

ABSTRACT
Galectin-3 (Gal3) can be expressed by many cells in the tumor microenvironment (TME), including cancer cells, cancer-associated fibroblasts, tumor-associated macrophages, and regulatory T cells (Tregs). In addition to immunosuppression, Gal3 expression has been connected to malignant cell transformation, tumor progression, and metastasis. In the present study, we found spontaneous T-cell responses against Gal3-derived peptides in PBMCs from both healthy donors and cancer patients. We isolated and expanded these Gal3-specific T cells in vitro and showed that they could directly recognize target cells that expressed Gal3. Finally, therapeutic vaccination with a long Gal3-derived peptide epitope, which induced the expansion of Gal3-specific CD8+ T cells in vivo, showed a significant tumor-growth delay in mice inoculated with EO771.LMB metastatic mammary tumor cells. This was associated with a significantly lower percentage of both Tregs and tumor-infiltrating Gal3+ cells in the non-myeloid CD45+CD11b− compartment and with an alteration of the T-cell memory populations in the spleens of Gal3-vaccinated mice. These results suggest that by activating Gal3-specific T cells by an immune-modulatory vaccination, we can target Gal3-producing cells in the TME, and thereby induce a more immune permissive TME. This indicates that Gal3 could be a novel target for therapeutic cancer vaccines.
KEYWORDS: Galectin-3, Gal3, tumor microenvironment, immune modulatory vaccine
Introduction
The tumor microenvironment (TME) harbors an ongoing, complex interplay between a variety of cell types, including immune cells, cancer cells, stromal cells, and extracellular matrix components, which can promote tumor proliferation, survival, and metastasis.1 Cancers employ various immune-evasive mechanisms to survive,1,2 including the expression of the immunosuppressive molecule Galectin-3 (Gal3). Gal3 is a polyfunctional protein with high binding affinity for β-galactosides and glycosylated molecules. It has been linked to the transformation, adhesion, and proliferation of malignant cells, tumor progression, metastasis, and suppression of the anti-cancer immune response.1,3,4 By oligomerization in the extracellular space, Gal3 can link glycosylated surface proteins, cells, or extracellular matrix molecules, which subsequently induces intracellular signal transduction.1 Elevated Gal3 protein levels have been associated with poor survival in several cancers such as leukemia, lymphoma, thyroid-and breast cancer.3 Furthermore, Gal3 is highly expressed by cancer cells and classic immunosuppressive cell types like macrophages and other myeloid cells, mesenchymal stromal cells (namely fibroblasts),1,3,5,6 and Tregs.7 Extracellularly, Gal3 has been linked to a reduction in T-cell receptor affinity for its cognate ligand8 and the induction of apoptosis in activated human and murine T cells,9–12 limiting the tumor-specific immune response. Several studies have linked the immunosuppressive mechanisms of Gal3 to its carbon-recognition domain (CRD).9–11 Furthermore, in vitro studies have shown that Gal3 diminishes T-cell cytotoxicity toward cancer cells, impairs interferon (IFN)γ-secreting capabilities, and negatively influences the expansion rate, cytotoxicity, and cytokine-secreting capabilities of human tumor-infiltrating lymphocytes (TILs).8,13–15 These findings suggest that targeting Gal3 could be beneficial in a cancer immunotherapeutic setting, as reducing Gal3 could enhance the survival of tumor-reactive T cells by removing the apoptotic functions of Gal3 in addition to improving T-cell IFNγ-secreting capabilities.
In solid cancers, the accumulation of TILs in the tumor is considered a good prognostic factor.16 Hence, new therapeutic strategies have focused on increasing the immune infiltration to turn the TME from a poorly infiltrated, “cold” phenotype into an immune-infiltrated, “hot” phenotype.17 However, the presence of immunosuppressive molecules like Gal3 in the TME directly influences the ability of infiltrating T cells to battle cancer cells. Targeting these molecules is therefore of utmost importance in order to achieve a clinically successful treatment.
In previous studies, we have shown that pro-inflammatory, self-reactive effector T cells, so-called anti-regulatory T cells (anti-Tregs),2 were spontaneously present in cancer patients. These anti-Tregs could specifically target immunosuppressive cells by recognizing epitopes derived from cornerstone immunoregulatory proteins including indoleamine 2,3-dioxygenase (IDO),18,19 programmed death-ligand 1 (PD-L1)20,21 and 2 (PD-L2),22 CCL22,23 arginase-1,24 arginase-2,25 and TGFβ.26 Based on those findings, in the present study we investigated whether healthy donors (HD) and cancer patients harbored a natural immune response against peptides derived from the CRD of Gal3. In addition, we aimed to determine whether Gal3-derived peptides could be used to activate and expand Gal3-specific T cells in vivo. Finally, we evaluated the therapeutic potential of Gal3-derived peptide cancer vaccination in a syngeneic murine model of breast cancer. We show that both healthy donors and cancer patients harbor a natural response against Gal3-derived peptides, and that these cells can be utilized in vitro to directly target Gal3-expressing cancer cells. In vivo, a therapeutic Gal3-derived peptide vaccine demonstrated pre-clinical efficacy and immune modulatory effects.
Materials and methods
Healthy donor and patient PBMCs
Buffy coats from healthy, anonymized donors were acquired from the Blood Bank at Copenhagen University Hospital, Rigshospitalet, Denmark. According to the Danish Law of Research Ethics §14 section 3, the usage of anonymized biological material does not require approval from an ethics committee. Patient samples were collected at the National Center for Cancer Immune Therapy (CCIT-DK), Copenhagen University Hospital Herlev, in accordance with the provisions of the Declarations of Helsinki and informed, written consent. Patients had been diagnosed with malignant melanoma, multiple myeloma, breast-, kidney-or prostate cancer, myeloproliferative neoplasms or Waldenström’s macroglobulinemia. Peripheral blood mononuclear cells (PBMCs) were isolated and processed as described by Martinenaite et al.23 PBMCs were cultured in X–vivo 15 (Lonza, Basel, Switzerland) supplemented with 5% human serum.
Peptides
Gal3Long1 was synthesized by Schafer-N ApS (Copenhagen, Denmark). Gal3Long1 included five 9–10-mer human leukocyte antigen (HLA)-A2-predicted peptides: Gal301, Gal305, Gal307, Gal308, and Gal310. Short Gal3 peptides were synthesized by K.J. Ross-Petersen (Klampenborg, Denmark). The murine version of the Gal3Long1 peptide, mGal3Long1, was synthetized by Schafer-N ApS. mGal3Long1 included six 9-mer Major Histocompatibility Complex (MHC)-I-predicted epitopes: mGal301, mGal302, mGal303, mGal304, mGal305, and mGal306, synthetized by Pepscan (Lelystad, The Netherlands). Short epitopes were predicted using the SYFPEITHI database (http://www.syfpeithi.de). Peptide sequences, purity and reconstitution information can be found in Table 1. The HLA-A2-restricted HIV peptide (ILKEPVHGV) and HLA-A3-restricted HIV peptide (RLRPGGKKK) were synthetized by TAG Copenhagen A/S (Copenhagen, Denmark), and reconstituted in 2 mM H2O. Peptides dissolved in H2O were filtered (0.22 µm) before use.
Table 1.
Amino acid (aa) sequences, length, purity (%) and reconstitution information of the long and short Gal3 peptides used in this study. Murine peptides and the differences in sequences between the murine and human peptides are indicated in bold. Dashes are inserted to ensure proper alignment
| Peptide | Sequence | Length | Purity | Reconstitution |
|---|---|---|---|---|
| Gal3Long1 | AGPLIVPYNLPLPGGVVPRMLITILGTV | 28aa | 97.1% | 10 mM DMSO |
| mGal3Long1 | AGPLTVPYDLPLPGGVMPRMLITIMGTV | 28aa | 90.4% | 10 mM DMSO |
| Gal301 | – – – – – – – – – – – – RMLITILGTV | 10aa | 84.70% | 10 mM DMSO |
| Gal305 | – – – – – – – – – –GVVPRMLITI– – – - | 10aa | 82.74% | 2 mM sterile H2O |
| Gal307 | – – – – – – – – – – – – – MLITILGTV | 9aa | 92.07% | 10 mM DMSO |
| Gal308 | – – – LIVPYNLPL– – – – – – – – – – - | 9aa | 86.61% | 10 mM DMSO |
| Gal310 | – – – – – – NLPLPGGVV– – – – – – – | 9aa | 91.08% | 2 mM sterile H2O |
| mGal301 | HRMKNLREI | 9aa | 95.4% | 2 mM sterile H2O |
| mGal302 | KVAVNDAHL | 9aa | 93.6% | 2 mM sterile H2O |
| mGal303 | – – – – – – – – – – VMPRMLITI– – – – - | 9aa | 94.9% | 2 mM sterile H2O |
| mGal304 | TVKPNANRI | 9aa | 95.7% | 2 mM sterile H2O |
| mGal305 | KPNANRIVL | 9aa | 97.5% | 2 mM sterile H2O |
| mGal306 | FNENNRRVI | 9aa | 98.5% | 2 mM sterile H2O |
Enzyme-linked ImmunoSPOT (ELISPOT)
PBMCs were stimulated with 10 µM peptide, and IL-2 (Novartis, Basel, Switzerland) to a final concentration of 120 U/ml was added on day two. In vitro culturing with IL-2 can sometimes result in an unspecific activation of T cells in the PBMC pool, thereby leading do a high background in the ELISPOT assay in the control wells. ELISPOT was set up on day seven with up to 0.5 × 106 PBMCs/well. Murine ELISPOTs were set up with up to 0-8-0.9 × 106 splenocytes or lymph node cells/well or 0.2 × 106 sorted CD4+ or CD8a+ splenocytes. The latter were seeded with bone marrow-derived dendritic cells (BMDCs) working as antigen presenting cells (APC) in a 1:2 (T cell: APC) ratio. In all setups, the cells were seeded in an ELISPOT plate (MultiScreen MAIP N45; Merck, Darmstadt, Germany) pre-coated with IFNγ-specific capture antibodies (Mabtech, Stockholm, Sweden), stimulated with peptide at a 5 µM working concentration and incubated for 14–18 h at 37°C, 5% CO2. For murine ELISPOTs, Concanavalin A (1 μg/well) was used as a positive control. Following the incubation, the plates were washed and secondary IFNγ-specific biotinylated antibody (Mabtech) was added. After a 2 h incubation, plates were washed and streptavidin-conjugated alkaline phosphatase (Mabtech) was added, followed by a 1 h incubation. Next, plates were washed and the BCIP/NBT substrate (Mabtech) was added. Developed ELISPOT plates were analyzed with a CTL Immunospot S6 Ultimate-V analyzer and ImmunoSpot software, version 5.1 (CTL Analyzers, Shaker Heights, OH, USA). Unless otherwise stated, all experiments were performed in at least triplicate. All ELISPOT well images were color corrected to grayscale using Microsoft Power Point (Microsoft, Redmond, WA, USA).
Generation of dendritic cells
Monocytes were isolated from human PBMCs by CD14+ enrichment (#130-050-201, Miltenyi Biotec, Bergisch Gladbach, Germany) and matured into dendritic cells (DCs) as previously described.23 For murine experiments, BMDCs were generated from bone marrow cells isolated from the tibia and femur of C57BL/6 mice as described by Jæhger and Hübbe et al.27
Establishment of antigen-specific T-cell cultures (CTL cultures)
PBMCs from a patient diagnosed with malignant melanoma were stimulated twice with irradiated, Gal307-loaded, autologous DCs, and three times with irradiated, Gal307-loaded PBMCs, one week apart. The day after DC stimulation or PBMC stimulation, the cultures received 40 U/ml IL-7 and 20 U/ml IL-12 (both from Preprotech, Cranbury, NJ, USA) or 120 U/ml IL-2, respectively. The purity of the culture was inspected by tetramer staining.
Rapid expansion protocol and enrichment of Gal3-specific T cells
Gal3-specific T cell cultures were cloned by limiting dilution in rapid expansion protocol (REP) media, consisting of 20 ml X–vivo 15 with 5% human serum, 20 × 106 irradiated feeder cells, 0.6 µg anti-CD3 (clone OKT3, eBioscience, ThermoFischer Scientific, Waltham, MA, USA), and IL-2 to a final concentration of 6000 U/ml. Twice per week, half of the medium was replaced with fresh medium, containing 3000 U/ml IL-2. The cells were cryopreserved or used in assays after 15 days of culture.
Flow cytometry analysis
PBMCs were re-stimulated with 0.25 mM peptide for 5 h in the presence of GolgiPlugTM (BD Biosciences, San José, CA, USA), which was added 1 h after peptide-stimulation. Cells were extracellularly and intracellularly stained as described by Weis-Banke et al.25 We used the following human antibodies, all from BD Biosciences: anti-CD4-FITC (#347413), anti-CD107a-PE (#555801), anti-CD8-PerCP (#345774), anti-CD3-APC-H7 (#560275), Fluorescent Viability Stain-510 (#564406), anti-HLA-A2-PE (#558570), anti-CD45-APC (#340910), anti-IFNγ-APC (#341117) and anti-tumor necrosis factor (TNF)α-BV421 (#562783). PE-and APC-linked HLA-tetramers (Tetramer Shop, Copenhagen, Denmark and in-house) were loaded with peptide and used for staining following the manufacturer’s instructions. Flow cytometry analyses of human cells were performed on a BD FACSCanto II. Data were analyzed with BD FACSDiva v.8.0.1. For murine samples, Fc receptors were blocked (FcR Blocking Reagent, Miltenyi Biotec). Cells were stained extracellularly, permeabilized and stained intracellularly with the following antibodies, which were all supplied by BioLegend, San Diego, CA, USA unless otherwise stated: anti-CD45-FITC (#103108), anti-CD25-PE-Cy7 (#101916), anti-MMR(CD206)-PE-Cy7 (#141720), anti-CD8a-APC (#100712), anti-Ly6G-APC (#560599, BD Biosciences), anti-CD44-APC-Cy7 (#103027), anti-CD11b-APC-Cy7 (#101226), anti-Ly6C-AF700 (#128024), anti-CD3-AF700 (#100216), anti-CD4-BV421 (#100216), anti-F4/80 (#123131), anti-CD8a-BV605 (#563152, BD Biosciences), anti-CD62 L-BV650 (#564108, BD Biosciences), Zombie Aqua (#423102), anti-Gal3-PE (#126706), and anti-FoxP3-APC (#17-5773-82, eBioscience). Flow cytometry analyses were performed on an ACEA NovoCyte Quanteon (Agilent Technologies, Santa Clara, CA, USA). Data were analyzed with FlowJo v.10.6.1 (BD Biosciences). Gating strategies can be found in Supplementary Figures S6–9.
Cancer cell lines
All human cell lines were cultured in RPMI-1640 supplemented with 10% fetal bovine serum (FBS) (from here on termed R10) or in the recommended medium. The following cell lines were used: T2, THP-1, and HS-5 (from American Type Culture Collection, Manassas, VA, USA); Set-2, OCI-AML2, OCI-AML3, and U2940 (from Deutsche Sammlung von Mikroorganismen und Zellkulturen, Braunschweig, Germany); PB2B and MAC-2A (a kind gift from Niels Ødum, University of Copenhagen, Denmark); K562 cells stably transduced with HLA-A2 or HLA-A3; K562-A2 and K562-A3 (a kind gift from Mariam Heemskerk, University Hospital Leiden, The Netherlands); Marimo (a kind gift from Steffen Koschmieder, Universitätsklinikum Aachen, Germany); and UKE-1 (a kind gift from W. Fiedler, University Hospital of Eppendorf, Germany). PB2B and MAC-2A were from cutaneous lymphoma; Marimo, OCI-AML2, OCI-AML-3, THP-1, Set-2, and UKE-1 from acute myeloid leukemia (AML); and U2960 from diffuse large B cell lymphoma. The murine cancer cell line EO771.LMB (a kind gift from Janine Erler, University of Copenhagen, Denmark, and originally described by Johnstone et al.28) was maintained in Dulbecco’s modified Eagle´s medium (DMEM) with 20% FBS, 20 mM HEPES, and penicillin and streptomycin (Pen/Strep) to a final concentration of 100 U/ml and 100 μg/ml, respectively. All cell lines were tested and confirmed negative for mycoplasma.
Chromium release assay
0.5 × 106 cancer cells (target cells) were labeled with peptide, loaded with 100μCi 51Chromium (51Cr, Perkin Elmer, Waltham, MA, USA) and incubated for 1 h at 37ᵒC. Excess 51Cr and peptide was removed by washing in R10. Next, cells were placed in 96-well plates with Gal3-specific T cells (effector cells), at various effector:target ratios, and incubated for 4 h. Then, 100 μl medium was aspirated and the 51Cr release (experimental) was counted in a Perkin Elmer Wizard2 2470 Automatic Gamma Counter. The maximum and spontaneous 51Cr release was determined in separate wells by adding 100 μl 10% Triton X-100 or R10 to target cells. Specific lysis was calculated by: lysis(%) = ((experimental 51Cr release−spontaneous 51Cr release)/(maximum51Cr release−spontaneous 51Cr release))×100.
Generation of a Gal3-transduced K562-A2 cell line
Gal3 cDNA (NM_002306.3) was synthesized and inserted into a third-generation lentiviral vector (pTRP-EGFP, generously provided by Dr. James L. Riley, University of Pennsylvania, Philadelphia, PA, USA) between 5′AvrII and 3′SalI restriction sites (GeneArt, ThermoFisher Scientific) to generate the lentiviral vector, pTRP-EGFP-T2A-Gal3. Lentivirus was produced by transfecting the construct into 293 T human embryonic kidney cells cultured in DMEM, 10% FBS, 100IU/ml penicillin, and 100 µg/ml streptomycin. Briefly, cells were seeded at 2.5 × 105 per well in a 6-well plate, 24 h before transfection. Cells were transfected with 1 µg pTRP-EGFP-T2A-Gal3 lentivirus and 0.5 µg packaging and envelope plasmids (pTRP-RSV.Rev, pTRP-GAG-Pol, and pTRP-VSVg) mixed with TurboFect Transfection Reagent (ThermoFisher Scientific). Cells were cultured in a humidified atmosphere with 5% CO2 for 48 h, before collecting the viral supernatant. K562-A2 cells were stably transduced by incubating with the lentivirus supernatant for 72 h before sorting using a BD FACSAria flow cytometer. The same protocol was used to generate control K562-A2 cells transduced with the lentiviral vector pTRP-EGFP. Purity was inspected by flow cytometry for GFP expression before each assay.
HS-5 co-culture
0.5 × 106 HS-5 cells were cultured until adherence was reached before the addition of 3 × 106 cells of different suspension cancer cell lines. After 48 h of co-culture, the suspension cancer cells were used directly in ELISPOT assays or CD45+-sorted (#130-045-801, Miltenyi Biotec), inspected for purity by flow cytometry and frozen as cell pellets at −80ᵒC for later use in Western blots.
Animals
Female C57BL/6 mice were obtained from the animal facility at the Department of Oncology, Copenhagen University Hospital Herlev, Denmark, or from Taconic M&B (Borup, Denmark). Mice were acclimatized for at least two weeks and used for experiments at 6–18 weeks of age. Experimental procedures were approved by the Danish Animal Experimentation Council (2016–15-0201-01020) and performed according to the Danish guidelines.
Murine tumor studies
0.5 × 106 EO771.LMB cells in 100 μl of DMEM were inoculated subcutaneously into the right flank. Mice were stratified into treatment groups with equal average tumor volume when the tumors became palpable. Mice were sacrificed upon signs of distress, ulceration on the tumor or when the tumor volume exceeded 1500 mm3. Tumors were measured by digital caliper and their volume calculated by: Volume = (length×width2)/2.
Peptide vaccination
Mice were vaccinated subcutaneously on the left flank with an emulsion containing 100 μg of peptide. The emulsions were generated by mixing the peptide solution and the clinical-grade incomplete Freund’s adjuvant, Montanide (Seppic Inc., Courbevoie, France) 1:1. Control vaccinations were generated as a emulsion of an equivalent amount of dimethyl sulfoxide (DMSO) as used for the peptide solution, which was then diluted in sterile water and finally emulsified 1:1 with Montanide. For preliminary evaluation of the peptide immunogenicity in vivo, mice were vaccinated once. For tumors studies, mice were vaccinated for the first time on the day of stratification into treatment groups and every 7 days until the end of the experiment.
Single-cell suspensions of spleen, lymph nodes, and tumor tissues
Spleen and lymph nodes (inguinal, caudal, lumbar) were forced through a 70 μm cell strainer and red blood cells were lysed using RBC Lysis Buffer (Qiagen, Hilden, Germany). Tumors were excised, weighed, and placed in digestion buffer (RPMI supplemented with 1% Pen/Strep, 2.1 mg/ml collagenase type I, 75 µg/ml DNase I, and 5 mM CaCl2) before being cut into pieces and incubated overnight in an end-over-end rotator at 4ᵒC. The following day, the tumors where forced through a 70 μm cell strainer and red blood cells were lysed like described above.
CD4+ and CD8+ T cell isolation from splenocytes
CD4+ and CD8+ T cells were isolated from splenocytes by positive selection using CD4 (L3T4) MicroBeads, mouse (130–117-043; Miltenyi Biotec) and CD8a (Ly-2) MicroBeads, mouse (130–117-044; Miltenyi Biotec), respectively, following the manufacturer’s instructions.
Statistical analysis
Statistical analyses of single ELISPOT responses were performed with the distribution-free resampling rule (DFR = p ≤ .05) described by Moodie et al29 and performed with RStudio (http://www.rstud io.com/). Difference between groups in ELISPOT responses and flow cytometry measurements were assessed with the non-parametric, unpaired Mann-Whitney test provided in GraphPad Prism software (v8.0.0, GraphPad Software, San Diego, CA, USA). The difference in average tumor growth between control and Gal3Long1-vaccinated mice was evaluated with a Mixed Effect model using GraphPad Prism. For all statistical analyses, p values ≤.05 were considered statistically significant.
Results
Natural T-cell responses against a long galectin-3-derived peptide in healthy donors and cancer patients
To determine whether healthy donors and cancer patients harbored a natural T-cell response against Gal3, PBMCs were stimulated with low-dose IL-2 (120 U/ml) and the 28-mer Gal3Long1 peptide (Table 1) for one week. Gal3Long1 is derived from the CRD of the Gal3 protein, that have previously been connected to the immunosuppressive mechanisms of Gal3 toward T cells.9–11 70-75% of screened individuals (15/20 healthy donors and 23/33 cancer patients (10 malignant melanoma, four myeloproliferative neoplasms, six prostate cancer, one kidney cancer, five multiple myeloma and seven Waldenström’s macroglobulinemia)) showed significant Gal3Long1-specific responses in an IFNγ ELISPOT assay (Figure 1a,b). We observed no difference in the frequency of Gal3Long1-specific responses among healthy donors and cancer patients (Figure 1c). We assessed the phenotype of the T cells that were activated upon stimulation with Gal3Long1 in one healthy donor and two patients diagnosed with malignant melanoma by intracellular cytokine staining for IFNγ and TNFα production of Gal3Long1-stimulated PBMCs. We detected CD4+ T-cell responses in all three samples (Figure 1d). In addition, we found CD8+ T-cell responses against Gal3Long1 in the healthy donor (Figure 1e), suggesting that Gal3Long1 contains both HLA class I and II T-cell epitopes.
Figure 1.
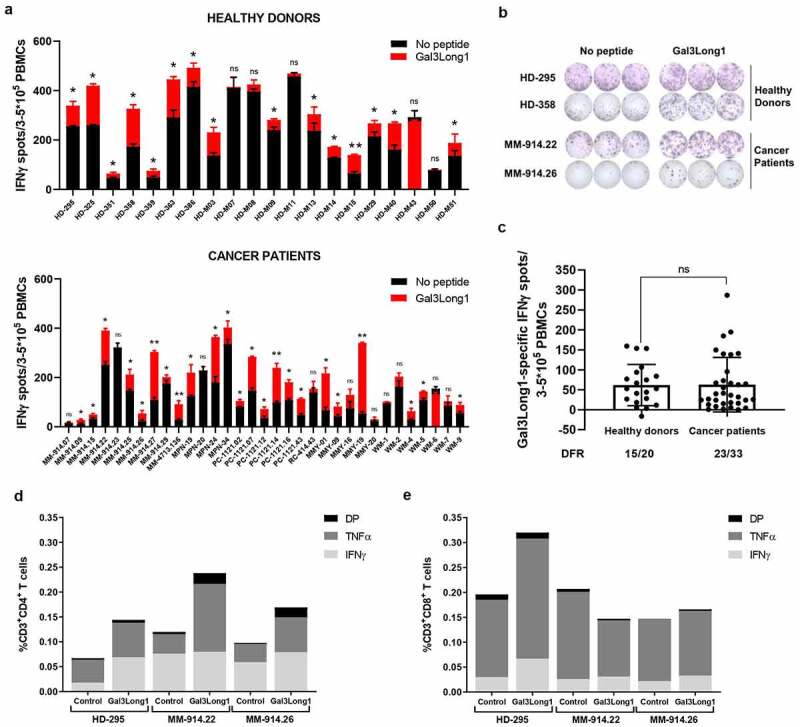
Gal3-reactive T cells are present in healthy donors and cancer patients. PBMCs from healthy donors and cancer patients were stimulated with low-dose IL-2 (120 U/ml) and Gal3Long1 for 7 days. (a) IFNγ ELISPOT responses against Gal3Long1 peptide in PBMCs from (A, top) healthy donors (n = 20) and (A, bottom) cancer patients (n = 33). Superimposed bars represent the mean number of spots per well in unstimulated (no peptide) or stimulated (Gal3Long1) conditions + SD. Experiments were performed in triplicates. Statistical analysis of ELISPOT responses was performed with the distribution-free resampling (DFR) model (ns = not significant, * = p ≤ .05, ** = p ≤ .01). (b) Representative example of ELISPOT responses against Gal3Long1 in samples shown in (a). (c) IFNγ ELISPOT specific responses against the Gal3Long1 peptide in healthy donors (n = 20) and patients with cancer (n = 33) shown in (a). Each dot represents the difference in mean counts of IFNγ-secreting cells between peptide-stimulated and control wells in ELISPOT. Data shown as mean -/+ SD. Statistical differences between groups were assessed with a Mann-Whitney test (ns = not significant). The number of significant responses of total assessed samples based on the DFR method is indicated below the figure. (d and e) One week after the initial stimulation, cells were re-stimulated with Gal3Long1 or no peptide (control) and assessed by an intracellular staining for TNFα and IFNγ production in (d) CD4+and (e) CD8+ T cells. HD = healthy donor, MM = malignant melanoma, MPN = myeloproliferative neoplasm, PC = prostate cancer, RC = kidney cancer, MMY = multiple myeloma, WM = Waldenström’s macroglobulinemia, DP = double positive for IFNγ and TNFα production.
Identification of HLA-A2-restricted galectin-3 epitopes
To characterize the Gal3Long1-specific CD8+ T-cell response, we screened four HLA-A2-matched healthy donors for responses against five 9–10-mer peptide epitopes selected based on their predicted affinity to HLA-A2 and contained within the Gal3Long1 sequence: Gal301, Gal305, Gal307, Gal308, and Gal310 (Table 1) in an IFNγ ELISPOT assay. We detected immune responses against three peptides: Gal301, Gal307 and Gal308 (Figure 2a). We then validated the Gal3-specific response against the two peptides that activated the highest responses, Gal307 and Gal308, in eight healthy donors and five cancer patients (four malignant melanoma and one breast cancer) for Gal307 (Figure 2b–d) or eight healthy donors and six cancer patients (five malignant melanoma and one breast cancer) for Gal308 (Figure 2e–g). The strongest responses were elicited by the Gal307 peptide (Figure 2b,c). Cancer patients appeared to harbor a stronger response against Gal307, especially two patients diagnosed with malignant melanoma, compared to healthy donors (Figure 2d).
Figure 2.
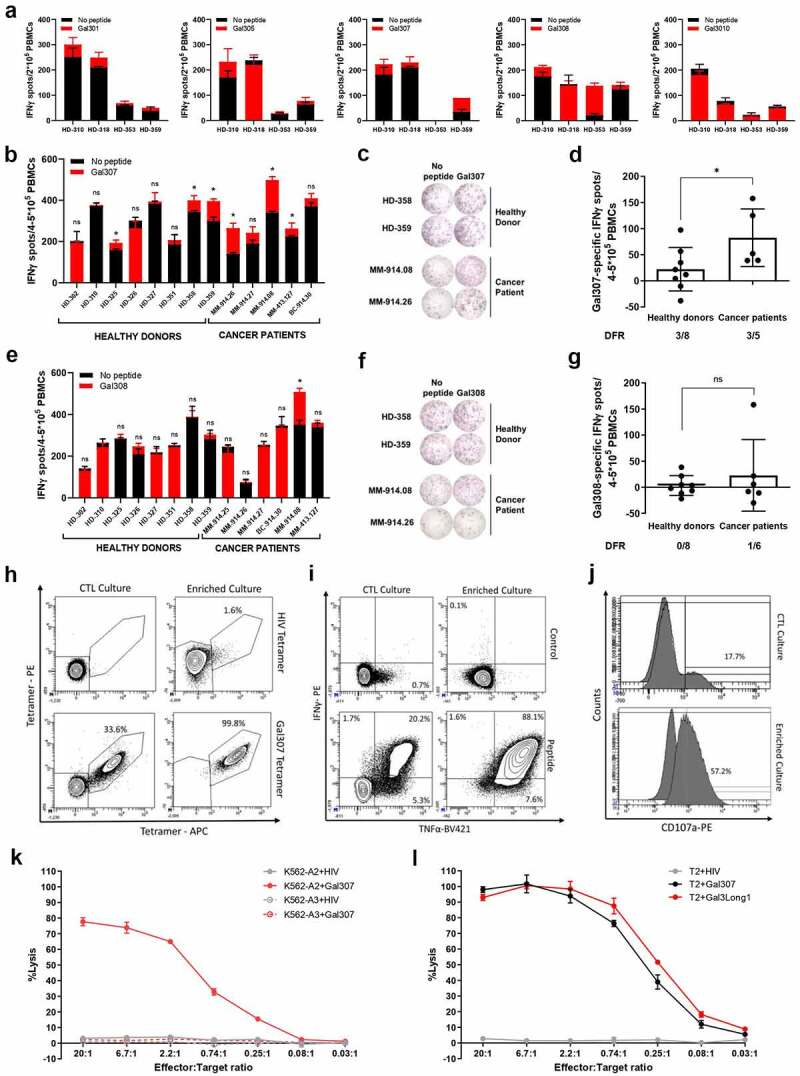
Healthy donors and cancer patients harbor a natural response against short Gal3 peptide epitopes, which results in the recognition and lysis of cancer cells pulsed with Gal3-derived peptides when Gal3-specific T cells are enriched and expanded. (a) IFNγ ELISPOT responses in PBMCs from healthy donors (n = 3–4) that were unstimulated or stimulated with short HLA-A2-predicted Gal3Long1-derived peptides (Gal301, Gal305, Gal307, Gal308, and Gal310). The experiment was run in duplicates. (b) IFNγ ELISPOT responses in PBMCs from healthy donors (n = 8) and cancer patients (n = 5) that were unstimulated or stimulated with Gal307. (c) Representative example of ELISPOT responses against Gal307 in samples shown in (b). (d) IFNγ ELISPOT specific responses against Gal307 peptide in healthy donors (n = 8) and patients with cancer (n = 5) shown in (b). (e) IFNγ ELISPOT responses in PBMCs from healthy donors (n = 8) and cancer patients (n = 6) that were unstimulated or stimulated with Gal308. (f) Representative example of ELISPOT responses against Gal308 in samples shown in (e). (g) IFNγ ELISPOT specific responses against Gal308 peptide in healthy donors (n = 8) and patients with cancer (n = 6) shown in (e). For (a), (b) and (e), superimposed bars represent the mean number of spots per well in unstimulated (no peptide) or stimulated (+peptide) conditions + SD. Experiments were performed in (a) duplicates or (b and e) triplicates. Statistical analysis of ELISPOT responses was performed with the distribution-free resampling (DFR) model (ns = not significant, * = p ≤ .05). For (d) and (g) each dot represents the difference in mean counts of IFNγ-secreting cells between peptide-stimulated and control wells in ELISPOT. Data shown as mean -/+ SD. Statistical differences between groups were assessed with a Mann-Whitney test (ns = not significant, * = p ≤ .05). The number of significant responses of total assessed samples based on the DFR method is indicated below the figure. HD = healthy donor, MM = malignant melanoma, BC = breast cancer. (h) Staining for HLA-A2-Gal307 tetramer+ cells in Gal307-specific (left) cytotoxic T-lymphocyte (CTL) cultures and (right) enriched cultures stimulated with (top) HIV Tetramer control or (bottom) Gal307 tetramers. (i) Intracellular cytokine staining for IFNγ and TNFα production in Gal307-specific (left) CTL cultures and (right) enriched cultures stimulated with (top) non-stimulated control (bottom) or Gal307. (j) Staining for CD107a in Gal307-specific (top) CTL cultures and (bottom) enriched culture. The histogram overlay shows the percentage upregulation of CD107a upon Gal307 stimulation compared to no-peptide control. (k) Percentage of lysis of HIV-derived peptide or Gal307-pulsed HLA-A2-transfected K562 (K562-A2) or HLA-A3-transfected K562 (K562-A3) cancer cell lines after exposure to enriched Gal3-specific T-cell cultures in a 51Cr release assay. Data points represent mean -/+ SD. (l) Percentage of lysis of HIV-derived peptide, Gal307 or Gal3Long1-pulsed T2 cancer cells after exposure to enriched Gal3-specific T-cell cultures in a 51Cr release assay. Data points represent mean -/+ SD.
Galectin-3-specific T cells recognize target cancer cells in a peptide and HLA-restricted manner
In order to characterize and validate the function of Gal3-specific T cells, we established a culture of Gal307-specific CD8+ T cells derived from a patient diagnosed with malignant melanoma by stimulating PBMCs five times with peptide-pulsed, autologous DCs and PBMCs (CTL culture). Next, we rapidly expanded the specific cells and cloned them by limiting dilution (enriched culture) before using them in downstream experiments. The expansion increased the specificity of the culture from 33.6% in the CTL culture to 99.8%, as assessed by HLA-A2-Gal307 tetramer+ cells (Figure 2h). The specificity and responsiveness of both cultures were confirmed by an increase in the percentage of TNFα+ and- IFNγ+ cells (Figure 2i), and cells positive for the degranulation marker CD107a (Figure 2j) following stimulation with Gal307 peptide.
We next assessed the ability of a Gal307-specific enriched culture to recognize and kill cancer cell lines pulsed with Gal3-derived peptides in a 51Cr release assay. We show that Gal307-specific T cells recognized and efficiently killed Gal307-pulsed K562 cancer cell line (Figure 2k). However, the killing was only seen when an HLA-A2-transduced cell line (K562-A2) was used, thereby confirming the HLA-A2 restriction of the Gal307 epitope. We lastly assessed whether Gal307-specific T cells could recognize and kill cancer cells pulsed with the longer Gal3-derived peptide, Gal3Long1. In order to do so, we used the TAP-deficient T2 cell line, which has previously been described to cross-present long peptides.23,30 We found that the Gal307-specific T-cell culture could lyse not only Gal307-loaded T2 cells but also T2 cells pulsed with the 28-mer Gal3Long1 (Figure 2l).
Galectin-3-specific T cells recognize and kill galectin-3-expressing target cells
Next, we investigated the ability of Gal3-specific T cells to recognize and kill Gal3-expressing cells. To that end, we generated a K562-A2 cancer cell line stably transduced with Gal3 and/or GFP as a control (Supplementary Figure S1). We confirmed by IFNγ ELISPOT that a Gal307-specific T-cell culture was able to recognize Gal3-expressing cells, as Gal307-specific T-cells secreted significantly more IFNγ upon stimulation with the Gal3 transduced cell line compared with a GFP transduced control (Figure 3a). Next, we sought to prove the recognition and killing of naturally Gal3-expressing cancer cell lines by Gal3-specific T cells. As Gal3 generally plays a role in supporting malignant cells in leukemias and lymphomas,3 we screened several HLA-A2+ hematopoietic cancer cell lines to see if our Gal3-specific T cells would recognize them. An IFNγ ELISPOT assay of Gal307-specific T cells with PB2B, Marimo, OCI-AML2, OCI-AML3, Set-2, U2960, THP-1 and UKE-1 cancer cell lines resulted in the detection of significant Gal307-specific IFNγ responses (Figure 3b), indicating that Gal307-specific T cells can recognize the mentioned cancer cell lines.
Figure 3.
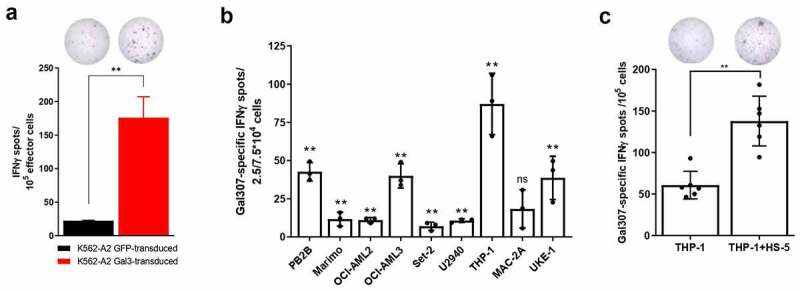
Gal3-specific T cells recognize Gal3-producing cancer cells. (a, top) Representative example of IFNγ ELISPOT response of (a, bottom) enriched Gal307-specific T-cell response against K562-A2 cells transduced with Gal3 or a GFP control. 105 effector cells were used for this experiment with a 3:1 effector to target ratio. Bars represent the mean number of spots per well + SD. The experiment was performed in triplicates. (b) IFNγ ELISPOT Gal307-specific T cell responses against different HLA-A2-matched cancer cell lines after exposure to enriched Gal307-specific T cells. 2.5 × 104 effector cells were used for THP-1, MAC-2A and UKE-1, while 7.5 × 104 effector cells were used for the rest of the target cell lines. Both used a 3:1 effector to target cell ratio. Each dot represents the difference in mean counts of IFNγ-secreting cells between Gal307-specific T cells assayed with or without different cancer cell lines. Bars represent the mean number of spots per well -/+ SD. (c, bottom) IFNγ ELISPOT Gal307-specific T-cell responses in Gal3-specific enriched T-cell cultures against THP-1 cells cultured with or without HS-5 cells for 48 h (n = 6). 105 effector cells were used for this experiment in a 3:1 effector to target cell ratio. Each dot represents the difference in mean counts of IFNγ-secreting cells between Gal307-specific T cells stimulated with THP-1 or THP-1 in co-culture with HS-5. Bars represent the mean number of spots per well -/+ SD. (d, top) Representative examples of IFNγ ELISPOT responses shown in (c, bottom). Data shown as mean -/+ SD. Statistical analysis of single ELISPOT responses in (a) and (b) was performed with the distribution-free resampling (DFR) model. Statistical differences between groups in (c) were assessed with a Mann-Whitney test. * = p ≤ .05, ** = p ≤ .01.
Yamamoto-Sugitani and colleagues previously showed that co-culturing chronic myelogenous leukemia (CML) cell lines with the HLA-A2− fibroblast cell line HS-5, of bone marrow stromal cell origin, led to Gal3 upregulation in the CML cells.31 We investigated in a similar setup, whether this upregulation could occur in the AML cell line THP-1,32 known for its phenotypic and functional plasticity,33 and whether that could affect the THP-1 recognition by Gal3-specific T cells. HS-5 cells were cultured until adherence was reached before addition of the suspension THP-1 cells. Following a 48 h co-culture, we set up an IFNγ ELISPOT assay with THP-1 cells alone or in co-culture with HS-5 to test whether Gal307-specific T cells could increase their recognition of the THP-1 cells. We observed a significantly higher Gal307-specific IFNγ response when THP-1 were co-cultured with HS-5 cells, compared to THP-1 cells alone in six independent experiments (Figure 3c). HS-5, as opposed to THP-1 cells, are HLA-A2− (Supplementary Figure S2A-B), therefore the specific IFNγ response by Gal307-specific T-cells is a direct result of recognition of Gal3-expressing THP-1 cells. To confirm this, we showed that Gal307-specific T cells do not secrete IFNγ upon interaction with HS-5 cells (Supplementary Figure S2C) and cannot lyse Gal307-pulsed HS-5 cells (Supplementary Figure S2D). Finally, we assessed whether the increased recognition of the co-cultured THP-1 cells was due to the HS-5-dependent induction of Gal3 expression in THP-1 cells. In order to do so, THP-1 cells were sorted from the co-culture based on CD45+ expression (Supplementary Figure S3A), yielding a CD45+ cell fraction with a 95.5% purity (Supplementary Figure S3B). We then performed Western blot analyses to evaluate Gal3 protein expression in cells of the enriched fraction. We found that the co-culture of THP-1 cells with HS-5-1 cells resulted in an increase in the levels of Gal3 protein in the THP-1 fraction (Supplementary Figure S3C). Notably, we did not detect any Gal3 upregulation in K562-A2, K562-A3, UKE-1 or Set-2 following HS-5 co-culture (Supplementary Figure S3D).
Therapeutic effect of Galectin-3-based cancer vaccination in vivo
To validate Gal3 as a target for therapeutic cancer vaccines in vivo, we assessed the immunogenicity of Gal3Long1 in mice. We detected a strong and significant Gal3Long1-specific response in both the spleen and in the draining lymph nodes (dLNs) of Gal3Long1-vaccinated mice (Figure 4a) one week following the initial vaccination. We next assessed the cross-reactivity of the Gal3Long1 peptide with its murine counterpart, mGal3Long1, which differs from the human sequence in four amino acids (Table 1). In IFNγ ELISPOT, we confirmed that cells from the spleen and dLN of mice vaccinated with Gal3Long1 also responds to mGal3Long1, and that this response is comparable to the one observed for Gal3Long1 (Figure 4b). Due to these results, we decided to keep working with the Gal3Long1 human peptide, as this would allow an easier translation into the clinic and a first-in-man clinical trial. We then evaluated the phenotype of Gal3Long1-specific T cells by sorting CD4+ and CD8+ T cells from splenocytes from Gal3Long1-vaccinated mice and assessing their reactivity against Gal3Long1 in an IFNγ ELISPOT. We found that Gal3Long1 peptide induced a CD8+ response in mice (Figure 4c). Next, we investigated six shorter 9-mer peptides (Table 1) with high predicted binding affinity for the MHC-I receptor H-2 Db in C57BL/6 mice. However, we detected very limited immunity toward these peptides (Supplementary Figure S4).
Figure 4.
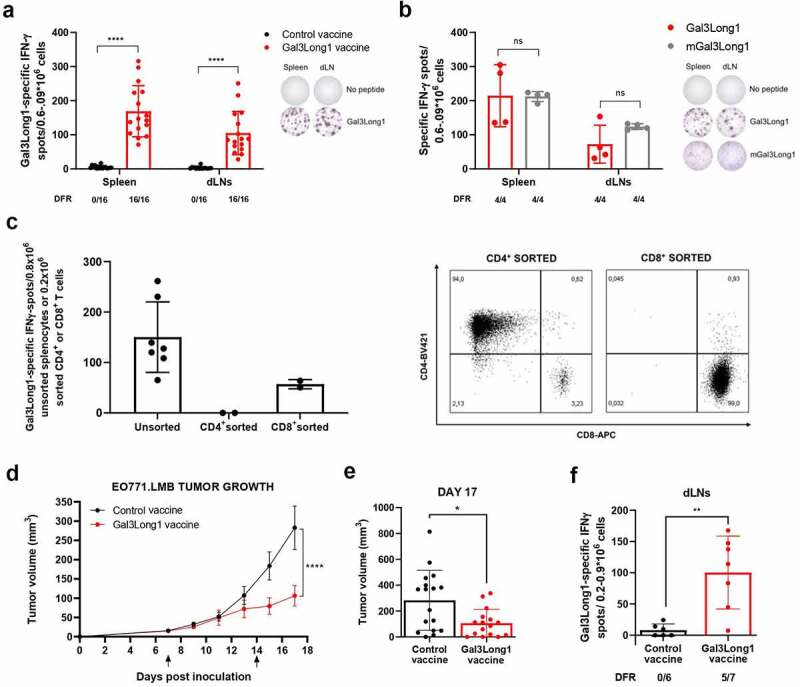
Gal3Long1 vaccination activates Gal3Long1-specific T cells and results in tumor growth delay in EO771.LMB-challenged mice. (a, left) Gal3Long1-specific IFNγ-secreting cells in the spleen and vaccine-draining lymph nodes (dLNs) of C57BL/6 mice (n = 16/group, assessed in n = 4 experiments) treated with a control or Gal3Long1 vaccine assayed by an IFNγ ELISPOT 7 days post vaccination. (a, right) Representative examples of IFNγ ELISPOT responses shown in (a, left). (b, left) Gal3Long1- and mGal3Long1-specific IFNγ-secreting cells in the spleen and dLNs of mice (n = 4/group) vaccinated with Gal3Long1 assayed by an IFNγ ELISPOT 7 days post vaccination. (b, right) Representative examples of IFNγ ELISPOT responses shown in (b, left). For (a) and (b) bars represent mean -/+ SD. Groups were compared with a Mann-Whitney test (****p ≤ .0001, ns = not significant). (c, left) Gal3Long1-specific IFNγ-secreting cells in unsorted splenocytes or CD4+ or CD8a+ T cells sorted from splenocytes of Gal3Long1-vaccinated mice co-cultured with bone marrow-derived DCs as antigen presenting cells (APC) in a 1:2 (T cell: APC) ratio assayed by an IFNγ ELISPOT. Bars represent mean -/+ SD. (c, right) Flow cytometry characterization of CD4+ and CD8+ T cells sorted from splenocytes from Gal3Long1-vaccinated mice. (d) Effect of Gal3Long1 vaccination on EO771.LMB tumor growth. Mice (n = 19/group) were inoculated with 0.5 × 106 EO771.LMB cells on day 0. After stratification into treatment groups on day 7, mice were vaccinated with Gal3Long1 or a DMSO control vaccine on day 7 and 14, as indicated by the arrows. The study was terminated on day 17 due to beginning of ulceration in the tumors. Data points represent mean -/+ SEM. Data is representative of n = 3 experiments with a minimum of 12 mice/group. Statistical significance was determined with a mixed-effects model (**** = p ≤ .0001). (e) Individual tumor volume at day 17. Bars represent mean -/+ SEM. Comparisons between groups were performed with an unpaired Mann-Whitney test (* = p ≤ .05). (f) Gal3Long1-specific IFNγ-secreting cells in the dLNs of EO771.LMB tumor-bearing mice (n = 6–7/group) vaccinated with Gal3Long1 assayed by an IFNγ ELISPOT. Bars represent mean -/+ SD. Statistical significance was assessed by a Mann-Whitney test (** = p ≤ .01). For (a), (b) and (f), each dot represents the difference in mean counts of IFNγ-secreting cells between peptide-stimulated and control wells in ELISPOT. Data shown as mean -/+ SD. Statistical analysis of ELISPOT responses was performed with the distribution-free resampling (DFR) model. The number of significant responses of total assessed samples is indicated below the figure.
Finally, we examined the anti-tumor efficacy of vaccination with Gal3-derived peptides in the syngeneic EO771.LMB murine tumor model, which has high Gal3 expression (unpublished data of EO771.LMB tumors by Daniel Hargbøl Madsen). The challenge of C57BL/6 mice with EO771.LMB tumor cells followed by Gal3Long1 vaccination on day 7 and 14 resulted in a significant tumor growth delay when compared to mice that received a control vaccination (Figure 4d,e). We confirmed that Gal3Long1-vaccinated mice developed an immune response against Gal3Long1 (figure 4f), which was comparable to that obtained in tumor-free mice (Figure 4a).
Vaccination with Galectin-3-derived peptide modulates the tumor microenvironment
To examine the impact of the Gal3Long1 vaccination on the myeloid and lymphoid composition of the TME, we conducted flow cytometric analysis of single-cell suspensions from tumors of the EO771.LMB tumor-bearing mice that were Gal3Long1 or control vaccinated. Gal3Long1 vaccinations did not significantly modify the percentage of tumor-infiltrating CD45+CD11b+ cells, granulocytic myeloid-derived suppressor cells (gMDSC), monocytic myeloid-derived suppressor cells (mMDSCs) or the intra-tumoral M1 to M2 ratio (Supplementary Figure S5A, 5C, 5E and 5G). In addition, the expression of Gal3 in these populations, represented by MFI, remained unchanged (Supplementary Figure S5B, 5D, 5F, 5H and 5I). Interestingly, even though the abundance of the non-myeloid CD45+CD11b− population in the tumor was not affected by Gal3Long1 vaccination (Figure 5a), a significant reduction in the percentage of Gal3+ cells was observed (Figure 5b). Regarding the lymphoid compartment, no significant differences were found in the percentage of tumor-infiltrating CD3+, CD8+ or CD4+ T cells in Gal3Long1-vaccinated mice (Supplementary Figure S5J-L), although a tendency toward a higher CD8+ T cell infiltration in the tumor of Gal3Long1-vaccinated mice was observed (Supplementary Figure S5K). Interestingly, Gal3Long1 vaccinations resulted in a significant reduction in the percentage of tumor-infiltrating CD25+FoxP3+ Tregs among CD4+ cells (Figure 5c).
Figure 5.
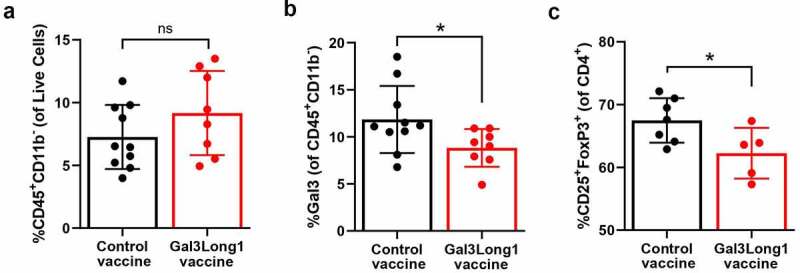
Gal3Long1 vaccination is associated with a reduction in tumor-infiltrating Gal3+ leukocytes and Tregs. Mice (n = 5–10/group) were inoculated with 0.5 × 106 EO771.LMB cells on day 0 and stratified into treatment groups on day 7. Mice received two DMSO control or Gal3Long1 vaccinations, one week apart, starting on day 7. On day 17 mice were sacrificed, tumors were harvested and flow cytometry was performed on tumor single-cell suspensions. (a) Percentage of CD45+CD11b− cells of total live cells. (b) Percentage of Gal3+ cells among the CD45+CD11b− population. (c) Percentage of CD25+FoxP3+ T cells (Tregs) of total CD4+ T cells. Each dot represents one mouse. Data are presented as mean -/+ SD. Statistical significance was assessed with a Mann-Whitney test (* = p ≤ .05, ns = not significant).
Galectin-3 based vaccination has an impact on the T-cell memory frequencies in the spleen
Next, we aimed at examining whether Gal3Long1 could induce a memory response and at studying the effect of Gal3-peptide-based vaccination on the T-cell memory populations in the spleen of EO771.LMB-challenged mice. Gal3Long1 vaccination significantly increased the frequency of both CD4+ and CD8+ central memory (TCM) T cells in the spleen (Figure 6a,c). This increase was accompanied by a significant decrease in the percentage of CD8+ effector memory (TEM) T cells in Gal3Long1-vaccinated mice (Figure 6b). In contrast, vaccination with the Gal3-derived peptide resulted in no significant difference in the CD4+ TEM population (Figure 6d).
Figure 6.
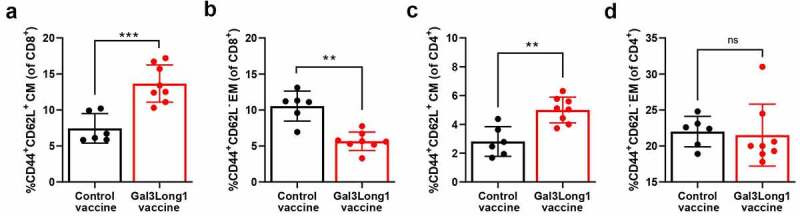
Gal3Long1 vaccination alters the composition of the T-cell memory populations in the spleen. Mice (n = 6–8/group) were inoculated with EO771.LMB cells on day 0. After stratification into treatment groups on day 10, mice were vaccinated with a DMSO control or Gal3Long1 vaccine on day 10 and 17. On day 24, mice were sacrificed, spleens were harvested and flow cytometry was performed. (a and c) Percentages of Central Memory (CM) T cells of total (a) CD8+ T cells and (c) CD4+ T cells. (b and d) Percentages of Effector Memory (EM) T cells of total (b) CD8+ T cells and (d) CD4+ T cells. CD8+ and CD4+ TEM cells were gated as CD44+ CD62L− of total CD8+ and CD4+ population, respectively; CD8+ and CD4+ TCM cells were gated as CD44+ CD62L+ of total CD8+ and CD4+ population, respectively. Each dot represents one mouse. Data are presented as the mean -/+ SD. Differences between groups were statistically evaluated with a Mann-Whitney test (** = p ≤ .01, *** = p ≤ .001, ns = not significant).
Discussion
The development of new, more efficient cancer treatment strategies is a field of growing interest. In the present study, we introduced Gal3 as a novel T-cell target. The reported findings shed new light on the use of Gal3-based cancer vaccination as a novel mean of targeting the TME. First, we designed a long Gal3 epitope, Gal3Long1, which had the potential to be presented on both HLA class I and II molecules, thereby stimulating both a CD8+ and CD4+ responses. Gal3Long1 was derived from the CRD of the Gal3 protein, which is believed to regulate the immunosuppressive effects of Gal3. Previous studies have shown that the apoptotic effect of extracellular Gal3 on T cells can be abrogated by the addition of a sugar ligand that binds to the Gal3 CRD with high affinity.9–11 Instead of directly targeting the extracellular protein, we propose a strategy based on targeting the Gal3-producing cells present in the TME by vaccine-induced T cells.
We identified the HLA-A2 restricted epitope, Gal307, within the Gal3Long1 peptide. In addition, we found naturally occurring CD4+ and CD8+ T-cell responses against Gal3Long1 in both healthy donors and cancer patients, suggesting that Gal3Long1 must contain several HLA-restricted epitopes. This finding is consistent with our earlier observations, which have shown that immune responses against other self-antigens, e.g. arginase and transforming growth factor (TGF)β, can be found in healthy donors.24,26 We further described that isolated and expanded Gal3-specific CD8+ T cells could directly recognize cancer cells that overexpressed Gal3. Further, we mimicked the interplay between cancer-associated fibroblasts and cancer cells in the TME by co-culturing the adherent bone marrow stromal cell-derived fibroblast cell line, HS-5, with various suspended cancer cell lines to upregulate Gal3 in the suspended cells. This approach has previously been described by Yamamoto-Sugitani et al.31 We confirmed that HS-5 induced the upregulation of Gal3 in the AML cell line, THP-1, which was associated with a significant increase in the recognition of THP-1 by Gal3-specific T cells. Interestingly, expression of the LGALS3 gene encoding Gal3 has previously been associated with poor overall survival in patients with AML,34 and high plasma Gal3 levels in patients with AML correlated with reduced overall survival.35 However, although we have strong functional evidence to conclude that HS-5 cells upregulate Gal3 expression in THP-1 cells, there were limitations to this experiment. The sorted THP-1 cell fraction had a 4% contamination of CD45− cells. Consequently, it remains to be determined whether this CD45− fraction comprised THP-1 cells that had downregulated their CD45 expression or contamination with HS-5 cells, which might be responsible for the increase in Gal3 expression.
Several studies have described a special link between Gal3 and breast cancer. For instance, Gal3 expression in the tumor stroma of patients with breast cancer has been associated with a high grade of malignancy.5 In addition, tumors from patients with triple-negative breast cancer (TNBC), have been shown to express higher Gal3 levels, when compared to other types of cancer.36,37 Furthermore, in murine models of TNBC, Gal3 knock-in and knock-out experiments have demonstrated that Gal3 plays a role as a pro-tumorigenic protein.36,38,39 Importantly, Gal3 does not induce apoptosis in TNBC cells, like it does in T cells.9 Due to the described role of Gal3 in breast cancer, we decided to evaluate the therapeutic potential of Gal3-derived peptide vaccination in the murine EO771.LMB tumor model, which has been described to have a triple-negative, basal-like phenotype,28 closely resembling TNBC. We tested the anti-tumor effect of Gal3Long1, a 28-mer, as long peptide epitopes have been shown to be superior to shorter peptides for inducing robust anti-tumor responses, when administered with incomplete Freund’s adjuvant.40,41 This study has demonstrated the ability of Gal3Long1 vaccination to generate Gal3-specific CD8+ T cells in vivo and to significantly delay tumor growth in EO771.LMB-challenged mice. The response was associated with a decrease in the percentage of tumor-infiltrating Tregs, whose levels in the control group were comparable to other studies that used the same EO771 model,42,43 suggesting that Gal3Long1 vaccination can modulate the TME toward a less suppressive environment. This finding is of high importance, as a high abundance of tumor-infiltrating Tregs is associated with poor prognosis in most cancers.44 Furthermore, Gal3Long1 vaccination resulted in a reduction in Gal3 expression in the tumor-infiltrating CD45+CD11b− population, which is in line with our initial hypothesis that vaccine-induced Gal3-specific T cells infiltrate the tumor and target Gal3-expressing cells. As Tregs have been described to express Gal3,7 we speculate that the reduction in Gal3 expression in the non-myeloid CD45+CD11b− population could be explained by the observed decrease in percentage of tumor-infiltrating Tregs. Nevertheless, further analyses are required to elucidate the precise mechanism of action of Gal3Long1 vaccination. For instance, assessing the efficacy of Gal3-derived peptide vaccination with a Gal3-knockout EO771.LMB cell line could shed light on whether the main targets for the vaccine-induced T cells are cancer cells and/or other cells in the TME. Finally, we found that the Gal3Long1 vaccination changed the composition of the splenic T-cell population. We observed a significant increase in the CD8+ and CD4+ TCM fraction together with a reduction in the CD8+ TEM fraction. These are interesting and positive results, as TCM cells have been linked to a higher anti-tumor effect when compared to TEM cells in cancer vaccines.45 In addition, TCM have been shown to have greater persistence in vivo,46 which is a vital characteristic for vaccine-induced specific T cells. These findings provide insight for future evaluation of the clinical efficacy of Gal3-derived peptide vaccination in additional murine tumor models.
Previously, our group described the concept of naturally present, auto-reactive and pro-inflammatory anti-Tregs.2,18–26 In phase I/II clinical trials, it has been shown that the novel therapeutic peptide vaccines that activate IDO-specific and PD-L1-specific anti-Tregs are safe and exert minimal toxicity.2,47–49 Moreover, pre-clinical studies in mouse models have shown that target the self-proteins arginase-1- and 2-derived peptide vaccines are also safe and did not induce toxicity.50 These vaccines are currently undergoing evaluations in clinical phase I/II trials (NCT04051307/NCT03689192), and to date, no severe toxicity has been reported. In the present study, we found no apparent signs of toxicity, no behavioral changes, and no weight loss in Gal3Long1-treated mice (data not shown). Although additional work would be necessary to confirm the safety of this approach, based on the evidence previously provided, we suggest that Gal3Long1 peptide vaccination is a safe and promising alternative strategy to target the immune-suppressive TME. In terms of directions for future research, further research should be undertaken to maximize the therapeutic effect of Gal3Long1 vaccinations. The most obvious strategy to explore is the combination with other forms of cancer therapies. Recently, two papers describing the synergy targeting the PD-1/PD-L1 pathway and extracellular Gal1/Gal3 has been published. In a phase I trial, the combination of anti-PD-1 and Gal1/Gal3 blockade was associated with an increased amount of effector memory CD4/CD8 + T cells, a limited expansion of mMDSCs and an objective response in a subset of patients diagnosed with malignant melanoma and head and neck squamous cell carcinoma.51 Moreover, in a humanized xenograft model of non-small cell lung cancer, a Gal3 inhibitor in combination with PD-L1 blockade had a synergistic effect on tumor growth.52 As Gal3-derived peptide vaccination is designed to target Gal3-producing cells rather than extracellular Gal3, further combination studies utilizing anti-PD-1 checkpoint inhibition or our novel PD-L1 vaccine would be of high relevance for future research.
In summary, we demonstrated that not only do Gal3-specific T cells exist as a natural part of the immune cell repertoire in both healthy donors and cancer patients, but they can also target and kill Gal3-expressing cancer cells. Next, we demonstrated that the activation of CD8+ Gal3-specific T cells by a therapeutic Gal3-derived peptide vaccine in the EO771.LMB tumor model resulted in a significant tumor-growth delay. We showed that the vaccine modulates the TME toward a less immunosuppressive phenotype, as exemplified by a reduction of Gal3+ cells in the non-myeloid CD45+CD11b− compartment and by a significant decrease in intra-tumoral Tregs. Therefore, our findings indicated that Gal3 could serve as a novel target for immune modulatory cancer vaccines.
Supplementary Material
Acknowledgments
We greatly appreciate the tremendous technical assistance from Merete Jonassen and Tina Seremet.
Funding Statement
This project was supported by Danish Health Authority grant “Empowering Cancer Immunotherapy in Denmark”, grant number 4-1612-236/8, the Copenhagen University Hospital Herlev and Gentofte, and the Danish Council for Independent Research. The funders had no role in the study design, data collection and analysis, decision to publish, or manuscript preparation.
Author contribution
SKB, MPP, MLH, EM, MOH, SEW-B, MAJ, SMA, MHA designed the experiments. SKB, MPP, MLH, EM, MOH, SEW-B, MAJ, SMA, MTL, AZJ, MC performed the experiments. NGDJ, KMJ, CF, NØ, ÖM, IMS, DHM provided critical reagents. SKB, MPP, MLH, EM, MOH, SEW-B, MAJ, SMA, MHA interpreted data. SKB, MPP, MLH, EM, MOH, MHA wrote the manuscript. All authors read and agreed on the final version of the submitted manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Farhad M, Rolig AS, Redmond WL.. The role of Galectin-3 in modulating tumor growth and immunosuppression within the tumor microenvironment. OncoImmunology. 2018;7:e1434467. doi: 10.1080/2162402X.2018.1434467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andersen MH. The balance players of the adaptive immune system. Cancer Res. 2018;78:1379–13. doi: 10.1158/0008-5472.CAN-17-3607. [DOI] [PubMed] [Google Scholar]
- 3.Ruvolo PP. Galectin 3 as a guardian of the tumor microenvironment. Biochim Biophys Acta - Mol Cell Res. 2016;1863:427–437. doi: 10.1016/j.bbamcr.2015.08.008. [DOI] [PubMed] [Google Scholar]
- 4.Fortuna-Costa A, Gomes AM, Kozlowski EO, Stelling MP, Pavão MSG. Extracellular galectin-3 in tumor progression and metastasis. Front Oncol. 2014. JUN;4. doi: 10.3389/fonc.2014.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moisa A, Fritz P, Eck A, Wehner H-D, Mürdter T, Simon W, Gabius H-J. Growth/adhesion-regulatory tissue lectin galectin-3: stromal presence but not cytoplasmic/nuclear expression in tumor cells as a negative prognostic factor in breast cancer. Anticancer Res. 2007;27:2131–2139. [PubMed] [Google Scholar]
- 6.Sato S, Hughes RC. Control of Mac‐2 surface expression on murine macrophage cell lines. Eur J Immunol. 1994;24:216–221. doi: 10.1002/eji.1830240134. [DOI] [PubMed] [Google Scholar]
- 7.Hsu DK, Chen H-Y, Liu F-T. Galectin-3 regulates T-cell functions. Immunol Rev. 2009;230:114–127. doi: 10.1111/j.1600-065X.2009.00798.x. [DOI] [PubMed] [Google Scholar]
- 8.Demotte N, Wieërs G, Van Der Smissen P, Moser M, Schmidt C, Thielemans K, Squifflet J-L, Weynand B, Carrasco J, Lurquin C, Courtoy PJ, Van Der Bruggen P. A galectin-3 ligand corrects the impaired function of human CD4 and CD8 tumor-infiltrating lymphocytes and favors tumor rejection in mice. Cancer Res. 2010;70:7476–7488. doi: 10.1158/0008-5472.CAN-10-0761. [DOI] [PubMed] [Google Scholar]
- 9.Fukumori T, Takenaka Y, Yoshii T, Kim HRC, Hogan V, Inohara H, Kagawa S, Raz A. CD29 and CD7 mediate Galectin-3-induced type II T-cell apoptosis. Cancer Res. 2003;63:8302–8311. [PubMed] [Google Scholar]
- 10.Stillman BN, Hsu DK, Pang M, Brewer CF, Johnson P, Liu F-T, Baum LG. Galectin-3 and Galectin-1 Bind distinct cell surface glycoprotein receptors to induce T cell death. J Immunol. 2006;176:778–789. doi: 10.4049/jimmunol.176.2.778. [DOI] [PubMed] [Google Scholar]
- 11.Stowell SR, Qian Y, Karmakar S, Koyama NS, Dias-Baruffi M, Leffler H, McEver RP, Cummings RD. Differential roles of Galectin-1 and Galectin-3 in regulating leukocyte viability and cytokine secretion. J Immunol. 2008;180:3091–3102. doi: 10.4049/jimmunol.180.5.3091. [DOI] [PubMed] [Google Scholar]
- 12.Xue J, Gao X, Fu C, Cong Z, Jiang H, Wang W, Chen T, Wei Q, Qin C. Regulation of galectin-3-induced apoptosis of Jurkat cells by both O-glycans and N-glycans on CD45. FEBS Lett. 2013;587:3986–3994. doi: 10.1016/j.febslet.2013.10.034. [DOI] [PubMed] [Google Scholar]
- 13.Kouo T, Huang L, Pucsek AB, Cao M, Solt S, Armstrong T, Jaffee E. Galectin-3 shapes antitumor immune responses by suppressing CD8 + T cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunol Res. 2015;3:412–423. doi: 10.1158/2326-6066.CIR-14-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Demotte N, Bigirimana R, Wieërs G, Stroobant V, Squifflet J-L, Carrasco J, Thielemans K, Baurain J-F, Van Der Smissen P, Courtoy PJ, Van Der Bruggen P. A short treatment with galactomannan GM-CT-01 corrects the functions of freshly isolated human tumor-infiltrating lymphocytes. Clin Cancer Res. 2014;20:1823–1833. doi: 10.1158/1078-0432.CCR-13-2459. [DOI] [PubMed] [Google Scholar]
- 15.Melief SM, Visser M, van der Burg SH, Verdegaal EME. IDO and galectin-3 hamper the ex vivo generation of clinical grade tumor-specific T cells for adoptive cell therapy in metastatic melanoma. Cancer Immunol Immunother. 2017;66:913–926. doi: 10.1007/s00262-017-1995-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wiëers G, Demotte N, Godelaine D, van der Bruggen P. Immune suppression in tumors as a surmountable obstacle to clinical efficacy of cancer vaccines. Cancers. 2011;3:2904–2954. doi: 10.3390/cancers3032904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, Caux C, Depil S. Cold tumors: a therapeutic challenge for immunotherapy. Front Immunol. 2019;10. doi: 10.3389/fimmu.2019.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baek Sørensen R, Hadrup SR, Svane IM, Hjortsø MC, Straten Pt, Andersen MH. Indoleamine 2,3-dioxygenase specific, cytotoxic T cells as immune regulators. Blood. 2011;117:2200–2010. doi: 10.1182/blood-2010-06-288498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munir S, Larsen SK, Iversen TZ, Donia M, Klausen TW, Svane IM, Straten PT, Andersen MH. Natural CD4+ T-cell responses against indoleamine 2,3-dioxygenase. PLoS One. 2012;7:e34568. doi: 10.1371/journal.pone.0034568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Munir S, Andersen GH, Met Ö, Donia M, Frøsig TM, Larsen SK, Klausen TW, Svane IM, Andersen MH. HLA-restricted CTL that are specific for the immune checkpoint ligand PD-L1 occur with high frequency in cancer patients. Cancer Res. 2013;73:1764–1776. doi: 10.1158/0008-5472.CAN-12-3507. [DOI] [PubMed] [Google Scholar]
- 21.Munir S, Andersen GH, Svane IM, Andersen MH. The immune checkpoint regulator PD-L1 is a specific target for naturally occurring CD4 + T cells. Oncoimmunology. 2013;2:e23991. doi: 10.4161/onci.23991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahmad SM, Martinenaite E, Holmström M, Jørgensen MA, Met Ö, Nastasi C, Klausen U, Donia M, Pedersen LM, Munksgaard L, Ødum N, Svane IM, Andersen MH. The inhibitory checkpoint, PD-L2, is a target for effector T cells: novel possibilities for immune therapy. Oncoimmunology. 2018;7:e1390641. doi: 10.1080/2162402X.2017.1390641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinenaite E, Ahmad SM, Hansen M, Met Ö, Westergaard MW, Larsen SK, Klausen TW, Donia M, Svane IM, Andersen MH. CCL22-specific T cells: modulating the immunosuppressive tumor microenvironment. Oncoimmunology. 2016;5:e1238541. doi: 10.1080/2162402X.2016.1238541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinenaite E, Ahmad SM, Hansen M, Met Ö, Westergaard MW, Larsen SK, Klausen TW, Donia M, Svane IM, Andersen MH. Frequent adaptive immune responses against arginase-1. Oncoimmunology. 2018;7:e1404215. doi: 10.1080/2162402X.2017.1404215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weis-Banke SE, Hübbe ML, Holmström MO, Jørgensen MA, Bendtsen SK, Martinenaite E, Carretta M, Svane IM, Ødum N, Pedersen AW, Met Ö, Madsen DH, Andersen MH. The metabolic enzyme arginase-2 is a potential target for novel immune modulatory vaccines. Oncoimmunology. 2020;9:1771142. doi: 10.1080/2162402X.2020.1771142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holmström MO, Mortensen REJ, Pavlidis AM, Martinenaite E, Weis-Banke SE, Aaboe-Jørgensen M, Bendtsen SK, Met Ö, Pedersen AW, Donia M, Svane IM, Andersen MH. Cytotoxic T cells isolated from healthy donors and cancer patients kill TGFβ-expressing cancer cells in a TGFβ-dependent manner. Cell Mol Immunol. 2021;18:415–426. doi: 10.1038/s41423-020-00593-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jæhger DE, Hübbe ML, Kræmer MK, Clergeaud G, Olsen AV, Stavnsbjerg C, Wiinholt MN, Kjær A, Henriksen JR, Hansen AE, Andresen TL. Enhancing adoptive CD8 T cell therapy by systemic delivery of tumor associated antigens. Sci Rep. 2021;11. doi: 10.1038/s41598-021-99347-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnstone CN, Smith YE, Cao Y, Burrows AD, Cross RSN, Ling X, Redvers RP, Doherty JP, Eckhardt BL, Natoli AL, Restall CM, Lucas E, Pearson HB, Deb S, Britt KL, Rizzitelli A, Li J, Harmey JH, Pouliot N, Anderson RL. Functional and molecular characterisation of EO771.LMB tumours, a new C57BL/6-mouse-derived model of spontaneously metastatic mammary cancer. DMM Dis Model Mech. 2015;8:237–251. doi: 10.1242/dmm.017830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moodie Z, Price L, Gouttefangeas C, Mander A, Janetzki S, Löwer M, Welters MJP, Ottensmeier C, van der Burg SH, Britten CM. Response definition criteria for ELISPOT assays revisited. Cancer Immunol Immunother. 2010;59:1489–1501. doi: 10.1007/s00262-010-0875-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gnjatic S, Atanackovic D, Matsuo M, Jäger E, Lee SY, Valmori D, Chen Y-T, Ritter G, Knuth A, Old LJ . Cross-Presentation of HLA class I epitopes from exogenous NY-ESO-1 polypeptides by nonprofessional APCs. J Immunol. 2003;170:1191–1196. doi: 10.4049/jimmunol.170.3.1191. [DOI] [PubMed] [Google Scholar]
- 31.Yamamoto-Sugitani M, Kuroda J, Ashihara E, Nagoshi H, Kobayashi T, Matsumoto Y, Sasaki N, Shimura Y, Kiyota M, Nakayama R, Akaji K, Taki T, Uoshima N, Kobayashi Y, Horiike S, Maekawa T, Taniwaki M. Galectin-3 (Gal-3) induced by leukemia microenvironment promotes drug resistance and bone marrow lodgment in chronic myelogenous leukemia. Proc Natl Acad Sci U S A. 2011;108:17468–17473. doi: 10.1073/pnas.1111138108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsuchiya S, Yamabe M, Yamaguchi Y, Kobayashi Y, Konno T, Tada K. Establishment and characterization of a human acute monocytic leukemia cell line (THP‐1). Int J Cancer. 1980;26:171–176. doi: 10.1002/ijc.2910260208. [DOI] [PubMed] [Google Scholar]
- 33.Genin M, Clement F, Fattaccioli A, Raes M, Michiels C. M1 and M2 macrophages derived from THP-1 cells differentially modulate the response of cancer cells to etoposide. BMC Cancer. 2015;15:577. doi: 10.1186/s12885-015-1546-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng C-L, Hou H-A, Lee M-C, Liu C-Y, Jhuang J-Y, Lai Y-J, Lin C-W, Chen H-Y, Liu F-T, Chou W-C, Chen C-Y, Tang J-L, Ming Y, Huang S-Y, Ko B-S, Wu S-J, Tsay W, Tien H-F. Higher bone marrow LGALS3 expression is an independent unfavorable prognostic factor for overall survival in patients with acute myeloid leukemia. Blood. 2013;121:3172–3180. doi: 10.1182/blood-2012-07-443762. [DOI] [PubMed] [Google Scholar]
- 35.Gao N, Yu WZ, Guo NJ, Wang XX, Sun JR. Clinical significance of galectin-3 in patients with adult acute myeloid leukemia: a retrospective cohort study with long-term follow-up and formulation of risk scoring system. Leuk Lymphoma. 2017;58:1394–1402. doi: 10.1080/10428194.2016.1243677. [DOI] [PubMed] [Google Scholar]
- 36.Chavez KJ, Garimella SV, Lipkowitz S, Eng-Wong J, Zujewski JA. Triple negative breast cancer cell lines: one tool in the search for better treatment of triple negative breast cancer. Breast Dis. 2010;32:35–48. doi: 10.3233/BD-2010-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang H, Liang X, Duan C, Liu C, Zhao Z. Galectin-3 as a marker and potential therapeutic target in breast cancer. PLoS One. 2014;9:e103482. doi: 10.1371/journal.pone.0103482 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nangia-Makker P, Thompson E, Hogan C, Ochieng J, Raz A. Induction of tumorigenicity by galectin-3 in a non-tumorigenic human breast carcinoma cell line. Int J Oncol. 1995;7:1079–1087. doi: 10.3892/ijo.7.5.1079. [DOI] [PubMed] [Google Scholar]
- 39.Honjo Y, Nangia-Makker P, Inohara H, Raz A. Down-regulation of galectin-3 suppresses tumorigenicity of human breast carcinoma cells. Clin Cancer Res. 2001;7:661–668. [PubMed] [Google Scholar]
- 40.Bijker MS, van Den Eeden SJF, Franken KL, Melief CJM, Offringa R, van der Burg SH. CD8+ CTL priming by exact peptide epitopes in incomplete Freund’s adjuvant induces a vanishing CTL response, whereas long peptides induce sustained CTL reactivity. J Immunol. 2007;179:5033–5040. doi: 10.4049/jimmunol.179.8.5033. [DOI] [PubMed] [Google Scholar]
- 41.Hailemichael Y, Dai Z, Jaffarzad N, Ye Y, Medina MA, Huang X-F, Dorta-Estremera SM, Greeley NR, Nitti G, Peng W, Liu C, Lou Y, Wang Z, Ma W, Rabinovich B, Schluns KS, Davis RE, Hwu P, Overwijk WW. Persistent antigen at vaccination sites induces tumor-specific CD8+ T cell sequestration, dysfunction and deletion. Nat Med. 2013;19:465–472. doi: 10.1038/nm.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crosby EJ, Wei J, Yang XY, Lei G, Wang T, Liu C-X, Agarwal P, Korman AJ, Morse MA, Gouin K, Knott SRV, Lyerly HK, Hartman ZC. Complimentary mechanisms of dual checkpoint blockade expand unique T-cell repertoires and activate adaptive anti-tumor immunity in triple-negative breast tumors. Oncoimmunology. 2018;7:e1421891. doi: 10.1080/2162402X.2017.1421891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Priceman SJ, Shen S, Wang L, Deng J, Yue C, Kujawski M, Yu H. S1PR1 is crucial for accumulation of regulatory T cells in tumors via STAT3. CellReports. 2014;6:992–999. doi: 10.1016/j.celrep.2014.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol. 2019;16:356–371. doi: 10.1038/s41571-019-0175-7. [DOI] [PubMed] [Google Scholar]
- 45.Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, Palmer DC, Antony PA, Hwang ST, Rosenberg SA, Waldmann TA, Restifo NP. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci. 2005;102:9571–9576. doi: 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118:294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kjeldsen JW, Iversen TZ, Engell-Noerregaard L, Mellemgaard A, Andersen MH, Svane IM. Durable Clinical responses and long-term follow-up of stage III–IV non-small-cell lung cancer (NSCLC) patients treated with IDO peptide vaccine in a phase I study—a brief research report. Front Immunol. 2018;9. doi: 10.3389/fimmu.2018.02145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jørgensen NG, Klausen U, Grauslund JH, Helleberg C, Aagaard TG, Do TH, Ahmad SM, Olsen LR, Klausen TW, Breinholt MF, Hansen M, Martinenaite E, Met Ö, Svane IM, Knudsen LM, Andersen MH. Peptide vaccination against PD-L1 with IO103 a novel immune modulatory vaccine in multiple myeloma: a phase I first-in-human trial. Front Immunol. 2020;11:595035. doi: 10.3389/fimmu.2020.595035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kjeldsen JW, Lorentzen CL, Martinenaite E, Ellebaek E, Donia D, Holmstroem RB, Klausen TW, Madsen CO, Ahmad SM, Weis-Banke SE, Holmström MO, Hendel HW, Ehrnrooth E, Zocca M-B, Pedersen AW, Andersen MH, Svane IM. A phase 1/2 trial of an immune-modulatory vaccine against IDO/PD-L1 in combination with nivolumab in metastatic melanoma. Nat Med. 2021;2021:1–12. doi: 10.1038/s41591-021-01544-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jørgensen MA, Ugel S, Hübbe ML, Carretta M, Perez-Penco M, Weis-Banke SE, Martinenaite E, Kopp K, Chapellier M, Adamo A, Sanctis FD, Frusteri C, Iezzi M, Zocca M-B, Madsen DH, Pedersen AW, Bronte V, Andersen MH. Arginase 1-based immune modulatory vaccines induce anti-cancer immunity and synergize with anti-PD-1 checkpoint blockade. Cancer Immunol Res. 2021. canimm.0280.2021. doi: 10.1158/2326-6066.CIR-21-0280. [DOI] [PubMed] [Google Scholar]
- 51.Curti BD, Koguchi Y, Leidner RS, Rolig AS, Sturgill ER, Sun Z, Wu Y, Rajamanickam V, Bernard B, Hilgart-Martiszus I, Fountain CB, Morris G, Iwamoto N, Shimada T, Chang S, Traber PG, Zomer E, Horton JR, Shlevin H, Redmond WR. Enhancing clinical and immunological effects of anti-PD-1 with belapectin, a galectin-3 inhibitor. J Immunother Cancer. 2021;9:e002371. doi: 10.1136/jitc-2021-002371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang H, Liu P, Zhang Y, Han L, Hu Z, Cai Z, Cai J. Inhibition of galectin-3 augments the antitumor efficacy of PD-L1 blockade in non-small-cell lung cancer. FEBS Open Bio. 2021;11:911–920. doi: 10.1002/2211-5463.13088. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.