

Abstract
Single‐cell RNA sequencing (scRNA‐seq) is a powerful technique for dissecting the complexity of normal and diseased tissues, enabling characterization of cell diversity and heterogeneous phenotypic states in unprecedented detail. However, this technology has been underutilized for exploring the interactions between the host cell and viral pathogens in latently infected cells. Herein, we use scRNA‐seq and single‐molecule sensitivity fluorescent in situ hybridization (smFISH) technologies to investigate host single‐cell transcriptome changes upon the reactivation of a human neurotropic virus, herpes simplex virus‐1 (HSV‐1). We identify the stress sensor growth arrest and DNA damage–inducible 45 beta (Gadd45b) as a critical antiviral host factor that regulates HSV‐1 reactivation events in a subpopulation of latently infected primary neurons. We show that distinct subcellular localization of Gadd45b correlates with the viral late gene expression program, as well as the expression of the viral transcription factor, ICP4. We propose that a hallmark of a “successful” or “aborted” HSV‐1 reactivation state in primary neurons is determined by a unique subcellular localization signature of the stress sensor Gadd45b.
Keywords: Gadd45b, herpes simplex virus‐1, HSV‐1, viral latency, viral reactivation
Subject Categories: Chromatin, Transcription & Genomics; Microbiology, Virology & Host Pathogen Interaction; Signal Transduction
This study employs single‐cell analyses to identify and characterize cellular host factors that regulate latent viral genome reactivation in primary neurons. Stress sensor Gadd54b is identified as an important viral reactivation determinant.
Introduction
Human α‐herpesviruses, which include herpes simplex virus types 1 and 2 (HSV‐1, HSV‐2), and varicella zoster virus (VZV), are pathogens that enter the peripheral nervous system to establish a lifelong infection termed latency (Baringer & Swoveland, 1973; Warren et al, 1978; Richter et al, 2009). By adulthood almost the entire human population is latently infected by at least one of these viruses. With age and/or waning immunity, infected individuals may experience mild to severe disease. Pathogenesis arises from periodic re‐emergence (reactivation) from this pool of latent virus resulting in painful surface lesions and potentially, ocular damage and persistent, often debilitating, neuropathic pain. Spread of reactivated virus into the central nervous system (CNS) may result in life‐threatening viral encephalitis or give rise to long‐term cognitive problems, and there is growing evidence that α‐herpesviruses may act as an accelerant for the onset of age‐associate dementias including Alzheimer’s disease (Mangold & Szpara, 2019). Clinically approved antiviral drugs are only active against the replicating virus, while efficacious vaccines have only been successfully deployed against VZV (Warren‐Gash et al, 2017). Neither drugs nor vaccine influence the latent or dormant pool sequestered in the nervous system. Understanding the molecular basis of latency and how reactivation is triggered when neurons are stressed/damaged remains a priority in the field.
Like HSV‐2 and VZV, HSV‐1 latency is characterized by incorporation of the circularized viral genome into transcriptionally non‐permissive heterochromatin within the neuronal nucleus that results in a highly restricted program of viral gene expression (Bloom, 2016). To ensure continued transmission, latent genomes periodically reactivate, wherein the expression of the full viral genome is restarted and results in the axonal transport of newly synthesized viral particles to surface tissues innervated by the latently infected ganglia. The molecular details of how HSV‐1 latency and reactivation are controlled are not fully understood, in part due to the limitations of prevailing live‐animal models to fully recapitulate human disease, difficulties manipulating vital signalling pathways, and cell‐type heterogeneity within isolated ganglia (Wagner & Bloom, 1997; Wilson & Mohr, 2012; Thellman & Triezenberg, 2017). However, in vitro infection models using cultured primary neurons isolated from dissociated ganglia of rodents (Wilcox & Johnson, 1988; Camarena et al, 2010; Cliffe et al, 2015) recapitulate many aspects of in vivo animal models and provide a powerful opportunity to dissect the molecular circuitry that impacts virus‐neuron interactions and latency. Importantly, studies from our group and others have discovered that continuous neuronal host‐cell signalling through the AKT‐mTORC1 axis is required to maintain HSV‐1 latency (Camarena et al, 2010; Kobayashi et al, 2012; Hu et al, 2019; Zhao et al, 2020). Using this in vitro infection model, we recently showed that maintenance of HSV‐1 latency in primary neurons involves a convergence of the extrinsic TrkA receptor–mediated neurotrophin signalling with a secondary intrinsic signal generated via the endogenous nuclear DNA damage response (DDR) that acts together to maintain AKT activation (Hu et al, 2019). Thus, transient inhibition of either the TrkA receptor–mediated signalling axis, the DDR, or the AKT‐mTORC1 pathway leads to HSV‐1 reactivation in these latently infected primary neurons. Whether there are additional neuronal factors that further regulate the efficiency of HSV‐1 reactivation remains to be determined.
Single‐cell RNA sequencing (scRNA‐seq) has gained wider usage in recent years, enabling the study of transcription in distinct cell types or in rare cell populations present in heterogenous tissue environments (Stubbington et al, 2017; Kinker et al, 2020; Shnayder et al, 2020). Compared to bulk RNA‐seq techniques that profile the average levels of gene expression across a potentially heterogeneous cell population, scRNA‐seq is capable of producing gene expression measurements both at the genome‐wide scale and at single‐cell resolution to reveal an underappreciated cellular heterogeneity in normal or diseased tissues. Likewise, the ability to visualize and image messenger RNA (mRNA) and long non‐coding RNA (lncRNA) at single‐molecule sensitivity provides a powerful means to quantify select gene expression changes, and to probe for its role in gene expression at the single‐cell level (Chao & Lionnet, 2018). Here, we have integrated scRNA‐seq and single‐molecule sensitivity fluorescent in situ hybridization (smFISH) technologies (Raj et al, 2008) to examine a dynamic host–viral pathogen relationship and identify new host regulators of HSV‐1 latency. Using an established cultured primary neuron infection model of HSV‐1 latency, we identified a unique host gene expression signature of a small population of primary neurons during induced HSV‐1 reactivation. By focusing on the transcriptome of just this small population of neurons undergoing reactivation, we identified and validated the stress sensors Gadd45b and components of heat shock response pathway as novel neuronal determinants of HSV‐1 reactivation. Intriguingly, we found that distinct subcellular localization of Gadd45b correlated with the viral late gene expression program, and expression of the viral transcription factor, ICP4. We propose that the localization of the stress sensor Gadd45b could be a predictive marker for HSV‐1–infected neurons undergoing “successful” or “aborted” reactivation. Thus, our study demonstrates the power of using single‐cell approaches for the identification of critical host factors that control viral transmission in a heterogeneous population of neurons.
Results
HSV‐1 establishes a latency‐like state in rat SCG‐derived neuron cultures
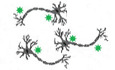
In previous studies, we developed an in vitro infection model for HSV‐1 latency by using dissociated superior cervical ganglia (SCG) neurons from embryonic day 21 (E21) rat embryos (Hu et al, 2020). These sympathetic neurons are dependent on the neurotrophic factor, nerve growth factor (NGF), for axon and neurite maintenance and survival, and importantly are amenable to small molecule and genetic perturbations critical for molecular dissection of signalling pathways required to maintain HSV‐1 latency (Camarena et al, 2010; Kobayashi et al, 2012; Wilson & Mohr, 2012; Linderman et al, 2017; Cuddy et al, 2020; Hu et al, 2020). In addition, the antiviral compound acyclovir (ACV, viral DNA polymerase inhibitor) is included at the time of infection to limit any unwanted viral replication that could potentially overwhelm the culture before control is established. While the establishment of HSV‐1 latency and the induction of reactivation in cultured SCG‐derived neurons recapitulates other in vitro, ex vivo, and in vivo animal infection studies, it is unclear whether non‐neuronal cells play a role in establishing or maintaining viral latency in the ganglia. To interrogate the possibility that other cell types contribute to HSV‐1 latency, we applied single‐cell transcriptome profiling to HSV‐1 latently infected primary rat SCG‐derived neuron cultures (Fig 1A). We performed scRNA‐seq on multiple latently infected and uninfected biological replicates using the 10× Genomics Chromium Single Cell 3’ v2 Reagent Kits (see Methods). Following sequencing and alignment of reads against the Rattus norvegicus transcriptome, we generated gene expression counts and imported these into Seurat (Butler et al, 2018; Stuart et al, 2019). Datasets were merged using canonical correction analysis, and this enabled the identification of four discrete cell clusters: satellite glial cells, Schwann cells, fibroblasts and peripheral sympathetic neurons (Fig 1B). Neurons were broadly classified by β‐tubulin 3 (Tubb3) and peripherin (Prph) expression and further stratified by neuropeptide Y (Npy) expression (Fig EV1A). While SCG‐derived cultures consist of a heterogeneous mix of cell types, those showing a neuronal transcriptome profile appeared to be relatively homogeneous and was the dominant cell type (60–85%) within the wider population (Fig EV1B). Unfortunately, due to inherent limitations in the sensitivity of the 10× Genomics v2 chemistry, we were unable to detect viral RNAs in either latently infected or reactivating cells. Similarly, this methodology precluded the capture and sequencing of the (abundant) HSV‐1 LAT intron lariat (a long non‐coding viral RNA), a well‐established signature of latent virus (Phelan et al, 2017).
Figure 1. The cellular composition of SCG ex vivo cultures that support HSV‐1 latency.
-
ASuperior cervical ganglia (SCG) excised from stage E21 embryos were dissociated and plated. Treatment with the anti‐mitotic agents fluorouracil (5‐FU) and aphidicolin over a period of five days allows further enrichment for non‐dividing cells (i.e. post‐mitotic neurons). SCG cultures were subsequently infected with HSV‐1 at a multiplicity of infection (MOI) of 1.5 in the presence of 100 μM acyclovir (ACV, an inhibitor of viral DNA replication) to allow the establishment of HSV‐1 latency. Following removal of ACV, latently infected cultures were maintained for up to two weeks prior to treatment with PI3K inhibitor LY2940002 (LY) to induce reactivation. Uninfected, latently infected, and reactivating SCG cultures subsequently processed for scRNA‐seq using the 10X genomics Chromium Single‐Cell 3’ v2 Reagent Kits.
-
B, CscRNA‐seq of eight discrete SCG cultures using v2 chemistry yielded 28,346 cells passing filter (also Dataset EV1, Appendix Table S2). Clustering of cell types by UMAP reveals four discrete cellular populations classified according to specified markers for neurons (Prph, Tubb3), fibroblasts (Col3a1, Fn1), satellite glial cells (Aif‐1, Cd68), and Schwann cells (Sox10, S100b).
-
DsmFISH images showing LAT RNA in Ntrk1‐positive neurons. SCG‐derived cultures were infected with HSV‐1 in the presence of ACV and allowed to establish latency over 7 days before being processed for smFISH using probes for HSV‐1 LAT (green) and neuronal marker Ntrk1 (TrkA) (red) or fibroblast marker Col3a1 (red). Signal intensities for the LAT probe were quantified in neurons and fibroblasts and plotted by ImageJ. The percentage of LAT‐positive cells were quantified and displayed as bar graphs with mean ± SEM. Left: 3 biological replicates, signal intensities of 50 cells were quantified for each replicate. Middle: 3 biological replicates, signal intensities of 30 cells were quantified for each replicate. Right: the blue and red dots represent the number of biological replicates done for quantification of each cell type. Scale bar, 10 μm.
Data information: P values ≤ 0.05 were considered significant, asterisks denote statistical significance (****P < 0.0001). P values are calculated using two‐tailed unpaired Student’s t test. P values > 0.05 were not significant (ns).
Figure EV1. scRNA‐seq analysis of the homogeneity of SCG‐derived neurons.
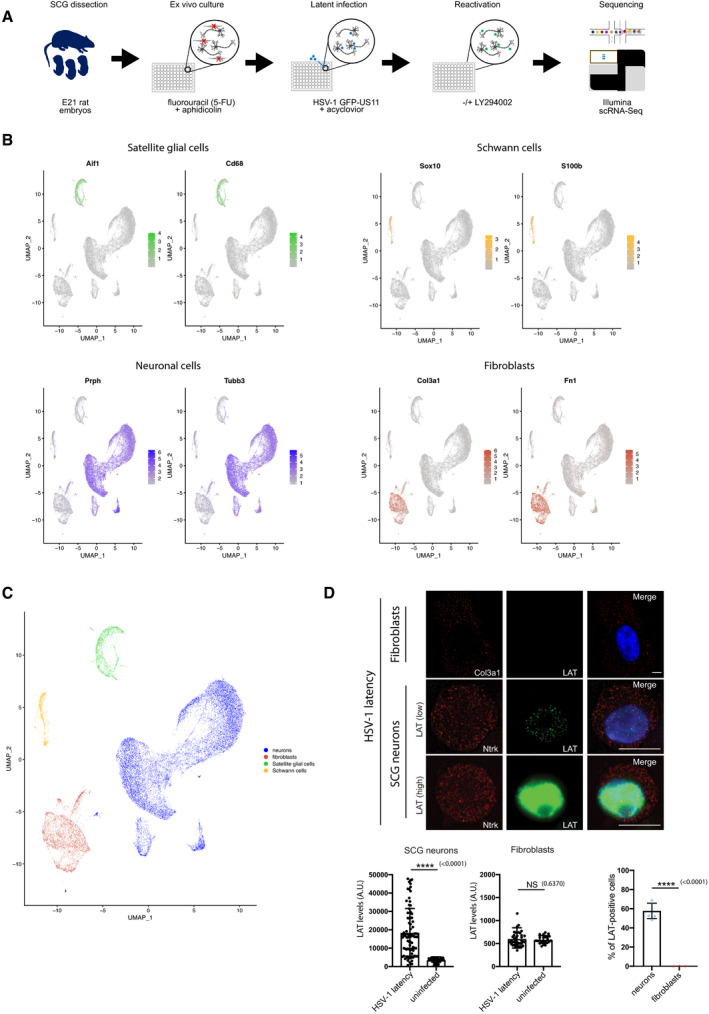
- Classification of sympathetic neurons as either Npy+/Dbh‐ and Npy‐/Dbh+, the latter correlating with cells that have comparatively higher proportions of sequence reads aligning to mitochondrial genes (a marker of cell stress). Note that all neurons were positive for tyrosine hydroxylase (Th+). Markers are as follows: Npy, neuropeptide Y; Th, tyrosine hydroxylase; and Dbh, dopamine beta (β)‐hydroxylase. Note that percent.mt indicates the proportion of sequence reads aligning to genes encoded by the mitochondrial genome.
- The relative proportions of four distinct cell types neurons (blue), fibroblasts (red), satellite glia (green) and Schwan cells (yellow), as detected by their transcriptomic signatures in SCG‐derived cultures remained essentially constant across biological replicates and the experimental conditions used in this study, with approximately 61–85% of the population consisting of sympathetic neurons.
- WAY‐150138 treatment prevents HSV‐1 spreading during lytic infection. SCG neurons were infected with HSV‐1 at MOI = 0.1 for productive infection for 3 days. The live‐cell images were taken using an epifluorescence microscopy. Scale bar, 100 μm.
- The Us11 viral mRNA expression levels were quantified at different time‐points and displayed as bar graphs with mean ± SEM. Latently infected cultures were untreated (latent) or treated with LY294002 and WAY‐150138 for indicated times (n = 3 biological replicates).
Data information: P values ≤ 0.05 were considered significant, asterisks denote statistical significance (**P < 0.01; ***P < 0.001; ****P < 0.0001). P values are calculated using two‐tailed unpaired Student’s t test.
To determine which cell type(s) harbour latent HSV‐1 genomes expressing LAT and to quantify expression levels of selected host and viral transcripts at the single cell level, we turned to a highly sensitive technique called single‐molecule fluorescent in situ hybridization (smFISH) (Femino et al, 1998; Raj et al, 2008). Although HSV‐1 latently infected SCG‐derived cultures contain a mixture of sympathetic neurons, fibroblasts, satellite glial cells, and Schwann cells (Fig 1B and C), only the neuronal population gave a detectable signal for the LAT intronic RNA (Fig 1D). Consistent with previous studies using human and murine HSV‐1–infected ganglia, LAT RNA was detected throughout the nucleoplasm with obvious nucleolar sparing (Arthur et al, 1993). Importantly, LAT was not detected in any Col3a1‐positive fibroblasts. We consistently identified roughly 60% of the SCG neurons as having detectable LAT expression (Fig 1D). Acknowledging that some latently infected neurons may possess insufficient amounts of LAT for detection, nevertheless, this analysis supports that in this infection model, latency is established in a majority of the neurons and therefore provides a robust model to study the molecular basis of latency establishment, maintenance and exit.
Capturing viral transcriptional changes at the single‐cell level by smFISH upon HSV‐1 reactivation
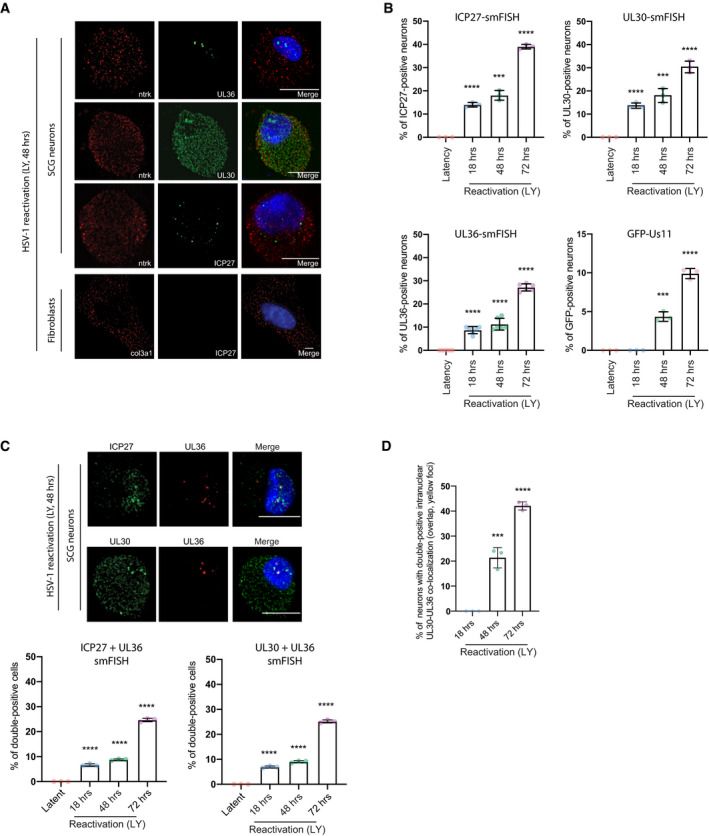
To measure the transition period to HSV‐1 reactivation in SCG neurons at the single‐cell level, we first established HSV‐1 latency in vitro using a wild‐type (WT) HSV‐1 strain expressing the enhanced green fluorescent protein (GFP) fused to the Us11 true‐late (γ2) protein (GFP‐Us11), which can be readily used to detect reactivation in living neurons (Camarena et al, 2010). To accomplish this, we treated the latently infected SCG cultures with LY294002 (LY, PI3‐kinase inhibitor) in a time‐course experiment to induce HSV‐1 reactivation. We also simultaneously treated the cells with WAY‐150138 (a small molecule inhibitor of viral genome packaging) to prevent viral spreading (Fig EV1C); this enables us to quantify productive reactivation events at the single‐cell level without any potential skewing of the results when infectious virus from a single reactivating cell infects neighbouring cells (van Zeijl et al, 2000). Using smFISH to detect viral RNA expression at the single‐molecule sensitivity and single‐cell level (Raj et al, 2008; Chou & Lionnet, 2018), we showed that only neurons (Ntrk1‐ or TrkA‐positive cells, neuronal marker), and not fibroblasts (Col3a1‐positive cells, fibroblast marker), expressed immediate‐early, early and late viral transcripts (ICP27, UL30 and UL36, respectively) upon treatment with LY (Fig 2A). Even though early and late (and true‐late) viral transcripts are already being produced after 18 h of LY treatment (Figs 2B and EV1D), the detection of a true‐late viral protein, GFP‐Us11, was only observed after 48 h (Fig 2B). This could be due to post‐transcriptional or translational control of viral mRNAs at the later time‐points; however, we do not rule out sensitivity differences between smFISH and immunofluorescence detection. Importantly, viral transcript expression levels were high at 72 h of LY‐induced HSV‐1 reactivation, similar to peak GFP‐Us11 protein expression. While the percentage of cells harbouring viral transcripts was 25–30% in the SCG cultures, the effective reactivation efficiency, as quantified by GFP‐Us11 signal, was only ~10% (Fig 2B). This suggests that not all viral transcriptional activation events result in HSV‐1 late gene protein production, implying that only a small fraction of latently infected cultured neurons are capable of progressing to the replication of viral genomes upon stimulation with LY. To determine whether there is some coordination of viral gene expression during the reactivation timeline, we simultaneously probed for two different viral transcripts by smFISH (Fig 2C) and found that there were more cells with at least two viral transcripts (double‐positive) being made at 72 h, compared to 48 h, suggesting that viral transcription initially proceeds asynchronously. Intriguingly, a subset of nuclear foci from these double‐positive neurons also presented with an overlap of two different signals (yellow nuclear foci displayed as a merged image), consistent with the possibility that the same episomal viral genomes could be transcribing two or more productive‐cycle genes (Fig 2C). Significantly, the increase in overlapping signal (yellow dots) in the nuclear foci correlates with the time point when a viral true‐late protein GFP‐Us11 becomes more detectable (48 and 72 h) (Fig 2B and D).
Figure 2. Detection of specific HSV‐1 productive cycle transcripts during reactivation by smFISH.
-
AsmFISH images showing selected viral mRNAs in reactivating neurons probed for ICP27 (immediate‐early), UL30 (early), UL36 (true‐late) or cellular markers, Ntrk (neurons), or Col3a1 (fibroblasts). Nuclear DNA was visualized using DAPI (blue). To induce reactivation, latently infected SCG‐derived cultures were simultaneously treated with LY294002 (20 μM) and WAY‐150138 (20 μg/ml) and cultured for 48 h prior to processing for smFISH. Scale bar, 10 μm.
-
BThe percentage of cells positive for a viral mRNA or for GFP fluorescence was quantified at different time points and displayed as bar graphs with mean ± SEM. Latently infected cultures were untreated (latent) or treated with LY294002 and WAY‐150138 for 18, 48, or 72 h. Each dot corresponds to the value from each biological replicate.
-
C, DReactivating neurons probed simultaneously for two separate viral productive cycle mRNAs. Signal overlap is evident as white or yellow nuclear foci in the merge (C). Bar, 10 μm. In (D), the percentage of positive nuclei with overlapping UL30 and UL36 smFISH signals were quantified as the mean ± SEM (n = 3 biological replicates).
Data information: P values ≤ 0.05 were considered significant, asterisks denote statistical significance (***P < 0.001; ****P < 0.0001). P values are calculated using two‐tailed unpaired Student’s t test.
Enhanced scRNA‐seq identifies differentially regulated host mRNAs in a subset of neurons containing HSV‐1 transcripts
Following the release of an improved 10× Genomics Chromium v3 Single‐Cell chemistry (see Methods), we repeated our scRNA‐seq analysis of HSV‐1 latently infected SCG ex vivo cultures at 20, 48, and 72 h post LY treatment (i.e. at multiple timepoints during viral reactivation) to determine whether we could detect host mRNA expression changes in neurons harbouring HSV‐1 productive cycle transcripts. Here, the increased sensitivity provided by the v3 chemistry enabled the identification of viral RNAs that increased in abundance over time of reactivation within a small subpopulation of neurons (Figs 3A and EV2A). Because most HSV‐1 RNAs share a common 3’ end sequence due to the shared cleavage and polyadenylation sites (CPAS), it is not possible to specifically ascribe individual 3’ scRNA‐seq reads to specific viral gene transcripts. Instead, we assigned reads mapping to the viral genome to a set of discrete transcription units (TU1‐20) (Appendix Table S3). When focusing on stratifying the cellular transcriptomes between this neuronal subpopulation and the main neuronal population, we examined 557 genes (i.e. genes for which there was sufficient expression for detection) and identified ~20 significantly upregulated host transcripts and ~3 significantly downregulated in neurons undergoing HSV‐1 reactivation (Fig 3B and C, Dataset EV1 and Appendix Table S2). Upregulated transcripts include mRNAs encoding the stress response factor Gadd45b and components of the heat shock response pathway (Fig 3B and C, Dataset EV1 and Appendix Table S2).
Figure 3. Identification of host mRNAs by scRNA‐seq that are regulated during HSV‐1 reactivation.
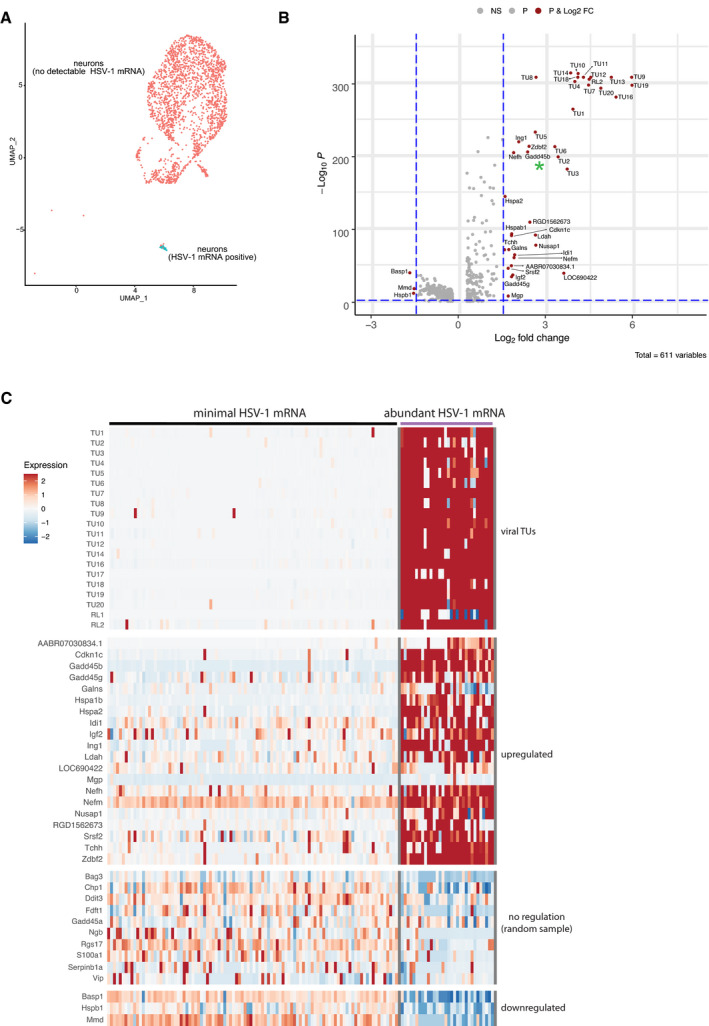
- scRNA‐seq profiling of SCG cultures using 10X Genomics Chromium Single‐Cell v3 Reagent Kits enabled the detection of abundant viral RNAs (HSV‐1 mRNA positive) in a small neuronal subpopulation (cyan) during reactivation (see also Fig EV2A).
- Differential gene expression analysis of cellular transcripts showed a significant upregulation of at least 19 viral transcript units (TU) and a subset of cellular genes in neurons undergoing reactivation (see also Appendix Table S3). Three host transcripts were also downregulated. Gadd45b is highlighted with a green asterisk. Vertical hashed blue lines indicate Log2 fold changes >1.5 and <−1.5, while the horizontal hashed blue line indicates an adjusted P‐value of 0.01. Significantly regulated genes are named and shown as magenta circles.
- Heatmap showing the relative levels of selected cellular and viral transcripts in neuronal subpopulations containing either minimal (no detectable) HSV‐1 mRNAs, indicative of latent or uninfected neurons, or with abundant HSV‐1 mRNA, indicating reactivation (see also Dataset EV1).
Figure EV2. scRNA‐seq profiling of SCG cultures using enhanced chemistry.
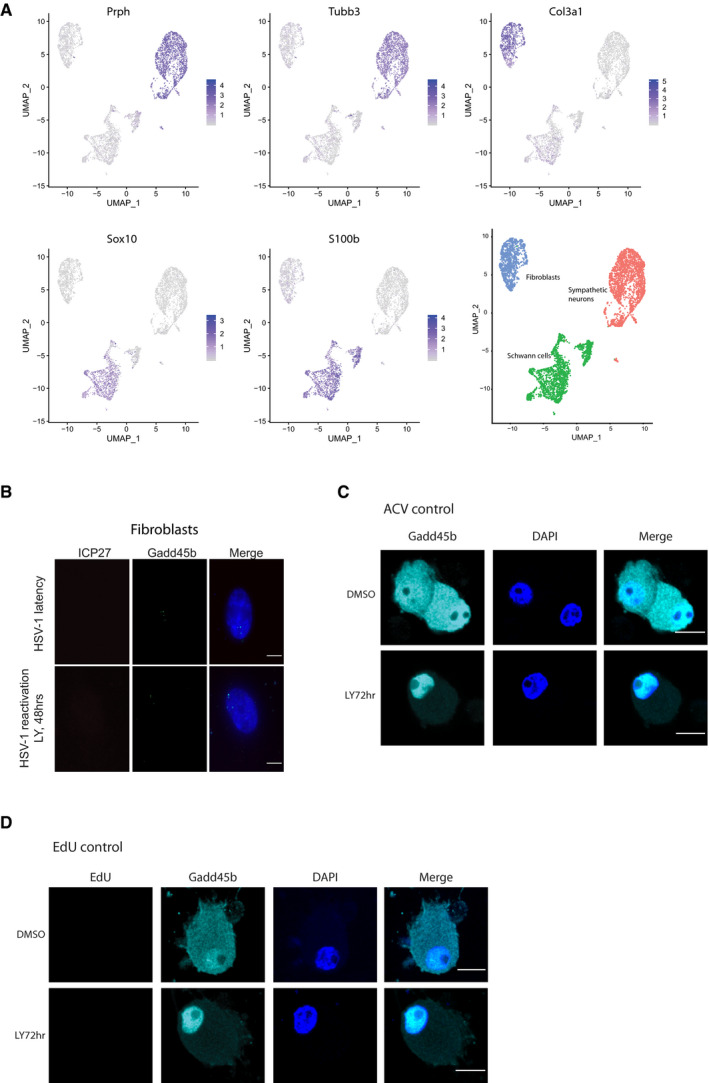
- scRNA‐seq profiling of SCG cultures using 10X genomics v3 chemistry identified three distinct cell populations and classified according to specified markers for neurons (Prph & Tubb3), fibroblasts (Col3a1) and Schwann cells (Sox10 and S100b). Signature transcripts correspond to: Prph, peripherin; Tubb3, beta III tubulin; Col3a1, Collagen Type III Alpha 1 Chain; Sox10, SRY‐Box Transcription Factor 10; S100b, S100 calcium‐binding protein B.
- smFISH images showing LY294002 treatment doesn’t affect Gadd45b mRNA localization and expression levels. Cells treated LY294002 and WAY‐150138 for 48 h and then smFISH was performed. Scale bar, 10 μm.
- ACV treatment by itself did not impact the subcellular localization of Gadd45b in uninfected neurons. Uninfected SCGs were treated with ACV for 7 days then treated with either DMSO or LY294002 for 72 h, and then cells were fixed and stained for Gadd45b and DAPI, and visualized by immunofluorescence. Scale bar, 10 μm.
- EdU treatment by itself did not impact Gadd45b localization or lead to EdU labeling in the uninfected, post‐mitotic neurons. Uninfected SCGs were treated with ACV for 7 days, followed by treatment with DMSO or LY294002 for 72 h. Cells were then subjected to EdU pulse‐labeling (6 h), followed by immunofluorescence staining of Gadd45b and EdU signal. Scale bar, 10 μm.
HSV‐1‐induced Gadd45b mRNA expression anti‐correlates with viral late gene expression in reactivating neurons
To validate the host transcripts that were upregulated in neurons supporting HSV‐1 reactivation, we employed smFISH to examine the association of the Gadd45b mRNA with representative viral transcripts at the single‐cell level. We found that single‐cell Gadd45b mRNA levels were increased in HSV‐1 reactivating neurons (LY, 48 h) when compared to the latent state (Fig 4A and C). However, single‐cell Gadd45b mRNA levels of uninfected cells (DMSO or LY‐treated neurons) remained similar to the latent state (Fig 4A and B, and C, and D). Similarly, single‐cell Gadd45b mRNA levels remain low in fibroblasts from reactivating SCG cultures (Fig EV2B), indicating that this response is specific to virus‐infected neurons. Gadd45b mRNA levels also correlated with detection of the UL36 viral late gene transcript in reactivating conditions (LY, 48 h) (Fig 4A). The increase in Gadd45b mRNA expression was not simply due to PI3‐kinase inhibition alone, as single‐cell Gadd45b mRNA levels were not elevated in uninfected neurons that were treated with LY for 48 h (Fig 4B and D). Higher expression of Gadd45b mRNA at the single‐cell level can be distinguished from basal expression due to its cytoplasmic localization of highly expressed Gadd45b mRNA, as shown by smFISH (Fig 4A and E). By contrast, low levels of Gadd45b mRNA localized strictly within neuronal nuclei with little or no detectable HSV‐1 transcripts during conditions of HSV‐1 latency or in uninfected cells (Fig 4A and B, and E, and F). Intriguingly, neurons with cytoplasmic localization of Gadd45b mRNA showed an anti‐correlation to true‐late UL36 transcript levels, but there was no correlation between Gadd45b mRNA levels with either the immediate‐early ICP27 or early UL30 viral transcripts (Fig 4A and G). This is also consistent with reduced Gadd45b mRNA levels found in GFP‐positive neurons that are expressing the true late gene GFP‐Us11 reporter upon LY‐induced reactivation (Fig 4H). Together these findings suggest that virally induced Gadd45b expression in LY‐treated neurons may be part of a neuronal cell intrinsic response that restricts viral late gene expression and thus act to suppress reactivation.
Figure 4. Single‐cell Gadd45b mRNA levels are altered during HSV‐1 reactivation.
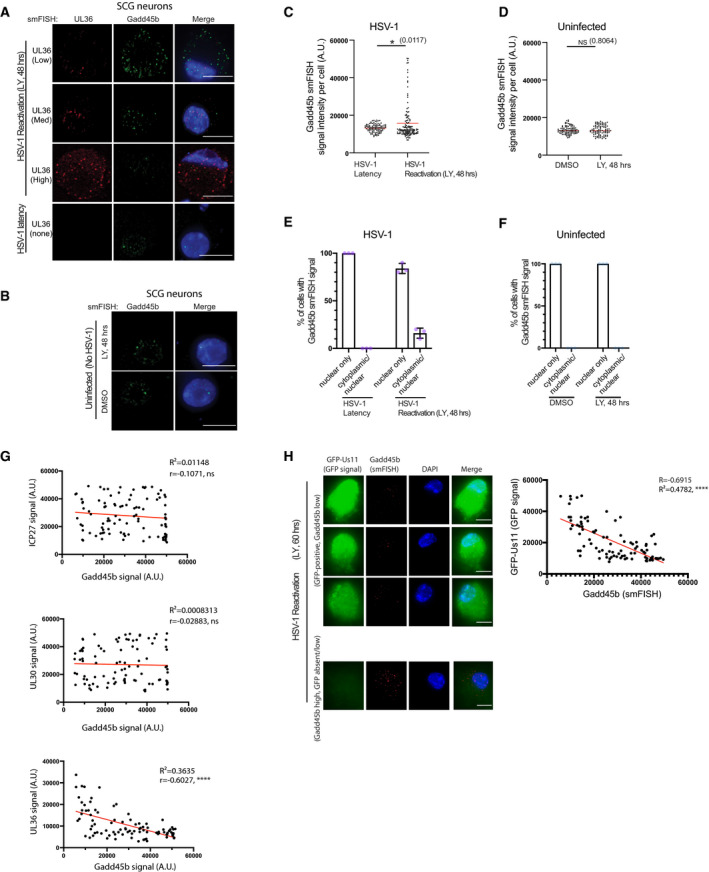
- smFISH images showing nuclear Gadd45b mRNA in latent (UL36‐negative) neurons and nuclear/cytoplasmic Gadd45b mRNA in viral UL36‐positive neurons. Latently infected SCG neurons treated with LY294002 and WAY‐150138 for 48 h were processed using smFISH to detect Gadd45b (green) and viral UL36 (red) mRNA levels and colocalization. Nuclei were stained with DAPI (blue). Scale bar, 10 μm.
- smFISH images processed as in (A) showing nuclear Gadd45b mRNA in uninfected neurons at 48 h post treatment with LY294002. Scale bar, 10 μm.
- Elevated levels of Gadd45b mRNA in reactivating neurons. Gadd45b signal intensity in HSV‐1 latently infected neurons either untreated (latent) or treated with LY294002 and WAY‐150138 for 48 h (reactivating) were quantified by ImageJ and plotted. Red line indicates the mean. Three biological replicates; 25–30 cells were quantified for each replicate.
- Quantification of Gadd45b mRNA signal intensity at 48 h post treatment with LY294002 or DMSO in uninfected neurons. The red line indicates the mean. 3 biological replicates, 25–30 cells were quantified for each replicate.
- The percentage of HSV‐1 latently infected neurons or reactivating neurons at 48 h post treatment with LY294002 and WAY‐150138 were scored for either predominantly nuclear or nuclear/cytoplasmic Gadd45b mRNA localization and displayed as mean ± SEM. Each purple dot represents a biological replicate experiment.
- Quantification of nuclear or nuclear/cytoplasmic Gadd45b mRNA in uninfected neurons at 48 h post treatment with either LY294002 or in DMSO. Each light blue dot represents a biological replicate experiment.
- Regression analysis showing that the signal intensity of Gadd45b mRNA in reactivating neurons at 48 h post treatment with LY294002 and WAY‐150138 was inversely related to the signal intensity of viral UL36 mRNA. No significant relationship was detected for ICP27 or UL30 mRNAs. Each dot represents a single reactivating neuron (n = 3 biological replicates).
- Combined smFISH and immunofluorescence (IF) images showing high GFP‐expressing neurons (GFP‐positive) or low GFP‐expressing (GFP absent) neurons simultaneously probed for Gadd45b mRNA. Reactivation was induced by treatment with LY294002 and WAY‐150138 for 60 h and processed for simultaneous smFISH and IF staining. The right panel shows quantification (as in G) of the smFISH signal intensity of Gadd45b mRNA and IF signal intensity of GFP protein at 48 h post treatment with LY294002 and WAY‐150138 (n = 3 biological replicates). The signal intensity was quantified, plotted, and analysed by ImageJ. Bar, 10 μm.
Data information: P values ≤ 0.05 were considered significant, asterisks denote statistical significance (*P < 0.05; ****P < 0.0001). (C and D): P values are calculated using the two‐tailed unpaired Student’s t test. P values > 0.05 were not significant (ns). (G and H): R and R square values were calculated using correlation analysis by Prism 8.
Gadd45b nuclear exclusion in neurons undergoing viral DNA synthesis
In non‐neuronal cells at least, robust transcription of the viral true‐late genes requires viral DNA replication (Jones & Roizman, 1979). Using a well‐established viral DNA polymerase inhibitor, phosphonoacetic acid (PAA), to block HSV‐1 DNA synthesis (Mao & Robishaw, 1975; Honess & Watson, 1977), we found that PAA treatment was capable of inhibiting LY‐induced Gadd45b mRNA expression in latently infected cultured SCG neurons (Fig 5A). This suggested that Gadd45b mRNA expression is upregulated in response to viral DNA synthesis. Next, we wanted to determine whether HSV‐1 DNA synthesis correlates with Gadd45b protein expression and localization. To address this, we utilized the nucleotide analogue, 5‐ethynyl‐2’‐deoxyuridine (EdU), for the pulse‐labelling of non‐dividing SCG neurons to capture and visualize LY‐induced viral DNA synthesis. The incorporation of EdU molecules into newly synthesized viral episomal DNA readily identifies the subset of neurons undergoing active viral DNA synthesis (Manska et al, 2020). Probing for the localization of the Gadd45b protein by immunofluorescence with an anti‐Gadd45b antibody, we found that Gadd45b was present in both nuclear and cytoplasmic compartments of uninfected and HSV‐1 latently infected cultured neurons/population (Fig 5B and C). Curiously, upon reactivation for 72 h with LY treatment, we observed distinct subcellular localization properties for Gadd45b in neurons undergoing viral DNA synthesis versus those that lacked a detectable EdU signal: (i) Gadd45b localized predominantly in the cytoplasm in EdU‐positive neurons (Fig 5B and D) and (ii) by contrast, Gadd45b localized to either nuclear foci/puncta (in ~9% of the cells) or as a pan‐nuclear‐enriched staining pattern in EdU‐negative neurons (Fig 5B and E). As a control, we observed similar pan‐nuclear‐enriched staining for Gadd45b, but importantly did not observe any nuclear foci/puncta staining pattern in LY‐treated, uninfected neurons (Fig 5C). Also, we did not observe any nuclear foci/puncta Gadd45b localization effects with either ACV or EdU treatment alone in uninfected SCG neurons (Fig EV2C and D). This suggests that the Gadd45b nuclear foci/puncta staining pattern coincides with latently infected cultured neurons/population in which some of the neurons fail to reactivate (following LY treatment) as defined by detection of active viral DNA replication. In agreement with this, we only found the Gadd45b nuclear foci/puncta staining pattern in LAT‐positive cells in neurons co‐stained by IF and smFISH (~100%) (Fig 5F). The percentage of LAT‐positive neurons showing Gadd45b nuclear puncta staining is ~18%, while cytoplasmic staining was ~40%. This suggests that the reactivation efficiency (as scored by proficient viral DNA synthesis) of latently infected neurons is ~40% if using Gadd45b subcellular localization changes as a biomarker. Also, neurons with Gadd45b nuclear puncta staining were not ICP4‐positive (~0%) (Fig 5G). In contrast, of the neurons showing a pan‐nuclear Gadd45b pattern, about 5% were ICP4‐positive (Fig 5G). This suggests that Gadd45b nuclear puncta, but not its pan‐nuclear localization, anti‐correlates with ICP4 nuclear expression in reactivating neurons.
Figure 5. Subcellular localization of Gadd45b protein during HSV‐1 viral DNA synthesis.
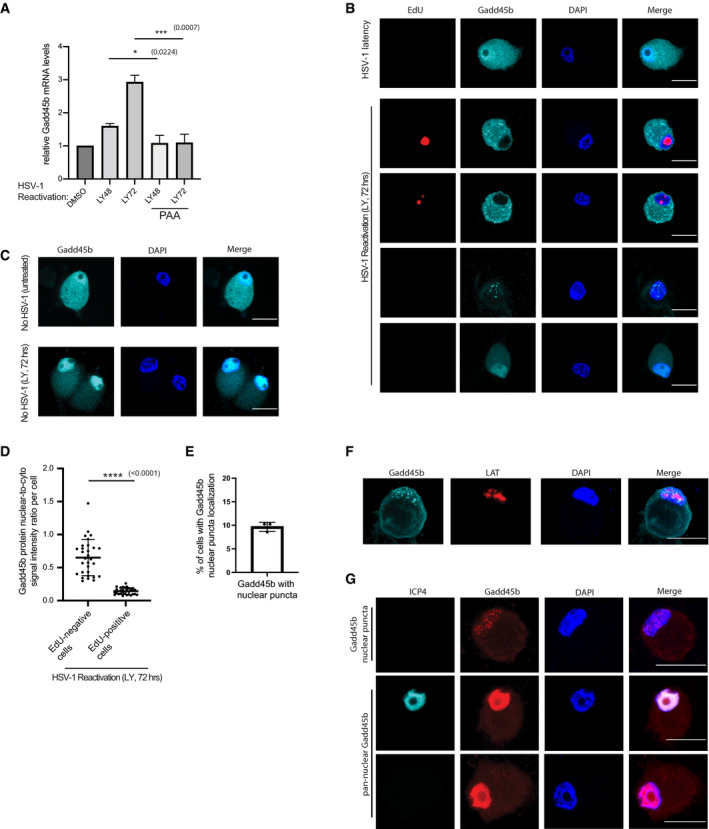
- Viral DNA polymerase inhibitor phosphonoacetic acid (PAA, 300 μg/ml) treatment suppressed Gadd45b mRNA expression. Latently infected SCG neurons were treated with LY294002 for 18 h to induce reactivation then treated with or without PAA until the termination time‐points indicated. RNA was then collected and levels of Gadd45b mRNA quantified by RT‐qPCR (n = 3 biological replicates, the bars and error bars are mean ± SD).
- Immunofluorescence images showing nuclear/cytoplasmic Gadd45b in latent neurons and cytoplasmic Gadd45b in viral DNA synthesis positive neurons. Latent SCG neurons treated with LY294002 and WAY‐150138 for 72 h and then performed EdU pulse‐labeling for detecting newly synthesized viral DNA followed by immunofluorescence staining probing with Gadd45b. The uninfected neurons served as control. Scale bar, 20 μm.
- Immunofluorescence images showing nuclear‐enriched Gadd45b in uninfected neurons at 72 h post treatment with LY294002. Scale bar, 20 μm.
- The signal intensity quantification showing the ratio of nuclear/cytoplasmic Gadd45b at 72 h post treatment with LY294002 and WAY‐150138 neurons. The signal intensity was quantified by ImageJ. Three biological replicates; 10 EdU positive cells and 10 EdU negative cells were quantified for each replicate. The horizonal line and error bars are mean ± SD.
- The percentage of Gadd45b nuclear puncta‐localized neurons were quantified from latent neurons treated with LY294002 and WAY‐150138 for 72 h. Each dot represents each biological replicate; 20 cells were quantified for each replicate. The bars and error bars are mean ± SD.
- Combined smFISH and immunofluorescence images showing Gadd45b nuclear puncta‐presenting neurons were also LAT‐positive. Reactivation was induced by treatment with LY294002 and WAY‐150138 for 72 h. Scale bar, 20 μm.
- Latent SCG neurons treated with LY294002 and WAY‐150138 for 72 h; the Gadd45b nuclear puncta‐presenting neurons were ICP4 negative (none observed in over 60 Gadd45b nuclear puncta‐positive neurons), but a small population (~5%) in Gadd45b nuclear‐enriched neurons were ICP4 positive. Scale bar, 20 μm.
Data information: P values ≤ 0.05 were considered significant, asterisks denote statistical significance (*P < 0.05; ***P < 0.001; ****P < 0.0001). P values are calculated using two‐tailed unpaired Student’s t test.
Depletion of Gadd45b promotes HSV‐1 reactivation independent of the AKT‐mTORC1‐JNK signalling pathway
To determine whether depletion of Gadd45b can impact HSV‐1 reactivation, we employed two different lentivirus‐delivered shRNAs against Gadd45b (Fig 6A). Using GFP‐Us11 protein expression as a reporter for HSV‐1 reactivation efficiency, we found that depletion of Gadd45b alone (no LY treatment) led to an increase in GFP‐Us11 expression in SCG neurons (Fig 6A). This was Gadd45 gene family member–specific because while Gadd45b knockdown also led to HSV‐1 reactivation, the reduction of Gadd45a had no effect (Fig EV3A and B). Importantly, at the single‐cell level (measuring UL36 smFISH signal), the depletion of Gadd45b was capable of inducing the expression of the viral late gene UL36 in the absence of a reactivation stimulus such as LY (Fig 6B). Thus, these results suggest that cellular expression of Gadd45b (and likely Gadd45g) during the virus latent state in neurons antagonizes the HSV‐1 late gene expression program in order to prevent spontaneous HSV‐1 reactivation.
Figure 6. Depletion of the stress sensor Gadd45b causes HSV‐1 reactivation.
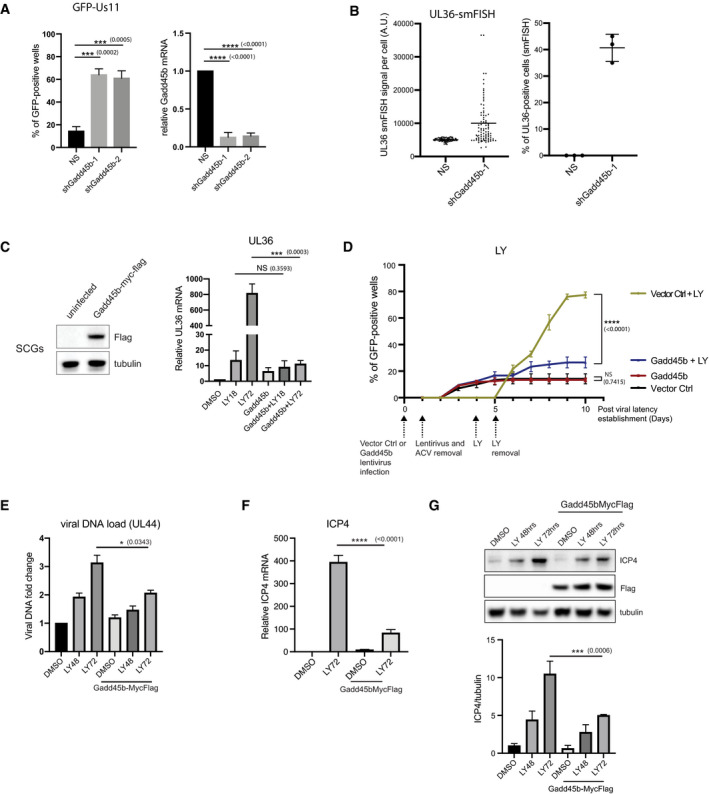
- Depletion of Gadd45b expression using lentiviral‐transduced shRNAs in latently infected SCG neurons case cause HSV‐1 reactivation. HSV1‐GFP‐Us11 latently infected cultures were infected with lentiviruses expressing either two different shRNAs against Gadd45b mRNA or a non‐silencing shRNA (NS). Reactivation was quantified by scoring the percentage of wells expressing GFP after 5 days. Knockdown efficiencies for individual shRNAs in latently infected SCG neurons were confirmed by RT‐qPCR using RNA collected in parallel. Reactivation rates were quantified from 30 wells for each condition, 3 biological replicates. RT‐qPCR were quantified from three biological replicates. The bars and error bars are mean ± SD.
- smFISH shows that depletion of Gadd45b leads to increased UL36 expression. smFISH was performed 3 days post‐lentiviral transduction. smFISH signals were quantified and plotted using ImageJ and Prism. Left: three biological replicates, 30 cells were quantified for each replicate. The horizonal lines are mean. Right: three biological replicates. The horizonal lines and error bars are mean ± SD.
- Ectopic expression of Gadd45b‐Myc‐Flag inhibits viral mRNA expression. Lentivirus‐delivered ectopic expression of Gadd45b‐Myc‐Flag in SCG neurons was confirmed by immunoblotting. Latently infected SCG neurons were transduced with Gadd45b‐Myc‐Flag for 3 days and then treated with LY294002 and WAY‐150138 to induce reactivation. RNA was collected at the indicated time‐points (LY treatment for 18 or 72 h) and levels of viral UL36 mRNA quantified by RT‐qPCR. Three biologicalreplicates. The bars and error bars are mean ± SD.
- Ectopic expression of Gadd45b‐Myc‐Flag antagonizes LY294002‐induced HSV‐1 reactivation. Latently infected SCG neurons were either transduced with vector control (Ctrl) or Gadd45b‐Myc‐Flag from lentivirus for 3 days prior to addition of LY294002. Reactivation was quantified by scoring the percentage of wells expressing GFP fluorescence at the indicated days. The percentage of wells expressing GFP‐positive was scored as the mean ± SEM (n = 3 biological replicates). Reactivation rates were quantified from 30 wells for each condition, three biological replicates.
- Ectopic expression of Gadd45b‐Myc‐Flag slightly inhibited LY294002‐induced HSV‐1 DNA synthesis. Latent cultures were transduced with Gadd45b for 3 days and then treated with LY294002 and WAY‐150138. Total DNA were collected at the indicated times (LY treatment for 18 or 72 h) post‐reactivation in the presence or absence of Gadd45b‐Myc‐Flag. The relative levels of viral genomic DNA were determined by qPCR using primers for the HSV‐1 UL44 gene. Input DNA was normalized by qPCR detection of the rat RPL19 gene. Three biological replicates. The bars and error bars are mean ± SD.
- Ectopic expression of Gadd45b‐Myc‐Flag suppressed viral ICP4 mRNA expression. Latently infected SCG neurons were transduced with Gadd45b for 3 days and then treated with LY294002 and WAY‐150138 for 72 h. Viral ICP4 mRNA was quantified by qRT‐PCR. Three biological replicates. The bars and error bars are mean ± SD.
- Immunoblots for viral ICP4 protein in latent SCG neurons treated with LY294002 and WAY‐150138 in the presence or absence of Gadd45b‐Myc‐Flag was quantified by ImageJ (n = 3 biological replicates).
Data information: P values ≤ 0.05 were considered significant, asterisks denote statistical significance (*P < 0.05; ***P < 0.001; ****P < 0.0001). P values are calculated using two‐tailed unpaired Student’s t test. P values > 0.05 were not significant (ns).
Source data are available online for this figure.
Figure EV3. Depletion of Gadd45b causes HSV‐1 reactivation independent of the AKT‐mTORC1‐JNK signaling pathway.
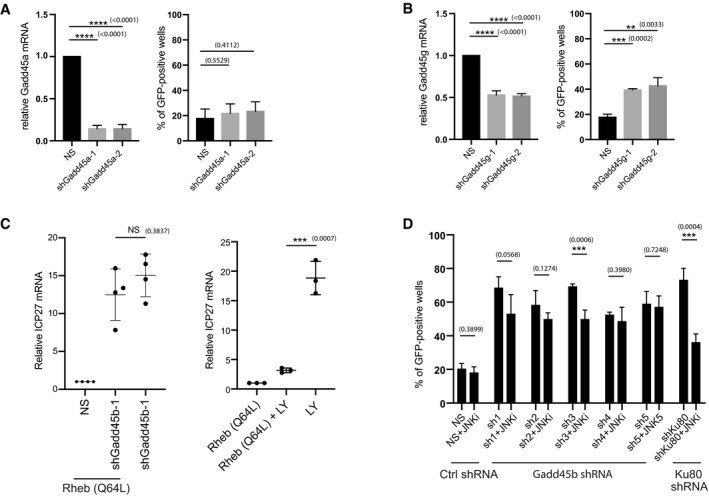
- Depletion of Gadd45a expression using lentiviral‐transduced shRNAs in latently infected SCG neurons does not induce HSV‐1 reactivation. HSV1‐GFP‐Us11 latently infected cultures were infected with lentiviruses expressing either two different shRNAs against Gadd45a mRNA or a non‐silencing shRNA (NS). Reactivation was quantified by scoring the percentage of wells expressing GFP after 5 days. Knockdown efficiencies for individual shRNAs in latently infected SCG neurons were confirmed by RT‐qPCR using RNA collected in parallel. Reactivation rates were quantified from 30 wells for each condition; three biological replicates. RT‐qPCR were quantified from three biological replicates. The bars and error bars are mean ± SD.
- Depletion of Gadd45g expression using lentiviral‐transduced shRNAs in latently infected SCG neurons induces HSV‐1 reactivation. Reactivation rates were quantified from 30 wells for each condition; three biological replicates. RT‐qPCR were quantified from three biological replicates. The bars and error bars are mean ± SD.
- HSV‐1 reactivation in response to Gadd45b depletion was independent of AKT‐mTORC1 signalling. Constitutively active Flag‐Rheb (Q64L) was introduced into HSV‐1 latently infected SCG neurons together with either of two independent shRNAs against Gadd45b or the non‐silencing shRNA (NS) and viral ICP27 mRNA levels were quantified by RT‐qPCR after 48 h (left panel). Four biological replicates. The bars and error bars are mean ± SD. The ability of Rheb (Q64L) to suppress LY‐induced reactivation serves as a positive control (right panle). Three biological replicates. The bars and error bars are mean ± SD.
- HSV‐1 reactivation induced by Gadd45b depletion (using 5 different shRNAs) was generally not inhibited by treatment with JNK inhibitor. Reactivation induced by shRNA‐mediated depletion of Ku80 served as a positive control for the JNK inhibitor. The percentage of wells expressing GFP‐positive was scored as the mean ± SEM (n = 3 biological replicates). The bars and error bars are mean ± SD.
Data information: P values ≤ 0.05 were considered significant, asterisks denote statistical significance (**P < 0.01; ***P < 0.001; ****P < 0.0001). Numbers next to asterisk above the bar graphs indicate exact P values. P values are calculated using two‐tailed unpaired Student’s t test. P values > 0.05 were not significant (ns).
Our previous work demonstrated that the AKT‐mTORC1 signalling axis promotes the maintenance of HSV‐1 latency in SCG neurons (Camarena et al, 2010; Kobayashi et al, 2012). Next, we wanted to determine whether Gadd45b was controlling HSV‐1 reactivation within the canonical AKT‐mTORC1 signalling pathway or whether it was functioning downstream and/or independent of mTORC1 activation. We utilized a Rheb Gln64 to Leu mutant (Q64L) that locks the Rheb guanosine‐5’triphosphate (GTP) hydrolase into a GTP‐bound state that behaves as a constitutive mTORC1 activator (Kobayashi et al, 2012). Thus, the Rheb Q64L mutant can bypass the requirement for AKT phosphorylation and/or its upstream regulators. Our previous work showed that Rheb Q64L expression is sufficient to inhibit LY‐mediated and DNA damage–induced HSV‐1 reactivation (Hu et al, 2019). Surprisingly, Rheb Q64L expression had no discernible effect on HSV‐1 reactivation due to Gadd45b shRNA knockdown (Fig EV3C). The early initiation (phase I, LY treatment for 18 h) of HSV‐1 gene expression upon reactivation by LY treatment (PI3‐K inhibition) requires a Jun N‐terminal kinase (JNK)–dependent methyl/phospho switch event on histone H3 surrounding the viral lytic gene promoters (Cliffe et al, 2015). In contrast to previous results that showed both LY‐ and DNA double‐strand break (DSB) repair knockdown–mediated HSV‐1 reactivation are blocked by JNK inhibition (Hu et al, 2019), HSV‐1 reactivation in response to Gadd45b shRNA‐mediated depletion was mostly insensitive to the same JNK inhibitor (Fig EV3D). Ku80 depletion was used as a positive control to verify the JNK inhibition sensitivity (Hu et al, 2019). This suggests that Gadd45b‐mediated control of HSV‐1 reactivation occurs downstream of the AKT‐mTORC1‐JNK signalling axis, as delineated previously by Cliffe and colleagues (Cliffe et al, 2015). So instead of controlling the early expression of HSV‐1 lytic genes during phase I (18 h), Gadd45b may instead selectively suppress viral late gene expression during phase II (48–72 h) of HSV‐1 reactivation, coincident with the onset of viral DNA replication and dependency on viral trans‐activators such as VP16 (Kim et al, 2012) as well as the recruitment and activity of host demethylases (Cliffe et al, 2015).
Ectopic expression of Gadd45b suppresses viral gene expression and prevents HSV‐1 reactivation
To determine whether Gadd45b can functionally inhibit viral gene expression during HSV‐1 reactivation, we ectopically over‐expressed Gadd45b in SCG neurons and assessed whether viral gene expression can be affected upon LY‐induced HSV‐1 reactivation (Fig 6C). Using RT‐qPCR, we found that Gadd45b ectopic expression inhibited reactivation‐induced viral IE, E, and L gene expression (ICP27, UL30, UL36 and Us11) between 40 and 80 fold at the late (72 h), but not at the early (18 h) reactivation time‐points (Figs 6C and EV4A and B). In concordance with the viral gene expression time‐course analysis, we also showed that ectopic expression of Gadd45b, but not vector control (Ctrl), inhibited LY‐induced HSV‐1 reactivation efficiency, as shown by the reduced number of GFP‐Us11–expressing neurons (Fig 6D). Significantly, Gadd45b also suppressed Mirin‐induced (DNA repair inhibitor that was shown to potently cause HSV‐1 reactivation) (Hu et al, 2019) HSV‐1 reactivation (Fig EV4C). Because the level of Gadd45b‐mediated suppression was similar between LY‐ and Mirin‐induced HSV‐1 reactivation, this suggests that Gadd45b operates mechanistically at a common point of these two induction pathways (likely downstream of AKT‐mTORC1‐JNK signalling axis) to suppress late viral gene expression. Surprisingly, the effect of Gadd45b on HSV‐1 gene expression is specific to reactivation events, as ectopic expression of Gadd45b had no appreciable effect on the viral gene expression program during lytic replication in SCG neurons (Fig EV4D).
Figure EV4. Gadd45b overexpression inhibits DNA damage response‐induced HSV‐1 reactivation.
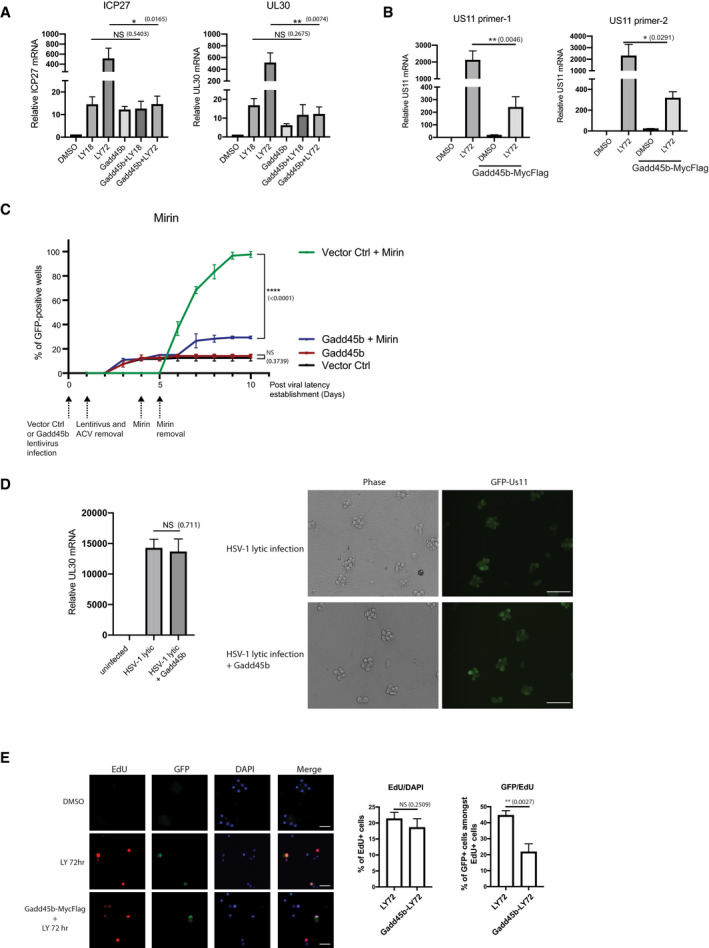
- Latently infected SCG neurons were transduced with Gadd45b‐Myc‐Flag for 3 days and then treated with LY294002 and WAY‐150138 to induce reactivation. RNA was collected at the indicated time‐points and levels of viral ICP27 and UL30 mRNA quantified by RT‐qPCR. three biological replicates. The bars and error bars are mean ± SD.
- Overexpression of Gadd45b‐Myc‐Flag suppressed viral true late Us11 gene mRNA expression. Latently infected SCG neurons were transduced with Gadd45b for 3 days and then treated with LY294002 and WAY‐150138 for 72 h. mRNAs were collected and quantified by qRT‐PCR for Us11 viral mRNA levels. Three biological replicates. The bars and error bars are mean ± SD.
- Ectopic expression of Gadd45b‐Myc‐Flag antagonizes Mirin‐induced HSV‐1 reactivation. Latently infected SCG neurons were either transduced with vector control (Ctrl) or Gadd45b‐Myc‐Flag from lentivirus for 3 days prior to addition of Mirin. Reactivation was quantified by scoring the percentage of wells expressing GFP fluorescence at the indicated days. The percentage of wells expressing GFP‐positive was scored as the mean ± SEM (n = 3 biological replicates). Reactivation rates were quantified from 30 wells for each condition, three biological replicates.
- Expression of Gadd45b‐Myc‐Flag does not affect productive replication (lytic infection) of HSV‐1 in SCG neurons, as measured by UL30 mRNA levels or visible GFP‐Us11 fluorescence. SCG neurons were transduced with Gadd45b‐Myc‐Flag for 3 days, then secondarily infected with HSV‐1 at MOI=1.5 in the absence of ACV to allow a productive infection. Levels of UL30 viral mRNA were quantified by RT‐qPCR (n = 3 biological replicates). The bars and error bars are mean ± SD. Scale bar, 100 μm.
- Gadd45b overexpression does not affect HSV‐1 DNA synthesis. Latent cultures were transduced with Gadd45b‐Myc‐Flag for 3 days and then treated with LY294002 and WAY‐150138 for 72 h. Cells were pulse‐labeled with EdU for 6 h prior to fixation and imaging of EdU‐positive cells and anti‐GFP immunofluorescence staining to detect GFP‐positive cells. The percentage of EdU‐positive cells and the percentage of GFP‐positive cells amongst EdU‐positive cells were quantified and displayed as bar graphs with mean ± SEM (n = 3 biological replicates). Scale bar, 50 μm.
Data information: P values ≤ 0.05 were considered significant, asterisks denote statistical significance (*P < 0.05; **P < 0.01; ****P < 0.0001). P values are calculated using two‐tailed unpaired Student’s t test. P values > 0.05 were not significant (ns).
How Gadd45b could be regulating viral late gene expression (or processes upstream of late transcription) to impact HSV‐1 reactivation is currently unknown. To assess whether Gadd45b ectopic expression can inhibit viral DNA replication, we first used qPCR to measure viral DNA load upon reactivation. Although there was some measurable decrease in viral DNA replication, the effect was not very dramatic (Fig 6E). Using another method to capture viral DNA synthesis levels with EdU pulse‐labelling, we showed that while the number of EdU‐positive cells was not appreciably reduced by Gadd45b ectopic expression (total EdU‐positive cells remains relatively similar) there was an appreciable reduction of GFP‐positive cells (~50% decrease of cells with GFP‐Us11 expression) within the EdU‐positive group of cells (Fig EV4E). This suggested to us that Gadd45b likely impacted the subsequent viral late gene transcriptional program, rather than by inhibiting viral DNA replication altogether. A recent report by Dremel and DeLuca, found that the viral transcription factor, ICP4, is continuously needed (in addition to viral DNA replication) for the activation of late gene transcriptional initiation (Dremel & DeLuca, 2019a, 2019b). Importantly, we found that Gadd45b ectopic expression reduced the expression of ICP4 upon LY‐induced reactivation at 72 h (Fig 6F). This also correlated with a reduction in ICP4 protein levels as measured by Western blot analysis (Fig 6G). These findings provide supportive evidence that Gadd45b restricts viral reactivation by downregulating ICP4 to hinder the viral late gene transcriptional program.
Based on our scRNA‐seq analysis, we also identified components of the heat shock response pathway, such as HSP70, to be transcriptionally upregulated in response to HSV‐1 reactivation (Fig 3B and C, Dataset EV1 and Appendix Table S1). HSP70 is a critical component of the heat shock response pathway and proteostasis (Li et al, 2017). HSP70 protein levels were observed to increase in HSV‐1–reactivating neurons and this coincided with GFP‐Us11 expression in HSV‐1–reactivating neurons at the single‐cell level (Fig EV5A and B). This HSP70 induction was selective for latently infected cultures induced to reactivate, as it was not detected in LY‐treated, uninfected neurons or in latently infected cultured neurons without LY treatment (Fig EV5B). Therefore, both Gadd45b and HSP70 are upregulated in a small subset of latently infected cultured neurons under conditions that stimulate HSV‐1 reactivation, as predicted by our scRNA‐seq analysis using the 10x Genomics v3 platform. Previous studies have linked heat shock factor 1 (HSF1) to the transcriptional induction of Gadd45b (Ueda et al, 2017). However, treatment of latently infected SCG neurons with an HSF1 inhibitor compound (KRIBB11) (Yoon et al, 2011) did not affect LY‐induced HSV‐1 reactivation in our system (Fig EV5C). Unexpectedly, treating latently infected cultured neurons with an HSF1 activator compound (HSF1A) (Neef et al, 2010) caused robust HSV‐1 reactivation, suggesting that the heat shock response pathway can promote reactivation at levels similar to LY treatment (Fig EV5C). This is in stark contrast to ectopic expression of the Gadd45b stress response factor, which inhibits HSV‐1 reactivation (Fig 6D). Understanding the molecular basis of these contrasting outcomes will require a more systematic analysis of these complex pathways. Regardless of the mechanism, two of the top host mRNA hits from our single‐cell transcriptome analysis have been independently validated and there are likely to be other critical host response factors that regulate HSV‐1 reactivation in a cell type–specific manner (Dataset EV1). Overall, these findings demonstrate the power of using scRNA‐seq and single‐cell technologies to identify host gene products that functionally impacts viral replication in a subset of reactivating neurons.
Figure EV5. Activation of the heat shock response pathway induces HSV‐1 reactivation.
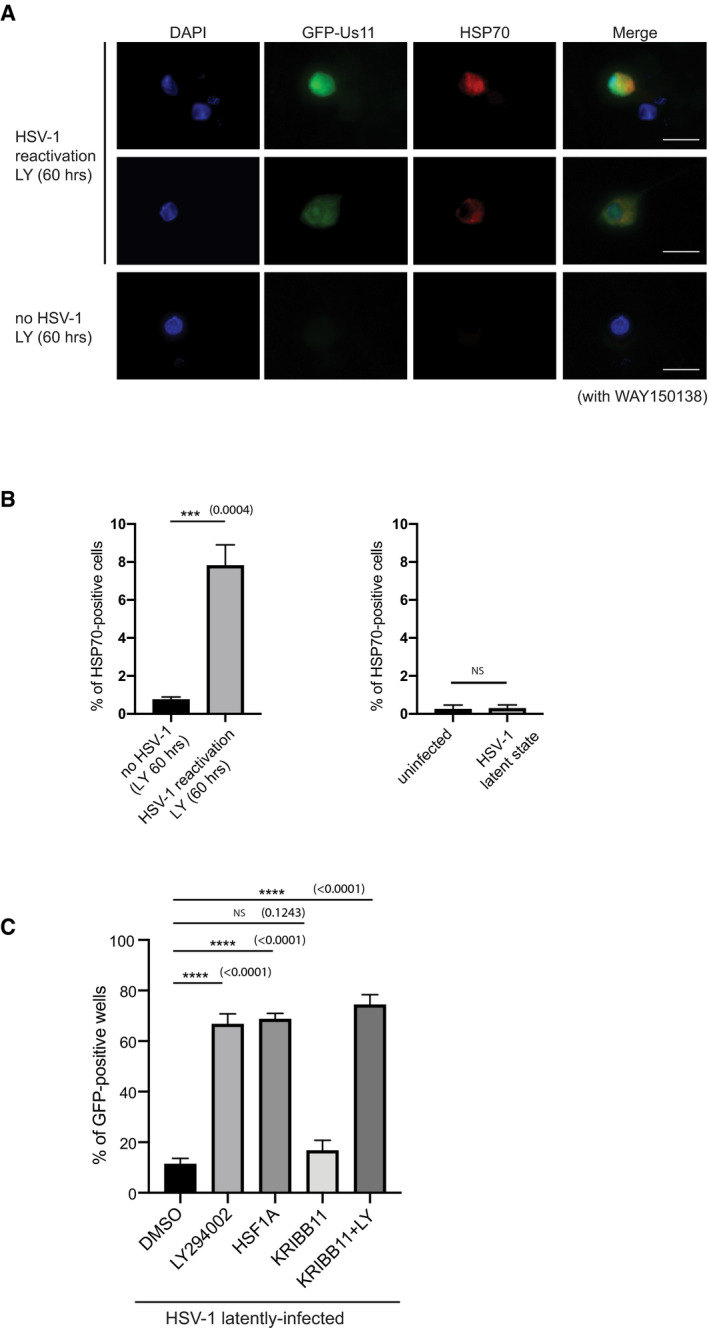
- Elevated HSP70 expression in reactivated SCG neurons. Latent or uninfected neurons treated with LY294002 and WAY‐150138 for 60 h and then immunofluorescence was performed probing with HSP70 and GFP. Scale bar, 20 μm.
- The percentage of cells with HSP70 in latent or uninfected neurons were quantified from (A) and displayed as bar graphs with mean ± SEM (n = 3 biological replicates).
- Activation of HSF1 induces HSV‐1 reactivation. Reactivation assay comparing latently infected SCG neuron cultures treated with LY294002 (20 μM), HSF1A (80 μM), KRIBB11 (30 μM), or DMSO. Reactivation rates were quantified by scoring the percentage of wells with visible GFP fluorescence after 3 days (n = 3 biological replicates). The bars and error bars are mean ± SD.
Data information: P values ≤ 0.05 were considered significant, asterisks denote statistical significance (***P < 0.001; ****P < 0.0001). P values are calculated using two‐tailed unpaired Student’s t test. P values > 0.05 were not significant (ns).
Discussion
Heterogeneity is an intrinsic but often overlooked property of biological systems including cell culture models (Lahav et al, 2004). Studies in a wide range of developmental and disease models have shown that initiating events culminating in profound biological outcomes often begin in just a minority of the cells. This can make studies of disease processes including host–pathogen interactions very difficult, especially at the molecular level. Previous studies of productive infection by HSV‐1 at high multiplicity have revealed unanticipated cell‐to‐cell differences in both the viral and cellular transcriptional responses, suggesting that heterogeneity is a fundamental property of α‐herpesvirus infections (Drayman et al, 2019; Wyler et al, 2019).
Here for the first time, we have combined two distinct but complementary single cell approaches to examine virus‐host interactions in the context of HSV‐1 latency and reactivation. First, we used single cell RNA sequencing (scRNA‐seq) to identify the cell types present in primary neuronal cultures established from dissociated sympathetic ganglia following a well‐established protocol. As expected, this showed that the cultures, regardless of the presence or absence of virus, contained a preponderance of neurons along with smaller populations of satellite glial cells, Schwann cells and fibroblasts (Amendola et al, 2015). This heterogeneity was confirmed by smFISH using probes to signature RNAs for each cell type. The presence of non‐neuronal cells is not altogether surprising, but not previously addressed with such precision. Peripheral ganglia are complex tissues, consisting of the cell bodies of neurons, various support cells, infiltrating immune cells and connective tissue. After dissociation and plating, cultures are treated with two mitotic poisons to eliminate any dividing cells, sparing the post‐mitotic neurons. Our analysis indicates that a fraction of non‐neuronal cells can survive this treatment, at least through the six days of drug exposure, resulting in a more diverse culture than generally assumed.
Importantly however, we showed, using a mix of smFISH probes, that detectable expression of the HSV‐1 latency‐associated transcript (LAT) was entirely restricted to neurons and was not detected in other cell types. This argues that neurons are unique in their ability to support a persistent HSV‐1 infection (viral latent state), consistent with previous studies and further validates the capacity of in vitro neuronal culture models to recapitulate fundamental molecular aspects of latency as originally defined in small animal models (Sawtell & Thompson, 2016). Additionally, we found that productive cycle (lytic) transcripts were restricted to neurons in reactivating cultures and were only detectable after exposure to LY. We conclude that in this in vitro model, HSV‐1 can establish viral latency in neurons but not in other cell types, and that with the exception of LAT (and associated miRNAs), the transcription of viral genes is largely suppressed.
By monitoring a limited set of viral transcripts, we and others have described a unique biphasic program of viral gene transcription through which the latent viral genomes transition from a transcriptionally repressed state into active replication culminating in the production of new infectious progeny (Du et al, 2011; Kim et al, 2012; Cliffe et al, 2015; Cliffe & Wilson, 2017; Cuddy et al, 2020). The same progression from Phase I (18 h) to Phase II (48 h) was evident at the single‐cell level but with only a modest increase in the number of viral mRNA‐positive neurons between 18 and 48 h detectable using smFISH. This suggests that the progression from Phase I to Phase II represents increased accumulation of viral RNAs rather than a change in the number of neurons undergoing reactivation. Interestingly, levels of GFP‐Us11 protein, which we have used extensively as a fluorescent readout of reactivation events, was essentially undetectable until 48 h, even though transcripts from the replication dependent true‐late genes UL36 and GFP‐Us11 were detect as early as 18 h. It is possible that transcript detection is more sensitive than GFP fluorescence or there is a lag in the translation and accumulation of the fusion protein to levels that are visible by fluorescence microscopy.
Previous studies in murine and human ganglia have found that latently infected neurons carry a widely variable number of viral genomes (Sawtell, 1997; Wang et al, 2005; Catez et al, 2012). We have previously estimated an average of about 25 genomes per neuron by quantitative PCR (Kim et al, 2012) but have not performed more direct measurements. Regardless of the system, there is very little information on how many latent genomes are capable of responding to reactivation stimuli. The signals from smFISH probes consisted of a small number of strong nuclear foci, which likely represent mRNAs that are still associated with individual viral genomes, and a larger number of cytoplasmic foci corresponding to exported and translating mRNAs (Chao & Lionnet, 2018). This suggests that only a subset of the viral genomes in a responsive neuron become transcriptionally active in response to LY treatment. Intriguingly, when two smFISH probes were used to detect two different viral mRNAs simultaneously, a subset of nuclear foci showed overlap of two different signals (yellow nuclear foci displayed as a merged image), consistent with the possibility that the same genomes were transcribing two or more productive‐cycle genes. The frequency of signal overlap within the nuclear foci correlates with more efficient reactivation conditions at later time‐points (Fig 2D). This supports our original contention that Phase I represent a general release from heterochromatin‐mediated transcriptional repression with transcription initiating at multiple viral promoters in Phase I, followed by the active recruitment of host demethylases and virus‐encoded transcriptional activators in Phase II (Kim et al, 2012; Cliffe et al, 2015).
Using an improved scRNA‐seq chemistry (see Methods), we were able to detect and profile neurons that contained multiple viral productive‐cycle transcripts indicative of reactivation. This has allowed us to compare the host transcriptomes of neurons that maintain the virus in a latent state and those undergoing LY‐induced reactivation. Strikingly, this revealed a consistent upregulation of cellular mRNAs associated with stress sensors, including genes encoding components of the heat shock response pathway and Gadd45 proteins. Previously, reactivation in response to transient hyperthermia achieved by raising the core body temperature of latently infected, restrained mice to 43°C has been reported (Sawtell & Thompson, 1992). Because this hyperthermia reactivation model involved live animals, it is impossible to know whether thermal stress operated at the level of the host neuron or was a secondary effect reflecting the action of thermal stress on one of many organ systems. Interestingly, Gadd45b was one of several host cell mRNAs found to be induced during productive infection of non‐neuronal cells (Taddeo et al, 2002). However in that study, evidence of whether Gadd45b regulated any functional aspect of HSV‐1 infection biology, including productive replication and latency/reactivation was not presented. Here, we establish that mRNA levels of both Gadd45b and Gadd45g were preferentially increased, whereas Gadd45a showed no consistent change. Although Gadd45b transcripts were detected at low levels in cells lacking viral transcripts, these were restricted to the nucleus, and thus are potentially not actively translated. In contrast, higher levels of Gadd45b transcripts could be detected in 10–15% of LY‐treated neurons at 48 h and showed an equivalent shift to cytoplasmic localization consistent with active translation. Studies in other cell types have established that Gadd45b and Gadd45g proteins confer protection against environmental stresses including DNA damage but independent of the p38/JNK pathway (Shaulian & Karin, 1999; Wang et al, 1999). Given that induction of Phase I requires phosphorylation of chromatin by JNK1/2, this observation suggests that Gadd45b expression might potentiate transcription of viral productive‐cycle RNAs. However, depletion of either Gadd45b (and to a lesser extent, Gadd45g) resulted in increased detection of GFP‐Us11, which is more consistent with a repressive role. Indeed, reactivation in response to Gadd45b depletion was relatively insensitive to a JNK inhibitor, arguing that activation of viral gene transcription following Gadd45b depletion is occurring independently of the JNK‐mediated phospho‐methyl switch. Further supporting an inhibitory role of Gadd45b in the control of viral productive‐cycle gene transcription, ectopic expression of Gadd45b in latently infected neurons was able to suppress ICP4 gene expression, viral late gene transcripts and GFP‐Us11 protein production. Importantly, the effects of Gadd45b expression on HSV‐1 reactivation was not restricted to LY‐induced reactivation conditions, as reactivation in Mirin‐treated neurons (Hu et al, 2019) was similarly suppressed by Gadd45b. Thus, we propose that nuclear Gadd45b may act as a natural anti‐viral restriction factor to limit neurotropic virus reactivation by counteracting viral late gene expression.
An intriguing study by Dremel and DeLuca provided new evidence that nuclear‐replicating DNA viruses, such as HSV‐1, employ a common strategy of coupling late gene transcription to genome replication (Dremel & DeLuca, 2019a, 2019b). They showed that the onset of viral DNA replication permanently altered genomic accessibility and, with the aid of the viral transcription factor, ICP4, DNA replication facilitated binding of initiation factors to previously silenced late gene promoters (Dremel & DeLuca, 2019a, 2019b). Therefore, we propose that by reducing the levels of ICP4, Gadd45b establishes a non‐permissive environment that prevents HSV‐1 late gene transcription and curtails virus reactivation at the single‐cell level. In our study, we discovered that upon HSV‐1 reactivation, Gadd45b protein localization changes dramatically in neurons (Fig 5B). In uninfected or latently infected cultured neurons, Gadd45b is present in both cytoplasmic and nuclear compartments (Fig 5B and C). However, in neurons undergoing viral DNA replication (EdU‐positive), Gadd45b is excluded from the nucleus (cytoplasmic staining) (Fig 5B and D). Interestingly, in LY‐induced HSV‐1 reactivating conditions, a small fraction of Gadd45b is localized to nuclear puncta (foci) in EdU‐negative cells (Fig 5B and E). This phenotype was not observed in LY‐treated, uninfected neurons, suggesting that the Gadd45b nuclear puncta‐representing neurons have responded to the reactivation stimulus but somehow failed to achieve detectable viral DNA synthesis. In support of this finding, we found that Gadd45b nuclear puncta‐presenting neurons strongly correlated with LAT‐positive signal (Fig 5F). Based on our Gadd45b ectopic expression data showing that Gadd45b expression leads to the reduction of both ICP4 and viral late gene expression (Fig 6C and D, and E, and F, and G), we propose that endogenous Gadd45b nuclear puncta localization represent infected neurons in which Gadd45b is actively suppressing ICP4 transcription and viral late gene expression (Fig 7). In support of this model, we do not observe Gadd45b nuclear puncta‐representing cells that are expressing ICP4 (Fig 5G). In contrast, when virus reactivation is occurring, Gadd45b is restricted to the cytoplasmic compartment by an unknown mechanism (Fig 7B).
Figure 7. Working model of how Gadd45b regulates HSV‐1 reactivation in the neuronal population at the single‐cell level.
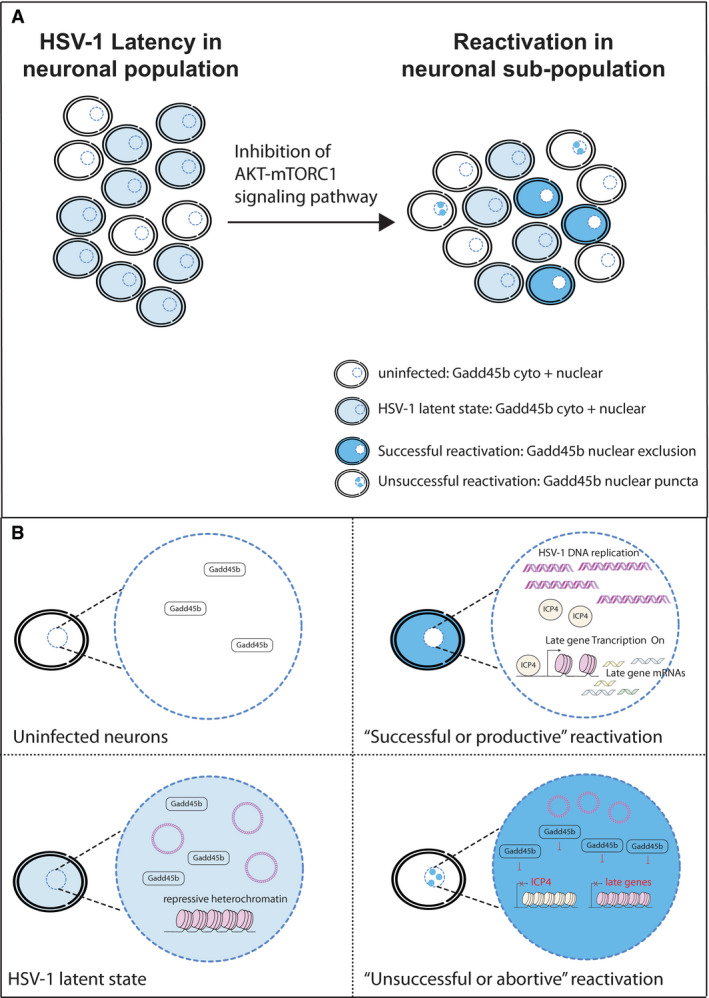
- A schematic representation of the heterogeneity within a neuronal population for HSV‐1 reactivation following inhibition of the canonical AKT‐mTORC1 signalling pathway. Dark blue neurons signifies cells undergoing “Successful” (productive) reactivation with Gadd45b localized in the cytoplasm (nuclear exclusion); light blue neurons shows cells remaining in the latent state with Gadd45b localized to both nuclear and cytoplasmic compartments; white‐labelled cells with dark blue nuclear puncta signifies “Unsuccessful” (abortive) reactivation with Gadd45b localized to nuclear foci/puncta; and white‐labelled neurons represent uninfected cells with low‐level Gadd45b localized to both nuclear and cytoplasmic compartments.
- Neurons that undergo “Unsuccessful” reactivation are conceived as those in which Gadd45b suppresses ICP4 mRNA transcription (and protein accumulation) and viral late gene expression. In contrast, neurons that can undergo “Successful” reactivation have relocalized or excluded Gadd45b protein from the nucleus to allow for ICP4 transcription and efficient viral late gene expression, and presumably culminating in synthesis of new viral progeny.
In summary, we establish that HSV‐1 reactivation can be counteracted through the expression and subcellular localization of cellular Gadd45b protein in a neuronal sub‐population (Fig 7A). As such, we predict that the likelihood of “successful” (or productive) reactivation is high in latently infected neurons when Gadd45b is excluded from the nucleus and low in latently infected neurons when Gadd45b accumulates within the nuclear puncta (Fig 7B). This helps to explain the heterogeneous reactivation response even though all of the neurons are exposed to the same reactivation stimulus. Cell‐to‐cell differences in the expression and subcellular localization of Gadd45b were not evident at the bulk population level but are only revealed by the use of single‐cell methodologies that can simultaneously monitor both viral and host gene transcript, protein and viral DNA synthesis levels. Little is known about the regulation of Gadd45 expression in neurons. In the future, it will interesting to explore how the subcellular localization and activity of Gadd45b are regulated and how this impacts the virus–host relationship in vivo and ultimately the ability of HSV‐1 and possibly other neurotrophic α‐herpesviruses to cause disease.
Materials and Methods
Reagents and Tools table
| Reagent/Resource | Reference or source | Identifier or catalog number | |
|---|---|---|---|
| Experimental models | |||
| Rat: embryonic day 21 primary SCG neurons | Charles River | SAS SD(400)‐ Sasco Sprague Dawley | |
| Human: 293LTV cell line | N/A | N/A | |
| Recombinant DNA | |||
| pLKO.1‐puro Non‐Target shRNA Control Plasmid | Sigma‐Aldrich | SHC016‐1EA | |
| shRNA: Gadd45a‐1 | Sigma‐Aldrich | TRCN0000054690 | |
| shRNA: Gadd45a‐2 | Sigma‐Aldrich | TRCN0000218764 | |
| shRNA: Gadd45b‐1 | Sigma‐Aldrich | TRCN0000088312 | |
| shRNA: Gadd45b‐2 | Sigma‐Aldrich | TRCN0000234401 | |
| shRNA: Gadd45b‐3 | Sigma‐Aldrich | TRCN0000234403 | |
| shRNA: Gadd45b‐4 | Sigma‐Aldrich | TRCN0000088311 | |
| shRNA: Gadd45b‐5 | Sigma‐Aldrich | TRCN0000088309 | |
| shRNA: Gadd45g‐1 | Sigma‐Aldrich | TRCN0000234425 | |
| shRNA: Gadd45g‐2 | Sigma‐Aldrich | TRCN0000234427 | |
| FLAG‐Rheb (Q64L)‐pcw107 | Addgene | Cat# 64607 | |
| Gadd45b‐Myc‐DDK‐tagged | Origene | CAT# RC213354L1 | |
| pLenti‐C‐Myc‐DDK Lentiviral Vector | Origene | CAT# PS100064 | |
| Antibodies | |||
| Rabbit polyclonal anti‐HSP70 | Cell Signaling Technology | Cat# 4872; RRID: AB_2279841 | |
| Mouse monoclonal anti‐GFP | Santa Cruz Biotechnology | Cat# sc‐9996; RRID: AB_627695 | |
| Mouse monoclonal anti‐FLAG (M2) | Sigma‐Aldrich | Cat# F1804; RRID: AB_262044 | |
| Mouse monoclonal anti‐ICP4 | Abcam | Cat# ab6514; RRID: AB_305537 | |
| Mouse monoclonal anti‐Gadd45b | Santa Cruz Biotechnology | Cat# sc‐377311 | |
| Rabbit polyclonal anti‐Gadd45b | Abcam | Cat# ab230646 | |
| Oligonucleotides and other sequence‐based reagents | |||
| ICP27‐F: 5′TTTCTCCAGTGCTACCTGAAGG‐3′ | Kim et al (2012) | N/A | |
| ICP27‐R: 5′‐TCAACTCGCAGACACGACTCG‐3′ | Kim et al (2012) | N/A | |
| UL30‐F: 5′‐CGCGCTTGGCGGGTATTAACAT‐3′ | Kim et al (2012) | N/A | |
| UL30‐R: 5′‐TGGGTGTCCGGCAGAATAAAGC‐3′ | Kim et al (2012) | N/A | |
| UL36‐F: 5′‐CGCTGCACGAATAGCATGGAATC‐3′ | This study | ||
| UL36‐R: 5′‐CCAGCTCCCCGGAACACATTTA‐3′ | This study | ||
| Us11‐F1: GGCGACCCAGATGTTTACTT | This study | ||
| Us11‐R1: CCCGCCGCTTGATCATTA | This study | ||
| Us11‐F2: CGATCTCGAAGCCATCTGAAG | This study | ||
| Us11‐R2: TCTGGGTTCCCTAGGTTCTC | This study | ||
| 18S‐F: 50′‐AGGAATTGACGGAAGGGCACCA‐3′ | Kobayashi et al (2012) | N/A | |
| 18S‐R: 5′TTATCGGAATTAACCAGACAAATCG‐3′ | Kobayashi et al (2012) | N/A | |
| Gadd45a‐F: 5′‐GAGAACGACAAGAGCCCC‐3′ | This study | N/A | |
| Gadd45a‐R: 5′‐ATCCATGTAGCGACTTTCCC‐3′ | This study | N/A | |
| Gadd45b‐F: 5′‐GAGAGCAGAGGCAATAACCAG‐3′ | This study | N/A | |
| Gadd45b‐R: 5′‐TGCTTTAGGTGTTTAGAGTGGG‐3′ | This study | N/A | |
| Gadd45g‐F: 5′‐TCCGCCAAAGTCCTGAATG‐3′ | This study | N/A | |
| Gadd45g‐R: 5′‐CGCCTGGATCAACGTAAAATG‐3′ | This study | N/A | |
| smFISH probes sequences for rat ntrk1: | This study | N/A | |
| agcatcaacgaagtcaccag | tggttttccacatagagctc | ||
| cactcttcacgatggttagg | ggggtgaaatggaaggcatc | ||
| ggagagattcaggtgactga | agagggactccaaagcattg | ||
| gtcaagtcctgtagggagag | accacaactgttgttgtgtc | ||
| atctggatcttcactgaggg | cacttccacagagtcattgg | ||
| cctggcactgcagaaaaacg | cagatctccagatttcttca | ||
| ggtgacattgaccagagtta | cacgtcacattcttcttgtt | ||
| aaggagacgctgacttggac | acagagaagggaatgcacca | ||
| cagagccgttgaagaaccag | gatgaagctggtctcattga | ||
| cgctgactccaagaactgag | tagttcccgttgttgacatg | ||
| cataaaggcagccatgatgg | tcctcagggttgaactcaaa | ||
| ctcttgatgtgctgttagtg | gagggcagaaaggaagaggg | ||
| ccacatttgttgagcactag | ggttgatcccaaatttgctc | ||
| atgaagtgtagggacatggc | gtaggggaaagagaactgcc | ||
| cactgaagtactgtgggttc | gcttgatatggtggacacag | ||
| tcccacttgagaatgatgtc | gaaagacctttccaaaggct | ||
| tcagaaggttgtagcactca | ttctcagatgtctccttcag | ||
| acgatggaagtcctgacgag | tacgatgtgttggtgctgta | ||
| ccgtgcagactccaaagaag | tgtactcgaagaccatgagc | ||
| cggaggaaacggttgaggtc | aagtctccaatcttgaccac | ||
| tagtctgtgctgtagatgtc | cactctcggtgctgaacttg | ||
| gctttccataggtgaagatc | cagtgttggagagctggtac | ||
| atgatggcgtagacatcagg | cagaacgtccaggtaactcg | ||
| cacatgctgagggtaagact | ctattatgatggatgctggc | ||
| smFISH probes sequences for rat col3a1: | This study | N/A | |
| agacatgagactctttgtgc | gggtgagaagaaaccaggtc | ||
| tggactgctgtgccaaaata | ttgcatcccaattcatctac | ||
| ctagactcataggactgacc | tatttgacatggttctggct | ||
| tcatcacagaggacagatcc | tggctcatcatcacacatta | ||
| gctgtggacatattgcacaa | atgacaggagcaggtgtaga | ||
| cttgaggtctattaccatca | agaataattctggccaccag | ||
| gactccagacttgacatcat | tatccaggagaaccaggaga | ||
| agatggaccaatagcaccag | atcatttcctttaggaccag | ||
| tatgccttgtaatccttgtg | ttccaggagaaccactgtta | ||
| aattccaagtggacctggag | aggagaaccattttcaccac | ||
| atgtcctttgatgccattag | acttgatccatcttttccag | ||
| aatatggtgaaaagccgcca | gaaatccattggatcatccc | ||
| atgatctcctcagtgttgat | gtccgttaacagacttgagt | ||
| agggctgataagactctcta | cagggttttttctagaacca | ||
| atttcaggtctctgcaattc | taggatcaacccagtattct | ||
| tttctccggtttccatatta | tcatgggactggcatttatg | ||
| gaactgaaagccaccattca | ggaagatcaggattgccata | ||
| gctgtacatcaaggacatct | ggctggaaagaagtctgagg | ||
| tatgtaatgttctgggaggc | catgtaggcaatgctgtttt | ||
| tgaattctccctcattggat | aatttgctatttccttcagc | ||
| ccatcctctagaactgtgta | aaagactgtcttgctccatt | ||
| tatgatggggagtctcatgg | caatgtcatagggtgcgata | ||
| ccaaattcttgatcaggacc | aagcaaacagggccaatgtc | ||
| gtttcagagagtttggctta | gcaccattgagacattatga | ||
| smFISH probes sequences for rat gadd45b: | This study | N/A | |
| agacggaggtgacacgga | ccccctcgaaattaatcc | ||
| gaggcggtgggacttact | cagaagtgtccgaggctt | ||
| gtaaaaaccttcccgcgc | cactcgtccagatccgaa | ||
| cacggaagatccccaagg | gtcacacttcacagcgga | ||
| ctcttccagggtcatgat | ctgcatcttctgaaccgc | ||
| ggtgaggcgatcctgacg | gggtccacattcatcagt | ||
| agcaggcacaagaccacg | tggatcagggtgaagtga | ||
| tgtcattgtcgcagcaga | ctttgcatgcctgatacc | ||
| cgtttgtgcctagagtct | gaggcagtgcaggtctcg | ||
| gtatgacagttcgtgacc | aagccttggcttttccag | ||
| agtaactggccacctcca | attgcctctgctctcttc | ||
| atataggggacccactgg | ctcagcgttcctccagag | ||
| ggggacagcaactcaaca | ttctggggaatgaggggg | ||
| agggtagcttttgaggga | ctcccggaaggtatcacg | ||
| tctcagtctcggtgactc | tcttggcgaccacctgtg | ||
| aggccttggctctaaagt | aggctggctgcatgattg | ||
| tgtccagaccgtctgctg | Cgctgagttacgcacagt | ||
| atgtgccttagctgcgaa | gatctgtcttgctcagca | ||
| smFISH probes sequences for viral ICP27: | This study | N/A | |
| atcaatgtcagtcgccat | aggccgaggtcaattagc | ||
| agatcgctgtcggagagg | ctgtccgattccaggtcg | ||
| gtcttccatgtcctcgtc | gagcggcgtcgagtatcg | ||
| gggtcttctggacgagac | cgtcggacgaggcgtctg | ||
| ggttgaggatcgttgggg | cgaccacacactgtgggg | ||
| tgctcgggggagcaagac | tttggtcggtgggggttg | ||
| ccgacaaaccatcggcag | agggttgcgattggttct | ||
| caatcgccagcgtcatta | gtggcgtcgatggtgtcg | ||
| atggagcgcaggaccaac | ctcgctgatgcggtcgac | ||
| tgtgcgctgcggccaaag | ccaaaggggtcgtgcatg | ||
| ctattcgcggcgggaaac | gtcaaagggccctccttg | ||
| caggagacccgtctggtc | cgtgggcgaccaaggttt | ||
| agtgcgatagaggctcgg | gatgcggcccgaggattg | ||
| attttcttggcgcagcac | tgcacatcttgcaccacg | ||
| cagcggcaggttgtggtg | cgataatggggtcctggg | ||
| gaggttatccagcacagc | agcactggagaaagggcc | ||
| aacacagttcgtccaggc | cttaatgtccgccagacg | ||
| gcctggccagaatgacaa | cacgctcgacgcggttgg | ||
| aagggtcgcgtagtcgat | atgcatcttctctccgac | ||
| aggatttcgatcaggccc | cgaacactcctggcggtg | ||
| cgtcaactcgcagacacg | ggggcgacgatgtgactg | ||
| smFISH probes sequences for viral UL30: | This study | N/A | |
| gcgaggtaggggttgtaaaa | atcgcattcgctatagtacg | ||
| gcggggcgatgaatcgaaat | gaaagacggtgatggtgggg | ||
| ttctccaggatgtcatacac | gtgtgatggcgtccataaac | ||
| cgtgccgtaaacgtgaacgg | ctcctccttgttcatgtaaa | ||
| tctcgtagtagtacacgtcg | cggacgtagacgcggtaaaa | ||
| agaagttgtcgcacaggtac | gccgtacagttaaactcgac | ||
| atcgaagcacatgagcttgt | acaggatatctggatgacca | ||
| accgagcgaaaacaggagga | tgtcgaattccagaaccacg | ||
| gccaacagcatctcgaattc | ctgtttcacaagggtcatga | ||
| gccagtcgaagttgatgatg | tgtaaatgtccgtcagcttg | ||
| acatgtcgatgttcaccatg | atcttgtcggttataatccc | ||
| ttgagcttgtagctcgagag | gaatcctgtatgcagtactc | ||
| taaaaaacagctggcccacc | agctccagatggggcaaaaa | ||
| tggtgcgggtgatgttaata | gtaaagacgcggatctgctg | ||
| agaataaagcccttctggtc | tcacgtgaaacccggaagtg | ||
| tacaggctggcaaagtcgaa | cgtgagccttgacgaagaac | ||
| gagttacacacgaccttgat | cgtagatgatgcgcatggaa | ||
| agcacaaatatggagtccgt | ttgatggggggcagaaacag | ||
| agcttggtgaacgtcttttc | cgatgtactttttcttggcg | ||
| ctggaggtgcggttgataaa | cgtcgtaaaacagcaggtcg | ||
| aggacaaagtcctggatgtc | ccatgagcttgtaatacacc | ||
| agtaatagtccgtgttcagg | caaacagggccttgaatgtc | ||
| aacagactctcggtgatctt | ccacacttcgggaataaacc | ||
| caacattcgacgagtttcct | gctagagtatcaaaggctct | ||
| smFISH probes sequences for viral UL36: | This study | N/A | |
| ctcgacgatcgaacacatgc | ctgggtactcgaggaagttg | ||
| gtatacgcgcgagaagacac | aatggttaggagcgtgtacg | ||
| gggtcaaaaaggtatgcggt | tcaggtatggatacatgtcc | ||
| gcacgaaaaacaccatggct | atgtcgtcgtcatcttcttc | ||
| ggatttcttcttggttttgg | cgttctcgatgagaaaggcg | ||
| catgttcagggtaaactcca | gaccgatcagagagttttcg | ||
| tagccacaaggatcagcttc | gaggcgtgtatacagattgg | ||
| tatacagaagcggttctggg | cccggtaaaagtactcctta | ||
| aagacaggcagggactcgag | tgttggtcgttaatgtcgcg | ||
| cctggagtttgtgcaagaac | agagaaacggagtctctggg | ||
| gcgtgatcggcgaagaactg | tatgggttaaacgcaagggc | ||
| gaaacatgtcggcgtacgtg | cctcgtggtagtcgataaac | ||
| aagtcttccgacaggtgtag | gcccttttgaaaaaacggca | ||
| tcacgaatgcgacgtagtcg | tcagcagggtcttcagattg | ||
| accagaatttcgacctgatc | tcaaaggtacgctgggcatg | ||
| cagggtgtcaaagttcatct | tggcgtacgcgctatataag | ||
| caaaaagctgggggcgaacg | tgatgacggtgaggacgtac | ||
| cgtccgaagagctgaaagtt | caaactcgcgcgtgagagtg | ||
| gcacgtcgttctcgttaaag | caggcgccaaaaggtgtaaa | ||
| tgagatgcatgtactcgtgt | ttctgaaacgcacgggggag | ||
| gctgcacgaatagcatggaa | aacatgcaggtccaaagagc | ||
| tcatactctgtgtagtcgtc | ggtcagaagggcgtcgaaac | ||
| aaacagcgtgagacacagct | cagacacgcgttttcgatcg | ||
| gtcatggagatgtccacgag | ggacgaagaagcggcagaag | ||
| smFISH probes sequences for viral LAT: | This study | N/A | |
| agaagcaggtgtctaaccta | ctggtgtgctgtaacacgag | ||
| tgggagggggggaaaagaac | gtcggacgggtaagtaacag | ||
| cttagataagggggcagttg | ttggagtacgtgggtcattg | ||
| cacaaaaacacaggcggggt | aagacccaagcatagagagc | ||
| actttagggggggtacacac | ccatatcctcgctttaggaa | ||
| cgtggggttggtgtgcaaag | atgctcggtggtatgaagga | ||
| tgttggttaccaacccacac | gaatcacaggagacgagggg | ||
| aaacaagaatgcggtgcagc | gggggagacagaaacaggaa | ||
| caacaaaggatgggaggggt | ggggggttaactacactata | ||
| ttgggtgtgggtgttaagtt | catggagccagaaacacagt | ||
| ctctggtgttaaccacagag | tctgtgttggatgtatcgcg | ||
| ttacttttgtatcttttccc | aactcaggggatttttgctg | ||
| ttttgtgttggccctaataa | aaagacacgggcaccacaca | ||
| cgtgtcggggaggtggaaag | aaaacaataagggacgcccc | ||
| tggggaacatgctgttcgac | tggaacagcctctggatgac | ||
| tggaccagacgggaaacatg | acatggagacggggaacatc | ||
| gccttacgtgaacaagacga | aggaagtgtgcccggaagac | ||
| ggagacaagaggaaacctcc | |||
| Chemicals, enzymes and other reagents | |||
| LY294002, PI3K inhibitor | Calbiochem | Cat# 440202 | |
| Mirin, MRE11 inhibitor | TOCRIS | Cat# 3190 | |
| JNK inhibitor (JNK Inhibitor II) | Calbiochem | Cat# 420128 | |
| HSF1A, HSF1 activator | Calbiochem | Cat# 5.33761 | |
| KRIBB11, HSF1 inhibitor | Calbiochem | Cat# 385570 | |
| Nerve growth factor | Envigo | Cat# B.5017 | |
| Acyclovir | Calbiochem | Cat# 114798 | |
| Neurobasal media | GIBCO | Cat# 12348017 | |
| NeuroCult™ SM1 Neuronal Supplement | STEMCELL Technologies | Cat# 05711 | |
| Phosphonoacetic acid (PAA) | Sigma‐Aldrich | Cat# 284270 | |
| WAY‐150138 | Kim et al (2012) | N/A | |
| Cy®5 Mono NHS Ester | GE Healthcare | Cat# PA15101 | |
| Cy®3 Mono NHS Ester | GE Healthcare | Cat# PA13101 | |
| Software | |||
| GraphPad Prism (version 9) | GraphPad Software | N/A | |
| ImageJ (version 2.0.0‐rc‐69/1.52p) | NIH | N/A | |
| Fiji | https://doi.org/10.1038/nmeth.2019 | ||
| Seurat 3.0 | https://satijalab.org/seurat/install.html | ||
| Cellranger v3.0.0 | https://support.10xgenomics.com/single‐cell‐gene‐expression/software/downloads/latest | ||
| R v4.0.0 | https://www.r‐project.org/ | ||
| Rstudio | https://rstudio.com/ | ||
| R markdown | https://rmarkdown.rstudio.com/ | ||
| Other | |||
| RNeasy Mini Kit | QIAGEN | Cat# 74104 | |
| SYBR Green Master reagent | Applied Biosystems | Cat# 4309155 | |
| Click‐iT® Plus EdU Imaging Kits | Life Technologies | Cat# C10640 | |
Methods and Protocols
Primary neuronal culture
Rat primary superior cervical ganglion (SCG) were excised from euthanized E21 Sprague‐Dawley rat embryos by dissection. All protocols for isolating SCGs were approved by the Institutional Animal Care and Use Committee (IACUC) at the NYU School of Medicine. Isolated SCGs were placed in Leibovitz’s L‐15 media (Leibovitz’s L‐15 with 0.4% D(+)‐glucose) and dissociated in trypsin (0.25%, Invitrogen) and collagenase (1 mg/ml, Sigma) at 37°C for 15 min. Cells were then washed in modified Eagle medium (MEM) supplemented with 0.4% D (+)‐glucose, 2 mM L‐glutamine, 10% FBS and passed through 21G and 23G needles followed by a 70‐μm cell strainer. Dissociated cells were plated onto rat‐tail collagen (0.66 mg/ml, Millipore) and laminin (2 mg/ml, Sigma)‐coated plates with MEM medium supplemented with 50 ng/ml 2.5S NGF. After 1 day of culture, the growth medium was replaced with NBM medium (neurobasal medium, 0.4% D (+)‐glucose, 2 mM L‐glutamine, NeuroCult SM1 Neuronal Supplement [Stem Cell Technologies], 50 ng/ml 2.5S NGF) supplemented with aphidicolin (5 μM, Calbiochem) and 5‐fluorouracil (20 μM, Sigma) to eliminate any proliferating cells.
HSV‐1 latency establishment and reactivation
To establish HSV‐1 latency in SCG‐derived neurons, six day‐old cultures were pre‐treated with acyclovir (ACV, 100 μM, Calbiochem) overnight. The next day, the neurons were infected with WT HSV‐1 (Patton strain expressing an EGFP‐Us11 fusion marker) at an MOI of 1.5 plaque forming units (pfu) per neuron (titre determined on Vero cells) in NBM medium for 2 hr and then changed to NBM medium supplemented with 100 μM ACV for at least 6 days to establish latency. For reactivation, ACV was removed and latently infected cultures treated with LY294002 (20 μg/ml, Calbiochem), Mirin (100 μg/ml, TOCRIS), or transduced with Gadd45b shRNAs (Sigma). Cells were cultured on 96‐well plates at a density of ~6,500 cells/well. The GFP‐positive cells were detected by inverted fluorescent microscope of living neurons. Designation of a GFP‐positive well is when a well contains three or more GFP‐positive neurons. Reactivation was quantified by counting the percentage of GFP‐positive wells. Typically at least 30 infected wells were analysed for individual treatments. HSV‐1 stocks were amplified and titered on Vero cell monolayers by standard methods (Hu et al, 2019).
Single‐molecule FISH
SCG‐derived neurons were plated onto 18‐mm glass coverslips coated with poly‐D‐lysine, collagen, and laminin at a density of ~1.2 × 105 cells per well. Infected cells were allowed to establish latency or induced to reactivate as described above except that WAY‐150138 (20 μg/ml) was included in the media to prevent spread of any newly synthesized infectious virus. Latent or reactivated cells were fixed with 4% paraformaldehyde (Electron Microscopy Sciences) in PBS for 10 min and washed with PBS for 10 min with gentle shaking. Cells were then permeabilized with 0.5% Triton X‐100 in PBS for 10 min, washed with PBS for 10 min, and incubated with pre‐hybridization buffer (10% deionized formamide [Invitrogen] in 2X saline‐sodium citrate [SSC] buffer for 10 min at room temperature). The hybridization was performed by adding FISH probes (60 ng for each gene) with hybridization buffer (10% deionized formamide, 100 μg/ml E. coli tRNA, 100 μg/ml salmon sperm ssDNA, 2 mg/ml BSA, 10% dextran sulfate in 2X SSC buffer) at 37°C overnight. All the probes were designed and ordered by Biosearch Technologies and then conjugated with Alexa Fluor 488, Cy3, or Cy5 NHS Ester amine reactive fluorescent dyes (GE Healthcare). After hybridization, cells were washed with pre‐hybridization buffer at 37°C for 20 min and washed three times with pre‐hybridization buffer at room temperature with gentle shaking. Cells were then washed once with PBS, and the nuclei were stained with DAPI (0.5 μg/ml) in PBS. Coverslips were mounted onto glass slides using prolong gold antifade mountant (Invitrogen), and images were acquired by Nikon ECLIPSE Ti inverted microscope. In brief, we performed a maximum intensity projection of the raw image stacks, then manually circled each cell and measured its average intensity in the smFISH channel using ImageJ software. smFISH probe sequences for specific gene products are listed in Reagents and Tools table.
RNA isolation and reverse‐transcription quantitative PCR (RT‐qPCR)
To deplete Gadd45b using shRNA‐mediated RNA interference, 10‐day‐old uninfected or latently infected SCG neuron cultures were transduced with Gadd45b shRNA lentiviral particles and maintained for 20 h in NBM medium. After three days, cells were harvested and processed for RNA isolation. To express a constitutively active Rheb, latently infected SCGs were transduced with a lentivirus encoding Flag‐tagged Rheb (Q64L) in the presence of 100 μM ACV. The next day, both the lentivirus and ACV were removed and the cultures were maintained in NBM medium for a further 48 h; then they were transduced with Gadd45b shRNA lentivirus for 20 h and then maintained in NBM medium for three days. For RNA isolation cells were rinsed once with PBS and lysed in 350 μl RLT buffer (RNeasy mini kit, QIAGEN) and centrifuged through a QIAshredder spin column for homogenization. Each sample was combined with 550 μl RNase‐free water and 450 μl 100% ethanol and mixed thoroughly to precipitated the RNA. The samples were then loaded to the RNeasy columns, washed as recommended, and then eluted with RNase‐free water. The RNA was treated with RNase‐free DNase I at 37°C for 10 min and then treated with 5 mM EDTA at 75°C for 10 min to inactivate the DNase I. The RNA was converted to cDNA using qScript cDNA SuperMix (QuantaBio). Real‐time quantitative PCR was performed using SYBR Green PCR Master Mix (Applied Biosystem) and a StepOnePlus Real‐Time PCR System (Applied Biosystem). Statistical calculations were made using the two‐tailed paired Student’s t‐test (Prism 8, Graphpad). Oligonucleotide sequences for RT‐qPCR for specific gene products are listed in Reagents and Tools table.
Single‐cell RNA sequencing and analysis
scRNA‐seq libraries were prepared using either Single Cell 3′ Library and Gel Bead Kit v2 (PN‐120237) or Single Cell 3ʹ GEM, Library and Gel Bead Kit v3 (PM‐1000075) according to manufacturer’s instructions. Libraries were sequenced in the paired‐end mode on either an Illumina HiSeq 4000 or Illumina NovaSeq 6000 (Appendix Table S2). We generated eight libraries using the Chromium Single Cell 3′ v2 chemistry and three libraries using the Chromium Single Cell 3′ v3 chemistry. Following sequencing, the 10x Genomics Cell Ranger Single‐Cell Software Suite was used to perform sample demultiplexing, barcode processing and single‐cell 3′ gene counting through alignment of sequence reads to a hybrid HSV‐1 and Rattus norvegicus (rn6) genome/transcriptome (Gibbs et al, 2004). Because multiple distinct HSV‐1 RNAs often share overlapping 3’ ends, sequence reads aligning to these cannot be accurately assigned to individual transcripts. Instead, these HSV‐1 RNAs were grouped into a set of 20 transcription units (TU1‐20) for the purposes of mapping and analysis (Fig 3 and Appendix Table S3). Aligned data were subsequently imported into Seurat v3.0 (Mayer et al, 2018) for additional quality filtering, identification of highly variable genes, data integration, dimensionality reduction, unsupervised clustering and integrated differential gene expression analysis using MAST (Finak et al, 2015). Full details of these analysis are recorded in R Markdown files supplied as Appendix Computer Code 1 and 2. Software and algorithms used for analyzing sequencing data are listed in the Reagents and Tools table.
Immunofluorescence staining
Neurons were seeded at a density of ~1.2 × 105 cells onto 18‐mm glass coverslips pre‐coated with poly‐D‐lysine, collagen, and laminin. After treatments, cultures were washed with PBS and fixed with 4% paraformaldehyde in PBS for 10 min at RT then permeabilized with 0.5% Triton X‐100 in PBS. After blocking in 2% BSA in PBS for 20 min, cells were incubated with primary antibodies overnight at 4°C and detected using anti‐mouse‐Alexa Fluor® 488, anti‐rabbit‐Alexa Fluor® 555, anti‐mouse‐Alexa Fluor® 647, or anti‐rabbit‐Alexa Fluor® 647 for 1.5 h at RT. Nuclei were stained with Hoechst 33258 (Life Technologies). Images were acquired by Zeiss LSM 700 confocal microscope software and were processed and analyzed using ImageJ. Primary antibodies used for immunofluorescence staining: anti‐GFP (Santa Cruz Biotechnology), anti‐HSP70 (Cell Signaling Technology), anti‐ICP4 (Abcam), anti‐Gadd45b (Abcam; Santa Cruz Biotechnology). Source of antibodies are listed in Reagents and Tools table.
Combined fluorescent viral DNA EdU label and immunofluorescence staining
SCG neurons were seeded at a density of ~1.2 × 105 cells onto 18‐mm glass coverslips pre‐coated with poly‐D‐lysine, collagen, and laminin. The EdU pulse‐labeling for viral DNA synthesis was performed for 6 h at 37°C incubator prior to fixation of cells. After EdU pulse‐labeling and treatments, cultures were fixed with 4% paraformaldehyde in PBS for 15 min at RT then permeabilized with 0.5% Triton X‐100 in PBS. Imaging of EdU‐labeled cells were done according to instructions from the Click‐iT® Plus EdU Imaging Kits (Life Technologies). After EdU labeling, cells were wash with PBS and then incubate with primary antibodies overnight. After secondary antibodies staining, nuclei were stained with Hoechst 33258 or DAPI (Life Technologies) and images were acquired by Zeiss LSM 700 confocal microscope software.
Western blot analysis
Total whole cell extracts from SCGs were collected by lysis in Laemmli buffer (Bio‐Rad) from approximately 50,000 neurons and fractionated on NuPAGE 4–12% Bis‐Tris Protein Gels (Life Technologies). The protein was then transferred to polyvinylidene difluoride (PVDF) membranes (Millipore), blocked with 5% milk in TBST for 1 h at room temperature and incubated with primary antibody overnight at 4°C. The signals were detected by HRP‐conjugated secondary antibodies and ECL reagent. Primary antibodies used for western blot analysis are anti‐ICP4 (Abcam), anti‐α‐tubulin (Millipore), anti‐Flag (Sigma‐Aldrich). Source of antibodies are listed in Appendix Table S5.
Statistical analysis
All statistical analysis was performed using Prism software (8.0). Number of biological replicates (using rat pups from different dissection days) of the experiments (n = X number) and statistical significance are indicated in the corresponding figures and figure legends. P values ≤ 0.05 were considered significant, asterisks denote statistical significance (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001). P values are calculated using two‐tailed unpaired Student’s t test. P values > 0.05 were not significant (ns).
Author contributions
H‐LH, ACW, DPD, and TTH designed the study. H‐LH performed all of the experiments with technical support from KPS, SW, and DPD; MVC and TL provided critical reagents and equipment for the study; and H‐LH, IM, TL, ACW, DPD, and TTH analyzed the data. H‐LH, IM, ACW, DPD, and TTH wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Source Data for Figure 6
Acknowledgements
The authors dedicate this study to the memory of our friend and colleague “Randy” (Randall Jay Cohrs), whose sudden departure leaves the α‐herpesvirus latency field all the poorer. The authors thank members of the Huang, Mohr, Wilson, Lionnet, and Chao labs for technical assistance and critical discussions. This work was supported by NIH grants: GM107257 and GM139610 (T.T.H.), AI103933, AI130618 and AI151358 (A.C.W.), AI073898 and GM056927 (I.M.), AI152543 (I.M. and D.P.D.), GM127538 (T.L.) and MH119136 and NS107616 (M.V.C). This work was also supported by the Melanoma Research Alliance grant 687306 (T.L.). M.V.C is a Simons Foundation Senior Fellow. Sequencing was performed at the NYU Genome Technology Center (RRID: SCR_017929). This shared resource is partially supported by the Cancer Center Support Grant P30CA016087 at the Laura and Isaac Perlmutter Cancer Center.
EMBO Reports (2022) 23: e53543.
Contributor Information
Angus C Wilson, Email: angus.wilson@nyulangone.org.
Daniel P Depledge, Email: depledge.daniel@mh-hannover.de.
Tony T Huang, Email: tony.huang@nyumc.org.
Data availability
All sequencing datasets generated as part of this study are available via the European Nucleotide Archive under the project accession PRJEB39022 (https://www.ebi.ac.uk/ena/browser/view/PRJEB39022).
References
- Amendola J, Boumedine N, Sangiardi M, El Far O (2015) Optimization of neuronal cultures from rat superior cervical ganglia for dual patch recording. Sci Rep 5: 14455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur J, Efstathiou S, Simmons A (1993) Intranuclear foci containing low abundance herpes simplex virus latency‐associated transcripts visualized by non‐isotopic in situ hybridization. J Gen Virol 74(Pt 7): 1363–1370 [DOI] [PubMed] [Google Scholar]
- Baringer JR, Swoveland P (1973) Recovery of herpes‐simplex virus from human trigeminal ganglions. N Engl J Med 288: 648–650 [DOI] [PubMed] [Google Scholar]
- Bloom DC (2016) Alphaherpesvirus latency: a dynamic state of transcription and reactivation. Adv Virus Res 94: 53–80 [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, Satija R (2018) Integrating single‐cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36: 411–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camarena V, Kobayashi M, Kim JY, Roehm P, Perez R, Gardner J, Wilson AC, Mohr I, Chao MV (2010) Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV‐1 latency in neurons. Cell Host Microbe 8: 320–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catez F, Picard C, Held K, Gross S, Rousseau A, Theil D, Sawtell N, Labetoulle M, Lomonte P (2012) HSV‐1 genome subnuclear positioning and associations with host‐cell PML‐NBs and centromeres regulate LAT locus transcription during latency in neurons. PLoS Pathog 8: e1002852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao JA, Lionnet T (2018) Imaging the life and death of mRNAs in single cells. Cold Spring Harb Perspect Biol 10: a032086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou YY, Lionnet T (2018) Single‐molecule sensitivity RNA FISH analysis of influenza virus genome trafficking. Methods Mol Biol 1836: 195–211 [DOI] [PubMed] [Google Scholar]
- Cliffe AR, Arbuckle JH, Vogel JL, Geden MJ, Rothbart SB, Cusack CL, Strahl BD, Kristie TM, Deshmukh M (2015) Neuronal stress pathway mediating a histone methyl/phospho switch is required for herpes simplex virus reactivation. Cell Host Microbe 18: 649–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffe AR, Wilson AC (2017) Restarting lytic gene transcription at the onset of herpes simplex virus reactivation. J Virol 91: e01419‐16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuddy SR, Schinlever AR, Dochnal S, Seegren PV, Suzich J, Kundu P, Downs TK, Farah M, Desai BN, Boutell C et al (2020) Neuronal hyperexcitability is a DLK‐dependent trigger of herpes simplex virus reactivation that can be induced by IL‐1. eLife 9: e58037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drayman N, Patel P, Vistain L, Tay S (2019) HSV‐1 single‐cell analysis reveals the activation of anti‐viral and developmental programs in distinct sub‐populations. eLife 8: e46339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dremel SE, DeLuca NA (2019a) Genome replication affects transcription factor binding mediating the cascade of herpes simplex virus transcription. Proc Natl Acad Sci U S A 116: 3734–3739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dremel SE, DeLuca NA (2019b) Herpes simplex viral nucleoprotein creates a competitive transcriptional environment facilitating robust viral transcription and host shut off. eLife 8: e51109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du T, Zhou G, Roizman B (2011) HSV‐1 gene expression from reactivated ganglia is disordered and concurrent with suppression of latency‐associated transcript and miRNAs. Proc Natl Acad Sci U S A 108: 18820–18824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Femino AM, Fay FS, Fogarty K, Singer RH (1998) Visualization of single RNA transcripts in situ. Science 280: 585–590 [DOI] [PubMed] [Google Scholar]
- Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M et al (2015) MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single‐cell RNA sequencing data. Genome Biol 16: 278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, Scott G, Steffen D, Worley KC, Burch PE et al (2004) Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 428: 493–521 [DOI] [PubMed] [Google Scholar]
- Honess RW, Watson DH (1977) Herpes simplex virus resistance and sensitivity to phosphonoacetic acid. J Virol 21: 584–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu HL, Shiflett LA, Kobayashi M, Chao MV, Wilson AC, Mohr I, Huang TT (2019) TOP2β‐dependent nuclear DNA damage shapes extracellular growth factor responses via dynamic AKT phosphorylation to control virus latency. Mol Cell 74: 466–480.e464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu HL, Srinivas KP, Mohr I, Huang TT, Wilson AC (2020) Using primary SCG neuron cultures to study molecular determinants of HSV‐1 latency and reactivation. Methods Mol Biol 2060: 263–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PC, Roizman B (1979) Regulation of herpesvirus macromolecular synthesis. VIII. The transcription program consists of three phases during which both extent of transcription and accumulation of RNA in the cytoplasm are regulated. J Virol 31: 299–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Mandarino A, Chao MV, Mohr I, Wilson AC (2012) Transient reversal of episome silencing precedes VP16‐dependent transcription during reactivation of latent HSV‐1 in neurons. PLoS Pathog 8: e1002540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinker GS, Greenwald AC, Tal R, Orlova Z, Cuoco MS, McFarland JM, Warren A, Rodman C, Roth JA, Bender SA et al (2020) Pan‐cancer single‐cell RNA‐seq identifies recurring programs of cellular heterogeneity. Nat Genet 52: 1208–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Wilson AC, Chao MV, Mohr I (2012) Control of viral latency in neurons by axonal mTOR signaling and the 4E‐BP translation repressor. Genes Dev 26: 1527–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahav G, Rosenfeld N, Sigal A, Geva‐Zatorsky N, Levine AJ, Elowitz MB, Alon U (2004) Dynamics of the p53‐Mdm2 feedback loop in individual cells. Nat Genet 36: 147–150 [DOI] [PubMed] [Google Scholar]
- Li J, Labbadia J, Morimoto RI (2017) Rethinking HSF1 in stress, development, and organismal health. Trends Cell Biol 27: 895–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linderman JA, Kobayashi M, Rayannavar V, Fak JJ, Darnell RB, Chao MV, Wilson AC, Mohr I (2017) Immune escape via a transient gene expression program enables productive replication of a latent pathogen. Cell Rep 18: 1312–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangold CA, Szpara ML (2019) Persistent infection with herpes simplex virus 1 and Alzheimer's disease‐a call to study how variability in both virus and host may impact disease. Viruses 11: 966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manska S, Octaviano R, Rossetto CC (2020) 5‐Ethynyl‐2'‐deoxycytidine and 5‐ethynyl‐2'‐deoxyuridine are differentially incorporated in cells infected with HSV‐1, HCMV, and KSHV viruses. J Biol Chem 295: 5871–5890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao JC, Robishaw EE (1975) Mode of inhibition of herpes simplex virus DNA polymerase by phosphonoacetate. Biochemistry 14: 5475–5479 [DOI] [PubMed] [Google Scholar]
- Mayer C, Hafemeister C, Bandler RC, Machold R, Batista Brito R, Jaglin X, Allaway K, Butler A, Fishell G, Satija R (2018) Developmental diversification of cortical inhibitory interneurons. Nature 555: 457–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neef DW, Turski ML, Thiele DJ (2010) Modulation of heat shock transcription factor 1 as a therapeutic target for small molecule intervention in neurodegenerative disease. PLoS Biol 8: e1000291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan D, Barrozo ER, Bloom DC (2017) HSV1 latent transcription and non‐coding RNA: a critical retrospective. J Neuroimmunol 308: 65–101 [DOI] [PubMed] [Google Scholar]
- Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S (2008) Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5: 877–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter ER, Dias JK, Gilbert JE, Atherton SS (2009) Distribution of herpes simplex virus type 1 and varicella zoster virus in ganglia of the human head and neck. J Infect Dis 200: 1901–1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell NM (1997) Comprehensive quantification of herpes simplex virus latency at the single‐cell level. J Virol 71: 5423–5431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell NM, Thompson RL (1992) Rapid in vivo reactivation of herpes simplex virus in latently infected murine ganglionic neurons after transient hyperthermia. J Virol 66: 2150–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell NM, Thompson RL 2016) Herpes simplex virus and the lexicon of latency and reactivation: a call for defining terms and building an integrated collective framework. F1000Res 5: 2038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaulian E, Karin M (1999) Stress‐induced JNK activation is independent of Gadd45 induction. J Biol Chem 274: 29595–29598 [DOI] [PubMed] [Google Scholar]
- Shnayder M, Nachshon A, Rozman B, Bernshtein B, Lavi M, Fein N, Poole E, Avdic S, Blyth E, Gottlieb D et al (2020) Single cell analysis reveals human cytomegalovirus drives latently infected cells towards an anergic‐like monocyte state. eLife 9: e52168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, Satija R (2019) Comprehensive integration of single‐cell data. Cell 177: 1888–1902.e1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbington MJT, Rozenblatt‐Rosen O, Regev A, Teichmann SA (2017) Single‐cell transcriptomics to explore the immune system in health and disease. Science 358: 58–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddeo B, Esclatine A, Roizman B (2002) The patterns of accumulation of cellular RNAs in cells infected with a wild‐type and a mutant herpes simplex virus 1 lacking the virion host shutoff gene. Proc Natl Acad Sci U S A 99: 17031–17036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thellman NM, Triezenberg SJ (2017) Herpes simplex virus establishment, maintenance, and reactivation: in vitro modeling of latency. Pathogens 6: 28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda T, Kohama Y, Kuge A, Kido E, Sakurai H (2017) GADD45 family proteins suppress JNK signaling by targeting MKK7. Arch Biochem Biophys 635: 1–7 [DOI] [PubMed] [Google Scholar]
- Wagner EK, Bloom DC (1997) Experimental investigation of herpes simplex virus latency. Clin Microbiol Rev 10: 419–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Lau TY, Morales M, Mont EK, Straus SE (2005) Laser‐capture microdissection: refining estimates of the quantity and distribution of latent herpes simplex virus 1 and varicella‐zoster virus DNA in human trigeminal Ganglia at the single‐cell level. J Virol 79: 14079–14087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Gorospe M, Holbrook NJ (1999) gadd45 is not required for activation of c‐Jun N‐terminal kinase or p38 during acute stress. J Biol Chem 274: 29599–29602 [DOI] [PubMed] [Google Scholar]
- Warren KG, Brown SM, Wroblewska Z, Gilden D, Koprowski H, Subak‐Sharpe J (1978) Isolation of latent herpes simplex virus from the superior cervical and vagus ganglions of human beings. N Engl J Med 298: 1068–1069 [DOI] [PubMed] [Google Scholar]
- Warren‐Gash C, Forbes H, Breuer J (2017) Varicella and herpes zoster vaccine development: lessons learned. Expert Rev Vaccines 16: 1191–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox CL, Johnson EM (1988) Characterization of nerve growth factor‐dependent herpes simplex virus latency in neurons in vitro . J Virol 62: 393–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AC, Mohr I (2012) A cultured affair: HSV latency and reactivation in neurons. Trends Microbiol 20: 604–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyler E, Franke V, Menegatti J, Kocks C, Boltengagen A, Praktiknjo S, Walch‐Rückheim B, Bosse J, Rajewsky N, Grässer F et al (2019) Single‐cell RNA‐sequencing of herpes simplex virus 1‐infected cells connects NRF2 activation to an antiviral program. Nat Commun 10: 4878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon YJ, Kim JA, Shin KD, Shin DS, Han YM, Lee YJ, Lee JS, Kwon BM, Han DC (2011) KRIBB11 inhibits HSP70 synthesis through inhibition of heat shock factor 1 function by impairing the recruitment of positive transcription elongation factor b to the hsp70 promoter. J Biol Chem 286: 1737–1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zeijl M, Fairhurst J, Jones TR, Vernon SK, Morin J, LaRocque J, Feld B, O'Hara B, Bloom JD, Johann SV (2000) Novel class of thiourea compounds that inhibit herpes simplex virus type 1 DNA cleavage and encapsidation: resistance maps to the UL6 gene. J Virol 74: 9054–9061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Zhu L, Wijesekera N, Jones C (2020) Specific Akt family members impair stress‐mediated transactivation of viral promoters and enhance neuronal differentiation: important functions for maintaining latency. J Virol 94: e00901‐20 [DOI] [PMC free article] [PubMed] [Google Scholar]