

Abstract
Objectives
Autologous chimeric antigen receptor (CAR) αβ T‐cell therapies have demonstrated remarkable antitumor efficacy in patients with haematological malignancies; however, not all eligible cancer patients receive clinical benefit. Emerging strategies to improve patient access and clinical responses include using premanufactured products from healthy donors and alternative cytotoxic effectors possessing intrinsic tumoricidal activity as sources of CAR cell therapies. γδ T cells, which combine innate and adaptive mechanisms to recognise and kill malignant cells, are an attractive candidate platform for allogeneic CAR T‐cell therapy. Here, we evaluated the manufacturability and functionality of allogeneic peripheral blood‐derived CAR+ Vδ1 γδ T cells expressing a second‐generation CAR targeting the B‐cell‐restricted CD20 antigen.
Methods
Donor‐derived Vδ1 γδ T cells from peripheral blood were ex vivo‐activated, expanded and engineered to express a novel anti‐CD20 CAR. In vitro and in vivo assays were used to evaluate CAR‐dependent and CAR‐independent antitumor activities of CD20 CAR+ Vδ1 γδ T cells against B‐cell tumors.
Results
Anti‐CD20 CAR+ Vδ1 γδ T cells exhibited innate and adaptive antitumor activities, such as in vitro tumor cell killing and proinflammatory cytokine production, in addition to in vivo tumor growth inhibition of B‐cell lymphoma xenografts in immunodeficient mice. Furthermore, CD20 CAR+ Vδ1 γδ T cells did not induce xenogeneic graft‐versus‐host disease in immunodeficient mice.
Conclusion
These preclinical data support the clinical evaluation of ADI‐001, an allogeneic CD20 CAR+ Vδ1 γδ T cell, and a phase 1 study has been initiated in patients with B‐cell malignancies (NCT04735471).
Keywords: adoptive cell therapy, B‐cell lymphoma, CD20, chimeric antigen receptor, γδ T cells
The findings of our study demonstrate preclinical proof of concept for a CD20 CAR+ Vδ1 γδ T‐cell product, manufactured at clinical scale, in the treatment of CD20+ B‐cell malignancies. Complemented by innate and adaptive antitumor immunity, CD20 CAR+ Vδ1 γδ T cells have a unique advantage as an allogeneic CAR T‐cell product platform and, as a consequence, have the potential to improve clinical responses in eligible cancer patients. A phase 1 clinical study evaluating ADI‐001, an allogeneic CD20 CAR+ Vδ1 γδ T‐cell product, in relapsed/refractory B‐cell NHL patients has been initiated (NCT04735471).

Introduction
Allogeneic CAR T‐cell products have the potential to expand patient access to CAR T‐cell therapy and to improve clinical outcomes for patients with relapsed or refractory B‐cell non‐Hodgkin lymphoma (R/R B‐cell NHL). Healthy donors can be selected based on T‐cell fitness and on successful and consistent manufacturability, ensuring that eligible R/R B‐cell NHL patients have immediate access to a high‐quality and uniform CAR T‐cell product. 1 , 2 , 3 , 4 , 5 Challenges do exist with allogeneic CAR T‐cell products, namely graft‐versus‐host disease (GvHD) and immunogenicity (i.e. host‐versus‐graft rejection) of the allogeneic CAR T‐cell product, both of which are consequences of human leucocyte antigen (HLA) incompatibility between donors and patients. 1 , 2 , 3 , 4 , 5 Common approaches for reducing the risk of GvHD are to perform gene editing to prevent surface expression of the αβ T‐cell antigen receptor (TCR), to manufacture a CAR T‐cell product using non‐alloreactive T cells (e.g. Epstein–Barr virus‐ or multivirus‐specific T cells) or to include a truncated form of CD3ζ in the CAR construct that, when expressed, will be incorporated into the endogenous TCR and inhibit its signalling. 1 , 3 , 4 , 5 , 6 , 7 Approaches to reduce the immunogenicity of allogeneic CAR T cells are to use preconditioning lymphodepletion regimens to deplete the patient’s potential alloreactive cells, to combine preconditioning with gene editing of donor T cells to silence or reduce expression of β2 microglobulin, a subunit of HLA class I required for its surface expression, or to match patient and donor HLA alleles using a bank of universal CAR T‐cell products. 1 , 3 , 4 , 5
Allogeneic CAR‐based cell therapy lends itself to using alternative cytotoxic effectors, such as γδ T cells, which play established roles in tumor surveillance and antitumor immunity. 3 , 8 , 9 γδ T cells represent the third lymphoid lineage, along with αβ T cells and B cells, which express somatically rearranged antigen receptors. 10 , 11 Antigen recognition by γδTCR and αβTCR differs, in that the γδTCR recognises native unprocessed antigens, many of which are induced or upregulated following cellular injury, infection or transformation, whereas the αβTCR recognises processed peptide antigens that are presented by major histocompatibility complex (MHC) molecules. 12 γδ T cells can also detect stress‐induced surface molecules through expression of non‐TCR molecules, such as the germline‐encoded natural killer cell receptors (NKRs). 12 The co‐expression of TCR and NKR molecules endows γδ T cells with the ability to monitor and maintain tissue integrity through recognition and destruction of stressed, infected or malignant cells. 12
γδ T cells are an attractive platform for allogeneic CAR T‐cell therapy for a number of reasons. First, γδ T cells have established roles in both tumor surveillance and antitumor immunity and are the immune cell population whose tumor infiltration has the most significant correlation with survival. 8 , 9 , 12 , 13 , 14 , 15 , 16 Second, there is clinical experience with allogeneic γδ T cells in the treatment of haematological malignancies. In leukaemia patients who received αβ T‐cell‐depleted bone marrow transplants from HLA‐mismatched donors, complete responses were observed in 24% of patients and correlated with elevated persistence of donor‐derived peripheral blood γδ T cells. 17 In addition, patients with elevated numbers of γδ T cells after stem cell transplantation had a significantly lower incidence of infections, particularly bacterial and fungal, than patients with normal‐to‐low numbers of γδ T cells. 18 These findings suggest that adoptively transferred γδ T cells can mediate not only antitumor activity but also protection against infections, a key functional attribute considering that infections are a serious adverse event observed with CAR T‐cell products targeting B‐cell malignancies. 18 , 19 , 20 Third, because γδ T cells recognise antigen in an MHC‐unrestricted manner, they are not associated with induction of GvHD and therefore do not require gene editing to disrupt TCR expression or TCR signalling. 9 , 12 , 14 , 15 Fourth, because γδ T cells predominantly reside in peripheral tissues, they are better adapted than circulating lymphocytes at functioning in a hypoxic environment, which is commonly associated with tumors. 14 , 21 Last, CAR+ γδ T cells can kill tumor cells through recognition of the target antigen by the CAR and through recognition of stress‐induced surface molecules by the γδTCR and NKRs. 12 , 14 , 15 , 16 , 21 The recognition of various antigens on the tumor cell surface by endogenous receptors on CAR+ γδ T cells may act to minimise tumor escape associated with CAR‐mediated target antigen loss.
Here, we evaluated the manufacturability and the functionality of allogeneic peripheral blood CAR+ Vδ1 γδ T cells expressing a second‐generation CAR containing a single‐chain variable fragment (scFv) derived from a novel, fully human anti‐CD20 monoclonal antibody (mAb). CAR‐dependent and CAR‐independent antitumor activities were assessed in both in vitro studies and in in vivo human xenograft models in NOD scid gamma (NSG) mice. In addition, the potential of CD20 CAR+ Vδ1 γδ T cells to induce xenogeneic GvHD and any acute, chronic or delayed‐onset toxicities was investigated in NSG mice. Together, the data demonstrate preclinical proof of concept for CD20 CAR+ Vδ1 γδ T cells in the treatment of CD20+ B‐cell malignancies.
Results
Targeting CD20 using a novel fully human anti‐CD20 mAb, clone 3H7
The antigen‐binding domain of the CD20 CAR consists of a scFv derived from the 3H7 mAb, a novel fully human anti‐CD20 mAb, that was generated using the VelocImmune™ human antibody mouse platform. 22 , 23 The binding properties of the 3H7 mAb were evaluated and compared with those of the therapeutic anti‐CD20 mAb rituximab using flow cytometry. The 3H7 mAb displayed concentration‐dependent binding to CD20+ Raji, Daudi and HEK293‐hCD20 cells with EC50 values that were similar to those of rituximab (Figure 1a). When biochemical off‐rates of 3H7 mAb and rituximab were compared by measuring the amount of antibody bound to Raji cells over a 3‐h time course by flow cytometry, the 3H7 mAb and rituximab exhibited similar decreases in binding to Raji cells over time (Figure 1b). Therefore, 3H7 mAb and rituximab have similar affinities for human CD20 (10−9 m range) and similar biochemical off‐rates.
Figure 1.
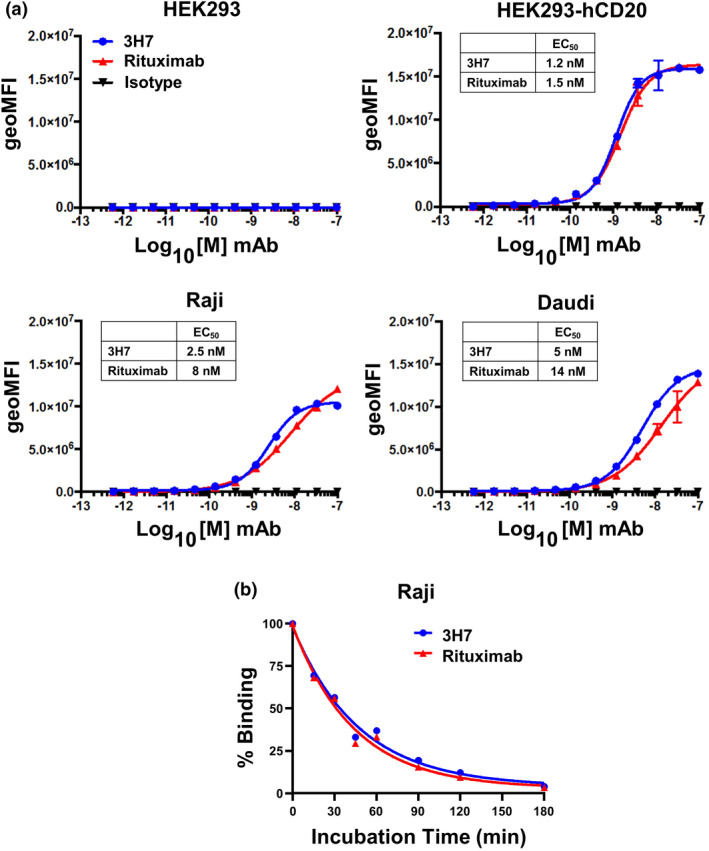
Binding properties of the fully human anti‐CD20 mAb clone 3H7. (a) Titration of 3H7 mAb ( ), therapeutic anti‐CD20 mAb rituximab (
), therapeutic anti‐CD20 mAb rituximab ( ) and human IgG1 control antibody (
) and human IgG1 control antibody ( ) binding to parental HEK293 cells, HEK293 cells stably expressing human CD20 (HEK293‐hCD20), and Raji and Daudi human B‐cell lymphoma cell lines. Cell‐surface binding of the antibodies over a range of concentrations (1.69 pm to 100 nm) was performed at 4°C, detected by flow cytometry using APC‐labelled anti‐human IgG antibody and reported as geometric mean fluorescence intensity (geoMFI). For each dose curve, staining with the APC‐labelled anti‐human IgG antibody alone is included in the graph as a continuation of the threefold serial dilution and is represented as the lowest dose. EC50 values of the binding curves for each anti‐CD20 mAb on each CD20+ cell line are shown. Data are representative of two independent experiments. (b) Biochemical off‐rates of 3H7 mAb () and rituximab () were determined by measuring the amount of cell‐bound antibody over the course of a 3‐h time period. Antibodies (2 μg mL−1 each) were incubated with Raji cells for 2 h at room temperature. The cells were then washed, resuspended in 1% serum containing medium and incubated at 37°C. At time 0, 15, 30, 45, 60, 90, 120 and 180 min, an aliquot of cells was removed, washed, stained with PE‐labelled anti‐human Fc antibody and analysed by flow cytometry.
) binding to parental HEK293 cells, HEK293 cells stably expressing human CD20 (HEK293‐hCD20), and Raji and Daudi human B‐cell lymphoma cell lines. Cell‐surface binding of the antibodies over a range of concentrations (1.69 pm to 100 nm) was performed at 4°C, detected by flow cytometry using APC‐labelled anti‐human IgG antibody and reported as geometric mean fluorescence intensity (geoMFI). For each dose curve, staining with the APC‐labelled anti‐human IgG antibody alone is included in the graph as a continuation of the threefold serial dilution and is represented as the lowest dose. EC50 values of the binding curves for each anti‐CD20 mAb on each CD20+ cell line are shown. Data are representative of two independent experiments. (b) Biochemical off‐rates of 3H7 mAb () and rituximab () were determined by measuring the amount of cell‐bound antibody over the course of a 3‐h time period. Antibodies (2 μg mL−1 each) were incubated with Raji cells for 2 h at room temperature. The cells were then washed, resuspended in 1% serum containing medium and incubated at 37°C. At time 0, 15, 30, 45, 60, 90, 120 and 180 min, an aliquot of cells was removed, washed, stained with PE‐labelled anti‐human Fc antibody and analysed by flow cytometry.
To investigate the 3H7 CAR’s potential for off‐target binding, a 3H7 minibody was used as a surrogate binding protein to stain CD20+ and CD20− cell lines and cryosections from a normal human tissue panel. The 3H7 minibody was constructed by fusing the 3H7 scFv to the hinge region and CH3 domain of human IgG1 to form a bivalent scFv molecule. As shown in Supplementary figure 1a, the 3H7 minibody produced moderate‐to‐intense membrane and cytoplasmic staining of Rat2‐hCD20 fibroblasts but no staining of parental Rat2 fibroblasts. Furthermore, staining of normal human tissues with the 3H7 minibody displayed expected reactivity with CD20 in follicles of secondary lymphoid tissues and in lymphoid structures or aggregates in various tissues such as colon, small intestine, stomach and lung (Supplementary figure 1b and c; data not shown). Therefore, the staining of the normal human tissue panel with the 3H7 minibody showed expected reactivity with its target antigen CD20 and no unanticipated cross‐reactivity.
Manufacturability of peripheral blood Vδ1 γδ T cells
Vδ1 γδ T cells are the predominant γδ T‐cell subset in lymphoid and mucosal tissues 12 ; however, in the blood, they represent 12–22% of total γδ T cells or 0.2–1% of peripheral blood mononuclear cells (PBMCs). 24 To develop a large‐scale method to activate, expand and engineer peripheral blood Vδ1 γδ T cells, we generated an agonistic mAb that selectively activates and expands Vδ1 γδ T cells in healthy donor‐derived PBMCs to clinically relevant numbers (Figure 2a). Once activated, Vδ1 γδ T cells were transduced with a self‐inactivating, replication‐incompetent gamma‐retroviral vector to express the CD20 CAR, consisting of the 3H7 scFv, CD8α hinge and transmembrane domains, and the 4‐1BB and CD3ζ signalling domains (Figure 2b). After transduction, γδ T cells were further expanded in culture, depleted of αβ T cells and then formulated for cryopreservation (Figure 2a). Small‐scale expansion methods were used for initial development prior to transition into a large‐scale manufacturing process. The large‐scale manufacturing process resulted in an average of a 23 983‐fold expansion (range 9569–34 321) of Vδ1 γδ T cells (Figure 2c), of which approximately 64% (range 40–86%) expressed the CD20 CAR (Figure 2d). The higher average fold expansion in the small‐scale (66 604, range 13 732–102 133) compared with that in the large‐scale process may be associated with the additional 3 days in culture, but across multiple runs, no significant difference was observed. During the expansion process, the average percentage of Vδ1 γδ T cells at Day 0 of culture was 0.6% (range 0.3–1.0%) and increased to 77% (range 68–84%) at Day 14 prior to αβ T‐cell depletion (Figure 2e). Flow cytometric analyses of three donor final cell products showed that, on average, 93% are Vδ1+ γδ T cells, 0.2% are Vδ2+ γδ T cells, 0.9% are Vδ1−Vδ2− γδ T cells, 1.8% are NK cells, and 0.03% are αβ T cells (Figure 2f). No B cells or monocytes were detected by flow cytometry in the final cell product.
Figure 2.
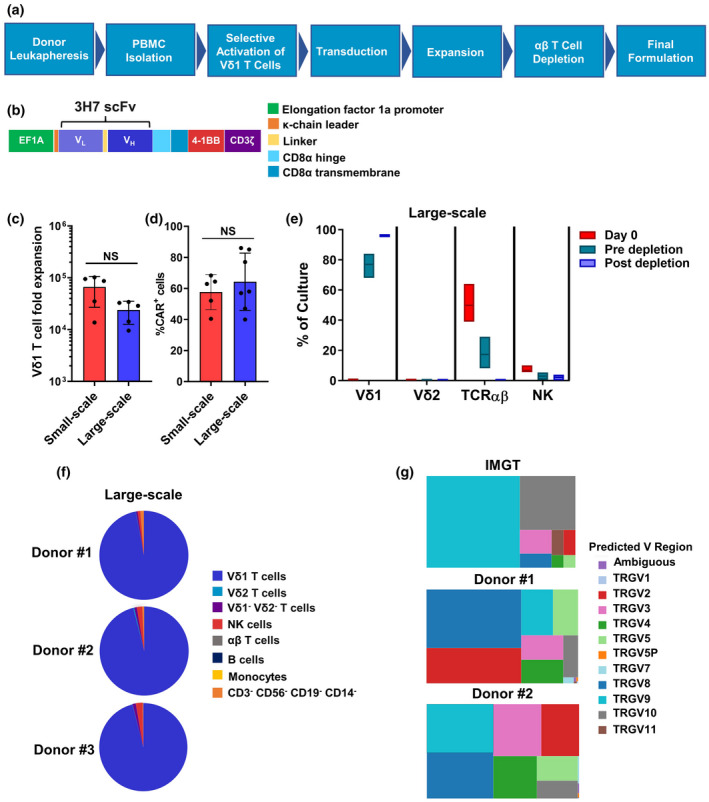
Overview of allogeneic CD20 CAR Vδ1 γδ T‐cell manufacturing process. (a) Flow chart highlighting the key steps in the manufacture of allogeneic CD20 CAR+ Vδ1 γδ T cells. Vδ1 γδ T cells present in healthy donor‐derived PBMCs are ex vivo‐activated and expanded with an agonistic anti‐Vδ1 mAb and then engineered to express the CD20 CAR by transduction with a self‐inactivating, replication‐incompetent gamma‐retroviral vector. After transduction, γδ T cells are further expanded in culture with human IL‐2 and are then enriched by αβ T‐cell depletion prior to formulation and cryopreservation. (b) Schematic diagram of the second‐generation CD20 CAR, which consists of the 3H7 scFv combined with CD8α hinge and transmembrane regions fused to the intracellular signalling domains of 4‐1BB and CD3ζ. CAR expression is regulated by the EF1a promoter. (c) The CD20 CAR+ Vδ1 γδ T‐cell manufacturing process results in a substantial fold expansion of Vδ1 γδ T cells. The mean ± standard deviation (SD) of five manufacturing runs using PBMCs from three different donors (large‐scale) and five different donors (small‐scale) are shown. (d) Average percentage of Vδ1 γδ T cells expressing the CD20 CAR from small‐scale and large‐scale manufacturing runs as measured by flow cytometry. The mean ± SD of five small‐scale manufacturing runs using PBMCs from five different donors and of seven large‐scale manufacturing runs using PBMCs from four different donors are shown. (e) Box plots of % cell composition using flow cytometric analysis of CD20 CAR+ Vδ1 T‐cell products derived from four different donors at Day 0, pre‐depletion and post‐depletion of αβ T‐cell time points (large scale). Box plot horizontal lines represent the mean, minimum and maximum values of each % cell type (Vδ1 T cells, Vδ2 T cells, αβ T cells and NK cells). (f) Cellular composition of final cell product was assessed by flow cytometry. Pie charts represent the composition of three representative donor‐derived CD20 CAR+ Vδ1 γδ T‐cell products. (g) Selective activation and expansion of Vδ1 γδ T cells with an agonistic anti‐Vδ1 mAb results in a diverse, not skewed, TCRγ repertoire. Treemaps of the TCRγ repertoire, which were created using Tableau software and are reported as TRGV (Vγ) usage in each functionally rearranged CDR3 sequence. Two donor‐derived CD20 CAR+ Vδ1 γδ T‐cell products are shown. The size of the square indicates its relative frequency in each sample. Relative frequencies of functionally rearranged TRGV (Vγ) sequences from ImMunoGeneTics (IMGT) are provided as a reference. Statistics were calculated using the paired t‐test; ns, not significant.
The natural TCR repertoire of antigen‐naïve Vδ1 γδ T cells at baseline is highly diverse but becomes clonally focused after immune challenge. 24 , 25 To investigate whether activation and expansion of Vδ1 γδ T cells with the agonistic anti‐Vδ1 mAb focuses the TCRγ repertoire, we analysed TCRγ diversity in cell products by immunosequencing. TCRγ repertoire analysis revealed a marked shift in the use of Vγ regions (e.g. Vγ2, Vγ4, Vγ5 and Vγ8) found in TCRγ chains that pair with the Vδ1 TCRδ chain (Figure 2g). 24 Importantly, we observed a diverse range of functional CDR3 sequences for each Vγ region, indicating no clonal focusing of the TCRγ repertoire in the final cell product. These data demonstrate that ex vivo activation and expansion of Vδ1 γδ T cells with an agonistic anti‐Vδ1 mAb retains a polyclonal TCRγ repertoire while enriching for TCRδ1 populations, which in turn preserves the adaptive targeting potential of γδTCRs on expanded CD20 CAR+ Vδ1 γδ T cells to contribute to tumor cell recognition and destruction.
The 3H7 CAR does not induce antigen‐independent tonic signalling
Engagement of CARs by target antigen is designed to lead to T‐cell activation and to subsequent differentiation into antitumor effectors. High CAR expression levels and scFv‐induced CAR aggregation on the T‐cell surface, however, can result in antigen‐independent tonic signalling, which may lead to T‐cell exhaustion and to reduced long‐term antitumor efficacy. 26 , 27 , 28 Accordingly, we evaluated the potential of the 3H7 CAR to induce antigen‐independent tonic signalling in the Jurkat‐Lucia NFAT reporter cell line. When the 3H7 CAR vector was transduced and expressed at high levels in Jurkat‐Lucia cells, target antigen engagement was required for NFAT activation (Figure 3a and b) and for IL‐2 production (data not shown), indicating that the 3H7 CAR itself does not induce antigen‐independent tonic signalling. To determine whether cellular context affects antigen‐independent tonic signalling, we first examined the propensity of 3H7 CARs to aggregate on Vδ1 γδ T cells by elucidating their surface structure. To this end, CD20 CAR+ Vδ1 γδ T cells were surface‐biotinylated and lysed in mild detergent, and their CAR complexes were immunoprecipitated, resolved by SDS‐PAGE and then analysed by Western blot analysis. When resolved under non‐reducing conditions, surface CARs existed as dimers and as higher‐order oligomers on the γδ T‐cell surface (Figure 3c, Supplementary figure 2a), suggesting that surface 3H7 CAR complexes aggregate as do surface TCR complexes. 29 Next, we determined whether such aggregation led to spontaneous CAR signalling by examining the phosphorylation status of CAR‐associated CD3ζ, specifically pTyr142, in unstimulated and stimulated CD20 CAR+ Vδ1 γδ T cells by Western blot analysis. Notably, we found that the CAR‐associated CD3ζ domain was phosphorylated in stimulated CD20 CAR+ Vδ1 γδ T cells but not in unstimulated CD20 CAR+ Vδ1 γδ T cell (Figure 3d, Supplementary figure 2b and c), demonstrating that the 3H7 CAR does not induce antigen‐independent tonic signalling in Vδ1 γδ T cells.
Figure 3.
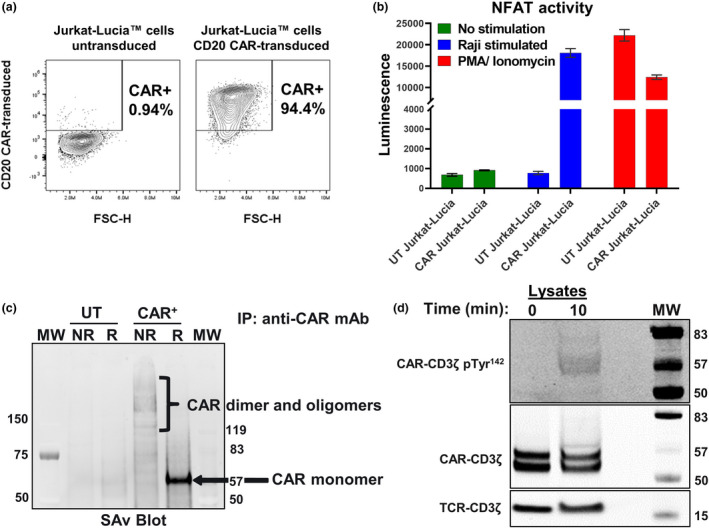
Structure and signalling capacity of the CD20 (3H7) CAR. (a) Transduction efficiency of Jurkat‐Lucia™ cells, which carry an NFAT‐inducible luciferase reporter, with the CD20 CAR gamma‐retroviral vector. Percentage of CAR‐transduced Jurkat‐Lucia cells expressing the CD20 CAR on their cell surface was measured by flow cytometry and is representative of three independent experiments. (b) The CD20 CAR, when expressed on the surface of Jurkat‐Lucia cells, does not induce antigen‐independent tonic signalling. CAR‐mediated signal transduction and activation were assessed in the presence or absence of target antigen by culturing parental (untransduced) and CAR‐transduced Jurkat‐Lucia cells either alone or at a 1:1 E:T ratio with CD20+ Raji cells. Stimulation with PMA (phorbol 12‐myristate 13‐acetate) and ionomycin was used as a positive control for NFAT activation. Duplicate samples were tested for each condition. Data are representative of three independent experiments. (c) The 3H7 CAR is expressed as dimers and higher‐order oligomers on the surface of Vδ1 γδ T cells. Untransduced (UT) or CAR‐transduced Vδ1 γδ T cells were surface‐biotinylated using EZ‐Link Sulfo‐NHS‐LC‐Biotin, lysed in Brij® O10 buffer containing a protease inhibitor cocktail and incubated with an anti‐CD20 CAR mAb coupled to Sepharose 4B beads. Immunoprecipitated proteins were resolved by non‐reducing (NR) and reducing SDS‐PAGE analysis and transferred to nitrocellulose membranes. Biotinylated CAR complexes were visualised by IRDye® 800CW Streptavidin and the Odyssey Imager. The positions of CAR dimers and oligomers (NR conditions) and monomers (R conditions) are marked. Data are representative of four independent experiments. (d) CAR‐associated phospho‐CD3ζ is only detected after CAR stimulation. CD20 CAR+ Vδ1 γδ T cells were unstimulated or stimulated on Protein L‐coated 96‐well plates for 10 min and then lysed in Milliplex® MAP Cell Signaling Universal Lysis Buffer. Lysates were resolved by reducing SDS‐PAGE analysis and then transferred to nitrocellulose membranes. Membranes were blocked and then incubated with antibodies against total CD3ζ or phospho‐CD3ζ (Tyr 142) overnight at 4°C on a shaker. CAR‐associated CD3ζ and phospho‐CD3ζ proteins were visualised using anti‐mouse IgG IR800 and the Odyssey Imager, and their positions are marked. In whole‐cell lysates, the CAR resolves as two bands: enzymatic deglycosylation of cell lysates revealed that the upper band is the glycosylated form, and that the lower band is the unglycosylated form (data not shown). Data are representative of three independent experiments.
CD20 CAR+ Vδ1 γδ T cells display a less‐differentiated memory cell phenotype, express multiple NKRs and chemokine receptors, and exist in a resting, non‐exhausted state
Phenotypic attributes of CAR αβ T cells have been identified that associate with improved clinical outcome. 30 , 31 , 32 For example, CAR T cells with a naïve‐like or less‐differentiated memory phenotype are associated with potent in vivo tumor control in mouse models and with objective responses in treated patients. 30 , 31 , 32 As γδ T cells differentiate from naïve T cells to terminally differentiated effector memory T cells, their expression of CD27 and CD45RA changes in this order: naïve‐like: CD27+ CD45RA+ → central memory (CM): CD27+ CD45RA− → effector memory (EM): CD27− CD45RA− → terminally differentiated effector memory (TEMRA): CD27− CD45RA+. 33 Phenotypic analysis of cell products from three donors revealed that > 50% of CD20 CAR+ Vδ1 γδ T cells possess a naïve‐like T‐cell memory phenotype, as they co‐express markers associated with both naïve (CD27, CD45RA and CD62L) and memory T cells (CD95 and CD45RO) (Figure 4a and b). Thus, the majority of CD20 CAR+ Vδ1 γδ T cells have the same naïve‐like memory cell phenotype as CAR αβ T‐cell products that display enhanced expansion and antitumor activity. 30 , 31 , 32
Figure 4.

Phenotypic analysis of CD20 CAR+ Vδ1 γδ T cells. (a, b) Flow cytometric analysis reveals that the majority of CAR+ Vδ1 γδ T cells in three donor‐derived CD20 CAR+ Vδ1 γδ T‐cell products exhibit a naïve‐like T‐cell memory phenotype. Expression levels of CCR7, CD62L, CD95 and CD45RO on gated naïve (CD27+ CD45RA+) CD4+ T cells from healthy donor‐derived PBMCs are shown as naïve T‐cell controls. Memory cell subsets were defined by the expression of CD27 and CD45RA: naïve‐like (CD27+ CD45RA+); central memory or TCM (CD27+ CD45RA−); effector memory or TEM (CD27− CD45RA−); and terminally differentiated effector memory or TEMRA (CD27− CD45RA+). (c) Heatmap showing percentages of CAR+ Vδ1 γδ T cells in three donor‐derived CD20 CAR+ Vδ1 γδ T‐cell products that express chemokine receptors, natural killer (NK) cell receptors and terminal differentiation markers. (d) Heatmap showing percentages of CAR+ Vδ1 γδ T cells in four donor‐derived CD20 CAR+ Vδ1 γδ T‐cell products that co‐express PD‐1 and another co‐inhibitory receptor, that is TIM‐3, LAG‐3 or TIGIT.
Efficient trafficking to the tumor site plus NKR‐mediated tumor cell killing are proposed mechanisms of action for γδ T‐cell‐mediated antitumor activity. 12 , 34 , 35 Expression of CCR4, CCR5, CCR8, CXCR3, CXCR4 and CXCR6, which are involved in the homing of lymphocytes to lymphoid and non‐lymphoid tumors and tissues, was detected on subpopulations of CAR+ Vδ1 γδ T cells (Figure 4c). The expression of multiple chemokine receptors may allow CD20 CAR Vδ1 γδ T cells to respond to a diverse array of tumor‐derived chemokines. In addition, the NK cell‐associated activating receptors, NKG2D and DNAM1, were expressed on > 90% of CD20 CAR+ Vδ1 γδ T cells (Figure 4c). This phenotypic profile is clinically relevant to treating B‐cell malignancies, given that CCL5, CCL17, CCL22 and CXCL12, which are ligands for several chemokine receptors expressed by CD20 CAR+ Vδ1 γδ T cells, are detected in the lymphoma microenvironment and that B‐cell lymphomas express an array of NKR ligands. 36 , 37 , 38
T‐cell activation is critical to mount an effective antitumor response, but antigen‐independent tonic signalling or chronic stimulation can lead to an exhausted phenotype with impaired effector functions. 26 , 27 , 28 We found that ≤ 1% of cells co‐expressed PD‐1 and another co‐inhibitory receptor (Figure 4d), a phenotype that marks exhausted T cells, including γδ T cells. 39 , 40 These results confirm that the 3H7 CAR does not induce antigen‐independent tonic signalling when expressed in γδ T cells and suggest that the CD20 CAR+ Vδ1 γδ T cells are not chronically stimulated during the manufacturing process.
CAR expression and signalling enhance the intrinsic antitumor activities of Vδ1 γδ T cells
Because γδ T cells have the potential to kill tumor cells through recognition of stress‐induced surface molecules, 12 we investigated the effects of CAR expression and signalling on the cytotoxic potential of Vδ1 γδ T cells. To accomplish this, we evaluated and compared the cytotoxic activities of small‐scale manufactured untransduced and CAR‐transduced Vδ1 γδ T cells against CD20+ target cells in a 48‐h IncuCyte® Immune Cell Killing Assay in which target cell death is monitored over time. As shown in Figure 5, untransduced Vδ1 γδ T cells controlled B‐cell tumor growth at 10:1 and 3.3:1 effector:target (E:T) ratios for the 48‐h assay duration. Similar cytotoxic activities were observed between untransduced and irrelevant CAR‐transduced Vδ1 γδ T‐cell controls (Supplementary figure 3). However, only CD20 CAR+ Vδ1 γδ T cells achieved durable tumor regression at all tested E:T ratios, indicating that CAR expression and signalling augment the intrinsic tumoricidal activity of Vδ1 γδ T cells against B‐cell tumor cells.
Figure 5.
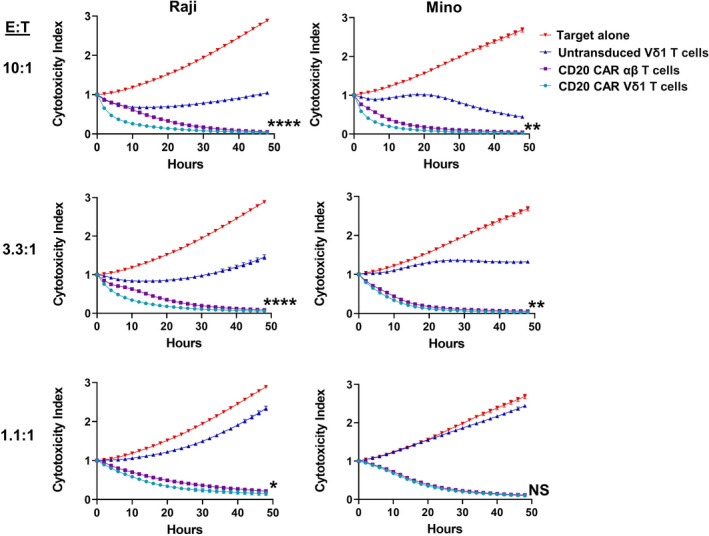
Comparison of the cytotoxic potentials of untransduced Vδ1 γδ T cells, CD20 CAR‐transduced Vδ1 γδ T cells and CD20 CAR‐transduced αβ T cells against CD20+ target cells. Cytotoxic potentials of untransduced Vδ1 γδ T cells, CD20 CAR‐transduced Vδ1 γδ T cells (71.5% CAR+) and CD20 CAR‐transduced αβ T cells (68% CAR+), all derived from the same healthy donor, were evaluated against CD20+ target cells in a 48‐h IncuCyte Immune Cell Killing Assay, in which T cells were co‐cultured with NucR‐expressing Raji or Mino target cells at E:T ratios of 10:1, 3:3:1 and 1.1:1. The viability of the NucR‐expressing targets, reported as cytotoxicity index, was monitored every 2 h over the course of 48 h with the IncuCyte Live‐Cell Analysis System. Please note that the lower the cytotoxicity index, the better the cytotoxic potential. Data are shown as mean ± SD of triplicates and are representative of three independent experiments using cell products from two different donors. The killing curves for each cell product were statistically analysed using the mixed model two‐way ANOVA. (****P < 0.0001, **P < 0.01 and *P < 0.05; NS, not significant).
Next, using the 48‐h IncuCyte Immune Cell Killing Assay, we sought to compare the cytotoxic activities of small‐scale manufactured CD20 CAR+ Vδ1 γδ T cells and CD20 CAR+ αβ T cells, both derived from the same donor and both ~70% CAR+. Although the cytotoxicity indexes were equivalent for CD20 CAR+ Vδ1 γδ T cells and CD20 CAR+ αβ T cells at the end of the 48‐h assay, CD20 CAR+ Vδ1 γδ T cells killed CD20+ target cells with significantly faster kinetics than did CD20 CAR+ αβ T cells, especially at higher E:T ratios (Figure 5). These results reveal a striking difference between CAR+ Vδ1 γδ T cells and CAR+ αβ T cells in their kinetics of tumor cell killing.
Robust antitumor activities of large‐scale manufactured CD20 CAR+ Vδ1 γδ T cells after in vitro target antigen engagement
CD20 CAR+ Vδ1 γδ T cells are anticipated to display multiple effector functions that may represent key functional attributes. Because CD20 expression levels vary per B‐cell lymphoma type, 41 we assessed the cytotoxic potential of large‐scale manufactured CD20 CAR+ Vδ1 γδ T cells against Mino cells (mantle cell lymphoma; high CD20), Raji cells (Burkitt lymphoma; intermediate CD20) and WILL‐2 cells (diffuse large B‐cell lymphoma; very low CD20) (Figure 6a). The ability of CD20 CAR+ Vδ1 γδ T cells to kill these target cells was both E:T ratio‐dependent and CD20 expression level‐dependent, with cytotoxic activity increasing as E:T ratios increased and as CD20 expression levels increased (Figure 6b and Supplementary figure 4a). Because CD20 CAR+ Vδ1 γδ T cells lysed WILL‐2 cells, which are derived from a patient who relapsed after eight courses of rituximab therapy, 42 these results highlight the potential of CD20 CAR+ Vδ1 γδ T cells to be effective in the treatment of B‐cell NHL patients exhibiting CD20 downregulation associated with rituximab‐resistant tumors.
Figure 6.
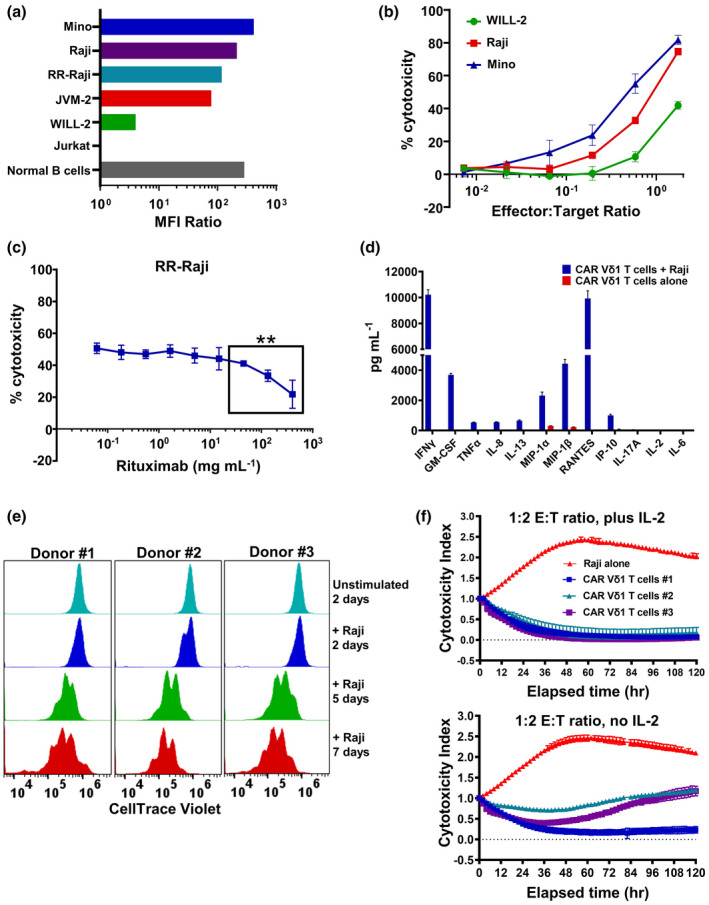
In vitro functional analyses of preclinical large‐scale manufactured CD20 CAR+ Vδ1 γδ T cells. (a) Relative CD20 expression levels on malignant B‐cell lines [Mino, Raji, JVM‐2, WILL‐2, RR‐Raji], normal peripheral blood B cells and the CD20− Jurkat T‐ALL cell line as determined by flow cytometry. Relative CD20 expression is reported as CD20 geoMFI/isotype control geoMFI (MFI ratio). The data are representative of two independent experiments. (b) Cytotoxic potential of CD20 CAR+ Vδ1 γδ T cells against B‐lymphoma cell lines expressing varying levels of CD20 in a short‐term cytotoxicity assay. Cytotoxic potential was assessed by titrating E:T ratios in an 18‐h cytotoxicity assay in which CD20 CAR+ Vδ1 γδ T‐cell effectors were co‐cultured with RFluc‐expressing target cells. Controls included CD20 CAR+ Vδ1 γδ T cells and B‐lymphoma cell lines cultured alone. Data are shown as mean ± SD of triplicates and are representative of two independent experiments using two different donor‐derived CD20 CAR+ Vδ1 γδ T‐cell products. (c) Effect of clinically relevant concentrations of rituximab on the ability of CD20 CAR+ Vδ1 γδ T cells to recognise and kill RFluc‐expressing RR‐Raji target cells. Cytotoxic potential of CD20 CAR+ Vδ1 γδ T cells against RR‐Raji cells at a fixed 3:1 ratio was assessed after pre‐incubation of target cells with increasing concentrations of rituximab (0–400 μg mL−1) in an 18‐h luciferase cytotoxicity assay. Data are shown as mean ± SD of triplicates and are representative of two different rituximab‐resistant B‐lymphoma cell lines [**P ≤ 0.005 (t‐test)]. (d) Cytokine and chemokine production by CD20 CAR+ Vδ1 γδ T‐cell effectors after an 18‐h co‐culture with Raji cells at fixed 1.75:1 ratio. Levels of IFN‐γ, GM‐CSF, TNF‐α, IL‐2, IL‐6, IL‐8, IL‐13, MIP‐1α, MIP‐1β, RANTES, IP‐10 and IL‐17A were measured in culture medium supernatants using the Luminex platform. Data are shown as mean ± SD of triplicates and are representative of two independent experiments using two different donor‐derived CD20 CAR+ Vδ1 γδ T‐cell products. (e) Proliferative potential of CD20 CAR+ Vδ1 γδ T cells following three rounds of target antigen exposure in a 7‐day culture period. Aliquots of cells harvested on Days 2, 5 and 7 were analysed by flow cytometry to assess cellular proliferation by dye dilution. Representative data from three independent experiments using three donor‐derived CD20 CAR+ Vδ1 γδ T‐cell products are shown. (f) Cytotoxic potential of CD20 CAR+ Vδ1 γδ T cells against Raji cells in a long‐term cytotoxicity assay. Cytotoxic potential was assessed at a 1:2 E:T ratio in a 120‐h cytotoxicity assay in which CD20 CAR+ Vδ1 γδ T cells were co‐cultured with NucR‐expressing Raji cells ± 20 IU of human IL‐2. The viability of the NucR‐expressing Raji cells was monitored every 2 h over the course of 120 h with the IncuCyte Live‐Cell Analysis System. Data are shown as mean ± SD of triplicates and represent three donor‐derived CD20 CAR+ Vδ1 γδ T‐cell products.
Given that R/R B‐cell NHL patients may receive maintenance rituximab therapy, we investigated the potential effects of residual serum rituximab on the cytotoxic potential of CD20 CAR+ Vδ1 γδ T cells. In the presence of physiological concentrations of rituximab (0–400 μg mL−1), 43 rituximab‐resistant Raji (RR‐Raji) cell killing was significantly impaired at rituximab concentrations between 44 and 400 μg mL−1, but only at the 400 μg mL−1 peak concentration was killing inhibited by > 50% (Figure 6c). The inhibitory effect of rituximab on tumor cell killing may be because of the blocking of CD20 CAR binding, as rituximab has been recently shown to interact with amino acid residues in both of CD20’s extracellular loops. 44
Within 18 h of target antigen engagement, CD20 CAR+ Vδ1 γδ T cells produced IFN‐γ, GM‐CSF, TNF‐α, IL‐8, IL‐13, MIP‐1α, MIP‐1β, RANTES and IP‐10 but not IL‐6, nor the tumor‐promoting IL‐17 cytokine (Figure 6d and Supplementary figure 4b). Notably, in vitro CAR‐activated allogeneic and autologous CD19 CAR αβ T cells also produce GM‐CSF, IFN‐γ, TNF‐α, IL‐8, IL‐10, IL‐13 and MIP‐1α, with elevated IFN‐γ, IL‐8 and MIP‐1α levels being produced by in vitro CAR‐activated autologous CD19 CAR αβ T cells from patients who respond to CAR T‐cell therapy. 45 , 46
Long‐term functional assays were also performed to evaluate the proliferative and cytotoxic potential of CD20 CAR+ Vδ1 γδ T cells when tumor burden is high. To evaluate proliferative potential, CellTrace Violet‐labelled CD20 CAR+ Vδ1 γδ T cells were stimulated in a 7‐day repetitive target antigen engagement assay. After the second and third rounds of Raji cell stimulation, dye dilution indicative of multiple rounds of cellular division was observed for the three donor‐derived cell products tested (Figure 6e). Compared with Raji cell repetitive stimulation, a further increase in cell division was observed using the higher CD20‐expressing Mino cell line (Supplementary figure 5). To assess cytotoxic potential in the presence of high tumor burden, CD20 CAR+ Vδ1 γδ T cells and NucR‐expressing Raji cells were co‐cultured at a low 1:2 ratio in a 120‐h IncuCyte Immune Cell Killing Assay in the presence or absence of 20 IU human IL‐2. Without the addition of exogenous IL‐2, cell products from Donors #2 and #3 controlled tumor growth for the 120‐h assay duration, but only the cell product from Donor #1 achieved durable tumor regression (Figure 6f). However, with the addition of exogenous IL‐2 to simulate the effects of lymphodepleting chemotherapy on homeostatic cytokine levels, the antitumor activities of the cell products from Donors #2 and #3 were enhanced and both achieved durable tumor regression (Figure 6f). These data suggest that the CD20 CAR+ Vδ1 γδ T cells in the presence of high tumor burden are capable of proliferating and, in the presence of a homeostatic cytokine, of achieving durable tumor growth inhibition.
CD20 CAR+ Vδ1 γδ T cells do not initiate xenogeneic GvHD in NSG mice
γδ T cells recognise antigens in an MHC‐unrestricted manner and therefore are not expected to initiate GvHD. 9 , 12 , 14 , 15 However, the small fraction of αβ T cells that remain in the cell product after αβ T‐cell depletion has the potential to be alloreactive and to induce GvHD in patients. Therefore, to assess the potential of the CD20 CAR+ Vδ1 γδ T‐cell product to induce xenogeneic GvHD, NSG mice were injected intravenously (IV) on Day 1 with 4 × 107 CD20 CAR+ Vδ1 γδ T cells, 4 × 107 PBMCs (positive control) or medium alone (negative control). Both CD20 CAR+ Vδ1 γδ T‐cell‐treated and negative control groups survived to Day 61, whereas mice in the PBMC‐treated group had a median survival of 19 days and showed signs of GvHD, including accelerated body weight loss, as early as Day 7 (Supplementary figure 6a and b; data not shown). PBMC‐treated mice were euthanised between Day 14 and Day 35, and their deaths were classified as GvHD‐related because of > 15% body weight loss (mean GvHD score 4.8 ± 0.5). By contrast, on Day 61, mean GvHD scores for mice treated with CD20 CAR+ Vδ1 γδ T cells were similar to those for mice treated with medium (2.6 ± 0.2 versus 2.0 ± 0.4) (Supplementary figure 6a and b; data not shown). Histopathological analysis of selected organs harvested at Day 61 from CD20 CAR+ Vδ1 γδ T‐cell‐treated mice showed no treatment‐related microscopic findings and no human CD3+ cells by IHC in any harvested tissues (Supplementary figure 6c). In PBMC‐treated mice, however, infiltrating human CD3+ cells were observed in pancreas, skin, whole lung, spleen, liver and kidneys, and treatment‐related necrosis was observed in the pancreas, skin, whole lung, spleen and liver (Supplementary figure 6c; data not shown). Therefore, neither the CD20 CAR+ Vδ1 γδ T cells nor the residual αβ T cells in the cell product induce xenogeneic GvHD or any acute, chronic or delayed‐onset toxicities in NSG mice.
To further investigate the lack of GvHD observed for CD20 CAR+ Vδ1 γδ T cells, we performed an ex vivo tissue analysis on Raji‐bearing NSG mice that received a single IV dose of either CellTrace Violet‐labelled CD20+ CAR‐transduced Vδ1 γδ or CellTrace Violet‐labelled CD20+ CAR‐transduced αβ T cells. At Day 6 post‐treatment, tumors, blood, bone marrow, spleen and liver were harvested, and T‐cell proliferation was assessed by flow cytometry. Ex vivo analysis showed robust proliferation of CD20 CAR+ αβ T cells in all analysed tissues, but robust proliferation of CD20 CAR+ Vδ1 T cells was observed only in the tumor (Supplementary figure 7). These data, combined with those from the in vivo GvHD study, demonstrate that CD20 CAR+ αβ T cells have significant xenoreactive proliferation in all non‐targeted murine tissues examined, whereas the CD20 CAR+ Vδ1 γδ T cells, and the respective manufacturing process, are associated with no measurable xenoreactive potential and, by extension, no apparent alloreactive potential.
CD20 CAR+ Vδ1 γδ T cells mediate in vivo tumor growth inhibition in B‐cell lymphoma xenografts in NSG mice
The in vivo efficacy of CD20 CAR+ Vδ1 γδ T cells was evaluated in subcutaneously (SC) implanted xenograft models using Raji and JVM‐2 cell lines, which express intermediate and low CD20 levels, respectively (Figure 6a). NSG mice with well‐established tumors (~200 mm3 in volume) received a single IV administration of CD20 CAR+ Vδ1 γδ T cells at three dose levels. Because of species specificity of key cytokines supporting T‐cell growth and differentiation, NSG mice do not possess the cytokine‐rich lymphodepleted microenvironment that is key to CAR T‐cell expansion, function and persistence in patients. 47 , 48 Therefore, to simulate the favorable cytokine profile induced by a lymphodepletion regimen, 48 CD20 CAR+ Vδ1 γδ T‐cell doses were supplemented with exogenous human IL‐2. Significant tumor growth inhibition was observed at Day 14 post‐treatment in Raji‐bearing mice (P = 0.0011; Figure 7a and Supplementary figure 8) and at Day 30 post‐treatment in JVM‐2‐bearing mice (P = 0.0062; Figure 7b). Importantly, the in vivo antitumor response was durable, with all dose levels controlling tumor growth until Day 36 in the Raji model and Day 55 in the JVM‐2 model. These data indicate that CD20 CAR+ Vδ1 γδ T cells significantly inhibit tumor cell growth in well‐established human xenograft tumors expressing different levels of CD20.
Figure 7.
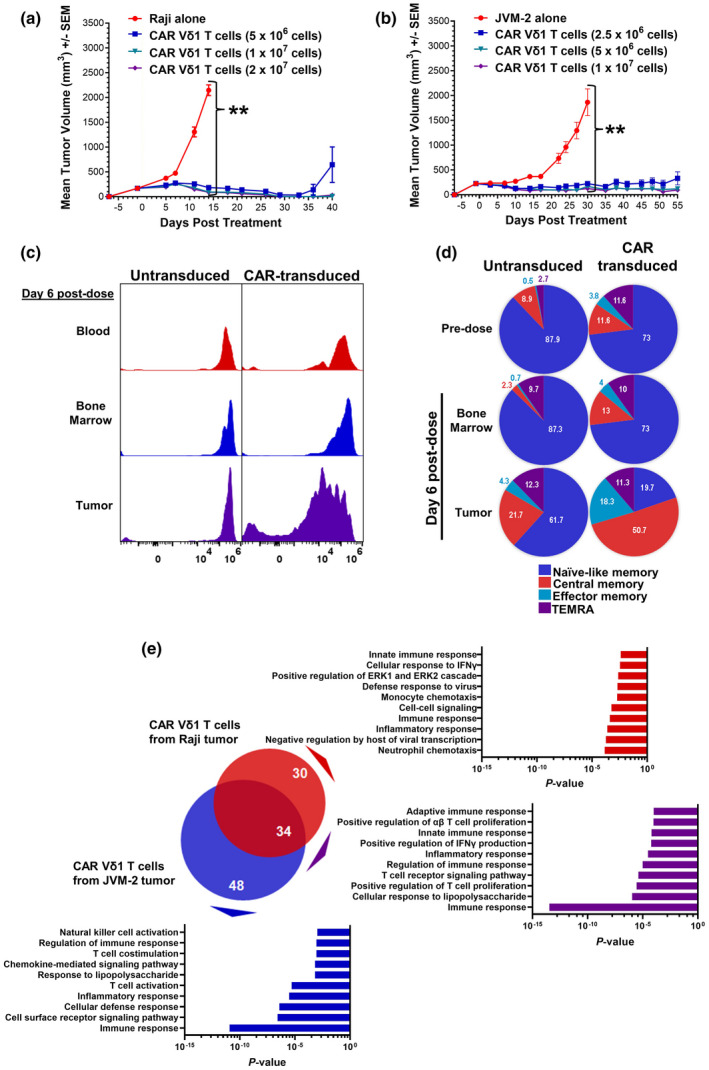
In vivo and ex vivo analyses of CD20+ tumor‐bearing NSG mice treated with CD20 CAR+ Vδ1 γδ T cells. (a) In vivo efficacy of three different doses (5 × 106, 1 × 107 and 2 × 107) of viable CD20 CAR+ Vδ1 γδ T cells in combination with 13 000 IU IL‐2 in a SC Raji Burkitt lymphoma model in NSG mice (n = 5 per group). The SC xenograft model was chosen because tumor control in this model is dependent on the ability of CAR T cells to traffic to the tumor, to proliferate and to lyse tumor cells. Tumor growth, body weight and animal health were monitored two times per week after tumor implantation. Tumor volume was calculated using the formula: Volume (mm3) = (length × width2)/2. The Kruskal–Wallis test was used to assess statistical significance among the groups on Day 14 (**P = 0.0011). Data are representative of three independent experiments using one donor‐derived CD20 CAR+ Vδ1 γδ T‐cell product. (b) In vivo efficacy of three different doses (2.5 × 106, 5 × 106 and 1 × 107) of viable CD20 CAR+ Vδ1 γδ T cells in combination with 13 000 IU IL‐2 in a SC JVM‐2 mantle cell lymphoma model in NSG mice (n = 5 per group). The Kruskal–Wallis test was used to assess statistical significance among the groups on Day 30 (**P = 0.0062). Data are representative of two independent experiments using one donor‐derived CD20 CAR+ Vδ1 γδ T‐cell product. (c) Proliferative potential of untransduced and CD20 CAR+ Vδ1 γδ T cells in blood, bone marrow and tumor harvested from Raji tumor‐bearing mice on Day 6 post‐treatment. Untransduced and CD20 CAR+ Vδ1 γδ T cells were labelled with CellTrace Violet prior to adoptive transfer at a dose of 8 × 106 cells in combination with IL‐2 (n = 3 per group). Data are representative of two independent experiments using one donor‐derived CD20 CAR+ Vδ1 γδ T‐cell product. (d) Differentiation potential of untransduced and CD20 CAR+ Vδ1 γδ T cells in bone marrow and tumor harvested from Raji tumor‐bearing mice on Day 6 post‐treatment. Memory cell subsets were defined by the expression of CD27 and CD45RA: naïve‐like (CD27+ CD45RA+); TCM (CD27+ CD45RA−); TEM (CD27− CD45RA−); and TEMRA (CD27− CD45RA+). Memory phenotypes of pre‐dose untransduced and CD20 CAR+ Vδ1 γδ T cells are shown for comparison. (e) Venn diagrams of DEGs in Raji tumors treated with 1 × 107 CD20 CAR+ Vδ1 γδ T cells (red circle) from two donors and in JVM‐2 tumors treated with 1 × 107 CD20 CAR+ Vδ1 γδ T cells (blue circle) from two donors in combination with IL‐2. The overlapping area represents DEGs that are shared between the two tumor models. Gene expression levels were quantitated using the nCounter® CAR T Characterization and Human Immunology Panels on the nCounter® SPRINT Profiler according to the manufacturer’s instructions. nSolver™ software was used to normalise the data and to identify DEGs (i.e. adjusted P ≤ 0.05 and log2 fold change ≥ 1) in tumors treated with CD20 CAR+ Vδ1 γδ T cells versus tumors treated with untransduced Vδ1 γδ T cells. Gene ontology analysis was performed using Database for Annotation, Visualization and Integrated Discovery (DAVID, https://david.ncifcrf.gov/) to identify the biological pathways associated with DEGs that are unique to each tumor model and that are shared by the two tumor models.
To elucidate the mechanisms of action that lead to tumor regression, ex vivo analysis of γδ T cells in tumor‐bearing mice was performed. Raji‐bearing NSG mice received a single IV dose of CellTrace Violet‐labelled untransduced or CD20 CAR‐transduced Vδ1 γδ T cells. At Day 6 post‐treatment, when initial tumor regression was observed, tumors, blood, and bone marrow were harvested, and proliferation and memory cell phenotype were assessed by flow cytometry. In the tumor, a marked dilution of the CellTrace Violet dye was observed in CAR‐transduced, but not untransduced, Vδ1 γδ T cells (Figure 7c), indicating that the CAR induced cellular proliferation after CD20 engagement. By contrast, minimal cell proliferation was observed in the blood and bone marrow, consistent with the lack of human CD20 expression in these tissues. No changes were observed in pre‐ and post‐dose memory cell phenotypes of untransduced or CAR‐transduced Vδ1 γδ T cells in the bone marrow (Figure 7d). As expected, intratumoral CAR‐transduced Vδ1 γδ T cells underwent differentiation, with dramatic increases in the percentages of TCM cells (11.6–50.7%) and TEM cells (3.8–18.3%) (Figure 7d). Although intratumoral untransduced Vδ1 γδ T cells failed to proliferate (Figure 7c), they underwent differentiation in response to recognition of Raji cells by their endogenous receptors, as evidenced by increases in the percentages of TCM cells (8.9–21.7%), TEM cells (0.5–4.3%) and TEMRA cells (2.7–12.3%) (Figure 7d). The data indicate that CAR signalling not only leads to robust proliferation but also enhances CAR‐independent T‐cell memory differentiation of intratumoral CD20 CAR+ Vδ1 γδ T cells.
To identify a gene signature associated with tumor regression, we performed immune‐related gene expression profiling of γδ T cells and tumor cells in Raji and JVM‐2 xenograft models. Total RNA was extracted from intact tumors harvested at the first appearance of tumor control from tumor‐bearing mice treated with CD20 CAR+ Vδ1 γδ T cells, untransduced Vδ1 γδ T cells or no cells. Gene expression profiling identified 34 differentially expressed genes (DEGs) shared by CD20 CAR+ Vδ1 γδ T‐cell‐treated Raji and JVM‐2 tumors (Figure 7e; Supplementary table 1). Gene ontology analysis revealed that the shared DEGs are involved in innate and adaptive immune responses, TCR signalling, T‐cell proliferation and positive regulation of IFN‐γ production. These results confirm our hypothesis that the in vivo antitumor activities of CD20 CAR+ Vδ1 γδ T cells combine innate and adaptive mechanisms.
cGMP‐compliant clinical‐scale manufacturing of CD20 CAR+ Vδ1 γδ T cells maintains in vitro and in vivo antitumor activities
Phenotypic and in vitro and in vivo functional analyses were performed to determine whether clinical‐scale manufacturing of CD20 CAR+ Vδ1 γδ T cells maintained a favorable memory cell phenotype and potent antitumor activities. Phenotypic analysis of clinical‐scale cell product revealed that 79% of CD20 CAR+ Vδ1 γδ T cells displayed the desirable, naïve‐like T‐cell memory phenotype (Figure 8a). Additionally, functional analyses of clinical‐scale CD20 CAR+ Vδ1 γδ T cells demonstrated E:T‐dependent tumor cell killing and IFN‐γ production after co‐incubation with Raji cells (Figure 8b), durable in vitro control of tumor growth of Raji cells in the 120‐h cytotoxicity assay (Figure 8c) and statistically significant in vivo tumor control in Raji‐bearing NSG mice (P = 0.0001; Figure 8d). Therefore, clinical‐scale CD20 CAR+ Vδ1 γδ T cells retained the key phenotypic and functional attributes of preclinical large‐scale CD20 CAR+ Vδ1 γδ T cells.
Figure 8.
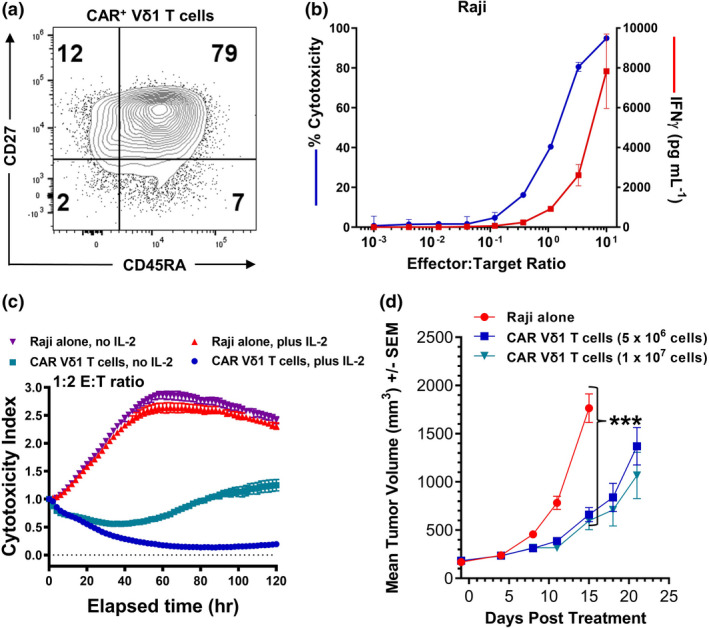
Phenotypic and functional analyses of clinical‐scale CD20 CAR+ Vδ1 γδ T cells. (a) Dot plot demonstrating that 79% of clinical‐scale CD20 CAR+ Vδ1 γδ T cells exhibit a naïve‐like T‐cell memory phenotype. Data are representative of two independent experiments. (b) Cytotoxic potential of, and IFN‐γ production by, clinical‐scale CD20 CAR+ Vδ1 γδ T cells (49% CAR+) against Raji cells in an 18‐h cytotoxicity assay. Controls included CD20 CAR+ Vδ1 γδ T cells and B‐lymphoma cell lines cultured alone. Levels of IFN‐γ in culture medium supernatants were measured using the Luminex platform. Data are shown as mean ± SD of triplicates and are representative of three independent experiments. (c) Cytotoxic potential of clinical CD20 CAR+ Vδ1 γδ T cells against Raji cells in a long‐term cytotoxicity assay. Cytotoxic potential was assessed at a 1:2 E:T ratio in a 120‐h cytotoxicity assay in which CD20 CAR+ Vδ1 γδ T‐cell effectors were co‐cultured with NucR‐expressing Raji cells in the presence or absence of 20 IU of human IL‐2. The viability of the NucR‐expressing Raji cells was monitored every 2 h over the course of 120 h with the IncuCyte Live‐Cell Analysis System. Data are shown as mean ± SD of triplicates and are representative of three independent experiments. (d) In vivo efficacy of two different doses (5 × 106 and 1 × 107) of viable clinical‐scale CD20 CAR+ Vδ1 γδ T cells in combination with 13 000 IU IL‐2 in a SC Raji Burkitt lymphoma model in NSG mice (n = 7 mice per cell treatment group and n = 10 mice in the tumor‐alone group). Tumor growth, body weight and animal health were monitored two times per week after tumor implantation. The Kruskal–Wallis test was used to assess statistical significance among groups on Day 15 (***P = 0.0001). The data represent one independent experiment.
Discussion
γδ T cells are an attractive candidate for CAR T‐cell therapy because of their ability to combine innate and adaptive mechanisms to recognise and kill malignant cells and because their prevalence in both haematological and solid tumors is associated with clinical benefit. 12 , 13 , 14 , 15 , 16 However, given that they are only a small fraction of PBMCs, it was necessary to develop a large‐scale manufacturing process that expands peripheral blood γδ T cells to clinically relevant numbers while maintaining their intrinsic antitumor properties. In the current study, we not only demonstrated that allogeneic peripheral blood Vδ1 γδ T cells can be manufactured as an off‐the‐shelf product but also identified phenotypic and functional attributes of CD20 CAR+ Vδ1 γδ T cells that are unique to γδ T cells and that are shared by effective CAR αβ T‐cell products. Phenotypic analyses revealed that CD20 CAR+ Vδ1 γδ T cells possess a naïve‐like T‐cell memory phenotype and exist in a resting, non‐exhausted state, both of which are phenotypes shared by effective CAR αβ T‐cell therapies. 26 , 27 , 28 , 30 , 31 , 32 Also, CD20 CAR+ Vδ1 γδ T cells express multiple chemokine receptors and NKRs, which may facilitate their homing to the tumor site and their innate recognition and killing of tumor cells. The latter phenotypic profile is clinically relevant to treating mature B‐cell malignancies, as multiple chemokines are detected in the lymphoma microenvironment and that B‐cell lymphomas express an array of NKR ligands. 36 , 37 , 38 CD20 CAR+ Vδ1 γδ T cells also exhibit antitumor activities such as in vitro tumor cell killing and proinflammatory cytokine production in addition to in vivo tumor growth inhibition, shown to be mediated through a combination of innate and adaptive mechanisms, in two xenograft models. Last, neither the CAR+ Vδ1 γδ T cells nor the residual αβ T cells in the cell product induced xenogeneic GvHD or any toxicities in the NSG mouse. Together, these preclinical data support the clinical development of allogeneic CD20 CAR+ Vδ1 γδ T cells.
Our study demonstrates that Vδ1 γδ T cells have a large capacity for proliferation. The Vδ1 γδ T‐cell subset, which represents less than 1% of PBMCs, 24 underwent a > 20 000‐fold expansion during the manufacturing process. Phenotypic analysis of preclinical‐ and clinical‐scale CD20 CAR+ Vδ1 γδ T‐cell products revealed, even with this sizeable expansion, that they display a naïve‐like T‐cell memory phenotype and exist in a resting, non‐exhausted state. Notably, critical differences in the memory cell phenotypes of Vδ1 γδ T‐cell‐ and Vδ2 γδ T‐cell‐based products have been reported, with untransduced and CAR‐transduced Vδ1 γδ T cells exhibiting a naïve‐like memory cell phenotype and untransduced and CAR‐transduced Vδ2 γδ T cells exhibiting a more‐differentiated TEM phenotype. 49 , 50 It is not known whether this phenotypic difference is because of differences in modes of cell activation and expansion or because of differences in the differentiation pathways of the two Vδ subsets. Nonetheless, to date, it is the Vδ1 γδ T‐cell‐based products that have the same memory cell phenotype as CAR αβ T‐cell products that display enhanced expansion and antitumor activity. 30 , 31 , 32 For memory αβ T cells, it is the less‐differentiated memory T‐cell phenotype that is associated with high proliferative potential after CAR activation. 51 , 52 Likewise, we have demonstrated that CD20 CAR+ Vδ1 γδ T cells have the capacity to undergo multiple rounds of proliferation in vitro in a repetitive stimulation assay and in vivo in implanted human B‐lymphoma xenografts. Together, these data demonstrate that our large‐scale manufacturing process produces clinically relevant numbers of CAR+ Vδ1 γδ T cells with a naïve‐like memory cell phenotype and with a high proliferative potential.
Because patients with B‐cell malignancies may have tumors that become resistant to rituximab‐mediated cytotoxicity or may receive maintenance rituximab therapy, we investigated whether rituximab‐resistant cell lines can be killed by CD20 CAR+ Vδ1 γδ T cells and whether clinically relevant concentrations of rituximab could affect CAR‐mediated killing by CD20 CAR+ Vδ1 γδ T cells. In the absence of rituximab, CD20 CAR+ Vδ1 γδ T cells were able to recognise and kill WILL‐2 and RR‐Raji cells, both of which are resistant to rituximab‐mediated cytotoxicity, 42 suggesting that the CD20 CAR+ Vδ1 γδ T‐cell product has the potential to be effective in the treatment of patients with rituximab‐resistant tumors. In the presence of rituximab, CAR‐mediated killing or RR‐Raji cell killing was largely retained, and only at a concentration of 400 μg mL−1 was killing inhibited by 50%. For reference, median serum rituximab concentrations in B‐cell NHL patients are 96.2 μg mL−1 (range 0–284.3 μg mL−1 in 124 patients) 1 month post‐rituximab treatment and 20.2 μg mL−1 (range 0–96.8 μg mL−1 in 104 patients) 3 months post‐rituximab treatment. 43 It is important to note that because rituximab does not penetrate all areas of a lymphoma, 53 the serum rituximab concentration may not adequately reflect its tumor concentration; therefore, the inhibitory effect of rituximab observed in the in vitro cytotoxicity assay may overestimate its inhibitory effect in vivo.
Although the NSG mouse is the preferred animal model in which to evaluate in vivo antitumor efficacy of candidate CAR T‐cell products, it is important to note that while NSG mice do have available lymphoid niches for CAR T‐cell engraftment, they do not have species‐specific homeostatic cytokines (e.g. IL‐2, IL‐7 and IL‐15) that support and maintain CAR T‐cell persistence. 47 , 48 For this reason, CD20 CAR+ Vδ1 γδ T cells were provided with exogenous human IL‐2, as cytokine support, to enhance their expansion and persistence in tumor‐bearing NSG mice. In the absence of such support, the in vivo antitumor response of CD20 CAR+ Vδ1 γδ T cells was not durable (data not shown). Notably, IL‐2 supplementation is not required when evaluating in vivo preclinical efficacy of non‐gene‐edited CAR+ αβ T‐cell products, as recognition of murine MHC by the human αβTCR provides these CAR+ αβ T cells with a secondary means of activation and consequently a proliferative advantage. Interestingly, in vivo preclinical studies evaluating gene‐edited CAR+ αβ T cells (e.g. knockout of endogenous TCR) or CAR+ NK cells, neither of which induce GvHD, demonstrated a requirement for human homeostatic cytokines in order to observe significant antitumor efficacy. 54 , 55 Alternative approaches to improve survival and persistence of allogeneic CAR products are the inclusion of the IL2 or IL15 gene into the CAR construct to provide cytokine support. 56 , 57 Engineering of CAR Vδ1 γδ T cells to secrete IL‐15 is currently being explored for use in solid tumors. 58 Together, these data highlight the importance of bioactive homeostatic cytokines in CAR‐mediated expansion, function and persistence of allogeneic products in NSG tumor models.
Our investigation of the molecular mechanisms of action of CD20 CAR+ Vδ1 γδ T cells revealed that the in vivo antitumor activities of CD20 CAR+ Vδ1 γδ T cells combine innate and adaptive mechanisms. We identified 34 DEGs that were shared by CD20 CAR+ Vδ1 γδ T‐cell‐treated Raji and JVM‐2 tumors, with notable shared genes associated with T‐cell activation (IL2RA, TNFRSF9 and CD45RO), cytotoxic function (GZMA, GMZB, PRF1 and CTSW), cytokines/chemokines (CCL1, XCL1/2, CLL5, CSF1, CSF2, IL32 and IFNG) and innate immunity (KLRK1, NKG7, KLRC1, KLRC2 and CD96). Interestingly, we also identified distinct sets of DEGs in CD20 CAR+ Vδ1 γδ T‐cell‐treated Raji and JVM‐2 tumors. These gene expression differences could be attributed to the response and adaptation of the CD20 CAR+ Vδ1 γδ T cells to the tumor microenvironment of Raji and JVM‐2 xenografts. Moreover, differences between Raji and JVM‐2 tumors in their respective expression levels of CD20, γδTCR/NKR ligands and co‐stimulatory/co‐inhibitory receptor ligands may also contribute to the unique transcriptional profiles. Understanding the molecular changes that occur within the CD20 CAR+ Vδ1 γδ T cells after in vivo CAR engagement within the tumor microenvironment will help identify an intratumoral immune gene signature that may serve as a guide to identify molecular biomarkers associated with good clinical outcome in patients with B‐cell malignancies receiving CD20 CAR+ Vδ1 T‐cell products.
The successful development of a clinical‐scale cGMP‐compliant manufacturing process for CAR T‐cell therapies is required to ensure a standard of quality is achieved (i.e. controlled, reproducible and robust) to maximise the efficacy for treated patients. The complexities and challenges associated with large‐scale manufacturing processes of autologous and gene‐edited CAR T cells can lead to functional and phenotypic changes potentially impacting their clinical activity. 59 , 60 We were able to demonstrate that CD20 CAR+ Vδ1 γδ T cells generated under a cGMP‐compliant clinical‐scale manufacturing process were associated with potent in vitro functional activity using short‐term functional assays (Figure 8a and b). Although significant in vivo tumor control was observed (Figure 8d), overall tumor growth inhibition was less robust than other large‐scale manufacturing runs (non‐GMP) tested (Figure 7a, Supplementary figure 8). Using the 120‐h long‐term cytotoxicity assay, we did observe a higher dependence on the presence of IL‐2 for increased killing of target cells (Figure 8c), which could influence tumor control in our in vivo animal models. Future investigations to understand which factor(s) (i.e. cellular fitness, persistence and phenotype) contribute to potential differences associated with tumor control between CD20 CAR+ Vδ1 γδ T cells derived from cGMP‐compliant clinical‐scale lots will be important to identify and monitor.
Conclusion
In summary, the findings of our study demonstrate preclinical proof of concept for a CD20 CAR+ Vδ1 γδ T‐cell product, manufactured at clinical scale, in the treatment of CD20+ B‐cell malignancies. By combining innate and adaptive antitumor immunity, CD20 CAR+ Vδ1 γδ T cells have a unique advantage as an allogeneic CAR T‐cell product platform and, as a consequence, have the potential to improve clinical responses in eligible cancer patients. A phase 1 clinical study evaluating ADI‐001, an allogeneic CD20 CAR+ Vδ1 γδ T‐cell product, in R/R B‐cell NHL patients has been initiated (NCT04735471).
Methods
Cell lines
HEK293, 239T, Sp2/0, Rat2, Jurkat, Raji, JVM‐2, Daudi and Mino cell lines were purchased from ATCC (Manassas, VA). The WILL‐2 cell line and the Vγ8/Vδ1+ BE‐13 cell line were purchased from DSMZ (Braunschweig, Germany), the Jurkat‐Lucia™ NFAT reporter cell line from InvivoGen (San Diego, CA), the Expi293F™ cells from Thermo Fisher (Waltham, MA) and the 293Vec‐RD114™ packaging cell line from Biovec Pharma (Québec, Canada).
Several CD20+ target cell lines were modified to stably express red‐shifted luciferase (RFluc) or NucLight Red (NucR) nuclear label by transduction with RediFect RFLuc Lentiviral Particles (PerkinElmer, Waltham, MA) or IncuCyte® NucR Lentivirus Reagent (Sartorius, Göttingen, Germany), respectively. RR‐Raji cells were established by culturing RFLuc‐expressing Raji cells with rituximab (Genentech, South San Francisco, CA) in medium containing human serum as a source of complement for a total of ten passages. 61 HEK293 cells and Rat2 cells were stably transfected to express human CD20 (hCD20) using a plasmid vector.
Expi293F™ cells (Thermo Fisher, Waltham, MA) were transiently transfected to express and secrete the human Vγ8Vδ1TCR‐Fc, Vγ9Vδ1TCR‐Fc, Vγ8Vδ2TCR‐Fc or Vγ9Vδ2TCR‐Fc fusion protein using a pCI‐neo expression vector (Promega, Madison, WI) carrying recombinant DNA encoding the extracellular domains of the specific human VγVδ TCR heterodimer fused to the Fc domain of human IgG. Expi293F™ cells were transiently transfected to express and secrete a soluble anti‐CD20 minibody using the pCI‐neo expression vector carrying recombinant DNA encoding the anti‐CD20 (clone 3H7) scFv fused to the CH3 domain of human IgG1.
Construction of the CD20 CAR gamma‐retroviral vector
CAR vector, consisting of the anti‐CD20 (clone 3H7) scFv, CD8α hinge and transmembrane domains, and 4‐1BB and CD3ζ signalling domains, was constructed by cloning a DNA fragment containing all CAR domains (Integrated DNA Technologies, Coralville, IA) into a self‐inactivating (SIN) Moloney murine leukaemia virus (MMLV) gamma‐retroviral plasmid. CAR vector (pRetroSIN‐CAR) was manufactured using the following stable and transient virus production systems.
Stable single‐producer cell lines were generated in the 293Vec‐RD114 gamma‐retroviral packaging cell line (Biovec Pharma) following transfection, blasticidin (Sigma Aldrich, St Louis, MO) selection, cell sorting using a BD FACSAria II cell sorter (BD Biosciences, San Jose, CA) and determination of viral titre production by flow cytometry. 62 Bulk viral harvests were collected at 24, 48 and 72 h post‐confluency.
Transient production utilised 293T cells transfected with three plasmids: (1) pRetroSIN‐CAR; (2) Gag/Pol plasmid encoding MMLV packaging proteins; and (3) RD114 plasmid encoding the feline endogenous retrovirus envelope protein. Bulk viral harvests were collected at 48 and 72 h post‐transfection.
The process to clarify and concentrate the vector was similar in both stable and transient virus production systems. Briefly, bulk viral harvests were clarified by filtration through a 0.45‐μm filter, pooled and then concentrated/diafiltered into 10 mm HEPES and 0.9% NaCl by tangential flow filtration (Repligen, Waltham, MA).
Generation of an agonistic anti‐Vδ1 monoclonal antibody
Soluble human VγVδ TCR‐Fc fusion proteins were harvested from supernatants of transiently transfected Expi293F™ cells and purified using Protein A affinity chromatography (Cytiva, Marlborough, MA). The Vγ8Vδ1 TCR‐Fc protein was emulsified with an equal volume of TITERMAX® Gold (Sigma Aldrich) or Imject Alum Adjuvant (Thermo Fisher) and injected into the footpads of female BALB/c mice (The Jackson Laboratory, Bar Harbor, ME) twice per week for 4 weeks. Lymphocytes from immunised mice were harvested and then fused with Sp2/0 cells using the Hybrimmune™ Hybridoma Production System (BTX, Holliston, MA) as per the manufacturer’s instructions. Once fused, cells were cultured for 6–7 days in high‐glucose DMEM (Thermo Fisher) supplemented with 15% HyClone FBS (Cytiva) and then single‐cell‐sorted into 384‐well flat‐bottom plates (Corning, Tewksbury, MA) using the FACSAria II cell sorter. After 10 days of growth, hybridoma supernatants were screened for Vδ1 reactivity by flow cytometry using the Vy8Vδ1 TCR+ BE‐13 cell line and for Vδ1 specificity using an ELISA, in which supernatants were incubated in wells coated with human Vγ8Vδ1TCR‐Fc, Vγ9Vδ1TCR‐Fc, Vγ8Vδ2TCR‐Fc or Vγ9Vδ2TCR‐Fc fusion protein, probed with HRP‐labelled goat anti‐mouse IgG (Thermo Fisher), incubated with TMB substrate solution (Thermo Fisher) and then read at OD 450 using the Cytation™ 5 plate reader (BioTek Instruments, Winooski, VT). The commercial anti‐Vδ1 TCR antibody clone TS1 (Thermo Fisher) was used as a positive control for both flow cytometry and ELISA assays. Wells with cells containing antibodies that bound to BE‐13 cells by flow cytometry and to only Vδ1+ VγVδ TCR‐Fc fusion proteins by ELISA were further tested for their capacity to selectively activate and expand Vδ1 γδ T cells in unfractionated PBMCs to clinically relevant numbers. The agonistic anti‐Vδ1 mAb that, across multiple donors, consistently induced proliferation in a high percentage of Vδ1 γδ T cells, high Vδ1 fold expansion and a high percentage of naïve‐like memory cells at the end of culture was then purified on a HiTrap™ MabSelect™ SuRe column (Cytiva), eluted using 100 mm glycine (pH 2.7) and then formulated in 50 mm histidine and 100 mm NaCl at a pH of 6.0.
Small‐scale production of CD20 CAR+ Vδ1 γδ T cells
Peripheral blood mononuclear cells were isolated from leucapheresis samples collected from eligible, cytomegalovirus (CMV)‐negative, healthy donors using density gradient separation and/or RBC lysis prior to cryopreservation (STEMCELL Technologies, Cambridge, MA). PBMCs from a single donor were thawed and plated (4 × 106–15 × 106 cells) onto immobilised agonistic anti‐Vδ1 mAb in culture plates or T‐flasks. Antibody‐activated PBMCs were transduced with the gamma‐retroviral CAR construct prepared from a stable single‐producer cell line in combination with RetroNectin® (Takara, Mountain View, CA). Following transduction, cells were expanded in X‐VIVO 15 medium (Lonza, Walkersville, MD) containing 10% HyClone FBS and 100 IU mL−1 human IL‐2 (Peprotech, Cranbury, NJ) in shake flasks on an orbital shaker with a speed between 100 and 125 RPM. Following expansion, the remaining αβ T cells were labelled with biotinylated anti‐TCRαβ antibody and anti‐biotin immunomagnetic beads (Miltenyi, Auburn, CA) and depleted using the autoMACS® Pro (Miltenyi) prior to filling into vials and cryopreserved in CryoStor® CS10 and CryoStor CSB freezing media (Biolife Solutions, Bothell, WA) on Day 18.
Small‐scale production of CD20 CAR+ αβ T cells
Peripheral blood mononuclear cells from a single donor (STEMCELL Technologies) were thawed, activated with 50 ng mL−1 soluble anti‐CD3 mAb (OKT3; Miltenyi) and 300 IU mL−1 human IL‐2 in a T25 flask, and then transduced on Day 2 with the gamma‐retroviral CD20 CAR construct in combination with RetroNectin®. Following transduction, cells were expanded in RPMI‐1640 medium containing 10% HyClone FBS and 300 IU mL−1 human IL‐2 in static T150 flasks and then cryopreserved on Day 11 in CryoStor® CS10 and CryoStor CSB freezing media.
Large‐scale production of CD20 CAR+ Vδ1 γδ T cells
Peripheral blood mononuclear cells (HemaCare, Northridge, CA) were enriched from apheresis material collected from eligible, CMV‐negative, healthy donors using a Ficoll–Paque gradient on a Sepax C‐Pro Cell Processing Instrument (Cytiva), prior to cryopreservation in a solution containing CryoStor® CS10 and CryoStor CSB freezing media. PBMCs from a single donor were thawed and plated (~1 × 108–5.0 × 108 cells) onto immobilised agonistic anti‐Vδ1 mAb in T‐flasks. Antibody‐activated PBMCs were transduced with the gamma‐retroviral CD20 CAR construct in transduction vessel coated with RetroNectin®. CD20 CAR vector from stable virus production was used in early large‐scale manufacturing, whereas vector from transient virus production was used in later stage large‐scale and in clinical‐scale manufacturing. Following transduction, cells were expanded in a Xuri™ perfusion bioreactor (Cytiva) in X‐VIVO™ 15 medium containing 10% HyClone FBS and human IL‐2 (Proleukin, Clinigen, Yardley, PA). Following expansion, the remaining αβ T cells were labelled with biotinylated anti‐TCRαβ antibody and anti‐biotin immunomagnetic beads (Miltenyi) and depleted using the CliniMACS Plus® (Miltenyi). Following depletion, cells were formulated in CryoStor CS10 and CryoStor CSB freezing media, filled into vials and cryopreserved on Day 15. Clinical‐scale cell product was manufactured at a cGMP‐compliant contract manufacturing organisation following the large‐scale manufacturing process using a vector from transient virus production.
Flow cytometry
Cellular composition was determined using a staining panel including antibodies against Vδ1, Vδ2, CD3, CD14, CD19, CD56, TCRαβ and TCRγδ. Extended phenotypic analysis incorporated antibodies against CD45, CD95, CD45RO, CD62L, CD27, CD45RA, CD57, KLRG1, NKG2D, DNAM1, CCR4, CCR5, CCR7, CCR8, CXCR3, CXCR4, CXCR6, PD‐1, TIM‐3, LAG‐3 and TIGIT. Binding properties of anti‐CD20 mAbs, clone 3H7 and rituximab (Genentech, Lot #3108505) were evaluated using human IgG1 isotype control and fluorochrome‐conjugated anti‐human Fc antibodies. Antibodies were purchased from BioLegend (San Diego, CA), BD Biosciences, Beckman Coulter (Brea, CA), Miltenyi or Jackson ImmunoResearch (West Grove, PA). Surface CAR expression was measured using an anti‐CAR idiotype antibody (Adicet Therapeutics, Menlo Park, CA) or biotinylated‐Protein L (Thermo Fisher) and BV421‐conjugated streptavidin (BioLegend). Samples were acquired on a BD FACSCanto™ II (BD Biosciences), a NovoCyte (Agilent, Santa Clara, CA) or an iQue® (Sartorius) flow cytometer. Data were analysed using FlowJo™ software (Tree Star, Ashland, OR) or iQue software (Sartorius).
Immunocytochemistry and immunohistochemistry
The 3H7 minibody was purified from supernatants of transiently transfected Expi293F™ cells using affinity chromatography on an FcXL™ (Thermo Fisher) packed column and was then biotinylated and polished to remove aggregates. The biotinylated soluble 3H7 minibody protein (3H7‐MB1‐Biotin) was used to stain CD20+ and CD20− cell lines by immunocytochemistry (ICC) and to stain cryosections from a full panel of normal human tissues by immunohistochemistry (IHC). For ICC, 5 μg mL−1 3H7‐MB1‐Biotin or 5 μg mL−1 biotinylated human IgG1 control antibody (Jackson ImmunoResearch) was incubated with parental Rat2 or with Rat2‐hCD20 cells that were cultured and fixed on slides. Binding of 3H7‐MB1‐Biotin was detected by VECTASTAIN ELITE ABC‐Peroxidase and DAB (3,3′‐diaminobenzidine) (Vector Laboratories, Burlingame, CA) substrate. For IHC, 5 μg mL−1 3H7‐MB1‐Biotin or 5 μg mL−1 biotinylated human IgG1 control antibody was incubated with formalin‐fixed tissue sections from various tissue samples harvested from three separate healthy donors. Binding of 3H7‐MB1‐Biotin was detected by VECTASTAIN ELITE ABC‐Peroxidase and DAB substrate (Vector Laboratories). ICC and IHC slides were visualised under light microscopy and examined and scored by a board‐certified veterinary pathologist.
TCRG repertoire
Immunosequencing of TCRG locus was performed using Archer® Immunoverse™ (ArcherDX, Boulder, CO). Treemaps of the TCRγ repertoire were created using Tableau software (Seattle, WA).
Protein analysis
A total of 10 million untransduced (UT) or 1.5 × 107 CD20 CAR+ Vδ1 γδ T cells were surface‐biotinylated using EZ‐Link Sulfo‐NHS‐LC‐Biotin (Thermo Fisher), lysed in Brij® O10 (Millipore Sigma, Burlington, MA) lysis buffer containing a protease inhibitor cocktail (Millipore Sigma) and then incubated with an anti‐CD20 CAR mAb either bound to Protein A‐coupled Sepharose 4B beads or coupled to CNBr‐activated Sepharose 4B beads (Cytiva). Immunoprecipitated proteins were resolved by non‐reducing (NR) and reducing SDS‐PAGE analysis and transferred to an iBlot™ 2 nitrocellulose membrane (Thermo Fisher). Biotinylated CAR complexes were visualised by IRDye® 800CW Streptavidin (Thermo Fisher) and the Odyssey® Imager (LI‐COR, Lincoln, NE).
A total of 100 000 CD20 CAR+ Vδ1 γδ T cells were unstimulated or stimulated on Protein L (Thermo Fisher)‐coated 96‐well plates for 10 min and then lysed in Milliplex® MAP Cell Signaling Universal Lysis Buffer (Millipore Sigma) at 1E7 cells mL−1. Lysates from replicate wells were pooled and were loaded based on equivalent amounts of total protein, resolved by reducing SDS‐PAGE analysis, and then transferred to iBlot™ 2 nitrocellulose membranes. Membranes were blocked with StartingBlock™ (TBS) Blocking Buffer (Thermo Fisher) and then incubated with antibodies against total CD3ζ (BD Biosciences) or phospho‐CD3ζ (Tyr 142; BD Biosciences) overnight at 4°C on a shaker. CAR‐associated CD3ζ and phospho‐CD3ζ proteins were visualised using anti‐mouse IgG IR800 (Advansta, San Jose, CA) and the Odyssey® Image.
Analysis of CAR‐mediated tonic signalling in Jurkat‐Lucia cells
Jurkat‐Lucia cells, which carry an NFAT‐inducible luciferase reporter, were transduced with the gamma‐retroviral CD20 CAR construct in combination with RetroNectin®, and the percentage of CAR‐transduced Jurkat‐Lucia cells expressing the CD20 CAR on their cell surface was measured by flow cytometry. CAR‐mediated signal transduction and activation were assessed in the presence or absence of target antigen by culturing parental and CAR‐transduced Jurkat‐Lucia cells either alone or at a 1:1 E:T ratio with CD20+ Raji cells. Stimulation with PMA (phorbol 12‐myristate 13‐acetate) and ionomycin (BioLegend) was used as a positive control for NFAT activation. At 24 h, an aliquot of supernatant was mixed with QUANTI‐Luc™ reagent (InvivoGen), and luciferase activity was detected using the Cytation™ 5 imaging plate reader. Duplicate samples were tested for each condition.
Short‐term cytotoxicity and cytokine release assays
Cytotoxic potential of CD20 CAR+ Vδ1 γδ T cells against RFluc‐expressing B‐lymphoma cell lines at titrated effector:target (E:T) ratios was assessed in an 18‐h luciferase cytotoxicity assay. Cytotoxic potential of CD20 CAR+ Vδ1 γδ T cells against RFluc‐expressing RR‐Raji cells at a fixed 3:1 ratio was assessed in an 18‐h luciferase cytotoxicity assay after pre‐incubation of target cells for at least 1 h with clinically relevant concentrations of rituximab (0–400 μg mL−1). 43 Controls included CD20 CAR+ Vδ1 γδ T cells and B‐lymphoma cell lines cultured alone. Luciferase activity was quantitated using the Steady‐Glo® Luciferase Assay System (Promega) and the Cytation™ 5 plate reader. Each culture condition was run in triplicate. % cytotoxicity was calculated using the following formula:
where Target OnlyLum refers to mean of luminescence values of target only, and SampleLum refers to mean of luminescence values of a given E:T ratio.
MILLIPLEX® Immunology Multiplex Assay Panels (Millipore Sigma) were used to measure cytokine and chemokine levels in culture supernatants on the FLEXMAP 3D® Instrument (Luminex®, Austin, TX).
Long‐term proliferation and cytotoxicity assays
Proliferation was assessed after repetitive target antigen engagement as previously described. 63 Briefly, CD20 CAR+ Vδ1 γδ T cells were labelled with CellTrace™ Violet (BioLegend) and then plated alone, with Raji or Mino cells at a 1:2 ratio. On Days 2 and 5, CD20 CAR+ Vδ1 γδ T cells were counted and replated in fresh X‐VIVO™ 15 medium containing 10% HyClone FBS at a 1:1 ratio with Raji or Mino cells. Aliquots of cells harvested on Days 2, 5 and 7 were analysed by flow cytometry to assess proliferation by dye dilution.
For the long‐term cytotoxicity assays, CD20 CAR+ Vδ1 γδ T cells were co‐cultured with NucR‐expressing Raji cells at a fixed 1:2 ratio ±20 IU of human IL‐2 or with NucR‐expressing Raji or Mino cells at titrated E:T ratios. Viable NucR‐expressing target cells were quantitated every 2 h over the course of 48 or 120 h using the IncuCyte® S3 system (Sartorius). The cytotoxicity index was calculated by dividing the total red object area (mm2/well) of all time points by the value at time = 0.
Mouse experiments
The fully human anti‐CD20 mAb, clone 3H7, was generated using the VelocImmune® human antibody mouse platform as previously described. 22 , 23
For in vivo efficacy studies, 7‐ to 9‐week‐old female NOD. Cg‐Prkdcscid IL2rgtm1Wjl /SzJ (NSG) mice (The Jackson Laboratory) were implanted subcutaneously with 1 × 106 Raji or 2 × 106 JVM‐2 cells mixed with Matrigel® (1:1 ratio) (Corning, Bedford, MA). When tumor volume reached ~200 mm3, mice were randomised and then injected IV with 2.5 × 106 to 2 × 107 CD20 CAR+ Vδ1 γδ T cells. Human IL‐2 (13 000 IU; Peprotech or Proleukin, Pharmaceutical Buyers, Inc., New Hyde Park, NY) was administered intraperitoneally immediately prior to cell treatment and then thrice weekly for the study duration. Tumor volume was measured twice weekly using callipers.
For ex vivo analysis of γδ T cells in tumor‐bearing mice, untransduced and CD20 CAR‐transduced Vδ1 γδ T cells were labelled with CellTrace Violet (BioLegend) and then injected IV into Raji‐bearing NSG mice. On Day 6 post‐treatment, mice were killed, and blood, bone marrow and tumors were harvested. Intact tumors were dissociated into single cells using the gentleMACS™ Dissociator (Miltenyi). Proliferation and memory cell phenotype were assessed by flow cytometry.
For tumor immune profiling, Raji‐ or JVM‐2‐bearing NSG mice were treated with untransduced or CD20 CAR‐transduced Vδ1 γδ T cells. On Days 4–6 post‐treatment, mice were killed and then perfused transcardially with 1X PBS. Tumors were harvested, and RNA was extracted using the Qiagen RNeasy Mini Kit (Germantown, MD). Gene expression levels were quantitated using nCounter® Panels (NanoString, Seattle, WA) on the nCounter SPRINT Profiler (NanoString), according to manufacturer’s instructions. nSolver™ software (NanoString) was used to normalise the data and to identify DEGs.
To evaluate the potential for xenogeneic GvHD, 9‐week‐old female NSG mice were injected IV on Day 1 with a single dose of X‐VIVO 15 medium, 4 × 107 CD20 CAR+ Vδ1 γδ T cells or 4 × 107 PBMCs (HemaCare). Mice were monitored thrice weekly and euthanised when body weight loss > 15% of initial body weight was observed or at study termination (Day 61). A GvHD scoring system was used that is based on the five categories of weight loss, activity, posture, fur texture and skin integrity. Each category had three scores: 0 is normal, 1 is mild GvHD, and 2 is moderate‐to‐severe GvHD. GvHD scores comprised the sum of the five categories, with a maximum possible score of 10 for each mouse. After euthanasia, tissues were collected for histopathological and immunohistochemistry analyses to assess histopathological changes and infiltration of human CD3+ cells. Slides were examined by a board‐certified veterinary pathologist.
All mouse experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals and followed all institutional and national guidelines.
Statistical analyses
Statistical analyses were performed using GraphPad Prism Software (San Diego, CA), and the tests selected to determine statistical significance, defined as a P‐value < 0.05, are stated in the figure captions.
Conflict of interest
KPN, TB, AA, AM, JMR, LB, MMB, JK‐W, HS, SP, AD, CM, NTH, HT, VDR, AG‐R, M Hoopes, DS, RS, M Herrman, SEA, BTA, ZA, SP and SMH are, or were, employees of Adicet Bio, Inc., and FD, KS and KB are employees of Regeneron Pharmaceuticals, Inc.
Author contributions
Kevin P Nishimoto: Conceptualisation; data curation; investigation; methodology; formal analysis; validation; writing – original draft; writing – review and editing. Taylor Barca: Data curation; formal analysis; investigation; methodology; validation; writing – review and editing. Aruna Azameera: Data curation; formal analysis; investigation; methodology; validation; writing – review and editing. Amani Makkouk: Methodology; investigation; writing – review and editing. Jason M Romero: Data curation; formal analysis; investigation; methodology; writing – review and editing. Lu Bai: Formal analysis; investigation; methodology; writing – review and editing. Mary M Brodey: Data curation; formal analysis; investigation; methodology; writing – review and editing. Jackie Kennedy‐Wilde: Data curation; formal analysis; investigation; methodology; writing – review and editing. Hui Shao: Investigation; methodology; writing – review and editing. Stephanie Papaioannou: Investigation; methodology; resource; writing – review and editing. Amy Doan: Investigation; methodology; resource; writing – review and editing. Cynthia Masri: Investigation; writing – review and editing. Ngoc T Hoang: Investigation; methodology; resources; writing – review and editing. Hayden Tessman: Investigation; methodology; resources; writing – review and editing. Vidhya Dhevi Ramanathan: Data curation; formal analysis; investigation; methodology; validation; writing – review and editing. Ana Giner‐Rubio: Data curation; formal analysis; investigation; methodology; validation; writing – review and editing. Frank Delfino: Supervision; methodology; formal analysis; writing – review and editing. Kriti Sharma: Data curation, formal analysis; investigation; methodology; writing – review and editing. Kevin Bray: Data curation, formal analysis; investigation; methodology; writing – review and editing. Matthew Hoopes: Methodology; resources; writing – review and editing. Daulet Satpayev: Supervision; investigation; writing – review and editing. Ranjita Sengupta: Supervision; methodology; writing – review and editing. Marissa Herrman: Data curation; investigation; methodology; resources; supervision; writing – review and editing. Stewart E Abbot: Supervision; writing – review and editing. Blake T Aftab: Guarantor; writing – review and editing. Zili An: Methodology; supervision; writing – review and editing. Swapna Panuganti: Investigation; methodology; resources; supervision; writing – review and editing. Sandra M Hayes: Conceptualisation; data curation; investigation; methodology; formal analysis; project administration; supervision; visualisation; writing – original draft; writing – review and editing.
Supporting information
Supplementary Material
Acknowledgments
The authors acknowledge Charles River Laboratories for performing the GvHD study and the GLP‐compliant cross‐reactivity study using the 3H7 minibody. In addition, we thank Michael Salum, Kevin Nguyen, Yih‐Pai Chu, Joel H Martin and Erin M Allison for their technical support. Last, we acknowledge Francesco Galimi, Lloyd Klickstein, Tamara Do, Ori Maller, John Lin and Gavin Thurston for critical review of the manuscript.
References
- 1. McCreedy BJ, Senyukov VV, Nguyen KT. Off the shelf T cell therapies for hematologic malignancies. Best Pract Res Clin Haematol 2018; 31: 166–175. [DOI] [PubMed] [Google Scholar]
- 2. Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol 2019; 16: 372–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ruella M, Kenderian SS. Next‐generation chimeric antigen receptor T‐cell therapy: going off the shelf. BioDrugs 2017; 31: 473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Graham C, Jozwik A, Pepper A, Benjamin R. Allogeneic CAR‐T cells: more than ease of access? Cells 2018; 7: 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aftab BT, Sasu B, Krishnamurthy J, Gschweng E, Alcazer V, Depil S. Toward “off‐the‐shelf” allogeneic CAR T cells. Adv Cell Gene Ther 2020; 3: 1–11. [Google Scholar]
- 6. Gilham DE, Michaux A, Breman E et al. TCR inhibitory molecule as a promising allogeneic NKG2D CAR‐T cell approach. J Clin Oncol 2018; 36(Suppl 15): Abstract e15042. [Google Scholar]
- 7. Shen RR, Pham CD, Min WUM, Munson DJ, Aftab BT. Functional demonstration of CD19 chimeric antigen receptor (CAR) engineered Epstein‐Barr virus (EBV) specific T cells: an off‐the‐shelf, allogeneic CAR T‐cell immunotherapy platform. Cancer Res 2019; 79(Suppl 13): Abstract 2310. [Google Scholar]
- 8. Gomes AQ, Martins DS, Silva‐Santos B. Targeting γδ T lymphocytes for cancer immunotherapy: from novel mechanistic insight to clinical application. Cancer Res 2010; 70: 10024–10027. [DOI] [PubMed] [Google Scholar]
- 9. Lamb LS, Musk P, Ye Z et al. Human γδ+ T lymphocytes have in vitro graft vs leukemia activity in the absence of an allogeneic response. Bone Marrow Transplant 2001; 27: 601–606. [DOI] [PubMed] [Google Scholar]
- 10. Hayday AC, Saito H, Gillies SD et al. Structure, organization, and somatic rearrangement of T cell gamma genes. Cell 1985; 40: 259–269. [DOI] [PubMed] [Google Scholar]
- 11. Brenner MB, McLean J, Dialynas DP et al. Identification of a putative second T‐cell receptor. Nature 1986; 322: 145–149. [PubMed] [Google Scholar]
- 12. Bonneville M, O’Brien RL, Born WK. γδ T cell effector functions: a blend of innate programming and acquired plasticity. Nat Rev Immunol 2010; 10: 467–478. [DOI] [PubMed] [Google Scholar]
- 13. Gentles AJ, Newman AM, Liu CL et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med 2015; 21: 938–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wu Y, Kyle‐Cezar F, Woolf RT et al. An innate‐like Vδ1+ γδ T cell compartment in the human breast is associated with remission in triple‐negative breast cancer. Sci Transl Med 2019; 11: eaax9364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lo Presti E, Dieli F, Meraviglia S. Tumor‐infiltrating γδ T lymphocytes: pathogenic role, clinical significance, and differential programing in the tumor microenvironment. Front Immunol 2014; 5: 607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Siegers GM, Lamb LS Jr. Cytotoxic and regulatory properties of circulating Vδ1+ γδ T cells: a new player on the cell therapy field? Mol Ther 2014; 22: 1416–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Deniger DC, Moyes JS, Cooper LJ. Clinical applications of gamma delta T cells with multivalent immunity. Front Immunol 2014; 5: 636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Perko R, Kang G, Sunkara A, Leung W, Thomas PG, Dallas MH. Gamma delta T cell reconstitution is associated with fewer infections and improved event‐free survival after hematopoietic stem cell transplantation for pediatric leukemia. Biol Blood Marrow Transplant 2015; 21: 130–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bertaina A, Roncarolo MG. Graft engineering and adoptive immunotherapy: new approaches to promote immune tolerance after hematopoietic stem cell transplantation. Front Immunol 2019; 10: 1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hill JA, Li D, Hay KA et al. Infectious complications of CD19‐targeted chimeric antigen receptor‐modified T‐cell immunotherapy. Blood 2018; 131: 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Siegers GM, Dutta I, Lai R, Postovit LM. Functional plasticity of gamma delta T cells and breast tumor targets in hypoxia. Front Immunol 2018; 9: 1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Murphy AJ, Macdonald LE, Stevens S et al. Mice with megabase humanization of their immunoglobulin genes generate antibodies as efficiently as normal mice. Proc Natl Acad Sci USA 2014; 111: 5153–5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Smith EJ, Olson K, Haber LJ et al. A novel, native‐format bispecific antibody triggering T‐cell killing of B‐cells is robustly active in mouse tumor models and cynomolgus monkeys. Sci Rep 2015; 5: 17943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Davey MS, Willcox CR, Joyce SP et al. Clonal selection in the human Vδ1 T cell repertoire indicates γδ TCR‐dependent adaptive immune surveillance. Nat Commun 2017; 8: 14760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Di Lorenzo B, Simões AE, Caiado F et al. Broad cytotoxic targeting of acute myeloid leukemia by polyclonal delta one T cells. Cancer Immunol Res 2019; 7: 552–558. [DOI] [PubMed] [Google Scholar]
- 26. Frigault MJ, Lee J, Basil MC et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol Res 2015; 3: 356–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Long AH, Haso WM, Shern JF et al. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 2015; 21: 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gomes‐Silva D, Mukherjee M, Srinivasan M et al. Tonic 4–1BB costimulation in chimeric antigen receptors impedes T cell survival and is vector‐dependent. Cell Rep 2017; 21: 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Blanco R, Alarcón B. TCR nanoclusters as the framework for transmission of conformational changes and cooperativity. Front Immunol 2012; 3: 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fraietta JA, Lacey SF, Orlando EJ et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med 2018; 24: 563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sommermeyer D, Hudecek M, Kosasih PL et al. Chimeric antigen receptor‐modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo . Leukemia 2016; 30: 492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Locke FL, Rossi J, Neelapu SS et al. Product characteristics associated with in vivo expansion of anti‐CD19 CAR T cells in patients treated with axicabtagene ciloleucel (axi‐cel). J Clin Oncol 2017; 35(Suppl 15): Abstract 3023. [Google Scholar]
- 33. Pizzolato G, Kaminski H, Tosolini M et al. Single‐cell RNA sequencing unveils the shared and distinct cytotoxic hall marks of human TCRVδ1 and TCRVδ2 γδ T lymphocytes. Proc Natl Acad Sci USA 2019; 116: 11906–11915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sebestyen Z, Prinz I, Déchanet‐Merville J, Santos‐Silva B, Kuball J. Translating gammadelta (γδ) T cells and their receptors into cancer cell therapies. Nat Rev Drug Discov 2020; 19: 169–184. [DOI] [PubMed] [Google Scholar]
- 35. Kabelitz D, Wesch D. Features and functions of γδ T lymphocytes: focus on chemokines and their receptors. Crit Rev Immunol 2003; 23: 339–370. [DOI] [PubMed] [Google Scholar]
- 36. Scott DW, Gascoyne RD. The tumour microenvironment in B cell lymphomas. Nat Rev Cancer 2014; 14: 517–534. [DOI] [PubMed] [Google Scholar]
- 37. Fowler NH, Cheah CY, Gascoyne RD et al. Role of the tumor microenvironment in mature B‐cell lymphoid malignancies. Haematologica 2016; 101: 531–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sarkar S, Sabhachandani P, Ravi D et al. Dynamic analysis of human natural killer cell response at single‐cell resolution in B‐cell non‐Hodgkin lymphoma. Front Immunol 2017; 8: 1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015; 15: 486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fisher J, Sharma R, Don DW et al. Engineering γδ T cells limits tonic signaling associated with chimeric antigen receptors. Sci Signal 2019; 12: eaax1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Prevodnik VK, Lavrencak J, Horvat M, Novakovic BJ. The predictive significance of CD20 expression in B cell lymphomas. Diagn Pathol 2011; 6: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sonoki T, Li Y, Miyanishi S et al. Establishment of a novel CD20 negative mature B‐cell line, WILL2, from a CD20 positive diffuse large B‐cell lymphoma patient treated with rituximab. Int J Hematol 2009; 89: 400–402. [DOI] [PubMed] [Google Scholar]
- 43. Berinstein NL, Grillo‐López AJ, White CA et al. Association of serum rituximab (IDEC‐C2B8) concentration and anti‐tumor response in the treatment of recurrent low‐grade or follicular non‐Hodgkin’s lymphoma. Ann Oncol 1998; 9: 995–1001. [DOI] [PubMed] [Google Scholar]
- 44. Rougé L, Chiang N, Steffek M et al. Structure of CD20 in complex with the therapeutic monoclonal antibody rituximab. Science 2020; 367: 1224–1230. [DOI] [PubMed] [Google Scholar]
- 45. Xue Q, Bettini E, Paczkowski P et al. Single‐cell multiplexed cytokine profiling of CD19 CAR‐T cells reveals a diverse landscape of polyfunctional antigen‐specific response. J Immunother Cancer 2017; 5: 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rossi J, Paczkowski P, Shen Y‐W et al. Preinfusion polyfunctional anti‐CD19 chimeric antigen receptor T cells are associated with clinical outcomes in NHL. Blood 2018; 132: 804–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol 2007; 7: 118–130. [DOI] [PubMed] [Google Scholar]
- 48. Neelapu SS. CAR‐T efficacy: is conditioning the key? Blood 2019; 133: 1799–1800. [DOI] [PubMed] [Google Scholar]
- 49. Almeida AR, Correia DV, Fernandes‐Platzgummer A et al. Delta one T cells for immunotherapy of chronic lymphocytic leukemia: clinical‐grade expansion/differentiation and preclinical proof of concept. Clin Cancer Res 2016; 22: 5795–5804. [DOI] [PubMed] [Google Scholar]
- 50. Capsomidis A, Benthall G, Van Acker HH et al. Chimeric antigen receptor‐engineered human gamma delta T cells: enhanced cytotoxicity with retention of cross presentation. Mol Ther 2018; 26: 354–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kaartinen T, Luostarinen A, Maliniemi P et al. Low interleukin‐2 concentration favors generation of early memory T cells over effector phenotypes during chimeric antigen receptor T‐cell expansion. Cytotherapy 2017; 19: 689–702. [DOI] [PubMed] [Google Scholar]
- 52. Xu Y, Zhang M, Ramos CA et al. Closely related T‐memory stem cells correlate with in vivo expansion of CAR.CD19‐T cells and are preserved by IL‐7 and IL‐15. Blood 2014; 123: 3750–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Daydé D, Ternant D, Ohresser M et al. Tumor burden influences exposure and response to rituximab: pharmacokinetic‐pharmacodynamic modeling using a syngeneic bioluminescent murine model expressing human CD20. Blood 2009; 113: 3765–3772. [DOI] [PubMed] [Google Scholar]
- 54. Fujisaki H, Kakuda H, Shimasaki N et al. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res 2009; 69: 4010–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sommer C, Boldajipour B, Kuo TC et al. Preclinical evaluation of allogeneic CAR T cells targeting BCMA for the treatment of multiple myeloma. Mol Ther 2019; 27: 1126–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liu E, Tong Y, Dotti G et al. Cord blood NK cells engineered to express IL‐15 and a CD19‐targeted CAR show long‐term persistence and potent antitumor activity. Leukemia 2018; 32: 520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen Y, Sun C, Landoni E, Metelitsa L, Dotti G, Savoldo B. Eradication of neuroblastoma by T cells redirected with an optimized GD2‐specific chimeric antigen receptor and interleukin‐15. Clin Cancer Res 2019; 25: 2915–2924. [DOI] [PubMed] [Google Scholar]
- 58. Makkouk A, Yang X, Barca T et al. Off‐the‐shelf Vδ1 gamma delta T cells engineered with glypican‐3 (GPC‐3)‐specific chimeric antigen receptor (CAR) and soluble IL‐15 display robust antitumor efficacy against hepatocellular carcinoma. J Immunother Cancer 2021; 9: e003441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Abou‐el‐Enein M, Elsallab M, Feldman SA et al. Scalable manufacturing of CAR T cells for cancer immunotherapy. Blood Cancer Discov 2021; 5: 408–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Eyles JE, Vessillier S, Jones A, Stacey G, Schneider CK, Price J. Cell therapy products: focus on issues with manufacturing and quality control of chimeric antigen receptor T‐cell therapies. Chem Technol Biotechnol 2019; 94: 1008–1016. [Google Scholar]
- 61. Czuczman MS, Olejniczak D, Gowda A et al. Acquirement of rituximab resistance in lymphoma cell lines is associated with both global CD20 gene and protein down‐regulation regulated at the pretranscriptional and posttranscriptional levels. Clin Cancer Res 2008; 14: 1561–1570. [DOI] [PubMed] [Google Scholar]
- 62. Zheng Z, Chinnasamy N, Morgan RA. Protein L: a novel reagent for the detection of chimeric antigen receptor (CAR) expression by flow cytometry. J Transl Med 2012; 10: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang D, Starr R, Alizadeh D, Yang X, Forman SJ, Brown CE. In vitro tumor cell rechallenge for predictive evaluation of chimeric antigen receptor T cell antitumor function. J vis Exp 2019; (144): e59275. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material