

Key Points
Question
What is the prevalence of familial disease among patients with idiopathic dilated cardiomyopathy (DCM) and the lifetime risk of DCM for their first-degree family members by race and ethnicity?
Findings
In this family-based, cross-sectional study of 1220 patients with DCM and their 1693 family members, the estimated familial DCM prevalence was 29.7% and the estimated DCM risk by age 80 years in family members was 19%.
Meaning
These findings suggest substantial prevalence of familial DCM among patients and elevated lifetime risk of DCM among their first-degree family members.
Abstract
Importance
Idiopathic dilated cardiomyopathy (DCM) aggregates in families, and early detection in at-risk family members can provide opportunity to initiate treatment prior to late-phase disease. Most studies have included only White patients, yet Black patients with DCM have higher risk of heart failure–related hospitalization and death.
Objective
To estimate the prevalence of familial DCM among DCM probands and the age-specific cumulative risk of DCM in first-degree relatives across race and ethnicity groups.
Design, Setting, and Participants
A family-based, cross-sectional study conducted by a multisite consortium of 25 US heart failure programs. Participants included patients with DCM (probands), defined as left ventricular systolic dysfunction and left ventricular enlargement after excluding usual clinical causes, and their first-degree relatives. Enrollment commenced June 7, 2016; proband and family member enrollment concluded March 15, 2020, and April 1, 2021, respectively.
Exposures
The presence of DCM in a proband.
Main Outcomes and Measures
Familial DCM defined by DCM in at least 1 first-degree relative; expanded familial DCM defined by the presence of DCM or either left ventricular enlargement or left ventricular systolic dysfunction without known cause in at least 1 first-degree relative.
Results
The study enrolled 1220 probands (median age, 52.8 years [IQR, 42.4-61.8]; 43.8% female; 43.1% Black and 8.3% Hispanic) and screened 1693 first-degree relatives for DCM. A median of 28% (IQR, 0%-60%) of living first-degree relatives were screened per family. The crude prevalence of familial DCM among probands was 11.6% overall. The model-based estimate of the prevalence of familial DCM among probands at a typical US advanced heart failure program if all living first-degree relatives were screened was 29.7% (95% CI, 23.5% to 36.0%) overall. The estimated prevalence of familial DCM was higher in Black probands than in White probands (difference, 11.3% [95% CI, 1.9% to 20.8%]) but did not differ significantly between Hispanic probands and non-Hispanic probands (difference, −1.4% [95% CI, −15.9% to 13.1%]). The estimated prevalence of expanded familial DCM was 56.9% (95% CI, 50.8% to 63.0%) overall. Based on age-specific disease status at enrollment, estimated cumulative risks in first-degree relatives at a typical US advanced heart failure program reached 19% (95% CI, 13% to 24%) by age 80 years for DCM and 33% (95% CI, 27% to 40%) for expanded DCM inclusive of partial phenotypes. The DCM hazard was higher in first-degree relatives of non-Hispanic Black probands than non-Hispanic White probands (hazard ratio, 1.89 [95% CI, 1.26 to 2.83]).
Conclusions and Relevance
In a US cross-sectional study, there was substantial estimated prevalence of familial DCM among probands and modeled cumulative risk of DCM among their first-degree relatives.
Trial Registration
ClinicalTrials.gov Identifier: NCT03037632
This study aims to estimate the prevalence of familial dilated cardiomyopathy (DCM) among DCM probands and the age-specific cumulative risk of DCM in first-degree relatives across race and ethnicity groups.
Introduction
Dilated cardiomyopathy is defined as systolic dysfunction and left ventricular enlargement.1,2 In heart failure clinical trials, patients are classified as having ischemic or nonischemic dilated cardiomyopathy, with the latter accounting for 40% to 50% of cases.1,2 Clinical causes of nonischemic dilated cardiomyopathy include valvular disease, endocrine disorders, exposure to myocardial toxins, and other causes,1,2 but no clinical cause can be identified for most patients. Such patients are classified as having idiopathic dilated cardiomyopathy (DCM). Accurate measures of the prevalence of DCM remain elusive.1,2 While studies from the 1980s suggested DCM was rare,3,4 more recent reports suggested that the prevalence of DCM may be 1 in 250.1,2,5 DCM also aggregates in families, suggesting genetic cause.1,5
This study sought to define the prevalence of familial DCM. An estimate of 23% was obtained by a meta-analysis based on 23 studies with individual estimates ranging from 2% to 65%.6 This study also sought to define familial DCM prevalence in Black and White patients, because DCM and consequent heart failure have been shown to be more common and associated with higher risk of hospitalization and death in Black individuals7,8,9,10 and attributed to hypertension or adverse social determinants of health.8,9,11 Despite these adverse outcomes in Black patients, most family-based DCM studies have included only White families.
This study also sought to define the age-specific cumulative risk of DCM for first-degree relatives using cross-sectional age-specific data on DCM status at enrollment. As the prevalence of DCM increases with age, such knowledge may be helpful for clinicians to motivate family members to engage in clinical screening. Information by race could also be especially helpful for the care of Black patients with DCM and their families.
Methods
Participants
The DCM Precision Medicine Study was a multisite, consortium-based, cross-sectional study of families at 25 US clinical sites (eFigure 1 in Supplement 1).12 Accrual of patients with DCM (probands) and family members occurred from June 7, 2016, to March 15, 2020 (probands), and April 1, 2021 (family members). The institutional review boards at The Ohio State University and all clinical sites approved the initial period of the study followed by single institutional review board oversight at the University of Pennsylvania. Written informed consent was obtained from all participants.
All probands met diagnostic criteria for DCM defined by left ventricular systolic dysfunction (LVSD; left ventricular ejection fraction <50%) and left ventricular enlargement (LVE) with other usual clinical causes excluded, as previously defined.12 A proband was defined as having familial DCM if at least 1 living first-degree relative had a DCM diagnosis meeting the same criteria. First-degree relatives who had either LVSD or LVE without known cause were referred to as having a partial DCM phenotype.13 Because familial DCM was assigned to probands based on clinical evidence in their first-degree relatives, an expanded definition of familial DCM, which required at least 1 living first-degree relative to have had DCM or either LVSD or LVE without known cause, was also considered. Both definitions were restricted to living first-degree relatives because the success of obtaining medical records to establish a conclusive DCM diagnosis in deceased individuals is highly variable.
At enrollment, study personnel collected a cardiovascular history and a pedigree in a standardized interview. Race and ethnicity were included because of their relevance for health outcomes and were self-reported by study participants using structured race (African American, Asian, Native American or Alaska Native, Native Hawaiian or Pacific Islander, White, more than 1 race, or unknown) and Hispanic ethnicity (yes, no, or unknown) categories. Probands were asked to inform all living first-degree relatives of the study and to seek their oral permission for contact from study personnel.12 Study staff then sought enrollment of first-degree relatives who provided oral permission for contact. For first-degree relatives without prior clinical cardiovascular screening, a research-based cardiovascular-directed history and examination, an electrocardiogram, and a transthoracic echocardiogram were obtained in most cases at a DCM Consortium site. Alternatively, the results of clinical screening accomplished by the family member’s physician as part of recommended clinical care were used. A structured medical record query form was used to abstract cardiovascular medical information for probands and family members.12
Clinical data from probands and family members were centrally adjudicated to establish whether DCM and partial phenotypes were present at or prior to enrollment.12 Central adjudication, including electrocardiogram interpretation, was performed by The Ohio State University site principal investigator (G.J.H.) without knowledge of family/pedigree relationships or genetic information. Research echocardiograms were interpreted at sites by study site personnel.
Statistical Analysis
Technical descriptions of the statistical methods have been provided elsewhere (eAppendix in Supplement 1). All analyses were performed in SAS/STAT version 15.2 software, Version 9.4 (TS1M7) of the SAS System for 64-bit Windows (SAS Institute Inc), and R version 4.0.2 (R Foundation). All statistical tests and confidence intervals were 2-sided with α = .05.
Prevalence of Familial DCM Among Probands
The crude familial DCM prevalence was estimated as the proportion of probands with 1 or more enrolled first-degree relatives having DCM for descriptive purposes. Because it was not possible to observe or confirm disease in unenrolled first-degree relatives, the crude prevalence of familial DCM likely underestimated the true familial DCM prevalence to an extent depending on first-degree relative participation (see eAppendix in Supplement 1). Estimating the true familial DCM prevalence if all living first-degree relatives were screened required modeling the relationship between the number of screened first-degree relatives with DCM and the possibly unobserved number of living first-degree relatives with DCM. This approach assumed that first-degree relative participation and disease risk depended on only proband or family characteristics, which was necessary in part due to lack of covariate data on unenrolled first-degree relatives.
The crude prevalence also depended on the composition of the sample due to heterogeneity in the patient populations across DCM Consortium sites. To account for both this heterogeneity and intrafamilial correlation in disease status, a beta-binomial mixed model was fit using maximum likelihood. This model expressed the marginal disease probability in a first-degree relative as a function of proband characteristics, including fixed effects for race and ethnicity group, enrollment age quartile, and sex, and random effects for site and race and ethnicity group within site. In addition to conditional odds ratios comparing groups at a particular site, this model fit was used to obtain marginally standardized estimates of familial DCM prevalence among probands at a typical US advanced heart failure program, as described below.
Age-Specific Cumulative Risk for First-degree Relatives
For each screened first-degree relative, it was possible to determine whether DCM or partial phenotypes were present by the age at enrollment. This observation scheme yielded current status data, a type of interval-censored survival data14,15 that can be used to model age-specific cumulative risks. To do this, the unobserved age at disease onset in first-degree relatives was assumed to have a marginal distribution with a Weibull baseline survivor function influenced by covariates and random effects through a proportional hazards model. Covariates included proband race and ethnicity group, age at diagnosis quartile, and sex and first-degree relative sex; random effects included site and race and ethnicity group within site. This model was fit as a generalized estimating equation–type generalized linear mixed model16,17 with a compound symmetry working correlation structure to account for intrafamilial correlation. Estimation within this framework assumed that, given covariates and site, first-degree relative participation did not depend on disease status and age at enrollment did not depend on the age at disease onset. In addition to conditional hazard ratios comparing groups at a particular site, this model fit was used to obtain marginally standardized estimates of age-specific cumulative risk among first-degree relatives at a typical US advanced heart failure program, as described below.
Marginally Standardized Estimates for a Typical US Advanced Heart Failure Program
For clinicians seeking to understand risks in different groups at their programs, conditional estimates of familial DCM prevalence and age-specific cumulative risk for a single advanced heart failure program in the US are most relevant.18,19 To facilitate broad application, conditional estimates for a typical advanced heart failure program in the US, defined as a program at the mean or mode of the random effects distribution describing the population of such programs,16,18,19,20,21 were presented. A more detailed explanation of this approach has been provided (eAppendix in Supplement 1).
Marginal standardization, which involved taking weighted averages of the conditional estimates for each combination of covariates according to an assumed covariate distribution, was used to obtain conditional estimates of familial DCM prevalence and age-specific cumulative risk in representative subpopulations.22 Detailed descriptions of the assumed covariate distributions have been provided in the corresponding table notes or figure legends. For familial DCM prevalence, which was a function of the number of living first-degree relatives, marginal standardization involved taking a weighted average over the assumed Poisson distribution of this number. The delta method23 was used to provide standard errors for all marginally standardized quantities.
Results
The study cohort comprised 1220 patients with DCM (probands), of whom 43.1% were Black, 8.3% were of Hispanic ethnicity, and 43.8% were female. Probands were enrolled at a median age of 52.8 years (IQR, 42.4-61.8) and had a median of 4 (IQR, 3-6) living first-degree relatives at enrollment. A median of 28% (IQR, 0%-60%) of these per family were enrolled and screened, which yielded 1693 first-degree relatives (eFigure 2 in Supplement 1), 59.8% of whom were female. Characteristics of probands and first-degree relatives by race and ethnicity are presented in Table 1.
Table 1. Characteristics of Probands and First-degree Relatives, by Proband Self-reported Race and Ethnicity.
| Characteristic | Median (IQR) [No.] | |||
|---|---|---|---|---|
| Hispanic (probands, n = 102; relatives, n = 159) | Non-Hispanic (probands, n = 1118; relatives, n = 1534) | |||
| Black (probands, n = 10; relatives, n = 13) | White (probands, n = 92; relatives, n = 146) | Black (probands, n = 516; relatives n = 492) | White (probands, n = 602; relatives, n = 1042) | |
| Age at enrollment, y | ||||
| Probands | 48.6 (34.8 to 56.4) | 49.0 (38.2 to 58.0) | 50.9 (41.7 to 59.5) | 55.4 (43.9 to 64.5) |
| Relatives | 28.4 (21.1 to 36.7) | 36.6 (23.6 to 48.8) | 40.6 (27.1 to 55.3) | 44.5 (28.1 to 60.1) |
| Sex | ||||
| Female, No. (%) | ||||
| Probands | 3 (30.0) | 39 (42.4) | 228 (44.2) | 264 (43.9) |
| Relatives | 5 (38.5) | 93 (63.7) | 325 (66.1) | 589 (56.5) |
| Male, No. (%) | ||||
| Probands | 7 (70.0) | 53 (57.6) | 288 (55.8) | 338 (56.2) |
| Relatives | 8 (61.5) | 53 (36.3) | 167 (33.9) | 453 (43.5) |
| No. of living first-degree relatives per proband | 3 (2 to 5) | 4 (3 to 6) | 4 (3 to 6) [n = 514] | 4 (3 to 6) |
| % Living first-degree relatives enrolled per proband | 50.0 (16.7 to 75.0) | 33.3 (0.0 to 70.8) | 16.7 (0.0 to 50.0) [n = 508] | 40.0 (11.1 to 66.7) |
| Probands with ≥1 enrolled first-degree relative, No. (%) | 8 (80.0) | 64 (69.6) | 292 (56.6) | 460 (76.4) |
| Relatives’ relationship to proband, No. (%) | ||||
| Child | 8 (61.5) | 76 (52.1) | 249 (50.6) | 491 (47.1) |
| Full sibling | 4 (30.8) | 42 (28.8) | 154 (31.3) | 335 (32.2) |
| Parent | 1 (7.7) | 28 (19.2) | 89 (18.1) | 216 (20.7) |
| Age at diagnosis of probands, y | 42.3 (34.3 to 52.4) | 39.7 (30.8 to 50.6) | 43.1 (33.3 to 52.4) | 46.0 (35.7 to 55.3) [n = 601] |
| Interventions for probands, No. (%) | ||||
| Left ventricular assist device | 1 (10.0) | 12 (13.0) | 136 (26.4) | 106 (17.6) |
| Heart transplant | 1 (10.0) | 26 (28.3) | 60 (11.6) | 96 (16.0) |
| Biventricular pacemaker | 2 (20.0) | 13 (14.4) [n = 90] | 50 (10.1) [n = 496] | 101 (17.3) [n = 583] |
| Implantable cardioverter defibrillator | 7 (70.0) | 63 (68.5) | 351 (68.6) [n = 512] | 390 (65.1) [n = 599] |
| Comorbidities of probands, No. (%) | ||||
| Hypertension | 5 (50.0) | 51 (55.4) | 346 (67.1) | 245 (40.7) |
| High cholesterol | 3 (30.0) | 34 (37.0) | 146 (28.3) | 149 (24.8) |
| Diabetes | 1 (10.0) | 25 (27.2) | 161 (31.2) | 115 (19.1) |
| Cancer | 0 | 5 (5.4) | 22 (4.3) | 39 (6.5) |
| Lung disease | 0 | 2 (2.2) | 24 (4.7) | 27 (4.5) |
| Relatives with DCM diagnosis prior to enrollment, No. (%) | 1 (7.7) | 8 (5.5) | 45 (9.2) | 61 (5.9) |
| DCM phenotype of relatives, No. (%) | ||||
| Dilated cardiomyopathy | 1 (7.7) | 14 (9.6) | 59 (12.0) | 76 (7.3) |
| Left ventricular enlargement only | 1 (7.7) | 12 (8.2) | 34 (6.9) | 84 (8.1) |
| Left ventricular systolic dysfunction only | 0 | 3 (2.1) | 25 (5.1) | 47 (4.5) |
| No left ventricular enlargement or left ventricular systolic dysfunction | 11 (84.6) | 117 (80.1) | 374 (76.0) | 835 (80.1) |
| Left ventricular ejection fraction, % | ||||
| Probands | 23 (10 to 37) | 20 (16 to 25) | 20 (15 to 25) [n = 515] | 23 (16 to 30) [n = 598] |
| Relatives | 59 (54 to 67) [n = 12] | 62 (55 to 65) [n = 144] | 57 (53 to 61) [n = 483] | 59 (55 to 63) [n = 1026] |
| Left ventricular internal diastolic dimension, mm | ||||
| Probands | 66 (62 to 70) | 66 (60 to 72) | 65 (60 to 71) [n = 514] | 64 (59 to 69) [n = 598] |
| Relatives | 47 (45 to 53) [n = 12] | 47 (43 to 51) [n = 143] | 47 (44 to 52) [n = 483] | 48 (44 to 52) [n = 1015] |
| Left ventricular internal diastolic dimension (z score)a | ||||
| Probands | 4.2 (2.9 to 5.8) | 4.7 (3.5 to 5.8) | 4.2 (3.1 to 5.6) [n = 514] | 3.9 (2.8 to 5.1) [n = 596] |
| Relatives | 0.1 (−0.7 to 1.0) [n = 10] | 0.1 (−1.4 to 1.1) [n = 140] | −0.1 (−1.1 to 1.3) [n = 474] | 0.0 (−1.1 to 1.1) [n = 994] |
Abbreviation: DCM, dilated cardiomyopathy.
Calculated based on sex and height24 for all study participants with heights of at least 152 cm (male) or 137 cm (female).
Familial DCM Prevalence by Race and Ethnicity
The crude prevalence of familial DCM and the expanded definition of familial DCM in the cohort were 11.6% and 24.1%, respectively (Table 2). Crude prevalences were 10.9% among non-Hispanic Black and 12.0% among non-Hispanic White probands for familial DCM, and 19.8% and 27.6% in these groups, respectively, for the expanded definition of familial DCM. As described in the Methods section, crude prevalences must be interpreted with caution, particularly because family member participation varied substantially between racial and ethnic subgroups (Table 1).
Table 2. Prevalence of Familial DCM in Probands, by Proband Self-reported Race and Ethnicity.
| Race and ethnicity group | Total | Familial DCM prevalence | |||
|---|---|---|---|---|---|
| Standard definitiona | Expanded definitiona | ||||
| Crude | Model-basedb | Crude | Model-basedb | ||
| No. (%) | % (95% CI) | No. (%) | % (95% CI) | ||
| All races and ethnicities | 1220 | 141 (11.6) | 29.7 (23.5-36.0) | 294 (24.1) | 56.9 (50.8-63.0) |
| All ethnicities | |||||
| Black | 526 | 57 (10.8) | 39.4 (29.9-48.8) | 104 (19.8) | 60.6 (52.4-68.7) |
| White | 694 | 84 (12.1) | 28.0 (21.4-34.6) | 190 (27.4) | 56.2 (49.7-62.7) |
| All races | |||||
| Hispanic | 102 | 13 (12.8) | 28.6 (14.1-43.1) | 26 (25.5) | 53.7 (40.0-67.3) |
| Non-Hispanic | 1118 | 128 (11.5) | 30.0 (23.7-36.2) | 268 (24.0) | 57.7 (51.4-63.9) |
| Hispanic | |||||
| Black | 10 | 1 (10) | 25.7 (0.0-68.3)c | 2 (20.0) | 45.0 (1.2-88.7) |
| White | 92 | 12 (13) | 28.8 (13.8-43.7) | 24 (26.1) | 54.1 (40.1-68.2) |
| Non-Hispanic | |||||
| Black | 516 | 56 (10.9) | 40.3 (30.8-49.9) | 102 (19.8) | 61.7 (53.7-69.7) |
| White | 602 | 72 (12.0) | 27.8 (21.2-34.5) | 166 (27.6) | 56.8 (50.1-63.5) |
Abbreviations: DCM, dilated cardiomyopathy; LVE, left ventricular enlargement; LVSD, left ventricular systolic dysfunction.
A proband has familial DCM if at least 1 living first-degree relative has DCM by the standard definition and if at least 1 living first-degree relative has DCM, LVSD, or LVE without known cause by the expanded definition.
Estimates from a beta-binomial mixed model (eTables 1 and 3 in Supplement 1; Supplement 2) were used to obtain marginally standardized estimates and delta method 95% CIs for familial DCM prevalence if all living first-degree relatives were screened. Probands without living or screened first-degree relatives provide no information on the parameters of interest in the likelihood; all model-based estimates were therefore derived from 822 probands (8 Hispanic Black, 64 Hispanic White, 290 non-Hispanic Black, 460 non-Hispanic White) with at least 1 screened first-degree relative. The reported estimates are for patients seen at a typical US advanced heart failure program, defined as a program at the mean or mode of the random effects distribution describing the population of such programs. Each patient subpopulation defined by race and ethnicity was assumed to be balanced across the 8 possible sex and age quartile combinations, and the number of living first-degree relatives was assumed to follow a Poisson distribution with a mean of 4.53, which was the mean number of living first-degree relatives per family in the cohort. In subpopulations containing multiple race and ethnicity groups, constituent groups were represented in proportion to 2019 US Census population estimates for the groups of interest (All races and ethnicities: 0.99% Hispanic Black, 13.97% non-Hispanic Black, 18.06% Hispanic White, 66.98% non-Hispanic White; All ethnicities, Black: 6.64% Hispanic, 93.36% non-Hispanic; All ethnicities, White: 21.24% Hispanic, 78.76% non-Hispanic; All races, Hispanic: 5.21% Black, 94.79% White; All races, non-Hispanic: 17.26% Black, 82.74% White).
The actual lower bound of the 95% CI was negative and truncated at zero.
The model-based estimate of the prevalence of familial DCM among patients with DCM at a typical US heart failure program if all living first-degree relatives were screened was 29.7% (95% CI, 23.5%-36.0%) overall. The estimated prevalence of familial DCM was higher in Black (39.4%) than in White (28.0%) probands (difference, 11.3% [95% CI, 1.9%-20.8%]) and was not significantly different in probands of Hispanic compared with non-Hispanic ethnicity (95% CI, −15.9% to 13.1%) (Table 2; eTable 2 in Supplement 1). First-degree relatives of non-Hispanic Black probands had greater odds of having DCM relative to first-degree relatives of non-Hispanic White probands (odds ratio, 1.67 [95% CI, 1.14-2.46]; eTable 1 in Supplement 1). An expanded definition of familial DCM was also considered that included first-degree relatives who had partial phenotypes in addition to first-degree relatives who had DCM. The estimated prevalence with the expanded definition of familial DCM was 56.9% (95% CI, 50.8%-63.0%), with the difference between Black probands and White probands attenuated to 4.3% (95% CI, −3.9% to 12.5%) (Table 2; eTable 4 in Supplement 1).
Age-Specific Cumulative Risks in First-degree Relatives of Patients With DCM
Model-based estimates of the age-specific cumulative risk of DCM for first-degree relatives of patients diagnosed as having DCM at a typical US advanced heart failure program were obtained from cross-sectional data on DCM status at enrollment. Estimated cumulative risk of a DCM diagnosis for a first-degree relative increased with age, reaching 19% (95% CI, 13%-24%) by age 80 years for DCM and 33% (95% CI, 27%-40%) when considering DCM and partial phenotypes (Figure, A). Estimated cumulative risk of DCM by age 80 years was 16% (95% CI, 11%-22%) for first-degree relatives of non-Hispanic White probands and 28% (95% CI, 18%-38%) for first-degree relatives of non-Hispanic Black probands (Figure, B). The DCM hazard was higher for first-degree relatives of non-Hispanic Black probands than for first-degree relatives of non-Hispanic White probands (hazard ratio, 1.89 [95% CI, 1.26-2.83]; Table 3). Moreover, the estimated cumulative risk of DCM by age 80 years was 13% (95% CI, 6%-19%) among first-degree relatives of probands in the oldest diagnosis age quartile and 26% (95% CI, 16%-36%) for first-degree relatives of probands in the youngest diagnosis age quartile (Figure, C). The DCM hazard was higher for first-degree relatives of probands in the youngest diagnosis age quartile than for the first-degree relatives of probands in the oldest age quartile (hazard ratio, 2.19 [95% CI, 1.19-4.00]; Table 3).
Figure. Model-Based Estimates of Age-Specific Cumulative Risk in First-degree Relatives of Patients With Dilated Cardiomyopathy (DCM).
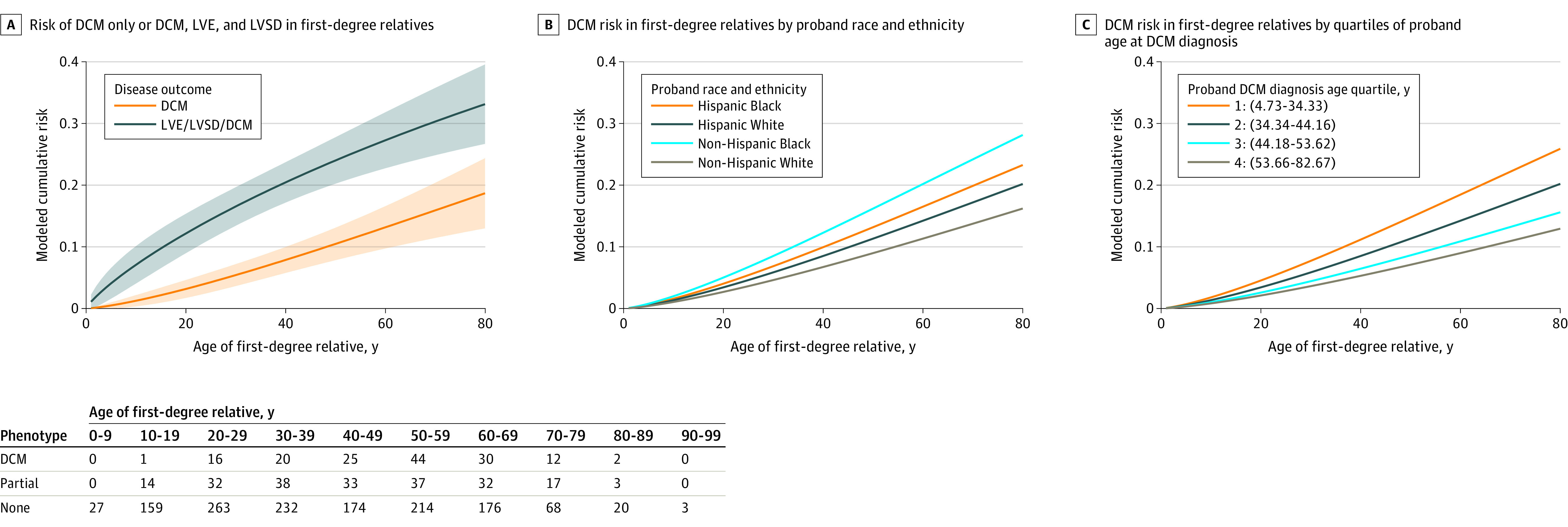
A Weibull proportional hazards model for age-specific cumulative risk was fit to cross-sectional data on disease status at enrollment from first-degree relatives. Each panel presents marginally standardized age-specific cumulative risks derived from this model fit that apply to first-degree relatives of patients seen at a typical US advanced heart failure program, defined as a program at the mean or mode of the random effects distribution describing the population of such programs. Marginally standardized age-specific cumulative risks were calculated by obtaining conditional estimates at a typical US advanced heart failure program in each first-degree relative subpopulation defined by a combination of the covariates and then taking the weighted average according to an assumed covariate distribution described for each panel. A, Each patient subpopulation defined by race and ethnicity was assumed to be balanced across the 16 possible sex (first-degree relative and proband) and proband DCM diagnosis age quartile combinations. These subpopulations, in turn, were represented in proportion to 2019 US Census population estimates (0.99% Hispanic Black, 13.97% non-Hispanic Black, 18.06% Hispanic White, 66.98% non-Hispanic White) in the overall marginally standardized age-specific cumulative risk. Bands represent delta method pointwise 95% CIs. Numbers of individuals within age strata by phenotype are provided in the table below the figure. B, Each patient subpopulation defined by race and ethnicity was assumed to be balanced across the 16 possible sex (first-degree relative and proband) and proband DCM diagnosis age quartile combinations. C, Each subpopulation of first-degree relatives defined by proband DCM diagnosis age quartile, race, and ethnicity was assumed to be balanced across the 4 possible sex (first-degree relative and proband) combinations. Marginally standardized estimates for a particular subpopulation defined by proband DCM diagnosis age quartile were then obtained by weighting race and ethnicity groups in proportion to 2019 US Census population estimates (0.99% Hispanic Black, 13.97% non-Hispanic Black, 18.06% Hispanic White, 66.98% non-Hispanic White). LVE indicates left ventricular enlargement; LVSD, left ventricular systolic dysfunction.
Table 3. Model-Based Hazard Ratios for DCM and Partial DCM Phenotypes in First-degree Relatives of Patients With DCM.
| Predictor | DCMa | DCM or partial phenotypesa | ||
|---|---|---|---|---|
| HR (95% CI) | P value | HR (95% CI) | P value | |
| Proband race and ethnicity | ||||
| Non-Hispanic | ||||
| White | 1 [Reference] | 1 [Reference] | ||
| Black | 1.89 (1.26-2.83) | .002 | 1.27 (0.99-1.63) | .06 |
| Hispanic | ||||
| White | 1.28 (0.64-2.54) | .48 | 1.04 (0.66-1.65) | .87 |
| Black | 1.51 (0.29-7.82) | .63 | 0.93 (0.24-3.61) | .92 |
| Proband age at diagnosis, quartile | ||||
| I (4.73-34.33 y) | 2.19 (1.19-4.00) | .01 | 1.67 (1.14-2.44) | .008 |
| II (34.34-44.16 y) | 1.64 (0.91-2.95) | .10 | 1.60 (1.09-2.35) | .02 |
| III (44.18-53.62 y) | 1.23 (0.65-2.31) | .53 | 1.28 (0.91-1.79) | .15 |
| IV (53.66-82.67 y) | 1 [Reference] | 1 [Reference] | ||
Abbreviations: DCM, dilated cardiomyopathy; GEE, generalized estimating equation; GLMM, generalized linear mixed model; HR, hazard ratio.
Within-program HRs and Wald 95% CIs for a Weibull proportional hazards survival model were obtained from a GEE-type GLMM fit to current disease status data (eTables 5 and 6 in Supplement 1; Supplement 3) and are adjusted for proband and first-degree relative sex and site in addition to other variables shown here; 1692 first-degree relatives contributed to this analysis (1 non-Hispanic White first-degree relative was excluded due to missing covariate data). Two-sided P values are for the null hypothesis that the HR is 1.
Discussion
In a US family-based, cross-sectional study, there was substantial estimated prevalence of familial DCM among DCM probands and modeled cumulative risk of DCM among their first-degree relatives. These findings are relevant to the care of the families of patients diagnosed as having DCM and provide an estimate for clinicians practicing at a US advanced heart failure program of the cumulative risks of DCM or partial phenotypes in first-degree relatives of their patients. The findings also suggest that first-degree relatives of non-Hispanic Black probands or probands diagnosed at a young age will be at higher risk of DCM.
While DCM is usually silent until late-phase disease, often presenting with heart failure, conventional medical treatment has been shown to mitigate asymptomatic DCM.25 These findings, therefore, substantiate cardiomyopathy guidelines: that at diagnosis probands should be counseled regarding familial risk and first-degree relatives should undergo clinical screening.26 Although the yield and possible clinical benefit of genetics and genetic testing have not been established for this study, the familial aggregation data support these phenotype- and family-based recommendations because clinical detection of DCM can prompt interventions in first-degree relatives regardless of possible genetic analysis.
The primary study end point, familial DCM, required only cardiovascular clinical data from living family members to establish the diagnosis. While in usual clinical genetics care the family history of DCM of deceased family members is appropriately integrated into risk assessments, the validation with medical records to support such DCM diagnoses frequently cannot be achieved. Thus, the familial DCM definition for this study, while more restrictive, provided a conservative but rigorous estimate of familial DCM prevalence that can be achieved in clinical practice. Moreover, a partial DCM phenotype was defined simply as LVSD or LVE in family members with no other known cardiovascular disease. This use of a partial DCM phenotype was in agreement with a prior evaluation of family members of patients with DCM.13 That study identified 15.5% of family members who had LVE without LVSD and 2.7% who had LVSD without LVE; of those, 20% progressed to DCM over a median follow-up of 57 months and only those 2 clinical findings were independently associated with risk of future DCM. A second study over an 11-year follow-up of patients without ischemic disease observed that an increase in left ventricular dimension of 1 SD was associated with a risk factor–adjusted heart failure hazard ratio of 1.47.27 Long-term follow-up of this cohort, which was much larger and had greater diversity, may provide additional insights into these prior findings.
The SOLVD prevention trial25 showed that patients with asymptomatic nonischemic DCM with an LV ejection fraction less than 35% randomized to treatment with angiotensin-converting enzyme inhibitor had reduced hospitalization and mortality. In the ensuing 30 years, a randomized study has not been undertaken for first-degree relatives receiving no medical therapy who have clinically detectable but asymptomatic very early DCM,28 or incipient DCM as indicated by a partial phenotype, to prevent the symptomatic phase of DCM. Such a study can now be considered, likely using advanced imaging (eg, LV strain with transthoracic echocardiography or advanced cardiac magnetic resonance) to detect very early disease.
This finding of greater risk to first-degree relatives of non-Hispanic Black probands was consonant with existing research on cardiovascular risk in this population. Black individuals with hypertension enrolled in HyperGEN were more likely to have had isolated reduction of LV ejection fraction.29 Moreover, incident heart failure has been observed more commonly in Black individuals before 50 years of age, particularly when systolic dysfunction is present.7,30 Nonetheless, the current study did not provide a clear explanation for this elevated risk. The central hypothesis of the DCM Precision Medicine Study is that most DCM, whether familial or nonfamilial, has a genetic basis.12 This finding could suggest additional genetic risk in non-Hispanic Black populations. At the same time, environmental factors beyond genetics and biology also affect disease presentation and progression. The observed increased risk of heart failure in Black individuals, for example, has been attributed to a greater incidence of hypertension, diabetes, and other adverse social determinants of health.11 But DCM is a fundamentally different diagnosis than heart failure even though heart failure is the most common presentation of DCM, and heart failure can occur from conditions other than DCM. Furthermore, disentangling risk of DCM from heart failure risk is complex, particularly when considering the initiation of the heart failure syndrome as a final end-stage progression from DCM.28 Moreover, from a genetics perspective, the start of heart failure could be considered akin to an added biological or environmental insult to the development or progression of DCM.
While increased risk of DCM was observed in Black compared with White first-degree relatives, this risk was less when family members with partial phenotypes were included. The explanation for this observation remains uncertain, although it is possible that the effect of adverse social determinants of health may have been attenuated due to a more equitable opportunity to identify early signs of DCM within this family-based research study.
Regardless of uncertainty of the underlying causes as noted above, the finding that first-degree relatives of non-Hispanic Black probands had increased risk of DCM underscores the importance of making clinical screening of at-risk family members available to this population. This is particularly relevant because the end stage of DCM, heart failure, has significantly greater morbidity and mortality among Black patients.7,31 In particular, Black patients were shown to have had increased risk of heart failure death, with relative risks of 1.25 to 1.36 in the SOLVD Treatment and Prevention trials, even after adjusting for disease severity and treatment.31
Limitations
This study has several limitations. First, the percentage of first-degree relatives enrolled was lower among non-Hispanic Black probands than other proband groups despite the use of standardized recruitment processes and procedures across all clinical sites, possibly due to differences in timing of enrollment or mistrust in medical research.32 This enrollment difference implied that the racial differences in crude prevalence of familial DCM likely did not reflect differences in the true prevalence with complete ascertainment of first-degree relatives. However, it was not likely to have affected racial differences in model-based estimates that accounted for enrollment, family size, and other factors.
Second, the clinical data revealed that non-Hispanic Black probands tended to be sicker than non-Hispanic White probands, while enrolled non-Hispanic Black family members were slightly more likely to have had a prior DCM diagnosis. Whether the severity of illness in probands or family members influences the likelihood of study enrollment is poorly understood. If these observations indicated preferential enrollment of sicker Black families, the estimated prevalence of familial DCM and age-specific cumulative risks for first-degree relatives among Black families could have been overestimated. However, these same observations could also be explained by representative sampling, reflecting the higher morbidity and prevalence of DCM among Black individuals,7,31 in which case there would have been no overestimation.
Third, this study was conducted solely among living probands and their first-degree relatives for better and consistent phenotyping. Because some first-degree relatives may have died with DCM prior to the study, the prevalence of familial DCM and age-specific cumulative risk in first-degree relatives would likely have been underestimated, and the true magnitude of disease burden may be larger. As a slightly higher percentage of non-Hispanic Black first-degree relatives were deceased prior to the study (eTable 7 in Supplement 1), the underestimation would likely be larger in this group, suggesting that the actual racial differences might be even larger.
Fourth, estimated age-specific cumulative risks were modeled using age-specific disease status data at enrollment instead of prospective cohort data. Long-term follow-up of this cohort is needed to validate and extend these findings.
Fifth, this study was conducted in multiple, geographically representative advanced heart failure and transplantation programs, and caution should be taken when generalizing the results to primary care settings. Studies need to include patients with DCM and families beyond academic medical centers to extend these findings.
Sixth, this analysis relied on statistical modeling and various assumptions. Factors such as family member self-selection bias, elevated polygenic background among DCM families evaluated at advanced heart failure programs, and nongenetic familial aggregation of social determinants of health could have influenced estimates. Also, choices for marginal standardization were not the only possible ones but were designed to obtain estimates with the broadest applicability.
Conclusions
In a US cross-sectional study, there was substantial estimated prevalence of familial DCM among DCM probands and modeled cumulative risk of DCM among their first-degree relatives.
eAppendix. Statistical Methods
eTable 1. Model Fit for the Marginal Probability of DCM in the Population of First-Degree Relatives of Probands with Specified Characteristics at a Particular US Advanced Heart Failure Program
eTable 2. Differences in Marginally Standardized Familial DCM Prevalences (Standard Definition) between Proband Subpopulations Defined by Self-Reported Ethnicity and Race at a Typical US Advanced Heart Failure Program
eTable 3. Model Fit for the Marginal Probability of DCM or Partial Phenotypes in the Population of First-Degree Relatives of Probands with Specified Characteristics at a Particular US Advanced Heart Failure Program
eTable 4. Differences in Marginally Standardized Familial DCM Prevalences (Expanded Definition) between Proband Subpopulations Defined by Self-Reported Ethnicity and Race at a Typical US Advanced Heart Failure Program
eTable 5. Model Fit for the Age-Specific Cumulative Risk of DCM in a First-Degree Relative of a Patient with DCM at a Particular US Advanced Heart Failure Program
eTable 6. Model Fit for the Age-Specific Cumulative Risk of LVE, LVSD, or DCM in a First-Degree Relative of a Patient with DCM at a Particular US Advanced Heart Failure Program
eTable 7. Vital Status of First-Degree Relatives at Proband Enrollment, by Proband Ethnicity and Race
eFigure 1. Clinical Sites of the DCM Consortium
eFigure 2. DCM Precision Medicine Study Participant Recruitment
eReferences
fam_prev_ests_cov.csv
fdr_risk_ests_cov.csv
References
- 1.McNally EM, Mestroni L. Dilated cardiomyopathy: genetic determinants and mechanisms. Circ Res. 2017;121(7):731-748. doi: 10.1161/CIRCRESAHA.116.309396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bozkurt B, Colvin M, Cook J, et al. ; American Heart Association Committee on Heart Failure and Transplantation of the Council on Clinical Cardiology; Council on Cardiovascular Disease in the Young; Council on Cardiovascular and Stroke Nursing; Council on Epidemiology and Prevention; and Council on Quality of Care and Outcomes Research . Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement from the American Heart Association. Circulation. 2016;134(23):e579-e646. doi: 10.1161/CIR.0000000000000455 [DOI] [PubMed] [Google Scholar]
- 3.Codd MB, Sugrue DD, Gersh BJ, Melton LJ III. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy: a population-based study in Olmsted County, Minnesota, 1975-1984. Circulation. 1989;80(3):564-572. doi: 10.1161/01.CIR.80.3.564 [DOI] [PubMed] [Google Scholar]
- 4.Manolio TA, Baughman KL, Rodeheffer R, et al. Prevalence and etiology of idiopathic dilated cardiomyopathy (summary of a National Heart, Lung, and Blood Institute workshop. Am J Cardiol. 1992;69(17):1458-1466. doi: 10.1016/0002-9149(92)90901-A [DOI] [PubMed] [Google Scholar]
- 5.Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10(9):531-547. doi: 10.1038/nrcardio.2013.105 [DOI] [PubMed] [Google Scholar]
- 6.Petretta M, Pirozzi F, Sasso L, Paglia A, Bonaduce D. Review and meta-analysis of the frequency of familial dilated cardiomyopathy. Am J Cardiol. 2011;108(8):1171-1176. doi: 10.1016/j.amjcard.2011.06.022 [DOI] [PubMed] [Google Scholar]
- 7.Bibbins-Domingo K, Pletcher MJ, Lin F, et al. Racial differences in incident heart failure among young adults. N Engl J Med. 2009;360(12):1179-1190. doi: 10.1056/NEJMoa0807265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bahrami H, Kronmal R, Bluemke DA, et al. Differences in the incidence of congestive heart failure by ethnicity: the multi-ethnic study of atherosclerosis. Arch Intern Med. 2008;168(19):2138-2145. doi: 10.1001/archinte.168.19.2138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loehr LR, Rosamond WD, Chang PP, Folsom AR, Chambless LE. Heart failure incidence and survival (from the Atherosclerosis Risk in Communities study). Am J Cardiol. 2008;101(7):1016-1022. doi: 10.1016/j.amjcard.2007.11.061 [DOI] [PubMed] [Google Scholar]
- 10.Carnethon MR, Pu J, Howard G, et al. ; American Heart Association Council on Epidemiology and Prevention; Council on Cardiovascular Disease in the Young; Council on Cardiovascular and Stroke Nursing; Council on Clinical Cardiology; Council on Functional Genomics and Translational Biology; and Stroke Council . Cardiovascular health in African Americans: a scientific statement from the American Heart Association. Circulation. 2017;136(21):e393-e423. doi: 10.1161/CIR.0000000000000534 [DOI] [PubMed] [Google Scholar]
- 11.Nayak A, Hicks AJ, Morris AA. Understanding the complexity of heart failure risk and treatment in black patients. Circ Heart Fail. 2020;13(8):e007264. doi: 10.1161/CIRCHEARTFAILURE.120.007264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kinnamon DD, Morales A, Bowen DJ, Burke W, Hershberger RE; DCM Consortium . Toward genetics-driven early intervention in dilated cardiomyopathy: design and implementation of the DCM Precision Medicine Study. Circ Cardiovasc Genet. 2017;10(6):e001826. doi: 10.1161/CIRCGENETICS.117.001826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mahon NG, Murphy RT, MacRae CA, Caforio AL, Elliott PM, McKenna WJ. Echocardiographic evaluation in asymptomatic relatives of patients with dilated cardiomyopathy reveals preclinical disease. Ann Intern Med. 2005;143(2):108-115. doi: 10.7326/0003-4819-143-2-200507190-00009 [DOI] [PubMed] [Google Scholar]
- 14.Lawless JF. Statistical Models and Methods for Lifetime Data. 2nd ed. John Wiley & Sons; 2011. [Google Scholar]
- 15.Jewell NP, van der Laan MJ. Current status data: review, recent developments and open problems, in UC Berkeley Division of Biostatistics Working Paper Series. 2002.
- 16.Stroup W. Generalized Linear Mixed Models: Modern Concepts, Methods, and Applications. CRC Press; 2012. [Google Scholar]
- 17.Vonesh E. Generalized Linear and Nonlinear Models for Correlated Data: Theory and Applications Using SAS. 1st ed. SAS Institute; 2012. [Google Scholar]
- 18.Falconieri N, Van Calster B, Timmerman D, Wynants L. Developing risk models for multicenter data using standard logistic regression produced suboptimal predictions: a simulation study. Biom J. 2020;62(4):932-944. doi: 10.1002/bimj.201900075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wynants L, Vergouwe Y, Van Huffel S, Timmerman D, Van Calster B. Does ignoring clustering in multicenter data influence the performance of prediction models? a simulation study. Stat Methods Med Res. 2018;27(6):1723-1736. doi: 10.1177/0962280216668555 [DOI] [PubMed] [Google Scholar]
- 20.Austin PC, Merlo J. Intermediate and advanced topics in multilevel logistic regression analysis. Stat Med. 2017;36(20):3257-3277. doi: 10.1002/sim.7336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Molenberghs G, Verbeke G, Models for Discrete Longitudinal Data. Springer; 2005. [Google Scholar]
- 22.US Census Bureau Population Division . Annual Estimates of the Resident Population by Sex, Race, and Hispanic Origin for the United States: April 1, 2010, to July 1, 2019 (NC-EST2019-SR11H). US Census Bureau; June 2020. [Google Scholar]
- 23.van der Vaart AW. Asymptotic Statistics: Cambridge Series in Statistical and Probabilistic Mathematics. Cambridge University Press; 1998: 443. [Google Scholar]
- 24.Vasan RS, Larson MG, Levy D, Evans JC, Benjamin EJ. Distribution and categorization of echocardiographic measurements in relation to reference limits: the Framingham Heart Study: formulation of a height- and sex-specific classification and its prospective validation. Circulation. 1997;96(6):1863-1873. doi: 10.1161/01.CIR.96.6.1863 [DOI] [PubMed] [Google Scholar]
- 25.Yusuf S, Pitt B, Davis CE, Hood WB Jr, Cohn JN; SOLVD Investigators . Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. N Engl J Med. 1992;327(10):685-691. doi: 10.1056/NEJM199209033271003 [DOI] [PubMed] [Google Scholar]
- 26.Musunuru K, Hershberger RE, Day SM, et al. ; American Heart Association Council on Genomic and Precision Medicine; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular and Stroke Nursing; and Council on Clinical Cardiology . Genetic testing for inherited cardiovascular diseases: a scientific statement from the American Heart Association. Circ Genom Precis Med. 2020;13(4):e000067. doi: 10.1161/HCG.0000000000000067 [DOI] [PubMed] [Google Scholar]
- 27.Vasan RS, Larson MG, Benjamin EJ, Evans JC, Levy D. Left ventricular dilatation and the risk of congestive heart failure in people without myocardial infarction. N Engl J Med. 1997;336(19):1350-1355. doi: 10.1056/NEJM199705083361903 [DOI] [PubMed] [Google Scholar]
- 28.Hershberger RE, Cowan J, Jordan E, Kinnamon DD. The complex and diverse genetic architecture of dilated cardiomyopathy. Circ Res. 2021;128(10):1514-1532. doi: 10.1161/CIRCRESAHA.121.318157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Devereux RB, Bella JN, Palmieri V, et al. ; Hypertension Genetic Epidemiology Network Study Group . Left ventricular systolic dysfunction in a biracial sample of hypertensive adults: the Hypertension Genetic Epidemiology Network (HyperGEN) Study. Hypertension. 2001;38(3):417-423. doi: 10.1161/01.HYP.38.3.417 [DOI] [PubMed] [Google Scholar]
- 30.Husaini BA, Mensah GA, Sawyer D, et al. Race, sex, and age differences in heart failure-related hospitalizations in a southern state: implications for prevention. Circ Heart Fail. 2011;4(2):161-169. doi: 10.1161/CIRCHEARTFAILURE.110.958306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dries DL, Exner DV, Gersh BJ, Cooper HA, Carson PE, Domanski MJ. Racial differences in the outcome of left ventricular dysfunction. N Engl J Med. 1999;340(8):609-616. doi: 10.1056/NEJM199902253400804 [DOI] [PubMed] [Google Scholar]
- 32.Smirnoff M, Wilets I, Ragin DF, et al. A paradigm for understanding trust and mistrust in medical research: the Community VOICES Study. AJOB Empir Bioeth. 2018;9(1):39-47. doi: 10.1080/23294515.2018.1432718 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eAppendix. Statistical Methods
eTable 1. Model Fit for the Marginal Probability of DCM in the Population of First-Degree Relatives of Probands with Specified Characteristics at a Particular US Advanced Heart Failure Program
eTable 2. Differences in Marginally Standardized Familial DCM Prevalences (Standard Definition) between Proband Subpopulations Defined by Self-Reported Ethnicity and Race at a Typical US Advanced Heart Failure Program
eTable 3. Model Fit for the Marginal Probability of DCM or Partial Phenotypes in the Population of First-Degree Relatives of Probands with Specified Characteristics at a Particular US Advanced Heart Failure Program
eTable 4. Differences in Marginally Standardized Familial DCM Prevalences (Expanded Definition) between Proband Subpopulations Defined by Self-Reported Ethnicity and Race at a Typical US Advanced Heart Failure Program
eTable 5. Model Fit for the Age-Specific Cumulative Risk of DCM in a First-Degree Relative of a Patient with DCM at a Particular US Advanced Heart Failure Program
eTable 6. Model Fit for the Age-Specific Cumulative Risk of LVE, LVSD, or DCM in a First-Degree Relative of a Patient with DCM at a Particular US Advanced Heart Failure Program
eTable 7. Vital Status of First-Degree Relatives at Proband Enrollment, by Proband Ethnicity and Race
eFigure 1. Clinical Sites of the DCM Consortium
eFigure 2. DCM Precision Medicine Study Participant Recruitment
eReferences
fam_prev_ests_cov.csv
fdr_risk_ests_cov.csv