

Abstract
SARS-CoV-2 (CoV-2) viral infection results in COVID-19 disease, which has caused significant morbidity and mortality worldwide. A vaccine is crucial to curtail the spread of SARS-CoV-2, while therapeutics will be required to treat ongoing and reemerging infections of SARS-CoV-2 and COVID-19 disease. There are currently no commercially available effective anti-viral therapies for COVID-19, urging the development of novel modalities. Here, we describe a molecular therapy specifically targeted to neutralize SARS-CoV-2, which consists of extracellular vesicles (EVs) containing a novel fusion tetraspanin protein, CD63, embedded within an anti-CoV-2 nanobody. These anti-CoV-2-enriched EVs bind SARS-CoV-2 spike protein at the receptor-binding domain (RBD) site and can functionally neutralize SARS-CoV-2. This work demonstrates an innovative EV-targeting platform that can be employed to target and inhibit the early stages of SARS-CoV-2 infection.
Keywords: extracellular vesicles, nanobody, SARS-CoV-2, COVID-19, therapeutic, neutralization, spike, CD63 fusion, VOC
Graphical abstract
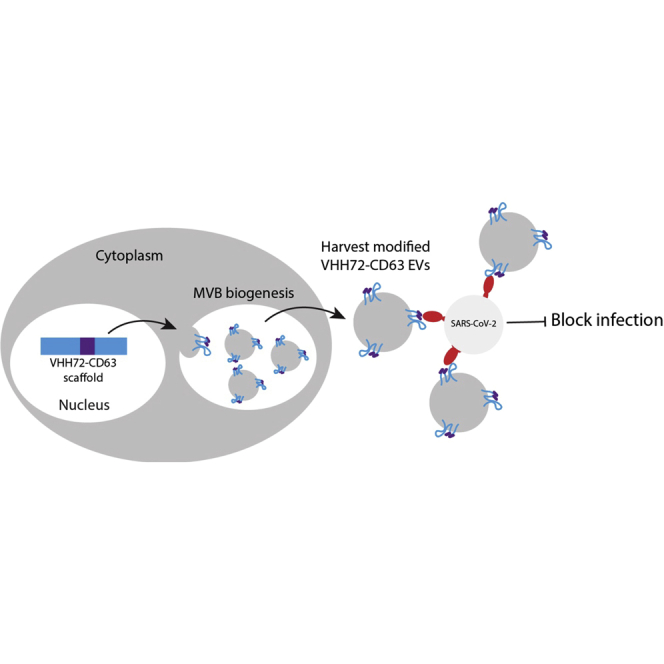
Described here is a modular extracellular vesicle (EV) platform that can redirect EVs to target virus envelope proteins and the ability of these virus-targeted EVs to broadly neutralize SARS-COV-2 variants of concern.
Introduction
SARS-CoV-2 (CoV-2) has emerged on the world stage as a highly infectious agent that can spread rapidly geographically, causing significant mortality globally.1 While current vaccines have proven useful at preventing severe infection; vaccine hesitancy, the rapid emergence of variants of concern (VOCs) that could escape pre-existing immunity, and the possibility of the endemic establishment of SARS-CoV-2, suggests that COVID-19 cases will continue to be observed into the foreseeable future, which will require the development of novel approaches that directly target and inhibit SARS-CoV-2 during infection.
Currently, there are no approved effective anti-viral treatments for COVID-19; however, there has been emergency use authorization (EUA) of traditional antiviral drugs like Remdesivir and monoclonal antibodies therapies like REGN-CoV-2. Recently, a protease inhibitor, PF-00835231, has shown promise in pre-clinical studies2 and new drugs, paxlovid and molnupiravir, have received EUA. SARS-CoV-2 intracellular entry is mediated through its spike protein consisting of S1 and S2 subunits, whereby the S1 contains the receptor-binding domain (RBD) that interacts with host angiotensin-converting enzyme 2 (ACE2) receptor on the surface of target cells to initiate infection.3 This binding results in the reorganization of the S2 subunit into a fusogenic state, which is further matured by transmembrane serine protease 2 (TMPRSS2).3 Although other target sites in the spike protein have been found to be important, the RBD is considered the major target for antibody-mediated neutralization,4 which blocks the interaction of this target site with ACE2, preventing infection and representing a site for targeted virus inactivation.
Extracellular vesicle (EV) is an umbrella term for a wide range of nano-size particles secreted from cells comprised of, but not limited to, the broadly defined multivesicular body-derived exosomes (30–150 nm), membrane-derived microvesicles (100–1,000 nm), and apoptotic bodies.5 These particles have lipid bilayer membranes, a feature that allows these systems to be engineered to present artificial targeting ligands on their surface or the incorporation of various payloads like RNA, DNA, and protein in the luminal compartment.6 They are biodegradable, potentially allogeneic , biocompatible, and safe.7 Importantly, EVs are emerging as a therapeutic platform with applications as anti-inflammatory,8 anti-viral,9 and anti-cancer agents.10
Artificial proteins have been incorporated onto EVs to inhibit SARS-CoV-2 intracellular entry, whereby a decoy hACE2 protein presented on the EV surface can inhibit the entry of spike pseudotyped virions.11 However, the impact of administrating EVs expressing ACE2, which is known to function in the regulation of the cardiovascular system, is currently unknown, and targeting the virus directly may prove more specific and therapeutically relevant. Furthermore, the EV surface can be engineered with various overexpressed receptors. Not all receptors are efficiently incorporated into EVs (e.g., the spike protein of SARS-CoV-2);12 therefore, using enriched scaffolds could be exploited for this purpose. EVs have a wide range of specifically enriched markers, most notably the tetraspanin protein family, including, CD63, CD9, and CD81, as well as other recently identified novel scaffolds,13 that could be explored for this purpose.
To determine the ability to embed virus-targeted modalities into the extracellular surface of EVs, we engineered a SARS-CoV-2 spike-specific nanobody and a control human immunodeficiency virus (HIV-1)-specific N6 antibody into the CD63 tetraspanin EV-specific receptor. We demonstrate the ability of these CD63-engineered EVs to functionally bind and inhibit HIV-1 and SARS-CoV-2. Furthermore, anti-SARS-CoV-2 EVs were shown to inhibit lentivirus pseudotyped with a range of spikes from VOCs as well as infectious SARS-CoV-2. Here, we report the characterization of this modality and its ability to inhibit SARS-CoV-2 infection, a novel proof-of-concept anti-viral for COVID-19.
Results
Characterization of VHH72-CD63-expressing EVs
EVs alone can be taken up by cells in a non-specific manner, but they have a predilection for being taken up by cells similar to their origin.14 One means to bias EV uptake to a particular cell type is by generating EVs that have a specific receptor agonist, single-chain variable fragment (scFv), or nanobodies, embedded into the extracellular membrane of the CD63 EV-associated protein. Toward this goal, we first determined the optimal extracellular loop and position within CD63 to embed the targeting protein. We utilized an scFv derived from the broadly neutralizing N6 antibody targeted to HIV-1 gp160 to create a series of N6-scFv fusion constructs15 (Figure 1A). These N6-CD63 fusion constructs were transfected into human embryonic kidney (HEK293) cells, and EVs were collected using established differential ultra-centrifugation procedures to isolate small EVs, containing the exosome fraction known to be enriched for CD63 expression.16 The optimal extra-cellular loop in CD63 to embed the N6 was determined to be in loop 2 (EC2) in the Ex2.4 configuration (Figure 1A), as this site demonstrated significant binding of N6-CD63 EVs to gp120 in bead-binding assays (Figure 1B). Inserting the N6 into loop 1 (EC1) as well as fusing a truncated CD4 protein (tCD4-D1D2), the GP160’s target receptor, to the N terminus also gave an enriched signal. However, the N6-CD63 was also fused to a nanoluciferase (Nluc) (Figure 1A), and uptake on stable gp160 HEK293 cells was assessed. There was high background signal due to the non-specific binding and possible uptake of EVs on the cells, but the EVs with the N6 scFv in the Ex2.4 configuration resulted in a low but significant increase in uptake signal (Figure 1C), highlighting this as the preferential format going forward. The morphology of EVs containing the N6-CD63 Ex2.4 format (referred to as N6-CD63 onward) was characterized by nanotracking analysis (NTA) and transmission electron microscopy (TEM), and the enrichment of EV markers and the N6-CD63 scaffold was confirmed by western blotting (Figures S1A–S1C). The functionality of the N6-CD63 EVs was then verified by performing a HIV-1 neutralization assay. The pre-incubation of the N6-CD63 EVs with a pNL4-Bal virus resulted in the significant inhibition of infection as measured by the reduction of luciferase signal in a HIV-responsive TZM-bl reporter cell line (Figure S1D).
Figure 1.

Characterization of targeted CD63 EVs
(A) Schematic of the CD63 receptor and the insertion sites of the N6 scFv or VHH72 nanobody (Ex1.1, Ex2.2, Ex2.3, or Ex2.4) or a truncated CD4 domains 1 and 2 attached to the N terminus of CD63 (tCD4-D1D2). EC1 and EC2 denote the two main loops of CD63, and cysteine disulfide bonds are highlighted. A Nluc was fused in-frame to the C -terminus. (B) The N6-CD63 EVs were bound to beads and then incubated with gp120, and binding was assessed by flow cytometry, made relative to the CD63 control set at 100%. Error bars represent standard deviation generated from samples treated in triplicate. The p values were generated using a one-way ANOVA compared with the control (∗p < 0.05, ∗∗p < 0.01). (C) HEK293 cells that stably express gp160 were treated with N6-CD63 EVs, and the levels of Nluc were assessed at 18 h post-addition. The Nluc levels were normalized to HEK293 WT cells and made relative to the CD63 control set at 100%. Error bars represent standard deviation generated from samples treated in triplicate. The p values were generated using an unpaired Student’s t test compared with the control (∗∗p < 0.01). (D) TEM and (E) NTA analysis for the CD63 control and VHH72-CD63 EVs. (F) EVs and cell lysates were assessed by western blot for known EV markers (TSG101, ALIX, CD81) and the components of the CD63 fusion protein (Nluc and CD63). GAPDH was included as a loading control. Ladder molecular weights are indicated.
Encouraged by these results for targeting HIV-1 gp160 with a scFv, we sought to determine whether the Ex2.4 locus was tolerant of other targeted approaches. As a result of the current global significance of SARS-CoV-2, a camelid-derived nanobody, which is a single-domain heavy-chain variable region (VHH) was embedded into the Ex2.4 site. A VHH requires only a single heavy chain to bind its cognate target, representing a simple and compact modality for generating fusion constructs. Recently, VHH72 was described to bind the spike of the beta-coronavirus’s RBD.17 Originally developed for SARS-CoV-1, VHH72 targets a conserved region that sterically blocks spike engagement with the ACE2 receptor and can neutralize SARS-CoV-2 when expressed in a bivalent format.
To determine the ability to target the spike of SARS-CoV-2, we inserted the bivalent VHH72 into the Ex2.4 locus of CD63 (Figure 1A), transiently transfected the vector into HEK293 cells, and purified EVs as described. The EVs were characterized by TEM and NTA, showing the characteristic cup-shaped morphology and similar in shape and size (∼130 nm) to EVs generated from cells transfected with the control CD63 vector (Figures 1D and 1E). Western blot analysis of the CD63 and VHH72-CD63 EVs showed enrichment of inclusion EV markers ALIX, CD81, and the recently described putative universal EV biomarker syntenin-118 and the absence of the cell-specific exclusion marker calnexin (Figures 1F and S2A). Notably, detection of CD63 or Nluc showed specific enrichment of VHH72-CD63 in EVs compared with cell lysates (Figure 1F). However, detection of the CD63 scaffolds showed different molecular weights in the cell lysates compared with the EVs. This observation may represent possible differences in post-translational modifications of CD63 present in EVs compared with cells, which has been observed by others using overexpressed CD63 vectors.16,19 Furthermore, Nluc activity was readily detectable in EV preparations, further verifying the presence of the CD63 scaffolds in the EVs (Figure S2B).
VHH72-CD63 EVs can bind to SARS-CoV-2 spike
To determine whether the VHH72 on the surface of EVs was able to bind to its cognate target, we performed an enrichment assay where beads were coated with an anti-spike antibody to the S1 ectodomain and then bound to the SARS-CoV-2 trimeric spike, which were subsequently incubated with VHH72-CD63 EVs. A Nluc signal was observed with the VHH72-CD63 EVs with increasing amounts of recombinant trimeric spike protein (Figures 2A, S2C and S2D). Notably, an enrichment was not observed with the control EVs. To further verify that the modified EVs were binding to the RBD, VHH72-CD63 EVs were bound to beads and then incubated with SARS-CoV-2 RBD before determining the levels of bound RBD by flow cytometry. A higher signal was observed with VHH72-CD63 EVs, demonstrating that the EVs were binding to the RBD (Figure 2B). Furthermore, TEM imaging revealed that the surface of VHH72-CD63 EVs bound to SARS-CoV-2 RBD when detected using gold-nanoparticle labeling, which often appeared as a cluster on the EV surface (Figures 2C and S2E). We also observed an increased uptake of VHH72-CD63 EVs in HEK293 cells transfected with a vector expressing the spike protein (Figures 2D and S2F). Lastly, to determine mechanistically whether the VHH72 containing EVs could block a RBD interaction with the ACE2 protein, an ACE2-blocking assay was performed. The VHH72-EVs were incubated at increasing doses with the SARS-CoV-2 RBD and then added to a plate coated with recombinant ACE2, and the amount of ACE2 bound to RBD was determined. A significant reduction in signal was observed with increasing amounts of VHH72-EVs compared with the control CD63 EVs (Figure 2E). Collectively, these data verified that VHH72-CD63 EVs were able to bind to the SARS-CoV-2 spike protein on the membrane surface and block its interaction with ACE2.
Figure 2.

VHH72-CD63 EVs can bind SARS-CoV-2 spike
(A) Beads were coated with an anti-SARS-CoV-2 spike antibody and bound to increasing concentrations of recombinant trimeric spike (0.1–10 μg/mL). The spike-bound beads were incubated with VHH72-CD63 EVs, and the levels of Nluc were assessed, which were made relative to the beads without spike, set at 100%. Error bars represent standard deviation generated from samples treated in duplicate. (B) The VHH72-CD63 EVs were bound to beads and incubated with SARS-CoV-2 RBD, and then flow cytometry was used to assess binding. Error bars represent standard deviation generated from samples treated in triplicate. The p values were generated using an unpaired Student’s t test compared with RBD-negative samples (∗p < 0.05). (C) TEM analysis of the VHH72-CD63 and CD63 containing EVs bound to gold-nanoparticle-labeled SARS-CoV-2 RBD. Red arrows highlight bound gold particles. Scale bar represents 200 nm. (D) HEK293 cells were transfected with a spike-expressing vector and treated with VHH72-CD63 EVs, and the levels of Nluc were assessed at 4 h post-addition. The Nluc levels were normalized to untransfected HEK293 cells and made relative to the CD63 control. Error bars represent standard deviation generated from samples treated in triplicate. The p values were generated using an unpaired Student’s t test compared with the control (∗∗∗p < 0.005). (E) A SARS-CoV-2 RBD fused to a HRP was pre-incubated with 2E9, 1E9, 5E8, and 1E8 total particles of the CD63 or VHH72-CD63 EVs, and the amount of RBD bound to ACE2 was assessed through a colorimetric assay. The percent inhibition was calculated from samples treated in duplicate. A recombinant VHH72 was included as a positive control.
Neutralization of SARS-CoV-2 pseudotyped lentivirus
To assess whether the VHH72-CD63 EVs were able to neutralize the SARS-CoV-2 spike, we utilized a pseudotyped lentiviral vector assay. We first optimized cellular and viral conditions to facilitate spike infection of the target cells. Lentiviral particles7 were packaged with a GFP-firefly luciferase reporter (GFP-Fluc) and pseudotyped with a WT spike protein. These pseudotyped lentiviral particles were able to transduce stable hACE2-HEK293 cells (Figures S3A and S4A), which was further increased in hACE2-hTMPRSS2-HEK293 cells in a dose-responsive manner (Figures S3B and S4B). The virus was unable to transduce WT HEK293 cells. VERO-E6 cells have been described to be SARS-CoV-2 permissive, but we proceeded with the HEK293 cells since the VERO-E6 cells showed lower transduction even with stable hACE2-hTMPRSS2 expression, possibly because of restriction factors in these cells (Figure S4C). Furthermore, we introduced the dominant D614G mutation present in circulating strains, which improves ACE2 binding, virus transmissibility, and infection,20, 21, 22 alone or in combination with a mutation in the furin cleavage site R682Q.23 The highest infection levels observed was with the D614G-R682Q combination (Figure S4D). Finally, we tested a C-terminal truncated spike protein, which has been shown to improve spike incorporation into lentiviral particles and virus transduction, but showed only modest improvements in transduction and was not explored further (Figure S4E). Furthermore, the truncated spike had increased non-specific entry into HEK293 cells, which may have resulted from the truncation increasing the spike’s fusogenic properties.24 A combination assay of the relevant individually optimized conditions is presented in Figure S4F.
Having established a reliable infection system with pseudotyped spike particles, the D614G-R682Q pseudotyped virions were incubated with VHH72-CD63 EVs followed by infection of hACE2-hTMPRSS2-HEK293 cells. The Fluc activity was assessed at 72 h post-infection, and a reduction in luciferase activity was observed in the VHH72-CD63 EVs relative to the CD63 control (Figure 3A, left graph). Simultaneous detection of Nluc in the transduced cells verified the inclusion of the EVs (Figure 3A, right graph). The neutralization of pseudotyped D614G-R682Q virus was dose dependent, which increased with a higher ratio of VHH72-CD63 EVs to viral particles (Figure 3B). COVID-19 convalescent plasma (CPP) from patients recovered from SARS-CoV-2 infection was able to reduce transduction by ∼50% (Figure S5A), which was comparable to the VHH72-CD63 EVs, suggesting effective inactivation of the D614G-R682Q spike. Of interest, the RBD-targeting S35 mAb was ineffective at neutralizing the D614G-R682Q pseudotyped virion.
Figure 3.

VHH72-CD63 EVs broadly neutralize SARS-CoV-2 pseudovirus
(A) The pseudotyped spike D614G-R682Q lentiviral particles were incubated with EVs and then transduced on HEK293-hACE2-hTMPRSS2 cells, and the levels of Fluc and Nluc were assessed at 72 h post-transduction. Error bars represent standard deviation generated from samples treated in triplicate. The p values were generated using an unpaired Student’s t -test compared with the CD63 control treated samples (∗∗p < 0.01). (B) The EVs were incubated at increasing concentrations with a set number of lentiviral particles (LP), and transductions were performed as described. Error bars represent standard deviation generated from samples treated in triplicate from two independent experiments. The p values were generated using a one-way ANOVA by comparing the means of the two experiments relative to the virus-only control (∗p < 0.05, ∗∗p < 0.01). (C) The pseudotyped lentiviral particles with the spike protein from the Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), and Epsilon (B.1.429) variants were incubated with 2E9 EVs, and then HEK293-hACE2-hTMPRSS2 cells were transduced. The levels of Fluc were assessed at 48 h post-transduction. The line represents the mean from samples treated in triplicate from two independent experiments. (D) The lentiviral particles pseudotyped with spikes from SARS-CoV-2 VOC were incubated with the S35 mAb (1.5 μg/mL) or CCP (P9K; 1:100 dilution), and then HEK293-hACE2-hTMPRSS2 cells were transduced. The line represents the mean from an experiment performed in triplicate. (E) VHH72-CD63 EVs were tested against lentivirus pseudotyped with the Delta (B.1.617.2) and Kappa (B.1.617.1) spike variants. The line represents the mean from samples treated in triplicate from two independent experiments, except for the S35- and recombinant VHH72-treated samples for the Kappa variant, which were generated from one independent experiment. The p values for (C), (D), and (E) were generated using a one-way ANOVA by comparing the means of the two experiments relative to the CD63 (C and E) or virus-only control (D) (∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
To assess whether the VHH72-CD63 EVs can inactivate spike proteins derived from SARS-CoV-2 VOCs, the EVs were incubated with lentivirus pseudotyped with the spike from Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), and Epsilon (B.1.429) variants. These preparations were then transduced onto the hACE2-hTMPRSS2-HEK293 cells, and high levels of neutralization were observed with the VHH72-CD63 EVs for all variants tested compared with the CD63 control (Figure 3C). Furthermore, the neutralization was comparable to a recombinant bivalent VHH72. A 10-fold lower number of EVs reduced the neutralization of the pseudotyped lentiviral variants by the VHH72-CD63, demonstrating a dosing effect (Figure S5B). Importantly, the anti-RBD S35 mAb and CPP (P9K) were unable to effectively neutralize the spikes from the Beta and Gamma variants, highlighting the conserved nature of the VHH72 target site (Figure 3D). Encouraged by these results, we tested the VHH72-CD63 EVs against lentivirus pseudotyped with the Kappa (B.1.617.1) and Delta (B.1.617.2) spikes, and likewise was able to potently neutralize these variants (Figure 3E), demonstrating broad activity against a wide range of emerging VOCs.
Neutralization of infectious SARS-CoV-2
Finally, to verify that the VHH72-CD63 EVs can affect SARS-CoV-2 infection in vitro, neutralization assays were performed in a live-virus challenge with an ancestral strain. Indeed, reduced infection was observed in a dose-dependent manner with the VHH72-CD63 EVs compared with the CD63 control (Figure 4A). The VHH72-CD63 EVs were subsequently tested in a live-virus challenge with VOCs: Delta, Beta, and Kappa (Figures 4B–4D, respectively). Effective neutralization with the ancestral strain was observed at >1x10^8 (1E8) total particles, and so a starting dose of 5x10^8 (5E8) EV particles was used, and effective neutralization of the VOCs was observed. However, higher doses of VHH72-CD63 EVs did not significantly improve inhibition. Collectively, these data show that the VHH72-CD63 EVs can inhibit live SARS-CoV-2 VOCs, mediated through the inhibition of the spike protein.
Figure 4.

VHH72-CD63 EVs neutralize SARS-CoV-2
(A–D) EVs were incubated at the described amounts with live SARS-CoV-2 from an ancestral strain (A) or VOC; Delta (B), Beta (C), Kappa (D), before infecting Vero E6 cells, and plaques were subsequently counted. A mAb targeted to the RBD, CB6, or 5309 were included as positive controls. Triplicate treated cells are shown with the standard error of the mean, and p values were determined by one-way ANOVA (Dunnett’s post-test) when compared against virus only (∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
Discussion
Here, we describe a novel EV fusion receptor that can be used to redirect EVs to viral targets. Specifically, we characterized its binding to SARS-CoV-2 spike protein and the inhibition of both pseudotyped and authentic virus. There are a number of methods to attach ligands pre- and post-production to EVs,25 but a feature of the lipid bilayer membrane allows for transmembrane protein incorporation, which can be leveraged to express artificial receptors during production, a powerful tool for EV retargeting. The EV CD63 scaffold has been used functionally in various ways: embedding inhibitory peptides to treat muscular dystrophy,26 fusing an apolipoprotein A-I (apo-A1) ligand to the C terminus to direct EVs to liver cancer cells,27 presenting antigens for vaccine augmentation,28 or real-time EV imaging through embedded reporters.16 This work adds to the growing versatility of tetraspanins by showing that antibody-derived fragments can be embedded into CD63 for functional receptor targeting. Furthermore, the EC1 of CD63 has been used for protein presentation,16,26 which was further corroborated in this study (Figure 1B). More extensive characterization of the loops suggested other regions may have improved features for protein presentation (Figures 1A and 1B), highlighting the versatility of CD63. Interestingly, recent work suggests that truncated variants of CD63 may still be functional in EVs, only requiring transmembrane 3 for incorporation into EVs,29 signifying CD63 as a highly malleable and compact system for enrichment of ligands.
Still, although effective neutralization of live SARS-CoV-2 was observed, complete neutralization compared with a mAb was not achieved (Figure 4). Neutralization may be improved by optimizing several features of the surface modified EVs. There are several challenges in using expressed receptors for pre-incorporation into EVs, which include receptor enrichment and product purity of the modified EVs. The pDisplay system has been used to target HIV-1’s GP160 built on the platelet-derived growth factor receptor (PDGFR) transmembrane domain,9 while other studies have used the lysosome-associated membrane protein 2 (Lamp2b) receptor, a known marker of EVs (reviewed in Dang et al.6). However, recent studies have shown that these are not highly enriched in HEK293-dervied EVs compared with other receptors including tetraspanins. A comparison of novel-enriched versus passive scaffolds showed improved functionality,13 encouraging the use of enriched scaffolds. Furthermore, Lamp2b has been shown to be proteolytically unstable, requiring additional optimization to produce stable versions,13,30 which is not the case for the ubiquitous, stable tetraspanins that are known to be enriched in EVs from various cell types, making tetraspanins an attractive scaffold for further development.
The TEM images presented here showed that not all EVs bound to gold-labeled RBD, and often the fusion proteins were found in clusters on the EV surface (Figures 2C and S2D). This observation may reflect binding to VHH72-CD63 in tetraspanin-enriched microdomains, with a similar phenomenon observed with GPI scaffolds associated with lipid rafts.31 Supporting this notion are observations that the pHluorin-CD63 reporter was found in heterogeneous clusters on EV particles and mainly enriched in EVs from the smaller range, preferentially in exosomes.16 Selective isolation methods will need to be explored to improve product purity of EVs modified with artificial scaffolds to ensure optimal products for therapeutic applications.
Furthermore, the N6-CD63 scaffold, although functional, had significantly reduced levels of expression (Figure S1C). To a lesser extent, the VHH72-CD63 had reduced expression compared with the CD63 control (Figure 1F), which highlights the need to identify stable ligands empirically, a common process in developing targeted vesicles. As a result of the simplicity of ligands that have single effector modules, such as nanobodies and darpins,32 these modalities may be attractive ligands for further development into tetraspanin scaffolds.
The R682Q furin mutation has been explored to optimize in vitro transduction with pseudotyped SARS-CoV-2 lentiviral vectors.23 Mutation of the furin domain emerges when serially passaging SARS-CoV-2, representing a cell culture adaptation,33 which may affect mAb neutralization.34 Although effective neutralization of the D614G-R682Q spike lentivirus was observed with the VHH72-CD63 EVs, suggesting conservation of the target epitope (Figure 3B), neutralization clearly affected the S35 mAb and CPP (Figure 1, Figure 2, Figure 3, Figure 45A) that readily inhibited several pseudotyped VOCs (Figure 3D). These data suggest possible structural changes in the spike because of the furin mutation, which impacts antibody binding, advising caution when using furin-mutated spikes for neutralization studies.
The emergence of VOCs has caused great concern, as these can escape from vaccinated and COVID-19 patient serum35 as well as from therapeutic monoclonal antibodies,36 resulting in the need for antibody cocktails to prevent escape.37 Alternatively, targeting conserved regions in betacoronavirus spike could counter this issue. The VHH72 targets a region adjacent to the ACE2 binding site, a conserved site, that can cross-neutralize SARS-CoV-1 and -2, and bind spike protein from a bat coronavirus, WIV1-CoV.38 The data presented here demonstrate that the VHH72-CD63 EVs can neutralize a range of VOCs (Figures 3C and 3E, Figure 4). Interestingly, anti-viral activity with recombinant VHH72 has been shown to improve against the N501K mutation,39 as this mutation re-established a positively charged amino acid present at the equivalent position in SARS-CoV-1,38 suggesting that VHH72-CD63 EVs may even have improved activity against some variants.
EVs represent a multi-functional system that could be adapted to inhibit the virus through mechanisms other than neutralization. EVs are able to transfer incorporated therapeutic payloads and, loaded with chemotherapeutic agents, have been shown to improve drug potency to cancer cells10 and deliver RNA interference effectors40 or gene-regulatory proteins.41 Spike-targeted EVs could be adapted to deliver anti-CoV-2 payloads to infected tissues in combination with neutralizing effects. Furthermore, inflammation is a crucial factor in severe COVID-19, and stem-cell-derived EVs have anti-inflammatory properties.42 Using anti-inflammatory EVs targeted to the sites of SARS-CoV-2 infection could be a potent therapeutic approach to augment COVID-19 disease progression and modulate a SARS-CoV-2-induced cytokine storm. The versatility of anti-viral SARS-CoV-2 EVs could be leveraged to offer unique properties to treat COVID-19 over other anti-viral modalities. Overall, we have demonstrated the engineering of a novel CD63 fusion receptor that can be enriched in EVs to target and inhibit SARS-CoV-2, representing an innovative therapeutic approach for the treatment of COVID-19.
Materials and methods
Vectors
The CD63 vectors that contained the N6 (Ex1.1, Ex2.2, Ex2.3, Ex2.4) or VHH72 (Ex2.4) were ordered as gBLOCKs (IDT, IA, USA) and cloned into a pDB30 vector digested with HindIII and BbvCI. The tCD4-D1D2 vector was generated by amplify the CD4 domains 1 and 2 from a CD4 expression vector (primers: tCD4-D1D2-CD63 F and R) and cloned into a HindIII-digested pDB30 vector. The pDB30 vector that contains a WT hCD63 fused by a GS linker to Nluc was kindly provided by Prof. M. Fussenegger.43 The WT spike mammalian expression vector (codon optimized) was obtained from Sino Biological (Beijing, China). To generate the mutant spike vectors, primers with the mutations were used to amplify the sequence by PCR. For the D614G vector, two fragments were amplified with one containing the D614G mutation (primers: Spike-F + D614G-R) and a second WT sequence fragment (primers: D614G-F and Spike-R). To generate the double mutation D614G-R682Q, an amplicon was generated with the R682Q mutation (primers: R682Q-F + Spike-R) and a second amplicon (primers: R614G-F + R682Q-R), which were both diluted down to 1:100 in sterile water, mixed at a 1:1 ratio, and then amplified in a fusion PCR (primers R614G-F + spike-R) to generate a single fragment with the R682Q mutation. The WT spike vector was digested with Bsu36I and BsrGI, and the D614G fragment was mixed either with the WT fragment or the R682Q fragment and cloned into the digested vector. The truncated spike vector was generated by PCR amplification of the spike sequence (Spike-F2 and spike-R2) and cloned into a EcoRI- and NotI-digested pCAGGS vector.
To generate the hACE2-T2A-puromycin and the hTMPRSS2-T2A-hygromycin vectors, the sequences were ordered as gBLOCKs (IDT, IA, USA) and were cloned in a XbaI- and XmaI-digested epHIV7-eGFP-Fluc vector, which removed the GFP-Fluc transgene and replaced it with the receptors. The primers and gBLOCK sequences for the CD63 and spike vectors are provided in Tables S1 and S2, respectively. Amino acid sequences of the CD63 vectors are provided in Table S3. All the fragments were amplified using Q5 Hot Start High-Fidelity 2X Master Mix (NEB, MA, USA) and were cloned into their respective digested vectors using the NEBuilder HiFi DNA Assembly master mix (NEB) according to the manufacturer’s instructions. The generated constructs were screened by digestion and confirmed by Sanger sequencing.
Cell lines and culture
HEK293 (ATCC CRL-1573), HEK293-GP160-92UG037.8 (kindly provided by Prof. Chen44), HEK293-hACE2 (NR-52512, BEI resources), HEK293-hACE2-hTMPRSS2, TZM-bl (NIH HIV Reagent Program, Division of AIDS, NIAID, NIH: ARP-8129, contributed by Drs. John C. Kappes and Xiaoyun Wu), and VERO-E6 cells (ATCC CRL-1586) were maintained in Dulbecco's modified Eagle’s medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific) at 37°C and 5% CO2. To generate the VERO-E6-hACE2, VERO-E6-hACE2-hTMPRSS2, or HEK293-hACE2-TMPRSS2 cell lines, lentiviral particles were generated using standard procedures that contained the epHIV7-hACE2-T2A-puromycin or epHIV7-hTMPRSS2-T2A-hygromycin transgenes; the cell lines were transduced and selected with either 1.5 μg/mL puromycin or 400 μg/mL hygromycin, respectively.
Detection of proteins in EVs and cell lysates
HEK293 EVs and producer cells were lysed in RIPA lysis buffer, and the protein concentration was determined by a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific) according to manufacturer protocols. Equal amounts of protein (10 μg) from each sample were then loaded onto 4%–20% Mini-PROTEAN TGX Precast Protein Gels (Bio-Rad, Hercules, CA, USA) and run in Tris-glycine/SDS electrophoresis buffer (Bio-Rad). Protein was transferred onto nitrocellulose membrane overnight at 4°C in Tris-glycine buffer (Bio-Rad). The membrane was rinsed and blocked using 2% BSA in TBS-T blocking buffer and then probed using the following primary antibodies: α-CD63 mouse antibody (cat. no. sc-5275, Santa Cruz Biotechnology, Santa Cruz, CA, USA), α-CD81 mouse antibody (cat. no. sc-7637, Santa Cruz Biotechnology), α-TSG101 mouse antibody (cat. no. sc-136111, Santa Cruz Biotechnology), α-ALIX mouse antibody (cat. no. 2171S, Cell Signaling, Waltham, MA, USA), α-GAPDH mouse antibody (cat. no. sc-47724, Santa Cruz Biotechnology), α-NanoLuc mouse antibody (cat. no. MAB100261, Novus Bio, Centennial, CO, USA), α-calnexin rabbit antibody (cat. no. ab92573, Abcam, Cambridge, UK), and α-syntenin rabbit antibody (cat. no. ab133267, Abcam). The primary antibodies were then bound with a horseradish peroxidase (HRP)-conjugated α-mouse IgG goat antibody (cat. no. 1705047, Bio-Rad) or Immun-Star goat anti-rabbit (GAR)-HRP conjugate (cat. no. 170546, Bio-Rad) and exposed using a Pierce SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific). The signal was detected on the Bio-Rad Chemidoc Touch Gel-Imaging System and analyzed on Bio-Rad Image Lab software v.6.0.0. Primary and secondary antibodies were diluted 1:1,000 in blocking buffer. The membranes were thoroughly washed and stripped with Restore PLUS Western Blot Stripping Buffer (Thermo Fisher Scientific), if necessary, between repeat probings.
To detect hACE, hTMPRSS2, and CoV-2 S proteins, the cells were pelleted and then lysed as described above and then loaded onto a Bio-Rad Mini Protean TGX 7.5% gel and run in TGS running buffer (Bio-Rad). Protein was transferred to nitrocellulose membranes using Bio-Rad’s TransBlot Turbo Transfer system (Bio-Rad). The membranes were blocked for 1 h in 1× TBS-T with 5% milk. Membranes were washed and then incubated overnight at 4°C with anti-hACE2 monoclonal antibody (OTI4D2; cat. no. MA5-26628, Thermo Fisher Scientific), anti-hTMPRSS2 antibody (H-4; cat. no. sc-515727, Santa Cruz Biotechnology), or anti-SARS-CoV-2 spike (T62; Sino Biological, cat. no. 40592-T62) diluted 1:1,000 in blocking buffer. The membranes were washed three times for 2 min each before incubation for 30 min at room temperature with the described secondary antibodies. Membranes were exposed with Pierce ECL Plus Western Blotting Substrate (Thermo Fisher Scientific) before imaging on the Bio-Rad Chemidoc Touch Gel-Imaging system. Images were analyzed on Bio-Rad Image Lab software v.6.0.0.
Immunoelectron microscopy
For immunogold labeling, 5 μL of specimen was absorbed to glow discharged carbon-coated Formvar grids for 2 min. After a rinse in PBS containing 0.05% bovine serum albumin (BSA), grids were incubated with biotinylated SARS-CoV-2 (COVID-19) S protein RBD, His,Avitag (cat. no. SPD-C82E9-25μg; Acrobiosystems, Newark, DE, USA) or MonoRab Rabbit Anti-Camelid VHH Cocktail Biotin (cat. no. A02015-200, Genscript ProBio, Piscataway, NJ, USA) at 1:400 dilution for 15 min. After a washing, grids were incubated with 10 nm gold particle-conjugated streptavidin from Streptomyces avidinii (cat. no. S9059-.25ML, Millipore Sigma, Burlington, MA, USA) at 1:50 dilution for 15 min. Finally, immunolabeled samples were negatively stained with 1% uranyl acetate for 20 s, and electron microscopy images were taken on an FEI Tecnai 12 transmission electron microscope equipped with a Gatan OneView CMOS camera.
Bead-binding assays
To assess enrichment of Nluc, 4 μM Aldehyde/Sulfate Latex Beads (Thermo Fisher Scientific, Carlsbad, CA, USA) were first washed with PBS and then incubated with recombinant anti-SARS-CoV-2 spike ectodomain antibody (cat. no. ab277512, Abcam) centrifuged at 3,000 × g for 10 min at 4°C. The conjugated beads were then blocked with blocking buffer containing 0.5% BSA in PBS, centrifuged at 3,000 × g for 10 min at 4°C and washed, and then SARS-CoV-2 (COVID-19) S-trimer (D614G) recombinant protein (cat. no. 92-748, ProSci Inc., Fort Collins, CO, USA) was incubated with the conjugated beads at increasing concentration (0–10 μg/mL). The bound recombinant protein was then centrifuged at 3000 × g for 10 min at 4°C, washed, and incubated with 5E8 EV particles. The bound EV particles were then centrifuged at 3,000 × g for 10 min at 4°C and washed, and the levels of Nluc were assessed using the Nano-Glo Luciferase Assay System (Promega, Madison, WI, USA) and read on the GloMax Discover MicroPlate Reader (Promega).
To assess EV binding to the Spike-RBD, Aldehyde/Sulfate Latex Beads (Thermo Fisher CA) were washed in PBS, spun at 3,000 × g for 10 min, and then incubated with an anti-hCD63 antibody in PBS for 1 h at 4°C and then centrifuged at 3000 × gg for 10 min and blocked in 1× PBS with 0.2% BSA for 30 min at 4°C. The sample was then centrifuged at 3,000 × gg for 10 min, and the VHH72-CD63 or control CD63 EVs were incubated with the sample in PBS for 1 h at 4°C. The beads were centrifuged again at 3,000 × g for 10 min and then incubated with 5 μg/mL SARS-CoV-2 RBD (cat. no. Z03479, Genscript), centrifuged 3,000 × g for 10 min, and then incubated with a secondary anti-His tag antibody conjugated to Alexa Fluor 488 (cat. no. 652509; Biolegend, San Diego, CA, USA) for 30 min at 4°C and then centrifuged at 3,000 × g for 10 min. The beads were resuspended in PBS, signal was detected on BD Accuri C6 Plus Flow Cytometer, and the data were analyzed the FlowJo software. For the N6-CD63 EVs, the recombinant HIV-1 (group M, subtype B, isolate BAL) gp120 protein (His Tag) (cat. no. 40404-V08H; Sino Biological, Beijing, China) was used in the assays.
Uptake assays
Ten thousand HEK293 cells were seeded in a 96-well plate and 24 h later were transfected with a control vector (empty) or a WT SARS-CoV-2 spike-expressing vector (Sino Biological, Beijing, China). At 48 h post-transfection, equal numbers of VHH72-CD63 or control CD63 EVs were added to the cells and incubated at 37°C and 5% CO2 for the described amount of time and washed with PBS, and luciferase activity was determined using the Nano-glo luciferase assay system (Promega). For the N6-CD63 EVs, HEK293-GP160 cells44 were seeded, and 24 h later the EVs were added and Nluc activity was assessed as described.
SARS-CoV-2 neutralization antibody detection assay (sVNT/ACE2 blocking assay)
To assess the ability of the VHH72-CD63 EVs to block a spike-ACE2 interaction, a SARS-CoV-2 neutralization antibody detection assay (RUO) was performed according to the manufacturer’s instructions (cat. no. L00847-A, Genscript). The CD63 or VHH72-CD63 EVs were incubated with a SARS-CoV-2 RBD fused to a HRP conjugate (HRP-RBD) at 37°C for 30 min, and then transferred to a 96-well plate pre-coated with recombinant ACE2. The recombinant VHH72 bivalent nanobody was incubated with the HRP-RBD as a positive control (cat. no. LMAB10541, R&D Systems, Minneapolis, MN, USA). The plate was incubated at 37°C for 15 min and washed, and TMB solution was added, starting a colorimetric substrate reaction incubated for 15 min at room temperature in the dark. The reaction was stopped and the absorbance measured (450 nm) on a GloMax Explorer Multimode Microplate Reader (Promega).
Negative-staining electron microscopy
EVs in a concentration range of 1–2 × 109/μL were absorbed to glow-discharged, carbon-coated 200 mesh Formvar grids. Samples were prepared by conventional negative staining in 1% (w/v) uranyl acetate. Electron microscopy images were taken on an FEI Tecnai 12 transmission electron microscope equipped with a Gatan OneView CMOS camera.
Lentivirus production and infection assays
The pseudotyped lentiviral particles were generated using standard procedures. Briefly, HEK293T cells were transfected with a pHIV7-GFP-ffluc packaging vector, pHIV-Gag-pol, pCMV-Rev2, and either pCoV2-S WT or mutant vectors at 15:10:1:2 ratio, and 72–96 h post-transfection the supernatant was centrifuged and then passed through a 0.45-μm filter (Thermo Fisher Scientific). The filtered supernatant was PEG precipitated at 4°C for 16 h and then ultracentrifuged at 20,000 × g at 4°C for 2 h. The pellet was resuspended in serum-free DMEM, and the physical titer of particles was determined by an p24 ELISA using the HIV-1 Gag p24 DuoSet ELISA (R&D Systems) according to the manufacturer’s instructions. For the infection assays, the described cells were seeded at 10,000 cells/well in a 96-well plate and then transduced with the described number of lentiviral particles with protamine sulfate (4 μg/mL), and the levels of luciferase were assessed post-transduction using the Bright-Glo Luciferase Assay System (Promega) and measured on a GloMax Explorer Multimode Microplate Reader (Promega).
EV production and characterization
Producer HEK293T cells were seeded in a 15-cm tissue culture plate and 24 h later transfected using Lipofectamine 3000 (Thermo Fisher Scientific) according to manufacturer instructions. Twenty-four hours post-transfection, the cells were washed with DMEM and the medium was replaced with 30 mL of DMEM+10% EV-depleted FBS (cat. no. A2720803, Thermo Fisher Scientific). Supernatant was collected at 48 and 96 h later and centrifuged at 300 × g for 10 min at 4°C. The viability of the cells was determined at the time of EV collection, which was greater than 90%. Supernatant was transferred to a new 50-mL conical tube and then centrifuged at 2000 × g for 20 min at 4°C, and then the supernatant was passed through a 0.45-μm filter (Millipore Sigma). The filtered supernatant was centrifuged at 100,000 × g for 120 min at 4°C using a SW32Ti rotor in a Beckman XL-900 ultracentrifuge. The supernatant was discarded, and EVs were resuspended in PBS and stored at −20°C until used. The EVs were quantified using Nanoparticle Tracking Analysis (NTA) on a NanoSight (Malvern Panalytical, Inc., Westborough, MA, USA). The NTA samples were run at a 1:4,000 dilution. A blue 488-nm laser was used to detect the extracellular vesicles, with a slide shutter level set to 1,259 and the slider gain set to 366, and the syringe pump speed set to 30, using a flow-cell top plate module. A threshold setting of 3 was used to determine particle count.
Neutralization assay
For the infectious HIV assays, a HIV-1 pNL4-Bal (MOI 0.2) was incubated with the described amount of EVs for 1 h at 37°C and added to 10,000 freshly seeded TZM-bl cells in a 96-well plate. Four-eight hours later, a luciferase assay was performed. TZM-bl cells contain a luciferase controlled by a HIV-responsive promoter with luciferase activity indicative of the levels of infection. The production of the infectious pNL4-Bal virus has been previously described.45 The 3BNC117 broadly neutralizing mAb targets the CD4binding site and is used to neutralize HIV-1 at a final concentration of 0.5 μg/mL, included as a positive control. The 3BNC117 mAb was obtained through the NIH HIV Reagent Program, Division of AIDS, NIAID, NIH: Anti-Human Immunodeficiency Virus (HIV)-1 gp120 Monoclonal Antibody (3BNC117), ARP-12474, contributed by Dr. Michel Nussenzweig.46
For the pseudotyped virus assays, HEK293-hACE2-hTMPRSS2 cells were seeded at 10,000 cells/per well in a 96-well plate, and 24 h later a total of 2.5E5 D614G-R682Q pseudotyped lentiviral particles were incubated with the described amount of EVs for 30 min at room temperature and then added to the cells. At 72 h post-transduction, the levels of Fluc and Nluc activity were assessed using a Nano-Glo Dual Reporter Assay (Promega
To perform the SARS-CoV-2 neutralization assay with the pseudotyped VOCs, the spike pseudotyped lentiviral vectors were obtained from a commercial source (BPS Bioscience, San Diego, CA, USA) including the Alpha (B.1.1.7, UK, cat. no. 78112-1), Beta (B.1.351, SA, cat. no. 78142-1), Gamma (P.1, Brazil, cat. no. 78144-1), Epsilon (B.1.429, cat. no. 78172-1), Kappa (B.1.617.1, cat. no. 78205-1), and Delta variants (B.1.617.2, cat. no. 78215-1). The described amount of EV particles was incubated with 1 μL of pseudotyped lentiviral vector (Alpha, Beta, Gamma = 1.2 E5 TU/mL; Epsilon = 1.5E5 TU/mL; Kappa = 7E5 TU/mL; Delta 3E5 TU/mL), and incubated at 37°C for 30 min, and then added to HEK293-hACE2-hTMPRSS2 cells. The recombinant VHH72 bivalent nanobody was included as a positive control. Virus-only controls were incubated in PBS. The S35 (cat. no. SAD-S35, Acrobiosystems) was added to the pseudotyped virus at the described final concentrations. The CPP was obtained from individuals previously exposed to SARS-CoV-2 (P9K and LE4) and incubated with the pseudotyped virus at the described dilutions. Plasma from a COVID-19 convalescent patient (P9KMAW8T) was used as reference. This plasma was collected under IRB-20204 (PI, Dr. John A. Zaia), with appropriate informed consent, upon confirmation of COVID-19 diagnosis. This plasma was collected at 94 days post-diagnosis and showed high titer of anti-CoV-2 IgG in a serological assay and potent RBD-neutralizing activity (>97% inhibition) in the sVNT assay as described above (data not shown). The cell final volume of media on the cells was 50 μL when the virus:EV mixture was added. At 48 h, the medium was removed and the Bright-Glo Luciferase Assay System was used to assess luciferase activity (Promega). The luciferase activity was measured on a GloMax Explorer Multimode Microplate Reader (Promega, WI).
To perform the SARS-CoV-2 neutralization assay, the ancestral (VIC1) strain,47 Beta variant (VIC18383), Kappa variant (VIC18447), and Delta variant (VIC18440) of SARS-CoV-2 were cultured in Vero E6 cells and originally obtained from the Peter Doherty Institute for Infection and Immunity and Melbourne Health, Victoria, Australia. Viral titer was determined by the viral immunoplaque assay as previously described.48 EVs or the mAb anti-SARS-CoV-2 RBD antibodies (clone nos. CB6 and 5309) were incubated with 250 PFUs of SARS-CoV-2 at the described concentrations for 30 min at room temperature before infecting Vero E6 cells for 1 h at 37°C. The virus was then removed, and the wells were layered with MC agar. The numbers of plaques were assessed 4 days after infection.
Statistical analysis
All statistical analyses were performed using the statistical software package GraphPad Prism 9, and the details of each analysis are described in the figure legends.
Acknowledgments
This project was supported by the National Institute of Mental Health (NIMH) R01 113407-01 and National Institute of Allergy and Infectious Diseases (NIAID) R56AI147684 to K.V.M. In addition, the research was made possible by a grant from the California Institute for Regenerative Medicine (Grant Number CLIN2COVID19-11775). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of CIRM or any other agency of the State of California. We would like to thank Dr. Chris McMillan and Dr. Naphak Modhiran from the University of Queensland for providing us with the mAb anti-SARS-CoV-2 RBD antibody (clone nos. CB6 and 5309). Thank you to Dr. John A. Zaia for reviewing the manucript and providing valued input.
Author contributions
T.A.S. and K.V.M. conceived the study; T.A.S., C.S., and N.G. designed and constructed the CD63 and spike vectors; T.A.S., C.S., and N.G. designed and executed the binding and uptake assays; S.S. performed the TEM experiments, T.A.S., A.C., C.S., and N.G. executed the pseudoviral infection and neutralization assays; R.R. designed and executed the N6-CD63 HIV-1 neutralization assays; A.I., A.S., G.K., and N.A.J.M. designed and performed the SARS-CoV-2 live viral experiments; T.A.S., N.A.J.M., and K.V.M. wrote and edited the manuscript.
Declaration of interests
K.V.M. and T.A.S. have submitted a patent application 048,440-749001WO based on this technology. All other authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.01.015.
Supplemental information
References
- 1.Ji Y., Ma Z., Peppelenbosch M.P., Pan Q. Potential association between COVID-19 mortality and health-care resource availability. The Lancet. Glob. Health. 2020;8:e480. doi: 10.1016/s2214-109x(20)30068-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boras B., Jones R.M., Anson B.J., Arenson D., Aschenbrenner L., Bakowski M.A., Beutler N., Binder J., Chen E., Eng H., et al. Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID19. Nat. Commun. 2021;12:6055. doi: 10.1038/s41467-021-26239-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang Y., Yang C., Xu X.-f., Xu W., Liu S.-w. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacologica Sinica. 2020;41:1141–1149. doi: 10.1038/s41401-020-0485-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brouwer P.J.M., Caniels T.G., van der Straten K., Snitselaar J.L., Aldon Y., Bangaru S., Torres J.L., Okba N.M.A., Claireaux M., Kerster G., et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science. 2020;369:643. doi: 10.1126/science.abc5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Niel G., D'Angelo G., Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018;19:213–228. doi: 10.1038/nrm.2017.125. [DOI] [PubMed] [Google Scholar]
- 6.Dang X.T.T., Kavishka J.M., Zhang D.X., Pirisinu M., Le M.T.N. Extracellular vesicles as an efficient and versatile system for drug delivery. Cells. 2020;9 doi: 10.3390/cells9102191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu X., Badawi M., Pomeroy S., Sutaria D.S., Xie Z., Baek A., Jiang J., Elgamal O.A., Mo X., Perle K.L., et al. Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from HEK293T cells. J. Extracell Vesicles. 2017;6:1324730. doi: 10.1080/20013078.2017.1324730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iyer S.S., Rojas M. Anti-inflammatory effects of mesenchymal stem cells: novel concept for future therapies. Expert Opin. Biol. Ther. 2008;8:569–581. doi: 10.1517/14712598.8.5.569. [DOI] [PubMed] [Google Scholar]
- 9.Zou X., Yuan M., Zhang T., Wei H., Xu S., Jiang N., Zheng N., Wu Z. Extracellular vesicles expressing a single-chain variable fragment of an HIV-1 specific antibody selectively target Env(+) tissues. Theranostics. 2019;9:5657–5671. doi: 10.7150/thno.33925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tian Y., Li S., Song J., Ji T., Zhu M., Anderson G.J., Wei J., Nie G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014;35:2383–2390. doi: 10.1016/j.biomaterials.2013.11.083. [DOI] [PubMed] [Google Scholar]
- 11.Cocozza F., Névo N., Piovesana E., Lahaye X., Buchrieser J., Schwartz O., Manel N., Tkach M., Théry C., Martin-Jaular L. Extracellular vesicles containing ACE2 efficiently prevent infection by SARS-CoV-2 Spike protein-containing virus. J. Extracell Vesicles. 2020;10:e12050. doi: 10.1002/jev2.12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuate S., Cinatl J., Doerr H.W., Uberla K. Exosomal vaccines containing the S protein of the SARS coronavirus induce high levels of neutralizing antibodies. Virology. 2007;362:26–37. doi: 10.1016/j.virol.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dooley K., McConnell R.E., Xu K., Lewis N.D., Haupt S., Youniss M.R., Martin S., Sia C.L., McCoy C., Moniz R.J., et al. A versatile platform for generating engineered extracellular vesicles with defined therapeutic properties. Mol. Ther. 2021;29:1729–1743. doi: 10.1016/j.ymthe.2021.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Colombo M., Moita C., van Niel G., Kowal J., Vigneron J., Benaroch P., Manel N., Moita L.F., Thery C., Raposo G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013;126:5553–5565. doi: 10.1242/jcs.128868. [DOI] [PubMed] [Google Scholar]
- 15.Huang J., Kang B.H., Ishida E., Zhou T., Griesman T., Sheng Z., Wu F., Doria-Rose N.A., Zhang B., McKee K., et al. Identification of a CD4-binding-site antibody to HIV that evolved near-Pan neutralization breadth. Immunity. 2016;45:1108–1121. doi: 10.1016/j.immuni.2016.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sung B.H., von Lersner A., Guerrero J., Krystofiak E.S., Inman D., Pelletier R., Zijlstra A., Ponik S.M., Weaver A.M. A live cell reporter of exosome secretion and uptake reveals pathfinding behavior of migrating cells. Nat. Commun. 2020;11:2092. doi: 10.1038/s41467-020-15747-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wrapp D., De Vlieger D., Corbett K.S., Torres G.M., Wang N., Van Breedam W., Roose K., van Schie L., Team V.-C.C.-R., Hoffmann M., et al. Structural basis for potent neutralization of betacoronaviruses by single-domain camelid antibodies. Cell. 2020;181:1436–1441. doi: 10.1016/j.cell.2020.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kugeratski F.G., Hodge K., Lilla S., McAndrews K.M., Zhou X., Hwang R.F., Zanivan S., Kalluri R. Quantitative proteomics identifies the core proteome of exosomes with syntenin-1 as the highest abundant protein and a putative universal biomarker. Nat. Cell Biol. 2021;23:631–641. doi: 10.1038/s41556-021-00693-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yao X., Lyu P., Yoo K., Yadav M.K., Singh R., Atala A., Lu B. Engineered extracellular vesicles as versatile ribonucleoprotein delivery vehicles for efficient and safe CRISPR genome editing. J. Extracell Vesicles. 2021;10:e12076. doi: 10.1002/jev2.12076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hou Y.J., Chiba S., Halfmann P., Ehre C., Kuroda M., Dinnon K.H., Leist S.R., Schäfer A., Nakajima N., Takahashi K., et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science. 2020;370:1464. doi: 10.1126/science.abe8499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou B., Thao T.T.N., Hoffmann D., Taddeo A., Ebert N., Labroussaa F., Pohlmann A., King J., Steiner S., Kelly J.N., et al. SARS-CoV-2 spike D614G change enhances replication and transmission. Nature. 2021;592:122–127. doi: 10.1038/s41586-021-03361-1. [DOI] [PubMed] [Google Scholar]
- 22.Daniloski Z., Jordan T.X., Ilmain J.K., Guo X., Bhabha G., tenOever B.R., Sanjana N.E. The Spike D614G mutation increases SARS-CoV-2 infection of multiple human cell types. eLife. 2021;10:e65365. doi: 10.7554/eLife.65365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson M.C., Lyddon T.D., Suarez R., Salcedo B., LePique M., Graham M., Ricana C., Robinson C., Ritter D.G. Optimized pseudotyping conditions for the SARS-COV-2 spike glycoprotein. J. Virol. 2020;94 doi: 10.1128/jvi.01062-20. e01062–01020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petit C.M., Melancon J.M., Chouljenko V.N., Colgrove R., Farzan M., Knipe D.M., Kousoulas K.G. Genetic analysis of the SARS-coronavirus spike glycoprotein functional domains involved in cell-surface expression and cell-to-cell fusion. Virology. 2005;341:215–230. doi: 10.1016/j.virol.2005.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liang Y., Duan L., Lu J., Xia J. Engineering exosomes for targeted drug delivery. Theranostics. 2021;11:3183–3195. doi: 10.7150/thno.52570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ran N., Gao X., Dong X., Li J., Lin C., Geng M., Yin H. Effects of exosome-mediated delivery of myostatin propeptide on functional recovery of mdx mice. Biomaterials. 2020;236:119826. doi: 10.1016/j.biomaterials.2020.119826. [DOI] [PubMed] [Google Scholar]
- 27.Liang G., Kan S., Zhu Y., Feng S., Feng W., Gao S. Engineered exosome-mediated delivery of functionally active miR-26a and its enhanced suppression effect in HepG2 cells. Int. J. Nanomedicine. 2018;13:585–599. doi: 10.2147/ijn.S154458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanuma T., Yamamoto T., Kobiyama K., Moriishi E., Masuta Y., Kusakabe T., Ozasa K., Kuroda E., Jounai N., Ishii K.J. CD63-Mediated antigen delivery into extracellular vesicles via DNA vaccination results in robust CD8(+) T cell responses. J. Immunol. 2017;198:4707–4715. doi: 10.4049/jimmunol.1600731. [DOI] [PubMed] [Google Scholar]
- 29.Curley N., Levy D., Do M.A., Brown A., Stickney Z., Marriott G., Lu B. Sequential deletion of CD63 identifies topologically distinct scaffolds for surface engineering of exosomes in living human cells. Nanoscale. 2020;12:12014–12026. doi: 10.1039/D0NR00362J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hung M.E., Leonard J.N. Stabilization of exosome-targeting peptides via engineered glycosylation. J. Biol. Chem. 2015;290:8166–8172. doi: 10.1074/jbc.M114.621383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kooijmans S.A.A., Aleza C.G., Roffler S.R., van Solinge W.W., Vader P., Schiffelers R.M. Display of GPI-anchored anti-EGFR nanobodies on extracellular vesicles promotes tumour cell targeting. J. Extracell. Vesicles. 2016;5:31053. doi: 10.3402/jev.v5.31053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frank A.M., Buchholz C.J. Surface-engineered lentiviral vectors for selective gene transfer into subtypes of lymphocytes. Mol. Ther. - Methods Clin. Dev. 2019;12:19–31. doi: 10.1016/j.omtm.2018.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sasaki M., Uemura K., Sato A., Toba S., Sanaki T., Maenaka K., Hall W.W., Orba Y., Sawa H. SARS-CoV-2 variants with mutations at the S1/S2 cleavage site are generated in vitro during propagation in TMPRSS2-deficient cells. PLOS Pathog. 2021;17:e1009233. doi: 10.1371/journal.ppat.1009233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson B.A., Xie X., Bailey A.L., Kalveram B., Lokugamage K.G., Muruato A., Zou J., Zhang X., Juelich T., Smith J.K., et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature. 2021;591:293–299. doi: 10.1038/s41586-021-03237-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou D., Dejnirattisai W., Supasa P., Liu C., Mentzer A.J., Ginn H.M., Zhao Y., Duyvesteyn H.M.E., Tuekprakhon A., Nutalai R., et al. Evidence of escape of SARS-CoV-2 variant B.1.351 from natural and vaccine-induced sera. Cell. 2021;184:2348–2361.e2346. doi: 10.1016/j.cell.2021.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Z., VanBlargan L.A., Bloyet L.-M., Rothlauf P.W., Chen R.E., Stumpf S., Zhao H., Errico J.M., Theel E.S., Liebeskind M.J., et al. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell host & microbe. 2021;29:477–488.e474. doi: 10.1016/j.chom.2021.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baum A., Fulton B.O., Wloga E., Copin R., Pascal K.E., Russo V., Giordano S., Lanza K., Negron N., Ni M., et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science. 2020;369:1014. doi: 10.1126/science.abd0831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wrapp D., De Vlieger D., Corbett K.S., Torres G.M., Wang N., Van Breedam W., Roose K., van Schie L., Hoffmann M., Pöhlmann S., et al. Structural basis for potent neutralization of betacoronaviruses by single-domain camelid antibodies. Cell. 2020;181:1004–1015.e1015. doi: 10.1016/j.cell.2020.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thomson E.C., Rosen L.E., Shepherd J.G., Spreafico R., da Silva Filipe A., Wojcechowskyj J.A., Davis C., Piccoli L., Pascall D.J., Dillen J., et al. Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell. 2021;184:1171–1187.e1120. doi: 10.1016/j.cell.2021.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alvarez-Erviti L., Seow Y., Yin H., Betts C., Lakhal S., Wood M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011;29:341–345. doi: 10.1038/nbt.1807. [DOI] [PubMed] [Google Scholar]
- 41.Villamizar O., Waters S.A., Scott T., Grepo N., Jaffe A., Morris K.V. Mesenchymal Stem Cell exosome delivered Zinc Finger Protein activation of cystic fibrosis transmembrane conductance regulator. J. Extracell. Vesicle. 2021;10:e12053. doi: 10.1002/jev2.12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jeon J.Y., An J.H., Kim S.U., Park H.G., Lee M.A. Migration of human neural stem cells toward an intracranial glioma. Exp. Mol. Med. 2008;40:84–91. doi: 10.3858/emm.2008.40.1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kojima R., Bojar D., Rizzi G., Hamri G.C.-E., El-Baba M.D., Saxena P., Ausländer S., Tan K.R., Fussenegger M. Designer exosomes produced by implanted cells intracerebrally deliver therapeutic cargo for Parkinson’s disease treatment. Nat. Commun. 2018;9:1305. doi: 10.1038/s41467-018-03733-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen J., Kovacs J.M., Peng H., Rits-Volloch S., Lu J., Park D., Zablowsky E., Seaman M.S., Chen B. HIV-1 ENVELOPE. Effect of the cytoplasmic domain on antigenic characteristics of HIV-1 envelope glycoprotein. Science (New York, N.Y.) 2015;349:191–195. doi: 10.1126/science.aaa9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Urak R.Z., Soemardy C., Ray R., Li S., Shevchenko G., Scott T., Lim L., Wang X., Morris K.V. Conditionally replicating vectors mobilize chimeric antigen receptors against HIV. Mol. Ther. - Methods Clin. Dev. 2020;19:285–294. doi: 10.1016/j.omtm.2020.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shingai M., Nishimura Y., Klein F., Mouquet H., Donau O.K., Plishka R., Buckler-White A., Seaman M., Piatak M., Jr., Lifson J.D., et al. Antibody-mediated immunotherapy of macaques chronically infected with SHIV suppresses viraemia. Nature. 2013;503:277–280. doi: 10.1038/nature12746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Caly L., Druce J., Roberts J., Bond K., Tran T., Kostecki R., Yoga Y., Naughton W., Taiaroa G., Seemann T., et al. Isolation and rapid sharing of the 2019 novel coronavirus (SARS-CoV-2) from the first patient diagnosed with COVID-19 in Australia. Med. J. Aust. 2020;212:459–462. doi: 10.5694/mja2.50569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Amarilla A.A., Modhiran N., Setoh Y.X., Peng N.Y.G., Sng J.D.J., Liang B., McMillan C.L.D., Freney M.E., Cheung S.T.M., Chappell K.J., et al. An optimized high-throughput immuno-plaque assay for SARS-CoV-2. Front Microbiol. 2021;12:625136. doi: 10.3389/fmicb.2021.625136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.