

ABSTRACT
A large number of microbes exist in the gut and they have the ability to process and utilize ingested food. It has been reported that their products are involved in colorectal cancer development. The molecular mechanisms which underlie the relationship between gut microbial products and CRC are still not fully understood. The role of some microbial products in CRC is particularly controversial. Elucidating the effects of gut microbiota products on CRC and their possible mechanisms is vital for CRC prevention and treatment. In this review, recent studies are examined in order to describe the contribution metabolites and toxicants which are produced by gut microbes make to CRC, primarily focusing on the involved molecular mechanisms.
Abbreviations: CRC: colorectal cancer; SCFAs: short chain fatty acids; HDAC: histone deacetylase; TCA cycle: tricarboxylic acid cycle; CoA: cytosolic acyl coenzyme A; SCAD: short chain acyl CoA dehydrogenase; HDAC: histone deacetylase; MiR-92a: microRNA-92a; KLF4: kruppel-like factor; PTEN: phosphatase and tensin homolog; PI3K: phosphoinositide 3-kinase; PIP2: phosphatidylinositol 4, 5-biphosphate; PIP3: phosphatidylinositol-3,4,5-triphosphate; Akt1: protein kinase B subtype α; ERK1/2: extracellular signal–regulated kinases 1/2; EMT: epithelial-to-mesenchymal transition; NEDD9: neural precursor cell expressed developmentally down-regulated9; CAS: Crk-associated substrate; JNK: c-Jun N-terminal kinase; PRMT1: protein arginine methyltransferase 1; UDCA: ursodeoxycholic acid; BA: bile acids; CA: cholic acid; CDCA: chenodeoxycholic acid; DCA: deoxycholic acid; LCA: lithocholic acid; CSCs: cancer stem cells; MHC: major histocompatibility; NF-κB: NF-kappaB; GPR: G protein-coupled receptors; ROS: reactive oxygen species; RNS: reactive nitrogen substances; BER: base excision repair; DNA: deoxyribonucleic acid; EGFR: epidermal growth factor receptor; MAPK: mitogen activated protein kinase; ERKs: extracellular signal regulated kinases; AKT: protein kinase B; PA: phosphatidic acid; TMAO: trimethylamine n-oxide; TMA: trimethylamine; FMO3: flavin-containing monooxygenase 3; H2S: Hydrogen sulfide; SRB: sulfate-reducing bacteria; IBDs: inflammatory bowel diseases; NSAID: non-steroidal anti-inflammatory drugs; BFT: fragile bacteroides toxin; ETBF: enterotoxigenic fragile bacteroides; E-cadherin: extracellular domain of intercellular adhesive protein; CEC: colonic epithelial cells; SMOX: spermine oxidase; SMO: smoothened; Stat3: signal transducer and activator of transcription 3; Th17: T helper cell 17; IL17: interleukin 17; AA: amino acid; TCF: transcription factor; CDT: cytolethal distending toxin; PD-L1: programmed cell death 1 ligand 1
KEYWORDS: Human gut microbiome, colorectal cancer, metabolism, short-chain fatty acids, secondary bile salts, bacterial toxin
1. Introduction
Colorectal cancer (CRC) is the world’s third most common malignancy and second leading cause of cancer death [1]. Although extensive research has been conducted, the exact cause and etiopathogenesis of CRC are yet to be fully clarified. Due to technological advancements, such as high-throughput sequencing, changes in human gut microbiome type and abundance in CRC patients have been identified [2,3]. There are approximately 38 trillion bacteria in the human body, and the intestine is the organ which is most densely colonized [4]. The colon also contains a minimum of two orders of magnitude more bacteria than any other organ [5]. The human gut microbiome is a complex community that is composed of bacteria, archaea, viruses, and eukaryotes [6]. This complex ecosystem contains approximately 500 different bacteria species [7]. These intestinal bacteria are mainly composed of bacteroidetes, firmicutes, actinomycetes, proteobacteria, and ruminococcaceae in addition to relatively few clostridium [8]. As well as the regulation of immunity and maintenance of human health, the human gut microbiome also mediates the occurrence and development of some diseases, which includes CRC [9,10].
It is commonly believed that intestinal microbiota products, including butyrate, H2S, and bacterial toxins, contribute to CRC’s development and progression [11–13]. Butyrate, for example, displays significant wellness promoting and anti-tumor characteristics. It is the prime energy resource for colon cells, maintaining epithelial integrity and inhibiting inflammation and cancer by the role it plays in immunity, gene expression, and epigenetic regulation [13]. However, the molecular mechanisms which underlie the effects of gut microbial products on CRC have not yet been fully clarified, and the effect of some products on CRC is still controversial. Extensive studies have explored the relationship between gut microbes, their products, and CRC, and their relationship will be explored based on recent studies. This review examines the contribution some products of the human gut microbiome have made to the development of CRC, in particular the molecular mechanisms between products and CRC, to explore methods of preventing and treating CRC using these controllable factors.
2. Factors inhibiting colorectal cancer
2.1. Butyrate inhibits the invasion and proliferation of CRC and promotes the apoptosis of cancer cells
Colon bacteria break down indigested dietary fibers and starches and produce short-chain fatty acids (SCFA), such as acetic acid, propionic acid, and butyric acid [14]. The aerobic glycolysis of SCFA provides colon cells with their main energy source [15], while also playing a part in the immunity and metabolism of the host intestine. SCFA content in CRC patients’ plasma decreases significantly, which proves that a decrease in SCFA promotes CRC progression [16].
Butyrate is the most studied short-chain fatty acid and it is mostly synthesized by glycolysis from hydrocarbons by two families of the firmicutes of the human colon, ruminococcaceae and lachnospiraceae [17]. As a histone deacetylase (HDAC) inhibitor, butyrate inhibits carcinoma cell multiplication while triggering cell death [18]. In normal colon cells, butyrate is β-oxidized by mitochondria and produces energy from the tricarboxylic acid cycle (TCA cycle) or cytosolic acyl coenzyme A(CoA). Otto Warburg et al. remarked that carcinoma cells have the ability to alter their metabolic modes even under oxygen, and they prefer to undergo a glycolytic pathway rather than an oxidative phosphorylation (OXPHOS) pathway, in order to transform the absorbing glucose mostly into lactate [19]. The transformation of glycolytic metabolism has been recognized as being a dominant characteristic of carcinoma cells; cancerous colon cells prefer glucose to butyrate as their preferred energy resource due to the Warburg effect pathway. As a result, cancerous colon cells accumulate a large amount of butyrate which acts as HDAC inhibitor [20]. Butyrate can enter the nucleus directly and inhibit histone deacetylase 1 and cause a reduction in short-chain acyl CoA dehydrogenase (SCAD) levels, which is the primary process in the catalyzation of mitochondrial butyrate oxidation [21]. This reduces the auto-oxidation of butyrate in CRC cells [22] and allows butyrate to accumulate in carcinoma cells, thereby restraining CRC development. This also explains why tumor cells have a greater sensitivity to histone deacetylase (HDAC) inhibitors than non-transformed cells [23].
The overexpression of microRNA-92a (MiR-92a) in CRC [24] facilitates CRC growth and invasion through the targeting of kruppel-like factor 4(KLF4) and downstream p21 [25], and a reduction in miR-92a can cause apoptosis of cancer cells [26]. MiR-92a also inhibits phosphatase and tensin homolog (PTEN) expression [24], which is a typical anti-oncogene that can be found in region 10q23 of chromosome 10 and is the foremost negative regulator of the phosphoinositide 3-kinase(PI3K) signaling pathway [27]. When faced with extracellular stimuli (including insulin, growth factors, and chemokines), activated PI3K converts PIP2 (phosphatidylinositol-4,5-bisphosphate) into PIP3 (phosphatidylinositol-3,4,5-trisphosphate) and this phosphorylates and activates Akt (protein kinase B). PTEN antagonizes PI3K by dephosphorylating PIP3 and forming PIP2 (thereby blocking the PI3K signaling cascade) [28]. Butyrate can down-regulate miR-92a expression via c-Myc, which reduces the proliferation of colon cancer cells and stimulating apoptosis [29]. Butyrate can also reduce the phosphorylation of Akt1 (protein kinase B subtype α) and ERK1/2 (Extracellular signal–regulated kinases 1/2) by blocking HDAC3 activity and inhibiting any subsequent cell movement, which ultimately impedes CRC cell metastasis and invasion [30].
MiR-203 expression levels are significantly reduced in CRC tissues and carcinoma cell lines, and this low expression relates to tumor size and pathologic staging (pTNM) [31]. Previous studies have demonstrated that in the early progression of cancer, epithelial cells are subjected to a procedure known as epithelial-mesenchymal transition (EMT). This is evidenced by the absence of E-calcium adhesion protein (the main ingredient of adhesion) and results in interrupted cell-to-cell contact. Hakai is an E3 ubiquitin ligase which binds and degrades E-cadherin in a phosphorylation-dependent way, regulating cell adhesion. Hakai expression is upregulated in colorectal adenocarcinomas and adenomas using differentiated TNM staging (I–IV) and compared with healthy human colon tissue. Hakai overexpression in epithelial cells also induces cell transformation, mesenchymal and invasive phenotypes, while inhibiting E-cadherin promotes increases proliferation and the oncogenic potential of the N-cadherin expression [32]. MiR-203 directly targets Hakai and lowers its level, thereby inhibiting cell proliferation. NEDD9 (neural precursor cell expressed developmentally down-regulated9), also known as HEF1 or cas-1, is a part of the crk-associated substrate (CAS) family and has a high level of expression in multiple carcinoma types, involving the adherence, migration, and invasion of cancer cells. NEDD9 promotes EMT in CRC through the JNK (c-Jun N-terminal kinase) pathway [33]. MiR-203 targets NEDD9 in order to downregulate, thereby inhibiting CRC cell multiplication, colonization, and invasion, and inducing apoptosis in CRC cells [34]. In addition to inhibiting tumor development and metastasis, miR-203 also inhibits CRC’s chemical resistance [35]. Butyrate upregulates miR-203, which inhibits CRC cell multiplication, colony formation, and invasion, and promotes CRC cell apoptosis [34](Figure 1).
Figure 1.
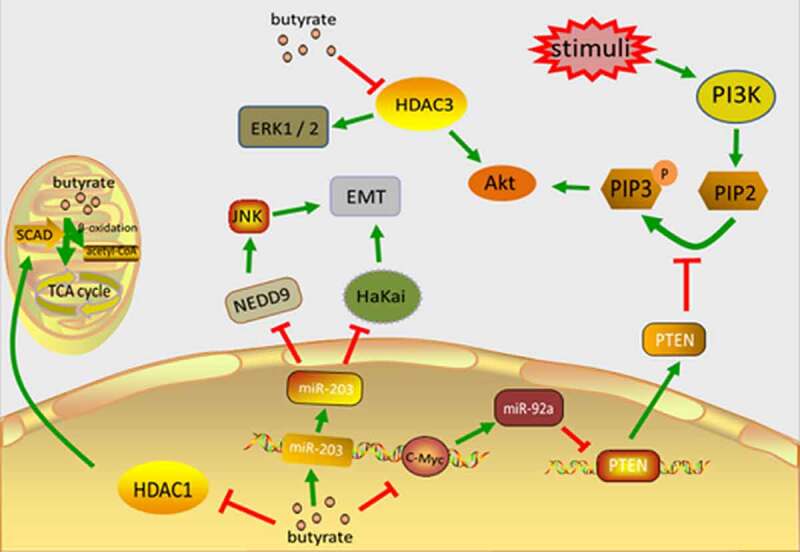
The main mechanism of butyrate inhibiting the occurrence and development of CRC. Butyrate directly enters the cell nucleus to inhibit HDAC1, reduces SCAD level, and reduces the self-oxidation of butyrate in carcinoma cells. Butyrate accumulates in cancer cells and inhibits their proliferation. Butyrate can block the activation of HDAC3, leading to decreased phosphorylation of Akt1 and erk1/2, thereby inhibiting cell motility and ultimately CRC cell migration and invasion. Butyric acid regulates the expression of c-Myc, inhibits the transcription of miR-92a, increases the expression of PTEN, and therefore antagonizes the effect of PI3K, thereby reducing the proliferation of colon cancer cells and stimulating apoptosis. Butyrate upregulates miR-203 which directly targets HaKai, reducing its level and inhibiting cell proliferation
The bacterium Propionibacterium, which is found in fibrous foods and dairy products, creates SCFAs, mainly propionates and acetate. Acetate inhibits CRC cell multiplication and triggers CRC cell apoptosis in a dose-dependent manner. However, the precise mechanism by which it transports across the CRC cell membrane is not completely understood [36,37]. Studies suggest that monocarboxylate transporter-1 (MCT1) and aquaporins play crucial roles in acetate uptake [38]. Acetate has the ability to trigger apoptosis and inhibit cell proliferation in CRC cells by DNA fragmentation and caspase-3 activation. However, acetate cam also induce the release of histone D into the cytoplasm, thereby protecting CRC cells from acetate induced apoptosis through the degradation of damaged mitochondria [39,40]. It is believed that propionate prevents colorectal cancer, but a lack of studies of its mechanism have been conducted. It is suggested by evidence that the protein arginine methyltransferase 1 (PRMT1) is overexpressed in early CRC and its high expression has an association with poor CRC patient prognosis. Recent studies suggest propionate induces PRMT1 downregulation and therefore apoptosis in CRC cells. Unfortunately, the exact mechanism which connects propionate and PRMT1 regulation remains unclear [41]. Further studies are required for the investigation of whether acetate and propionate protect against CRC and the magnitude of their inhibitory effect on CRC, particularly the mechanisms of how they are involved in CRC development.
2.2. Ursodeoxycholic acid (UDCA) inhibits CRC by modulating inflammatory responses and enhancing immune surveillance
Bile acids (BAs) are made in the liver from cholesterol and are then transported into the intestine by bile which promotes the intestine’s uptake of fat. Two main BAs, cholic acid (CA) and chenodeoxycholic acid (CDCA), are produced in the human liver by the ‘classical’ pathway [42]. More than 90% of intestinal BAs are reabsorbed in the ileum and then transported through the portal vein to the liver, where they are processed by hepatocytes and secreted again into the bile [43]. Bacteria in the gut, including Clostridium, Enterococcus, Bifidobacterium, and Lactobacillus, convert unabsorbed BAs into hydrophobic secondary bile salts. For example, gut bacteria transform CA into deoxycholic acid (DCA), and CDCA into lithocholic acid (LCA) [44–46].
UDCA is a secondary bile acid which is produced by Clostridium species, including Clostridium absonum, and Clostridium baratii. It has a chemical structure that is quite similar to that of DCA, but unlike the hydrophobic bile acid DCA, UDCA has been proven to impede colon cancer occurrence [47,48]. Patients with colorectal adenoma who have taken UDCA for a long period of time are less likely to relapse following the removal of the colorectal adenoma, and the proliferation of colonic epithelium is significantly reduced [49]. UDCA can also significantly reduce atypical adenoma’s recurrence rate [50]. UDCA can inhibit CRC in several ways, including by increasing the hydrophilicity of the bile pool, decreasing the concentration of hydrophobic BA [51], and regulating oxidative stress in colon cancer cells and colon cancer stem cells (CSC) [52]. Furthermore, UDCA up-regulates colonic major histocompatibility(MHC) expression, which enhances the immune surveillance of tumors [53], suppresses cox-2 in CRC [47], and inhibits NF-kappaB (NF-κB) activated IL-1 and deoxycholic acid induced Aβ and AP-1 in human CRC cells [54]. However, some studies have suggested that UDCA does not have a preventive effect on CRC [55]. In addition, high doses of oral UDCA are linked to a higher risk of CRC [56]. The impact UDCA has on CRC is still controversial and further studies are required in order to prove its function.
2.3. Niacin acts on G protein-coupled receptors (GRA) and prostaglandin receptors to inhibit colonic inflammation and carcinogenesis
In addition to being obtained from food, vitamin B is produced by the intestinal microbiota, for example, Lactobacillus acidophilus. Niacin, which is also called nicotinic acid or vitamin B3, acts as a precursor to coenzymes, including nicotinamide adenine dinucleotide (NAD) and nicotinamide adenine dinucleotide phosphate (NADP), and its presence is indispensable for viable cells [57]. In addition to its hypolipidemic effects, it is believed that niacin has anti-inflammatory effects [58]. Niacin signals through GPR109a, and GPR109a signaling enhances the anti-inflammatory effects of colonic macrophages and dendritic cells, allowing them to induce Treg cell and IL-10-producing T cell differentiation. Animal studies suggest that niacin can prevent colitis and colon cancer in mice through the activation of GPR109a, although the exact molecular mechanism of this remains unclear [59]. Some experiments have suggested that niacin can protect the intestinal mucosa by reducing the level of TNF-α through GPR109a [60]; whereas others have implied that niacin achieves its protective effect on the intestinal mucosa through the mediation of the release of prostaglandin D2 through GPR109a [61]. CSC intervene in tumor development and sustainment; the cells are chemically resistant and characterized by self-replenishing, multipotency, flexibility, and diversification. The elimination of CSC may increase patient survival rate [62]. Niacin has also shown effects on tumor stem cells, with small doses favoring cell proliferation in colon CSCs, and high doses inducing cell death [63]. However, as of yet, no studies have been conducted on the mechanism of this phenomenon. Many gaps remain regarding whether niacin can prevent CRC and the way in which it protects the intestinal mucosa from inflammation and CRC.
3. Factors promoting colorectal cancer
3.1. Different secondary BAs have different effects on CRC by causing oxidative stress, activating MAPK cascade, and other mechanisms
Those who follow high-fat diets generate a greater amount of secondary BA, mostly DCA and LCA, and have a higher incidence of CRC [64,65]. Cholesterol is a component of the lipid membrane which is essential and causes hardening of the membranes [66]. The secondary BA is a cholesterol derivative with washing characteristics, so when they are present in high levels, it is possible that they cause the destruction of cell membranes and local disruption to the intestinal epithelium [67]. This injury then stimulates the repair mechanism that is involved in the inflammatory response and the over-proliferation of undifferentiated cells. The over-proliferation of colonic mucosa is considered as being one of the initial steps in CRC development. Additionally, serum DCA levels have been found to be correlated with the rate of hyperplasia of the colonic mucosa [68]. Hydrophobic bile acids can produce reactive oxygen species (ROS) and reactive nitrogen substances (RNS), which cause oxidative stress, damage to DNA and proteins, and destruction of the base excision repair (BER) pathway [69]. The BER pathway can address DNA oxidation injuries facilitated by ROS. DNA repairs defects that are caused by oxidative damage as a CRC risk factor [70]. BA can also cause genomic instability via the oxidative damage pathway [67]. In the carcinogenic methane peroxide-induced rat tumor model system, DCA has been proven to raise the rate of CRC, and tumors in K-ras point mutual mutations [70].
Epidermal growth factor receptor (EGFR) is a tyrosine kinase receptor, an ErbB family protein which promotes proliferation [67], invasion, or metastasis of various tumors, including CRC, by mutation or overexpression [71,72]. Through ligand stimulation, EGFR is dimerized, and the dimerization of EGFR is followed by receptor internalization and autophosphorylation, serving as the binding site for recruiting signal transducers and intracellular signal transduction cascade activators. EGFR-linked activation of the mitogen-activated protein kinase (MAPK) cascade facilitates the regulation of downstream molecules ERKs and Akt [73]. Of all the diverse subfamilies of the MAPK pathway, ERK1/2 promote cells differentiation, division, and block apoptosis, whereas p38 MAPK and SAPK/JNK1/2 induce apoptosis. Therefore, the balance of these pathways’ dynamics is a key factor for the determination of cell fate and processes. Abnormal activation of MAPK triggers colorectal mucosal overgrowth, which leads to the formation of colorectal tumors [74]. Phosphatidic acid (PA) is one of the crucial components of EGFR signaling nanodomains on cell surfaces. Secondary bile acid DCA significantly enhances the local spatial aggregation of phospholipid acid and induces co-localization between PA and EGFR, which promotes EGFR dimerization/oligomerization, and stimulates EGFR-MARK signaling [75]. DCA stimulates cell proliferation in addition to inducing EGFR phosphorylation in ligand-dependent manner [76], meaning that the participation of natural ligand (EGF) is required for EGFR activation by DCA. However, it has also been discovered that DCA regulates MAPK activation through calcium signaling [77]. In addition, BA, particularly DCA and LCA, trigger colon cancer development through the regulation of M3R and Wnt/β-catenin signaling in order to make normal colon epithelial cells convert into CSC [78](Figure 2).
Figure 2.
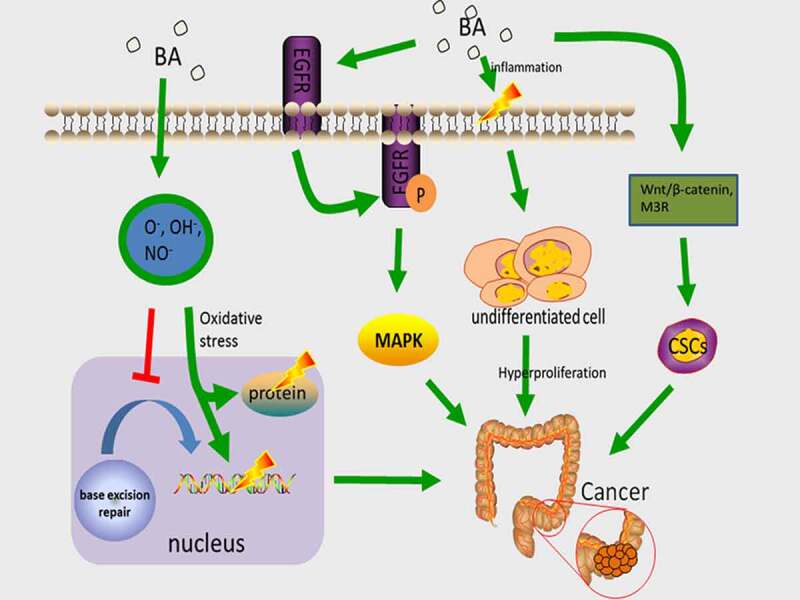
The main mechanism of BA carcinogenesis. Hydrophobic bile acids produce ROS and RNS, damage DNA and proteins, and damage BER, increasing the incidence of mutations. DCA induces PA co-localization with ERGF, promoting EGFR dimerization/multimerization and activating the MAPK cascade. activation of MAPK triggers colonic mucosal hyperproliferation, causing the development of colorectal tumors. Bile acids regulate M3R and Wnt/beta-catenin signaling and induce CSC in colonic epithelial cells, thereby inducing colon carcinogenesis
3.2. Trimethylamine n-oxide (TMAO) is related to CRC by unknown underlying mechanisms
Trimethylamine N – oxide is a Trimethylamine (TMA) oxidation product, belonging to the amine oxide family [79]. TMA consists of dietary choline and phospholipids (lecithin) under the effect of the human gut microbiome [80,81]. The host absorbs TMA and it is then processed by flavin-containing monooxygenases (mainly FMO3) in the liver to TMAO and excreted via the kidneys [82,83]. A prospective cohort study including 835 CRC cases and 835 matched controls discovered that increased plasma levels of TMAO are related to a greater CRC risk [84]. Another study discovered a significant association between TMAO and CRC, and noted that TMAO engaged in a number of genetic pathways with an apparent association to carcinomas, in particular colon cancer [85]. Although there is evidence suggesting elevated TMAO levels are related to an increased cancer risk, it remains uncertain whether elevated TMAO levels are a reason for or a result of cancer [86], so the role of TMAO in promoting cancer remains controversial. Currently, evidence suggests that inflammation is a potential contributor to the connection between TMAO and cancer [87], but further evidence is required in order to validate this.
3.3. Hydrogen sulfide (H2S) regulates the growth or death of cells and promotes the proliferation of CRC
H2S is created by Sulfate-reducing bacteria (SRB) metabolizing sulfates in food, and other sulfur-containing compounds, including taurine [88]. There is evidence suggesting that the level of H2S in CRC subjects’ feces is higher than in the control group without tumors [89]. Another study discovered significantly higher fecal H2S levels in patients with colon tumors and sigmoid surgery compared to healthy individuals of a similar age [90], meaning that the ability of colon detoxification H2S is also reduced in colon cancer patients [91]. It can therefore be suggested that H2S will likely work in the pathogenesis of intestinal diseases, inflammatory bowel diseases (IBDs,) and CRC [92]. Studies on the role played by H2S in CRC have reached different conclusions. Some studies believe that H2S promotes CRC due to its ability to promote inflammation and genotoxicity at physiological concentrations [93,94], in addition to being able to inhibit butyric acid oxidation and promote cell proliferation in vitro [95,96]. The pro-inflammatory effect of H2S appears to be related to the disruption of disulfide bonds in the double layer of mucus in the colonic wall by H2S, which leads to the epithelium being exposed to bacteria and toxins [97]. Interestingly, some studies suggest that H2S can protect the mucus layer and reconstitute the already disrupted mucus layer, thereby preventing inflammation [98,99]. Several studies have confirmed this using new non-steroidal anti-inflammatory drugs (NSAID) which release H2S [100,101]. H2S-releasing compounds exhibit potent anticancer effects through inhibition of the proliferation and/or inducing apoptosis in several types of cancer cell, including CRC, but the mechanism that is involved remains unknown and could be related to H2S inhibiting nuclear factor-κB (NF-κB) signaling and increasing intracellular Ca2+ concentration, which leads to cell cycle arrest [102,103]. Generally, the role of H2S in CRC is controversial and further study of the mechanistic pathways is required.
3.4. Bacterial toxins can cause DNA damage, promote inflammation, and regulate tumor microenvironment for promotion of CRC occurrence and invasion
In addition to transforming nutrients, bacteria also affect CRC by producing carcinogenic metabolites or toxic factors. These toxins can be characterized by genotoxicity, pro-inflammatory, and epithelial infiltration, and can induce and promote CRC occurrence. The commonly held belief is that there is a complex interaction between bacterial toxins and CRC occurrence.
B.fragilis toxin (BFT), which is the main virulence factor Enterotoxigenic bacteria fragilis (ETBF) produces, is a 20kDa zinc-dependent metalloproteinase toxin with three isotypes (BFT-1, BFTt-2, and BFT-3), all of which demonstrate similar biological activity [104]. BFT induce lysis of the extracellular domain of intercellular adhesive protein (E-cadherin) in vitro, cause a loss of epithelial integrity and increase the permeability of the single layer of colonic epithelial cells (CEC) [105], while triggering the activation of Wnt by β-catenin localization (inducing transcription and translation of the proto-oncogene c-Myc, which promotes CEC proliferation) [106]. BFT also activates NF-κB signaling, causing CEC to secrete cytokines, and potentially promoting mucosal inflammation. Enhanced NF-κB signaling can assist in CEC’s oncogenic effects [107]. Bacterially produced BFT up-regulates the spermidine oxidase (SMOX) gene that is expressed in human healthy CEC [108], and SMOX codified SMO (smooth) protein is essential for the normal metabolism of polyamines. SMO promotes the conversion of spermine to spermidine, and generates hydrogen peroxide and aldehydes, which causes DNA damage and apoptosis, and promotes cancer growth [109].
Stat3(signal transducer and activator of transcription 3) is a multifunctional transcription factor in which affects the pathogenesis of autoimmunity by binding to numerous genes that are associated with Th17 cell division, activation, and multiplication, while mediating expression and epigenetic alterations [110]. BFT can activate Stat3 for the regulation of Th17 cells [111]. IL-17 is mostly produced by Th17 cells [112,113], and endogenous IL-17 is tumorigenic, which directly affects CEC signaling, cell survival, and proliferation [114]. In addition, activated NF-κB has been observed in IL-17-stimulated cells, whereas the activation of Stat3 maintains the activity of NF-κB [115]. ETBF can selectively activate Stat3 in the colon, inducing Th17 cell infiltration to cause colon cancer. After blocking IL-17, ETBF can inhibit colon tumor which is induced by ETBF [116](Figure 3).
Figure 3.
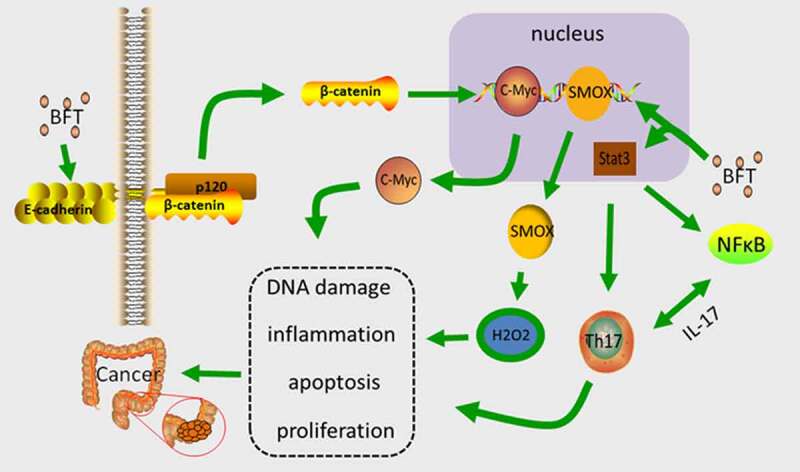
The main mechanism of BFT carcinogenesis. BFT up-regulates the expression level of SMOX and increases the synthesis of SMO, which promotes the conversion of spermine, which produces hydrogen peroxide causing DNA damage and apoptosis, and promotes the progression of cancer. BFT can induce cleavage of E-cadherin, trigger activation of Wnt by β-catenin nuclear localization, induce transcription and translation of c-Myc, and promote proliferation of CEC. BFT activates Stat3 to regulate Th17 cells, produce IL-17, and promote tumor development
The cell surface protein FadA is a key poison factor for fusobacterium nucleatum (Fn), regulating the adhesion and invasions of bacteria. The expression of FadA gene was obviously increased in CRC patient specimens in comparison to normal tissues [117]. It exists in two forms, a non-secretory pre-FadA which consists of 129 amino acid (AA) residues, and a secretory maturing FadA (mFadA) which consists of 111AA and has a signal sequence of 18 AA [118]. Intrinsic FadA and mFadA precomplexes secure Fn binding and invade host epithelial cells [119]. The binding of FadA to host endothelial receptors and vascular endothelial globulin (CDH5) is essential for Fn adhesion and cell invasion and results in the detachment of CDH5 from the cellular junction, which increases endothelial permeability and allows bacteria to pass through the loose junctions [120]. FadA can also be bound to E-cadherin in vitro. E-cadherin exists in epithelial cells, including non-cancerous HEK293, and CRC cells with the exception of RKO. FadA mediates Fn adhesion and the invasion of CRC cells through E-cadherin [117]. FadAc specifically binds to E-cadherin, which leads to phosphorylation and the internalization of E-cadherin on the membrane, thereby inhibiting its tumor suppressor activity. This results in elevated β-catenin-regulated transcription and triggers an inflammatory response, which increases the gene expression of the transcription factor NF-κB and Wnt pathways, and promotes CRC cell proliferation [121].
The main toxins that intestinal bacteria produce are colistin and CDT (cytolethal distending toxin), which are made by Escherichia coli (E. coli) and other gram-negative bacteria in the large intestine and directly damage DNA [116,122,123]. Colibactin is a heterogeneous ketone compound/non-ribosomal peptide complex that is produced by a complicated biosynthetic mechanism [124]. Certain strains which produce myxomycetin often have an association with CRC [125,126]. Colibactin can cause the breakage of double-strand DNA, chromosome instability, and cell senescence in eukaryotic cells [122,126–128]. Bacteria which produce E. coli can modify the tumor microenvironment, which leads to cellular aging and can also influence tumor progression through the secretion of growth factors [129]. CDT are bacterial protein family toxins that are produced by a number of gram-negative bacteria, including E. coli, actinomycetes, shigella dysentery, and helicobacter pylori [130]. The genotoxin CDT consists of three subunits CdtA, CdtB, and CdtC. CdtB is similar to DNAase I and can cause damage to host DNA [131]. CdtA and CdtC subunits are required proteins which mediate the combination of toxins with target cytoplasmic membranes and allow the internalization of essential active subunit CdtB [132]. CDT can trigger a DNA damage response, which leads to the arrest of the cell cycle and causes cellular senescence or death [133–135]. CDT has a critical effect on the carcinogenic effect that campylobacter jejuni induces. CDT-derived campylobacter jejuni cause injury to host cell DNA, and promote colorectal tumorigenesis by triggering cell multiplication and the enhancing of nuclear translocation of β-catenin protein [131].
4. Outlook and conclusion
Colorectal cancer is a multifactorial disease and microbial dysbiosis in the human gut that has been identified as a danger factor for CRC [136–138]. Although the underlying mechanisms of the role of microbial products in CRC is not fully understood, the use of dietary regulation or probiotics in CRC control has been investigated. The gnotobiotic mouse model discovered that dietary fiber and human gut microbiome can regulate the colon lumen of butyric acid salt level, and therefore the structure and colonic crypt in the presence of stromal cells, allowing the development of its role of inhibiting colon cancer in the body. These findings suggest that probiotics and/or prebiotics can be used in order to elevate the endogenous HDAC inhibitors’ content and reduce tumor development, without the adverse reactions similar to those caused by the use of synthetic HDAC(e.g., vorinostat/SAHA) in chemotherapy [139]. Gut microbiome can influence cancer chemotherapy’s efficacy and toxicity [140]. In an animal model, the co-administration of bifidobacterium long and bifidobacterium short can improve cancer control, significantly reduce tumor progression and enhance the PD-L1 blocking antibody’s anticancer effect [141]. It has also been demonstrated that gut microbiota disorders can lead to the reduced antitumor efficacy of 5-Fluorouracil (5-FU) [142]. Although these results cannot be directly applied to the treatment of human cancer, they offer the potential to use microorganisms for the prevention of CRC.
To summarize, gut microbes’ products can play an important role in the prevention of CRC (Table 1) or the promotion of CRC progression (Table 2) using various mechanisms. Limited studies have been conducted which explore the relationship between gut microbiota and CRC. Further evidence is required to support a causal relationship between human gut microbiome in CRC, and more clinical data is required to support the feasibility of microbial prevention and treatment of CRC.
Table 1.
Factors inhibiting CRC
| Origins | Products | Potential microbe | Mechanism | References | |
|---|---|---|---|---|---|
| dietary fibers and starches | SCFA | Butyrate |
ruminococcaceae lachnospiraceae |
Function as HDAC inhibitor | 20,23 |
| Down-regulate miR-92a expression via c-Myc | 29 | ||||
| Upregulates miR-203 | 34 | ||||
| Acetate | Propionibacterium | DNA fragmentation | 39,40 | ||
| Caspase-3 activation | 39,40 | ||||
| Propionates | Propionibacterium | Induces PRMT1 downregulation | 41 | ||
| BA | UDCA | Clostridium absonum, Clostridium baratii, | Decreasing the concentration of hydrophobic BA | 51 | |
| Regulate oxidative stress | 52 | ||||
| Up-regulates colonic MHC expression, enhancing immune surveillance | 53 | ||||
| Inhibits NF-κB activated | 54 | ||||
| Unknown | Niacin | Lactobacillus acidophilus | Enhances anti-inflammatory effects by activating GPR109a | 59 | |
| Effect on CSC | 63 | ||||
| Sulfates and other sulfur-containing compounds | H2S | Sulfate-reducing bacteria | Inhibit NF-κB signaling and increase intracellular Ca2+ concentration, leading to cell cycle arrest | 101,102 | |
CRC: colorectal cancer; SCFAs: short chain fatty acids
HDAC: histone deacetylase;
MiR-92a: microRNA-92a;
BA: bile acids
UDCA: ursodeoxycholic acid
MHC: major histocompatibility
NF-κB: NF-kappaB
GPR: G protein-coupled receptors
CSCs: cancer stem cells
H2S: Hydrogen sulfide
Table 2.
Factors promoting CRC
| Origins | Products | Potential microbe | Mechanism | References |
|---|---|---|---|---|
| BA | DCA and LCA | Clostridium, Enterococcus, Bifidobacterium, Lactobacillus | produce ROS and RNS, causing oxidative stress, and damaging DNA | 69 |
| K-ras point mutual mutations | 70 | |||
| Stimulate EGFR-MARK signaling | 70 | |||
| Regulate M3R and Wnt/β-catenin signaling | 78 | |||
| Choline and phospholipids | TMAO | Unknown | Promotes inflammation | 87 |
| Sulfates and other sulfur-containing compounds | H2S | Sulfate-reducing bacteria | Promotes inflammation, genotoxicity, inhibits butyric acid oxidation | 93–96 |
| Bacterial toxins | B.fragilis toxin (BFT) | Enterotoxigenic bacteria fragilis (ETBF) | Induces E-cadherin lysis, resulting in loss of epithelial integrity | 105 |
| Triggers activation of Wnt, induces c-Myc expression and promotes CEC proliferation | 106 | |||
| Activates NF-κB signaling and promotes inflammation | 107 | |||
| Upregulates the expression of SMOX genes, leading to DNA damage | 108,109 | |||
| Activates Stat3 to regulate Th17 cells and maintain NF-κB activity | 112,116 | |||
| FadA | Fusobacterium nucleatum (Fn) | Binds to CDH5, increases endothelial permeability, and allows bacteria to pass through loose junctions | 117 | |
| Binds to E-cadherin and mediates Fn adhesion and invasion of CRC cells | 117 | |||
| Binding to E-cadherin inhibits its tumor suppressor activity and increases the expression of NF-κB and Wnt pathways | 121 | |||
| Colibactin | E. coli and other gram-negative bacteria | Leads to double-stranded DNA breaks, chromosomal instability and cellular senescence | 122,126–128 | |
| Cytolethal distending toxin (CDT) | Causes DNA damage, leading to cell cycle arrest and cellular senescence or death | 133–135 |
CRC: colorectal cancer
BA: bile acids
DCA: deoxycholic acid
LCA: lithocholic acid
ROS: reactive oxygen species
RNS: reactive nitrogen substances
EGFR: epidermal growth factor receptor
MAPK: mitogen activated protein kinase
TMAO: trimethylamine n-oxide
H2S: Hydrogen sulfide
BFT: fragile bacteroides toxin
ETBF: enterotoxigenic fragile bacteroides
CEC: colonic epithelial cells
SMOX: spermine oxidase
NF-κB: NF-kappaB
Fn: Fusobacterium nucleatum
CDT: cytolethal distending toxin
Funding Statement
Social Development - Health Care Project of Yangzhou, jiangsu Province, (No. YZ2018087). High-level talent ‘six one projects’ top talent scientific research project of jiangsu Province, (No. LGY2019034).
Disclosure statement
The authors declare that there is no conflict of interest regarding the publication of this paper.
References
- [1].Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries.. CA Cancer J Clin. 2018;68(6):394–424. [DOI] [PubMed] [Google Scholar]
- [2].Xu K, Jiang B.. Analysis of mucosa-associated microbiota in colorectal cancer. Med Sci Monit. 2017;23:4422–4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Yu J, Feng Q, Wong SH, et al. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut. 2017;66(1):70–78. [DOI] [PubMed] [Google Scholar]
- [4].Dang AT, Marsland BJ.. Microbes, metabolites, and the gut–lung axis. Mucosal Immunol. 2019;12(4):843–850. [DOI] [PubMed] [Google Scholar]
- [5].Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016;14(8):e1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ringel Y, Maharshak N, Ringel-Kulka T, et al. High throughput sequencing reveals distinct microbial populations within the mucosal and luminal niches in healthy individuals. Gut Microbes. 2015;6(3):173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Morgillo F, Dallio M, Della Corte CM, et al. Carcinogenesis as a result of multiple inflammatory and oxidative hits: a comprehensive review from tumor microenvironment to gut microbiota. Neoplasia. 2018;20(7):721–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Shukla SD, Budden KF, Neal R, et al. Microbiome effects on immunity, health and disease in the lung. Clin Transl Immunology. 2017;6(3):e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Feng Q, Chen W-D, Wang Y-D. Gut microbiota: an integral moderator in health and disease. Front Microbiol. 2018;9:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dai Z, Zhang J, Wu Q, et al. The role of microbiota in the development of colorectal cancer. Int J Cancer. 2019;145(8):2032–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Guo -F-F, Yu T-C, Hong J, et al. Emerging roles of hydrogen sulfide in inflammatory and neoplastic colonic diseases. Front Physiol. 2016;7:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wu X, Wu Y, He L, et al. Effects of the intestinal microbial metabolite butyrate on the development of colorectal cancer. J Cancer. 2018;9(14):2510–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].O’Keefe SJD. Diet, microorganisms and their metabolites, and colon cancer. Nature Reviews Gastroenterology & Hepatology. 2016;13(12):691–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].van de Wouw M, Boehme M, Lyte JM, et al. Short-chain fatty acids: microbial metabolites that alleviate stress-induced brain-gut axis alterations. J Physiol. 2018;596(20):4923–4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang G, Yu Y, Wang Y-Z, et al. Role of SCFAs in gut microbiome and glycolysis for colorectal cancer therapy. J Cell Physiol. 2019;234(10):17023–17049. [DOI] [PubMed] [Google Scholar]
- [16].Yusuf F, Adewiah S, Fatchiyah F. The level short chain fatty acids and HSP 70 in colorectal cancer and non-colorectal cancer. Acta informatica medica: AIM: journal of the Society for Medical Informatics of Bosnia & Herzegovina: casopis Drustva za medicinsku informatiku BiH. 2018;26:160–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Louis P, Flint HJ. Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol. 2017;19(1):29–41. [DOI] [PubMed] [Google Scholar]
- [18].Medina V, Edmonds B, Young GP, et al. Induction of caspase-3 protease activity and apoptosis by butyrate and trichostatin A (inhibitors of histone deacetylase): dependence on protein synthesis and synergy with a mitochondrial/cytochrome c-dependent pathway.. Cancer Res. 1997;57(17):3697–3707. [PubMed] [Google Scholar]
- [19].Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nature Reviews Cancer. 2011;11(5):325–337. [DOI] [PubMed] [Google Scholar]
- [20].Eslami M, Sadrifar S, Karbalaei M, et al. Importance of the microbiota inhibitory mechanism on the warburg effect in colorectal cancer cells. J Gastrointest Cancer. 2020;51(3):738–747. [DOI] [PubMed] [Google Scholar]
- [21].Astbury SM, Corfe BM. Uptake and metabolism of the short-chain fatty acid butyrate, a critical review of the literature. <![CDATA[Current Drug Metabolism]]>. 2012;13(6):815–821. [DOI] [PubMed] [Google Scholar]
- [22].Han A, Bennett N, Ahmed B, et al. Butyrate decreases its own oxidation in colorectal cancer cells through inhibition of histone deacetylases. Oncotarget. 2018;9(43):27280–27292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Dashwood RH, Myzak MC, Ho E. Dietary HDAC inhibitors: time to rethink weak ligands in cancer chemoprevention? Carcinogenesis. 2006;27(2):344–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang G, Zhou H, Xiao H, et al. MicroRNA-92a functions as an oncogene in colorectal cancer by targeting PTEN. Dig Dis Sci. 2014;59(1):98–107. [DOI] [PubMed] [Google Scholar]
- [25].Lv H, Zhang Z, Wang Y, et al. MicroRNA-92a promotes colorectal cancer cell growth and migration by inhibiting KLF4. Oncology Research Featuring Preclinical and Clinical Cancer Therapeutics. 2016;23(6):283–290. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tsuchida A, Ohno S, Wu W, et al. miR-92 is a key oncogenic component of the miR-17-92 cluster in colon cancer. Cancer Sci. 2011;102(12):2264–2271. [DOI] [PubMed] [Google Scholar]
- [27].Milella M, Falcone I, Conciatori F, et al. PTEN: multiple functions in human malignant tumors. Front Oncol. 2015;5:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Molinari F, Frattini M. Functions and Regulation of the PTEN Gene in Colorectal Cancer.. Front Oncol. 2013;3:326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hu S, Liu L, Chang EB, et al. Butyrate inhibits pro-proliferative miR-92a by diminishing c-Myc-induced miR-17-92a cluster transcription in human colon cancer cells. Mol Cancer. 2015;14(1):180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Li Q, Ding C, Meng T, et al. Butyrate suppresses motility of colorectal cancer cells via deactivating Akt/ERK signaling in histone deacetylase dependent manner. J Pharmacol Sci. 2017;135(4):148–155. [DOI] [PubMed] [Google Scholar]
- [31].Chiang Y, Song Y, Wang Z, et al. Aberrant expression of miR-203 and its clinical significance in gastric and colorectal cancers. J Gastrointest Surg. 2011;15(1):63–70. [DOI] [PubMed] [Google Scholar]
- [32].Castosa R, Martinez-Iglesias O, Roca-Lema D, et al. Hakai overexpression effectively induces tumour progression and metastasis in vivo. Sci Rep. 2018;8(1):3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Meng H, Wu J, Huang Q, et al. NEDD9 promotes invasion and migration of colorectal cancer cell line HCT116 via JNK/EMT.. Oncol Lett. 2019;18(4):4022–4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Han R, Sun Q, Wu J, et al. Sodium butyrate upregulates miR-203 expression to exert anti-proliferation effect on colorectal cancer cells. Cell Physiol Biochem. 2016;39(5):1919–1929. [DOI] [PubMed] [Google Scholar]
- [35].Deng B, Wang B, Fang J, et al. MiRNA-203 suppresses cell proliferation, migration and invasion in colorectal cancer via targeting of EIF5A2. Sci Rep. 2016;6(1):28301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Jan G, Belzacq A-S, Haouzi D, et al. Propionibacteria induce apoptosis of colorectal carcinoma cells via short-chain fatty acids acting on mitochondria. Cell Death Differ. 2002;9(2):179–188. [DOI] [PubMed] [Google Scholar]
- [37].Lan A, Lagadic-Gossmann D, Lemaire C, et al. Acidic extracellular pH shifts colorectal cancer cell death from apoptosis to necrosis upon exposure to propionate and acetate, major end-products of the human probiotic propionibacteria. Apoptosis. 2007;12(3):573–591. [DOI] [PubMed] [Google Scholar]
- [38].Ferro S, Azevedo-Silva J, Casal M, et al. Characterization of acetate transport in colorectal cancer cells and potential therapeutic implications. Oncotarget. 2016;7(43):70639–70653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Oliveira CS, Pereira H, Alves S, et al. Cathepsin D protects colorectal cancer cells from acetate-induced apoptosis through autophagy-independent degradation of damaged mitochondria. Cell Death Dis. 2015;6(6):e1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Marques C, Oliveira CSF, Alves S, et al. Acetate-induced apoptosis in colorectal carcinoma cells involves lysosomal membrane permeabilization and cathepsin D release. Cell Death Dis. 2013;4(2):e507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ryu TY, Kim K, Son M-Y, et al. Downregulation of PRMT1, a histone arginine methyltransferase, by sodium propionate induces cell apoptosis in colon cancer.. Oncol Rep. 2019;41(3):1691–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Dawson PA, Karpen SJ. Intestinal transport and metabolism of bile acids. J Lipid Res. 2015;56:1085–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lin C-H, Kohli R. Bile acid metabolism and signaling: potential therapeutic target for nonalcoholic fatty liver disease. Clin Transl Gastroenterol. 2018;9(6):164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Maillette de Buy Wenniger L, Beuers U. Bile salts and cholestasis. Dig Liver Dis. 2010;42(6):409–418. [DOI] [PubMed] [Google Scholar]
- [45].Molinero N, Ruiz L, Sánchez B, et al. Intestinal bacteria interplay with bile and cholesterol metabolism: implications on host physiology. Front Physiol. 2019;10:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Li T, Chiang JYL, Ma Q. Bile acid signaling in metabolic disease and drug therapy. Pharmacol Rev. 2014;66(4):948–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Khare S, Mustafi R, Cerda S, et al. Ursodeoxycholic acid suppresses Cox-2 expression in colon cancer: roles of Ras, p38, and CCAAT/enhancer-binding protein. Nutr Cancer. 2008;60(3):389–400. [DOI] [PubMed] [Google Scholar]
- [48].Im E, Martinez JD. Ursodeoxycholic acid (UDCA) can inhibit deoxycholic acid (DCA)-induced apoptosis via modulation of EGFR/Raf-1/ERK signaling in human colon cancer cells. J Nutr. 2004;134(2):483–486. [DOI] [PubMed] [Google Scholar]
- [49].Serfaty L, et al. Ursodeoxycholic acid therapy and the risk of colorectal adenoma in patients with primary biliary cirrhosis: an observational study. Hepatology. 2003;38(1):203–209. [DOI] [PubMed] [Google Scholar]
- [50].Alberts DS, Martínez ME, Hess LM, et al. Phase III trial of ursodeoxycholic acid to prevent colorectal adenoma recurrence. JNCI: Journal of the National Cancer Institute. 2005;97(11):846–853. [DOI] [PubMed] [Google Scholar]
- [51].Ridlon JM, Bajaj JS. The human gut sterolbiome: bile acid-microbiome endocrine aspects and therapeutics. Acta Pharmaceutica Sinica B. 2015;5(2):99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kim E-K, Cho JH, Kim E, et al. Ursodeoxycholic acid inhibits the proliferation of colon cancer cells by regulating oxidative stress and cancer stem-like cell growth. PloS One. 2017;12(7):e0181183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Rigas B, Tsioulias GJ, Allan C, et al. The effect of bile acids and piroxicam on MHC antigen expression in rat colonocytes during colon cancer development.. Immunology. 1994;83(2):319–323. [PMC free article] [PubMed] [Google Scholar]
- [54].Shah SA, Volkov Y, Arfin Q, et al. Ursodeoxycholic acid inhibits interleukin beta 1 and deoxycholic acid-induced activation of NF-κB and AP-1 in human colon cancer cells. Int J Cancer. 2006;118(3):532–539. [DOI] [PubMed] [Google Scholar]
- [55].Pearson T, Caporaso JG, Yellowhair M, et al. Effects of ursodeoxycholic acid on the gut microbiome and colorectal adenoma development. Cancer Med. 2019;8(2):617–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Eaton JE, Silveira MG, Pardi DS, et al. High-dose ursodeoxycholic acid is associated with the development of colorectal neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Am J Gastroenterol. 2011;106(9):1638–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Peterson CT, Rodionov DA, Osterman AL, et al. B Vitamins and their role in immune regulation and cancer. Nutrients. 2020;12(11):3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zeman M, Vecka M, Perlík F, et al. Pleiotropic effects of niacin: current possibilities for its clinical use. Acta Pharm. 2016;66(4):449–469. [DOI] [PubMed] [Google Scholar]
- [59].Singh N, Gurav A, Sivaprakasam S, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014;40(1):128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Salem HA, Wadie W. Effect of niacin on inflammation and angiogenesis in a murine model of ulcerative colitis. Sci Rep. 2017;7(1):7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Li J, Kong D, Wang Q, et al. Niacin ameliorates ulcerative colitis via prostaglandin D 2 -mediated D prostanoid receptor 1 activation. EMBO Mol Med. 2017;9(5):571–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kharkar PS. Cancer stem cell (CSC) inhibitors: a review of recent patents (2012-2015). Expert Opin Ther Pat. 2017;27(7):753–761. [DOI] [PubMed] [Google Scholar]
- [63].Sen U, Shenoy PS, Bose B. Opposing effects of low versus high concentrations of water soluble vitamins/dietary ingredients Vitamin C and niacin on colon cancer stem cells (CSCs). Cell Biol Int. 2017;41(10):1127–1145. [DOI] [PubMed] [Google Scholar]
- [64].Bajor A, Gillberg P-G, Abrahamsson H. Bile acids: short and long term effects in the intestine. Scand J Gastroenterol. 2010;45(6):645–664. [DOI] [PubMed] [Google Scholar]
- [65].O’Neill AM, Burrington CM, Gillaspie EA, et al. High-fat Western diet–induced obesity contributes to increased tumor growth in mouse models of human colon cancer. Nutr Res. 2016;36(12):1325–1334. [DOI] [PubMed] [Google Scholar]
- [66].Ajouz H, Mukherji D, Shamseddine A. Secondary bile acids: an underrecognized cause of colon cancer. World J Surg Oncol. 2014;12(1):164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Payne CM. Hydrophobic bile acids, genomic instability, Darwinian selection, and colon carcinogenesis. Clin Exp Gastroenterol. 2008;1:19–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Ochsenk�hn T, Bayerd�rffer E, Meining A, et al. Colonic mucosal proliferation is related to serum deoxycholic acid levels. Cancer. 1999;85(8):1664–1669. . [PubMed] [Google Scholar]
- [69].Nguyen TT, Ung TT, Kim NH, et al. Role of bile acids in colon carcinogenesis. World J Clin Cases. 2018;6(13):577–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Bernstein H, Bernstein C, Payne CM, et al. Bile acids as endogenous etiologic agents in gastrointestinal cancer. World J Gastroenterol. 2009;15(27):3329–3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Zhang T, et al. Hsa-miR-875-5p exerts tumor suppressor function through down-regulation of EGFR in colorectal carcinoma (CRC). Oncotarget. 2016;7(42225–42240). DOI: 10.18632/oncotarget.9944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Kato S, et al. Revisiting epidermal growth factor receptor (EGFR) amplification as a target for anti-EGFR therapy: analysis of cell-Free circulating tumor DNA in patients with advanced malignancies. JCO Precis Oncol. 2019;3. DOI: 10.1200/po.18.00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Zhou S, Li Y, Lu J, et al. Nuclear factor-erythroid 2-related factor 3 (NRF3) is low expressed in colorectal cancer and its down-regulation promotes colorectal cancer malignance through activating EGFR and p38/MAPK.. Am J Cancer Res. 2019;9(3):511–528. [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [74].Kundu S, Kumar S, Bajaj A. Cross-talk between bile acids and gastrointestinal tract for progression and development of cancer and its therapeutic implications. IUBMB Life. 2015;67(7):514–523. [DOI] [PubMed] [Google Scholar]
- [75].Liang H, Estes MK, Zhang H, et al. Bile acids target proteolipid nano-assemblies of EGFR and phosphatidic acid in the plasma membrane for stimulation of MAPK signaling. PloS One. 2018;13(8):e0198983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Werneburg NW, Yoon J-H, Higuchi H, et al. Bile acids activate EGF receptor via a TGF-α-dependent mechanism in human cholangiocyte cell lines. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2003;285(1):G31–36. [DOI] [PubMed] [Google Scholar]
- [77].Centuori SM, Gomes CJ, Trujillo J, et al. Deoxycholic acid mediates non-canonical EGFR-MAPK activation through the induction of calcium signaling in colon cancer cells. Biochimica Et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 2016;1861(7):663–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Farhana L, Nangia-Makker P, Arbit E, et al. Bile acid: a potential inducer of colon cancer stem cells. Stem Cell Res Ther. 2016;7(1):181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Ufnal M, Zadlo A, Ostaszewski R. TMAO: a small molecule of great expectations. Nutrition. 2015;31(11–12):1317–1323. [DOI] [PubMed] [Google Scholar]
- [80].Kaysen GA, Johansen KL, Chertow GM, et al. Associations of trimethylamine N-Oxide with nutritional and inflammatory biomarkers and cardiovascular outcomes in patients new to dialysis. Journal of renal nutrition: the official journal of the Council on Renal Nutrition of the National Kidney Foundation. 2015;25(4):351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Tang WH, Wang Z, Kennedy DJ, et al. Gut microbiota-dependent trimethylamine N -Oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res. 2015;116(3):448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Tang WH, Hazen SL. The contributory role of gut microbiota in cardiovascular disease. J Clin Invest. 2014;124(10):4204–4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Koeth RA, Levison B, Culley M, et al. γ-Butyrobetaine Is a Proatherogenic Intermediate in Gut Microbial Metabolism of L -Carnitine to TMAO. Cell Metab. 2014;20(5):799–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Bae S, Ulrich CM, Neuhouser ML, et al. Plasma Choline Metabolites and Colorectal Cancer Risk in the Women’s Health Initiative Observational Study. Cancer Res. 2014;74(24):7442–7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Xu R, Wang Q, Li L. A genome-wide systems analysis reveals strong link between colorectal cancer and trimethylamine N-oxide (TMAO), a gut microbial metabolite of dietary meat and fat. BMC Genomics. 2015;16(Suppl S7):S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Chan CWH, Law BMH, Waye MMY, et al. Trimethylamine-N-oxide as one hypothetical link for the relationship between intestinal microbiota and cancer - where we are and where shall we go? J Cancer. 2019;10(23):5874–5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Rohrmann S, Linseisen J, Allenspach M, et al. Plasma concentrations of trimethylamine-N-oxide are directly associated with dairy food consumption and low-grade inflammation in a german adult population. J Nutr. 2016;146(2):283–289. [DOI] [PubMed] [Google Scholar]
- [88].Kimura H. Production and physiological effects of hydrogen sulfide. Antioxid Redox Signal. 2014;20(5):783–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].O’Keefe SJD, Li JV, Lahti L, et al. Fat, fibre and cancer risk in African Americans and rural Africans. Nat Commun. 2015;6(1):6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Kanazawa K, Konishi F,Mitsuoka T, et al. Factors influencing the development of sigmoid colon cancer. Bacteriologic and biochemical studies. Cancer. 1996;77:1701–1706. [DOI] [PubMed] [Google Scholar]
- [91].Ramasamy S, Singh S, Taniere P, et al. Sulfide-detoxifying enzymes in the human colon are decreased in cancer and upregulated in differentiation. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2006;291(2):G288–296. [DOI] [PubMed] [Google Scholar]
- [92].Carbonero F, Benefiel AC, Alizadeh-Ghamsari AH, et al. Microbial pathways in colonic sulfur metabolism and links with health and disease. Front Physiol. 2012;3:448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Fan X, Jin Y, Chen G, et al. Gut Microbiota dysbiosis drives the development of colorectal cancer. Digestion. 2020;1–8. doi: 10.1159/000508328 [DOI] [PubMed] [Google Scholar]
- [94].Attene-Ramos MS, Wagner ED, Gaskins HR, et al. Hydrogen sulfide induces direct radical-associated DNA damage. Mol Cancer Res. 2007;5(5):455–459. [DOI] [PubMed] [Google Scholar]
- [95].Alhinai EA, Walton W, Commane C. The role of the gut microbiota in colorectal cancer causation. Int J Mol Sci. 2019;20(21):5295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Han S, Zhuang J, Wu Y, et al. <p>Progress in Research on Colorectal Cancer-Related Microorganisms and Metabolites. <![cdata[cancer Management and Research]]>. 2020;12:8703–8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Ijssennagger N, van der Meer R, van Mil SWC. Sulfide as a mucus barrier-breaker in inflammatory bowel disease? Trends Mol Med. 2016;22(3):190–199. [DOI] [PubMed] [Google Scholar]
- [98].Motta J-P, Flannigan KL, Agbor TA, et al. Hydrogen sulfide protects from colitis and restores intestinal microbiota biofilm and mucus production. Inflamm Bowel Dis. 2015;21(5):1006–1017. [DOI] [PubMed] [Google Scholar]
- [99].Wallace JL, Motta J-P, Buret AG. Hydrogen sulfide: an agent of stability at the microbiome-mucosa interface. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2018;314(2):G143–g149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Gemici B, Elsheikh W, Feitosa KB, et al. H2S-releasing drugs: anti-inflammatory, cytoprotective and chemopreventative potential. Nitric Oxide. 2015;46:25–31. [DOI] [PubMed] [Google Scholar]
- [101].Kashfi K, Olson KR. Biology and therapeutic potential of hydrogen sulfide and hydrogen sulfide-releasing chimeras. Biochem Pharmacol. 2013;85(5):689–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Faris P, Ferulli F, Vismara M, et al. Hydrogen Sulfide-Evoked Intracellular Ca2+ Signals in Primary Cultures of Metastatic Colorectal Cancer Cells. Cancers (Basel). 2020;12(11):3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Kodela R, Nath N, Chattopadhyay M, et al. Hydrogen sulfide-releasing naproxen suppresses colon cancer cell growth and inhibits NF-κB signaling.. Drug Des Devel Ther. 2015;9:4873–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Rhee K-J, Wu S, Wu X, et al. Induction of persistent colitis by a human commensal, enterotoxigenic Bacteroides fragilis, in wild-type C57BL/6 mice. Infect Immun. 2009;77(4):1708–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Wu S, Rhee K-J, Zhang M, et al. Bacteroides fragilis toxin stimulates intestinal epithelial cell shedding and -secretase-dependent E-cadherin cleavage. J Cell Sci. 2007;120(11):1944–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Wu S, Morin PJ, Maouyo D, et al. Bacteroides fragilis enterotoxin induces c-Myc expression and cellular proliferation. Gastroenterology. 2003;124(2):392–400. [DOI] [PubMed] [Google Scholar]
- [107].Sears CL. Enterotoxigenic Bacteroides fragilis: a rogue among symbiotes. Table of Contents Clin Microbiol Rev. 2009;222:349–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Goodwin AC, Shields CED, Wu S, et al. Polyamine catabolism contributes to enterotoxigenic Bacteroides fragilis-induced colon tumorigenesis. Proc Natl Acad Sci U S A. 2011;108(37):15354–15359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Snezhkina AV, Krasnov GS, Lipatova AV, et al. The Dysregulation of Polyamine Metabolism in Colorectal Cancer Is Associated with Overexpression of c-Myc and C/EBP β rather than Enterotoxigenic Bacteroides fragilis Infection. Oxid Med Cell Longev. 2016;2016:2353560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Durant L, Watford WT, Ramos HL, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity. 2010;32(5):605–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Wick EC, Rabizadeh S, Albesiano E, et al. Stat3 activation in murine colitis induced by enterotoxigenic Bacteroides fragilis. Inflamm Bowel Dis. 2014;20(5):821–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Guéry L, Hugues S. Th17 cell plasticity and functions in cancer immunity. Biomed Res Int. 2015;2015:314620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Wu D, Wu P, Huang Q, et al. Interleukin-17: a promoter in colorectal cancer progression. Clin Dev Immunol. 2013;2013:436307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Geis AL, Fan H, Wu X, et al. Regulatory T-cell Response to Enterotoxigenic Bacteroides fragilis Colonization Triggers IL17-Dependent Colon Carcinogenesis. Cancer Discov. 2015;5(10):1098–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].De Simone V, Franzè E, Ronchetti G, et al. Th17-type cytokines, IL-6 and TNF-α synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene. 2015;34(27):3493–3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Wu S, Rhee K-J, Albesiano E, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15(9):1016–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Rubinstein MR, Wang X, Liu W, et al. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe. 2013;14(2):195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Han YW, Ikegami A, Rajanna C, et al. Identification and characterization of a novel adhesin unique to oral fusobacteria. J Bacteriol. 2005;187(15):5330–5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Xu M, Yamada M, Li M, et al. FadA from Fusobacterium nucleatum utilizes both secreted and nonsecreted forms for functional oligomerization for attachment and invasion of host cells. J Biol Chem. 2007;282(34):25000–25009. [DOI] [PubMed] [Google Scholar]
- [120].Fardini Y, Wang X, Témoin S, et al. Fusobacterium nucleatum adhesin FadA binds vascular endothelial cadherin and alters endothelial integrity. Mol Microbiol. 2011;82(6):1468–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Sun C-H, Li -B-B, Wang B, et al. The role of Fusobacterium nucleatum in colorectal cancer: from carcinogenesis to clinical management. Chronic Dis Transl Med. 2019;5(3):178–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Cuevas-Ramos G, Petit CR, Marcq I, et al. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci U S A. 2010;107(25):11537–11542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Ge Z, Schauer DB, Fox JG. In vivo virulence properties of bacterial cytolethal-distending toxin. Cell Microbiol. 2008;10(8):1599–1607. [DOI] [PubMed] [Google Scholar]
- [124].Taieb F, Petit C, Nougayrède J-P, et al. The enterobacterial genotoxins: cytolethal distending toxin and colibactin. EcoSal Plus. 2016;7(1). DOI: 10.1128/ecosalplus.ESP-0008-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Buc E, Dubois D, Sauvanet P, et al. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PloS One. 2013;8(2):e56964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Arthur JC, Perez-Chanona E, Mühlbauer M, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338(6103):120–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Nougayrède J-P, et al. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science. 2006;313(5788):848–851. [DOI] [PubMed] [Google Scholar]
- [128].Arthur JC, Gharaibeh RZ, Mühlbauer M, et al. Microbial genomic analysis reveals the essential role of inflammation in bacteria-induced colorectal cancer. Nat Commun. 2014;5(1):4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Dalmasso G, Cougnoux A, Delmas J, et al. The bacterial genotoxin colibactin promotes colon tumor growth by modifying the tumor microenvironment. Gut Microbes. 2014;5(5):675–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Guerra L, Cortes-Bratti X, Guidi R, et al. The biology of the cytolethal distending toxins. Toxins (Basel). 2011;3(3):172–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].He Z, Gharaibeh RZ, Newsome RC, et al. Campylobacter jejuni promotes colorectal tumorigenesis through the action of cytolethal distending toxin. Gut. 2019;68(2):289–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Li G, Niu H, Zhang Y, et al. Haemophilus parasuis cytolethal distending toxin induces cell cycle arrest and p53-dependent apoptosis. PloS One. 2017;12(5):e0177199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Bezine E, Vignard J, Mirey G. The cytolethal distending toxin effects on Mammalian cells: a DNA damage perspective. Cells. 2014;3(2):592–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Asakura M, Hinenoya A, Alam MS, et al. An inducible lambdoid prophage encoding cytolethal distending toxin (Cdt-I) and a type III effector protein in enteropathogenic Escherichia coli. Proc Natl Acad Sci U S A. 2007;104(36):14483–14488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Ge Z, Rogers AB, Feng Y, et al. Bacterial cytolethal distending toxin promotes the development of dysplasia in a model of microbially induced hepatocarcinogenesis. Cell Microbiol. 2007;9(8):2070–2080. [DOI] [PubMed] [Google Scholar]
- [136].Gagnière J, et al. Gut microbiota imbalance and colorectal cancer. World J Gastroenterol. 2016;22(2):501–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Gao R, Gao Z, Huang L, et al. Gut microbiota and colorectal cancer. Eur J Clin Microbiol Infect Dis. 2017;36(5):757–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Lucas C, Barnich N, Nguyen HTT. Microbiota, Inflammation and Colorectal Cancer.. Int J Mol Sci. 2017;18(6). DOI: 10.3390/ijms18061310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Bultman SJ. The microbiome and its potential as a cancer preventive intervention. Semin Oncol. 2016;43(1):97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Mima K, Ogino S, Nakagawa S, et al. The role of intestinal bacteria in the development and progression of gastrointestinal tract neoplasms. Surg Oncol. 2017;26(4):368–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Sivan A, Corrales L, Hubert N, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350(6264):1084–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Yuan L, et al. The influence of gut microbiota dysbiosis to the efficacy of 5-Fluorouracil treatment on colorectal cancer. Biomed Pharmacothe. 2018;108(184–193). DOI: 10.1016/j.biopha.2018.08.165 [DOI] [PubMed] [Google Scholar]