

Abstract
Background
There is a growing interest in using gut commensal bacteria as “next generation” probiotics. However, this approach is still hampered by the fact that there are few or no strains available for specific species that are difficult to cultivate. Our objective was to adapt flow cytometry and cell sorting to be able to detect, separate, isolate, and cultivate new strains of commensal species from fecal material. We focused on the extremely oxygen sensitive (EOS) species Faecalibacterium prausnitzii and the under-represented, health-associated keystone species Christensenella minuta as proof-of-concept.
Results
A BD Influx® cell sorter was equipped with a glovebox that covered the sorting area. This box was flushed with nitrogen to deplete oxygen in the enclosure. Anaerobic conditions were maintained during the whole process, resulting in only minor viability loss during sorting and culture of unstained F. prausnitzii strains ATCC 27766, ATCC 27768, and DSM 17677. We then generated polyclonal antibodies against target species by immunizing rabbits with heat-inactivated bacteria. Two polyclonal antibodies were directed against F. prausnitzii type strains that belong to different phylogroups, whereas one was directed against C. minuta strain DSM 22607. The specificity of the antibodies was demonstrated by sorting and sequencing the stained bacterial fractions from fecal material. In addition, staining solutions including LIVE/DEAD™ BacLight™ Bacterial Viability staining and polyclonal antibodies did not severely impact bacterial viability while allowing discrimination between groups of strains. Finally, we combined these staining strategies as well as additional criteria based on bacterial shape for C. minuta and were able to detect, isolate, and cultivate new F. prausnitzii and C. minuta strains from healthy volunteer’s fecal samples.
Conclusions
Targeted cell-sorting under anaerobic conditions is a promising tool for the study of fecal microbiota. It gives the opportunity to quickly analyze microbial populations, and can be used to sort EOS and/or under-represented strains of interest using specific antibodies, thus opening new avenues for culture experiments.
Video abstract
Supplementary Information
The online version contains supplementary material available at 10.1186/s40168-021-01206-7.
Keywords: Microbiota, Faecalibacterium, Christensenella, Anaerobic, Flow cytometry, Sorting, Culturomics
Background
With the availability of next-generation sequencing technologies that allow high-throughput analysis of the composition and function of complex microbial ecosystems, the field of microbiome research has grown rapidly in recent years, and countless associations have been reported between microbiota composition and specific health conditions. This is especially true for the human gut ecosystem, for which microbial signatures have been associated with metabolic syndrome, inflammatory bowel diseases (IBD), and response to cancer immunotherapy to mention just a few. This offers new fundamental and applied research avenues, with the ultimate goal to develop new, complementary tools for treating these conditions [1–3]. In particular, 16S rRNA gene amplicon or shotgun metagenomic analysis conducted on fecal samples collected from cohorts of patients vs. controls highlighted decreased occurrence of several commensal bacterial species in pathological conditions [4]. There is thus a growing interest in using cultured, well-characterized strains to complement deficiencies in the gut microbiota, referred to as “next-generation probiotics” (NGP) [5].
This is the case for Faecalibacterium prausnitzii, which accounts for about 5‑10% of dominant microbial communities within the healthy gut microbiota [6], and has been associated with a number of favorable outcomes in various pathologies including lower risk of postoperative recurrence of ileal Crohn’s disease [7] and an improved response to immune check point blockers [8, 9]. The phylogeny of F. prausnitzii is complex, comprising at least 3 different phylogroups, and possibly represents several species that remain to be described taxonomically [10–12]. Relative proportions of the different phylogroups in one same individual seem to vary depending on specific disease condition, with phylogroup IIb strains being depleted in Crohn’s disease patients [13, 14]. It has consequently been proposed to use corresponding relative abundances as disease biomarker [15].
Other NGP candidates can be found within the family Christensenellaceae [16]. Relative abundancy of these heritable bacteria is inversely correlated to host body mass index and the type species Christensenella minuta has been demonstrated to reduce weight gain in germ-free mice colonized with fecal microbiota collected from obese individuals [17]. Recently, it has also been reported that C. minuta DSM33407 protected from diet-induced obesity and regulated associated metabolic markers such as glycemia and leptin in a diet-induced obesity mouse model [18].
In this context, and knowing that specific biological properties of gut bacteria, including host beneficial properties, can vary significantly from one strain to another [19], there is a major interest in building collections of different commensal strains of target species of interest identified via NGS studies. However, this approach is still hampered by the fact that retrieving target species from clinical samples (usually fecal material) can be difficult. Extreme oxygen sensitivity (EOS) (F. prausnitzii) or under-representation of the target species in the community (C. minuta) can be important limitations, with the addition of specific nutritional requirements rendering target species difficult to cultivate in synthetic media. Flow cytometry (FCM) coupled with cell-sorting has the potential to circumvent most if not all these limitations. With constantly increasing technological performances, FCM can be used for bacterial or even viral cell populations’ analysis with or without subsequent sorting [20, 21]. With the objective to use FCM and cell-sorting to analyze, sort, and cultivate bacterial species of interest from fecal samples, we adapted a cell sorter and associated workflow to conduct sorting experiments under strictly anaerobic conditions. We then evaluated the impact of sorting as well as non-specific and specific staining methods on the viability of several representative strains of the EOS species F. prausnitzii. To test the feasibility of the specific targeting of bacteria using anaerobic sorting, we isolated new F. prausnitzii strains from fecal samples using antibodies raised against available reference strains. Finally, we applied this method to sort and cultivate novel strains of C. minuta, which is more tolerant to oxygen exposure but usually found at low abundance (in contrast to F. prausnitzii), demonstrating the potential of this approach.
Results
Evaluation of polyclonal antibodies against reference strains of F. prausnitzii and Christensenella spp.
A recent publication suggested that the species F. prausnitzii comprises at least two phylogroups whose contribution to human health might be different [10]. Therefore, we used representatives of both phylogroups to generate polyclonal antibodies: strain DSM 17677 (A2-165, phylogroup IIb) and a mix of the closely related strains ATCC 27766 and ATCC 27768 (phylogroup I). Antibodies raised against the ATCC strains were consistently effective in detecting > 90% of target bacteria. They showed cross-reactivity with strain A2-165 but the intensity of the staining was much lower, thus allowing the definition of a phylogroup-specific gating region (Fig. 1 and Additional files 1 and 3). Staining efficiency was more variable for antibodies directed against strain A2-165, with the proportion of stained bacteria ranging from 50% to > 90% depending on the experiment. These antibodies also slightly reacted with the ATCC strains but here again, a phylogroup-specific gating region could easily be defined (Fig. 1 and Additional files 1 and 3). We did not observe any fluorescence of unstained F. prausnitzii cells when exciting with the Violet 405 (450/50 nm) or Red 670 (670/30 nm) lasers whereas significant auto-fluorescence was observed for the 3 different strains when exciting with the Blue 540/30 nm laser (Additional file 1). However, in this case, the signal was still 1 to 2 log lower than the signal used for gating live cells after staining with SYTO 9 contained in the LIVE/DEAD™ kit (Additional file 3), so auto-fluorescence in that channel is not likely to interfere with the viability staining.
Fig. 1.

Representative staining of F. prausnitzii reference strains. Fresh cultures of the three strains: DSM-17677 (A2-165), ATCC-27766, and ATCC-27768 were analyzed after staining with polyclonal antibodies. The measurements were based on gated events corresponding to the vast majority of bacteria in the suspensions (A). Proportions of bacteria stained with polyclonal antibodies generated with strain A2-165 (B), or with polyclonal antibodies generated with both ATCC strains (C) were then analyzed. These experiments were repeated 3 times
We also generated polyclonal antibodies by injecting rabbits with heat-inactivated bacteria of the publicly available C. minuta strain DSM 22607. These antibodies were then tested against a pure culture of the parental strain and the related species “Christensenella massiliensis” and “Christensenella timonensis.” Nearly all cells within the pure culture of C. minuta were indeed stained (Fig. 2 and Additional files 2 and 3). Limited cross-reactivity was observed with “C. massiliensis,” for which, depending on the experiment, 1.2 to 14.3% of the bacteria were stained by C. minuta antibodies (Fig. 2 and Additional file 2). There was no cross-reactivity with “C. timonensis.”
Fig. 2.

Representative staining of Christensenellaceae reference strains. Fresh cultures of Christensenella minuta DSM 22607, “Christensenella massiliensis” DSM 102344 and “Christensenella timonensis” DSM 102800 were analyzed. The measurements were made only with gated events corresponding to the vast majority of bacteria in the suspensions (A). Proportions of bacteria stained with polyclonal antibodies generated with C. minuta DSM 22607 strain were then analyzed (B). These experiments were repeated 3 times
Impact of staining and anaerobic sorting on F. prausnitzii and C. minuta viability
We then tested the effect of the sorting procedures on viability of the EOS species F. prausnitzii [22] by comparing two conditions: (i) sorting performed under anaerobic conditions, (ii) sorting performed under normal atmosphere [23]. The recovery of unlabeled F. prausnitzii after sorting under anaerobic conditions was approximately 20% for the three tested strains, while no colony could be observed if the sorting was performed under normal atmosphere (Fig. 3).
Fig. 3.

Impact of sorting under anaerobic or atmospheric conditions on F. prausnitzii recovery. Fresh cultures of the 3 F.prausnitzii reference strains were sorted on mGAM plates in anaerobic (left) or atmospheric (right) conditions. Increasing (1 to 1000) amounts of bacteria were sorted and plated on each spot. Colonies were stained with crystal violet for better visualization of tiny colonies (black arrows). Experiments were repeated at least 3 times
We then evaluated the effect of different staining methods (SYTO 9 and propidium iodide in the LIVE/DEAD™ BacLight™ Bacterial Viability Kit and the 4 strain specific antibodies) on cultivability compared to unstained bacteria (see sorting gates in Additional file 3). These experiments showed no significant impact of any of the tested staining on F. prausnitzii cultivability during sorting in anaerobic conditions (Fig. 4). Several colonies were still observed after anaerobic sorting and cultivation of the propidium iodide (PI)-stained fraction, corresponding to approximately 0.1 to 1% of cultivable bacteria in this fraction (Fig. 4). The cultivability of C. minuta was very high, and remained unaffected after LIVE/DEAD™ and antibody staining.
Fig. 4.

Fractions of bacteria recovered in culture after various staining. Fresh cultures of the three F. prausnitzii and C. minuta reference strains were stained with SYTO 9 and propidium iodide, and with specific antibodies and were then sorted in increasing amounts on mGAM plates to calculate the percentage of cultivability. Experiments were performed two times in triplicates
Unstained and pre-immune controls
We did not detect any auto-fluorescence in the Red 670/30 nm channel for samples collected from healthy volunteers (HV) 1 to 4. Additional signal observed when the same samples were stained with the pre-immune fluorescent antibodies was limited, with percentages of stained events ranging from 0.01 to 0.21% (Additional file 4). The signal detected after staining with Alexa Fluor™ 647-antibodies directed against the ATCC strains of F. prausnitzii was clearly distinguishable from the background (Additional file 4, HV1 and HV4). The situation was markedly different in the 450/50 nm channel, with auto-fluorescent events being clearly detected as distinct populations accounting for 0.03 to 1.96% of the events in the “bacteria” gate (Additional file 4). However, samples in which events were detected after staining with Alexa Fluor™ 405-antibodies directed against F. prausnitzii A2-165 displayed populations that could clearly be distinguished from the background auto-fluorescence from unstained bacteria (Additional file 4, HV1 and HV4).
Evaluation of antibodies’ specificity and enrichment rates for fractions sorted after staining with polyclonal antibodies and for the 450 nm-auto-fluorescent fraction
As shown in Fig. 5A, we observed a very substantial enrichment in the sorted fractions, with 91.6% of the 16S rRNA gene amplicon sequences being annotated as F. prausnitzii in the fraction sorted using the antibody directed against strains ATCC 27766 and ATCC 27768, and 75.4% in the fraction sorted using the antibody directed against strain A2-165. The enrichment rates calculated on the basis of the normalized sequences were of the order of 100 for both antibodies and there was no significant enrichment of other zOTUs in the F. prausnitzii-sorted fractions (Fig. 6A and B).
Fig. 5.

Selective enrichment and 16S rRNA gene amplicon analysis of bacterial populations sorted after staining of pooled fecal samples HV2 + HV4 with polyclonal antibodies directed against F. prausnitzii ATCC 27766 + 27768 and F. prausnitzii A2-165 (A), and with polyclonal antibodies directed against C. minuta DSM 22607 (B). Bacteria presenting auto-fluorescence in the 450/50 nm channel after excitation with the 405 nm laser were also sorted and analyzed by sequencing (A). One million bacteria were sorted for each population to be sequenced, and a 100% identity threshold corresponding to the zero radius OTU (zOTU) definition was used to delineate OTUs [24]. These experiments were performed once for F. prausnitzii (A) and once for C. minuta (B)
Fig. 6.

Enrichment rates of specific phylogenetic groups in the populations sorted after staining with the polyclonal antibodies directed against F. prausnitzii ATCC 27766 + ATCC 277668 (A), F. prausnitzii A2-165 (B), C. minuta DSM 22607 (C), and in bacterial population presenting auto-fluorescence in the 450/50 nm channel after excitation with the 405 nm laser (D). Rates were calculated by dividing normalized numbers of sequences affiliated to each zOTU in the sorted fractions (dark gray) by normalized numbers of sequences affiliated to the same zOTU in the raw material (light gray). Only groups with a least 0.05% affiliated sequences in the 16S rRNA gene amplicon repertoire of pooled fecal samples and for which enrichment rate is ≥ 1 were considered, and a maximum of 15 most enriched groups are shown for each antibody. These experiments were performed once for F. prausnitzii and autofluorescent bacteria (A, B, and D), and once for C. minuta (C)
Since auto-fluorescent events were detected in samples collected from HV4 when exciting with the 405 nm laser, we also sorted and then performed 16S rRNA gene amplicon sequencing and analysis of the sorted fraction from HV2+HV4 mix. When excluding zOTUs to which less than 0.05% of the normalized sequences were affiliated in the original fecal material, the most enriched bacteria in the sorted fraction were affiliated to the Methanobacteriaceae (Figs. 5A and 6D). Several zOTUs affiliated to genera Bacteroides, Eubacterium, and Faecalibacterium, and to the Christensenellaceae R-7 group were also enriched by more than 5 times in the sorted fraction compared to the original pooled material (Fig. 6D).
For the fraction sorted from the pool of 2 samples spiked at approx. 2% with the C. minuta collection strain (see Additional file 5 for sorting gates), 71.7% of the sequences were indeed annotated as C. minuta (Fig. 5B). The enrichment rate calculated on the basis of the normalized sequences was of 16.8, and it was of 16.7 for a zOTU affiliated to the genus Faecalibacterium (Fig. 6C), suggesting that these antibodies can potentially cross-react with species other than C. minuta.
Use of polyclonal antibodies for sorting and cultivation of new F. prausnitzii strains from fecal material
Based on the results observed with pure cultures of F. prausnitzii collection strains, we chose to combine LIVE/DEAD™ staining with specific polyclonal antibodies to perform sorting and cultivation experiments from frozen fecal material. In this series of experiments with fecal samples collected from 5 healthy volunteers, Live (i.e., SYTO 9-positive and PI-negative) bacteria ranged from 27.5 to 36.5%. Similarly to what was observed in preliminary experiments, auto-fluorescent bacteria accounted for 0.22 to 1.18% of the events for 3 of the 5 volunteers when exciting with the 405 nm laser (Fig. 7 and Table 1). Interestingly, these 3 volunteers were also those for which OTUs affiliated to the Methanobacteriaceae family were detected in the 16S rRNA gene amplicon repertoire (Fig. 8). As for preliminary experiments, we did not observe any significant auto-fluorescence when exciting with the Red 640 nm laser (data not shown) and there was only little staining with the pre-immune antibodies conjugated with Alexa Fluor™ 647 (Table 1).
Fig. 7.

Flow cytometry analysis of fecal microbiota collected from 5 healthy volunteers (HV5 to HV9). A first gate was defined to select events that likely correspond to bacteria (A). The second gate (B) was defined to exclude events that were auto-fluorescent in the 450/50 nm channel after excitation with the 405 nm laser. The third gate (C) was defined to select bacteria considered as live after staining with the LIVE/DEAD™ BacLight™ Bacterial Viability Kit. Among them, 240 events stained with anti-F. prausnitzii A2-165 (D) or anti-F. prausnitzii ATCC 27766 + ATCC 27768 (E) antibodies were selected for sorting and cultivation. These experiments were performed once for each of the 5 healthy volunteers
Table 1.
Results of FCM analysis, sorting and cultivation when targeting F. prausnitzii in samples HV5 to HV9

Fig. 8.

16S rRNA gene amplicons analysis of fecal samples used to sort and cultivate F. prausnitzii (HV5 to HV9) or C. minuta (HV10 to HV17). Only the genera with which the species we focused on in this study are affiliated are shown for clarity
The percentage of bacteria individually stained with the anti-F. prausnitzii A2-165 antibodies represented up to 0.69% of total bacteria for HV5, falling to 0.38% when taking into account only live bacteria (Table 1). Percentages were higher for anti-F. prausnitzii ATCC 27766 +27768 antibodies, with up to 3.04% of total and 1.29% of live bacteria being stained for HV7.
For every fecal sample, 240 events stained with anti-F. prausnitzii A2-165 or anti-F. prausnitzii ATCC 27766 + ATCC 27768 antibodies were sorted and plated on mGAM-CRI plates, resulting in variable numbers of colonies depending on the fecal samples and on the gated regions. Forty-two colonies presenting morphologies compatible with F. prausnitzii (i.e., exclusion of large colonies that appeared in less than 48 h) were screened with species-specific primers, resulting in a total of 10 PCR-positive colonies isolated from 4 different donors in the Ab 66/68-gated events and 5 PCR-positive colonies isolated from 3 different donors in the Ab A2-165-gated events (Table 1). To assess the heterogeneity of the new isolates, we screened the colonies that appeared positive for species specific PCR using the RAPD, which allows discrimination of closely related strains of the same species [25]. We were able to distinguish 7 isolates presenting different RAPD profiles that were further analyzed using 16S rRNA gene sequencing to confirm their identity. Six of seven isolates were confirmed as F. prausnitzii whereas the unique isolate collected from HV6 was assigned as Ruthenibacterium lactatiformans, which belongs to the Oscillospiraceae family along with F. prausnitzii (Additional files 6 and 7).
Use of polyclonal antibodies for sorting and cultivation of new C. minuta strains from fecal material
Since C. minuta is usually present in only very low amounts in fecal material compared to F. prausnitzii [6, 16], we first performed an experiment in which C. minuta was spiked in different amounts for better delineation of the sorting gate. Taking advantage of the very small size of cells from this bacterial species, we adjusted the gating strategy that consisted in selecting antibodies-stained bacteria among Live bacteria presenting FSC/SSC parameters similar to those of C. minuta DSM 22607 (Fig. 9). Twenty-five of 30 (83%) and 105 of 107 (98%) colonies recovered from the 0.01% and 0.1% spiked material, respectively, were confirmed as C. minuta by species-specific PCR, thus validating the selection strategy. We therefore used it to isolate C. minuta strains from fresh fecal samples from 8 healthy volunteers (HV10‑HV17). As expected, events that potentially correspond to C. minuta were scarce, ranging between nearly 0 and 1.0% of the Live bacteria among different samples. Colonies with C. minuta-compatible morphology (Fig. 9) that developed 48 to 72 h after plating were subjected to PCR analysis using species-specific primers. Between 13 and 64% of these colonies from 4 out of 8 samples were indeed positive in this assay (Table 2). Ten different RAPD profiles were observed but three of the isolates actually corresponded to species other than C. minuta, as demonstrated by partial 16S sequencing. We therefore ended with 7 different C. minuta isolates collected from 3 different donors and presenting 6 different RAPD profiles (Additional files 8 and 9). It should be noted that 16S gene amplicon repertoire sequencing detected C. minuta only for HV10 and HV17 fecal samples, representing 0.67 and 0.01% of OTUs, respectively (Fig. 8).
Fig. 9.

Development and validation of the C. minuta enrichment and sorting strategy. Original fecal sample was spiked with 0.1% or 0.01% C. minuta DSM 22607 (A). Bacteria considered as live after staining with the LIVE/DEAD™ BacLight™ Bacterial Viability Kit were selected (B) and among them only events presenting FSC/SSC values similar to those of C. minuta DSM 22607 (as determined in panel D) were considered (C, black circle). Bacteria stained with anti-C. minuta antibodies were then selected from these regions and directly sorted on cultivation plates (E and F, antibodies-stained frequencies relate to total events). Colonies circled in green tested positive for C. minuta-specific PCR. These experiments were performed once for each spiked dilution
Table 2.
Results of FCM analysis, sorting and cultivation when targeting C. minuta in samples HV10 to HV17

Correlating sequencing results with 405 nm auto-fluorescence results
In order to better assess the relevance of auto-fluorescence measurement for detecting archaea, we compared relative abundance of auto-fluorescent events detected in cytometry to relative abundance of Methanobacteriaceae calculated from 16S rRNA-encoding genes sequencing results for the 13 donors (HV5 to HV 17) for which both data sets were available. This resulted in a R2 value of 0.68, with a systematic under-estimation of sequencing compared to FCM results (Fig. 10).
Fig. 10.
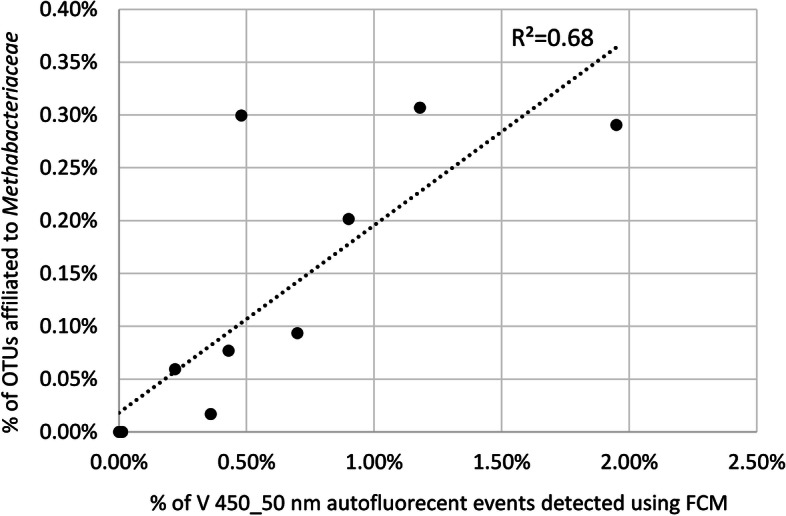
Correlation between relative abundance of autofluorescent events detected in the 450/50 nm channel when exciting with the 405 nm laser and relative abundance of Methanobacteriaceae calculated from 16S rRNA-encoding genes sequencing results for donors HV5 to HV 17. P value calculated with the Pearson method is 5.1 × 10−4
Discussion
The recent description of FCM and bacterial cell sorting under anaerobic conditions [23] prompted us to explore this technology for targeted enrichment and culture of species of interest from fecal material. We first focused on the commensal species F. prausnitzii which is usually found in significant numbers in fecal samples, but is very sensitive to oxygen exposure. A first batch of antibodies was raised against the type strain A2-165 that belongs to F. prausnitzii phylogroup IIb, and a second batch was raised against a mix of the closely related strains ATCC 27766 and ATCC 27768 that belong to phylogroup I recently proposed as “Faecalibacterium moorei” [10]. Interestingly, polyclonal antibodies generated with type strains from one phylogroup presented only limited reactivity against type strains of the other phylogroup. This could be due to lower affinity binding, or to the limited presence of common epitopes. In addition, the presence of extracellular compounds that mask specific epitopes cannot be excluded [26]. The presence of such an extracellular matrix may depend on the growth phase, which could also explain the observed variations in staining efficiency against strain A2-165. In a similar way, polyclonal antibodies generated using type strain C. minuta DSM 22607 were only poorly reactive or even completely unreactive against type strains of “C. massiliensis” and “C. timonensis,” respectively. Both for the three F. prausnitzii strains and the C. minuta strain, bacteria sorted after antibody staining remained cultivable and LIVE/DEAD™ staining correlated well with cultivability, with only a few colonies obtained from bacteria stained by PI. We recently reported similar cultivability results after LIVE/DEAD™ staining for a limited number of anaerobic commensal species, suggesting that this labeling has real value for enriching anaerobic commensal bacteria to be sorted for cultivation [27]. However, one should remember that exceptions have also been reported [28] and that staining efficacy can potentially be affected if bacteria form endospores, which is the case for a variety of gut commensal species [29].
Since the ultimate goal of the sorting experiments was to cultivate stained bacteria rather than to measure their relative abundancy in the samples, we chose to use a stringent gating strategy that was common to all samples. Sorting and sequencing experiments confirmed the good species specificity of polyclonal antibodies directed against the two F. prausnitzii phylogroups, for which nonspecific enrichment was almost absent. The fact that we were not able to culture F. prausnitzii strains from fecal samples collected on HV5 and HV6 for which F. prausnitzii OTUs were detected by sequencing can be due to the specificity of the two antibodies, which probably do not cover the whole variety of possible strains. Since stained events were detected with both antibodies especially for HV5, another explanation could be that some strains require specific nutrients that were not present in our complemented mGAM medium. Concerning the two pairs of F. prausnitzii strains that were isolated from donors HV7 (strains 281 and 282) and HV8 (strains 275 and 276) using the two different polyclonal antibodies, it was interesting to note that they presented different but still related RAPD profiles (Additional Fig. 6) but that they presented 100% 16S rRNA-encoding gene identities with each other (Additional Fig. 7). Whether differences observed in RAPD profiles reflect evolution of one commensal strain through gain and/or loss or several genes [30] or are due to technical biases will be confirmed when sequencing the complete genomes. These results also call into question the specificity of our polyclonal antibodies. It cannot be excluded that phylogroup-specificity observed with polyclonal antibodies directed against type strains will be challenged by new strains that will react with both antibodies.
Autofluorescence of methanogenic archaea has already been reported: it is due to the redox cofactor F420 and has been proved useful for fast and reliable quantification of methanogenic archaea in biogas digesters using flow cytometry [31]. Although performed with a limited number of samples, our work tends to confirm that it could also be used to monitor methanogenic archaea in fecal samples. This could be of interest for the development of microbiota-based biomarkers since methanogenic archaea are considered major contributors to carbohydrate metabolism and their absence or presence in various amounts has been reported to be associated with several phenotypes, including severe acute malnutrition [32] or a lean phenotype [33, 34], to mention just a few. Interestingly, the presence of members of the Christensenellaceae family in the gut microbiota was reported to be associated with the presence of methanogenic archaea [17], with both groups being late colonizers of the gut ecosystem [35], which could be due to syntrophy via interspecies hydrogen transfer between Christensenella and Methanobrevibacter species [36]. In our limited number of samples, we did not observe any correlation between the presence of Christensenellaceae and the presence of methanogenic archaea when evaluated by FCM or 16S repertoire sequencing. C. minuta has been reported as a keystone species that comprises on average 0.01% of the fecal microbiota [16]. This low abundancy could explain why culture studies that used non-specific methods to cultivate a large diversity of gut commensal species did not succeed at cultivating C. minuta strains [11], thus highlighting the need for methods that can enrich cultivated fractions with specific species of interest. This includes antibodies, but also a number of additional stains such as for instance fluorescent analogs of glucose that have recently been shown to be taken up by members of the Christensenellaceae-R7 group [37].
In conclusion, this proof-of-concept study confirms that FCM is well adapted for complex bacterial microbiota studies. When used in conjunction with appropriate staining and associated controls, it gives a general overview of microbiota composition and variations in longitudinal studies [38], including bacterial load which is an important piece of information [39]. In addition, the use of more specific staining such as antibodies is a promising strategy to target, sort, and cultivate species of interest from these complex ecosystems. Recent studies demonstrated that these antibodies can be generated using a reverse genomics approach [40], which opens important avenues since approximately 70% of the gut microbial species still lack cultured representatives [41]. Due to the lack of detailed knowledge of their reactivity, it remains difficult to use polyclonal antibodies such as those described in this study for an analysis of the relative abundance of specific species of interest. However, in the future, better characterized monoclonal antibodies or antibody cocktails may offer an interesting alternative to molecular biology-based methods for longitudinal monitoring of commensal species of interest.
This should be accompanied by specific technological developments in the field of FCM to allow simple, commercially available solutions enabling routine sorting experiments in controlled atmosphere conditions, which will be of strong interest for commensal bacteria but also for cellular biology applications necessitating oxygen conditions that are close to in vivo conditions [42, 43].
Methods
Bacterial strains and growth conditions
The following reference strains were used in this study: Faecalibacterium prausnitzii DSM-17677 (A2-165), ATCC-27766, and ATCC-27768, Christensenella minuta DSM 22607. They were cultured using modified Gifu Anaerobic Medium (mGAM, HyServe 05426) complemented with 30% bovine rumen, cellobiose (1 mg/ml), and inulin (1 mg/ml) with (plates) or without (broth) 1.5% agar (mGAM-CRI). All media and reagents were reduced for at least 48 h in the BACTRON600 (Sheldon) anaerobic chamber before use, and cultures were incubated at 37 °C in the chamber.
Polyclonal antibodies
Rabbit polyclonal antibodies (pAb) were produced in New Zealand rabbits using a standard 53 days protocol (Covalab). Rabbits were immunized with a 50/50 mix of phylogenetically related strains F. prausnitzii ATCC-27766 and ATCC-27768, or pure cultures of F. prausnitzii A2-165 and C. minuta DSM 22607. At day 0, animals received intradermal injection of 0.5 ml of a 1 × 109 suspension of heat-inactivated bacteria + 0.5 ml of complete Freund's Adjuvant. At day 14 and day 39, they received sub-cutaneous injection of 0.5 ml of a 2x109 suspension of heat-inactivated bacteria + 0.5 ml of incomplete Freund's Adjuvant. Serum samples collected at day 39 were tested for immune-reactivity. In case of good reactivity (which was the case for F. prausnitzii A2-165), animals were sacrificed at day 53 for serum collection. If reactivity of day 39 sera was too low (F. prausnitzii ATCC 27766 + ATCC 27768 and C. minuta DSM 22607), animals were boosted at days 60, 74 and 88 with sub-cutaneous injection of 0.5 ml of a 2x109 suspension of heat-inactivated bacteria + 0.5 ml of incomplete Freund’s adjuvant before sacrifice at day 108. After rabbit bleeding, sera were harvested and IgGs were purified on protein A and labeled with Alexa Fluor™ 647 or Alexa Fluor™ 405 using the protein labeling kit (Thermo Fisher Scientific) as recommended by the manufacturer. IgGs from a non-immunized rabbit were also coupled with Alexa Fluor™ 647 as negative control.
Viability staining procedure
Viability staining was performed using the LIVE/DEAD™ BacLight™ Bacterial Viability Kit (SYTO 9/Propidium Iodide, Thermo Fisher Scientific) as recommended by the manufacturer. After staining, bacteria were washed in PBS and then analyzed within 30 min using an Influx® (Becton-Dickinson) cell sorter equipped with a 200 mW-488 nm laser, a 120 mW-640 nm laser, and a 100 mW-405 nm laser.
Anaerobic sorting of single strains
The BD Influx® cell sorter used for anaerobic sorting has been described by Thompson et al. [23]. Briefly, anaerobic sorting was achieved by eliminating oxygen from the sort stream and cell deposition areas of the cell sorter by using a customized glove box. Paraffin oil was used to cover samples in both conditions to prevent oxygen entry to the sample during the transfer from the anaerobic chamber, where the samples were prepared, to the flow cytometer. Durations of the sorting steps were normalized to 30 min. Oxygen concentration in the glove box was monitored using a ToxiRAE PRO detector (RAE, France). For sorting experiments, reduced mGAM-CRI plates were transferred from the anaerobic chamber to the cell sorter glove box using sealed bags. Nitrogen was then injected and anaerobic sorting experiments were started when the oxygen concentration was measured below 0.7%. Analysis and sorting followed by cultivation of stained and unstained bacteria was used to evaluate the impact of the process on F. prausnitzii and C. minuta cultivability. Bacteria used for sorting experiments were anaerobically cultivated for 48 h at 37 °C on mGAM-CRI plates. One colony was then sub-cultivated in mGAM-CRI broth for 24 h at 37 °C. One milliliter of bacterial cultures were then washed in reduced PBS and diluted 1:100 before staining with LIVE/DEAD™ BacLight™ Bacterial Viability Kit as described above. Polyclonal antibodies generated using the mix of F. prausnitzii ATCC 27766 and ATCC 27768 bacteria were conjugated with Alexa Fluor™ 647 whereas those generated with F. prausnitzii A2-165 were conjugated with Alexa Fluor™ 405. Polyclonal antibodies generated using C. minuta DSM 22607 bacteria were conjugated with Alexa Fluor™ 647. Staining with 1/100th antibodies (final concentration: 10 μg/ml) and viability kit was performed in anaerobic conditions for 30 min in the dark and then bacteria were washed in reduced PBS before analysis. After washing, stained bacteria were suspended in reduced PBS containing 0.5 mg/l resazurin, 2.1 mM soldium sulfure, and 2.8 mM L-cystein HCl, and the suspensions were covered with 750 μl of paraffin oil to prevent oxygen exposure. The tubes were taken out of the anaerobic chamber and bacteria were analyzed and sorted within 30 min. Sorting speed was adjusted at 1000 events per second and four series of 1, 3, 10, 30, 100, 300, or 1000 events were sorted on one single spot for each tested condition. Once sorting experiments were achieved, plates were re-introduced in sealed bags and transferred in the anaerobic chamber where they were incubated at 37 °C for 2 to 3 days before observation. Percentages of recovery were calculated by taking into account the smallest number of sorted bacteria resulting in the growth of colonies visible to the naked eye. The following formula was then applied: percentage of recovery = (n/N × 1/B) × 100
n: number of colonies counted per row (or for 2 rows when only one bacterium was deposited)
N: number of sorted spots (8 for the first 2 rows for which only one bacterium was deposited per spot, 4 for the other rows for which higher numbers of bacteria were deposited)
B: number of bacteria sorted on each spot.
Unstained and pre-immune controls
We performed control experiments by analyzing a series of 4 fecal frozen samples collected from healthy volunteers (HV) 1 to 4. Samples were left unstained to evaluate potential auto-fluorescence, or stained with pre-immune antibodies conjugated with Alexa Fluor™ 647 dye to evaluate non-specific staining. The same 4 samples were also stained with both anti-F. prausnitzii antibodies in a separate tube. Briefly, washed bacteria were incubated in PBS with SYTO 9/PI added with 1/100th conjugated antibodies dilution (final concentration: 10 μg/ml) for 30 min. Bacteria were then washed once with reduced PBS, covered with paraffin oil to protect them from oxygen and analyzed by flow cytometry.
Sorting and DNA extraction from antibodies-enriched fractions
To test the specificity of the F. prausnitzii antibodies, we sorted antibodies-stained fractions collected from the pool of frozen fecal samples HV2 and HV4. Bacteria stained by the polyclonal antibodies directed against C. minuta being almost undetected in the mix, we decided to spike the same pool of 2 fecal samples with C. minuta DSM 22607 at approx. 2% relative to the bacterial counts measured by flow cytometry. This allowed sorting of sufficient material for subsequent sequencing and evaluation of antibodies specificity. Events presenting auto-fluorescence when exciting with the 405 nm laser were also sorted for further identification. One million events were sorted for each of the 3 antibodies as well as for the auto-fluorescent events. DNA was extracted from the sorted cells using the mericon™ DNA Bacteria Kit (QIAGEN) with adjustments as follows. Bacterial cell pellets were resuspended in 20‑40 μl Fast Lysis Buffer, depending on the volume of the pellet, by brief, vigorous vortexing. The samples were placed into a thermal shaker (800 rpm) set to 100 °C for 10 min. Samples were then allowed to cool at room temperature for 2 min, before centrifugation (13,000 × g, 5 min). 20‑40 μl of the supernatant were transferred to a 1.5 ml microcentrifuge tube and purified using the NucleoSpin™ Gel and PCR Clean-up Kit (Macherey-Nagel) according to manufacturer’s instructions.
Library preparation and 16S rRNA gene amplicon data analysis
For native fecal samples, library preparation and sequencing were performed with 24 ng template DNA as described in detail previously [44] using a robotized platform (Biomek400, Beckman Coulter). For the samples extracted with the mericon™ DNA Bacteria Kit (QIAGEN), 1‑8 μL template DNA were used for PCR. The V3-V4 region of 16S rRNA genes was amplified in duplicates for 25 cycles with DNA from fecal samples, or with 35 cycles with DNA from cell sorted samples, following a two-step protocol [45] using primers 341F-785R [46]. After purification using the AMPure XP system (Beckman Coulter), sequencing was carried out with pooled samples spiked with 25% (v/v) PhiX standard library in paired-end modus (PE300) using a MiSeq system (Illumina, Inc.) according to the manufacturer’s instructions. Raw reads were processed using an in-house developed pipeline (www.imngs.org) [47] based on UPARSE [48]. In brief, sequences were demultiplexed and trimmed to the first base with a quality score < 3. Pairing, chimera filtering and operational taxonomic units (OTUs) clustering (97% sequence identity) was done using USEARCH 11.0 [49]. Sequences with less than 350 and more than 500 nucleotides and with an expected error > 2 were excluded from the analysis. Remaining reads were trimmed by ten nucleotides on each end to avoid GC bias and non-random base composition. Only OTUs occurring at a relative abundance > 0.25% in at least one sample were kept. To highlight differences in specificities of the antibodies raised against F. prausnitzii that includes different phylogroups, a zero-radius approach [24] using the UNOISE algorithm (USEARCH 11.0) [50] was chosen, increasing the taxonomic resolution with which the molecular species in sorted bacterial populations were delineated. Sequence alignment and taxonomic classification was conducted with SINA v1.6.1, using the taxonomy within the SILVA release 128 [51]. Downstream analysis was performed in the R programming environment using Rhea (https://lagkouvardos.github.io/Rhea/) [52]. Normalization of OTU and ZOTU tables was performed by dividing through sample size and subsequent multiplication by the number of reads in the smallest sample, to account for differences in sequencing depth.
Isolation of bacterial DNA from stool samples
DNA was isolated using a modified protocol according to Godon et al. [53]. Snap frozen samples were mixed with 600 μl stool DNA stabilizer (Stratec biomedical), thawed, and transferred into autoclaved 2-ml screw-cap tubes containing 500 mg 0.1 mm-diameter silica/zirconia beads. Next, 250 μl 4 M guanidine thiocyanate in 0.1 M Tris (pH 7.5) and 500 μl 5% N-lauroyl sarcosine in 0.1 M PBS (pH 8.0) were added. Samples were incubated at 70 °C and 700 rpm for 60 min. A FastPrep® instrument (MP Biomedicals) fitted with a 24 × 2 ml cooling adaptor filled with dry ice was used for cell disruption. The program was run 3 times for 40 s at 6.5 M/s. After each run, the cooling adapter was refilled with dry ice. An amount of 15 mg polyvinylpyrrolidone (PVPP) was added and samples were vortexed, followed by 3 min centrifugation at 15.000 × g and 4 °C. Approximately, 650 μl of the supernatant were transferred into a new 2 ml tube, which was centrifuged again for 3 min at 15.000 × g and 4 °C. Subsequently, 500 μl of the supernatant was transferred into a new 2 ml tube and 50 μg of RNase was added. After 20 min at 37 °C and 700 rpm, gDNA was isolated using the NucleoSpin® gDNA Clean-up Kit from Macherey-Nagel. Isolation was performed according to the manufacturer’s protocol. DNA was eluted from columns twice using 40 μl elution buffer and concentration was measured with NanoDrop® (Thermo Scientific). Samples were stored at −20 °C before being processed as described above for 16S repertoire analysis.
Anaerobic sorting and cultivation from fecal material
Fecal samples were collected at home by healthy volunteers and immediately transferred in plastic pouches that were then sealed after adding an AnaeroGen™ sachet (Oxoid). Samples were shipped to the lab within 2 h at ambient temperature. All subsequent steps were performed in the anaerobic chamber. For each sample, 1 g of fecal material was suspended in 10 ml PBS and homogenized by vortexing using 2.4 mm glass beads. Suspensions were then 10-fold diluted in PBS and filtered through a 70-μm cell strainer (Biologix). After an additional 100-fold dilution in PBS, bacteria were numbered by FCM using counting beads (LIVE/DEAD™ BacLight™ Bacterial Viability and Counting Kit, Thermo Fisher Scientific). LIVE/DEAD™ staining was used to select live bacteria whereas polyclonal antibodies were used to enrich with target bacterial species. Staining was performed in anaerobic conditions for 30 min in the dark and then bacteria were washed in reduced PBS before analysis. After washing, stained bacteria were suspended in reduced PBS and the suspensions were covered with 500 μl of paraffin oil to prevent oxygen exposure. The tubes were taken out of the anaerobic chamber and bacteria were analyzed and sorted within 30 min. Bacteria were gated based on FSC/SSC parameters. Among them, live ones were selected according to SYTO 9/PI fluorescence and 240 (F. prausnitzii) to 1152 (C. minuta) events collected from the antibodies-stained gates were sorted on mGAM-CRI plates. Plates were then incubated for 5 days at 37 °C in anaerobic conditions. Colonies were then visually selected based on morphologies that were compatible with the target species: small, flat colonies encrusted in the agar plates for F. prausnitzii, and tiny bulging colonies for C. minuta. They were then screened using previously described PCR primers: Fprau 02 (5′-GAG CCT CAG CGT CAG TTG GT-3′) and Fprau 07 (5′-CCA TGA ATT GCC TTC AAA ACT GTT-3′) for F. prausnitzii [7], Yso F (5′-CCC ACC AAG TCAA CGA TGG G-3′), and ChrisM-R1 (5′-CCC TCT CCT GTA CTC AAG TC-3′) for C. minuta [54] and this study.
Identification of cultured isolates
To distinguish potentially different strains isolated from the same sample, Random Amplified Polymorphic DNA (RAPD) analysis was conducted. Briefly, candidate F. prausnitzii or C. minuta colonies were subcultivated on mGAM plates and then several colonies were resuspended in 100 μl DNA-free water and DNA was isolated using InstaGene™ Matrix (BioRad) kit according to the manufacturer guidelines. This DNA preparation served as a template for PCR reaction using primer D9355 for F. prausnitzii candidates and primer D14307 for C. minuta candidates, and a PCR program described by Akopyanz et al. [25]. One isolate corresponding to each specific RAPD profile was then chosen for further identification by sequencing the almost complete 16S rRNA-encoding genes.
For phylogenetic analysis, 16S rRNA-encoding genes were aligned using Muscle [55] integrated in MEGA7 [56] with default parameters. The phylogenetic tree was inferred using the Maximum Likelyhood method based on the Kimura 2-parameters model with 1000 bootstrap replicates [19].
Supplementary Information
Additional file 1. Unstained, pre-immune-stained and antibodies-stained controls for F. prausnitzii strains A2-165 (phylogroup IIb), ATCC 27766 (phylogroup I) and ATCC 27768 (phylogroup I).
Additional file 2. Unstained, pre-immune-stained and antibodies-stained controls for C. minuta DSM 22607, ‘C. massiliensis’ DSM 102344 and ‘C. timonensis’ DSM 102800.
Additional file 3. Gates used to select, sort and culitvate F. prausnitzii and C. minuta bacteria after staining with the LIVE/DEAD™ kit and with the specific antibodies.
Additional file 4. Preliminary FCM controls. (A) events gated as bacteria, (B) auto-fluorescence of fecal material in the Violet 450/50 nm and Red 670/30 nm channels, (C) potential staining with polyclonal antibodies collected from pre-immune serum and conjugated with Alexa Fluor™ 647, and (D) staining with anti-F. prausnitzii A2-165 and anti-F. prausnitzii ATCC 27766 + 27768 antibodies conjugated with Alexa Fluor™ 405 and Alexa Fluor™ 647, respectively.
Additional file 5. Gates that were used to sort and then sequence the 16S rRNA gene amplicon repertoire of bacteria stained with the C. minuta-antibodies.
Additional file 6. Random Amplified Polymorphism DNA profiles obtained with newly isolated F. prausnitzii strains using primer D9355 [25].
Additional file 7. Phylogenetic tree representing newly isolated F. prausnitzii strains. The phylogenetic tree was inferred from Muscle alignment of partial 16S rRNA-encoding gene sequences using the Maximum Likelihood method based on the Kimura 2-parameters model with 1,000 bootstrap replicates. Branch values < 50% are not displayed. The tree was built using reference sequences and outgroups described by [10]. Colonies aspects (bar: 0.5 cm) as well as Gram-stains (100x objective lens) are reported for the strains used in this study. Previously described strains with demonstrated anti-inflammatory activities are indicated with a red arrow. Strains highlighted in red were isolated with the polyclonal antibodies directed against F. prausnitzii ATCC 27766 + 27768, strains highlighted in violet were isolated with the polyclonal antibodies directed against F. prausntizii A2-165.
Additional file 8. Random Amplified Polymorphism DNA profiles obtained with newly isolated C. minuta strains using primer D14307 [25].
Additional file 9. Phylogenetic tree representing newly isolated C. minuta strains. The phylogenetic tree was inferred from Muscle alignment of partial 16S rRNA-encoding gene sequences using the Maximum Likelihood method based on the Kimura 2-parameters model with 1,000 bootstrap replicates. Branch values < 50% are not displayed. The tree was built using reference sequences and outgroups described in [57].
Acknowledgements
We thank Ntana Kousetzi (University Hospital of RWTH Aachen) for outstanding technical support with 16S rRNA gene amplicon sequencing.
Authors’ contributions
VT and SB conceptualized and designed the experiments. JB and GE contributed to the technical setup of the anoxic cell sorter. MN, AD, MA, and SB performed the staining and sorting experiments, and contributed to strains identification using specific PCR and 16S rRNA gene sequencing. IB performed the RAPD assay. WM contributed to C. minuta strains identification. SB and VT wrote the manuscript. NT and TC helped with 16S rRNA gene amplicon sequencing. NT performed data analysis. NT and TC interpreted data. TC provided access to essential infrastructure. All authors read and approved the final manuscript.
Funding
This work has received, through BIOASTER investment, funding from the French Government through the Investissement d’Avenir program (Grant No. ANR-10-AIRT-03). TC received funding from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), Project-ID 403224013 – SFB 1382 “Gut-liver axis.” Part of this work corresponds to contract research established between BIOASTER and YSOPIA Bioscience to isolate C. minuta strains.
Availability of data and materials
16S gene amplicon sequencing data generated from fecal samples collected from healthy volunteers HV5-HV17 and from sorted fractions were deposited in Sequence Read Archive under accession number PRJNA748004. Partial 16S-rRNA encoding gene sequences of the new F. prausnitzii and C. minuta isolates described in this study are available under GenBank accession numbers MZ577583 to MZ577588, and MZ577589 to MZ577595, respectively.
Declarations
Ethics approval and consent to participate
Stool samples were anonymously collected from healthy volunteers. As required by the French regulation, this collection was declared to the French ministry of Higher Education, Research and Innovation (n°DC-2015-2513) and received an approval by the ethical committee in March 2016. Each volunteer received an information and signed a consent before participating to the study.
Consent for publication
Not applicable.
Competing interests
WM reports personal fees from YSOPIA Bioscience as employee, during the conduct of the study.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Routy B, Gopalakrishnan V, Daillere R, Zitvogel L, Wargo JA, Kroemer G. The gut microbiota influences anticancer immunosurveillance and general health. Nat Rev Clin Oncol. 2018;15(6):382–396. doi: 10.1038/s41571-018-0006-2. [DOI] [PubMed] [Google Scholar]
- 2.Plichta DR, Graham DB, Subramanian S, Xavier RJ. Therapeutic opportunities in inflammatory bowel disease: mechanistic dissection of host-microbiome relationships. Cell. 2019;178(5):1041–1056. doi: 10.1016/j.cell.2019.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kolodziejczyk AA, Zheng D, Shibolet O, Elinav E. The role of the microbiome in NAFLD and NASH. EMBO Mol Med. 2019;11(2):e9302. [DOI] [PMC free article] [PubMed]
- 4.Wang J, Jia H. Metagenome-wide association studies: fine-mining the microbiome. Nat Rev Microbiol. 2016;14(8):508–522. doi: 10.1038/nrmicro.2016.83. [DOI] [PubMed] [Google Scholar]
- 5.O’Toole PW, Marchesi JR, Hill C. Next-generation probiotics: the spectrum from probiotics to live biotherapeutics. Nat Microbiol. 2017;2:17057. doi: 10.1038/nmicrobiol.2017.57. [DOI] [PubMed] [Google Scholar]
- 6.Wu S, Sun C, Li Y, Wang T, Jia L, Lai S, Yang Y, Luo P, Dai D, Yang YQ, et al. GMrepo: a database of curated and consistently annotated human gut metagenomes. Nucleic Acids Res. 2020;48(D1):D545–D553. doi: 10.1093/nar/gkz764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A. 2008;105(43):16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, Prieto PA, Vicente D, Hoffman K, Wei SC, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359(6371):97–103. doi: 10.1126/science.aan4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C, Boselli L, Routier E, Cassard L, Collins M, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol. 2017;28(6):1368–1379. doi: 10.1093/annonc/mdx108. [DOI] [PubMed] [Google Scholar]
- 10.Fitzgerald CB, Shkoporov AN, Sutton TDS, Chaplin AV, Velayudhan V, Ross RP, Hill C. Comparative analysis of Faecalibacterium prausnitzii genomes shows a high level of genome plasticity and warrants separation into new species-level taxa. BMC Genomics. 2018;19(1):931. doi: 10.1186/s12864-018-5313-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poyet M, Groussin M, Gibbons SM, Avila-Pacheco J, Jiang X, Kearney SM, Perrotta AR, Berdy B, Zhao S, Lieberman TD, et al. A library of human gut bacterial isolates paired with longitudinal multiomics data enables mechanistic microbiome research. Nat Med. 2019;25(9):1442–1452. doi: 10.1038/s41591-019-0559-3. [DOI] [PubMed] [Google Scholar]
- 12.Benevides L, Burman S, Martin R, Robert V, Thomas M, Miquel S, Chain F, Sokol H, Bermudez-Humaran LG, Morrison M, et al. New insights into the diversity of the genus Faecalibacterium. Front Microbiol. 2017;8:1790. doi: 10.3389/fmicb.2017.01790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopez-Siles M, Martinez-Medina M, Abella C, Busquets D, Sabat-Mir M, Duncan SH, Aldeguer X, Flint HJ, Garcia-Gil LJ. Mucosa-associated Faecalibacterium prausnitzii phylotype richness is reduced in patients with inflammatory bowel disease. Appl Environ Microbiol. 2015;81(21):7582–7592. doi: 10.1128/AEM.02006-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang X, Deeke SA, Ning Z, Starr AE, Butcher J, Li J, Mayne J, Cheng K, Liao B, Li L, et al. Metaproteomics reveals associations between microbiome and intestinal extracellular vesicle proteins in pediatric inflammatory bowel disease. Nat Commun. 2018;9(1):2873. doi: 10.1038/s41467-018-05357-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopez-Siles M, Duncan SH, Garcia-Gil LJ, Martinez-Medina M. Faecalibacterium prausnitzii: from microbiology to diagnostics and prognostics. ISME J. 2017;11(4):841–852. doi: 10.1038/ismej.2016.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Waters JL, Ley RE. The human gut bacteria Christensenellaceae are widespread, heritable, and associated with health. BMC Biol. 2019;17(1):83. doi: 10.1186/s12915-019-0699-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, Beaumont M, Van Treuren W, Knight R, Bell JT, et al. Human genetics shape the gut microbiome. Cell. 2014;159(4):789–799. doi: 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mazier W, Le Corf K, Martinez C, Tudela H, Kissi D, Kropp C, Coubard C, Soto M, Elustondo F, Rawadi G, et al. A new strain of Christensenella minuta as a potential biotherapy for obesity and associated metabolic diseases. Cells. 2021;10(4):823. doi: 10.3390/cells10040823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin R, Miquel S, Benevides L, Bridonneau C, Robert V, Hudault S, Chain F, Berteau O, Azevedo V, Chatel JM, et al. Functional characterization of novel Faecalibacterium prausnitzii strains isolated from healthy volunteers: a step forward in the use of F. prausnitzii as a next-generation probiotic. Front Microbiol. 2017;8:1226. [DOI] [PMC free article] [PubMed]
- 20.Lippe R. Flow virometry: a powerful tool to functionally characterize viruses. J Virol. 2018;92(3):e01765–17. [DOI] [PMC free article] [PubMed]
- 21.Muller S, Nebe-von-Caron G. Functional single-cell analyses: flow cytometry and cell sorting of microbial populations and communities. FEMS Microbiol Rev. 2010;34(4):554–587. doi: 10.1111/j.1574-6976.2010.00214.x. [DOI] [PubMed] [Google Scholar]
- 22.Duncan SH, Flint HJ: Faecalibacterium. In: Bergey’s Manual of Systematics of Archaea and Bacteria. Edited by M.E. Trujillo SD, P. DeVos, B. Hedlund, P. Kämpfer, F.A. Rainey and W.B. Whitman; 2015: 1-6.
- 23.Thompson AW, Crow MJ, Wadey B, Arens C, Turkarslan S, Stolyar S, Elliott N, Petersen TW, van den Engh G, Stahl DA, et al. A method to analyze, sort, and retain viability of obligate anaerobic microorganisms from complex microbial communities. J Microbiol Methods. 2015;117:74–77. doi: 10.1016/j.mimet.2015.07.009. [DOI] [PubMed] [Google Scholar]
- 24.Edgar RC. Updating the 97% identity threshold for 16S ribosomal RNA OTUs. Bioinformatics. 2018;34(14):2371–2375. doi: 10.1093/bioinformatics/bty113. [DOI] [PubMed] [Google Scholar]
- 25.Akopyanz N, Bukanov NO, Westblom TU, Kresovich S, Berg DE. DNA diversity among clinical isolates of Helicobacter pylori detected by PCR-based RAPD fingerprinting. Nucleic Acids Res. 1992;20(19):5137–5142. doi: 10.1093/nar/20.19.5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rossi O, Khan MT, Schwarzer M, Hudcovic T, Srutkova D, Duncan SH, Stolte EH, Kozakova H, Flint HJ, Samsom JN, et al. Faecalibacterium prausnitzii strain HTF-F and its extracellular polymeric matrix attenuate clinical parameters in DSS-induced colitis. PLoS ONE. 2015;10(4):e0123013. doi: 10.1371/journal.pone.0123013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duquenoy A, Bellais S, Gasc C, Schwintner C, Dore J, Thomas V. Assessment of gram- and viability-staining methods for quantifying bacterial community dynamics using flow cytometry. Front Microbiol. 2020;11(1469):1469. doi: 10.3389/fmicb.2020.01469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi L, Gunther S, Hubschmann T, Wick LY, Harms H, Muller S. Limits of propidium iodide as a cell viability indicator for environmental bacteria. Cytometry A. 2007;71(8):592–598. doi: 10.1002/cyto.a.20402. [DOI] [PubMed] [Google Scholar]
- 29.Browne HP, Forster SC, Anonye BO, Kumar N, Neville BA, Stares MD, Goulding D, Lawley TD. Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation. Nature. 2016;533(7604):543–546. doi: 10.1038/nature17645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garud NR, Good BH, Hallatschek O, Pollard KS. Evolutionary dynamics of bacteria in the gut microbiome within and across hosts. PLoS Biol. 2019;17(1):e3000102. doi: 10.1371/journal.pbio.3000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lambrecht J, Cichocki N, Hubschmann T, Koch C, Harms H, Muller S. Flow cytometric quantification, sorting and sequencing of methanogenic archaea based on F420 autofluorescence. Microb Cell Fact. 2017;16(1):180. doi: 10.1186/s12934-017-0793-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Camara A, Konate S, Tidjani Alou M, Kodio A, Togo AH, Cortaredona S, Henrissat B, Thera MA, Doumbo OK, Raoult D, et al. Clinical evidence of the role of Methanobrevibacter smithii in severe acute malnutrition. Sci Rep. 2021;11(1):5426. doi: 10.1038/s41598-021-84641-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Million M, Angelakis E, Maraninchi M, Henry M, Giorgi R, Valero R, Vialettes B, Raoult D. Correlation between body mass index and gut concentrations of Lactobacillus reuteri, Bifidobacterium animalis, Methanobrevibacter smithii and Escherichia coli. Int J Obes. 2013;37(11):1460–1466. doi: 10.1038/ijo.2013.20. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Million M, Maraninchi M, Henry M, Armougom F, Richet H, Carrieri P, Valero R, Raccah D, Vialettes B, Raoult D. Obesity-associated gut microbiota is enriched in Lactobacillus reuteri and depleted in Bifidobacterium animalis and Methanobrevibacter smithii. Int J Obes. 2012;36(6):817–825. doi: 10.1038/ijo.2011.153. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Roswall J, Olsson LM, Kovatcheva-Datchary P, Nilsson S, Tremaroli V, Simon MC, Kiilerich P, Akrami R, Kramer M, Uhlen M, et al. Developmental trajectory of the healthy human gut microbiota during the first 5 years of life. Cell Host Microbe. 2021;29(5):765–776. doi: 10.1016/j.chom.2021.02.021. [DOI] [PubMed] [Google Scholar]
- 36.Ruaud A, Esquivel-Elizondo S, de la Cuesta-Zuluaga J, Waters JL, Angenent LT, Youngblut ND, Ley RE. Syntrophy via interspecies H2 transfer between Christensenella and Methanobrevibacter underlies their global cooccurrence in the human gut. mBio. 2020;11(1):e03235–19. [DOI] [PMC free article] [PubMed]
- 37.Tao J, McCourt C, Sultana H, Nelson C, Driver J, Hackmann TJ. Use of a fluorescent analog of glucose (2-NBDG) to identify uncultured rumen bacteria that take up glucose. Appl Environ Microbiol. 2019;85(7):e03018–18. [DOI] [PMC free article] [PubMed]
- 38.Zimmermann J, Hubschmann T, Schattenberg F, Schumann J, Durek P, Riedel R, Friedrich M, Glauben R, Siegmund B, Radbruch A, et al. High-resolution microbiota flow cytometry reveals dynamic colitis-associated changes in fecal bacterial composition. Eur J Immunol. 2016;46(5):1300–1303. doi: 10.1002/eji.201646297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vandeputte D, Kathagen G, D'Hoe K, Vieira-Silva S, Valles-Colomer M, Sabino J, Wang J, Tito RY, De Commer L, Darzi Y, et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature. 2017;551(7681):507–511. doi: 10.1038/nature24460. [DOI] [PubMed] [Google Scholar]
- 40.Cross KL, Campbell JH, Balachandran M, Campbell AG, Cooper SJ, Griffen A, Heaton M, Joshi S, Klingeman D, Leys E, et al. Targeted isolation and cultivation of uncultivated bacteria by reverse genomics. Nat Biotechnol. 2019;37(11):1314–1321. doi: 10.1038/s41587-019-0260-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Almeida A, Nayfach S, Boland M, Strozzi F, Beracochea M, Shi ZJ, Pollard KS, Sakharova E, Parks DH, Hugenholtz P, et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat Biotechnol. 2021;39(1):105–14. [DOI] [PMC free article] [PubMed]
- 42.Llufrio EM, Wang L, Naser FJ, Patti GJ. Sorting cells alters their redox state and cellular metabolome. Redox Biol. 2018;16:381–387. doi: 10.1016/j.redox.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Monceaux V, Chiche-Lapierre C, Chaput C, Witko-Sarsat V, Prevost MC, Taylor CT, Ungeheuer MN, Sansonetti PJ, Marteyn BS. Anoxia and glucose supplementation preserve neutrophil viability and function. Blood. 2016;128(7):993–1002. doi: 10.1182/blood-2015-11-680918. [DOI] [PubMed] [Google Scholar]
- 44.Lagkouvardos I, Klaring K, Heinzmann SS, Platz S, Scholz B, Engel KH, Schmitt-Kopplin P, Haller D, Rohn S, Skurk T, et al. Gut metabolites and bacterial community networks during a pilot intervention study with flaxseeds in healthy adult men. Mol Nutr Food Res. 2015;59(8):1614–1628. doi: 10.1002/mnfr.201500125. [DOI] [PubMed] [Google Scholar]
- 45.Berry D, Ben Mahfoudh K, Wagner M, Loy A. Barcoded primers used in multiplex amplicon pyrosequencing bias amplification. Appl Environ Microbiol. 2011;77(21):7846–7849. doi: 10.1128/AEM.05220-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glockner FO. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013;41(1):e1. doi: 10.1093/nar/gks808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lagkouvardos I, Joseph D, Kapfhammer M, Giritli S, Horn M, Haller D, Clavel T. IMNGS: A comprehensive open resource of processed 16S rRNA microbial profiles for ecology and diversity studies. Sci Rep. 2016;6:33721. doi: 10.1038/srep33721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10(10):996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 49.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 50.Edgar RC. UNOISE 2: improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv. 2016:081257.
- 51.Pruesse E, Peplies J, Glockner FO. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics. 2012;28(14):1823–1829. doi: 10.1093/bioinformatics/bts252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lagkouvardos I, Fischer S, Kumar N, Clavel T. Rhea: a transparent and modular R pipeline for microbial profiling based on 16S rRNA gene amplicons. PeerJ. 2017;5:e2836. doi: 10.7717/peerj.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Godon JJ, Zumstein E, Dabert P, Habouzit F, Moletta R. Molecular microbial diversity of an anaerobic digestor as determined by small-subunit rDNA sequence analysis. Appl Environ Microbiol. 1997;63(7):2802–2813. doi: 10.1128/aem.63.7.2802-2813.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jinatham V, Kullawong N, Kespechara K, Gentekaki E, Popluechai S. Comparison of gut microbiota between lean and obese adult Thai individuals. Microbiology and Biotechnology Letters. 2018;46(3):277–287. [Google Scholar]
- 55.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zou Y, Xue W, Lin X, Hu T, Liu SW, Sun CH, Luo G, Lv M, Dai Y, Kristiansen K et al: Taxonomic description and genome sequence of Christensenella intestinihominis sp. nov., a novel cholesterol-lowering Bacterium isolated from human gut. Front Microbiol 2021, 12:632361. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Unstained, pre-immune-stained and antibodies-stained controls for F. prausnitzii strains A2-165 (phylogroup IIb), ATCC 27766 (phylogroup I) and ATCC 27768 (phylogroup I).
Additional file 2. Unstained, pre-immune-stained and antibodies-stained controls for C. minuta DSM 22607, ‘C. massiliensis’ DSM 102344 and ‘C. timonensis’ DSM 102800.
Additional file 3. Gates used to select, sort and culitvate F. prausnitzii and C. minuta bacteria after staining with the LIVE/DEAD™ kit and with the specific antibodies.
Additional file 4. Preliminary FCM controls. (A) events gated as bacteria, (B) auto-fluorescence of fecal material in the Violet 450/50 nm and Red 670/30 nm channels, (C) potential staining with polyclonal antibodies collected from pre-immune serum and conjugated with Alexa Fluor™ 647, and (D) staining with anti-F. prausnitzii A2-165 and anti-F. prausnitzii ATCC 27766 + 27768 antibodies conjugated with Alexa Fluor™ 405 and Alexa Fluor™ 647, respectively.
Additional file 5. Gates that were used to sort and then sequence the 16S rRNA gene amplicon repertoire of bacteria stained with the C. minuta-antibodies.
Additional file 6. Random Amplified Polymorphism DNA profiles obtained with newly isolated F. prausnitzii strains using primer D9355 [25].
Additional file 7. Phylogenetic tree representing newly isolated F. prausnitzii strains. The phylogenetic tree was inferred from Muscle alignment of partial 16S rRNA-encoding gene sequences using the Maximum Likelihood method based on the Kimura 2-parameters model with 1,000 bootstrap replicates. Branch values < 50% are not displayed. The tree was built using reference sequences and outgroups described by [10]. Colonies aspects (bar: 0.5 cm) as well as Gram-stains (100x objective lens) are reported for the strains used in this study. Previously described strains with demonstrated anti-inflammatory activities are indicated with a red arrow. Strains highlighted in red were isolated with the polyclonal antibodies directed against F. prausnitzii ATCC 27766 + 27768, strains highlighted in violet were isolated with the polyclonal antibodies directed against F. prausntizii A2-165.
Additional file 8. Random Amplified Polymorphism DNA profiles obtained with newly isolated C. minuta strains using primer D14307 [25].
Additional file 9. Phylogenetic tree representing newly isolated C. minuta strains. The phylogenetic tree was inferred from Muscle alignment of partial 16S rRNA-encoding gene sequences using the Maximum Likelihood method based on the Kimura 2-parameters model with 1,000 bootstrap replicates. Branch values < 50% are not displayed. The tree was built using reference sequences and outgroups described in [57].
Data Availability Statement
16S gene amplicon sequencing data generated from fecal samples collected from healthy volunteers HV5-HV17 and from sorted fractions were deposited in Sequence Read Archive under accession number PRJNA748004. Partial 16S-rRNA encoding gene sequences of the new F. prausnitzii and C. minuta isolates described in this study are available under GenBank accession numbers MZ577583 to MZ577588, and MZ577589 to MZ577595, respectively.