

Abstract
Cholesterol is a multifaceted metabolite known to modulate processes in cancer, atherosclerosis, and autoimmunity. A common denominator between these diseases appears to be the immune system, in which many cholesterol-associated metabolites are seen to impact both adaptive and innate immunity. Many cancers display altered cholesterol metabolism, and recent studies demonstrate that manipulating systemic cholesterol metabolism may be useful in improving immunotherapy responses. However, cholesterol can have both proinflammatory and anti-inflammatory roles in mammals, acting via multiple immune cell types, and depending on context. Gaining mechanistic insight on various cholesterol-related metabolites can improve our understanding of their functions and extensive effects on the immune system and ideally inform the design of future therapeutic strategies against cancer and/or other pathologies.
Keywords: Cholesterol, oxysterol, LXR, immune, statins
Cholesterol metabolism can impact the immune system and influence anti-tumor immune responses
Cholesterol serves as a structural component of plasma membranes, where it regulates membrane fluidity, acts as a solubilizer of other lipids, and serves as a signaling mediator [1]. Accordingly, cholesterol metabolism plays a crucial role in regulating anti-tumor immune responses by acting on a variety of immune cells involved in innate and adaptive immune responses. For example, cholesterol depletion is a key feature of tumor-associated macrophages in human lung adenocarcinoma [2] and a large-macrophage morphology associated with enriched cholesterol metabolism has been associated with poor survival in colorectal cancer patients [3]. Of note, cholesterol in the tumor microenvironment (TME) has induced CD8+ T-cell ‘exhaustion’ in a murine melanoma model [4]. Studies have also demonstrated the role of cholesterol in weakening anti-tumor immune responses [4]. However, many recent studies report conflicting results regarding the role of cholesterol and statins in regulating survival and or responses to immunotherapies. Thus, an improved understanding of the regulation of anti-tumor immune responses by both adaptive and innate components of the immune system via cholesterol may guide the development of new putative therapies against malignancies.
While some aspects of cholesterol-mediated regulation of mammalian immune responses are well known, many are not. In addition, how cholesterol metabolism in different immune cell components orchestrates the overall immune response against tumor cells has remained a longstanding knowledge gap. Elucidating the biological mechanisms underlying the regulation of immune activity in response to perturbations in cholesterol metabolism and exploiting that for effective anti-tumor immune responses might lead to novel candidate combination therapies against cancer. This review discusses a brief overview of cholesterol metabolism, as well as new evidence of the effects of cholesterol on adaptive and innate immunity. The article then focuses on recent advances concerning cholesterol-mediated regulation of anti-tumor immune responses by different immune cells, including the demonstration that some of the genetic and pharmacological agents perturbing cholesterol metabolism can effectively regulate anti-tumor immunity. Overall, this review aims to improve our understanding of cholesterol metabolism in immunity and to highlight outstanding questions and areas of focus that might inform the development of putative therapeutics against cancer and/or other diseases.
The cholesterol pathway: regulation, in immunity, and anti-tumor immunity
The Mevalonate pathway
Cholesterol synthesis begins in the mevalonate pathway with the combination of two metabolites of acetyl-CoA, which are further catalyzed by 3-hydroxy-3-methylglutaryl-CoA synthase and 3-hydroxy-3-methylglutaryl-CoA reductase, the latter of which is a highly regulated enzyme targeted by statin medications [5,6]. The subsequent product, mevalonic acid, is catalyzed by mevalonate kinase, which is highly regulated downstream by geranyl diphosphate (GPP) and farnesyl diphosphate (FPP). Mutations in mevalonate kinase resulting in its reduced activity can cause hyperimmunoglobulinemia D syndrome and mevalonic aciduria [5,7]. Both conditions can clinically present with a fever and increased serum immunoglobulin titers in human patients relative to healthy controls [7,8]. In regulatory B cells, mevalonate kinase deficiency due to inherited mutations diminishes systemic geranylgeranyl diphosphate concentrations, which results in inadequate production of IL-10 from human B cells [9]. Towards the downstream end of the mevalonate pathway, farnesyl pyrophosphate synthase (FPPS) converts geranyl diphosphate into farnesyl pyrophosphate (FPP). Subsequently, FPP is converted into geranylgeranyl pyrophosphate (GGPP) by geranylgeranyl pyrophosphate synthase [5,6,10]. Both FPP and GGPP contribute to posttranslational modifications of proteins such as adding lipid moieties, also known as prenylation [5,10]. Protein prenylation such as protein farnesylation and geranylgeranylation control the localization and activity of proteins in multiple biological processes, which has been discussed elsewhere [11].
The drug class statins, which are hydroxymethylglutaryl-CoA reductase inhibitors, not only reduce cholesterol concentrations, but can also alter the prenylation arm of the mevalonate pathway in human B cells [9]. In the TME, statins or amino-bisphosphonate FPPS inhibitors can reduce tumor cell proliferation, promote apoptosis, induce autophagy, decrease migration and invasion, and promote anti-inflammatory immunomodulation by affecting key proteins such as Ras, RhoA/C, Rac, and Rab [5,12].
The Sterol pathway
Squalene, a polyunsaturated hydrocarbon, is an essential intermediate for the endogenous synthesis of cholesterol. Squalene synthase combines two FPP molecules from the mevalonate pathway together to generate squalene in a two-step reaction and commits the entry of squalene into the sterol synthesis pathway [6]. Subsequently, squalene is converted into different sterols, including cholesterol, through a series of oxidation and reduction steps [13,14]. 7-dehydrocholesterol reductase (DHCR7) catalyzes the penultimate step in cholesterol synthesis for the Bloch pathway and the last step in the Kandutsch-Russell pathway [6,13,14]. Germline variants in the cholesterol pathway and altered cholesterol metabolites have been associated with impaired immunity (box 1). Moreover, cholesterol is frequently dysregulated in cancer, a concept that has been extensively reviewed (box 2).
Box 1: Sterol and oxysterol pathway alterations in human immunity.
Patients who suffer from Smith-Lemli-Opitz syndrome have an inactivating mutation in dehydrocholesterol reductase, causing a deficiency in cholesterol and accumulation of the precursors 7-dehydrocholesterol (7-DHC) and desmosterol [83]. These patients suffer from an array of abnormalities, including immune dysfunctions, such as increased allergies and inflammation that can be resolved with increased dietary cholesterol. Mast cells with DHCR7 knockout exhibit increased degranulation and cytokine production as a result of lipid raft disruption relative to controls; decreasing cholesterol and/or accumulation of 7-DHC has increased cytokine production and degranulation in mast cells [83]. Inactivating mutation in sterol-C4-methyl oxidase-like gene (SC4MOL) also causes a cholesterol biosynthesis defect [84]. The inactivation of SC4MOL in humans results in a deficiency in sterol-C4-methyl oxidase (SMO) and accumulation of C4-Methylsterols (MASs) [84]. MASs regulate lipid transport in the epidermis and play a key role in innate and adaptive immunity. Of note, SMO deficient patients exhibit increased cell proliferation in skin and blood and display an altered immunophenotype, evident from the elevated proportion of activated CD16+ granulocytes in circulation relative to healthy controls. Of note, decreasing sterol biosynthesis enzymes and metabolites occurs naturally in response to viruses and LPS-positive bacteria, as seen in macrophages and other tissues, including the liver [15,16,85].
Box 2: Cholesterol is crucial for cancer growth.
Cancer requires extensive metabolic reprogramming to support its unyielding growth and cell division. The reprogramming includes increased glycolysis, protein synthesis, nucleotide biosynthesis, and fatty acid and cholesterol synthesis or uptake, which must be elevated to support the biosynthesis of organelles and the plasma membrane [86]. To achieve the same, certain cancers -- including pancreatic ductal adenocarcinoma, prostate cancer, hepatocellular carcinoma, breast cancer stem cells, triple-negative breast cancer, colorectal cancer, clear-cell renal cell carcinoma, and esophageal squamous cell carcinoma -- increase cellular cholesterol concentrations through increased uptake, decreased export, or de novo synthesis, usually through the upregulation SREBP2 [87–98]. Cholesterol impinges upon several growth-promoting signaling pathways in cancer cells. Notably, the invasive effects of mutated TP53 -- a genetic alteration observed in nearly 50% of all cancers -- are mediated by the modulation of cholesterol biosynthesis, as shown in breast cancer cell lines [99]. Furthermore, mutant p53 can coimmunoprecipitate with SREBP2 and localize to the sterol regulatory elements in promoters of sterol synthesizing genes. Many cancers may upregulate cholesterol accumulation as a result of SREBP2 activity being driven by extracellular pH [94]. AKT can also stimulate the production of cholesterol through an AKT–PCK1–INSIG1/2–SREBP axis [100]. Further widespread potential can be seen with RAR-related orphan receptor gamma (RORγ) -- revealed as a crucial stem cell regulator, also known to activate SREBP2 to promote transcription of cholesterol synthesizing genes [96,101]. This finding is interesting given the role of RORγ in modulating inflammation and cytokines, as oxysterols act as agonists for RORγ, and RORγ inhibition in pancreatic cancer can promote apoptosis while hindering tumor initiation, growth, and propagation [24,101,102]. Furthermore, the mammalian target of rapamycin complex 1 (mTORC1) is frequently utilized by various cancers, relaying environmental and growth cues to growth-promoting mechanisms. Accordingly, the FDA has approved drugs to inhibit mTORC1 in certain cancers [103]. Of note, mTORC1 is also positively regulated by lysosomal cholesterol and increased mTORC1 activity has correlated with increased SREBP2 [104–107]. Lastly, cholesterol metabolism can play a key role in the oncogenic hedgehog signaling pathway and is mandatory for the activation of Smoothened [108]. Thus, cholesterol typically accumulates in cancer cells, where it can serve as a pivotal component accelerating the proliferation of malignant cells.
Sterol regulatory element-binding proteins (SREBPs) are master regulators of cholesterol synthesis. SREBP2 controls the transcription of sterol synthesizing enzymes [15,16]. Upon recognition of a viral or bacterial infection, SREBP2 and its targeted genes are decreased to prevent cholesterol synthesis and to promote the production of interferon-β (IFN-β) in human and mouse macrophages [16]. Of note, the response with IFN-β can be altered by modulating membrane stability, such as by replenishing depleted cholesterol or by adding 7-DHC to mouse macrophages [15]. Additionally, increased expression of cholesterol-25-hydroxylase plays a prominent role in viral infections -- enveloped viruses including VSV, HSV, HIV-1, and MHV68, and acutely pathogenic EBOV, RVFV, RSSEV, and Nipah viruses -- by increasing 25-hydroxycholesterol (25-HC) in mouse macrophages and B cells [17]. In the context of tumors, the impact of tumor-derived 25-HC can decrease cholesterol synthesizing genes by inhibiting SREBP2 and activating the liver X receptor (LXR) in human THP-1 monocyte cell line-derived macrophages [18]. The lipid 25-HC can also inhibit HCV infections through LXR-independent mechanisms [19]. 25-HC has been reported to impede membrane fusion and viral replication through several mechanisms, including decreasing web biogenesis in the human liver cancer cell line Huh-7.5 [19]. 25-HC can also induce immunity against Listeria monocytogenes and Shigella flexneri infections by activating acyl-CoA:cholesterol acyltransferase (ACAT), and hence, reducing the available cholesterol in the plasma membrane in HEK293A human fetal kidney cells [20]. In the context of cancer, tumor-derived extracellular vesicles (TEVs) precondition THP-1 monocytes by downregulating cholesterol 25-hydroxylase (Ch-25-HC), which results in the attenuated production of 25-HC relative to controls [21]. Ch-25-HC inhibits the uptake of TEVs by normal cells and limits the formation of pre-metastatic niches in melanoma mouse tumor models [21]. Recently, studies have shown downmodulation of 27-HC with SARS-CoV-2 infection; in one report, SARS-CoV-2 patients exhibited significantly decreased amounts of 27-HC but not 25-or 24-HC in the blood compared to the matched control group [22]. Also, 2HP-bCD:27OHC (27OHC complexed with 2-hydroxypropyl-β-cyclodextrin) demonstrated antiviral activity against SARS-CoV-2 and the human coronavirus HCoV-OC-43 pathogenic strains of CoVs in Vero E6 African green monkey kidney cells and Huh77 human hepatocellular carcinoma cells [22]. By contrast, in adipose tissue, 27HC treatment amplified the inflammatory response by increasing the accumulation of “M1-like” or classically activated macrophages in white adipose tissues [23]. Thus, in various settings, hindering sterol synthesis and modulating oxysterol concentrations can trigger certain immune responses or inflammatory events.
Cholesterol metabolism is tightly regulated to promote proper function while avoiding toxicity. Elevated amounts of cholesterol can be further fluxed into esterified cholesterol or be converted into oxygenated sterols [24,25]. SREBP cleavage-activating protein (SCAP) responds to a decreased concentration of endoplasmic reticulum cholesterol to promote active SREBP isoforms -1a, -1c, and -2 [26]. Oxygenated sterols, such as 25-HC, can inhibit cholesterol synthesis by hindering the ability of SCAP to promote active SREBP and by activating the liver X receptor (LXR) [24,25,27]. LXR serves as a cholesterol sensor by monitoring increased oxysterol concentrations; it also promotes transcriptional mechanisms to reduce cholesterol concentrations, such as those resulting in increased efflux [24]. Furthermore, oxysterols can promote the degradation of the rate-limiting enzyme HMG-CoA reductase (HMGR) and increase oxysterol flux into esterified cholesterol by allosterically promoting acyl-CoA:cholesterol acyltransferase and increasing the accessibility of plasma membrane cholesterol [25]. Oxysterols are therefore crucial regulators of cholesterol metabolism.
Impact of cholesterol and derivatives on the adaptive immune system
Changes in cholesterol metabolism can have a profound impact on T cell activity. Of note, different T cell subsets exhibit different cholesterol metabolism. Cholesterol, by associating with the TCRβ chain, enhances T-cell receptor nanoclustering and signaling as well as a more efficient formation of immunological synapses on CD8+ T cells [28]. This was evidenced from the enhanced effector function and increased proliferation of CD8+ but not CD4+ T cells upon inhibition of cholesterol esterification in T cells -- via genetic ablation or pharmacological inhibition of ACAT1, a key cholesterol esterification enzyme -- in C57BL/6J mice; this resulted in increased plasma membrane cholesterol concentrations in CD8+ T cells relative to controls [28]. By contrast, cholesterol can also negatively regulate TCR signaling. Specifically, a recent study utilized synthetic molecules to show that cholesterol and its metabolites could regulate the allosteric transitions of the TCR in human CD4+ Jurkat T cells, stabilizing the TCR in the resting state, and preventing its phosphorylation [29]. Another study used T cells derived from lymph nodes of mice deficient in a major enzyme responsible for sulfating cholesterol, Sult2b1−/− B6 (CD45.2+), or control mice, to demonstrate that cholesterol sulfate --a cholesterol metabolite -- disrupted the binding of cholesterol to the TCR-CD3 complex and broke down TCR nanoclusters into monomers; hence, the findings demonstrated that cholesterol sulfate could operate as a specific negative regulator of T cell signaling through the TCR–CD3 complex, and played an important role in regulating thymocyte sensitivity during maturation [30]. Also, tyrosine phosphatase CD45, which limits TCR signaling activation, is mostly pre-excluded from the tips of microvilli (MV) on human and mouse CD4+ and CD8+ T cells and Tregs prior to antigen encounter [31]. These seemingly contradictory pieces of evidence suggest that cholesterol promotes the potential for signaling while simultaneously preventing accidental signaling that could lead to autoimmunity (Figure 1). Furthermore, the role of cholesterol in T cell signaling is contextual. Notably, cholesterol metabolism is differentially regulated in γδ T cells, as compared to αβ T cells from C57BL/6J mice. Many genes involved in free cholesterol esterification (Acat1, 2, and Lcat) or utilization (Cyp39a1, Cyp46a1, Trerf1) are upregulated in γδ T cells, compared to αβ T cells [32]. In addition, γδ T cells from C57BL/6 mice carry more cholesteryl ester and lipid rafts that αβ T cells [32]. Furthermore, cholesterol depletion with methyl-β-cyclodextrin in splenocytes from C57BL/6J mice stimulated with T cell activating CD3/CD28 beads for 4 hours with or without cholesterol depletion, showed that cholesterol depletion reduced the activated phenotype of γδ T cells [32]. Therefore, further studies are warranted to investigate if T cell signaling and activation responses in vivo are modulated in hypercholesteremic patients or other patients that have received therapeutic treatments with statins.
Figure 1. Examples of immune system alterations associated with cholesterol metabolism in mammals.
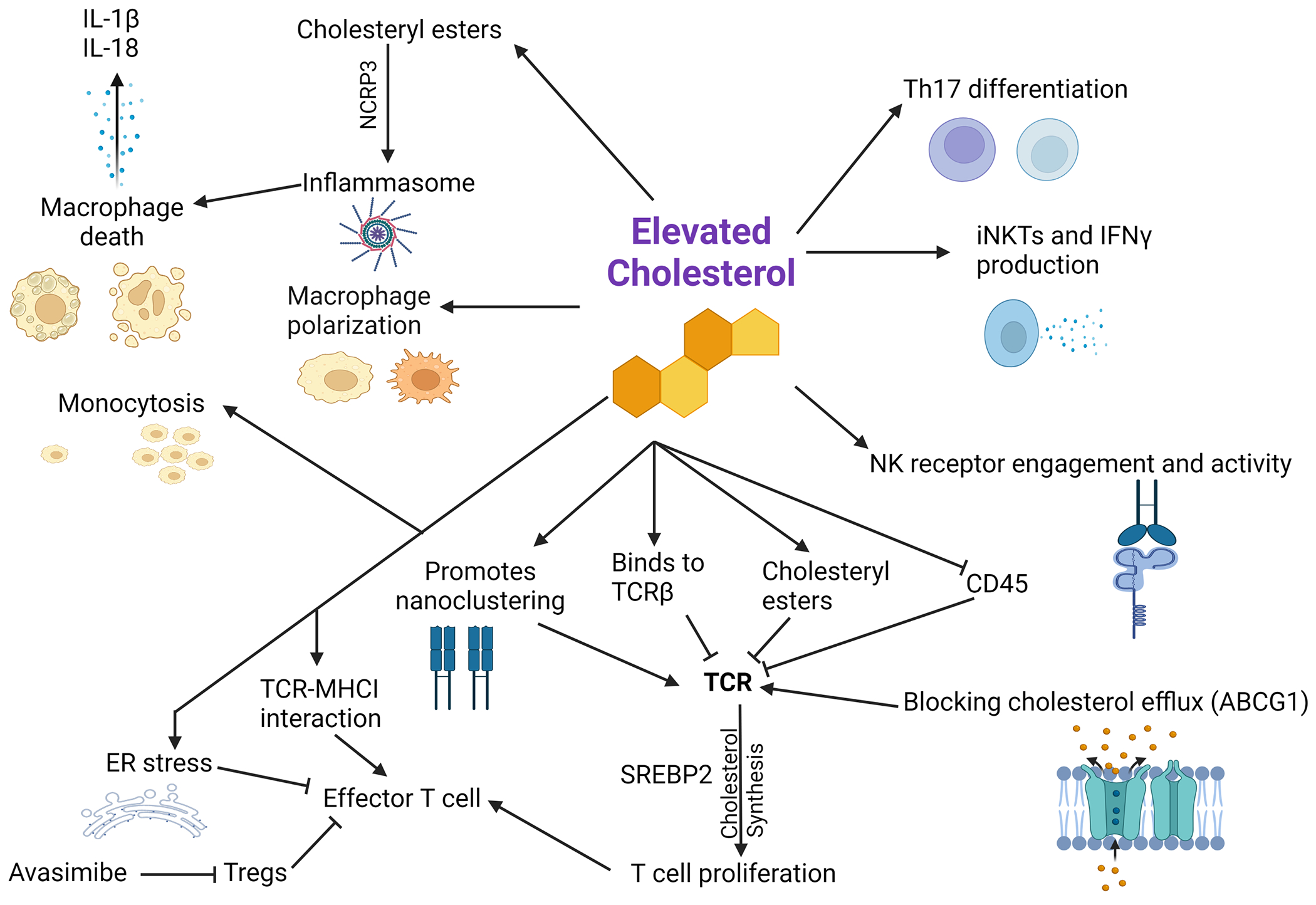
Depicted are key immunomodulatory roles of the cholesterol pathway in mammalian adaptive and innate immunity. Cholesterol can impact the T cell receptor (TCR) by direct binding or through indirect mechanisms, thereby regulating T cell signaling [29–31,112]. When a signal is present and TCR signaling occurs, T cells upregulate cholesterol production – which is needed for cell proliferation – through the sterol regulatory element-binding protein 2 (SREBP2) [18,26,34]. However, too much cholesterol can result in endoplasmic reticulum stress, thereby diminishing T cell function [4]. Avasimibe, which can inhibit cholesterol esterification, can inhibit regulatory T cells (Tregs) that are known to inhibit effector T cells [113]. Decreased cholesterol efflux through ATP-binding cassette transporter G1 (ABCG1) is associated with increased proliferation and TCR signaling [36]. Cholesterol can also play a variety of roles in macrophages, for example through monocytosis and inflammasome induction through cholesteryl ester accumulation, thereby triggering NLR family pyrin domain-containing 3 (NCRP3) to cause cell death and the release of IL-1β and IL-18 [54–56,58,114–116]. Cholesterol can also contribute to type 17 T helper (Th17) cell differentiation and interferon γ (IFN-γ) production by invariant natural killer T (iNKT) cells [63,117]. Cholesterol-rich lipid rafts enhance the co-localization of NK engagement receptors and are associated with increased NK activity [64]. Illustration created with BioRender.com. Abbreviations: ER, endoplasmic reticulum; MHCI, major histocompatibility complex class I; NK, natural killer; TCR, T cell receptor.
Cholesterol in the T cell membrane has another effect in the protection against perforin autolysis. Specifically, CD8+ T cells from BL/6 OTI transgenic mice are resistant to perforin binding and lysis in 51Cr release cytotoxicity assays, when compared to EL4 (CD8−) target cells [33]. Further, highly ordered sphingomyelin and cholesterol domains prevent perforin binding, as measured by atomic force microscopy, whereas oxysterol 7-ketocholesterol can promote disorder and perforin sensitivity in model membranes comprising lipid mixtures [33]. TCR activation in murine splenic and lymph node T cells with CD3+/−CD28, and phorbol 12-myristate 13-acetate with ionomycin, increased cholesterol and fatty acid synthesis gene expression, which could be abrogated by PI3K inhibitor LY294002 or mTOR inhibitor rapamycin [34]. Depleting SREBP cleavage-activating protein (SCAP), which is required for the mature forms of SREBPs, by using T cell-specific SCAP-deficient (Cd4-Cre-Scapfl/fl) mice, drastically inhibited the proliferation of CD8+ T cells in response to anti-CD3/CD28 antibody stimulation relative to controls [34]. Although SCAP impacted SREBP1/2, the supplementation of cholesterol alone significantly restored survival and proliferation of CD8+T cells in this model [26,34]. T cell dysfunction has also been observed, and is deemed to occur because of the cholesterol-rich TME in human colon cancer and myeloma tumors [4]. Of note, this study also showed that CD8+ T cells in the TME expressed elevated exhaustion markers PD-1, 2B4, TIM-3, and LAG-3 [4]. The cholesterol-specific findings from the previous studies [26,35] could also be recapitulated in this study by adding tumor culture supernatants from subcutaneously grown B16 melanoma tumors from C57BL/6 mice to CD8+ T cells [4]. This showed that tumor cell-secreted cholesterol upregulated the expression of the immune checkpoints. Moreover, the presence of cholesterol in cultures decreased CD8+ T cell proliferation, Ki67 expression, T cell migration, as well as cytokine and granzyme production, while promoting apoptosis in a dose-dependent mechanism in vitro [4]. Depleting cholesterol from the supernatant in vitro or modulating cholesterol amounts in vivo by using knockdown of HMG-CoA reductase (the rate-limiting enzyme in the cholesterol biosynthesis pathway) in B16 cells or through simvastatin (an HMG-CoA reductase inhibitor clinically used to lower cholesterol), reduced the expression of PD-1 and 2B4 exhaustion markers on CD8+ T cells, and diminished the B16 tumor burden in C57BL6/J mice [4]. Ultimately, this study revealed that T cell endoplasmic reticulum stress via cholesterol was responsible for this phenotype, in which the X-linked binding protein 1 (XBP1) was upregulated and could bind to the promoters of genes encoding PD-1 and 2B4 exhaustion markers to drive their gene expression [4]. These studies established a role for cholesterol in contributing to CD8+ T cell exhaustion via endoplasmic reticulum stress.
In addition to cholesterol, oxygenated cholesterol metabolites, also known as oxysterols, can also have a profound effect on T cell function (Figure 2). Oxysterols, as well as downstream LXR signaling, can negatively affect T cell activation [35]. Hence, tumor-derived oxysterols in the TME can impair anti-tumor immunity, as evidenced from decreased oxysterol amounts and subsequent LXR inhibition during T cell activation relative to controls[35]. Accordingly, LXR signaling inhibits the proliferation of purified mouse and human CD4+ and CD8+ T cells in vitro [35]. LXR promotes cholesterol efflux through ATP-binding cassette transporter G1 (ABCG1). Relative to wildtype mice, ABCG1-deficient T cells from Abcg1−/− C57BL6/J mice exhibit increased TCR signaling and demonstrate increased proliferation in vivo and in vitro upon CD3/CD28 stimulation [36]. However, oxysterols and LXR signaling can promote T cell activation indirectly, depending on the environment. LXR activation has been reported to enhance the effects of therapies such as immune checkpoint-blockade and adoptive T cell transfer, promoting robust responses [37]. Stimulating LXR can also inhibit myeloid-derived suppressor cells (MDSCs) that have been reported to inhibit T cells [38]. Inhibiting cholesterol esterification through ACAT can also enhance the proliferation and function of CD8+ T cells [28,37]. For instance, avasimibe -- a specific acyl coenzyme A-cholesterol acyltransferase 1 (ACAT1) inhibitor -- was recently found to improve Kras-cancer vaccination in a lung cancer mouse model, which was associated with increased tumor-infiltrating CD8+ T cells and decreased regulatory T cells (Tregs) relative to controls [39]. Avasimibe was chosen to help facilitate the TCR-major histocompatibility complex class I (MHCI) interaction based on the premise that cholesterol modulated TCR localization [18,39]. MHCI was also recently shown to play a key role in CD8+ T cell infiltration in the TME via PCSK9, a key cholesterol regulator; specifically, PCSK9 inhibition (either through genetic knockout in C57BL/6J mice or by using PCSK9 antibodies), caused a significant increase in tumor cell surface MHC I expression, promoting intratumoral infiltration of CD8+ T-cells [40]. Of note, PCSK9 regulates cholesterol metabolism by limiting the recycling of LDL receptors through lysosomal degradation [40]. Moreover, PCSK9 targeting enhanced the therapeutic efficacy of immune checkpoint blockade, as evidenced from the increased anti-tumor efficacy of mouse anti-PD-1 antibody treatment in syngeneic mice inoculated with PCSK9-deficient B16F10, MC38, 4T1, and CT26 tumor cells. The study further reported that PCSK9 could disrupt MHC I surface expression and recycling by physically binding to it, promoting its relocation and lysosome degradation [40]. Thus, these data showed that cholesterol, and its regulatory genes, played an important role in modulating immune responses in this context.
Figure 2. Examples of immune system alterations associated with oxysterols in mammals.
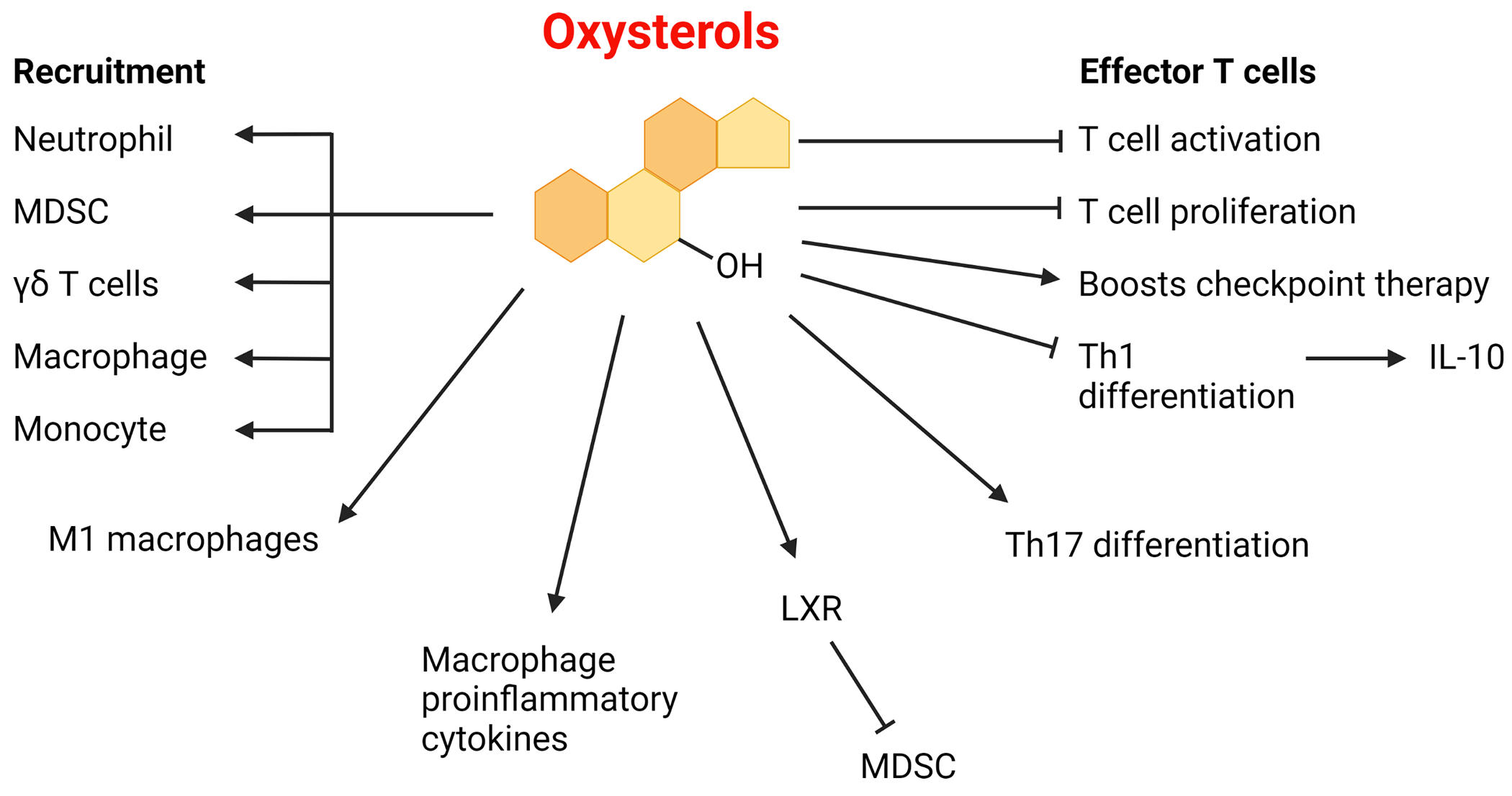
Oxysterols can act as recruiting factors for several types of immune cells [18,37,68,70]. Furthermore, oxysterols can induce macrophage inflammatory pathways and other immunoregulatory mechanisms [23,69]. Oxysterols can act as agonists for liver X receptor (LXR) to inhibit myeloid-derived suppressor cells (MDSCs), which can help enable helper T cell activation [38]. Although oxysterols can help boost checkpoint immunotherapy, they have also been reported to limit T cell proliferation [18,37,132]. Oxysterols also inhibit type 1 T helper (Th1) cell switching and can inhibit the switch from interferon-γ (IFNγ) production to IL-10 production [45]. Oxysterols can also promote Th17 differentiation in humans and mouse models, though the reverse outcome was observed in a mouse model of experimental autoimmune encephalomyelitis [102,131]. Oxysterols have been observed to promote a pro-inflammatory state in macrophages by enhancing the M1-like phenotype and promoting pro-inflammatory M2-like macrophages [137–139]. Illustration created with BioRender.com.
Like oxysterols, statins can inhibit cholesterol synthesis; however, statins can inhibit many other downstream pathways unrelated to cholesterol, such as prenylation and ubiquinone synthesis. Statins are seen to act as a double-edged sword as both T cells and cancer cells need functional cholesterol homeostasis and prenylation. However, knowing the relative sensitivity of different cell types in the TME toward statins is key to understanding the overall impact of statins in cancer (Figure 3). On the one hand, if the molecular profile of the cancer cell is geared towards enhanced cholesterol accumulation, it is possible that the cancer cell may have greater resistance or a greater dependency on cholesterol synthesis compared to endogenous proliferation or effector cues in T cells. Accumulating reports suggest that statins can cause cancer cell apoptosis though a wide variety of signaling pathways, depending on the cancer cell line [12]. Notably, phagocytosis of apoptotic PROb rat cancer cells by monocyte-derived antigen presenting cells can cause increased release of neoantigens, and increased MHC I and MHC II presentation, which triggered adaptive immune responses in rats [41]. Thus, statins may boost anti-tumor immune response via cancer cell apoptosis. Statins block mevalonate production and can decrease the downstream production of cholesterol precursor metabolites, including FPP, which is used by squalene synthase to generate squalene in the first committed step of the sterol synthesis pathway [42]. Farnesyl-diphosphate farnesyltransferase 1 (FDFT1), also known as squalene synthase, was identified in a recent CRISPR-Cas9 metabolic screen and found that Fdft1 was necessary to induce the proliferation of mouse spontaneous pancreatic tumor-derived cell lines and in vivo tumor growth in C57BL/6J mice in 3D cultures [43]. Of note, Fdft1 knockdown tumor cell grew slower in immunocompetent C57BL/6J mice than in athymic nude mice, suggesting a putative immune component to targeting squalene. Indeed, Fdft1 knockout tumors or tumors treated with FDFT1 inhibitor TAK-475, were associated with increased intratumoral CD8+ T cells relative to controls[43]. The presence of statins can also promote a more favorable environment for increasing CD8+ T cell infiltration by reducing M2-like immunosuppressive macrophages in FVB/N-Tg(MMTVneu) mice [44]. Recently, a functional cholesterol biosynthesis pathway was reported to be necessary for Th1 differentiation in human CD4+ T cells and for the production of immunosuppressive IL-10, which could be decreased by lipophilic statins or 25-FIC induced downregulation of cholesterol synthesis enzymes [45]. Furthermore, statins can elevate IFN-γ in vitro in human NK, CD4+, and CD8+ T cells, which may aid in the presentation of antigens by the MHC complex to be targeted [46–48]. Statins can promote infiltration of Tregs in human colorectal tumors, as shown via immunohistochemical analysis [49]. The mevalonate pathway can also serve to inhibit prenylation and synthesis of cholesterol needed by cancer cells, an effect which is being examined in ongoing and recently completed clinical trials in various cancers (NCT00572468i, NCT02569645ii, NCT03358017iii and NCT03134157iv). Overall, cholesterol, including its derivatives and precursors, can play key roles in modulating T cell phenotypes and functions.
Figure 3. Examples of immune system alterations associated with statins and nitrogen-containing bisphosphonates in mammals.

Statins and NBPs lower the production of cholesterol and decrease the production of downstream metabolites, with a potential increase in upstream metabolites; this can cause decreased prenyl pyrophase, acting as a danger signal, potentially leading to macrophage and dendritic cell (DC) death and the release of IL-1β and IL-18 [12,50–53,118–120]. γδ T cells can sense this danger signal directly through the Vγ9Vδ2 receptor, causing γδ T cell activation [118,119]. Statins can impair the function of natural killer (NK) cells through a variety of mechanisms, although IL-1β and IL-18 can also promote NK cell functions [12,58,60,61]. When cotreated with IL-2, simvastatin and mevastatin can increase IFN-γ production, thereby promoting major histocompatibility complex I (MHC I) presentation on nearby cells [46,47,121–123]. Furthermore, statins can increase the expression of MICA in melanoma, which allows targeting by NK cells [135]. Statins can also hinder the ability of antigen-presenting cells (APCs) to process and present antigens, in addition to stimulating effector cells, and can block proinflammatory type 1 T helper (Th1) cell differentiation into anti-inflammatory cells [45,124–128]. Statins can increase T cell activation, decrease inhibitory checkpoint molecules and decrease T cell exhaustion in some cancer mouse models [4,118,119,126]. Statin treatment can increase IFN production in NK cells and CD4+ and CD8+ T cells [46,48], and can also enhance cancer cell apoptosis, which may allow the presentation of neoantigens that can then be targeted by T cells [12]. Statins can induce regulatory T cells (Tregs) and may stimulate the production of TGF-β and IL-10 depending on the environment [49,129,136]. NBPs can inhibit myeloid-derived suppressor cells (MDSCs) and decrease M2-like to M1-like polarization [44,130]. Statins inhibit antigen processing and presentation, APC binding to T cells, and co-stimulatory molecules and cytokines; however, the latter has been observed in DC cells to depend on if the DC cell has already been stimulated [133,134]. Illustration created with BioRender.com.
Impact of cholesterol and derivatives on the innate immune system
Statins, cholesterol, and oxysterols can also affect innate immune cells (Figures 1,2,3). One similar characteristic linking the cholesterol pathway with the innate immune system is the induction and response to the inflammasome. The inflammasome is comprised of a large cluster of proteins used to activate caspase-1 to generate bioactive interleukin-1β (IL-1β) and IL-18 and ultimately induce cell death through pyroptosis [50–52]. The inflammasome can be triggered by decreased prenyl pyrophosphate and geranylgeranylation -- a mechanism mediated by statins. However, elevated amounts of cholesterol can lead to crystal formation and cellular stress, which can promote activation of NLR Family Pyrin Domain Containing 3 (NLRP3) to trigger the inflammasome in bone marrow-derived macrophages[53–58]. Cholesterol inhibitor (simvastatin) treatment of human peripheral blood mononuclear cells (PBMC) can enhances IFN-γ production by NK cells in the presence of IL-2 treatment [12,46]. Notably, simvastatin- and IL-2-treated human NK cells demonstrate improved cytotoxic killing of human kidney tumor cells in vitro, in a mechanism that depends on induced IFN-γ release by NK cells [12,46,59–61]. Of note, statin treatment can increase the expression of MHC class I chain-related protein A (MICA) on human melanoma cell lines, allowing them to be targeted by NK cells in vitro [12,47,62]. In contrast, lipophilic statins and hydrophilic fluvastatin can impair NK cytotoxicity through a prenylation-dependent mechanism, independently of cholesterol [61]. Of note, PPARγ-induces cholesterol synthesis in invariant natural killer T (iNKT) cells, which in turn induces their IFN-γ production [63]. iNKT specific Pparg deletion in PLZF-cre Ppargfl/fl mice, attenuates thymus accumulation of iNKT cells. Additionally, Pparg-deleted iNKT from these mice exhibit reduced IFN-γ production in ex vivo assays relative to wildtype controls [63]. Furthermore, in mice fed a high cholesterol diet, NK cells exhibited increased cholesterol accumulation in lipid rafts at the plasma membrane relative to control mice [64]. Also, enhanced co-localization of NK receptors in cholesterol-rich lipid rafts was associated with increased downstream signaling and NK cell activity against lung tumors in high cholesterol diet-fed mice relative to controls [64].
Other alterations in the cholesterol pathway impacting the innate immune system include examples such as ovarian cancer which can utilize >100 kDa hyaluronic acid oligomers to promote cholesterol efflux in macrophages, resulting in the subsequent depletion of cholesterol in mouse bone marrow-derived macrophages (BMDMs) [65]. As a result, mouse BMDMs exhibited upregulated Arg1 gene expression in response to IL-4, while downregulating Nos2 gene expression in response to IFN-γ [65]. These data suggested that cancer might potentially control macrophage polarization by influencing cholesterol metabolism. This was indeed recently observed in a study in which PD-1 knockout in myeloid cells in LysM-cre Pdcd1fl/fl mice directed the fate and phenotype of myeloid cells [66]. These myeloid cells avoided MDSC differentiation and instead, favored macrophage and dendritic cell (DC) differentiation in a mechanism that utilized mTORC1-induced cholesterol accumulation through SREBP activation in the mutant mice compared with wildtype mice [66]. This study also supported the role of cholesterol in promoting proinflammatory myeloid cell differentiation during hematopoiesis. Another study reported that targeting Leukocyte Immunoglobulin Like Receptor B2 (LILRB2), which promoted M1-like macrophage polarization while inhibiting M2-like macrophage differentiation, resulted in a change in cholesterol/lipid metabolism in human monocytes. In this study, anti-LILRB2 antibody treatment significantly attenuated the transcripts of genes associated with cholesterol pathways in human monocytes treated with IL-4, but not in LPS-derived macrophages. [67]. From another angle, one product of cholesterol oxidation, oxysterol 25-HC, has been documented to stimulate the migration of macrophages and monocytes [37,68]. Additionally, RNA-seq data of BMDMs derived from Ch25h−/− mice suggested that 25-HC could amplify macrophage inflammatory signaling and serve as a key mediator of innate immunity [69]. Another study showed that a T cell lymphoma cell line produced 27-HC oxysterol, fostering tumor growth by inducing neutrophil migration in a CXCR2-dependent mechanism in vitro and in vivo [18,70]. Also, LXR can recognize and binds oxysterols [18,25]. Thus, a previous study showed that LXRβ agonist treatment of mice could decrease tumor burden in multiple models including, lung, glioblastoma, ovarian, renal cell, triple negative breast, melanoma, and colon cancer models, which also correlated with a decreased frequency of intratumoral MDSCs in tumor-bearing mice [38].
Modulating statin and oxysterol concentrations.
Cholesterol modulation in cancers is a prevalent feature (box 3), and fortunately, FDA-approved statin inhibitors of cholesterol exist. Statins have numerous beneficial effects in combating cancer in multiple preclinical models, including pancreatic cancer, kidney cancer and prostate cancer [12]. A recent clinical study showed that initiation of statin therapy in the 12 months following diagnosis was associated with improved overall survival (OS) and breast cancer-specific survival (BCSS) benefits in I, II, and III Triple-negative breast cancer (TNBC) patients [71]. However, cholesterol has a complex role, with many facets and associations which depend on the environment and remain unexplored. Observational clinical studies have revealed that the impact of statins on tumors can be highly variable depending on the contextual environment, such as the type of cancer, type of statin (hydrophilic or lipophilic), if the statin was taken before diagnosis, the duration of statin use, and smoking status [12,72–74]. Other confounding issues have appeared in observational studies involving statins. For example, patients receiving statins may have presented with high cholesterol or have been pre-conditioned to exhibiting elevated cholesterol, cholesterol-associated comorbidities, cholesterol-independent benefits of statins, and statin-associated toxicities when combined with chemotherapies. As an example, a recent study demonstrated that familial hypercholesterolemia patients with long-term exposure to an enriched cholesterol environment harbored proinflammatory monocytes that persisted after statin treatment and which normalized cholesterol serum concentrations [75]. These results suggest that cholesterol might influence long-term metabolite-induced memory. Statins can also impact the production of vitamin D, ubiquinone, and protein prenylation, which may have an immunomodulatory impact that is largely independent from cholesterol [76–78].
Box 3: Examples of other cholesterol effects in cancer mouse models.
The cholesterol pathway can also impact key components of prenylation, dolichols, vitamin D, and ubiquinone. Cholesterol can also potentially affect other important factors, such as hormones, being a precursor of five major classes of steroid hormones [109]. Steroidogenesis is a process through which cholesterol is converted to steroids [110]. Further, CYP11A1 catalyzes the rate-limiting step for the conversion of cytoplasmic cholesterol into steroids. Thus, CD4+ T helper cells from Cyp11a1-mCherry reporter mice display increased expression of CYP11A1 upon activation in vitro [111]. Additionally, tumor-infiltrating T cells but not peripheral T cells display de novo steroidogenesis in melanoma and breast tumor-bearing- Cyp11a1-mCherry reporter mice. Notably, secreted steroids can impact other immune cells, as evidenced by increased M1-like/M2-like macrophage ratios in the TME of T cell-specific Cyp11a1 knockout mice [111]. Furthermore, in the TME, Tim-3+PD-1+ CD8+ T cells display increased expression of glucocorticoid receptors in colon cancer and melanoma-tumor-bearing mice. Also, this study shows that glucocorticoid treatments increase checkpoint inhibitor expression and dampen effector responses in murine and human CD8+ T cells in ex vivo assays [111]. Of note, low Cyp11a1 mRNA expression has correlated with a substantial survival benefit in colon adenocarcinoma and stomach adenocarcinoma in mouse models[111].
Concluding remarks
Multiple lines of evidence show that changes in the cholesterol pathway can modulate the immune system via various mechanisms and can exert a wide array of responses. This modulation is relevant for different diseases, including cancer, atherosclerosis, autoimmunity, and genetic disorders. To understand the impact of cholesterol modulation, we need to consider where in the cholesterol pathway it is occurring and in what environment. Depending on the environment, blocking the upstream production of cholesterol through statins might result in either immunosuppressive or immunostimulatory effects [4,5,12,46,47,53,59–61,79]. However, cholesterol, whether increased or decreased, can inhibit T cell functions. Oxysterols are also modulated by cancer to alter the immune system. This review highlights the significance of cholesterol alterations centered mostly around malignancies. Many more potential roles of cholesterol-related products have been observed to impact the immune system (box 3). The mechanisms and potential of cholesterol modulation are far-reaching. While many benefits have can exist in targeting the cholesterol metabolism, many questions remain (see Outstanding Questions). With the diverse and immunoregulatory functions of statins, how various combinations with statins might be utilized to yield putative clinical benefit, remains to be addressed. As discussed, the cholesterol-rich TME can lead to T cell exhaustion and upregulation of checkpoint inhibitors [4]. While simvastatin can downregulate immune checkpoint inhibitor expression, whether T cell functionality can be restored, remains an open question. The modulation of checkpoint inhibitors brings into question what possible combinations might be tested with different cholesterol pathway modifying drugs. Another question is whether checkpoint inhibitor drugs might truly bypass the presumed exhaustion brought on by cholesterol and in what contexts; alternatively, are these drugs simply treating a symptom rather than a cause for dysfunctional anti-tumor immunity. From a different perspective, recent studies have linked hypercholesterolemia with improved survival in certain advanced cancer patients that were treated with checkpoint immunotherapy [80–82]. If cholesterol inhibitors could also block checkpoint receptors, it is reasonable to speculate that potential synergistic effects might be achieved with other therapeutic approaches such as via adoptively transferred T cells, ex vivo pulsed DC cells, or augmenting NK cell numbers in tumors bearing low MHC I expression. Some success has been reported in targeting cholesterol through Pcsk9 inhibition combined with anti-PD1 immunotherapy for certain malignancies [40]. However, it remains to be determined which cancers can upregulate or downregulate cholesterol amounts, and which ones might respond positively to combination therapies. The immunoregulatory properties of the cholesterol pathway might show great promise when testing how effective it might eventually be across different cancer types and patient settings.
Outstanding Questions Box:
What are the differences in cholesterol metabolites in circulation and in the tumor microenvironment? It is known that cancer tissues have higher esterified cholesterol amounts than non-malignant tissues. The relative contributions of uptake versus synthesis in immune cells in different tumor models may hold an answer to finding optimal targets for manipulating anti-tumor immune responses.
What is the nature and relative effect of tumor cell-secreted factors on cholesterol metabolism in various immune cells? What would be some of the optimal targets for modulating cholesterol metabolism to induce tumor cell killing by cytotoxic immune cells? The modulation of such factors may be useful in identifying metabolic vulnerabilities, when aiming to selectively facilitate anti-tumor immunity while not supporting tumor growth.
What epigenetic factors/signaling pathways regulate various enzymes in cholesterol metabolism? Do such factors present actionable targets for eliciting anti-tumor immune response? How are such factors regulated by the somatic mutations in tumor cells and the heterogeneity of the tumor microenvironment? A better understanding of such factors may lead to novel candidate agents for precision medicine.
Given the heterogeneity of reports on the effect of targeting cholesterol metabolism in various immune cells, what is the net effect of targeting cholesterol on anti-tumor immune response? What factors govern the net outcome in tumors of diverse origins? Understanding such responses and factors may facilitate tumor cell-directed therapies against various tumor types.
What is the impact of elevated dietary cholesterol on the tumor microenvironment? Do popular diets, such as the ketogenic or western diet impact such outcomes? Identifying optimal diets to perturb the immune cell cholesterol uptake/metabolism might lead to novel insights in the field of immunology and immuno-oncology.
Highlights.
Frequent alterations in cholesterol homeostasis have been classically thought to support the production of building blocks for growth. However, the immunomodulatory properties of cholesterol, oxysterols, statins, and related metabolites can significantly impact the mammalian immune system.
Targeting cholesterol imbalances might be exploited to hinder tumor growth and restore immune functions in certain malignancies.
Excessive cholesterol can lead to CD8+ T cell exhaustion in certain models. Recent evidence suggests that targeting the cholesterol pathway might be a therapeutic approach that might synergize with PD-1 checkpoint blockade immunotherapy in certain cancer models.
Immune cells require cholesterol for activation. Therefore, excessive targeting or depletion of cholesterol and subsequent putative effects on immune cells should be major considerations.
Targeting the cholesterol pathway might constitute a promising approach in anti-cancer therapies. However, careful consideration must be given to the tumor microenvironment and the locations and contexts in which the pathway can be targeted.
Acknowledgments
This work was supported in part by the funding from National Institutes of Health (R01 CA216853, R01CA210439, R01CA163649, and P01 CA217798, NCI) to PKS.
Glossary
- Bloch pathway
One of the parallel metabolic pathways of cholesterol synthesis downstream of lanosterol; lanosterol produces cholesterol via sequential conversions into desmosterol, which requires a final desaturation by 24-dehydrocholesterol reductase to produce cholesterol.
- T cell Exhaustion
A stepwise and progressive loss of T-cell effector functions associated with chronic antigen exposure, e.g. in cancer and chronic infections.
- Hyperimmunoglobulinemia D
Rare, autosomal-recessive genetic disorder caused by mutations in the mevalonate kinase (MVK) gene; characterized by recurrent febrile episodes typically associated with lymphadenopathy, abdominal pain, and elevated serum polyclonal immunoglobulin D (IgD) titers.
- Immune checkpoint blockade
Immunotherapy drugs that block immune checkpoint proteins from binding to their partner proteins. Immune checkpoints include key pathways in the immune system that signal via molecules/receptors (e.g. PD-1, CTLA-4, etc.) to modify cell functions. In the case of the examples listed, these inhibitory receptors counter activation, and at stead-state, some of their functions include limiting immune cell toxicity (e.g to avoid fratricide) and overactivation, and contribute to establishing tolerance.
- Inflammasome
Multiprotein complex that acts as an innate immunity sensor and contributes to regulating inflammatory pathways; it activates caspase-1 to induce inflammation and cell death through pyroptosis.
- Kandutsch-Russell pathway
One of the parallel metabolic pathways of cholesterol synthesis; downstream conversions of lanosterol cause desaturation carried out by 24-dehydrocholesterol reductase to produce cholesterol.
- Liver X receptor (LXR)
Receptor belong to the nuclear receptor family of transcription factors; controls the production of cholesterol, among others. Oxysterols activate LXR to decrease intracellular cholesterol.
- M1-like macrophage
A proinflammatory macrophage known for its immune response and lytic capabilities.
- M2-like macrophage
An anti-inflammatory macrophage known for its role in wound healing and tissue repair.
- Mevalonic aciduria
An inborn error of metabolism caused by mevalonate kinase mutations and characterized by dysmorphology, psychomotor retardation, progressive cerebellar ataxia, and recurrent febrile crises.
- Myeloid-derived suppressor cell (MDSC)
Immature myeloid cell with strong immunosuppressive properties; can inhibit adaptive and innate immune responses either directly or indirectly.
- Natural killer (NK) cells
A cytotoxic innate immune cell known for killing without priming, but instead, by a balance of inhibitory and activating signals.
- Regulatory B cells
B cell subset exerting immunoregulatory functions
- RAR-related orphan receptor gamma (RORγ)
Transcription factor that can be activated by oxysterols to promote SREBP2 downstream genes, including those in the cholesterol pathway.
- Sterol regulatory element-binding protein (SREBP)
Denotes transcription factors regulating lipid and fatty acid synthesis (SREBP1) and cholesterol concentrations (SREBP2).
- SREBP cleavage-activating protein (SCAP)
Monitors cholesterol concentration in the endoplasmic reticulum. If the concentration is low, it acts as a chaperone for SREBPs to migrate to the Golgi where SREBPs are cleaved, allowing the migration to the nucleus to promote the synthesis of target genes.
- γδ (gamma delta) T cells
Unconventional T cell possessing a T-cell receptor (TCR) containing gamma and delta chains. γδ T cells play a role in recognizing lipid antigens and responding to danger signals; play important roles in mucosal tissues.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest: The authors disclose no conflicts of interest
Resources
This study is listed at https://clinicaltrials.gov/ct2/show/NCT00572468
This study is listed at https://clinicaltrials.gov/ct2/show/NCT02569645
This study is listed at https://clinicaltrials.gov/ct2/show/NCT03358017
This study is listed at https://clinicaltrials.gov/ct2/show/NCT03134157
References
- 1.Kawakami LM et al. (2017) Understanding How Sterols Regulate Membrane Remodeling in Supported Lipid Bilayers. Langmuir 33, 14756–14765. 10.1021/acs.langmuir.7b03236 [DOI] [PubMed] [Google Scholar]
- 2.Hoppstadter J et al. (2021) Dysregulation of cholesterol homeostasis in human lung cancer tissue and tumour-associated macrophages. EBioMedicine 72, 103578. 10.1016/j.ebiom.2021.103578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Donadon M et al. (2020) Macrophage morphology correlates with single-cell diversity and prognosis in colorectal liver metastasis. J Exp Med 217, 103578. 10.1084/jem.20191847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma X et al. (2019) Cholesterol Induces CD8(+) T Cell Exhaustion in the Tumor Microenvironment. Cell Metab 30, 143–156 e145. 10.1016/j.cmet.2019.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buhaescu I and Izzedine H (2007) Mevalonate pathway: a review of clinical and therapeutical implications. Clin Biochem 40, 575–584. 10.1016/j.clinbiochem.2007.03.016 [DOI] [PubMed] [Google Scholar]
- 6.Rondini EA et al. (2015) Transcriptional Regulation of Cytosolic Sulfotransferase 1C2 by Intermediates of the Cholesterol Biosynthetic Pathway in Primary Cultured Rat Hepatocytes. J Pharmacol Exp Ther 355, 429–441. 10.1124/jpet.115.226365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bekkering S et al. (2018) Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell 172, 135–146 e139. 10.1016/j.cell.2017.11.025 [DOI] [PubMed] [Google Scholar]
- 8.Frey T et al. (2019) Monocyte Production of IFN-gamma Is Interleukin-12 Dependent in a Model of Mevalonate Kinase Deficiency. J Interferon Cytokine Res 39, 364–374. 10.1089/jir.2018.0126 [DOI] [PubMed] [Google Scholar]
- 9.Bibby JA et al. (2020) Cholesterol metabolism drives regulatory B cell IL-10 through provision of geranylgeranyl pyrophosphate. Nat Commun 11, 3412. 10.1038/s41467-020-17179-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kavanagh KL et al. (2006) The crystal structure of human geranylgeranyl pyrophosphate synthase reveals a novel hexameric arrangement and inhibitory product binding. J Biol Chem 281, 22004–22012. 10.1074/jbc.M602603200 [DOI] [PubMed] [Google Scholar]
- 11.Wang M and Casey PJ (2016) Protein prenylation: unique fats make their mark on biology. Nat Rev Mol Cell Biol 17, 110–122. 10.1038/nrm.2015.11 [DOI] [PubMed] [Google Scholar]
- 12.Pisanti S et al. (2014) Novel prospects of statins as therapeutic agents in cancer. Pharmacol Res 88, 84–98. 10.1016/j.phrs.2014.06.013 [DOI] [PubMed] [Google Scholar]
- 13.Prabhu AV et al. (2016) DHCR7: A vital enzyme switch between cholesterol and vitamin D production. Prog Lipid Res 64, 138–151. 10.1016/j.plipres.2016.09.003 [DOI] [PubMed] [Google Scholar]
- 14.Mazein A et al. (2013) A comprehensive machine-readable view of the mammalian cholesterol biosynthesis pathway. Biochem Pharmacol 86, 56–66. 10.1016/j.bcp.2013.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao J et al. (2020) Targeting 7-Dehydrocholesterol Reductase Integrates Cholesterol Metabolism and IRF3 Activation to Eliminate Infection. Immunity 52, 109–122 e106. 10.1016/j.immuni.2019.11.015 [DOI] [PubMed] [Google Scholar]
- 16.York AG et al. (2015) Limiting Cholesterol Biosynthetic Flux Spontaneously Engages Type I IFN Signaling. Cell 163, 1716–1729. 10.1016/j.cell.2015.11.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu SY et al. (2013) Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 38, 92–105. 10.1016/j.immuni.2012.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fessler MB (2016) The Intracellular Cholesterol Landscape: Dynamic Integrator of the Immune Response. Trends Immunol 37, 819–830. 10.1016/j.it.2016.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anggakusuma et al. (2015) Interferon-inducible cholesterol-25-hydroxylase restricts hepatitis C virus replication through blockage of membranous web formation. Hepatology 62, 702–714. 10.1002/hep.27913 [DOI] [PubMed] [Google Scholar]
- 20.Abrams ME et al. (2020) Oxysterols provide innate immunity to bacterial infection by mobilizing cell surface accessible cholesterol. Nat Microbiol 5, 929–942. 10.1038/s41564-020-0701-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ortiz A et al. (2019) An Interferon-Driven Oxysterol-Based Defense against Tumor-Derived Extracellular Vesicles. Cancer Cell 35, 33–45 e36. 10.1016/j.ccell.2018.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marcello A et al. (2020) The cholesterol metabolite 27-hydroxycholesterol inhibits SARS-CoV-2 and is markedly decreased in COVID-19 patients. Redox Biol 36, 101682. 10.1016/j.redox.2020.101682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asghari A et al. (2019) 27-Hydroxycholesterol Promotes Adiposity and Mimics Adipogenic Diet-Induced Inflammatory Signaling. Endocrinology 160, 2485–2494. 10.1210/en.2019-00349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duc D et al. (2019) Oxysterols in Autoimmunity. Int J Mol Sci 20, 4522. 10.3390/ijms20184522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bielska AA et al. (2012) Oxysterols as non-genomic regulators of cholesterol homeostasis. Trends Endocrinol Metab 23, 99–106. 10.1016/j.tem.2011.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moon YA (2017) The SCAP/SREBP Pathway: A Mediator of Hepatic Steatosis. Endocrinol Metab (Seoul) 32, 6–10. 10.3803/EnM.2017.32.1.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Radhakrishnan A et al. (2007) Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc Natl Acad Sci U S A 104, 6511–6518. 10.1073/pnas.0700899104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang W et al. (2016) Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature 531, 651–655. 10.1038/nature17412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swamy M et al. (2016) A Cholesterol-Based Allostery Model of T Cell Receptor Phosphorylation. Immunity 44, 1091–1101. 10.1016/j.immuni.2016.04.011 [DOI] [PubMed] [Google Scholar]
- 30.Wang F et al. (2016) Inhibition of T cell receptor signaling by cholesterol sulfate, a naturally occurring derivative of membrane cholesterol. Nat Immunol 17, 844–850. 10.1038/ni.3462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jung Y et al. (2021) CD45 pre-exclusion from the tips of T cell microvilli prior to antigen recognition. Nat Commun 12, 3872. 10.1038/s41467-021-23792-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng HY et al. (2013) Increased cholesterol content in gammadelta (gammadelta) T lymphocytes differentially regulates their activation. PLoS One 8, e63746. 10.1371/journal.pone.0063746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rudd-Schmidt JA et al. (2019) Lipid order and charge protect killer T cells from accidental death. Nat Commun 10, 5396. 10.1038/s41467-019-13385-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kidani Y et al. (2013) Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol 14, 489–499. 10.1038/ni.2570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bensinger SJ et al. (2008) LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell 134, 97–111. 10.1016/j.cell.2008.04.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Armstrong AJ et al. (2010) ATP-binding cassette transporter G1 negatively regulates thymocyte and peripheral lymphocyte proliferation. J Immunol 184, 173–183. 10.4049/jimmunol.0902372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang B et al. (2020) Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nature Metabolism 2, 132–141. 10.1038/s42255-020-0174-0 [DOI] [PubMed] [Google Scholar]
- 38.Tavazoie MF et al. (2018) LXR/ApoE Activation Restricts Innate Immune Suppression in Cancer. Cell 172, 825–840 e818. 10.1016/j.cell.2017.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pan J et al. (2019) Potentiation of Kras peptide cancer vaccine by avasimibe, a cholesterol modulator. EBioMedicine 49, 72–81. 10.1016/j.ebiom.2019.10.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu X et al. (2020) Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature 588, 693–698. 10.1038/s41586-020-2911-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Henry F et al. (1999) Antigen-presenting cells that phagocytose apoptotic tumor-derived cells are potent tumor vaccines. Cancer Res 59, 3329–3332 [PubMed] [Google Scholar]
- 42.Mahboobnia K et al. (2021) PCSK9 and cancer: Rethinking the link. Biomed Pharmacother 140, 111758. 10.1016/j.biopha.2021.111758 [DOI] [PubMed] [Google Scholar]
- 43.Biancur DE et al. (2021) Functional Genomics Identifies Metabolic Vulnerabilities in Pancreatic Cancer. Cell Metab 33, 199–210 e198. 10.1016/j.cmet.2020.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mira E et al. (2013) A lovastatin-elicited genetic program inhibits M2 macrophage polarization and enhances T cell infiltration into spontaneous mouse mammary tumors. Oncotarget 4, 2288–2301. 10.18632/oncotarget.1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perucha E et al. (2019) The cholesterol biosynthesis pathway regulates IL-10 expression in human Th1 cells. Nat Commun 10, 498. 10.1038/s41467-019-08332-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gruenbacher G et al. (2010) IL-2 costimulation enables statin-mediated activation of human NK cells, preferentially through a mechanism involving CD56+ dendritic cells. Cancer Res 70, 9611–9620. 10.1158/0008-5472.CAN-10-1968 [DOI] [PubMed] [Google Scholar]
- 47.Tilkin-Mariame AF et al. (2005) Geranylgeranyl transferase inhibition stimulates anti-melanoma immune response through MHC Class I and costimulatory molecule expression. FASEB J 19, 1513–1515. 10.1096/fj.04-3482fje [DOI] [PubMed] [Google Scholar]
- 48.Coward WR et al. (2006) Statin-induced proinflammatory response in mitogen-activated peripheral blood mononuclear cells through the activation of caspase-1 and IL-18 secretion in monocytes. J Immunol 176, 5284–5292. 10.4049/jimmunol.176.9.5284 [DOI] [PubMed] [Google Scholar]
- 49.Al-Husein BA et al. (2018) Immunomodulatory effect of statins on Regulatory T Lymphocytes in human colorectal cancer is determined by the stage of disease. Oncotarget 9, 35752–35761. 10.18632/oncotarget.26293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fernandes-Alnemri T et al. (2007) The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ 14, 1590–1604. 10.1038/sj.cdd.4402194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghayur T et al. (1997) Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature 386, 619–623. 10.1038/386619a0 [DOI] [PubMed] [Google Scholar]
- 52.Martinon F et al. (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10, 417–426. 10.1016/s1097-2765(02)00599-3 [DOI] [PubMed] [Google Scholar]
- 53.Thurnher M et al. (2013) Regulation of mevalonate metabolism in cancer and immune cells. Biochim Biophys Acta 1831, 1009–1015. 10.1016/j.bbalip.2013.03.003 [DOI] [PubMed] [Google Scholar]
- 54.Akula MK et al. (2016) Control of the innate immune response by the mevalonate pathway. Nat Immunol 17, 922–929. 10.1038/ni.3487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liao YH et al. (2013) HMG-CoA reductase inhibitors activate caspase-1 in human monocytes depending on ATP release and P2X7 activation. J Leukoc Biol 93, 289–299. 10.1189/jlb.0812409 [DOI] [PubMed] [Google Scholar]
- 56.Montero MT et al. (2004) Geranylgeraniol regulates negatively caspase-1 autoprocessing: implication in the Th1 response against Mycobacterium tuberculosis. J Immunol 173, 4936–4944. 10.4049/jimmunol.173.8.4936 [DOI] [PubMed] [Google Scholar]
- 57.Duewell P et al. (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361. 10.1038/nature08938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Park YH et al. (2016) Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat Immunol 17, 914–921. 10.1038/ni.3457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Poggi A et al. (2013) Selective role of mevalonate pathway in regulating perforin but not FasL and TNFalpha release in human Natural Killer cells. PLoS One 8, e62932. 10.1371/journal.pone.0062932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Raemer PC et al. (2009) Statins inhibit NK-cell cytotoxicity by interfering with LFA-1-mediated conjugate formation. Eur J Immunol 39, 1456–1465. 10.1002/eji.200838863 [DOI] [PubMed] [Google Scholar]
- 61.Tanaka T et al. (2007) Lipophilic statins suppress cytotoxicity by freshly isolated natural killer cells through modulation of granule exocytosis. Int Immunol 19, 163–173. 10.1093/intimm/dxl133 [DOI] [PubMed] [Google Scholar]
- 62.Pich C et al. (2013) Statins Reduce Melanoma Development and Metastasis through MICA Overexpression. Front Immunol 4, 62. 10.3389/fimmu.2013.00062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fu S et al. (2020) Impaired lipid biosynthesis hinders anti-tumor efficacy of intratumoral iNKT cells. Nat Commun 11, 438. 10.1038/s41467-020-14332-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Qin WH et al. (2020) High Serum Levels of Cholesterol Increase Antitumor Functions of Nature Killer Cells and Reduce Growth of Liver Tumors in Mice. Gastroenterology 158, 1713–1727. 10.1053/j.gastro.2020.01.028 [DOI] [PubMed] [Google Scholar]
- 65.Goossens P et al. (2019) Membrane Cholesterol Efflux Drives Tumor-Associated Macrophage Reprogramming and Tumor Progression. Cell Metab 29, 1376–1389 e1374. 10.1016/j.cmet.2019.02.016 [DOI] [PubMed] [Google Scholar]
- 66.Strauss L et al. (2020) Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci Immunol 5. 10.1126/sciimmunol.aay1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen HM et al. (2018) Blocking immunoinhibitory receptor LILRB2 reprograms tumor-associated myeloid cells and promotes antitumor immunity. J Clin Invest 128, 5647–5662. 10.1172/JCI97570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eibinger G et al. (2013) On the role of 25-hydroxycholesterol synthesis by glioblastoma cell lines. Implications for chemotactic monocyte recruitment. Exp Cell Res 319, 1828–1838. 10.1016/j.yexcr.2013.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gold ES et al. (2014) 25-Hydroxycholesterol acts as an amplifier of inflammatory signaling. Proc Natl Acad Sci U S A 111, 10666–10671. 10.1073/pnas.1404271111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Raccosta L et al. (2013) The oxysterol-CXCR2 axis plays a key role in the recruitment of tumor-promoting neutrophils. J Exp Med 210, 1711–1728. 10.1084/jem.20130440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nowakowska MK et al. (2021) Association of statin use with clinical outcomes in patients with triple-negative breast cancer. Cancer 127, 4142–4150. 10.1002/cncr.33797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee HS et al. (2016) Statin Use and Its Impact on Survival in Pancreatic Cancer Patients. Medicine (Baltimore) 95, e3607. 10.1097/MD.0000000000003607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nielsen SF et al. (2012) Statin use and reduced cancer-related mortality. N Engl J Med 367, 1792–1802. 10.1056/NEJMoa1201735 [DOI] [PubMed] [Google Scholar]
- 74.Han JY et al. (2011) A phase 2 study of irinotecan, cisplatin, and simvastatin for untreated extensive-disease small cell lung cancer. Cancer 117, 2178–2185. 10.1002/cncr.25790 [DOI] [PubMed] [Google Scholar]
- 75.Bekkering S et al. (2019) Treatment with Statins Does Not Revert Trained Immunity in Patients with Familial Hypercholesterolemia. Cell Metab 30, 1–2. 10.1016/j.cmet.2019.05.014 [DOI] [PubMed] [Google Scholar]
- 76.Morck C et al. (2009) Statins inhibit protein lipidation and induce the unfolded protein response in the non-sterol producing nematode Caenorhabditis elegans. Proc Natl Acad Sci U S A 106, 18285–18290. 10.1073/pnas.0907117106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Perez-Castrillon JL et al. (2007) Effects of Atorvastatin on vitamin D levels in patients with acute ischemic heart disease. Am J Cardiol 99, 903–905. 10.1016/j.amjcard.2006.11.036 [DOI] [PubMed] [Google Scholar]
- 78.Watts GF et al. (1993) Plasma coenzyme Q (ubiquinone) concentrations in patients treated with simvastatin. J Clin Pathol 46, 1055–1057. 10.1136/jcp.46.11.1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sarrabayrouse G et al. (2010) Melanoma cells treated with GGTI and IFN-gamma allow murine vaccination and enhance cytotoxic response against human melanoma cells. PLoS One 5, e9043. 10.1371/journal.pone.0009043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Perrone F et al. (2020) The Prognostic Role of High Blood Cholesterol in Advanced Cancer Patients Treated With Immune Checkpoint Inhibitors. J Immunother 43, 196–203. 10.1097/CJI.0000000000000321 [DOI] [PubMed] [Google Scholar]
- 81.Tong J 3rd et al. (2021) Baseline Serum Cholesterol Levels Predict the Response of Patients with Advanced Non-Small Cell Lung Cancer to Immune Checkpoint Inhibitor-Based Treatment. Cancer Manag Res 13, 4041–4053. 10.2147/CMAR.S304022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Karayama M et al. (2021) Increased serum cholesterol and long-chain fatty acid levels are associated with the efficacy of nivolumab in patients with non-small cell lung cancer. Cancer Immunol Immunother. 10.1007/s00262-021-02979-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kovarova M et al. (2006) Cholesterol deficiency in a mouse model of Smith-Lemli-Opitz syndrome reveals increased mast cell responsiveness. J Exp Med 203, 1161–1171. 10.1084/jem.20051701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.He M et al. (2011) Mutations in the human SC4MOL gene encoding a methyl sterol oxidase cause psoriasiform dermatitis, microcephaly, and developmental delay. J Clin Invest 121, 976–984. 10.1172/JCI42650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Blanc M et al. (2011) Host defense against viral infection involves interferon mediated down-regulation of sterol biosynthesis. PLoS Biol 9, e1000598. 10.1371/journal.pbio.1000598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vander Heiden MG et al. (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. 10.1126/science.1160809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ehmsen S et al. (2019) Increased Cholesterol Biosynthesis Is a Key Characteristic of Breast Cancer Stem Cells Influencing Patient Outcome. Cell Rep 27, 3927–3938 e3926. 10.1016/j.celrep.2019.05.104 [DOI] [PubMed] [Google Scholar]
- 88.Che L et al. (2020) Cholesterol biosynthesis supports the growth of hepatocarcinoma lesions depleted of fatty acid synthase in mice and humans. Gut 69, 177–186. 10.1136/gutjnl-2018-317581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Guillaumond F et al. (2015) Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc Natl Acad Sci U S A 112, 2473–2478. 10.1073/pnas.1421601112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Krycer JR et al. (2012) A key regulator of cholesterol homoeostasis, SREBP-2, can be targeted in prostate cancer cells with natural products. Biochem J 446, 191–201. 10.1042/BJ20120545 [DOI] [PubMed] [Google Scholar]
- 91.Kim J et al. (2019) Uptake of HDL-cholesterol contributes to lipid accumulation in clear cell renal cell carcinoma. Biochim Biophys Acta Mol Cell Biol Lipids 1864, 158525. 10.1016/j.bbalip.2019.158525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhong C et al. (2019) SREBP2 is upregulated in esophageal squamous cell carcinoma and cooperates with cMyc to regulate HMGCR expression. Mol Med Rep 20, 3003–3010. 10.3892/mmr.2019.10577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang B et al. (2018) Phospholipid Remodeling and Cholesterol Availability Regulate Intestinal Stemness and Tumorigenesis. Cell Stem Cell 22, 206–220 e204. 10.1016/j.stem.2017.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kondo A et al. (2017) Extracellular Acidic pH Activates the Sterol Regulatory Element-Binding Protein 2 to Promote Tumor Progression. Cell Rep 18, 2228–2242. 10.1016/j.celrep.2017.02.006 [DOI] [PubMed] [Google Scholar]
- 95.Gebhard RL et al. (1987) Abnormal cholesterol metabolism in renal clear cell carcinoma. J Lipid Res 28, 1177–1184 [PubMed] [Google Scholar]
- 96.Cai D et al. (2019) RORgamma is a targetable master regulator of cholesterol biosynthesis in a cancer subtype. Nat Commun 10, 4621. 10.1038/s41467-019-12529-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jun SY et al. (2021) Reduction of Squalene Epoxidase by Cholesterol Accumulation Accelerates Colorectal Cancer Progression and Metastasis. Gastroenterology 160, 1194–1207 e1128. 10.1053/j.gastro.2020.09.009 [DOI] [PubMed] [Google Scholar]
- 98.Wen YA et al. (2018) Downregulation of SREBP inhibits tumor growth and initiation by altering cellular metabolism in colon cancer. Cell Death Dis 9, 265. 10.1038/s41419-018-0330-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Freed-Pastor WA et al. (2012) Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 148, 244–258. 10.1016/j.cell.2011.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Xu D et al. (2020) The gluconeogenic enzyme PCK1 phosphorylates INSIG1/2 for lipogenesis. Nature 580, 530–535. 10.1038/s41586-020-2183-2 [DOI] [PubMed] [Google Scholar]
- 101.Lytle NK et al. (2019) A Multiscale Map of the Stem Cell State in Pancreatic Adenocarcinoma. Cell 177, 572–586 e522. 10.1016/j.cell.2019.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Soroosh P et al. (2014) Oxysterols are agonist ligands of RORgammat and drive Th17 cell differentiation. Proc Natl Acad Sci U S A 111, 12163–12168. 10.1073/pnas.1322807111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang Y et al. (2017) A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/mTOR Pathway Alterations. Cancer Cell 31, 820–832 e823. 10.1016/j.ccell.2017.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lim CY et al. (2019) ER-lysosome contacts enable cholesterol sensing by mTORC1 and drive aberrant growth signalling in Niemann-Pick type C. Nat Cell Biol 21, 1206–1218. 10.1038/s41556-019-0391-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Eid W et al. (2017) mTORC1 activates SREBP-2 by suppressing cholesterol trafficking to lysosomes in mammalian cells. Proc Natl Acad Sci U S A 114, 7999–8004. 10.1073/pnas.1705304114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Castellano BM et al. (2017) Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science 355, 1306–1311. 10.1126/science.aag1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Triki M et al. (2020) mTOR Signaling and SREBP Activity Increase FADS2 Expression and Can Activate Sapienate Biosynthesis. Cell Rep 31, 107806. 10.1016/j.celrep.2020.107806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Canterini S et al. (2017) Shortened primary cilium length and dysregulated Sonic hedgehog signaling in Niemann-Pick C1 disease. Hum Mol Genet 26, 2277–2289. 10.1093/hmg/ddx118 [DOI] [PubMed] [Google Scholar]
- 109.Berg JM et al. (2002) Biochemistry 5 edn), W. H. Freeman and Company [Google Scholar]
- 110.Cain DW and Cidlowski JA (2017) Immune regulation by glucocorticoids. Nat Rev Immunol 17, 233–247. 10.1038/nri.2017.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mahata B et al. (2020) Tumors induce de novo steroid biosynthesis in T cells to evade immunity. Nat Commun 11, 3588. 10.1038/s41467-020-17339-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Martinez-Martin N et al. (2009) Cooperativity between T cell receptor complexes revealed by conformational mutants of CD3epsilon. Sci. Signal 2, ra43. [DOI] [PubMed] [Google Scholar]
- 113.Huang B et al. (2020) Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nat. Metab 2, 132–141 [DOI] [PubMed] [Google Scholar]
- 114.Lee MK et al. (2016) High-density lipoprotein inhibits human M1 macrophage polarization through redistribution of caveolin-1. Br. J. Pharmacol 173, 741–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Murphy AJ et al. (2011) ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J. Clin. Invest 121, 4138–4149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Seijkens T et al. (2014) Hypercholesterolemia-induced priming of hematopoietic stem and progenitor cells aggravates atherosclerosis. FASEB J 28, 2202–2213 [DOI] [PubMed] [Google Scholar]
- 117.Webb LM et al. (2020) Protein arginine methyltransferase 5 promotes cholesterol biosynthesis-mediated Th17 responses and autoimmunity. J. Clin. Invest 130, 1683–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gober HJ et al. (2003) Human T cell receptor gammadelta cells recognize endogenous mevalonate metabolites in tumor cells. J. Exp. Med 197, 163–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li J et al. (2009) Reduced expression of the mevalonate pathway enzyme farnesyl pyrophosphate synthase unveils recognition of tumor cells by Vgamma9Vdelta2 T cells. J. Immunol 182, 8118–8124 [DOI] [PubMed] [Google Scholar]
- 120.Nussbaumer O et al. (2011) DC-like cell-dependent activation of human natural killer cells by the bisphosphonate zoledronic acid is regulated by gammadelta T lymphocytes. Blood 118, 2743–2751 [DOI] [PubMed] [Google Scholar]
- 121.Rodriguez T et al. (2007) Distinct mechanisms of loss of IFNgamma mediated HLA class I inducibility in two melanoma cell lines. BMC Cancer 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Svane IM et al. (1997) Interferon-gamma-induced MHC class I expression and defects in Jak/Stat signalling in methylcholanthrene- induced sarcomas. Scand. J. Immunol 46, 379–387 [DOI] [PubMed] [Google Scholar]
- 123.Zhang S (2019) Systemic interferon-gamma increases MHC class I expression and T-cell infiltration in cold tumors: results of a phase 0 clinical trial. Cancer Immunol. Res 7, 1237–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Greenwood J et al. (2006) Statin therapy and autoimmune disease: from protein prenylation to immunomodulation. Nat. Rev. Immunol 6, 358–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Khan MA et al. (2009) Statins impair CD1d-mediated antigen presentation through the inhibition of prenylation. J. Immunol 182, 4744–4750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Okoye I et al. (2017) Atorvastatin downregulates co-inhibitory receptor expression by targeting Ras-activated mTOR signalling. Oncotarget 8, 98215–98232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hechinger AK et al. (2013) Inhibition of protein geranylgeranylation and farnesylation protects against graftversus- host disease via effects on CD4 effector T cells. Haematologica 98, 31–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Youssef S et al. (2002) The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature 420, 78–84 [DOI] [PubMed] [Google Scholar]
- 129.Meng X et al. (2012) Statins induce the accumulation of regulatory T cells in atherosclerotic plaque. Mol. Med 18, 598–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Coscia M et al. (2010) Zoledronic acid repolarizes tumour-associated macrophages and inhibits mammary carcinogenesis by targeting the mevalonate pathway. J. Cell Mol. Med 14, 2803–2815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cui G et al. (2011) Liver X receptor (LXR) mediates negative regulation of mouse and human Th17 differentiation. J. Clin. Invest 121, 658–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tavazoie MF et al. (2018) LXR/ApoE activation restricts innate immune suppression in cancer. Cell 172, 825–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Greenwood J et al. (2006) Statin therapy and autoimmune disease: from protein prenylation to immunomodulation. Nat. Rev. Immunol 6, 358–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Khan MA et al. (2009) Statins impair CD1d-mediated antigen presentation through the inhibition of prenylation. J. Immunol 182, 4744–4750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pich C et al. (2013) Statins reduce melanoma development and metastasis through MICA overexpression. Front. Immunol 4, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rodríguez-Perea AL et al. (2015) Statins increase the frequency of circulating CD4+FOXP3+ regulatory T cells in healthy individuals. J. Immunol. Res 2015, 762506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Buttari B et al. (2013) 7-Oxo-cholesterol potentiates pro-inflammatory signaling in human M1 and M2 macrophages. Biochem. Pharmacol 86, 130–137 [DOI] [PubMed] [Google Scholar]
- 138.Buttari B et al. (2014) Resveratrol counteracts inflammation in human M1 and M2 macrophages upon challenge with 7-oxocholesterol: potential therapeutic implications in atherosclerosis. Oxid. Med. Cell. Longev 2014, 257543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gargiulo S et al. (2015) Relation between TLR4/NF-κB signaling pathway activation by 27-hydroxycholesterol and 4-hydroxynonenal, and atherosclerotic plaque instability. Aging Cell 14, 569–581 [DOI] [PMC free article] [PubMed] [Google Scholar]