

ABSTRACT
Although osteosarcoma (OS) is the most common malignant tumor among juvenile bone tumors, its treatment plan and clinical outcome have not improved significantly in recent decades. Tetrandrine (TET), a Chinese medicine that is usually used in the therapy of silicosis, hypertension and arthritis, has been confirmed by many studies to possess potent antitumour growth properties, but there are different limitations when describing specific mechanisms. Here, we found that TET can obviously prevent the proliferation, migration and invasion of both 143B and MG63 cells and promote their apoptosis in vitro. Our results for luciferase reporter and Western blotting assays show that TET may exert its antitumour activity by regulating multiplex signaling pathways, including the MAPK/Erk, PTEN/Akt, Juk and Wnt signaling pathways. Furthermore, the regulatory effect of TET on OS cells and related signaling pathways was verified again in vivo. Our findings suggest that the anticancer function of TET on human OS may be mediated by its targeting of multiple signaling pathways and that TET may be used as a single drug or in combination with other drugs during the treatment of OS.
KEYWORDS: Osteosarcoma, tetrandrine, cell growth, anticancer effect, multiple signaling pathways
Introduction
Osteosarcoma (OS) is a primary malignant bone tumor that is usually observed in adolescents, and the outcomes of patients who only undergo amputation or disarticulation are unsatisfactory, especially for patients with distant organ metastasis diagnosed before the 1970s [1,2]. The current treatment strategy is based on diagnostic surgery and resection combined with polychemotherapy, and the long-term survival rate has considerably improved from less than 20% to approximately 55%-75% [3–5]. Unfortunately, the current treatment options have stagnated in terms of their effectiveness; the survival rate of patients has not improved significantly in recent years, and resistance to cytotoxic drugs may be the main reasons for chemotherapy failure [6]. The development of malignant cancer is more likely a result of the cooperation of multiple biological pathways, such as the PI3K/mTOR [7], Wnt/β-catenin [8], MAPK/Erk [9,10], and RANKL/NF-κ-B [11] pathways. Thus, it is necessary now to identify a new and efficacious treatment that can target multiple pathogenic signaling pathways.
TET is a bisbenzylisoquinoline alkaloid that can be extracted from a variety of Chinese herbal medicines, such as Cyclea barbata, menispermaceous plants, and Stephania cepharantha Hayata, and is usually used in Chinese medicine as a calcium channel blocker to treat hypertension, silicosis, angina pectoris and arrhythmias [12,13]. The available evidence suggests that TET can inhibit the growth of a variety of tumors, including human colorectal cancer, gliomas, bladder cancer, leukemia, hepatocellular carcinoma, and so on [14–18]. Previous results have shown that TET exhibits a significant anticancer effect at lower doses than other conventional chemotherapy drugs and exhibits synergistic anticancer activity with other drugs [19]. In addition, TET has a remarkably vital role in reversing tumor drug resistance; therefore, it has great potential as a new cancer drug [19]. Furthermore, some studies have reported that TET can affect specific signaling pathways involved in regulating cell cycle progression, apoptosis, migration and cytotoxicity capabilities of tumors [14–16,19,20]. TET has the characteristics of universality, low dose requirement, multiple effects, and reversal of drug resistance in the process of antitumour growth, but since the specific mechanism is still unclear, further research is necessary.
To clarify the effect of TET on OS growth and the specific mechanism, in this study, we explore the antitumour mechanism of TET in cell and animal experiments. Our outcomes suggest that TET can markedly inhibit proliferation, invasion and migration and significantly promote apoptosis in the human OS lines MG63, 143B and SaOS2. Among them, 143B cells had the most obvious response to TET, followed by MG63 cells. Therefore, we used 143B cells as the experimental object in subsequent experiments. Further studies found that the expression of proliferation-, apoptosis- and migration-related proteins was significantly influenced by TET. Animal experiments also confirmed that TET obviously suppressed the growth of OS cells, and its inhibitory effect was enhanced with increasing concentration. In addition, our results show that TET influences various signaling pathways, including the MAPK/Erk, PTEN/Akt and Wnt pathways, which are also closely associated with the abnormal behaviors of tumors [7,10,19,21,22]. Our results indicated that TET exerts its antitumour effect by acting on multiple signaling pathways and that the response of OS cells to TET is possibly determined by the activation of multiple signaling pathways. Nonetheless, TET may be used as a single drug or in combination with other drugs during the treatment of OS in the future, but further investigation is required.
Materials and methods
Reagents. TET (purity, 98%) was purchased from Herbpurify (Chendu Herbpurify CO., LTD, Chendu, China). Dulbecco’s modified Eagle’s medium (DMEM) and fetal bovine serum (FBS) were both obtained from Gibco (Grand Island, NY, USA). All antibodies were purchased from Cell Signaling Technology (CST; Danvers, MA, USA), except for the β-actin antibody, which was obtained from Abcam (MA, USA). Unless otherwise indicated, all chemicals were purchased from Sigma-Aldrich (Saint Louis, USA).
Cell culture and drug dilution. The human OS lines MG63, 143B and SAOZ-2 cells, which were provided by Dr Tongchuan He (University of Chicago, USA), originated from the America Type Culture Collection (ATCC, USA) and were verified by STR analysis. Cells were cultured at 37°C in complete DMEM supplemented with 10% FBS, 1% penicillin, and 1% streptomycin in a 5% CO2 incubator under a humidified atmosphere. The original TET (purity>98%; 6.23 mg) was dissolved in 2 ml DMSO, shaken and mixed uniformly, and then filtered through a 0.22 µm needle filter. The filtered stock was stored at −20°C and diluted to the specified concentration with DMEM when needed.
Crystal violet viability assay. A crystal violet assay was conducted in accordance with a previous study [19]. OS cells were seeded in 24-well plates and treated with TET when they reached 30–40% confluence. At 24 h, 48 h and 72 h after treatment, the cells were repeatedly washed three times with cold PBS and fixed for 10 minutes with 4% paraformaldehyde at 37°C. The paraformaldehyde was discarded, and the cells were washed three times with phosphate-buffered saline (PBS) and stained with Giemsa dye for 10 minutes. The stained cells were gently rinsed with running water and air dried to obtain macrographic images. For quantitative measurement, the stained cells were dissolved in 10% acetic acid (1 ml per well for 24-well plates) at room temperature for 20 minutes with shaking. A 500 μl portion was taken and added to 2 ml of double-distilled H2O. Absorbance was measured at 570 to 590 nm [23].
Hoechst staining test. Cells were seeded in 24-well plates and treated with TET at the indicated concentration when they were 60–70% confluent. After 24 h of treatment, the cells were repeatedly washed three times with cold PBS and fixed for 10 minutes with 4% paraformaldehyde at 37°C. The paraformaldehyde was discarded, and the cells were washed three times with cold PBS and stained with Hoechst 33,258 dye for 5 minutes in the dark. The dye was discarded, and the cells were repeatedly washed with PBS as before. Nuclear morphology was observed under a fluorescence microscope and photo preservation.
MTT assay. Cell viability was tested by the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetr azolium bromide] colorimetric assay. A total of 3000 cells per well were seeded in 96-well plates and cultured for 0, 24, 48, and 72 h after treatment with various concentrations of TET or DMSO. Additionally, 20 µL MTT reagent (5 mg/mL; Sigma-Aldrich, St. Louis, MO, USA) was added to each well, and the plate was incubated for 4 h. Thereafter, 150 µL dimethyl sulfoxide was added to each well for 15 minutes after the cell culture medium was discarded. Finally, the absorbance was measured at 492 nm with the use of a microplate reader.
Flow cytometry. Both 143B and MG63 cells in logarithmic growth phase were seeded in 6-well plates, treated with TET for 24 h, digested, repeatedly washed with PBS and fixed overnight with 70% ethanol at 4°C. Next, the cells were incubated with PI/Triton X-100 and stained for 15 minutes, and the cell cycle and cell apoptotic distribution were examined by flow cytometry as described previously [24].
Cell apoptosis assay. MG63 and 143B cells were inoculated into 6-well plates and treated with different concentrations of TET for 24 h. The cells were then collected, washed and resuspended in PBS three times, and 500 µl of PBS was added to resuspend the cells. The apoptosis rate was determined by flow cytometry according to the process provided by the Annexin V-FITC/PI double labeling staining kit (Beyotime Biotechnology, Shanghai, China), and both early and late cell apoptosis were counted.
Transwell migration assay. Approximately 20,000 cells per well were seeded into the upper Transwell chamber with 200 µL of 0% FBS culture medium. In addition, 600 µL of 10% FBS culture medium that had been treated with TET was transferred into the Transwell lower chamber. The cells were cultured for 24 h, fixed with 4% paraformaldehyde and stained with 0.1% crystal violet.
Matrigel invasion assays. Matrigel (5 mg/ml to 1 mg/ml) was diluted in serum-free cold cell culture medium (DMEM), 100 µl of diluted Matrigel was placed into the upper chamber of a 24-well Transwell, and then, the Transwell was incubated at 37°C for at least 2 to 3 h for gelling. The follow-up operation was the same as that described above for the Transwell migration assay.
Colony formation test. In addition, 1000 cells per well were seeded into 6-well plates with 1% FBS medium after treatment with TET (when the treatment conditions had a toxic effect on the cells, because the number of cells was small and the early post-adherence state was poor, the cells could not survive due to the toxicity of drugs). The same situation was encountered in this experiment. After high concentrations of TET were used to directly treat OS cells, the cells could not survive. Therefore, it was essential to choose a lower concentration for drug treatment in the colony formation experiment, and the final treatment concentrations were 0 μM, 0.2 μM, 0.4 μM, 0.6 μM, 0.8 μM, and 1 μM, respectively. The treatment was performed for 8 days. At the indicated time, the cells were washed three times with PBS and fixed for 10 minutes with 4% paraformaldehyde at 37°C. The paraformaldehyde was discarded, and the cells were washed three times with PBS and stained with Giemsa dye for 10 minutes; the cells were gently rinsed with running water and air dried. The cells were examined under a microscope, and a cluster of more than 50 cells was considered a colony.
Wound healing test. The cells were seeded into a 6-well plate and treated with TET when they reached 90 confluence. A wound was made with a 10 µL pipette tip, and the cells were cultured for 24 h with serum-free medium. The specific wound area was photographed at 0, 12, and 24 h. The wound healing rate was calculated as follows: (0 h width – 12 h or 24 h width)/0 h width × 100%.
Transfection and luciferase reporter assay. A firefly luciferase reporter assay was carried out as described previously [25,26]. In brief, 143B cells were seeded in 25-cm2 culture flasks and transfected with 3.0 g per flask of the BMP/Smad-Luc, NFκ-B-Luc, Erk-Luc, CREB-Luc, RBPjk-Luc, and HIF-Luc luciferase reporters using LipofectamineTM 2000 (Invitrogen, Gaithersburg, MD). At 24 h after transfection, the cells were replated in 24-well plates and treated with various concentrations of TET or DMSO control. After 24 h, the culture supernatant was collected and subjected to luciferase activity assays using a Gaussia Luciferase Assay Kit (New England Biolabs). Each assay condition was tested in triplicate. Luciferase activity was normalized to total cellular protein concentrations among the samples.
Western blotting. Cells were washed three times with cold PBS and lysed in RIPA lysis buffer (Beyotime, Haimen, China) in the presence of protease and phosphatase inhibitors. The protein concentration was measured with a bicinchoninic acid (BCA) protein assay kit (Beyotime, Haimen, China). Equivalent amounts of protein were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to polyvinylidene difluoride (PVDF) membranes. The PVDF membranes were blocked with 5% bovine serum albumin (BSA; Solarbio, Beijing, China) in Tris-buffered saline/Tween 20 (TBST) for 2 h at 37°C and then incubated at 4°C overnight with primary antibodies against PCNA (1:1000; rabbit, no. 13,110), Cyclin D1 (1:1000; rabbit, no. 2978), c-Myc (1:1000; rabbit, no. 13,987), E-Cadherin (1:1000; mouse, no. 14,472), N-Cadherin (1:1000; mouse, no. 13,116), MMP7 (1:1000; rabbit, no. 3801), MMP9 (1:1000; rabbit, no. 13,667), Snail (1:1000; mouse, no. 3895), Vimentin (1:1000; rabbit, no. 5741), Bad (1:1000; rabbit, no. 9239), Bcl-2 (1:1000; mouse, no. 15,071), Capase-3 (1:1000; rabbit, no. 9665), Cleaved Capase-3 (1:1000; rabbit, no. 9664), PARP (1:1000; rabbit, no. 9542), Cleaved PARP (1:1000; rabbit, no.5 625), p38MAPK (1:1000; rabbit, no. 9212), p-p38MAPK (1:1000; rabbit, no. 4511), Erk1/2 (1:1000; rabbit, no. 4695), SAPK/JNK (1:1000; rabbit, no. 9252), Pten (1:1000; rabbit, no. 9552s), p-Pten (1:1000; rabbit, no. 9551s), Akt (1:1000; rabbit, no. 9272s), p-Akt (1:1000; rabbit, no. 9275s), GSK-3β (1:1000; rabbit, no. 12,456), p-GSK-3β (1:1000; rabbit, no. 5558), β-Catenin (1:1000; rabbit, no. 8480), and β-Actin (1:1000; rabbit, no. 3700); all the antibodies were purchased from CST. The membranes were washed 3 times with TBST and incubated with either an anti-mouse or anti-rabbit secondary antibody (1:5000; ZhongShanGolden Bridge, Beijing, China) for 1 h at 37°C. Specific bands were visualized using BeyoECL Plus (Beyotime, Haimen, China) as described previously [24]. All experiments were repeated three times.
Establishment of an ectopic OS tumor animal model. Fifty microliters (2 × 107 cells/ml) of 143B cells suspended in medium was injected into the proximal tibial periosteum of 4- to 6-week-old female nude mice. Then, the animals were treated with different doses of TET (2, 6 or 10 mg/kg) or sodium carboxymethyl cellulose (CMC-Na), as a control, once every 2 days. The tumor length and width were measured every 2 days after the first week, and the animals were killed by cervical dislocation at 21 days after injection. Death was confirmed when there was no respiration or heartbeat. The tumor volume was calculated according to the following formula: 0.5 × L× W2 (where L is the tumor length, and W is the tumor width; all the recovered tumors were smaller than 2 cm). Tumor tissue specimens were collected for follow-up tests. All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Chongqing Medical University.
Immunohistochemistry (IHC). Immunofluorescence was employed to show PCNA, Bcl-2, Erk1/2, and Akt expression in tumor tissue. Tumor tissue sections were blocked and immunostained with antibodies targeting PCNA (1:100), Bcl-2 (1:100), Erk1/2 (1:50), and Akt (1:100). Images were captured using a light microscope. The mean integrated optical density was detected by Image-Pro Plus 6.0 software in three random fields.
Statistical analysis. All data were analyzed using SPSS 18.0 statistical software (SPSS Inc., Chicago, IL, USA). The results are expressed as the mean ± SD or mean ± SEM. One-way ANOVA followed by Tukey’s post hoc test was used to compare data from different groups. P < 0.05 was considered to indicate a statistically significant difference.
Results
Cytotoxic effects of TET on cultured OS cells. Studies have shown that TET can affect the growth of tumors [20]. For this reason, we tested the effect of TET on the proliferation of OS cells through a series of experiments. In this study, TET markedly impeded the proliferation of 143B, MG63 or SAOS-2 OS cells, as determined through crystal violet staining (Figure 1(a-b)), and similar results were obtained from the MTT assay (Figure 1(c)). The optimal concentration of TET was approximately 6 µM, which was lower than that of other chemotherapeutic drugs [19], so 2 µM, 6 µM, 8 µM and 10 µM were chosen as treatment concentrations in the subsequent experiments. Moreover, flow cytometry results revealed a significant increase in the proportion of G1/S-phase cells after treatment with TET (Figure 1(d-e)). We also found that the expression of the proliferation-associated proteins PCNA, cyclin D1 and c-Myc was significantly suppressed after treatment with TET (Figure 1(f-g)). The above experimental results indicate that TET can markedly suppress the proliferation of OS cells and that the effect of TET on relative cell viability was both concentration and time dependent.
Figure 1.
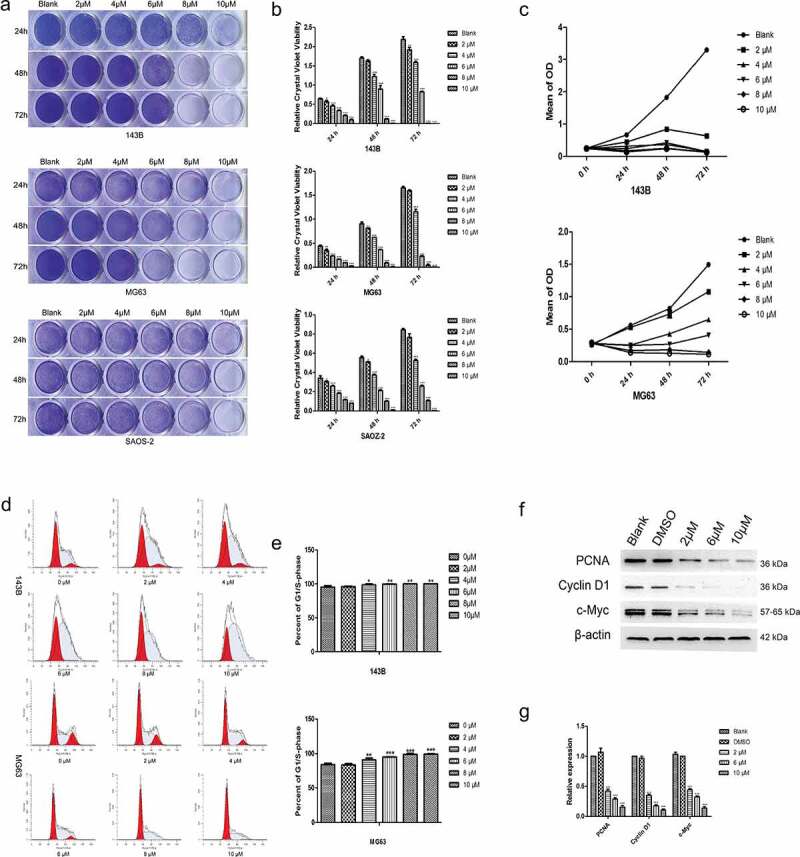
Cytotoxic effects of TET on OS cells. (a), (b) representative images of crystal violet staining and the quantitative crystal violet staining results. three kinds of OS cells were treated with TET at different concentrations for the indicated times, analyzed by crystal violet staining and subjected to viability assays. (c) MTT assays were used to detect the influence of TET on cell proliferation at the indicated times. (d), (e) flow cytometry was used to detect the cell cycle after treatment with TET for 36 h. (f), (g) The expression of the cell cycle-related proteins PCNA, c-Myc and Cyclin D1 was detected by western blotting of 143B cells after treatment with TET for 3 days. each experiment was effectively repeated. the data were normally distributed; *p < 0.05, ** p < 0.01, *** p < 0.001 vs. blank
TET suppresses the colony formation and migration of OS cells. The capacities to maintain infinite proliferation potential and undergo distant migration are indispensable for malignant cancer cells and to examine the changes in the abovementioned ability after TET treatment of OS cells through various methods. The colony formation assay results showed that TET-treated cells formed significantly fewer and smaller colonies than control cells (Figure 2(a)), and this phenomenon also showed that TET inhibits the proliferation of OS cells. The migration capacity was considerably inhibited in TET-treated OS cells, as shown by Transwell migration assays and wound healing tests (Figure 2(b-c)). These results suggest that TET suppresses the migration and proliferation of OS cells and may be a potential chemotherapeutic drug.
Figure 2.
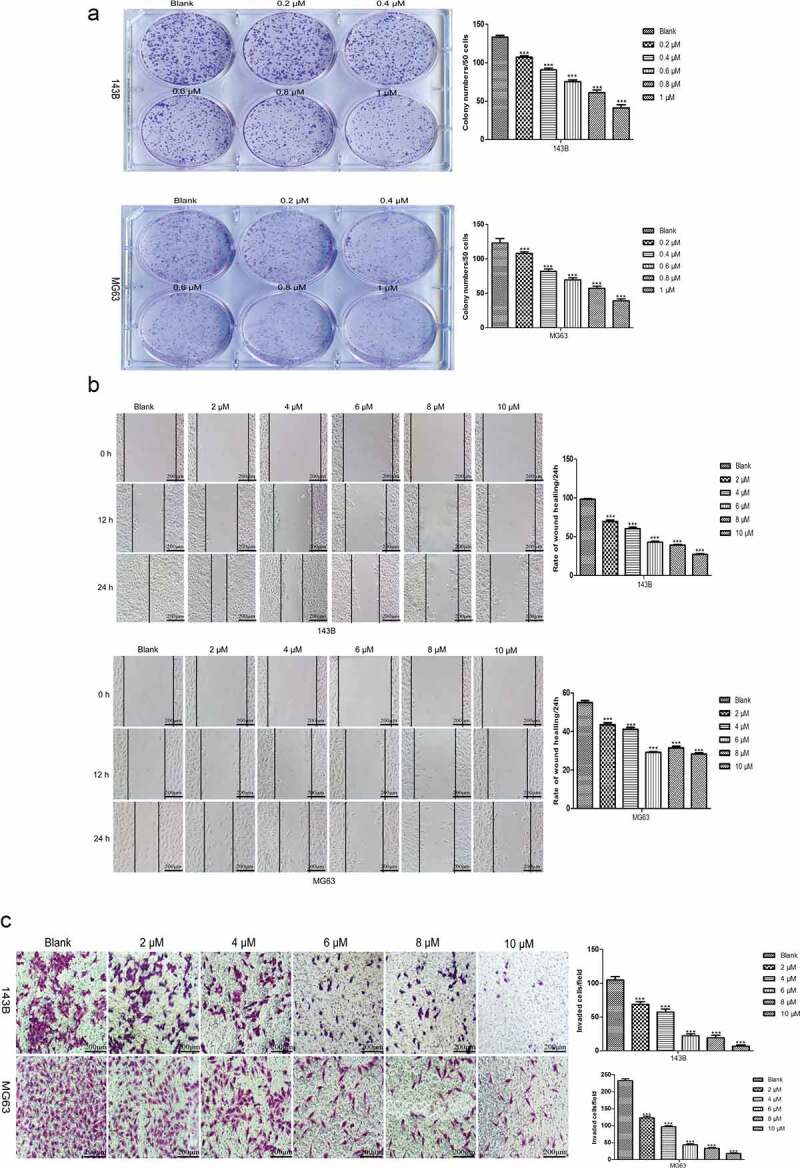
TET suppresses the colony formation and migration abilities of OS cells. (a) the effect of TET on the colony formation capacity was detected by colony formation assays after treatment for 8 days (1 × 103 cells/well). typical images are shown, and the treatment concentration was one tenth of the selected concentrations. (b) wound healing tests detected the capability of cell migration at the indicated times after treatment with TET. magnification, ×100. (c) transwell migration assays detected the capability of cell migration after treatment with TET for 24 h. cells were stained with 0.1% crystal violet. magnification, ×100. each experiment was effectively repeated. the data were normally distributed; *p < 0.05, ** p < 0.01, *** p < 0.001 vs. blank
TET inhibits the invasion and related protein expression of cancer cells. Next, we studied the influence of TET on OS cell invasion and related protein expression by applying Matrigel invasion assays and Western blotting. The outcomes indicated that the invasion capacity was markedly reduced by TET in both 143B and MG63 cells (Figure 3(a-b)). Moreover, the expression of invasion-related proteins was significantly inhibited by TET, which is consistent with the above results (Figure 3(c)). The results of these experiments strongly suggest that TET can exhibit an anticancer effect by preventing cancer cell invasion.
Figure 3.
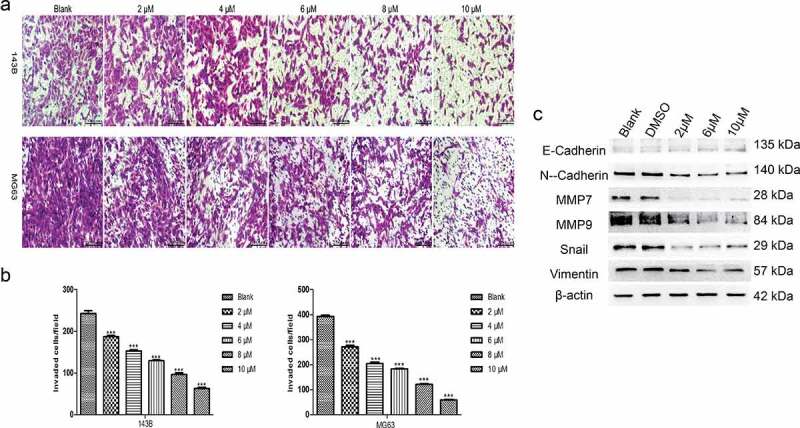
TET suppresses cancer cell invasion and invasion-related protein expression. (a), (b) the invasion capacity was analyzed by matrigel invasion assays after treatment with TET for 24 h. magnification, ×100. (c) migration- and invasion-related protein expression was detected by western blotting after treatment for 3 days in 143B cells. each experiment was effectively repeated. the data were normally distributed; *p < 0.05, ** p < 0.01, *** p < 0.001 vs. blank
TET induces the apoptosis of cancer cells. The influence of TET on OS cell apoptosis was also detected. Hoechst staining results showed that apoptosis was markedly induced by TET in both 143B and MG63 cells (Figure 4(a-b)), and similar results were detected by flow cytometry (Figure 4(c-d)). It is clear that the apoptosis-inducing effect of TET is highly concentration dependent. Furthermore, the expression of apoptosis-related proteins, such as Bad, caspase-3 and PARP, was markedly increased, but the expression of Bcl-2, a negative regulator of apoptosis, was correspondingly decreased, as indicated by Western blotting (Figure 4(e)). These results show that TET could effectively induce cancer cell apoptosis, which is consistent with previous studies.
Figure 4.
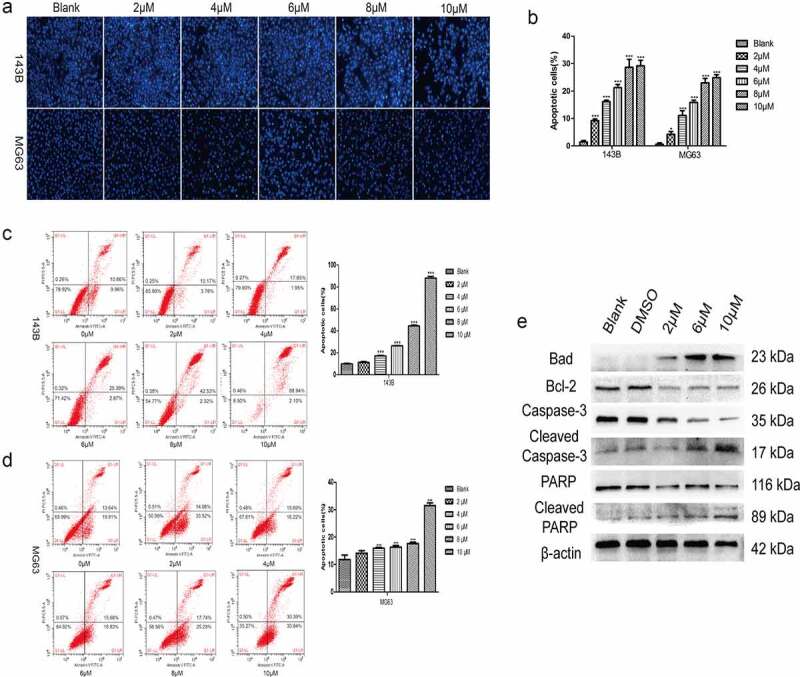
TET promotes apoptosis of OS cells. (a), (b) hoechst staining detected the apoptosis of cells after treatment with TET for 24 h. magnification, ×100. (c), (d) flow cytometry detected the apoptosis of cells after treatment with TET for 36 h. (e) western blotting detected apoptosis-related protein expression after treatment of 143B cells with TET for 3 days. each experiment was effectively repeated. the data were normally distributed; *p < 0.05, ** p < 0.01, *** p < 0.001 vs. blank
Multiple signaling pathways participate in the anticancer effect of TET. To further study the mechanism behind the TET-mediated anticancer effect, we assessed the activity of several luciferase reporters, including BMP/Smad-Luc, NF-κB-Luc, Erk-Luc, CREB-Luc, RBPjk-Luc, and HIF-Luc, in 143B cells after treatment with TET. The activities of the NF-κB-Luc and Erk-Luc reporters were considerably inhibited by TET; the CREB-Luc and HIF-Luc reporters were activated by TET, but the BMP/Smad-Luc and RBPjk-Luc reporters showed no obvious change in activity (Figure 5(a)). In addition, the expression of some key proteins of PTEN/Akt signaling, MAPK/Erk signaling, and Wnt signaling was markedly inhibited by TET, which is consistent with the luciferase reporter results (Figure 5(b-d)). These results indicate that multiple signaling pathways are involved in the anticancer effect of TET.
Figure 5.
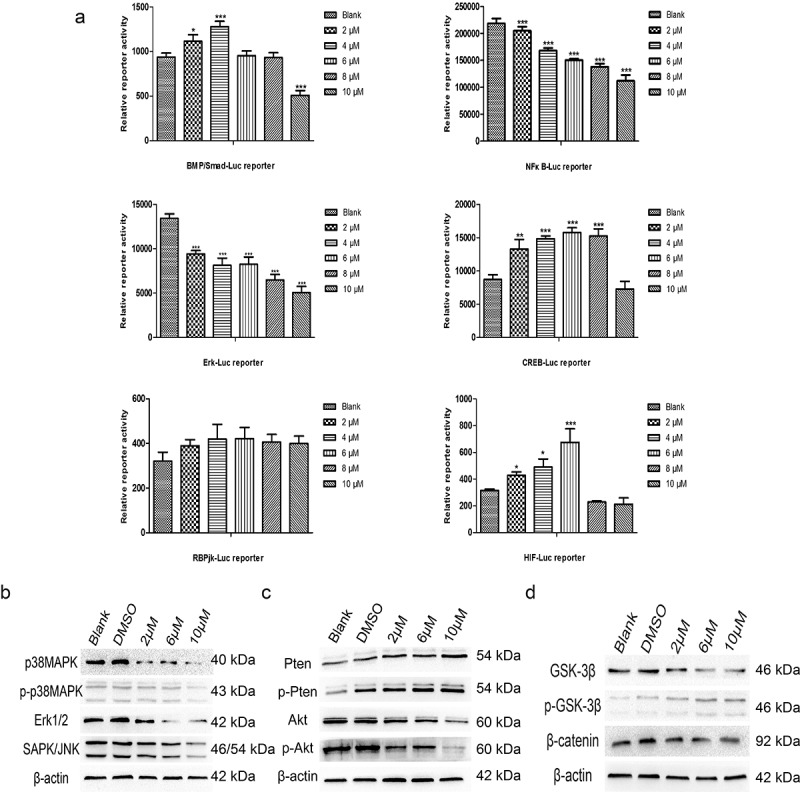
Multiple signaling pathways participate in the anticancer effect of TET. (a) the activities of the BMP/Smad-Luc, NF-κB-Luc, Erk-Luc, CREB-Luc, RBPjk-Luc, and HIF-Luc reporters were detected in 143B cells after treatment with TET for 24 h. (b), (c), (d) the expression of some key proteins of the PTEN/Akt signaling, MAPK/Erk signaling, and Wnt signaling pathways was detected after treatment with TET for 3 days. each experiment was effectively repeated. the data were normally distributed; *p < 0.05, ** p < 0.01, *** p < 0.001 vs. blank
TET suppresses the growth of OS cells in vivo. To investigate whether TET has similar anticancer effects in animals, we established an animal model of OS with 143B cells. The outcomes showed that tumor growth was inhibited with increasing TET dosage (Figure 6(a-b)). The immunohistochemical outcomes also showed that the expression of PCNA, Bcl-2, Erk1/2, and Akt decreased after TET treatment (Figure 6(c-d)). The above results suggest that TET can inhibit the growth of OS cells in vivo by regulating multiple signaling pathways.
Figure 6.
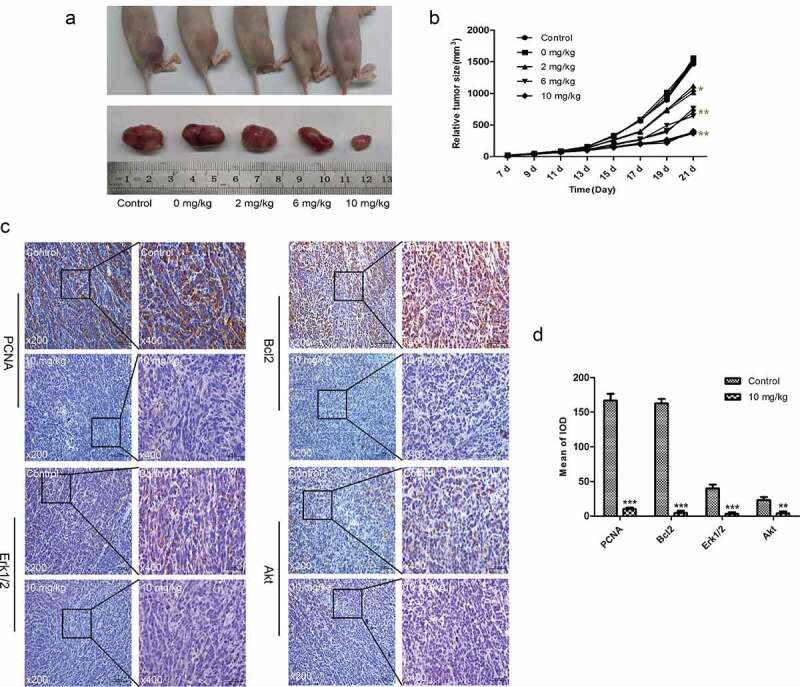
TET suppresses the growth of OS cells in vivo. (a) macrographic images of ectopic xenograft tumor tissues. (b) the TET treatment group had smaller tumor volumes than the control group. (c, d) PCNA, Bcl-2, Erk1/2, and Akt were detected by immunohistochemistry in tumor tissues. magnification, ×200 and ×400. the data were normally distributed; *p < 0.05, ** p < 0.01, *** p < 0.001 vs. blank
Discussion
It is imperative to find new therapeutic approaches to improve the survival prognosis of OS patients, and previous studies have indicated that TET exhibits anticancer activity in a variety of tumors. Some studies have also proven that TET can regulate specific signaling pathways and target genes or suppress antiangiogenic activity to achieve tumor inhibition [14–16,19,20]. However, the specific mechanism of its antitumour activity is still unclear. In this study, we used additional OS cell types and experimental methods to further analyze the influence of TET on the growth of OS cells and the specific mechanism.
In this study, we studied the specific mechanism underlying the anticancer activity of TET in OS cells. A previous study demonstrated that the half-maximal inhibitory concentration (IC50) for TET was approximately 5 µM among many tumor cell lines [19]. Our crystal violet viability assay results showed that TET could notably inhibit cell viability at low concentrations in three OS cell lines. The MTT assay yielded an analogous result, showing that TET has a potential anticancer effect, which is consistent with previous studies [14–20,27]. OS cells can invade adjacent tissues and induce some complications, such as bone destruction or outward expansion, which usually cause local fractures and pain. Therefore, some experiments were used to detect the invasion ability of OS cells, and our results showed that OS cell invasion was notably inhibited by TET, and there was a corresponding decrease in the expression of migration- and invasion-related proteins, such as MMP7, MMP9, N-cadherin, vimentin, and Snail.
In addition, the expression of E-cadherin, a negative regulator of cell invasion, increased after treatment with TET, suggesting a potential mechanism by which TET may activate certain anticancer factors. Moreover, infinite proliferation and abnormal apoptosis are important for the malignancy status of tumors, and it has been reported that these features are closely related to the abnormal activation of some signaling pathways or cancer suppressor genes, such as the Wnt signaling pathway or caspase-3, Bcl-2 and GSK-3β [16,19,28,29]. Previous studies by Tao et al analyzed only the effect of TET on OS through simple cell experiments and did not verify it in animal experiments or conduct in-depth mechanistic studies. Our outcomes indicated that TET can significantly promote cancer cell apoptosis and considerably decrease the expression of apoptosis-related proteins, including caspase-3, PPAR, Bcl-2 and GSK-3β. Our outcomes also suggest that suppressing Wnt/β-catenin signaling might be involved in the anticancer effects of TET, which is consistent with previous studies. Moreover, we observed an interesting phenomenon: the expression of MMP2, which is a transfer-related protein, increased significantly after treatment with TET (data not shown), but the expression of MMP7 and MMP9 decreased, which may indicate that TET exerts its anticancer effect by initiating the escape mechanism of cancer cells.
To further investigate the influence of TET on cell proliferation, the cell cycle was analyzed by flow cytometry, and the results indicated that most cells were blocked at the Gl/S phase and that proliferation-associated protein expression was markedly decreased after treatment with TET, especially cyclin D1 [27]. Previous studies have demonstrated that specific blockade of P38/MAPK signaling markedly inhibits the proliferation of cancer in cell and animal experiments and can also influence drug resistance and intratumoural blood and lymphatic vessel formation [21,22,30]. However, Tian et al found that TET may activate the MAPK/Erk pathway to upregulate PENT activity, thereby inhibiting OS proliferation and promoting apoptosis. The results of our experiment suggest that the activities of MAPK/Erk were strongly suppressed by TET, and the proliferation and colony formation ability of tumor cells were correspondingly decreased, which is consistent with previous research results. Some reports have found that activating PI3K/Akt signaling could promote tumorigenesis, tumor growth and metastasis by suppressing the expression of PTEN [7,31]. PTEN can inhibit the activity of the PI3K-Akt pathway, and our results also demonstrated that the migration and invasion ability of cancer cells were notably inhibited by TET; PTEN expression was markedly increased, but the activity of PI3K/Akt signaling was significantly decreased. Finally, the animal experiment results showed that TET suppressed the growth of OS cells in vivo, and the immunohistochemical results indicated that proliferation-related protein expression and the activities of the PTEN/Akt signaling and MAPK/Erk signaling pathways were measurably inhibited after treatment with TET, which is consistent with previous cell experiments.
In conclusion, our results indicate that TET could dramatically suppress the proliferation, migration and invasion but promote the apoptosis of OS cells in both cell and animal experiments. It is likely that multiple signaling pathways, including the MAPK/Erk, PTEN/Akt and Wnt signaling pathways, participate in the negative regulation of TET in OS cells, and some of these results are consistent with previous studies [32]. These outcomes confirm that TET suppresses the growth of OS cells, and it can be a potential drug for use in the biomedical industry or in combination with other drugs during the treatment of OS.
Conclusion
In summary, our study demonstrates that TET can dramatically suppress the growth of OS cells and that multiple signaling pathways, including the p38/MAPK, PTEN/Akt and Wnt signaling pathways, may participate in the anticancer effect of TET. These results suggest that TET may be used as a potential single drug or in combination with other drugs during the treatment of OS in the future, but whether there is a difference in the effect and mechanism of TET on different OS cells was not discussed in our study. Future studies will focus on finding the target proteins of TET and verifying the role of TET in different cells, which would help us to clarify the specific mechanism underlying the anticancer activity of TET.
Funding Statement
This study was funded by the National Natural Science Foundation of China (81873998) and the Natural Science Foundation Project of Chongqing Science and Technology Commission (No. cstc 2017jcyjAX).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Availability of data and material
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Authors’ contributions
* Contributed equally
The study was designed and conceived by Xiao-Ji Luo and Jin Chen. The experiments were conducted by Wang Nan, Sheng-dong Yang, Yang-mei Chen, Tao Tan, Yanran Huang. The article was written by Wang Nan. The experimental data were analysed by Chao-qun Dong.
References
- [1].Rosen G, Tan C, Sanmaneechai A, et al. The rationale for multiple drug chemotherapy in the treatment of osteogenic sarcoma. Cancer. 2015;35(S3):936–945. [DOI] [PubMed] [Google Scholar]
- [2].Sandberg AA, Bridge JA.. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: osteosarcoma and related tumors. Cancer Genet Cytogenet. 2003;14(1–2):1–30. [PubMed] [Google Scholar]
- [3].Jaffe N, Bruland OS, Bielack S (2010). [cancer treatment and research] pediatric and adolescent osteosarcoma volume 152 || osteosarcoma: review of the past, impact on the future. the american experience. 10.1007/978-1-4419-0284-9(Chapter 12), 239-262. [DOI] [PubMed] [Google Scholar]
- [4].Rosen G, Caparros B, Huvos AG, et al. Preoperative chemotherapy for osteogenic sarcoma: selection of postoperative adjuvant chemotherapy based on the response of the primary tumor to preoperative chemotherapy. Cancer. 1982;49(6):1221–1230. [DOI] [PubMed] [Google Scholar]
- [5].Benjamin RS. Adjuvant and neoadjuvant chemotherapy for osteosarcoma: a historical perspective. Adv Exp Med Biol. 2020;1257:1–10. [DOI] [PubMed] [Google Scholar]
- [6].Bajelan E, Haeri A, Vali AM, et al. Co-delivery of doxorubicin and PSC 833 (Valspodar) by stealth nanoliposomes for efficient overcoming of multidrug resistance. J Pharm Pharm Sci. 2012;15(4):568–582. [DOI] [PubMed] [Google Scholar]
- [7].Lee MS, Jeong MH, Lee HW, et al. PI3K/AKT activation induces PTEN ubiquitination and destabilization accelerating tumourigenesis. Nat Commun. 2015;6(1):7769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chew MH, Tan WS, Liu Y, et al. Genomics of Hereditary Colorectal Cancer: lessons Learnt from 25 Years of the Singapore Polyposis Registry. Ann Acad Med Singap. 2015. Aug;44(8):290–296. [PubMed] [Google Scholar]
- [9].Xiao F, Liu B, Zhu QX. c-Jun N-terminal kinase is required for thermotherapy-induced apoptosis in human gastric cancer cells. World J Gastroenterol. 2012. Dec 28;18(48):7348–7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tang C, Liang J, Qian J, et al. Opposing role of JNK-p38 kinase and ERK1/2 in hydrogen peroxide-induced oxidative damage of human trophoblast-like JEG-3 cells. Int J Clin Exp Pathol. 2014. Feb 15;7(3):959–968. [PMC free article] [PubMed] [Google Scholar]
- [11].Lee JA, Jung JS, Kim DH, et al. RANKL expression is related to treatment outcome of patients with localized, high-grade osteosarcoma. Pediatr Blood Cancer. 2011;56(5):738–743. [DOI] [PubMed] [Google Scholar]
- [12].Liu T, Liu X, Li W. Tetrandrine, a Chinese plant-derived alkaloid, is a potential candidate for cancer chemotherapy. Oncotarget. 2016. Jun 28;7(26):40800–40815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Jj L, Bao JL, Chen XP, et al. Alkaloids isolated from natural herbs as the anticancer agents. Evid Based Complement Alternat Med. 2012;2012:485042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shi DX, Ma LM, Lu YJ, et al. [Apoptosis-inducing effect of tetrandrine and imatinib on K562/G01 cells and its related mechanism]. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2014. Jun;22(3):723–728. [DOI] [PubMed] [Google Scholar]
- [15].Wu JM, Chen Y, Chen JC, et al. Tetrandrine induces apoptosis and growth suppression of colon cancer cells in mice. Cancer Lett. 2010;287(2):187–195. [DOI] [PubMed] [Google Scholar]
- [16].Li X, Su B, Liu R, et al. Tetrandrine induces apoptosis and triggers caspase cascade in human bladder cancer cells. J Surg Res. 2011;166(1):e45–51. [DOI] [PubMed] [Google Scholar]
- [17].Liu C, Gong K, Mao X, et al. Tetrandrine induces apoptosis by activating reactive oxygen species and repressing Akt activity in human hepatocellular carcinoma. Int J Cancer. 2011. Sep 15;129(6):e45–e51. [DOI] [PubMed] [Google Scholar]
- [18].Luan F, He X, Zeng N. Tetrandrine: a review of its anticancer potentials, clinical settings, pharmacokinetics and drug delivery systems. J Pharm Pharmacol. 2020. Nov;72(11):1491–1512. [DOI] [PubMed] [Google Scholar]
- [19].He BC, Gao JL, Zhang BQ, et al. Tetrandrine inhibits Wnt/beta-catenin signaling and suppresses tumor growth of human colorectal cancer. Mol Pharmacol. 2011;79(2):211–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gao JL, Ji X, He TC, et al. Tetrandrine Suppresses Cancer Angiogenesis and Metastasis in 4T1 Tumor Bearing Mice. Evid Based Complement Alternat Med. 2013;2013:265061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gupta J, del Barco Barrantes I, Igea A, et al. Dual function of p38alpha MAPK in colon cancer: suppression of colitis-associated tumor initiation but requirement for cancer cell survival. Cancer Cell. 2014;25(4):484–500. [DOI] [PubMed] [Google Scholar]
- [22].Grossi V, Peserico A, Tezil T, et al. p38alpha MAPK pathway: a key factor in colorectal cancer therapy and chemoresistance. World J Gastroenterol. 2014;20(29):9744–9758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ishiyama M, Tominaga H, Shiga M, et al. A combined assay of cell viability and in vitro cytotoxicity with a highly water-soluble tetrazolium salt, neutral red and crystal violet. Biol Pharm Bull. 1996;19(11):1518–1520. [DOI] [PubMed] [Google Scholar]
- [24].Wang N, Liu W, Tan T, et al. Notch signaling negatively regulates BMP9-induced osteogenic differentiation of mesenchymal progenitor cells by inhibiting JunB expression. Oncotarget. 2017;8(65):109661–109674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Luo Q, Kang Q, Si W, et al. Connective tissue growth factor (CTGF) is regulated by Wnt and bone morphogenetic proteins signaling in osteoblast differentiation of mesenchymal stem cells. J Biol Chem. 2004;279(53):55958–55968. [DOI] [PubMed] [Google Scholar]
- [26].Zhou L, An N, Jiang W, et al. Fluorescence-based functional assay for Wnt/beta-catenin signaling activity. BioTechniques. 2002;33(5):1126–1128. 1130, 1132 passim. [DOI] [PubMed] [Google Scholar]
- [27].Xu H, Li X, Kong H, et al. Characterization of the Uptake Efficiency and Cytotoxicity of Tetrandrine-Loaded Poly(N-vinylpyrrolidone)-Block-Poly(ε-caprolactone) (PVP-b-PCL) Nanoparticles in the A549 Lung Adenocarcinoma Cell Line. J Biomed Nanotechnol. 2016. Aug;12(8):1699–1707. [DOI] [PubMed] [Google Scholar]
- [28].McCubrey JA, Steelman LS, Bertrand FE, et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget. 2014;5(10):2881–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhao P, Li Q, Shi Z, et al. GSK-3beta regulates tumor growth and angiogenesis in human glioma cells. Oncotarget. 2015;6(31):31901–31915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Leelahavanichkul K, Amornphimoltham P, Molinolo AA, et al. A role for p38 MAPK in head and neck cancer cell growth and tumor-induced angiogenesis and lymphangiogenesis. Mol Oncol. 2014;8(1):105–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Conley-lacomb MK, Saliganan A, Kandagatla P, et al. PTEN loss mediated Akt activation promotes prostate tumor growth and metastasis via CXCL12/CXCR4 signaling. Mol Cancer. 2013;12(1):85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Heymann MF, Brown HK, Heymann D. Drugs in early clinical development for the treatment of osteosarcoma. Expert Opin Investig Drugs. 2016;25(11):1265–1280. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.