

Abstract
RASSF1A promoter methylation has been correlated with tumor dedifferentiation and aggressive oncogenic behavior. Nevertheless, the underlying mechanism of RASSF1A‐dependent tumor dedifferentiation remains elusive. Here, we show that RASSF1A directly uncouples the NOTCH‐HES1 axis, a key suppressor of differentiation. Interestingly, the crosstalk of RASSF1A with HES1 occurs independently from the signaling route connecting RASSF1A with the Hippo pathway. At the molecular level, we demonstrate that RASSF1A acts as a scaffold essential for the SUMO‐targeted E3 ligase SNURF/RNF4 to target HES1 for degradation. The reciprocal relationship between RASSF1A and HES1 is evident across a wide range of human tumors, highlighting the clinical significance of the identified pathway. We show that HES1 upregulation in a RASSF1A‐depleted environment renders cells non‐responsive to the downstream effects of γ‐secretase inhibitors (GSIs) which restrict signaling at the level of the NOTCH receptor. Taken together, we report a mechanism through which RASSF1A exerts autonomous regulation of the critical Notch effector HES1, thus classifying RASSF1A expression as an integral determinant of the clinical effectiveness of Notch inhibitors.
Keywords: cancer stemness, Hippo‐Notch signaling crosstalk, RASSF1A‐HES1‐SNURF/RNF4 complex, γ‐secretase inhibitors (GSIs)
Subject Categories: Cancer, Post-translational Modifications & Proteolysis, Signal Transduction
The RASSF1A tumor suppressor uncouples the NOTCH‐HES1 axis by triggering SNURF/RNF4‐mediated HES1 ubiquitination. Loss of RASSF1A promotes cancer stemness via NOTCH‐independent HES1 stabilization and confers resistance to γ‐secretase inhibitors.
Introduction
RASSF1A epigenetic inactivation correlates with poor clinicopathological characteristics in the vast majority of human solid malignancies (Grawenda & O'Neill, 2015). RASSF1A is a key mediator of Hippo pathway activity and facilitates transcription factor selection for the Hippo effector YAP (Hamilton et al, 2009; van der Weyden et al, 2012; Pefani et al, 2016). In addition to its tumor‐suppressive role, RASSF1A undergoes stringent epigenetic regulation throughout early embryonic development to allow smooth transition from pluripotency to differentiation (Papaspyropoulos et al, 2018).
Tumorigenesis and somatic cell reprogramming are governed by similar mechanisms, as tumor progression is the outcome of dedifferentiation processes (Friedmann‐Morvinski & Verma, 2014). Tumor dedifferentiation has been linked with upregulation of stem cell (SC) markers, such as OCT4, NANOG, and SOX2 and their target genes, which are normally enriched in embryonic stem cells (ESCs) (Friedmann‐Morvinski & Verma, 2014; Hepburn et al, 2019).
Current findings support that Notch signaling holds a crucial role in inducing tumor dedifferentiation. Activation of the NOTCH1‐HES1 axis in solid tumors results in increased epithelial‐to‐mesenchymal transition (EMT) accompanied by upregulation of stemness genes (Reedijk et al, 2008; Fender et al, 2015; Jin et al, 2017; Fendler et al, 2020). The NOTCH‐HES1 axis can be effectively disrupted by γ‐secretase inhibitors (GSIs) preventing the proteolytic cleavage of the NOTCH receptor and subsequent translocation of its intracellular domain (NCID) to the nucleus, thereby abrogating both Notch signaling and SC marker expression (Chu et al, 2013).
In this study, we present a novel mechanism through which RASSF1A prevents tumor dedifferentiation by triggering HES1 degradation in a Hippo‐independent manner. The significance of the identified pathway is depicted in a wide spectrum of human malignancies, whereas employing NOTCH inhibitors defines the precise clinical setting for potential therapeutic intervention.
Results and Discussion
RASSF1A levels inversely correlate with HES1 expression across human tumor types
We previously showed that NOTCH signaling negatively correlates with RASSF1A in human tumors such as breast and lung (Pefani et al, 2016). The Notch target HES1 is a basic helix‐loop‐helix (bHLH) transcriptional regulator (Artavanis‐Tsakonas et al, 1999) which promotes CSC self‐renewal (Gao et al, 2014). To investigate a potential RASSF1A‐HES1 regulation, we analyzed RNA sequencing (RNA‐seq) data from The Cancer Genome Atlas (TCGA) and the Genotype‐Tissue Expression (GTEx) portal deriving from 32 different human tumor types arising in different organs (n = 9,527 samples). Interestingly, we found that RASSF1A and HES1 are reciprocally expressed across whole patient cohorts for tumors with both high (Fig 1A and Dataset EV1) and low (Fig 1B and Dataset EV1) levels of RASSF1A. Along the same lines, a correlative analysis implementing again RNA‐seq data from the TCGA and GTEx cancer databases demonstrated a statistically significant (P < 0.01) negative correlation of RASSF1A with HES1 levels across all tumor types (Fig 1C and Dataset EV1). In line with our clinical data (Fig 1D), upon silencing of RASSF1A, HES1 levels were significantly increased in both human osteosarcoma (U2OS) and cervical cancer (HeLa) cells (Fig 1E and F), implying that RASSF1A may functionally regulate HES1. RASSF1A and HES1 Transcripts Per Million (TPMs) in normal tissues (Fig 1D) were only included to provide a rough estimation of how deregulated the expression of each marker may be in cancer compared to normal tissue. A limitation to this estimation may derive from the small sample size of two normal tissue types (the respective of cervical cancer‐CESC and sarcoma‐SARC) which relates to their poor representation by the TCGA database.
Figure 1. The RASSF1A expression pattern is reciprocal to HES1 across human tumor types.
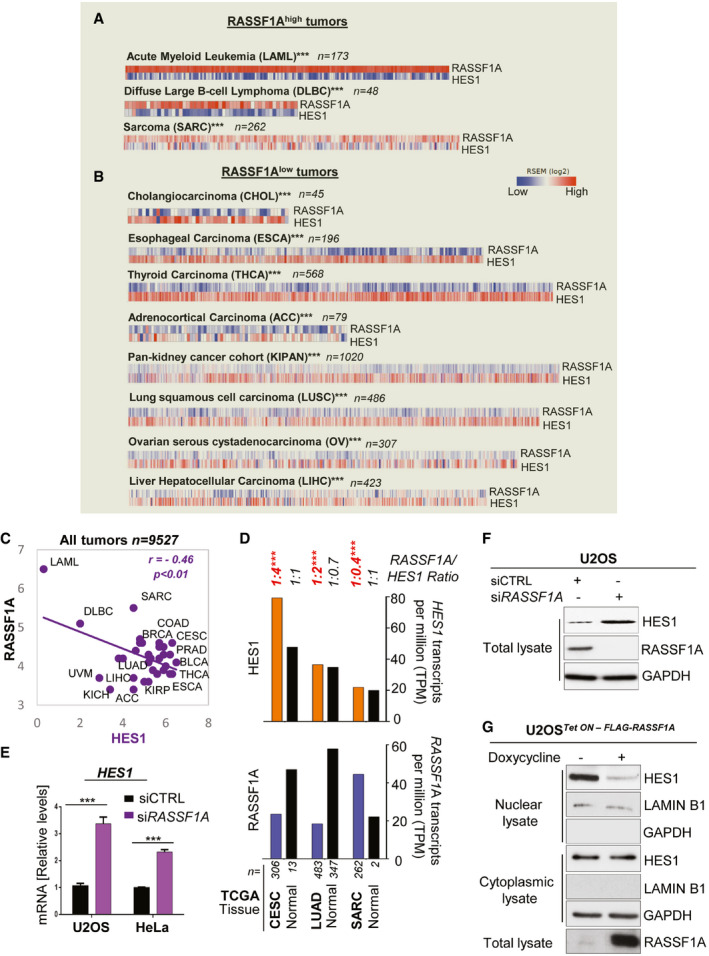
- Correlation heatmaps depicting an opposite RASSF1A‐HES1 expression pattern across the tumors with highest expression of RASSF1A. RNA seq data were retrieved from the TCGA database. Of the 32 tumor types of the TCGA database included in the analysis, tumor types with an average of > 4.2 (median) RASSF1A Transcripts per million (TPM) were classified as RASSF1Ahigh, whereas tumor types with an average of < 4.2 RASSF1A TPM were classified as RASSF1Alow. See also Dataset EV1.
- Same as A, for the TCGA tumors with the lowest expression of RASSF1A. See also Dataset EV1.
- RASSF1A (bottom) and HES1 (top) transcripts per million (TPM) in the indicated types of human tumors and respective normal tissue. Data were extracted from the TCGA and GTEx databases using the GEPIA/GEPIA2 online tool for normal and cancer gene expression profiling and interactive analyses. The number of patients is indicated in each tumor type. See also Dataset EV1.
- qPCR for HES1 mRNA levels in siCTRL (non‐targeting) and siRASSF1A‐transfected U2OS and HeLa cells.
- U2OS cells were transfected with either siCTRL or siRASSF1A and lysates were immunoblotted for HES1 levels.
- U2OS cells Tet‐On inducibly expressing FLAG‐RASSF1A were treated with doxycycline at a concentration of 0.5 μg/ml for 24 h. The cells were fractionated in order to acquire the nuclear and cytoplasmic extracts, which were subsequently Western blotted and probed with the indicated antibodies.
Data information: Tumor type abbreviations: ACC: Adrenocortical carcinoma; BLCA: Breast invasive carcinoma; BRCA: Breast invasive carcinoma; CESC: Cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL: Cholangiocarcinoma; COAD: Colon adenocarcinoma; DLBC: Lymphoid Neoplasm Diffuse Large B‐cell Lymphoma; ESCA: Esophageal carcinoma; KICH: Kidney Chromophobe; KIRC: Kidney renal clear cell carcinoma; KIRP: Kidney renal papillary cell carcinoma; KIPAN: Pan‐kidney cohort (KIRP + KIRC + KICH); LAML: Acute Myeloid Leukemia; LIHC: Liver hepatocellular carcinoma; LUSC: Lung squamous cell carcinoma; OV: Ovarian serous cystadenocarcinoma; PRAD: Prostate adenocarcinoma; SARC: Sarcoma; THCA: Thyroid carcinoma; UVM: Uveal melanoma. ***P < 0.001 of Student’s t‐test. Error bars indicate s.e.m. Data shown are representative of three biological replicates (n = 3).
Source data are available online for this figure.
To identify potential RASSF1A‐dependent regulators of HES1, we analyzed TCGA data from clinical samples expressing high and low RASSF1A levels (Fig EV1A and Dataset EV1). One of the top hits significantly correlating with RASSF1A levels was the transcription factor GATA1 (P < 0.00001; Fig EV1A), a HES1 transcriptional repressor (Ross et al, 2012). Indeed, GATA1 depletion led to HES1 upregulation in our system (Fig EV1B). Interestingly, GATA1 levels were found decreased upon silencing of the Hippo pathway component Mst1/2 in Xenopus embryos (Nejigane et al, 2013), likely indicating dependency on RASSF1A which functions as an MST2 activator (Matallanas et al, 2007). In support of this, RASSF1A depletion and overexpression in U2OS cells resulted in GATA1 reduction and increase, respectively (Fig EV1C and D), indicating that GATA1 may be responsible for HES1 regulation by RASSF1A at the transcriptional level.
Figure EV1. Transcriptional regulation of HES1 by RASSF1A.
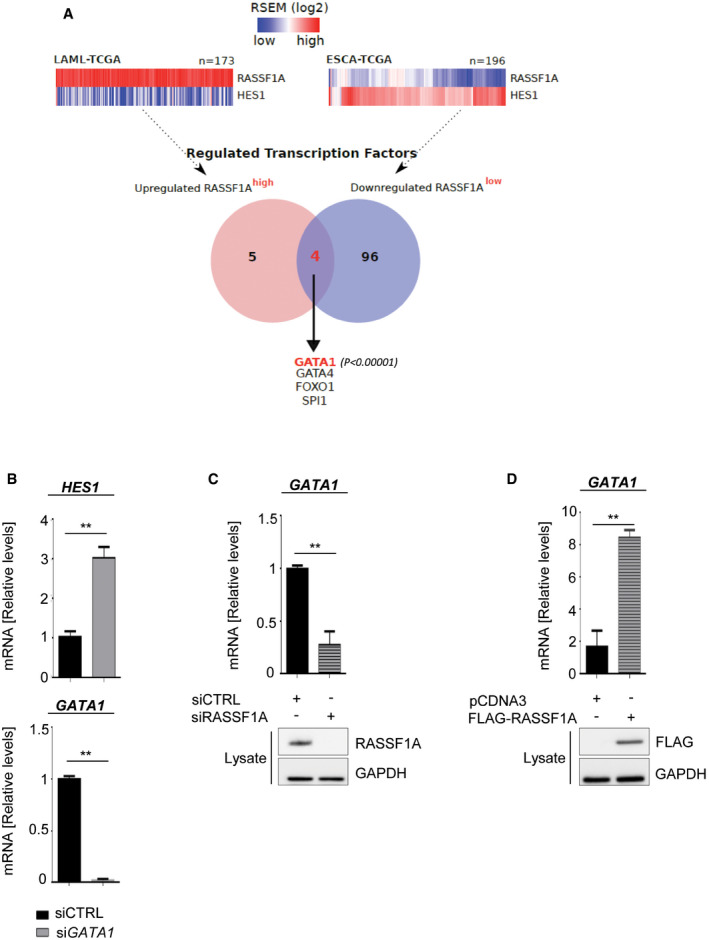
- Analysis of TCGA RNA‐seq data from clinical samples expressing highest (LAML) and lowest (ESCA) RASSF1A levels to identify transcription factors following RASSF1A expression. One of the most prominent hits was the HES1 repressor GATA1 (P < 0.00001).
- Top: qPCR for HES1 levels in U2OS cells transfected with non‐targeting (siCTRL) or GATA1‐targeting (siGATA1) siRNA. Bottom: qPCR for GATA1 levels verifying successful KD.
- qPCR for GATA1 levels in U2OS cells transfected with either siCTRL or RASSF1A‐targeting (siRASSF1A) siRNA. Western blotting for RASSF1A levels demonstrates knockdown efficiency.
- qPCR for GATA1 levels in U2OS cells transfected with pCDNA3 or FLAG‐RASSF1A. Western blotting demonstrates FLAG‐RASSF1A induction.
Data information: Tumor type abbreviations: LAML: Acute Myeloid Leukemia; ESCA: Esophageal carcinoma. **P < 0.01, of Student’s t‐test. Error bars indicate s.e.m. Data shown are representative of three biological replicates (n = 3).
Source data are available online for this figure.
The observation that HES1 mRNA levels follow the HES1 protein stabilization pattern after RASSF1A modulation (Fig 1E and F) is in keeping with the presence of deeply characterized feedback loops in the case of HES1, where the amount of HES1 protein or its targets in the cell determine HES1 gene transcription rate (Kobayashi et al, 2009; Roese‐Koerner et al, 2017; Duan et al, 2019). Similarly to HES1, which is expressed in cells and tissues in an oscillatory fashion (Kobayashi et al, 2009; Roese‐Koerner et al, 2017), RASSF1A levels have also been described to oscillate through the cell cycle via degradation by SCF E3 ligase (Song et al, 2008) suggesting that these regulatory processes may be linked.
Given that HES1 can be both nuclear and cytoplasmic (Sturrock et al, 2014), we next asked whether RASSF1A‐HES1 regulation may differentially affect HES1 protein levels in the nuclear and cytoplasmic fraction. We employed a U2OS Tet‐On cell line inducibly expressing FLAG‐RASSF1A (U2OS Tet ON – FLAG‐RASSF1A ) upon doxycycline treatment and assessed HES1 protein levels following subcellular fractionation. Interestingly, consistent with a recently described RASSF1A function in nucleocytoplasmic protein transfer and nuclear RASSF1A localization (Chatzifrangkeskou et al, 2019), RASSF1A expression reduced only the nuclear HES1 pool while cytoplasmic HES1 levels remained unaffected (Figs 1G and EV2A).
Figure EV2. RASSF1A‐mediated ubiquitination of HES1.
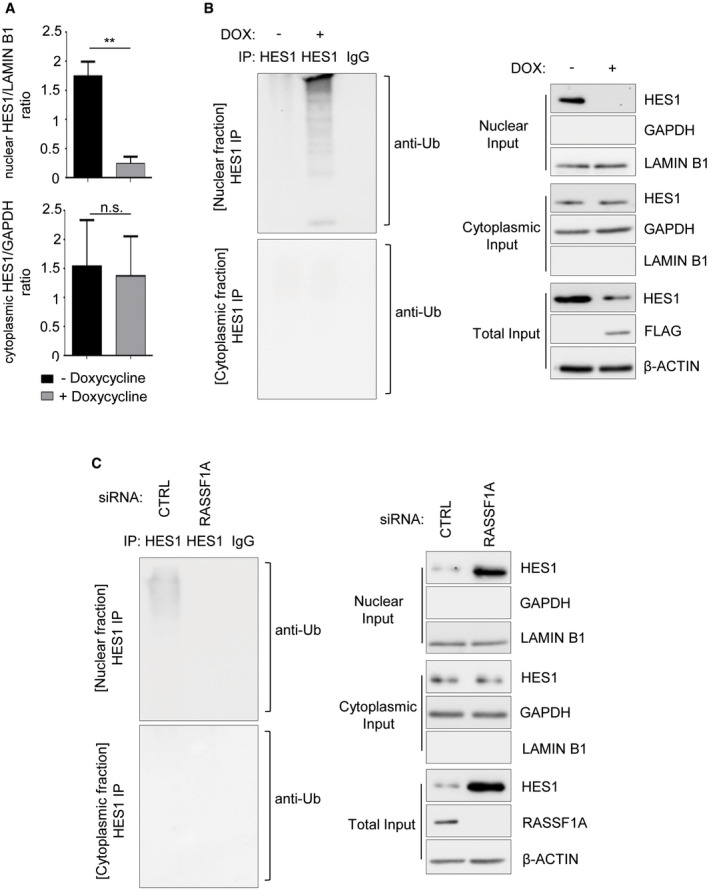
- Densitometry on Fig 1G showing that only nuclear HES1 levels are significantly reduced upon RASSF1A induction, based on the nuclear HES1/LAMIN B1 ratio. Cytoplasmic HES1 levels remain unaffected based on the cytoplasmic HES1/GAPDH ratio.
- In vivo ubiquitination assay in HES1 immunoprecipitates from nuclear and cytoplasmic fractions of U2OS cells Tet‐On inducibly expressing FLAG‐RASSF1A versus Control. Doxycycline (DOX) was used at a concentration of 0.5 μg/ml for 24 h. Immunoprecipitates and Input lysates are probed with displayed antibodies.
- In vivo ubiquitination assay in HES1 immunoprecipitates from nuclear and cytoplasmic fractions of U2OS cells transfected with either siCTRL or siRASSF1A. Immunoprecipitates and Input lysates are probed with displayed antibodies.
Data information: **P < 0.01, of Student’s t‐test. Error bars indicate s.e.m. Data shown are representative of three biological replicates (n = 3).
Source data are available online for this figure.
RASSF1A stabilizes the E3 ligase RNF4 to inactivate HES1 through formation of a RASSF1A‐RNF4‐HES1 complex
Apart from RASSF1A regulating HES1 expression, a proteomic screen for RASSF1A binding partners surprisingly identified HES1 itself as a potential interactor (Dataset EV2), suggesting that RASSF1A‐HES1 regulation may expand to the post‐translational level thereby affecting HES1 stability. In our proteomic screen (Dataset EV2), we also identified the E3 ubiquitin ligase SNURF/RNF4, whose Drosophila ortholog Degringolade (Dgrn) was shown to target HES proteins for SUMO‐independent ubiquitination in vitro and in vivo (Abed et al, 2011; Barry et al, 2011). Additionally, a Sleeping Beauty genetic screen carried out in Rassf1a −/− mice identified RNF4 among the potentially most frequently inactivated genes synergizing with RASSF1A loss in tumorigenesis (van der Weyden et al, 2012).
HES1 has been found to be degraded by additional E3 ligases in different contexts. For example, the E3 ubiquitin ligase SCFFBXL14 complex targets HES1 for proteolysis, thereby enabling neuronal differentiation (Chen et al, 2017). As previously published reports (Abed et al, 2011; Barry et al, 2011; van der Weyden et al, 2012) and our proteomic data strongly pointed to RNF4 as a potential player affecting RASSF1A‐HES1 regulation in cancer cells, we were urged to explore a possible connection.
Interestingly, RASSF1A expression in U2OS cells led to significant stabilization of RNF4 protein levels (Fig 2A). HES1 and RNF4 were both identified as co‐immunoprecipitating in U2OS cells supporting the regulatory connection, while the HES1/RNF4 ratio was decreased in the presence of FLAG‐RASSF1A, suggesting the interaction is supported by RASSF1A (Fig 2B). Moreover, HES1 was found associated with both RNF4 and RASSF1A, confirming the presence of a RASSF1A‐RNF4‐HES1 complex, concomitant with a reduction of HES1 protein levels (Fig 2B). To confirm that RNF4 was directly responsible for the decrease of HES1 upon RASSF1A activation, we overexpressed RASSF1A in the presence or absence of an RNF4‐targeting siRNA and assessed HES1 ubiquitination levels in HES1 immunoprecipitates (Fig 2C). In accordance with data above, RASSF1A overexpression stabilized RNF4 with a concomitant reduction in HES1 levels due to increased HES1 ubiquitination (Fig 2C). However, depletion of RNF4 rescued HES1 despite RASSF1A overexpression (Fig 2C), confirming that RNF4 is required for RASSF1A‐mediated HES1 regulation. In keeping with previous results suggesting that RASSF1A‐HES1 regulation is nuclear, we found that HES1 was ubiquitinated predominantly in the nuclear fraction of U2OS cells, upon RASSF1A induction (Fig EV2B and C). Our observations in cells were further validated in an in vitro ubiquitination assay showing increased RNF4‐mediated HES1 ubiquitination in the presence of RASSF1A (Fig 2D). Taken together, our data uncover a novel mechanism through which RASSF1A regulates the NOTCH effector HES1, via the E3 ligase RNF4 (Fig 2E).
Figure 2. RASSF1A‐dependent upregulation of E3 ligase SNURF/RNF4 destabilizes HES1 through the formation of a RASSF1A‐RNF4‐HES1 complex.
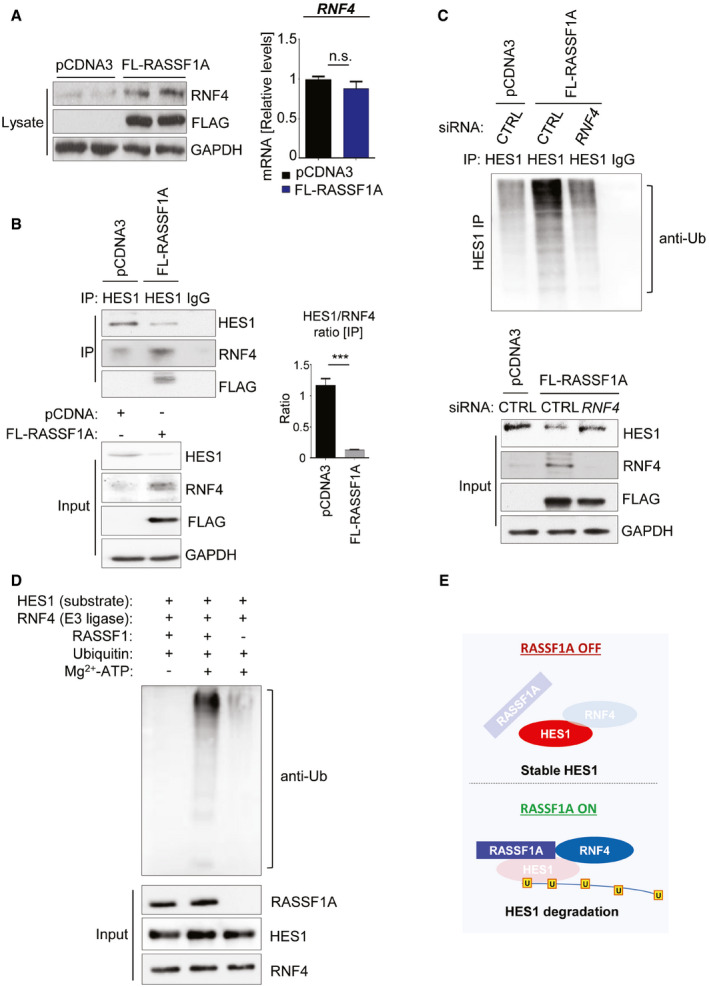
- U2OS cells were transfected with either pCDNA3 or FLAG‐RASSF1A. Cell extracts were collected and Western blotted with the indicated antibodies. qPCR from the same extracts demonstrated no significant differences in RNF4 mRNA levels.
- HES1 immunoprecipitation in U2OS cells treated with either pCDNA3 or FLAG‐RASSF1A and immunoblotting with the indicated antibodies. Densitometry shows the HES1/RNF4 ratio is inversed upon RASSF1A induction. A RASSF1A‐RNF4 complex destabilizes HES1 through direct binding.
- In vivo ubiquitination assay in HES1 immunoprecipitates from U2OS cells transfected with either pCDNA3/siCTRL, FLAG‐RASSF1A/siCTRL, or FLAG‐RASSF1A/siRNF4 to assess Ub chain incorporation. Immunoprecipitates and Input lysates are probed with displayed antibodies. In the absence of RNF4, HES1 levels remain stable despite RASSF1A expression. See also Fig EV2B and C.
- In vitro ubiquitination assay using purified proteins for HES1, RASSF1A, and RNF4. RNF4 served as the E3 ligase of the reaction.
- Schematic illustrating the RASSF1A‐mediated regulation of HES1 stability via the E3 ligase RNF4.
Data information: ***P < 0.001 of Student’s t‐test; n.s., non‐significant. Error bars indicate s.e.m. Data shown are representative of three biological replicates (n = 3).
Source data are available online for this figure.
RASSF1A regulates HES1 activity in a Hippo pathway‐independent manner
RASSF1A epigenetic inactivation has been found to directly induce the expression of the core pluripotency gene Pou5f1/Oct4 in mouse ESCs and pre‐implantation embryogenesis through the Hippo pathway effector YAP (Papaspyropoulos et al, 2018). Although mouse models have proven vital tools in recapitulating human disorders, substantial differences occur between the two species, rendering confirmation in human settings essential (Perlman, 2016). Therefore, we sought to investigate whether RASSF1A may exert a similar regulation of pluripotency genes in human cancer cells. To this end, we silenced RASSF1A in HeLa and U2OS cells and found that the core pluripotency markers were upregulated at the protein (Fig EV3A, B and D) and mRNA (Fig EV3C) level. To verify that those effects were RASSF1A‐mediated, we induced RASSF1A expression in U2OS Tet ON – FLAG‐RASSF1A cells and confirmed that RASSF1A induction was indeed sufficient to restrict endogenous expression of all core pluripotency markers (Fig EV3E).
Figure EV3. RASSF1A regulates the core pluripotency markers in cancer.
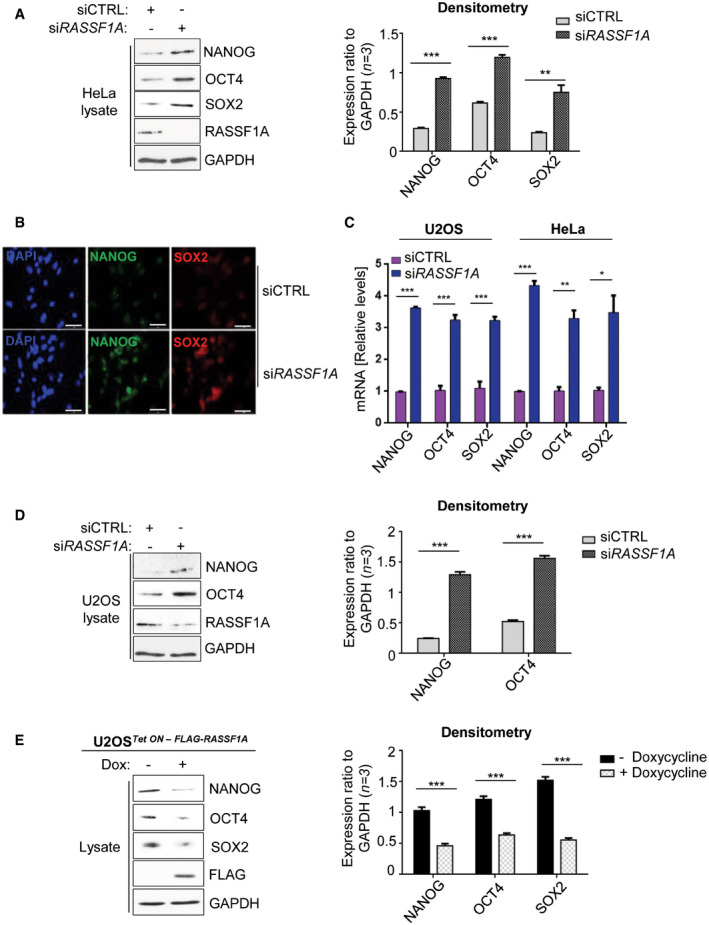
- Western blotting and densitometry for core pluripotency marker expression in HeLa cells transfected with either non‐targeting siRNA (siCTRL) or siRNA against RASSF1A (siRASSF1A).
- Immunofluorescence for core pluripotency marker expression in HeLa cells transfected with either siCTRL or siRASSF1A.
- qPCR for core stem cell marker levels in U2OS and HeLa cells in response to siRASSF1A versus siCTRL.
- Immunoblotting and densitometry for core pluripotency marker expression in U2OS cells transfected with either siCTRL or siRASSF1A.
- U2OS cells Tet‐On inducibly expressing FLAG‐RASSF1A were treated with 0.5 μg/ml of doxycycline for 24 h. Cells were subsequently lysed and lysates were immunoblotted with the indicated antibodies. Densitometry is provided for displayed Western blot images.
Data information: Scale bars: 20 μm. *P < 0.05, **P < 0.01, and ***P < 0.001, of Student’s t‐test. Error bars indicate s.e.m. Data shown are representative of three biological replicates (n = 3).
Source data are available online for this figure.
In accordance with our previous findings, ectopic expression of RASSF1A in a non‐small cell lung carcinoma cell line (H1299) subjected to a two‐dimensional (2D) differentiation assay, completely abrogated SC marker expression and forced cell colonies to lose their round shape, indicating acquisition of a differentiated phenotype (Fig EV4A and B). The RASSF1A‐mediated differentiation phenotype was additionally accompanied by loss of HES1 expression, while RNF4 was markedly stabilized (Fig EV4C).
Figure EV4. RASSF1A induction confers a differentiation phenotype accompanied by HES1 inactivation in cancer cells.
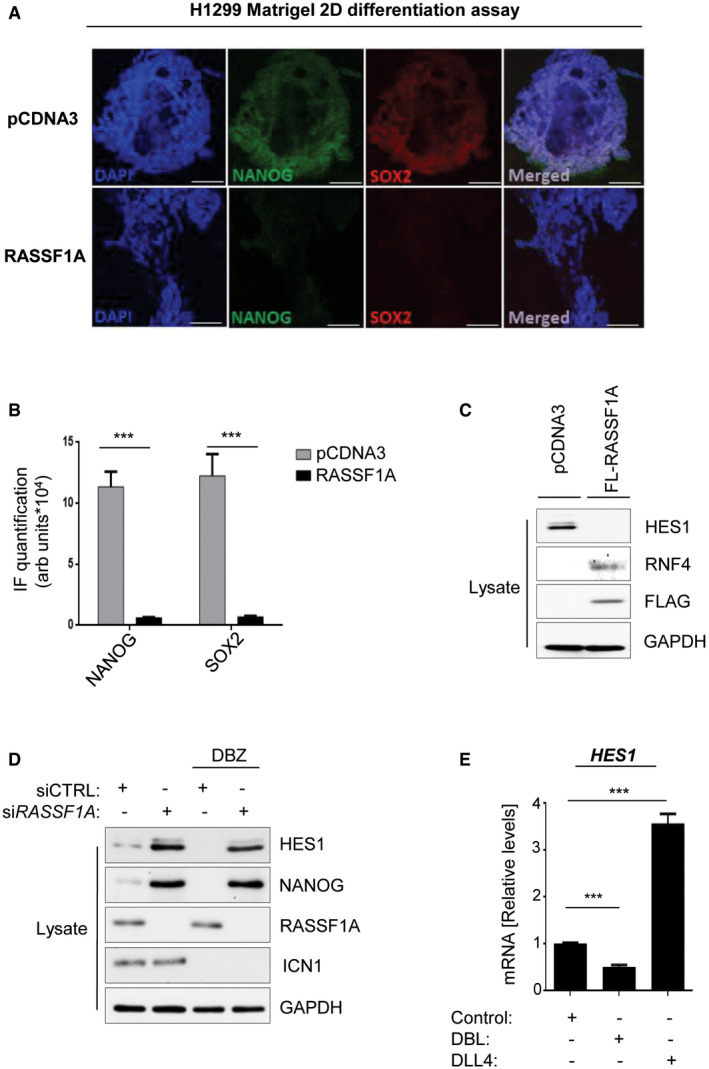
- H1299 cells transfected with either pCDNA3 or FLAG‐RASSF1A and subjected to Matrigel 2D differentiation assay. RASSF1A expression induces differentiation as indicated by changes in the round shape and reduction of stem cell markers assessed by immunofluorescence.
- Quantification of NANOG and SOX2 signal intensities from (A).
- H1299 cells transfected with either pCDNA3 or FLAG‐RASSF1A were lysed and probed with the indicated antibodies confirming a reciprocal HES1/RNF4 regulation in response to RASSF1A.
- HeLa cells transfected with either siCTRL or siRASSF1A were subjected or not to treatment with the GSI DBZ (100 nM) and their lysates were immunoblotted against indicated antibodies. Cleaved Notch (ICN1) levels were used as readout of DBZ treatment.
- qPCR for HES1 mRNA levels in ESC in response to the γ‐secretase inhibitor DBZ (100 nM) or the Notch ligand DLL4 (1 μg/ml).
Data information: Scale bars: 50 μm. ***P < 0.001, of Student’s t‐test. Error bars indicate s.e.m. Data shown are representative of three biological replicates (n = 3).
Source data are available online for this figure.
Our previous work in ESCs and pre‐implantation embryonic development revealed that forced RASSF1A silencing aberrantly activated the core pluripotency network, but to no obvious developmental defects in the mouse embryo. Consistently, Rassf1a −/− mice appear to develop normally and be fertile; however, they are prone to spontaneous tumor formation (Tommasi et al, 2005; van der Weyden et al, 2005). Our current data showing that RASSF1A drives tumor differentiation through downregulation of core pluripotency genes provide a potential explanation for the pronounced tumor predisposition observed in Rassf1a −/− mice.
In order to determine whether RASSF1A‐HES1 regulation may act independently from the Hippo pathway, we evaluated HES1 level changes in response to RASSF1A depletion, upon additional silencing of the Hippo effectors YAP/TAZ. While RASSF1A depletion expectedly increased HES1 levels and destabilized RNF4, additional single knockdowns of YAP or TAZ or dual YAP/TAZ depletion were not accompanied by differences in HES1 or RNF4 levels, confirmed in two independent cell lines (Fig EV5A). Conversely, HES1 reduction in response to RASSF1A was not perturbed by additional induction of active YAP or its phosphorylation‐defective mutant YAP S127A (Fig EV5B). Thus, our data strongly indicate that RASSF1A may control HES1 activity away from its role as a Hippo pathway scaffold.
Figure EV5. HES1 activates core pluripotency marker expression in cancer cells and is regulated by RASSF1A independently of the Hippo pathway.
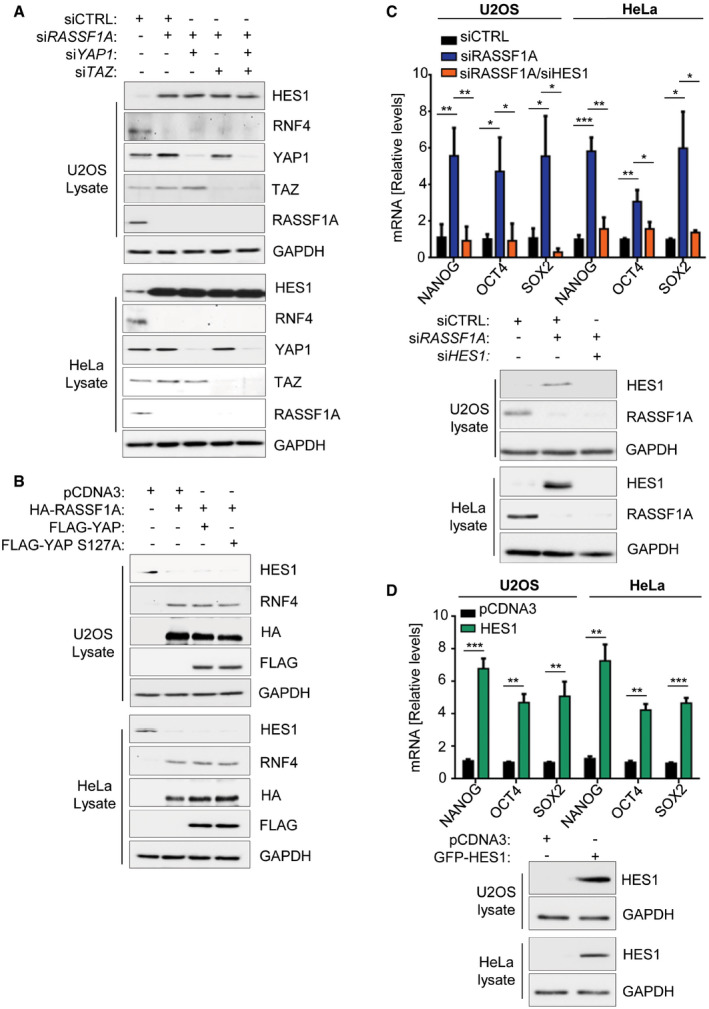
- U2OS cells or HeLa cells transfected with the indicated siRNAs were lysed and probed with shown antibodies. HES1 levels increase in response to RASSF1A loss regardless of YAP/TAZ signaling.
- U2OS cells or HeLa cells transfected with the indicated plasmids were lysed and Western blotted with displayed antibodies. HES1 levels decrease in response to RASSF1A induction regardless of YAP activity.
- qPCR for core pluripotency markers in U2OS and HeLa cells transfected with siCTRL, siRASSF1A, or siRASSF1A/siHES1. HES1 silencing reverses pluripotency marker increase observed upon RASSF1A loss. Immunoblotting from U2OS and HeLa cell lysates using the indicated antibodies demonstrates KD efficiencies.
- qPCR for core pluripotency markers in U2OS and HeLa cells transfected with either pCDNA3 or GFP‐HES1. HES1 induction increases all core pluripotency marker levels. Western blotting from U2OS and HeLa cell lysates using the indicated antibodies demonstrates efficient HES1 induction.
Data information: ***P < 0.001, **P < 0.01, and *P < 0.05, of Student’s t‐test. Error bars indicate s.e.m. Data shown are representative of three biological replicates (n = 3).
Source data are available online for this figure.
RASSF1A overrides Notch signaling to autonomously regulate the NOTCH downstream effector HES1 and to alter response to treatment
We next interrogated whether the RASSF1A‐mediated regulation of HES1 could occur through the NOTCH‐HES1 axis. As HES1 has been directly implicated in SC marker upregulation in various cancer settings (Reedijk et al, 2008; Gao et al, 2014; Fender et al, 2015; Jin et al, 2017; Fendler et al, 2020), we used NANOG expression as a readout for SC marker expression. To address the potential involvement of RASSF1A in NOTCH signaling, we employed collagen 3D‐HeLa spheroid assays to mimic the in vivo tumor microenvironment. Supporting this model with the NOTCH ligand Delta‐like Canonical Notch Ligand 4 (DLL4) resulted in an elevation of HES1 and cancer stemness (NANOG) (Fig 3A and B, DLL4 vs Control). Intriguingly, we find that reduction of RASSF1A levels not only increased baseline HES1 in the absence of ligand but also rendered HES1 and NANOG levels insensitive to the downstream effects of the GSI dibenzazepine (DBZ) which prevents NOTCH cleavage (Figs 3A and B, and EV4D). This suggests that Notch activation and RASSF1A loss independently stabilize HES1 levels and that RASSF1A controls HES1 and NANOG levels even when Notch receptor cleavage is perturbed (Fig 3B). To confirm this finding, we additionally implemented DAPT as an independent GSI in our system, and again found that RASSF1A loss rendered cells irresponsive to the downstream effects of the treatment, as indicated by the unaltered HES1 levels (Fig 3C).
Figure 3. Loss of RASSF1A stabilizes HES1 independently of the NOTCH‐HES1 axis and confers resistance to GSIs.
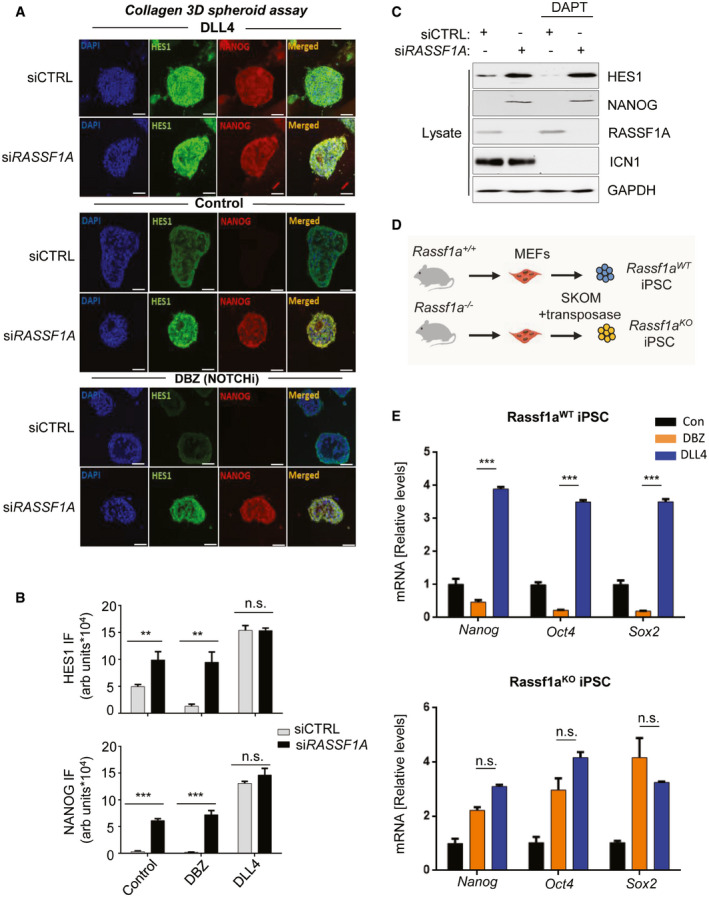
- Top panel: HeLa cells were transfected with either siCTRL or siRASSF1A and subjected to collagen 3D spheroid assay, in the additional presence of the Notch ligand DLL4 (1 μg/ml), increasing HES1 and NANOG expression as assessed by immunofluorescence. Middle panel: Same as top panel, without the presence of DLL4. Bottom panel: Same as top panel, in the additional presence of the GSI DBZ (100 nM) showing upregulation of both NANOG and HES1 upon RASSF1A depletion despite NOTCH‐HES1 inhibition. See also Fig EV4D.
- Quantification of HES1 and NANOG signal intensities displayed in A.
- HeLa cells transfected with either siCTRL or siRASSF1A were subjected or not to treatment with the GSI DAPT (50 μM) and their lysates were immunoblotted against indicated antibodies. Cleaved Notch (ICN1) levels were used as readout of DAPT treatment.
- Schematic depicting iPSC derivation from Rassf1aWT and Rassf1a −/− fibroblasts isolated from the respective mouse strains.
- Top: qPCR for core SC marker mRNA expression in Rassf1aWT iPSC upon DBZ or DLL4 treatment for 24 h. See also Fig EV5E. Bottom: qPCR for core SC marker mRNA expression in Rassf1aKO iPSC upon DBZ or DLL4 treatment for 24 h. RASSF1A loss desensitizes cells to the downstream effects of DBZ treatment.
Data information: Scale bars: 50 μm. **P < 0.01 and ***P < 0.001, respectively, of Student’s t‐test; n.s. stands for non‐significant. Error bars indicate s.e.m. Data shown are representative of three biological replicates (n = 3).
Source data are available online for this figure.
In order to validate NOTCH‐HES1‐dependent regulation of pluripotency markers in response to RASSF1A, we depleted HES1 in U2OS and HeLa cells lacking RASSF1A and found that sufficient to rescue SC marker increase (Fig EV5C). Conversely, ectopic expression of HES1 led to upregulation of all core pluripotency markers (Fig EV5D). To address our findings in a genetically clean system constantly expressing core SC genes, we employed Rassf1aWT or Rassf1aKO induced pluripotent stem cells (iPSC) deriving from mouse embryonic fibroblasts (MEFs) as previously described (van der Weyden et al, 2012; Papaspyropoulos et al, 2018) (Fig 3D). By utilizing mouse E14Tg2a ESCs and our Rassf1a‐proficient iPSC lines (Rassf1aWT iPSC), we confirmed that all core pluripotency markers decreased upon DBZ treatment and increased upon addition of DLL4, following Hes1 regulation, as expected (Figs 3E and EV4E). However, genetic ablation of Rassf1a (Rassf1aKO iPSC) rendered iPSCs insensitive to the downstream effects of DBZ treatment, while DLL4 had a redundant effect with regards to SC marker expression (Fig 3E), thereby corroborating our findings in tumor cells. Taken together, our data strongly imply that Notch‐independent, HES1 stabilization in response to RASSF1A loss may actively contribute to increased SC marker expression.
HES1 has been implicated in chemotherapy resistance, deriving from its capacity to promote cancer stem cell self‐renewal (Liu et al, 2015). HES1‐mediated multidrug resistance has been widely attributed to STAT3 upregulation and p21 suppression (Liu et al, 2015). HES1 has been found to activate STAT3 (Kamakura et al, 2004; Zhou et al, 2013), which in turn induces Nanog and Oct4 transcription in ESC and iPSC through direct binding at the distal enhancers of the genes (Do et al, 2013). Interestingly, although a link between RASSF1A and STAT3 has not been identified yet, RASSF1A was found to upregulate p21 in cancer cells (Thaler et al, 2009), which may contribute to the removal of the drug resistance barrier in RASSF1A‐expressing tumors. Thus, besides RASSF1A itself, clinical patient response may be further stratified based on p21 levels; however, this warrants further investigation. Along the same lines, the expression levels of pluripotency genes may help stratify patient response based on our data demonstrating that core stem cell marker levels are directly reciprocal to RASSF1A activity in cancer cells.
In the present study, for the first time, we provide evidence of a RASSF1A‐mediated regulation of the NOTCH effector HES1, independent of the RASSF1A role as a Hippo pathway scaffold. Our results provide mechanistic insight into RASSF1A‐HES1 regulation, by identifying the E3 ligase SNURF/RNF4 as a critical component of HES1 stability leading to its ubiquitination upon RASSF1A activation. Moreover, we demonstrate that GSIs are likely inefficient in tumors lacking RASSF1A, as HES1 levels remain stabilized regardless of upstream Notch signals. We additionally show that reciprocal RASSF1A‐HES1 regulation is observed in the vast majority of human malignancies, indicating that our identified mechanism may play a critical role in the dedifferentiation process of all tumor types (Fig 4).
Figure 4. Model.
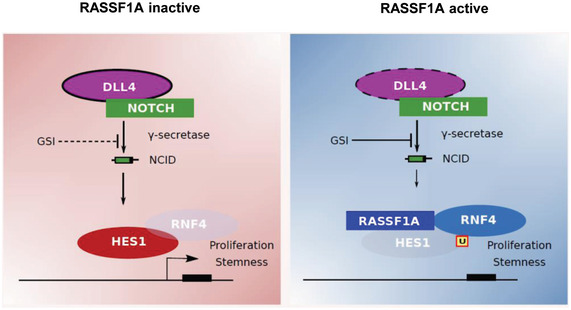
RASSF1A loss leads to HES1 stabilization in a Hippo pathway‐independent mechanism involving the E3 ligase SNURF/RNF4. In the absence of RASSF1A, HES1 levels remain unaffected by GSIs such as DBZ and DAPT. RASSF1A activation induces SNURF/RNF4‐mediated HES1 degradation via targeted HES1 ubiquitination. GSIs may further destabilize HES1 in the presence of RASSF1A. The RASSF1A status may constitute a robust biomarker of the effectiveness of Notch inhibitors in precision oncology.
Different types of oncogenic stimuli can have a varying impact on tumor suppressor activation and subsequent phenotypes (Velimezi et al, 2013; Galanos et al, 2016; Gorgoulis et al, 2019; Lagopati et al, 2021; Zampetidis et al, 2021). RASSF1A is a pleiotropic tumor suppressor protein, which, in response to different signals, is involved in a plethora of biological functions such as cell cycle control, cellular metabolism, and differentiation (Dubois et al, 2019). Our results provide a mechanistic explanation for the role of RASSF1A in tumor dedifferentiation, verified clinically through the analysis of a wide range of human tumor types, consisting of large patient cohorts. In the last few years, GSIs have been in the spotlight of cancer research, exploring their potential clinical implementation in numerous clinical trials (Ran et al, 2017; Cook et al, 2018; Pine, 2018). In our study, we demonstrated that GSIs may be inefficient in tumors lacking RASSF1A; however, in tumors maintaining RASSF1A expression, GSI‐based treatments would likely further contribute to HES1 inactivation. In line with our model, previous research has shown that in tumor types maintaining high levels of RASSF1A such as leukemias (Fig 1A), treatment of patient xenografts with GSIs in vivo decreases tumor burden and prolongs overall survival (Habets et al, 2019; Sohani et al, 2021). Conversely, clinical trials in small cohorts of lung cancer patients or wider cohorts of ovarian cancer patients where RASSF1A levels are traditionally low (Fig 1B), demonstrated insufficient activity of GSIs against cancer progression (Krop et al, 2012; Diaz‐Padilla et al, 2015; Messersmith et al, 2015).
Taken together, our data identify RASSF1A as a clinical biomarker predicting individual patient response to therapy with potential inhibitors of the NOTCH‐HES1 axis, thereby delineating the clinical setting where treatment with such inhibitors might be a productive therapeutic avenue against cancer progression at the patient level.
Materials and Methods
Cell lines and reagents
U2OS, HeLa, and H1299 cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum (Gibco). U2OS Tet‐On cells (Clontech) inducibly expressing FLAG‐RASSF1A upon doxycycline administration were established as described in the manufacturer's protocol (Yee & O'Neill, 2010). E14Tg2a mouse embryonic stem cells and iPS cells were maintained in culture as previously described (Papaspyropoulos et al, 2018). All cell lines were provided from the American Type Culture Collection (ATCC) and Clontech and were maintained in culture for no longer than 2 months. Cell counting was carried out using C‐Chip disposable counting chambers (NE63508‐01) (Wiener et al, 2018).
For Notch stimulation or inhibition, Delta like ligand 4 (DLL4; R&D Systems) and γ‐secretase inhibitors DBZ (R&D Systems) and DAPT were, respectively, used. DLL4 was either added directly in cell medium at 1 μg/ml οr used at the same concentration in coating gelatinized plates in 0.1% gelatin for 2 h. Plates were washed with PBS before cells were seeded. DBZ was added in the media at 100 nM and DAPT at 50 μM. Cells were harvested 24 h after treatment.
Co‐immunoprecipitation
For lysate preparation, cells were lysed in ice‐cold 1% (v/v) Nonidet P40 lysis buffer (20 mM Hepes pH 7.5, 1% NP40 (Roche), 150 mM NaCl, 0.5 mM EDTA, 50 mM NaF, 10 mM β‐glycerophosphate, 0.5 mM Na3VO4, and 1 × EDTA‐free protease inhibitors (Roche)). Lysates were centrifuged at 4°C at 10,000 g for 10 min and were subsequently subjected to sonication for 15 s at 20% amplitude. Samples were normalized with Bradford protein assay and equal protein volumes (1–2 mg) were used per reaction in each experiment. Immunocomplexes were precipitated with Protein G sepharose (Sigma) or Protein G Dynabeads (Life Technologies). The antibodies used were as follows: anti‐YAP (H‐125; sc‐15404), anti‐YAP (clone 63.7;sc‐101199), anti‐TAZ (H‐70; sc‐48805), anti‐TAZ (Ab118373), and anti‐HES1 (AB5702). IPs were incubated on an end‐to‐end rotator for 3 h at 4°C. The beads were then washed three times with NP40 wash buffer. IP samples were resuspended in 50 μl 1 × Laemmli sample buffer (12.5% glycerol (v/v), 2% SDS (w/v), 78 mM Tris pH 6.8, 720 mM β‐mercaptoethanol, bromophenol blue) and boiled (100°C; 5 min).
Immunoblotting
Immunoblotting from immunoprecipitates and cell lysates was carried out as previously described (Pefani et al, 2014; Tzekaki et al, 2021a,b). The following primary antibodies were used at a concentration of 1:1,000; β‐CATENIN (sc‐7199), p73 (ep436Y), YAP (sc‐15404 and sc‐101199), HES1 (AB5702), GAPDH (Abcam;2251‐1; 1/10,000), TAZ (sc‐48805 and Ab118373), FLAG (M2; Agilent; 200472‐21), RASSF1A (sc‐58470), NANOG (Cell Signaling; 4893S), OCT4 (ab19857), SOX2 (MAB4423; 1/500), LAMIN B1 (Ab16048; 1/10,000), RNF4 (Novus Biologicals), TEAD1 (BD Transduction Laboratories; 610922), p73 (EP436Y; ½,000), Ub (Cell Signaling; 3936), HA (Cell Signaling; 3724S), ICN1 (Cell Signaling; 4147S), β‐ACTIN (Cell Signaling; 4967L), and HRP‐conjugated anti‐mouse and anti‐rabbit secondary antibodies were used at a concentration of 1:5,000 (Jackson Immunoresearch).
Nuclear/cytoplasmic fractionation
Cell fractionation was performed using the NE‐PER Nuclear and Cytoplasmic Extraction Reagents Kit (Thermo Scientific). Briefly, 5 × 106 cells were harvested with trypsin‐EDTA and centrifuged at 500 g for 5 min in a 1.5‐ml microcentrifuge tube. The cells were then rinsed with PBS and centrifuged at 500 g for 3 min. The supernatant was carefully removed and the cell pellet was resuspended in ˜ 100 μl ice‐cold CER I reagent (depending on the size of the cell pellet), vortexed, and incubated on ice for 10 min. Following that, 5 μl ice‐cold CER II reagent was added to the tube, which was vortexed and incubated on ice for 1 min. After vortexing again, the tube was centrifuged for 5 min at maximum speed and the supernatant (cytoplasmic extract) was immediately transferred to a pre‐chilled tube, which could be stored at −20°C. The insoluble fraction (pellet) was then suspended in ˜ 50 μl ice‐cold NER reagent and was vortexed for 15 s every 10 min for 40 min in total. Lastly, the tube was centrifuged at maximum speed for 10 min and the supernatant (nuclear extract) could be stored at −80°C in a microcentrifuge tube.
Mass spectrometry
Samples were prepared according to the co‐immunoprecipitation protocol from whole cell lysates with a few modifications. Only Protein G Dynabeads (Life Technologies) were used. After incubation on an end‐to‐end rotator for 3 h, the immunoprecipitates were washed four times, the two first being with NP40 lysis buffer and the last two only with filtered PBS. Tubes were subsequently left uncapped to air‐dry for approximately 15 min. Mass spectrometry analysis was performed using an Orbitrap mass spectrometer. The CRAPome database (Mellacheruvu et al, 2013) was used to discard false‐positive targets frequently found to precipitate with FLAG.
In vivo ubiquitination assay
U2OS cells were transfected with non‐targeting siRNA (siCTRL)/pCDNA3, siCTRL/FLAG‐RASSF1A, and siRNF4/FLAG‐RASSF1A using Lipofectamine 2000 (Invitrogen). 48 h following transfection, cells were treated with MG‐132 proteasome inhibitor (C2211, Sigma‐Aldrich) for 3 h at 30 µM and subsequently lysed in RIPA buffer (Tris–HCl at pH 8.0, 50 mM, NaCl 150 mM, SDS 0.1%, sodium deoxycholate 1%, Triton X‐100 1% and protease (#58440, Pierce, Thermo Scientific), phosphatase (#78420, Pierce, Thermo Scientific) inhibitor cocktail and N‐Ethylmaleimide deubiquitinase inhibitor (04259, Sigma‐Aldrich)). U2OS Tet ON – FLAG‐RASSF1A cells with and without treatment with 0.5 μg/ml of Doxycycline, as well as U2OS cells transfected with siCTRL and siRASSF1A were subjected to nuclear/cytoplasmic fractionation prior to in vivo ubiquitination assay. Protein lysates were precleared with protein G agarose beads (#16‐266, Millipore) for 1 h and then incubated with G‐protein beads bound to HES1 antibody (AB5702) for 2 h at 4°C. Cell lysates and immunoprecipitates were analyzed in Western blotting.
In vitro ubiquitination assay
The assay was carried out in 1× E3 ligase Reaction Buffer (R&D Systems; B‐71) with the addition of the following reaction components up to a final volume of 25–50 μl: 100 nM Recombinant Human His6‐Ubiquitin E1 Enzyme (UBE1, R&D Systems; E‐304), 1 µM E2 Enzyme UBCH5a/UBE2D1 (R&D Systems; E2‐616), 100 µM Recombinant Human Ubiquitin (R&D Systems; U‐100H), and 10 mM Mg2+‐ATP solution (R&D Systems; B‐20). 5 μΜ of purified HES1 (Origene; TP311709), 1 μM of purified RNF4 (Origene; AR50585PU‐S), and/or 5 μΜ of purified RASSF1 (Origene; AR50639PU‐S) were added to the reaction mix. All reactions were incubated at 37°C for 1 h. As a negative control, no Mg2+‐ATP was added to the reaction mix. Reactions were terminated with the addition of 2× SDS‐PAGE sample buffer. After boiling at 100°C for 5 min, the reactions were subjected to Western blotting.
qPCR analysis
Quantitative Real‐Time PCR was carried out according to the Power SYBR Green Cells‐to‐Ct kit protocol (Applied Biosystems), as previously described (Papaspyropoulos et al, 2018). For qRT–PCR following ChIP and ChIP seq, the Input samples were at a concentration of 2.5 ng/μl. The following primers were used for human genes: HES1 FW: ATTCCTCGTCCCCGGTGGCT and REV: GCTTGGAATGCCGCGAGCTATCTTT, NANOG FW: GGTGTGACGCAGAAGGCCTCA and REV: CCCAGTCGGGTTCACCAGGCA, OCT4 FW: CCTGAAGCAGAAGAGGATCACC and REV: AAAGCGGCAGATGGTCGTTTGG, SOX2 FW: GAGCTTTGCAGGAAGTTTGC and REV: GCAAGAAGCCTCTCCTTGAA, RNF4 FW: AAGTGGGTTGCTGTTGAGGC and REV: CCAGTATTTTCAAGTCCGCTGC, GATA1 FW: CACGACACTGTGGCGGAGAAAT and REV: TTCCAGATGCCTTGCGGTTTCG, 18S FW: AGTCCCTGCCCTTTGTACACA and REV: GATCCGAGGGCCTCACTAAAC and OCT4 promoter (Cell Signaling; 4641). The following primers were used for mouse genes: Hes1 FW: CAACACGACACCGGACAAAC and REV: GGAATGCCGGGAGCTATCTT, Nanog FW: AGGGTCTGCTACTGAGATGCTCTG and REV: CAACCACTGGTTTTTCTGCCACCG, Oct4 FW: CTGAGGGCCAGGCAGGAGCACGAG and REV: CTGTAGGGAGGGCTTCGGGCACTT, Sox2 FW: CAGGAGTTGTCAAGGCAGAGA and REV: CTTAAGCCTCGGGCTCCAAA, Gapdh FW: CTCCACTCACGGCAAATTCA and REV: CGCTCCTGGAAGATGGTGAT.
Immunofluorescence
Cells were seeded and cultured either on coverslips or collagen‐coated wells with defined stiffness of collagen matrix as previously described (Pankova et al, 2019). Cell mounting was done onto microscope slides using Prolong Gold antifade reagent with DAPI (Life Technologies). Images of the cells were acquired using a Nikon 90i microscope with the NIS elements software, a Leica DM IRBE microscope with Simple PCI6 software or a Zeiss LSM710 confocal microscope with the Zeiss ZEN software. The following primary (dilution 1:100) and secondary (dilution 1:500) antibodies were used: NANOG (Cell Signaling; 4893S), SOX2 (MAB4423), HES1 (AB5702), Alexa fluor goat anti‐rabbit and anti‐mouse (Invitrogen).
Immunofluorescence staining in 3D collagen
HeLa multicellular tumor spheroids were generated according to the hanging‐drop method as previously described (Foty, 2011; Pankova et al, 2019). In brief, HeLa cells were detached with 2 mM EDTA and re‐suspended in medium containing methylcellulose (20%, Sigma‐Aldrich) and Matrigel (1%, Corning, Growth factor reduced). Cells were incubated as hanging droplets (25 μl) containing 2,000 cells for 48 h to generate multicellular aggregates. For 3D spheroid staining, collagen–spheroid gels were washed with PBS, fixed with 4% PFA, and cross linked in sodium azide solution overnight. Spheroids were incubated in primary antibodies (see Immunofluorescence section, all diluted 1: 100), diluted in Triton and 10% NGS overnight, washed, and incubated in secondary antibodies (see Immunofluorescence section, dilution 1:1,000) overnight at 4°C. Spheroids–collagen gels were washed with PBS, mounted with DAPI, and following gel polymerization, cells were stained using the Immunofluorescence protocol above. Images were obtained by using Nikon 20×/0.30 Ph1 objectives.
Three‐dimensional Matrigel differentiation assay
H1299 multicellular tumor spheroids were generated using the hanging‐drop method. Aggregates were washed and cultured on Matrigel (8 mg/ml, Corning, Reduced growth factors) for 24 h. Images were obtained every 30 min at 37°C using a motorized inverted Nikon Ti microscope (4×/0.10 NA air objective lens) connected to a Nikon camera.
DNA constructs and siRNA experiments
For siRNA‐mediated silencing and transient plasmid expression in all cell lines, Lipofectamine 2000 (Invitrogen) was used according to manufacturer instructions. The following oligonucleotides were used for siRNA‐mediated silencing: non‐targeting (CTRL): UAAGGUAUGAAGAGAUAC (Dharmacon), RASSF1A: GACCUCUGUGGCGACUUCA, YAP1: CUGGUCAGAGAUACUUCUUtt (Eurofins MWG). siRNAs to TAZ (GS25937), RNF4 (GS6047), HES1 (GS3280), and GATA1 (GS2623) were purchased from Qiagen. The following plasmids were purchased from Addgene: p2xFLAGhYAP1 (#17791) and p2xFLAGhYAP1‐S127A (#17790). The GFP‐HES1 plasmid vector was purchased from Origene (#RG211709).
Chromatin immunoprecipitation
ChIP was performed according to the ChIP‐IT High Sensitivity kit (Active Motif) as previously described (Papaspyropoulos et al, 2018). Binding to promoter regions was assessed via qPCR. YAP (clone 63.7;sc‐101199) and TAZ (sc‐48805) antibodies were used for the immunoprecipitations. An IgG antibody (Cell Signalling; 2729 S) served as control.
Bioinformatics analyses
Raw RNA‐seq read count data for 34 publicly available tumors and available adjacent normal tissues were downloaded from The Cancer Genome Atlas (TCGA) database (https://www.cancer.gov/tcga) (Cancer Genome Atlas Research Network et al, 2013). For correlation heatmaps, the average RSEM (log2) value per gene of interest was calculated from all replicates per tumor. Spearman's correlation was performed after calculating Eucledian distance on the ranked values of the dataset. Hierarchical clustering was applied in columns and rows. Heatmaps were constructed in R using the ggplot2 package (https://ggplot2.tidyverse.org.) (Wickham, 2016). In order to categorize the expression of genes as high and low, the average RSEM(log2) from all the available replicates per cancer was calculated, and afterward, the median value was selected as a threshold (> median → high, < median → low). All values can be found in Appendix Table S1.
Statistical analysis
For all experiments reported in this manuscript, at least three biological replicates were used and statistical significance was determined by Student’s t‐test.
Author contributions
AP, EON, and VG designed the study, analyzed, and interpreted data. AP, AA, IM, DP, and KT performed experiments. AikP performed bioinformatics analyses. SL, NL, AK, and KE assisted with experiments and manuscript preparation. AAP and EC contributed toward data interpretation. AP, EON, and VG wrote the manuscript and supervised the project.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Source Data for Expanded View and Appendix
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Acknowledgements
This work was supported by CRUK A19277, MRC, Kidani Memorial Trust, the Federal Agency for Scientific Organizations (FASO Russia grant No. 0324‐2016‐0008), the Wellcome Trust and the Science Foundation Ireland (grant No. 15/CDA/3495). This research was also co‐financed by Greece and the European Union (European Social Fund‐ ESF) through the Operational Program «Human Resources Development, Education and Lifelong Learning» in the context of the project “Reinforcement of Postdoctoral Researchers ‐ 2nd Cycle” (MIS‐5033021), implemented by the State Scholarships Foundation (ΙΚΥ); the Hellenic Foundation for Research and Innovation (HFRI) and the General Secretariat for Research and Innovation (GSRI), under the grant agreement No 775, entitled “Hippo pathway in genome and epigenome maintenance – HIPPO”; the HFRI and the GSRΙ, under the grant agreement No 3782, entitled “Interrogating the Pathophysiological Consequences of deregulated Replication Licensing – PaCoReL”; the European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska‐Curie grants agreement no. 722729 (SYNTRAIN); the National Public Investment Program of the Ministry of Development and Investment / General Secretariat for Research and Technology, in the framework of the Flagship Initiative to address SARS‐CoV‐2 (2020ΣΕ01300001); the Welfare Foundation for Social & Cultural Sciences (KIKPE), Athens, Greece; the H. Pappas donation; the NKUA‐SARG grants 70/3/8916 and 70/3/12128.
EMBO Reports (2022) 23: e51287.
Contributor Information
Angelos Papaspyropoulos, Email: a.papaspyropoulos@med.uoa.gr.
Eric O’Neill, Email: eric.oneill@oncology.ox.ac.uk.
Vassilis Gorgoulis, Email: vgorg@med.uoa.gr.
Data availability
No primary datasets have been generated and deposited. The authors declare that all data supporting the findings of this study are available within the article and its supplementary information files or from the corresponding authors upon reasonable request.
References
- Abed M, Barry KC, Kenyagin D, Koltun B, Phippen TM, Delrow JJ, Parkhurst SM, Orian A (2011) Degringolade, a SUMO‐targeted ubiquitin ligase, inhibits Hairy/Groucho‐mediated repression. EMBO J 30: 1289–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artavanis‐Tsakonas S, Rand MD, Lake RJ (1999) Notch signaling: cell fate control and signal integration in development. Science 284: 770–776 [DOI] [PubMed] [Google Scholar]
- Barry KC, Abed M, Kenyagin D, Werwie TR, Boico O, Orian A, Parkhurst SM (2011) The Drosophila STUbL protein Degringolade limits HES functions during embryogenesis. Development 138: 1759–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network , Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM (2013) The Cancer Genome Atlas Pan‐Cancer analysis project. Nat Genet 45: 1113–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzifrangkeskou M, Pefani DE, Eyres M, Vendrell I, Fischer R, Pankova D, O'Neill E (2019) RASSF1A is required for the maintenance of nuclear actin levels. EMBO J 38: e101168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Zhang C, Wu H, Ma Y, Luo X, Gong X, Jiang F, Gui Y, Zhang H, Lu F (2017) The E3 ubiquitin ligase SCF(FBXL14) complex stimulates neuronal differentiation by targeting the Notch signaling factor HES1 for proteolysis. J Biol Chem 292: 20100–20112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Q, Orr BA, Semenkow S, Bar EE, Eberhart CG (2013) Prolonged inhibition of glioblastoma xenograft initiation and clonogenic growth following in vivo Notch blockade. Clin Cancer Res 19: 3224–3233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook N, Basu B, Smith DM, Gopinathan A, Evans J, Steward WP, Palmer D, Propper D, Venugopal B, Hategan M et al (2018) A phase I trial of the gamma‐secretase inhibitor MK‐0752 in combination with gemcitabine in patients with pancreatic ductal adenocarcinoma. Br J Cancer 118: 793–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz‐Padilla I, Wilson MK, Clarke BA, Hirte HW, Welch SA, Mackay HJ, Biagi JJ, Reedijk M, Weberpals JI, Fleming GF et al (2015) A phase II study of single‐agent RO4929097, a gamma‐secretase inhibitor of Notch signaling, in patients with recurrent platinum‐resistant epithelial ovarian cancer: A study of the Princess Margaret, Chicago and California phase II consortia. Gynecol Oncol 137: 216–222 [DOI] [PubMed] [Google Scholar]
- Do DV, Ueda J, Messerschmidt DM, Lorthongpanich C, Zhou YI, Feng BO, Guo G, Lin PJ, Hossain MZ, Zhang W et al (2013) A genetic and developmental pathway from STAT3 to the OCT4‐NANOG circuit is essential for maintenance of ICM lineages in vivo . Genes Dev 27: 1378–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan B, Liu Y, Hu HE, Shi F‐G, Liu Y‐L, Xue H, Yun X‐Y, Yan M‐Y, Han X‐R, Chen A‐F et al (2019) Notch1‐ADAM8 positive feed‐back loop regulates the degradation of chondrogenic extracellular matrix and osteoarthritis progression. Cell Commun Signal 17: 134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois F, Bergot E, Zalcman G, Levallet G (2019) RASSF1A, puppeteer of cellular homeostasis, fights tumorigenesis, and metastasis‐an updated review. Cell Death Dis 10: 928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fender AW, Nutter JM, Fitzgerald TL, Bertrand FE, Sigounas G (2015) Notch‐1 promotes stemness and epithelial to mesenchymal transition in colorectal cancer. J Cell Biochem 116: 2517–2527 [DOI] [PubMed] [Google Scholar]
- Fendler A, Bauer D, Busch J, Jung K, Wulf‐Goldenberg A, Kunz S, Song K, Myszczyszyn A, Elezkurtaj S, Erguen B et al (2020) Inhibiting WNT and NOTCH in renal cancer stem cells and the implications for human patients. Nat Commun 11: 929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foty R (2011) A simple hanging drop cell culture protocol for generation of 3D spheroids. J Vis Exp 51: 2720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann‐Morvinski D, Verma IM (2014) Dedifferentiation and reprogramming: origins of cancer stem cells. EMBO Rep 15: 244–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanos P, Vougas K, Walter D, Polyzos A, Maya‐Mendoza A, Haagensen EJ, Kokkalis A, Roumelioti F‐M, Gagos S, Tzetis M et al (2016) Chronic p53‐independent p21 expression causes genomic instability by deregulating replication licensing. Nat Cell Biol 18: 777–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F, Zhang Y, Wang S, Liu Y, Zheng L, Yang J, Huang W, Ye Y, Luo W, Xiao D (2014) Hes1 is involved in the self‐renewal and tumourigenicity of stem‐like cancer cells in colon cancer. Sci Rep 4: 3963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, Campisi J, Collado M, Evangelou K, Ferbeyre G et al (2019) Cellular senescence: defining a path forward. Cell 179: 813–827 [DOI] [PubMed] [Google Scholar]
- Grawenda AM, O'Neill E (2015) Clinical utility of RASSF1A methylation in human malignancies. Br J Cancer 113: 372–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habets RA, de Bock CE, Serneels L, Lodewijckx I, Verbeke D, Nittner D, Narlawar R, Demeyer S, Dooley J, Liston A et al (2019) Safe targeting of T cell acute lymphoblastic leukemia by pathology‐specific NOTCH inhibition. Sci Transl Med 11: eaau6246 [DOI] [PubMed] [Google Scholar]
- Hamilton G, Yee KS, Scrace S, O'Neill E (2009) ATM regulates a RASSF1A‐dependent DNA damage response. Curr Biol 19: 2020–2025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepburn AC, Steele RE, Veeratterapillay R, Wilson L, Kounatidou EE, Barnard A, Berry P, Cassidy JR, Moad M, El‐Sherif A et al (2019) The induction of core pluripotency master regulators in cancers defines poor clinical outcomes and treatment resistance. Oncogene 38: 4412–4424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Vu T, Yuan G, Datta PK (2017) STRAP promotes stemness of human colorectal cancer via epigenetic regulation of the NOTCH pathway. Cancer Res 77: 5464–5478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamakura S, Oishi K, Yoshimatsu T, Nakafuku M, Masuyama N, Gotoh Y (2004) Hes binding to STAT3 mediates crosstalk between Notch and JAK‐STAT signalling. Nat Cell Biol 6: 547–554 [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Mizuno H, Imayoshi I, Furusawa C, Shirahige K, Kageyama R (2009) The cyclic gene Hes1 contributes to diverse differentiation responses of embryonic stem cells. Genes Dev 23: 1870–1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krop I, Demuth T, Guthrie T, Wen PY, Mason WP, Chinnaiyan P, Butowski N, Groves MD, Kesari S, Freedman SJ et al (2012) Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK‐0752 in adult patients with advanced solid tumors. J Clin Oncol 30: 2307–2313 [DOI] [PubMed] [Google Scholar]
- Lagopati N, Belogiannis K, Angelopoulou A, Papaspyropoulos A, Gorgoulis V (2021) Non‐canonical functions of the ARF tumor suppressor in development and tumorigenesis. Biomolecules 11: 86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZH, Dai XM, Du B (2015) Hes1: a key role in stemness, metastasis and multidrug resistance. Cancer Biol Ther 16: 353–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matallanas D, Romano D, Yee K, Meissl K, Kucerova L, Piazzolla D, Baccarini M, Vass JK, Kolch W, O'Neill E (2007) RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol Cell 27: 962–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellacheruvu D, Wright Z, Couzens AL, Lambert J‐P, St‐Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY et al (2013) The CRAPome: a contaminant repository for affinity purification‐mass spectrometry data. Nat Methods 10: 730–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messersmith WA, Shapiro GI, Cleary JM, Jimeno A, Dasari A, Huang B, Shaik MN, Cesari R, Zheng X, Reynolds JM et al (2015) A Phase I, dose‐finding study in patients with advanced solid malignancies of the oral gamma‐secretase inhibitor PF‐03084014. Clin Cancer Res 21: 60–67 [DOI] [PubMed] [Google Scholar]
- Nejigane S, Takahashi S, Haramoto Y, Michiue T, Asashima M (2013) Hippo signaling components, Mst1 and Mst2, act as a switch between self‐renewal and differentiation in Xenopus hematopoietic and endothelial progenitors. Int J Dev Biol 57: 407–414 [DOI] [PubMed] [Google Scholar]
- Pankova D, Jiang Y, Chatzifrangkeskou M, Vendrell I, Buzzelli J, Ryan A, Brown C, O'Neill E (2019) RASSF1A controls tissue stiffness and cancer stem‐like cells in lung adenocarcinoma. EMBO J 38: e100532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaspyropoulos A, Bradley L, Thapa A, Leung CY, Toskas K, Koennig D, Pefani D‐E, Raso C, Grou C, Hamilton G et al (2018) RASSF1A uncouples Wnt from Hippo signalling and promotes YAP mediated differentiation via p73. Nat Commun 9: 424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pefani DE, Latusek R, Pires I, Grawenda AM, Yee KS, Hamilton G, van der Weyden L, Esashi F, Hammond EM, O'Neill E (2014) RASSF1A‐LATS1 signalling stabilizes replication forks by restricting CDK2‐mediated phosphorylation of BRCA2. Nat Cell Biol 16: 962–971, 961–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pefani DE, Pankova D, Abraham AG, Grawenda AM, Vlahov N, Scrace S, O'Neill E (2016) TGF‐beta targets the hippo pathway scaffold RASSF1A to facilitate YAP/SMAD2 nuclear translocation. Mol Cell 63: 156–166 [DOI] [PubMed] [Google Scholar]
- Perlman RL (2016) Mouse models of human disease: an evolutionary perspective. Evol Med Public Health 2016: 170–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pine SR (2018) Rethinking Gamma‐secretase inhibitors for treatment of non‐small‐cell lung cancer: is notch the target? Clin Cancer Res 24: 6136–6141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran Y, Hossain F, Pannuti A, Lessard CB, Ladd GZ, Jung JI, Minter LM, Osborne BA, Miele L, Golde TE (2017) gamma‐Secretase inhibitors in cancer clinical trials are pharmacologically and functionally distinct. EMBO Mol Med 9: 950–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reedijk M, Odorcic S, Zhang H, Chetty R, Tennert C, Dickson BC, Lockwood G, Gallinger S, Egan SE (2008) Activation of Notch signaling in human colon adenocarcinoma. Int J Oncol 33: 1223–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roese‐Koerner B, Stappert L, Brustle O (2017) Notch/Hes signaling and miR‐9 engage in complex feedback interactions controlling neural progenitor cell proliferation and differentiation. Neurogenesis 4: e1313647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross J, Mavoungou L, Bresnick EH, Milot E (2012) GATA‐1 utilizes Ikaros and polycomb repressive complex 2 to suppress Hes1 and to promote erythropoiesis. Mol Cell Biol 32: 3624–3638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohani M, Rostami S, Azad M, Hojjatipour T, Chahardouli B, Alizadeh S (2021) Promoter methylation status and expression levels of RASSF1A gene in different phases of acute lymphoblastic leukemia (ALL). Int J Hematol Oncol Stem Cell Res 15: 7–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MS, Song SJ, Kim SJ, Nakayama K, Nakayama KI, Lim DS (2008) Skp2 regulates the antiproliferative function of the tumor suppressor RASSF1A via ubiquitin‐mediated degradation at the G1‐S transition. Oncogene 27: 3176–3185 [DOI] [PubMed] [Google Scholar]
- Sturrock M, Hellander A, Aldakheel S, Petzold L, Chaplain MA (2014) The role of dimerisation and nuclear transport in the Hes1 gene regulatory network. Bull Math Biol 76: 766–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z (2017) GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res 45: W98–W102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Kang B, Li C, Chen T, Zhang Z (2019) GEPIA2: an enhanced web server for large‐scale expression profiling and interactive analysis. Nucleic Acids Res 47: W556–W560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaler S, Hahnel PS, Schad A, Dammann R, Schuler M (2009) RASSF1A mediates p21Cip1/Waf1‐dependent cell cycle arrest and senescence through modulation of the Raf‐MEK‐ERK pathway and inhibition of Akt. Cancer Res 69: 1748–1757 [DOI] [PubMed] [Google Scholar]
- Tommasi S, Dammann R, Zhang Z, Wang Y, Liu L, Tsark WM, Wilczynski SP, Li J, You M, Pfeifer GP (2005) Tumor susceptibility of Rassf1a knockout mice. Can Res 65: 92–98 [PubMed] [Google Scholar]
- Tzekaki EE, Geromichalos G, Lavrentiadou SN, Tsantarliotou MP, Pantazaki AA, Papaspyropoulos A (2021a) Oleuropein is a natural inhibitor of PAI‐1‐mediated proliferation in human ER‐/PR‐ breast cancer cells. Breast Cancer Res Treat 186: 305–316 [DOI] [PubMed] [Google Scholar]
- Tzekaki EE, Papaspyropoulos A, Tsolaki M, Lazarou E, Kozori M, Pantazaki Alpha A (2021b) Restoration of BMI1 levels after the administration of early harvest extra virgin olive oil as a therapeutic strategy against Alzheimer's disease. Exp Gerontol 144: 111178 [DOI] [PubMed] [Google Scholar]
- Velimezi G, Liontos M, Vougas K, Roumeliotis T, Bartkova J, Sideridou M, Dereli‐Oz A, Kocylowski M, Pateras IS, Evangelou K et al (2013) Functional interplay between the DNA‐damage‐response kinase ATM and ARF tumour suppressor protein in human cancer. Nat Cell Biol 15: 967–977 [DOI] [PubMed] [Google Scholar]
- van der Weyden L, Papaspyropoulos A, Poulogiannis G, Rust AG, Rashid M, Adams DJ, Arends MJ, O'Neill E (2012) Loss of RASSF1A synergizes with deregulated RUNX2 signaling in tumorigenesis. Cancer Res 72: 3817–3827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Weyden L, Tachibana KK, Gonzalez MA, Adams DJ, Ng BL, Petty R, Venkitaraman AR, Arends MJ, Bradley A (2005) The RASSF1A isoform of RASSF1 promotes microtubule stability and suppresses tumorigenesis. Mol Cell Biol 25: 8356–8367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2016) ggplot2: elegant graphics for data analysis. New York, NY: Springer‐Verlag; [Google Scholar]
- Wiener DJ, Basak O, Asra P, Boonekamp KE, Kretzschmar K, Papaspyropoulos A, Clevers H (2018) Establishment and characterization of a canine keratinocyte organoid culture system. Vet Dermatol 29: 375 [DOI] [PubMed] [Google Scholar]
- Yee KS, O'Neill E (2010) YAP1–friend and foe. Cell Cycle 9: 1447–1448 [DOI] [PubMed] [Google Scholar]
- Zampetidis CP, Galanos P, Angelopoulou A, Zhu Y, Polyzou A, Karamitros T, Kotsinas A, Lagopati N, Mourkioti I, Mirzazadeh R et al (2021) A recurrent chromosomal inversion suffices for driving escape from oncogene‐induced senescence via subTAD reorganization. Mol Cell 81: 4907–4923.e8 [DOI] [PubMed] [Google Scholar]
- Zhou X, Smith AJ, Waterhouse A, Blin G, Malaguti M, Lin CY, Osorno R, Chambers I, Lowell S (2013) Hes1 desynchronizes differentiation of pluripotent cells by modulating STAT3 activity. Stem Cells 31: 1511–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]