

Abstract
A proteomics tool capable of identifying single proteins would be important for cell biology research and applications. Here, we demonstrate a nanopore-based single-molecule peptide reader sensitive to single-amino-acid substitutions within individual peptides. A DNA-peptide conjugate was pulled through the biological nanopore MspA by the DNA helicase Hel308. Reading the ion current signal through the nanopore enabled discrimination of single-amino-acid substitutions in single reads. Molecular dynamics simulations showed these signals to result from size exclusion and pore binding. We also demonstrate the capability to ‘rewind’ peptide reads, obtaining numerous independent reads of the same molecule, yielding an error rate <10−6 in single amino acid variant identification. These proof-of-concept experiments constitute a promising basis for the development of a single-molecule protein fingerprinting and analysis technology.
One-sentence summary:
This paper presents proof-of-concept experiments and simulations of a nanopore-based approach for linearly reading individual peptides with sensitivity to single amino acid substitutions.
Genetic sequences are a key source of information about protein primary sequence. However, because they do not directly encode information about protein abundance or about post-translational modification and splicing of proteins, neither the DNA genome nor the RNA transcriptome fully describe the protein phenotype. A robust method for directly identifying proteins and detecting post translational modifications at the single-molecule level would greatly benefit proteomics research(2), enabling quantification of low-abundance proteins as well as distributions and correlations of post-translational modifications (PTMs), all at a single-cell level. Here, we provide proof-of-concept data for a nanopore-based approach that can discriminate single peptides at single-amino-acid sensitivity with high fidelity and potential for high throughput. Although it is not presently capable of de novo protein sequencing, this nanopore peptide reader provides site-specific information about the peptide’s primary sequence that may find applications in single-molecule protein fingerprinting and variant identification.
Recently, biological nanopores have been used as the basis of a single-molecule DNA sequencing technology(3) that is capable of long reads and detection of epigenetic markers in a portable platform with minimal cost(4). In such experiments, single-stranded DNA is slowly moved step-by-step through a protein nanopore embedded in a thin membrane, partially blocking an electrical current carried by ions through the nanopore. The DNA stepping is accomplished using a DNA-translocating motor enzyme which moves DNA through the pore in discrete steps, yielding a series of steps in the ion current. Each ion current level characterizes the bases residing in the pore at that step, and the sequence of levels can be decoded into the DNA base sequence.
It has been hypothesized that nanopores can also be used for protein fingerprinting or sequencing(5, 6). Methods in which small peptide fragments freely translocate through a pore have shown sensitivity to single amino acids(7–9), but lack a method for determining the order of amino acids and reconstructing the sequence of single proteins. Using a ClpX protein unfoldase to pull a peptide through a nanopore yielded signals that effectively distinguished between different peptides(10), but these reads were difficult to interpret, in part due to the irregular stepping behavior of ClpX(11). Here, we instead applied the precise stepwise control of a DNA-translocating motor(12–14) to pull a peptide through a nanopore, similarly to simultaneous work by Yan et al(15) but presenting several key advances: the use of a helicase that pulls the polymer through MspA in smaller, half-nucleotide steps, the ability to identify single-amino-acid substitutions, and the capability to obtain high-fidelity signals by re-reading the same single molecule multiple times.
We developed a system in which a DNA-peptide conjugate is pulled through a biological nanopore by a helicase that is walking on the DNA section (Fig. 1). The conjugate strand consisted of an 80-nucleotide DNA strand that was covalently linked to a 26-amino-acid synthetic peptide by a DBCO click linker on the 5’ end of the DNA connecting to an azide modification at the C-terminus of the peptide (Supplemental Text §1, Fig. S1). A negatively charged peptide sequence of mostly aspartic acid (D) and glutamic acid (E) residues was chosen so that the electrophoretic force assisted in pulling the peptide into the pore. We used the mutant nanopore M2 MspA(16) with a cup-like shape that separates the helicase by ~10 nm from the constriction of the pore where the blockage of ion current occurs (17). For the DNA-translocating motor enzyme, we used Hel308 DNA helicase (i) because it pulls single-stranded DNA through MspA in half-nucleotide ~0.33 nm observable steps(14), which are close to single-amino acid steps, (ii) because it is a stable and processive helicase that tolerates high salt concentrations(17), and (iii) its >50 pN pulling force(17) is likely to denature any secondary structure in target peptides.
Fig. 1: Reading peptides with a nanopore.
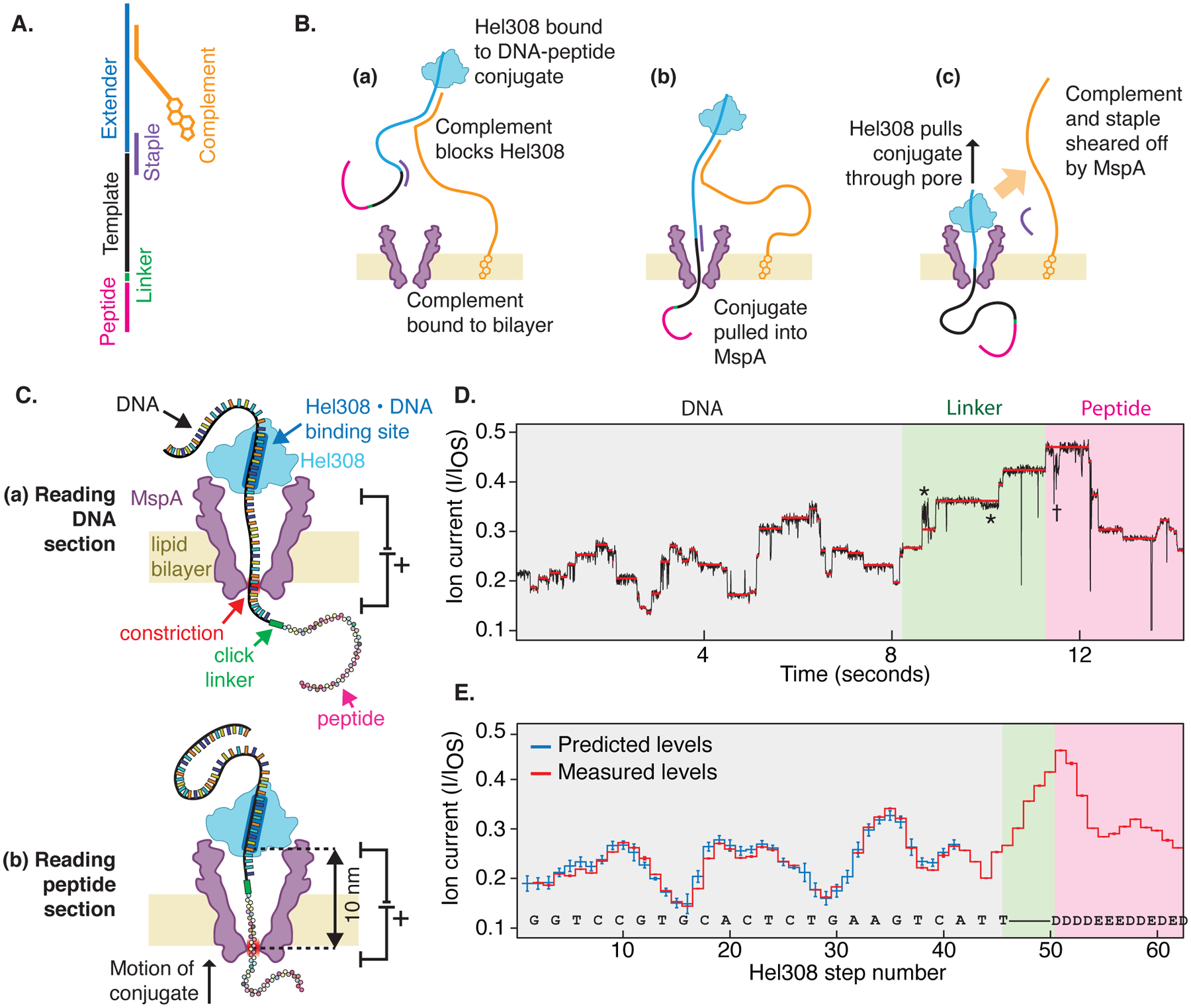
(A) The DNA-peptide conjugate consists of a peptide (pink) attached via a click linker (green) to an ssDNA strand (black). This DNA-peptide conjugate is extended with a typical nanopore adaptor comprised of an extender that acts as a site for helicase loading (blue) and a complementary oligo with a 3’ cholesterol modification (gold). (B) The cholesterol associates with the bilayer as shown in (a), increasing the concentration of analyte near the pore. The complementary oligo blocks the helicase, until it is pulled into the pore (b), causing the complementary strand to be sheared off (c), whereupon the helicase starts to step along DNA. (C) As the helicase walks along the DNA, it pulls it up through the pore, resulting in (a) a read of the DNA portion followed by (b) a read of the attached peptide. (D) Typical nanopore read of a DNA-peptide conjugate (black), displaying step-like ion currents (identified in red). The asterisks * indicate a spurious level not observed in most reads and therefore omitted from further analysis. The dagger † indicates a helicase backstep. (E) Consensus sequence of ion current steps (red), which for the DNA section is closely matched by the predicted DNA sequence (blue). The linker and peptide sections are identified by counting half-nucleotide steps over the known structural length of the linker. Error bars in the measured ion current levels are errors in the mean value, often too small to see. Error bars in the prediction are standard deviations of the ion current levels that were used to build the predictive map in previous work(19).
We found that, similarly to nanopore reads of DNA, ratcheting a peptide through the nanopore generated a distinct step-like pattern in the ion current (Fig. 1C). Durations of ion current steps varied from read to read, but the sequence of levels was highly reproducible (Fig. S2). The progression of ion current steps was accurately identified using custom software (Supplemental Text §2, Fig. S3) and further analysis was performed on the sequence of the median values of ion current for each step (Fig. 1D).
This sequence of ion current levels first closely tracked the sequence expected for the template strand of DNA, which can be predicted using a DNA-sequence-to-ion-current map developed previously(18, 19) (Supplemental Text §3). After the end of the DNA crossed to the cis side of MspA’s constriction, we continued to observe stepping over the linker (a length of ~2 nm, or six Hel308 steps), and subsequently over the peptide. The stepping of the peptide through the MspA constriction produced distinguishable ion current steps, much like those from DNA, but with a higher average ion current. While individual reads may contain a varying number of steps due to helicase backstepping and errors in step segmentation, we identified these features by cross-comparison of several independent reads, producing a “consensus” ion current sequence free of helicase mis-steps or step-segmentation errors (Supplemental Text §4). By counting the steps in these consensus sequence traces, we determined the parts of the traces that corresponded to the linker (the first six steps after the DNA) and the peptide (all steps thereafter) in the MspA constriction. We confirmed this analysis by altering the peptide sequence at a selected site and observing the location of the resulting change in the ion current stepping sequence, as discussed below. We restricted further analysis to reads containing both DNA and peptide sections (Supplemental Text §5, Fig. S4).
Our approach allowed us to discriminate peptide variants that differed by only a single amino acid. We obtained reads (N=211) of three different DNA-peptides in nineteen different pores, where the peptide sequences consisted of a mixture of negatively D and E residues, with a single variation, i.e., D, glycine (G), or tryptophan (W), placed four amino acids away from the C-terminus that connects to the linker (Table S1 for full sequences). The three variants showed a reproducible difference at the site of the substituted amino acid, which could be seen by comparing the consensus sequences of ion current levels (Figs. 2A and B). As is typical of nanopore experiments, a single-site variation was found to affect several ion current steps, because an “8-mer” of amino acids around the pore constriction of MspA affect the ion current blockage level (12, 18) due to the finite constriction height and stochastic displacements of the strand up and down through the nanopore(20). The center of the differing region in the ion current sequence was at the expected site: about 10 helicase steps away from the end of the DNA section (6 half-nucleotide steps for the linker and 4 more along the peptide to the variant site). The signals varied by several standard deviations over multiple sequential levels, demonstrating that variations as small as a single-amino-acid substitution could be resolved. The differences of the ion currents for the W- and G-substituted variants from the D-substituted variant (Fig. 2B) showed an interesting behavior: when G, which has merely a hydrogen atom as a side chain, occupied the nanopore constriction, we saw higher ion current levels, as expected from a smaller amino acid volume. But when the bulky W variant moved through the constriction, the ion current first decreased and then, counterintuitively, increased relative to the medium-sized D variant.
Fig. 2: Detection of single amino acid substitutions in single peptides.
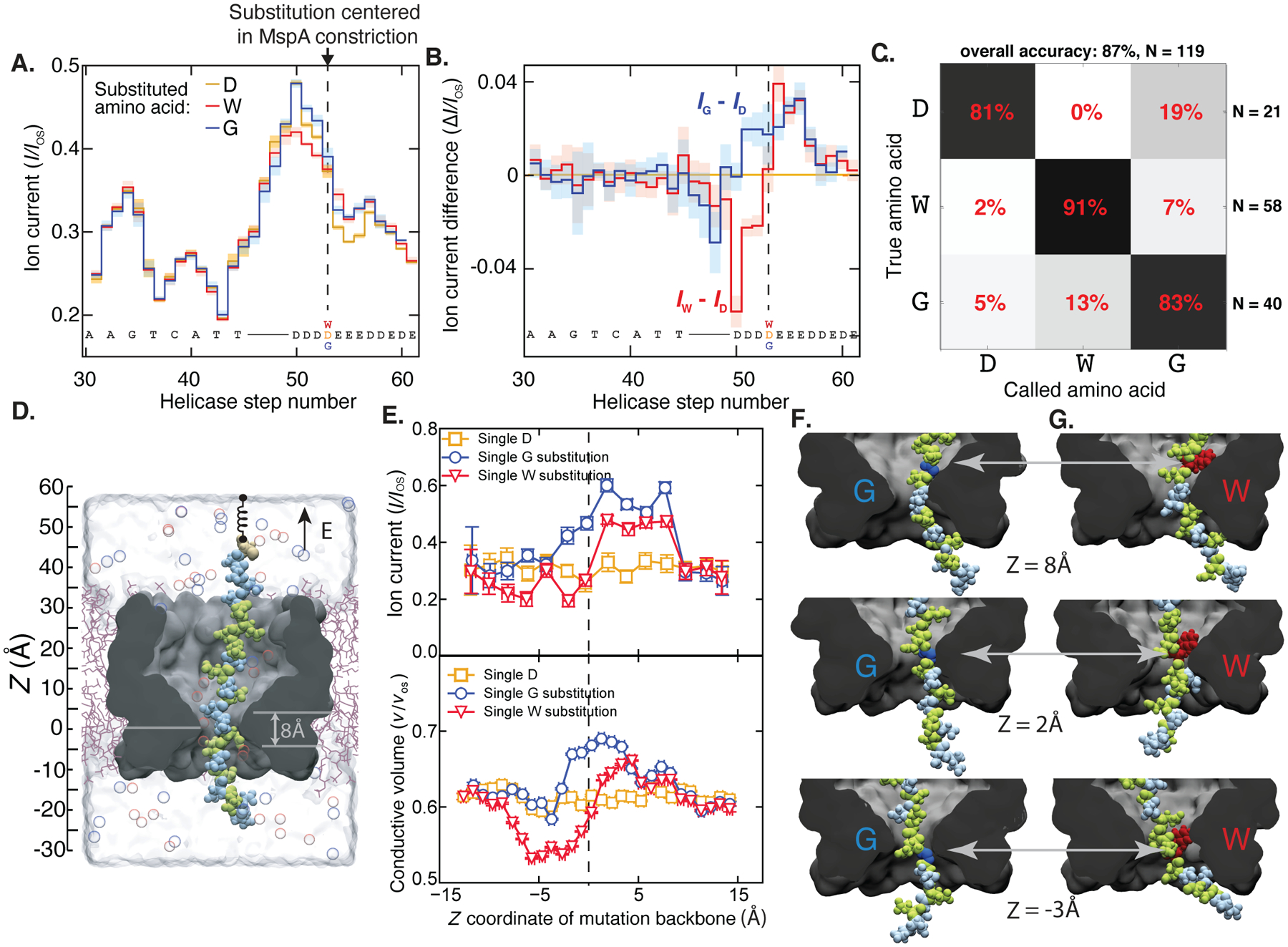
(A) Consensus ion current sequences for each of the three measured variants (D, gold; W, red; G, blue), which differ significantly at the site of the amino acid substitution. (B) Difference in ion current between the W (red) and G (blue) variants and the D variant. Error bars are standard deviations. (C) Confusion matrix showing error modes of a blind classifier in identifying variants of reads, demonstrating an 87% single-read accuracy. (D) All-atom model where a reduced-length MspA pore (grey) confines a polypeptide chain (Glu: green, Asp: light blue; Cys: beige). The top end of the peptide is anchored using a harmonic spring potential, representing the action of the helicase at the rim of a full-length MspA. Water and ions are shown as semitransparent surface and spheres, respectively. (E) Top: Ionic current in MspA constriction versus z coordinate of the mutated residue backbone from MD simulations. Bottom: Fraction of nanopore construction volume available for ion transport. Vertical and horizontal error bars denote standard errors and standard deviations, respectively. (G,H) Representative molecular configurations observed in MD simulations of peptide variants. Glycine and tryptophane residues are shown in dark blue and red, respectively. Significant peptide/pore surface interactions are observed.
To understand the origin of these patterns, we performed all-atom molecular dynamics simulations measuring the ion current with peptide variants at varying positions within the MspA constriction. In a typical simulation, a polypeptide chain was threaded through a reduced-length model of MspA nanopore that was embedded in a lipid bilayer and surrounded by 0.4M KCl electrolyte (Fig. 2C). Peptides with either one W or G substitution in a mixed D/E sequence were examined under a +200 mV bias at various locations relative to the MspA constriction (see Supplemental Text §6 and Figs. S5–S8 for details). Patterns of ionic current blockades resulted in Fig. 2E (top panel), matching the counterintuitive blockade current patterns that were experimentally measured for G and W substitutions (cf. Fig. 2A,B). Furthermore, the ion current correlated with the nanopore constriction volume that was available for ion transport near the pore mouth, Fig. 2E (bottom panel), with the latter quantity being more accurately characterized by the all-atom MD method (20). In the case of a G residue, its upward motion was accompanied by an increase of the nanopore volume (Fig. 2E, bottom), that subsided as the residue left the nanopore constriction (Fig. 2F), in sync with the blockade current (Fig. 2E, top). A W residue, however, reduced the nanopore constriction volume when it was located below the constriction (Fig. 2E, top), but increased the volume at and above the constriction. The latter counterintuitive effect could be traced back to a binding of the W side chain to the nanopore surface above the constriction (Fig. 2G). Thus, a glycine substitution merely increases the nanopore volume as the residue passes through the constriction, whereas the tryptophan residue decreases the volume when its side chain enters the constriction, and subsequently increases the volume when its side chain binds to the inner nanopore surface (Fig. S9).
To quantitatively assess the distinguishability of peptide variants, we computed a so-called confusion matrix (Fig. 2D). Using a hidden Markov model, we quantified the relative likelihoods of the alignments to the three consensus sequences for 119 reads withheld from the consensus sequence generation, finding that we could identify the correct variant with an average of 87% accuracy (Supplemental Text §7). This high rate of correct single substitution identification compares favorably to early nanopore experiments, that identified single-nucleotide variants with significantly lower accuracy(18). Still, the limited single-read accuracy is an ongoing challenge in developing nanopore sequence analysis approaches, requiring the implementation of strategies to increase sequencing fidelity to acceptable levels (19, 21). The largest error modes in nanopore reads are due to random effects as enzymes step stochastically both forwards and backwards, and sometimes step too quickly to be clearly resolved, resulting in incorrect step identifications. In DNA sequencers, this random error is typically addressed by obtaining 20x coverage or more, averaging many independent reads of different molecules. However, for a truly single-molecule technology, single-read accuracy is essential.
The identification fidelity of our nanopore protein reader can be greatly increased by obtaining many independent re-readings of the same individual molecule with a succession of controlling helicases, eliminating the random errors that lead to inaccuracies in nanopore reads. At a very high concentration of helicase, on the order of 1 μM, the DNA in the pore nearly always had a second helicase queued up behind the one controlling its motion (Fig. 3A)(22). When the first helicase reached the linker at the end of the DNA section, it could no longer process the molecule and subsequently fell off. The DNA-peptide conjugate was then immediately pulled back into the nanopore such that the queued helicase, which was still bound to the DNA, took control as the new DNA-pulling enzyme. This ‘rewound’ the system and initiated a new and independent read of the peptide. The numbers of re-reads on the same single peptide can be very large: Fig. 3A shows an example of a raw data trace with 117 re-reads on a single peptide containing the G-substitution. This event was purposefully ended by the reversal of voltage to eject the DNA-peptide conjugate from the pore. We observed a typical rewinding distance of approximately 17 helicase steps, commensurate with a rewinding by a distance of ~17 amino acids, a number that is consistent with the ~9 DNA bases that are bound within the controlling helicase(17). Of the 117 re-reads in Fig. 3B, 45 re-reads stepped back far enough to provide a re-read of the variant site.
Fig. 3: Re-reading of a single peptide.
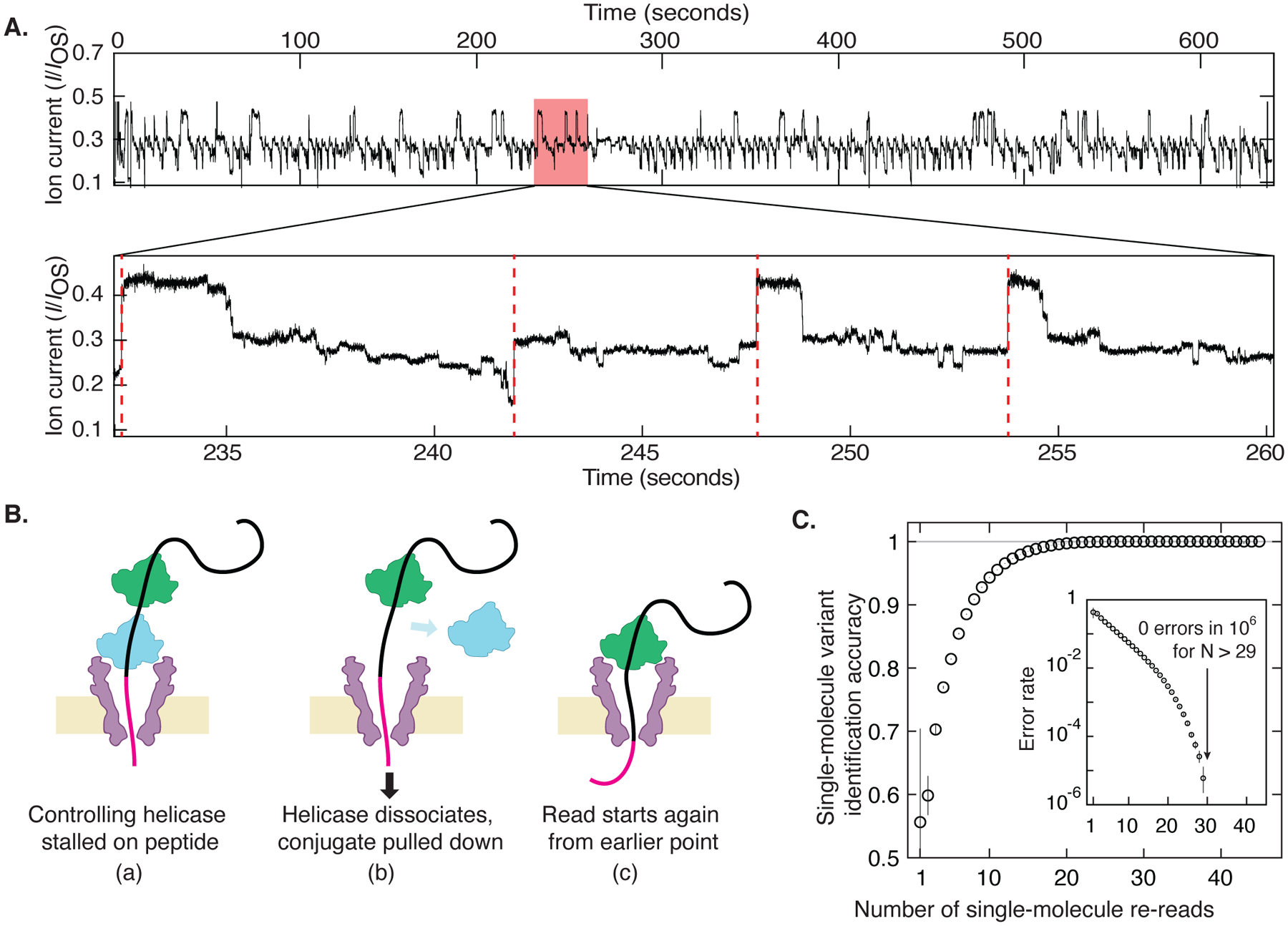
(A) Highly repetitive ion current signal corresponding to numerous re-reads of the same section of an individual peptide (in this case, the G-substituted variant). The expanded plot below shows a region that contains four rewinding events (red dashed lines), where the trace jumps back to level 52 ± 2 of the consensus displayed in Fig. 2A. (B) Re-reading is facilitated by helicase queueing, where (a) a second helicase binds behind the primary helicase that controls the DNA-peptide conjugate, re-reading starts when (b) the primary helicase dissociates, and (c) the secondary one becomes the primary helicase that drives a new round of reading. (C) By using information from multiple re-reads of the same peptide, the identification accuracy can be raised to very high levels of fidelity. These results indicate that with sufficient numbers of re-reads, random error can be eliminated and single-molecule error rate can be pushed lower than 1 in 106 even with poor single-pass accuracy. Inset is a logarithmic plot of the error rate = 1 - accuracy.
We observed significant improvement of the read accuracy with an increasing number of re-reads (Fig. 3C). To quantify the increase in the accuracy of the readings as a function of the number of re-readings, we randomly chose subsets of the 45 measured re-reads and computed the identification accuracy using N re-reads as the fraction of subsets containing N re-reads that yielded the correct consensus identification (Supplemental Text §8). Even when single reads were limited to as low as ~50% identification accuracy due to only partial coverage of the variant site, the re-reading method allowed single molecules to be identified at high levels of confidence. As the inset to Fig.3C shows, the error rate decreased with the number of re-reads, yielding an undetectably low error rate (< 1 in 106) when using more than ~30 re-reads of an individual peptide. Analysis on re-read traces from other variants yielded similar results (Fig. S10).
The method described here provides an approach for reading single proteins with sensitivity to single-amino-acid changes, which is particularly powerful because of the re-reading mode of operation that reduces the stochastic error. Transforming this into a technology capable of de novo protein sequencing remains a substantial challenge. With any of 20 amino acids at each position along the protein sequence and a read-head width(18) of ~8 amino acids, the number of measurements required to build an ion-current-to-amino-acid map is impractically large. However, many proteomics applications do not require de novo sequencing, but instead concern other forms of sequence analysis that rely on a priori knowledge of candidate sequences before decoding. These include identifying or “fingerprinting” proteins even in heterogeneous mixtures, mapping post-translational modifications, and measurements of small samples, which all involve comparing single-molecule measurements to reference signals of known proteins and interesting variants.
Our methodology has several limitations, but these may be addressed experimentally. While the pore is capable of translocating heterogeneously charged peptides with neutral polar, nonpolar, negative, and positive amino acids (Supplemental Text §9; sample reads shown in Fig. S11), highly positively charged peptides may not efficiently be translocated through the pore. Fortunately, analysis of the human proteome reveals that negatively charged stretches of protein sequence are more common than positively charged stretches(23), particularly in alkaline pH conditions like those used in our experiments. If needed, the MspA pore can be engineered to provide stronger electro-osmotic forces, which can exceed electrophoretic forces and translocate analytes regardless of charge(8, 24). The read length intrinsic to the technique, approximately 25 amino acids depending on the length of the DNA-peptide linker, does allow application of this method to many biologically relevant short peptides, such as 8–12 amino acid MHC-binding peptides(25). Additionally, this finite read length still represents an improvement over the <10 amino acid long peptide fragments used in mass spectrometry(26), and protein fragmentation and shotgun sequencing methods similar to those used in traditional protein sequencing can naturally be applied to this new technique. Technical modifications such as using a variable-voltage control scheme(19) have been shown to improve the accuracy of DNA sequencing, and the physical principle of this is equally applicable to peptide sequencing (Supplemental Text §10 and Fig. S12).
Reads of DNA-peptide conjugates like those presented here could be measured in high throughput with any existing commercially available nanopore sequencing hardware capable of accommodating MspA (e.g. the commercial MinION system) without requiring any re-engineering of the device, changing only the sample preparation and data analysis. Furthermore, our methodology retains the features that enabled the success of nanopore DNA sequencing: low overhead cost, physical rather than chemical sensitivity to small changes in single molecules, and the flexibility to be re-engineered to target specific applications. Overall, our findings comprise a promising first step towards a low-cost method capable of single-cell proteomics at the ultimate limit of sensitivity to concentration, with a wide range of applications in both fundamental biology and the clinic.
Supplementary Material
Acknowledgements
We thank Prof. Jens Gundlach and his lab members at University of Washington for providing the MspA nanopore and for sharing key pieces of software, and we thank Foteini Mentzou and Xin Shi for assistance with data collection, Eli van der Sluis for Hel308 purification, and Jaco van der Torre for his helpful advice on DNA construct preparation. We also acknowledge supercomputer time on the Blue Waters at UIUC, Expanse at UCSD and Frontera at TACC.
Funding
Dutch Research Council (NWO) NWO-I680 (SMPS) (CD)
Dutch Research Council (NWO) / Ministry of Education, Culture and Science (OCW) Gravitation programs NanoFront (CD)
European Research Council Advanced Grant 883684 (CD)
European Commission Marie Skłodowska-Curie action Individual Fellowship 897672 (HB)
European Molecular Biology Organization Short-Term Fellowship 8968 (AK)
National Institutes of Health grant R21-HG011741 (AA)
Extreme Science and Engineering Discovery environment allocation MCA05S028 (AA)
Leadership Resource Allocation MCB20012 on Frontera of the Texas Advanced Computing Center (AA)
Footnotes
Competing Interests
TU Delft has filed a patent application (PCT/NL2020/050814) on technologies described herein, with H.B. and C.D. listed as inventors.
Supplementary Materials
The PDF file includes:
Data and Materials Availability
All data and custom code used in this paper are available for download online(1).
References and Notes
- 1.Brinkerhoff H, Kang ASW, Liu J, Aksimentiev A, Dekker C. Code and Data for “Multiple rereads of single proteins at single-amino-acid resolution using nanopores” [Data set]. Zenodo (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alfaro JA et al. , The emerging landscape of single-molecule protein sequencing technologies. Nature Methods 18, 604–617 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kasianowicz JJ, Brandin E, Branton D, Deamer DW, Characterization of individual polynucleotide molecules using a membrane channel. Proceedings of the National Academy of Sciences 93, 13770 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deamer D, Akeson M, Branton D, Three decades of nanopore sequencing. Nature Biotechnology 34, 518–524 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nivala J, Marks DB, Akeson M, Unfoldase-mediated protein translocation through an α-hemolysin nanopore. Nature Biotechnology 31, 247 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodriguez-Larrea D, Bayley H, Multistep protein unfolding during nanopore translocation. Nature Nanotechnology 8, 288–295 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Piguet F et al. , Identification of single amino acid differences in uniformly charged homopolymeric peptides with aerolysin nanopore. Nature Communications 9, 966 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Restrepo-Pérez L, Wong CH, Maglia G, Dekker C, Joo C, Label-Free Detection of Post-translational Modifications with a Nanopore. Nano letters 19, 7957–7964 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ouldali H et al. , Electrical recognition of the twenty proteinogenic amino acids using an aerolysin nanopore. Nature Biotechnology 38, 176–181 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nivala J, Mulroney L, Li G, Schreiber J, Akeson M, Discrimination among Protein Variants Using an Unfoldase-Coupled Nanopore. Acs Nano 8, 12365–12375 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Cordova Juan C. et al. , Stochastic but Highly Coordinated Protein Unfolding and Translocation by the ClpXP Proteolytic Machine. Cell 158, 647–658 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Manrao EA et al. , Reading DNA at single-nucleotide resolution with a mutant MspA nanopore and phi29 DNA polymerase. Nature Biotechnology 30, 349 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cherf GM et al. , Automated forward and reverse ratcheting of DNA in a nanopore at 5-Å precision. Nature Biotechnology 30, 344 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Derrington IM et al. , Subangstrom single-molecule measurements of motor proteins using a nanopore. Nature Biotechnology 33, 1073 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan S et al. , Single Molecule Ratcheting Motion of Peptides in a Mycobacterium smegmatis Porin A (MspA) Nanopore. Nano Lett 21, 6703–6710 (2021). [DOI] [PubMed] [Google Scholar]
- 16.Butler TZ, Pavlenok M, Derrington IM, Niederweis M, Gundlach JH, Single-molecule DNA detection with an engineered MspA protein nanopore. Proceedings of the National Academy of Sciences 105, 20647 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Craig JM et al. , Determining the effects of DNA sequence on Hel308 helicase translocation along single-stranded DNA using nanopore tweezers. Nucleic Acids Research 47, 2506–2513 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laszlo AH et al. , Decoding long nanopore sequencing reads of natural DNA. Nature Biotechnology 32, 829 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noakes MT et al. , Increasing the accuracy of nanopore DNA sequencing using a time-varying cross membrane voltage. Nature Biotechnology 37, 651–656 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhattacharya S, Yoo J, Aksimentiev A, Water Mediates Recognition of DNA Sequence via Ionic Current Blockade in a Biological Nanopore. ACS Nano 10, 4644–4651 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tyler AD et al. , Evaluation of Oxford Nanopore’s MinION Sequencing Device for Microbial Whole Genome Sequencing Applications. Scientific Reports 8, 10931 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rand AC, doctoral thesis. University of California, Santa Cruz, Santa Cruz, CA, United States: (2017). [Google Scholar]
- 23.Requião RD et al. , Protein charge distribution in proteomes and its impact on translation. PLOS Computational Biology 13, e1005549 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoogerheide DP, Gurnev PA, Rostovtseva TK, Bezrukov SM, Mechanism of α-synuclein translocation through a VDAC nanopore revealed by energy landscape modeling of escape time distributions. Nanoscale 9, 183–192 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wieczorek M et al. , Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Frontiers in Immunology 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cox J, Hubner NC, Mann M, How Much Peptide Sequence Information Is Contained in Ion Trap Tandem Mass Spectra? Journal of the American Society for Mass Spectrometry 19, 1813–1820 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Wiggins PA, An information-based approach to change-point analysis with applications to biophysics and cell biology. Biophys J 109, 346–354 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Phillips JC et al. , Scalable molecular dynamics on CPU and GPU architectures with NAMD. The Journal of Chemical Physics 153, 044130 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hart K et al. , Optimization of the CHARMM additive force field for DNA: Improved treatment of the BI/BII conformational equilibrium. J Chem Theory Comput 8, 348–362 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klauda JB et al. , Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B 114, 7830–7843 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML, Comparison of simple potential functions for simulating liquid water. The Journal of Chemical Physics 79, 926–935 (1983). [Google Scholar]
- 32.Beglov D, Roux B, Finite representation of an infinite bulk system: Solvent boundary potential for computer simulations. The Journal of Chemical Physics 100, 9050–9063 (1994). [Google Scholar]
- 33.Yoo J, Aksimentiev A, New tricks for old dogs: improving the accuracy of biomolecular force fields by pair-specific corrections to non-bonded interactions. Physical Chemistry Chemical Physics 20, 8432–8449 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andersen HC, Rattle: A “velocity” version of the shake algorithm for molecular dynamics calculations. Journal of Computational Physics 52, 24–34 (1983). [Google Scholar]
- 35.Miyamoto S, Kollman PA, Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. Journal of Computational Chemistry 13, 952–962 (1992). [Google Scholar]
- 36.Darden T, York D, Pedersen L, Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. The Journal of Chemical Physics 98, 10089–10092 (1993). [Google Scholar]
- 37.Martyna GJ, Tobias DJ, Klein ML, Constant pressure molecular dynamics algorithms. The Journal of Chemical Physics 101, 4177–4189 (1994). [Google Scholar]
- 38.Brünger AT, Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475 (1992). [DOI] [PubMed] [Google Scholar]
- 39.Aksimentiev A, Schulten K, Imaging alpha-hemolysin with molecular dynamics: ionic conductance, osmotic permeability, and the electrostatic potential map. Biophys J 88, 3745–3761 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gumbart J, Khalili-Araghi F, Sotomayor M, Roux B, Constant electric field simulations of the membrane potential illustrated with simple systems. Biochim Biophys Acta 1818, 294–302 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and custom code used in this paper are available for download online(1).