

Abstract
Macrophages play crucial roles in many human disease processes. However, obtaining large numbers of primary cells for study is often difficult. We describe 2D and 3D methods for directing human induced pluripotent stem cells (hiPSCs) into macrophages (iMACs). iMACs generated in 2D culture showed functional similarities to human primary monocyte-derived M2-like macrophages, with and could be successfully polarized into a M1-like phenotype. Both M1- and M2-like iMACs showed phagocytic activity and reactivity to endogenous or exogenous stimuli. In contrast, iMACs generated by a 3D culture system showed mixed M1- and M2-like functional characteristics. 2D-iMACs from patients with fibrodysplasia ossificans progressiva (FOP), an inherited disease with progressive heterotopic ossification driven by inflammation, showed prolonged inflammatory cytokine production and higher Activin A production after M1-like polarization, resulting in dampened responses to additional LPS stimulation. These results demonstrate a simple and robust way of creating hiPSC-derived M1- and M2-like macrophage lineages, while identifying macrophages as a source of Activin A that may drive heterotopic ossification in FOP.
Graphical Abstract
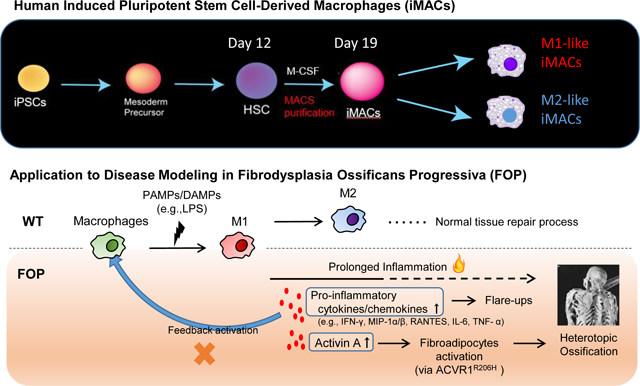
Summary
A simple 2D method creates M1-like and M2-like macrophages from human induced pluripotent stem cells. Applying this method to FOP, a rare bone disease where primary samples are difficult to obtain, revealed increased macrophage inflammatory responses caused by ACVR1 overactivity.
Introduction
Macrophages play crucial roles in normal tissue homeostasis and repair. Macrophage dysfunction is a major contributor to disease pathology. Innate immune cells, including neutrophils, monocytes, and macrophages, are recruited in response to injury, infections, or other hazards (Eming et al., 2017; Oishi and Manabe, 2018). During the early phases of tissue repair, these innate immune cells help clear the necrotic debris or infectious organisms and produce various chemokines to trigger inflammation. In the intermediate phase, acute inflammatory responses mainly caused by pro-inflammatory macrophages (M1-like macrophages, or classically activated macrophages), which then transition into a resolution phase where macrophages shift their function into anti-inflammatory or reparative roles (M2-like macrophages, or alternatively activated macrophages) (Ivashkiv, 2011). Tissue homeostasis is eventually restored once inflammatory cells are eliminated from the site of injury or infection.
Although inflammation helps drive the normal tissue repair process, excessive or dysregulated inflammatory responses result in pathological conditions (Wynn and Barron, 2010). This includes promotion of fibrosis, which impairs normal tissue functions (Wynn and Vannella, 2016); changes to cellular function, such as interleukin-1β (IL-1β)-, Toll-like receptor 2 (TLR2)-, or TLR4-driven islet cell dysfunction in diabetes (Ehses et al., 2007; Richardson et al., 2009); and changes to insulin resistance driven by lipid accumulation in adipocytes leading to production of tumor necrosis factor-α (TNF-α) and IL-1β from activated macrophages (Donath, 2014). Infections such as SARS-Cov-2 induced COVID-19 also highlight how a dysfunctional innate immune response can be a major contributor to acute lung and end organ damage (Mathew et al., 2020; Merad and Martin, 2020; Schulte-Schrepping et al., 2020). Thus, understanding how macrophages function and become dysregulated is of major importance for many fields of biomedical research.
One emerging area is the role of macrophages in bone diseases. Bone homeostasis is closely linked to the immune system (Matsuo et al., 2019). In addition to supporting bone marrow for hematopoiesis, bone stromal cells are known to have major impact on the hematopoietic process (Anthony and Link, 2014). Less well understood is how the hematopoietic system affects bone. Osteoclasts, which break down bone, originate from the monocyte/macrophage lineage (Udagawa, 2003). In addition, osteal macrophages appear to promote the formation of osteoblasts and support anabolic bone formation, as macrophage-deficient mice show a significant reduction in bone density and impaired ability of mesenchymal stem cells to differentiate into osteoblasts (Vi et al., 2015). Dysfunction of immune cells can also lead to abnormal bone homeostasis. Heterotopic ossification (HO), which is a debilitating process of excessive bone formation at non-bone sites, is linked to immunological dysfunction (Kaplan et al., 2012; McCarthy and Sundaram, 2005) and often occurs in settings of severe inflammation such as trauma, some rheumatologic diseases, or burns (Hurvitz et al., 1992; Kocic et al., 2010; Leung et al., 2005; McCarthy and Sundaram, 2005; Potter et al., 2007). In addition, depletion of macrophages can mitigate HO formation in mice (Convente et al., 2018).
One critical barrier to elucidating the tissue effects of macrophages is the limited ability to obtain large numbers of characterized human cells for detailed research. Induced pluripotent stem cells (iPSCs) hold potential for addressing this challenge by theoretically providing an almost unlimited starting source for creating different cell types. Although several protocols to create macrophages from mouse (Senju et al., 2009; Takata et al., 2017) and human (Lachmann et al., 2015; Muffat et al., 2016; Nenasheva et al., 2020; Takata et al., 2017; van Wilgenburg et al., 2013) iPSCs have been described, these protocols remain challenging because of limited direct comparisons to primary cells (Cao et al., 2019; Senju et al., 2009) and limited demonstration of applicability to known disease states, particularly in the skeletal system and in conditions where primary cells are difficult to obtain.
Here, we describe methods to generate human iPSC-derived macrophages (iMACs) by comparing two different protocols, detailing differences in cytokine production, and comparing to human primary monocyte-derived macrophage lineages. This method is then applied to understand how aberrant activation of the BMP pathway affects macrophage function. We use the prototypical disease fibrodysplasia ossificans progressiva (FOP), a rare inherited disease with progressive heterotopic ossification caused by an ACVR1R206H mutation that is associated with pro-inflammatory changes (Barruet and Hsiao, 2018) and macrophage dysfunction (Chakkalakal et al., 2012; Convente et al., 2018; Wang et al., 2018), to compare the iMACs to primary monocyte-derived macrophage lineages in a human disease of heterotopic ossification. In addition, obtaining primary cells from patients with FOP is risky or impossible, as even minor procedures can induce enough trauma to drive heterotopic ossification (Kaplan et al., 2012). Thus, having a generalizable method of creating robust iMACs is crucial for elucidating disease pathology in FOP and has implications for the study of other human conditions.
Materials and Methods
Cell culture.
Human iPSCs lines were derived from control (WTC11, WT1323) and FOP patients (FOP1–1, FOP3–2), as previously described (Matsumoto et al., 2013; Miyaoka et al., 2014). Cells were cultured on feeder layers of irradiated SNL (mouse fibroblast STO cell line transformed with neomycin resistance and murine LIF genes) in mTeSR1 or mTeSR plus (StemCell Technologies) (McMahon and Bradley, 1990). Cells were passaged every 3–5 days using Accutase (StemCell Technologies). ROCK inhibitor Y-27632 (10 μM, StemCell Technologies) dissolved in 100 % DMSO was added to mTeSR1 or mTeSR plus at the time of passaging and removed on the following day. To remove SNL, hiPSCs were passaged on growth-factor-reduced Matrigel-coated plates (Corning, 150–300 ug/ml, 40 min coating) at least twice before use in differentiation into macrophages.
Macrophage differentiation (3D culture method).
All cell culture was performed under hypoxic conditions (5% O2). EBs were formed from hiPSCs and cultured in the combination of 10% mTeSR1 and 90% of aggregation media, consisted of Stem-Pro-34 (Invitrogen) supplemented with 1% GlutaMAX (Thermo Fisher Scientific), 150 mg/ml transferrin (Roche), 50ug/ml ascorbic acid (Sigma), and 450 μM monothioglycerol (Sigma-Aldrich) on 10cm ultra-low attachment plate (Corning). On the first day, EBs were cultured in the combination media with 12 ng/ml BMP4 (Peprotech) and 1 μg/ml ROCK inhibitor Y-27632 (StemCell Technologies). On the following day, media including floating cells were collected and the supernatant was aspirated after the pellet was settled down at the bottom of a conical tube without centrifugation. Cells were gently mixed and cultured for 3 days in the combination medium supplemented with 12 ng/ml BMP4 (Peprotech) and 5 ng/ml human basic fibroblast growth factor (bFGF, Peprotech). On day 4, medium were removed in the same way as described above and cultured on a new low-attachment plate in medium consisting of PromoCell basal media (PromoCell) supplemented with Cytokine Mix E (PromoCell), 10ng/ml VEGF (Calbiochem), 10ng/ml bFGF (Peprotech), 25ng/ml IL-6 (Peprotech), and 10ng/ml IL-11 (Peprotech). Medium were changed on day 7 and EBs were cultured until day10. Then, EBs were collected, washed with PBS and dissociated using Accutase. EBs were incubated with 5 ml Accutase for 10 min in a conical tube with intermittent vortexing. After centrifugation, supernatant was aspirated and cells were mixed well in IMDM (Thermo Fisher Scientific) supplemented with 10% FBS and 1% penicillin/streptomycin (Thermo Fisher Scientific). Aggregates were removed by filtering through 70μm nylon mesh. Single cells were seeded on a normal attachment tissue culture plate and cultured for 5–7 days in IMDM supplemented with 10% FBS, 1% penicillin/streptomycin (Thermo Fisher Scientific), 50ng/ml M-CSF (Peprotech) and 25ng/ml IL-3 (Peprotech). Once the attached cell layer generated floating cells, the cell suspension in a dish was collected and centrifuged. Supernatant was removed and the cell pellet was mixed with RPMI (Corning) supplemented with 10% FBS, 1% penicillin/streptomycin (Thermo Fisher Scientific) and 50 ng/ml M-CSF (Peprotech). Cells were cultured for at least 7 days until they were attached to the plate and showed typical microscopic phenotype. Once matured, the 3D-iMACs were maintained in hypoxic conditions in the same media that was changed every 3–4 days.
Macrophage differentiation (2D culture method).
HSCs were generated from hiPSCs using STEMdiff™ Hematopoietic Kit (StemCell Technologies), according to the manufacturer’s instructions. All cell culture in this method was performed under normoxic conditions. Cells harvested on day 12 were differentiated into iMACs based on either of following protocols: HSCs underwent magnetic sorting (MACS) using anti-CD34-coated beads (Miltenyi Biotec) and cultured for 7 days in RPMI (Corning) supplemented with 10% FBS, 1% penicillin/streptomycin (Thermo Fisher Scientific), and 100 ng/ml M-CSF (Peprotech). Harvested cells underwent further differentiation in the same media described above and were followed by MACS sorting using anti-CD45-coated beads (Miltenyi Biotec) on day 19. As the quality of HSCs harvested on day 12 was the most critical for generating well matured iMACs on day 19, we defined a differentiation as successful when both the harvested cell number on day 12 was more than 500,000 per well in a 12-well plate, and the cell viability was more than 70%. Once matured, 2D-iMACs were maintained under the same condition as described in 3D culture method or polarized into M1-like phenotype by culturing for 24 hours with the combination of 20 ng/ml IFN-γ (Peprotech) and 10 ng/ml LPS (MilliporeSigma).
Primary monocyte isolation and differentiation into macrophages.
Primary monocytes from control subjects and patients with FOP were isolated from carefully-collected peripheral blood samples, as we previously described (Barruet et al., 2018) and following protocols approved by the UCSF Institutional Review Board including written consent. Blood samples were obtained either directly into BD Vacutainer CPT Cell preparation tubes (BD Biosciences, catalog 362761) with sodium citrate or were retrieved from a TRIMA leukoreduction filter (Vitalant, Trima 50ml MNC enriched). Following gradient centrifugation to isolate the buffy coat, cells were purified by MACS using anti-CD14-coated beads (Miltenyi Biotec) to yield the final primary monocyte preparations. Monocytes were cultured for 7 days in RPMI (Corning) supplemented with 10% FBS, 1% penicillin/streptomycin (Thermo Fisher Scientific) and 50 ng/ml M-CSF (Peprotech), then differentiated into M2-like macrophages. These M2-like cells were polarized into a M1-like phenotype by culturing for 24 hours with the combination of 20 ng/ml IFN-γ (Peprotech) and 10 ng/ml LPS (MilliporeSigma).
Flow cytometry.
Cells were stained with anti-human CD34-FITC, CD45-PE, CD14-PE-Cyanine7, CD11b-FITC, CD11b-APC, CD163-PE, CD206-APC, CD80-PerCP-eFlour 710, or CD3-APC (Supplementary Table 1). Analysis was performed using a BD LSR II (BD Biosciences), according to the manufacturer’s instructions. Each percentage shown in figures are percent of living cells that fall within the gate shown. Sytox blue (Thermo Fisher Scientific) was used as a viability marker.
Quantitative real-time PCR (qRT-PCR).
Total RNA was prepared using TRI Reagent (MilliporeSigma, catalog T9424) and processed with the Arcturus PicoPure RNA isolation kit (Applied Biosystems, catalog KIT0204) as previously described (Schepers et al., 2012). RNA (0.2–0.5 μg) was reverse transcribed into cDNA with the Maxima H minus cDNA synthesis kit (Thermo Scientific, catalog K1682). cDNA pre-amplified with GE PreAmp Master Mix (Fluidigm Inc., catalog 100–5580). Quantitative PCR (qPCR) was analyzed in technical triplicates with TaqMan™ Universal Master Mix II, with UNG (Applied Biosystems, catalog 4440038) or by Sybr Green on a Viia7 Real-Time PCR System (Life Technologies) according to manufacturers’ instructions. Taqman and Sybr Green primers are listed in Supplemental Table 2. Levels of mRNA expression were normalized to those of a housekeeping gene, β-actin or GAPDH, as indicated.
Macrophage stimulation with LPS, HMGB1, and S100A8/A9.
After the polarization into M1- or M2-like cells, primary macrophages and iMACs were cultured for an additional 3 days in RPMI (Corning) supplemented with 10% FBS, 1% penicillin/streptomycin (Thermo Fisher Scientific), and 50 ng/ml M-CSF (Peprotech) to wash out the effects of the low dose LPS stimulation used to drive M1 differentiation. Culture medium was changed just before the stimulation. Cells were then stimulated with LPS (0.1, 1, or 10 ng/ml, MilliporeSigma L4391) for 24 hours, 5 ug/ml HMGB1 (R&D Systems 1690-HMB-050) for 6 hours, or 2 ug/ml S100A8/A9 (R&D Systems 8226-S8–050) for 6 hours for the analysis of their gene expression. To analyze their cytokine production in response to DAMPs or PAMPs, cells were stimulated with 10 ng/ml LPS, 5 μg/ml HMGB1, or 2 ug/ml S100A8/A9 for 24 hours.
Cytokine analysis.
Primary macrophages and iMACs were seeded at 10,000 cells per well in a 48-well plate. 200 μl of media were used in each well. Cells were stimulated as described above and the medium was collected and stored at −80°C before analysis. Samples were sent to Eve Technologies (Canada) for analysis with Human Cytokine/Chemokine 42-, 48-, or 65-Plex panels (HD42, HD48, or HD65). The concentration of Activin A was quantified by Quantikine Elisa from R&D Systems. For generating heatmaps, fluorescence intensity was used when all samples are analyzed in the same run. When samples were analyzed in different runs, cytokine concentrations were used.
Anti-human/mouse/rat Activin A antibody and SB431542 treatment.
After M1-like polarization of iMACs, culture medium were changed and 100 ng/ml anti-human/mouse/rat Activin A antibody (R&D Systems) or 10 mM SB431542 (Tocris Bioscience) was added every 24 hours. After 3 days of culture, mediaum was collected for cytokine analysis as above.
Phagocytosis assays.
Polarized macrophages were plated at 10,000 cells per well in a 48-well plate. Culture medium was removed, and a bacterial phagocytosis assay was performed using pHrodo Green E. coli BioParticles conjugate (Life Technologies, catalog #P35366) following the manufacturer’s instructions. Images were obtained using a fluorescent microscope.
Statistical analysis.
All data are expressed as mean ± standard error of the mean (SEM). The normality of the data was confirmed by Shapiro-Wilk test for all comparisons of two groups. For comparison of two groups, statistical analysis was performed using a two-tailed unpaired Student’s t-test. For comparison of more than three groups with normal distribution, statistical analysis was performed by one-way analysis of variance (ANOVA) followed by Dunnett’s test (vs. control group) or Tukey-Kramer test. For more than three groups with non-normal distribution, Shirley-Williams test (vs. control group) or Steel-Dwass test was used. The software R was used for generating heatmaps. Data were considered significant (P < 0.05) or highly significant (P < 0.01).
Results
A 3D embryoid-body culture method creates macrophages similar to primary M2-like macrophages
Multiple 2D and 3D methods have been proposed to generate iMACs (Table 1). Several reports previously suggested that cells within embryoid body (EB) differentiations can contain mesodermal precursors which can generate hematopoietic cells (Muffat et al., 2016; Takata et al., 2017). We attempted to generate macrophages from two independent and established undifferentiated hiPSCs (cell lines WT1323 and WTC11) (Matsumoto et al., 2013; Miyaoka et al., 2014) via EBs (Fig. 1A) in hypoxic (5% O2) conditions. EBs were formed by suspension culture for 4 days with bone morphogenetic protein (BMP)-4 and basic fibroblast growth factor (bFGF), followed by hematopoietic specification with 6 days of culture in thrombopoietin (TPO), stem cell factor (SCF), vascular endothelial growth factor (VEGF), bFGF, Flt3 ligand (FLT3LG), IL-3, IL-6, and IL-11 (see Methods). After transition to adherent culture with IL-3 and macrophage colony-stimulating factor (M-CSF) on day 10, the hemogenic endothelial monolayer began to produce floating cells. These cells were harvested on day 15–17, then matured into macrophages with M-CSF (referred to as 3D-iMACs). Adherent cells showed a microscopic phenotype consistent with human primary macrophages (Fig. 1B). FACS analysis demonstrated CD45, CD11b, CD14, CD206, and CD163 expression (Fig. 1C; Supplementary Fig. 1). Cytokine production patterns in untreated (NT) or LPS-stimulated (LPS) conditions showed similar patterns to human primary M2-like macrophages (Fig. 1D). Differentiated iMACs also showed phagocytic capacity (Fig. 1E). However, this EB-based method suffered from consistently low efficiency and reproducibility between differentiation experiments (10% success rate, n=20 attempts on WTC11 and WT1323) in creating M2-like macrophages. Thus, we examined other methods for creating hiPSC derived macrophages.
Table 1.
Recent reports for generating human iPSC-derived macrophages
| Study | Cultural dimension | Culture period (days) |
Surface marker profiling | Direct comparison to primary cells (surface marker) |
Cytokine profiling | Direct comparison to primary cells (cytokine) |
Disease application |
|---|---|---|---|---|---|---|---|
| Wilgenburg et al., 2013 | 3D | 21 | CD14, CD16, CD163, CD86, MHCⅡ, CD206 | Yes | Yes (36 kinds) |
No | No |
| Yanagi-machi et al., 2013 | 2D | 23–35 | CD14, CD68, CX3CR1, CD86, CD80, CD163, CD206, HLA-DR | Yes | Yes (2 kinds) |
Yes | No |
| Lachmann et al., 2015 | 3D | 22–30 | CD14, CD45, CD11b, CD163, CD86, TRA-1–60, CD34, CD19, CD66b | Yes | Yes (12 kinds) |
Yes | No |
| Zhang et al., 2015 | 3D | 22 | CD43,CD34,CD18,CD45,CD11b/c,CD14,CD16,CD115,CX3CR1,CCR2,CD1a,CD83,CD3,CD19 | Yes | Yes (36 kinds) |
Yes | Yes (Mendelian disease) |
| Takata et al., 2017 | 2D | 26 | CD14, CD45, CD11b, CD163, CX3CR1 | No | Yes (16 kinds) |
Yes (only to disease model) |
Yes (familial Mediterranean fever) |
| Hong et al., 2018 | 3D | 35–40 | CD11b, CD14, CD40, CD68, MHCⅡ | No (only to THP-1 cell line) |
No | No | Yes (Tuberculosis infection) |
| Cao et al., 2019 | 2D | 19 | CD45, CD11b, CD18, CD163, CD80, CD206 | Yes | Yes (11 kinds) |
Yes | No |
| Nenasheva et al., 2020 | 3D | 22–27 | CD45, CD11b, CD64, CD16, CD80, CD86, HLA-DR, CD195, CD163, CD206 | Yes | Yes (41 kinds) |
Yes | No |
| This report | 2D/3D | 19/22 | CD45, CD34, CD11b, CD14, CD163, CD80, CD206 | Yes | Yes (65 kinds) |
Yes | Yes (FOP) |
Figure 1. Differentiation of 3D-iMACs and their functional analysis.
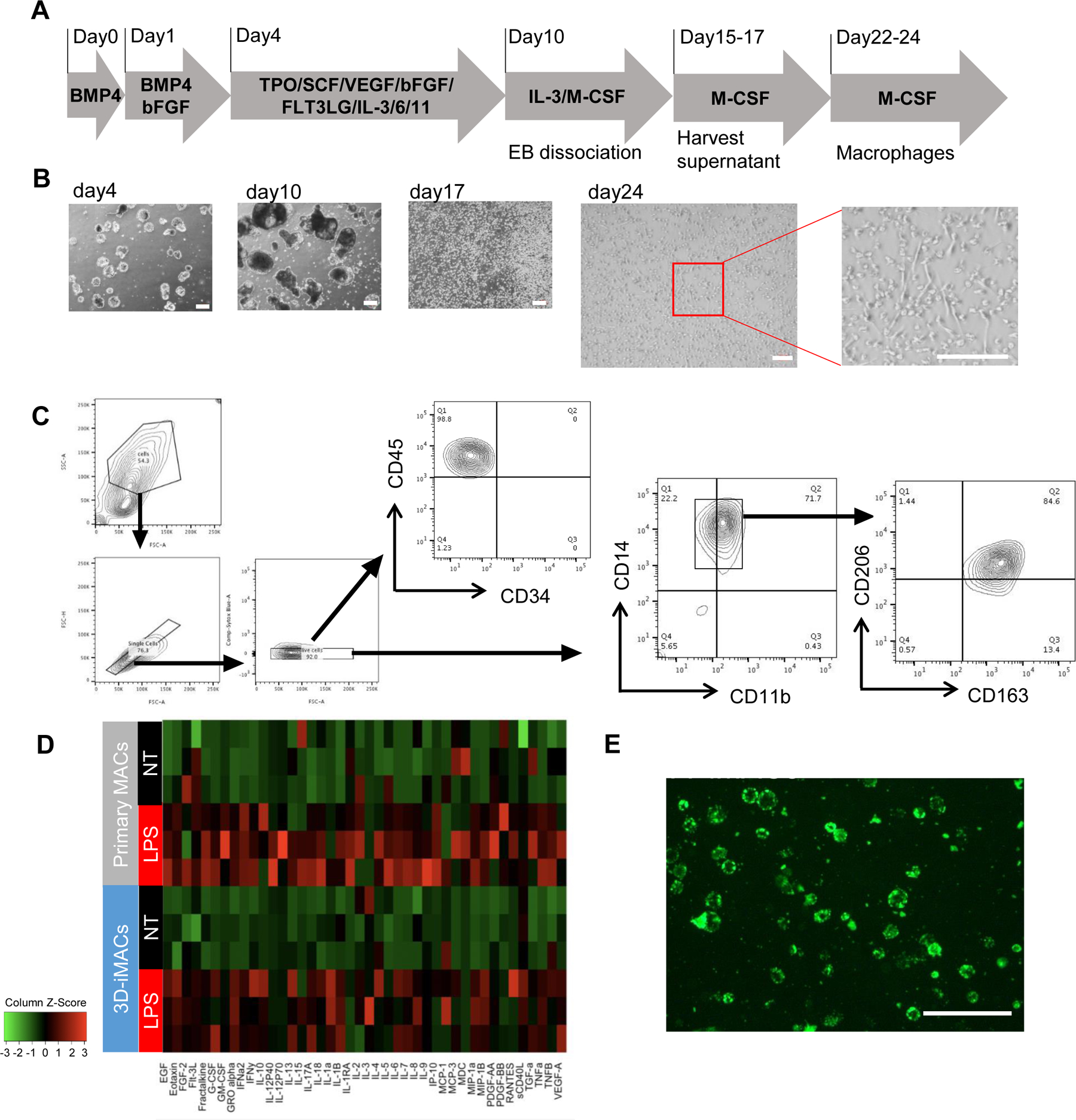
A. Schematic representation of the protocol for generating 3D-iMACs. All cell culture was performed under hypoxic (5% O2) conditions. B. Bright-filed images at day 4, 10,17, and 24. Scale bar, 200μm. C. Surface marker expressions of 3D-iMACs at day 24. Most cells express both of CD163 and CD206. D. Comparison of cytokine production profiles between 3D-iMACs and primary M2-like macrophages. They were not treated (NT) or stimulated with 10ng/ml LPS for 24 hours (LPS). n=3 biological replicates. iMACs and primary MACs showed similar cytokine profiles with or without LPS stimulation. E. Phagocytic activities of 3D-iMACs were confirmed under a fluorescent microscope. E. coli BioParticles conjugated with pHrodo Green appear bright green when they are taken up by macrophages. Scale bar, 200μm.
A 2D culture method can generate both M1- and M2-like macrophages, similar to primary macrophages
Although our 3D-iMACs could form M2-like human macrophages based on cell surface marker and cytokine profiling, we tested a 2D culture system to see if it could address some of the limitations of the EB-based 3D culture differentiation method. Hematopoietic stem cells (HSCs) were first created using a commercially available kit (STEMdiff™ Hematopoietic Kit, StemCell Technologies) and then directed into the monocyte/macrophage lineages with M-CSF (Fig. 2A). The iMACs generated by our 2D culture method (2D-iMACs) showed the appropriate morphology (Fig. 2B), and FACS analysis showed expression of CD45, CD11b, and CD14 on day 19 (Fig. 2C). These cells were positive for CD68, and negative for CD3 by FACS (Supplementary Fig. 2), and showed levels of CD3, CD56, and CD19 expression consistent with M1-like and M2-like macrophages derived from primary monocytes (Supplementary Fig. 2C). The cells were maintained in M-CSF (Fig. 2D), showed gene expression patterns consistent with M2-like cells (Fig. 2E, blue bars) (Huang et al., 2014; Mantovani et al., 2002), and expressed higher levels of CD163. The overall success rate for generating 2D-iMACs was approximately 70%.
Figure 2. Differentiation of 2D-iMACs and their functional analysis.
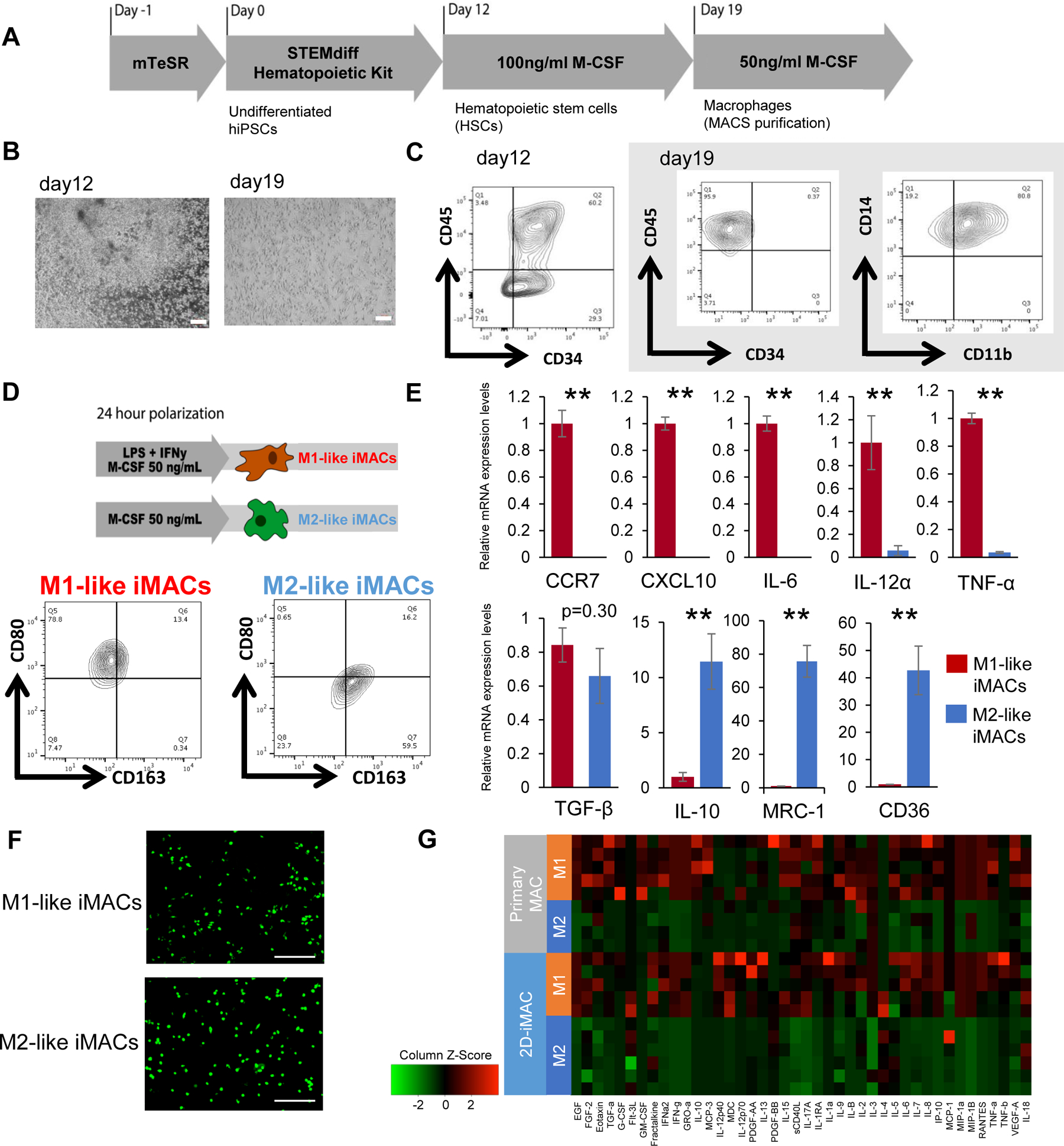
A. Schematic representation of the protocol for generating 2D-iMACs. B. Bright-field images at days 12 and 19. Scale bar, 200μm. C. Surface marker expression of 2D-iMACs at days 12 and 19 (WT1323). They are positive for CD45, CD14, and CD11b. D. Polarization protocol and their subsequent surface maker expressions. While M2-like iMACs showed high CD163 and low CD80 positivity, the expression patterns were opposite in M1-like iMACs. E. mRNA expression levels of macrophage-related genes in M1-like and M2-like 2D-iMACs. M1-related and M2-related genes are shown in the upper and lower respectively. They showed the expected expression patterns except for TGF-β. Expression levels are normalized to levels of a housekeeping gene, β-actin. Student’s t-test was used for comparison of two groups. **p<0.01. Data represent mean ± SEM of four independent experiments with technical triplicates. F. Phagocytic activities of M1-like and M2-like 2D-iMACs were confirmed (WTC11). E. coli BioParticles taken up by macrophages appear bright green. Scale bar, 200μm. G. Comparison of cytokine production profiles between 2D-iMACs and primary macrophages. M1-like and M2-like 2D-iMACs showed similarities to primary M1- and M2-like macrophages respectively. n=4–6 biological replicates.
As granulocyte-macrophage colony-stimulating factor (GM-CSF) is known to polarize human primary macrophages into a M1-like phenotype (Hamilton, 2008; Jaguin et al., 2013; Sierra-Filardi et al., 2010), we first tested if culturing with GM-CSF instead of M-CSF could generate robust M1-like iMACs. Although GM-CSF increased CD80 expression, a marker of M1-like macrophages, our cell number yield remained low (Supplementary Fig. 3).
As an alternative, the combination of interferon-γ (IFN-γ) and lipopolysaccharide (LPS) has been used to polarize primary monocyte-derived macrophages, THP-1 cells, and iMACs cells into a M1-like phenotype (Cao et al., 2019; Genin et al., 2015; Mommert et al., 2020). Treatment of our 2D-iMACs with 20ng/ml IFN-γ and 10ng/ml LPS increased CD80 expression and decreased CD163 expression (Fig. 2D) as expected for M1-like cells. qPCR analysis showed that these IFN-γ/LPS treated 2D-iMACs expressed high levels of M1-related pro-inflammatory cytokine genes, while unpolarized 2D-iMACs showed higher expression of M2-related genes (Fig. 2E). Although we tested if IFN-γ alone could polarize these cells into the M1-like phenotype, as indicated previously (Gan et al., 2017; Mantovani et al., 2004), the combination of IFN-γ and LPS was more effective (Supplementary Fig. 4). Our results show a strong similarity of 2D-iMACs prior to addition of polarization stimuli to primary “M2-like” macrophages (monocyte-derived macrophages cultured with M-CSF alone in our manuscript), which indicates that our iMAC protocol generates M2-like polarized iMACs as the starting point. Addition of IL-4 to the 2D-iMACs had only minimal effects on surface marker and gene expression in the 2D-iMACs (Supplementary Fig. 5.), despite other approaches that used IL-4 for M2-like polarization (Murray et al., 2014). Thus, we define our M1-like macrophages as ones polarized with IFN/LPS, and M2-like macrophages as iMACs that continue to be cultured with M-CSF alone, respectively. Both types of iMACs showed intact phagocytic activity (Fig. 2F).
To definitively confirm the M1-like and M2-like characteristics of our differentiated iMACs, we compared their spectrum of cytokine production with that of human primary monocyte-derived macrophages polarized by the same methods. Both M1- and M2-like iMACs showed strong similarities in cytokine production compared to their respective M1- and M2-like human primary macrophages (Fig. 2G). Several differences between the iMACs and primary macrophages were identified: the concentration of IL-12p40 was significantly higher in M1-like iMAC media, while GM-CSF, IL-10, and IL-1RA were significantly higher in primary M1-like macrophage media (Supplementary Fig. 6A). The concentration of IL-4 was significantly higher in M2-like iMACs, while IL-1RA, MCP-3, IL-8, MIP-1α, MIP-1β, and TNF-α were higher in primary M2-like macrophages (Supplementary Fig. 6B). However, no major differences were identified in the other 32 cytokines assayed.
Comparison of 2D vs. 3D derived iMACs
The results from our 2D and 3D iMAC differentiation strategies suggested that both protocols favored a M2-like phenotype by default. Comparing the M2-like cells from these two methods showed that they had equivalent expression levels of CD14 and CD206, but that M2-like 3D-iMACs had slightly higher expression of CD163 and CD80 compared to M2-like 2D-iMACs (Fig. 3A). Compared with macrophages derived from primary cells, both types of iMACs showed detectable but lower expression levels of CD11b, a gene that mediates leukocyte adhesion and migration (Solovjov et al., 2005).
Figure 3. Comparison between primary macrophages, 3D-iMACs and 2D-iMACs.
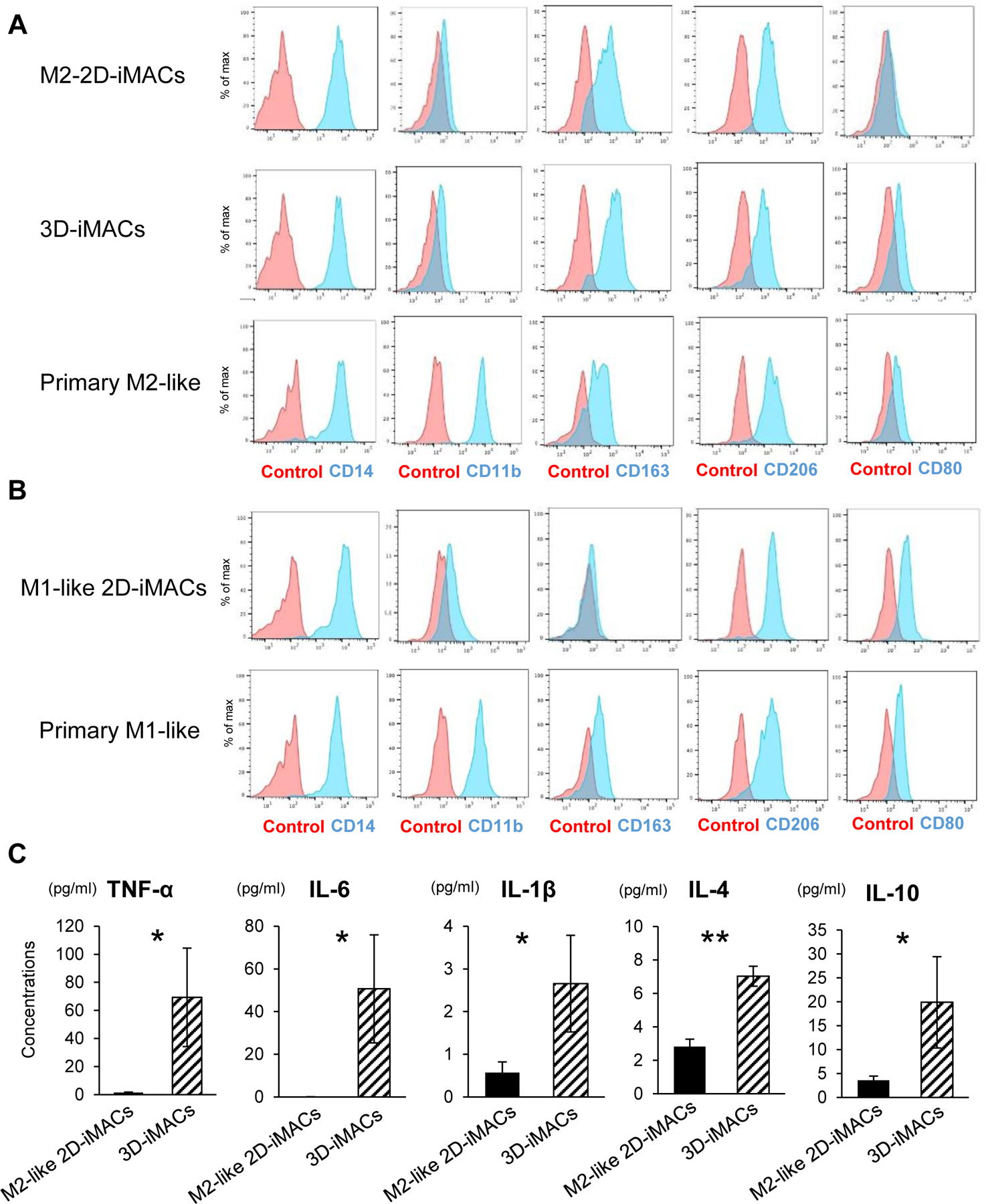
A. Surface marker expression of primary M2-like macrophages, 3D-iMACs, and 2D-M2-like iMACs. Each type of macrophages and unstained cells as negative controls are shown in blue or red respectively. CD163 and CD80 expressions were slightly higher in 3D-iMACs compared with M2-like 2D-iMACs. B. Surface marker expression of primary M1-like macrophages and M1-like 2D-iMACs. They showed similar profiles except CD11b. C. Concentrations of key pro- and anti-inflammatory cytokines secreted by 3D- and M2-like 2D-iMACs. Cytokines levels shown here were significantly higher in 3D-iMACs compared with M2-like 2D-iMACs. Student’s test was used for comparison. *p<0.05, **p<0.01. Data represent mean ± SEM of six independent experiments with technical triplicates.
M1-like 2D-iMACs showed similar expression patterns compared to primary M1-like macrophages, but also showed lower CD11b (Fig. 3B), as has been described previously (Lachmann et al., 2015), and slightly lower levels of CD163 compared to primary M1-like macrophages. CD163 levels were lower than the M2-like macrophages, consistent with polarization into the M1-like lineage. Despite multiple attempts using GM-CSF to encourage M1-like differentiation, we were unable to get sufficient yield of M1-like macrophages from the 3D-iMACs.
Comparison of the cytokine production between the M2-like 3D-iMACs and M2-like 2D-iMACs showed strong similarities. Notably, several key pro- and anti-inflammatory cytokines, including TNF-α, IL-6, IL-1β, IL-4, and IL-10, were significantly higher in M2-like 3D iMACs compared with M2-like 2D-iMACs (Fig. 3C). Several of these cytokines are associated with M1-like macrophages, although the absolute concentrations were much lower than M1-like 2D-iMACs. Combined with the finding that 3D-iMACs showed a higher expression of CD80, a M1 macrophage marker, than M2-like 2D-iMACs, the comparison of the phenotypes between M2-like 3D-iMACs and M2-like 2D-iMACs suggests that the 3D-iMAC protocol may produce macrophages that have mixed M1/M2-like characteristics, and thus unable to be redirected into a M1-like lineage possibly due to a developmental or technical block in the 3D approach.
2D-iMACs showed response to DAMPs and PAMPs stimulation
The M1-like and M2-like 2D-iMACs showed good correlation of cytokine production in the unstimulated state with primary M1-like and M2-like macrophages. Macrophages respond to triggers that can cause inflammation in response to trauma or infections, which are typically classified into two main groups. Damage-associated molecular patterns (DAMPs) are derived from host cells including dead cells (Newton and Dixit, 2012; Tang et al., 2012). Pathogen-associated molecular patterns (PAMPs) are derived from microorganisms. LPS from the outer membrane of gram-negative bacteria, was used as a representative PAMP (Mogensen, 2009).
M1-like and M2-like iMACs were cultured for 3 days after their polarization (Fig. 4A) to wash out the effects of the initial low dose LPS stimulation, because continuous stimulation can induce macrophage exhaustion and dampen the response to acute LPS exposure (Hamaidia et al., 2019). For DAMP stimulation, we tested high mobility group box 1 protein (HMGB1), which is a nuclear protein found in the cytoplasm of cells undergoing necrosis and known to mediate inflammation (Bianchi, 2007; Jube et al., 2012); and S100A8/A9, which are Ca2+ binding proteins released from damaged cells and act as alarmins in various inflammatory diseases (Schaefer, 2014). To test responses to PAMP stimulation, M1-like and M2-like 2D-iMACs were treated with different concentrations of LPS for 24 hours. Heatmaps of their cytokine production patterns showed iMACs can produce the comparable amounts of cytokines to primary monocyte-derived macrophages in response to LPS (Fig. 4B and 4C). Surprisingly, iMACs appeared to have higher responses to HMGB1 than primary cells. Responses to S100A8/A9 were identifiable but low in both iMACs and primary cells.
Figure 4. Response to PAMP (LPS) and DAMPs (HMGB1, S100A8/A9) stimulations in 2D-iMACs.
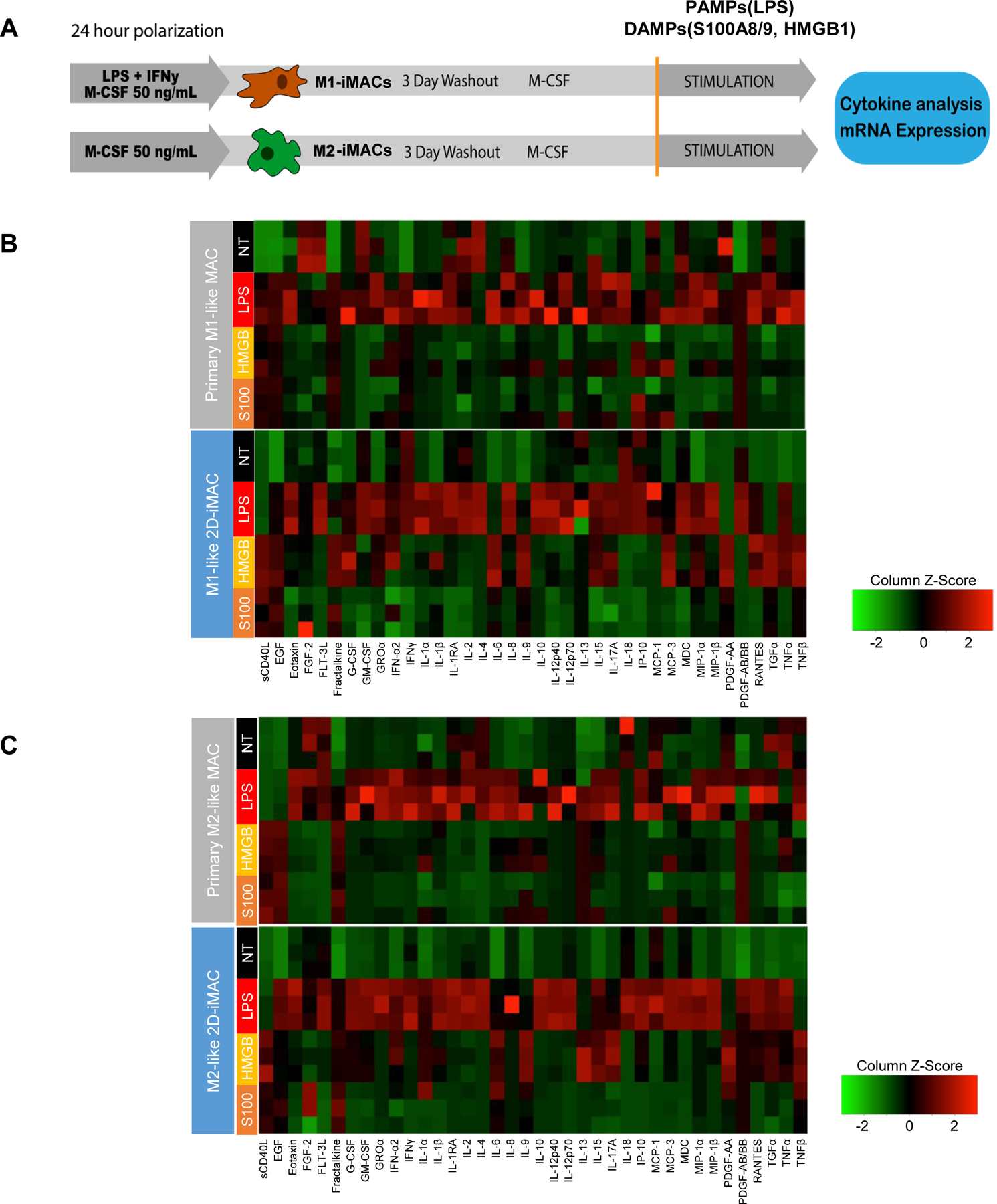
A. Protocol for testing the response to PAMPs and DMAPs. B. Comparison of cytokine production profiles in response to a PAMP (LPS) and DAMPs (HMGB1, S100A8/A9) between primary M1-like macrophages and M1-like 2D iMACs. Primary cells and iMACs showed similarities regarding their responses to LPS. NT, HMGB, and S100 labels refer to untreated, stimulated with HMGB1, and stimulated with S100A8/A9, respectively. Primary cells and iMACs showed strong similarities regarding their responses to LPS. C. Comparison of cytokine production profiles in response to PAMP (LPS) and DAMPs (HMGB1, S100A8/A9) between primary M2-like macrophages and M2-like 2D iMACs. n=2–3 biological replicates for each condition.
qPCR analysis to identify gene expression changes showed significant upregulation of various pro-inflammatory cytokines including IL-6 and TNF-α both in M1-like and M2-like iMACs. Similar trends were confirmed with each type of stimulation (Supplementary Fig. 7A and 7B). The HMGB1 and S100A8/A9 DAMPs induced very similar responses. Concerning the CCR7 and IL-10 expression, similar trends were observed with other DAMPs, although no statistical difference was detected likely due to the low number of replicates. Pro-inflammatory cytokines were also significantly elevated both in M1-like and M2-like iMACs in response to LPS, except for significant downregulation of IL-10 in M1-like iMACs.
Unstimulated WT- and FOP-iMACs showed similar cytokine gene expression
Genetic disorders of abnormal macrophage activity can result in significant medical complications. One such disorder is FOP, a rare inherited disease with progressive heterotopic ossification (Barruet and Hsiao, 2018) and increased BMP signaling activity. The ACVR1R206H mutation associated with FOP induces a significant elevation of cytokine levels, including IL-3, IL-7, IL-8, and GM-CSF in the serum and by primary monocyte-derived macrophages from patients with FOP (Barruet et al., 2018). Other data in mouse models of FOP suggest that depletion of macrophages can decrease the severity of HO formation (Convente et al., 2018). However, detailed investigation of primary human macrophages with the FOP mutation are limited since collection of primary samples from patients with FOP can induce inflammatory “flare-ups” with significant long term consequences (Baujat et al., 2017).
Macrophages from FOP patient-derived hiPSCs (FOP-iMACs) were created using the 2D-iMAC protocol. Gene expression and surface marker analyses showed no significant differences between WT- and FOP-2D-iMACs immediately after polarization (Supplementary Fig. 8).
FOP-M1-like iMACs but not FOP-M2-like iMACs showed prolonged inflammatory cytokine production
Our prior studies suggested that FOP macrophages may be “primed” to respond to an acute inflammatory event, as reflected by increased responses to low doses of LPS (Barruet et al., 2018). Because our protocol to create M1-like iMACs uses a low level of stimulation with LPS, we hypothesized that this prior stimulation would result in earlier exhaustion of the FOP M1-like iMACs compared to control WT-like iMACs. Unlike the prior human primary monocyte-derived M1-like and M2-like macrophages (Barruet et al., 2018), which did not undergo chronic low level LPS stimulation to generate M1-like cells, we found that the lowest dose of LPS (0.1 ng/ml) showed an almost equivalent level of response to the highest dose of LPS (10 ng/ml) (Supplementary Fig. 9). This result was not because of differences in TLR4 expression levels (Supplementary Fig. 10). Comparison of the cytokine production profiles between WT- and FOP-iMACs with or without LPS stimulation (Fig. 5A) showed a qualitatively different pattern in M1-like iMACs. This notion is supported by pairwise comparisons using Student’s t-test between conditions to identify the number of cytokines that are significantly different (13 cytokines at baseline are significantly higher in FOP-M1 than WT-M1; 26 cytokines after LPS stimulation are significantly higher in WT-M1 than FOP-M1). In contrast, M2-like iMACs showed strong similarity between WT and FOP, except for higher MIP-1α at baseline; and higher RANTES, IP-10, TRAIL, and MIP-1δ when stimulated (Supplementary Fig. 11). We observed differences between WT and FOP in cytokine responses that were similar to our prior results in primary FOP cells (Barruet et al., 2018). In FOP-M1-like iMACs, several key pro-inflammatory cytokines including TNF-α, IL-1α, IL-6, IFN-γ, MIP-1α/β, and RANTES in the nontreated (NT) group were significantly higher than WT-M1-like iMACs at their baseline state (Fig. 5B). In contrast, pro-inflammatory cytokines including TNF-α, IL-6, and IL-1β in FOP-M1-like iMACs were lower in response to LPS compared with WT-M1-like iMACs (Fig. 5C). This suggests that FOP-M1-like iMACs have a prolonged inflammatory response that persists even after 3 days of wash out culture, and likely leads to exhaustion and decreased responsiveness to an acute LPS stimulation.
Figure 5. Comparison between WT-iMACs and FOP-iMACs.
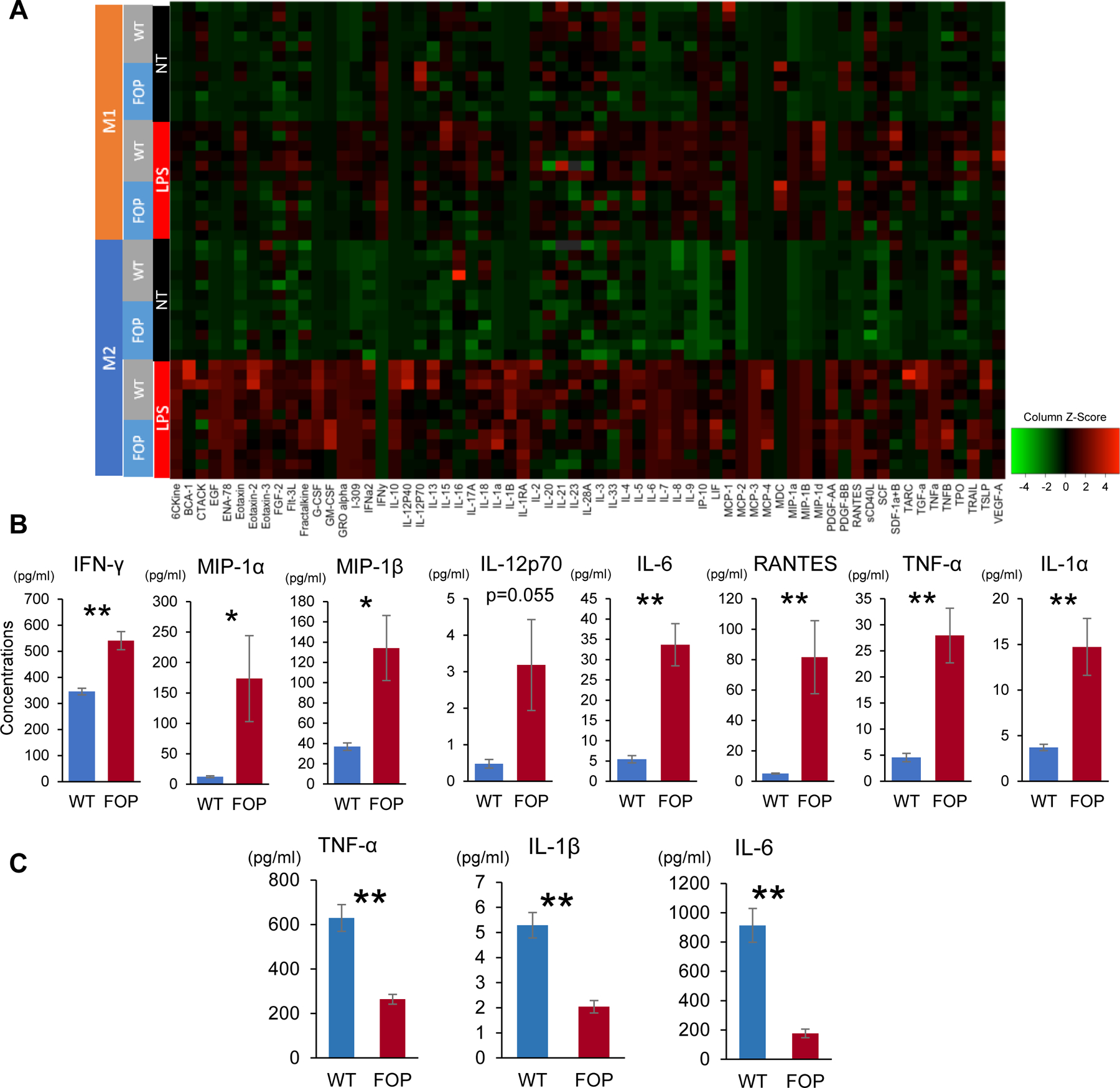
A. Comparison of cytokine production profiles between WT and FOP-2D-iMACs. Cells were not treated (NT) or stimulated with 10ng/ml LPS for 24 hours (LPS) based on the protocol shown in Figure 4A. n=6 biological replicates. Differences between WT- and FOP-iMACs were more apparent in M1-like iMACs. B. Cytokines that showed higher production in FOP-M1-like iMACs compared with WT-M1-like iMACs at their baseline state (nontreated, NT). Student’s t-test was used for comparison of two groups. *p<0.05, **p<0.01. Data represent mean ± SEM of six independent experiments with technical triplicates. C. Cytokine concentrations showing significant differences between WT- and FOP-M1-like iMACs stimulated with 10ng/ml LPS. Key pro-inflammatory cytokine concentrations were higher in WT-M1-like iMACs when stimulated with 10ng/ml LPS. Student’s t-test was used for comparison of two groups. **p< 0.01Data represent mean ± SEM of six independent experiments with technical triplicates.
FOP-M1-like and M2-like iMACs showed similarities to WT-iMACs regarding their responses to DAMPs stimulation
Because we found significant differences in the responses to LPS, we next tested if the FOP iMACs could also respond to DAMPs using a similar stimulation protocol previously used in Fig. 4B. FOP-M1-like and M2-like iMACs showed significant upregulation of pro-inflammatory cytokine genes including CXCL10, IL-6, TNF- α, and IL-1β in response to both DAMPs. Regarding CCR7 and IL-10 expression, similar trends towards upregulation were observed in all conditions, although some did not meet statistical significance. Taken together, the FOP iMACs showed similar mRNA cytokine responses to DAMP stimulation except for the IL-10 response in M1-like iMACs, as compared to control iMACs.
FOP-M1-like iMACs showed a significantly higher production of Activin A
Prior studies showed that Activin A is a driver of the ossification in FOP (Hino et al., 2017; Hino et al., 2015). The specific source and function of Activin A has remained unclear in FOP, although prior studies have shown that Activin A is produced by M1-like macrophages and Activin A can promote the macrophage inflammation response (Sierra-Filardi et al., 2011). Activin A concentrations in the untreated FOP-M1-like iMACs (NT group) were significantly higher than in WT-M1-like iMACs (Fig. 6A). The levels of Activin A production were not significantly increased in NT or LPS-stimulated FOP-M2-like iMACs versus WT-M2-like iMACs. The response to 10 ng/ml LPS was also blunted by the presence of Activin A, similar to other pro-inflammatory cytokines as shown above and suggesting macrophage exhaustion. Blockade of Activin A by a neutralizing antibody or by treatment with the BMP pathway inhibitor SB431542 on FOP-M1-like iMACs did not significantly decrease pro-inflammatory cytokine production compared with the control groups (Fig. 6B). In addition, we confirmed that ACVR1 expression in FOP-iMACs was not significantly different from WT-iMACs (Supplementary Fig. 13) and Egln3, which is an Activin A dependent pro-inflammatory macrophage marker, was downregulated in FOP-iMACs, indicating that the pro-inflammatory responses in FOP-iMACs was not caused via Activin A signaling. Our results suggest that in FOP, M1-like macrophages are likely a key source of Activin A, and that the production is likely a result of a generalized inflammatory response. The lack of effect on proinflammatory cytokine production by blockade of Activin A suggests Activin A may not have a self-amplifying signaling feedback loop within the macrophage lineages in FOP.
Figure 6. Activin A concentrations in iMACs and cytokine concentrations in FOP-M1-like iMACs treated with inhibitors of activin A pathways.
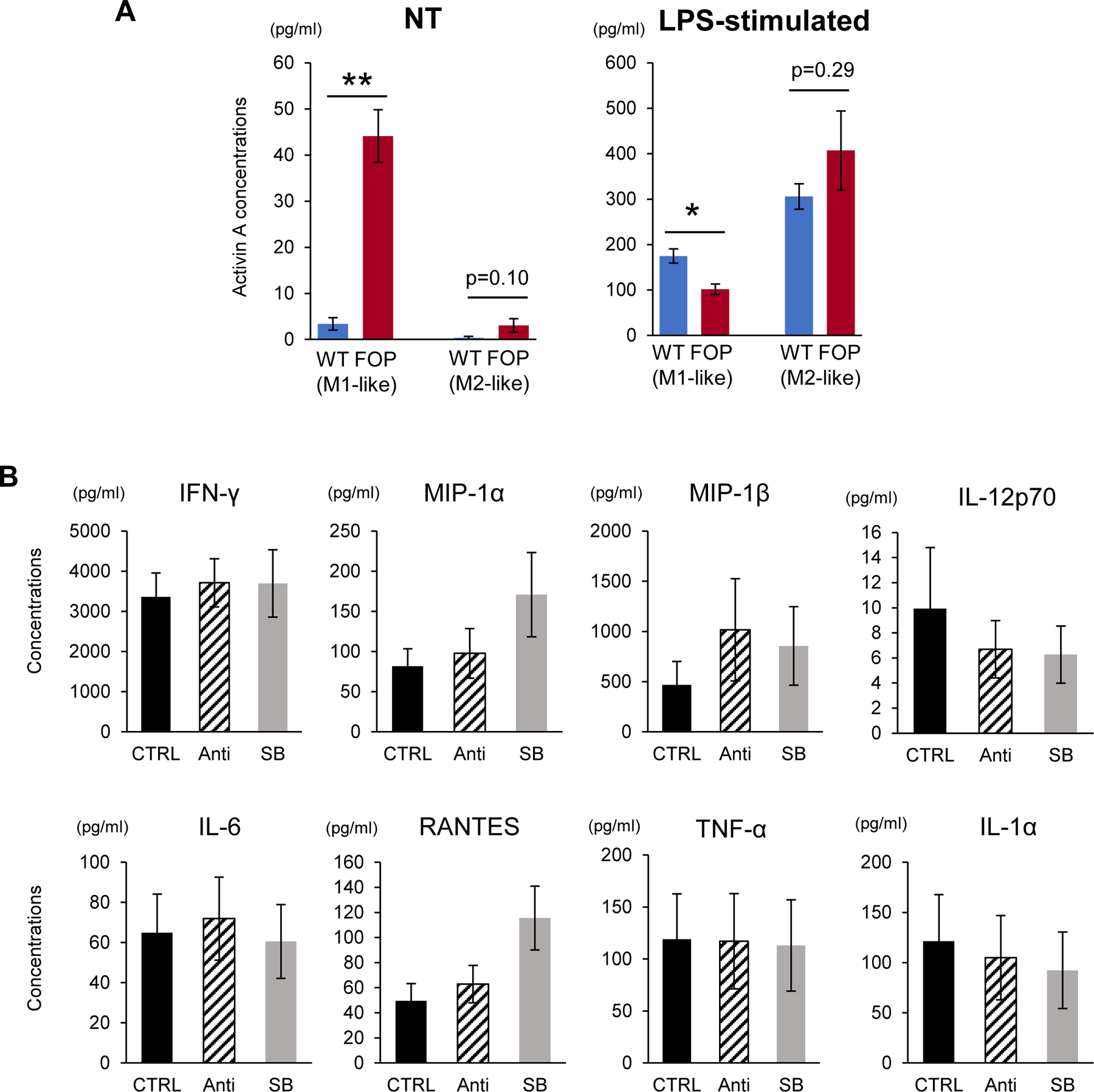
A. Activin A concentrations in NT and LPS-stimulated group were analyzed using Human/Mouse/Rat Activin A Quantikine ELISA Kit. Analyzed samples were the same as those used in Fig. 5A. The concentrations of FOP-M1-like iMACs were significantly higher than WT-M1-like iMACs in NT group, while their response to LPS was downregulated. Student’s t-test was used for comparison of two groups. *p<0.05, **p<0.01. Data represent mean ± SEM of six independent experiments with technical triplicates. B. FOP-M1-like iMACs were treated with 100 ng/ml anti-human/mouse/rat activin A antibody (Anti) or 10 mM SB431542 (SB) after M1 polarization. Each inhibitor was added every 24 hours. After 3 days culture, supernatants were collected and analyzed. There were no significant differences in key pro-inflammatory cytokines that were found elevated in FOP-M1-like iMACs as shown in Fig. 5C. Dunnett’s test was used to compare each group to control (CTRL). Data represent mean ± SEM of five independent experiments with technical triplicates.
Discussion
Creating macrophages from hiPSCs to model human skeletal disease remains challenging. Multiple protocols are available but there is significant variability of the potential macrophage phenotypes that can be formed (Table 1). Multiple cytokines are important for generating M1- and M2-like macrophages in vivo, including the use of GM-CSF or M-CSF as factors that can drive cells into M1- or M2-like lineages respectively (Hamilton, 2008; Jaguin et al., 2013; Sierra-Filardi et al., 2010).
Our study compared two different methods to generate macrophages from hiPSCs: a 3D embryoid body-based method or a 2D monolayer culture method. Both differentiation methods led to iMACs with typical microscopic phenotypes. Furthermore, the polarized M1-like and M2-like 2D-iMACs showed cytokine production and phagocytic activities comparable to primary human monocyte-derived M1-like and M2-like macrophages. Surprisingly, the iPSCs-derived 2D- and 3D-iMACs showed low but detectable levels of the cell surface marker CD11b, although levels of CD14, CD80, CD206, and CD163 were as expected. Lower levels of CD11b were previously reported in a different iMAC differentiation protocol (Lachmann et al., 2015), and lower expression of CD11b has been identified in several tissue macrophage populations in mice (Ghosn et al., 2010; Haldar and Murphy, 2014). The phenotypes and origins of human tissue-resident macrophages remain incompletely defined (Davies and Taylor, 2015). The finding of lower CD11b expression levels may indicate that our hiPSC-derived macrophages share similarities with tissue-resident macrophages, while retaining functional characteristics of monocyte-derived macrophages.
Other groups have previously described protocols for generating iMACs via 3D EB strategies (Lachmann et al., 2015; Muffat et al., 2016; Nenasheva et al., 2020; Takata et al., 2017; van Wilgenburg et al., 2013). These differentiation methods are thought to mimic aspects of the physiological embryogenesis of early erythro-myeloid progenitors (EMP) in yolk-sac, and EB-derived iMACs can show gene expression profiles consistent with yolk-sac-derived macrophages, marked by absence of HOXA expression and MYB independent differentiation pathways (Lee et al., 2018). Our 3D-iMACs showed a pattern of cytokine production typical of primary M2-like macrophages, but different levels as compared to primary monocyte-derived M2-like cells and the 2D-iMACs. In addition, the cell surface markers suggested an intermediate phenotype based on the elevated expression of both M1 (CD80) and M2 (CD163) markers, which is also indicated in a recent report (Nenasheva et al., 2020).
Considering that macrophages can have profound plasticity and variability of surface marker expression profiles in each inflammatory phase (Wu et al., 2013), these findings, combined with those of other EB-derived methods (Lachmann et al., 2015; Muffat et al., 2016; Takata et al., 2017; van Wilgenburg et al., 2013), suggest that the 3D EB approach may generate macrophages that are functionally distinct from those made by 2D methods. In our hands, however, the low reproducibility and high variability between cell lines limited the application of the 3D EB method for further experimentation. Although EB sizes are reported to affect their subsequent differentiation and cystic spheroids are likely to generate hematopoietic lineages (Hong et al., 2010; Miyamoto and Nakazawa, 2016), attempts to control the different EB sizes or cell numbers in EBs could not overcome the reproducibility concern. Moreover, tissue-specific microenvironmental stimuli are required to replicate the functions and phenotypes of macrophages in tissues (Nenasheva et al., 2020). This suggests that the 3D EB method may be highly cell line dependent, and pose challenges for using it to compare cells derived from different cell lines.
In contrast, the 2D approach has been used to direct undifferentiated hiPSCs into mesodermal lineages (Cao et al., 2019; Niwa et al., 2011; Yanagimachi et al., 2013). We utilized a commercially available kit that allowed us to make HSCs easily and quickly, and this method showed higher reproducibility. 1 week of culture after the HSC stage was sufficient to allow iMAC maturation in our protocol, producing cells with cytokine production equivalent to human primary macrophages.
We found that polarization of the iMACs into M1-like cells required the combination of IFN-γ and LPS treatment. While some previous reports defined macrophages polarized with IFN-γ alone as M1-like (Gan et al., 2017; Mantovani et al., 2004), we found that IFN-γ alone increased expression of both CD80 and CD163, which are M1- and M2-specific markers respectively. In contrast, qPCR analysis showed that the combination of LPS and IFN-γ more effectively upregulated IL-6 and suppressed IL-10, suggesting that the combination strategy is more effective at driving M1-like differentiation.
Our comparison with primary cells also showed that M1-like 2D-iMACs polarized with IFN-γ and LPS showed significantly higher IL-12p40 production, but lower IL-10 and IL-1RA, which are anti-inflammatory cytokines. This suggests that hiPSC-derived macrophages may be more likely to acquire a pro-inflammatory phenotype consistent with previous reports (Cao et al., 2019). In contrast, the default iMACs formed after culture with M-CSF appears to adopt a M2-like phenotype even in the absence of IL-4 or IL-13, which have previously been used to drive a M2-like phenotype (Murray et al., 2014). IL-4 was insufficient in our hands to drive more M2-like polarization (Supplementary Fig. 3), which differed from other literature (Shapouri-Moghaddam et al., 2018). However, the higher production of IL-4 by our M2–2D-iMACs might explain the baseline M2-like phenotype and the lack of additional polarization with extra IL-4 treatment.
Functionally, primary macrophages and iMACs showed strong similarities in their responses to LPS at both protein and gene expression levels. In contrast, iMACs tended to show increased responsiveness to the HMGB1 DAMP. Differences between the measured cytokine levels and mRNA analyses likely result from the different assessment timepoints (24 hrs for cytokines and earlier for mRNA) and from the different mechanisms of cytokine release (new synthesis vs. release of intracellular stores). Considering that mRNA levels were analyzed after 6 hrs of stimulation for both in HMGB1 and S100A8/A9, our results may indicate that the peak of upregulated gene expression in HMGB1 may occur in a later phase compared to S100A8/A9. This may also contribute to the differences in measures of total cytokine concentrations of HMGB1 which were higher at 24 hrs as shown in the heatmap. In addition, the effect of DAMP stimulation in M1-polarized macrophages may also be affected by the pre-treatment with LPS used as part of the M1 differentiation protocol. This could occur since HMGB1 is secreted during macrophage activation and may in turn modulate the macrophage inflammatory response (El Gazzar, 2007). To define the DAMP response further, more specific protocols to remove the interactions between each stimulation would be desired. However, our findings still showed interesting mRNA expression results, including an upregulation in TGF-β, which is categorized as an anti-inflammatory M2-like cytokine, while IL-10, which is also an M2-related cytokine, was downregulated when stimulated with LPS. TGF-β may have a pleiotropic role with both potent regulatory and inflammatory activity (Li and Flavell, 2008a; Li and Flavell, 2008b; Sanjabi et al., 2009). These findings support the need for future analysis of the DAMP response by iMACs and provided the justification for using PAMP stimulation when applying our system to FOP.
One unique aspect of our study is the application to understand the immunological responses in FOP. FOP is a rare inherited disease characterized by progressive heterotopic ossification (Barruet and Hsiao, 2018). FOP is caused by activating mutations in ACVR1 (Shore et al., 2006). The highly recurrent single point mutation (ACVR1R206H) results in constitutive activation of BMP signaling pathways (Shore et al., 2006). Activin A also acts as a neo-ligand for ACVR1R206H, causing aberrant signaling through SMAD1/5/9 pathway in FOP (Aykul and Martinez-Hackert, 2016; Hatsell et al., 2015; Hino et al., 2015; Olsen et al., 2015). We previously reported significant elevations of cytokine levels, including IL-3, IL-7, IL-8, and granulocyte-macrophage colony-stimulating factor (GM-CSF) in FOP (Barruet et al., 2018). TGF-β, one cytokine secreted by M2-like macrophages, was increased in the sera of FOP patients, compatible with a previous study in mice (Wang et al., 2018).
Using the 2D-iMACs, we found higher pro-inflammatory cytokine production in unstimulated FOP-M1-like iMACs, consistent with our prior findings of a pro-inflammatory state in FOP serum and in primary monocyte-derived M1-like macrophages (Barruet et al., 2018). MIP-1α levels were elevated in both M1-like and M2-like iMACs, consistent with the findings in our previous report showing FOP-patient derived primary monocytes produced higher level of MIP-1α in response to LPS stimulation. In addition, significantly higher IP-10 (CXCL10) and RANTES (CCL5) in FOP-M2-like iMACs were also consistent with the result of FOP-patient derived primary monocytes in our previous report. These findings support a crucial role for macrophage dysfunction in driving excessive inflammation in FOP, however, additional studies are needed to determine the precise mechanisms of how trauma ultimately causes flare-ups and subsequent HO in patients with FOP (Baujat et al., 2017).
Surprisingly, we found evidence that cytokine production was saturated in the M1-like 2D-iMACs when stimulated with the lowest concentration of LPS stimulation. qPCR analysis showed that the expression levels of TLR4 were not significantly different between 2D-iMACs and primary macrophages. However, FOP-M1-like iMACs showed higher inflammatory cytokine production, although the difference was not present immediately after the M1-like polarization steps. The difference in timing suggests that the 3 days washout period of LPS/IFN-γ after polarization can revert control cells towards a more responsive phenotype (ie more rapid recovery to further LPS stimulation), while the FOP-iMACs retained their LPS-activated pro-inflammatory nature for a longer period of time, leading to desensitization to further LPS activation and the seemingly late paradoxical decrease in cytokine production of FOP M1-like iMACs. This finding in FOP M1-like iMACs was consistent with our prior experiment with primary FOP samples showing that LPS stimulation of naive FOP monocytes (never previously exposed to LPS) have prolonged activation of NF-kb compared to control monocytes (Barruet et al., 2018).
The ACVR1R206H mutation confers an intriguing neoceptor capacity to the ACVR1 receptor, leading to abnormal sensitivity to Activin A (Hino et al., 2015). Although we previously did not find changes in serum levels of Activin A (Barruet et al., 2018), we found higher Activin A production by FOP-M1-like iMACs compared to controls, and that this response could not be blocked by anti-Activin A or BMP inhibitory treatments. This suggests that local tissue macrophages may be an important source of Activin A to drive ossification in FOP. This also suggests the decrease in HO induced by blockade of Activin A after injury in mouse models of FOP (Hatsell et al., 2015) involves mechanisms that do not directly target macrophages, but rather decrease ligand availability to the ACVR1R206H neoceptor. Our inhibitor studies indicated that the higher Activin A production and higher pro-inflammatory nature of the FOP-iMACs were not changed by the inhibition of Activin A pathways. Thus, while Activin A may play a role in driving abnormal BMP pathway activation in ACVR1R206H containing cells, Activin A production in macrophages is not directly driven by ACVR1R206H. This is consistent with recent findings suggesting that Activin A inhibition does not block non-FOP HO formation (Hwang et al., 2020) and consistent with a proposed mechanism in which inflammation-induced Activin A functions on ACVR1R206H receptor expressed on stem cells in skeletal muscle tissue, such as fibroadipocytes (Lees-Shepard et al., 2018), to cause the heterotopic ossification.
These findings using our iMAC method provide new information about the pathogenesis of heterotopic ossification in FOP (Fig. 7). FOP M1-like macrophages in particular show a mild increased sensitivity to certain ligands, such as LPS, and produce higher levels of proinflammatory cytokines and a tendency towards prolonged activation. This likely contributes to the dramatic inflammatory symptoms of the flare ups. Surprisingly, we did not see a large difference in the cytokine production responses to the two DAMPs that might be released after tissue injury, suggesting that other trigger molecules released during injury still need to be identified. The FOP M1-like macrophages also showed increased Activin A production, providing a source of the neoligand for the ACVR1R206H receptor. The Activin A production does not appear to be directly driven by an Activin A feedback loop, but rather is a result of the increased activation response of FOP macrophages. These results suggest that the two pathways of the inflammatory flareups and the increased Activin A driven heterotopic ossification can be separated, and may account for the clinical reports of patients with FOP forming heterotopic bone without classical inflammatory flares.
Figure 7. Working model of our findings with WT- and FOP-iMACs.
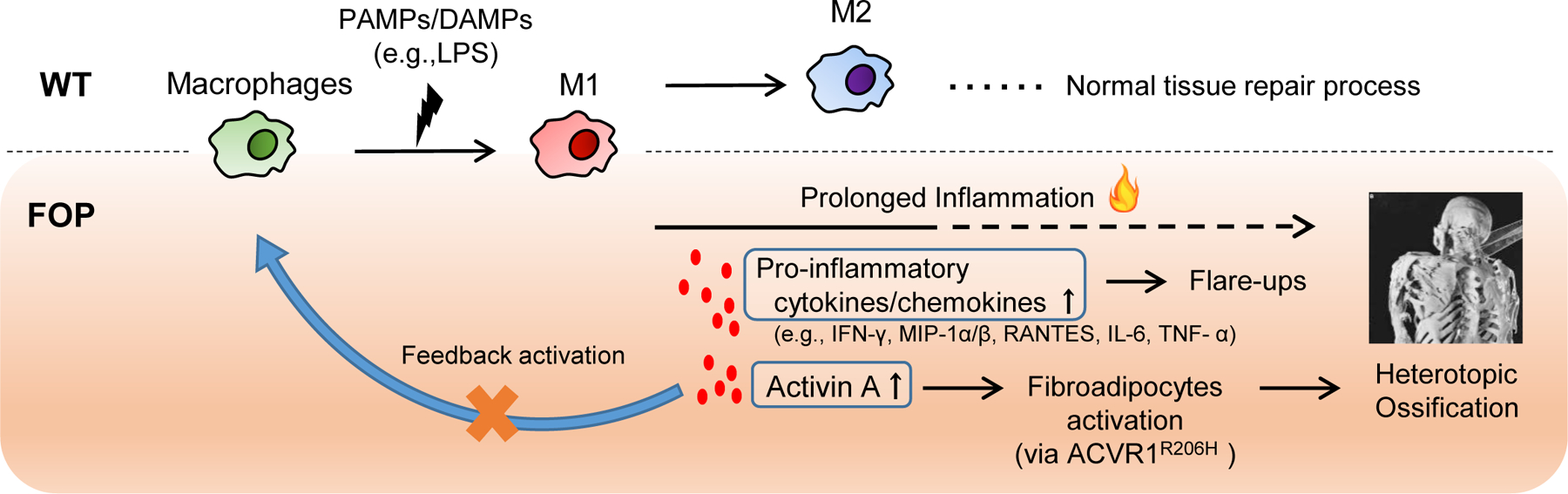
FOP macrophages showed a prolonged activation compared with controls. Activated macrophages produce higher pro-inflammatory cytokines/chemokines, resulting in flare-ups in patients. Also, the higher production of Activin A by FOP activated macrophage causes fibroadipocytes activation via ACVR1 mutation, leading into heterotopic ossifications. Activin A blockade showed no feedback activation of macrophages by activin A.
Our studies have several important limitations. First, our experience shows that detailed phenotyping using cell surface marker expression, gene expression, and cytokine production, at baseline and of the stimulated state of hiPSC-derived macrophages, is important for defining the specific cell subtypes and potential responses, particularly for disease modeling. The specific differentiation protocols can produce subtle but important differences, including the yet unexplained lower levels of CD11b in iMACs relative to primary macrophages. Second, differences between individual hiPSC lines can be a significant confounder, particularly since primary cells also show individual variability in gene expression and cytokine production. Comparison to primary cells provides an important validation for the hiPSC derived lineages. Finally, cellular immune responses remain highly complex, and so complementary in vitro and in vivo studies are very valuable for understanding disease phenotypes.
In summary, we established two different methods for generating macrophages from hiPSCs. Macrophages differentiated from FOP patient-derived hiPSCs showed a prolonged inflammatory nature that is present in human primary FOP cells, indicating that our iMACs model can recapitulate aspects of the human disease state for further study. In addition, we found that human macrophages may be a major source of inflammation-mediated Activin A production in humans. These findings demonstrate the utility of a robust iPSC derived macrophage protocol to provide novel insight into the key role of macrophages in humans for driving the formation of HO in patients with FOP.
Supplementary Material
Acknowledgements:
We graciously appreciate the grant support to ECH from the National Institutes of Health (AR066735, AR073015, DK17035, and AR075055), the Doris Duke Charitable Fund Clinical Scientist Development Award (2014099), the March of Dimes (1-FY14-211), the UCSF-Stanford CERSI Program (Grant#31), the UCSF Academic Senate, and the Robert L Kroc Chair in Rheumatic and Connective Tissue Diseases III; to MN from Department of Veterans Affairs Biomedical Laboratory R&D Merit Award (BX002994), the National Institute of Arthritis and Musculoskeletal and Skin Diseases Awards (R01AR066735 and P30AR070155), and the Russell/Engelman Rheumatology Research Center at UCSF; to RDC from AR072778-S1; to KM from the Mochida Memorial Foundation for Medical and Pharmaceutical Research and the Uehara Memorial Foundation; and to AT from the UCSF Summer Explore Fellowship and the Alpha Omega Alpha Carolyn Kuckein Student Research Fellowship. We appreciate the technical support and discussions from Kelly Wentworth.
Conflict of Interest Statement:
ECH serves without compensation on the Medical Registry Advisory Board of the International Fibrodysplasia Ossificans Progressiva Association, the International Clinical Council on FOP, and on the Fibrous Dysplasia Foundation Medical Advisory Board. ECH receives clinical trials research support from Clementia Pharmaceuticals Inc., an Ipsen company, to support clinical trials of palovarotene in FOP. ECH received prior support from Regeneron Pharmaceuticals and Neurocrine Biosciences for clinical trials related activities. These pose no conflicts for this study.
References
- Anthony BA, and Link DC. 2014. Regulation of hematopoietic stem cells by bone marrow stromal cells. Trends Immunol 35:32–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aykul S, and Martinez-Hackert E. 2016. Transforming Growth Factor-beta Family Ligands Can Function as Antagonists by Competing for Type II Receptor Binding. J Biol Chem 291:10792–10804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barruet E, and Hsiao EC. 2018. Application of human induced pluripotent stem cells to model fibrodysplasia ossificans progressiva. Bone 109:162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barruet E, Morales BM, Cain CJ, Ton AN, Wentworth KL, Chan TV, Moody TA, Haks MC, Ottenhoff TH, Hellman J, Nakamura MC, and Hsiao EC. 2018. NF-kappaB/MAPK activation underlies ACVR1-mediated inflammation in human heterotopic ossification. JCI Insight 3: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baujat G, Choquet R, Bouee S, Jeanbat V, Courouve L, Ruel A, Michot C, Le Quan Sang KH, Lapidus D, Messiaen C, Landais P, and Cormier-Daire V. 2017. Prevalence of fibrodysplasia ossificans progressiva (FOP) in France: an estimate based on a record linkage of two national databases. Orphanet J Rare Dis 12:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi ME 2007. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol 81:1–5. [DOI] [PubMed] [Google Scholar]
- Cao X, Yakala GK, van den Hil FE, Cochrane A, Mummery CL, and Orlova VV. 2019. Differentiation and Functional Comparison of Monocytes and Macrophages from hiPSCs with Peripheral Blood Derivatives. Stem Cell Reports 12:1282–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakkalakal SA, Zhang D, Culbert AL, Convente MR, Caron RJ, Wright AC, Maidment AD, Kaplan FS, and Shore EM. 2012. An Acvr1 R206H knock-in mouse has fibrodysplasia ossificans progressiva. J Bone Miner Res 27:1746–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Convente MR, Chakkalakal SA, Yang E, Caron RJ, Zhang D, Kambayashi T, Kaplan FS, and Shore EM. 2018. Depletion of Mast Cells and Macrophages Impairs Heterotopic Ossification in an Acvr1(R206H) Mouse Model of Fibrodysplasia Ossificans Progressiva. J Bone Miner Res 33:269–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies LC, and Taylor PR. 2015. Tissue-resident macrophages: then and now. Immunology 144:541–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath MY 2014. Targeting inflammation in the treatment of type 2 diabetes: time to start. Nat Rev Drug Discov 13:465–476. [DOI] [PubMed] [Google Scholar]
- Ehses JA, Perren A, Eppler E, Ribaux P, Pospisilik JA, Maor-Cahn R, Gueripel X, Ellingsgaard H, Schneider MK, Biollaz G, Fontana A, Reinecke M, Homo-Delarche F, and Donath MY. 2007. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 56:2356–2370. [DOI] [PubMed] [Google Scholar]
- El Gazzar M 2007. HMGB1 modulates inflammatory responses in LPS-activated macrophages. Inflamm Res 56:162–167. [DOI] [PubMed] [Google Scholar]
- Eming SA, Wynn TA, and Martin P. 2017. Inflammation and metabolism in tissue repair and regeneration. Science 356:1026–1030. [DOI] [PubMed] [Google Scholar]
- Gan ZS, Wang QQ, Li JH, Wang XL, Wang YZ, and Du HH. 2017. Iron Reduces M1 Macrophage Polarization in RAW264.7 Macrophages Associated with Inhibition of STAT1. Mediators Inflamm 2017:8570818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genin M, Clement F, Fattaccioli A, Raes M, and Michiels C. 2015. M1 and M2 macrophages derived from THP-1 cells differentially modulate the response of cancer cells to etoposide. BMC Cancer 15:577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosn EE, Cassado AA, Govoni GR, Fukuhara T, Yang Y, Monack DM, Bortoluci KR, Almeida SR, Herzenberg LA, and Herzenberg LA. 2010. Two physically, functionally, and developmentally distinct peritoneal macrophage subsets. Proceedings of the National Academy of Sciences of the United States of America 107:2568–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldar M, and Murphy KM. 2014. Origin, development, and homeostasis of tissue-resident macrophages. Immunol Rev 262:25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaidia M, Gazon H, Hoyos C, Hoffmann GB, Louis R, Duysinx B, and Willems L. 2019. Inhibition of EZH2 methyltransferase decreases immunoediting of mesothelioma cells by autologous macrophages through a PD-1-dependent mechanism. JCI Insight 4: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton JA 2008. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol 8:533–544. [DOI] [PubMed] [Google Scholar]
- Hatsell SJ, Idone V, Wolken DM, Huang L, Kim HJ, Wang L, Wen X, Nannuru KC, Jimenez J, Xie L, Das N, Makhoul G, Chernomorsky R, D’Ambrosio D, Corpina RA, Schoenherr CJ, Feeley K, Yu PB, Yancopoulos GD, Murphy AJ, and Economides AN. 2015. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci Transl Med 7:303ra137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hino K, Horigome K, Nishio M, Komura S, Nagata S, Zhao C, Jin Y, Kawakami K, Yamada Y, Ohta A, Toguchida J, and Ikeya M. 2017. Activin-A enhances mTOR signaling to promote aberrant chondrogenesis in fibrodysplasia ossificans progressiva. J Clin Invest 127:3339–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hino K, Ikeya M, Horigome K, Matsumoto Y, Ebise H, Nishio M, Sekiguchi K, Shibata M, Nagata S, Matsuda S, and Toguchida J. 2015. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proceedings of the National Academy of Sciences of the United States of America 112:15438–15443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong D, Ding J, Li O, He Q, Ke M, Zhu M, Liu L, Ou WB, He Y, and Wu Y. 2018. Human-induced pluripotent stem cell-derived macrophages and their immunological function in response to tuberculosis infection. Stem cell research & therapy 9:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SH, Werbowetski-Ogilvie T, Ramos-Mejia V, Lee JB, and Bhatia M. 2010. Multiparameter comparisons of embryoid body differentiation toward human stem cell applications. Stem Cell Res 5:120–130. [DOI] [PubMed] [Google Scholar]
- Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, Beatty W, Love-Gregory L, Lam WY, O’Neill CM, Yan C, Du H, Abumrad NA, Urban JF Jr., Artyomov MN, Pearce EL, and Pearce EJ. 2014. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol 15:846–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurvitz EA, Mandac BR, Davidoff G, Johnson JH, and Nelson VS. 1992. Risk factors for heterotopic ossification in children and adolescents with severe traumatic brain injury. Arch Phys Med Rehabil 73:459–462. [PubMed] [Google Scholar]
- Hwang C, Pagani CA, Das N, Marini S, Huber AK, Xie L, Jimenez J, Brydges S, Lim WK, Nannuru KC, Murphy AJ, Economides AN, Hatsell SJ, and Levi B. 2020. Activin A does not drive post-traumatic heterotopic ossification. Bone 138:115473. [DOI] [PubMed] [Google Scholar]
- Ivashkiv LB 2011. Inflammatory signaling in macrophages: transitions from acute to tolerant and alternative activation states. Eur J Immunol 41:2477–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaguin M, Houlbert N, Fardel O, and Lecureur V. 2013. Polarization profiles of human M-CSF-generated macrophages and comparison of M1-markers in classically activated macrophages from GM-CSF and M-CSF origin. Cell Immunol 281:51–61. [DOI] [PubMed] [Google Scholar]
- Jube S, Rivera ZS, Bianchi ME, Powers A, Wang E, Pagano I, Pass HI, Gaudino G, Carbone M, and Yang H. 2012. Cancer cell secretion of the DAMP protein HMGB1 supports progression in malignant mesothelioma. Cancer Res 72:3290–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan FS, Chakkalakal SA, and Shore EM. 2012. Fibrodysplasia ossificans progressiva: mechanisms and models of skeletal metamorphosis. Dis Model Mech 5:756–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocic M, Lazovic M, Mitkovic M, and Djokic B. 2010. Clinical significance of the heterotopic ossification after total hip arthroplasty. Orthopedics 33:16. [DOI] [PubMed] [Google Scholar]
- Lachmann N, Ackermann M, Frenzel E, Liebhaber S, Brennig S, Happle C, Hoffmann D, Klimenkova O, Luttge D, Buchegger T, Kuhnel MP, Schambach A, Janciauskiene S, Figueiredo C, Hansen G, Skokowa J, and Moritz T. 2015. Large-scale hematopoietic differentiation of human induced pluripotent stem cells provides granulocytes or macrophages for cell replacement therapies. Stem Cell Reports 4:282–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CZW, Kozaki T, and Ginhoux F. 2018. Studying tissue macrophages in vitro: are iPSC-derived cells the answer? Nat Rev Immunol 18:716–725. [DOI] [PubMed] [Google Scholar]
- Lees-Shepard JB, Yamamoto M, Biswas AA, Stoessel SJ, Nicholas SE, Cogswell CA, Devarakonda PM, Schneider MJ Jr., Cummins SM, Legendre NP, Yamamoto S, Kaartinen V, Hunter JW, and Goldhamer DJ. 2018. Activin-dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nat Commun 9:471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung C, Casey AT, Goffin J, Kehr P, Liebig K, Lind B, Logroscino C, and Pointillart V. 2005. Clinical significance of heterotopic ossification in cervical disc replacement: a prospective multicenter clinical trial. Neurosurgery 57:759–763; discussion 759–763. [DOI] [PubMed] [Google Scholar]
- Li MO, and Flavell RA. 2008a. Contextual regulation of inflammation: a duet by transforming growth factor-beta and interleukin-10. Immunity 28:468–476. [DOI] [PubMed] [Google Scholar]
- Li MO, and Flavell RA. 2008b. TGF-beta: a master of all T cell trades. Cell 134:392–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, and Locati M. 2004. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 25:677–686. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sozzani S, Locati M, Allavena P, and Sica A. 2002. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23:549–555. [DOI] [PubMed] [Google Scholar]
- Mathew D, Giles JR, Baxter AE, Oldridge DA, Greenplate AR, Wu JE, Alanio C, Kuri-Cervantes L, Pampena MB, D’Andrea K, Manne S, Chen Z, Huang YJ, Reilly JP, Weisman AR, Ittner CAG, Kuthuru O, Dougherty J, Nzingha K, Han N, Kim J, Pattekar A, Goodwin EC, Anderson EM, Weirick ME, Gouma S, Arevalo CP, Bolton MJ, Chen F, Lacey SF, Ramage H, Cherry S, Hensley SE, Apostolidis SA, Huang AC, Vella LA, Unit UPCP, Betts MR, Meyer NJ, and Wherry EJ. 2020. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science 369: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto Y, Hayashi Y, Schlieve CR, Ikeya M, Kim H, Nguyen TD, Sami S, Baba S, Barruet E, Nasu A, Asaka I, Otsuka T, Yamanaka S, Conklin BR, Toguchida J, and Hsiao EC. 2013. Induced pluripotent stem cells from patients with human fibrodysplasia ossificans progressiva show increased mineralization and cartilage formation. Orphanet J Rare Dis 8:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo K, Chavez RD, Barruet E, and Hsiao EC. 2019. Inflammation in Fibrodysplasia Ossificans Progressiva and Other Forms of Heterotopic Ossification. Curr Osteoporos Rep 17:387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy EF, and Sundaram M. 2005. Heterotopic ossification: a review. Skeletal Radiol 34:609–619. [DOI] [PubMed] [Google Scholar]
- McMahon AP, and Bradley A. 1990. The Wnt-1 (int-1) proto-oncogene is required for development of a large region of the mouse brain. Cell 62:1073–1085. [DOI] [PubMed] [Google Scholar]
- Merad M, and Martin JC. 2020. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol 20:355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto D, and Nakazawa K. 2016. Differentiation of mouse iPS cells is dependent on embryoid body size in microwell chip culture. J Biosci Bioeng 122:507–512. [DOI] [PubMed] [Google Scholar]
- Miyaoka Y, Chan AH, Judge LM, Yoo J, Huang M, Nguyen TD, Lizarraga PP, So PL, and Conklin BR. 2014. Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nat Methods 11:291–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogensen TH 2009. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 22:240–273, Table of Contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mommert S, Huer M, Schaper-Gerhardt K, Gutzmer R, and Werfel T. 2020. Histamine up-regulates oncostatin M expression in human M1 macrophages. Br J Pharmacol 177:600–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muffat J, Li Y, Yuan B, Mitalipova M, Omer A, Corcoran S, Bakiasi G, Tsai LH, Aubourg P, Ransohoff RM, and Jaenisch R. 2016. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat Med 22:1358–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, Locati M, Mantovani A, Martinez FO, Mege JL, Mosser DM, Natoli G, Saeij JP, Schultze JL, Shirey KA, Sica A, Suttles J, Udalova I, van Ginderachter JA, Vogel SN, and Wynn TA. 2014. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nenasheva T, Gerasimova T, Serdyuk Y, Grigor’eva E, Kosmiadi G, Nikolaev A, Dashinimaev E, and Lyadova I. 2020. Macrophages Derived From Human Induced Pluripotent Stem Cells Are Low-Activated “Naive-Like” Cells Capable of Restricting Mycobacteria Growth. Front Immunol 11:1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, and Dixit VM. 2012. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol 4: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa A, Heike T, Umeda K, Oshima K, Kato I, Sakai H, Suemori H, Nakahata T, and Saito MK. 2011. A novel serum-free monolayer culture for orderly hematopoietic differentiation of human pluripotent cells via mesodermal progenitors. PLoS One 6:e22261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi Y, and Manabe I. 2018. Macrophages in inflammation, repair and regeneration. Int Immunol 30:511–528. [DOI] [PubMed] [Google Scholar]
- Olsen OE, Wader KF, Hella H, Mylin AK, Turesson I, Nesthus I, Waage A, Sundan A, and Holien T. 2015. Activin A inhibits BMP-signaling by binding ACVR2A and ACVR2B. Cell Commun Signal 13:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter BK, Burns TC, Lacap AP, Granville RR, and Gajewski DA. 2007. Heterotopic ossification following traumatic and combat-related amputations. Prevalence, risk factors, and preliminary results of excision. J Bone Joint Surg Am 89:476–486. [DOI] [PubMed] [Google Scholar]
- Richardson SJ, Willcox A, Bone AJ, Foulis AK, and Morgan NG. 2009. Islet-associated macrophages in type 2 diabetes. Diabetologia 52:1686–1688. [DOI] [PubMed] [Google Scholar]
- Sanjabi S, Zenewicz LA, Kamanaka M, and Flavell RA. 2009. Anti-inflammatory and pro-inflammatory roles of TGF-beta, IL-10, and IL-22 in immunity and autoimmunity. Curr Opin Pharmacol 9:447–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer L 2014. Complexity of danger: the diverse nature of damage-associated molecular patterns. J Biol Chem 289:35237–35245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schepers K, Hsiao EC, Garg T, Scott MJ, and Passegue E. 2012. Activated Gs signaling in osteoblastic cells alters the hematopoietic stem cell niche in mice. Blood 120:3425–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte-Schrepping J, Reusch N, Paclik D, Bassler K, Schlickeiser S, Zhang B, Kramer B, Krammer T, Brumhard S, Bonaguro L, De Domenico E, Wendisch D, Grasshoff M, Kapellos TS, Beckstette M, Pecht T, Saglam A, Dietrich O, Mei HE, Schulz AR, Conrad C, Kunkel D, Vafadarnejad E, Xu CJ, Horne A, Herbert M, Drews A, Thibeault C, Pfeiffer M, Hippenstiel S, Hocke A, Muller-Redetzky H, Heim KM, Machleidt F, Uhrig A, Bosquillon de Jarcy L, Jurgens L, Stegemann M, Glosenkamp CR, Volk HD, Goffinet C, Landthaler M, Wyler E, Georg P, Schneider M, Dang-Heine C, Neuwinger N, Kappert K, Tauber R, Corman V, Raabe J, Kaiser KM, Vinh MT, Rieke G, Meisel C, Ulas T, Becker M, Geffers R, Witzenrath M, Drosten C, Suttorp N, von Kalle C, Kurth F, Handler K, Schultze JL, Aschenbrenner AC, Li Y, Nattermann J, Sawitzki B, Saliba AE, Sander LE, and Deutsche C-OI. 2020. Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell 182:1419–1440 e1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senju S, Haruta M, Matsunaga Y, Fukushima S, Ikeda T, Takahashi K, Okita K, Yamanaka S, and Nishimura Y. 2009. Characterization of dendritic cells and macrophages generated by directed differentiation from mouse induced pluripotent stem cells. Stem Cells 27:1021–1031. [DOI] [PubMed] [Google Scholar]
- Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, Seifi B, Mohammadi A, Afshari JT, and Sahebkar A. 2018. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol 233:6425–6440. [DOI] [PubMed] [Google Scholar]
- Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M, Morhart R, Rogers JG, Smith R, Triffitt JT, Urtizberea JA, Zasloff M, Brown MA, and Kaplan FS. 2006. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nature genetics 38:525–527. [DOI] [PubMed] [Google Scholar]
- Sierra-Filardi E, Puig-Kroger A, Blanco FJ, Nieto C, Bragado R, Palomero MI, Bernabeu C, Vega MA, and Corbi AL. 2011. Activin A skews macrophage polarization by promoting a proinflammatory phenotype and inhibiting the acquisition of anti-inflammatory macrophage markers. Blood 117:5092–5101. [DOI] [PubMed] [Google Scholar]
- Sierra-Filardi E, Vega MA, Sanchez-Mateos P, Corbi AL, and Puig-Kroger A. 2010. Heme Oxygenase-1 expression in M-CSF-polarized M2 macrophages contributes to LPS-induced IL-10 release. Immunobiology 215:788–795. [DOI] [PubMed] [Google Scholar]
- Solovjov DA, Pluskota E, and Plow EF. 2005. Distinct roles for the alpha and beta subunits in the functions of integrin alphaMbeta2. J Biol Chem 280:1336–1345. [DOI] [PubMed] [Google Scholar]
- Takata K, Kozaki T, Lee CZW, Thion MS, Otsuka M, Lim S, Utami KH, Fidan K, Park DS, Malleret B, Chakarov S, See P, Low D, Low G, Garcia-Miralles M, Zeng R, Zhang J, Goh CC, Gul A, Hubert S, Lee B, Chen J, Low I, Shadan NB, Lum J, Wei TS, Mok E, Kawanishi S, Kitamura Y, Larbi A, Poidinger M, Renia L, Ng LG, Wolf Y, Jung S, Onder T, Newell E, Huber T, Ashihara E, Garel S, Pouladi MA, and Ginhoux F. 2017. Induced-Pluripotent-Stem-Cell-Derived Primitive Macrophages Provide a Platform for Modeling Tissue-Resident Macrophage Differentiation and Function. Immunity 47:183–198 e186. [DOI] [PubMed] [Google Scholar]
- Tang D, Kang R, Coyne CB, Zeh HJ, and Lotze MT. 2012. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev 249:158–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udagawa N 2003. The mechanism of osteoclast differentiation from macrophages: possible roles of T lymphocytes in osteoclastogenesis. J Bone Miner Metab 21:337–343. [DOI] [PubMed] [Google Scholar]
- van Wilgenburg B, Browne C, Vowles J, and Cowley SA. 2013. Efficient, long term production of monocyte-derived macrophages from human pluripotent stem cells under partly-defined and fully-defined conditions. PLoS One 8:e71098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vi L, Baht GS, Whetstone H, Ng A, Wei Q, Poon R, Mylvaganam S, Grynpas M, and Alman BA. 2015. Macrophages promote osteoblastic differentiation in-vivo: implications in fracture repair and bone homeostasis. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 30:1090–1102. [DOI] [PubMed] [Google Scholar]
- Wang X, Li F, Xie L, Crane J, Zhen G, Mishina Y, Deng R, Gao B, Chen H, Liu S, Yang P, Gao M, Tu M, Wang Y, Wan M, Fan C, and Cao X. 2018. Inhibition of overactive TGF-beta attenuates progression of heterotopic ossification in mice. Nat Commun 9:551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu AC, Raggatt LJ, Alexander KA, and Pettit AR. 2013. Unraveling macrophage contributions to bone repair. Bonekey Rep 2:373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, and Barron L. 2010. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis 30:245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, and Vannella KM. 2016. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 44:450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagimachi MD, Niwa A, Tanaka T, Honda-Ozaki F, Nishimoto S, Murata Y, Yasumi T, Ito J, Tomida S, Oshima K, Asaka I, Goto H, Heike T, Nakahata T, and Saito MK. 2013. Robust and highly-efficient differentiation of functional monocytic cells from human pluripotent stem cells under serum- and feeder cell-free conditions. PLoS One 8:e59243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Xue C, Shah R, Bermingham K, Hinkle CC, Li W, Rodrigues A, Tabita-Martinez J, Millar JS, Cuchel M, Pashos EE, Liu Y, Yan R, Yang W, Gosai SJ, VanDorn D, Chou ST, Gregory BD, Morrisey EE, Li M, Rader DJ, and Reilly MP. 2015. Functional analysis and transcriptomic profiling of iPSC-derived macrophages and their application in modeling Mendelian disease. Circ Res 117:17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.