

ABSTRACT
Vitamin B-12 is a water-soluble vitamin that plays important roles in intermediary metabolism. Vitamin B-12 deficiency has many identifiable causes, including autoimmune and other gastrointestinal malabsorption disorders, dietary deficiency, and congenital defects in genes that are involved in vitamin B-12 trafficking and functions. Another putative cause of vitamin B-12 deficiency is the high-folate–low vitamin B-12 interaction, first suspected as the cause for observed relapse and exacerbation of the neurological symptoms in patients with pernicious anemia who were prescribed high oral doses of folic acid. We propose that this interaction is real and represents a novel cause of vitamin B-12 depletion with specific etiology. We hypothesize that excessive intake of folic acid depletes serum holotranscobalamin (holoTC), thereby decreasing active vitamin B-12 in the circulation and limiting its availability for tissues. This effect is specific for holoTC and does not affect holohaptocorrin, the inert form of serum vitamin B-12. Depletion of holoTC by folic acid in individuals with already low vitamin B-12 status further compromises the availability of vitamin B-12 coenzymes to their respective enzymes, and consequently a more pronounced state of biochemical deficiency. This hypothesis is drawn from evidence of observational and intervention studies of vitamin B-12–deficient patients and epidemiological cohorts. The evidence also suggests that, in a depleted state, vitamin B-12 is diverted to the hematopoietic system or the kidney. This most likely reflects a selective response of tissues expressing folate receptors with high affinity for unmetabolized folic acid (UMFA; e.g., hematopoietic progenitors and renal tubules) compared with those tissues (e.g., liver) that only express the reduced folate carrier, which is universally expressed but has poor affinity for UMFA. The biochemical and physiological mechanisms underlying this interaction require elucidation to clarify its potential public health significance.
Keywords: vitamin B-12, folic acid, folate, holotranscobalamin, homocysteine, methylmalonic acid, pernicious anemia
Statement of Significance: There is significant circumstantial evidence that excess folic acid consumption exacerbates vitamin B-12 insufficiency, but a biochemical/physiological mechanism has not yet been identified. Herein it is hypothesized that excessive intake of folic acid depletes serum holotranscobalamin, thereby decreasing active vitamin B-12 in the circulation and limiting its availability for tissues.
Introduction
Vitamin B-12 is a water-soluble vitamin that plays important, yet diverse, roles in intermediary metabolism via complex physiology. Vitamin B-12 is the source of coenzymes for the respective methylation of homocysteine by 5-methyltetrahydrofolate (5-methylTHF) to form methionine and for the mitochondrial conversion of methylmalonyl-CoA to succinyl-CoA (Figure 1). The physiology of vitamin B-12 is a multistep process that is characterized by multiple trafficking events, associations and dissociations among several binding/carrier proteins, specific and multiligand receptor-mediated endocytosis, intracellular trafficking into different cellular compartments, and protein-specific controlled exit (1, 2). Interference with any of these steps can lead to a state of vitamin B-12 deficiency, and thus deficiency of this vitamin is multicausal. Nutritional deficiency is caused by a lack of the vitamin in the diet, whereas acquired deficiency occurs when the intestinal absorption of the vitamin is interfered with by diseases and conditions such as pernicious anemia (an autoimmune disorder affecting intrinsic factor protein produced by gastric parietal cells), chronic atrophic gastritis, Crohn's and celiac diseases, gastric bypass surgery, and medications such as metformin, gastric acid secretion blockers (i.e., proton-pump inhibitors and H2-receptor antagonists), and others (2, 3). Congenital defects in any of the 15 genes that are involved in trafficking and metabolism of vitamin B-12 also cause vitamin B-12 deficiency, as does chronic exposure to nitrous oxide (2).
FIGURE 1.

Vitamin B-12–dependent reactions. Vitamin B-12 serves as a cofactor in 2 metabolic reactions: (A) as methylcobalamin in the conversion of homocysteine and methyltetrahydrofolate to methionine and tetrahydrofolate by methionine synthase and (B) as adenosylcobalamin in the conversion of methylmalonyl CoA to succinyl CoA by methylmalonyl CoA mutase. MMA, methylmalonic acid.
Advances in pathology, serology, biochemical methodology, and cellular and molecular biology provide many of the tools to identify these deficiency states and often their causes. One exception is our poor understanding of the nature and existence of what has been referred to as the high-folate–low-vitamin B-12 interaction. This interaction was first suspected as the cause for the observed relapse and exacerbation of the neurological symptoms in pernicious anemia patients who were prescribed high oral doses of folic acid (4–6).
The use of folic acid for the treatment of pernicious anemia patients began with the availability of synthetic folic acid in 1945 (7–9). In 1947, Leon M Meyer described the results of his study on a small number of pernicious anemia patients with positive hematological responses resulting from the intake of very high doses of folic acid (20–50 mg/d) (10). According to Meyer, the high doses of folic acid were necessary to treat the hematological irregularities in patients with pernicious anemia. This practice continued for the next 4–5 y using manufactured packages of this new “Elixir,” which contained 5–20 mg folic acid (11). In 1951 Conley and Krevans from the Johns Hopkins Hospital challenged the practice, raising the possibility that the neurological manifestations in some “new” pernicious anemia patients may be linked to previously prescribed high oral doses of folic acid (20–28 mg/d) (12). This suspicion was reinforced as the number of suspected incidental cases of relapse or exacerbation of the neural symptoms in pernicious anemia patients on oral folic acid increased in the following years (4). By 1971, this practice had been discontinued, in part because it was realized that folic acid is ineffective in treating pernicious anemia, and that vitamin B-12 administration is the proper approach to treat this disease (11).
Discontinuation of this practice left unresolved the nature and the meaning of the potential harmful effect of folic acid when administered to pernicious anemia patients or individuals with vitamin B-12 deficiency. This issue, however, was rekindled and became a subject of debate with the 1996 approval by the US FDA of mandatory folic acid fortification of staple food products for the prevention of neural tube defect–affected pregnancies (13). Concern was raised that this measure could be potentially harmful to those with vitamin B-12 deficiency caused by the expected increase in folic acid intake (14–17). Others maintained that folic acid is safe at all levels and that the case studies could be interpreted other than that of harmful outcome due to folic acid intake (11, 18–20).
The full implementation of folic acid fortification in 1998 in the United States, and in Canada shortly thereafter (and now in >80 countries globally), resulted in remarkable population-wide increases in overall folate status. In the postfortification period, serum folate concentrations were 2.5-fold higher and RBC folate concentrations were >50% higher than those in the prefortification period (21). However, at the same time that folate deficiency was largely eliminated in these populations, the prevalence of intake exceeding the Tolerable Upper Intake Level of folic acid intake (1 mg/d) increased substantially (6, 22), although this was attributed to the concurrent use of supplements and fortified ready-to-eat cereals rather than to fortification alone. During the same period, population serum vitamin B-12 concentrations did not change (21).
The unresolved issue of the high-folate–low-vitamin B-12 interaction reemerged due to challenging but inconclusive findings from population studies in the postfortification era. A cross-sectional study of elderly participants (n >1300; mean age: 70 ± 0.3 y) in the 1999–2002 NHANES provided the first evidence of a possible high-folate–low-vitamin B-12 interaction in an epidemiological setting. The data showed higher prevalence of anemia and cognitive impairment in those participants with vitamin B-12 deficiency [defined as serum vitamin B-12 concentration <148 pmol/L or plasma methylmalonic acid (MMA) concentration ≥210 nmol/L] than in those with normal vitamin B-12 status, as expected. However, the OR for these 2 clinical manifestations of vitamin B-12 deficiency was significantly higher when the deficiency was accompanied by high serum folate (≥59 nmol/L) (23). A second study from Australia (where flour is also fortified with folic acid) made a similar observation using data from 3 elderly cohorts: low vitamin B-12 status (serum vitamin B-12 <250 pmol/L) in combination with elevated RBC folate (>1594 nmol/L) was associated with lower Mini-Mental State Examination (MMSE) scores than seen with low vitamin B-12 status in combination with nonelevated RBC folate (24). Interestingly, this study also found that cognitive performance was impaired in participants that had high RBC folate and serum vitamin B-12 within the low-normal range (median value = 383 pmol/L). Similar findings were observed in older adults (age >65 y) in another country, Chile, in which flour is fortified with folic acid: increased risk of low scores on a modified version of the MMSE were observed with high serum folate concentrations (>84.9 nmol/L), particularly in subjects with the lowest vitamin B-12 concentration (<148 pmol/L) (25).
The NHANES study summarized above (23) has been criticized (26), invoking the argument that those individuals with high folate status and low vitamin B-12 status were likely those who had been taking vitamin B-12–containing vitamin supplements, and that the low vitamin B-12 status is a consequence of vitamin B-12 malabsorption. Also, a study conducted in elderly individuals from the United Kingdom (where folic acid fortification was not mandatory) found no significant correlation between high-folate–low-vitamin B-12 status and cognitive impairment (OR: 1.50; 95% CI: 0.91, 2.46) (27). However, it has been pointed out (28) that the cutoff value used to define elevated serum folate in the latter study (>30 nmol/L) was relatively low compared with other studies [e.g., ≥59 nmol/L in (23) and >89.9 nmol/L in (24)], which may have reduced the likelihood of detecting a significant association of high-folate–low vitamin B-12 status with cognitive impairment. Notably, when a higher cutoff value for elevated serum folate was used in this study (≥60 nmol/L), the OR associated with high folate–low vitamin B-12 increased in magnitude [2.46 (0.90, 6.71)], although it did not reach statistical significance likely because the sample size of participants with cognitive impairment was only n = 7 (23). Altogether, these observations resurrected the issue of high-folate–low-vitamin B-12 interaction as a potential public health concern in the postfortification era—especially in populations where supplement use is prevalent.
In considering the possibility of a high-folate–low-vitamin B-12 interaction, it is important not to conflate the metabolic and public health aspects of this issue. First, we must determine whether the interaction exists as a genuine metabolic and physiologic phenomenon, and define the dose at which excess folate produces the effect. Only then can we resolve the nature of the interaction, its clinical consequences, and who might be susceptible to its effects. In this synthesis of existing observations, we approach this issue through the examination of reported biomarker responses, which are universally recognized as quantitative metrics of vitamin B-12 status (29–35). The evidence suggests that an interaction between high folate and low vitamin B-12 does in fact exist, that the biochemical response to this interaction is paradoxical, and that it represents a novel (acquired) vitamin B-12 deficiency state with a specific etiology.
Current Status of Knowledge
A biochemical paradox
Reference to the relation between high folate and low vitamin B-12 as a biochemical paradox was first made by the late Rene Malinow, who expressed concern at the time of the planning for the Vitamin Intervention for Stroke Prevention (VISP) Study, a randomized controlled trial that was designed to assess the effects of high-dose folic acid, vitamin B-12, and vitamin B-6 supplementation on recurrent stroke (36, 37). Malinow's concern was based on the result of our study of the Framingham Offspring cohort (38), which showed that, in subjects who were taking vitamin supplements, folic acid fortification was associated with significantly higher serum total homocysteine (tHcy) concentrations than at baseline (Figure 2). This is unlike those who did not take vitamin supplements where fortification with folic acid was, as expected, associated with lower serum tHcy concentrations than at baseline. Subsequently, Malinow and colleagues performed an intervention study with >300 volunteers who were given 1–2 mg folic acid/d for 3 wk, which showed that, although there was a significant increase in serum folate concentrations, the change in tHcy serum concentrations was variable, with ∼20% of the subjects exhibiting paradoxical increases in serum tHcy from baseline (Figure 3) (39).
FIGURE 2.
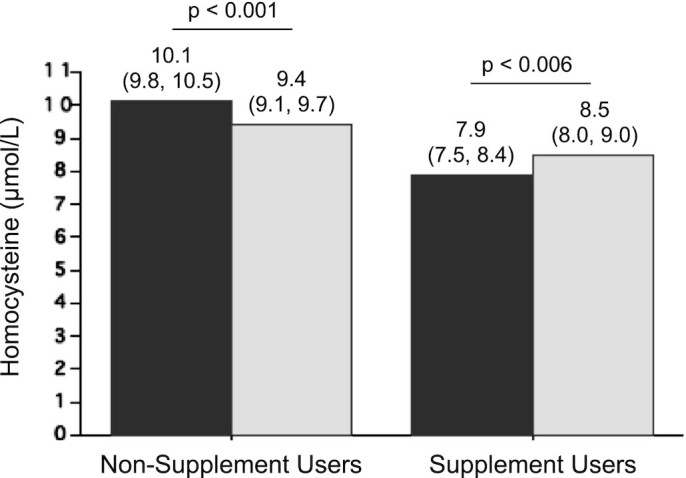
Homocysteine concentrations before and after folic acid fortification in the Framingham Offspring Cohort by use of B-vitamin supplements. In the Framingham Offspring Cohort, serum total homocysteine concentrations decreased in non–supplement users, but paradoxically increased in supplement users after the initiation of folic acid fortification. Dark-gray bars: prefortification; light-gray bars: postfortification. Non–supplement users: n = 248; supplement users: n = 102. Values are geometric means with 95% CIs. Significant differences (P < 0.05 by generalized linear model procedure). Graph created based on data reported in reference (38).
FIGURE 3.

Net changes in homocysteine concentrations in subjects (n = 304) receiving 1–2 mg folic acid for 3 wk. Although the mean net change in homocysteine was a decrease of 1.24 μmol/L (9.2% decrease from baseline), the highest quintile (∼20%) of the subjects exhibited an increase in homocysteine (mean increase = 1.46 μmol/L; 16.3% increase from baseline). Bars represent mean differences ± SDs. Q, quartile. Reproduced from reference 39 with permission.
In vitamin B-12 deficiency, elevated folate is associated with increased concentrations of tHcy and MMA
Observational studies
In a first study of its kind (40), some of us assessed the relations between serum folate and vitamin B-12 concentrations and 2 functional indicators of vitamin B-12 status, tHcy and MMA. We examined this interaction among adult participants in phase 2 of the pre–folic acid fortification NHANES III (1991–1994) and in the postfortification NHANES 1999–2002. We used data on serum folate and vitamin B-12 concentrations from 4940 NHANES III participants and 5473 NHANES 1999–2002 participants who were over the age of 20 y. We divided the NHANES III and NHANES 1999–2002 participants into 2 serum vitamin B-12 groups: vitamin B-12-deficient (vitamin B-12 <148 pmol/L) and nondeficient (vitamin B-12 ≥148 pmol/L) and 3 serum folate groups for NHANES III participants and 4 folate groups for NHANES 1999–2002 participants (Table 1). Of note is the difference between the surveys with respect to folate concentrations: median serum folate ranged from 7 to 23 nmol/L from the lowest to highest categories in NHANES III participants compared with 16 to 44 nmol/L in the NHANES 1999–2002 participants. The data revealed significant interactions between serum folate and serum vitamin B-12 in relation to circulating concentrations of both serum tHcy and serum MMA. Specifically, geometric mean tHcy decreased across increasing serum folate categories as expected for individuals with serum vitamin B-12 ≥148 pmol/L in both surveys (Ptrend < 0.001 for both NHANES III and NHANES 1999–2002) (Figure 4 and Table 1). However, we observed an interaction between folate and vitamin B-12 status among the NHANES 1999–2002 participants who were studied after fortification (Pinteraction = 0.005). Among the vitamin B-12–deficient participants, tHcy demonstrated a paradoxical association with folate, increasing significantly across the 4 serum folate categories represented in the NHANES 1999–2002 cohort (Ptrend = 0.003) (Figure 4 and Table 1).
TABLE 1.
tHcy and MMA concentrations by serum vitamin B-12 and folate categories in NHANES III (1991–1994) and NHANES (1999–2002)1
| tHcy | MMA | |||||
|---|---|---|---|---|---|---|
| Serum vitamin B-12 | Serum vitamin B-12 | |||||
| Folate | <148 pmol/L | ≥148 pmol/L | P | <148 pmol/L | ≥148 pmol/L | P |
| 1991–1994 | ||||||
| 7 nmol/L | 15.5 | 10.9 | <0.001 | 347 | 202 | 0.194 |
| (13.2, 18.1) | (10.5, 11.3) | (178, 676) | (180, 228) | |||
| 11 nmol/L | 12.2 | 9.2 | <0.001 | 290 | 191 | 0.140 |
| (10.4, 14.2) | (8.8, 9.2) | (155, 543) | (176, 206) | |||
| 23 nmol/L | 11.9 | 7.6 | 0.009 | 523 | 191 | 0.002 |
| (8.7, 16.3) | (7.5, 7.8) | (333, 821) | (180, 202) | |||
| 1999–2002 | ||||||
| 16 nmol/L | 9.9 | 9.2 | 0.222 | 175 | 138 | 0.005 |
| (8.8, 11.1) | (9.0, 9.4) | (149, 204) | (133, 145) | |||
| 23 nmol/L | 10.8 | 8.0 | <0.001 | 265 | 137 | <0.001 |
| (9.4, 12.4) | (7.8, 8.2) | (218, 322) | (132, 142) | |||
| 30 nmol/L | 10.5 | 7.5 | 0.001 | 265 | 132 | <0.001 |
| (9.1, 12.0) | (7.4, 7.7) | (194, 363) | (127, 137) | |||
| 44 nmol/L | 11.8 | 7.1 | <0.001 | 314 | 128 | <0.001 |
| (10.1, 13.8) | (7.0, 7.2) | (234, 421) | (125, 130) | |||
Serum folate values are category medians. Values for tHcy (μmol/L) and MMA (nmol/L) are least-square geometric means (95% CI). MMA, methylmalonic acid; tHcy, total homocysteine. Adapted from reference 40. Copyright (2007) National Academy of Sciences.
FIGURE 4.

Association of serum folate concentrations with serum homocysteine and serum methylmalonic acid concentrations in older adults from the NHANES 1999–2002 cohort. Serum homocysteine (A) and serum methylmalonic acid (B) concentrations decreased with increasing serum folate in vitamin B-12–replete subjects (serum vitamin B-12 ≥148 pmol/L), but increased with increasing serum folate in vitamin B-12–deficient subjects (serum vitamin B-12 <148 pmol/L). Symbols represent geometric means. vitamin B-12 <148 pmol/L: n = 32, 33, 32, and 33 for lowest to highest folate categories, respectively; vitamin B-12 ≥148 pmol/L: n = 1032, 1152, 892, and 2267 for lowest to highest folate categories, respectively. B-12, vitamin B-12. Reproduced from reference 40 with permission. Copyright (2007) National Academy of Sciences.
Serum folate also interacted significantly with serum vitamin B-12 in relation to serum MMA concentrations in the NHANES 1999–2002 participants (Pinteraction = 0.003) (Figure 4 and Table 1). Among participants in that survey with serum vitamin B-12 <148 pmol/L, geometric mean serum MMA increased significantly across categories of increasing serum folate (Ptrend = 0.008), whereas a modest but significant decrease in MMA with increasing serum folate was observed among subjects with serum vitamin B-12 ≥148 pmol/L (Ptrend < 0.001). Although no significant trends in MMA concentrations were detected across serum folate categories for participants in either serum vitamin B-12 category in the NHANES III cohort (perhaps due to small sample sizes that limited the ability to detect important trends in MMA concentrations), we did observe that vitamin B-12-deficient participants had higher MMA than non–vitamin B-12-deficient participants when serum folate concentrations were in the highest tertile (median folate = 23 nmol/L), while no significant differences in MMA were observed between vitamin B-12–deficient and non–vitamin B-12-deficient participants when serum folate was in the middle (median folate = 11 nmol/L) or lowest (median folate = 7 nmol/L) tertile. This suggests that, even prior to folic acid fortification, higher folate status may have affected MMA concentrations in people with low vitamin B-12 status (Figure 4 and Table 1).
Taken together, these data on the interaction of vitamin B-12 and folate suggest that, in vitamin B-12 deficiency, serum folate greater than ∼20 nmol/L is associated with higher serum concentrations of both tHcy and MMA. While these associations were seen in the US population, particularly after folic acid fortification, these data also suggest the presence of an interaction before fortification.
Further support for the existence of this biochemical paradox derives from a cross-sectional study of 1535 participants (age ≥60 y) in the Sacramento Area Latino Study on Aging (SALSA) (41). Subjects were divided into 4 groups on the basis of plasma concentrations of vitamin B-12 (< or ≥148 pmol/L) and folate (< or ≥45.3 nmol/L). Plasma tHcy and MMA concentrations were determined for each group. Findings from this study (Figure 5) showed that individuals with normal vitamin B-12 and elevated folate have, as expected, lower tHcy and no difference in MMA concentrations than in the group with normal vitamin B-12 and nonelevated folate. In contrast, while tHcy and MMA concentrations were higher in both vitamin B-12 deficiency groups, they were highest in the low vitamin B-12/elevated folate group.
FIGURE 5.

Plasma homocysteine and methylmalonic acid concentrations by vitamin B-12 and folate status in older adults from the Sacramento Area Latino Study on Aging. Elevation of homocysteine (A) and methylmalonic acid (B) concentrations associated with low vitamin B-12 status (<148 pmol/L) were more pronounced in subjects with elevated plasma folate (>45.3 nmol/L) than in those with nonelevated plasma folate (≤45.3 nmol/L). Bars represent geometric means and 95% CIs. Sample sizes are low vitamin B-12/nonelevated folate: n = 78; low vitamin B-12/elevated folate: n = 22; non-low vitamin B-12/nonelevated folate: n = 1055; and non-low/elevated folate: n = 380. In both panels A and B, different letters indicate statistically significant differences (P ≤ 0.001 by 2-factor ANOVA followed by Scheffe's test controlling for age, sex, education, supplement use, and creatinine). B-12, vitamin B-12. Reproduced from reference 41 with permission.
A third study (42) involved 230 ambulatory nondiabetic individuals with normal renal function and normal serum vitamin B-12 concentrations who were evaluated to determine whether high serum folate concentrations contribute to changes in plasma tHcy and serum MMA concentrations. Data showed that older adults (≥60 y) with low-normal vitamin B-12 concentration (154–230 pmol/L) and elevated folate (>45 nmol/L) had higher MMA but lower tHcy concentrations than when folate was not elevated. Importantly, this study also showed that the higher MMA concentrations were only seen in elderly individuals with high folate and not in younger individuals, irrespective of folate concentration. A possible age dependency of the interaction is consistent with the study by Mills et al. (43) of 2507 Irish university students that failed to detect an association of high serum folate with higher plasma tHcy or serum MMA concentrations among students with serum vitamin B-12 concentrations <148 pmol/L. Notably, these observations may reflect exacerbation of the age dependency of serum MMA concentrations, irrespective of vitamin B-12 status, which has been previously reported (44).
A fourth study (45) of 1112 older adults (>60 y; including 768 healthy controls, 133 patients with mild cognitive impairment, and 211 Alzheimer disease patients) found a U-shaped relation between RBC folate and plasma tHcy, such that higher tHcy was observed with the lowest and highest RBC folate concentrations compared with intermediate concentrations. Notably, this U-shaped curve was observed irrespective of plasma vitamin B-12 concentrations. This finding is consistent with the Australian study described above (24) that found cognitive performance was impaired in participants who had high RBC folate and serum vitamin B-12 within the low-normal range.
Intervention study
While the studies cited above are largely consistent with the existence of a high-folate–low-vitamin B-12 interaction affecting both tHcy and MMA metabolism, data are limited on the effect of high-dose folic acid supplements on these 2 biomarkers in vitamin B-12–deficient individuals. The data in Figure 6 represent the results of vitamin B-12–deficient patients who were mistakenly prescribed daily doses of folic acid (0.4–1.0 mg) for 12 to 67 d before measurement of tHcy and MMA, followed by subsequent correction by vitamin B-12 administration. Three of the 5 patients experienced increases in both serum tHcy and serum MMA, which were reduced significantly upon vitamin B-12 administration (46).
FIGURE 6.

Effect of folic acid supplementation (0.4–1.0 mg/d) for 12–67 d on serum homocysteine and methylmalonic acid in 5 vitamin B-12–deficient patients prior to administration of vitamin B-12. Three of 5 patients exhibited increases in both homocysteine (A) and methylmalonic acid (B) concentrations after folic acid supplementation. Letters (a, b, c, d, and e) represent individual patients. Reproduced from reference 46 with permission.
In vitamin B-12 deficiency, elevated folate is associated with a lower rate of restoration of normal biomarker concentrations after vitamin B-12 administration
In 1999, Chile instituted mandatory folic acid fortification of wheat flour at a higher level than in the United States and Canada (220 mg/100 g vs. 140 mg/100 g, respectively), and this raised concern of increased folic acid–related vitamin B-12 imbalance in older adults (47). In an attempt to assess the impact of this fortification, Brito and colleagues (29) assessed the effect of a single intramuscular injection of 10 mg vitamin B-12 (plus 100 mg vitamin B-6 and 100 mg thiamin) in 51 Chilean asymptomatic vitamin B-12–deficient older adults on changes in vitamin B-12 status determined by a combined–vitamin B-12 (c-B12) index, a calculated measure that combines the 4 blood biomarkers of vitamin B-12 status: total vitamin B-12, holotranscobalamin (holoTC), tHcy, and MMA (48). Also investigated (29) was the effect of this intervention on neurophysiologic function that was assessed on the basis of peripheral nerve conductivity. Treatment with vitamin B-12 increased serum total vitamin B-12, serum holoTC, and c-B12, and reduced plasma tHcy and serum MMA (P < 0.001 for all vitamin B-12 biomarkers). Vitamin B-12 administration also produced consistent improvement in the conduction of myelinated peripheral nerves.
Of interest is that a secondary analysis of the Brito et al. (29) data showing that the response to vitamin B-12 treatment was dependent on baseline folate concentrations. Response to vitamin B-12 administration determined as c-B12 after 4 mo was significantly lower in those with baseline folate concentrations greater than or equal to the median (≥33.9 nmol/L) compared with those with baseline folate lower than the median (<33.9 nmol/L) (Figure 7). After the 4-mo intervention, c-B12 values did not reach nondeficient levels in those with high folate and low vitamin B-12 at baseline. Taken together, these findings suggest that replenishment of vitamin B-12 into body stores is compromised by high folate status, although it cannot be excluded that individuals with the high-folate/low–vitamin B-12 phenotype have altered capacity to metabolize folic acid or vitamin B-12 due to undetermined genetic, epigenetic, or physiologic impairments.
FIGURE 7.

Association of serum folate with response to vitamin B-12 treatment in asymptomatic, vitamin B-12–deficient Chilean elderly individuals. Increase in the combined indicator of vitamin B-12 status (cB12) was attenuated in subjects with high serum folate (≥33.9 nmol/L) compared with those with low serum folate (<33.9 nmol/L). Median values: cB12 = −0.88; serum folate = 33.9 nmol/L. Symbols represent means ± SEs. Significant differences (P < 0.05 by 2-factor repeated-measures ANOVA followed by Tukey's honestly significant difference test). NS, not statistically significant (P > 0.05). B12, vitamin B-12. Reproduced from reference 29 with permission.
In vitamin B-12 deficiency, elevated folate is associated with serum depletion of active vitamin B-12 (holoTC)
Observational study
HoloTC is referred to as the active form of serum vitamin B-12 because of its role in the transport of the vitamin to all tissues of the body (49). It is also a sensitive and early indicator of vitamin B-12 depletion. HoloTC typically represents 20–30% of total serum vitamin B-12, with the remaining vitamin B-12 bound to a second transport protein, haptocorrin, which is essentially inert, except perhaps being involved in enterohepatic circulation (50, 51). In the SALSA cohort described above (41), holoTC concentration also appears to be subject to the high-folate–low-vitamin B-12 interaction similar to the 2 functional biomarkers, tHcy and MMA, described previously. Specifically, elevated folate among the vitamin B-12–deficient participants was associated with a >50% lower holoTC concentration compared with vitamin B-12–deficient participants with nonelevated folate (Figure 8A). Importantly, in vitamin B-12 deficiency, high folate was associated with 16% lower total vitamin B-12 concentration than the state of vitamin B-12 deficiency with nonelevated folate status. This lower concentration of total vitamin B-12 can be totally accounted for by a high-folate–associated decrease in holoTC concentration. In support of this idea, when holoTC concentrations were subtracted from total vitamin B-12 concentrations, the remaining vitamin B-12, represented by the holohaptocorrin (holoHC) fraction of serum vitamin B-12, was unaffected by the concentration of plasma folate (Figure 8B).
FIGURE 8.

Plasma holotranscobalamin and holohaptocorrin concentrations by vitamin B-12 and folate status in older adults from the Sacramento Area Latino Study on Aging. Reduction in holotranscobalamin (A) concentrations associated with low vitamin B-12 status (<148 pmol/L) was more pronounced in subjects with elevated plasma folate (>45.3 nmol/L) than in those with nonelevated plasma folate (≤45.3 nmol/L). In contrast, no difference in holohaptocorrin (B) concentration was observed in subjects with low vitamin B-12 status between those with elevated and nonelevated folate. Bars represent geometric means and 95% CIs. Sample sizes are low vitamin B-12/nonelevated folate: n = 78; low vitamin B-12/elevated folate: n = 22; non-low vitamin B-12/nonelevated folate: n = 1055; and non-low/elevated folate: n = 380. In both panels A and B, different letters indicate statistically significant differences (P ≤ 0.001 by 2-factor ANOVA followed by Scheffe's test controlling for age, sex, education, supplement use, and creatinine). B12, vitamin B-12. (A) Reproduced from reference 41 with permission. (B) New figure from previously unpublished data.
Intervention studies
There are no intervention studies in individuals with vitamin B-12 insufficiency or deficiency that have assessed the effect of folic acid supplementation on serum holoTC concentrations, but 2 studies from the 1950s suggest there is an effect. In the study shown in Figure 9, pernicious anemia patients in remission (n = 31) who were treated routinely with monthly vitamin B-12 injections were selected among other patients because of their low hemoglobin status (<12.5 g/dL) (52). For the first 6 mo (period 1), patients were each given biweekly intramuscular injections of 30 μg crystalline vitamin B-12 or purified liver extract containing 20 μg vitamin B-12. (The specific cobalamin forms of vitamin B-12 used in this study were not specified.) In the next 6 mo (period 2), patients received the same biweekly intramuscular injections of vitamin B-12 plus daily oral doses of 5 mg folic acid. The data show (Figure 9) that, after the first period, serum vitamin B-12 increased significantly from a mean of 190 pg/mL (95% CI: 102, 275 pg/mL; 140 pmol/L; 95% CI: 75, 203 pmol/L) to a mean of 449 pg/mL (95% CI: 308, 591 pg/mL; 331 pmol/L; 95% CI: 244, 436 pmol/L; P < 0.001). Increases in serum vitamin B-12 concentrations occurred in close to 80% of the patients and ranged from several-fold to only a small percentage increase. Importantly, the inclusion of daily 5-mg oral doses of folic acid with the biweekly intramuscular injections of vitamin B-12 (period 2) resulted in dramatic decreases in serum vitamin B-12 to a mean of 277 pg/mL (95% CI: 221, 334 pg/mL; 204 pmol/L; 95% CI: 180, 246 pmol/L; P < 0.02), a concentration that is only slightly higher than that observed at baseline. After the first period, hemoglobin concentration increased slightly among some of the patients from a mean of 11.6 g/dL (95% CI: 11.3, 12.0 g/dL) to a mean of 12.5 g/dL (95% CI: 12.0, 13.0 g/dL). The mean hemoglobin concentration of 12.7 g/dL (95% CI: 12.3, 13.1 g/dL) did not significantly change at period 2 when oral folic acid was included with the vitamin B-12 injections.
FIGURE 9.

Effect of high-dose oral folic acid (5 mg/d) on serum vitamin B-12 in 35 patients with pernicious anemia in remission being treated with intramuscular vitamin B-12 injections (20–30 μg 2×/wk). Subjects were treated with only intramuscular vitamin B-12 for the first 6 mo, then with oral folic acid in addition to the intramuscular vitamin B-12 for the next 6 mo. Response of serum vitamin B-12 concentrations for each subject (A) and the mean serum vitamin B-12 responses (B) are shown. Serum vitamin B-12 concentrations increased after the first 6 mo, but decreased 6 mo after addition of oral folic acid to the treatment regimen. In panel B, different letters indicate statistically significant differences (P < 0.05 by ANOVA followed by Scheffe's test). Conversion factor for serum vitamin B-12: pg/mL divided by 1.355 = pmol/L. B12, vitamin B-12. Graphs created based on data from reference 52.
In the study shown in Figure 10, 13 pernicious anemia patients experiencing a relapse of anemia with no sign of neurological manifestations were administered daily doses of 15 mg folic acid for 5–8 d (53). Vitamin B-12 concentration and hemopoietic (reticulocyte) responses were determined before and after folic acid administration for 11 patients. Results show that folic acid treatment was associated with a mean reduction in serum vitamin B-12 concentrations of 23.3 pg/mL (17.2 pmol/L) (P = 0.003) (Figure 10A, C). This decrease in serum vitamin B-12 concentration appeared to be accompanied by corresponding increases in reticulocyte response (quantified as maximal % increase of reticulocytes over RBC count) (R2 = 0.49, P < 0.001) (Figure 10C), an indication that anemia had been reversed.
FIGURE 10.

Effect of short-term folic acid supplementation on serum vitamin B-12 concentration and reticulocyte response in 13 relapsed pernicious anemia patients. Subjects were treated with oral folic acid (18 mg/d) for 5–8 d. Response of serum vitamin B-12 concentrations for each subject (A), the mean serum vitamin B-12 responses (B), and the correlation between % reticulocyte response and % decrease in serum vitamin B-12 (C) are shown. Serum vitamin B-12 concentrations decreased in most subjects after folic acid treatment and the % reticulocyte response was directly correlated with % decrease in serum vitamin B-12. In panel B, different letters indicate statistically significant differences (P = 0.003 by paired t test). In panel C, r and P values based on simple regression. Conversion factor for serum vitamin B-12: pg/mL divided by 1.355 = pmol/L. B12, vitamin B-12; FA, folic acid. Graphs created based on data from reference 53.
Importantly, although holoTC was not measured in either intervention study summarized above (52, 53), it is reasonable to assume that the decreases in serum total vitamin B-12 observed in the pernicious anemia patients after high-dose folic-acid supplementation were reflective of decreases in serum holoTC.
Discussion
Resolving the high-folate–low-vitamin B-12 interaction and the biochemical paradox
Based on the evidence presented above, we propose that “the high-folate–low-vitamin B-12 interaction” represents a genuine cause of vitamin B-12 depletion. We hypothesize that this is the consequence of excessive intake of folic acid (rather than merely exposure to high folate status) that results in a specific decrease in the active form of the transportable vitamin B-12 in serum, holoTC. In vitamin B-12 deficiency, this decrease in holoTC concentration would further compromise the availability of vitamin B-12 coenzymes for enzyme action, and consequently exacerbate the deficiency. Moreover, we hypothesize that this effect is specific to oral folic acid, which, when consumed in a high-enough dose, will enter the circulation and selectively bind to tissues expressing the high affinity folate receptor (FR-α). This, in turn, leads to sequestration of holoTC in hematopoietic tissue and altered reabsorption in the kidney. A summary illustration of this hypothesis is provided in Figure 11.
FIGURE 11.

Hypothesized effects of excess folic acid on serum holoTC and metabolic and physiological functions in vitamin B-12 insufficiency/deficiency. (A) HoloTC (TC-B12) delivers vitamin B-12 to all tissues of the body, primarily by receptor-mediated endocytosis via the TC-B12 receptor, CD320 (green squares). In addition, in the kidney, TC-B12 is filtered through the renal glomeruli and then taken up by renal proximal tubule cells via the megalin/cubilin/amnionless receptor complex (blue square), thus retaining vitamin B-12 by limiting its excretion in the urine. Within cells, the TC-B12 complex enters lysosomes in which the vitamin B-12 is released from TC and then processed to serve as a cofactor in either of 2 metabolic reactions: 1) as methyl-B12 in the cytosolic remethylation of homocysteine to methionine, and 2) as deoxyadenosyl-B12 (ado-B12) in the mitochondrial conversion of methylmalonyl CoA (MMCoA) to succinyl CoA. In vitamin B-12 insufficiency, serum TC-B12 concentrations are low, thus leading to low cellular vitamin B-12 uptake, impairment of the 2 vitamin B-12–dependent metabolic reactions, and increases in serum Hcy and MMA. As insufficiency progresses to deficiency, the classical pathophysiological consequences include impaired hematopoiesis leading to reduced reticulocyte synthesis and neurodegeneration in the central and peripheral nervous systems. (B) It is hypothesized that exposure to excess folic acid (via supplements and fortified foods) causes exacerbation of vitamin B-12 deficiency by binding to folic acid receptors (FR; red triangles) in the bone marrow and the renal proximal tubule cells. This leads to diversion of the limited supply of serum TC-B12 (depicted by thick black arrows) to the bone marrow to support folic acid–mediated hematopoiesis and reticulocyte formation, or into the urine by possibly interfering with TC-B12 uptake via the megalin/cubilin/amnionless receptor complex in the renal proximal tubule cells or by some other mechanism yet to be elucidated. The TC-B12 is diverted away from other tissues (e.g., liver and brain; depicted by thin dashed arrows). This leads to accentuated elevations in Hcy and MMA concentrations in serum (also depicted by thick black arrows) and exacerbation of neurodegeneration. B12, vitamin B-12; Hcy, homocysteine; holoTC, holotranscobalamin; MMA, methylmalonic acid; THF, tetrahydrofolate..
As discussed above, this hypothesis explains the findings from 2 large cross-sectional studies, NHANES and SALSA (40, 41), that show the classic association of vitamin B-12 biomarkers (tHcy and MMA) with vitamin B-12 status is paradoxical when a vitamin B-12 insufficiency state is accompanied by high folate (Figures 4 and 5).
Further support for the hypothesis comes from the study described above in which folic acid supplements were mistakenly prescribed to vitamin B-12–deficient patients (Figure 6) (46). This was a part of a larger study whose aim was to establish the measurement of both tHcy and MMA as diagnostic tools for folate and vitamin B-12 deficiencies, as well as to establish efficacy when patients with these deficiencies were being treated with the corresponding vitamin. Showing that administering folic acid to vitamin B-12–deficient patients resulted in increased serum tHcy and MMA concentrations is of importance for 2 reasons. First, it mirrors the experimental conditions of older case reports where folic acid was administered to pernicious anemia patients. These conditions in the older studies might well have led to similar results had assays for tHcy and MMA measurement been available at the time. Second, despite the small number of patients, this study is an intervention design, and hence the observed increases in the 2 functional indicators can be considered a direct consequence of folic acid administration and exacerbation of the deficiency as evident by the abolishment of the higher tHcy and MMA concentrations when vitamin B-12 was subsequently administered.
The study in vitamin B-12–deficient Chilean elderly individuals is noteworthy for several reasons (Figure 7) (29). It is an intervention study with the objective of administering vitamin B-12 to treat the neurological symptoms of this deficiency, and as a secondary analysis assessing rates of replenishment of vitamin B-12 status (determined as c-B12). The latter analysis suggested that the extent of vitamin B-12 replenishment is significantly lower among those with higher serum folate at baseline than in those with lower serum folate. This interaction mirrors those described above, except that rather than accelerating (or exacerbating) the process of vitamin B-12 depletion, the interaction manifests as limited restoration of vitamin B-12 status. Of note is that this is 1 of 2 studies in this review where the high-folate–low-vitamin B-12 interaction was observed in subjects who were administered vitamin B-12 by intramuscular injection [the other being the study by Lear and Castle (Figure 9) (52) in which vitamin B-12-deficient subjects receiving high-dose intramuscular injections of vitamin B-12 exhibited paradoxical reductions in serum vitamin B-12 after initiation of high-dose folic-acid supplements]. This suggests that the high-folate–low-vitamin B-12 interaction occurs outside the gastrointestinal tract.
The high-folate–related increase in serum concentrations of both tHcy and MMA in vitamin B-12 deficiency states reflects increasingly impaired activity of both vitamin B-12–dependent enzymes, presumably due to a more limited supply of the 2 vitamin B-12 coenzymes: methylcobalamin for tHcy metabolism and 5-deoxyadenosylcobalamin for MMA metabolism (Figure 1). The cause of the limited supply can be either a consequence of vitamin B-12 deficiency due to impaired intestinal absorption or interference with the serum transport and/or subsequent cellular processing of the vitamin. Considering the former possibility, folate and vitamin B-12 are absorbed by the intestine by entirely different mechanisms at different sites and the chance that the absorption of one interferes with the other is improbable. The alternative is that this interaction occurs outside the intestine, as suggested by the Chilean elderly study (Figure 7) (29) and the pernicious anemia patient study (Figure 9) (52), where this interaction was seen after intramuscular injection of vitamin B-12. Most important, however, are the data from the SALSA study, which showed that, in addition to elevated tHcy and MMA, the combination of vitamin B-12 deficiency and high folate is associated with a substantially lower serum holoTC concentration than that seen in vitamin B-12 deficiency with nonelevated folate or in any other combination of vitamin B-12 and folate status (Figure 8A) (41). Subsequent analysis of the SALSA data shows that this relation between high folate status and lower holoTC concentrations is specific to this fraction of serum vitamin B-12, and is unrelated to holoHC, the other fraction of serum vitamin B-12 (Figure 8B).
Because holoTC functions as the serum transporter of vitamin B-12 to all cells, a lower concentration of this form of vitamin B-12 in the presence of (general) vitamin B-12 deficiency would further compromise the supply of vitamin B-12 coenzymes, hence exacerbating the deficiency. Thus, all of the other functional changes associated with the high-folate–low-vitamin B-12 interaction could be consequences of reduced holoTC. As 1 of the 2 constituents of serum vitamin B-12, a lower concentration of holoTC is reflected by an equivalent numerical decrease in the amount of serum total vitamin B-12. The data from the SALSA study support the hypothesis that, in vitamin B-12 deficiency, high folate is associated with depletion in that fraction of serum vitamin B-12 which is bound to transcobalamin (41). In this respect, the studies from the late 1950s by Lear and Castle (52) and by Bok et al. (53) (Figures 9 and 10) showing that excessive intake of folic acid in pernicious anemia patients resulted in decreased serum concentrations of vitamin B-12 can be considered as representing the “intervention” equivalent of the observation in the SALSA study. HoloTC is the active form of serum vitamin B-12, with corresponding receptors (CD320, megalin) widely distributed among peripheral cells and possessing a serum half-life of ∼2 h. This is in contrast to holoHC, which has a half-life of ∼10 d, is taken up by only the liver via nonspecific asialoglycoprotein receptors, and is practically inert. It is, therefore, quite reasonable to assume that these losses of serum vitamin B-12 reported by Lear and Castle (52) and by Bok and colleagues (53) represent losses of holoTC.
Thus, we are proposing that the high-folate–low-vitamin B-12 interaction seen in observational and interventional studies does, in fact, exist, and can be explained by a physiological mechanism whereby consumption of folic acid above a certain threshold under limiting conditions of general or holoTC deficiency results in a functional impairment of vitamin B-12–dependent metabolism. This interaction manifests as the exacerbation of the deficiency, which is evident in lower serum holoTC and total vitamin B-12 concentrations, increased serum tHcy and MMA concentrations, and perhaps lower assimilation of administered vitamin B-12 (irrespective of the administration route; i.e., oral vs. intramuscular injection). Yet to be determined is the threshold of vitamin B-12 status and folic acid intake at which these phenomena are no longer biochemically or clinically noticeable. Of importance, the term “paradoxical” was used due to the observation in the Framingham Offspring cohort in which those who had been taking a vitamin supplement had higher serum tHcy concentrations after folic acid fortification than at baseline before fortification (Figure 2) (38). This observation, which was later supported by an actual intervention study showing that folic acid administration resulted in higher tHcy concentrations in ∼20% of the subjects (Figure 3) (39), can readily be interpreted on the basis of this high-folate–low-vitamin B-12 interaction. According to this hypothesis, the additional intake of folic acid is associated with a subsequent vitamin B-12 deficiency due to a decrease in holoTC concentration. This possibility also addresses the criticism by Berry et al (26), who argued that subjects who have high folate and low vitamin B-12 may be users of vitamin supplements containing both folic acid and vitamin B-12, but also are vitamin B-12 malabsorbers, thus explaining the coexistence of high folate and low vitamin B-12. As pointed out above, 2 of the studies in this review depicting the presence of the high-folate–low-vitamin B-12 interaction (Figures 7 and 9) (29, 52) relied on the metabolism or fate of intramuscular injection of vitamin B-12, thus supporting the notion that this interaction occurs outside the gastrointestinal tract (irrespective of whether malabsorption is the primary cause of the deficiency). The presence of this interaction is also suggested by the data of Solomon (42), showing that older adults (>60 y) with low normal serum vitamin B-12 concentration (154–230 pmol/L) and elevated serum folate (>45 nmol/L) had higher serum MMA but lower plasma tHcy concentration than when serum folate was lower.
The high-folate–low-vitamin B-12 interaction and UMFA
Data on the hematological response of 2 groups of pernicious anemia patients to folic acid imply that depleted vitamin B-12 may be diverted to different tissues dependent on the clinical state of these patients. The study of pernicious anemia patients with relapse of anemia (Figure 10) (53) has shown that the depletion of serum vitamin B-12 after folic acid administration is accompanied by a proportional increase in reticulocyte counts, suggesting that serum vitamin B-12 is diverted into the hematopoietic system to provide the necessary vitamin B-12 coenzyme for methionine production. This is consistent with the methyl folate trap hypothesis (54), which predicts that, in vitamin B-12 deficiency, the inhibition of DNA synthesis and cell proliferation is removed upon administration of high doses of folic acid because of increased availability of nonmethylated folate. The data by Bok et al. (53) imply that the diversion of holoTC into the hematopoietic system provides the necessary vitamin B-12 for methionine synthesis. Moreover, this diversion would be reflected by a reduction in serum holoTC and hence reduction in total serum vitamin B-12.
Here it is noteworthy that serum depletion of nutrients is known to occur in macrocytic anemia patients upon initiation of treatment. Both serum iron and potassium can decrease significantly upon treatment of these patients with high-dose folic acid or vitamin B-12, presumably due to utilization of these 2 minerals for hematopoiesis (55, 56). For both iron and potassium, the resulting decline in serum concentrations may be large enough to induce clinical deficiency states, including increased risk of death from cardiac failure due to hypokalemia (56).
However, in Lear and Castle's study of pernicious anemia patients in remission, the depletion of serum vitamin B-12 after treatment with folic acid occurred in the absence of any noticeable change in hematopoiesis (Figure 9) (52). In fact, the great majority of the early case reports suggest that the improvement in hematopoiesis seen in pernicious anemia patients after folic acid treatment is temporary, and in time they regress and relapse (4–6, 12, 15). Furthermore, the more recent observational data suggesting that the combination of high folate and low vitamin B-12 is associated with the worsening of both clinical outcomes and elevations in functional biomarkers of vitamin B-12 (23, 40, 41) imply that depletion of holoTC is attained even without stimulating hematopoiesis. The possibility that sustained folic acid intake would deplete vitamin B-12 independent of hematopoiesis suggests a plausible alternative mechanism based on the known details of the renal handling of holoTC, whereby folic acid might interfere with holoTC reuptake or cause its sequestration in the kidney, promoting the excretion of holoTC and further depleting metabolically active vitamin B-12 during vitamin B-12 deficiency. With a molecular weight of 45 kDa, holoTC is readily filtered through the renal glomeruli, and then re-internalized by multiple receptor-dependent endocytic processes (via the megalin/cubilin/amnionless receptor complex) (57, 58). The internalized complex reaches the lysosome where it dissociates, with the receptor protein recycled into the apical membrane while the transcobalamin protein is degraded and the vitamin B-12 released. The vitamin B-12 exits the lysosomes for recycling into the blood, remains in the lysosomes, or is excreted in the urine (57, 58). Whether high folic-acid intake or high folate status causes altered renal handling of holoTC and increased urinary excretion of vitamin B-12 remains to be investigated. However, we note that the folic acid and vitamin B-12 handling by the kidney is tightly coordinated: The expression of the high-affinity folate receptors (i.e., FR-α and FR-β) on the epithelial membrane coincides with that of megalin and cubulin (58–60); megalin binds and mediates the internalization of folate receptors in the megalin-cubulin complex (61), and binding of folic acid to folate receptors results in rapid internalization and relocation of the receptor complex in renal proximal tubule epithelia (62), which may be inhibited by receptor binding (63). Moreover, the expression of folate receptors is highly sensitive to micronutrient status (64). It is therefore entirely plausible that chronic saturation of this receptor complex by UMFA could interfere with the reuptake and retention of their respective ligands, including holoTC and vitamin B-12, and this effect could be accentuated in individuals with renal insufficiency.
This leads to an important question, which is whether the high-folate–low-vitamin B-12 interaction is indeed specific to folic acid (as opposed to reduced forms of folate). The answer may lie in differences in affinities of folate transporters and receptors for folic acid compared with reduced folates. Although folic acid absorption by the intestine is catalyzed by the proton-coupled folate transporter (PCFT), which has similar affinity for both folic acid and reduced folates (Table 2) (65, 66), the affinity for folic acid of the reduced folate carrier (RFC), which is ubiquitously expressed in all tissues, is 50- to 150-fold lower than that for 5-methylTHF and other reduced folates (Table 2) (65, 66). This is different for folate receptors for which the affinity for folic acid is several-fold higher than for 5-methylTHF (65, 66). Importantly, unlike RFC, the folate receptors are not expressed in liver. In contrast, at least 1 of the folate receptors, FR-α, is expressed in hematopoietic cells and the kidney proximal tubules, among other tissues (Table 2). When consumed in excess, it is known that some folic acid is converted into reduced folates within the intestinal cell, but some is absorbed intact and enters the portal circulation as UMFA (67). This occurs because the enzyme, dihydrofolate reductase, has limited capacity to reduce folic acid in mammalian cells, particularly in human cells (68). Therefore, unlike with reduced folates where intestinal absorption is followed by direct incorporation into the liver, some of the UMFA in the portal blood likely bypasses the liver to subsequently bind to folate receptors on peripheral tissues such as the kidney (58).
TABLE 2.
Folate transport and receptor proteins1
| Protein | Expression | Substrate affinity |
|---|---|---|
| Reduced folate carrier (RFC1) | All normal adult tissues, tissue culture cells, some tumor cells, some fetal tissues | Reduced folates: Kt = 3 μM; folic acid: Kt = 150–200 μM; methotrexate: similar to reduced folates |
| Proton coupled folate transporter (PCFT) | Small intestine (jejunum, duodenum), kidney, liver, placenta, brain | Reduced folates: Kt = 0.5–0.8 μM; folic acid: Kt = 0.5–0.8 μM; methotrexate: poor affinity |
| Folate receptor α (FR-α) | Kidney proximal tubules, choroid plexus, placenta, erythropoietic cells, vas deferens, ovaries, fallopian tubes, uterine epididymis, lung alveolar, acinar cells of the breast, submandibular salivary and bronchial cells, retinal pigment epithelial cells, breast milk, fetal and adult cells, some tumors | Reduced folates: Kd = 1–10 nM; folic acid: Kd = 0.1 nM; methotrexate: poor affinity |
| Folate receptor β (FR-β) | Placenta, spleen, thymus, thymus, some monocytes, thyroid gland (low), gut mucosal cells (very low), during myelopoiesis, acute myelogenous leukemia blasts, fetal and adult cells | Reduced folates: Kd = 1–10 nM; folic acid: Kd = 0.1 nM; methotrexate: poor affinity |
The response of the hematopoietic system to high-dose oral folic acid in pernicious anemia patients is very likely the result of entry of UMFA into the hematopoietic progenitor cells via FR-α. Moreover, the proposed diversion of vitamin B-12, and consequent depletion of vitamin B-12 in the serum, may be in direct response to preferential uptake of UMFA. Notably, this suggests that the hematopoietic response to folic acid in pernicious anemia would not occur if folic acid had been converted to 5-methylTHF in the process of intestinal absorption. Precisely how bound and/or internalized UMFA would affect hematopoietic progenitor cells is uncertain. The observational evidence clearly suggests that, in some cases, it can stimulate hematopoiesis, albeit unsustainably. Theoretically, the intracellular assimilation of folic acid via dihydrofolate reductase might enhance thymidylate synthetase by bypassing the methylfolate trap, thereby allowing cellular proliferation to progress temporarily. Alternatively, or concomitantly, high-affinity binding of UMFA to FR-α may also elicit the induction of noncanonical signaling pathways, as has been shown to occur in other tissues (69–75). These hypothetical mechanisms are amenable to experimental verification.
A similar line of reasoning involves the kidney in the manifestations of the high-folate–low-vitamin B-12 interaction: depletion of serum vitamin B-12 may also be the result of preferential uptake of folic acid into the renal proximal tubule cells via FR-α. This then may lead to altered renal handling of holoTC and increased urinary excretion of vitamin B-12, as hypothesized above.
FR-α is also present in the choroid plexus, which has the important function of supplying folate to the central nervous system (76, 77). However, there is good evidence that, although folic acid binds avidly to this receptor in the choroid plexus, it does not cross the opposite, apical membrane to enter the cerebrospinal fluid (CSF) (78). In fact, injection of 3H-folic acid into rabbit CSF resulted in the transport of the label back into blood (78). This is unlike 5-methylTHF, which is readily transported into the CSF (79). Moreover, experimental data show that the 5-methylTHF–FR-α complex is secreted by the choroid plexus as an exosome with several other proteins for uptake from the CSF by the brain parenchyma (80).
Thus, by nature of its different affinity for the reduced folate carrier compared with folate receptors, the binding and transport of UMFA may selectively affect those tissues with exposed folate receptors that facilitate cellular entry and possibly signaling. This is unlike 5-methylTHF whose transport across all membranes is universal. Notably, this proposed selective tissue entry of UMFA is the principle upon which folic acid conjugates have been exploited as probes for diagnostics and drug delivery in cancerous and inflammatory diseased tissues that contain folate receptors (81–87). The success of this approach is, in part, due to the high binding specificity of folic acid to these receptors versus the poor affinity of RFC for folic acid.
Presuming that the putative high-folate–low-vitamin B-12 interaction on serum holoTC and the metabolic markers of vitamin B-12 status is due to excess folic acid, an important consideration is at what level of exposure might these effects become metabolically and clinically relevant. The early reports of relapse or exacerbation of neural symptoms in pernicious anemia patients treated with folic acid describe dose levels ranging from 20 to 50 mg/d (7–12). Today, such extreme dosing and exposure are highly unlikely. However, the data summarized herein from cross-sectional observational and treatment studies showing interactive associations of high folate–low vitamin B-12 with tHcy, MMA, holoTC, and c-B12 were obtained in cohorts with significant proportions of individuals consuming >1 mg folic acid/d from concurrent consumption of supplements, ready-to-eat cereals, and other fortified foods (29, 40–42, 46). Thus, the interactive metabolic effects of high folate–low vitamin B-12 may occur at levels of exposure to folic acid that are regularly achieved in populations consuming folic-acid–fortified foods and supplements. It bears repeating here that the Tolerable Upper Intake Level set by the Institute of Medicine for folic acid is 1 mg/d (6). Moreover, because some studies have observed significant associations between high folate status and both elevated homocysteine and cognitive impairment, even in subjects with low-normal vitamin B-12 status (24, 45), the definition of high-folate/low-vitamin B-12 status in this context may include more than just individuals with outright vitamin B-12 deficiency (defined as serum vitamin B-12 <148 pmol/L).
Conclusions
We hypothesize that the high-folate–low vitamin B-12 interaction represents a novel cause of vitamin B-12 depletion caused by high folic-acid intake and consequent reduction in serum holoTC (Figure 11). Our hypothesis focuses primarily on the putative metabolic consequences of this interaction (i.e., depletion of serum holoTC and elevations in tHcy and MMA). The hypothesis provides tenable predictions that may be tested in rigorously designed experiments in vitro and in vivo. As examples, some of these predictions are stated here:
Under conditions of B12 insufficiency, excess folic acid intake will cause the following:
Diversion of holoTC from serum to hemopoietic cells to support reticulocyte proliferation
Loss of holoTC in urine due to interference of its reuptake by the megalin-receptor complex in the renal tubules
Reduced availability of holoTC to the liver, brain, and other tissues due to its diversion to the hemopoietic system and loss in the urine; this, in turn, leads to accentuated elevations in blood concentrations of tHcy and MMA, and exacerbation of neurological sequelae due to vitamin B-12 deficiency in the brain and peripheral nervous system
To test these and other predictions, in addition to human studies, it will be important to develop better animal models with closer similarity to the physiologic and clinical features of vitamin B-12 deficiency than rodent models currently provide. It will also be important to determine if this interaction is specific for excess exposure to folic acid or UMFA, or is a consequence of excess exposure to all forms of folate because of the widespread use of folic acid for food fortification and vitamin supplements.
The clinical and public health implications of the interaction remain uncertain. It is important to avoid conflating evidence of biological interaction with evidence of public health harms. Concern about excessive intake and potential adverse effects of folic acid in addition to other health outcomes has been the subject of NIH-sponsored workshops and meetings of experts (88, 89). Our critical re-evaluation of the literature and analysis of the potential implications of transport selectivity of UMFA suggest a new explanation for some of the observed associations between high folic-acid intake and adverse health outcomes. In the study of the elderly participants of NHANES 1999–2002, the combination of high circulating UMFA and vitamin B-12 deficiency was related to lower cognitive test scores (90). These findings have recently been extended in an analysis of the NHANES 2011–2014 cohort, in which the combination of low vitamin B-12 and high UMFA was again associated with poorer performance on specific subdomains of cognitive function (91). The hypothesis proposed herein relies heavily upon the studies that focused primarily on older adults consuming large amounts of folic acid. In addition to experimental studies of the proposed mechanisms that underpin this hypothesis, there is a need for further epidemiological studies to identify population groups that may be susceptible to this interaction.
ACKNOWLEDGEMENTS
The authors’ responsibilities were as follows—JS: conceived of and drafted the first and subsequent drafts of the manuscript; JWM, AMT, JBM, and PFJ: contributed to specific sections and editing of the manuscript; and all authors: read and approved the final manuscript.
Notes
Supported by the NIH National Institute for Aging (R01 AG059011).
Author disclosures: JWM has been a consultant to Church & Dwight Co., Inc. (Princeton, NJ), and was an invited speaker by Nu Skin International (Provo, UT). JBM serves on the scientific advisory board of Care/of (New York, NY). The other authors report no conflicts of interest.
Perspective articles allow authors to take a position on a topic of current major importance or controversy in the field of nutrition. As such, these articles could include statements based on author opinions or point of view. Opinions expressed in Perspective articles are those of the author and are not attributable to the funder(s) or the sponsor(s) or the publisher, Editor, or Editorial Board of Advances in Nutrition. Individuals with different positions on the topic of a Perspective are invited to submit their comments in the form of a Perspectives article or in a Letter to the Editor.
Abbreviations used: c-B12, combined vitamin B-12; CSF, cerebrospinal fluid; FR, folate receptor; holoTC, holotranscobalamin; MMA, methylmalonic acid; MMSE, Mini-Mental State Examination; RFC, reduced folate carrier; SALSA, Sacramento Area Latino Study on Aging; tHcy, total homocysteine; UMFA, unmetabolized folic acid; 5-methylTHF, 5-methyltetrahydrofolate.
Contributor Information
Jacob Selhub, Tufts–USDA Human Nutrition Research Center on Aging, Boston, MA, USA.
Joshua W Miller, Department of Nutritional Sciences, Rutgers University, New Brunswick, NJ, USA.
Aron M Troen, School of Nutritional Sciences and Institute of Biochemistry Food Science and Nutrition, The Robert H Smith Faculty of Agriculture Food and Environment, The Hebrew University of Jerusalem, Jerusalem, Israel.
Joel B Mason, Tufts–USDA Human Nutrition Research Center on Aging, Boston, MA, USA.
Paul F Jacques, Tufts–USDA Human Nutrition Research Center on Aging, Boston, MA, USA.
References
- 1. Nielsen MJ, Rasmussen MR, Andersen CB, Nexo E, Moestrup SK. Vitamin B12 transport from food to the body's cells—a sophisticated, multistep pathway. Nat Rev Gastroenterol Hepatol. 2012;9(6):345–54. [DOI] [PubMed] [Google Scholar]
- 2. Green R, Allen LH, Bjørke-Monsen AL, Brito A, Guéant JL, Miller JW, Molloy AM, Nexo E, Stabler S, Toh BH et al. Vitamin B(12) deficiency. Nat Rev Dis Primers. 2017;3:17040. [DOI] [PubMed] [Google Scholar]
- 3. Miller JW. Proton pump inhibitors, H2-receptor antagonists, metformin, and vitamin B-12 deficiency: clinical implications. Adv Nutr. 2018;9:511S–8S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Savage DG, Lindenbaum J. Folate cobalamin interactions. In: Bailey LB, editor. Folate in health and disease. New York: Marcel Dekker, Inc; 1995. p. 237–85. [Google Scholar]
- 5. Will JJ, Mueller JF, Brodine C, Kiely CE, Friedman B, Hawkins VR, Dutra J, Vilter RW. Folic acid and vitamin B12 in pernicious anemia; studies on patients treated with these substances over a ten year period. J Lab Clin Med. 1959;53(1):22–38. [PubMed] [Google Scholar]
- 6. Institute of Medicine . Dietary Reference Intakes for thiamin, riboflavin, niacin, vitamin B6, folate, vitamin B12, pantothenic acid, biotin, and choline. Washington (DC): The National Academies Press; 1998. [PubMed] [Google Scholar]
- 7. Castle WB, Ross JB, Davidson CS, Burchenal JH, Fox HJ, Ham TH. Extrinsic factor in pernicious anemia: ineffectiveness of purified casein and of identified components of the vitamin B complex. Science. 1944;100(2587):81–3. [DOI] [PubMed] [Google Scholar]
- 8. Vilter CF, Vilter RW, Spies TD. The occurrence of combined system disease in persons with pernicious anemia during treatment with the L. casei factor (folic acid). Proc Ann Meet Central Soc Clin Res US. 1946;19:26. [PubMed] [Google Scholar]
- 9. Vilter CF, Vilter RW, Spies TD. The treatment of pernicious and related anemias with synthetic folic acid; observations on the maintenance of a normal hematologic status and on the occurrence of combined system disease at the end of one year. J Lab Clin Med. 1947;32(3):262–73. [PubMed] [Google Scholar]
- 10. Meyer LM. Folic acid in the treatment of pernicious anemia. Blood. 1947;2:50–62. [PubMed] [Google Scholar]
- 11. Berry RJ. Lack of historical evidence to support folic acid exacerbation of the neuropathy caused by vitamin B12 deficiency. Am J Clin Nutr. 2019;110(3):554–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Conley CL, Krevans JR. Development of neurological manifestations of pernicious anemia during multivitamin therapy. N Engl J Med. 1951;245:529–31. [DOI] [PubMed] [Google Scholar]
- 13. Junod SW. Folic acid fortification: fact and folly. [Internet]. [Accessed 2021 Feb 20]. Available from: https://www.fda.gov/media/110471/download.
- 14. Reynolds E. Fortification of flour with folic acid. Fortification has several potential risks. BMJ. 2002;324(7342):918. [PMC free article] [PubMed] [Google Scholar]
- 15. Reynolds EH. The risks of folic acid to the nervous system in vitamin B12 deficiency: rediscovered in the era of folic acid fortification policies. J Neurol Neurosurg Psychiatry. 2017;88(12):1097–8. [DOI] [PubMed] [Google Scholar]
- 16. Mills JL, Molloy AM, Reynolds EH. Do the benefits of folic acid fortification outweigh the risk of masking vitamin B12 deficiency?. BMJ. 2018;360:k724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tucker KL, Mahnken B, Wilson PW, Jacques P, Selhub J. Folic acid fortification of the food supply: potential benefits and risks for the elderly population. JAMA. 1996;276(23):1879–85. [DOI] [PubMed] [Google Scholar]
- 18. Dickinson CJ. No reliable evidence that folate is harmful in B-12 deficiency. BMJ. 1995;311(7010):949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Oakley GP Jr. Let's increase folic acid fortification and include vitamin B-12. Am J Clin Nutr. 1997;65(6):1889–90. [DOI] [PubMed] [Google Scholar]
- 20. Wald NJ, Law M, Jordan R. Folic acid food fortification to prevent neural tube defects. Lancet. 1998;351(9105):834. [DOI] [PubMed] [Google Scholar]
- 21. Pfeiffer CM, Caudill SP, Gunter EW, Osterloh J, Sampson EJ. Biochemical indicators of B vitamin status in the US population after folic acid fortification: results from the National Health and Nutrition Examination Survey 1999-2000. Am J Clin Nutr. 2005;82(2):442–50. [DOI] [PubMed] [Google Scholar]
- 22. Choumenkovitch SF, Selhub J, Wilson PW, Rader JI, Rosenberg IH, Jacques PF. Folic acid intake from fortification in United States exceeds predictions. J Nutr. 2002;132(9):2792–8. [DOI] [PubMed] [Google Scholar]
- 23. Morris MS, Jacques PF, Rosenberg IH, Selhub J. Folate and vitamin B-12 status in relation to anemia, macrocytosis, and cognitive impairment in older Americans in the age of folic acid fortification. Am J Clin Nutr. 2007;85(1):193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moore EM, Ames D, Mander AG, Carne RP, Brodaty H, Woodward MC, Boundy K, Ellis KA, Bush AI, Faux NG et al. Among vitamin B12 deficient older people, high folate levels are associated with worse cognitive function: combined data from three cohorts. J Alzheimers Dis. 2014;39(3):661–8. [DOI] [PubMed] [Google Scholar]
- 25. Castillo-Lancellotti C, Margozzini P, Valdivia G, Padilla O, Uauy R, Rozowski J, Tur JA. Serum folate, vitamin B12 and cognitive impairment in Chilean older adults. Public Health Nutr. 2015;18:2600–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Berry RJ, Carter HK, Yang Q. Cognitive impairment in older Americans in the age of folic acid fortification. Am J Clin Nutr. 2007;86(1):265–9. [DOI] [PubMed] [Google Scholar]
- 27. Clarke R, Sherliker P, Hin H, Molloy AM, Nexo E, Ueland PM, Emmens K, Scott JM, Evans JG. Folate and vitamin B12 status in relation to cognitive impairment and anaemia in the setting of voluntary fortification in the UK. Br J Nutr. 2008;100(5):1054–9. [DOI] [PubMed] [Google Scholar]
- 28. Smith AD, Refsum H. Homocysteine, B vitamins, and cognitive impairment. Annu Rev Nutr. 2016;36:211–39. [DOI] [PubMed] [Google Scholar]
- 29. Brito A, Verdugo R, Hertrampf E, Miller JW, Green R, Fedosov SN, Shahab-Ferdows S, Sanchez H, Albala C, Castillo JL et al. Vitamin B-12 treatment of asymptomatic, deficient, elderly Chileans improves conductivity in myelinated peripheral nerves, but high serum folate impairs B-12 status response assessed by the combined indicator B-12 status. Am J Clin Nutr. 2016;103(1):250–7. [DOI] [PubMed] [Google Scholar]
- 30. Quadros EV, Lai SC, Nakayama Y, Sequeira JM, Hannibal L, Wang S, Jacobsen DW, Fedosov S, Wright E, Gallagher RC et al. Positive newborn screen for methylmalonic aciduria identifies the first mutation in TCblR/CD320, the gene for cellular uptake of transcobalamin-bound vitamin B(12). Hum Mutat. 2010;31(8):924–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hannibal L, Lysne V, Bjorke-Monsen AL, Behringer S, Grunert SC, Spiekerkoetter U, Jacobsen DW, Blom HJ. Biomarkers and algorithms for the diagnosis of vitamin B12 deficiency. Front Mol Biosci. 2016;3:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Herbert V. The 1986 Herman award lecture. Nutrition science as a continually unfolding story: the folate and vitamin B-12 paradigm. Am J Clin Nutr. 1987;46(3):387–402. [DOI] [PubMed] [Google Scholar]
- 33. Lindenbaum J, Savage DG, Stabler SP, Allen RH. Diagnosis of cobalamin deficiency: II. Relative sensitivities of serum cobalamin, methylmalonic acid, and total homocysteine concentrations. Am J Hematol. 1990;34(2):99–107. [DOI] [PubMed] [Google Scholar]
- 34. Selhub J, Jacques PF, Dallal G, Choumenkovitch S, Rogers G. The use of blood concentrations of vitamins and their respective functional indicators to define folate and vitamin B12 status. Food Nutr Bull. 2008;29(2 Suppl):S67–73. [DOI] [PubMed] [Google Scholar]
- 35. Stabler SP, Lindenbaum J, Allen RH. The use of homocysteine and other metabolites in the specific diagnosis of vitamin B-12 deficiency. J Nutr. 1996;126(4 Suppl):1266S–72S. [DOI] [PubMed] [Google Scholar]
- 36. Spence JD, Howard VJ, Chambless LE, Malinow MR, Pettigrew LC, Stampfer M, Toole JF. Vitamin Intervention for Stroke Pevention (VISP) trial: rationale and design. Neuroepidemiology. 2001;20(1):16–25. [DOI] [PubMed] [Google Scholar]
- 37. Toole JF, Malinow MR, Chambless LE, Spence JD, Pettigrew LC, Howard VJ, Sides EG, Wang CH, Stampfer M. Lowering homocysteine in patients with ischemic stroke to prevent recurrent stroke, myocardial infarction, and death: the Vitamin Intervention for Stroke Prevention (VISP) randomized controlled trial. JAMA. 2004;291(5):565–75. [DOI] [PubMed] [Google Scholar]
- 38. Jacques PF, Selhub J, Bostom AG, Wilson PW, Rosenberg IH. The effect of folic acid fortification on plasma folate and total homocysteine concentrations. N Engl J Med. 1999;340(19):1449–54. [DOI] [PubMed] [Google Scholar]
- 39. Malinow MR, Duell PB, Williams MA, Kruger WD, Evans AA, Anderson PH, Block PC, Hess DL, Upson BM, Graf EE et al. Short-term folic acid supplementation induces variable and paradoxical changes in plasma homocyst(e)ine concentrations. Lipids. 2001;36(Suppl):S27–32. [DOI] [PubMed] [Google Scholar]
- 40. Selhub J, Morris MS, Jacques PF. In vitamin B12 deficiency, higher serum folate is associated with increased total homocysteine and methylmalonic acid concentrations. Proc Natl Acad Sci USA. 2007;104(50):19995–20000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Miller JW, Garrod MG, Allen LH, Haan MN, Green R. Metabolic evidence of vitamin B-12 deficiency, including high homocysteine and methylmalonic acid and low holotranscobalamin, is more pronounced in older adults with elevated plasma folate. Am J Clin Nutr. 2009;90(6):1586–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Solomon LR. Advanced age as a risk factor for folate-associated functional cobalamin deficiency. J Am Geriatr Soc. 2013;61(4):577–82. [DOI] [PubMed] [Google Scholar]
- 43. Mills JL, Carter TC, Scott JM, Troendle JF, Gibney ER, Shane B, Kirke PN, Ueland PM, Brody LC, Molloy AM. Do high blood folate concentrations exacerbate metabolic abnormalities in people with low vitamin B-12 status?. Am J Clin Nutr. 2011;94(2):495–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mineva EM, Sternberg MR, Zhang M, Aoki Y, Storandt R, Bailey RL, Pfeiffer CM. Age-specific reference ranges are needed to interpret serum methylmalonic acid concentrations in the US population. Am J Clin Nutr. 2019;110(1):158–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Faux NG, Ellis KA, Porter L, Fowler CJ, Laws SM, Martins RH, Pertile KK, Rembach A, Rowe CC, Rumble RL et al. Homocysteine, vitamin B12, and folic acid levels in Alzheimer's disease, mild cognitive impairment, and healthy elderly: baseline characteristics in subjects of the Australian Imaging Biomarker Lifestyle Study. J Alzheimers Dis. 2011;27:909–22. [DOI] [PubMed] [Google Scholar]
- 46. Allen RH, Stabler SP, Savage DG, Lindenbaum J. Diagnosis of cobalamin deficiency I: usefulness of serum methylmalonic acid and total homocysteine concentrations. Am J Hematol. 1990;34(2):90–8. [DOI] [PubMed] [Google Scholar]
- 47. Hertrampf E, Cortes F. Folic acid fortification of wheat flour: Chile. Nutr Rev. 2004;62(Suppl 1):S44–S8. [DOI] [PubMed] [Google Scholar]
- 48. Fedosov SN, Brito A, Miller JW, Green R, Allen LH. Combined indicator of vitamin B12 status: modification for missing biomarkers and folate status and recommendations for revised cut-points. Clin Chem Lab Med. 2015;53(8):1215–25. [DOI] [PubMed] [Google Scholar]
- 49. Herzlich B, Herbert V. Depletion of serum holotranscobalamin. II. An early sign of negative vitamin B12 balance. Lab Invest. 1988;58(3):332–7. [PubMed] [Google Scholar]
- 50. Kanazawa S, Herbert V, Herzlich B, Drivas G, Manusselis C. Removal of cobalamin analogue in bile by enterohepatic circulation of vitamin B12. Lancet. 1983;1(8326 Pt 1):707–8. [DOI] [PubMed] [Google Scholar]
- 51. Alpers DH. Absorption and blood/cellular transport of folate and cobalamin: pharmacokinetic and physiological considerations. Biochimie. 2016;126:52–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lear AA, Castle WB. Supplemental folic acid therapy in pernicious anemia: the effect on erythropoiesis and serum vitamin B12 concentrations in selected cases. J Lab Clin Med. 1956;47(1):88–97. [PubMed] [Google Scholar]
- 53. Bok J, Nieweg HO, Kroese WF, Faber JG, De Vries A. [Vitamin B12 content of blood during folic acid therapy of untreated patients with pernicious anemia]. Ned Tijdschr Geneeskd. 1958;102(16):795–8. [PubMed] [Google Scholar]
- 54. Scott JM, Weir DG. The methyl folate trap: a physiological response in man to prevent methyl group deficiency in kwashiorkor (methionine deficiency) and an explanation for folic-acid induced exacerbation of subacute combined degeneration in pernicious anaemia. Lancet. 1981;2(8242):337–40. [DOI] [PubMed] [Google Scholar]
- 55. Hawkins CF. Value of serum iron levels in assessing effect of haematinics in the macrocytic anaemias. Br Med J. 1955;1(4910):383–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lawson DH, Murray RM, Parker JLW, Hay G. Hypokalemia in megaloblastic anaemias. Lancet. 1970;2(7673):588–90. [DOI] [PubMed] [Google Scholar]
- 57. Birn H, Willnow TE, Nielsen R, Norden AGW, Bönsch C, Moestrup SK, Nexø E, Christensen EI. Megalin is essential for renal proximal tubule reabsorption and accumulation of transcobalamin-B(12). Am J Physiol Renal Physiol. 2002;282(3):F408–16. [DOI] [PubMed] [Google Scholar]
- 58. Birn H. The kidney in vitamin B12 and folate homeostasis: characterization of receptors for tubular uptake of vitamins and carrier proteins. Am J Physiol Renal Physiol. 2006;291(1):F22–36. [DOI] [PubMed] [Google Scholar]
- 59. Eshbach ML, Weisz OA. Receptor-mediated endocytosis in the proximal tubule. Annu Rev Physiol. 2017;79:425–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Christensen EI, Birn H. Megalin and cubilin: synergistic endocytic receptors in renal proximal tubule. Am J Physiol Renal Physiol. 2001;280:F562–73. [DOI] [PubMed] [Google Scholar]
- 61. Birn H, Zhai X, Holm J, Hansen SI, Jacobsen C, Christensen EI, Moestrup SK. Megalin binds and mediates cellular internalization of folate binding protein. FEBS J. 2005;272:4423–30. [DOI] [PubMed] [Google Scholar]
- 62. Selhub J, Nakamura S, Carone FA. Renal folate absorption and the kidney folate binding protein. II. Microinfusion studies. Am J Physiol. 1987;252(4 Pt 2):F757–60. [DOI] [PubMed] [Google Scholar]
- 63. Birn H, Selhub J, Christensen EI. Internalization and intracellular transport of folate-binding protein in rat kidney proximal tubule. Am J Physiol. 1993;264(2 Pt 1):C302–10. [DOI] [PubMed] [Google Scholar]
- 64. da Costa M, Rothenberg SP, Sadasivan E, Regec A, Qian L. Folate deficiency reduces the GPI-anchored folate-binding protein in rat renal tubules. Am J Physiol Cell Physiol. 2000;278:C812–21. [DOI] [PubMed] [Google Scholar]
- 65. Shane B. Folate chemistry and metabolism. In: Bailey LB, editor. Folate in health and disease. 2nd ed. Boca Raton (FL): CRC Press; 2010. p. 1–24. [Google Scholar]
- 66. Bailey LB, da Silva V, West AA, Caudill MA. Zempleni J, Suttie JW, Gregory JF, Stover PJ, editors. Handbook of vitamins. 5th ed. Boca Raton (FL): CRC Press; 2014. p. 421–46. [Google Scholar]
- 67. Patanwala I, King MJ, Barrett DA, Rose J, Jackson R, Hudson M, Philo M, Dainty JR, Wright AJA, Finglas PM et al. Folic acid handling by the human gut: implications for food fortification and supplementation. Am J Clin Nutr. 2014;100(2):593–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bailey W, Ayling JE. The extremely slow and variable activity of dihydrofolate reductase in human liver and its implications for high folic acid intake. Proc Natl Acad Sci USA. 2009;106(36):15424–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Frigerio B, Bizzoni C, Jansen G, Leamon CP, Peters GJ, Low PS, Matherly LH, Figini M. Folate receptors and transporters: biological role and diagnostic/therapeutic targets in cancer and other diseases. J Exp Clin Cancer Res. 2019;38(1):125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mohanty V, Siddiqui MR, Tomita T, Mayanil CS. Folate receptor alpha is more than just a folate transporter. Neurogenesis (Austin). 2017; 4(1):e1263717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hansen MF, Greibe E, Skovbjerg S, Rohde A, Kristensen ACM, Jensen TR, Stentoft C, Kjær KH, Kronberg CS, Martensen PM. Folic acid mediates activation of the pro-oncogene STAT3 via the folate receptor alpha. Cell Signal. 2015;27(7):1356–68. [DOI] [PubMed] [Google Scholar]
- 72. Kuo C-T, Chang C, Lee W-S. Folic acid inhibits COLO-205 colon cancer cell proliferation through activating the FRα/c-SRC/ERK1/2/NFκB/TP53 pathway: in vitro and in vivo studies. Sci Rep. 2015;5:11187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhang X-M, Huang G-W, Tian Z-H, Ren D-L, Wilson JX. Folate stimulates ERK1/2 phosphorylation and cell proliferation in fetal neural stem cells. Nutr Neurosci. 2009;12(5):226–32. [DOI] [PubMed] [Google Scholar]
- 74. Chaudhari SN, Mukherjee M, Vagasi AS, Bi G, Rahman MM, Nguyen CQ, Paul L, Selhub J, Kipreos ET. Bacterial folates provide an exogenous signal for C. elegans germline stem cell proliferation. Dev Cell. 2016;38(1):33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Qiu W, Gobinath A, Wen Y, Austin J, Galea LAM. Folic acid, but not folate, regulates different stages of neurogenesis in the ventral hippocampus of adult female rats. J Neuroendocrinol. 2019;31(10):e12787. [DOI] [PubMed] [Google Scholar]
- 76. Spector R. Affinity of folic acid for the folate-binding protein of choroid plexus. Arch Biochem Biophys. 1979;194(2):632–4. [DOI] [PubMed] [Google Scholar]
- 77. Suleiman SA, Spector R. Purification and characterization of a folate binding protein from porcine choroid plexus. Arch Biochem Biophys. 1981;208(1):87–94. [DOI] [PubMed] [Google Scholar]
- 78. Levitt M, Nixon PF, Pincus JH, Bertino JR. Transport characteristics of folates in cerebrospinal fluid; a study utilizing doubly labeled 5-methyltetrahydrofolate and 5-formyltetrahydrofolate. J Clin Invest. 1971;50(6):1301–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Spector R, Lorenzo AV. Folate transport by the choroid plexus in vitro. Science. 1975;187(4176):540–2. [DOI] [PubMed] [Google Scholar]
- 80. Grapp M, Wrede A, Schweizer M, Hüwel S, Galla HJ, Snaidero N, Simons M, Bückers J, Low PS, Urlaub H et al. Choroid plexus transcytosis and exosome shuttling deliver folate into brain parenchyma. Nat Commun. 2013;4:2123. [DOI] [PubMed] [Google Scholar]
- 81. Leamon CP, Low PS. Delivery of macromolecules into living cells: a method that exploits folate receptor endocytosis. Proc Natl Acad Sci USA. 1991;88(13):5572–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hilgenbrink AR, Low PS. Folate receptor-mediated drug targeting: from therapeutics to diagnostics. J Pharm Sci. 2005;94(10):2135–46. [DOI] [PubMed] [Google Scholar]
- 83. Kraus VB, McDaniel G, Huebner JL, Stabler TV, Pieper CF, Shipes SW, Petry NA, Low PS, Shen J, McNearney TA et al. Direct in vivo evidence of activated macrophages in human osteoarthritis. Osteoarthritis Cartilage. 2016;24(9):1613–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Low PS, Henne WA, Doorneweerd DD. Discovery and development of folic-acid-based receptor targeting for imaging and therapy of cancer and inflammatory diseases. Acc Chem Res. 2008;41(1):120–9. [DOI] [PubMed] [Google Scholar]
- 85. Low PS, Kularatne SA. Folate-targeted therapeutic and imaging agents for cancer. Curr Opin Chem Biol. 2009;13(3):256–62. [DOI] [PubMed] [Google Scholar]
- 86. Lu Y, Sega E, Leamon CP, Low PS. Folate receptor-targeted immunotherapy of cancer: mechanism and therapeutic potential. Adv Drug Deliv Rev. 2004;56(8):1161–76. [DOI] [PubMed] [Google Scholar]
- 87. Paulos CM, Turk MJ, Breur GJ, Low PS. Folate receptor-mediated targeting of therapeutic and imaging agents to activated macrophages in rheumatoid arthritis. Adv Drug Deliv Rev. 2004;56(8):1205–17. [DOI] [PubMed] [Google Scholar]
- 88. Maruvada P, Stover PJ, Mason JB, Bailey RL, Davis CD, Field MS, Finnell RH, Garza C, Green R, Gueant JL et al. Knowledge gaps in understanding the metabolic and clinical effects of excess folates/folic acid: a summary, and perspectives, from an NIH workshop. Am J Clin Nutr. 2020;112(5):1390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Boyles AL, Yetley EA, Thayer KA, Coates PM. Safe use of high intakes of folic acid: research challenges and paths forward. Nutr Rev. 2016;74(7):469–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Morris MS, Jacques PF, Rosenberg IH, Selhub J. Circulating unmetabolized folic acid and 5-methyltetrahydrofolate in relation to anemia, macrocytosis, and cognitive test performance in American seniors. Am J Clin Nutr. 2010;91(6):1733–44. [DOI] [PubMed] [Google Scholar]
- 91. Bailey RL, Jun S, Murphy L, Green R, Gahche JJ, Dwyer JT, Potischman N, McCabe GP, Miller JW. High folic acid or folate combined with low vitamin B-12 status: potential but inconsistent association with cognitive function in a nationally representative cross-sectional sample of US older adults participating in the NHANES. Am J Clin Nutr. 2020;112(6):1547–57. [DOI] [PMC free article] [PubMed] [Google Scholar]