

Abstract
Cancer stem-like cells (CSC) induce aggressive tumor phenotypes such as metastasis formation, which is associated with poor prognosis in triple-negative breast cancer (TNBC). Repurposing of FDA-approved drugs that can eradicate the CSC subcompartment in primary tumors may prevent metastatic disease, thus representing an effective strategy to improve the prognosis of TNBC. Here, we investigated spheroid-forming cells in a metastatic TNBC model. This strategy enabled us to specifically study a population of long-lived tumor cells enriched in CSCs, which show stem-like characteristics and induce metastases. To repurpose FDA-approved drugs potentially toxic for CSCs, we focused on pyrvinium pamoate (PP), an anthelmintic drug with documented anticancer activity in preclinical models. PP induced cytotoxic effects in CSCs and prevented metastasis formation. Mechanistically, the cell killing effects of PP were a result of inhibition of lipid anabolism and, more specifically, the impairment of anabolic flux from glucose to cholesterol and fatty acids. CSCs were strongly dependent upon activation of lipid biosynthetic pathways; activation of these pathways exhibited an unfavorable prognostic value in a cohort of breast cancer patients, where it predicted high probability of metastatic dissemination and tumor relapse. Overall, this work describes a new approach to target aggressive CSCs that may substantially improve clinical outcomes for patients with TNBC, who currently lack effective targeted therapeutic options.
Introduction
TNBC is an aggressive breast cancer subtype that does not respond to targeted therapies and has a poor prognosis (1). Patients with TNBC develop resistance and relapse after chemotherapy, ultimately succumbing to metastatic disease (2, 3). The aggressiveness of TNBC is mediated, at least partially, by a subset of rare, long-lived cells characterized by the expression of stemness markers, self-renewal capacity, anchorage-independent growth, and high tumorigenicity (4), which are referred to as cancer stem-like cells (CSC). In TNBC, CSCs make critical contributions to disease metastasis (2, 5, 6), tumor relapse, and resistance to anticancer therapeutics (7, 8); thus, it is anticipated that drugs targeting CSCs may substantially improve the prognosis for patients with TNBC. Unfortunately, the identification of molecular vulnerabilities of CSC has proven difficult due to the limited number of CSCs in the bulk tumor. Three-dimensional (3D) culture models to generate spheroid-like multicellular structures, followed by in vivo serial transplantation, have enabled the study of CSCs in functional assays to determine their capacity for self-renewal and in vivo tumor formation (9, 10), and for drug screening; however, these efforts have not yet identified any treatments to selectively eradicate CSCs in a clinical setting.
The repurposing of FDA-approved drugs for novel therapeutic indications is an attractive drug development approach to reduce the risks and costs associated with delivering a therapeutic option to patients. Because approved drugs have been extensively characterized with regard to pharmacokinetic, pharmacodynamic, and toxicity properties, repurposing of approved drugs also shortens the duration between preclinical proof of concept for a new indication and approval to evaluate the therapy in the clinic (11). Repurposing drugs that can effectively eradicate CSCs and control metastatic disease in TNBC may thus represent an effective strategy to rapidly improve the prognosis for patients diagnosed with this disease. Several reports have identified the ability of pyrvinium pamoate (PP), an FDA-approved anthelminthic drug, to kill cancer cells and, particularly, CSCs (12). PP (CAS: 3546–41-6, Fig. 1A) was initially identified as an inhibitor of multiple mitochondria electron transport chain (ETC) complexes, wherein it targets both complexes I and II (13, 14), resulting in reduced mitochondria fitness. The antitumoral activity of PP in cancer cells or CSCs has previously been linked to the reduction of WNT- and Hedgehog-dependent signaling pathways (12, 15, 16), as well as to the inhibition of mitochondria respiration (17, 18). Moreover, PP can negatively regulate the nutrient-sensor pathway, PI3K/S6, in the context of oncogenic PIK3CA mutations (19). Thus, PP is an attractive candidate for repurposing to target CSCs (20), but the specific metabolic consequences of PP therapy and their implications for the cytotoxic effects of PP against CSCs and metastasis formation have not been investigated.
Figure 1.
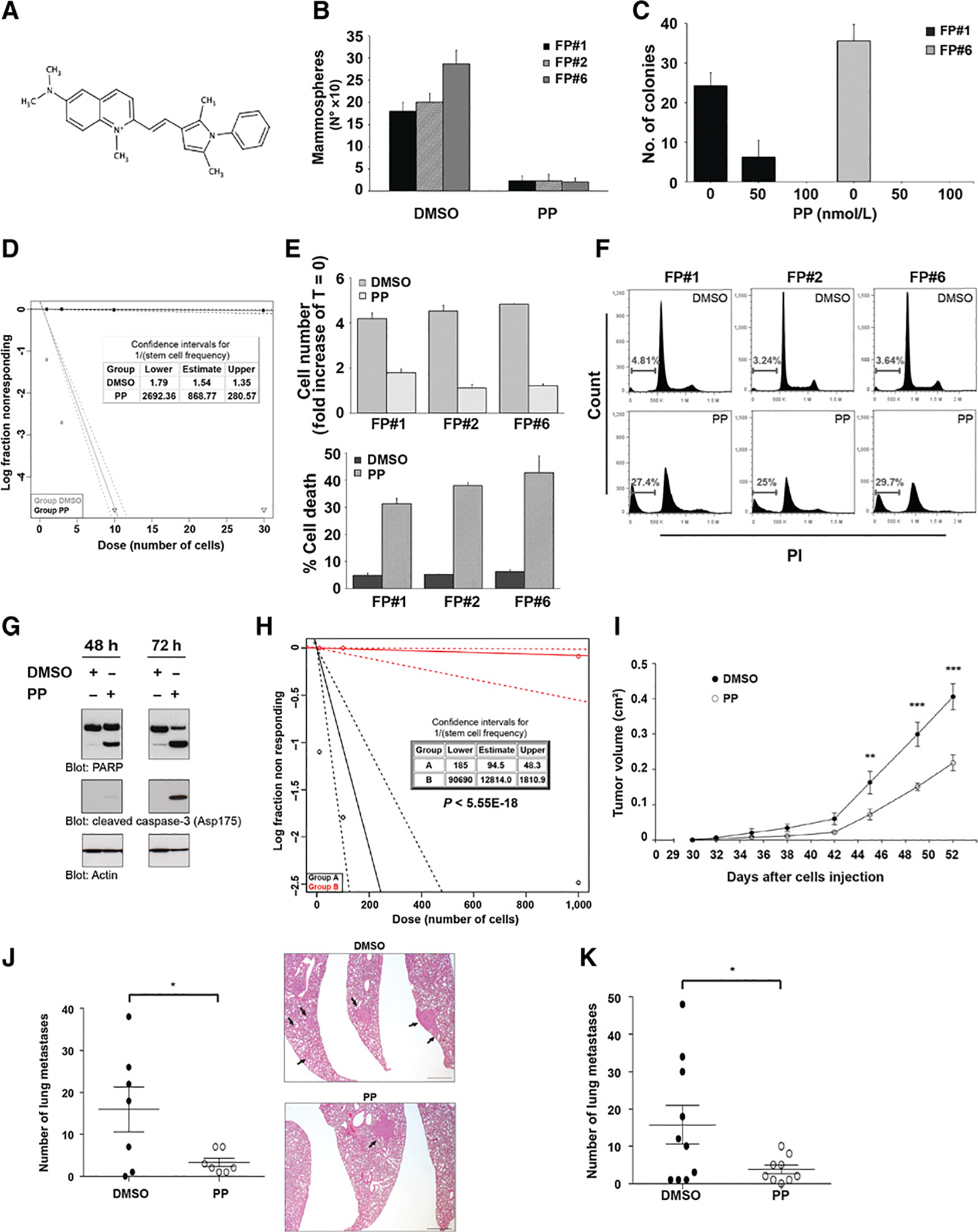
PP treatment inhibits tumor growth and metastasis in vivo. A, Structure of the anthelmintic drug, pyrvinium. B, FP-CSC clones, FP#1, FP#2, and FP#6, were treated with 100 nmol/L PP or DMSO, cultured for 72 hours, and the number of mammospheres was evaluated by counting spheroids. Data represent the mean ± SD of at least three experiments in replicates. Cellular proliferation assays involving CSCs were performed in mammospheres formation medium (see Materials and Methods). C, Histograms show the number of colonies/100 plated cells of FP#1 and FP#6 in a soft agar assay under the indicated treatments. Mean ± SD of three independent experiments is shown. D, LDA of FP#1. Spheres were dissociated into single cells and seeded in the presence of DMSO or PP 100 nmol/L. LDA was done on day 10 using ELDA software. E, FP#1, FP#2, and FP#6 cells were treated with 100 nmol/L PP or DMSO and cultured for 72 hours. Cell number (fold increase of T = 0; top) and percentage of cell death (bottom) were evaluated using the Trypan blue exclusion method. F, Cell death was measured by PI staining and flow cytometry. The percentage of sub-G1 events is shown in representative dot plots. Data represent the mean ± SD of three independent experiments performed in triplicates. G, Immunoblot analysis of total lysates from FP#6 upon treatment for 48 or 72 hours with 100 nmol/L PP or DMSO. Cell extracts were analyzed with PARP and cleaved caspase-3 antibodies. Actin served as the loading control. Data represent three independent experiments. H, FP#1 cells were grown as spheroids and treated with DMSO (group A) or PP 100 nmol/L (group B) for 72 hours, then mammospheres were dissociated and an equal number of viable cells from the two groups was used for in vivo LDA (see Materials and Methods). Table depicts stem cell frequency in FP#1 in group A and B, and the P value of differences in stem cell frequency between groups. LDA was performed by serial dilution of dissociated MFCs cells implanted orthotopically in the mammary FP of NSG mice. I, Tumor growth kinetics of mice injected with FP#1 into the mammary FP and treated with daily intraperitoneal injection of DMSO (black) or PP (white; detailed dosing regimen is provided in Materials and Methods). Data are mean ± SEM. Differences in tumor volume were evaluated using a two-way ANOVA, followed by Bonferroni multiple comparison post hoc tests. **, P < 0.01; ***, P < 0.001. J, The number of lung metastases (left) and representative images of hematoxylin and eosin staining of lung sections (right) from mice treated with DMSO or PP until tumor resection. Data are mean ± SEM. *, P < 0.05. Arrows, lung metastases. Scale bar, 800 μm. K, The number of lung metastases following tumor resection from mice treated intraperitoneally for three weeks with PP as adjuvant therapy. Mean ± SEM are reported. *, P < 0.05.
A large body of evidence has indicated that CSCs of TNBC that harbor amplifications of the MYC oncogene display high oxidative phosphorylation (OXPHOS) activity and dependency on mitochondrial fatty acid oxidation (mFAO; refs. 21, 22). These findings have motivated the use of mitochondria ETC complex I inhibitors as well as the mFAO inhibitor, etomoxir, as potential strategies to impair tumor bioenergetics and inhibit CSC growth (23, 24), but toxicity issues and metabolic plasticity have thus far limited the translational relevance of such therapeutics. In addition, the effects of complex I inhibition by drugs such as metformin can be rescued through other nutrient oxidation pathways (25, 26), and they confer in vitro antitumor effects only at concentrations precluding their safe use in a translational setting. Thus, alternative strategies to target the metabolic dependencies of TNBC CSCs are needed.
The causal role of fatty acids (FA) and cholesterol metabolism in cancer progression and aggressiveness has received attention due to their link to the propagation of CSCs (27). Specifically, FA uptake and metabolism have been reported to modulate the proliferation of aggressive metastasis-initiating cells (21, 28, 29). The desaturation of FAs represents a key metabolic activity in CSCs, supporting both metabolism and signaling pathways relevant to stemness (30, 31). In addition, cancer cells are known to fine-tune lipolytic enzyme activity, to regulate lipid anabolic metabolism, and to optimize growth and metastatic progression (32). The role of cholesterol in breast cancer has also been thoroughly investigated, mainly due to the well-established link between obesity, breast cancer risk, and mortality (33). Notably, breast cancer mouse models fed with a high-fat diet that induced increased plasma cholesterol levels had an increased frequency of tumor formation, and those tumors were characterized by high histologic grade and increased metastasis to the lung (34). However, although elevated plasma cholesterol levels are recognized as a risk factor for breast cancer development and progression to metastatic disease (35, 36), the utility of statin drugs to prevent or control disease remains controversial due to varied reports on whether a statin treatment regimen correlates with improved clinical outcomes (37–40). Cell-intrinsic functions of cholesterol have also been demonstrated to promote cancer (41, 42). Indeed, intracellular cholesterol has recently emerged as a promoter of stem cell proliferation and tumor development (43). Finally, 27-hydroxycholesterol, which is an oxidated metabolite of cholesterol, has been shown to play a role in promoting metastases of breast cancer through its effects on immune cell surveillance (44). Of note, mutation of the tp53 tumor suppressor, which is frequent in TNBC, has been demonstrated to be both necessary and sufficient to disrupt the acinar morphology of breast tissue architecture via SREBP activation (45), while the tumor-suppressive function of wild-type p53 has been shown to suppress the mevalonate pathway responsible for cholesterol biosynthesis (46). Overall, a compelling body of evidence implicates FA and cholesterol metabolism as rational targets for anticancer drugs.
In this work, we demonstrate a potentially new therapeutic intervention for TNBC through drug repurposing. Our data demonstrate that PP limited the survival and the metastatic potential of TNBC CSCs by inhibiting the anabolic metabolism of FAs and cholesterol, both of which are essential to CSC survival. PP impaired anabolic flux from glucose to FA and cholesterol synthesis in CSCs, which directly contributed to its cytotoxic effects. Taken together, our data demonstrate that lipid anabolism is a metabolic vulnerability in TNBC stem-like cells that can be exploited to reduce metastatic potential in this disease that currently lacks effective targeted therapeutic approaches.
Materials and Methods
Cell lines
SUM159, SUM149T, HCC38, and BT-549 TNBC cell lines were obtained from Dr. G. Blandino (“Regina Elena”- National Cancer Institute, Rome, Italy) and grown according to manufacturer’s protocols. Mammary epithelial hTERT-HME (HME) and MCF-10A cell lines were obtained from Prof. A. Bardelli (Candiolo Cancer Institute IRCCS, Torino, Italy) and were cultured as described in Supplementary Methods. All cell lines not directly obtained from ATCC were tested to confirm their identity using an AmpFLSTR Identifier Plus PCR Amplification Kit (Applied Biosystems) with a 3500 Genetic Analyzer (Applied Biosystems) and analyzed by GeneMapper Software v4.1 for genomic matching. All cell lines were regularly tested for Mycoplasma contamination using the Mycoprobe Mycoplasma Detection Kit (R&D Systems). All cells were cultured in Heracell (Thermo Fisher Scientific) humidified incubators at 37°C and 5% CO2.
Mammosphere formation
Adherent cells were replated in ultralow attachment flasks (Corning) and cultured in mammosphere-forming medium (MFM) composed of serum-free DMEM/F12-modified medium containing 20 ng/mL EGF (PeproTech, 100–15), 10 ng/mL bFGF (PeproTech, 100–18B), 50 μg/L insulin (Sigma, 91077C), and supplemented as described in Supplementary Methods. For second- and third-generation spheroids, mammospheres were dissociated to single cells, replated, and grown for 7 days. Mammospheres (>60 μm in size) were counted on day 7.
Gene expression profiling
One-hundred nanograms of total RNA was labelled and hybridized to the Affymetrix GeneChip1.0ST array (Affymetrix) following the manufacturer’s instructions. Data analyses were performed by comparing tool classes of BRB-Array Tools.
Seahorse metabolic flux analysis
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured simultaneously at baseline and following the addition of reagents for indicated times using an XF-extracellular Analyzer (Seahorse Bioscience). FP-CSC were seeded in poly-l-lysine–coated XFp cell culture miniplates at a density of 16,000 cells/well in unbuffered base XF DMEM supplemented with 1 mmol/L pyruvate, 2 mmol/L glutamine, and 10 mmol/L glucose prewarmed to 37°C. Cells were incubated in a CO2-free incubator for 1 hour. All media was adjusted to pH 7.4 on the day of the assay. Data analysis was performed using Wave 2.4 (Seahorse Bioscience). All data were normalized to cell density.
XF Cell Mito stress test
Experimental design followed the protocol provided by Seahorse Bioscience. Following three baseline measurements of OCR and ECAR, cells were metabolically perturbed by the sequential injection of different mitochondrial inhibitors: oligomycin (1 μmol/L), FCCP 1 μmol/L, and a mix of complex I inhibitor rotenone and complex III inhibitor antimycin A (A/R) (0.5 μmol/L working concentration). The decrease in OCR upon injection of oligomycin represents the portion of basal respiration used to drive ATP production, which was expressed as a percentage of baseline value and calculated as 100× [(OCR phase 1 − OCR phase 2)/Basal OCR]. The proton leakage represents residual basal respiration not coupled to ATP production.
Limiting dilution assay
Single-cell suspensions were serially diluted 1:3 from 10 cells to 1 cell in ultra-low adhesion 96-well plates (Corning) under standard growth conditions in the presence of DMSO or indicated drugs to investigate the frequency of sphere formation. After 10 to 20 days, vital colonies were counted. Clonogenic capability was assessed visually under the microscope and the frequency was calculated using the Extreme Limiting Dilution Analysis (ELDA) “limdil” function (http://bioinf.wehi.edu.au/software/elda/index.html).
Cell migration
Cell migration was assessed in 24-well transwell Boyden chambers (Costar Scientific Corporation) following treatment with atorvastatin and cholesterol. Cholesterol pretreated and untreated cells (8 × 104) were seeded in a 6-well plate for 24 hours. Subsequently, cells were suspended in complete growth medium containing DMSO, cholesterol 10 μmol/L, atorvastatin 10 μmol/L, or atorvastatin 10 μmol/L + cholesterol 10 μmol/L (pretreatment or posttreatment) and placed into top chambers. After 24 hours of incubation, migrated cells were stained with Coomassie Brilliant Blue and counted under the microscope.
In vivo experiment in immunodeficient mice
NOD scid gamma (NSG) mice were purchased from Jackson Laboratory and bred in the animal facility of ISS and IRE (Rome, Italy). Animal care and experimental procedures were approved by the Ethics Committee for Animal Experimentation of the institute according to Italian law. Seven-week-old female NSG mice were injected unilaterally into the fourth mammary fat pad with 5 × 104 FP#1-CSC mixed 1:3 with Matrigel (BD Biosciences). Mice were treated intraperitoneally once a day with vehicle (DMSO) or PP after palpable lesions were present (about 30 days after cell injection), using a dose-escalation regimen, starting from 0.3 mg/kg in the first week through 1.2 mg/kg in the fourth week. Tumor growth was monitored biweekly using calipers, and tumor volume was calculated as 0.5 × d12 × d2, where d1 and d2 are the smaller and larger diameters, respectively. Animal health status was monitored daily and body weight was measured three times during the treatment.
For metastasis induction experiments, orthotopic tumors were generated in 7-week-old female NSG mice by unilaterally injecting 5 × 104 FP#1-CSC mixed 1:3 with Matrigel (BD Biosciences) into the fourth mammary fat pad. In one protocol, mice were treated when palpable lesions were present and until the surgical removal of tumors at defined tumor volume (0.4 cm3). In the second protocol, mice were treated following surgical removal of early tumors at defined tumor volume (0.2 cm3). Primary tumors and organs were fixed in 10% neutral buffered formalin, paraffin-embedded, sectioned, and stained with hematoxylin and eosin (BioOptica). To optimize the detection of microscopic metastases and ensure systematic uniform and random sampling, lungs were cut transversally, to the trachea, into 2.0-mm thick parallel slabs with a random position of the first cut in the first 2 mm of the lung, resulting in 5 to 8 slabs per lung. The slabs were then embedded cut surface down and sections were stained with hematoxylin and eosin. Two pathologists independently evaluated slides.
Statistical analysis
Results were expressed as mean ± SD, and the number of individual replicates is detailed in figure legends. Statistical significance of the mean values was established by two-tailed distribution Student t test. Where appropriate, the use of different statistical approaches has been specifically described in the figure legend.
Supplementary methods
This section includes additional cellular and molecular methods, plus computational and metabolomics methods.
Results
PP induces apoptosis and inhibits metastatic disease in a TNBC CSC model
We derived mammosphere-forming cells (MFC) from a metastatic TNBC model to study specifically the effects of PP on the subset of long-lived tumor cells characterized by stem-like properties (CSCs). Spheroid-forming cells were derived from SUM159, a TNBC primary tumor cell line with metastatic potential. SUM159 harbors a MYC amplification and mutations in PIK3CA and TP53, which are commonly cooccuring mutations in metastatic breast tumors (47). The 3D-spheroid-based culturing of MFCs followed by in vivo tumors promotes the expansion of undifferentiated cancer stem-like and progenitor cells. Thus, to generate a tumor model enriched in CSCs derived from primary tumor cells and able to generate metastases, SUM159-derived mammospheres expressing a Luc/EFGP reporter were implanted into the mammary fat pads of mice. Following tumor growth and mastectomy, distant metastases appeared. EGFP-positive tumor cells were isolated from fat pad (FP) orthotopic tumors and from the corresponding metastatic lesions (met), and they were further propagated in vitro as mammospheres, based on the intrinsic self-renewal ability of MFCs (Supplementary Fig. S1A–S1C; Materials and Methods).
Phenotypic and functional assays were performed to characterize the cellular heterogeneity of CSCs derived from FP mammosphere-forming cell (FP-MFC) tumors. Flow cytometry indicated that almost 90% of cells in spheroids generated from FP-MFCs were characterized by expression of CD44high/CD24low markers, with no detection of more differentiated CD44high/CD24high-expressing cells. Notably, when grown as a monolayer (SUM159–2D), SUM159 cells had a high percentage of CD44high/CD24low, consistent with an extremely high sphere-forming capacity. However, almost 30% of SUM159–2D cells in culture were CD44high/CD24high (Supplementary Fig. S1D). Propagation of SUM159 cells as spheroids for three generations (SUM159–3D) increased the percentage of CD44high/CD24low cells and reduced the percentage of CD44high/CD24high cells (Supplementary Fig. S1D). To better characterize and quantify the effective stem-like cell population of FP-MFCs, we examined ALDH activity, a relevant stemness marker for breast CSCs. Almost 75% of FP-MFCs showed high ALDH enzymatic activity, which was markedly higher compared with ALDH levels in either SUM159–2D (9%) or SUM159–3D (20%) cultures (Supplementary Fig. S1E). Moreover, evaluation of FP-MFCs in a limiting dilution assay (LDA) demonstrated that almost 70% of FP-MFCs possessed clonogenic capacity in vitro (Supplementary Fig. S1F). Most importantly, FP-MFCs were enriched in tumor-forming cells 180-fold compared with SUM159–2D cells, as demonstrated by in vivo LDA (Supplementary Fig. S1G). The percentage of CD44high/CD24low-expressing cells in metastasis-derived MFC (met-MFC) clones was similar to what we observed in FP-MFCs (Supplementary Fig. S2). Mutational analysis of the most frequently mutated or amplified cancer genes demonstrated that the MFC clones did not genetically diverge compared with the reported genotype of the parental SUM159 cell line (Supplementary Tables S1 and S2; ref. 47), demonstrating that our in vitro/in vivo protocol to enrich for CSCs did not result in the emergence of additional genetic mutations. Taken together, our data indicate that FP-MFCs isolated from this primary tumor model were highly enriched in cells possessing molecular and functional phenotypes consistent with CSCs and, hereafter, this mammosphere model will be referenced as FP-CSCs.
We next investigated the effect of PP (Fig. 1A) on the viability of FP-CSCs. Exposure to 100 nmol/L PP suppressed mammosphere formation and the ability of CSCs to grow in soft agar (Fig. 1B and C), and it completely suppressed the clonogenic growth of FP-CSCs (Fig. 1D). The growth inhibition following PP treatment was associated with a strong cytotoxic effect (Fig. 1E and F). Specifically, activation of molecular mediators of apoptosis, such as caspase-3 and PARP cleavage, demonstrated that PP induced cell death through the activation of apoptosis (Fig. 1G). Notably, PP induced only a cytostatic effect on the nontransformed mammary epithelial cell lines HME and MCF10A, with no overt signs of cytotoxicity, even when applied at a micromolar concentration (Supplementary Fig. S3A and S3B). Cellular toxicity was linked to pyrvinium, as we demonstrated that the pamoate salt alone had no effect on cell viability or proliferation (Supplementary Fig. S4). Most importantly, in vivo LDA demonstrated that treating FP-CSCs with PP reduced the viability of tumor-initiating cells by more than 100-fold, confirming effective killing of CSCs by PP (Fig. 1H). PP also abrogated the growth of third-generation mammospheres generated from a panel of TNBC cells derived from primary tumors in soft agar (Supplementary Fig. S5), indicating that the antiproliferative effects of PP are not restricted to SUM159 cells.
To investigate the effects of PP on tumor growth and metastasis in vivo, we generated tumors via orthotropic injection of FP-CSCs into the mammary FP. Tumor and metastasis formation was evaluated in a neoadjuvant-like setting wherein we initiated daily dosing of PP or vehicle control by intraperitoneal injection 30 days after tumor implantation. The dose of PP was increased each week from 0.3 mg/kg up to 1.2 mg/kg during the fourth week of treatment. PP significantly inhibited tumor growth compared with vehicle treatment (Fig. 1I) and was well tolerated at these doses, as assessed by body weight, behavioral, and histologic analyses of tissues from treated mice (Supplementary Fig. S6A–S6C). We also evaluated the effect of this dosing regimen on disease dissemination by dosing mice as described with PP until tumors reached a volume of 0.4 mm3 and were surgically removed by mastectomy. Treatment with PP significantly reduced the number of lung metastases, a prime metastatic site in this model, compared with vehicle-treated mice (median of 2 and 18 metastases in PP- vs. vehicle-treated mice, respectively), indicating that PP has the potential to robustly reduce metastatic spread of TNBC (Fig. 1J). To evaluate the potential of PP to inhibit metastasis in an adjuvant-like therapeutic setting, we treated mice with PP following early surgical removal of the tumor (0.2 mm3). PP significantly reduced the number of lung metastases (Fig. 1K), providing additional support for the ability of PP to work as an antimetastatic agent in a preclinical model of TNBC CSC-derived metastases.
In addition, we investigated the effects of PP in combination with standard-of-care chemotherapeutics prescribed for TNBC. The cytotoxicity PP dosed concurrently with docetaxel or paclitaxel was evaluated using the combination index (CI) method (Supplementary Fig. S7A and S7B; ref. 48). For all concentrations tested (ranging from 0.25× to 4× of the IC50), combination therapy yielded moderate synergistic effects on FP-CSC proliferation compared with single-agent therapy.
PP inhibits multiple ETC complexes and induces a metabolic crisis
Next, we investigated the mechanisms underlying PP cytotoxicity in three different FP-CSC clones. Because PP inhibits the activity of mitochondrial ETC complexes, and based on the strong dependency of TNBC CSCs on OXPHOS, we first evaluated the effects of PP on mitochondrial respiration. FP-CSCs were grown as described (see Materials and Methods) in the presence or absence of 100 nmol/L PP for 12 hours and analyzed using a Seahorse XF analyzer. In cells treated with PP, basal mitochondria OCR was completely inhibited even at oligomycin-insensitive levels, and ECAR was increased, indicating upregulated glycolysis (Fig. 2A). Moreover, PP limited more than 80% of stress-induced mitochondrial OCR (Fig. 2B–D), thus confirming that PP targets mitochondria activity in CSCs. A time-course analysis of OCR and ECAR in FP-CSCs treated with PP or DMSO control demonstrated that PP directly blocks mitochondrial OCR, ruling out the possibility of indirect, posttranscriptional mechanisms (Fig. 2E). These effects were specific to pyrvinium, as we confirmed that the pamoate salt alone did not exert any effect on OCR or ECAR measurements (Supplementary Fig. S8). To confirm the mitochondrial targets of PP, we next assayed the effects of PP on OCR in FP-CSCs before and after injection of the selective mitochondrial substrates of OXPHOS, pyruvate, and succinate. PP directly inhibited both pyruvate- and succinate-dependent OCR, indicating that PP completely impaired function of both ETC complexes I and II (Fig. 2F and G). We also demonstrated that PP completely inhibited the direct oxidation of palmitoyl-carnitine (PC), a substrate for mFAO (Fig. 2H). Furthermore, OCR measurements of FP-CSCs in the presence of antimycin A before and after injection with TMPD, a synthetic substrate that provides electrons for cytochrome C substrate oxidation, revealed that PP also blocked cytochrome C–linked complex IV activity (Fig. 2I). Taken together, our data confirm that PP inhibits multiple ETC complexes and, accordingly, we observed that treating CSCs with PP for 12 hours reduced total cellular ATP content in FP-CSCs (Fig. 2J).
Figure 2.
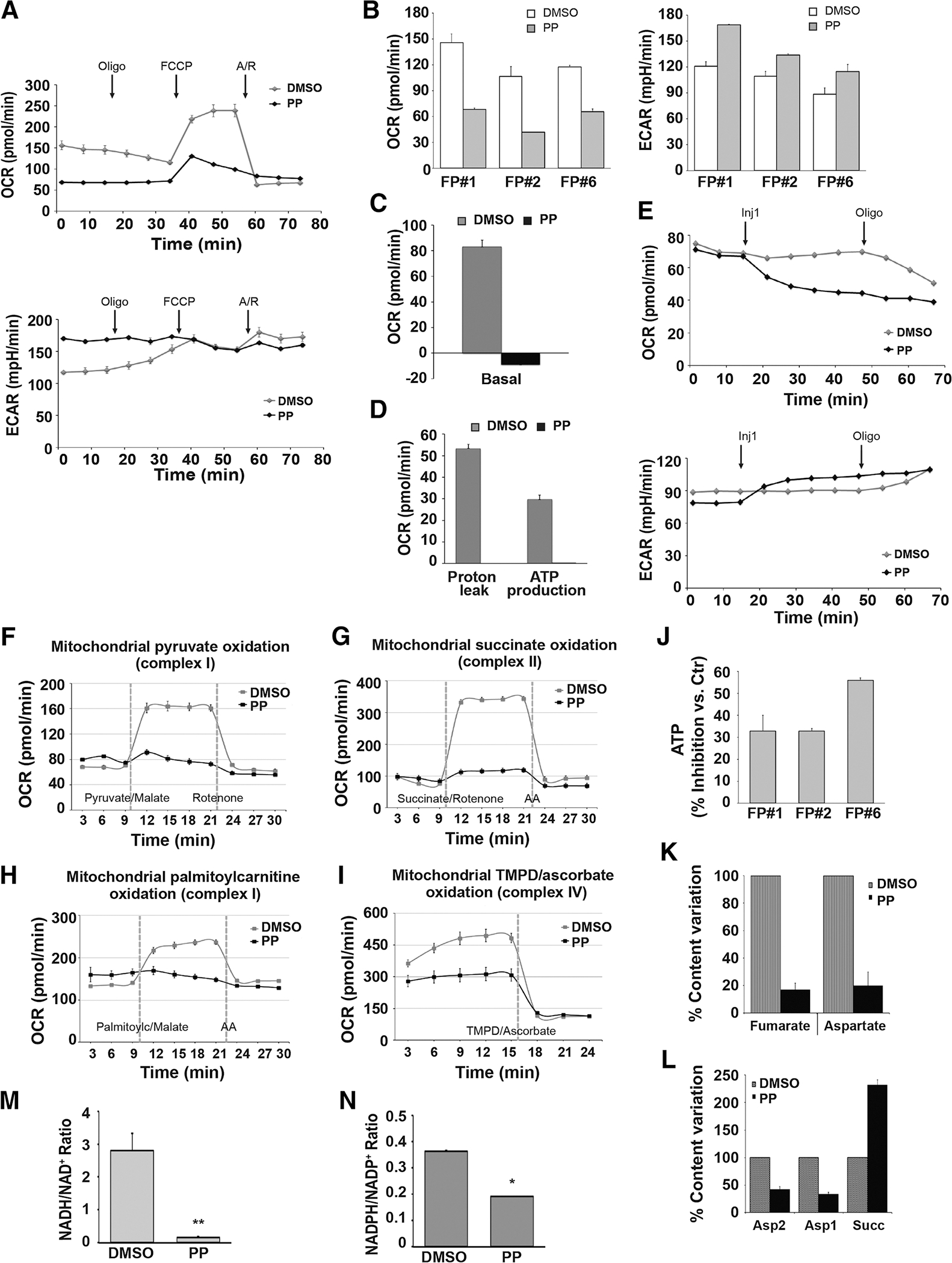
PP acts as an inhibitor of multiple ETC complexes and generates a metabolic crisis. A–D, Cellular respiration and glycolysis of FP#1, FP#2, and FP#6 CSC clones treated with 100 nmol/L PP or DMSO for 12 hours and analyzed by mitochondrial stress test. OCR and ECAR were measured before and after sequential injections of 1 μmol/L oligomycin, 1 μmol/L FCCP, and combination of antimycin A/rotenone (A/R), 0.5 μmol/L each. A, Representative plots for measurements in FP#1 clones are shown. B, Bar histograms show basal respiration and ECAR of PP- and DMSO-treated cells. Quantitative analysis demonstrates differences in basal response (measurements 1–3). Proton leak (C) and ATP production (D). All experiments were performed at least twice, and each data point represents the mean ± SD, n = 3. Representative graphs (C and D), relative to FP#1 CSCs, are shown. E, OCR (top) and ECAR (bottom) were measured in in FP#1 by a mitochondrial stress test before and after sequential injections of 400 nmol/LPP or DMSO (Inj1 = measurements 4–8) and oligomycin as indicated (oligomycin = measurement 9–11). F–I, FP#1 CSCs treated with 100 nmol/L PP or DMSO for 12 hours were permeabilized with the XF Plasma Membrane Permeabilizer. Specific substrate oxidation was examined by measuring OCR changes. Each data point represents the mean ± SD, n = 3. Representative graphs of at least two experiments are shown. Data analysis was performed using Wave 2.4 (Seahorse Bioscience). F, Oxidation of complex I-linked substrates. OCR was measured before and after sequential injections of pyruvate with malate (measurements 4–7) and rotenone. G, To measure complex II activity, OCR was measured before and after sequential injections of succinate with rotenone (measurements 4–7) and antimycin A. H, OCR was measured prior to and after sequential injections of palmitoyilcarnitine (measurements 4–7) and antimycin A. I, Cytochrome C oxidase activity: OCR was measured after injection of TMPD with ascorbate (measurements 5–7; Materials and Methods). J, FP#1, FP#2, and FP#6 cells cultured with 100 nmol/L PP or DMSO for 12 hours and ATP levels were measured. The percent inhibition of ATP content compared with control cells was calculated as the mean ± SD of three independent experiments, performed using multiple replicates. K and L, FP#1 cells treated with DMSO or 100 nmol/L PP were cultured for 12 hours and analyzed by nuclear magnetic resonance spectroscopy (see Materials and Methods). K, Fumarate and aspartate signal intensities in DMSO- and PP-treated cell spectra (as a percentage of/normalized to DMSO-treated sample value). L, Aspartate and succinate signal intensities from DMSO and PP-treated PCA extracts spectra (as a percentage of/normalized to DMSO-treated sample value). Data are the mean ± SD of three different experiments. FP#1 cells treated with DMSO or 100 nmol/L PP were cultured for 24 hours and the NADH/NAD+ ratio (M) and the NADPH/NADP+ ratio (N) were measured. *, P < 0.05; **, P < 0.01.
Because PP inhibits ETC complex II, implying the down-regulation of succinate dehydrogenase activity, we hypothesized that PP would also reduce fumarate. Indeed, nuclear magnetic resonance analysis revealed that FP-CSCs incubated in the presence of 100 nmol/L PP produced approximately 80% less fumarate compared to DMSO-treated cells. We also observed a strong PP-induced decrease in aspartate levels (Fig. 2K and L), which was expected due to the demonstrated ETC complex I inhibition by PP (26, 49). Importantly, PP affected the overall reducing power homeostasis, thus reducing the NADH/NAD+ ratio almost 15-fold (Fig. 2M) and the NADPH/NADP+ ratio by 2-fold (Fig. 2N).
Our data confirmed extensive dysregulation of mitochondria metabolism induced by the inhibition of ETC complexes’ activity and of the oxidation of major substrates; thus, we posited that the effects of PP on oxidative metabolism may result in the cytotoxic effects we observed in PP-treated FP-CSCs. To test this, we repeated in vitro experiments to measure growth of FP-CSCs and nontransformed mammary cells, as described above, in conditions where the oxidation of major substrates such as FAs, pyruvate, or glutamine were limiting, as well as in the presence of ETC complex I inhibitors, rotenone and metformin. None of these mechanisms of mitochondria inhibition resulted in any significant effect on the proliferation or survival of FP-CSCs (Supplementary Fig. S9A). Also, the inhibition of mitochondria ETC complex I by rotenone or metformin treatments had no effects on the cellular proliferation and survival of FP-CSCs, while these drugs did impair the growth and survival of nontransformed cell models (Supplementary Fig. S9B and S9C). Thus, inhibition of OXPHOS of mitochondrial substrates by PP is not the molecular mechanism underlying the growth and survival phenotype observed in FP-CSCs, suggesting that PP might target an as-yet unidentified metabolic vulnerability of PF-CSCs.
GSSM and metabolomic analyses demonstrate that FA and cholesterol anabolism are highly active pathways in FP-CSCs
The prediction of the major activated metabolic pathways in FP-CSCs would represent an advantage in reasoning the metabolic impact of PP. This prediction can be achieved through genome-scale metabolic models (GSMM). GSMMs decipher a cell’s metabolic status within a given context to discover active metabolic pathways based on transcriptomic data (50). We generated GSMMs from FP-CSCs and from corresponding met-CSCs grown in similar conditions to help rule out any metabolic phenotypes specifically linked to CSC model generation and subcloning (Supplementary Fig. S1A–S1C; Materials and Methods). The predictive value of these GSMM-based studies was supported by a principal component analysis for gene expression profiles, which revealed significant differences between FP- versus met-CSCs (Supplementary Fig. S10A).
To identify metabolic pathways specifically active and relevant to FP-CSCs, we applied the metabolic transformation algorithm (MTA; ref. 51), which we employed here to predict metabolic reactions that are transcriptionally active in the FP-CSCs (the “source state”) and no longer active in a met-CSCs (the “target state”). The inputs to MTA were transcriptomic measurements of the two states, and its output was a ranked list of metabolic reactions specifically active in the source state (FP-CSCs; Materials and Methods). MTA assigned the highest scores to enzymes involved in FA metabolism, particularly mFAO, FA synthase (FAS), FA activation, FA elongation, and cholesterol biosynthesis, indicating that these pathways were predicted to be transcriptionally active in FP-CSC metabolism (Fig. 3A). To evaluate this result experimentally, we conducted 13C6-glucose tracer metabolomics, which confirmed that FP-CSCs rely on glucose to fuel cholesterol, palmitate, and stearate synthesis, consistent with active lipid anabolism pathways that were uncovered by GSMM analysis. Moreover, in FP-CSCs, we detected M+2 and M+5 enrichment in citrate and M+3 enrichment in malate, aspartate, and fumarate, suggesting that glucose was entering the TCA cycle through pyruvate carboxylase (PC) and pyruvate dehydrogenase (PDH; Fig. 3B). Activation of the cholesterol biosynthetic pathway in FP-CSCs was mediated by the sterol regulatory element-binding protein (SREBP), as demonstrated by qRT-PCR analysis of SREBP target genes following treatment with betulin, an SREBP-specific inhibitor (Supplementary Fig. S10B).
Figure 3.
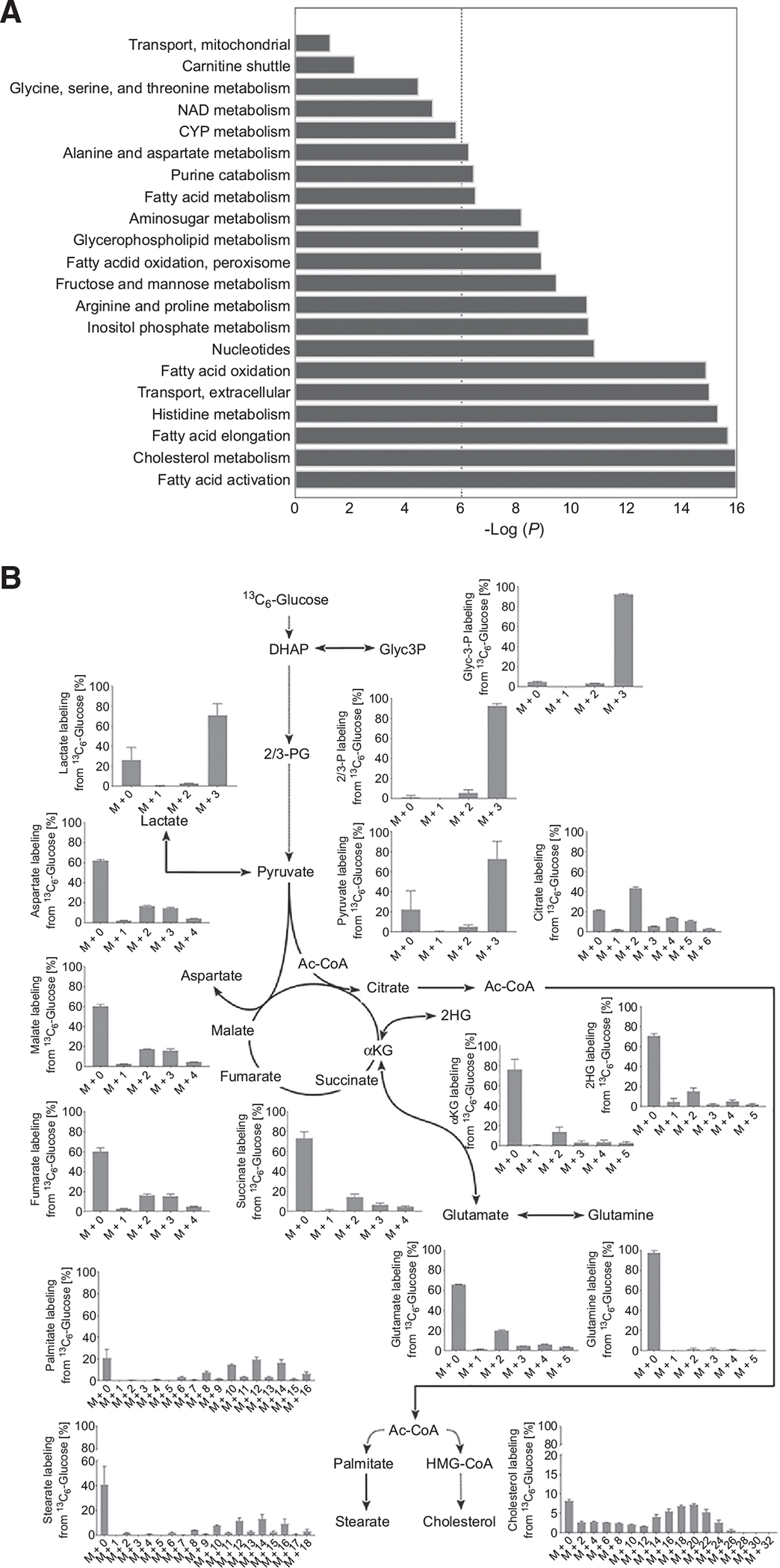
GSSM and metabolomics analyses demonstrate that FAs and cholesterol anabolism are major active pathways in FP-CSCs. A, Bar plots showing hyper geometric P values for pathway enrichment of the reactions robustly predicted by MTA to be elevated in FP-CSCs. The dashed line represents a significance threshold of 0.05 (corrected for multiple hypotheses testing). B, Metabolic map representing the distribution of carbons derived from 13C6 glucose in metabolites of glycolysis, TCA metabolism, cholesterol, and FAs synthesis. FP#1 CSCs were incubated with 13C6-glucose for 96 hours. Each graph represents the mass distribution vector (MDV) from 13C6 glucose; the error bars show the mean ± SD from three independent samples. Dashed lines indicate multiple enzymatic steps. DHAP, dihydroxyacetone phosphate; Glyc3P, glyceraldehyde 3-phosphate; 2/3PG, 2/3-phosphoglycerate; ac-CoA, acetyl-CoA; αKG, α-ketoglutarate; HMG-CoA, hydroxymethylglutaryl-CoA.
PP significantly inhibits lipid anabolism in CSCs
Because we identified activation of FAs and cholesterol anabolism, we monitored the effect of PP treatment on lipid metabolism. Quantitative GC-MS analysis of FP-CSCs grown for 24 hours in the presence of 100 nmol/L PP or DMSO control demonstrated that PP caused a 50% decrease in cholesterol levels (Fig. 4A), which corresponded with increases in the concentrations of intermediate metabolites in the cholesterol biosynthetic pathway (Fig. 4B–E; i.e., S1: cholesta-8,14-dien-3β-ol, S2: 7-dehydrocholesterol, S3: cholesta-8,14,24-trien-3β-ol, and S4: 7-dehydrodesmosterol; Materials and Methods; Supplementary Fig. S11A). This effect on cholesterol levels was not the result of inhibition of SREBP activity, as revealed by analysis of target gene expression. Rather, key SREBP target genes involved in cholesterol biosynthesis were upregulated following PP treatment (Supplementary Fig. S11B). In addition to reduced cholesterol levels, we measured a robust drop in cholesteryl esters (CE) levels (Fig. 4F) in PP-treated FP-CSCs. PP treatment also downregulated levels of most of the analyzed free FAs (FFA), particularly myristic, palmitic, and oleic acids (Fig. 4G).
Figure 4.
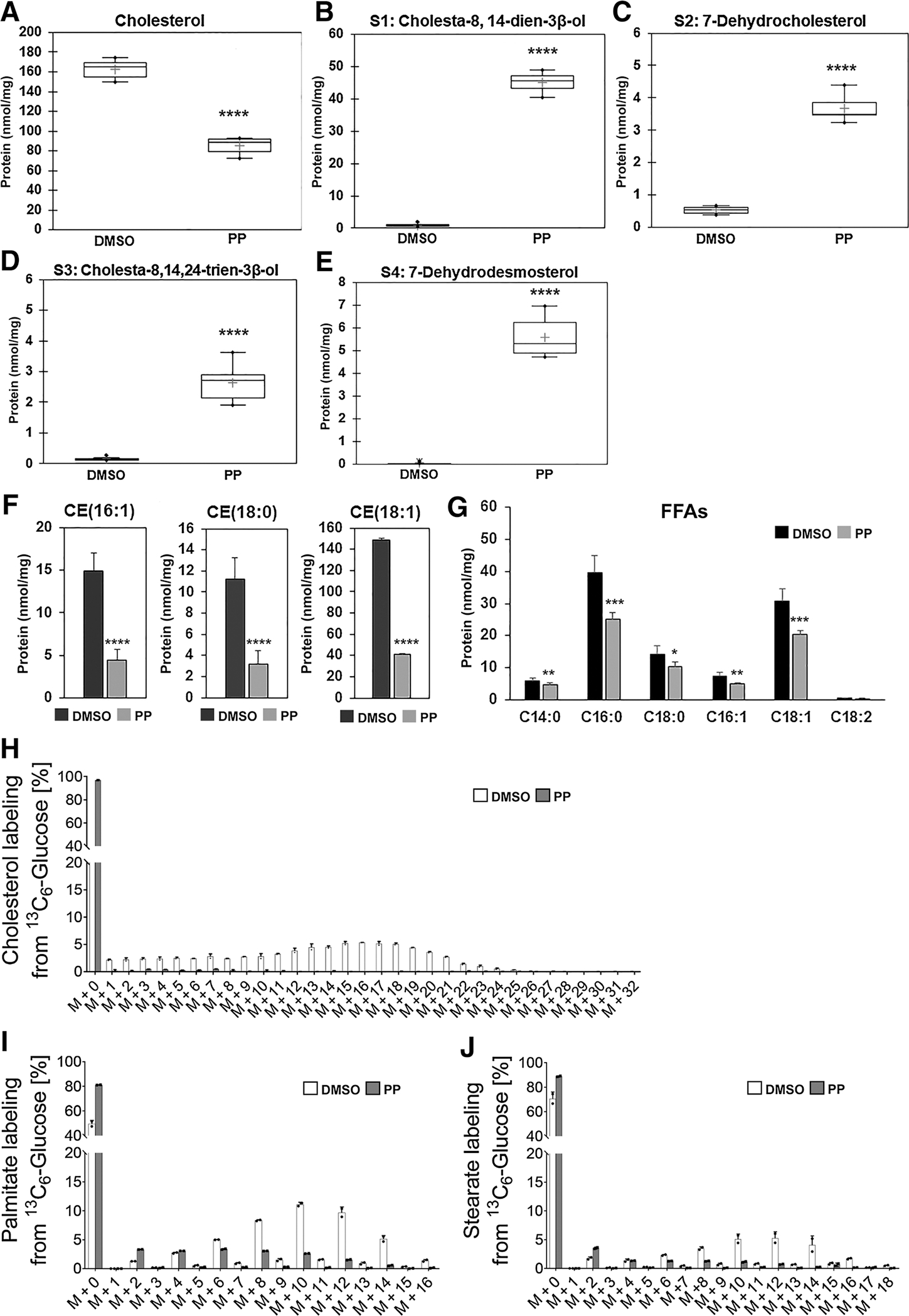
PP suppresses the anabolic metabolism of FAs and cholesterol in CSCs. Quantitative assessments of cholesterol (A), intermediates of cholesterol biosynthesis (B–E), esters of cholesterol (F), and free FAs (G) in FP#1 cells treated with 100 nmol/L PP or DMSO for 24 hours. Results were obtained from six independent experiments and are expressed as nmol/mg protein. Values are expressed as the mean ± SD. In the box plots (A–E), means are marked with the + symbol. *, P ≤ 0.05; **, P ≤ 0.001; ***, P ≤ 0.0005; ****, P ≤ 0,0001. H–J, 13C enrichment of palmitate, stearate, and cholesterol from 13C6 glucose in control (DMSO, white bars) versus PP-treated (gray bars) FP#1 cells incubated with 13C6-glucose for 48 hours. Error bars show the mean ± SD from three independent biological samples. *, P < 0.05.
To evaluate the effect of PP on lipid synthesis, we performed a 13C6-glucose tracer analysis in FP-CSCs incubated in 100 nmol/L PP for 48 hours. PP treatment significantly decreased the transfer of glucose-derived carbons to cholesterol, palmitate, and stearate (Fig. 4H–J), which suggests that PP inhibits de novo synthesis of these FAs, as well as de novo cholesterol synthesis.
To further determine the impact of PP treatment on overall lipid homeostasis, we extracted neutral lipids from PP- or DMSO- treated cells and subjected the samples to RPLC-MS in positive-ion mode (+ESI). The analysis revealed several species that were significantly increased (244 species) or decreased (77 species) by PP treatment (P ≤ 0,05; fold change, FC ≥1.5). Among the identified species, levels of triglycerides (TG), sterols, and phospholipids were the most markedly regulated in PP-treated cells (Supplementary Fig. S12A and S12B), with a significant decrease in total TGs levels (Supplementary Fig. S12C).
Overall, our results are consistent PP inducing a significant downregulation of lipid homeostasis and impairing the anabolic flux of glucose toward FFA and cholesterol biosynthesis in TNBC FP-CSCs.
PP exerts cytotoxic effects on FP-CSCs through inhibition of cholesterol biosynthesis
To evaluate whether there may be a mechanistic link between the observed reduction in lipid anabolism and the cytotoxic effect of PP on TNBC FP-CSCs, we focused on cholesterol biosynthesis, and we performed a set of cholesterol add-back experiments. FP-CSCs were grown for 72 hours in medium containing DMSO, 100 nmol/L PP, or 100 nmol/L PP plus 10 μmol/L cholesterol, and cultures were assayed by trypan blue exclusion to determine cell proliferation and death. Addition of cholesterol in the presence of PP effectively prevented more than 50% of PP-induced cell death, while moderately rescuing the cytostatic effects of PP (Fig. 5A), likely due to the persistent OXPHOS inhibition. We next performed quantitative GC-MS analysis of lipids, which demonstrated that cholesterol addition was able to restore intracellular cholesterol levels in PP-treated cells to levels comparable with DMSO-treated controls (Fig. 5B). Importantly, levels of CEs were also restored in cholesterol-supplemented PP cultures (Fig. 5C). On the basis of these data, inhibition of cholesterol synthesis and CEs contributes to PP-induced cell death.
Figure 5.
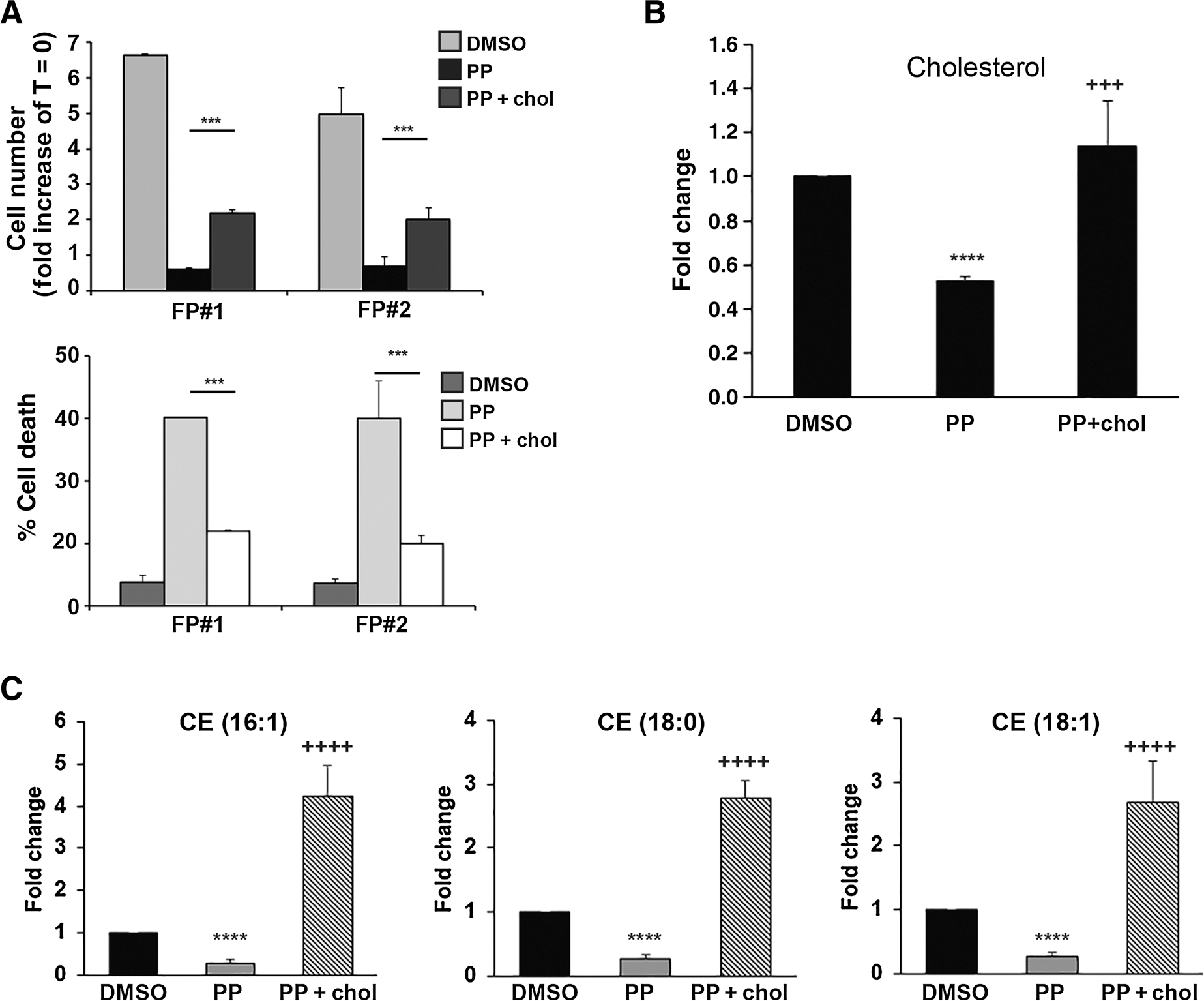
The cytotoxic activity of PP on FP-CSCs is dependent on the inhibition of cholesterol biosynthesis. A, FP#1 and FP#2 CSCs were treated with DMSO, 100 nmol/L PP, or PP in combination with 10 μmol/L cholesterol (PP + chol) for 72 hours. The cell number (fold increase of T = 0; top) and the percentage of cell death (bottom) were evaluated by the Trypan blue exclusion method. B and C, Quantitative assessments of cholesterol (B) and esters of cholesterol (C) in FP#1 cells treated with PP alone or PP in combination with cholesterol. Results were obtained from six independent experiments and are expressed as fold change over DMSO. Values are expressed as means ± SD. ****, P ≤ 0,0001: PP vs. DMSO; +++, P ≤ 0,001; ++++, P ≤ 0,0001: PP+chol vs. PP.
The cytotoxic activity of PP on FP-CSCs is not primarily dependent on inhibition of the WNT/β-catenin pathway
The activity of PP against cancer cells has been linked to inhibition of the WNT/β-catenin pathway, based on observations of inhibition of β-catenin–activated target gene expression (52–54). To elucidate the role of WNT/β-catenin inhibition to kill PP-treated FP-CSCs, we measured the change in β-catenin target-gene expression upon PP treatment. As a control, we used ICRT3, a well-established inhibitor of β-catenin that interferes with β-catenin binding with the TCF coactivator, so that we may monitor the effects of specific inhibition of nuclear β-catenin. Treatment of FP-CSCs with 100 nmol/L PP resulted in no or mild effects on expression of β-catenin target genes (average inhibition: 20%–30%). Treatment of FP-CSCs with a low dose of ICRT3 (25 μmol/L) confirmed that the expression of these genes was under the control of β-catenin, and it mirrored the effect of PP. A higher dose of ICRT3 (75 μmol/L) showed a stronger inhibitory effect on target genes expression (average inhibition: 40%–80%; Supplementary Fig. S13A). However, treating cells with a low dose of ICRT3 had no cytotoxic effects, suggesting that the moderate downregulation of β-catenin activity in FP-CSC, per se, was not sufficient to induce cytotoxicity. In contrast, PP, although showing comparable or a lesser degree of β-catenin inhibition, had a remarkably cytotoxic effect (Supplementary Fig. S13B). Increasing the concentration of ICRT3 up to 75 μmol/L resulted in strong cytotoxic activity but, importantly, this was not rescued by cholesterol supplementation (Supplementary Fig. S13C), supporting the conclusion that β-catenin inhibition–induced cell death through a different mechanism compared with PP.
Cholesterol biosynthesis is essential for FP-CSC survival and migration
To strengthen the functional relevance of the cholesterol biosynthetic pathway as a vulnerability in FP-CSCs, we tested whether directly targeting cholesterol levels might be sufficient to kill CSCs. Because CSCs are grown in medium lacking serum and exogenous cholesterol, de novo biosynthesis is the only source of cholesterol in these models, and pharmacologic inhibition of cholesterol synthesis by statins, which inhibit an early step in cholesterol biosynthesis, can directly reduce intracellular cholesterol levels. Atorvastatin 10 μmol/L treatment for 72 hours strongly impaired FP-CSC proliferation and survival (Fig. 6A and B). Notably, cholesterol addition partially rescued atorvastatin effects, confirming that cytotoxicity induced by atorvastatin depends on modulation of cholesterol levels below an essential threshold (Fig. 6A). LDA demonstrated that atorvastatin completely suppressed the clonogenic activity of FP-CSCs (Fig. 6C). Gene expression analysis of genes involved in cholesterol biosynthesis pathways identified in MTA studies showed that the SQLE and MVD genes were heavily up-regulated in FP-CSCs (Supplementary Fig. S14A). Thus, we evaluated the sensitivity of FP-CSCs to terbinafine or 6-fluoromevalonate, inhibitors of SQLE and MVD, respectively. As anticipated, survival assays demonstrated that these inhibitors robustly induced cell death, which could be prevented by adding cholesterol (Fig. 6D and E). Atorvastatin treatment for 24 hours, while not affecting cell viability, significantly abrogated the migratory capacity of FP-CSCs, and this effect was again completely recovered by pretreating cells with extracellular cholesterol (Fig. 6F). Thus, both the survival and the migration of FP-CSCs require sufficient intracellular cholesterol.
Figure 6.
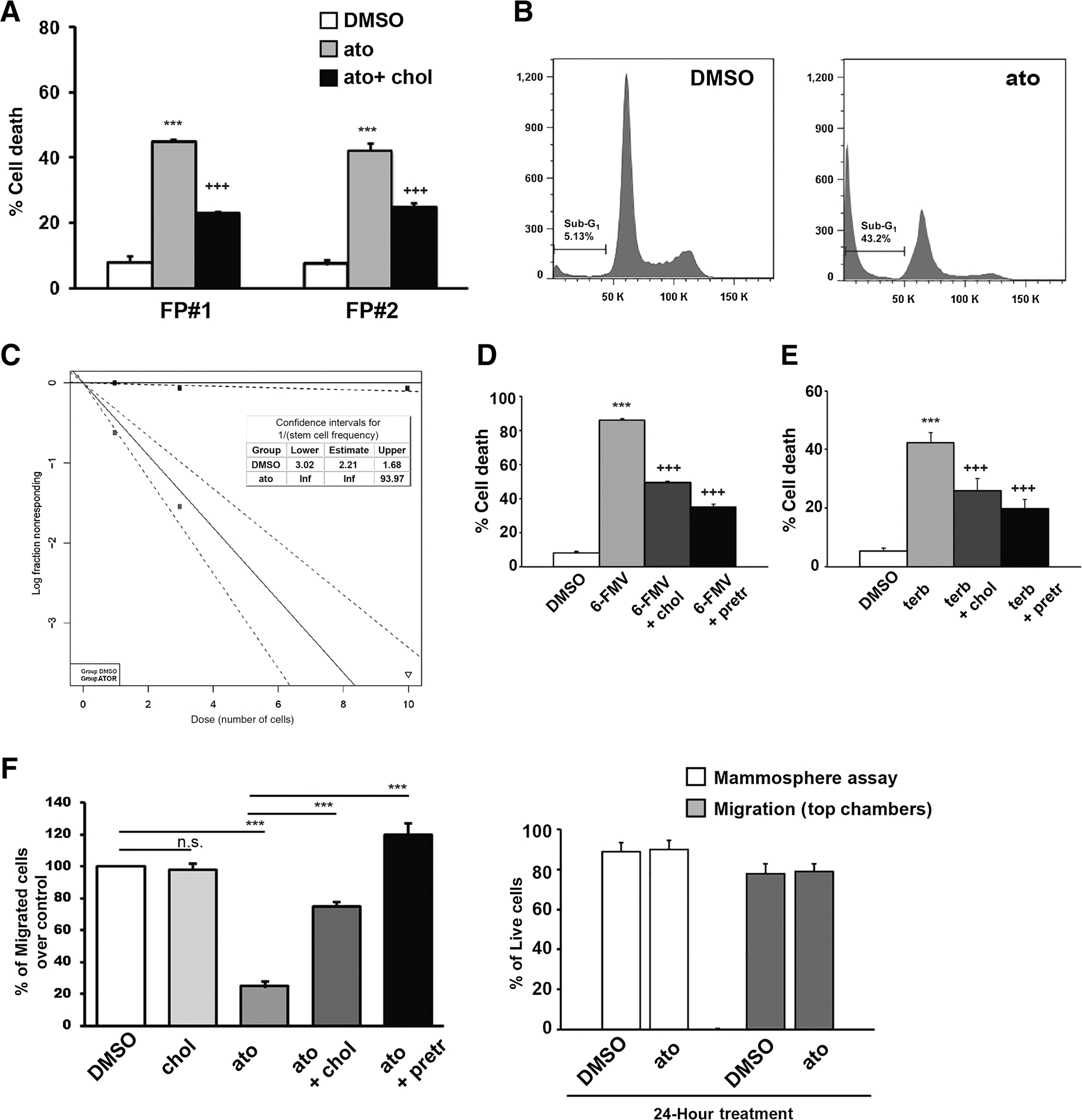
Cholesterol is essential to FP-CSC survival and migration. A, FP#1 and FP#2 CSCs were treated with DMSO, 10 μmol/L atorvastatin (ato), or atorvastatin combined with 10 μmol/L cholesterol and cultured for 72 hours. The percentage of cell death was evaluated by the trypan blue exclusion method. B, Cell death in FP#1 CSCs was also measured by PI staining and flow cytometry. The percentage of SubG1 events is shown in a representative dot plot of three independent experiments performed in triplicate. C, FP#1 spheres were dissociated into single cells and seeded in the presence of DMSO or 10 μmol/L atorvastatin and analyzed by LDA. Log-fraction plot of dilution model fitted to the data was generated using ELDA software. The slope of the line is the log-active cell fraction. The dotted lines indicate 95% confidence interval. The data value with zero negative response at corresponding dose is represented by a down-pointing triangle. Table showing the stem cell frequency in cells treated with DMSO (gray line) or atorvastatin (ato; black line). D, FP#1 CSCs were treated with DMSO, 50 μmol/L 6-Fluoromevalonate (6-FMV), or combination of 6-FMV plus 10 μmol/L cholesterol (+chol) or pretreated for 12 hours with 10 μmol/L cholesterol (+pretr) before adding 6-FMV and cultured for 72 hours. The percentage of cell death was evaluated by Trypan blue exclusion method. E, FP#1 CSCs were treated with DMSO or 60 μmol/L terbinafine (terb) or terb plus 10 μmol/L cholesterol (+chol) or pretreated for 12 hours with 10 μmol/L cholesterol (+pretr) before adding terbinafine, and cultured for 72 hours. The percentage of cell death was evaluated by trypan blue exclusion method. Histograms in D and E indicate the mean ± SD of at least three independent experiments. Drug vs. DMSO: ***, P < 0.001; Drug+cholesterol versus drug alone: +++, P < 0.001. F, Left, In vitro migration assay of FP#1 cells treated with 10 μmol/L atorvastatin alone or in combination with 10 μmol/L cholesterol or pretreated for 12 hours with cholesterol (+pretr) before adding atorvastatin. Histogram quantifies relative migratory capacities. The results shown in the figure are the mean ± SD of 5 independent experiments. ***, P < 0.001. Right, cell viability of FP#1 after 24 hours of atorvastatin treatment. White bars, cells were treated with DMSO or 10 μmol/L atorvastatin and cultured in mammosphere assay. Gray bars, cells from the top part of migration chambers were collected after 24 hours of DMSO or atorvastatin treatment. Cell viability was evaluated by FACS analysis of 7-ADD–negative cells. Mean ± SD of three independent experiments is shown. n.s., nonsignificant.
Activation of de novo cholesterol biosynthesis is associated with aggressive disease
Active cholesterol biosynthesis emerged as a major cellular target for efficacious repurposing of PP against CSCs. To further evaluate cholesterol biosynthesis as a clinically-relevant target, we queried the prognostic value of genes involved in de novo cholesterol synthesis in breast cancer patient databases, with a focus on the correlation between gene expression and metastases and tumor relapse. We found that upregulation of several genes in the cholesterol biosynthesis pathway significantly correlated with poor clinical outcomes in breast cancer cohorts (Supplementary Fig. S14B). Notably, SQLE and MVD, the two enzymes that were significantly upregulated in FP-CSCs, showed the most significant negative prognostic value (Fig. 7A). Moreover, in a TNBC cohort, high expression was associated with a lower probability of relapse-free survival and of distal metastasis-free survival (DMFS) at the gene (SQLE and MVD) and protein (SQLE) expression levels (Fig. 7B–D), linking this pathway with aggressive disease in TNBC. High expression of either single or combined SQLE and MVD genes also had a significant negative prognostic value for disease relapse and metastasis occurrence in multiple breast cancer cohorts (Fig. 7E–G; Supplementary Fig. S14C). To strengthen the causal link between cholesterol pathway activation and prognosis, we searched for a correlation with metabolites of de novo cholesterol biosynthesis in breast tumor samples, and we found that high levels of squalene, the first intermediate of the squalenic phase, significantly correlated with poor clinical outcomes (Fig. 7H). These data strongly support that activation of de novo cholesterol biosynthesis is a clinically relevant target with a prognostic value to predict tumor relapse and metastatic progression in TNBC. Moreover, elevated expression of MVD and SQLE were associated with poor outcomes in overall breast cancer, suggesting that activation of cholesterol biosynthesis could be relevant to all breast cancer subtypes with aggressive and metastatic potential.
Figure 7.
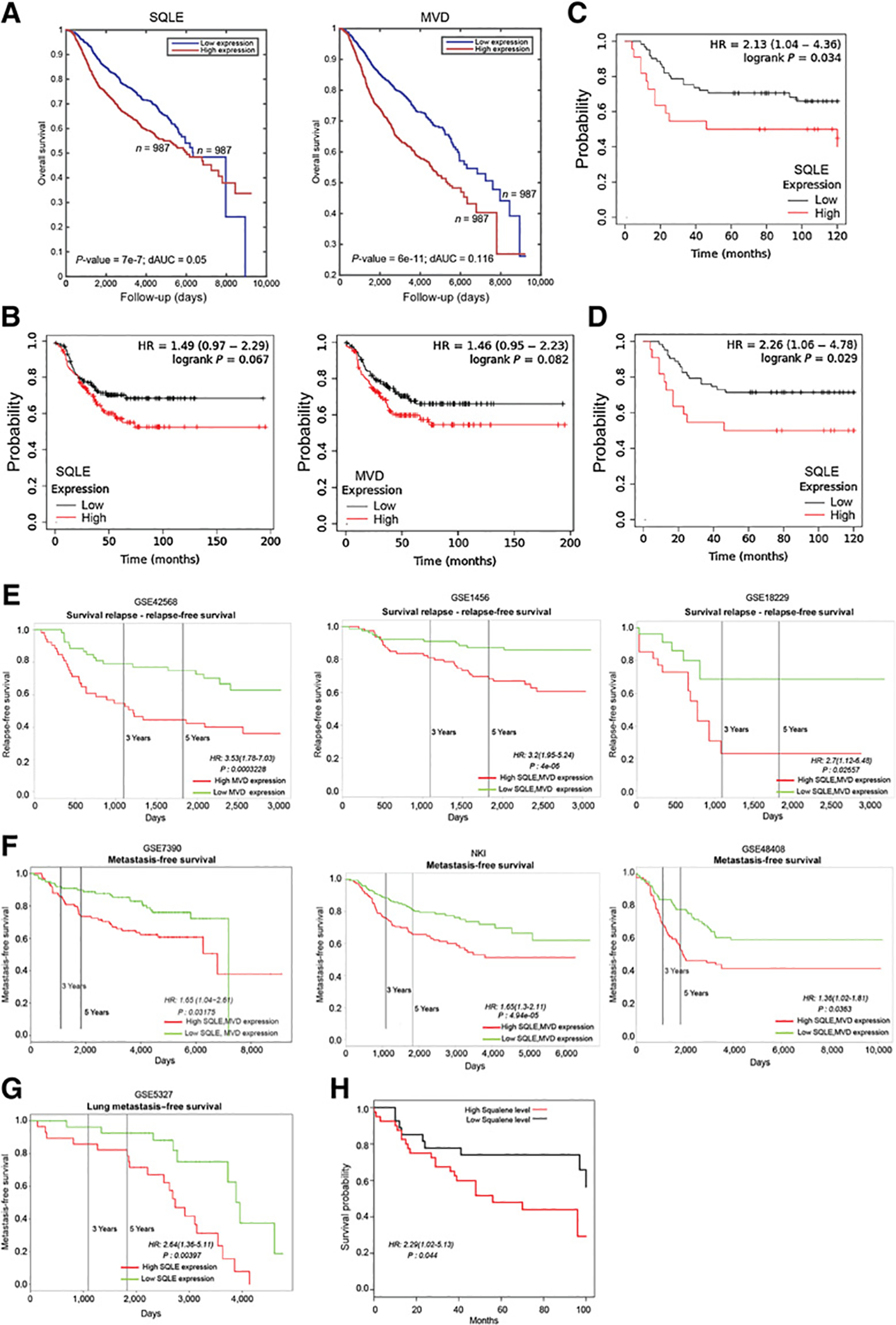
Activation of de novo cholesterol biosynthesis is associated with metastatic disease and tumor relapse in breast cancer. A, Kaplan–Meier survival curves for SQLE or MVD gene expression (top and bottom 0.5 quartiles) when considering all METABRIC dataset samples (n = 1,974). B, Kaplan–Meier relapse-free survival curves for SQLE (left) or MVD (right) gene expression when considering TNBC patients in the Kaplan–Meier plotter dataset samples (n = 210). Kaplan–Meier curve for relapse-free survival (C) and distant metastasis-free survival (D) for SQLE protein expression when considering patients with TNBC in the Kaplan–Meier plotter dataset samples (n = 124). Kaplan–Meier curves for combined or single SQLE and MVD gene expression for metastasis-free survival (D) and relapse-free survival (E) when considering different (indicated) datasets. F, Kaplan–Meier curves for squalene abundance associated with breast cancer overall survival. G, GSE5327 database showed a significant association between SQLE gene upregulation and lung metastases. H, Kaplan–Meier curves for squalene abundance associated with breast cancer overall survival.
Discussion
TNBC is associated with a poor prognosis, and suitable targeted therapeutic approaches are lacking. At the core of TNBC aggressiveness is the development of chemoresistance and metastasis formation, both of which depend on the subpopulation CSCs. On the basis of the hypothesis that targeting CSCs in primary tumors can prevent metastasis, we performed a drug repurposing study to identify therapeutics that can kill CSCs and reduce metastasis toward identifying a new therapeutic intervention for patients with TNBC.
Several reports have described the ability of the anthelmintic drug PP to kill CSCs, although the mechanisms by which it exerts antitumor and antimetastatic effects have remained unclear. We showed that PP induces a metabolic dysregulation, specifically attenuating lipid anabolism, and that this effect is essential for its cytotoxic activity against metastatic TNBC CSCs. To our knowledge, this is the first study reporting altered metabolic and lipidomic profiles of CSCs upon PP treatment, which demonstrated that PP blocks the anabolic metabolism of glucose toward lipids. With this approach, we identified a previously unrecognized mode of action of PP that supports its repurposing against lipid-addicted tumor cells.
Previous studies have linked the antitumor activity of PP to inhibition of the WNT/β-catenin–dependent signaling pathway. However, the molecular targets of PP have been debated: while the ability of PP to act as CK1α agonist appeared controversial, PP was demonstrated to inhibit the AKT/GSK3β axis (55) and the PI3K/S6-dependent pathway (19). It is conceivable that the profound metabolic crisis induced by PP may downregulate metabolic homeostasis-sensor pathways, including PI3K/AKT/S6 and others. Noteworthy, the WNT and Hedgehog pathways require FAs (56, 57) or FAs and cholesterol (58), respectively, to be fully activated. Thus, due to the requirement for lipid components in the posttranslational modification of many proteins, pleiotropic effects of PP are expected on multiple signaling pathways.
To identify the mechanism(s) causing cell death in PP-treated cells, we explored lipid-mediated effects in TNBC CSCs. Our data convincingly demonstrated that cholesterol supplementation effectively limited PP-induced cytotoxicity by restoring levels of both free and bound cholesterol (CEs). This indicates that the depletion of cholesterol below an essential threshold significantly contributes as a primary mechanism of PP-induced cytotoxicity in FP-CSCs, although a contribution from other pathways cannot be excluded by our experiments. The mechanistic link was further supported by our demonstration that multiple, specific inhibitors of cholesterol biosynthesis exerted cytotoxic effects in FP-CSCs. Conversely, inhibition of β-catenin by ICRT3 induced cell death, and cholesterol addition was insufficient to reverse this effect. Furthermore, at a low dose of ICRT3 that exerted similar repression of β-catenin signaling to PP treatment, no cell killing was observed. Overall, these data argue against β-catenin as the primary PP-regulated pathway that results in cytotoxicity.
We observed antitumor and antimetastatic effects of PP against CSCs in preclinical models of TNBC at doses that were well tolerated upon intraperitoneal injection. Daily doses up to 1.2 mg/kg PP result in a peak plasma concentration of 150 nmol/L (59), which approximates the cytotoxic concentration we used in our in vitro studies. Thus, our data and other characterization of PP provide a preclinical rationale to evaluate PP in drug repurposing phase I clinical studies of TNBC, and suggest that there may be a therapeutic window to target aggressive TNBC CSCs at tolerated doses.
We found that PP halted the anabolic flux from glucose to both FAs and cholesterol by a metabolic mechanism yet to be specifically identified. However, our studies highlighted the role of glucose-derived lipids, such as cholesterol and FAs, as necessary for TNBC CSC migration and survival. Cholesterol is an important component of cellular membranes and serves as a precursor for steroid hormones and vitamin D. Moreover, isoprenoids are used for the synthesis of important biomolecules, such as ubiquinone and coenzyme Q. Hydrophobic chains represent key mediators of signal transduction pathways, and cholesterol controls key pathways necessary for the migration of cancer cells (60, 61). During the revision of this study, other researchers reported that the activation of cholesterol biosynthesis was a key characteristic of breast CSCs relevant to patient outcomes. These studies strengthen our observations and, particularly, the relevance of addressing cholesterol biosynthesis as an anticancer strategy against aggressive stem-like cells (62).
Consistent with our data that cholesterol promotes the survival and migration of aggressive CSCs, we found epidemiologic studies indicating the prognostic value of genes involved in cholesterol anabolism for patients with TNBC. The negative prognostic value of the SQLE gene in breast cancer was reported previously (63, 64). However, our results expand our understanding of SQLE and cholesterol biosynthesis upregulation as a prognostic biomarker specifically for disease metastasis and relapse, two endpoints that were not specifically investigated in previous studies. Moreover, we determined the prognostic relevance of both SQLE and MVD alone or in combination. This aspect was important to correlate the prognostic value of the metabolic pathway independently from MYC overexpression, which frequently cooccurs with SQLE overexpression, as they both reside on 8q24.
The relevance of the cholesterol biosynthetic pathway as a therapeutic target is consistent with clinical evidence indicating the benefit of statin treatment in patients with breast cancer to reduce recurrence and mortality rate (39, 40, 65). An intimate molecular link between the tumor suppressor function of wild-type p53 and the cholesterol biosynthetic pathway has been reported (45, 46). Because Tp53 is the most frequently mutated tumor suppressor gene, it suggests that effective approaches to target cholesterol biosynthetic pathway may have broad applications in many tumors. PP, which disrupts the homeostasis of lipid anabolism at multiple levels, is likely to be more effective in tumor cells addicted to lipids, where it may effectively thwart adaptive metabolic reprogramming.
In conclusion, our characterization of the effects of PP in a model of highly aggressive TNBC CSCs prompts drug repurposing clinical studies of PP in both adjuvant and neoadjuvant settings to reduce disease progression and relapse. Other tumor types characterized by similar dependency on lipid metabolism may also respond to targeted therapy with PP.
Supplementary Material
Significance:
These findings provide preclinical evidence that a drug repurposing approach to prevent metastatic disease in TNBC exploits lipid anabolism as a metabolic vulnerability against CSCs in primary tumors.
Acknowledgments
We are grateful to G. Loreto, Dr. P. Di Matteo, Dr. A. Rosi, Dr. A. Palma (ISS, Rome, Italy), Dr. M. Ludovici (ISG, Rome, Italy), and S. Barresi for technical support. We thank and Dr. Angela K. Deem (UT MD Anderson Cancer Center, Houston, TX) for English editing of the manuscript. The project was partially funded by the Italian Ministry of Health, research grant numbers GR-2011–02351749 and 5 × 1000 IRE. G. Doglioni is supported by a Kom op tegen Kanker PhD fellowship. S.-M. Fendt acknowledges funding from FWO – Research Grants and Projects, and KU Leuven – Methusalem Co-Funding.
Disclosure of Potential Conflicts of Interest
R. De Maria reports grants from Hibercell advisory board and research grant, other funding from Kiromic advisory board, and personal fees from Exiris advisory board outside the submitted work. S.-M. Fendt reports grants from FWO, grants from ERC, grants from Fonds Baillet Latour, and grants from KU Leuven Methusalem during the conduct of the study; other fees from Bayer (fee for service), other fees from Merck (fee for service), other fees from Black Belt Therapeutics (fee for service), and other funding from funds + consulting outside the submitted work. No potential conflicts of interest were disclosed by the other authors.
Footnotes
Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
References
- 1.Oakman C, Santarpia L, Di Leo A. Breast cancer assessment tools and optimizing adjuvant therapy. Nat Rev Clin Oncol 2010;7:725–32. [DOI] [PubMed] [Google Scholar]
- 2.Li F, Tiede B, Massague J, Kang Y. Beyond tumorigenesis: cancer stem cells in metastasis. Cell Res 2007;17:3–14. [DOI] [PubMed] [Google Scholar]
- 3.Liedtke C, Mazouni C, Hess KR, Andre F, Tordai A, Mejia JA, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol 2008;26:1275–81. [DOI] [PubMed] [Google Scholar]
- 4.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature 2001;414:105–11. [DOI] [PubMed] [Google Scholar]
- 5.Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 2015;526:131–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oskarsson T, Batlle E, Massague J. Metastatic stem cells: sources, niches, and vital pathways. Cell Stem Cell 2014;14:306–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miyoshi Y, Shien T, Ogiya A, Ishida N, Yamazaki K, Horii R, et al. Differences in expression of the cancer stem cell marker aldehyde dehydrogenase 1 among estrogen receptor-positive/human epidermal growth factor receptor type 2-negative breast cancer cases with early, late, and no recurrence. Breast Cancer Res 2016;18:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells - what challenges do they pose? Nat Rev Drug Discov 2014;13:497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grimshaw MJ, Cooper L, Papazisis K, Coleman JA, Bohnenkamp HR, Chiapero-Stanke L, et al. Mammosphere culture of metastatic breast cancer cells enriches for tumorigenic breast cancer cells. Breast Cancer Res 2008;10:R52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pastrana E, Silva-Vargas V, Doetsch F. Eyes wide open: a critical review of sphere-formation as an assay for stem cells. Cell Stem Cell 2011;8:486–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Papapetropoulos A, Szabo C. Inventing new therapies without reinventing the wheel: the power of drug repurposing. Br J Pharmacol 2018;175:165–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu L, Zhang L, Hu C, Liang S, Fei X, Yan N, et al. WNT pathway inhibitor pyrvinium pamoate inhibits the self-renewal and metastasis of breast cancer stem cells. Int J Oncol 2016;48:1175–86. [DOI] [PubMed] [Google Scholar]
- 13.Ishii I, Harada Y, Kasahara T. Reprofiling a classical anthelmintic, pyrvinium pamoate, as an anti-cancer drug targeting mitochondrial respiration. Front Oncol 2012;2:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tomitsuka E, Kita K, Esumi H. An anticancer agent, pyrvinium pamoate inhibits the NADH-fumarate reductase system–a unique mitochondrial energy metabolism in tumour microenvironments. J Biochem 2012;152:171–83. [DOI] [PubMed] [Google Scholar]
- 15.Li B, Fei DL, Flaveny CA, Dahmane N, Baubet V, Wang Z, et al. Pyrvinium attenuates Hedgehog signaling downstream of smoothened. Cancer Res 2014;74: 4811–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thorne CA, Hanson AJ, Schneider J, Tahinci E, Orton D, Cselenyi CS, et al. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1alpha. Nat Chem Biol 2010;6:829–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamb R, Ozsvari B, Lisanti CL, Tanowitz HB, Howell A, Martinez-Outschoorn UE, et al. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: treating cancer like an infectious disease. Oncotarget 2015;6:4569–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiang W, Cheong JK, Ang SH, Teo B, Xu P, Asari K, et al. Pyrvinium selectively targets blast phase-chronic myeloid leukemia through inhibition of mitochondrial respiration. Oncotarget 2015;6:33769–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carrella D, Manni I, Tumaini B, Dattilo R, Papaccio F, Mutarelli M, et al. Computational drugs repositioning identifies inhibitors of oncogenic PI3K/AKT/P70S6K-dependent pathways among FDA-approved compounds. Oncotarget 2016;7:58743–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Momtazi-Borojeni AA, Abdollahi E, Ghasemi F, Caraglia M, Sahebkar A. The novel role of pyrvinium in cancer therapy. J Cell Physiol 2018;233: 2871–81. [DOI] [PubMed] [Google Scholar]
- 21.Camarda R, Zhou AY, Kohnz RA, Balakrishnan S, Mahieu C, Anderton B, et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat Med 2016;22:427–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park JH, Vithayathil S, Kumar S, Sung PL, Dobrolecki LE, Putluri V, et al. Fatty acid oxidation-driven Src links mitochondrial energy reprogramming and oncogenic properties in triple-negative breast cancer. Cell Rep 2016;14: 2154–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009;138:645–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol 2015;11:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gui DY, Sullivan LB, Luengo A, Hosios AM, Bush LN, Gitego N, et al. Environment dictates dependence on mitochondrial complex I for NAD+ and aspartate production and determines cancer cell sensitivity to metformin. Cell Metab 2016;24:716–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 2015;162:552–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer 2013;13:227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elia I, Doglioni G, Fendt SM. Metabolic hallmarks of metastasis formation. Trends Cell Biol 2018;28:673–84. [DOI] [PubMed] [Google Scholar]
- 29.Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017;541:41–5. [DOI] [PubMed] [Google Scholar]
- 30.Li J, Condello S, Thomes-Pepin J, Ma X, Xia Y, Hurley TD, et al. Lipid desaturation is a metabolic marker and therapeutic target of ovarian cancer stem cells. Cell Stem Cell 2017;20:303–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mancini R, Noto A, Pisanu ME, De Vitis C, Maugeri-Sacca M, Ciliberto G. Metabolic features of cancer stem cells: the emerging role of lipid metabolism. Oncogene 2018;37:2367–78. [DOI] [PubMed] [Google Scholar]
- 32.Baenke F, Peck B, Miess H, Schulze A. Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Dis Model Mech 2013;6:1353–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuzu OF, Noory MA, Robertson GP. The role of cholesterol in cancer. Cancer Res 2016;76:2063–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Llaverias G, Danilo C, Mercier I, Daumer K, Capozza F, Williams TM, et al. Role of cholesterol in the development and progression of breast cancer. Am J Pathol 2011;178:402–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Danilo C, Frank PG. Cholesterol and breast cancer development. Curr Opin Pharmacol 2012;12:677–82. [DOI] [PubMed] [Google Scholar]
- 36.Nelson ER, Chang CY, McDonnell DP. Cholesterol and breast cancer pathophysiology. Trends Endocrinol Metab 2014;25:649–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ahern TP, Lash TL, Damkier P, Christiansen PM, Cronin-Fenton DP. Statins and breast cancer prognosis: evidence and opportunities. Lancet Oncol 2014;15: e461–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Undela K, Srikanth V, Bansal D. Statin use and risk of breast cancer: a meta-analysis of observational studies. Breast Cancer Res Treat 2012;135:261–9. [DOI] [PubMed] [Google Scholar]
- 39.Cardwell CR, Hicks BM, Hughes C, Murray LJ. Statin use after diagnosis of breast cancer and survival: a population-based cohort study. Epidemiology 2015;26: 68–78. [DOI] [PubMed] [Google Scholar]
- 40.Liu B, Yi Z, Guan X, Zeng YX, Ma F. The relationship between statins and breast cancer prognosis varies by statin type and exposure time: a meta-analysis. Breast Cancer Res Treat 2017;164:1–11. [DOI] [PubMed] [Google Scholar]
- 41.Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr. Cellular fatty acid metabolism and cancer. Cell Metab 2013;18:153–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yue S, Li J, Lee SY, Lee HJ, Shao T, Song B, et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab 2014;19:393–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang B, Rong X, Palladino END, Wang J, Fogelman AM, Martin MG, et al. Phospholipid remodeling and cholesterol availability regulate intestinal stemness and tumorigenesis. Cell Stem Cell 2018;22:206–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baek AE, Yu YA, He S, Wardell SE, Chang CY, Kwon S, et al. The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat Commun 2017;8:864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012;148:244–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moon SH, Huang CH, Houlihan SL, Regunath K, Freed-Pastor WA, Morris JPt, et al. p53 represses the mevalonate pathway to mediate tumor suppression. Cell 2019;176:564–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barnabas N, Cohen D. Phenotypic and molecular characterization of MCF10DCIS and SUM breast cancer cell lines. Int J Breast Cancer 2013; 2013:872743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 2010;70:440–6. [DOI] [PubMed] [Google Scholar]
- 49.Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 2015;162:540–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yizhak K, Chaneton B, Gottlieb E, Ruppin E. Modeling cancer metabolism on a genome scale. Mol Syst Biol 2015;11:817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yizhak K, Gabay O, Cohen H, Ruppin E. Model-based identification of drug targets that revert disrupted metabolism and its application to ageing. Nat Commun 2013;4:2632. [DOI] [PubMed] [Google Scholar]
- 52.Xu F, Zhu Y, Lu Y, Yu Z, Zhong J, Li Y, et al. Anthelmintic pyrvinium pamoate blocks Wnt/beta-catenin and induces apoptosis in multiple myeloma cells. Oncol Lett 2018;15:5871–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu W, Lacerda L, Debeb BG, Atkinson RL, Solley TN, Li L, et al. The antihelmintic drug pyrvinium pamoate targets aggressive breast cancer. PLoS One 2013;8:e71508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang C, Zhang Z, Zhang S, Wang W, Hu P. Targeting of Wnt/beta-catenin by anthelmintic drug pyrvinium enhances sensitivity of ovarian cancer cells to chemotherapy. Med Sci Monit 2017;23:266–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Venerando A, Girardi C, Ruzzene M, Pinna LA. Pyrvinium pamoate does not activate protein kinase CK1, but promotes Akt/PKB down-regulation and GSK3 activation. Biochem J 2013;452:131–7. [DOI] [PubMed] [Google Scholar]
- 56.Takada R, Satomi Y, Kurata T, Ueno N, Norioka S, Kondoh H, et al. Monounsaturated fatty acid modification of Wnt protein: its role in Wnt secretion. Dev Cell 2006;11:791–801. [DOI] [PubMed] [Google Scholar]
- 57.Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T, et al. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 2003; 423:448–52. [DOI] [PubMed] [Google Scholar]
- 58.Blassberg R, Jacob J. Lipid metabolism fattens up hedgehog signaling. BMC Biol 2017;15:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jones JO, Bolton EC, Huang Y, Feau C, Guy RK, Yamamoto KR, et al. Non-competitive androgen receptor inhibition in vitro and in vivo. Proc Natl Acad Sci U S A 2009;106:7233–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mullen PJ, Yu R, Longo J, Archer MC, Penn LZ. The interplay between cell signalling and the mevalonate pathway in cancer. Nat Rev Cancer 2016;16: 718–31. [DOI] [PubMed] [Google Scholar]
- 61.Murai T The role of lipid rafts in cancer cell adhesion and migration. Int J Cell Biol 2012;2012:763283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ehmsen S, Pedersen MH, Wang G, Terp MG, Arslanagic A, Hood BL, et al. Increased cholesterol biosynthesis is a key characteristic of breast cancer stem cells influencing patient outcome. Cell Rep 2019;27:3927–38. [DOI] [PubMed] [Google Scholar]
- 63.Brown DN, Caffa I, Cirmena G, Piras D, Garuti A, Gallo M, et al. Squalene epoxidase is a bona fide oncogene by amplification with clinical relevance in breast cancer. Sci Rep 2016;6:19435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Helms MW, Kemming D, Pospisil H, Vogt U, Buerger H, Korsching E, et al. Squalene epoxidase, located on chromosome 8q24.1, is upregulated in 8q+ breast cancer and indicates poor clinical outcome in stage I and II disease. Br J Cancer 2008;99:774–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Murtola TJ, Visvanathan K, Artama M, Vainio H, Pukkala E. Statin use and breast cancer survival: a nationwide cohort study from Finland. PLoS One 2014; 9:e110231. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.