

Abstract
Chimeric antigen receptor (CAR)-modified T cells have exhibited impressive anti-tumor effects in both B cell malignancies and some types of solid tumors. However, single-chain variable fragment (scFv) of a murine monoclonal antibody will induce immune responses, limit CAR-T cell persistence, and thus increase the risk of relapse. This study successfully constructed a CAR-targeting interleukin-13 receptor α2 (IL-13Rα2) according to a murine antibody, and then humanized the scFv sequence to generate another CAR. T cells expressing any of these two CARs demonstrated superior tumor inhibitory effects in vitro and in two xenograft mouse models. However, T cells transduced with humanized CAR have an increased expansion and reduced cytokines, including interleukin-6 and interferon-γ. The top expressed genes clustered in leukocyte-mediated cytotoxicity, and T cell migration and immunological synapse formation contributed to the anti-glioblastoma (GBM) activity of the humanized CAR. In conclusion, we successfully generated a humanized third-generation CAR-targeting IL-13Rα2 and confirmed its anti-GBM efficacy, which provide a candidate method for clinical GBM treatment.
Keywords: interleukin-13 receptor α2, chimeric antigen receptor-modified T cells, glioblastoma, adoptive T cell therapy, expansion, cytokine, high-throughput sequencing, real-time cell analysis
Graphical abstract
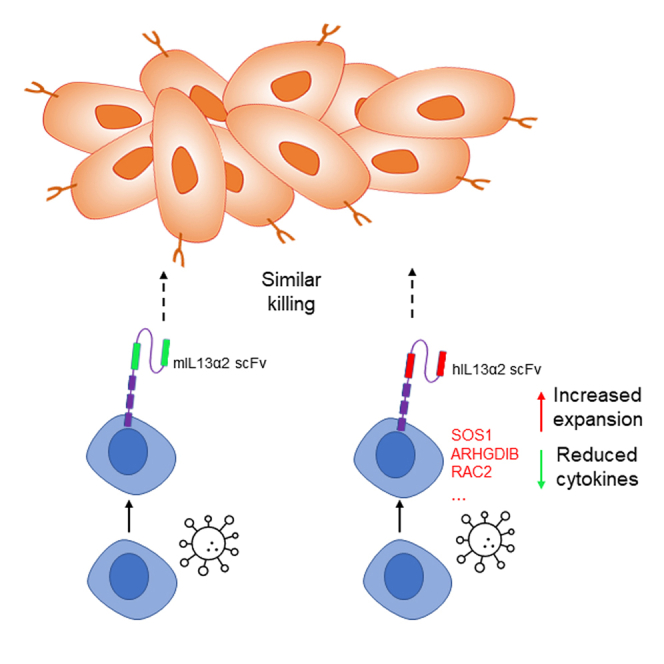
Chang Xu and colleagues generated two CARs targeting IL-13Rα2. They demonstrated that IL-13Rα2 CAR-T cells could kill GBM cells in vitro and in xenograft mouse models. T cells expressing humanized CAR had increased expansion, reduced cytokine production, and similar anti-tumor efficacy when compared with murine-base CAR-T cells.
Introduction
Glioblastoma (GBM) is the most common and aggressive primary brain malignancy in adults.1 Due to its high morbidity, high mortality, and low cure rates, GBM has caused a great social and medical burden worldwide, and is still incurable with conventional therapies.2 Therefore, developing novel therapies for GBM is necessary to improve the outcome and prognosis of GBM patients.
Adoptive T cell immunotherapy is a promising strategy for cancer treatment, especially chimeric antigen receptor (CAR)-modified T cell therapy.3 CARs are synthetic molecules that redirect T cells to eradicate tumors through specific recognition of surface proteins expressed on tumor cells, which consist of an extracellular tumor antigen-binding domain linked to hinge, transmembrane, and intracellular signaling domains.4, 5, 6 T cells expressing CAR (CAR-T cells) can directly identify tumor-associated antigens through single-chain variable fragment (scFv) of the extracellular domain and then, activated by intracellular signal transduction, release multiple cytokines, such as perforin, granzyme, interferon-γ (IFN-γ), and tumor necrosis factor (TNF), and induce tumor cell apoptosis.7,8 Therefore, CAR-T cells act in an MHC-independent manner and ingeniously combine the abilities of antibody-specific recognition of antigens and cytotoxicity of T lymphocytes.
CAR-T cell therapy was first applied in treatment of hematologic B cell malignancies and exhibited effective and encouraging results.9,10 Two products (Kymrial and Yescata) have been available for the treatment of hematologic malignancies in the United States and Europe.11,12 However, CAR-T cell treatment for solid tumors have shown limited anti-tumor activity and is still in the experimental stage. Until recently, several groups have developed different CARs for GBM treatment, including CARs targeting IL-13 receptor subunit α2 (IL-13Rα2), human epidermal growth factor receptor 2 (HER2), or epidermal growth factor receptor variant III (EGFRvIII), and these have exhibited encouraging anti-tumor effects.13, 14, 15
IL-13Rα2 is a monomeric high-affinity interleukin-13 (IL-13) receptor, which is overexpressed by the majority of GBM tumors (>60%) and not expressed at significant levels on normal brain tissue.16, 17, 18 IL-13Rα2 expression is closely associated with that of the mesenchymal GBM subtype and is a prognostic indicator of poor patient survival.19 This favorable expression profile provides the rationale for continued development of CAR-T cells targeting IL-13Rα2.20,21 Previous CARs interacting with IL-13Rα2 were mostly derived from murine antibodies.22 However, murine-derived CARs could induce human anti-mouse antibody responses, which further limit the persistence of the CAR-T cells and increase the risk of relapse.23
In this study, we generated a CAR derived from a murine antibody targeting IL-13Rα2 and humanized the sequence to form a humanized anti-IL-13Rα2 CAR. We hypothesized that T cells expressing humanized CAR-targeting IL-13Rα2 would exhibit similar tumor cell killing activity, but better expansion and lower cytokine production. We evaluated the expansion, anti-GBM efficacy, and cytokine release of T cells transfected with these two CARs in vitro and in xenograft mouse models.
Results
T cells expressing the humanized third-generation CAR-targeting IL-13Rα2 inhibit GBM cell growth and have a better expansion
To develop GBM-specific CARs for adoptive T cells therapy, we first confirmed the expression of IL-13Rα2 on the GBM cell surface by flow cytometry. We observed that more than 91.4% U251 and more than 80.9% U373 cells were IL-13Rα2 positive (Figure 1A). Subsequently, we generated the humanized IL-13Rα2 CAR using a typical third-generation design (Figure 1B). The scFv derived from mouse monoclonal antibody targeting IL-13Rα2 or the humanized scFv was connected with the CD28 transmembrane domain. The intracellular signaling domain contains the co-stimulation domain consisting of CD28 and 41BB, and the CD3ζ domain (Figure 1B). The CAR transduction was processed using retrovirus, and the efficiency was confirmed by immunoblotting (Figure 1C).
Figure 1.

Developing IL-13Rα2-specific CAR-T cells and evaluating the anti-GBM activity in vitro
(A) Flow cytometry examining the cell surface IL-13Rα2 expression of GBM cells. (B) Schematic diagram of the CAR constructions. (C) Immunoblotting examined the CAR transduction. (D) T cell expansion after co-culturing with GBM cells at E:T = 1:1. (E) In vitro killing assay at E:T = 1:1. The tumor cell lysis was examined by detecting luciferase activity. (F) Tumor cell killing activity of CAR-T cells was evaluated by real-time cell analysis. Results were analyzed by Student’s t test. Statistical significance was set at p < 0.05. ∗p < 0.05, ∗∗p < 0.01.
T cell expansion was examined via calculating the cell number after co-culturing with IL-13Rα2-positive GBM cells. As shown in Figure 1D, T cells transduced with humanized CAR-targeting IL-13Rα2 had a 12.3- or 24.6-fold expansion after being co-cultured with U251 or U373 cells separately, compared with mIL-13Rα2 CAR-T cells that had an 8.1- or 11.6-fold expansion. Subsequent in vitro kill assays verified the anti-GBM activity of humanized IL-13Rα2 CAR-T cells. As shown in Figure 1E, both kinds of IL-13Rα2 CAR-T cells lysed more than 30% tumor cells compared with NT cells that lysed 12.1% U251 cells and 2.82% U373 cells at an effector to target (E:T) cell ratio = 1:1. The GBM cell killing activities were similar between these two CAR-T cells at four different E:T ratios (Figure S1). The complete tumor cell killing processes were recorded using a real-time cell growth monitoring system (Figure 1F). The tumor cell growth was repressed by both kinds of CAR-T cells, but not by NT cells. Significantly increased IFN-γ, TNF-α, and IL-6 levels were found in the medium from the IL-13Rα2 CAR-T cells compared with NT cells (Figure 2), whereas hIL-13Rα2 CAR-T cells released relatively lower levels of IFN-γ and IL-6, when compared with mIL-13Rα2 CAR-T cells (Figure 2).
Figure 2.

T cells expressing humanized CAR-targeting IL-13Rα2 release reduced cytokines
The in vitro killing assay was processed at E:T = 1:1 and 2.5:1 for 8 h and the medium was subjected to ELISA for cytokine detection. Results were analyzed by Student’s t test. Statistical significance was set at p < 0.05. ∗p < 0.05, ∗∗p < 0.01.
T cells expressing CAR-targeting IL-13Rα2 inhibit GBM growth in xenograft mouse models
To further examine the anti-GBM activity of humanized IL-13Rα2 CAR-T cells in vivo, we generated the GBM xenograft mouse model with subcutaneous injection of U251-eGFP-Luc into non-obese diabetic/severe combined immunodeficient (NOD-SCID) mice with peritumoral administration of T cells 9 days later (Figure 3A). The tumor growth was then monitored and quantified by using an in vivo imaging system (IVIS). As shown in Figures 3B and 3C, the tumor growth was significantly repressed by IL-13Rα2 CAR-T cells compared with the NT group. Meanwhile, IL-13Rα2 CAR-T cell treatment significantly prolonged the survival of tumor-bearing mice (p = 0.006). Although T cells expressing humanized CAR have better tumor repressing activity, the results were not statistically significant (p = 0.13) (Figures 3B–3D).
Figure 3.

T cells expressing humanized CAR-targeting IL-13Rα2 exhibit impressive anti-GBM efficacy in a subcutaneous xenograft mouse model
(A) Schematic diagram of the generation of GBM xenograft mouse model and the ministration of CAR-T cells. (B) A xenograft mouse model was generated via U251-eGFP-Luc cells subcutaneous injection and followed by the treatment of T cells. The tumor growth was monitored by using the IVIS system. (C) The luciferase signal was quantified by using Living Image® 3.2 software. (D) The overall survival was measured using Kaplan-Meier method, with Cox proportional hazard regression analysis for group comparison. Statistical significance was set at p<0.05.
Subsequently, we evaluated the anti-GBM activity of CAR-T cells by using an intracranial GBM mouse model via U251-eGFP-Luc intracranial implantation, followed by intravenous T cell administration (Figure 4). IL-13Rα2 CAR-T cell treatment repressed the tumor growth (Figures 4B and 4C) and significantly prolonged the survival of tumor-bearing mice (Figure 4D). A similar phenotype has been observed in a U373 cell-generated GBM mouse model (Figure S2A). Intravenous administration of IL-13Rα2 CAR-T cells inhibits the tumor growth in the first 40 days (Figures S2B and S2C). However, the tumors relapsed after day 40, and the survival of tumor-bearing mice was not significantly improved (Figure S2D).
Figure 4.

T cells expressing humanized CAR-targeting IL-13Rα2 exhibit impressive anti-GBM efficacy in an intracranial mouse model
A xenograft mouse model was generated via U251-eGFP-Luc cell intracranial injection. Tumor growth was monitored using the IVIS system. The overall survival was measured using the Kaplan-Meier method, with Cox proportional hazard regression analysis for group comparison. Statistical significance was set at p < 0.05.
Top expressed genes in the humanized third-generation IL-13Rα2 CAR-T cells were functionally enriched in T cell proliferation, migration, and cytotoxicity
To understand the origin of powerful anti-GBM activity of IL-13Rα2 CAR-T cells, we examined the gene expression profile by high-throughput sequencing. There were 23,577 detectable genes in IL-13Rα2 CAR-T cells. The top 100 expressed genes were subjected to GO analysis for three categories: biological process, molecular function, and cellular component. As shown in Figure 5A, five genes (SOS1, CALM1, IL2RB, IL2RG, and UBC) were enriched in MAPK cascade, five genes (CD74, SOS1, ITGAL, MSN, and MYH9) were enriched in leukocyte migration and ten genes were enriched in immune response (CD74, ETS1, TNFRSF1B, GZMA, IL2RG, IL32, LTB, HLA-B, HLA-E, and HLA-DRA). We also observed that five genes (ARL4C, ARHGDIB, EEF1A1, EEF2, and RAC2) functionally enriched in GTPase activity that may contribute to activated T cell activity (Figure 5B). The subcellular localization analysis indicated that proteins encoded by these genes evenly distributed in the cell nucleus, cytosol, extracellular matrix, and on the cell membrane (Figure 5C).
Figure 5.

GO analysis of the top 100 expressed genes in CAR-T cells
T cells transfected with humanized CAR-targeting IL-13Rα2 were subjected to high-throughput sequencing to examine the gene expression profile. The top 100 expressed genes in CAR-T cells were subjected to GO analysis using an online bioinformatics tool: DAVID Bioinformatics Resources 6.8: https://david.ncifcrf.gov/. Fisher’s exact test was used for the gene-enrichment analysis. The GO terms of the three categories, BP(A), CC(B) and MF(C) were shown separately. BP, biological process; CC, cellular components; MF, molecular function.
Subsequently, we analyzed the interrelations of different pathways in immune system processes. We observed that genes enriched in T cell proliferation, activation, toxicity, migration, and synapse formation pathways (Figure 6). While genes, such as MSN, HLA-E, SOS1, CD74, PTPRC, and RAC2, were key hubs connected these pathways (Figure 6). Finally, we analyzed the interaction of these top expressed genes, and found four main clusters (Figure 7). These clusters include cytoskeleton genes (cluster 1), cytotoxicity-related genes (cluster 2), protein folding and modification genes (cluster 3), and mitochondrial function genes (cluster 4).
Figure 6.

ClueGO enrichment analysis of top expressed genes in humanized CAR-T cells
The top 100 expressed genes were subjected to ClueGO plugin of Cytoscape software for immune process-related pathway interrelation analysis. Functionally grouped network with terms as nodes were linked based on their kappa score level (≥0.3), where only the label of the most significant term per group is shown. The node size represents the term enrichment significance. Functionally related groups partially overlap. The color gradient shows the gene proportion of each cluster associated with the term. The node size represents the term enrichment significance.
Figure 7.

rotein-protein interaction analysis of the top 100 expressed genes
Protein-protein interaction analysis of the top 100 expressed genes in CAR-T cells.
Discussion
GBM is the most aggressive primary brain malignancy in adults, and is still incurable with conventional therapies.1,2 Recently, the emerging immune-based technologies exhibited encouraging anti-GBM activity including adoptive T cell therapies. Brown and colleagues reported for the first time that a second-generation autologous CAR-T cells targeting IL-13Rα2 mediated a transient complete response in a patient with recurrent multifocal glioblastoma, with dramatic improvements in quality of life.13 In this study, we generated a humanized third-generation CAR-targeting IL-13Rα2 and then examined the anti-GBM efficacy in vitro and in xenograft mouse models. To our knowledge, this is the first research of the humanized third-generation CAR-targeting IL-13Rα2 for GBM treatment that provides a candidate tool for clinical use.
The first-generation CAR without a co-stimulatory signal domain was first developed to treat cancer, but T cells transduced with these CARs were found to be poorly persistent in vivo, which promoted the later incorporation of co-stimulatory signal domains from CD28 or 4-1BB into the CAR intracellular structure. Since the two domains determine different functional properties of CAR-T cells, the third-generation of CARs having these two different functional properties were established. Because mouse antibody-derived CARs will induce immune responses, limit CAR-T cell persistence, and increase the risk of relapse, in this study we developed the humanized third-generation CAR for GBM treatment. Our in vitro cell killing result indicated that this humanized third-generation CAR-targeting IL-13Rα2 did not secrete increased IL-6 and expressed dramatic low levels of IL-10. Since IL-6 was identified to be a key driver of CRS,24 while IL-10 was found to induce T cell dysfunction,25 our results suggest a powerful anti-GBM potential in patients.
One key reason for lack of progress in the treatment of glioblastoma is extensive intra- and inter-tumor heterogeneity.26 In this study, the humanized third-generation CAR-targeting IL-13Rα2 had an impressive anti-GBM efficacy in U251 cell-generated mouse models but did not exhibit significant anti-tumor effect in a U373-generated mouse model. This may be a result of lower IL-13Rα2 expression that will escape from the T cell surveillance.
CAR-T cell therapy has been successfully applied in the treatment of hematologic B cell malignancies. However, CAR-T cell treatment for solid tumors has limited anti-tumor activity. One of the main causes is the suppressive tumor microenvironment, because activated T cells must adapt their metabolism to meet the energetic demands associated with rapid proliferation and effector function. The aberrant metabolic milieu in tumor microenvironments, a result of the high metabolic activity of tumor cells and dysfunctional tumor vasculature, inhibits T cell effector functions and promotes a defective T cell state.27,28 The blood-brain barrier (BBB) is also a key factor that limits the efficacy of CAR-T cells. In this study, both U251 and U373 generated intracranial tumors relapsed 40 days after treatment, which may be the result of tumor microenvironment and reduced T cell persistence. So, further study will be focused on optimizing the CAR structure to increase the persistence of T cells and combination with other treatments to overcome the BBB for GBM treatment.
High-throughput sequence data unveiled the origin of powerful anti-GBM activity of this kind of humanized third-generation CAR-T cells. Genes consisting of a cytoskeleton, contributing to cytotoxicity, mitochondrial function, and protein folding, were found highly expressed in CAR-T cells, such as MYH9 and FLNA. MYH9 was the sole class II myosin expressed in T cells and required for T cell motility but is dispensable for synapse formation.29 FLNA provides an important link between the integrin and the actin cytoskeleton, which is required for T cell adhesion and trafficking.30 These highly expressed genes represent the high migration ability of T cells. The initiation of T cell activation results in the reprogramming of cellular metabolic processes to facilitate proliferation, differentiation, and production of effector molecules, such as cytokines.31 The highly expressed mitochondrial genes and protein folding-related genes contribute to the activation of T cells.
In conclusion, we successfully constructed a humanized third-generation CAR-targeting IL-13Rα2 and then examined its anti-GBM activity in vitro and in xenograft mouse models, which provide a candidate tool for clinical GBM treatment.
Materials and methods
Cell lines
The human GBM cell lines U251 and U373 were kindly provided by Dr W.K. Cavenee. The retrovirus packaging cell line PG13 was purchased from the American Type Culture Collection. U251 and U373 cells expressing eGFP and firefly luciferase were generated by retroviral infection. All these cells were maintained in Dulbecco’s modified Eagle’s medium (Lonza), containing 10% fetal bovine serum (Biosera) and 10,000 IU/mL penicillin/10,000 μg/mL streptomycin (EallBio Life Sciences).
Generation of retroviral vectors encoding humanized IL13-BBζ CAR-T cells
IL-13Rα2-scFv coding sequence was synthesized by GeneArt (Invitrogen) and then subcloned into the SFG retroviral vector. Transfection experiments were performed using a Calcium Phosphate Transfection Kit (Sigma) according to the instructions with minor modifications.
Generation of CAR-T cells
Human peripheral blood mononuclear cells (PBMCs) from healthy donors were isolated by gradient centrifugation using Ficoll solution (GE Healthcare). T cells in PBMCs were stimulated with anti-CD3/CD28 T cell Activator Dynabeads (Invitrogen). After 48 h of bead activation, T cells were transduced with retroviral supernatants by centrifugation on Retronectin (Takara)-coated plates. On day 7, the transduction efficiency was measured through detecting CD3ζ level by immunoblotting. T cells were cultured in X-VIVO-15 medium with 5% human AB serum (Sigma), 100 U/mL IL-2, 100 U/mL penicillin, and 100 μg/mL streptomycin (EallBio Life Sciences). This study was approved by the institutional review committee of Beijing Shijitan Hospital and informed consent was obtained from all participants.
Xenograft mouse model with subcutaneous glioma cells injection
Six- to 8-week-old NOD-SCID mice were purchased from Charles River Laboratories. The right flanks of female NOD-SCID mice were injected with 5 × 106 U251-eGFP-Luc cells to construct the xenograft mouse model. Ten days after tumor cell injection, 3 × 107 CAR-T cells were injected directly into the tumor. Tumor development was monitored using IVIS (IVIS, Xenogen, Alameda, CA, USA), and the mice were sacrificed when the diameter of the tumor reached 20 mm.
Xenograft mouse model with glioma cells intracranial implantation
Six- to 8-week-old NOD-SCID mice were anesthetized with a ketamine/xylazine cocktail solution. Animals were secured in a stereotaxic head frame, a 1 cm midline scalp incision was made, and 2 × 105 U373-eGFP-Luc cells in 5 μL PBS were injected into the left striatum (coordinates: 2.5 mm lateral and 0.5 mm posterior to the bregma) through a burr hole in the skull using a 10-μL BD syringe to deliver tumor cells to a 3.5-mm intraparenchymal depth. The burr hole in the skull was sealed with bone wax, and the incision was closed using medical adhesive glue (COMPONT). Two weeks after the tumor cells injection, 3 × 107 CAR-T cells were injected through the tail vein and tumor growth was monitored using the IVIS in vivo imaging system (IVIS, Xenogen). All experiments with mice were approved by the Beijing Shijitan Hospital institutional review board.
Flow cytometry analysis
Flow cytometry was performed on a FACSCanto Plus instrument (BD Biosciences) and FlowJo v.10 (FlowJo, LLC) was used for data analysis.
Antibody list: APC-conjugated CD3 antibody (BD Pharmingen 555340); V450-conjugated CD4 antibody (BD Pharmingen 560650); PE-Cy7-conjugated CD8 antibody (BD Pharmingen 560960); FITC-labeled goat anti-mouse IgG(H+L) antibody (Sigma); FITC-conjugated IL13Ra antibody (BD Biosciences); and APC-labeled anti-CD107a-APC antibody (BD Biosciences).
Cytotoxicity assay
IL-13Rα2 CAR-T cells were co-cultured with GBM cells at an E:T ratio from 1:1 to 10:1 in a 24-well plate. Non-transduced T cells were used as negative control. After 24 h, cells were collected and stained with the tumor cell surface marker to evaluate the cytotoxicity of CAR-T cells by flow cytometry.
Cytokine production assays
CAR-T cells were co-cultured with the human GBM cell lines at an E:T = 1:1 for 24 h. The expression of human IFN-γ, TNF-α, IL-6, and IL-10 in the supernatant from co-culture cells was assessed using commercial Cytometric Bead Array Kits (BD Biosciences) according to the procedure of the manufacturer. The level of human IFN-γ was also evaluated using DuoSet ELISA kits (R&D, Minneapolis, MN, USA) following the manufacturer’s instructions.
For intracellular cytokine staining assay, effect T cells were co-cultured with target cells at a 10:1 ratio in 24-well microplates. Every well contained 1 mL T cell medium and 1 μL Golgi Stop (BD cat no. 554724). After 6 h, the cells were harvested and incubated with anti-human IFN-γ (BD Pharmingen, clone B27) antibodies and then measured through flow cytometry analysis.
Real-time cell analysis
The xCELLigence real-time cell analysis (RTCA) system (Roche Applied Science, Basel, Switzerland) was used to evaluate the proliferation/cytotoxicity of CAR-T cells. This system is based on electrical impedance readings of a gold plate sensor electrode at the bottom of cytotoxicity plate (E-16 plate). Human GBM cells, U251 and U373, were seeded in an E-plate at a density of 10,000 cells per well. After 24 h, 100,000 CAR-T cells were added in the E-plate to incubate with the human GBM cells and monitored every 15 min to obtain the cell index for 48 h. Every independent experiment was performed in triplicate. RTCA software was used to automatically calculate the interval slope and evaluate the change rate of the cell index. To demonstrate the effect of treatments, the cell index was normalized to an equal value at the normalization time point.
Immunoblotting
Cells were washed with PBS three times and then the protein was extracted using RIPA buffer. The protein samples were quantified using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific), and then denatured in sodium dodecyl sulfate (SDS)/β-mercaptoethanol sample buffer. Samples (10 μg) were separated on a 15% SDS-polyacrylamide gel and blotted onto polyvinylidene fluoride membranes (Millipore) by electrophoretic transfer. The membrane was incubated with mouse anti-human CD3ζ chain antibody (BD Biosciences, cat. no. 556366) overnight at 4°C, and then the specific protein-antibody complex was detected using HRP-conjugated goat anti-mouse secondary antibody (Santa Cruz Biotechnology). Detection of the chemiluminescence reaction was carried out using an ECL kit (Thermo Fisher Scientific). The experiment was repeated at least three times.
Statistically analysis
All experiments were performed at least in triplicate and GraphPad Prism version 8.0.2 (GraphPad Software) was used for statistical analysis. Data are presented as mean ± standard deviation. The differences between means were tested using appropriate tests. Overall survival of mice with GBM xenografts was measured using the Kaplan-Meier method, with Cox proportional hazard regression analysis for group comparison. A p value less than 0.05 was considered significant.
Ethics approval and consent to participate
This research was approved by the Beijing Shijitan Hospital institutional review board.
Consent for publication
Written informed consent was obtained from all the participants.
Acknowledgments
This research was supported by the Beijing Municipal Science and Technology Commission, Brain Science Research Fund (Z16110000021636).
Author contributions
Conception and design, X.Z.; data acquisition, C.X., Z.A., Y.H., Y.B., and C.Z.; data analysis, C.X., Y.H., and Y.B.; funding acquisition, X.Z.; resources, C.X.; software, Y.H. and Z.A.; writing – original draft, C.X. and Y.H.; writing – review & editing, X.Z.; final approval, C.X., Y.B., Y.H., C.Z., Z.A., and X.Z.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2022.01.002.
Supplemental information
References
- 1.Omuro A., DeAngelis L.M. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013;310:1842–1850. doi: 10.1001/jama.2013.280319. [DOI] [PubMed] [Google Scholar]
- 2.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 3.Bielamowicz K., Khawja S., Ahmed N. Adoptive cell therapies for glioblastoma. Front. Oncol. 2013;3:275. doi: 10.3389/fonc.2013.00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sadelain M., Brentjens R., Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3:388–398. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fesnak A.D., June C.H., Levine B.L. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat. Rev. Cancer. 2016;16:566–581. doi: 10.1038/nrc.2016.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gacerez A.T., Arellano B., Sentman C.L. How chimeric antigen receptor design affects adoptive T cell therapy. J. Cell. Physiol. 2016;231:2590–2598. doi: 10.1002/jcp.25419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martinez-Lostao L., Anel A., Pardo J. How do cytotoxic lymphocytes kill cancer cells? Clin. Cancer Res. 2015;21:5047–5056. doi: 10.1158/1078-0432.CCR-15-0685. [DOI] [PubMed] [Google Scholar]
- 8.Eshhar Z., Waks T., Gross G., Schindler D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. U S A. 1993;90:720–724. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kochenderfer J.N., Wilson W.H., Janik J.E., Dudley M.E., Stetler-Stevenson M., Feldman S.A., Maric I., Raffeld M., Nathan D.A., Lanier B.J., et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brentjens R.J., Riviere I., Park J.H., Davila M.L., Wang X., Stefanski J., Taylor C., Yeh R., Bartido S., Borquez-Ojeda O., et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheridan C. First approval in sight for Novartis’ CAR-T therapy after panel vote. Nat. Biotechnol. 2017;35:691–693. doi: 10.1038/nbt0817-691. [DOI] [PubMed] [Google Scholar]
- 12.Brudno J.N., Kochenderfer J.N. Chimeric antigen receptor T-cell therapies for lymphoma. Nat. Rev. Clin. Oncol. 2018;15:31–46. doi: 10.1038/nrclinonc.2017.128. [DOI] [PubMed] [Google Scholar]
- 13.Brown C.E., Alizadeh D., Starr R., Weng L., Wagner J.R., Naranjo A., Ostberg J.R., Blanchard M.S., Kilpatrick J., Simpson J., et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. New Engl. J. Med. 2016;375:2561–2569. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahmed N., Brawley V., Hegde M., Bielamowicz K., Kalra M., Landi D., Robertson C., Gray T.L., Diouf O., Wakefield A., et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. 2017;3:1094–1101. doi: 10.1001/jamaoncol.2017.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O'Rourke D.M., Nasrallah M.P., Desai A., Melenhorst J.J., Mansfield K., Morrissette J.J.D., Martinez-Lage M., Brem S., Maloney E., Shen A., et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017;9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Debinski W., Gibo D.M., Hulet S.W., Connor J.R., Gillespie G.Y. Receptor for interleukin 13 is a marker and therapeutic target for human high-grade gliomas. Clin. Cancer Res. 1999;5:985–990. [PubMed] [Google Scholar]
- 17.Thaci B., Brown C.E., Binello E., Werbaneth K., Sampath P., Sengupta S. Significance of interleukin-13 receptor alpha 2-targeted glioblastoma therapy. Neuro Oncol. 2014;16:1304–1312. doi: 10.1093/neuonc/nou045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jarboe J.S., Johnson K.R., Choi Y., Lonser R.R., Park J.K. Expression of interleukin-13 receptor alpha2 in glioblastoma multiforme: implications for targeted therapies. Cancer Res. 2007;67:7983–7986. doi: 10.1158/0008-5472.CAN-07-1493. [DOI] [PubMed] [Google Scholar]
- 19.Brown C.E., Warden C.D., Starr R., Deng X., Badie B., Yuan Y.C., Forman S.J., Barish M.E. Glioma IL13Ralpha2 is associated with mesenchymal signature gene expression and poor patient prognosis. PLoS One. 2013;8:e77769. doi: 10.1371/journal.pone.0077769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krenciute G., Krebs S., Torres D., Wu M.F., Liu H., Dotti G., Li X.N., Lesniak M.S., Balyasnikova I.V., Gottschalk S. Characterization and functional analysis of scFv-based chimeric antigen receptors to redirect T cells to IL13Ralpha2-positive glioma. Mol. Ther. 2016;24:354–363. doi: 10.1038/mt.2015.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown C.E., Starr R., Aguilar B., Shami A.F., Martinez C., D’Apuzzo M., Barish M.E., Forman S.J., Jensen M.C. Stem-like tumor-initiating cells isolated from IL13Ralpha2 expressing gliomas are targeted and killed by IL13-zetakine-redirected T cells. Clin. Cancer Res. 2012;18:2199–2209. doi: 10.1158/1078-0432.CCR-11-1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim J.W., Young J.S., Solomaha E., Kanojia D., Lesniak M.S., Balyasnikova I.V. A novel single-chain antibody redirects adenovirus to IL13Ralpha2-expressing brain tumors. Sci. Rep. 2015;5:18133. doi: 10.1038/srep18133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maus M.V., Haas A.R., Beatty G.L., Albelda S.M., Levine B.L., Liu X., Zhao Y., Kalos M., June C.H. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol. Res. 2013;1:26–31. doi: 10.1158/2326-6066.CIR-13-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giavridis T., van der Stegen S.J.C., Eyquem J., Hamieh M., Piersigilli A., Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018;24:731–738. doi: 10.1038/s41591-018-0041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rivas J.R., Liu Y., Alhakeem S.S., Eckenrode J.M., Marti F., Collard J.P., Zhang Y., Shaaban K.A., Muthusamy N., Hildebrandt G.C., et al. Interleukin-10 suppression enhances T-cell antitumor immunity and responses to checkpoint blockade in chronic lymphocytic leukemia. Leukemia. 2021;35:3188–3200. doi: 10.1038/s41375-021-01217-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perrin S.L., Samuel M.S., Koszyca B., Brown M.P., Ebert L.M., Oksdath M., Gomez G.A. Glioblastoma heterogeneity and the tumour microenvironment: implications for preclinical research and development of new treatments. Biochem. Soc. Trans. 2019;47:625–638. doi: 10.1042/BST20180444. [DOI] [PubMed] [Google Scholar]
- 27.Scharping N.E., Menk A.V., Moreci R.S., Whetstone R.D., Dadey R.E., Watkins S.C., Ferris R.L., Delgoffe G.M. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity. 2016;45:374–388. doi: 10.1016/j.immuni.2016.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turley S.J., Cremasco V., Astarita J.L. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol. 2015;15:669–682. doi: 10.1038/nri3902. [DOI] [PubMed] [Google Scholar]
- 29.Jacobelli J., Chmura S.A., Buxton D.B., Davis M.M., Krummel M.F. A single class II myosin modulates T cell motility and stopping, but not synapse formation. Nat. Immunol. 2004;5:531–538. doi: 10.1038/ni1065. [DOI] [PubMed] [Google Scholar]
- 30.Savinko T., Guenther C., Uotila L.M., Llort Asens M., Yao S., Tojkander S., Fagerholm S.C. Filamin A is required for optimal T cell integrin-mediated force transmission, flow adhesion, and T cell trafficking. J. Immunol. 2018;200:3109–3116. doi: 10.4049/jimmunol.1700913. [DOI] [PubMed] [Google Scholar]
- 31.Chapman N.M., Boothby M.R., Chi H. Metabolic coordination of T cell quiescence and activation. Nat. Rev. Immunol. 2020;20:55–70. doi: 10.1038/s41577-019-0203-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.