

Abstract
Since Waller and Cajal in the nineteenth and early twentieth centuries, laboratory traumatic peripheral nerve injury studies have provided great insight into cellular and molecular mechanisms governing axon degeneration and the responses of Schwann cells, the major glial cell type of peripheral nerves. It is now evident that pathways underlying injury-induced axon degeneration and the Schwann cell injury-specific state, the repair Schwann cell, are relevant to many inherited and acquired disorders of peripheral nerves. This review provides a timely update on the molecular understanding of axon degeneration and formation of the repair Schwann cell. We discuss how nicotinamide mononucleotide adenylyltransferase 2 (NMNAT2) and sterile alpha TIR motif containing protein 1 (SARM1) are required for axon survival and degeneration, respectively, how transcription factor c-JUN is essential for the Schwann cell response to nerve injury and what each tells us about disease mechanisms and potential therapies. Human genetic association with NMNAT2 and SARM1 strongly suggests aberrant activation of programmed axon death in polyneuropathies and motor neuron disorders, respectively, and animal studies suggest wider involvement including in chemotherapy-induced and diabetic neuropathies. In repair Schwann cells, cJUN is aberrantly expressed in a wide variety of human acquired and inherited neuropathies. Animal models suggest it limits axon loss in both genetic and traumatic neuropathies, whereas in contrast, Schwann cell secreted Neuregulin-1 type 1 drives onion bulb pathology in CMT1A. Finally, we discuss opportunities for drug-based and gene therapies to prevent axon loss or manipulate the repair Schwann cell state to treat acquired and inherited neuropathies and neuronopathies.
Supplementary Information
The online version contains supplementary material available at 10.1007/s13311-021-01125-3.
Keywords: Wallerian degeneration, Programmed axon death, Repair Schwann cell, NMNAT2, SARM1, C-JUN, Regeneration, Neuregulin
Introduction
Traumatic injury has long been used to study peripheral nerve degeneration and regeneration [1, 2], in part because morphological similarities with peripheral neuropathies and other nerve disorders suggested similar mechanisms and the potential to inform therapies [3]. More recently, the advent of molecular biology and the use of spontaneous and induced mutants in mice and other species have revealed similar molecular mechanisms underlying injury and disease. The central theme of this review is that both the axon-intrinsic degeneration mechanism after nerve injury and the Schwann cell response leading to nerve repair have been revealed mostly by nerve injury and genetic modification studies but are proving highly relevant also in peripheral nerve disorders, including many that do not involve traumatic injury. Good drug targets and genetic therapies have emerged in both fields that promise to underlie future advances in the treatment of peripheral neuropathies.
A transected nerve degenerates by Wallerian degeneration [2]. For the first 150 years after Wallerian degeneration was first described, we knew very little of its molecular mechanism, but in the last 20 years, this has changed dramatically. Lubinska in the 1970s and early 1980s had moved beyond Waller’s original concept that injured axons die passively because they lack “nourishment” by the soma, suggesting instead a specific, soma-derived inhibitor of axon degeneration and of the Schwann cell response. She proposed that this inhibitor is delivered by anterograde axonal transport and becomes depleted distal to a site of injury [4, 5]. Today, the best known match for this inhibitor is NAD-synthesizing enzyme nicotinamide mononucleotide adenylyltransferase 2 (NMNAT2) [6], because it is essential for axon growth and survival [7], quickly depleted distal to an axon injury [6] and functionally similar to a much more stable, aberrant fusion protein that strongly delays Wallerian degeneration [8]. Further understanding of the degenerative mechanism that NMNAT2 blocks has been largely driven by Drosophila genetics followed by confirmation in mammals [9–11]. It seems Wallerian degeneration is a remarkably well-conserved process involving enzymes that are functionally interchangeable between mammals and flies. The central execution step involves Toll-like receptor (TLR) adapter protein sterile alpha and TIR motif containing protein 1 (SARM1) [10], which has unexpected, intrinsic NADase and other enzyme activities required for its prodegenerative role [12]. The existence of a protein whose activated form kills axons but which is inhibited in healthy axons through the actions of other proteins has led to the concept of a programmed axon death mechanism (Fig. 1) that underlies both Wallerian degeneration after nerve injury and axon loss in a wide range of inherited, toxic and metabolic disorders.
Fig. 1.
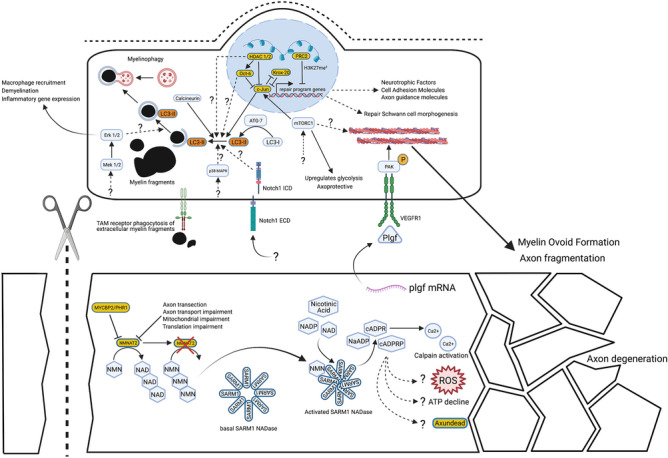
Overview of the molecular mechanisms within the axon and the Schwann cell during Wallerian degeneration. Upon nerve transection, the axonal transport of NMNAT2 is interrupted, and NMNAT2 already present in axons is degraded in a PHR1- and proteasome-dependent manner. Conversion of NMN to NAD by NMNAT2 is halted so NMN builds up inside the axon. NMN binds the SARM1 octamer, causing a conformational change and its activation. SARM1 activity generates cyclic ADP-ribose (cADPR) from NAD but also other products from nicotinamide adenine dinucleotide phosphate (NADP) and other substrates, such as nicotinic acid adenine dinucleotide phosphate (NaADP) and 2′-phospho-cyclic ADP-ribose (cADPRP). It is incompletely understood how SARM1 activation leads to further downstream steps in the axonal degeneration pathway, such as calcium release, ROS generation, ATP decline and the role of the molecule Axundead. The timings of the activation of the molecular pathways involved in the Schwann cell injury response in relation to those that regulate the axon degeneration machinery have not been fully delineated. It is likely that the majority of the Schwann cell injury response occurs during or slightly after axon degeneration has been executed. During axon degeneration, placental growth factor (Plgf) is released from axons and activates VEGF receptors leading to constriction of actin filaments in the Schwann cell, which helps break up axon fragments. It is possible that mTORC1 activation contributes to this process. Within the nucleus, c-JUN upregulation mediates a substantial amount of the Schwann cell response to nerve injury, especially repair program gene expression, cell shape change forming repair Schwann cells, upregulation of myelinophagy to aid in myelin sheath removal and repression of the myelin program through inhibition of Krox-20 function. Other pathways that aid myelin clearance include calcineurin, MEK-ERK, Notch and P38 MAPKinase pathway activation, though their full mechanism is not completely understood. Furthermore, TAM receptor phagocytosis also contributes to myelin clearance. Within the nucleus, both OCT6 and HDAC1/2 repress c-JUN function and the polycomb repressive complex 2 (PRC2) represses a number of other repair program genes. Broken lines with question marks highlight a hypothetical association or an unknown quality. Created with BioRender.com
Ramon y Cajal demonstrated the regenerative ability of peripheral nerves and postulated that it was likely some property of the distal stump that attracted axons to regenerate through it. Cajal further described in great detail the fragmentation of myelin sheaths, Schwann cell proliferation and lateral infiltration of Schwann cell tubes with haematogenous macrophages [1]. Progress was made in the 1960s with the application of electron microscopy, which allowed detailed visualisation of the cellular events that occur after nerve injury. However, up until the last two decades, there has been much debate over three questions. Firstly, do Schwann cells actively respond to nerve injury through a controlled molecular mechanism or do they passively revert to an immature phenotype when they lose axonal-derived differentiation signals following axon degeneration? Secondly, do Schwann cells contribute to myelin clearance or is all myelin and axonal debris removed by macrophages and other immune cells? Thirdly, how important are Schwann cells for axon regeneration and functional nerve repair? Through use of mouse conditional knockout technology, we now know that Schwann cells actively respond to nerve injury and this process is regulated by a number of genes, but expression of the transcription factor c-JUN is crucial [13, 14] (Fig. 1). We also know that Schwann cells do not revert to an immature phenotype after injury but are instead reprogrammed to an injury specialised cellular state, termed, repair Schwann cells [13, 15]. Repair Schwann cells use a form of macroautophagy, termed myelinophagy to clear myelin debris after injury, alongside myelin clearance by haematogenous macrophages [16, 17]. Finally, if repair Schwann cell formation is disrupted, then sensory and motor axon regeneration is significantly slowed, a substantial proportion of neurons die and the PNS repair process is permanently arrested [13].
In the last decade, there have been substantial advances in the understanding of the cellular and molecular mechanisms that govern axon degeneration and the Schwann cell injury response. Furthermore, there has been recent progress in linking some of these underlying mechanisms to neurological disease and to developing therapies to both protect from axon loss and to promote axons to regenerate once they have been damaged. In this review, we will highlight the current understanding of the signals that govern the axon intrinsic mechanism of degeneration and the Schwann cell response to PNS injury and repair. This includes what is known from studies using rodents, fish and flies about the molecular pathways governing axon degeneration, those regulating repair Schwann cells, demyelination and the axon extrinsic mechanisms of regeneration. We will not discuss the axon intrinsic mechanisms of regeneration, which are reviewed elsewhere [18–20]. It is important to remember that many additional cell types play significant roles in PNS injury and repair too, including cells of the innate and adaptive immunity, satellite glia in the dorsal root ganglion (DRG), perineurial glia, endoneurial fibroblasts/tactocytes and endothelial cells. These topics are beyond the scope of this review and are discussed in detail elsewhere [21–26]. We will then comment upon recent research linking genes involved in regulating axon degeneration and repair Schwann cells to PNS diseases. Finally, we will discuss the current translation of these fundamental biological mechanisms into therapies to both protect against axon loss and promote axon regrowth in the diseased or injured PNS, and some immediate, outstanding questions for these fields to answer.
Basic Biology of Injury-Induced Axon Degeneration
Today’s detailed knowledge of the molecular mechanism of programmed axon death stems from the discovery in 1989 of an overtly normal strain of mice in which a spontaneous mutation delayed the axon degeneration distal to a nerve injury by tenfold [27]. Instead of the normal latent phase of around 36 h, during which substantial Schwann cell morphological responses discussed below begin, the distal stump remains intact for 2–3 weeks [28]. Both PNS and CNS axons of these Wallerian degeneration slow (WLDS mice are protected; they remain functionally competent for much of their extended survival time if an action potential is artificially evoked in the distal stump [8, 27], and the molecular mechanism is clearly distinct from programmed cell death by apoptosis [29, 30]. However, the principle of a self-destructive mechanism blocked by upstream regulators and activated by multiple, diverse stimuli does indeed mirror that of apoptosis. Why it should be evolutionarily beneficial to have a self-destruct mechanism for rapid axon loss remains unclear but possible explanations including preventing spread of pathogens around the nervous system by axonal transport [31] and promoting subsequent nerve repair. This seems consistent with the slower degeneration in the mammalian CNS, although there are indications that this mostly reflects slower removal of myelin debris [32], while axon degeneration itself is only marginally slower [33].
The discovery that the WLDS mutant gene encodes an NAD synthesising enzyme [8] of the NMNAT family that is partially targeted into axons [34] began a series of findings, still going on today, of how closely axon survival is linked to NAD-related metabolism (Fig. 1). Overexpression of other NMNAT isoforms was found to protect injured axons too, at least if these are axonally targeted and sufficiently stable [35–38]. However, when endogenous NMNATs are removed, the only one whose loss causes an axonal phenotype is NMNAT2 [7, 39, 40] ([41] p. 3).
The remarkable finding that murine WLDS protects injured axons when ectopically expressed in Drosophila validated the use of Drosophila as an experimental organism for further genetic analysis [42, 43]. This led to the identification of dSarm, the Drosophila orthologue of SARM1, as a protein required for injured axons to undergo rapid axon degeneration, and confirmation that mammalian SARM1 is also an essential effector of Wallerian degeneration [10]. An RNAi-based screen in mouse neurons subsequently confirmed this finding [44], and further research revealed an unexpected NAD degrading enzyme activity of SARM1 [12, 45]. SARM1 is also an nicotinamide adenine dinucleotide phosphate glycohydrolase (NADPase) and has base exchange activities that are sometimes even dominant over its NADase activity [46, 47]. Any one of these activities could drive or contribute to axon degeneration, although most attention has so far focussed on NADase. NMNAT2 was found to be a upstream negative regulator of SARM1, in fact Sarm1 deletion in mice completely rescues the otherwise perinatal lethal Nmnat2 null phenotype in which long axons fail to grow [48, 49]. This, and a similar, recent finding of complete protection by Sarm1 deletion from a neurotoxin [50, 51], shows the full protective capacity that could be achieved by effective targeting of SARM1, at least in disorders where programmed axon death is activated very specifically.
Considerable recent progress has begun to reveal how NMNAT2 holds the SARM1 enzyme activity at basal levels. Loss of NMNAT2 from injured axons leads to accumulation of its substrate, NMN, which was found to promote axon degeneration [52]. Sequestering NMN using an ectopically expressed bacterial enzyme, NMN deamidase, is highly protective [53]. For several years, this led to competing hypotheses regarding whether axons die from NAD depletion or from accumulation of its precursor NMN to toxic levels [54] until these were unified by the exciting discovery that NMN is an activator of SARM1 NADase [47] (Fig. 1). Thus, accumulation of NMN after NMNAT2 loss does not just accompany NAD depletion due to loss of its synthetic enzyme; it actually drives NAD depletion even faster by increasing NAD degradation. In the latest developments, NAD has been found to oppose the activating effect of NMN through binding of the same allosteric site in the inhibitory ARM domain, countering one another at physiological levels of each [46, 55]. Another, more potent activator has also been identified [50, 51]. Vacor mononucleotide (VMN), an analogue of NMN and a metabolic product of the disused neurotoxin vacor, was found to bind and activate SARM1 with around twice the potency of NMN, killing neurons and their axons, suggesting this is the likely basis of vacor toxicity [56]. With all three structures now available [50, 51, 55, 57], these findings greatly facilitate rational drug design targeting SARM1 regulation.
Some additional progress has been made upstream of NMNAT2 and downstream of SARM1. NMNAT2 is targeted to axonal transport vesicles by palmitoylation, which unexpectedly lowers its stability and its capacity to protect injured axons [35, 36]. A partial explanation is that NMNAT2 also exists in a separate, soluble pool [35, 36, 58] and that turnover of the vesicular and soluble proteins is regulated by different proteins. The MYCBP2(PHR1)/FBXO45/SKP1A ubiquitin ligase complex regulates turnover of the vesicular form, and kinases DLK and LZK regulate the half-life of the soluble form. Interestingly, inhibition of these proteins, or corresponding gene deletion, is also protective [9, 59]. Stathmin-2 (STMN2) has an as-yet undefined role as another, albeit weaker inhibitor of programmed axon death [60] which may be important in the context of ALS (see below). Its many similarities to NMNAT2, including being targeted by palmitoylation to the same vesicle population and being turned over by the same enzymes [58], suggest this is the most likely point at which it impacts the pathway, although this remains to be determined.
Downstream of SARM1, the loss of NAD, and subsequently of ATP, is not the only important consequence. The loss of NADP, and consequently of NADPH, is likely to limit the capacity for reactive oxygen species (ROS) buffering, especially as SARM1 NADPase is also activated by NMN [46], and there is SARM1-dependent accumulation of calcium [61] that may drive degeneration through calpains [62], likely due to the several calcium mobilising products of NAD cyclisation and base exchange (Fig. 1). The Drosophila protein Axundead also has a poorly understood but essential role downstream of dSarm [63] that may yet fit with any of these mechanisms (Fig. 1).
The Schwann Cell Response to Nerve Injury
Myelinating and non-myelinating Schwann cells have major roles in both the degeneration and regeneration phase of nerve injury. Schwann cells react early to nerve injury with changes in gene expression; however, it is still not clear whether these changes occur before or at the time of axon degeneration [14, 64]. Several studies, conducted first in zebrafish and later in mice, have shown that Schwann cells participate in the breakup of the axon during the process of axon degeneration [65–70]. This process involves the formation of constricting actomyosin spheres and partially requires placental growth factor signalling from the axon, the vascular endothelial growth factor receptor (VEGFR) on Schwann cells, activation of mechanistic target of rapamycin (mTOR) and potentially calcineurin B in Schwann cells [65, 69, 71, 72]. Furthermore, it has recently been shown that Schwann cells upregulate glycolysis after injury and that this process may actually help to protect axons for a short period after injury prior to axon degeneration [65].
During or slightly after the process of axon degeneration, though the exact timing is still unknown, Schwann cells undergo a remarkable biochemical and morphological transformation into repair Schwann cells [13–15]. This conversion can be described as a reversible injury-induced change of cellular state, termed adaptive cellular reprogramming. This is similar to other adult mammalian cellular responses to injury, such as fibroblast to myofibroblast conversion in wound healing, as well as the PNS neuronal upregulation of an axon regeneration program [73]. As part of this transition, repair Schwann cells ingest a proportion of their own myelin sheaths using a form of macroautophagy, termed myelinophagy [17]. Lipidated LC3 (LC3-II), a marker of autophagosomes, is strongly expressed in demyelinating Schwann cells, in vitro and in vivo, in addition to many autophagy machinery genes, such as Atg7. When Atg7 is specifically inactivated in Schwann cells, autophagy and thus myelin clearance after nerve injury is significantly perturbed [17, 74, 75]. Additionally, Schwann cells also use phagocytosis through TAM receptors and the necroptosis pathway to clear myelin debris [74, 76] (Fig. 1). The formation of repair Schwann cells also involves substantial morphological changes to myelinating and non-myelinating Schwann cells transforming into vastly longer, bipolar, branched repair Schwann cells that partially overlap with neighbouring cells within their basal lamina tubes, forming the bands of Büngner [15]. Correct formation of the bands of Büngner likely underlies efficient axon regeneration [13]. This potentially explains why PNS regeneration is more efficient after nerve crush compared to a full nerve transection as a crush injury maintains continuity of the Schwann cell basal lamina tubes between proximal and distal sites [77]. On the contrary, when a nerve is fully transected a multicellular bridge is formed from Schwann cells, fibroblasts, perineurial cells, blood vessels, macrophages and regenerating axons [22]. A number of factors have specific roles in modulating the Schwann cell phenotype specifically in the nerve bridge,these include SOX2, TGFβ1, Robo signalling and ephrin-B/EphB2 signalling. These are reviewed elsewhere [22, 78].
Repair Schwann cell formation involves large-scale changes in gene expression. Genes involved in myelin differentiation are suppressed, and instead there is upregulation of genes involved in a repair program [79]. The repair program broadly comprises of (1) re-expression of some developmentally expressed genes such as N-cadherin, Sox-2, c-Jun, p75ngfr and Gfap, which are normally repressed in myelinating Schwann cells; (2) expression of cytokines and chemokines, such as tumour necrosis factor-α (TNF-α), interleukin-6 (IL-6), IL-1α/β, leukaemia inhibitory factor (LIF) and monocyte chemoattractant protein-1/CCL2 (MCP-1); and (3) upregulation of genes and proteins that are important in promoting axon guidance and neuron survival, such as GDNF, BDNF, artemin, NT3, sonic hedgehog (SHH), semaphorins (e.g. Sema4F) and ephrins (e.g. Epha5) in addition to cell adhesion and matrix molecules such as integrins (e.g. Itgb2), collagens (e.g. Col18a1) and matrix metalloproteins (e.g. Mmp17) [13, 14, 64, 80].
The transition of a myelinating Schwann cell into a repair Schwann cell shares many similarities with the process of epithelial-mesenchymal transition (EMT) [14]. Myelinating Schwann cells represent an epithelial-like cell, since they have tight junctions, a basement membrane, cell polarity with an adaxonal and abaxonal membrane and express epithelial proteins such as E-cadherin, claudin-19, occludin and the polarity protein, PAR3 [81–85]. Their injury induced conversion into repair cells involves the formation of a more motile, proliferative and invasive cellular state, similar to cells of mesenchymal origin and express a number of EMT-enriched genes such as vimentin, snail, Tgf-β1, Wt1, Met, Hmga2, mir221 and mir222 [64, 86].
Molecular Signals that Regulate Repair Schwann Cells, Remyelination and the Non-cell Autonomous Regulation of Axon Regeneration
We will briefly summarise the major transcription factors, signalling pathways and epigenetic factors that regulate repair Schwann cells, but this topic has been comprehensively reviewed elsewhere [14] (Figs. 1 and 2). The transcription factor, c-JUN, was identified as a central regulator of repair Schwann cells, controlling demyelination/myelinophagy, expression of many repair program genes, such as Shh, Bdnf and Gdnf, axon regeneration, motor and sensory neuron survival and functional recovery [13, 17, 87] (Fig. 1). C-JUN expression is suppressed in Schwann cells in the adult nerve and is strongly upregulated after nerve injury [88]. C-JUN acts as an inhibitor of myelination and re-myelination, and its timely O-GlcNAcylation is necessary to inhibit its activity and promote remyelination of regenerated axons [88–90]. Additionally, the POU domain transcription factor, OCT-6 is upregulated by Schwann cells after injury and appears to repress c-JUN induction and delay demyelination and axon regeneration [91] (Fig. 1). Another important transcription factor is STAT3 which promotes the long-term survival of Schwann cells after nerve injury, in addition to maintaining the expression of c-JUN and other repair program genes [92].
Fig. 2.

Overview of the molecular mechanisms of Schwann cell remyelination. Schwann cell remyelination is promoted by axonal signals centred around NRG1 type III and basal lamina signalling via the g-protein coupled receptor, GPR126, similar to myelination during development. One distinct molecular difference from development is that Schwann cell derived soluble NRG1 type I also contributes to remyelination. Certainly macroscopically, remyelination leads to thinner myelin sheaths and shorter internodal distances compared to developmental myelination. C-JUN is an inhibitor of remyelination and OGT, through direct O-GlcNAcylation, represses c-JUN function to allow remyelination to proceed. YAP/TAZ is required for remyelination, in addition to other important regulators of myelination, such as CTCF, ZEB2, HDAC1/2 and HDAC3. SLI = Schmidt-Lanterman incisure. Broken lines with question marks highlight a hypothetical association or an unknown quality. Created with BioRender.com
The transcription factor ZEB2 and two transcriptional activators of the Hippo signalling pathway, YAP and TAZ, are required for Schwann cell remyelination after injury but not for the initial formation of repair Schwann cells, myelin clearance or c-JUN upregulation (Fig. 2). ZEB2-deficient Schwann cells fail to remyelinate, and it is postulated that this is due to maintained expression of myelination inhibitors, such as Sox2, Hey2 and Id2. While it is uncertain whether physiological levels of YAP/TAZ directly regulate c-JUN in Schwann cells, in vivo, YAP/TAZ does appear to be required for eventual c-JUN downregulation during remyelination [93–96]. Furthermore, the tumour suppressor protein, merlin, is important for timely c-JUN upregulation though not for demyelination and is crucial for adequate axonal regeneration and proper remyelination after injury. In the absence of merlin, YAP is aberrantly overexpressed after nerve injury, and deletion of Yap is sufficient to rescue the regenerative deficit in merlin null mice [97].
Epigenetic factors such as chromatin remodelling enzymes, non-coding RNAs and DNA methylation also regulate the Schwann cell injury phenotype [14, 98]. Nerve injury induces demethylation of the repressive histone mark, H3K27 trimethylation, at enhancers of a number of repair program genes (Fig. 1). Importantly activity of the histone demethylases, KDM6B/KDM6A, appears not to be involved [99, 100]. Concurrently, repair program gene promoters gain the active H3K4 methylation mark, which is also postulated to drive their expression [101]. The polycomb repressive complex 2 (PRC2) acts as a histone methyltransferase and is responsible for repressing expression of a number of Schwann cell injury genes such as Fgf5, Shh, Sema4f, EphA5, Olig1, Runx2, Nrg1 and Mmp17, likely through H3K27 trimethylation [102] (Fig. 1). Deletion of the Eed subunit of PRC2 in Schwann cells leads to aberrant upregulation of a subset of repair program genes in the uninjured state and early after injury. However, Eed appears to be largely dispensable for repair Schwann cell formation, since demyelination, c-JUN expression and remyelination are normal, and there is only a temporary delay in axonal regeneration, in its absence [102]. These findings suggest that H3K27 demethylation of enhancers may work in parallel with upregulation of c-JUN-dependent genes in Schwann cells after nerve injury.
The chromatin remodelling enzymes, histone deacetylases (HDACs) have been shown to have several roles in Schwann cells after injury. Deletion of Hdac1/2 in Schwann cells leads to accelerated myelin clearance and axon regeneration but impaired remyelination, whereas Hdac3 deletion has no effect on demyelination or regeneration but leads to exaggerated remyelination [91, 103]. Hdac 1/2 deletion impairs OCT-6 upregulation after injury and causes exaggerated levels of c-JUN expression, which may explain why there is faster demyelination and axon regeneration [91] (Fig. 1). HDAC2 has been shown to de-acetylate eukaryotic translation elongation factor 1 alpha 1 (eEF1A1), inactivating its ability to shuttle SOX10 out of the nucleus, thus allowing remyelination to proceed [104] (Fig. 1). HDAC4 is known to supress expression of c-JUN in Schwann cells in development, but it’s function has not yet been studied after nerve injury [105]. Additionally, the CCCTC-binding factor (CTCF) has recently been shown to be crucial for both PNS myelination and remyelination after injury, likely through modifying the chromatin accessibility of myelin gene promoters and enhancers, in particular, Krox-20 [230]. MicroRNAs appear to help repress the expression of some repair program genes during development and also promote remyelination [106, 107], whereas the role of long non-coding RNAs remains largely unknown [14, 231]. Finally, there are no global changes in the DNA methylome after nerve injury arguing against a major role for CpG methylation in regulating the repair cell phenotype. It is unknown whether DNA hydroxymethylation or hemimethylation play roles in Schwann cells however [64, 108].
A number of signalling pathways are activated in Schwann cells soon after injury [14]. The mTOR pathway is activated rapidly after nerve trauma, and, conditional removal of mTORC1 in Schwann cells leads to a delay in demyelination, slower c-JUN upregulation and impaired upregulation of glycolysis [65, 109] (Fig. 1). The Notch pathway has a major role in controlling the rate of demyelination in injured nerves though it remains unknown whether it regulates repair Schwann cell formation, c-JUN expression and axon regeneration [110] (Fig. 1). The Raf-MEK-ERK mitogen-activated protein kinase pathway (MAPK) is important in regulating cytokine and chemokine expression by Schwann cells, macrophage influx into the nerve, demyelination after injury and may also play a role in regulating blood-nerve barrier integrity, in addition to axon regeneration rates [111–114] (Fig. 1). It remains uncertain whether the Raf-MEK-ERK pathway works in parallel to or mainly upstream of c-JUN controlled events within repair Schwann cells. Furthermore, ERK1/2 also has differing roles in myelination and remyelination, which is reviewed in more detail elsewhere [14]. The p38MAPK and c-Jun-N-terminal kinase (JNK) pathways are also both activated after nerve injury [88, 115] (Fig. 1). Inactivation of p38α, the major p38MAPK isoform expressed in the PNS, demonstrated slower myelin clearance after injury but no effect on macrophage influx or axonal regeneration, whereas the role of the JNK pathway in Schwann cells in vivo has not been studied yet [116]. Additionally, Schwann cell RalGTPases have been shown to regulate remyelination and motor neuron reinnervation and calcineurin B has recently been shown to regulate myelinophagy, independently of c-JUN expression [72, 117].
Neuregulin-1 (NRG-1) has 15 different isoforms and multifaceted roles in PNS development and repair [118]. Membrane bound NRG-1 type III is expressed on axons, though Schwann cells upregulate expression of NRG-1 type I after injury [119, 120]. Axonal NRG-1 and the erbB receptor complex expressed on Schwann cells appear largely dispensable for demyelination and repair Schwann cell formation [121–123]. Axonal NRG-1 and Schwann cell erbB signalling does play a role in influencing the rate of axonal regeneration, and both axonal NRG-1 type III and Schwann cell produced NRG-1 type I regulate remyelination after nerve injury [120, 122–124] (Fig. 2). Additionally, the transmembrane protease, BACE1, which cleaves NRG1, and the ERBB2 binding protein, Erbin, are both required for proper Schwann cell remyelination [125, 126]. The g-protein coupled receptor GPR126, expressed in Schwann cells, is important for both myelination and remyelination (Fig. 2). It also regulates, non-cell autonomously, macrophage influx into the injured nerve, axonal regeneration and terminal Schwann cell morphology as well as reinnervation of the neuromuscular junction, though its function in nerve injury does not require its prion protein ligand [127–129, 232]. GPR126 is dispensable for c-JUN upregulation and repair Schwann cell formation, however [128]. Schwann cells also upregulate many extracellular matrix (ECM) proteins and neurotrophic factors after nerve injury [130, 131]. Laminins, collagens, fibronectin and tenascins are all expressed by repair Schwann cells [131, 132]. Deletion of laminin γ1, Col18a1 and tenascin C in the PNS all lead to defective axon regeneration [133–135]. Interestingly, post-translational modification of collagen by lysyl-hydroxylase 3 in Schwann cells in zebrafish underlies target selective regeneration [136].
Repair Schwann cells express a number of neurotrophins after injury, these include NGF which binds TrkA and p75NGFR receptors; BDNF and NT-4/5 which bind TrkB receptors; GDNF and artemin, which bind GDNF receptors; and the neuropoietic cytokines, CNTF and LIF, which bind their cognate receptors and signal through gp130 [130, 137]. Creation of a pan-neurotrophin mouse, which expressed a chimeric neurotrophin under the endogenous BDNF promoter, which is only active in repair Schwann cells after injury, accelerated motor and sensory axon regeneration [13, 138]. Use of NGF blocking antibodies demonstrated no reduction in nociceptor regeneration rates but did identify a role for NGF in injury-induced collateral sprouting [139–141]. Blocking BDNF function after injury led to reduced axon regeneration rates and a substantial reduction in myelinated axons numbers, however, BDNF haploinsufficiency is sufficient for normal sensory axon regeneration rates [142, 143]. Importantly, regeneration rates of Thy1.1 YFP labelled, sensory axons in NT-4/5 heterozygous and homozygous nerve grafts were substantially reduced [142]. GDNF and artemin are required for adequate motor neuron survival and regeneration, in addition to promoting the survival and function of a subset of large and small fibre DRG neurons [13, 87, 144–146]. Regarding the neuropoietic cytokines, CNTF acts as a lesion factor, released from myelinating Schwann cells, promoting the survival of motor neurons after injury, and LIF promotes the regeneration of sensory axons [147]. Finally, there are two additional factors produced by Schwann cells that have been shown to act in a similar way to the neurotrophins, the first is pleiotrophin and the other is sonic hedgehog, which promote PNS motor and sensory axon regeneration, respectively [13, 106, 148, 149].
Relevance of Molecular Mechanisms Regulating Axon Degeneration and Repair Schwann Cell for PNS Diseases
A common approach to developing therapies for inherited and acquired peripheral nerve disorders is to identify the genetic and environmental causes and then find ways to block them. Examples of success include over 70 known genes for Charcot-Marie-Tooth disease [150] and the neurotoxicity of cancer chemotherapeutics such as vincristine, paclitaxel, bortezomib and oxaliplatin in chemotherapy-induced peripheral neuropathy [151, 152]. However, fully understanding and preventing the mechanisms these genes and toxins activate is more challenging. The well-characterised programmed axon death pathway, which can already be blocked, at least in animal models, offers an alternative approach of identifying specific human diseases and patients in which this contributes to axon loss. Animal model studies have strongly validated this approach [153], and human genetics now provides important opportunities for translation by identifying and functionally characterising naturally occurring human mutations in programmed axon death genes and testing for association with disease.
Programmed Axon Death and Disease
The concept that degeneration after nerve injury may inform us about how axons degenerate in disease dates right back to Waller, who wrote of his observations on transected nerves: “it is particularly with reference to nervous diseases that it will be most desirable to extend these researches” [2]. When the discovery of delayed Wallerian degeneration in WldS mice raised the prospect of molecular understanding of the process in 1989, part of the impetus to identify the underlying genetic cause was a similar thinking, that this could have therapeutic implications for non-injury disorders. Indeed, the concept that toxins could produce an effective “chemical transection” of axons had recently been proposed by Bouldin and Cavanagh [154], and if that was their mode of action, then the degenerative mechanisms may be similar.
Confirmation that axons can die through the same WldS-sensitive pathway without physical injury first came in experiments with the cancer chemotherapeutics vincristine and paclitaxel [155, 156] in cell culture and mouse models of chemotherapy-induced peripheral neuropathies. Protection of axons exposed to these toxins by the WldS mutation clearly showed that physical injury was not necessary to activate this degenerative mechanism, strongly supporting the clinical relevance of understanding and blocking it. More extensive mechanistic similarities were later indicated by observations that Sarm1 deletion also protects axons and alleviates the pain responses in these models [157, 158] and that axons expressing lower than normal NMNAT2 levels show enhanced vulnerability to vincristine [159]. The findings that WldS protects axons and alleviates symptoms in models of toxic disorders were quickly followed by reports of similar protection in mouse of genetic disorders, such as Charcot-Marie-Tooth 1B (CMT1B) involving myelin protein zero [160] and progressive encephalopathy with distal spinal muscular atrophy (SMA) involving biallelic mutation of tubulin chaperone E (TBCE) [161, 162]. Models of many other PNS and CNS disorders were also found to be alleviated, involving other neurotoxins [163–165], mutations [166], metabolic perturbations [51, 158] and non-transecting physical forces such as raised intraocular pressure modelling a major risk factor for glaucoma [167, 168]. However, axons are not or not strongly protected in all disease models by blocking programmed axon death, suggesting these are predominantly driven by other mechanisms. These include SOD1 transgene models of ALS [169, 170], SMA models [171, 172] and some other causes of CMT [173]. Full summaries of which models show protective responses are provided in earlier reviews [153, 174].
The ability to alleviate many disease models by blocking programmed axon death indicates that this pathway contributes to axon loss in these models, not necessarily as an initiating event, nor in isolation from other mechanisms, but at least at some level. However, it is also now clear that aberrant activation of programmed axon death can initiate some axonopathies or increase axon vulnerability to stresses that would not normally kill them. To illustrate this in animal models, Nmnat2 null mice fail to grow long axons and consequently die at birth with respiratory failure [7, 40]. Mice expressing only 30% as much NMNAT2 as C57BL/6 controls have axons that are more vulnerable to stresses such as vincristine, mitochondrial uncoupling and normal ageing [50, 51, 159]. Importantly, both of these mouse genotypes have human counterparts. Biallelic NMNAT2 null mutation is associated with a similar, even more severe stillbirth phenotype in humans [175]. Partial NMNAT2 loss-of-function occurs in an inherited polyneuropathy with neuropathic pain [176], and NMNAT2 expression level shows wide variation in the human population [177]. Taken together with the observations in mice, this suggests a spectrum of intrinsic axon vulnerability in humans.
Aberrant activation can also occur directly through SARM1. In an exciting, recent development, SARM1 gain-of-function has been shown to be a statistically significant risk factor in sporadic ALS and to associate with hereditary spastic paraplegia and other motor nerve disorders [178, 179]. Taken together, these studies report twelve different missense or microdeletion variants in 17 patients, altering the inhibitory, N-terminal ARM domain of the protein. All constitutively hyperactivate SARM1 basal NADase activity, and at least five of them to a level 20-fold higher than the wild-type enzyme [179]. Remarkably, this activity in this assay exceeds even that of the NMN-activated wild-type protein many times over, raising important questions about how these individuals survive at all, some with age of onset as late as 70 years [179]. Moreover, their occurrence in sporadic rather than familial cases suggests interaction with other risk factors to produce disease. Nevertheless, neurons expressing these gain-of-function variants are more sensitive to stress in primary culture and die in vivo [178, 179], further indicating their pathogenic role. It will be important now to determine whether SARM1 mediates the apparent contribution of STMN2 depletion to ALS [180, 181] and whether the previously reported GWAS linkage to the SARM1 locus on chromosome 17 is mediated by SARM1 gene expression level [182, 183].
There is also evidence of toxic hyperactivation of SARM1 in human disease. A downstream metabolite of vacor, a disused rodenticide and nicotinamide analogue, is a potent and direct activator of SARM1 that causes SARM1-dependent axon and neuronal death [50, 51]. Before vacor was banned, individuals who used it in suicide attempts and survived often developed widespread neurological deficits and peripheral nerve axon loss within hours or days [56]. It is extremely likely that SARM1 activation was the major cause of this rapid-onset neuropathy. Although these same individuals also often developed diabetes, the onset of neuropathy within hours suggests it was prior rather than secondary to diabetes, although the later may well have sustained the problem.
Table 1 summarises the accumulating evidence of aberrant activation of programmed axon death in specific human diseases. Together with the more widespread alleviation of animal models of peripheral neuropathies, motor nerve disorders and other conditions [153, 174], this suggests that blocking this pathway will be beneficial in at least a subset of patients with activating genetics or environment in multiple disorders.
Table 1.
Highlighting the relevance of programmed axon death to human peripheral nerve diseases. For additional summary of animal model data, see Conforti et al. [153]
| Disease type | Details | References |
|---|---|---|
| Fetal akinesia deformation sequence | Stillbirth with complete absence of skeletal muscle, likely of neurogenic origin, and hydrocephalus, associated with biallelic null mutation of Nmnat2 | [175] |
| Polyneuropathy with erythromelalgia | Distal sensory and motor axon loss, painful episodes of erythromelalgia in distal limbs especially following infection, associated with biallelic hypomorphic mutation of Nmnat2 | [176] |
| ALS | Late-onset (40–71 y) sporadic ALS, spinal or bulbar onset, often though not always progressing quickly. Associated with monoallelic constitutive hyperactivation of SARM1 | [178, 179] |
| Upper and lower motor nerve disorder | Middle age onset with unilateral leg weakness and wasting, slowly progressing over 25 y with later mild hand weakness and lower limb spasticity. Associated with monoallelic constitutive hyperactivation of SARM1 | [179] |
| Vacor neuropathy | Rapid onset (2 h-3d) lower limb weakness and numbness, ataxia, areflexia, following vacor ingestion. Often associated with additional CNS phenotypes and diabetes. Neurotoxic effect completely dependent on SARM1 | [50, 51, 56] |
Repair Schwann Cell Molecular Mechanisms in PNS Diseases
While nerve regeneration in lower vertebrates and small mammals is fairly efficient, the regenerative capacity of human nerves is much poorer. Less than 50% of patients undergoing surgical repairs of injured median or ulnar nerves regain adequate motor or sensory function, in the long term [184]. Furthermore, regenerative functional outcomes deteriorate with increasing age above 40 years; a more proximal lesion site, and thus increasing regenerative distance; and delaying surgical repair for greater than 6 months after trauma [184]. The reason for the deterioration in repair capacity of peripheral nerves appears to be in large part down to the response of Schwann cells [185]. Using nerve grafting experiments in mice, age-related decline in repair capacity of the PNS was found to be due to the age of the nerve graft and not the host, suggesting that the Schwann cell and not the neuronal or inflammatory cell response was responsible [186]. Aged Schwann cells have a reduced capacity to activate myelinophagy and upregulate repair program genes, especially the transcription factor c-JUN [186, 187]. The regenerative decline after delaying nerve repair is also largely due to a loss of regeneration support by repair Schwann cells in the distal stump, rather than a reduction in the intrinsic neuronal regeneration capacity [187–189]. Chronic denervation in mouse and human nerves leads to unfavourable changes in Schwann cells, with downregulation of c-JUN overtime, leading to senescence and eventually cell death [187, 190, 191] (Table 2).
Table 2.
Highlighting the relevance of the Schwann cell injury response to peripheral nerve diseases
| Disease type | Details | References |
|---|---|---|
| Chronic denervation after traumatic injury |
c-JUN is downregulated in chronic denervation in mouse and human nerves after traumatic injury. This downregulation is correlated with Schwann cell death and regenerative decline Transgenically augmenting c-JUN levels in mouse nerves in chronic denervation rescues regeneration potential |
[187, 190, 191] |
| Inflammatory neuropathies | c-JUN is expression is upregulated in Schwann cells in patients with Guillain–Barre syndrome, chronic inflammatory demyelinating neuropathy and peripheral nerve vasculitis | [192, 193] |
| Genetic neuropathies |
c-JUN is upregulated in Schwann cells in CMT1A patients Deletion of Schwann cell c-Jun in a CMT1A mouse model worsens the phenotype, suggesting it is protective for sensory axons NRG1 type 1 is upregulated in postnatal nerves of a CMT1A rodent model Prolonged NRG1 type 1 signalling in a CMT1A mouse model is responsible for onion bulb formation SOX2 and ID2 are upregulated in Schwann cells in a CMT1B mouse model and modulate ER stress |
[192, 194–197] |
| Compression neuropathy | A GWAS of patients with carpal tunnel identified Adamsts17 as a risk gene. Adamsts17 is strongly upregulated after nerve injury | [198] |
The Schwann cell injury response is also activated in genetic and acquired neuropathies [199] (Table 2), similar to the situation described above for programmed axon death. In particular, c-JUN expression has been identified in Schwann cells in nerves of patients with a form of inherited neuropathy, Charcot-Marie-Tooth disease 1A (CMT1A) and inflammatory neuropathies such as chronic inflammatory demyelinating polyradiculoneuropathy (CIDP), Guillain-Barré syndrome (GBS) and vasculitic neuropathy [192, 193]. Additionally, a recent genome wide association study of carpal tunnel patients revealed that single nucleotide polymorphisms in the Adamts17 gene, which is upregulated by repair Schwann cells after injury, appears to confer risk to the development of carpal tunnel syndrome, a very commonly occurring compression neuropathy of the median nerve at the wrist [198]. The major question arising from all these findings is whether a partial Schwann cell injury response in the context of neuropathy is a broadly protective or deleterious reaction?
Interestingly, deletion of c-JUN in Schwann cells in a mouse model of CMT1A leads to a more severe phenotype with greater sensory axonal loss [197]. This suggests that c-JUN is upregulated in Schwann cells partially as a protective response in the context of neuropathy. Additionally, both SOX2 and ID2, two inhibitors of myelin differentiation that are also upregulated in repair Schwann cells after injury and in the context of neuropathy, appear to play a protective role. Deletion of either Sox2 or Id2, specifically in Schwann cells, in the mouse model of CMT1B, increases endoplasmic reticulum stress markers and worsens the dysmyelination phenotype [196]. The idea that Schwann cells utilise a partial injury reaction as an initial protective response in neuropathy is further exemplified by the role of NRG-1 type I in CMT1A [194, 199]. NRG-1 type 1 is upregulated by Schwann cells in CMT1A nerves in postnatal development where it helps promote myelination and ameliorate the disease phenotype [194]. Transgenic overexpression of axonally derived NRG-1 or supplementation with soluble NRG-1 in early postnatal development was sufficient to improve the myelination status of axons and compound motor action potentials (CMAPs) on neurophysiological testing in CMT1A rodent models [195]. However, despite the beneficial effects of NRG-1, prolonged Schwann cell NRG-1 type I paracrine signalling in CMT1A actually drives pathological hypermyelination and onion bulb formation. The Schwann cell specific deletion of Nrg1 in a CMT1A mouse model led to a better clinical phenotype, with improved neurophysiological and neuromuscular function [194]. Thus NRG-1 has a complex role in inherited demyelinating neuropathies, but these studies demonstrate that there may be a therapeutic window for exogenous NRG-1 early on in the disease course for genetic neuropathies such as CMT1A.
The Schwann cell injury response is also relevant to PNS tumours. Dysregulation of crucial pathways that regulate repair Schwann cells appears to be important in the formation of malignant peripheral nerve sheath tumours (MPNST), which are highly aggressive and invasive tumours that originate from the Schwann cell lineage [200]. Interestingly, despite repair Schwann cells adopting an EMT-like gene signature after nerve injury, lineage tracing studies in mice have shown that they remain lineage restricted and are not multipotent [14, 201]. This suggests that molecular regulators of repair Schwann cells likely prevent tumour formation. In these tumours, the HIPPO-TAZ/YAP pathway is hyperactivated, PRC2 is inactivated, and there is a complete loss of the H3K27 trimethylation mark along with inactivation of the Ink4a/Arf locus promoting unrestricted proliferation and malignant transformation [202–206]. Thus, deranged activation of key pathways and molecular regulators in repair Schwann cells plays a central role in tumourigenesis.
Future Treatment Strategies to Protect Against Axon Loss and Promote Axon Regeneration in the PNS
Therapeutic Opportunities in Programmed Axon Death
The ability of NMNAT overexpression or SARM1 deletion to delay axon loss and symptoms in animal models of widely varying neurological disorders [153, 174] has long suggested therapeutic potential if drugs could be developed to mimic these effects in patients. While the prospect of enhancing NMNAT activity or expression, or the NMNAT2 stability, requires some novel approaches to drug discovery, the discovery of proteins such as SARM1 and MYCBP2/PHR1 whose activities are required for axons to degenerate presents a seemingly more feasible way to block programmed axon death using inhibitors [9, 10, 12, 207]. Knockdown of SARM1 provides another route to protecting axons [208]. The discovery of SARM1’s NADase activity [12] and the serine-linked ubiquitylation activity of MCYBP2/PHR1 [207], together with relevant structural information for each protein [207, 209], further increase their attractiveness as drug targets. Potential adverse effects of blocking drugs include interference with innate immunity for SARM1 and axon growth effects for MYCBP2/PHR1, but mouse data suggest that with careful targeting and/or timing, these risks could be largely avoided. Other points for intervention in the pathway include MAPK inhibition [210] and supplementation with NAD precursors such as nicotinamide [211], nicotinamide riboside [212] or nicotinic acid riboside.
A very important question is the degree of axon protection that may be feasible with such approaches. For some years, animal model data suggested this may be partial and temporary but applying one single, major stress, such as an overexpressed mutant transgene or a high dose of a toxin, to genetically homogeneous mice does not represent what happens in most human disease. Sporadic disease results from a combination of multiple genetic and environmental risks, and in patients where genetic or toxic activation of programmed axon death is one of these, there is a realistic likelihood that by fully removing that risk pharmacologically, there could be substantial protection. This is particularly demonstrated by the lifelong rescue of the otherwise early lethal phenotype of Nmnat2 null mice when SARM1 is removed [48] and the full rescue of vacor-treated Sarm1 null neurons [50, 51]. It follows that it will be important to identify the specific diseases and patients in whom programmed axon death is most activated by genetic or toxic mechanisms as these are the individuals likely to respond best to drugs blocking SARM1 or other pathway components. The recent discovery of such genetic or toxic activation mechanisms [50, 51, 178, 179] are important first steps towards this. Additional opportunities arise in disorders such as chemotherapy-induced peripheral neuropathies (CIPN) where the axonal stress is anyway temporary, so supporting axons through this period could allow full prevention or recovery [213]. Thus, in CIPN, ALS, rare, inherited polyneuropathies or any other disorders in which programmed axon death activation is identified, there are good prospects for long-term beneficial effects.
Another important consideration is whether SARM1 would need to be fully inactivated for a protective effect or only partially inactivated. When its role in axon degeneration was first identified, transected homozygous null axons in mouse sciatic nerve were found to survive for 2–3 weeks, but those of hemizygous nulls were not protected at 5 days [10]. Now, however, it has become clear that hemizygosity, and similar degrees of knockdown achieved with antisense oligonucleotides, is also partially protective against multiple axon stresses including vincristine toxicity and axotomy [213, 214]. Thus, it is reasonable to expect that partial inhibition or silencing of SARM1 in the right patients for the right disease could still be profoundly protective.
Therapeutic Opportunities in the Schwann Cell Response
There are two substantial problems that need to be addressed in order to successfully promote axon regeneration and functional recovery after human traumatic neuropathy. Firstly, if a nerve is fully transected, a regeneration gap is formed that needs to be bridged either by an autologous nerve graft or by an artificial conduit, if a nerve graft is not suitable. Secondly, repair Schwann cell numbers decline over time in the denervated distal stump, and this adversely affects functional outcomes [190, 191, 215]. One way to overcome these issues is to culture, expand and transplant a patient’s own Schwann cells back into the injured nerve and/or nerve graft/conduit. Two cases of young adults with high sciatic nerve injuries and large regeneration gaps, that normally have poor functional outcomes when treated with sural nerve graft alone, have been treated with combined autologous sural nerve graft and autologous Schwann cell transplant with good regenerative outcomes over a 3-year follow up period [216]. Thus, autologous Schwann cell transplant may afford some promise in improving surgical repair outcomes from traumatic neuropathies.
However, use of autologous Schwann cell transplants does remain constrained by the fact that the Schwann cell supply must come from the patient’s own nerves and that these transplanted Schwann cells, similar to endogenous Schwann cells, will, once transplanted, still lose repair-promoting potential over time and perish. In order to solve the first problem, Schwann cells can be cultured directly from human skin samples [217], or cell reprogramming technology has been used to generate human Schwann cells directly from skin fibroblasts [218–221]. The second problem requires boosting the repair capacity and/or survival of Schwann cells in the injured nerve. One way to achieve this is through neurotrophic factor administration. Application of exogenous GDNF and BDNF protein to various peripheral nerve experimental animal injury models has generally shown positive outcomes. However, this approach, as a treatment strategy in patients, is limited as neurotrophins have short half-lives and penetrate tissues poorly [222]. Gene therapy can be used to abrogate these issues, however, continuous supply of GDNF to a regenerating peripheral nerve through lentiviral delivery results in aberrant sprouting and axon trapping, impairing functional outcomes [223]. Thus delivery of lentiviral expressed GDNF using an immune evasive tetracycline inducible switch, allowing for a pulsed supply of GDNF to the injured nerve, can not only protect motor neurons from injury induced death but also promote long range motor axon regeneration in a spinal root avulsion model [146]. The transcription factor c-JUN is another attractive candidate to improve the PNS repair response in situations where axon regeneration is impaired. These include long-distance regeneration across an artificial conduit to bridge proximal and distal stumps; in chronic denervation; ageing; and to encourage regeneration after secondary axonal loss in inherited and acquired neuropathies [185]. In this regard, Schwann cells transduced with a tetracycline-inducible c-JUN lentiviral construct and then transplanted into a nerve conduit were able to enhance axonal regeneration across a 10 mm gap in rats [224]. Furthermore, overexpression of one allele of c-JUN, specifically in Schwann cells, is able to rescue the decline in axon regeneration rates in aged animals and also after chronic denervation, without appearing to be tumourigenic in mice [89, 187].
It is likely a combination of the above strategies will be required in order to achieve adequate human peripheral nerve regeneration. Furthermore, cell transplant and gene therapy approaches will need to be applied alongside improved engineering of nerve graft conduits, a topic which is reviewed elsewhere [225].
Future Perspectives: Outstanding Questions
The genetic association of NMNAT2 and SARM1 with polyneuropathies and ALS and other motor nerve disorders [175, 176, 178, 179] raises the question of whether coding or gene expression variants in programmed axon death genes influences also other disorders. Certainly, animal model studies suggest so. SARM1 deletion alleviates chemotherapy-induced peripheral neuropathy with vincristine, paclitaxel, bortezomib, cisplatin and oxaliplatin and neuropathy in a type 2 diabetes model using a high fat diet [157, 158, 208, 214, 226, 227], all of which suggest relevance to more common disorders of peripheral nerve. Association with idiopathic peripheral neuropathy, inflammatory neuropathies and other toxic neuropathies, building on findings with vacor as well as cancer chemotherapy drugs will also be important to explore. SARM1 may also have a wider role in ALS than the rare coding variants so far identified, considering the reported GWAS linkage [182] and involvement of programmed axon death regulator STMN2 in this disease [180, 181].
The Schwann cell injury phenotype does appear to vary between motor and sensory nerves and between myelinating and non-myelinating Schwann cells [234, 233]. One future research area will be detailing the transcriptomic and proteomic variation, potentially with single cell resolution, in the repair Schwann cell response between motor and sensory nerves, and between myelinated and unmyelinated fibres, in addition to the effect of different anatomical locations, including root, various different peripheral nerves and end organ associated Schwann cells. This may also start to shed some light upon the reason why the many subtypes of inherited or acquired neuropathy present with involvement of specific and unique patterns of affected nerves, limbs or modalities.
Another part of the Schwann cell injury response that may show promise for development of future therapies for neuropathy is manipulation of Schwann cell myelinophagy. Many acquired and inherited neuropathies are characterised by primary demyelination, and in the Trembler J and C22 mice that model inherited demyelinating neuropathies due to Pmp22 point mutation and overexpression, there is evidence that modulating the level of autophagy in Schwann cells alters pathogenesis [228, 229].
While future cell and gene therapies may show early promise for encouraging axon regeneration after traumatic neuropathy, the next significant challenge will be extending these types of treatments to other acquired (inflammatory and vasculitis) and inherited neuropathies, where there can be substantial secondary axon loss. Any treatment designed to promote axon regeneration will still need to be used in combination with disease modifying treatments, such as the use of immunosuppressive agents in acquired neuropathies and potentially the future use of new genetic therapies for inherited neuropathies.
Supplementary Information
Below is the link to the electronic supplementary material.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Funding
Peter Arthur-Farraj is supported by the Wellcome Trust (Clinical Research Career Development Fellowship 206634). Michael Coleman is supported by the John and Lucille van Geest Foundation. Figures created with BioRender.com.
Footnotes
The original online version of this article was revised to correct ref. 232 Henzi, A., Aguzzi, A., (2021) The prion protein is not required for peripheral nerve de- and remyelination after crush injury. PLoS One, 16(1) e0245944. https://doi.org/10.1371/ journal.pone.0245944.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
11/29/2021
A Correction to this paper has been published: 10.1007/s13311-021-01162-y
Contributor Information
Peter Arthur-Farraj, Email: pja47@cam.ac.uk.
Michael P. Coleman, Email: mc469@cam.ac.uk
References
- 1.Ramon y Cajal, S. (1928). Degeneration and regeneration of the nervous system. Clarendon Press.
- 2.Waller A. Experiments on the section of the glossopharyngeal and hypoglossal nerves of the frog, and observations of the alterations produced thereby in the structure of their primative fibres. Philosophical Transactions of the Royal Society of London. 1850;140:423–429. doi: 10.1098/rstl.1850.0021. [DOI] [Google Scholar]
- 3.Blakemore WF, Cavanagh JB. NEUROAXONAL DYSTROPHY” OCCURRING IN AN EXPERIMENTAL, “DYING BACK. PROCESS IN THE RAT. Brain. 1969;92(4):789–804. doi: 10.1093/brain/92.4.789. [DOI] [PubMed] [Google Scholar]
- 4.Lubińska L. Early course of wallerian degeneration in myelinated fibres of the rat phrenic nerve. Brain Research. 1977;130(1):47–63. doi: 10.1016/0006-8993(77)90841-1. [DOI] [PubMed] [Google Scholar]
- 5.Lubińska, L. (1982). Patterns of Wallerian degeneration of myelinated fibres in short and long peripheral stumps and in isolated segments of rat phrenic nerve. Interpretation of the role of axoplasmic flow of the trophic factor. Brain Research, 233(2), 227–240. 10.1016/0006-8993(82)91199-4 [DOI] [PubMed]
- 6.Gilley J, Coleman MP. Endogenous Nmnat2 Is an Essential Survival Factor for Maintenance of Healthy Axons. PLoS Biology. 2010;8(1):e1000300. doi: 10.1371/journal.pbio.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gilley J, Adalbert R, Yu G, Coleman MP. Rescue of Peripheral and CNS Axon Defects in Mice Lacking NMNAT2. Journal of Neuroscience. 2013;33(33):13410–13424. doi: 10.1523/JNEUROSCI.1534-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mack TGA, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Fernando FS, Tarlton A, Andressen C, Addicks K, Magni G, Ribchester RR, Perry VH, Coleman MP. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nature Neuroscience. 2001;4(12):1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- 9.Babetto E, Beirowski B, Russler EV, Milbrandt J, DiAntonio A. The Phr1 Ubiquitin Ligase Promotes Injury-Induced Axon Self-Destruction. Cell Reports. 2013;3(5):1422–1429. doi: 10.1016/j.celrep.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, Sheehan AE, Avery MA, Hackett R, Logan MA, MacDonald JM, Ziegenfuss JS, Milde S, Hou Y-J, Nathan C, Ding A, Brown RH, Conforti L, Coleman M, Freeman MR. DSarm/Sarm1 Is Required for Activation of an Injury-Induced Axon Death Pathway. Science. 2012;337(6093):481–484. doi: 10.1126/science.1223899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiong X, Hao Y, Sun K, Li J, Li X, Mishra B, Soppina P, Wu C, Hume RI, Collins CA. The Highwire Ubiquitin Ligase Promotes Axonal Degeneration by Tuning Levels of Nmnat Protein. PLoS Biology. 2012;10(12):e1001440. doi: 10.1371/journal.pbio.1001440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Essuman K, Summers DW, Sasaki Y, Mao X, DiAntonio A, Milbrandt J. The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD + Cleavage Activity that Promotes Pathological Axonal Degeneration. Neuron. 2017;93(6):1334–1343.e5. doi: 10.1016/j.neuron.2017.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arthur-Farraj PJ, Latouche M, Wilton DK, Quintes S, Chabrol E, Banerjee A, Woodhoo A, Jenkins B, Rahman M, Turmaine M, Wicher GK, Mitter R, Greensmith L, Behrens A, Raivich G, Mirsky R, Jessen KR. C-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron. 2012;75(4):633–647. doi: 10.1016/j.neuron.2012.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jessen KR, Arthur-Farraj P. Repair Schwann cell update: Adaptive reprogramming, EMT, and stemness in regenerating nerves. Glia. 2019;67(3):421–437. doi: 10.1002/glia.23532. [DOI] [PubMed] [Google Scholar]
- 15.Gomez-Sanchez JA, Pilch KS, van der Lans M, Fazal SV, Benito C, Wagstaff LJ, Mirsky R, Jessen KR. After Nerve Injury, Lineage Tracing Shows That Myelin and Remak Schwann Cells Elongate Extensively and Branch to Form Repair Schwann Cells, Which Shorten Radically on Remyelination. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2017;37(37):9086–9099. doi: 10.1523/JNEUROSCI.1453-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barrette B, Hébert M-A, Filali M, Lafortune K, Vallières N, Gowing G, Julien J-P, Lacroix S. Requirement of myeloid cells for axon regeneration. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2008;28(38):9363–9376. doi: 10.1523/JNEUROSCI.1447-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gomez-Sanchez JA, Carty L, Iruarrizaga-Lejarreta M, Palomo-Irigoyen M, Varela-Rey M, Griffith M, Hantke J, Macias-Camara N, Azkargorta M, Aurrekoetxea I, De Juan VG, Jefferies HBJ, Aspichueta P, Elortza F, Aransay AM, Martínez-Chantar ML, Baas F, Mato JM, Mirsky R, Jessen KR. Schwann cell autophagy, myelinophagy, initiates myelin clearance from injured nerves. The Journal of Cell Biology. 2015;210(1):153–168. doi: 10.1083/jcb.201503019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bradke F, Fawcett JW, Spira ME. Assembly of a new growth cone after axotomy: The precursor to axon regeneration. Nature Reviews. Neuroscience. 2012;13(3):183–193. doi: 10.1038/nrn3176. [DOI] [PubMed] [Google Scholar]
- 19.Mahar M, Cavalli V. Intrinsic mechanisms of neuronal axon regeneration. Nature Reviews Neuroscience. 2018;19(6):323–337. doi: 10.1038/s41583-018-0001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rishal I, Fainzilber M. Axon-soma communication in neuronal injury. Nature Reviews. Neuroscience. 2014;15(1):32–42. doi: 10.1038/nrn3609. [DOI] [PubMed] [Google Scholar]
- 21.Avraham O, Deng P-Y, Jones S, Kuruvilla R, Semenkovich CF, Klyachko VA, Cavalli V. Satellite glial cells promote regenerative growth in sensory neurons. Nature Communications. 2020;11(1):4891. doi: 10.1038/s41467-020-18642-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cattin A-L, Lloyd AC. The multicellular complexity of peripheral nerve regeneration. Current Opinion in Neurobiology. 2016;39:38–46. doi: 10.1016/j.conb.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 23.Chen P, Piao X, Bonaldo P. Role of macrophages in Wallerian degeneration and axonal regeneration after peripheral nerve injury. Acta Neuropathologica. 2015;130(5):605–618. doi: 10.1007/s00401-015-1482-4. [DOI] [PubMed] [Google Scholar]
- 24.Davies, A. J., Rinaldi, S., Costigan, M., & Oh, S. B. (2020). Cytotoxic Immunity in Peripheral Nerve Injury and Pain. Frontiers in Neuroscience, 14. 10.3389/fnins.2020.00142 [DOI] [PMC free article] [PubMed]
- 25.Kucenas, S. (2015). Perineurial Glia. Cold Spring Harbor Perspectives in Biology, 7(6). 10.1101/cshperspect.a020511 [DOI] [PMC free article] [PubMed]
- 26.Stierli S, Imperatore V, Lloyd AC. Schwann cell plasticity-roles in tissue homeostasis, regeneration, and disease. Glia. 2019;67(11):2203–2215. doi: 10.1002/glia.23643. [DOI] [PubMed] [Google Scholar]
- 27.Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of Wallerian Degeneration does not Hinder Regeneration in Peripheral Nerve. European Journal of Neuroscience. 1989;1(1):27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- 28.Beirowski B, Adalbert R, Wagner D, Grumme DS, Addicks K, Ribchester RR, Coleman MP. No title found. BMC Neuroscience. 2005;6(1):6. doi: 10.1186/1471-2202-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deckwerth TL, Johnson EM. Neurites Can Remain Viable after Destruction of the Neuronal Soma by Programmed Cell Death (Apoptosis) Developmental Biology. 1994;165(1):63–72. doi: 10.1006/dbio.1994.1234. [DOI] [PubMed] [Google Scholar]
- 30.Finn JT, Weil M, Archer F, Siman R, Srinivasan A, Raff MC. Evidence That Wallerian Degeneration and Localized Axon Degeneration Induced by Local Neurotrophin Deprivation Do Not Involve Caspases. The Journal of Neuroscience. 2000;20(4):1333–1341. doi: 10.1523/JNEUROSCI.20-04-01333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsunoda I. Axonal degeneration as a self-destructive defense mechanism against neurotropic virus infection. Future Virology. 2008;3(6):579–593. doi: 10.2217/17460794.3.6.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vargas ME, Barres BA. Why Is Wallerian Degeneration in the CNS So Slow? Annual Review of Neuroscience. 2007;30(1):153–179. doi: 10.1146/annurev.neuro.30.051606.094354. [DOI] [PubMed] [Google Scholar]
- 33.Beirowski B, Nógrádi A, Babetto E, Garcia-Alias G, Coleman MP. Mechanisms of Axonal Spheroid Formation in Central Nervous System Wallerian Degeneration. Journal of Neuropathology & Experimental Neurology. 2010;69(5):455–472. doi: 10.1097/NEN.0b013e3181da84db. [DOI] [PubMed] [Google Scholar]
- 34.Babetto E, Beirowski B, Janeckova L, Brown R, Gilley J, Thomson D, Ribchester RR, Coleman MP. Targeting NMNAT1 to Axons and Synapses Transforms Its Neuroprotective Potency In Vivo. Journal of Neuroscience. 2010;30(40):13291–13304. doi: 10.1523/JNEUROSCI.1189-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Milde S, Fox AN, Freeman MR, Coleman MP. Deletions within its subcellular targeting domain enhance the axon protective capacity of Nmnat2 in vivo. Scientific Reports. 2013;3(1):2567. doi: 10.1038/srep02567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Milde S, Gilley J, Coleman MP. Subcellular Localization Determines the Stability and Axon Protective Capacity of Axon Survival Factor Nmnat2. PLoS Biology. 2013;11(4):e1001539. doi: 10.1371/journal.pbio.1001539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sasaki Y, Vohra BPS, Baloh RH, Milbrandt J. Transgenic Mice Expressing the Nmnat1 Protein Manifest Robust Delay in Axonal Degeneration In Vivo. Journal of Neuroscience. 2009;29(20):6526–6534. doi: 10.1523/JNEUROSCI.1429-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yahata N, Yuasa S, Araki T. Nicotinamide Mononucleotide Adenylyltransferase Expression in Mitochondrial Matrix Delays Wallerian Degeneration. Journal of Neuroscience. 2009;29(19):6276–6284. doi: 10.1523/JNEUROSCI.4304-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Conforti L, Janeckova L, Wagner D, Mazzola F, Cialabrini L, Di Stefano M, Orsomando G, Magni G, Bendotti C, Smyth N, Coleman M. Reducing expression of NAD+ synthesizing enzyme NMNAT1 does not affect the rate of Wallerian degeneration: NMNAT1 gene inactivation and axon degeneration. FEBS Journal. 2011;278(15):2666–2679. doi: 10.1111/j.1742-4658.2011.08193.x. [DOI] [PubMed] [Google Scholar]
- 40.Hicks AN, Lorenzetti D, Gilley J, Lu B, Andersson K-E, Miligan C, Overbeek PA, Oppenheim R, Bishop CE. Nicotinamide Mononucleotide Adenylyltransferase 2 (Nmnat2) Regulates Axon Integrity in the Mouse Embryo. PLoS One. 2012;7(10):e47869. doi: 10.1371/journal.pone.0047869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hikosaka K, Ikutani M, Shito M, Kazuma K, Gulshan M, Nagai Y, Takatsu K, Konno K, Tobe K, Kanno H, Nakagawa T. Deficiency of Nicotinamide Mononucleotide Adenylyltransferase 3 (Nmnat3) Causes Hemolytic Anemia by Altering the Glycolytic Flow in Mature Erythrocytes. Journal of Biological Chemistry. 2014;289(21):14796–14811. doi: 10.1074/jbc.M114.554378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoopfer ED, McLaughlin T, Watts RJ, Schuldiner O, O’Leary DDM, Luo L. Wlds Protection Distinguishes Axon Degeneration following Injury from Naturally Occurring Developmental Pruning. Neuron. 2006;50(6):883–895. doi: 10.1016/j.neuron.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 43.MacDonald JM, Beach MG, Porpiglia E, Sheehan AE, Watts RJ, Freeman MR. The Drosophila Cell Corpse Engulfment Receptor Draper Mediates Glial Clearance of Severed Axons. Neuron. 2006;50(6):869–881. doi: 10.1016/j.neuron.2006.04.028. [DOI] [PubMed] [Google Scholar]
- 44.Gerdts J, Summers DW, Sasaki Y, DiAntonio A, Milbrandt J. Sarm1-Mediated Axon Degeneration Requires Both SAM and TIR Interactions. Journal of Neuroscience. 2013;33(33):13569–13580. doi: 10.1523/JNEUROSCI.1197-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gerdts J, Brace EJ, Sasaki Y, DiAntonio A, Milbrandt J. SARM1 activation triggers axon degeneration locally via NAD+ destruction. Science. 2015;348(6233):453–457. doi: 10.1126/science.1258366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Angeletti C, Amici A, Gilley J, Loreto A, Trapanotto AG, Antoniou C, Coleman MP, Orsomando G. Programmed axon death executor SARM1 is a multi-functional NAD(P)ase with prominent base exchange activity, all regulated by physiological levels of NMN, NAD, NADP and other metabolites [Preprint] Biochemistry. 2021 doi: 10.1101/2021.07.14.451805. [DOI] [Google Scholar]
- 47.Zhao ZY, Xie XJ, Li WH, Liu J, Chen Z, Zhang B, Li T, Li SL, Lu JG, Zhang L, Zhang L, Xu Z, Lee HC, Zhao YJ. A Cell-Permeant Mimetic of NMN Activates SARM1 to Produce Cyclic ADP-Ribose and Induce Non-apoptotic Cell Death. IScience. 2019;15:452–466. doi: 10.1016/j.isci.2019.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gilley J, Ribchester RR, Coleman MP. Sarm1 Deletion, but Not Wld S, Confers Lifelong Rescue in a Mouse Model of Severe Axonopathy. Cell Reports. 2017;21(1):10–16. doi: 10.1016/j.celrep.2017.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gilley J, Orsomando G, Nascimento-Ferreira I, Coleman MP. Absence of SARM1 Rescues Development and Survival of NMNAT2-Deficient Axons. Cell Reports. 2015;10(12):1974–1981. doi: 10.1016/j.celrep.2015.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loreto A, Angeletti C, Gilley J, Arthur-Farraj P, Merlini E, Amici A, Desrochers LM, Wang Q, Orsomando G, Coleman MP. Potent activation of SARM1 by NMN analogue VMN underlies vacor neurotoxicity [Preprint] Neuroscience. 2020 doi: 10.1101/2020.09.18.304261. [DOI] [Google Scholar]
- 51.Loreto A, Hill CS, Hewitt VL, Orsomando G, Angeletti C, Gilley J, Lucci C, Sanchez-Martinez A, Whitworth AJ, Conforti L, Dajas-Bailador F, Coleman MP. Mitochondrial impairment activates the Wallerian pathway through depletion of NMNAT2 leading to SARM1-dependent axon degeneration. Neurobiology of Disease. 2020;134:104678. doi: 10.1016/j.nbd.2019.104678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Di Stefano M, Nascimento-Ferreira I, Orsomando G, Mori V, Gilley J, Brown R, Janeckova L, Vargas ME, Worrell LA, Loreto A, Tickle J, Patrick J, Webster JRM, Marangoni M, Carpi FM, Pucciarelli S, Rossi F, Meng W, Sagasti A, Conforti L. A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration. Cell Death & Differentiation. 2015;22(5):731–742. doi: 10.1038/cdd.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Di Stefano M, Loreto A, Orsomando G, Mori V, Zamporlini F, Hulse RP, Webster J, Donaldson LF, Gering M, Raffaelli N, Coleman MP, Gilley J, Conforti L. NMN Deamidase Delays Wallerian Degeneration and Rescues Axonal Defects Caused by NMNAT2 Deficiency In Vivo. Current Biology. 2017;27(6):784–794. doi: 10.1016/j.cub.2017.01.070. [DOI] [PubMed] [Google Scholar]
- 54.Sasaki Y, Nakagawa T, Mao X, DiAntonio A, Milbrandt J. NMNAT1 inhibits axon degeneration via blockade of SARM1-mediated NAD+ depletion. ELife. 2016;5:e19749. doi: 10.7554/eLife.19749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Figley MD, Gu W, Nanson JD, Shi Y, Sasaki Y, Cunnea K, Malde AK, Jia X, Luo Z, Saikot FK, Mosaiab T, Masic V, Holt S, Hartley-Tassell L, McGuinness HY, Manik MK, Bosanac T, Landsberg MJ, Kerry PS, Ve T. SARM1 is a metabolic sensor activated by an increased NMN/NAD+ ratio to trigger axon degeneration. Neuron. 2021;109:1118–1136. doi: 10.1016/j.neuron.2021.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.LeWitt PA. Neurotoxicity of the rat poison vacor. New England Journal of Medicine. 1980;302:73–77. doi: 10.1056/NEJM198001103020202. [DOI] [PubMed] [Google Scholar]
- 57.Jiang Y, Liu T, Lee C-H, Chang Q, Yang J, Zhang Z. The NAD+-mediated self-inhibition mechanism of pro-neurodegenerative Sarm1. Nature. 2020 doi: 10.1038/s41586-020-2862-z. [DOI] [PubMed] [Google Scholar]
- 58.Summers DW, Milbrandt J, DiAntonio A. Palmitoylation enables MAPK-dependent proteostasis of axon survival factors. Proceedings of the National Academy of Sciences. 2018;115(37):E8746–E8754. doi: 10.1073/pnas.1806933115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yamagishi Y, Tessier-Lavigne M. An Atypical SCF-like Ubiquitin Ligase Complex Promotes Wallerian Degeneration through Regulation of Axonal Nmnat2. Cell Reports. 2016;17(3):774–782. doi: 10.1016/j.celrep.2016.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shin JE, Miller BR, Babetto E, Cho Y, Sasaki Y, Qayum S, Russler EV, Cavalli V, Milbrandt J, DiAntonio A. SCG10 is a JNK target in the axonal degeneration pathway. Proceedings of the National Academy of Sciences. 2012;109(52):E3696–E3705. doi: 10.1073/pnas.1216204109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Loreto A, Di Stefano M, Gering M, Conforti L. Wallerian Degeneration Is Executed by an NMN-SARM1-Dependent Late Ca 2+ Influx but Only Modestly Influenced by Mitochondria. Cell Reports. 2015;13(11):2539–2552. doi: 10.1016/j.celrep.2015.11.032. [DOI] [PubMed] [Google Scholar]
- 62.Yang J, Weimer RM, Kallop D, Olsen O, Wu Z, Renier N, Uryu K, Tessier-Lavigne M. Regulation of Axon Degeneration after Injury and in Development by the Endogenous Calpain Inhibitor Calpastatin. Neuron. 2013;80(5):1175–1189. doi: 10.1016/j.neuron.2013.08.034. [DOI] [PubMed] [Google Scholar]
- 63.Neukomm LJ, Burdett TC, Seeds AM, Hampel S, Coutinho-Budd JC, Farley JE, Wong J, Karadeniz YB, Osterloh JM, Sheehan AE, Freeman MR. Axon Death Pathways Converge on Axundead to Promote Functional and Structural Axon Disassembly. Neuron. 2017;95(1):78–91.e5. doi: 10.1016/j.neuron.2017.06.031. [DOI] [PubMed] [Google Scholar]
- 64.Arthur-Farraj PJ, Morgan CC, Adamowicz M, Gomez-Sanchez JA, Fazal SV, Beucher A, Razzaghi B, Mirsky R, Jessen KR, Aitman TJ. Changes in the Coding and Non-coding Transcriptome and DNA Methylome that Define the Schwann Cell Repair Phenotype after Nerve Injury. Cell Reports. 2017;20(11):2719–2734. doi: 10.1016/j.celrep.2017.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Babetto E, Wong KM, Beirowski B. A glycolytic shift in Schwann cells supports injured axons. Nature Neuroscience. 2020;23(10):1215–1228. doi: 10.1038/s41593-020-0689-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Catenaccio A, Llavero Hurtado M, Diaz P, Lamont DJ, Wishart TM, Court FA. Molecular analysis of axonal-intrinsic and glial-associated co-regulation of axon degeneration. Cell Death & Disease. 2017;8(11):e3166–e3166. doi: 10.1038/cddis.2017.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rosenberg AF, Isaacman-Beck J, Franzini-Armstrong C, Granato M. Schwann Cells and Deleted in Colorectal Carcinoma Direct Regenerating Motor Axons Towards Their Original Path. The Journal of Neuroscience. 2014;34(44):14668–14681. doi: 10.1523/JNEUROSCI.2007-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rosenberg AF, Wolman MA, Franzini-Armstrong C, Granato M. In vivo nerve-macrophage interactions following peripheral nerve injury. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2012;32(11):3898–3909. doi: 10.1523/JNEUROSCI.5225-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vaquié A, Sauvain A, Duman M, Nocera G, Egger B, Meyenhofer F, Falquet L, Bartesaghi L, Chrast R, Lamy CM, Bang S, Lee S-R, Jeon NL, Ruff S, Jacob C. Injured Axons Instruct Schwann Cells to Build Constricting Actin Spheres to Accelerate Axonal Disintegration. Cell Reports. 2019;27(11):3152–3166.e7. doi: 10.1016/j.celrep.2019.05.060. [DOI] [PubMed] [Google Scholar]
- 70.Villegas R, Martin SM, O’Donnell KC, Carrillo SA, Sagasti A, Allende ML. Dynamics of degeneration and regeneration in developing zebrafish peripheral axons reveals a requirement for extrinsic cell types. Neural Development. 2012;7:19. doi: 10.1186/1749-8104-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chaballe L, Close P, Sempels M, Delstanche S, Fanielle J, Moons L, Carmeliet P, Schoenen J, Chariot A, Franzen R. Involvement of placental growth factor in Wallerian degeneration. Glia. 2011;59(3):379–396. doi: 10.1002/glia.21108. [DOI] [PubMed] [Google Scholar]
- 72.Reed CB, Frick LR, Weaver A, Sidoli M, Schlant E, Feltri ML, Wrabetz L. Deletion of Calcineurin in Schwann Cells Does Not Affect Developmental Myelination, But Reduces Autophagy and Delays Myelin Clearance after Peripheral Nerve Injury. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2020;40(32):6165–6176. doi: 10.1523/JNEUROSCI.0951-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jessen KR, Mirsky R, Arthur-Farraj P. The Role of Cell Plasticity in Tissue Repair: Adaptive Cellular Reprogramming. Developmental Cell. 2015;34(6):613–620. doi: 10.1016/j.devcel.2015.09.005. [DOI] [PubMed] [Google Scholar]
- 74.Brosius Lutz A, Chung W-S, Sloan SA, Carson GA, Zhou L, Lovelett E, Posada S, Zuchero JB, Barres BA. Schwann cells use TAM receptor-mediated phagocytosis in addition to autophagy to clear myelin in a mouse model of nerve injury. Proceedings of the National Academy of Sciences of the United States of America. 2017;114(38):E8072–E8080. doi: 10.1073/pnas.1710566114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jang SY, Shin YK, Park SY, Park JY, Lee HJ, Yoo YH, Kim JK, Park HT. Autophagic myelin destruction by Schwann cells during Wallerian degeneration and segmental demyelination. Glia. 2016;64(5):730–742. doi: 10.1002/glia.22957. [DOI] [PubMed] [Google Scholar]
- 76.Ying Z, Pan C, Shao T, Liu L, Li L, Guo D, Zhang S, Yuan T, Cao R, Jiang Z, Chen S, Wang F, Wang X. Mixed Lineage Kinase Domain-like Protein MLKL Breaks Down Myelin following Nerve Injury. Molecular Cell. 2018;72(3):457–468.e5. doi: 10.1016/j.molcel.2018.09.011. [DOI] [PubMed] [Google Scholar]
- 77.Nguyen QT, Sanes JR, Lichtman JW. Pre-existing pathways promote precise projection patterns. Nature Neuroscience. 2002;5(9):861–867. doi: 10.1038/nn905. [DOI] [PubMed] [Google Scholar]
- 78.Min Q, Parkinson DB, Dun X-P. Migrating Schwann cells direct axon regeneration within the peripheral nerve bridge. Glia. 2021;69(2):235–254. doi: 10.1002/glia.23892. [DOI] [PubMed] [Google Scholar]
- 79.Jessen KR, Mirsky R. The repair Schwann cell and its function in regenerating nerves. The Journal of Physiology. 2016;594(13):3521–3531. doi: 10.1113/JP270874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rotshenker S. Wallerian degeneration: The innate-immune response to traumatic nerve injury. Journal of Neuroinflammation. 2011;8(1):109. doi: 10.1186/1742-2094-8-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Alanne MH, Pummi K, Heape AM, Grènman R, Peltonen J, Peltonen S. Tight Junction Proteins in Human Schwann Cell Autotypic Junctions. Journal of Histochemistry and Cytochemistry. 2009;57(6):523–529. doi: 10.1369/jhc.2009.951681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bunge RP, Bunge MB. Interrelationship between Schwann cell function and extracellular matrix production. Trends in Neurosciences. 1983;6:499–505. doi: 10.1016/0166-2236(83)90235-7. [DOI] [Google Scholar]
- 83.Chan, J. R., Jolicoeur, C., Yamauchi, J., Elliott, J., Fawcett, J. P., Ng, B. K., & Cayouette, M. (2006). The polarity protein Par-3 directly interacts with p75NTR to regulate myelination. Science (New York, N.Y.), 314(5800), 832–836. 10.1126/science.1134069 [DOI] [PubMed]
- 84.Crawford AT, Desai D, Gokina P, Basak S, Kim HA. E-cadherin expression in postnatal Schwann cells is regulated by the cAMP-dependent protein kinase a pathway. Glia. 2008;56(15):1637–1647. doi: 10.1002/glia.20716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Miyamoto T, Morita K, Takemoto D, Takeuchi K, Kitano Y, Miyakawa T, Nakayama K, Okamura Y, Sasaki H, Miyachi Y, Furuse M, Tsukita S. Tight junctions in Schwann cells of peripheral myelinated axons. The Journal of Cell Biology. 2005;169(3):527–538. doi: 10.1083/jcb.200501154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Clements MP, Byrne E, Camarillo Guerrero LF, Cattin A-L, Zakka L, Ashraf A, Burden JJ, Khadayate S, Lloyd AC, Marguerat S, Parrinello S. The Wound Microenvironment Reprograms Schwann Cells to Invasive Mesenchymal-like Cells to Drive Peripheral Nerve Regeneration. Neuron. 2017;96(1):98–114.e7. doi: 10.1016/j.neuron.2017.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fontana X, Hristova M, Da Costa C, Patodia S, Thei L, Makwana M, Spencer-Dene B, Latouche M, Mirsky R, Jessen KR, Klein R, Raivich G, Behrens A. C-Jun in Schwann cells promotes axonal regeneration and motoneuron survival via paracrine signaling. The Journal of Cell Biology. 2012;198(1):127–141. doi: 10.1083/jcb.201205025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Parkinson DB, Bhaskaran A, Arthur-Farraj P, Noon LA, Woodhoo A, Lloyd AC, Feltri ML, Wrabetz L, Behrens A, Mirsky R, Jessen KR. C-Jun is a negative regulator of myelination. Journal of Cell Biology. 2008;181(4):625–637. doi: 10.1083/jcb.200803013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fazal SV, Gomez-Sanchez JA, Wagstaff LJ, Musner N, Otto G, Janz M, Mirsky R, Jessen KR. Graded Elevation of c-Jun in Schwann Cells In Vivo: Gene Dosage Determines Effects on Development, Remyelination, Tumorigenesis, and Hypomyelination. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2017;37(50):12297–12313. doi: 10.1523/JNEUROSCI.0986-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kim S, Maynard JC, Strickland A, Burlingame AL, Milbrandt J. Schwann cell O-GlcNAcylation promotes peripheral nerve remyelination via attenuation of the AP-1 transcription factor JUN. Proceedings of the National Academy of Sciences. 2018;115(31):8019–8024. doi: 10.1073/pnas.1805538115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brügger V, Duman M, Bochud M, Münger E, Heller M, Ruff S, Jacob C. Delaying histone deacetylase response to injury accelerates conversion into repair Schwann cells and nerve regeneration. Nature Communications. 2017;8(1):14272. doi: 10.1038/ncomms14272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Benito C, Davis CM, Gomez-Sanchez JA, Turmaine M, Meijer D, Poli V, Mirsky R, Jessen KR. STAT3 Controls the Long-Term Survival and Phenotype of Repair Schwann Cells during Nerve Regeneration. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2017;37(16):4255–4269. doi: 10.1523/JNEUROSCI.3481-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Grove M, Lee H, Zhao H, Son Y-J. Axon-dependent expression of YAP/TAZ mediates Schwann cell remyelination but not proliferation after nerve injury. ELife. 2020;9:e50138. doi: 10.7554/eLife.50138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jeanette H, Marziali LN, Bhatia U, Hellman A, Herron J, Kopec AM, Feltri ML, Poitelon Y, Belin S. YAP and TAZ regulate Schwann cell proliferation and differentiation during peripheral nerve regeneration. Glia. 2021;69(4):1061–1074. doi: 10.1002/glia.23949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Quintes S, Brinkmann BG, Ebert M, Fröb F, Kungl T, Arlt FA, Tarabykin V, Huylebroeck D, Meijer D, Suter U, Wegner M, Sereda MW, Nave K-A. Zeb2 is essential for Schwann cell differentiation, myelination and nerve repair. Nature Neuroscience. 2016;19(8):1050–1059. doi: 10.1038/nn.4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wu LMN, Wang J, Conidi A, Zhao C, Wang H, Ford Z, Zhang L, Zweier C, Ayee BG, Maurel P, Zwijsen A, Chan JR, Jankowski MP, Huylebroeck D, Lu QR. Zeb2 recruits HDAC–NuRD to inhibit Notch and controls Schwann cell differentiation and remyelination. Nature Neuroscience. 2016;19(8):1060–1072. doi: 10.1038/nn.4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mindos T, Dun X-P, North K, Doddrell RDS, Schulz A, Edwards P, Russell J, Gray B, Roberts SL, Shivane A, Mortimer G, Pirie M, Zhang N, Pan D, Morrison H, Parkinson DB. Merlin controls the repair capacity of Schwann cells after injury by regulating Hippo/YAP activity. The Journal of Cell Biology. 2017;216(2):495–510. doi: 10.1083/jcb.201606052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ma KH, Svaren J. Epigenetic Control of Schwann Cells. The Neuroscientist: A Review Journal Bringing Neurobiology, Neurology and Psychiatry. 2018;24(6):627–638. doi: 10.1177/1073858417751112. [DOI] [PubMed] [Google Scholar]
- 99.Duong, P., Ma, K. H., Ramesh, R., Moran, J. J., Won, S., Svaren, J. (2021) H3K27 demethylases are dispensable for activation of Polycomb-regulated injury response genes in peripheral nerve. The Journal of Biological Chemistry, 100852. 10.1016/j.jbc.2021.100852 [DOI] [PMC free article] [PubMed]
- 100.Hung HA, Sun G, Keles S, Svaren J. Dynamic regulation of Schwann cell enhancers after peripheral nerve injury. The Journal of Biological Chemistry. 2015;290(11):6937–6950. doi: 10.1074/jbc.M114.622878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ma KH, Hung HA, Svaren J. Epigenomic Regulation of Schwann Cell Reprogramming in Peripheral Nerve Injury. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2016;36(35):9135–9147. doi: 10.1523/JNEUROSCI.1370-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ma KH, Duong P, Moran JJ, Junaidi N, Svaren J. Polycomb repression regulates Schwann cell proliferation and axon regeneration after nerve injury. Glia. 2018;66(11):2487–2502. doi: 10.1002/glia.23500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.He X, Zhang L, Queme LF, Liu X, Lu A, Waclaw RR, Dong X, Zhou W, Kidd G, Yoon S-O, Buonanno A, Rubin JB, Xin M, Nave K-A, Trapp BD, Jankowski MP, Lu QR. A histone deacetylase 3–dependent pathway delimits peripheral myelin growth and functional regeneration. Nature Medicine. 2018;24(3):338–351. doi: 10.1038/nm.4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Duman M, Vaquié A, Nocera G, Heller M, Stumpe M, Siva Sankar D, Dengjel J, Meijer D, Yamaguchi T, Matthias P, Zeis T, Schaeren-Wiemers N, Hayoz A, Ruff S, Jacob C. EEF1A1 deacetylation enables transcriptional activation of remyelination. Nature Communications. 2020;11(1):3420. doi: 10.1038/s41467-020-17243-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gomis-Coloma C, Velasco-Aviles S, Gomez-Sanchez JA, Casillas-Bajo A, Backs J, Cabedo H. Class IIa histone deacetylases link cAMP signaling to the myelin transcriptional program of Schwann cells. The Journal of Cell Biology. 2018;217(4):1249–1268. doi: 10.1083/jcb.201611150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lin H-P, Oksuz I, Hurley E, Wrabetz L, Awatramani R. Microprocessor complex subunit DiGeorge syndrome critical region gene 8 (Dgcr8) is required for schwann cell myelination and myelin maintenance. The Journal of Biological Chemistry. 2015;290(40):24294–24307. doi: 10.1074/jbc.M115.636407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Viader A, Chang L-W, Fahrner T, Nagarajan R, Milbrandt J. MicroRNAs modulate Schwann cell response to nerve injury by reinforcing transcriptional silencing of dedifferentiation-related genes. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2011;31(48):17358–17369. doi: 10.1523/JNEUROSCI.3931-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Arthur-Farraj P, Moyon S. DNA methylation in Schwann cells and in oligodendrocytes. Glia. 2020;68(8):1568–1583. doi: 10.1002/glia.23784. [DOI] [PubMed] [Google Scholar]
- 109.Norrmén C, Figlia G, Pfistner P, Pereira JA, Bachofner S, Suter U. MTORC1 Is Transiently Reactivated in Injured Nerves to Promote c-Jun Elevation and Schwann Cell Dedifferentiation. Journal of Neuroscience. 2018;38(20):4811–4828. doi: 10.1523/JNEUROSCI.3619-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Woodhoo A, Alonso MBD, Droggiti A, Turmaine M, D’Antonio M, Parkinson DB, Wilton DK, Al-Shawi R, Simons P, Shen J, Guillemot F, Radtke F, Meijer D, Feltri ML, Wrabetz L, Mirsky R, Jessen KR. Notch controls embryonic Schwann cell differentiation, postnatal myelination and adult plasticity. Nature Neuroscience. 2009;12(7):839–847. doi: 10.1038/nn.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Boerboom A, Reusch C, Pieltain A, Chariot A, Franzen R. KIAA1199: A novel regulator of MEK/ERK-induced Schwann cell dedifferentiation. Glia. 2017;65(10):1682–1696. doi: 10.1002/glia.23188. [DOI] [PubMed] [Google Scholar]
- 112.Cervellini I, Galino J, Zhu N, Allen S, Birchmeier C, Bennett DL. Sustained MAPK/ERK Activation in Adult Schwann Cells Impairs Nerve Repair. The Journal of Neuroscience. 2018;38(3):679–690. doi: 10.1523/JNEUROSCI.2255-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ishii A, Furusho M, Bansal R. Sustained Activation of ERK1/2 MAPK in Oligodendrocytes and Schwann Cells Enhances Myelin Growth and Stimulates Oligodendrocyte Progenitor Expansion. The Journal of Neuroscience. 2013;33(1):175–186. doi: 10.1523/JNEUROSCI.4403-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Napoli I, Noon LA, Ribeiro S, Kerai AP, Parrinello S, Rosenberg LH, Collins MJ, Harrisingh MC, White IJ, Woodhoo A, Lloyd AC. A central role for the ERK-signaling pathway in controlling Schwann cell plasticity and peripheral nerve regeneration in vivo. Neuron. 2012;73(4):729–742. doi: 10.1016/j.neuron.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 115.Yang DP, Kim J, Syed N, Tung Y-J, Bhaskaran A, Mindos T, Mirsky R, Jessen KR, Maurel P, Parkinson DB, Kim HA. P38 MAPK activation promotes denervated Schwann cell phenotype and functions as a negative regulator of Schwann cell differentiation and myelination. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2012;32(21):7158–7168. doi: 10.1523/JNEUROSCI.5812-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Roberts SL, Dun X-P, Dee G, Gray B, Mindos T, Parkinson DB. The role of p38alpha in Schwann cells in regulating peripheral nerve myelination and repair. Journal of Neurochemistry. 2017;141(1):37–47. doi: 10.1111/jnc.13929. [DOI] [PubMed] [Google Scholar]
- 117.Galino J, Cervellini I, Zhu N, Stöberl N, Hütte M, Fricker FR, Lee G, McDermott L, Lalli G, Bennett DLH. RalGTPases contribute to Schwann cell repair after nerve injury via regulation of process formation. The Journal of Cell Biology. 2019;218(7):2370–2387. doi: 10.1083/jcb.201811002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Birchmeier C, Bennett DLH. Neuregulin/ErbB Signaling in Developmental Myelin Formation and Nerve Repair. Current Topics in Developmental Biology. 2016;116:45–64. doi: 10.1016/bs.ctdb.2015.11.009. [DOI] [PubMed] [Google Scholar]
- 119.Carroll SL, Miller ML, Frohnert PW, Kim SS, Corbett JA. Expression of neuregulins and their putative receptors, ErbB2 and ErbB3, is induced during Wallerian degeneration. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 1997;17(5):1642–1659. doi: 10.1523/JNEUROSCI.17-05-01642.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Stassart RM, Fledrich R, Velanac V, Brinkmann BG, Schwab MH, Meijer D, Sereda MW, Nave K-A. A role for Schwann cell-derived neuregulin-1 in remyelination. Nature Neuroscience. 2013;16(1):48–54. doi: 10.1038/nn.3281. [DOI] [PubMed] [Google Scholar]
- 121.Atanasoski S, Scherer SS, Sirkowski E, Leone D, Garratt AN, Birchmeier C, Suter U. ErbB2 signaling in Schwann cells is mostly dispensable for maintenance of myelinated peripheral nerves and proliferation of adult Schwann cells after injury. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2006;26(7):2124–2131. doi: 10.1523/JNEUROSCI.4594-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fricker, F. R., Antunes-Martins, A., Galino, J., Paramsothy, R., La Russa, F., Perkins, J., Goldberg, R., Brelstaff, J., Zhu, N., McMahon, S. B., Orengo, C., Garratt, A. N., Birchmeier, C., & Bennett, D. L. H. (2013). Axonal neuregulin 1 is a rate limiting but not essential factor for nerve remyelination. Brain: A Journal of Neurology, 136(Pt 7), 2279–2297. 10.1093/brain/awt148 [DOI] [PMC free article] [PubMed]
- 123.Fricker FR, Lago N, Balarajah S, Tsantoulas C, Tanna S, Zhu N, Fageiry SK, Jenkins M, Garratt AN, Birchmeier C, Bennett DLH. Axonally derived neuregulin-1 is required for remyelination and regeneration after nerve injury in adulthood. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2011;31(9):3225–3233. doi: 10.1523/JNEUROSCI.2568-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Han SB, Kim H, Lee H, Grove M, Smith GM, Son Y-J. Postinjury Induction of Activated ErbB2 Selectively Hyperactivates Denervated Schwann Cells and Promotes Robust Dorsal Root Axon Regeneration. Journal of Neuroscience. 2017;37(45):10955–10970. doi: 10.1523/JNEUROSCI.0903-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hu X, He W, Diaconu C, Tang X, Kidd GJ, Macklin WB, Trapp BD, Yan R. Genetic deletion of BACE1 in mice affects remyelination of sciatic nerves. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 2008;22(8):2970–2980. doi: 10.1096/fj.08-106666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Liang C, Tao Y, Shen C, Tan Z, Xiong W-C, Mei L. Erbin is required for myelination in regenerated axons after injury. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2012;32(43):15169–15180. doi: 10.1523/JNEUROSCI.2466-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jablonka-Shariff A, Lu C-Y, Campbell K, Monk KR, Snyder-Warwick AK. Gpr126/Adgrg6 contributes to the terminal Schwann cell response at the neuromuscular junction following peripheral nerve injury. Glia. 2020;68(6):1182–1200. doi: 10.1002/glia.23769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mogha A, Harty BL, Carlin D, Joseph J, Sanchez NE, Suter U, Piao X, Cavalli V, Monk KR. Gpr126/Adgrg6 Has Schwann Cell Autonomous and Nonautonomous Functions in Peripheral Nerve Injury and Repair. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2016;36(49):12351–12367. doi: 10.1523/JNEUROSCI.3854-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Monk, K. R., Naylor, S. G., Glenn, T. D., Mercurio, S., Perlin, J. R., Dominguez, C., Moens, C. B., & Talbot, W. S. (2009). A G Protein-Coupled Receptor is Essential for Schwann Cells to Initiate Myelination. Science (New York, N.Y.), 325(5946), 1402–1405. 10.1126/science.1173474 [DOI] [PMC free article] [PubMed]
- 130.Boyd JG, Gordon T. Neurotrophic factors and their receptors in axonal regeneration and functional recovery after peripheral nerve injury. Molecular Neurobiology. 2003;27(3):277–324. doi: 10.1385/MN:27:3:277. [DOI] [PubMed] [Google Scholar]
- 131.Gonzalez-Perez, F., Udina, E., & Navarro, X. (2013). Chapter Ten—Extracellular Matrix Components in Peripheral Nerve Regeneration. In S. Geuna, I. Perroteau, P. Tos, & B. Battiston (Eds.), International Review of Neurobiology (Vol. 108, pp. 257–275). Academic Press. 10.1016/B978-0-12-410499-0.00010-1 [DOI] [PubMed]
- 132.Nieuwenhuis B, Haenzi B, Andrews MR, Verhaagen J, Fawcett JW. Integrins promote axonal regeneration after injury of the nervous system. Biological Reviews. 2018;93(3):1339–1362. doi: 10.1111/brv.12398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chen Z-L, Strickland S. Laminin gamma1 is critical for Schwann cell differentiation, axon myelination, and regeneration in the peripheral nerve. The Journal of Cell Biology. 2003;163(4):889–899. doi: 10.1083/jcb.200307068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Guntinas-Lichius O, Angelov DN, Morellini F, Lenzen M, Skouras E, Schachner M, Irintchev A. Opposite impacts of tenascin-C and tenascin-R deficiency in mice on the functional outcome of facial nerve repair. The European Journal of Neuroscience. 2005;22(9):2171–2179. doi: 10.1111/j.1460-9568.2005.04424.x. [DOI] [PubMed] [Google Scholar]
- 135.Sakimoto T, Kim T, Ellenberg D, Fukai N, Jain S, Azar DT, Chang J-H. Collagen XVIII and corneal reinnervation following keratectomy. FEBS Letters. 2008;582(25):3674–3680. doi: 10.1016/j.febslet.2008.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Isaacman-Beck J, Schneider V, Franzini-Armstrong C, Granato M. The lh3 Glycosyltransferase Directs Target-Selective Peripheral Nerve Regeneration. Neuron. 2015;88(4):691–703. doi: 10.1016/j.neuron.2015.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gordon T. The role of neurotrophic factors in nerve regeneration. Neurosurgical Focus. 2009;26(2):E3. doi: 10.3171/FOC.2009.26.2.E3. [DOI] [PubMed] [Google Scholar]
- 138.Funakoshi H, Risling M, Carlstedt T, Lendahl U, Timmusk T, Metsis M, Yamamoto Y, Ibáñez CF. Targeted expression of a multifunctional chimeric neurotrophin in the lesioned sciatic nerve accelerates regeneration of sensory and motor axons. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(9):5269–5274. doi: 10.1073/pnas.95.9.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Diamond J, Foerster A, Holmes M, Coughlin M. Sensory nerves in adult rats regenerate and restore sensory function to the skin independently of endogenous NGF. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 1992;12(4):1467–1476. doi: 10.1523/JNEUROSCI.12-04-01467.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Diamond J, Holmes M, Coughlin M. Endogenous NGF and nerve impulses regulate the collateral sprouting of sensory axons in the skin of the adult rat. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 1992;12(4):1454–1466. doi: 10.1523/JNEUROSCI.12-04-01454.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rich KM, Yip HK, Osborne PA, Schmidt RE, Johnson EM. Role of nerve growth factor in the adult dorsal root ganglia neuron and its response to injury. The Journal of Comparative Neurology. 1984;230(1):110–118. doi: 10.1002/cne.902300110. [DOI] [PubMed] [Google Scholar]
- 142.English AW, Meador W, Carrasco DI. Neurotrophin-4/5 is required for the early growth of regenerating axons in peripheral nerves. European Journal of Neuroscience. 2005;21(10):2624–2634. doi: 10.1111/j.1460-9568.2005.04124.x. [DOI] [PubMed] [Google Scholar]
- 143.Zhang JY, Luo XG, Xian CJ, Liu ZH, Zhou XF. Endogenous BDNF is required for myelination and regeneration of injured sciatic nerve in rodents. The European Journal of Neuroscience. 2000;12(12):4171–4180. [PubMed] [Google Scholar]
- 144.Bennett DLH, Boucher TJ, Michael GJ, Popat RJ, Malcangio M, Averill SA, Poulsen KT, Priestley JV, Shelton DL, McMahon SB. Artemin has potent neurotrophic actions on injured C-fibres. Journal of the Peripheral Nervous System: JPNS. 2006;11(4):330–345. doi: 10.1111/j.1529-8027.2006.00106.x. [DOI] [PubMed] [Google Scholar]
- 145.Bennett DL, Michael GJ, Ramachandran N, Munson JB, Averill S, Yan Q, McMahon SB, Priestley JV. A distinct subgroup of small DRG cells express GDNF receptor components and GDNF is protective for these neurons after nerve injury. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 1998;18(8):3059–3072. doi: 10.1523/JNEUROSCI.18-08-03059.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Eggers, R., de Winter, F., Hoyng, S. A., Hoeben, R. C., Malessy, M. J. A., Tannemaat, M. R., & Verhaagen, J. (2019). Timed GDNF gene therapy using an immune-evasive gene switch promotes long distance axon regeneration. Brain: A Journal of Neurology, 142(2), 295–311. 10.1093/brain/awy340 [DOI] [PubMed]
- 147.Sendtner M, Götz R, Holtmann B, Thoenen H. Endogenous ciliary neurotrophic factor is a lesion factor for axotomized motoneurons in adult mice. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 1997;17(18):6999–7006. doi: 10.1523/JNEUROSCI.17-18-06999.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Martinez JA, Kobayashi M, Krishnan A, Webber C, Christie K, Guo G, Singh V, Zochodne DW. Intrinsic facilitation of adult peripheral nerve regeneration by the Sonic hedgehog morphogen. Experimental Neurology. 2015;271:493–505. doi: 10.1016/j.expneurol.2015.07.018. [DOI] [PubMed] [Google Scholar]
- 149.Mi R, Chen W, Höke A. Pleiotrophin is a neurotrophic factor for spinal motor neurons. Proceedings of the National Academy of Sciences. 2007;104(11):4664–4669. doi: 10.1073/pnas.0603243104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Inherited Neuropathy Consortium. Bis-Brewer DM, Gan-Or Z, Sleiman P, Hakonarson H, Fazal S, Courel S, Cintra V, Tao F, Estiar MA, Tarnopolsky M, Boycott KM, Yoon G, Suchowersky O, Dupré N, Cheng A, Lloyd TE, Rouleau G, Schüle R, Züchner S. Assessing non-Mendelian inheritance in inherited axonopathies. Genetics in Medicine. 2020;22(12):2114–2119. doi: 10.1038/s41436-020-0924-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Argyriou, A., Kyritsis, A., Makatsoris, T., & Kalofonos, H. (2014). Chemotherapy-induced peripheral neuropathy in adults: A comprehensive update of the literature. Cancer Management and Research, 135. 10.2147/CMAR.S44261 [DOI] [PMC free article] [PubMed]
- 152.Cashman CR, Höke A. Mechanisms of distal axonal degeneration in peripheral neuropathies. Neuroscience Letters. 2015;596:33–50. doi: 10.1016/j.neulet.2015.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Conforti L, Gilley J, Coleman MP. Wallerian degeneration: An emerging axon death pathway linking injury and disease. Nature Reviews Neuroscience. 2014;15(6):394–409. doi: 10.1038/nrn3680. [DOI] [PubMed] [Google Scholar]
- 154.Bouldin, T.W. & Cavanagh, J.B. (1979). Organophosphorous Neuropathy: I.A Teased-Fiber Study of the Spatio-Temporal Spread of Axonal Degeneration. American Journal of Pathology, 94(2), 241–252. [PMC free article] [PubMed]
- 155.Wang MS, Davis AA, Culver DG, Glass JD. Wlds mice are resistant to paclitaxel (taxol) neuropathy. Annals of Neurology. 2002;52(4):442–447. doi: 10.1002/ana.10300. [DOI] [PubMed] [Google Scholar]
- 156.Wang M-S, Wu Y, Culver DG, Glass JD. The Gene for Slow Wallerian Degeneration (Wlds) Is Also Protective against Vincristine Neuropathy. Neurobiology of Disease. 2001;8(1):155–161. doi: 10.1006/nbdi.2000.0334. [DOI] [PubMed] [Google Scholar]
- 157.Geisler S, Doan RA, Strickland A, Huang X, Milbrandt J, DiAntonio A. Prevention of vincristine-induced peripheral neuropathy by genetic deletion of SARM1 in mice. Brain. 2016;139(12):3092–3108. doi: 10.1093/brain/aww251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Turkiew E, Falconer D, Reed N, Höke A. Deletion of Sarm1 gene is neuroprotective in two models of peripheral neuropathy: Deletion of Sarm1 gene is neuroprotective in two models of peripheral neuropathy. Journal of the Peripheral Nervous System. 2017;22(3):162–171. doi: 10.1111/jns.12219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Gilley J, Mayer PR, Yu G, Coleman MP. Low levels of NMNAT2 compromise axon development and survival. Human Molecular Genetics. 2019;28(3):448–458. doi: 10.1093/hmg/ddy356. [DOI] [PubMed] [Google Scholar]
- 160.Samsam M, Mi W, Wessig C, Zielasek J, Toyka KV, Coleman MP, Martini R. The Wld s Mutation Delays Robust Loss of Motor and Sensory Axons in a Genetic Model for Myelin-Related Axonopathy. The Journal of Neuroscience. 2003;23(7):2833–2839. doi: 10.1523/JNEUROSCI.23-07-02833.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ferri A, Sanes JR, Coleman MP, Cunningham JM, Kato AC. Inhibiting Axon Degeneration and Synapse Loss Attenuates Apoptosis and Disease Progression in a Mouse Model of Motoneuron Disease. Current Biology. 2003;13(8):669–673. doi: 10.1016/S0960-9822(03)00206-9. [DOI] [PubMed] [Google Scholar]
- 162.Sferra A, Baillat G, Rizza T, Barresi S, Flex E, Tasca G, D’Amico A, Bellacchio E, Ciolfi A, Caputo V, Cecchetti S, Torella A, Zanni G, Diodato D, Piermarini E, Niceta M, Coppola A, Tedeschi E, Martinelli D, Bertini E. TBCE Mutations Cause Early-Onset Progressive Encephalopathy with Distal Spinal Muscular Atrophy. The American Journal of Human Genetics. 2016;99(4):974–983. doi: 10.1016/j.ajhg.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hasbani D, Omalley K. WldS mice are protected against the Parkinsonian mimetic MPTP. Experimental Neurology. 2006;202(1):93–99. doi: 10.1016/j.expneurol.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 164.Sajadi A, Schneider BL, Aebischer P. Wlds-Mediated Protection of Dopaminergic Fibers in an Animal Model of Parkinson Disease. Current Biology. 2004;17:326–330. doi: 10.1016/j.cub.2004.01.053. [DOI] [PubMed] [Google Scholar]
- 165.Summers DW, Frey E, Walker LJ, Milbrandt J, DiAntonio A. DLK Activation Synergizes with Mitochondrial Dysfunction to Downregulate Axon Survival Factors and Promote SARM1-Dependent Axon Degeneration. Molecular Neurobiology. 2019 doi: 10.1007/s12035-019-01796-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Mi W. The slow Wallerian degeneration gene, WldS, inhibits axonal spheroid pathology in gracile axonal dystrophy mice. Brain. 2004;128(2):405–416. doi: 10.1093/brain/awh368. [DOI] [PubMed] [Google Scholar]
- 167.Beirowski B, Babetto E, Coleman MP, Martin KR. The WldS gene delays axonal but not somatic degeneration in a rat glaucoma model. European Journal of Neuroscience. 2008;28(6):1166–1179. doi: 10.1111/j.1460-9568.2008.06426.x. [DOI] [PubMed] [Google Scholar]
- 168.Howell GR, Libby RT, Jakobs TC, Smith RS, Phalan FC, Barter JW, Barbay JM, Marchant JK, Mahesh N, Porciatti V, Whitmore AV, Masland RH, John SWM. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. The Journal of Cell Biology. 2007;179(7):1523–1537. doi: 10.1083/jcb.200706181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Fischer LR, Culver DG, Davis AA, Tennant P, Wang M, Coleman M, Asress S, Adalbert R, Alexander GM, Glass JD. The WldS gene modestly prolongs survival in the SOD1G93A fALS mouse. Neurobiology of Disease. 2005;19(1–2):293–300. doi: 10.1016/j.nbd.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 170.Peters OM, Lewis EA, Osterloh JM, Weiss A, Salameh JS, Metterville J, Brown RH, Freeman MR. Loss of Sarm1 does not suppress motor neuron degeneration in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Human Molecular Genetics. 2018;27(21):3761–3771. doi: 10.1093/hmg/ddy260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kariya S, Mauricio R, Dai Y, Monani UR. The neuroprotective factor Wlds fails to mitigate distal axonal and neuromuscular junction (NMJ) defects in mouse models of spinal muscular atrophy. Neuroscience Letters. 2009;449(3):246–251. doi: 10.1016/j.neulet.2008.10.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rose FF, Meehan PW, Coady TH, Garcia VB, Garcia ML, Lorson CL. The Wallerian degeneration slow (Wld) gene does not attenuate disease in a mouse model of spinal muscular atrophy. Biochemical and Biophysical Research Communications. 2008;375(1):119–123. doi: 10.1016/j.bbrc.2008.07.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Stum M, McLaughlin HM, Kleinbrink EL, Miers KE, Ackerman SL, Seburn KL, Antonellis A, Burgess RW. An assessment of mechanisms underlying peripheral axonal degeneration caused by aminoacyl-tRNA synthetase mutations. Molecular and Cellular Neuroscience. 2011;46(2):432–443. doi: 10.1016/j.mcn.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Coleman MP, Hoke A. Programmed axon degeneration: From mouse to mechanism to medicine. Nature Reviews Neuroscience. 2020;21:183–196. doi: 10.1038/s41583-020-0269-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lukacs M, Gilley J, Zhu Y, Orsomando G, Angeletti C, Liu J, Yang X, Park J, Hopkin RJ, Coleman MP, Zhai RG, Stottmann RW. Severe biallelic loss-of-function mutations in nicotinamide mononucleotide adenylyltransferase 2 (NMNAT2) in two fetuses with fetal akinesia deformation sequence. Experimental Neurology. 2019;320:112961. doi: 10.1016/j.expneurol.2019.112961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Huppke P, Wegener E, Gilley J, Angeletti C, Kurth I, Drenth JPH, Stadelmann C, Barrantes-Freer A, Brück W, Thiele H, Nürnberg P, Gärtner J, Orsomando G, Coleman MP. Homozygous NMNAT2 mutation in sisters with polyneuropathy and erythromelalgia. Experimental Neurology. 2019;320:112958. doi: 10.1016/j.expneurol.2019.112958. [DOI] [PubMed] [Google Scholar]
- 177.Ali YO, Allen HM, Yu L, Li-Kroeger D, Bakhshizadehmahmoudi D, Hatcher A, McCabe C, Xu J, Bjorklund N, Taglialatela G, Bennett DA, De Jager PL, Shulman JM, Bellen HJ, Lu H-C. NMNAT2:HSP90 Complex Mediates Proteostasis in Proteinopathies. PLOS Biology. 2016;14(6):e1002472. doi: 10.1371/journal.pbio.1002472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Bloom AJ, Mao X, Strickland A, Sasaki Y, Milbrandt J, DiAntonio A. Constitutively active SARM1 variants found in ALS patients induce neuropathy [Preprint] Neuroscience. 2021 doi: 10.1101/2021.04.16.439886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Gilley J, Jackson O, Pipis M, Estiar MA, Gan-Or Z, Goutman SA, Harms MB, Kaye J, Lima L, Genomics QS, Ravits J, Rouleau GA, Züchner S, Reilly MM, Coleman MP. Enrichment of SARM1 alleles encoding variants with constitutively hyperactive NADase in patients with ALS and other motor nerve disorders [Preprint] Neurology. 2021 doi: 10.1101/2021.06.17.21258268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Klim JR, Williams LA, Limone F, Juan Guerra San, I., Davis-Dusenbery, B. N., Mordes, D. A., Burberry, A., Steinbaugh, M. J., Gamage, K. K., Kirchner, R., Moccia, R., Cassel, S. H., Chen, K., Wainger, B. J., Woolf, C. J., & Eggan, K. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nature Neuroscience. 2019;22(2):167–179. doi: 10.1038/s41593-018-0300-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Melamed Z, López-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, Freyermuth F, McMahon MA, Beccari MS, Artates JW, Ohkubo T, Rodriguez M, Lin N, Wu D, Bennett CF, Rigo F, Da Cruz S, Ravits J, Lagier-Tourenne C, Cleveland DW. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nature Neuroscience. 2019;22(2):180–190. doi: 10.1038/s41593-018-0293-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Fogh, I., Ratti, A., Gellera, C., Lin, K., Tiloca, C., Moskvina, V., Corrado, L., Soraru, G., Cereda, C., Corti, S., Gentilini, D., Calini, D., Castellotti, B., Mazzini, L., Querin, G., Gagliardi, S., Del Bo, R., Conforti, F. L., Siciliano, G., … Zheng, J. G. (2014). A genome-wide association meta-analysis identifies a novel locus at 17q11.2 associated with sporadic amyotrophic lateral sclerosis. Human Molecular Genetics, 23(8), 2220–2231. 10.1093/hmg/ddt587 [DOI] [PMC free article] [PubMed]
- 183.SLAGEN Consortium. van Rheenen W, van Rheenen W, Dekker AM, McLaughlin RL, Diekstra FP, Pulit SL, van der Spek RAA, Võsa U, de Jong S, Robinson MR, Yang J, Fogh I, van Doormaal PT, Tazelaar GHP, Koppers M, Blokhuis AM, Sproviero W, Jones AR, Veldink JH. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nature Genetics. 2016;48(9):1043–1048. doi: 10.1038/ng.3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Ruijs, A. C. J., Jaquet, J.-B., Kalmijn, S., Giele, H., & Hovius, S. E. R. (2005). Median and ulnar nerve injuries: A meta-analysis of predictors of motor and sensory recovery after modern microsurgical nerve repair. Plastic and Reconstructive Surgery, 116(2), 484–494; discussion 495–496. 10.1097/01.prs.0000172896.86594.07 [DOI] [PubMed]
- 185.Jessen KR, Mirsky R. The Success and Failure of the Schwann Cell Response to Nerve Injury. Frontiers in Cellular Neuroscience. 2019;13:33. doi: 10.3389/fncel.2019.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Painter MW, Brosius Lutz A, Cheng Y-C, Latremoliere A, Duong K, Miller CM, Posada S, Cobos EJ, Zhang AX, Wagers AJ, Havton LA, Barres B, Omura T, Woolf CJ. Diminished Schwann cell repair responses underlie age-associated impaired axonal regeneration. Neuron. 2014;83(2):331–343. doi: 10.1016/j.neuron.2014.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wagstaff, L. J., Gomez-Sanchez, J. A., Fazal, S. V., Otto, G. W., Kilpatrick, A. M., Michael, K., Wong, L. Y., Ma, K. H., Turmaine, M., Svaren, J., Gordon, T., Arthur-Farraj, P., Velasco-Aviles, S., Cabedo, H., Benito, C., Mirsky, R., Jessen, K. R. (2021) Failures of nerve regeneration caused by aging or chronic denervation are rescued by restoring Schwann cell c-Jun. ELife, 10. 10.7554/eLife.62232 [DOI] [PMC free article] [PubMed]
- 188.Fu SY, Gordon T. Contributing factors to poor functional recovery after delayed nerve repair: Prolonged axotomy. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 1995;15(5 Pt 2):3876–3885. doi: 10.1523/JNEUROSCI.15-05-03876.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Fu SY, Gordon T. Contributing factors to poor functional recovery after delayed nerve repair: Prolonged denervation. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 1995;15(5 Pt 2):3886–3895. doi: 10.1523/JNEUROSCI.15-05-03886.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ebenezer, G. J., McArthur, J. C., Thomas, D., Murinson, B., Hauer, P., Polydefkis, M., & Griffin, J. W. (2007). Denervation of skin in neuropathies: The sequence of axonal and Schwann cell changes in skin biopsies. Brain: A Journal of Neurology, 130(Pt 10), 2703–2714. 10.1093/brain/awm199 [DOI] [PubMed]
- 191.Wilcox MB, Laranjeira SG, Eriksson TM, Jessen KR, Mirsky R, Quick TJ, Phillips JB. Characterising cellular and molecular features of human peripheral nerve degeneration. Acta Neuropathologica Communications. 2020;8(1):51. doi: 10.1186/s40478-020-00921-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Hutton EJ, Carty L, Laurá M, Houlden H, Lunn MPT, Brandner S, Mirsky R, Jessen K, Reilly MM. c-Jun expression in human neuropathies: A pilot study. Journal of the Peripheral Nervous System: JPNS. 2011;16(4):295–303. doi: 10.1111/j.1529-8027.2011.00360.x. [DOI] [PubMed] [Google Scholar]
- 193.Joshi AR, Holtmann L, Bobylev I, Schneider C, Ritter C, Weis J, Lehmann HC. Loss of Schwann cell plasticity in chronic inflammatory demyelinating polyneuropathy (CIDP) Journal of Neuroinflammation. 2016;13(1):255. doi: 10.1186/s12974-016-0711-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Fledrich R, Akkermann D, Schütza V, Abdelaal TA, Hermes D, Schäffner E, Soto-Bernardini MC, Götze T, Klink A, Kusch K, Krueger M, Kungl T, Frydrychowicz C, Möbius W, Brück W, Mueller WC, Bechmann I, Sereda MW, Schwab MH, Stassart RM. NRG1 type I dependent autoparacrine stimulation of Schwann cells in onion bulbs of peripheral neuropathies. Nature Communications. 2019;10(1):1467. doi: 10.1038/s41467-019-09385-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Fledrich R, Stassart RM, Klink A, Rasch LM, Prukop T, Haag L, Czesnik D, Kungl T, Abdelaal TAM, Keric N, Stadelmann C, Brück W, Nave K-A, Sereda MW. Soluble neuregulin-1 modulates disease pathogenesis in rodent models of Charcot-Marie-Tooth disease 1A. Nature Medicine. 2014;20(9):1055–1061. doi: 10.1038/nm.3664. [DOI] [PubMed] [Google Scholar]
- 196.Florio F, Ferri C, Scapin C, Feltri ML, Wrabetz L, D’Antonio M. Sustained Expression of Negative Regulators of Myelination Protects Schwann Cells from Dysmyelination in a Charcot–Marie–Tooth 1B Mouse Model. The Journal of Neuroscience. 2018;38(18):4275–4287. doi: 10.1523/JNEUROSCI.0201-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Hantke, J., Carty, L., Wagstaff, L. J., Turmaine, M., Wilton, D. K., Quintes, S., Koltzenburg, M., Baas, F., Mirsky, R., & Jessen, K. R. (2014). C-Jun activation in Schwann cells protects against loss of sensory axons in inherited neuropathy. Brain: A Journal of Neurology, 137(Pt 11), 2922–2937. 10.1093/brain/awu257 [DOI] [PMC free article] [PubMed]
- 198.Wiberg A, Ng M, Schmid AB, Smillie RW, Baskozos G, Holmes MV, Künnapuu K, Mägi R, Bennett DL, Furniss D. A genome-wide association analysis identifies 16 novel susceptibility loci for carpal tunnel syndrome. Nature Communications. 2019;10(1):1030. doi: 10.1038/s41467-019-08993-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Stassart RM, Woodhoo A. Axo-glial interaction in the injured PNS. Developmental Neurobiology. 2020 doi: 10.1002/dneu.22771. [DOI] [PubMed] [Google Scholar]
- 200.Farid M, Demicco EG, Garcia R, Ahn L, Merola PR, Cioffi A, Maki RG. Malignant peripheral nerve sheath tumors. The Oncologist. 2014;19(2):193–201. doi: 10.1634/theoncologist.2013-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Stierli, S., Napoli, I., White, I. J., Cattin, A.-L., Monteza Cabrejos, A., Garcia Calavia, N., Malong, L., Ribeiro, S., Nihouarn, J., Williams, R., Young, K. M., Richardson, W. D., & Lloyd, A. C. (2018). The regulation of the homeostasis and regeneration of peripheral nerve is distinct from the CNS and independent of a stem cell population. Development (Cambridge, England), 145(24). 10.1242/dev.170316 [DOI] [PMC free article] [PubMed]
- 202.De Raedt T, Beert E, Pasmant E, Luscan A, Brems H, Ortonne N, Helin K, Hornick JL, Mautner V, Kehrer-Sawatzki H, Clapp W, Bradner J, Vidaud M, Upadhyaya M, Legius E, Cichowski K. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature. 2014;514(7521):247–251. doi: 10.1038/nature13561. [DOI] [PubMed] [Google Scholar]
- 203.Gomez-Sanchez, J. A., Gomis-Coloma, C., Morenilla-Palao, C., Peiro, G., Serra, E., Serrano, M., & Cabedo, H. (2013). Epigenetic induction of the Ink4a/Arf locus prevents Schwann cell overproliferation during nerve regeneration and after tumorigenic challenge. Brain: A Journal of Neurology, 136(Pt 7), 2262–2278. 10.1093/brain/awt130 [DOI] [PubMed]
- 204.Lee W, Teckie S, Wiesner T, Ran L, Prieto Granada CN, Lin M, Zhu S, Cao Z, Liang Y, Sboner A, Tap WD, Fletcher JA, Huberman KH, Qin L-X, Viale A, Singer S, Zheng D, Berger MF, Chen Y, Chi P. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nature Genetics. 2014;46(11):1227–1232. doi: 10.1038/ng.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Röhrich M, Koelsche C, Schrimpf D, Capper D, Sahm F, Kratz A, Reuss J, Hovestadt V, Jones DTW, Bewerunge-Hudler M, Becker A, Weis J, Mawrin C, Mittelbronn M, Perry A, Mautner V-F, Mechtersheimer G, Hartmann C, Okuducu AF, Reuss DE. Methylation-based classification of benign and malignant peripheral nerve sheath tumors. Acta Neuropathologica. 2016;131(6):877–887. doi: 10.1007/s00401-016-1540-6. [DOI] [PubMed] [Google Scholar]
- 206.Zhang M, Wang Y, Jones S, Sausen M, McMahon K, Sharma R, Wang Q, Belzberg AJ, Chaichana K, Gallia GL, Gokaslan ZL, Riggins GJ, Wolinksy J-P, Wood LD, Montgomery EA, Hruban RH, Kinzler KW, Papadopoulos N, Vogelstein B, Bettegowda C. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nature Genetics. 2014;46(11):1170–1172. doi: 10.1038/ng.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Pao K-C, Wood NT, Knebel A, Rafie K, Stanley M, Mabbitt PD, Sundaramoorthy R, Hofmann K, van Aalten DMF, Virdee S. Activity-based E3 ligase profiling uncovers an E3 ligase with esterification activity. Nature. 2018;556(7701):381–385. doi: 10.1038/s41586-018-0026-1. [DOI] [PubMed] [Google Scholar]
- 208.Gould SA, White M, Wilbrey AL, Pór E, Coleman MP, Adalbert R. Protection against oxaliplatin-induced mechanical and thermal hypersensitivity in Sarm1−/− mice. Experimental Neurology. 2021;338:113607. doi: 10.1016/j.expneurol.2021.113607. [DOI] [PubMed] [Google Scholar]
- 209.Horsefield, S., Burdett, H., Zhang, X., Manik, M. K., Shi, Y., Chen, J., Qi, T., Gilley, J., Lai, J.-S., Rank, M. X., Casey, L. W., Gu, W., Ericsson, D. J., Foley, G., Hughes, R. O., Bosanac, T., Rathjen, J. P., Nanson, J. D., Boden, M., … Kobe, B. (2019). NAD+ cleavage activity by animal and plant TIR domains in cell death pathways. 8. [DOI] [PubMed]
- 210.Walker LJ, Summers DW, Sasaki Y, Brace E, Milbrandt J, DiAntonio A. MAPK signaling promotes axonal degeneration by speeding the turnover of the axonal maintenance factor NMNAT2. ELife. 2017;6:e22540. doi: 10.7554/eLife.22540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Hui F, Tang J, Williams PA, McGuinness MB, Hadoux X, Casson RJ, Coote M, Trounce IA, Martin KR, van Wijngaarden P, Crowston JG. Improvement in Inner Retinal Function in Glaucoma in Response to Nicotinamide (Vitamin B 3 Supplementation: A Crossover Randomized Clinical Trial [Preprint] Ophthalmology. 2020 doi: 10.1101/2020.01.28.20019075. [DOI] [PubMed] [Google Scholar]
- 212.Trammell SAJ, Weidemann BJ, Chadda A, Yorek MS, Holmes A, Coppey LJ, Obrosov A, Kardon RH, Yorek MA, Brenner C. Nicotinamide Riboside Opposes Type 2 Diabetes and Neuropathy in Mice. Scientific Reports. 2016;6(1):26933. doi: 10.1038/srep26933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Bosanac, T., Hughes, R. O., Engber, T., Devraj, R., Brearley, A., Danker, K., Young, K., Kopatz, J., Hermann, M., Berthemy, A., Boyce, S., Bentley, J., & Krauss, R. (2021). Pharmacological SARM1 inhibition protects axon structure and function in paclitaxel-induced peripheral neuropathy. Brain, awab184. 10.1093/brain/awab184 [DOI] [PMC free article] [PubMed]
- 214.Gould, S.A., Gilley, J., Ling, K., Jaffar-Nejad P., Ringo, F., & Coleman, M.P. (2021b). Sarm1 Haploinsufficiency and Low Expression Levels after Antisense Oligonucleotides Delays Programmed Axon Degeneration. Cell Reports, Cell Press Sneak Peek. [DOI] [PMC free article] [PubMed]
- 215.Höke A. Mechanisms of Disease: What factors limit the success of peripheral nerve regeneration in humans? Nature Clinical Practice. Neurology. 2006;2(8):448–454. doi: 10.1038/ncpneuro0262. [DOI] [PubMed] [Google Scholar]
- 216.Gersey ZC, Burks SS, Anderson KD, Dididze M, Khan A, Dietrich WD, Levi AD. First human experience with autologous Schwann cells to supplement sciatic nerve repair: Report of 2 cases with long-term follow-up. Neurosurgical Focus. 2017;42(3):E2. doi: 10.3171/2016.12.FOCUS16474. [DOI] [PubMed] [Google Scholar]
- 217.Stratton, J. A., Kumar, R., Sinha, S., Shah, P., Stykel, M., Shapira, Y., Midha, R., & Biernaskie, J. (2017). Purification and Characterization of Schwann Cells from Adult Human Skin and Nerve. ENeuro, 4(3). 10.1523/ENEURO.0307-16.2017 [DOI] [PMC free article] [PubMed]
- 218.Kim YJ, Lim H, Li Z, Oh Y, Kovlyagina I, Choi IY, Dong X, Lee G. Generation of multipotent induced neural crest by direct reprogramming of human postnatal fibroblasts with a single transcription factor. Cell Stem Cell. 2014;15(4):497–506. doi: 10.1016/j.stem.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 219.Kitada M, Murakami T, Wakao S, Li G, Dezawa M. Direct conversion of adult human skin fibroblasts into functional Schwann cells that achieve robust recovery of the severed peripheral nerve in rats. Glia. 2019;67(5):950–966. doi: 10.1002/glia.23582. [DOI] [PubMed] [Google Scholar]
- 220.Mazzara PG, Massimino L, Pellegatta M, Ronchi G, Ricca A, Iannielli A, Giannelli SG, Cursi M, Cancellieri C, Sessa A, Del Carro U, Quattrini A, Geuna S, Gritti A, Taveggia C, Broccoli V. Two factor-based reprogramming of rodent and human fibroblasts into Schwann cells. Nature Communications. 2017;8:14088. doi: 10.1038/ncomms14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Thoma EC, Merkl C, Heckel T, Haab R, Knoflach F, Nowaczyk C, Flint N, Jagasia R, Jensen Zoffmann S, Truong HH, Petitjean P, Jessberger S, Graf M, Iacone R. Chemical conversion of human fibroblasts into functional Schwann cells. Stem Cell Reports. 2014;3(4):539–547. doi: 10.1016/j.stemcr.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Eggers R, Tannemaat MR, De Winter F, Malessy MJA, Verhaagen J. Clinical and neurobiological advances in promoting regeneration of the ventral root avulsion lesion. The European Journal of Neuroscience. 2016;43(3):318–335. doi: 10.1111/ejn.13089. [DOI] [PubMed] [Google Scholar]
- 223.Tannemaat MR, Eggers R, Hendriks WT, de Ruiter GCW, van Heerikhuize JJ, Pool CW, Malessy MJA, Boer GJ, Verhaagen J. Differential effects of lentiviral vector-mediated overexpression of nerve growth factor and glial cell line-derived neurotrophic factor on regenerating sensory and motor axons in the transected peripheral nerve. The European Journal of Neuroscience. 2008;28(8):1467–1479. doi: 10.1111/j.1460-9568.2008.06452.x. [DOI] [PubMed] [Google Scholar]
- 224.Huang L, Xia B, Shi X, Gao J, Yang Y, Xu F, Qi F, Liang C, Huang J, Luo Z. Time-restricted release of multiple neurotrophic factors promotes axonal regeneration and functional recovery after peripheral nerve injury. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 2019;33(7):8600–8613. doi: 10.1096/fj.201802065RR. [DOI] [PubMed] [Google Scholar]
- 225.Vijayavenkataraman S. Nerve guide conduits for peripheral nerve injury repair: A review on design, materials and fabrication methods. Acta Biomaterialia. 2020;106:54–69. doi: 10.1016/j.actbio.2020.02.003. [DOI] [PubMed] [Google Scholar]
- 226.Cetinkaya-Fisgin A, Luan X, Reed N, Jeong YE, Oh BC, Hoke A. Cisplatin induced neurotoxicity is mediated by Sarm1 and calpain activation. Scientific Reports. 2020;10(1):21889. doi: 10.1038/s41598-020-78896-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Geisler S, Doan RA, Cheng GC, Cetinkaya-Fisgin A, Huang SX, Höke A, Milbrandt J, DiAntonio A. Vincristine and bortezomib use distinct upstream mechanisms to activate a common SARM1-dependent axon degeneration program. JCI Insight. 2019;4(17):e129920. doi: 10.1172/jci.insight.129920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Fortun J, Dunn WA, Joy S, Li J, Notterpek L. Emerging Role for Autophagy in the Removal of Aggresomes in Schwann Cells. Journal of Neuroscience. 2003;23(33):10672–10680. doi: 10.1523/JNEUROSCI.23-33-10672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Fortun J, Go JC, Li J, Amici SA, Dunn WA, Notterpek L. Alterations in degradative pathways and protein aggregation in a neuropathy model based on PMP22 overexpression. Neurobiology of Disease. 2006;22(1):153–164. doi: 10.1016/j.nbd.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 230.Wang, J., Wang, J., Lijun, Yang, L., Chuntao, Zhao Laiman Natalie, Wu Lingli, Xu Feng, Zhang Qinjie, Weng Michael, Wegner Q. Richard, Lu (2020) CTCF-mediated chromatin looping in EGR2 regulation and SUZ12 recruitment critical for peripheral myelination and repair. Nature Communications11(1). 10.1038/s41467-020-17955-2 [DOI] [PMC free article] [PubMed]
- 231.Yao, C., Yu, B. (2019) Role of Long Noncoding RNAs and Circular RNAs in Nerve Regeneration. Frontiers in Molecular Neuroscience12. 10.3389/fnmol.2019.00165 [DOI] [PMC free article] [PubMed]
- 232.Henzi, A., Aguzzi, A. (2021) The prion protein is not required for peripheral nerve de- and remyelination after crush injury. PLoS One, 16(1) e0245944. 10.1371/journal.pone.0245944 [DOI] [PMC free article] [PubMed]
- 233.Cheepudomwit T, Güzelsu E, Zhou C, Griffin JW, Höke A. Comparison of cytokine expression profile during Wallerian degeneration of myelinated and unmyelinated peripheral axons. Neuroscience Letters. 2008;430(3):230–235. doi: 10.1016/j.neulet.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 234.Brushart, T. M., Aspalter, M., Griffin, J.W., Redett, R., Hameed, H., Zhou, C., Wright, M., Vyas, A., & Höke, A. (2013) Schwann cell phenotype is regulated by axon modality and central–peripheral location and persists in vitro. Experimental Neurology 247, 272-281. 10.1016/j.expneurol.2013.05.007 [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.