positions of the sub-bands are fixed. P-value is provided for each Spearman’s correlation coefficient (r).
In summary, the values of the sub-peak ratios are not equivalent across several common deconvolution methods even though excellent fits of the amide I band can be achieved regardless of the method selected. Moreover, one deconvolution method is not a surrogate for another deconvolution method such that one cannot expect sub-peak ratios from one method to correlate with sub peak ratios from another technique, especially if the location of these peaks are not fixed. At this time, it is difficult to recommend one deconvolution method that is the most sensitive to differences in the structure of collagen type I. Towards that end, research is needed to validate the sub-peak ratios of the amide I band as being indicators of certain chemical moieties such as reducible or irreducible enzymatic crosslinks or different degrees of helicity. Until then, we caution against the use of sub-band fitting (i.e., deconvolution) and assessments of so-called collagen crosslink ratios using the area at 1660 cm−1 per area at 1690 cm−1 because of the uncertainty in what the shape and position of the deconvoluted bands mean. Lastly, reporting findings in the literature (Table 6) may be unique to the deconvolution method that was used.
6. Clinical assessment of bone matrix composition by probe-based Raman spectroscopy
RS holds great potential for the assessment of a patient’s bone matrix quality as a way to improve fracture risk prediction. For in vivo measurements of physiochemical properties of bone, custom-built, probe-based RS instruments (Probe RS) are generally required (Fig. 11). The major difference in design between commercial RS instruments, which are integrated with a microscope and provide confocality, and Probe RS instruments is the optics delivering the laser onto the tissue and then transferring the scattered photons from the tissue to the spectrometer (Fig. 1 and 11). This difference provides several distinct advantages for Probe RS over commercial Raman micro-spectroscopy. In Probe RS, the polarization bias is not an issue since the fiber optics do not preserve the orientation of light from the laser to the bone nor from the bone to the spectrometer. Moreover, the laser illuminates the tissue surface by a fiber optic cable, which has a greater diameter than an objective lens, resulting in the ability of using higher laser power than is possible with commercial RS instruments. In a Probe RS instrument, fibers also provide flexible laser deliver and photon collection without the need of alignment as required in commercial RS instruments, but background Raman signals generated by the fiber optics material itself during transfer of the photons may interfere with the Raman spectrum of the sample analyzed19,21.
Figure 11. A schematic depiction of a probe-based RS (Probe RS) instrument.
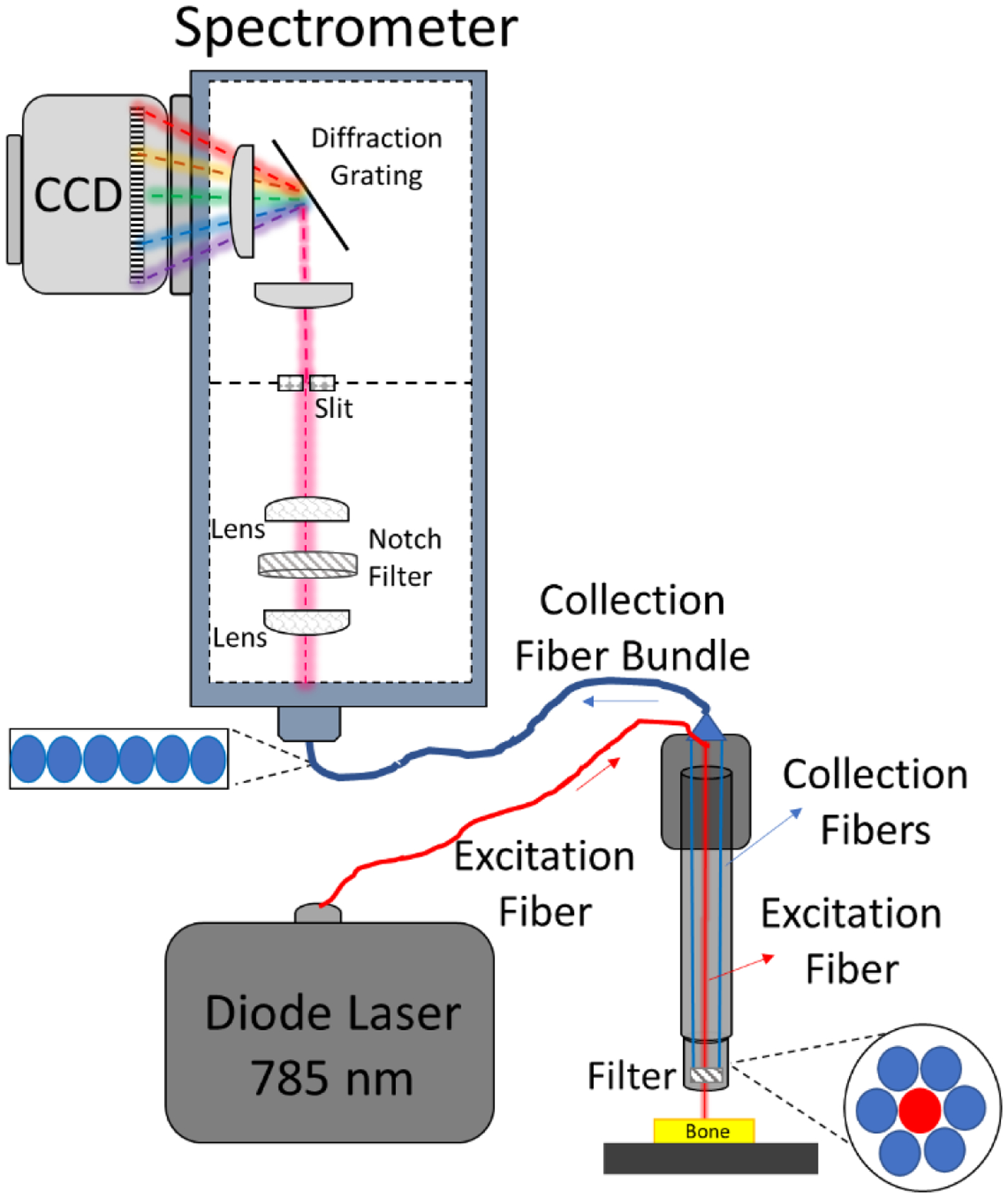
Probe RS instrument include fiber optics in a cable that passes the light from the excitation laser to the material of interest and the scattered light to the detector.
6.1. Spatially offset Raman spectroscopy
Probe RS generally involves one fiber optic (diameter = 200–500 μm) to deliver the laser onto the sample and multiple collection fiber optics of similar size to direct Raman photons to a spectrometer (Fig. 11). While the collection fibers can be distributed in a ring around the excitation fiber without any spatial gap, collection fibers can also be located at an offset of 2 mm to 14 mm from the excitation fiber as either a row of fiber optics at a fixed distance from the excitation source or as a ring of fiber optics with excitation source in the center, providing the collection of Raman photons from deeper regions of the tissue or overlying tissues. This method is known as spatially offset Raman spectroscopy (SORS).
In fact, collecting transcutaneous Raman spectra of bone tissue in vivo with SORS has been an active area of investigation for the past decade and half11,84–88. In early attempts to collect transcutaneous Raman bone spectrum, the researchers utilized an ultrafast Kerr-gated, time-resolved RS which was originally developed to suppress fluorescence in Raman spectrum89. Although transcutaneous Raman spectra of animal bone was successfully collected with this method, the laser power used in such a setup is much higher than the safety illumination limits90. Later on, the SORS concept has been successfully utilized to collect in vivo transcutaneous Raman spectra of animal and human bone tissues85,91.
Several configurations of SORS have emerged during the past decade, including inverse-SORS (where Raman is collected at the center of the probe and the laser is delivered at the spatially-offset rings from the center)90, Raman optical diffuse tomography (collecting Raman signal using a circular fiber-optic array involving multiple fiber optic probes)92, and digital micromirror devices (DMD)-based SORS (the mirror elements within the DMD replace the optic-fibers)88. However, the majority of the studies published on transcutaneous Raman spectra of bone tissue have focused on developing an efficient SORS set-up, optimizing spectra collection parameters, and developing a mathematical method to extract bone spectra from the combined spectra involving the Raman signal contribution from both soft and bone tissues (e.g., band-target entropy minimization, multivariate curve resolution, and parallel factor analysis). More recently, two studies have further showed that in vivo transcutaneous Raman analysis can predict mechanical properties of murine bone93. Nonetheless, only two pilot studies have investigated the feasibility of in vivo, non-invasive clinical SORS analysis of bone in osteoporosis and other bone diseases (i.e., osteogenesis imperfecta or OI) research. These two pilot studies used either principal component analysis (PCA)11 or ν1PO4/Amide III ratio94 to detect chemical compositional differences associated with diseases compared to the healthy subject.
Although SORS is a promising method for clinical Raman analysis of bone, several technical drawbacks still hinder it from being actively implemented in the field. For example, the spectra collected via fiber-optic SORS exhibit distortions in band shapes and position of spectra collected from different layers due to imaging imperfections present at some level with any spectrograph90. Such distortions restrain the extraction of true pure spectrum belong to individual layers within a sample, even after using multivariate decomposition methods. Secondly, the possible penetration depth in current SORS methods with a good SNR is up to a couple of millimeters95,96, resulting in implementing the SORS on a limited area of the human body (e.g., tibia or phalanges). With the advances in RS and optics technology, such technical drawbacks could be overcome soon, making the SORS method widely used in the bone research area.
6.2. High-wavenumber RS
While the vast majority of bone studies utilizing Raman spectroscopy have primarily been focused on fingerprint spectral range (300–1800 cm−1), the potential of high-wavenumber spectral region (generally, 2500–4500 cm−1) in Probe RS and Raman micro-spectroscopy has been also under exploration for physicochemical analysis of bone matrix, especially for water compartments14. This high-wavenumber region has distinct and strong spectral features of lipids, proteins, collagen, and water. Unal et al.14 developed a novel method of measurement different water content in bone using a custom-built RS instrument with an objective lens (10X, NA = 0.4) in the spectral range between 2500 cm−1 and 4000 cm−1. They further reported associations of RS-based water content measurement with biomechanical properties of bovine97 and human cortical bone98 and radiation sterilization of human cortical bone99. However, collecting Raman spectra over a high wavelength region poses different requirements on RS instrument such as different gratings, filters, and more importantly detector. Commercial RS instruments with NIR lasers are currently not capable of collecting data at this spectral range due to the rapid efficiency reduction of CCDs at this spectral range (discussed in Section 2.1.3). Alternatively, Probe RS with correct filter and laser (e.g., 680 nm) with a CCD optimized for 785 nm can acquire Raman signals in the high wavenumber region100. Thus, it is possible to cover the fingerprint regions and the high wavenumber region when assessing bone spectra to measure all three main components of bone (mineral, collagen, and water).
7. Other techniques and opportunities in the analysis of Raman spectra of bone
Recent developments in the analysis of Raman spectra can bring new insight to the assessment of bone composition and facilitate the implementation of RS to address unanswered questions. Such developments include quantitative analysis, complex mixture analysis, and Raman imaging/mapping. Qualitative analysis determines concentrations of different biological molecules and substrates33 with partial least-squares (PLS) models such as the multivariate calibration that was used to quantify the concentrations of glucose, lactate, and urea101. In quantitative analysis, the intensity of an individual peak and the intensity or integrated area ratios of two peaks are calibrated against known concentrations of a certain substrate using a linear regression analysis102,103 or/and PLS regression (PLSR)101,104 to obtain a calibration curve and equation. Such curve and equation are then used to predict the amount of a substrate when using the same RS device. In the bone field, so far, such an approach has been implemented in three studies. In first study, the amount of pyridinoline, a trivalent collagen crosslink, was analyzed quantitatively using the area of amide I sub bands at 1660 cm−1 83. In second study, type B carbonated apatite with different concentrations was analyzed quantitatively using the peak area ratios of 1071/960 cm−1 55. In the third study, the amount of bone mineral in the matrix using ν2PO4/Amide III was calibrated against the amount of mineral as measured by qBEI105.
The shape and height of the fluorescence background depend on both the sample being analyzed and the components of the RS instrument being used. As such, for the same RS instrument and type of bone samples, the degree and shape of the fluorescence background could potentially provide insight into bone quality such as the quantification of AGEs. When AGEs accumulate following glycation of bone, the amount of fluorescence background also elevates drastically23 indicating background fluorescence in RS is a marker of AGE content. Yet, to the best of our knowledge, there has been no study investigating fluorescence background of a Raman spectrum to extract meaningful information for bone or other biological tissues.
Raman imaging or mapping is another technique with potential to investigate the role of heterogeneity in bone fragility. This technique creates images of chemical moieties based on the spatial acquisition of Raman spectra in a selected region of interest. To create a Raman map, spectra can be acquired by either point-by-point mapping or line-focus mapping. In the point-by-point approach, the laser is focused on a spot and then after the spectrum is collected, a motorized microscope stage moves the sample under the laser to another spot whereupon another spectrum is collected. This is done sequentially in the defined region of interest. In line-focus mapping approach, the laser illuminates a line on the sample enabling the collection of spectra simultaneously from multiple positions at a single acquisition. The latter can raster across a region and saves time. Generating a Raman map can take couple minutes up to a couple days depending on the size of the area being mapped. There are multiple studies in the literature investigating bone matrix composition and orientation using Raman imaging/mapping techniques36,106,107, but unlike FTIR imaging108, Raman maps have yet to be acquired to determine whether compositional heterogeneity is associated with osteoporosis. With technical advances in RS technology and in the field of artificial intelligence and machine learning, Raman mapping has potential to investigate heterogeneity in bone matrix composition as well as collagen orientation within a short time-period.
Combining RS or surface-enhanced Raman (SERS) with different chemometric and curve fitting techniques, overlapping features in Raman spectra can be extracted. As an example, such an approach enabled label-free analysis and monitoring of osteogenic differentiation of human mesenchymal stem cells109–111. The identified features can then identify and characterize bone cells112–116. Beyond the scope of the present review is the use of machine learning algorithms in RS to extract information pertinent to bone when the spectra contain signals from soft tissue such as skin and muscle. For those interested multivariate techniques to assess bone characteristics, we refer readers to the work of Berger and co-workers87,93.
8. Recommendations for the use of Raman spectroscopy in bone studies
Since the quality and background fluorescence of Raman spectra of bone depends on the RS instrument, reproducibility and comparisons across studies require the reporting of key information about the components of the instrument, pre-processing techniques, and property determination. Moreover, the sample characteristics should be also provided, including whether the bone specimens were polished, dehydrated, or embedded as well as anatomical locations since the quality and fluorescence background of Raman spectra are also dependent on the characteristics of the bone sample.
With respect to the instrument, we recommend reporting the following parameters: laser wavelength (nm), laser power (mW) as measured by a laser meter placed at the surface of bone being analyzed, magnification (e.g., 20X) and numerical aperture of the objective lens (e.g., NA = 0.5), scan time of 1 spectrum (s or min), number of accumulations, the actual wavenumber range of the Raman shift (e.g., 360 cm−1 to 1780 cm−1) prior to truncating the ends (if applicable), the nature of confocality (e.g., slit/pinhole size = 10 μm to 200 μm), the approximate SNR values of at least two bands, preferably the ν1 phosphate and amide I, and the spectral resolution (cm−1). There is not a standard method for assessing SNR, and so if reporting it, a description of how SNR was determined may be necessary. Alternatively, investigators can provide representative spectra of bone samples.
It is important to note that removing background fluorescence does not guarantee that the true spectral shape of the bone tissue is being analyzed because the polynomial or piecewise baseline fit to the spectrum may not be the true baseline of many overlapping Raman peaks. Therefore, obtaining meaningful physiochemical properties of bone depends on consistently implementing such subtraction methods within a study. For any method used to remove background fluorescence, we suggest overlaying the raw and subtracted spectra and searching for intensities that are negative or appear exaggerated (i.e., higher than indicated by the raw spectrum due to overfitting) (Fig. S10). Once the selected fitting parameters (polynomial order, wavenumber range, and base points) have been established to not cause negative intensities nor exaggerated peak heights, they should be used to subtract background fluorescence from all spectra in a study. We recommend reporting the method that was used to remove background fluorescence and whether any additional ‘corrections’ were applied to define the baseline of peaks.
The optimal parameters of the smoothing process of Raman spectra (i.e., noise filtering) are not as easy to identify, especially since they depend on the SNR or spectral quality of the Raman signals being collected. We suggest a limited use of digital filtering when determining primary Raman properties of bone. If filtering is necessary to identify peaks, we recommend first selecting a higher polynomial order (e.g., 4th degree vs. 2nd degree) and narrower window size (e.g., 11) because this preserves much of original signals (it takes a light touch to noise suppression). If noise is still obscuring the location of peaks, the window size can be increased followed by a higher order polynomial (5th degree vs. 4th degree). However, selecting a wide window size (e.g., >20 for spectral resolution of 1 cm−1) and/or high polynomial order (e.g., >4th degree) for the S-G filter is risky because of the likelihood of masking small peaks and shoulders (i.e., distorting the shape of the original spectrum). Regardless of the parameters chosen, we strongly recommend using the same filtering parameters for all spectra within a given acquisition protocol and reporting the noise filtering process.
To minimize potential discrepancies among studies of bone using RS, we also suggest clearly stating the method by which Raman properties were determined. This includes baseline definition of each band of interest and the method for quantifying each peak of interest (maximum intensity or peak height, integrated area, or the use of fitted sub-bands). Reporting wavenumber location of each peak intensity, wavenumber range of the integrated area, and specifics on the deconvolution procedure are all useful information for making comparisons among different studies and ensuring reproducibility.
Because excellent fits of the sub-bands to the amide I band can be achieved for different deconvolution methods, we recommend using direct peak height ratios in which the location of the sub-peaks is determined by the local minima of the second derivative spectrum. Regardless of using the direct approach or an indirect deconvolution method, we highly recommend reporting i) the wavenumber range of the amide I band, ii) any baseline corrections or additional noise filtering, iii) the number of sub-bands that were fit (or used in the direct determination) and the reason for that number (e.g., 2nd derivative spectrum consistently had 4 minima below zero), iv) the functions being fit (e.g., Gaussian, Lorentzian, or blend of the two), and v) whether the positions of the sub-bands were fixed or allowed to move. To provide transparency, examples of the 2nd derivative spectrum, fitted sub-bands with R2 values, and the average wavenumber position of each sub-band can be provided in supplemental information, if not in the main body of the manuscript. In our experience, the wavenumber position of each sub-band varies within ~3 cm−1 of the local minima, and so we prefer to label our sub-peak ratios with 3 significant digits (e.g., 1610 cm−1 instead of 1605 cm−1 and 1670 cm−1 instead of 1668 cm−1). Even though the obtained peak-fitting solution is mathematically correct, the deconvolution is not a unique solution. Moreover, deconvolution of the amide I band into sub-bands is not necessarily attributable to the physicochemical properties of bone117.
9. Conclusions
When information about the composition of bone at the ultrastructural level of organization is desired without destroying the hydrated bone sample (e.g., because it is required for mechanical testing), Raman spectroscopy can provide spatial assessments of mineral-to-matrix ratio (mineralization), carbonate-to-phosphate ratio (Type B substitution), width of a phosphate peak (crystallinity), and other compositional characteristics. The sensitivity of RS to differences in these characteristics depends on the components of the instrument, acquisition parameters, sample preparation, and the methods for processing the spectra and then determining peak ratios. Therefore, users must ensure that the RS instrument is calibrated, the laser power is maximized without harming the tissue, optics of the instrument focus the light at the site of interest and are properly aligned to deliver the maximum number of photons to the spectrometer, and the balance between acquisition time and number of acquisitions provides spectra with minimal noise without being impractical.
Diode lasers in RS emit light with a preferred orientation, even without added optics, and so the orientation of bone samples must be consistent during the collection of Raman spectra. Collecting multiple spectra distributed throughout the specimen also minimizes polarization bias (i.e., peak ratios depend on bone orientation) and minimizes noise if the spectra are averaged into one spectrum prior to processing. Because bone contains fluorophores, subtracting background fluorescence is often necessary. Ideally, the quality or signal-to-noise of the Raman spectra is sufficient to preclude the use of a digital noise filter because it may mask subtle, but important Raman peaks of interest.
After pre-processing the acquired Raman spectra, using peak intensity ratios may provide better sensitivity to differences in compositional properties of bone than integrated area ratios because there is much less uncertainty in wavenumber location of peaks than in wavenumber range of corresponding bands and because peak heights are less sensitive than band areas to residual fluorescence (i.e., true baseline). For noisy spectra, integrated area ratios may however be the more sensitive method. Regardless, reporting how spectra are pre-processed and how peak ratios are determined ensures reproducibility and comparisons of findings across RS studies of bone.
Maximizing the inherent quality of the spectra that a given RS instrument can provide and then judiciously selecting pre-processing methods based on this spectral quality improves the ability of RS to identify differences in bone composition and organization among experimental groups. Furthermore, the potential for RS to provide new information about the physiochemical nature of bone and its contribution to fracture resistance can be improved if future studies report key details about the wavelength laser, the laser power density at the bone surface, sample preparation, numerical aperture of the objective lens (or the size and number of fiber optics), background fluorescence subtraction, and noise filtering.
Supplementary Material
Acknowledgements
The authors are grateful for funding from the National Institute of Arthritis and Musculoskeletal and Skin Diseases and the VA Office of Research and Development. Without such funding, our work involving Raman spectroscopy would not be possible. The content herein is of course the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or other funding agencies.
Grant Supporters:
NIAMS, VA
References
- 1.Walton AG, Deveney MJ and Koenig JL, Calc Tiss Res, 1970, 6, 162–167. [DOI] [PubMed] [Google Scholar]
- 2.Dendramis AL, Poser JW and Schwinn EW, Biochim Biophys Acta, 1983, 742, 525–529. [DOI] [PubMed] [Google Scholar]
- 3.Gevorkian BZ, Arnotskaia NE and Fedorova EN, Biofizika+, 1984, 29, 1046–52. [PubMed] [Google Scholar]
- 4.Ohsaki K, Yamashita S, Fujita A, Masuda Y, Ueda S, Sugiura T, Tamura K and Shibata A, Am J Otolaryng, 1988, 9, 117–126. [DOI] [PubMed] [Google Scholar]
- 5.Carden A and Morris MD, J Biomed Opt, 2000, 5, 259–268. [DOI] [PubMed] [Google Scholar]
- 6.Matousek P and Morris MD, Eds., Emerging Raman Applications and Techniques in Biomedical and Pharmaceutical Fields, Springer, 2010. [Google Scholar]
- 7.Goodyear SR and Aspden RM, Methods Mol Biology, 2012, 816, 527–534. [DOI] [PubMed] [Google Scholar]
- 8.Mandair GS and Morris MD, Bonekey Reports, 2015, 4, 620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paschalis EP, Gamsjaeger S and Klaushofer K, Osteoporosis Int, 2017, 28, 2275–2291. [DOI] [PubMed] [Google Scholar]
- 10.Taylor EA and Donnelly E, Bone, 2020, 139, 115490. [DOI] [PubMed] [Google Scholar]
- 11.Buckley K, Kerns JG, Vinton J, Gikas PD, Smith C, Parker AW, Matousek P and Goodship AE, J Raman Spectrosc, 2015, 46, 610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shipp DW, Sinjab F and Notingher I, Adv Opt Photonics, 2017, 9, 315. [Google Scholar]
- 13.Schrof S, Varga P, Hesse B, Schöne M, Schütz R, Masic A and Raum K, Acta Biomaterialia, 2016, 44, 51–64. [DOI] [PubMed] [Google Scholar]
- 14.Unal M, Yang S and Akkus O, Bone, 2014, 67, 228–236. [DOI] [PubMed] [Google Scholar]
- 15.Morris MD and Mandair GS, Clin Orthop Relat Res, 2011, 469, 2160–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Penel G, Leroy G, Rey C and Bres E, Calcified Tissue Int, 1998, 63, 475–481. [DOI] [PubMed] [Google Scholar]
- 17.Querido W, Ailavajhala R, Padalkar M and Pleshko N, Appl Spectrosc, 2018, 72, 1581–1593. [DOI] [PubMed] [Google Scholar]
- 18.McCreery RL, Raman spectroscopy for chemical analysis, Wiley-Interscience, 2000, vol. 157. [Google Scholar]
- 19.Smith E and Dent G, Modern Raman Spectroscopy, Wiley, 2nd edn., 2019. [Google Scholar]
- 20.Mahadevan-Jansen A, Patil CA and Pence IJ, in Biomedical Photonics Handbook, ed. Vo-Dinh T, 2014. [Google Scholar]
- 21.Pence I and Mahadevan-Jansen A, Chem Soc Rev, 2016, 45, 1958–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Butler HJ, Ashton L, Bird B, Cinque G, Curtis K, Dorney J, Esmonde-White K, Fullwood NJ, Gardner B, Martin-Hirsch PL, Walsh MJ, McAinsh MR, Stone N and Martin FL, Nat Protoc, 2019, 11, 664–687. [DOI] [PubMed] [Google Scholar]
- 23.Unal M, Uppuganti S, Leverant CJ, Creecy A, Granke M, Voziyan P and Nyman JS, J Biophotonics, 2018, 11, e201700352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patil CA, Pence IJ, Lieber CA and Mahadevan-Jansen A, Opt Lett, 2014, 39, 303. [DOI] [PubMed] [Google Scholar]
- 25.Lerner JM, Cytom Part A, 2006, 69A, 712–734. [DOI] [PubMed] [Google Scholar]
- 26.Battey DE, Slater JB, Wludyka R, Owen H, Pallister DM and Morris MD, Appl Spectrosc, 2016, 47, 1913–1919. [Google Scholar]
- 27.Creasey D, Akkus O and Yang S, Spectroscopy, 2017, 32, 46–54. [Google Scholar]
- 28.Yeni YN, Yerramshetty J, Akkus O, Pechey C and Les CM, Calcified Tissue Int, 2006, 78, 363–371. [DOI] [PubMed] [Google Scholar]
- 29.Nyman JS, Makowski AJ, Patil CA, Masui TP, O’Quinn EC, Bi X, Guelcher SA, Nicollela DP and Mahadevan-Jansen A, Calcified Tissue Int, 2011, 89, 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Esmonde-White FWL, Schulmerich MV, Esmonde-White KA and Morris MD, P Soc Photo-opt Ins, 2009, 7166, 716605–716605–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bocklitz TW, Dörfer T, Heinke R, Schmitt M and Popp J, Spectrochimica Acta Part Mol Biomol Spectrosc, 2015, 149, 544–549. [DOI] [PubMed] [Google Scholar]
- 32.Ray KG and McCreery RL, Appl Spectrosc, 1997, 51, 108–116. [Google Scholar]
- 33.Pelletier MJ, Appl Spectrosc, 2003, 57, 20A–42A. [DOI] [PubMed] [Google Scholar]
- 34.Makowski AJ, Patil CA, Mahadevan-Jansen A and Nyman JS, J Biomed Opt, 2013, 18, 055005–055005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Makowski AJ, Pence IJ, Uppuganti S, Zein-Sabatto A, Huszagh MC, Mahadevan-Jansen A and Nyman JS, J Biomed Opt, 2014, 19, 117008–117008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kazanci M, Roschger P, Paschalis EP, Klaushofer K and Fratzl P, J Struct Biol, 2006, 156, 489–496. [DOI] [PubMed] [Google Scholar]
- 37.Kazanci M, Wagner HD, Manjubala NI, Gupta HS, Paschalis E, Roschger P and Fratzl P, Bone, 2007, 41, 456–461. [DOI] [PubMed] [Google Scholar]
- 38.Raghavan M, Sahar ND, Wilson RH, Mycek M-A, Pleshko N, Kohn DH and Morris MD, J Biomed Opt, 2010, 15, 037001–037001–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bocklitz T, Walter A, Hartmann K, Rösch P and Popp J, Anal Chim Acta, 2011, 704, 47–56. [DOI] [PubMed] [Google Scholar]
- 40.Engel J, Gerretzen J, Szymańska E, Jansen JJ, Downey G, Blanchet L and Buydens LMC, Trac Trends Anal Chem, 2013, 50, 96–106. [Google Scholar]
- 41.Gautam R, Vanga S, Ariese F and Umapathy S, Epj Techniques Instrum, 2015, 2, 8. [Google Scholar]
- 42.Nyman JS, Uppuganti S, Unal M, Leverant CJ, Adabala S, Granke M, Voziyan P and Does MD, JBMR Plus, 2019, 3, e10135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Penel G, Leroy G and Bres E, Appl Spectrosc, 1998, 52, 312–313. [Google Scholar]
- 44.Chen T-C, Shea DA and Morris MD, Appl Spectrosc, 2002, 56, 1035–1037. [Google Scholar]
- 45.Shea DA and Morris MD, Appl Spectrosc, 2002, 56, 182–186. [Google Scholar]
- 46.Golcuk K, Mandair GS, Callender AF, Sahar N, Kohn DH and Morris MD, Biochim Biophys Acta, 2006, 1758, 868–873. [DOI] [PubMed] [Google Scholar]
- 47.Lieber CA and Mahadevan-Jansen A, Appl Spectrosc, 2003, 57, 1363–1367. [DOI] [PubMed] [Google Scholar]
- 48.Zhao J, Lui H, McLean DI and Zeng H, Appl Spectrosc, 2007, 61, 1225–1232. [DOI] [PubMed] [Google Scholar]
- 49.Cao A, Pandya AK, Serhatkulu GK, Weber RE, Dai H, Thakur JS, Naik VM, Naik R, Auner GW, Rabah R and Freeman DC, J Raman Spectrosc, 2007, 38, 1199–1205. [Google Scholar]
- 50.Wartewig S, in IR and Raman Spectroscopy: Fundamental Processing, Wiley-VCH Verlag GmbH & Co., 2003, pp. 75–122. [Google Scholar]
- 51.Unal M, Uppuganti S, Timur S, Mahadevan-Jansen A, Akkus O and Nyman JS, Sci Rep, 2019, 9, 7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Savitzky A and Golay MJE, Anal Chem, 1964, 36, 1627–1639. [Google Scholar]
- 53.Feuerstein D, Parker KH and Boutelle MG, Anal Chem, 2009, 81, 4987–4994. [DOI] [PubMed] [Google Scholar]
- 54.Bromba MUA and Ziegler H, Anal Chem, 1981, 53, 1583–1586. [Google Scholar]
- 55.Awonusi A, Morris MD and Tecklenburg MMJ, Calcified Tissue Int, 2007, 81, 46–52. [DOI] [PubMed] [Google Scholar]
- 56.Wang M, Qian R, Bao M, Gu C and Zhu P, Mater Lett, 2018, 210, 203–206. [Google Scholar]
- 57.Creecy A, Uppuganti S, Girard MR, Schlunk SG, Amah C, Granke M, Unal M, Does MD and Nyman JS, Bone, 2020, 130, 115126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gamsjaeger S, Klaushofer K and Paschalis EP, J Raman Spectrosc, 2014, 45, 794–800. [Google Scholar]
- 59.Akkus O, Adar F and Schaffler MB, Bone, 2004, 34, 443–453. [DOI] [PubMed] [Google Scholar]
- 60.Rux DR, Song JY, Pineault KM, Mandair GS, Swinehart IT, Schlientz AJ, Garthus KN, Goldstein SA, Kozloff KM and Wellik DM, J Bone Miner Res, 2017, 32, 1750–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Massie C, Knapp E, Chen K, Berger AJ and Awad HA, J Biomech, 2021, 116, 110243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Khan AF, Awais M, Khan AS, Tabassum S, Chaudhry AA and Rehman IU, Appl Spectrosc Rev, 2013, 48, 329–355. [Google Scholar]
- 63.Buckley K, Matousek P, Parker AW and Goodship AE, J Raman Spectrosc, 2012, 43, 1237–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martinez MG, Bullock AJ, MacNeil S and Rehman IU, Appl Spectrosc Rev, 2019, 54, 1–34. [Google Scholar]
- 65.Movasaghi Z, Rehman S and Rehman IU, Appl Spectrosc Rev, 2007, 42, 493–541. [Google Scholar]
- 66.Talari ACS, Movasaghi Z, Rehman S and ur Rehman I, Appl Spectrosc Rev, 2014, 50, 46–111. [Google Scholar]
- 67.Unal M, Jung H and Akkus O, J Bone Miner Res, 2016, 31, 1015–1025. [DOI] [PubMed] [Google Scholar]
- 68.Viguet-Carrin S, Garnero P and Delmas PD, Osteoporosis Int, 2006, 17, 319–336. [DOI] [PubMed] [Google Scholar]
- 69.Garnero P, Bonekey Reports, 2012, 1, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vashishth D, Curr Osteoporos Rep, 2007, 5, 62–66. [DOI] [PubMed] [Google Scholar]
- 71.Garnero P, Calcified Tissue Int, 2015, 97, 229–240. [DOI] [PubMed] [Google Scholar]
- 72.Saito M and Marumo K, Calcified Tissue Int, 2015, 97, 242–261. [DOI] [PubMed] [Google Scholar]
- 73.Bandekar J, Biochim Biophys Acta, 1992, 1120, 123–143. [DOI] [PubMed] [Google Scholar]
- 74.Paschalis EP, Verdelis K, Doty SB, Boskey AL, Mendelsohn R and Yamauchi M, J Bone Miner Res, 2001, 16, 1821–1828. [DOI] [PubMed] [Google Scholar]
- 75.Paschalis EP, Gamsjaeger S, Tatakis DN, Hassler N, Robins SP and Klaushofer K, Calcified Tissue Int, 2015, 96, 18–29. [DOI] [PubMed] [Google Scholar]
- 76.Krafft C, Codrich D, Pelizzo G and Sergo V, J Biophotonics, 2008, 1, 154–169. [DOI] [PubMed] [Google Scholar]
- 77.Kimura-Suda H and Ito T, J Oral Biosci, 2017, 59, 142–145. [DOI] [PubMed] [Google Scholar]
- 78.Paschalis EP, Shane E, Lyritis G, Skarantavos G, Mendelsohn R and Boskey AL, J Bone Miner Res, 2004, 19, 2000–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gourion-Arsiquaud S, Faibish D, Myers E, Spevak L, Compston J, Hodsman A, Shane E, Recker RR, Boskey ER and Boskey AL, J Bone Miner Res, 2009, 24, 1565–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Khalid M, Bora T, Ghaithi AA, Thukral S and Dutta J, Sci Rep, 2018, 8, 9417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tchanque-Fossuo CN, Gong B, Poushanchi B, Donneys A, Sarhaddi D, Gallagher KK, Deshpande SS, Goldstein SA, Morris MD and Buchman SR, Bone, 2012, 52, 712–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McNerny EM, Gong B, Morris MD and Kohn DH, J Bone Miner Res, 2015, 30, 455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gamsjaeger S, Robins SP, Tatakis DN, Klaushofer K and Paschalis EP, Calcified Tissue Int, 2017, 100, 565–574. [DOI] [PubMed] [Google Scholar]
- 84.Matousek P, Clark IP, Draper ERC, Morris MD, Goodship AE, Everall N, Towrie M, Finney WF and Parker AW, Appl Spectrosc, 2005, 59, 393–400. [DOI] [PubMed] [Google Scholar]
- 85.Schulmerich MV, Dooley KA, Vanasse TM, Goldstein SA and Morris MD, Appl Spectrosc, 2007, 61, 671–678. [DOI] [PubMed] [Google Scholar]
- 86.Keller MD, Majumder SK and Mahadevan-Jansen A, Opt Lett, 2009, 34, 926–8. [DOI] [PubMed] [Google Scholar]
- 87.Maher JR, Inzana JA, Awad HA and Berger AJ, J Biomed Opt, 2013, 18, 077001–077001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cui H, Glidle A and Cooper JM, Ieee Access, 2020, 8, 62905–62911. [Google Scholar]
- 89.Morris MD, Matousek P, Towrie M, Parker AW, Goodship AE and Draper ERC, J Biomed Opt, 2005, 10, 014014–0140147. [DOI] [PubMed] [Google Scholar]
- 90.Matousek P, Appl Spectrosc, 2006, 60, 1341–1347. [DOI] [PubMed] [Google Scholar]
- 91.Schulmerich MV, Cole JH, Kreider JM, Esmonde-White F, Dooley KA, Goldstein SA and Morris MD, Applied spectroscopy, 2009, 63, 286–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Demers J-LH, Esmonde-White FWL, Esmonde-White KA, Morris MD and Pogue BW, Biomed Opt Express, 2015, 6, 793–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen K, Massie C and Berger AJ, J Biophotonics, 2020, 13, jbio.202000256–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Buckley K, Kerns JG, Gikas PD, Birch HL, Vinton J, Keen R, Parker AW, Matousek P and Goodship AE, Ibms Bonekey, 2014, 11, 602. [Google Scholar]
- 95.Maher JR and Berger AJ, Appl Spectrosc, 2009, 64, 61–65. [DOI] [PubMed] [Google Scholar]
- 96.Sowoidnich K, Churchwell JH, Buckley K, Goodship AE, Parker AW and Matousek P, J Raman Spectrosc, 2016, 47, 240–247. [Google Scholar]
- 97.Unal M and Akkus O, Bone, 2015, 81, 315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Unal M, J Biomech, 2021, 119, 110342. [DOI] [PubMed] [Google Scholar]
- 99.Flanagan CD, Unal M, Akkus O and Rimnac CM, J Mech Behav Biomed, 2017, 75, 314–321. [DOI] [PubMed] [Google Scholar]
- 100.Masson LE, O’Brien CM, Pence IJ, Herington JL, Reese J, van Leeuwen TG and Mahadevan-Jansen A, Analyst, 2018, 143, 6049–6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ren M and Arnold MA, Anal Bioanal Chem, 2007, 387, 879–888. [DOI] [PubMed] [Google Scholar]
- 102.Li B, Yang S and Akkus O, Analyst, 2013, 139, 823–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Okotrub KA, Surovtsev NV, Semeshin VF and Omelyanchuk LV, Cytom Part A, 2015, 87, 68–73. [DOI] [PubMed] [Google Scholar]
- 104.Rohleder D, Kiefer W and Petrich W, Analyst, 2004, 129, 906–911. [DOI] [PubMed] [Google Scholar]
- 105.Roschger A, Gamsjaeger S, Hofstetter B, Masic A, Blouin S, Messmer P, Berzlanovich A, Paschalis EP, Roschger P, Klaushofer K and Fratzl P, J Biomed Opt, 2014, 19, 065002–065002. [DOI] [PubMed] [Google Scholar]
- 106.Timlin JA, Carden A, Morris MD, Rajachar RM and Kohn DH, Anal Chem, 2000, 72, 2229–2236. [DOI] [PubMed] [Google Scholar]
- 107.Schrof S, Varga P, Galvis L, Raum K and Masic A, J Struct Biol, 2014, 187, 266–275. [DOI] [PubMed] [Google Scholar]
- 108.Boskey AL, Donnelly E, Boskey E, Spevak L, Ma Y, Zhang W, Lappe J and Recker RR, J Bone Miner Res, 2016, 31, 1070–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.McManus LL, Burke GA, McCafferty MM, O’Hare P, Modreanu M, Boyd AR and Meenan BJ, Analyst, 2011, 136, 2471–2481. [DOI] [PubMed] [Google Scholar]
- 110.Hung P-S, Kuo Y-C, Chen H-G, Chiang H-HK and Lee OK-S, Plos One, 2013, 8, e65438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Moura CC, Tare RS, Oreffo ROC and Mahajan S, J Roy Soc Interface, 2016, 13, 20160182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Smith SJ, Emery R, Pitsillides A, Clarkin CE and Mahajan S, Analyst, 2017, 142, 1962–1973. [DOI] [PubMed] [Google Scholar]
- 113.Notingher I, Gough JE and Hench LL, Key Eng Mat, 2004, 254–256, 769–772. [DOI] [PubMed] [Google Scholar]
- 114.McManus LL, Bonnier F, Burke GA, Meenan BJ, Boyd AR and Byrne Hugh. J., Analyst, 2012, 137, 1559–1569. [DOI] [PubMed] [Google Scholar]
- 115.Brauchle E and Schenke-Layland K, Biotechnol J, 2013, 8, 288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jung GB, Kang IS, Lee YJ, Kim D, Park H-K, Lee G-J and Kim C, Current Optics and Photonics, 2017, 4, 412–420. [Google Scholar]
- 117.Unal M, Connect Tissue Res, 2020, 61, 1–2. [DOI] [PubMed] [Google Scholar]
- 118.Taylor EA, Lloyd AA, Salazar-Lara C and Donnelly E, Appl Spectrosc, 2017, 71, 2404–2410. [DOI] [PubMed] [Google Scholar]
- 119.Gourion-Arsiquaud S, Burket JC, Havill LM, DiCarlo E, Doty SB, Mendelsohn R, van der Meulen MC and Boskey AL, J Bone Miner Res, 2009, 24, 1271–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Turunen MJ, Saarakkala S, Rieppo L, Helminen HJ, Jurvelin JS and Isaksson H, Appl Spectrosc, 2011, 65, 595–603. [DOI] [PubMed] [Google Scholar]
- 121.Donnelly E, Boskey AL, Baker SP and van der Meulen MCH, J Biomed Mater Res A, 2010, 92A, 1048–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hammond MA, Gallant MA, Burr DB and Wallace JM, Bone, 2014, 60, 26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Creecy A, Uppuganti S, Merkel AR, O’Neal D, Makowski AJ, Granke M, Voziyan P and Nyman JS, Calcified Tissue Int, 2016, 99, 289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Raghavan M, Sahar ND, Kohn DH and Morris MD, Bone, 2012, 50, 942–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Uppuganti S, Granke M, Makowski AJ, Does MD and Nyman JS, Bone, 2016, 83, 220–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yerramshetty JS, Lind C and Akkus O, Bone, 2006, 39, 1236–1243. [DOI] [PubMed] [Google Scholar]
- 127.Freeman JJ, Wopenka B, Silva MJ and Pasteris JD, Calcified Tissue Int, 2001, 68, 156–162. [DOI] [PubMed] [Google Scholar]
- 128.Tanaka Y, Yajima N, Okada M, Matsumoto T, Higuchi Y, Miyazaki S, Yamato H and Hirata T, Analyst, 2017, 142, 4265–4278. [DOI] [PubMed] [Google Scholar]
- 129.Iwasaki Y, Kazama JJ, Yamato H and Fukagawa M, Bone, 2011, 48, 1260–1267. [DOI] [PubMed] [Google Scholar]
- 130.Rubin MR, Paschalis EP, Poundarik A, Sroga GE, McMahon DJ, Gamsjaeger S, Klaushofer K and Vashishth D, Plos One, 2016, 11, e0154700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Burke MV, Atkins A, Akens M, Willett TL and Whyne CM, J Orthop Res, 2016, 34, 2126–2136. [DOI] [PubMed] [Google Scholar]
- 132.Gamsjaeger S, Brozek W, Recker R, Klaushofer K and Paschalis EP, J Bone Miner Res, 2014, 29, 608–617. [DOI] [PubMed] [Google Scholar]
- 133.Misof BM, Gamsjaeger S, Cohen A, Hofstetter B, Roschger P, Stein E, Nickolas TL, Rogers HF, Dempster D, Zhou H, Recker R, Lappe J, McMahon D, Paschalis EP, Fratzl P, Shane E and Klaushofer K, J Bone Miner Res, 2012, 27, 2551–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bi X, Patil CA, Lynch CC, Pharr GM, Mahadevan-Jansen A and Nyman JS, J Biomech, 2011, 44, 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bi X, Grafe I, Ding H, Flores R, Munivez E, Jiang MM, Dawson B, Lee B and Ambrose CG, J Bone Miner Res, 2016, 32, 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yerramshetty JS and Akkus O, Bone, 2008, 42, 476–482. [DOI] [PubMed] [Google Scholar]
- 137.Inzana JA, Maher JR, Takahata M, Schwarz EM, Berger AJ and Awad HA, J Biomech, 2013, 46, 723–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Makowski AJ, Granke M, Ayala OD, Uppuganti S, Mahadevan-Jansen A and Nyman JS, Appl Spectrosc, 2017, 71, 2385–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Carretta R, Stüssi E, Müller R and Lorenzetti S, Biomed Res Int, 2015, 2015, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Donnelly E, Chen DX, Boskey AL, Baker SP and van der Meulen MCH, Calcified Tissue Int, 2010, 87, 450–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Burket JC, Brooks DJ, MacLeay JM, Baker SP, Boskey AL and van der Meulen MCH, Bone, 2013, 52, 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kim G, Cole JH, Boskey AL, Baker SP and van der Meulen MCH, Calcified Tissue Int, 2014, 95, 125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ojanen X, Isaksson H, Töyräs J, Turunen MJ, Malo MKH, Halvari A and Jurvelin JS, J Biomech, 2015, 48, 269–275. [DOI] [PubMed] [Google Scholar]
- 144.Meganck JA, Begun DL, McElderry JD, Swick A, Kozloff KM, Goldstein SA, Morris MD, Marini JC and Caird MS, Bone, 2013, 56, 204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gong B, Oest ME, Mann KA, Damron TA and Morris MD, Bone, 2013, 57, 252–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Orkoula MG, Vardaki MZ and Kontoyannis CG, Vib Spectrosc, 2012, 63, 404–408. [Google Scholar]
- 147.Timchenko EV, Zherdeva LA, Timchenko PE, Volova LT and Ponomareva UV, Physcs Proc, 2015, 73, 221–227. [Google Scholar]
- 148.Fiedler IAK, Casanova M, Keplinger T, Busse B and Müller R, J Biomed Opt, 2018, 23, 116504. [DOI] [PubMed] [Google Scholar]
- 149.Ng AH, Omelon S, Variola F, Allo B, Willett TL, Alman BA and Grynpas MD, J Bone Miner Res, 2016, 31, 369–379. [DOI] [PubMed] [Google Scholar]
- 150.Taylor EA, Donnelly E, Yao X, Johnson ML, Amugongo SK, Kimmel DB and Lane NE, Calcified Tissue Int, 2019, 106, 303–314. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.