

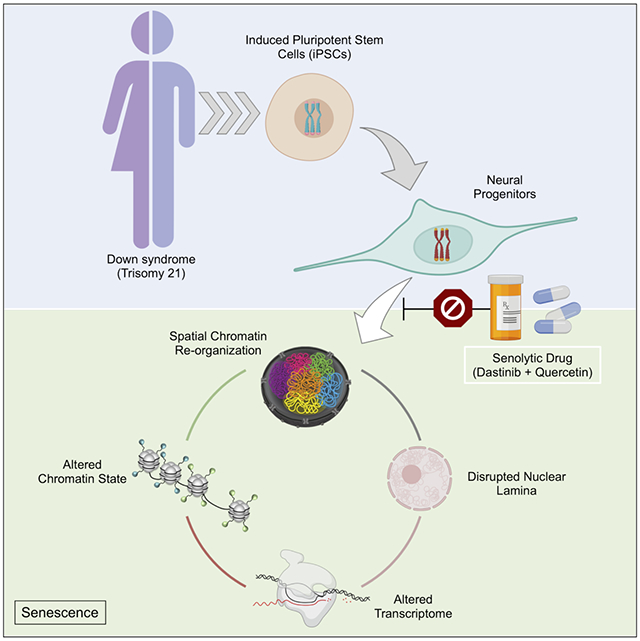
Summary
Down syndrome (DS) is a genetic disorder driven by the triplication of chromosome 21 (T21) that is characterized by a wide-range of neurodevelopmental and physical disabilities. Transcriptomic analysis of tissue samples from individuals with DS has revealed that T21 induces a genome-wide transcriptional disruption. However, the consequences of T21 on the nuclear-architecture and its interplay with the transcriptome remain unknown. In this study, we find that unlike human induced pluripotent stem cells (iPSCs), iPSC-derived neural progenitor cells (NPCs) exhibit genome-wide “chromosomal introversion”, disruption of lamina-associated domains, and global chromatin-accessibility changes in response to T21, consistent with the transcriptional and nuclear-architecture changes characteristic of senescent cells. Treatment of T21-harboring NPCs with senolytic drugs alleviates the transcriptional, molecular and cellular dysfunctions associated with DS. Our findings provide a mechanistic link between T21 and global transcriptional disruption, and indicate that senescence-associated phenotypes may play a key role in the neurodevelopmental pathogenesis of DS.
Keywords: Down syndrome, Aneuploidy, 3D-Genome, Epigenome, Transcriptome, Lamina-associated domains (LADs), Senescence, Senolytics, Neurodevelopment, Cell-Proliferation, Cell-Migration, Hi-C, ATAC-seq, RNA-seq, ChIP-seq
Graphical Abstract
eTOC Blurb
In this issue of Cell Stem Cell, Meharena et. al. (2021) show that Down syndrome or trisomy 21 disrupts the 3D-genome, epigenome and transcriptome of neural progenitors similar to those observed in senescent cells, and find that the senolytic drug combination of dasatinib and quercetin ameliorates these disruptions.
Introduction
Down syndrome (DS) is a genetic disorder caused by the triplication of chromosome 21 (T21) and recent whole-genome transcriptional profiling of various tissue samples from individuals with DS and DS mouse models have revealed that T21 induces genome-wide transcriptional disruption in addition to the dosage-dependent upregulation of a subset of expressed genes on chromosome 21, similar to those observed in other whole chromosome aneuploidies (Bianco et al., 2016; Durrbaum et al., 2014; FitzPatrick et al., 2002; Gonzales et al., 2018; Lejeune et al., 1959; Letourneau et al., 2014; Olmos-Serrano et al., 2016; Sheltzer et al., 2012; Tyler and Haydar, 2013; Walus et al., 2016; Wangsa et al., 2019). While the extra copy of chromosome 21 can explain upregulation of genes present on that chromosome, the mechanisms by which T21 induces genome-wide transcriptional disruption remain unknown.
The precise transcriptional output of a gene is orchestrated by transcription factors (TF) that bind locally to the gene promoter and distally to regulatory elements that are often separated from their target promoters by large genomic distances (Furlong and Levine, 2018). Epigenetic modifications of chromatin determine TF-binding site-accessibility at gene promoters and regulatory elements, and the folding of chromatin within the interphase nucleus brings these two regions into spatial proximity (McLaughlin et al., 2019; Pombo and Dillon, 2015). This three-dimensional (3D) genome organization of gene promoters and regulatory elements determines the transcriptional levels of specific genes within a cell (Schoenfelder and Fraser, 2019). Thus, both local and large-scale 3D-genome organization defined by A/B compartments, topologically associating domains (TADs), and enhancer-promoter interactions are necessary for the precise epigenetic regulation of the transcriptome required for proper embryonic development, neurogenesis, lineage commitment and stem cell differentiation (Battle et al., 2019; Dekker et al., 2002; Dixon et al., 2015; Lieberman-Aiden et al., 2009; Nora et al., 2012; Zheng and Xie, 2019). Furthermore, disease-associated mutations of genes involved in the organization of the 3D-genome, such as LMNB1, CTCF and cohesin, have been identified in individuals with neurodevelopmental disorders characterized by intellectual disability and craniofacial features similar to those observed in DS (Konrad et al., 2019; Yuan et al., 2015). However, the influence of T21 on the 3D-genome organization and the role of the epigenome in the global transcriptional disruption observed in DS remain unknown.
While individuals with DS exhibit wide-spread disease etiologies in different organs of the body, intellectual disability associated with abnormal brain morphogenesis is the most prevalent and highly penetrant pathology observed (Benda, 1940; Down, 1995; Pinter et al., 2001; Weijerman and de Winter, 2010). These brain abnormalities have been associated with dysfunctional neural progenitor cells (NPCs), the multipotent stem cells of the developing brain that differentiate into neurons, astrocytes or oligodendrocytes (Stagni et al., 2018; Tyler and Haydar, 2013). Fetal brain tissue derived from individuals with DS and brains of DS mouse models exhibit abnormal morphogenesis with accompanying deficits in cell proliferation and cell migration, which has been linked to the cognitive deficits observed in DS (Aldridge et al., 2007; Chakrabarti et al., 2007; Guidi et al., 2011; Moldrich et al., 2009; Olson et al., 2004). Furthermore, these abnormalities persist into adulthood and impact adult hippocampal neurogenesis, which is associated with learning and memory deficits in DS mouse models (Sylvester, 1983; Toda and Gage, 2018). From these studies and others, it is clear that NPC dysfunction is central to the pathogenesis of DS, however, the molecular mechanisms governing NPC dysfunction in DS remain unclear.
To explore the consequence of T21 on the molecular and cellular changes observed in DS, we established human-derived induced pluripotent stem cells (iPSCs) and iPSC-derived forebrain NPCs from multiple individuals and interrogated the consequences of T21 on the 3D-genome organization, epigenome and transcriptome (Buenrostro et al., 2013; Chambers et al., 2009; Rao et al., 2014; Topol et al., 2015; Weick et al., 2013). Our findings show that T21 induces chromosomal introversion, disrupts lamina-associated domains (LADs) and alters the genome-wide chromatin-accessibility of NPCs but not iPSCs. While the overall organization of A/B compartments and location of TAD-boundaries are conserved in NPCs harboring T21, we observe global loss of chromatin-accessibility within the A-compartment that is associated with transcriptional downregulation, and increased long-range chromatin interactions in the B-compartment that is associated with transcriptional upregulation. We find that these architectural changes are similar to those observed in senescent cells, and our transcriptional analysis confirms that differentially expressed genes (DEGs) identified in NPCs harboring T21 are highly correlated with DEGs identified in oxidative stress induced senescent cells. Finally, we demonstrate that the senolytic drug combination of dasatinib and quercetin alleviates the genome-wide transcriptional disruption, as well as deficits in cellular migration and proliferation observed in NPCs harboring T21.
Results
T21 induces chromosomal introversion in NPCs
To determine the consequences of T21 on the global 3D-genome organization we performed chromosome conformation capture (Hi-C) on an isogenic pair (euploid: Iso-E and trisomic: Iso-T cells derived from the same individual) of iPSCs and NPCs (Maclean et al., 2012), and identified ~2.5 billion unique interactions per cell type (Figure 1A-D; Table S1). We first sought to interrogate the consequences of T21 on the 3D-genome reorganization that occurs during differentiation of iPSCs to NPCs (Dixon et al., 2015). We observe significant gain and loss of both cis- and trans-chromosomal interactions in NPCs as compared to iPSCs in Iso-E during neural differentiation (Figure 1E), however, in Iso-T we observe significant genome-wide reduction of trans-chromosomal interactions in NPCs as compared to iPSCs (Figure 1F). While T21 minimally impacted the global trans-chromosomal interactions in iPSCs (Iso-E=19.82%±1.0 and Iso-T=21.72%±2.8, p-value= 3.4x10−1) (Figure S1A and S1B), we observe a significant decrease in the fraction of trans-chromosomal interactions in NPCs harboring T21 (Iso-E=21.55%±0.8 and Iso-T=15.85%±1.2, p-value= 2.2x10−3) (Figure 1G and H; Table S2). To evaluate whether the altered distribution of chromosomal interactions induced by T21 in NPCs is conserved across individuals with DS, we established iPSCs from an additional pair of male individuals with euploid (Ma-E) and trisomic (Ma-T) karyotypes and differentiated the cells into NPCs (Figure S1C). Consistent with our findings from the isogenic pair, we observed that NPCs harboring T21 exhibit reduced trans-chromosomal interactions as compared to NPCs harboring a euploid karyotype (Ma-E=19.17%±0.5, Ma-T=16.19%±0.5, p-value=1x10−4) (Figure S1D and E). As expected, we observed increased signal from chromosome 21 (HSA21) in both iPSCs and NPCs due to its triplication, however when normalized for the number of chromosomes we observed that loss of trans-chromosomal interactions in NPCs is distributed across all chromosomes, including HSA21 (Figure S1F; Table S3).
Figure 1 – T21 induces genome-wide chromosomal introversion in NPCs but not iPSCs.
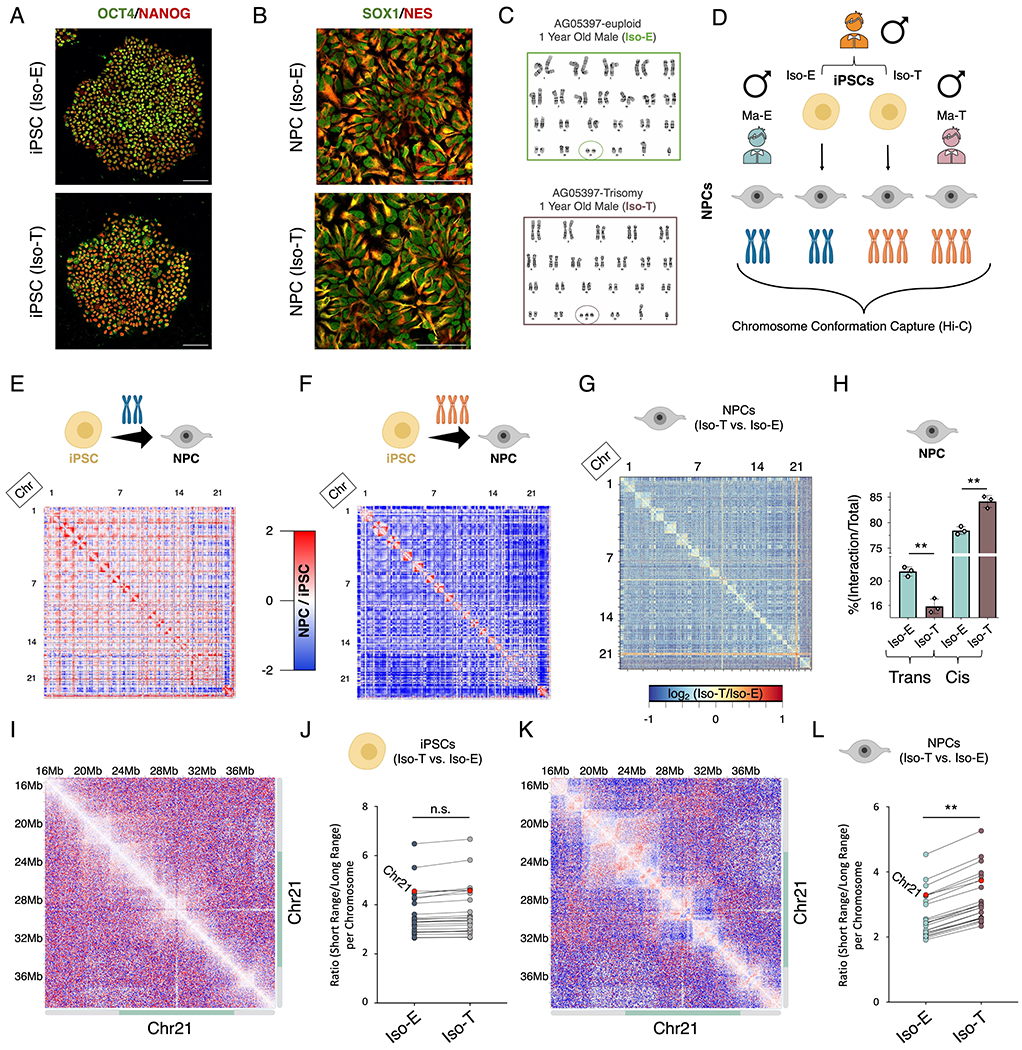
Immunofluorescence staining of (A) iPSCs (OCT4 (green), NANOG (red)) and (B) NPCs (SOX1 (green), Nestin (red)) from the isogenic pair (Iso-E (top) and Iso-T (bottom) as well as (C) respective G-band karyotyping of NPCs. (D) Schematic of experimental design to assess the 3D-genome of the isogenic pair iPSCs and NPCs as well as the euploid (Ma-E) and trisomic (Ma-T) male NPCs. (E-H) Comparative Hi-C analysis of Iso-E and Iso-T iPSCs and NPCs. Genome-wide differential (NPC/iPSC) Hi-C interaction maps for Iso-E (E) and Iso-T (F). (G) Genome-wide differential (Iso-T/Iso-E) Hi-C interaction maps for NPCs. (H) Percent distribution of trans- and cis-chromosomal interactions for the isogenic pair of NPCs. (I-L) Representative images of chromosome 21 (chr21) differential cis-chromosomal interaction maps for the isogenic pair of iPSCs (I) and NPCs (K). Dot plots representing the ratio of short-range (<1Mb) to long-range (>1Mb) interactions for the isogenic pair of iPSCs (J) and NPCs (L). Each dot represents a chromosome and chr21 is represented as a red dot. Wilcoxon rank-sum test shows that euploid and trisomic iPSCs were not statistically different (p-value=0.536) whereas NPCs show significantly increased short range interactions in the trisomic NPCs (p-value=0.004).
Decay of cis-chromosomal interaction frequency with distance is significantly altered in NPCs while remaining unchanged in iPSCs as a consequence of T21 (Figure S1l). T21 harboring NPCs exhibited reduced longer-range interactions (>~1Mb) and increased shorter-range interactions (<~1Mb), whereas in the isogenic pair of iPSCs, the distribution of cis-chromosomal interactions remain unchanged. This reduction of longer-range cis chromosomal interactions and increase of shorter-range interactions was observed across all chromosomes including HSA21 of T21 NPCs but not iPSCs (Figure 1I–L and Figure S1J). On average we observe an ~20% increase of shorter-range (< 1MB) interactions in T21 NPCs as compared to euploid NPCs (Table S4). To ensure that the 3D-genome reorganization observed in NPCs is not confounded due to the normalization of T21, we repeated the analysis with the chromosome 21 reads removed and found similar genome-wide chromosomal introversion in T21-NPCs (Figure S1K–N). In conclusion, we observe that T21 induces loss of trans-chromosomal interactions and increased shorter-range (<1MB) cis-chromosomal interactions in NPCs but not in iPSCs.
T21 induces reorganization of intra-TAD interactions and chromosomal looping
The genome is organized into two compartments within the nucleus: the active (A) compartment which is localized towards the core of the nucleus and the inactive (B) compartment localized towards the nuclear periphery on the nuclear lamina (Lieberman-Aiden et al., 2009). We found that T21 minimally impacts the classification of A/B compartments in both iPSCs and NPCs (Figure S2A–B; Table S5). A/B compartments are composed of multiple self-interacting structural units of the genome known as topologically associating domains (TADs) (Dixon et al., 2012; Nora et al., 2012; Rao et al., 2014) and we identified 5,559 and 4,731 in iPSC and NPC respectively (Table S6). These self-interacting domains facilitate the long-range interactions between promoters and distal regulatory elements. While T21 did not impact the intra-TAD interaction density (ID) of iPSCs, the intra-TAD ID of NPCs was significantly reorganized in trisomic (Iso-T) NPCs as compared to the euploid (Iso-E) NPCs, where 1,800 (~38%) TADs had significantly higher-ID and 389 (~8%) TADs had significantly reduced-ID (Figure 2A-D). Next, we performed an enrichment analysis (10 million randomized permutations) to assess whether the differentially interacting TADs preferentially localized to a specific compartment. We found that TADs with increased-ID were enriched in the B-compartment and unchanged TADs were predominantly localized in the A-compartment, however, the reduced-ID TADs were not enriched in either compartment (Figure 2E; Table S7). While the intra-TAD ID of ~46% of TADs were significantly altered, the directionality index and insulation score remained unchanged as a consequence of T21 in both iPSCs and NPCs, indicating a general maintenance of global chromatin architecture and TAD-boundaries (Figure S2C).
Figure 2 – T21 induced reorganization of TADs and loops are segregated into A/B compartments.
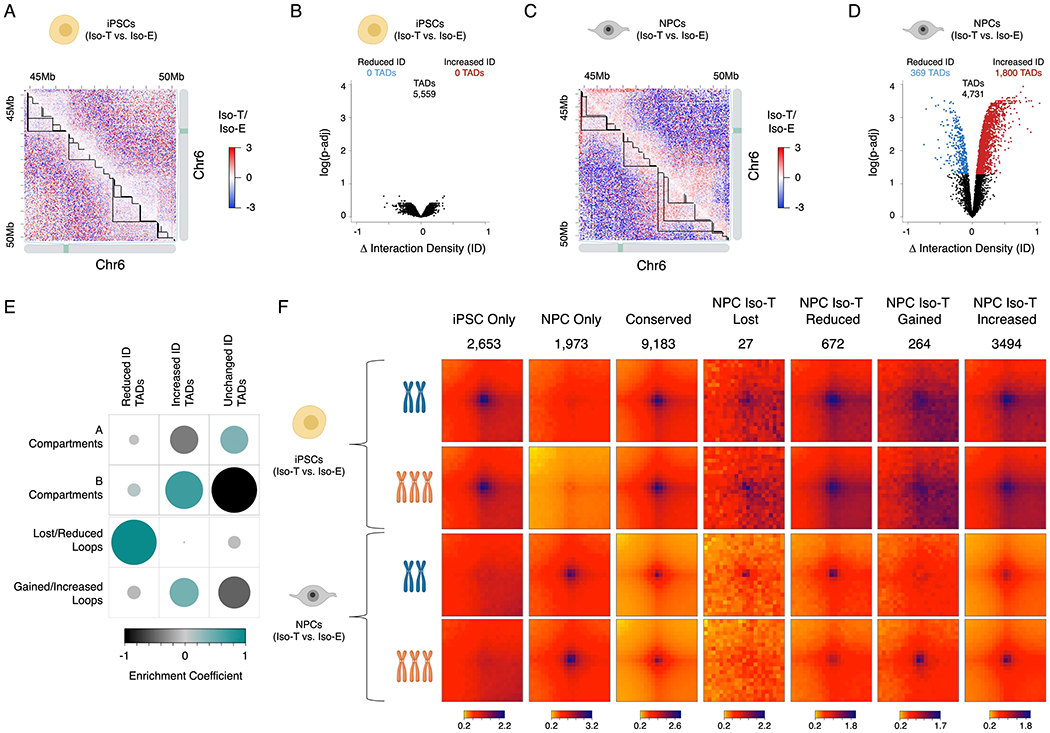
Representative images of differential Hi-C interaction maps (Iso-T/Iso-E) of a region on chromosome 6 (chr6) in the isogenic pair of iPSCs (A) and NPCs (C). Volcano plot of differential interaction density (ID) within TADs in the isogenic pair of iPSCs (B) and NPCs (D). (E) Randomized permutation test analysis between the differential interaction density (ID) TADs with A/B compartments and differential chromosomal loops in NPCs. Color intensity (positive correlations are displayed in green and negative correlations in black) and the size of the circle are proportional to the enrichment coefficients of A/B compartments with differentially interacting TADs identified as a consequence of T21 in NPCs. (F) Aggregate Peak Analysis (APA) analysis of loops uniquely identified in iPSCs (top rows), NPCs (bottom rows) in both Iso-E and Iso-T.
To interrogate the consequences of T21 on chromosomal looping we utilized arrowhead to identify all contact domains in iPSCs and NPCs. We identified 12,053 loops in iPSCs and 11,373 loops in NPCs: of these, 2,653 and 1,973 loops are unique to iPSCs and NPCs respectively (Figure 2F and S2D; Table S8). Differential chromosomal looping analysis revealed significant genome-wide reorganization of looping events in NPCs as a consequence of T21. We identified ~700 loops that were significantly reduced or lost in NPCs harboring T21 as compared to euploid NPCs, where 27 loops were only identified in euploid NPCs (z-score Iso-E = 17.75 and Iso-T = 0.84) and 672 were significantly reduced in T21 NPCs (z-score Iso-E = 64.46 and Iso-T = 32.46). Conversely, we identified ~3,800 loops significantly enhanced in NPCs harboring T21, with the appearance of 264 de novo loops in Iso-T NPCs (z-score Iso-E = 2.19 and Iso-T = 33.97) and 3,494 loops were significantly increased in T21 harboring NPCs (z-score Iso-E = 30.75 and Iso-T = 62.56). Additionally, enrichment analysis revealed that the chromosomal loops that were lost/reduced are predominantly localize to TADs with reduced-ID (p-value = 0.00596, 10 million randomized permutations), and the de novo/increased loops were enriched in TADs with increased-ID (p-value = 0.00539, 10 million randomized permutations) (Figure 2E; Table S7). In summary, we find that T21-induced introversion is predominantly localized within the inactive B-compartment.
Disruption of nuclear lamina is associated with increased intra-TAD interactions
Reorganization of the nuclear-architecture in T21 NPCs, specifically the gain of shorter-range interactions associated with chromosomal introversion, significantly impacts the B-compartment. The B-compartment of the genome is localized towards the nuclear periphery on the nuclear lamina and is enriched for the heterochromatin histone modification H3K9me3 (Shevelyov and Ulianov, 2019). To interrogate the consequences of T21 on the organization of the nuclear lamina we utilized immunofluorescence to label LMNB1 and H3K9me3. While we observed an overall maintenance of the nuclear envelope and heterochromatin organization in T21 iPSCs (Figure S3A-C; Table S9), we observed decreased levels of LMNB1 in NPCs harboring T21 (Iso-T) as compared to the euploid (Iso-E) NPCs (Figure 3A and 3B). The global intensity of H3K9me3 was unchanged in both iPSCs and NPCs harboring T21 (Figure S3C and S3D), however, we observed an increased number of H3K9me3 aggregates within the nuclei of Iso-T NPCs as compared to Iso-E NPCs, and this was unchanged in iPSCs (Figure S3E and S3F).
Figure 3 – T21 induced disruption of lamina associated domains (LADs) is associated with chromosomal introversion.
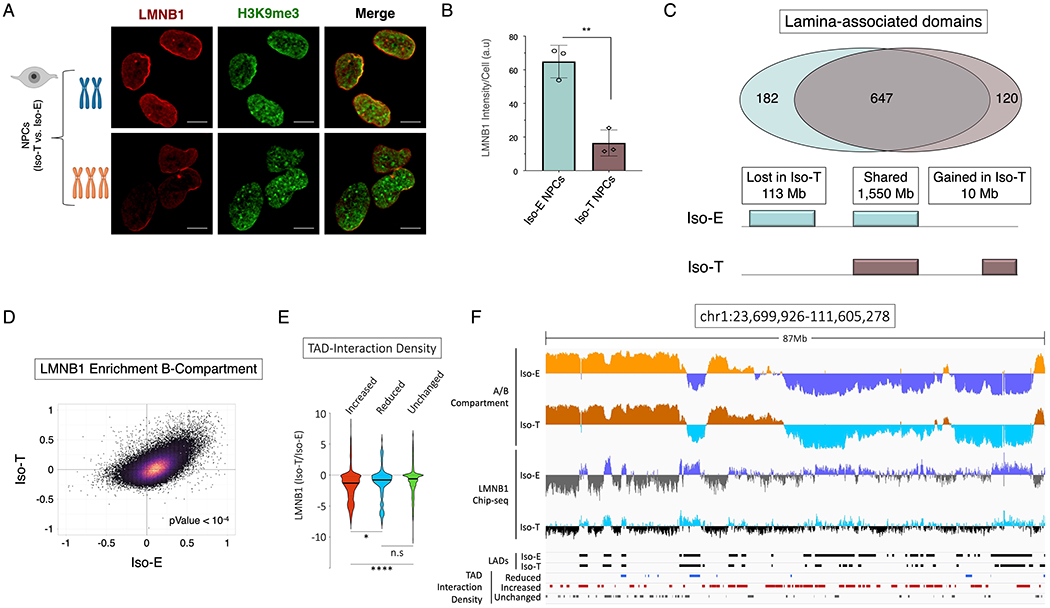
(A) Immunofluorescence of LMNB1 (red) and H3K9me3 (green) for the isogenic pair to assess LADs and heterochromatin distribution in NPCs. (B) Quantification of LMNB1 staining intensity in the isogenic pair of NPCs. Each dot on the histogram represents a replicate experiment of ~440 total nuclei analyzed for the isogenic pair of NPCs. (C) Venn diagram of LAD overlap between the euploid (Iso-E) and T21 (Iso-T) NPCs (intersect of replicates); bottom, schematic representation of genome coverage (base pairs) by LADs gained or lost as a consequence of T21 relative to euploid. (D) Scatter plot of LMNB1 enrichment over input in Iso-E (x-axis) and Iso-T (y-axis) of the genomic region in the B-compartment. (E) Violin plot of fold change of LMNB1 (Iso-T/Iso-E) of the differential (increased-ID (red), reduced-ID (blue) and unchanged-ID (green)) interaction density TADs. (F) IGV plot of A-compartment (Iso-E (yellow) and Iso-T (orange)) and B-compartment (Iso-E (blue) and Iso-T (light-blue)), LMNB1 ChIP-seq of Iso-E (blue) and Iso-T (light-blue), as well as the location of LADs (black) and the differentially interacting TADs (reduced-ID (blue), increased-ID (red) and unchanged-ID (gray)) an 87Mb region of chromosome 1.
To interrogate whether B-compartment TAD-introversion is associated with disruption of the nuclear lamina we performed ChIP-seq for LMNB1 in the isogenic pair of NPCs (Figure S3G). We identified 949 lamina-associated domains (LADs), which associated with ~ 53% of the genome, and from these we observed ~7% of the LADs (113Mb) are lost and <1% (10Mb) are gained in Iso-T NPCs harboring as compared Iso-E NPCs (Figure 3C and S3H; Table S10). As expected, enrichment analysis of LAD intensity shows that the A-compartment is depleted for LMNB1 association in both Iso-E and Iso-T NPCs (r = 0.4134) (Figure S3I), whereas the B-compartment was highly enriched for regions of the genome that are associated with LMNB1 in both Iso-E and Iso-T NPCs (r = 0.4267) (Figure 3D; Table S11). Additionally, LMNB1 occupancy was significantly (p-value < 104) reduced in the B-compartment of Iso-T NPCs as compared to Iso-E NPCs. Next, we sought to interrogate if differentially interacting TADs have altered LAD enrichment and found that TADs with increased-ID have significantly lower LMNB1 enrichment as compared to TADs with reduced-ID or unchanged-ID genome-wide (Figure 3E-F and S3J-O; Table S12). In conclusion we observe that the TADs within the B-compartment that have increased intrachromosomal interactions have a significant reduction of LMNB1 association in Iso-T NPCs as compared to Iso-E NPCs.
T21 induced transcriptional upregulation is associated with chromosomal introversion
To examine the relationship between T21 associated nuclear-architecture reorganization and the transcriptome we performed RNA-sequencing of iPSCs (isogenic pair) and NPCs derived from the isogenic pair as well as NPCs derived from individuals with DS (female (Fe-T) and male (Ma-T)) and euploid controls (female (Fe-E) and male (Ma-E)) (Figure 4A and Figure S1C). Principal component analysis showed that transcriptionally, NPCs derived from the different individuals segregate by karyotype on the first principal component (Figure S4A). We observed that T21 had a greater impact on the NPC transcriptome than the transcriptome of iPSCs: NPCs exhibited 1,869 downregulated and 2,724 upregulated differentially expressed genes (DEGs) (FDR < 0.01, log2 fold change > 0.5 or < −0.5), while iPSCs exhibited 102 downregulated and 423 upregulated DEGs (Figure 4B and C; Table S13 and S14). Comparison of the DEGs identified in NPCs derived from the different individuals (pairwise comparisons of Iso-T vs. Iso-E, Fe-T vs. Fe-E, and Ma-T vs. Ma-E) showed a significant concordance of the upregulated and downregulated genes identified as a consequence of T21 after running 10 million randomized permutations of the expression data (Figure S4B; Table S15 and S16). To ensure that the genome-wide disruption observed in NPCs is not due to normalization of T21, we repeated our differential analysis of the isogenic pair after removing all reads originating from chromosome 21 and observed genome-wide transcriptional disruption similar to what we observe in the presence of chromosome 21 (Figure S4C). While both iPSCs and NPCs exhibited upregulation of ~25% of the genes expressed on HSA21, NPCs showed disruption of ~20% (8.5% downregulation and 11.6% upregulation) of expressed genes on all of the other chromosomes. In contrast, iPSCs only exhibited alterations of ~3% (0.6% downregulation and 2.6% upregulation) of the expressed genes on all of the other chromosomes (Figure S4D and E).
Figure 4 – T21 DEGs are distinctly separated into the A/B compartments in NPCs.
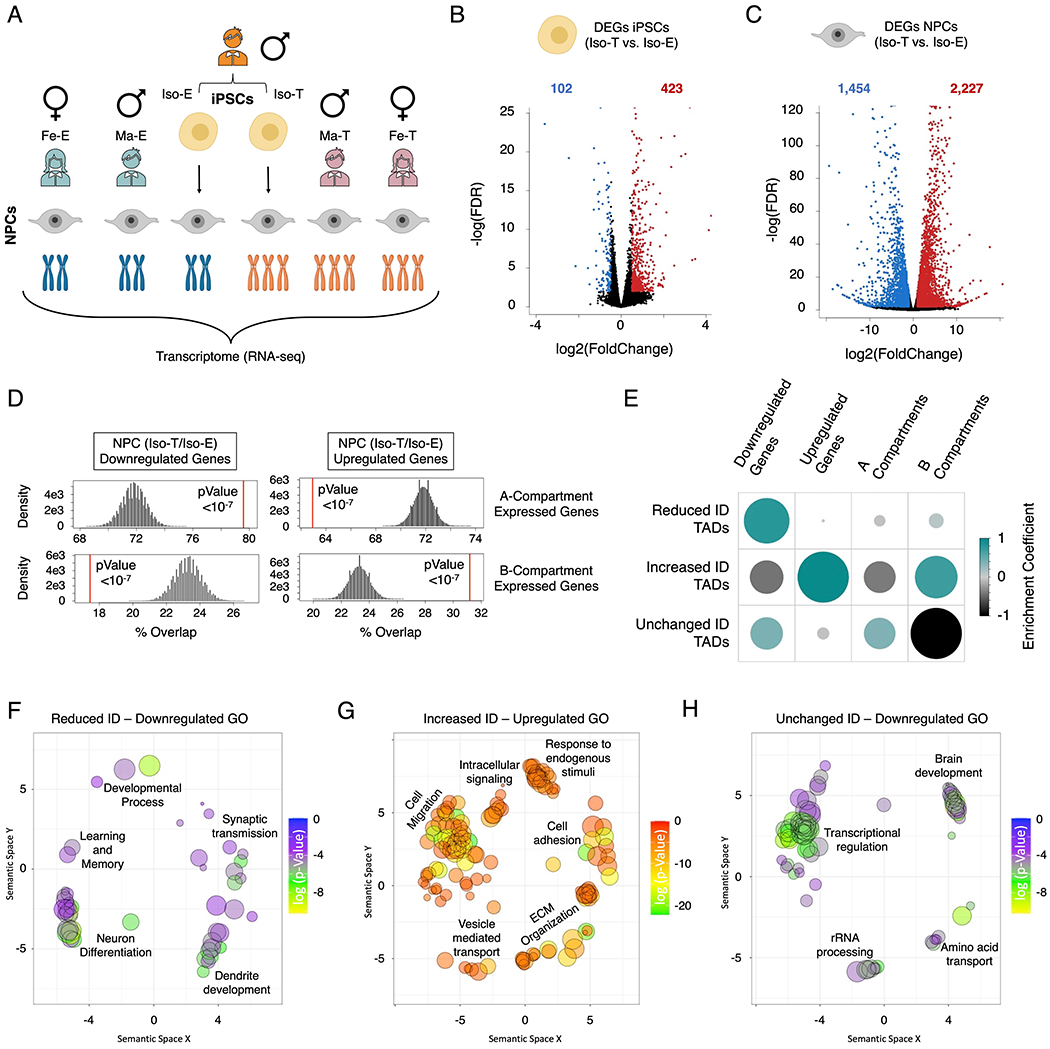
(A) Schematic of experimental design to assess the transcriptional consequences of T21 on the isogenic pair (Iso-E and Iso-T) of iPSCs and NPCs as well as NPCs derived from euploid (female (Fe-E) and male Ma-E)) and trisomic (female (Fe-T) and male (Ma-T)) individuals. (B, C) Volcano plots of the differentially expressed genes (DEGs) identified in the isogenic pair of iPSCs (B) and NPCs. (C). (D) Randomized permutation test between the downregulated and upregulated DEGs induced by T21 in NPCs with A/B compartments. Histogram represents the range of expected overlap percentage and the red line represents the observed percentage overlap. (E) Corrplot of the randomized permutation test between TADs with varying interaction densities (ID) with DEGs and A/B compartments. Color intensity (positive correlations are displayed in green and negative correlations in black) and the size of the circle are proportional to the enrichment coefficients. (F-H) Gene ontology visualized in semantic similarity-based scatterplots for downregulated genes associated with reduced-ID TADs (F), upregulated genes associated with increased-ID TADs (G), and downregulated genes localized within unchanged-ID TADs (H).
In both iPSCs and NPCs we found that the A and B compartments are composed of similar genome sizes and as expected, the majority of expressed genes are located in the A-compartment (~80% and ~76% of expressed genes in iPSCs and NPCs respectively) while the remaining ~20% (iPSCs) and ~24% (NPCs) of expressed genes are located in the B-compartment (Table S17). However, we found that the DEGs identified in NPCs were distinctly segregated into the two compartments, with downregulated genes predominantly located in the A-compartment and upregulated genes enriched in the B-compartment (Figure 4D; Table S18 and S19). We found that TADs with increased interaction densities and increased looping events are enriched for upregulated genes and are predominantly located in the B-compartment (randomized permutations p-value<1x10−07) (Figure 4E and S4F-G; Table S20-22). Additionally, we find that the regions with reduced nuclear lamina association as a consequence of T21 are enriched for upregulated genes (Figure S4F). Conversely, TADs with reduced-ID in T21 NPCs are enriched for downregulated genes and are not enriched in either compartment (randomized permutations p-value<1x10−07). Although the A-compartment is predominantly enriched for downregulated genes we observed that this compartment is predominantly enriched for TADs with unchanged-ID (randomized permutations p-value <1x10−07), suggesting that the downregulation of genes observed in the A-compartment are independent of altered long-range promoter-enhancer interactions.
Gene ontology analysis of iPSC DEGs showed enrichment of downregulated genes involved in biological processes such as GPCR signaling, receptor binding regulation and cellular migration, while upregulated genes were enriched for extracellular matrix (ECM) organization, regulation of developmental processes and immune activation (Figure S4I and S4J; Table S23). In NPCs, we observed downregulated genes were enriched for nuclear associated biological process (chromosome organization, cell cycle regulation, DNA-damage response, telomere maintenance, nuclear transport and mRNA-splicing), as well as apoptosis, cell proliferation and differentiation (Figure S4K). Conversely, upregulated genes were enriched for biological process associated with ECM organization, cell migration, cell adhesion, secretion by cell, response to endogenous stimuli and intracellular signaling associated biological processes (Figure S4L). Next, we focused on those DEGs localized within differential ID TADs and found that downregulated genes within reduced-ID TADs were enriched for brain development associated processes, whereas upregulated genes localized within increased-ID TADs were predominantly enriched for biological processes involving cell migration, cell adhesion and ECM organization (Figure 4F and G). While the downregulated genes within unchanged-ID TADs were associated with transcriptional regulation and brain development (Figure 4H), we do not capture a significant proportion of the downregulated biological processes in T21 NPCs, such as those associated with nuclear processes like chromosome organization and cell cycle regulation (Figure S4K). In summary, upregulated genes were enriched in B-compartment TADs with increased-ID and TADs with reduced-ID were enriched for downregulated genes located primarily in the A-compartment. However, the mechanism underlying the downregulation of genes localized within unchanged-ID TADs remains unknown.
T21 induced reduction of chromatin-accessibility is associated with dysfunction of nuclear processes
Posttranslational modifications of histone H3 are tightly linked to the regulation of gene expression, and past mutagenesis studies have indicated that dysfunction in this pathway causes cell proliferation and migration defects resulting in abnormal brain development (Hsu et al., 2000; Kang et al., 2007; Montgomery et al., 2009; Sun et al., 2018; Whetstine et al., 2005; Zhang et al., 2014a; Zinovyeva et al., 2006). Our transcriptional analysis showed a significant number of the downregulated genes in trisomic NPCs impact chromatin modifications on histone H3, including histone deacetylases (HDAC1 and HDAC2), polycomb-group proteins (EZH2 and EED), histone acetyltransferase (KAT2A), as well as the eukaryotic protein kinases AURKB and VRK1 (Figure S5A). To assess the post-translational modification landscape of the histone H3 residues, we performed global chromatin profiling (GCP), a targeted mass-spectrometry based assay that quantifies histone post-translational modifications in bulk chromatin (Figure S5B; Table S24) (Jaffe et al., 2013). Comparison of the isogenic pair of NPCs (Iso-E and Iso-T) showed a significant reduction of the mitotic marker H3 serine-10 phosphorylation (H3S10p), consistent with previous studies indicating reduced cellular proliferation in DS (Contestabile et al., 2009; Contestabile et al., 2007; Guidi et al., 2011; Moldrich et al., 2009). While we did not identify any significant changes to chromatin states that typically demarcate promoters or enhancers (H3 lysine-4 mono-methylation (H3K4me) and tri-methylation (H3K4me3) respectively), we observed reduction in H3 lysine-23 acetylation (H3K23ac) and increase in H3 lysine-27 acetylation (H3K27ac), H3 residues associated with chromatin and transcriptional activity, potentially indicating a relationship between the observed genome-wide transcriptional changes and overall chromatin state (Allis and Jenuwein, 2016; Klemm et al., 2019; Lane et al., 2014; Liu et al., 2016; Lu et al., 2015).
To assess the consequences of T21 on the genome-wide chromatin state, we utilized the assay for transposase-accessible chromatin using sequencing (ATAC-seq) on the isogenic pair of iPSCs and NPCs (Buenrostro et al., 2013). We found that while T21 minimally impacts the chromatin state of iPSCs (~1.6% of the accessible chromatin), ~20% of the accessible chromatin is altered as a consequence of T21 in NPCs with an FDR < 0.01 (Figure 5A and B; Table S25), a level of chromatin state alteration that has recently been observed in astroglia derived from individuals with DS (Bally et al., 2020). We identified 18,304 differentially accessible regions (DARs) in NPCs harboring T21 (14,814 more accessible and 3,490 less accessible). As expected, due to the chromosomal triplication, HSA21 is more permissive to transposase in both T21 iPSCs and NPCs (Figure S5C and D). Additionally, in T21-NPCs we observed that all other chromosomes harbor regions with both increased and decreased chromatin-accessibility, comparable to the distribution of DEGs across the chromosomes (Figure S4D and E). To confirm this genome-wide disruption of chromatin-accessibility is not associated with normalization of T21 we repeated the normalization and differential analysis in the absence of chromosome 21 and we observed 3,419 regions with reduced accessibility and 14,605 of with increased accessibility, FDR < 0.01 (Figure S5E). Both differential gene expression and differential accessibility analysis of NPCs indicate increased transcriptional activity in T21-NPCs harboring (Figure 4C and 5B). To asses if the transcriptional upregulation and increased accessibility is associated with increased nascent RNA expression we labeled nascent transcripts in live NPCs via a 5 min EUTP pulse and quantified EU-RNA fluorescence. We observe an ~20% increase in nascent RNA levels in Iso-T as compared to Iso-E NPCs (Figure S5F and G; Table S27).
Figure 5 – T21-induced genome-wide disruption of chromatin state in NPCs is associated with transcriptional downregulation.
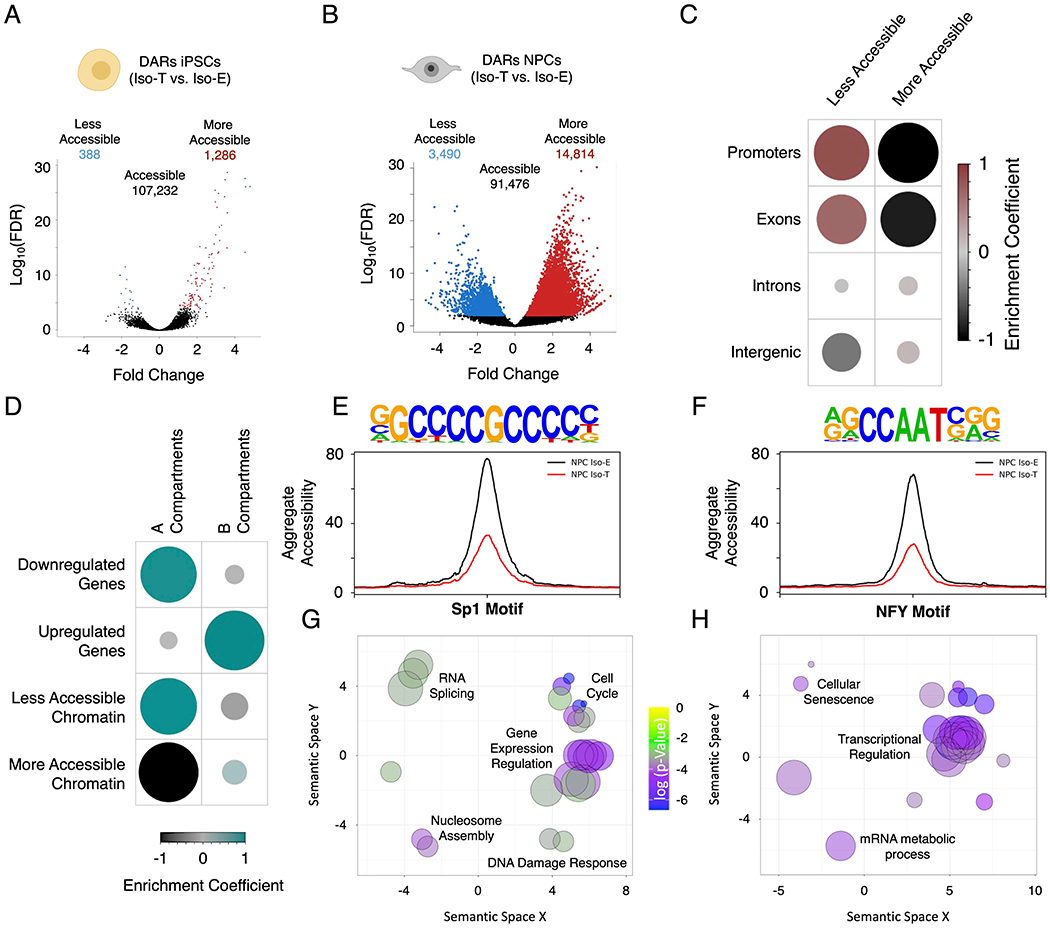
(A, B) Volcano plots of differentially accessible regions (DARs) identified by ATAC-sequencing in the isogenic pair of iPSCs (A) and NPCs (B). (C) Enrichment heatmap of the randomized permutation test between the DARs on promoters, exons, introns and intergenic regions with accessible regions genome-wide. Color intensity (positive correlations are displayed in red and negative correlations in black) and the size of the circle are proportional to the enrichment coefficients. (D) Enrichment heatmap of the randomized permutation test between the A/B compartments and DEGs and DARs. Color intensity (positive correlations are displayed in green and negative correlations in black) and the size of the circle are proportional to the enrichment coefficients of the specific genetic features with the A/B compartments. (E, F) Aggregate plots of less accessible regions harboring SP1 (E) and NFY (F) motifs. (G, H) Gene ontology visualized in semantic similarity-based scatterplots for downregulated genes with less accessible promoters harboring SP1 (G) and NFY (H) motifs.
To gain an understanding of the histone modifications associated with the DARs identified in T21 NPCs, we utilized published ChIP-seq data from H1-derived NPCs deposited in ENCODE (Hawkins et al., 2010). While less accessible regions are equally distributed on H3K4me3, H3K4me, H3K27ac, H3K23ac and regions not-marked by any of these modifications, the more accessible regions are predominantly localized on regions with H3K4me and regions that are not-marked (Figure S5H; Table S28). However, global enrichment analysis (10 million randomized permutations) of DARs on promoters, exons, introns and intergenic regions (GENCODE GRCh37) revealed that less accessible regions are significantly enriched on promoters and exons, whereas, the more accessible regions are significantly depleted on promoters and exons, and neither the less or more accessible DARs are significantly enriched on introns and intergenic regions (Figure 5C; Table S28). Consistent with these findings we observe that the less accessible regions are enriched for the promoter marker H3K4me3 as well as chromatin activity associated markers such as H3K27ac and H3K23ac (Figure S5H and S5I; Table S28). We found a significant concordance between the less accessible promoters and downregulated genes and globally we observe that less accessible regions are predominantly localized to the A-compartment (Figure 5D and S5J; Table S29 and 30). The biological processes associated with these downregulated genes with reduced promoter-accessibility includes cell cycle regulation, transcriptional regulation, mRNA-splicing and chromatin organization, the biological processes not associated with the 3D-genome re-organization (Figure S5K). While we observe a significantly higher number of more accessible regions genome-wide this increased chromatin-accessibility is not specific to any annotated genomic regions.
Motif analysis of the less accessible regions in Iso-T NPCs revealed significant enrichment of TF-binding motifs for SP1 (p-Value = 1e−44) and NFY (p-Value = 1e-41) (Figure 5E and F). NFY and SP1 are transcription factors involved in the regulation of cell cycle and proliferation (Benatti et al., 2011; Grinstein et al., 2002; Oh et al., 2007; Zhang et al., 2014b). While the accessibility of regions harboring the SP1 and NFY motifs are reduced, the transcript levels of SP1 and the different isoforms of NFY (NFYA, NFYB and NFYC) remain unchanged in T21-NPCs (Figure S5L). The downregulated gene promoters with less accessible SP1 motifs are enriched for biological processes involved in cell cycle, gene expression regulation, RNA splicing, nucleosome assembly and DNA damage response, whereas the downregulated genes with less accessible promoters harboring the NFY motifs are associated with transcriptional regulation, mRNA metabolic processes and cellular senescence (Figure 5G and H). Overall, our findings indicate that the biological process disrupted by T21 are driven by three disruptions in chromatin organization: 1) brain development associated downregulated genes are predominantly localized within reduced-ID TADs, 2) cell migration associated upregulated genes are localized within increased-ID TADs, and 3) cell proliferation, transcriptional regulation and chromosomal organization related downregulated genes are associated with reduction of promoter-accessibility (Figure 4F, 4G and S5K).
T21 induces NPC senescence
Chromosomal introversion, disruption of LADs, and genome-wide alterations of the transcriptome and chromatin state have been identified as key features of senescent cells, similar to our observations in NPCs harboring T21 (Criscione et al., 2016; Hernandez-Segura et al., 2017; Lenain et al., 2017; Parry et al., 2018; Sati et al., 2020; Zirkel et al., 2018). Senescence is a stress response that quenches the proliferative capacity of cells and is predominantly associated with cellular aging (Hayflick and Moorhead, 1961). Studies have shown increased senescent amniocytes and trophoblasts in placental samples from pregnancies with DS (Biron-Shental et al., 2015) ), however T21-induced senescence in the developing brain of individuals with DS remain unknown. To assess whether NPCs harboring T21 undergo senescence we performed senescence associated β-galactosidase staining and observed a ~15-fold increase (p-value = 0.001) in the number of senescent cells in Iso-T NPCs as compared to Iso-E NPCs (Figure 6A and B; Table S32). Additionally, we observed increased immunofluorescence of HP1-alpha and decreased levels of HMGB1 and of the H3K27me3 similar to what has been observed in senescent cells (Figure S6A and B; Table S33) (Zirkel et al., 2018). To confirm that T21-NPCs do not undergo premature differentiation we compared our RNA-seq data from iPSCs and NPCs with previously published datasets from iPSC-derived neurons, and NPC derived astrocytes and oligodendrocytes. We observed that NPCs harboring euploid and T21 transcriptionally cluster together and separate from all other cell types (Figure S6C) (Garcia-Leon et al., 2018; Lin et al., 2018; Tcw et al., 2017). Additionally, we stained for the astrocytic marker GFAP, a senescence marker p16INK4a and the intermediate filament marker vimentin. We observed that NPCs harboring both euploid and T21 do not express GFAP as compared to NPC-derived astrocytes, and morphologically these NPCs are markedly different from NPCs which is shown with Vimentin staining (Figure S6D and E). However, T21-NPCs have a significantly higher level of p16INK4a, indicating that T21-NPCs that express senescence markers do not undergo premature differentiation.
Figure 6 – T21 induces senescence in NPCs.
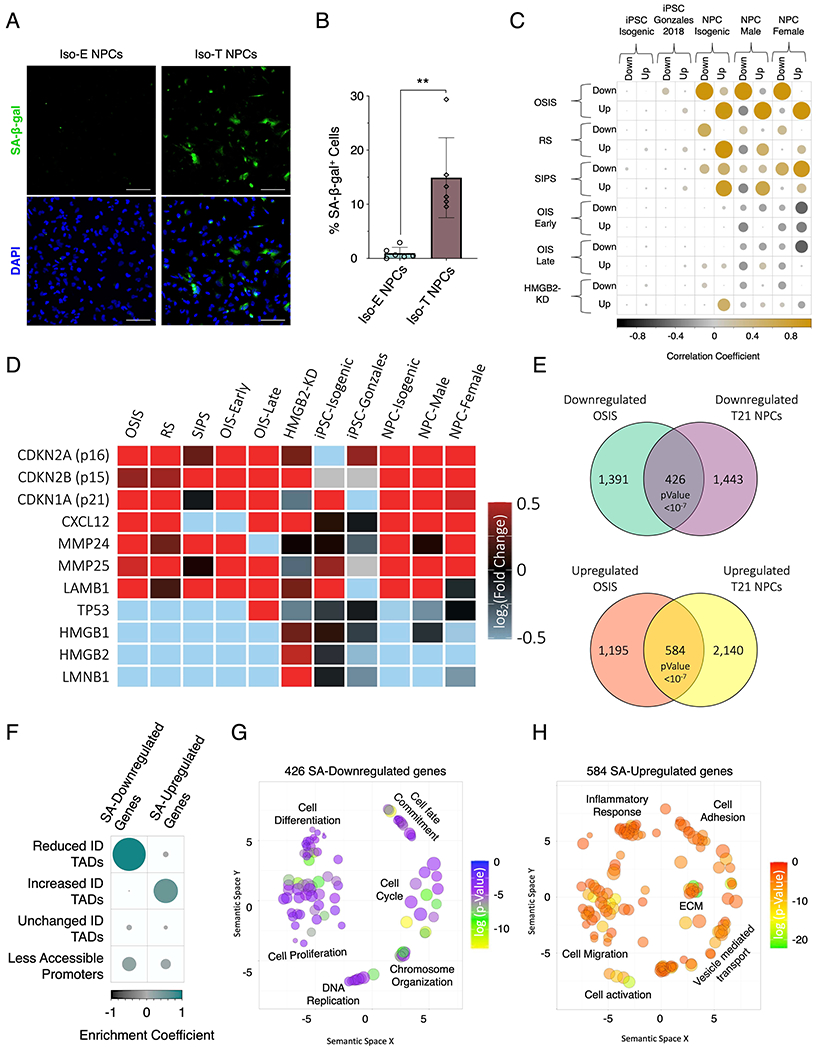
(A) Senescence associate β-galactosidase staining (SA-β-gal, green) marking senescent NPCs in euploid NPCs (left) and T21 (right). (B) Quantification of SA-β-gal staining intensity in the isogenic pair of NPCs. Each dot on the histogram represents a replicate experiment of ~200 cells analyzed for the isogenic pair of NPCs. (C) Correlation heatmap of the randomized permutation test of DEGs identified in isogenic, male, and female control and T21 iPSCs and NPCs as well as DEGs identified from a previous published iPSC data set (Gonzales), compared to previously published DEGs identified in various modes of senescence induction (oxidative stress induced senescence (OSIS), replicative senescence (RS), stress induced senescence (SIPS), oncogene induced senescence (OIS) at early and late timepoints, as well as senescence entry induced by HMGB2 knock down (HMGB2-KD). Color intensity (positive correlations are displayed in yellow and negative correlations in black) and the size of the circle are proportional to the enrichment coefficients. (D) Gene expression heatmap of senescence marker genes as fold change of T21/E21 or senescent/control. (E) Venn diagram of overlapping downregulated (top) and upregulated (bottom) identified in NPCs harboring T21 and compared to DEGs identified in OSIS induced senescent cells. (F) Enrichment heatmap of the randomized permutation test between DEGs identified in both T21-NPCs and OSIS-induced senescent cells with differential ID TADs as well as reduced promoter-accessibility. Color intensity (positive correlations are displayed in green and negative correlations in black) and the size of the circle are proportional to the enrichment coefficients. (G, H) Gene ontology visualized in semantic similarity-based scatterplots for downregulated (G) and upregulated (H) overlapping genes identified in both T21-NPCs and OSIS-induced senescent cells.
To assess the global transcriptional alteration similarities between T21 and senescent cells, we utilized previously published transcriptomic data where senescence was induced through oxidative stress (oxidative stress induced senescence (OSIS)), replicative stress (replicative senescence (RS)), radiation ionization (stress-induced premature senescence (SIPS)), overexpression of RAS (oncogene induced senescence (OIS)) and HMGB2 knockdown induced senescence (HMGB2-KD) (Alspach et al., 2014; Borras-Fresneda et al., 2016; Casella et al., 2019; Crowe et al., 2016; Herranz et al., 2015; Marthandan et al., 2014; Nelson et al., 2016; Sati et al., 2020; Zirkel et al., 2018). Correlation analysis (10 million randomized permutations) of DEGs identified in T21 iPSCs from our study (Iso-E and Iso-T) as well as from previously published dataset (Gonzales et al., 2018) showed a lack of correlation between DEGs identified in iPSCs and the various forms of senescence induction (Figure 6C; Table S34 and S35). In contrast, the DEGs identified in T21-NPCs (isogenic pair, female pair and male pair) had the highest correlation with the DEGs identified OSIS (10 million randomized permutations p-value < 1x10−07) and to a lesser extent with RS and SIPS. We found that T21 induced a marked expression increase of the senescence-associated cyclin-dependent kinase inhibitors (p16, p15 and p21), upregulation of senescence-associated secretory phenotype (SASP) genes (CXCL12, MMP24, MMP25 and LAMB1) and decreased expression of nuclear markers of senescence (LMNB1, P53, HMGB1 and HMGB2) similar to senescent cells induced through the various mechanisms, particularly OSIS and RS (Figure 6D; Table S34).
To identify the DEGs associated with senescence in T21-NPCs we focused our analysis on the DEGs identified through OSIS. We observe a significant overlap (randomized permutations p-value<1×10”07) between DEGs identified in T21-NPCs and OSIS, where 426 genes were downregulated and 584 genes are upregulated in both and hereafter referred to as senescence-associated (SA) DEGs (Figure 6E; Table S34). Next, we performed an enrichment analysis of the SA-DEGs identified in T21-NPCs and identified a significant enrichment of SA-downregulated genes in reduced-ID TADs (log2(obs/exp) = 1.17, p-value < 1x10−7), SA-upregulated genes in increased-ID TADs (log2(obs/exp) = 0.62, p-value < 1x10−7) and a modest but significant enrichment (log2(obs/exp) = 0.32, p-value = 0.00083) of SA-downregulated genes with less accessible promoters (Figure 6F; Table S35). The biological process associated with the SA-downregulated genes are predominantly involved in cell differentiation, proliferation and chromosome organization, whereas the SA-upregulated genes are associated with cell migration, vesicle mediated transport and inflammatory response (Figure 6G and H). The downregulated genes uniquely identified in T21-NPCs are involved in biological process associated with embryonic development, cell signaling and transcriptional regulation, conversely the downregulated genes uniquely identified in OSIS are associated with cell division, cell adhesion, chromosome organization, signal transduction and axon guidance (Figure S6G and H). Conversely, the upregulated genes uniquely identified in T21-NPCs are involved in cell differentiation, cell morphogenesis and chemotaxis, and the upregulated genes uniquely identified in OSIS are associated endoplasmic stress, cytokine signaling, phosphorylation, vesicle mediated transport and immune response (Figure S6I and J). In summary, our findings indicate that T21-NPCs exhibit features of senescent cells such as expression of senescent markers and a global transcriptional signature similar to OSIS.
Senolytics ameliorate the T21 induced phenotype in NPCs
Lifespan extension studies have identified senolytic drugs that could potentially reverse, delay or prevent aging associated diseases through selective removal of senescent cells (Serrano and Barzilai, 2018; Xu et al., 2018; Yousefzadeh et al., 2018; Zhu et al., 2017; Zhu et al., 2015). Early clinical trials utilizing a senolytic drug combination of a protein kinase inhibitor and an antioxidant, dasatinib and quercetin, have shown positive results in ameliorating the senescence-associated features in individuals with diabetic kidney disease (Hickson et al., 2019). Furthermore, studies indicate that senolytic drugs not only selectively remove senescent cells but also alleviate the transcriptional and heterochromatin architecture changes induced during senescence (Geng et al., 2019). Whole chromosome aneuploidies associated with aging and cancer, have been shown to induce premature cellular senescence (Andriani et al., 2016; Czarkwiani and Yun, 2018; Santaguida et al., 2017), however, T21-induced senescence and the beneficial impact of senolytic drugs remain unexplored. Thus, we investigated the consequences of treating human iPSC-derived NPCs harboring T21 with the senolytic drug combination of dasatinib and quercetin (DQ). The isogenic pair of NPCs were treated with either DQ (400nM of dasatinib and 15μM of quercetin) or vehicle (DMSO) for 5 days with daily replacement of culture media supplemented with the senolytic cocktail or vehicle (Figure 7A). First, we assessed the level of p16INK4a, a key marker of cellular senescence, in the isogenic pair of NPCs after treatment with DQ or DMSO (Figure 7B and C; Table S37). We observed that DQ treatment significantly reduced the elevated level of p16INK4a observed in the vehicle-treated Iso-T NPCs to those observed in vehicle-treated euploid NPCs (ANOVA p-value=1.2x10−2). Next, we interrogated the consequence of senolytic drugs on heterochromatin architecture in NPCs harboring T21 (Figure 7D). We found that 5 days of DQ treatment restored the heterochromatin distribution to similar levels observed in the euploid NPCs, as measured by the colocalization of H3K9me3 and LMNB1 (ANOVA p-value= 4.7x10−3) (Figure 7E; Table S38). Moreover, we observed that DQ treatment of Iso-T NPCs increased the LMNB1 levels to similar levels as observed in the Iso-E NPCs (ANOVA p-value= 2.4x10−4) (Figure 7F; Table S39).
Figure 7 – Senolytic drug alleviates the transcriptional and cellular dysfunctions induced by T21 in NPCs.
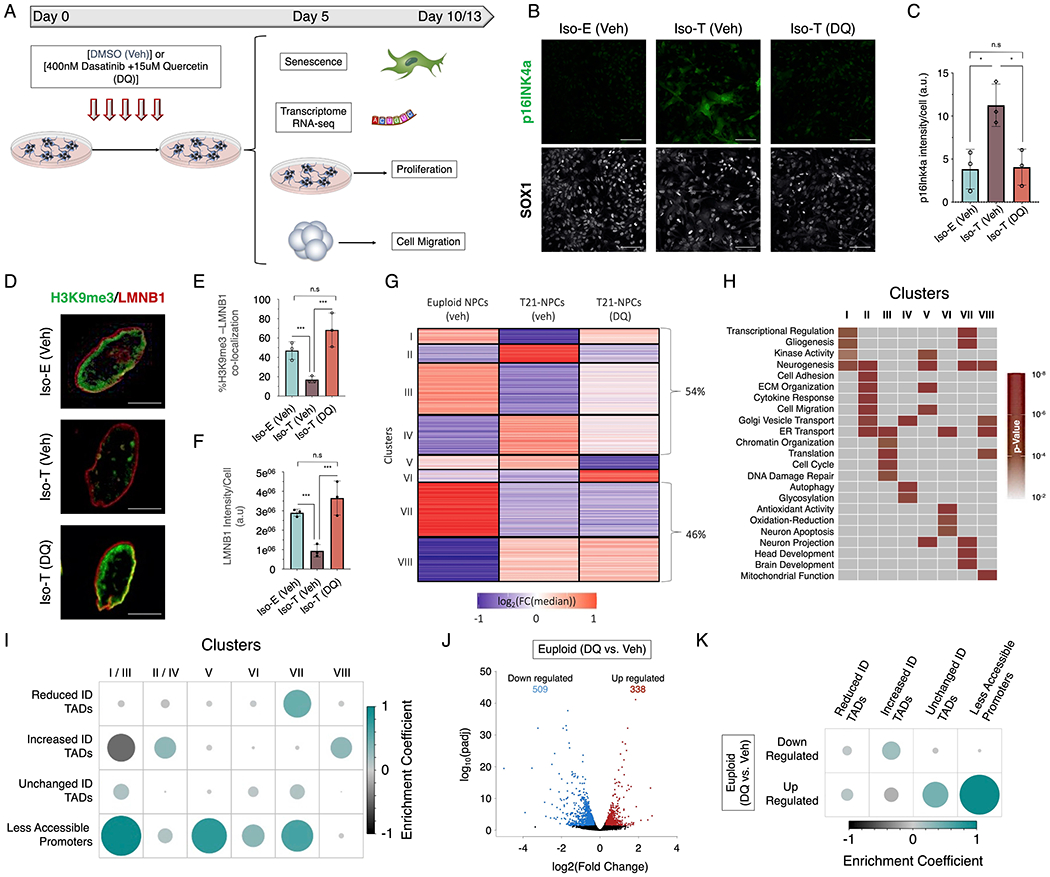
(A) Schematic of experimental design to assess the transcriptional and cellular consequences of the senolytic drug (DQ) treatment in isogenic T21 NPCs. (B) Immunofluorescence of p16INK4a (green) in euploid (Iso-E) and trisomic (Iso-T) NPCs (marked by SOX1, white) treated with vehicle or DQ. (C) Quantification of p16INK4a staining intensity in euploid (Iso-E) and trisomic (Iso-T, vehicle or DQ treatment) NPCs. (D) Immunofluorescence of H3K9me3 (green) and LMNB1 (red) in euploid (Iso-E) and trisomic (Iso-T) NPCs treated with vehicle or DQ. (E) Quantification of co-localization between H3K9me3 and LMNB1 in euploid (Iso-E) and trisomic (Iso-T) NPCs and (F) quantification of LMNB1 staining intensity of NPCs treated with vehicle or DQ. Each dot on the histogram represents a replicate and ~200 nuclei were analyzed per condition. (G) K-means clustering of the transcriptomic analysis of euploid (Iso-E) and trisomic (Iso-T) NPCs (treated with vehicle or DQ) identified 8 distinct clusters. Values in heatmap are plotted as the log2 fold change from the median. (H) Gene ontology of biological process associated with the 8 transcriptional clusters identified in response to senolytic drug treatment. (I) Enrichment plot of the randomized permutation test between the genes identified in the 8 different clusters with differential ID TADs as well as less accessible promoters. Genes from cluster I and III as well clusters II and IV were merged for this analysis. Color intensity (positive correlations are displayed in green and negative correlations in black) and the size of the circle are proportional to the enrichment coefficients. (J) Volcano plots of the differentially expressed genes (DEGs) identified in euploid NPCs treated with vehicle (DMSO) or DQ. (K) Enrichment plot of the randomized permutation test between DEGs identified in DEGs identified after DQ treatment with differential ID TADs as well as reduced promoter-accessibility. Color intensity (positive correlations are displayed in green and negative correlations in black) and the size of the circle are proportional to the enrichment coefficients.
Subsequently, we sought to explore the impact of senolytic treatment on the transcriptome of NPCs harboring T21. RNA-sequencing revealed that ~54% of the DEGs induced by T21 in NPCs were ameliorated after 5 days of DQ treatment of trisomic NPCs (Figure S7A; Table S40). Clustering of the DEGs identified 8 clusters: 2 clusters where gene expression was rescued to the same transcriptional levels as the euploid NPCs (clusters I and II), 2 clusters where gene expression changes were mitigated (cluster III and IV), 2 clusters with de novo changes in transcriptional levels (cluster V and VI), and 2 clusters that remained unchanged after DQ treatment (cluster VII and VIII) (Figure 5G). The de novo upregulated genes (cluster VI) represent a small percentage of the transcriptional changes induced by DQ treatment (<9%) and include a transcriptional response to the antioxidant activity of quercetin as well as neuronal apoptosis in response to the senolytic activity of DQ, indicating that NPCs harboring T21 are responsive to senolytic drug treatment. Analysis of apoptotic cells after 6hrs of DQ treatment revealed that only Iso-T NPCs have a significant increase of apoxin positive cells (p-value = 0.0004) (Figure S7B; Table S41).
We also observed amelioration of a significant number of genes associated with cell cycle regulation upon DQ treatment of T21 NPCs (cluster III). Notably, cell cycle dysfunction during neurodevelopment is a hallmark of DS (Contestabile et al., 2009; Contestabile et al., 2007). To examine cell cycle dynamics in T21 NPCs, we utilized a 2-hour BrdU incorporation to label newly replicated/synthesized DNA (a marker of S-phase cells) coupled with Ki-67 staining, a marker of all cycling cells irrespective of cell cycle phase (Figure S7C and D). After DQ treatment of trisomic NPCs, the proportion of proliferative (Ki-67+ and SOX1+) NPCs was rescued to the same levels as euploid NPCs (Iso-E(veh)=62.1%, Iso-T(veh)=44.9% and Iso-T(DQ)=73.1%, ANOVA p-value= 2.9x10−) (Figure S7E; Table S41). Furthermore, analysis of the NPCs that have traversed through S-phase (BrdU+ and SOX1+) in the 2 hour BrdU incorporation window showed that cycling NPCs harboring T21 had a significant reduction in S-phase cells (Iso-E(veh)=25.3% and Iso-T(veh)=13.4%, Tukey’s padj<1.0x10−4), similar to what has previously been observed in the developing brain of individuals with DS (Fig S7F; Table S42) (Contestabile et al., 2009; Contestabile et al., 2007). However, DQ treatment rescued the percentage of Iso-T NPCs that traversed through S-phase (Iso-T(veh)=26.7%) to comparable levels as those observed in euploid NPCs (Tukey’s padj=1.0x10−4).
Senolytic treatment also rescued the transcriptional levels of genes upregulated in NPCs harboring T21 that are involved in processes associated with ECM organization, cell-adhesion and cell migration, to similar levels as the euploid NPCs (cluster II). Further, we observed that DQ treatment resulted in de novo downregulation of genes involved in ECM organization and cell migration that were not previously altered as a consequence of DS (cluster V) (Figure 7G). Thus, we explored the impact of senolytic treatment on the migratory abilities of NPCs harboring T21, a process disrupted in DS as well as in senescent cells (Huo et al., 2018; Romanov et al., 2010). To interrogate the consequences of senolytics on the cellular migration of NPCs harboring T21, we treated the isogenic pair of NPCs with either vehicle or DQ for 5 days in a monolayer culture system (Figure S7G). Next, NPCs were allowed to self-aggregate into neurospheres for an additional 5 days without vehicle or DQ (Day 5-10), before embedding them into 3D-Matrigel scaffold (Figure S7H). After 3 days (Day 13) the embedded neurospheres were stained for the NPC marker SOX1 and the number of migratory cells as well as the distance traveled by each cell from the edge of the neurosphere were assessed (Figure S7I). We found DQ treatment of NPCs harboring T21 rescued both the average number of migratory cells per neurosphere (ANOVA p-value < 1.0x10−4), as well as the average distance migrated by NPCs away from the edge of the neurosphere (ANOVA p-value < 1.0x10−4) (Figure S7J and K; Table S43).
Next, we sought to characterize the genes impacted by senolytic treatment in terms of chromatin-accessibility and TAD organization. We identified that the genes downregulated in NPCs as a consequence of T21 and upregulated by DQ (clusters I and III) were enriched for the genes with reduced promoter-accessibility (log2(obs/exp) = 1.28, p-value < 1×10−7), whereas genes upregulated as a consequence of T21 and downregulated after treatment with DQ (clusters II and IV) are predominantly localized in increased-ID TADs (log2(obs/exp) = 0.37, p-value < 1x10−7) (Figure 7I; Table S44). The de novo downregulated (cluster V) and upregulated (cluster VI) genes as a consequence of treating with DQ were both enriched for genes with reduced promoter-accessibility as a consequence of T21. The upregulated genes as a consequence of T21 and that remained upregulated after treating with DQ (cluster VIII) were localized in increased-ID TADs (log2(obs/exp) = 0.42, p-value < 1x10−7), whereas the downregulated genes that were not impacted by DQ (Cluster VII) were enriched in reduced-ID TADs (log2(obs/exp) = 0.71, p-value < 1x10−7) and genes with less accessible promoters (log2(obs/exp) = 0.8, p-value < 1x10−7). Moreover, we observe that ~20% of downregulated (clusters I/III) and ~30% of upregulated genes (clusters II/IV) ameliorated by senolytic drug treatment are SA-DEGs (Figures S7L; Table S45). The ameliorated SA-DEGs are involved in biological process predominantly associated with cell cycle regulation (clusters I/III) and cell migration (clusters II/IV) (Table S45).
To interrogate whether senolytics directly impact the transcriptome of non-senescent cells we treated euploid NPCs with DQ for 5 days and performed RNA-seq. We identified 338 upregulated and 509 downregulated genes in Iso-E NPCs treated with DQ (Figure 7J; Table S46). We observed that the upregulated genes identified in DQ-treated Iso-E NPCs were predominantly enriched for biological processes associated with response to oxidative stress which is expected due to the antioxidant activity of quercetin (Figure S7M). Conversely, we observed that many of the key biological processes associated with genes upregulated in NPCs harboring T21 were downregulated in euploid NPCs treated with DQ, such as ECM organization, cell migration, cell differentiation and chemotaxis (Figure S7N) (Xu et al., 2016). Accordingly, DQ treatment of Iso-E NPCs significantly increased the average number of migratory cells per neurosphere as well as the average distance migrated by Iso-E NPCs away from the edge of the neurosphere (Figure S7J and K; Table S43). Finally, we observed that DQ treatment of euploid NPCs induced downregulation of genes localized to increased-ID TADs in T21 NPCs (log2(obs/exp) = 0.34, p-value = 8.3x10−5), the region of the genome that harbors many of the genes found to be upregulated in T21-NPCs (Figure 7J; Table S46). Whereas the genes upregulated in DQ-treated euploid NPCs are predominantly enriched in the TADs with unchanged-ID (log2(obs/exp) = 0.4, p-value < 1x10−7) and that exhibit reduced promoter-accessibility in T21 NPCs (log2(obs/exp) = 0.95, p-value < 1x10−7). In summary, senolytic drug treatment of NPCs harboring T21 reduced the number of senescent cells, alleviated the heterochromatin architecture changes, ameliorated the genome-wide transcriptional disruption, mitigated the cell migration deficits and increased the population of proliferative cells.
Discussion
Triplication of chromosome 21 (T21) impacts the brain development of nearly every individual with DS and currently it is estimated that more than 400,000 individuals live with this condition in United States alone (Joslyn et al., 2019; Presson et al., 2013). Studies utilizing human derived tissue samples and DS mouse models, leveraging next generation sequencing have established that T21 induces genome-wide transcriptional disruption in addition to the gene dosage dependent transcriptional upregulation (Bianco et al., 2016; Gonzales et al., 2018; Letourneau et al., 2014; Olmos-Serrano et al., 2016; Tyler and Haydar, 2013; Walus et al., 2016). However, the underlying molecular mechanisms contributing to an altered global transcriptional profile as a consequence of T21 were unknown. Here we utilized human iPSCs and iPSC-derived forebrain NPCs to decode the molecular mechanisms underlying these genome-wide transcriptional changes. While both iPSCs and NPCs exhibit a similar level of dosage-dependent transcriptional upregulation of a subset of expressed genes on HSA21, unexpectedly, we find that T21 has a distinct genome-wide impact on the transcriptome, epigenome and 3D-genome of NPCs that is absent in iPSCs. Even though a cell-type specific response to T21 is expected from the organ specific etiologies observed in DS (Weijerman and de Winter, 2010), the mechanisms governing this cell-type specific response remain unclear and require further studies to fully elucidate this distinct response to T21.
Globally, we observe chromosomal introversion in NPCs harboring T21, where every chromosome has reduced interactions with other chromosomes and increased self-interactions as compared to euploid NPCs. Furthermore, this increase in chromosomal self-interactions occurs within TADs, where promoter-enhancer interactions are typically observed (Robson et al., 2019). The genome is organized into two compartments: the active (A) compartments which is predominantly localized towards the core of nucleus, and the inactive (B) compartment which is anchored to the nuclear lamina on the nuclear periphery (Lieberman-Aiden et al., 2009). In T21 NPCs, the chromosomal introversion is predominantly observed in regions of the genome associated with the nuclear lamina (B-compartment) and this feature is associated with disruption of LADs which leads to intra-nuclear B-compartment bundles. In NPCs harboring T21, we find that the genome-wide transcriptional upregulation is enriched within TADs in the B-compartment that have increased chromosomal interaction densities. Conversely, in the A-compartment we observed that T21 reduced the chromatin-accessibility on promoters harboring SP1 and NFY motifs which are associated with the observed transcriptional downregulation in NPCs harboring T21. We find that the biological processes disrupted in T21 NPCs are defined by distinct structural alterations. While downregulated genes involved brain-development associated biological processes are predominantly localized within TADs with reduced-ID, the upregulated genes involved in cell migration are localized within TADs with increased-ID, and the downregulated genes involved in cell proliferation and chromosome organization are associated with loss of chromatin-accessibility.
Currently, there are no therapeutic interventions for individuals with DS and recent studies in the field of aging research have established promising therapeutic avenues targeting senescent cells (Kirkland et al., 2017). Senolytic drugs not only eliminate senescent cells but also restore key cellular and molecular hallmarks associated with senescence in various diseases including Alzheimer’s disease, a comorbidity in individuals with DS (Geng et al., 2019; Head et al., 2012; Zhang et al., 2019). Chromosomal introversion and disruption of the nuclear lamina are key features of senescence (Criscione et al., 2016; Zirkel et al., 2018), and our transcriptional analysis from three different individuals with DS revealed that NPCs harboring T21 exhibit a transcriptional profile that resembles senescence, and display several key senescence-associated phenotypes such as elevated levels of SA-β-galactosidase, p16INK4a and HP1a. Furthermore, similar to what has been observed with other senescence induction mechanisms we observe heterogeneity in the NPC population entering deep senescence, which may indicate a temporal propagation of senescence and eliminating these cells may have beneficial consequences (Bourgeron et al., 2015; Kirschner et al., 2020; Kumari and Jat, 2021; Xu and Teixeira, 2019). We find that treating NPCs harboring T21 with the senolytic drug combination of dasatinib and quercetin ameliorated the senescence-associated genome-wide transcriptional and heterochromatin architecture disruptions. Moreover, treatment with senolytics also restored the migratory and proliferative capacity in T21 NPCs, which are hallmark dysfunctions of the developing brain in individuals with DS (Chakrabarti et al., 2007). Our studies show that in addition to the elimination of senescent cells, senolytic drugs directly impact the transcriptome vulnerable to T21, indicating that senolytic drugs have the potential to serve as therapeutic opportunities for individuals with DS.
Recent studies have shown that senescence is a common feature of normal development (Munoz-Espin et al., 2013; Storer et al., 2013; Vasey et al., 2008), however, our findings suggest that excessive NPC-senescence induced by T21 could be a key feature driving the neurodevelopmental abnormalities observed in individuals with DS. Studies have shown that the most impacted regions of the developing brain in individuals with DS are the hippocampus and cerebellum (Contestabile et al., 2009; Contestabile et al., 2007; Guidi et al., 2011).). In both the cerebellum and hippocampus, neurogenesis continues to occur in the very early postnatal period, and additionally, hippocampal neurogenesis continues at a slower rate throughout life, providing a window of opportunity for therapeutic intervention (Stagni et al., 2015). Furthermore, studies have shown that neurons and glial cells are susceptible to senescence during aging and Alzheimer’s disease, and individuals with DS are prone to premature aging and developing early-onset Alzheimer’s disease (Bussian et al., 2018; Horvath et al., 2015; Martinez-Cue and Rueda, 2020; Zhang et al., 2019). These findings indicate that senolytics may have beneficial therapeutic properties for adults with DS as well, however, further studies are required to explore T21-induced senescence in more mature cell types of the adult human brain.
In summary, our findings indicate that senescence may play a key role in the neurodevelopmental pathogenesis of DS and we show that senolytics provide an exciting therapeutic avenue for treating individuals with DS by restoring NPC-dysfunctions induced by T21. Furthermore, the identification of the molecular mechanisms governing senescence-associated genome-wide transcriptional disruption opens potential opportunities for the identification of therapeutic interventions targeting senescent cells in numerous disorders.
Limitations of study
The iPSCs and NPCs generated for this study originated from individuals that deceased at a very young age, indicating the severity of trisomy 21 in these individuals and thus samples derived from older or non-deceased individuals may yield a milder phenotype than described in this study. There are currently several approaches for generating iPSCs and NPCs as well as identifying chromatin accessibility, lamina-associated domains and spatial chromatin organization and thus utilizing these different approaches or sampling at different time points may yield varying results to those described in this manuscript. While we compared NPCs derived from both male and female patients, the relatively small number of patients sampled also represents a limitation of our study. Further, patient iPSCs and iPSC-derived NPCs are a valuable tool to model the neurodevelopmental aspects of Down syndrome in-vitro, however they may not fully recapitulate in-vivo disease pathogenesis.
STAR Methods
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Li-Huei Tsai (lhtsai@mit.edu).
Materials availability
Unique resources and reagents generated in this study are available from the Lead Contact with a completed Material Transfer Agreement.
Data and code availability
Genomic sequencing data have been deposited at Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| OCT-4 | Cell Signaling Technology | Cat# 2890, RRID:AB_2167725 |
| Nanog | Cell Signaling Technology | Cat# 4893, RRID:AB_10548762 |
| SOX1 | R&D Systems | Cat# AF3369, RRID:AB_2239879 |
| Nestin | Millipore Sigma | Cat# MAB5326, RRID:AB_2251134 |
| PAX6 | R&D Systems | Cat# AF8150, RRID:AB_2827378 |
| H3K9me3 | Abcam | Cat# ab176916, RRID:AB_2797591 |
| HP1a | Cell Signaling Technology | Cat# 2616, RRID:AB_2070987 |
| H3K27me3 | Abcam | Cat# ab6002, RRID:AB_305237 |
| HMGB1 | Thermo Fisher Scientific | Cat# PA1-16926, RRID:AB_2248274 |
| LMNB1 | Abcam | Cat# ab16048, RRID:AB_10107828 |
| p16INK4a | Thermo Fisher Scientific | Cat# MA5-32133, RRID:AB_2809424 |
| KI-67 | Abcam | Cat# ab15580, RRID:AB_443209 |
| Vimentin | Abcam | Cat# ab24525, RRID:AB_778824 |
| GFAP | Millipore | Cat# AB5804, RRID:AB_2109645 |
| BrdU | Cell Signaling Technology | Cat# 5292, RRID:AB_10548898 |
| Alexa-Fluor 488 (Donkey anti-Goat) | Thermo Fisher Scientific | Cat# A-11015, RRID:AB_2534082 |
| Alexa-Fluor 488 (Donkey anti-Rabbit) | Thermo Fisher Scientific | Cat# A-21206, RRID:AB_2535792 |
| Alexa-Fluor 488 (Donkey anti-Mouse) | Thermo Fisher Scientific | Cat# A-21202, RRID:AB_141607 |
| Alexa-Fluor 488 (Donkey anti-Sheep) | Thermo Fisher Scientific | Cat# A-11015, RRID:AB_2534082 |
| Alexa-Fluor 555 (Donkey anti-Goat) | Thermo Fisher Scientific | Cat# A-21432, RRID:AB_2535853 |
| Alexa-Fluor 555 (Donkey anti-Rabbit) | Thermo Fisher Scientific | Cat# A-31572, RRID:AB_162543 |
| Alexa-Fluor 555 (Donkey anti-Mouse) | Thermo Fisher Scientific | Cat# A-31570, RRID:AB_2536180 |
| Alexa-Fluor 555 (Donkey anti-Sheep) | Thermo Fisher Scientific | Cat# A-21099, RRID:AB_2535753 |
| Alexa-Fluor 647 (Donkey anti-Goat) | Thermo Fisher Scientific | Cat# A-21447, RRID:AB_2535864 |
| Alexa-Fluor 647 (Donkey anti-Rabbit) | Thermo Fisher Scientific | Cat# A-31573, RRID:AB_2536183 |
| Alexa-Fluor 647 (Donkey anti-Mouse) | Thermo Fisher Scientific | Cat# A-31571, RRID:AB_162542 |
| Alexa-Fluor 647 (Donkey anti-Sheep) | Thermo Fisher Scientific | Cat# A-21448, RRID:AB_2535865 |
| DAPI | Thermo Fisher Scientific | Cat# D1306, RRID:AB_2629482 |
| Biological Samples | ||
| Iso-E and Iso-T iPSCs (AG05397) | Coriell (Maclean et al., 2012) | Cat# AG05397 |
| Ma-E iPSCs | Cohen, McLean, HMS (Raja et al., 2016) | Cat# CS-0020-01 |
| Ma-T Fibroblast | Coriell | Cat# AG06922 |
| Fe-E iPSCs | WiCell | Cat# WA09 |
| Fe-T Fibroblasts | Coriell | Cat# GM04616 |
| CF1 Mouse embryonic fibroblasts, irradiated | Thermo Fisher Scientific | Cat# A34181 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Fluoromount-G | Electron Microscopy Sciences | Cat# 17984-25 |
| PBS, pH 7.4 | Thermo Fisher Scientific | Cat# 10010049 |
| Triton X-100 | Sigma-Aldrich | Cat# T8787-100ML |
| Paraformaldehyde | Electron Microscopy Sciences | Cat# 15714-S |
| 5-Bromo-2′-deoxyuridine | Sigma-Aldrich | Cat# B5002-5G |
| Corning® Matrigel® hESC-Qualified Matrix | VWR International | Cat# 354277 |
| Corning® Matrigel® Basement Membrane Matrix | VWR International | Cat# 356254 |
| DMEM/F12 Glutamax | Life Technologies | Cat# 10565-042 |
| Knockout Serum Replacement | Life Technologies | Cat# 10828028 |
| MEM non-Essential amino acid solution | Sigma-Aldrich | Cat# M7145 |
| Glutamax | Thermo Fisher Scientific | Cat# 35050-079 |
| recombinant human FGF-basic | PeproTech | Cat# 100-18B |
| 2-mercaptoethanol | Sigma-Aldrich | Cat# M6250 |
| mTeSR1 medium | Stemcell technologies | Cat# 85850 |
| ReLeSR™ | Stemcell Technology | Cat# 05872 |
| AggreWell™800 | Stemcell Technology | Cat# 34811 |
| Neurobasal | Thermo Fisher Scientific | Cat# 21103049 |
| N-2 Supplement | Invitrogen | Cat# 17502-048 |
| B-27 Serum-Free Supplement | Gibco | Cat# 12587010 |
| insulin | Thermo Fisher Scientific | Cat# 41400045 |
| Pen/strep | Gemini Bio-Products | Cat# 400-109 |
| Dorsomorphin | PeproTech | Cat# 8666430 |
| SB431542 | Tocris | Cat# 1614 |
| STEMdiff™ Neural Rosette Selection Reagent | Stemcell technologies | Cat# 05832 |
| Sulfuric acid | Sigma-Aldrich | Cat# 339741-500ML |
| Trichloroacetic acid | BDH | Cat# BDH0310-500G |
| Trypsin | Promega | Cat# V5113 |
| C18 Sep-Pak cartridges | Waters | Cat# 186002318 |
| TrypLE | Thermo Fisher Scientific | Cat# 12605028 |
| Igepal CA-630 (NP-40) | Sigma-Aldrich | Cat# I8896 |
| AMPure beads | Beckman Coulter | Cat# A63880 |
| Critical Commercial Assays | ||
| CytoTune-iPS Sendai Reprogramming kit | Thermofisher scientific | Cat# A16518 |
| Kapa mRNA hyperprep kit for standard RNA libraries | Roche | Cat# KK8540 |
| Nextera DNA Library Prep Kit | Illumina | Cat# FC-121-1030 |
| Qiagen MinElute PCR Purification Kit | Qiagen | Cat# 28006 |
| Arima Hi-C kit | Arima Genomics | N/A |
| Kappa Hyperprep Kit | Roche | Cat# KK8502 |
| Deposited Data | ||
| Hi-C | This paper | GEO: GSE185192 |
| LMNB1 ChIP-seq | This paper | GEO: GSE185192 |
| RNA-seq | This paper | GEO: GSE185192 |
| ATAC-seq | This paper | GEO: GSE185192 |
| Senescence RNA-seq | Casella et al., 2019; Crowe et al., 2016; Alspach et al., 2014; Sati et al., 2020; Zirkel et al., 2018; Borras-Fresneda et al., 2016; | GEO: GSE130727, GSE58910, GSE56293, GSE130306, GSE98448, GSE80207, |
| RNA-seq of iPSC derived from Down syndrome and control individuals | Gonzales et al., 2018 | GSE101942 |
| RNA-seq of iPSC derived neurons, astrocytes and OPCs | Garcia-Leon et al., 2018; Lin et al., 2018; Tcw et al., 2017 | GEO: GSE106984, GSE102956, GSE97904 |
| ChIP-seq of Histone Modifications | Hawkins et al., 2010 | GEO:GSM956008 |
| Experimental Models: Cell Lines | ||
| Iso-E and Iso-T (AG05397) | Coriell Maclean et al., 2012) | Cat# AG05397 |
| Ma-E | Cohen, McLean, HMS (Raja et al., 2016) | Cat# CS-0020-01 |
| Ma-T | Coriell | Cat# AG06922 |
| Fe-E | WiCell | Cat# WA09 |
| Fe-T | Coriell | Cat# GM04616 |
| CF1 Mouse embryonic fibroblasts, irradiated | Thermo Fisher Scientific | Cat# A34181 |
| Software and Algorithms | ||
| Imarisx64 9.2.1 | Bitplane | http://www.bitplane.com/Default.aspx |
| ZEN imaging software | Carl Zeiss | https://www.zeiss.com/microscopy/int/products/microscope-software/zen.html |
| STAR (v.2.6.1a) | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| HTSeq | Anders et al., 2015 | https://htseq.readthedocs.io/en/master/ |
| RUVSeq | Risso et al., 2014 | https://bioconductor.org/packages/release/bioc/html/RUVSeq.html |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| Homer | Heinz et al., 2010 | (http://homer.ucsd.edu/homer/interactions2/index.html |
| MACS2.1.0 | Zhang et al., 2008 | https://github.com/taoliu/MACS |
| R | Chen et al., 2009 | https://www.r-project.org/ |
| Corrplot | Taiyun Wei and Viliam Simko (2017) | https://github.com/taiyun/corrplot |
| Diffbind v1.16.3 | Ross-Innes et al., 2012 | https://bioconductor.org/packages/release/bioc/html/DiffBind.html |
| Burrows-Wheeler Aligner (BWA) | Li and Durbin, 2009 | http://bio-bwa.sourceforge.net/ |
| Juicer | Rao et al., 2014 | https://github.com/aidenlab/juicer/wiki |
| Graphpad Prism 8 | Graphpad | https://www.graphpad.com/ |
| Distiller | Abdennur and Mirny, 2020 | https://github.com/mirnylab/distiller-nf)) |
| Hi-C explorer | Ramirez et al., 2018 | https://hicexplorer.readthedocs.io/en/latest/content/list-of-tools.html |
| BEDTools | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| deepTools | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ |
Experimental Model and Subject Details
Induced pluripotent stem cell (iPSC) generation and neural progenitor cell (NPC) differentiation
iPSCs -
The isogenic pair (Iso-E and Iso-T) of induced pluripotent stem cells (iPSCs) were provided by Dr. Stuart Orkin, Dana-Farber Cancer Institute, which have been described previously (Coriell, AG05397) (Maclean et al., 2012). The DS-iPSCs from the male individual (Ma-T, AG06922) purchased from Coriell and the female control iPSC (Fe-E, H9 WA09) purchased from WiCell. Fibroblasts of the derived from a female with DS, purchased from Coriell (GM04616) and fibroblasts from the male control (CS-0020-01) were utilized to generate iPSCs using Sendai virus to overexpress OCT4, SOX2, KLF4, and c-MYC, utilizing the CytoTune-iPS Sendai Reprogramming kit (Thermofisher scientific, A16518) as previously described (Raja et al., 2016). iPSCs were cultured on irradiated mouse embryonic fibroblasts (MEFs, Thermo Fisher Scientific, A34181) in DMEM/F12 media supplemented with knockout serum replacement (KSR, 20% v/v), MEM non-Essential amino acid solution (Sigma-Aldrich M7145), Glutamax (Thermo Fisher Scientific 35050-079), 8 ng/μl recombinant human FGF-basic (FGF2, PeproTech, 100-18B) and 71.5μM 2-mercaptoethanol (Sigma-Aldrich M6250). The quality of cells was monitored daily and differentiated cells were mechanically removed under a light microscope in a biosafety hood. iPSCs were cultured to 80% confluence and non-differentiated colonies were transferred to a feeder-free system and grown on Matrigel hESC-Qualified Matrix coated plates (VWR, 354277) using mTeSR1 medium (Stemcell technologies, 85850). iPSCs for RNA-seq, ATAC-seq, Hi-C and immunofluorescence imaging were grown under feeder-free conditions.
NPCs –
NPCs were differentiated as previously described with minor adjustments (Chambers et al., 2009). iPSCs were grown to 80% confluence on feeder-free conditions in mTeSR1 medium. iPSCs were then dissociated into single cell suspension using ReLeSR™ (Stemcell Technology, 05872) following the manufacturers guidelines. 3 million cells were then transferred into a single well of AggreWell™800 (Stemcell Technology, 34811) to form 300 embryoid bodies of 10,000 iPSCs each in NPC-differentiation medium [1:1 of DMEM/F12 Glutamax and Neurobasal (Life Technologies 10565-042 and Thermo Fisher Scientific 21103049), N-2 Supplement (Invitrogen,17502-048), B-27 Serum-Free Supplement (Gibco, 12587010), 2.5pg/l insulin (Thermo Fisher Scientific, 41400045), Glutamax (Thermo Fisher Scientific, 35050-079), MEM non-Essential amino acid solution (Sigma-Aldrich, M7145), 71.5μM 2-mercaptoethanol, Pen/strep (Gemini Bio-Products, 400-109), Dorsomorphin (PeproTech, 8666430) and SB431542 (Tocris,1614)]. Half NPC-induction medium replacement was performed daily for 5 days and on day 6 EBs were washed and transferred onto Matrigel coated plates. Complete NPC-induction medium replacement was performed daily for 7 days, and on day 12 STEMdiff™ Neural Rosette Selection Reagent (Stemcell technologies, 05832) was utilized to isolate NPC-rosettes and re-plated on onto fresh Matrigel coated plates in NPC-induction medium. Complete NPC-induction medium replacement was performed for 7 days and on day 20 NPCs were transferred onto fresh Matrigel coated plates in NPC-expansion medium [1:1 of DMEM/F12 Glutamax and Neurobasal (Life Technologies 10565-042 and Thermo Fisher Scientific 21103049), N-2 Supplement (Invitrogen, 17502-048), B-27 Serum-Free Supplement (Gibco, 12587010), 2.5μg/l insulin (Thermo Fisher Scientific, 41400045), Glutamax (Thermo Fisher Scientific, 35050-079), MEM non-Essential amino acid solution (Sigma-Aldrich, M7145), 71,5μM 2-mercaptoethanol, Pen/strep (Gemini Bio-Products, 400-109) and 8 ng/μl recombinant human FGF-basic (FGF2, PeproTech, 100-18B)]. NPCs were passaged once a week and NPCs for RNA-seq, ATAC-seq, Hi-C, senolytic drug treatment and immunofluorescence imaging were used on passage 3-5.
Method Details
Immunofluorescence imaging and analysis
iPSCs and NPCs grown on coverslips were fixed with 4% paraformaldehyde in 1X PBS for 15 minutes, then incubated overnight at 4°C in primary antibody in 1X PBS with 0.3% Triton X-100. Primary antibodies used were OCT-4 (CST C52G3), Nanog (CST 1E6C4), SOX1 (R&D Systems AF3369), Nestin (Millipore Sigma MAB5326), PAX6 (R&D Systems AF8150), H3K9me3 (Abcam 176916), LMNB1 (Abcam 16048), p16INK4a (Thermo Fisher Scientific MA5-32133), KI-67 (Abcam 15580) and BrdU (CST 5292S) 1:500 dilution overnight at 4oC. Coverslips were washed three times with PBS and incubated with Alexa-Fluor 488, Alexa-Flour 555 and/or Alex-Fluor 647 secondary antibodies (Thermo Fisher Scientific) 1:1,000 dilution, and DAPI (Invitrogen D1306) 1:5,000 dilution for two hours at room temperature. The coverslips were mounted on microscope slides with Fluoromount-G (Electron Microscopy Sciences 17984-25). The slides were imaged using a confocal microscope (LSM 710 and 880, Zeiss) with a 5X, 20x or 63x objective with identical settings for all matched images.
Heterochromatin architecture –
iPSCs and NPCs grown on coverslips were fixed with 4% paraformaldehyde in 1X PBS for 12 minutes, then incubated overnight at 4°C in primary antibody in 1X PBS with 0.3% Triton X-100. Primary antibodies used were anti-LMNB1 (1:500) and anti-H3K9Me3 (1:1,000). Primary antibody targets were visualized with Alexa-Fluor 488 and Alex-Fluor 647 secondary antibodies, and cell nuclei were stained with DAPI. The coverslips were mounted on microscope slides with Fluoromount-G. The slides were imaged using a confocal microscope (LSM 880, Zeiss) with a 63x objective and identical settings were used for all coverslips. Images were quantified using Imarisx64 9.2.1 (Bitplane, Zurich, Switzerland), to determine the number of heterochromatin aggregates (as labelled with H3K9Me3) and degree of co-localization with LMNB1 for the isogenic euploid, trisomic, and drug-treated trisomic cells. For each image, first, a “generous” surface was created on the LMNB1 staining and used to mask the H3K9Me3 channel, to eliminate general extranuclear and background signal. The “generous” LMNB1 surface had surface detail of 0.6, the Imaris auto-generated surface threshold, and a volume gate of greater than 40 μm3. Then, a “stringent” LMNB1 surface was created, to hone in on the areas of the nuclei with the strongest LMNB1 signal. These “stringent” surfaces had surface detail of 0.2, surface threshold of 105, and no volume gate. Finally, spots were created on the H3K9Me3 channel, with diameter 0.65 μm and quality threshold 15.6. To quantify the co-localization, the “Find spots close to surface” command was used between the H3K9Me3 spots and the “stringent” LMNB1 surface, with a threshold of 0.35 μm. For each of the images, a total number was determined for both H3K9Me3 spots co-localized with LMNB1 and intra-nuclear H3K9Me3 aggregates.
Proliferation –
NPCs were treated with 10μM BrdU for 2hrs before fixing the cells with 4% paraformaldehyde in 1X PBS for 12 minutes. DNA hydrolysis was performed by treating NPCs with 1.5M HCl for 10mins at room temperature and washed 3x with 1X PBS. Cells were then permeabilized and primary antibodies was used against BrdU (1:500), KI-67 (1:1,000) and SOX1 (1:100). Primary antibody targets were visualized with Alexa-Fluor 488, Alexa-Fluor 555 and Alex-Fluor 647 secondary antibodies, and cell nuclei were stained with DAPI. The coverslips were mounted on microscope slides with Fluoromount-G. The slides were imaged using a confocal microscope (LSM 880, Zeiss) with a 20x objective and identical settings for all coverslips. Images were quantified using Imarisx64 9.2.1 (Bitplane, Zurich, Switzerland), to determine the proportion of proliferative NPCs. SOX1 surface were used to identify all the NPCs in the images with a setting of 0.75μm surface grain size. Then BrdU and KI-67 positive cells under the SOX1 surface were quantified with a mean intensity of >18 and >10, respectively.
Cell Migration –
Matrigel embedded neurospheres in 96-well glass bottom plates were fixed with 4% paraformaldehyde in 1X PBS for 15 minutes, then incubated overnight at 4°C in primary antibody in 1X PBS with 0.3% Triton X-100 with primary SOX1 (1:100) and Nestin (1:500). SOX1 was visualized with Alexa-Fluor 488 and Nestin was visualized with Alexa-Fluor 555 secondary, and nuclei were stained with DAPI. Fluoromount-G was then added onto Matrigel embedded neurospheres. The Matrigel embedded neurospheres were imaged using a confocal microscope (LSM 710, Zeiss) with a 5x objective and identical settings were used for all images. Images were then quantified using Imarisx64 9.2.1 (Bitplane, Zurich, Switzerland), to determine how far individual NPCs had migrated out of the neurospheres for the isogenic pair of euploid, trisomic and drug-treated trisomic neurospheres. For each image, first, a DAPI sphere surface was created to demarcate the boundaries of the neurosphere core. This surface had surface detail of 2, surface threshold of the Imaris auto-generated threshold divided by two, and a volume gate such that all but the largest surface is eliminated. Next, the DAPI sphere surface was used to mask the SOX1 channel, to isolate the signal from NPCs that had migrated out of the primary neurosphere. Finally, spots were created on the masked SOX1 channel, with diameter 15 μm and quality threshold 5. Then, the distance between the SOX1 spots and the DAPI sphere surface was calculated and a list of individual distance values for each migrated NPC was generated for each neurosphere.
Global Chromatin Profiling (GCP)
The Global Chromatin Profiling (GCP) assay was performed as previously described (Creech et al., 2015; Litichevskiy et al., 2018). Briefly, three biological and three technical replicates of neural progenitor cells (NPCs), derived from isogenic-pair (AG-005397 (iso-E and Iso-T)), were grown in 6-well plates, washed in ice cold PBS 2x, scraped, and collected by centrifugation. After nuclei were isolated using mild lysis conditions (0.3% NP40), histones were extracted using sulfuric acid (Sigma-Aldrich, 339741-500ML), and precipitated using trichloroacetic acid (BDH, BDH0310-500G). Samples (10μg each) were propionylated, desalted and digested by trypsin (Promega, V5113) overnight following standard protocols. A second round of propionylation followed by desalting using C18 Sep-Pak cartridges (Waters, 186002318) was employed. A mix of isotopically labeled synthetic peptides for histone H3 was added as a reference to each sample prior to mass-spectrometric (MS) analysis. Peptides were separated on a C18 column (EASY-nLC 1000, Thermo Scientific) and analyzed by mass spectrometry (MS) in a parallel reaction monitoring mode (QExactiveTM Plus, Thermo Scientific) as previously described(Creech et al., 2015). Detailed protocols of sample preparation steps can be found in https://panoramaweb.org/labkey/wiki/LINCS/Overview%20Information/page.view?name=sops.
Library preparation and analysis
RNA-seq -
Biological replicates (n=3) of the isogenic pair (Iso-E and Iso-T) of iPSCs and NPCs as well as the male (Ma-E and Ma-T) and female (Fe-E and Fe-T) pairs were used for library preparation. RNA quality was checked using RNA Integrity number on an Agilent Bioanalyzer 2100, and samples with a RIN score of >9 were used for library preparation. 150 ng of RNA was used to prepare libraries using the Kapa mRNA hyperprep kit for standard RNA libraries (Roche, KK8540) on a TecanEvo 150s (Tecan). >50 million 75-bp paired-end reads were acquired for each replicate using the NextSeq 500 platform at the MIT BioMicro Center. FASTQ paired-end reads were aligned using STAR (v.2.6.1a) to GRCh37 reference genome (GENCODE 19) (Dobin et al., 2013). Transcripts were quantified using HTSeq, data was normalized utilizing RUV-seq and differential gene-expression analysis was performed through DESeq2 (Anders et al., 2015; Love et al., 2014; Risso et al., 2014). Significant differentially expressed genes (DEGs) were called with an FDR < 0.01, log2 fold change ≥ 0.5 or ≤ −0.5. Gene ontology analysis was performed using GOrilla with two unranked lists of genes (target and background lists) followed by REViGO a program that can takes a long list of Gene Ontology terms and removes redundant GO terms and generates a scatterplot with an unbiased reduction of the GO terms in semantic space (x, y) (Eden et al., 2009; Supek et al., 2011). Correlation analysis of the pairwise comparisons was performed using corrplot (https://github.com/taiyun/corrplot), utilizing the log2 fold overlap enrichment and significance p-value generated through randomized permutation test. Randomized permutation test was done in R using custom scripts. Briefly, a sample set of equal size as the observed data was randomly selected from the background set and then the statistic test was calculated for each randomly sampled data (s* = s1, s2,…sN) exactly as for the observed data (sObs). This was iterated N=107 times with replacement to create the null distribution and the p-value was calculated using the formula.
If sObs was greater than or less than all values in the null distribution, the p-value was set as < < 1E-07.
ChIP-seq -
Libraries were generated from 2 million iPSC derived NPCs using the ChIP-IT® Express Kit and Sonication Shearing Kit (Active Motif) with Spike-in Chromatin from Drosophila (Active Motif) following to the manufacturer’s instructions without any modifications (Egan et al., 2016). Briefly, the ChIP-seq library preparation consists of 8 steps: (i) cells were crosslinked with 1% formaldehyde, (ii) chromatin was sheared (Covaris E220 Focused-ultrasonicator) after lysing cells, (iii) digested chromatin was incubated with 2ug of LMNB1 antibody (Abcam, ab8982), samples were also incubated without any antibody for input and Spike-in chromatin was incubated with Spike-in antibody (Active motif). (iv) same amount of Spike-in chromatin/antibody was added to each sample. (V) antibody-bound DNA/protein complexes and input samples were captured and washed using Protein G magnetic beads, (vi) cross-links were reversed and protein was digested using proteinase K. (vii) DNA was purified utilizing QIAquick PCR Purification Kit (Qiagen, 28104) (viii) libraries were prepared utilizing the Kappa Hyperprep Kit (Roche, KK8502). Quality control was performed using the Agilent Bioanalyzer 2100. ~30 million 40-bp single-end reads were acquired for each replicate on the NextSeq 500 platform at the MIT BioMicro Center. FASTQ reads for each replicate were aligned using Bowtie 2.0 (bowtie2 --very-sensitive -k 30) to hg19 (human) and dm6 (drosophila) (Langmead and Salzberg, 2012). Samtools was used to sort (samtools sort), remove duplicates (samtools markdup), remove mitochondrial reads (samtools view) and index BAM files (samtools index) (Li et al., 2009). Bigwigs were generated from BAM files adjusted to the scale-factor for each sample using the percentage of mapped reads from the drosophila genome (dm6) to total reads over input utilizing deepTools (3.5.0) bamCoverage (Egan et al., 2016; Ramirez et al., 2016). Peak regions were identified using homer (4.11), with the function findPeaks utilizing the reads generated from input samples as background. Scatter plots and violin plots were generated utilizing the R package ggplot2.
ATAC-seq -
Libraries were prepared as previously described (Buenrostro et al., 2015). 50,000 cells from the biological replicates (n=3) of the isogenic pair of iPSCs and NPCs were used for library preparation. Cells were harvested after treatment for 4 mins with ReLeSR™ (Stemcell Technology, 05872) for iPSCs and TrypLE (Thermo Fisher Scientific, 12605028) for NPCs were then counted and 50,000 cells were spun down at 500×g for 5 min, 4°C. Cells were with 50μl PBS and spun down at 500×g for 5 min, 4°C. Cells were then resuspended in fresh 50ul cold lysis buffer (0.15% igepal CA-630 (NP-40)) and left on ice for 10 min and spun down at 500×g for 5 min, 4°C. Next, tagmentation (Nextera FC-121-1030, Illumina) was carried out in 50ul volume (25 μl TD, 2.5 μl TDE1, 22.5 μl nuclease-free water), for 30 min at 37°C with gentle rocking. Transposed DNA fragments were purified using the Qiagen MinElute PCR Purification Kit (28006) and were barcoded, amplified (PCR reaction: 1 cycle; 5min at 72°C, 30sec at 98°C, 8 cycles; 15sec at 98°C, 30sec at 60°C, 3min at 72°C) and purified using AMPure beads (Beckman Coulter, A63880). ATAC-seq library fragment size distribution was assessed on an Agilent Bioanalyzer 2100. >50 million 40-bp paired-end reads were acquired for each replicate on the NextSeq 500 platform at the MIT BioMicro Center. FASTQ paired-end reads for each replicate were aligned using Bowtie 2.0 (bowtie2 --very-sensitive -k 30) to hg19 (Langmead and Salzberg, 2012). Samtools was used to sort (samtools sort), remove duplicates (samtools markdup), remove mitochondrial reads (samtools view), index BAM files (samtools index), and BAM files were shifted by +4bp and −5bp using an in-house algorithm (Li et al., 2009). Accessible chromatin peak calling was performed using Model-based Analysis of ChIP-Seq (macs2 callpeak -f BAMPE -g hs -q 0.05) (Zhang et al., 2008). Differential peaks analysis between the isogenic pairs was performed using Diffbind v1.16.3 (Ross-Innes et al., 2012). Correlation between differential accessibility of gene promoters (−2,000bp and +500bp of TSS) with differential expression was computed by first calculating the overlap percentage and significance of enrichment/depletion using a randomized permutation test then data was plotted as a histogram.
In situ Hi-C -
Hi-C libraries were generated from 2 million iPSCs and NPCs using the Arima Hi-C kit (Arima Genomics, San Diego) following to the manufacturer’s instructions without any modifications. Briefly, the Arima Hi-C kit consists of 6 steps: (i) cells were crosslinked with 1% formaldehyde, (ii) DNA was digested using the Arima restriction enzyme cocktail within intact permeabilized nuclei, (iii) ends were filled and biotinylated, (iv) blunt ends were ligated, (v) ligated fragments were purified, sheared and a biotin pull down was performed using streptavidin, and (vi) adapters were ligated, barcoded and amplified using the Kappa Hyperprep Kit (Roche, KK8502). Quality control was performed after shearing and library-prep using the Agilent Bioanalyzer 2100. Finally, >1.5 billion 40-bp paired-end reads were acquired for each replicate on the NextSeq 500 platform at the MIT BioMicro Center. FASTQ paired-end reads were processed through Juicer as previously described (Rao et al., 2014). In Brief, paired-end read pairs were aligned separately to the human genome (hg19) using BWA, then, duplicates/invalid pairs were removed, and the remaining reads were filtered based on mapping quality score of 30 (Li and Durbin, 2009). Interaction matrices were generated at resolutions of 2.5Mb, 1Mb, 500kb, 250kb, 100kb, 50kb, 25kb, 10kb, and 5kb and normalized interactions matrices were produced at all resolutions using Knight and Ruiz (~/juicer/scripts/CPU/juicer.sh -D ~/juicer/ -y ~/juicer/restriction_sites/hg19_Arima.txt -s Arima).
Trans-chromosomal interactions stats were generated through Juicer (Table S1) and a t-test was run comparing euploid and trsiomic iPSCs and NPCs. Differential trans-interaction maps were generated through Hi-C Explorer utilizing the default setting (Ramirez et al., 2018). In brief, interaction matricies generated in juicer (.hic) were converted to cooler and then to h5 (hicConvertFormat -m matrix.hic --inputFormat hic --outputFormat cool -o matrix.cool --resolutions 1000000 -> hicConvertFormat -m matrix.cool --inputFormat cool --outputFormat h5 -o matrix.h5). Next, a matrix containing the normalized log2ratio values of trisomy/euploid was generated (hicCompareMatrices -m Trisomy.h5 Euploid.h5 --operation log2ratio -o Trisomy_Euploid.h5) and plotted (hicPlotMatrix -m Trisomy_Euploid.h5 --clearMaskedBins--vMin −1 --vMax 1 -o Trisomy_Euploid.png). Distribution of normalized counts was generated using distiller pipeline on nextflow (nextflow run distiller.nf -params-file project.yml) as previously described (https://github.com/mirnylab/distiller-nf) (Abdennur and Mirny, 2020). Trans-chromosomal interaction statistics generated by distiller was used to identify the distribution of the trans-chromosomal interaction for each chromosome and calculated as the percentage of total interactions per sample. A statistical t-test comparison of euploid and trisomic cells was performed for each chromosome after normalizing for the number of chromosomes, where all chromosomes were divided by 2 with the exception of chromosome 21 which was divided by two in euploid samples and divided by 3 in trisomic samples (Figure S1E; Table S3).
Cis-chromosomal interactions stats were generated through Juicer (Table S1) and a t-test was run comparing euploid and trisomic iPSCs and NPCs. Differential trans-interaction maps were generated using juicebox, and represents the foldchange of trisomic over euploid cells. Global decay plots were generated using an in-house algorithm. In brief, decay plots were made by plotting the “Fraction of total paired-end tags” vs “Distance between the paired-end tags” generated by homer Hi-C analysis pipeline (Heinz et al., 2018). Fold enrichment values for each distance intervals were calculated as log2(mean of trisomy replicates/ mean of euploid replicates).The FDR p-values for the enrichment between the tree replicates for trisomy and disomy was calculated using t-test (two-tailed) and corrected using p.adjust function in the R library qvalue using standard methods (Benjamini & Hochberg). Shorth-range (<1Mb) to long-range (>1Mb) interaction ratios were generated using Hi-C explorer utilizing the default settings (hicPlotSVL -m Euploid_10kb.cool Trisomy_10kb.cool --distance 1000000) and a Wilcoxon rank-sum test was run between euploid and trisomic samples and represented as dot plots for each chromosome.
A/B compartments were analyzed utilizing the HOMER Hi-C analysis pipeline (http://homer.ucsd.edu/homer/interactions2/index.html) utilizing the default settings (Heinz et al., 2018). In brief, FASTQ paired reads were aligned separately using BWA (bwa mem -t8 -A1 -B4 -E50 -L0 ~/BWA_Index/hg19.fa), then tag directories were generated (makeTagDirectory sample_R1.sam,sample_R2.sam -tbp 1 -removePEbg -removeSpikes 10000 5). Principal component analysis of the Hi-C interaction matrices was used to reveal the active and inactive compartments of the genome (runHiCpca.pl auto sample_TagDir/ -res 25000 -window 50000 -genome hg19). Regions were annotated (annotatePeaks.pl) and edgeR/limma was used to identify the differential compartments (getDiffExpression.pl). Comparative analysis was performed to define the concordance between first principal component-based compartments identified through HOMER and the first principal component of the Pearson’s matrix (eigenvector) identified through Juicer using bedtools intersect. Enrichment analysis with differentially expressed genes (DEGs) and differentially accessible regions (DARs) with A and B compartments was performed by first calculating the overlap percentage of DEGs and DARs with the different compartments and the significance of enrichment/depletion was calculated using a randomized permutation test (p-Value). Data was plotted as histogram or as corrplot on R.
Loops and Topologically associating domains (TADs) -
Loops were called using the default setting of HICCUPS on juicer at 5Kb or 10Kb resolution (java -Xmx125g -jar ~/juicer/juicer_tools.jar hiccups). Differential looping between euploid and trisomic cells was identified using the default setting of HiCCUPS Diff on juicer using a graphics processing unit (GPU) (java -Xmx125g -jar ~/juicer/juicer_tools.jar hiccupsdiff). Aggregate plot analysis was performed using the APA command tool on juicer using the default settings (java -Xmx125g -jar ~/juicer/juicer_tools.jar apa) and genome-wide aggregate plots were generated from the normalized APA matrices on R. TADs boundaries, insulation scores and directionality index was computed using the default setting on HOMER (findTADsAndLoops.pl find TagDir/ -cpu 5 -res 3000 -window 15000 -genome hg19 -p badRegions.bed). For differential intra-tad interaction density analysis TAD files for the biological replicates per cell-type were merged (merge2Dbed.pl) and then interaction densities were quantified across replicates (findTADsAndLoops.pl score -tad). Finally, edgeR/limma was utilized to identify the differentially interacting TADs (getDiffExpression.pl). Enrichment analysis with differentially expressed genes was performed between down and upregulated genes with differentially interacting TADs and unchanged TADs, by first calculating the overlap percentage and significance of enrichment/depletion (randomized permutation test, p-Value). Data was plotted using corrplot on R.
Quantification and Statistical Analysis
Data was analyzed using GraphPad Prism version 8. Replicate sizes and error bars definitions are indicated in the figure legends. Correct application of standard parametric procedures was confirmed and stated in the supplemental data. Two-tailed Student’s t test were performed for datasets with two groups. ANOVA analyses were used for comparisons of data with more than two groups. Post hoc group comparisons were performed and stated in the figure legends and supplemental tables.
Differences were considered statistically significant at * (p < 0.05), ** (p < 0.01), *** (p < 0.001), and **** (p < 0.0001).
Supplementary Material
Table S1 - T21 induces genome-wide chromosomal introversion in NPCs, Related to Figure 1
Table S2 - T21 induced reorganization of TADs and loops are segregated into A/B compartments, Related to Figure 2
Table S3 - T21 induced disruption of lamina associated domains (LADs) , Related to Figure 3
Table S4 - T21 DEGs are distinctly separated into the A/B compartments in NPCs, Related to Figure 4
Table S5 - T21-induced genome-wide disruption of chromatin state in NPCs, Related to Figure 5
Table S6 - T21 induces senescence in NPCs, Related to Figure 6
Table S7 - Senolytic drug alleviates the transcriptional and cellular dysfunctions induced by T21 in NPCs, Related to Figure 7
Highlights.
Trisomy 21 disrupts nuclear architecture and transcriptome of neural progenitors
Trisomy 21 harboring neural progenitors display signatures of cellular senescence
Senolytic drugs ameliorate trisomy 21 associated molecular and cellular dysfunctions
Acknowledgments:
This work was supported by grants from the Alana foundation (Received by L-HT), LuMind Foundation (Received by HSM), Burroughs Wellcome Fund (Received by HSM), UNCF-Merck postdoctoral fellowship (Received by HSM) and the National institute of health (3U54HG008097-05S1, received by JDJ and L-HT). We would like to thank Dr. Jay Penney, Dr. Scarlett Barker, Dr. Matheus Victor, Dr. Ravikiran Raju, Dr. Priyanka Narayan, Dr. Ping-Chieh Pao, Dr. Emily Niederst, and the members of Tsai Lab for all the constructive discussions and feedback on the manuscript. We would also like to thank Ying Zhou and Dr. Jennie Young of the Tsai lab, as well as Dr. Stuart Levine and his entire team at the MIT BioMicro Center. We would also like to acknowledge that cartoon illustrations were created with BioRender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Abdennur N, and Mirny LA (2020). Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics 36, 311–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldridge K, Reeves RH, Olson LE, and Richtsmeier JT (2007). Differential effects of trisomy on brain shape and volume in related aneuploid mouse models. Am J Med Genet A 143A, 1060–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allis CD, and Jenuwein T (2016). The molecular hallmarks of epigenetic control. Nat Rev Genet 17, 487–500. [DOI] [PubMed] [Google Scholar]
- Alspach E, Flanagan KC, Luo X, Ruhland MK, Huang H, Pazolli E, Donlin MJ, Marsh T, Piwnica-Worms D, Monahan J, et al. (2014). p38MAPK plays a crucial role in stromal-mediated tumorigenesis. Cancer Discov 4, 716–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andriani GA, Almeida VP, Faggioli F, Mauro M, Tsai WL, Santambrogio L, Maslov A, Gadina M, Campisi J, Vijg J, et al. (2016). Whole Chromosome Instability induces senescence and promotes SASP. Sci Rep 6, 35218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bally BP, Farmer WT, Jones EV, Jessa S, Kacerovsky JB, Mayran A, Peng H, Lefebvre JL, Drouin J, Hayer A, et al. (2020). Human iPSC-Derived Down Syndrome Astrocytes Display Genome-Wide Perturbations in Gene Expression, an Altered Adhesion Profile, and Increased Cellular Dynamics. Hum Mol Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battle SL, Doni Jayavelu N, Azad RN, Hesson J, Ahmed FN, Overbey EG, Zoller JA, Mathieu J, Ruohola-Baker H, Ware CB, et al. (2019). Enhancer Chromatin and 3D Genome Architecture Changes from Naive to Primed Human Embryonic Stem Cell States. Stem Cell Reports 12, 1129–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benatti P, Dolfini D, Vigano A, Ravo M, Weisz A, and Imbriano C (2011). Specific inhibition of NF-Y subunits triggers different cell proliferation defects. Nucleic Acids Res 39, 5356–5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benda CE (1940). Growth disorder of the skull in mongolism. Am J Pathol 16, 71–86 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco K, Gormley M, Farrell J, Zhou Y, Oliverio O, Tilden H, McMaster M, and Fisher SJ (2016). Placental transcriptomes in the common aneuploidies reveal critical regions on the trisomic chromosomes and genome-wide effects. Prenat Diagn 36, 812–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biron-Shental T, Liberman M, Sharvit M, Sukenik-Halevy R, and Amiel A (2015). Amniocytes from aneuploidy embryos have enhanced random aneuploidy and signs of senescence - can these findings be related to medical problems? Gene 562, 232–235. [DOI] [PubMed] [Google Scholar]
- Borras-Fresneda M, Barquinero JF, Gomolka M, Hornhardt S, Rossler U, Armengol G, and Barrios L (2016). Differences in DNA Repair Capacity, Cell Death and Transcriptional Response after Irradiation between a Radiosensitive and a Radioresistant Cell Line. Sci Rep 6, 27043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeron T, Xu Z, Doumic M, and Teixeira MT (2015). The asymmetry of telomere replication contributes to replicative senescence heterogeneity. Sci Rep 5, 15326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109, 21 29 21–21 29 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, and Baker DJ (2018). Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562, 578–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casella G, Munk R, Kim KM, Piao Y, De S, Abdelmohsen K, and Gorospe M (2019). Transcriptome signature of cellular senescence. Nucleic Acids Res 47, 11476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti L, Galdzicki Z, and Haydar TF (2007). Defects in embryonic neurogenesis and initial synapse formation in the forebrain of the Ts65Dn mouse model of Down syndrome. J Neurosci 27, 11483–11495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, and Studer L (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol 27, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contestabile A, Fila T, Bartesaghi R, and Ciani E (2009). Cell cycle elongation impairs proliferation of cerebellar granule cell precursors in the Ts65Dn mouse, an animal model for Down syndrome. Brain Pathol 19, 224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contestabile A, Fila T, Ceccarelli C, Bonasoni P, Bonapace L, Santini D, Bartesaghi R, and Ciani E (2007). Cell cycle alteration and decreased cell proliferation in the hippocampal dentate gyrus and in the neocortical germinal matrix of fetuses with Down syndrome and in Ts65Dn mice. Hippocampus 17, 665–678. [DOI] [PubMed] [Google Scholar]
- Creech AL, Taylor JE, Maier VK, Wu X, Feeney CM, Udeshi ND, Peach SE, Boehm JS, Lee JT, Carr SA, et al. (2015). Building the Connectivity Map of epigenetics: chromatin profiling by quantitative targeted mass spectrometry. Methods 72, 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criscione SW, De Cecco M, Siranosian B, Zhang Y, Kreiling JA, Sedivy JM, and Neretti N (2016). Reorganization of chromosome architecture in replicative cellular senescence. Sci Adv 2, e1500882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe EP, Tuzer F, Gregory BD, Donahue G, Gosai SJ, Cohen J, Leung YY, Yetkin E, Nativio R, Wang LS, et al. (2016). Changes in the Transcriptome of Human Astrocytes Accompanying Oxidative Stress-Induced Senescence. Front Aging Neurosci 8, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czarkwiani A, and Yun MH (2018). Out with the old, in with the new: senescence in development. Curr Opin Cell Biol 55, 74–80. [DOI] [PubMed] [Google Scholar]
- Dekker J, Rippe K, Dekker M, and Kleckner N (2002). Capturing chromosome conformation. Science 295, 1306–1311. [DOI] [PubMed] [Google Scholar]
- Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, et al. (2015). Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Down JL (1995). Observations on an ethnic classification of idiots. 1866. Ment Retard 33, 54–56. [PubMed] [Google Scholar]
- Durrbaum M, Kuznetsova AY, Passerini V, Stingele S, Stoehr G, and Storchova Z (2014). Unique features of the transcriptional response to model aneuploidy in human cells. BMC Genomics 15, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, and Yakhini Z (2009). GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan B, Yuan CC, Craske ML, Labhart P, Guler GD, Arnott D, Maile TM, Busby J, Henry C, Kelly TK, et al. (2016). An Alternative Approach to ChIP-Seq Normalization Enables Detection of Genome-Wide Changes in Histone H3 Lysine 27 Trimethylation upon EZH2 Inhibition. PLoS One 11, e0166438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FitzPatrick DR, Ramsay J, McGill NI, Shade M, Carothers AD, and Hastie ND (2002). Transcriptome analysis of human autosomal trisomy. Hum Mol Genet 11, 3249–3256. [DOI] [PubMed] [Google Scholar]
- Furlong EEM, and Levine M (2018). Developmental enhancers and chromosome topology. Science 361, 1341–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Leon JA, Kumar M, Boon R, Chau D, One J, Wolfs E, Eggermont K, Berckmans P, Gunhanlar N, de Vrij F, et al. (2018). SOX10 Single Transcription Factor-Based Fast and Efficient Generation of Oligodendrocytes from Human Pluripotent Stem Cells. Stem Cell Reports 10, 655–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng L, Liu Z, Zhang W, Li W, Wu Z, Wang W, Ren R, Su Y, Wang P, Sun L, et al. (2019). Chemical screen identifies a geroprotective role of quercetin in premature aging. Protein Cell 10, 417–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales PK, Roberts CM, Fonte V, Jacobsen C, Stein GH, and Link CD (2018). Transcriptome analysis of genetically matched human induced pluripotent stem cells disomic or trisomic for chromosome 21. PLoS One 13, e0194581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinstein E, Jundt F, Weinert I, Wernet P, and Royer HD (2002). Sp1 as G1 cell cycle phase specific transcription factor in epithelial cells. Oncogene 21, 1485–1492. [DOI] [PubMed] [Google Scholar]
- Guidi S, Ciani E, Bonasoni P, Santini D, and Bartesaghi R (2011). Widespread proliferation impairment and hypocellularity in the cerebellum of fetuses with down syndrome. Brain Pathol 21, 361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins RD, Hon GC, Lee LK, Ngo Q, Lister R, Pelizzola M, Edsall LE, Kuan S, Luu Y, Klugman S, et al. (2010). Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell 6, 479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayflick L, and Moorhead PS (1961). The serial cultivation of human diploid cell strains. Exp Cell Res 25, 585–621. [DOI] [PubMed] [Google Scholar]
- Head E, Powell D, Gold BT, and Schmitt FA (2012). Alzheimer’s Disease in Down Syndrome. Eur J Neurodegener Dis 1, 353–364. [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Texari L, Hayes MGB, Urbanowski M, Chang MW, Givarkes N, Rialdi A, White KM, Albrecht RA, Pache L, et al. (2018). Transcription Elongation Can Affect Genome 3D Structure. Cell 174, 1522–1536 e1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Segura A, de Jong TV, Melov S, Guryev V, Campisi J, and Demaria M (2017). Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr Biol 27, 2652–2660 e2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, Raguz S, Acosta JC, Innes AJ, Banito A, et al. (2015). mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol 17, 1205–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, Herrmann SM, Jensen MD, Jia Q, Jordan KL, et al. (2019). Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 47, 446–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, Garagnani P, Bacalini MG, Pirazzini C, Salvioli S, Gentilini D, Di Blasio AM, Giuliani C, Tung S, Vinters HV, et al. (2015). Accelerated epigenetic aging in Down syndrome. Aging Cell 14, 491–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu JY, Sun ZW, Li X, Reuben M, Tatchell K, Bishop DK, Grushcow JM, Brame CJ, Caldwell JA, Hunt DF, et al. (2000). Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell 102, 279–291. [DOI] [PubMed] [Google Scholar]
- Huo HQ, Qu ZY, Yuan F, Ma L, Yao L, Xu M, Hu Y, Ji J, Bhattacharyya A, Zhang SC, et al. (2018). Modeling Down Syndrome with Patient iPSCs Reveals Cellular and Migration Deficits of GABAergic Neurons. Stem Cell Reports 10, 1251–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe JD, Wang Y, Chan HM, Zhang J, Huether R, Kryukov GV, Bhang HE, Taylor JE, Hu M, Englund NP, et al. (2013). Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat Genet 45, 1386–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joslyn N, Berger H, and Skotko BG (2019). Geospatial Analyses of Accessibility to Down Syndrome Specialty Care. J Pediatr. [DOI] [PubMed] [Google Scholar]
- Kang TH, Park DY, Choi YH, Kim KJ, Yoon HS, and Kim KT (2007). Mitotic histone H3 phosphorylation by vaccinia-related kinase 1 in mammalian cells. Mol Cell Biol 27, 8533–8546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland JL, Tchkonia T, Zhu Y, Niedernhofer LJ, and Robbins PD (2017). The Clinical Potential of Senolytic Drugs. J Am Geriatr Soc 65, 2297–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner K, Rattanavirotkul N, Quince MF, and Chandra T (2020). Functional heterogeneity in senescence. Biochem Soc Trans 48, 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm SL, Shipony Z, and Greenleaf WJ (2019). Chromatin accessibility and the regulatory epigenome. Nat Rev Genet 20, 207–220. [DOI] [PubMed] [Google Scholar]
- Konrad EDH, Nardini N, Caliebe A, Nagel I, Young D, Horvath G, Santoro SL, Shuss C, Ziegler A, Bonneau D, et al. (2019). CTCF variants in 39 individuals with a variable neurodevelopmental disorder broaden the mutational and clinical spectrum. Genet Med 21, 2723–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari R, and Jat P (2021). Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front Cell Dev Biol 9, 645593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane AA, Chapuy B, Lin CY, Tivey T, Li H, Townsend EC, van Bodegom D, Day TA, Wu SC, Liu H, et al. (2014). Triplication of a 21q22 region contributes to B cell transformation through HMGN1 overexpression and loss of histone H3 Lys27 trimethylation. Nat Genet 46, 618–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lejeune J, Gauthier M, and Turpin R (1959). [Human chromosomes in tissue cultures]. C R Hebd Seances Acad Sci 248, 602–603. [PubMed] [Google Scholar]
- Lenain C, de Graaf CA, Pagie L, Visser NL, de Haas M, de Vries SS, Peric-Hupkes D, van Steensel B, and Peeper DS (2017). Massive reshaping of genome-nuclear lamina interactions during oncogene-induced senescence. Genome Res 27, 1634–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letourneau A, Santoni FA, Bonilla X, Sailani MR, Gonzalez D, Kind J, Chevalier C, Thurman R, Sandstrom RS, Hibaoui Y, et al. (2014). Domains of genome-wide gene expression dysregulation in Down’s syndrome. Nature 508, 345–350. [DOI] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YT, Seo J, Gao F, Feldman HM, Wen HL, Penney J, Cam HP, Gjoneska E, Raja WK, Cheng J, et al. (2018). APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 98, 1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litichevskiy L, Peckner R, Abelin JG, Asiedu JK, Creech AL, Davis JF, Davison D, Dunning CM, Egertson JD, Egri S, et al. (2018). A Library of Phosphoproteomic and Chromatin Signatures for Characterizing Cellular Responses to Drug Perturbations. Cell Syst 6, 424–443 e427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Wang L, Perna F, and Nimer SD (2016). Beyond transcription factors: how oncogenic signalling reshapes the epigenetic landscape. Nat Rev Cancer 16, 359–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Chen X, Sanders D, Qian S, and Zhong X (2015). High-resolution mapping of H4K16 and H3K23 acetylation reveals conserved and unique distribution patterns in Arabidopsis and rice.Epigenetics 10, 1044–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maclean GA, Menne TF, Guo G, Sanchez DJ, Park IH, Daley GQ, and Orkin SH (2012). Altered hematopoiesis in trisomy 21 as revealed through in vitro differentiation of isogenic human pluripotent cells. Proc Natl Acad Sci U S A 109, 17567–17572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marthandan S, Priebe S, Hemmerich P, Klement K, and Diekmann S (2014). Long-term quiescent fibroblast cells transit into senescence. PLoS One 9, e115597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Cue C, and Rueda N (2020). Cellular Senescence in Neurodegenerative Diseases. Front Cell Neurosci 14, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin K, Flyamer IM, Thomson JP, Mjoseng HK, Shukla R, Williamson I, Grimes GR, Illingworth RS, Adams IR, Pennings S, et al. (2019). DNA Methylation Directs Polycomb-Dependent 3D Genome Re-organization in Naive Pluripotency. Cell Rep 29, 1974–1985 e1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldrich RX, Dauphinot L, Laffaire J, Vitalis T, Herault Y, Beart PM, Rossier J, Vivien D, Gehrig C, Antonarakis SE, et al. (2009). Proliferation deficits and gene expression dysregulation in Down’s syndrome (Ts1Cje) neural progenitor cells cultured from neurospheres. J Neurosci Res 87, 3143–3152. [DOI] [PubMed] [Google Scholar]
- Montgomery RL, Hsieh J, Barbosa AC, Richardson JA, and Olson EN (2009). Histone deacetylases 1 and 2 control the progression of neural precursors to neurons during brain development. Proc Natl Acad Sci U S A 106, 7876–7881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Espin D, Canamero M, Maraver A, Gomez-Lopez G, Contreras J, Murillo-Cuesta S, Rodriguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M, et al. (2013). Programmed cell senescence during mammalian embryonic development. Cell 155, 1104–1118. [DOI] [PubMed] [Google Scholar]
- Nelson DM, Jaber-Hijazi F, Cole JJ, Robertson NA, Pawlikowski JS, Norris KT, Criscione SW, Pchelintsev NA, Piscitello D, Stong N, et al. (2016). Mapping H4K20me3 onto the chromatin landscape of senescent cells indicates a function in control of cell senescence and tumor suppression through preservation of genetic and epigenetic stability. Genome Biol 17, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. (2012). Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh JE, Han JA, and Hwang ES (2007). Downregulation of transcription factor, Sp1, during cellular senescence. Biochem Biophys Res Commun 353, 86–91. [DOI] [PubMed] [Google Scholar]
- Olmos-Serrano JL, Kang HJ, Tyler WA, Silbereis JC, Cheng F, Zhu Y, Pletikos M, Jankovic-Rapan L, Cramer NP, Galdzicki Z, et al. (2016). Down Syndrome Developmental Brain Transcriptome Reveals Defective Oligodendrocyte Differentiation and Myelination. Neuron 89, 1208–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson LE, Roper RJ, Baxter LL, Carlson EJ, Epstein CJ, and Reeves RH (2004). Down syndrome mouse models Ts65Dn, Ts1Cje, and Ms1Cje/Ts65Dn exhibit variable severity of cerebellar phenotypes. Dev Dyn 230, 581–589. [DOI] [PubMed] [Google Scholar]
- Parry AJ, Hoare M, Bihary D, Hansel-Hertsch R, Smith S, Tomimatsu K, Mannion E, Smith A, D’Santos P, Russell IA, et al. (2018). NOTCH-mediated non-cell autonomous regulation of chromatin structure during senescence. Nat Commun 9, 1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinter JD, Eliez S, Schmitt JE, Capone GT, and Reiss AL (2001). Neuroanatomy of Down’s syndrome: a high-resolution MRI study. Am J Psychiatry 158, 1659–1665. [DOI] [PubMed] [Google Scholar]
- Pombo A, and Dillon N (2015). Three-dimensional genome architecture: players and mechanisms. Nat Rev Mol Cell Biol 16, 245–257. [DOI] [PubMed] [Google Scholar]
- Presson AP, Partyka G, Jensen KM, Devine OJ, Rasmussen SA, McCabe LL, and McCabe ER (2013). Current estimate of Down Syndrome population prevalence in the United States. J Pediatr 163, 1163–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raja WK, Mungenast AE, Lin YT, Ko T, Abdurrob F, Seo J, and Tsai LH (2016). Self-Organizing 3D Human Neural Tissue Derived from Induced Pluripotent Stem Cells Recapitulate Alzheimer’s Disease Phenotypes. PLoS One 11, e0161969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Bhardwaj V, Arrigoni L, Lam KC, Gruning BA, Villaveces J, Habermann B, Akhtar A, and Manke T (2018). High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat Commun 9, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risso D, Ngai J, Speed TP, and Dudoit S (2014). Normalization of RNA-seq data using factor analysis of control genes or samples. Nat Biotechnol 32, 896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson MI, Ringel AR, and Mundlos S (2019). Regulatory Landscaping: How Enhancer-Promoter Communication Is Sculpted in 3D. Mol Cell 74, 1110–1122. [DOI] [PubMed] [Google Scholar]
- Romanov VS, Abramova MV, Svetlikova SB, Bykova TV, Zubova SG, Aksenov ND, Fornace AJ Jr., Pospelova TV, and Pospelov VA (2010). p21(Waf1) is required for cellular senescence but not for cell cycle arrest induced by the HDAC inhibitor sodium butyrate. Cell Cycle 9, 3945–3955. [DOI] [PubMed] [Google Scholar]
- Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. (2012). Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Richardson A, Iyer DR, M’Saad O, Zasadil L, Knouse KA, Wong YL, Rhind N, Desai A, and Amon A (2017). Chromosome Mis-segregation Generates Cell-Cycle-Arrested Cells with Complex Karyotypes that Are Eliminated by the Immune System. Dev Cell 41, 638–651 e635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sati S, Bonev B, Szabo Q, Jost D, Bensadoun P, Serra F, Loubiere V, Papadopoulos GL, Rivera-Mulia JC, Fritsch L, et al. (2020). 4D Genome Rewiring during Oncogene-Induced and Replicative Senescence. Mol Cell 78, 522–538 e529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfelder S, and Fraser P (2019). Long-range enhancer-promoter contacts in gene expression control. Nat Rev Genet 20, 437–455. [DOI] [PubMed] [Google Scholar]
- Serrano M, and Barzilai N (2018). Targeting senescence. Nat Med 24, 1092–1094. [DOI] [PubMed] [Google Scholar]
- Sheltzer JM, Torres EM, Dunham MJ, and Amon A (2012). Transcriptional consequences of aneuploidy. Proc Natl Acad Sci U S A 109, 12644–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevelyov YY, and Ulianov SV (2019). The Nuclear Lamina as an Organizer of Chromosome Architecture. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagni F, Giacomini A, Emili M, Guidi S, and Bartesaghi R (2018). Neurogenesis impairment: An early developmental defect in Down syndrome. Free Radic Biol Med 114, 15–32. [DOI] [PubMed] [Google Scholar]
- Stagni F, Giacomini A, Guidi S, Ciani E, and Bartesaghi R (2015). Timing of therapies for Down syndrome: the sooner, the better. Front Behav Neurosci 9, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J, et al. (2013). Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155, 1119–1130. [DOI] [PubMed] [Google Scholar]
- Sun B, Chang E, Gerhartl A, and Szele FG (2018). Polycomb Protein Eed is Required for Neurogenesis and Cortical Injury Activation in the Subventricular Zone. Cereb Cortex 28, 1369–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Bosnjak M, Skunca N, and Smuc T (2011). REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6, e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylvester PE (1983). The hippocampus in Down’s syndrome. J Ment Defic Res 27 (Pt 3), 227–236. [DOI] [PubMed] [Google Scholar]
- Tcw J, Wang M, Pimenova AA, Bowles KR, Hartley BJ, Lacin E, Machlovi SI, Abdelaal R, Karch CM, Phatnani H, et al. (2017). An Efficient Platform for Astrocyte Differentiation from Human Induced Pluripotent Stem Cells. Stem Cell Reports 9, 600–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda T, and Gage FH (2018). Review: adult neurogenesis contributes to hippocampal plasticity. Cell Tissue Res 373, 693–709. [DOI] [PubMed] [Google Scholar]
- Topol A, Tran NN, and Brennand KJ (2015). A guide to generating and using hiPSC derived NPCs for the study of neurological diseases. J Vis Exp, e52495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler WA, and Haydar TF (2013). Multiplex genetic fate mapping reveals a novel route of neocortical neurogenesis, which is altered in the Ts65Dn mouse model of Down syndrome. J Neurosci 33, 5106–5119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasey DB, Wolf CR, MacArtney T, Brown K, and Whitelaw CB (2008). p21-LacZ reporter mice reflect p53-dependent toxic insult. Toxicol Appl Pharmacol 227, 440–450. [DOI] [PubMed] [Google Scholar]
- Walus M, Kida E, Rabe A, Albertini G, and Golabek AA (2016). Widespread cerebellar transcriptome changes in Ts65Dn Down syndrome mouse model after lifelong running. Behav Brain Res 296, 35–46. [DOI] [PubMed] [Google Scholar]
- Wangsa D, Braun R, Stuelten CH, Brown M, Bauer KM, Emons G, Weston LA, Hu Y, Yang HH, Vila-Casadesus M, et al. (2019). Induced Chromosomal Aneuploidy Results in Global and Consistent Deregulation of the Transcriptome of Cancer Cells. Neoplasia 21, 721–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weick JP, Held DL, Bonadurer GF 3rd, Doers ME, Liu Y, Maguire C, Clark A, Knackert JA, Molinarolo K, Musser M, et al. (2013). Deficits in human trisomy 21 iPSCs and neurons. Proc Natl Acad Sci U S A 110, 9962–9967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weijerman ME, and de Winter JP (2010). Clinical practice. The care of children with Down syndrome. Eur J Pediatr 169, 1445–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whetstine JR, Ceron J, Ladd B, Dufourcq P, Reinke V, and Shi Y (2005). Regulation of tissue-specific and extracellular matrix-related genes by a class I histone deacetylase. Mol Cell 18, 483–490. [DOI] [PubMed] [Google Scholar]
- Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, Inman CL, Ogrodnik MB, Hachfeld CM, Fraser DG, et al. (2018). Senolytics improve physical function and increase lifespan in old age. Nat Med 24, 1246–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Tchkonia T, and Kirkland JL (2016). Perspective: Targeting the JAK/STAT pathway to fight age-related dysfunction. Pharmacol Res 111, 152–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, and Teixeira MT (2019). The many types of heterogeneity in replicative senescence. Yeast 36, 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M, Ling YY, Melos KI, Pirtskhalava T, Inman CL, et al. (2018). Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 36, 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan B, Pehlivan D, Karaca E, Patel N, Charng WL, Gambin T, Gonzaga-Jauregui C, Sutton VR, Yesil G, Bozdogan ST, et al. (2015). Global transcriptional disturbances underlie Cornelia de Lange syndrome and related phenotypes. J Clin Invest 125, 636–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ji F, Liu Y, Lei X, Li H, Ji G, Yuan Z, and Jiao J (2014a). Ezh2 regulates adult hippocampal neurogenesis and memory. J Neurosci 34, 5184–5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JP, Zhang H, Wang HB, Li YX, Liu GH, Xing S, Li MZ, and Zeng MS (2014b). Down-regulation of Sp1 suppresses cell proliferation, clonogenicity and the expressions of stem cell markers in nasopharyngeal carcinoma. J Transl Med 12, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Kishimoto Y, Grammatikakis I, Gottimukkala K, Cutler RG, Zhang S, Abdelmohsen K, Bohr VA, Misra Sen J, Gorospe M, et al. (2019). Senolytic therapy alleviates Abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat Neurosci 22, 719–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, and Xie W (2019). The role of 3D genome organization in development and cell differentiation. Nat Rev Mol Cell Biol 20, 535–550. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Doornebal EJ, Pirtskhalava T, Giorgadze N, Wentworth M, Fuhrmann-Stroissnigg H, Niedernhofer LJ, Robbins PD, Tchkonia T, and Kirkland JL (2017). New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging (Albany NY) 9, 955–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer AK, Ikeno Y, Hubbard GB, Lenburg M, et al. (2015). The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14, 644–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinovyeva AY, Graham SM, Cloud VJ, and Forrester WC (2006). The C. elegans histone deacetylase HDA-1 is required for cell migration and axon pathfinding. Dev Biol 289, 229–242. [DOI] [PubMed] [Google Scholar]
- Zirkel A, Nikolic M, Sofiadis K, Mallm JP, Brackley CA, Gothe H, Drechsel O, Becker C, Altmuller J, Josipovic N, et al. (2018). HMGB2 Loss upon Senescence Entry Disrupts Genomic Organization and Induces CTCF Clustering across Cell Types. Mol Cell 70, 730–744 e736. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 - T21 induces genome-wide chromosomal introversion in NPCs, Related to Figure 1
Table S2 - T21 induced reorganization of TADs and loops are segregated into A/B compartments, Related to Figure 2
Table S3 - T21 induced disruption of lamina associated domains (LADs) , Related to Figure 3
Table S4 - T21 DEGs are distinctly separated into the A/B compartments in NPCs, Related to Figure 4
Table S5 - T21-induced genome-wide disruption of chromatin state in NPCs, Related to Figure 5
Table S6 - T21 induces senescence in NPCs, Related to Figure 6
Table S7 - Senolytic drug alleviates the transcriptional and cellular dysfunctions induced by T21 in NPCs, Related to Figure 7
Data Availability Statement
Genomic sequencing data have been deposited at Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| OCT-4 | Cell Signaling Technology | Cat# 2890, RRID:AB_2167725 |
| Nanog | Cell Signaling Technology | Cat# 4893, RRID:AB_10548762 |
| SOX1 | R&D Systems | Cat# AF3369, RRID:AB_2239879 |
| Nestin | Millipore Sigma | Cat# MAB5326, RRID:AB_2251134 |
| PAX6 | R&D Systems | Cat# AF8150, RRID:AB_2827378 |
| H3K9me3 | Abcam | Cat# ab176916, RRID:AB_2797591 |
| HP1a | Cell Signaling Technology | Cat# 2616, RRID:AB_2070987 |
| H3K27me3 | Abcam | Cat# ab6002, RRID:AB_305237 |
| HMGB1 | Thermo Fisher Scientific | Cat# PA1-16926, RRID:AB_2248274 |
| LMNB1 | Abcam | Cat# ab16048, RRID:AB_10107828 |
| p16INK4a | Thermo Fisher Scientific | Cat# MA5-32133, RRID:AB_2809424 |
| KI-67 | Abcam | Cat# ab15580, RRID:AB_443209 |
| Vimentin | Abcam | Cat# ab24525, RRID:AB_778824 |
| GFAP | Millipore | Cat# AB5804, RRID:AB_2109645 |
| BrdU | Cell Signaling Technology | Cat# 5292, RRID:AB_10548898 |
| Alexa-Fluor 488 (Donkey anti-Goat) | Thermo Fisher Scientific | Cat# A-11015, RRID:AB_2534082 |
| Alexa-Fluor 488 (Donkey anti-Rabbit) | Thermo Fisher Scientific | Cat# A-21206, RRID:AB_2535792 |
| Alexa-Fluor 488 (Donkey anti-Mouse) | Thermo Fisher Scientific | Cat# A-21202, RRID:AB_141607 |
| Alexa-Fluor 488 (Donkey anti-Sheep) | Thermo Fisher Scientific | Cat# A-11015, RRID:AB_2534082 |
| Alexa-Fluor 555 (Donkey anti-Goat) | Thermo Fisher Scientific | Cat# A-21432, RRID:AB_2535853 |
| Alexa-Fluor 555 (Donkey anti-Rabbit) | Thermo Fisher Scientific | Cat# A-31572, RRID:AB_162543 |
| Alexa-Fluor 555 (Donkey anti-Mouse) | Thermo Fisher Scientific | Cat# A-31570, RRID:AB_2536180 |
| Alexa-Fluor 555 (Donkey anti-Sheep) | Thermo Fisher Scientific | Cat# A-21099, RRID:AB_2535753 |
| Alexa-Fluor 647 (Donkey anti-Goat) | Thermo Fisher Scientific | Cat# A-21447, RRID:AB_2535864 |
| Alexa-Fluor 647 (Donkey anti-Rabbit) | Thermo Fisher Scientific | Cat# A-31573, RRID:AB_2536183 |
| Alexa-Fluor 647 (Donkey anti-Mouse) | Thermo Fisher Scientific | Cat# A-31571, RRID:AB_162542 |
| Alexa-Fluor 647 (Donkey anti-Sheep) | Thermo Fisher Scientific | Cat# A-21448, RRID:AB_2535865 |
| DAPI | Thermo Fisher Scientific | Cat# D1306, RRID:AB_2629482 |
| Biological Samples | ||
| Iso-E and Iso-T iPSCs (AG05397) | Coriell (Maclean et al., 2012) | Cat# AG05397 |
| Ma-E iPSCs | Cohen, McLean, HMS (Raja et al., 2016) | Cat# CS-0020-01 |
| Ma-T Fibroblast | Coriell | Cat# AG06922 |
| Fe-E iPSCs | WiCell | Cat# WA09 |
| Fe-T Fibroblasts | Coriell | Cat# GM04616 |
| CF1 Mouse embryonic fibroblasts, irradiated | Thermo Fisher Scientific | Cat# A34181 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Fluoromount-G | Electron Microscopy Sciences | Cat# 17984-25 |
| PBS, pH 7.4 | Thermo Fisher Scientific | Cat# 10010049 |
| Triton X-100 | Sigma-Aldrich | Cat# T8787-100ML |
| Paraformaldehyde | Electron Microscopy Sciences | Cat# 15714-S |
| 5-Bromo-2′-deoxyuridine | Sigma-Aldrich | Cat# B5002-5G |
| Corning® Matrigel® hESC-Qualified Matrix | VWR International | Cat# 354277 |
| Corning® Matrigel® Basement Membrane Matrix | VWR International | Cat# 356254 |
| DMEM/F12 Glutamax | Life Technologies | Cat# 10565-042 |
| Knockout Serum Replacement | Life Technologies | Cat# 10828028 |
| MEM non-Essential amino acid solution | Sigma-Aldrich | Cat# M7145 |
| Glutamax | Thermo Fisher Scientific | Cat# 35050-079 |
| recombinant human FGF-basic | PeproTech | Cat# 100-18B |
| 2-mercaptoethanol | Sigma-Aldrich | Cat# M6250 |
| mTeSR1 medium | Stemcell technologies | Cat# 85850 |
| ReLeSR™ | Stemcell Technology | Cat# 05872 |
| AggreWell™800 | Stemcell Technology | Cat# 34811 |
| Neurobasal | Thermo Fisher Scientific | Cat# 21103049 |
| N-2 Supplement | Invitrogen | Cat# 17502-048 |
| B-27 Serum-Free Supplement | Gibco | Cat# 12587010 |
| insulin | Thermo Fisher Scientific | Cat# 41400045 |
| Pen/strep | Gemini Bio-Products | Cat# 400-109 |
| Dorsomorphin | PeproTech | Cat# 8666430 |
| SB431542 | Tocris | Cat# 1614 |
| STEMdiff™ Neural Rosette Selection Reagent | Stemcell technologies | Cat# 05832 |
| Sulfuric acid | Sigma-Aldrich | Cat# 339741-500ML |
| Trichloroacetic acid | BDH | Cat# BDH0310-500G |
| Trypsin | Promega | Cat# V5113 |
| C18 Sep-Pak cartridges | Waters | Cat# 186002318 |
| TrypLE | Thermo Fisher Scientific | Cat# 12605028 |
| Igepal CA-630 (NP-40) | Sigma-Aldrich | Cat# I8896 |
| AMPure beads | Beckman Coulter | Cat# A63880 |
| Critical Commercial Assays | ||
| CytoTune-iPS Sendai Reprogramming kit | Thermofisher scientific | Cat# A16518 |
| Kapa mRNA hyperprep kit for standard RNA libraries | Roche | Cat# KK8540 |
| Nextera DNA Library Prep Kit | Illumina | Cat# FC-121-1030 |
| Qiagen MinElute PCR Purification Kit | Qiagen | Cat# 28006 |
| Arima Hi-C kit | Arima Genomics | N/A |
| Kappa Hyperprep Kit | Roche | Cat# KK8502 |
| Deposited Data | ||
| Hi-C | This paper | GEO: GSE185192 |
| LMNB1 ChIP-seq | This paper | GEO: GSE185192 |
| RNA-seq | This paper | GEO: GSE185192 |
| ATAC-seq | This paper | GEO: GSE185192 |
| Senescence RNA-seq | Casella et al., 2019; Crowe et al., 2016; Alspach et al., 2014; Sati et al., 2020; Zirkel et al., 2018; Borras-Fresneda et al., 2016; | GEO: GSE130727, GSE58910, GSE56293, GSE130306, GSE98448, GSE80207, |
| RNA-seq of iPSC derived from Down syndrome and control individuals | Gonzales et al., 2018 | GSE101942 |
| RNA-seq of iPSC derived neurons, astrocytes and OPCs | Garcia-Leon et al., 2018; Lin et al., 2018; Tcw et al., 2017 | GEO: GSE106984, GSE102956, GSE97904 |
| ChIP-seq of Histone Modifications | Hawkins et al., 2010 | GEO:GSM956008 |
| Experimental Models: Cell Lines | ||
| Iso-E and Iso-T (AG05397) | Coriell Maclean et al., 2012) | Cat# AG05397 |
| Ma-E | Cohen, McLean, HMS (Raja et al., 2016) | Cat# CS-0020-01 |
| Ma-T | Coriell | Cat# AG06922 |
| Fe-E | WiCell | Cat# WA09 |
| Fe-T | Coriell | Cat# GM04616 |
| CF1 Mouse embryonic fibroblasts, irradiated | Thermo Fisher Scientific | Cat# A34181 |
| Software and Algorithms | ||
| Imarisx64 9.2.1 | Bitplane | http://www.bitplane.com/Default.aspx |
| ZEN imaging software | Carl Zeiss | https://www.zeiss.com/microscopy/int/products/microscope-software/zen.html |
| STAR (v.2.6.1a) | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| HTSeq | Anders et al., 2015 | https://htseq.readthedocs.io/en/master/ |
| RUVSeq | Risso et al., 2014 | https://bioconductor.org/packages/release/bioc/html/RUVSeq.html |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| Homer | Heinz et al., 2010 | (http://homer.ucsd.edu/homer/interactions2/index.html |
| MACS2.1.0 | Zhang et al., 2008 | https://github.com/taoliu/MACS |
| R | Chen et al., 2009 | https://www.r-project.org/ |
| Corrplot | Taiyun Wei and Viliam Simko (2017) | https://github.com/taiyun/corrplot |
| Diffbind v1.16.3 | Ross-Innes et al., 2012 | https://bioconductor.org/packages/release/bioc/html/DiffBind.html |
| Burrows-Wheeler Aligner (BWA) | Li and Durbin, 2009 | http://bio-bwa.sourceforge.net/ |
| Juicer | Rao et al., 2014 | https://github.com/aidenlab/juicer/wiki |
| Graphpad Prism 8 | Graphpad | https://www.graphpad.com/ |
| Distiller | Abdennur and Mirny, 2020 | https://github.com/mirnylab/distiller-nf)) |
| Hi-C explorer | Ramirez et al., 2018 | https://hicexplorer.readthedocs.io/en/latest/content/list-of-tools.html |
| BEDTools | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| deepTools | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ |