

ABSTRACT
C-Myc overexpression is a common finding in pancreatic cancer and predicts the aggressive behavior of cancer cells. It binds to the promoter of different genes, thereby regulating their transcription. C-Myc is downstream of KRAS and interacts with several oncogenic and proliferative pathways in pancreatic cancer. C-Myc enhances aerobic glycolysis in cancer cells and regulates glutamate biosynthesis from glutamine. It provides enough energy for cancer cells’ metabolism and sufficient substrate for the synthesis of organic molecules. C-Myc overexpression is associated with chemoresistance, intra-tumor angiogenesis, epithelial-mesenchymal transition (EMT), and metastasis in pancreatic cancer. Despite its title, c-Myc is not “undruggable” and recent studies unveiled that it can be targeted, directly or indirectly. Small molecules that accelerate c-Myc ubiquitination and degradation have been effective in preclinical studies. Small molecules that hinder c-Myc-MAX heterodimerization or c-Myc/MAX/DNA complex formation can functionally inhibit c-Myc. In addition, c-Myc can be targeted through transcriptional, post-transcriptional, and translational modifications.
KEYWORDS: C-myc, pancreatic cancer, BET inhibitor, chemoresistance, KRAS
Introduction
Pancreatic cancer constitutes 1.8% of all cancers and is responsible for 4.6% of cancer-related death. Its incidence, prevalence, and mortality markedly increased during recent years.1 Pancreatic ductal adenocarcinoma is the most common form of pancreatic cancer and accounts for 90% of all exocrine pancreatic cancers.2 Pancreatic ductal adenocarcinoma has a low 5-year survival rate ranging from 4.2% to 17.4% among patients of all stages.3 Despite great advances in chemotherapeutic methods, yet non-surgical treatments remain less effective for pancreatic ductal adenocarcinoma.3 There are several types of pancreatic cancer such as pancreatic ductal carcinoma, mucinous tumors, and pancreatic neuroendocrine tumors. Pancreatic cancer in this review is considered pancreatic ductal adenocarcinoma unless otherwise mentioned.
According to the latest studies, six months of gemcitabine and capecitabine or FOLFIRINOX regimen consisting of folinic acid, fluorouracil, irinotecan, and oxaliplatin are the first-line adjuvant chemotherapeutic management for pancreatic cancer. Likewise, nab-paclitaxel can be used for higher stages of this cancer. Despite many efforts, the chemotherapeutic response for pancreatic cancer remains weak and limited.4–7 Regarding the importance of pancreatic cancer and limited achievements in its treatment, new chemotherapeutic agents are needed to improve its management. Herein, in this review, a new target for chemotherapy with pronounced involvement in pancreatic cancer is discussed.
C-Myc oncoprotein has been implicated in the pathogenesis of several cancers such as pancreatic cancer, hepatocellular carcinoma, and breast cancer.8–10 Stabilized c-Myc can reprogram key genes expression involved in the metabolism, mitochondrial and ribosomal biogenesis, differentiation, apoptosis, and proliferation of cancer cells.8,11 C-Myc is located on human chromosome 8 and regulates genes expression by binding to their enhancer box (E-box).12 C-Myc is downstream of KRAS and its overexpression is found in 43.5% of primary pancreatic cancers.13,14 KRAS proto-oncogene is frequently found in tumor samples of patients with pancreatic cancer with more prevalence among patients with higher stages of the tumor. Even, the presence of KRAS mutation prognosticates shorter disease-free survival in patients with localized tumor.14 Recently, it was observed that activation of c-Myc is enough to convert indolent pancreatic intraepithelial neoplasm into pancreatic cancer in mice.15 Suppression of c-Myc in resistant pancreatic cancer could improve response to ordinary chemotherapeutic agents in preclinical studies.10
In this review, it is discussed how c-Myc is involved in the biology and progression of pancreatic cancer. Furthermore, this review attempts to illuminate the interaction between c-Myc and other molecules in pancreatic cancer. Finally, possible mechanisms to target c-Myc are briefly discussed.
The effect of c-Myc on cancer cells metabolism
C-Myc helps tumor cells to increase their glucose uptake and accelerate their lactate production instead of using the Krebs cycle for energy metabolism, even in the presence of a sufficient amount of oxygen. This effect is known as the Warburg effect or aerobic glycolysis.16 Glycolysis can provide a large amount of lactate that can be used for the anabolic processes of cancer cells.17 C-Myc can enhance pyruvate kinase isoenzyme type M2 (PKM2) expression to improve tumor cell glycolysis.18 Further, c-Myc-induced overexpression of PKM2 was shown to augment tumor cells proliferation, invasion, and chemoresistance.19 Similarly, it was shown that c-Myc, through repression of a lncRNA, IDH1-AS1, can downregulate isocitrate dehydrogenase 1 (IDH1). Increased activity of IDH1 increases α-ketoglutarate and downregulates HIF-1α. Hence, c-Myc through inhibition of IDH1 can upregulate HIF-1α and lactate production.20 Consistently, it was shown that c-Myc downregulation leads to inhibition of HIF-1α signaling in pancreatic cancer.21 In response to hypoxia, HIF-1α provokes glycolysis in pancreatic cancer cells to provide enough ATP and lactate.17 The collaboration between c-Myc and HIF-1α is responsible for the Warburg effect and lactate production in tumor cells.20 C-Myc also promotes the expression of lactate dehydrogenase A (LDHA) to convert pyruvate to lactate at the end of glycolysis.22 Inhibition of c-Myc downregulates LDHA expression in cancer cells.23 LDHA and PKM2 overexpression is a common finding in pancreatic cancer and enhances cancer cells proliferation and poor outcome.24,25 Consistently, treatment with either LDHA or PMK2 inhibitor could improve the efficacy of gemcitabine in pancreatic cancer, promote the apoptosis of pancreatic cancer cells and suppress their invasive behavior.26–28
C-Myc downregulates the expression of thioredoxin-interacting protein (TXNIP), which is a tumor suppressor and a potent inhibitor of glucose consumption.29 Repression of TXNIP expression strengthens the proliferative and metastatic capacity of pancreatic cancer.30 The inhibitory effect of TXNIP on glycolysis weakens the metastatic capacity of tumor cells.31 In addition, c-Myc promotes the expression of glucose transporters (GLUTs) such as GLUT1.9,32 GLUT1 overexpression predicts worse 3-year and 5-year overall survival and disease-free survival of solid tumors.33 By enhancing glucose uptake during glucose starvation, it can switch cellular metabolism from β-oxidation to glycolysis. Even, this property was shown to be protective against myocardial ischemia/reperfusion in an animal study. Moreover, it was observed that c-Myc can downregulate PGC-1α which is the major regulator of mitochondria biogenesis and negatively regulates glycolysis and Warburg effect.34–37 Consistently, it was shown that higher KRAS and c-Myc expression are associated with increased glucose uptake among patients with pancreatic cancer. Indeed, KRAS negatively regulates F-box/WD repeat-containing protein 7 (FBW7), a tumor suppressor gene. FBW7 also negatively regulates c-Myc in pancreatic cancer. Upregulation of KRAS abrogates the inhibitory effect of FBW7 on c-Myc and increases glucose uptake.38 Increased expression of GLUTs in cancer cells lets them uptake a higher amount of glucose in their microenvironment. This characteristic improves their survival during glucose starvation and provides enough energy for their rapidly progressive cell cycle. C-Myc is a master regulator of tumor cells metabolism which increases glucose consumption in the tumor microenvironment to provide enough energy and produce a sufficient amount of lipids, proteins, and nucleic acids for tumor cells proliferation.39 Interestingly, Sato et al. uncovered that hyperglycemia upregulates c-Myc expression in pancreatic cancer cells and expedites pancreatic cancer growth in mice.40
Gao et al. uncovered that c-Myc represses miR-23a and miR-23b to increase the expression of mitochondrial glutaminase in human cancer cells. The enzyme converts glutamine to glutamate which will be finally used for ATP production through the tricarboxylic acid (TCA) cycle. This function of c-Myc provides energy for cancer cells during energy depletion and protects them against oxidative stress.41 Cancer cells rely on glutamine for their anabolic process. Enough amount of glutamine guarantees cancer cells survival and proliferation. In addition, glutamine is critically involved in the uptake of essential amino acids and sustained activation of mTOR.42 Interestingly, it was uncovered that c-Myc also promotes the gene transcription of glutamine synthetase via demethylation of its promoter. In the presence of a sufficient amount of glucose in tumor cells’ microenvironment, glutamine synthetase increases de novo production of glutamine from glutamate and ammonia. Increased expression of glutamine synthetase reverses glutamine deficiency, improves tumor cells viability, and enhances nucleotide and amino acid transport.43 Similarly, it was observed that higher blood glutamine uptake by pancreatic cancer cells is associated with their invasive behavior and progression from pancreatic intraepithelial lesions to invasive pancreatic cancer.44 C-Myc reprograms metabolic pathways of pancreatic cancer cells to overcome their limitations (Figure 1).
Figure 1.
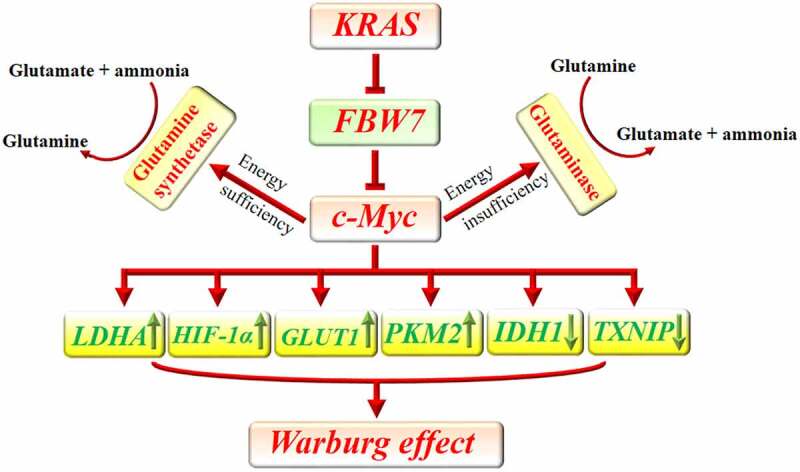
Implication of c-Myc in metabolic reprogramming of pancreatic cancer. KRAS downregulates FBW7 to prevent its inhibitory effect on c-Myc. C-Myc can increase the uptake of glucose by increasing the expression of GLUT1 and decreasing the expression of TXNIP. Furthermore, c-Myc promotes the expression of LDHA and PKM2 and decreases IDH1 to enhance glycolysis. Additionally, c-Myc potentiates HIF-1α which can similarly accelerate glycolysis. In the presence of energy insufficiency, c-Myc can promote glutaminase expression which subsequently increases glutamate bioavailability as a source of energy. In the presence of sufficient source of energy, c-Myc promotes the expression of glutamine synthetase and increases the available amount of glutamine which can be used for anabolic purposes in cancer cells.
C-Myc contributes to tumor angiogenesis, metastasis, and chemoresistance
Angiogenesis is an indispensable necessity for tumor growth, invasion, and metastasis. Normally, tumor cells enhance the expression of angiogenic factors such as VEGF to improve angiogenesis. The increased vascular network provides more blood supply for tumor cells proliferation and lets them migrate to other organs.45 C-Myc protects against endothelial dysfunction and enhances angiogenesis.46 It is also critically involved in tumor angiogenesis. C-Myc deletion is associated with decreased vascularization within the tumor. It can positively regulate the expression of VEGF and other molecules involved in angiogenesis.47,48 However, Dews et al. claimed that the effect of c-Myc on angiogenesis is not dependent on VEGF. Instead, it mainly downregulates the expression of anti-angiogenic factors such as thrombospondin-1 (Tsp1) and connective tissue growth factor (CTGF). C-Myc increases the expression of miR-17-92 and subsequently prevents the expression of Tsp1 and CTGF.49 Interestingly, Chen et al. revealed that c-Myc can post-transcriptionally promote the expression of HIF-1α and potentiate HIF-1α/VEGF axis to improve angiogenesis.50 In response to hypoxia, HIF-1α stimulates VEGF expression and accelerates angiogenesis.51 Targeting c-Myc as a positive regulator of angiogenesis can impair tumor cells’ metabolism and decrease their metastatic potential.
It was observed that fibroblast growth factor (FGF) binding protein 1 (FGFBP1) plays a pivotal role in the proliferation and metastasis of pancreatic cancer cells and negatively correlates with the survival of patients with pancreatic cancer. It was shown that FBW7 through a c-Myc-dependent manner downregulates FGFBP1. Also, downregulation of FBW7 results in c-Myc/FGFBP1 axis overactivity.52 FGFBP1 can contribute to angiogenesis which facilitates cancer cells migration.53
C-Myc enhances the expression of several transcription factors such as zinc finger E-box binding homeobox (ZEB) 1, ZEB2, Snail1, Snail2, and Twist to accelerate EMT.54 These downstream transcription factors can promote the transcription of EMT-associated genes such as MMPs, N-cadherin, and fibronectin. They also repress the transcription of E-cadherin, plakophilin, occludin, and cytokeratin to accelerate EMT of cancer cells.55 Consistently, it was shown that c-Myc increases matrix metallopeptidase (MMP) expression and decreases E-cadherin expression to augment EMT of pancreatic cancer cells.56–58 Increased expression of MMP and decreased expression of cell adhesion molecules can enhance cancer cells’ mobility.59 Therefore, c-Myc can enhance angiogenesis and accelerate EMT to facilitate cancer cells metastasis.
Pancreatic cancer poorly responds to chemotherapy and chemoresistance is the main obstacle for pancreatic cancer chemotherapy.59,60 Alteration of 1metabolic profile of cancer cells and activation of compensatory pathways seems to be involved in such behavior. C-Myc overexpression can strongly enhance pancreatic cancer cells viability against chemotherapy agents.10,61 Meanwhile, c-Myc downregulation or inhibition can improve chemoresistance and increase the efficacy of several chemotherapeutic agents such as nab-paclitaxel and gemcitabine.61–63 Therefore, simultaneous inhibition of c-Myc contributes to overcoming pancreatic cancer chemoresistance.
The correlation between c-Myc and oncogenic signaling pathways in pancreatic cancer
Aberrant activation of different signaling pathways is involved in the pathogenesis of pancreatic cancer.64 Additionally, inhibition of these proliferative signals showed satisfactory efficacy in preclinical studies of pancreatic cancer.64 As mentioned previously, KRAS mutation is a major propellant for the initiation and progression of pancreatic cancer. Mutant KRAS activates several signaling pathways in the pancreas to reprogram pancreatic acini into pancreatic cancer.65 Transgenic overexpression of KRAS resulted in the development of invasive pancreatic cancer in mice. Ablation or inhibition of c-Myc led to marked tumor shrinkage in these KRAS-mutated mice and increased cancer cells apoptosis.66 Likewise, downregulation of c-Myc inhibited KRAS-mediated pancreatic cancer cells growth in cell culture.67
KRAS activates phosphatidylinositol-3-kinase (PI3K) and WNT signaling pathways in pancreatic cancer.68–70 PI3K/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) signaling pathway is a major oncogenic pathway in pancreatic cancer which promotes tumor cells proliferation, epithelial-mesenchymal transition, and angiogenesis.71 Interestingly, it was shown that pan PI3K inhibitor or combinatorial PI3K/mTOR inhibitor can significantly decrease c-Myc expression in pancreatic cancer and inhibit cancer progression.71,72 In addition, c-Myc inhibition could significantly abrogate PI3K/AKT/mTOR-mediated proliferative response in pancreatic cancer.73 It was shown that protein phosphatase 2A (PP2A), a tumor suppressor gene, can downregulate c-Myc expression in pancreatic cancer and improve pancreatic cancer response to mTOR inhibitor. Meanwhile, it was shown that c-Myc overexpression can increase therapeutic resistance to mTOR inhibition. Further, decreased c-Myc expression improves cancer cells’ response to mTOR inhibition.74,75 C-Myc enhances mTOR-mediated 4EBP1 phosphorylation in the first stages of tumorigenesis.76 C-Myc blocks the inhibitory effect of tuberous sclerosis complex 2 (TSC2) on mTOR.77 In exchange, mTOR/S6/eIF4B regulates the translation of c-Myc in pancreatic cancer.78 The collaborative function of c-Myc and mTOR increases vascular endothelial growth factor (VEGF) expression in pancreatic neuroendocrine tumor which enhances intra-tumor vascular network and has been associated with lymph node metastasis.79 Furthermore, low expression of phosphatase and tensin homologue (PTEN) and liver kinase B1 (LKB1) in pancreatic neuroendocrine tumors resulted in decreased sensitivity to mTOR inhibitors and increased expression of c-Myc. LKB1 can inhibit mTOR/c-Myc through activation of AMPK/TSC2 signaling.80 Since AMPK negatively regulates mTOR and mTOR positively regulates c-Myc, AMPK activation and c-Myc inhibition both abrogated resistance to mTOR inhibitor.80 PTEN inhibits AKT/mTOR signaling to prevent cancer cells proliferation.80 It was shown that PTEN downregulates AKT/mTOR signaling pathway to decrease VEGF expression, intra-tumor angiogenesis, and tumor cells migration in pancreatic cancer.81 Mutual interaction between c-Myc and mTOR can promote ribosomal protein synthesis in tumor cells and provide their metabolic need for proliferation.82
Dysregulation of WNT/β-catenin signaling pathway leads to proliferative response, invasion, metastasis, and chemoresistance in pancreatic cancer.83,84 KRAS/WNT/β-catenin pathway increases c-Myc expression in pancreatic cancer to promote cancer cell proliferation.85,86 C-Myc overexpression provokes ribosomal biogenesis and c-Myc inhibition can partly reverse the effect of WNT/β-catenin pathway in pancreatic cancer.85 WNT/β-catenin signaling pathway relies on c-Myc for parts of its oncogenic function and inhibition of this signaling pathway suppresses the expression of c-Myc in pancreatic cancer.87–89 Inhibition of WNT/β-catenin signaling pathway seems to be an effective method to prevent the oncogenic effects of c-Myc in pancreatic cancer.90 It was shown that c-Myc can increase the transcription of WNT-related genes such as lymphoid enhancer-binding factor 1(LEF1) and augment WNT/β-catenin signaling pathway in cancer cells.91,92
Extracellular signal-regulated kinases (ERK) inhibition leads to growth arrest in KRAS-mutant pancreatic cancer which is partly mediated through c-Myc degradation.93 Additionally, it was shown that lncRNAs can modulate ERK function to regulate c-Myc and pancreatic cancer cells proliferation. For instance, LINC00261 inhibits ERK-mediated c-Myc expression and suppresses pancreatic cancer cells proliferation.94 Inhibition of ERK and attenuation of ERK-mediated c-Myc overexpression improved gemcitabine-induced cytotoxicity in pancreatic cancer.62
As mentioned previously, KRAS mutation is a major propellant for the initiation and progression of pancreatic cancer. Mutant KRAS activates several signaling pathways in the pancreas to reprogram pancreatic acini into pancreatic cancer.65 In addition to PI3K/AKT/mTOR and WNT/β-catenin, Mutant KRAS activates Raf/MEK/ERK signaling pathway to upregulate c-Myc.93,95 ERK was shown to be involved in the epidermal growth factor (EGF)/EGF receptor-mediated increase in c-Myc expression which has been associated with development, growth, and invasion of pancreatic cancer.96,97 EGF receptor also activates PI3K pathway to stimulate cancer cells proliferation.98 Interestingly, it has been elucidated that KRAS promotes EGFR signaling to increase c-Myc signaling in pancreatic cancer. However, EGFR signaling inhibition was not sufficient to inhibit EKR and PI3K signaling after KRAS activation.99
Inhibition of each one of these oncogenic pathways in pancreatic cancer can provide a limited benefit or is associated with chemoresistance through upregulation of other pathways, while, their simultaneous inhibition, for example, concurrent inhibition of PI3K and MEK pathways can conceivably improve treatment efficacy.93,100 Targeting c-Myc as the common downstream of several oncogenic pathways in pancreatic cancer may bring more favorable outcomes than targeting each one of them, alone (Figure 2).
Figure 2.
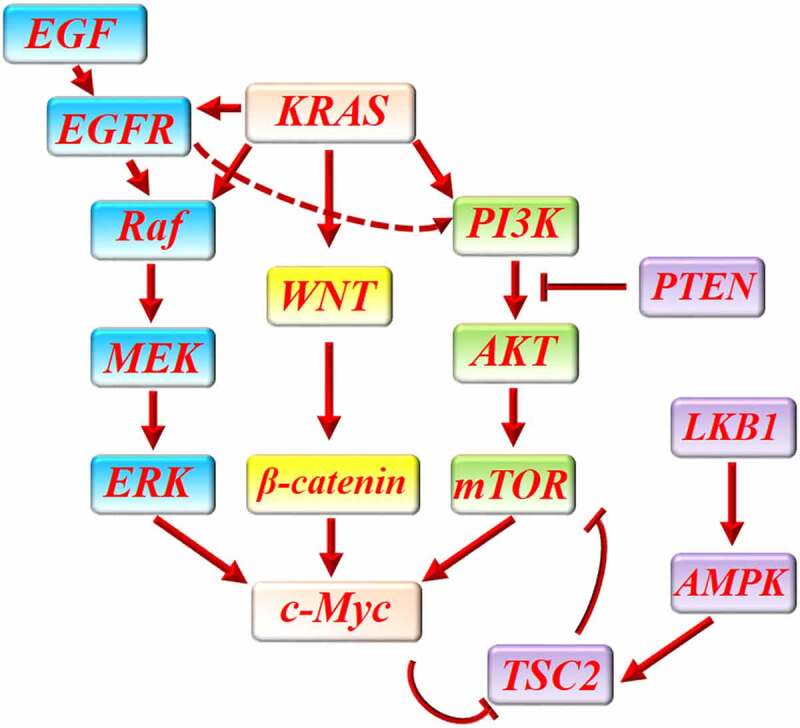
The interaction between c-Myc and main proliferative signaling pathways in pancreatic cancer. KRAS activates several signaling pathways in pancreatic cancer to promote cancer cells proliferation. WNT/β-catenin pathway, PI3K/AKT/mTOR pathway and Raf/MEK/ERK are three major proliferative pathways which are activated by KRAS in pancreatic cancer. In addition, KRAS enhances EGF/EGF receptor-mediated activation of PI3K/AKT/mTOR and Raf/MEK/ERK pathway. All of these pathways can finally increase the expression of c-Myc to promote cancer cells proliferation. LKB1/AMPK/TSC2 and PTEN can protect against pancreatic cancer through downregulation of PI3KAKT/mTOR pathway. C-Myc can increase mTOR function by abrogating the inhibitory effect of LKB1/AMPK/TSC2 on mTOR.
C-Myc downregulation makes pancreatic cancer cells susceptible to oxidative stress
Uncontrolled release of reactive oxygen species (ROS) is immensely involved in the anti-cancer effect of radiotherapy and chemotherapeutic agents.101 An excessive amount of ROS damages cancer cells organelles and biomacromolecules and endangers their DNA integrity leading to cancer cells death.101 Likewise, it has been found that cancer cells particularly pancreatic cancer cells recruit specific mechanisms to augment their defense mechanisms against oxidative stress.102 This capability has been implicated in the chemoresistance of pancreatic cancer cells.102 In addition, it was found that attenuation of these self-defense mechanisms can vigorously abrogate the chemoresistance of pancreatic cancer cells to routine chemotherapeutic agents such as 5-fluorouracil and gemcitabine.102 Regarding the effect of c-Myc on aerobic glycolysis and mitochondrial respiration, c-Myc upregulation is a major defense mechanism against oxidative stress in pancreatic cancer.103 Impaired mitochondrial oxidative phosphorylation results in oxidative burst and leads to cancer cells death.104 It was found that c-Myc downregulation contributes to mitochondrial respiration, accelerates oxidative stress, and contributes to overcoming chemoresistance.105 Furthermore, c-Myc binds to the promoter site of nuclear factor erythroid 2–related factor 2 (Nrf2) and enhances its expression.106 Nrf2 is the main transcription factor for numerous endogenous antioxidants.106 The interaction between c-Myc and Nrf-2 was shown to be crucially involved in the survival of pancreatic cancer cells.106 These findings show that c-Myc overexpression not only expedites tumor cells proliferation but also protects them against stress conditions.
C-Myc interacts with cyclin-dependent kinase (CDK) to enhance pancreatic cancer cells proliferation
CDKs are serine/threonine kinases that interact with cyclins to regulate cell cycle progression. Dysregulation and overactivity of CKDs are characterized by excessive proliferation and are a hallmark of cancers.107 Interestingly, it was uncovered that tumor cells use c-Myc overexpression as a resistance mechanism against CDK inhibitors. For instance, CDK4/6 inhibition has been associated with increased mTOR signaling and led to overexpression of c-Myc. These alterations reprogrammed cancer cells’ metabolism and improved glycolysis.108 In return, c-Myc can potentiate the cyclin-CDK system in pancreatic cancer cells by increasing the expression of its members such as cyclin D1, CDK1, and cyclin B1, and decreasing the expression of P21, cyclin-dependent kinase inhibitor 1.109 Besides, it has been shown that some CDKs such as CDK9 are involved in the expression of c-Myc and their inhibition will result in decreased c-Myc abundance in pancreatic cancer.67 These CKDs possess a pivotal role in the transcription initiation and elongation of c-Myc mRNA.110,111 Therefore, c-Myc can interact with the cyclin-CDK system to accelerate cell cycle progression and induce chemoresistance.
Targeting c-Myc improves immunotherapy in pancreatic cancer
Immune checkpoint blockade has shown acceptable efficacy in different cancers but not in pancreatic cancer.112 The presence of a markedly immunosuppressive and immune-privileged microenvironment and the absence of an effective level of T cells infiltration in pancreatic cancer have been implicated in the weak response of pancreatic cancer to immune checkpoint blockade.112 Hence, finding new solutions to overcome the immune-evasion characteristic of pancreatic cancer can greatly help its treatment.112
Previously, it has been found that c-Myc contributes to cancer cells’ immune evasion. C-Myc overexpression prevents immune cells from attacking tumor cells.113 Interestingly, the expression of c-Myc and programmed death ligand 1 (PD-L1) was positively correlated in the tissue samples obtained from patients with esophageal cancer.113 Furthermore, c-Myc overexpression, silencing, and inhibition replicated the same pattern on the expression of PD-L1 in cell culture of esophageal cancer, suggesting that c-Myc is a major regulator of PD-L1.113 Likewise, downregulation of c-Myc suppressed PD-L1 expression in non-small cell lung carcinoma and attenuated tumor cells immune-evasion.114 PD-L1 is vigorously expressed by tumor cells and acts as a defense mechanism.115 It binds to programmed death 1 (PD-1) on immune cells particularly on T cells surface and instigates their death.115 Inhibition of c-Myc enhanced the abundance of CD4+ cells, CD8+ cells, B cells, dendritic cells, and natural killer cells in the tumor microenvironment, in a mice model of prostate cancer.116 Furthermore, c-Myc inhibitor potentiated the anti-cancer efficacy of anti-PD-1 immunotherapy.116 Similarly, it was found that c-Myc expression and PD-L1 expression are positively correlated in the tissue specimens obtained from 87 patients with pancreatic cancer.117 In addition, activation of c-Myc in the mice model of pancreatic intraepithelial neoplasm has been associated with decreased abundance of infiltrating T cells and higher incidence of pancreatic cancer.15 Consistently, inhibition of c-Myc in mice or silencing it in cell culture downregulated the expression of PD-L1 in pancreatic cancer.117 Moreover, inhibition of c-Myc and PD-L1 blockade exerted a synergistic effect on the mice model of pancreatic cancer.117 C-Myc is a major transcription factor for PD-L1 in pancreatic cancer and c-Myc knockdown profoundly decreases PD-L1 expression in pancreatic cancer.118
C-Myc also increases CD47 expression in pancreatic cancer.119 C-Myc increases the transcription of CD47 in cancer cells. CD47 is located on the surface of cancer cells and interacts with signal-regulatory protein alpha (SIRP-α) to induce tolerance in immune cells, prevent cancer cells phagocytosis, and prolong cancer cells survival in pancreatic cancer.120,121 CD47 blockade enhanced the antitumor function of CD8 + T cells and macrophages in the mice model of pancreatic cancer.122 Likewise, immunotherapy with CD47-CAR-T cells significantly reduced pancreatic cancer cells population and prevent pancreatic cancer growth in mice.123
Interestingly, c-Myc is involved in the homeostasis and competent function of T regulatory cell which acts as the main player in tumor cells immune-evasion.124,125 Downregulation of c-Myc can impair the immunosuppressive function of T regulatory cells, thereby enhancing the anti-cancer capacity of effector T cells.124 These findings reveal that c-Myc is widely involved in the immune cells-tumor cells cross-talk and downregulation of c-Myc can enormously improve immunotherapy in pancreatic cancer.
Interaction between c-Myc and long non-coding RNAs in pancreatic cancer
LncRNAs are not translated into proteins but can modulate physiological processes such as growth, metabolism, and aging. They exert epigenetic modifications and regulate gene expression during and after transcription.126 Several lncRNAs were shown to be correlated with the prognosis of pancreatic cancer. Aberrant expression of these lncRNAs was shown to be involved in pancreatic cancer cells proliferation, invasion, and metastasis. Additionally, they interact with intracellular signaling pathways and modulate the expression of several proto-oncogenes and tumor-suppressor genes.127–129 A great number of these lncRNAs act through c-Myc. They can promote or inhibit the transcription of c-Myc, hence regulating its oncogenic effects. For instance, it was shown that LINC00261, a methylation-mediated lncRNA, is downregulated in pancreatic cancer. Increased methylation of the promoter of LINC00261 in pancreatic cancer decreases its transcription and is a sign of poor prognosis in pancreatic cancer. LINC00261 decreases c-Myc transcription by methylation of its promoter and inhibits its biologic function.94,127 LINC00346 is another lncRNA that promotes tumorigenesis, pancreatic cancer cells proliferation, migration, and invasion and is negatively associated with overall survival (OS) and disease-free survival in pancreatic cancer. It was revealed that LINC00346 is a positive regulator of c-Myc. Indeed, LINC00346 binds to CCCTC-binding factor (CTCF), a transcriptional repressor of c-Myc, to increase the transcription of c-Myc.130 LncRNA XLOC_006390 can similarly enhance the biological function of c-Myc by protecting it against ubiquitination and subsequent degradation.131 LncRNA HULC is overexpressed in pancreatic cancer which enhances pancreatic cancer cells proliferation and invasion and augments c-Myc expression.132 Furthermore, P53 can increase the expression of lncRNA Pvt1 to suppress the transcription of c-Myc and prevent tumor growth.133
The relationship between c-Myc and non-coding RNAs seems to be mutual and c-Myc can also regulate the expression of several non-coding RNAs involved in the pathogenesis of pancreatic cancer.128 For instance, CCAT1 is a lncRNA that is highly expressed in pancreatic cancer and promotes pancreatic cancer cells proliferation and migration. CCAT1 silencing has been associated with pancreatic cancer cell cycle arrest and decreased cyclin D1 expression. It was revealed that c-Myc increases the expression of CCAT1 by targeting its promoter. Consistently, downregulation of c-Myc has been associated with decreased CCAT1 expression.128
Interestingly, it was uncovered that dysregulated expression of these lncRNAs is also involved in the chemoresistance of pancreatic cancer cells. For instance, it was shown that overexpression of lncRNA GSTM3TV2 is associated with overexpression of c-Myc in pancreatic cancer cells and gemcitabine resistance.134 The interaction between lncRNAs and c-Myc has been implicated in the metastasis and chemoresistance of several cancers.135 Besides, it was shown that these non-coding RNAs can interact with c-Myc to regulate cancer cells metabolism.136 Herein, it was shown that XLOC_006390, a lncRNA, promotes c-Myc-mediated expression of glutamate dehydrogenase 1 to increase glutamate metabolism in pancreatic cancer.131 Regarding the interaction between c-Myc and lncRNAs in pancreatic cancer and the importance of these lncRNAs in pancreatic cancer, pharmacological inhibition of c-Myc can attenuate the deleterious effect of numerous lncRNAs in pancreatic cancer (Table 1). Also, lncRNAs mimics or inhibitors can be used to regulate the function of c-Myc. This will improve chemoresistance and inhibit tumor cells proliferation, invasion, and metastasis.
Table 1.
Interaction of different lncRNAs and microRNAs with c-Myc in pancreatic cancer
| Author | None-coding RNA | Micro-/lnc-RNA | Finding |
|---|---|---|---|
| Liu et al. Zhai et al. | LINC00261 | lncRNA | LINC00261 decreases c-Myc transcription via methylation of its promoter. |
| Peng et al. | LINC00346 | lncRNA | LINC00346 binds to CCCTC-binding factor (CTCF) to attenuate its inhibitory effect on c-Myc expression. |
| He et al. | XLOC_006390 | lncRNA | XLOC_006390 stabilizes c-Myc by preventing its ubiquitination and subsequent degradation. |
| Ou et al. | HULC | lncRNA | HULC promotes c-Myc expression to increase pancreatic cancer cells proliferation. |
| Yu et al. | CCAT1 | lncRNA | C-Myc increases the transcription of CCAT1 in pancreatic cancer. Subsequently, CCAT1 enhances the expression of cyclin D1 to increase pancreatic cancer proliferation. |
| Xiong et al. | GSTM3TV2 | lncRNA | GSTM3TV2 promotes the expression of c-Myc in pancreatic cancer and is associated with chemoresistance. |
| Olivero et al. | Pvt1 | lncRNA | P53 promotes the expression of Pvt1. Subsequently, Pvt1 suppresses the expression of c-Myc to prevent tumor growth. |
| He et al. | XLOC_006390 | lncRNA | XLOC_006390 increases glutamate bioavailability in cancer cells through upregulation of c-Myc. |
| Dey et al. | miR-29a | microRNA | C-Myc decreases the expression of miR-29a to prevent its inhibitory effect on the expression of several oncogenes such as LOXL2, MYBL2 and HGK. |
| Azmi et al. | MiR-145 | microRNA | MiR-145 downregulates the expression of several proto-oncogenes such as EGFR, MMP1 and c-Myc. |
| Liu et al. | MiR-494 | microRNA | MiR-494 decreases pancreatic cancer cells proliferation and accelerates their apoptosis by suppressing the expression of c-Myc and SIRT1. |
| Sureban et al. | let-7a | microRNA | Let-7a is a tumor suppressor that can downregulate c-Myc expression in pancreatic cancer. |
Interaction between c-Myc and microRNAs in pancreatic cancer
MicroRNAs are non-coding RNAs that can post-transcriptionally regulate genes expression, hence, they can be used to modulate specific genes expression.137 Yet several microRNAs were shown to interact with c-Myc. C-Myc represses the expression of miR-29a to attenuate its inhibitory effect on the expression of several oncogenes such as lysyl oxidase-like 2 (LOXL2), MYB proto-oncogene like 2 (MYBL2), hematopoietic progenitor kinase/germinal center kinase-like kinase (HGK), and neuroblastoma RAS viral oncogene homolog (NRAS). These genes particularly LOXL2 are overexpressed in pancreatic cancer and the competent function of miR-29a can downregulate the expression of these oncogenes and decrease pancreatic cancer growth.138
MiR-145 is downregulated in pancreatic cancer. It negatively regulates pancreatic cancer cells growth and migration. MiR-145 downregulates the expression of several proto-oncogenes such as epidermal growth factor receptor (EGFR), MMP1, and c-Myc.139 Similar to miR-145, miR-494 is downregulated in pancreatic cancer cells. MiR-494 suppresses the expression of c-Myc and SIRT1 to inhibit pancreatic cancer cells proliferation and stimulate their apoptosis.140 Moreover, decreased expression of miR-494 has been associated with larger tumor size, lymphatic involvement, distant metastasis, and poor prognosis in patients with pancreatic cancer.140 A miR-34a mimic in combination with polo-like kinase 1 (PLK1) oncogene silencing could effectively decrease the expression of c-Myc and decrease pancreatic cancer cells viability.141 It was shown that c-Myc promotes the expression of Lin-28B RNA binding proteins to decrease the expression of let-7 family in pancreatic cancer.142,143 Interestingly, let-7a can also downregulate c-Myc expression, therefore, their interaction can be mutual.143–145
In this section, some interactions between c-Myc and microRNAs were mentioned in pancreatic cancer (Table 1). Although, there are many more found in other types of tumor that can be replicated in pancreatic cancer. Expanding our knowledge surrounding these interactions helps to find new methods of cancer chemotherapy.
Different strategies to target c-Myc
Direct targeting of c-Myc
Han et al. developed small molecule c-Myc inhibitors such as MYCi361 and MYCi975 which can directly target c-Myc inside the cells and promote its phosphorylation on threonine-58 and thereby stimulate its ubiquitination and degradation.116 MYCi361 (initially at 50 mg/kg bid for 2 days, then 70 mg/kg/day for 9 days or 55 mg/kg/day, 3 consecutive days a week for 2 weeks) and MYCi975 (50 mg/kg bid for 2 or 3 days) suppressed tumor growth in mice and enhanced intra-tumor immune cells infiltration. Moreover, the compound improved tumor cells sensitivity to anti-PD1 immunotherapy.116
Because of structural characteristics, c-Myc needs to make a heterodimer with Myc-associated factor X (MAX) and thereby bind to DNA.146 KSI-3716 is a c-Myc inhibitor that hinders c-Myc/MAX heterodimer binding to the promoter of target genes. This property could effectively inhibit c-Myc-mediated overexpression of different genes such as cyclin D2, CDK4. Intravesical administration of KSI-3716 at a dose of 5 mg/kg could significantly inhibit bladder tumor growth in murine model.147 KSI-1449 and KSI-2302 can similarly inhibit the formation of c-Myc/Max/DNA complex, thereby preventing the biologic activity of c-Myc.148
Similar to KSI-3716, sAJM589 (with an IC50 of 1.8 ± 0.03 μM) is another small molecule that can dose-dependently hamper c-Myc/MAX heterodimer formation. sAJM589 could effectively decrease the expression of c-Myc target genes and even decrease the bioavailability of c-Myc in Burkitt lymphoma cell model.149 C-Myc/MAX interaction is necessary for binding to the E-box of target genes and without this heterodimer, c-Myc remains non-functional.149 Mycro3, another inhibitor of c-Myc/MAX dimerization, could significantly promote pancreatic cancer cells apoptosis.66 Mycro3 (up to 100 mg/kg/day for 2 weeks) also lead to a drastic decrease in tumor cells proliferation and shrinkage in tumor size.66
Another inhibitor of c-Myc/MAX dimerization, 10,058-F4, could increase apoptosis in pre-B cell ALL cell line and improve cancer cells sensitivity to other drugs such as dexamethasone and vincristine.150,151 The same compound could also improve the sensitivity of acute promyelocytic leukemia (APL) to arsenic trioxide.152 Treatment with 10,058-F4 also reduced pancreatic cancer cells viability by accelerating their apoptosis. It partly inhibited glycolysis and led to pancreatic cancer cell cycle arrest at G1/S transition. Also, treatment with 10,058-F4 (with an IC50 of 17 and 25 μM in different cell cultures) markedly improved the effect of gemcitabine on pancreatic cancer.153
Taken together, there are three mechanisms to directly target c-Myc: 1- Targeting c-Myc itself and accelerating its degradation. 2- Prevention of c-Myc/MAX heterodimerization. 3- Prevention of c-Myc/MAX/DNA complex formation.
Indirect targeting of c-Myc
Direct targeting of c-Myc with a potent inhibitor seems to be difficult because of structural complexity and nuclear localization.154 C-Myc lacks a fixed structure and has no binding pocket for drugs.155 Also, c-Myc possesses a wide range of interactions with different components of intracellular signaling pathways, transcriptional and translational complexes, and non-coding RNAs. Indirect inhibition of c-Myc can be achieved by epigenetic silencing of its gene, transcriptional, post-transcriptional, and translational alterations during its expression. Even, different mechanisms have been proposed to indirectly weaken c-Myc stabilization or prevent its effect on target genes.
Bromodomain and extraterminal (BET) inhibitors
BET proteins such as BRD2, BRD3, and BRD4, are nuclear proteins that regulate protein-protein interaction and modify gene transcription by exerting epigenetic effects after binding to acetylated lysine residues on histones and main transcriptional factors.156 BET proteins such as BRD2 and BRD3 upregulate c-Myc expression and BET inhibitors such as JQ1 and OTX015 vigorously downregulate the expression of c-Myc.157,158 BET inhibitors have been proposed as a new therapeutic option for c-Myc-dependent tumors and have shown promising results in primary studies.159 BET inhibitors showed satisfying efficacy in the treatment of pancreatic cancer in cell culture and mice model of pancreatic cancer.117,160,161 JQ1 (50 mg/kg intraperitoneal twice weekly), a BET inhibitor, markedly decreased tumor volume in mice with pancreatic cancer transplant.160 Their effect on pancreatic cancer is associated with the downregulation of c-Myc. Consistently, c-Myc upregulation reverts the therapeutic effect of BET inhibitors on pancreatic cancer and is responsible for resistance to BET inhibitors.160–162 BET inhibitors such as I-BET762 could lead to pancreatic cancer cell cycle arrest in G0/G1 phase and inhibit the proliferation and metastasis of pancreatic cancer cells. Besides, I-BET762 could promote the sensitivity of pancreatic cancer cells to gemcitabine.163 I-BET762 (30 mg/kg/day for 13 days) also decreased tumor size in mice with pancreatic cancer xenograft and markedly enhanced the effect of gemcitabine on pancreatic cancer xenograft.163 BET inhibitors can suppress several oncogenic pathways other than c-Myc-dependent pathways which can increase their efficacy as chemotherapeutic agents.164
PP2A
PP2A, a serine/threonine phosphatase, is a tumor suppressor which can dephosphorylate c-Myc and decrease its stabilization. The regulatory subunit of PP2A, B56α, plays a critical role in PP2A-mediated c-Myc destabilization and its following ubiquitination and degradation. Consistently, B56α knockdown is associated with c-Myc overexpression and overactivity.165–167 Interestingly, Farrington et al. revealed that the use of small-molecule activators of PP2A (SMAPs) in different cancers such as Burkitt lymphoma, KRAS-driven non–small cell lung cancer, and triple-negative breast cancer has been associated with decreased c-Myc bioavailability and diminished tumor cells proliferation.168 Similarly, B56α subunit knockdown has been associated with deregulated c-Myc activity and spontaneous tumor development in the liver of mice.169 Endogenous inhibitors of PP2A and cancerous inhibitors of PP2A are markedly overexpressed in pancreatic cancer and enhance the expression and stabilization of c-Myc in human pancreatic cancer.167 Also, inhibition or knockdown of these inhibitors of PP2A attenuated the tumorigenic potential of pancreatic cancer cells and hindered tumor growth in cell culture and animal model.167 Additionally, pharmacological activation of PP2A with FTY-720 (3 mg/kg/day intraperitoneal) notably decreased c-Myc protein levels and significantly decreased pancreatic cancer growth in both cell culture and mice model.170 Hence, SMAPs can be alternatively used to indirectly inhibit c-Myc in pancreatic cancer which resulted in acceptable efficacy in vitro and in vivo.74
Targeting c-Myc by inhibiting its upstream signaling pathways
As noted, c-Myc is downstream of three major proliferative pathways. Blocking each one of these pathways can partly downregulate c-Myc expression. For instance, ERK2 inhibition in lung cancer cells could decrease c-Myc expression. Attenuation of ERK/c-Myc axis in lung cancer resulted in reduced expression of proteins involved in G1-S progression and led to G1 phase arrest.171 In particular, targeting PI3K/AKT/mTOR pathway seems to be more effective. mTOR controls different molecules such as 4EBP1 and S6 which are crucial for the initiation and elongation phase of ribosomal translation of c-Myc mRNA.172 Furthermore, mTOR regulates the translation of translational factors themselves and many other proteins that their presence is indispensable for tumor cells proliferation.172 PI3K inhibitors, AKT inhibitors, mTOR inhibitors, and combinatorial PI3K/mTOR inhibitors have developed and are passing their clinical trials.173 Perhaps, use of these compounds can improve the management of pancreatic cancer.
Histone deacetylase (HDAC) inhibitors
By deacetylating the chromatin, HDACs can reprogram the expression of several proto-oncogenes and tumor-suppressor genes.174 Dysregulation of HDACs has been implicated in the pathogenesis of pancreatic cancer.175 HDAC inhibitors showed promising results in pancreatic cancer and could markedly inhibit tumor growth, prevent tumor cells migration and improve chemoresistance.175,176 HDAC inhibitors can promote the expression of tumor suppressor genes, hence, repressing tumor growth.177 It was unveiled that HDAC1 inhibition vigorously attenuates c-Myc-induced tumorigenesis.177 C-Myc and HDAC both are involved in the regulation of gene transcription and proliferation. Likewise, it was uncovered that c-Myc enhances the expression of HDAC2 in pancreatic cancer and thereby suppresses the expression of cyclin G2 (CCNG2), an inhibitor of tumor growth.178 Zhu et al. unveiled that HDAC7 can increase the transcription of c-Myc to increase the proliferation of colon cancer cells.179 Likewise, HDAC6 inhibition led to downregulation of c-Myc and hindered pancreatic cancer cells proliferation.180 Also, HDAC inhibition could strongly decrease c-Myc expression in lymphoma cells and non-small cell lung carcinoma.181,182 Liu et al. revealed that c-Myc can increase the expression of SIRT2, histone deacetylase class III, in pancreatic cancer, subsequently, SIRT2 reinforces c-Myc expression and enhances pancreatic cancer cells proliferation.183 HDACs and c-Myc have a mutual relationship and promote the expression of each other, hence, pharmacological inhibition of one of them can also partly attenuate the biologic function of the other one.
Nuclear factor activated T cells (NFAT) inhibition
NFATs are a group of calcineurin-responsive transcription factors that promote the transcription of c-Myc, thereby promoting cancer cells proliferation. NFATs particularly NFAT1c is excessively expressed in pancreatic cancer. Inhibition of calcineurin signaling or knockdown of NFAT1c led to G1 arrest and could drastically decrease pancreatic cancer cells proliferation.184,185 NFAT1c binds to serum responsive element in the proximal promoter and prepares local chromatin structure for Ets-like gene 1 (ELK1) activity. ELK1 vigorously increases the transcription of c-Myc and promotes its expression.185 Buchholz et al. revealed that NFATc1 is a major inducer of c-Myc expression, thereby increasing pancreatic cancer cells proliferation.184 Induction of NFAT/c-Myc pathway promoted pancreatic cancer growth and NFAT depletion reduced c-Myc expression and decreased pancreatic cancer growth in vitro and in vivo.184,185 Inhibition of NFAT can be a promising method to suppress the transcription of c-Myc and decrease its oncogenic effects.
GSKβ inhibition
KRAS promotes the gene expression of GSKβ in pancreatic cancer. GSKβ is heavily involved in cancer cells proliferation, survival, and chemoresistance. Additionally, inhibition of GSKβ was shown to be an effective mechanism to treat pancreatic cancer.186 Also, PI3K can upregulate c-Myc through a GSKβ-dependent manner. PI3K/GSKβ/c-Myc is crucial for pancreatic cancer cell cycle progression.187 GSKβ inhibition decreases the expression of NF-κB and NF-κB -mediated expression of c-Myc in pancreatic cancer, thereby reducing tumor growth.188
Insulin-like growth factor-2 mRNA-binding protein 1 (IGF2BP1 or IMP1) inhibitor
IMP1 is overexpressed in pancreatic cancer and higher expression of IMP1 predicts a poor prognosis of pancreatic cancer. Decreased expression of miR-494 leads to overexpression of IMP1 in pancreatic cancer.189 Meanwhile, downregulation of IMP1 could decrease pancreatic cancer cell growth in cell culture and mice model.189 IMP1 binds to mRNA of oncogenes such as c-Myc which stabilizes them. BTYNB, a potent and selective inhibitor of IMP1, significantly decreased the expression of c-Myc and suppressed the proliferation of IMP1-containing ovarian cancer and melanoma cells.190 IMP1 protects the coding region instability sequence of c-Myc mRNA against endonucleolytic cleavage.191 Similarly, IMP1 can bind to KRAS mRNA and regulate its expression. Deletion of IMP1 significantly reduced KRAS expression.192 Consistently, Zhai et al. uncovered that IMP1 binds to and stabilizes c-Myc mRNA in pancreatic cancer, thereby augmenting cancer cells proliferation.94 In addition, impaired function of IMP1 was associated with decreased c-Myc expression and reduced tumor cells proliferation in pancreatic cancer cells.94 Regarding the effect of IMP1 on the stabilization of c-Myc and KRAS mRNA, IMP1 inhibitors are ideal choices to indirectly target both c-Myc and KRAS in pancreatic cancer.
Stabilization of MAX/MAX homodimerization
Previously it was discussed how c-Myc-MAX heterodimerization is involved in binding to E-box of the promoter of target genes. Further, it was mentioned that disruption of c-Myc-MAX interaction can inhibit the oncogenic effects of c-Myc. In this regard, Struntz et al. used KI-MS2-008, an asymmetric polycyclic lactam that can stabilize MAX homodimer, thereby preventing its heterodimerization with c-Myc. This new mechanism could markedly downregulate c-Myc-driven transcription and reduce tumor size in mice-bearing hepatocellular carcinoma (0.24 mg/kg/day intraperitoneal) or T cell All (0.06 mg/kg/day intravenous).193
Oxidized low-density lipoprotein receptor 1 (OLR1) inhibition
OLR1 stimulation increases the expression of c-Myc. Consistently, OLR1 activation plays an important role in the pathogenesis of different cancers particularly pancreatic cancer.194,195 OLR1, through upregulation of c-Myc, can promote pancreatic cancer cells proliferation, metastasis, and chemoresistance to gemcitabine. OLR1 is overexpressed in pancreatic cancer. OLR1, through c-Myc, promotes the expression of high mobility group A2 (HMGA2). Similarly, overactivity of OLR1/c-Myc/ HMGA2 axis is associated with poor prognosis among patients with pancreatic cancer.134,195,196 HMGA2 is a transcription factor that binds to AT-rich sequences to change the structure of chromatin. It has been implicated in tumorigenesis, EMT, and chemoresistance.197 As mentioned, HMGA2 is markedly expressed in pancreatic cancer and higher expression of HMGA2 is associated with lymph node metastasis in pancreatic cancer.198 Regarding these findings, pharmacological attenuation of OLR1 can indirectly inhibit the deleterious effects of c-Myc on pancreatic cancer.
LncRNAs and microRNAs mimics and inhibitors
It has been previously discussed how different lncRNAs and microRNAs can modulate c-Myc expression possibly through epigenetic, transcriptional, and post-transcriptional modifications. Reinforcement of lncRNAs and microRNAs that can negatively regulate c-Myc expression such as LINC00261, Pvt1, MiR-145, MiR-494, and Let-7a seems to be a new and effective method to inhibit tumor cells proliferation and prevent cancer progression. Besides, inhibition of lncRNAs and microRNAs those can enhance c-Myc expression such as LINC00346, XLOC_006390, HULC, GSTM3TV2, and XLOC_006390 can be useful in the same way. However, non-coding RNAs have been the main focus of a notable proportion of recent investigations, their clinical application still did not receive much attention. They can target c-Myc expression in different stages of gene expression and help to overcome drug-resistant tumors. Interestingly, non-coding RNAs can indirectly affect c-Myc by regulating PP2A and other molecules which are in close relationship with c-Myc.199 Even, microRNA-22 can disrupt c-Myc/E-box interaction to attenuate the effect of c-Myc on target genes transcription.12 However, some c-Myc-interacting lncRNAs and microRNAs were found by previous studies, a greater number remain to be discovered by future studies.200 Their mimics or inhibitors are passing their preclinical studies and will constitute a major part of future clinical practice.137
Targeted therapy for c-Myc: when, how, and where?
C-Myc is normally expressed in non-cancerous tissues and is involved in several physiological processes such as metabolism, growth, apoptosis, and differentiation.201,202 C-Myc inhibition may exert serious adverse effects on normal tissues as it was shown that c-Myc null mutation was lethal in homozygote mice and decreased fertility in female heterozygote mice.203 Although, systemic inhibition of c-Myc in RAS-induced lung adenocarcinoma in mice showed that the effect of c-Myc inhibitors on regenerative tissue is tolerable and completely reversible.204 Various mutations that directly affect c-Myc gene or indirectly change its expression by epigenetic modulation can lead to dysregulation and overexpression of c-Myc, impair normal cells growth and contribute to neoplastic transformation.201,202 However, c-Myc is a transcription factor whose function is exerted mainly in the nucleus, it has been found that cytoplasmic protein levels of c-Myc independently and negatively correlate with patients survival in pancreatic cancer.205 As mentioned throughout the text, c-Myc inhibition showed benefit in different stages of pancreatic cancer as it is involved in pancreatic cancer growth, metabolism, angiogenesis, metastasis, and immune evasion. Therefore, inhibition of c-Myc can be beneficial in different stages of pancreatic cancer. Furthermore, it has been shown that c-Myc overexpression is significantly and positively correlated with histological tumor grade in pancreatic cancer, hence, c-Myc inhibition may provide greater benefit in advanced pancreatic cancer.13
To confine the adverse effects of c-Myc inhibitors can be delivered by nanoparticle systems which allow the clinicians to administer the lower doses of drugs and deliver them to the desired site of action.206 This method of drug delivery can change the pharmacokinetics of drugs, prolong their duration of action, and remove the unwanted effects of c-Myc inhibition in normal tissues.206 Furthermore, as c-Myc interacts with several signaling pathways and is involved in different cellular processes, simultaneous administration of c-Myc inhibitors and immune-checkpoint inhibitors or other chemotherapeutic agents can exert a synergistic effect on pancreatic cancer and contribute to overcoming chemoresistance.117,153 Therefore, c-Myc inhibitors in combination with other chemotherapeutic agents and in form of nanoparticulated drugs can provide the utmost benefits particularly for those having higher levels of c-Myc expression in their cancer cells.
Conclusion and future direction
C-Myc is a transcription for numerous oncogenes. In addition, the effect of several oncoproteins converges on c-Myc and they exert their oncogenic effects by upregulating c-Myc. Hence, inhibition of c-Myc can disrupt a series of oncogenic transformations. C-Myc overexpression enhances proliferation, invasion, metastasis, angiogenesis and immune evasion in pancreatic cancer and c-Myc inhibition attenuates chemoresistance and enhances immunotherapy. During recent years several methods developed to target the “undruggable” c-Myc. Yet, there is not any clinical trial attempting to measure the effect of c-Myc in pancreatic cancer. Regarding the poor response of pancreatic cancer to current chemotherapy protocols, the prevalence of c-Myc deregulation in pancreatic cancer, and the promising results of preclinical studies, it is worth investigating the effect of different inhibitors of c-Myc in clinical trials of pancreatic cancer.
Acknowledgments
There is nothing to convey in acknowledgment.
Funding Statement
This article received no funds or other kinds of financial support.
Footnotes
Figure 1 and Figure 2 are misplaced. Figure 1 must be marked as Figure 2 and Figure 2 must be inserted as figure 1. The caption for Figure 1 (implication of c-Myc in metabolic reprogramming ....) is correctly marked as Figure 1 but its associated figure is misplaced. Also, the caption for Figure 2 is correct but its associated Figure is wrongly marked as Figure 1. In summary, Figures but not their captions must be reordered. Their current cations do not belong to them.
Author contribution statement
Moein Ala conceived this article, performed literature search, wrote the manuscript, and edited it.
Ethics statement
Not applicable for a review article.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
References
- 1.Lippi G, Mattiuzzi C.. The global burden of pancreatic cancer. Arch Med Sci AMS. 2020;16(4):820. doi: 10.5114/aoms.2020.94845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cowgill SM, Muscarella P. The genetics of pancreatic cancer. Am J Surg. 2003;186(3):279–286. doi: 10.1016/S0002-9610(03)00226-5. [DOI] [PubMed] [Google Scholar]
- 3.Bengtsson A, Andersson R, Ansari D. The actual 5-year survivors of pancreatic ductal adenocarcinoma based on real-world data. Sci Rep. 2020;10(1):1–9. doi: 10.1038/s41598-020-73525-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Springfeld C, Jäger D, Büchler MW, Strobel O, Hackert T, Palmer DH, Neoptolemos JP. Chemotherapy for pancreatic cancer. Presse Med. 2019;48(3):e159–e74. doi: 10.1016/j.lpm.2019.02.025. [DOI] [PubMed] [Google Scholar]
- 5.Neoptolemos JP, Palmer DH, Ghaneh P, Psarelli EE, Valle JW, Halloran CM, Faluyi O, O’Reilly DA, Cunningham D, Wadsley J, et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): a multicentre, open-label, randomised, phase 3 trial. Lancet. 2017;389(10073):1011–1024. doi: 10.1016/S0140-6736(16)32409-6. [DOI] [PubMed] [Google Scholar]
- 6.Jones RP, Psarelli -E-E, Jackson R, Ghaneh P, Halloran CM, Palmer DH, Campbell F, Valle JW, Faluyi O, O’Reilly DA, et al. Patterns of recurrence after resection of pancreatic ductal adenocarcinoma: a secondary analysis of the ESPAC-4 randomized adjuvant chemotherapy trial. JAMA Surg. 2019;154(11):1038–1048. doi: 10.1001/jamasurg.2019.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leonhardt CS, Traub B, Hackert T, Klaiber U, Strobel O, Büchler MW, Neoptolemos JP. Adjuvant and neoadjuvant chemotherapy in pancreatic ductal adenocarcinoma. J Pancreatol. 2020;3(1):1–11. doi: 10.1097/JP9.0000000000000040. [DOI] [Google Scholar]
- 8.Lin C-P, Liu C-R, Lee C-N, Chan T-S, Liu HE. Targeting c-Myc as a novel approach for hepatocellular carcinoma. World J Hepatol. 2010;2(1):16. doi: 10.4254/wjh.v2.i1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jain S, Wang X, Chang -C-C, Ibarra-Drendall C, Wang H, Zhang Q, Brady SW, Li P, Zhao H, Dobbs J, et al. Src inhibition blocks c-Myc translation and glucose metabolism to prevent the development of breast cancer. Cancer Res. 2015;75(22):4863–4875. doi: 10.1158/0008-5472.CAN-14-2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parasido E, Avetian GS, Naeem A, Graham G, Pishvaian M, Glasgow E, Mudambi S, Lee Y, Ihemelandu C, Choudhry M, et al. The sustained induction of c-MYC drives nab-paclitaxel resistance in primary pancreatic ductal carcinoma cells. Mol Cancer Res. 2019;17(9):1815–1827. doi: 10.1158/1541-7786.MCR-19-0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dang CV. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med. 2013;3(8):a014217. doi: 10.1101/cshperspect.a014217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiong J, Du Q, Liang Z. Tumor-suppressive microRNA-22 inhibits the transcription of E-box-containing c-Myc target genes by silencing c-Myc binding protein. Oncogene. 2010;29(35):4980–4988. doi: 10.1038/onc.2010.241. [DOI] [PubMed] [Google Scholar]
- 13.Schleger C, Verbeke C, Hildenbrand R, Zentgraf H, Bleyl U. c-MYC activation in primary and metastatic ductal adenocarcinoma of the pancreas: incidence, mechanisms, and clinical significance. Mod Pathol. 2002;15(4):462–469. doi: 10.1038/modpathol.3880547. [DOI] [PubMed] [Google Scholar]
- 14.Allenson K, Castillo J, San Lucas F, Scelo G, Kim D, Bernard V, Davis G, Kumar T, Katz M, Overman MJ, et al. High prevalence of mutantKRAS in circulating exosome-derived DNA from early-stage pancreatic cancer patients. Ann Oncol. 2017;28(4):741–747. doi: 10.1093/annonc/mdx004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sodir NM, Kortlever RM, Barthet VJ, Campos T, Pellegrinet L, Kupczak S, Anastasiou P, Swigart LB, Soucek L, Arends MJ, et al. Myc instructs and maintains pancreatic adenocarcinoma phenotype. Cancer Discov. 2020;10(4):588–607. doi: 10.1158/2159-8290.CD-19-0435. [DOI] [PubMed] [Google Scholar]
- 16.Warburg O, Wind F, Neglers E. 1930. The metabolism of tumors. London: Constable & Co. Ltd. [Google Scholar]
- 17.Yan L, Raj P, Yao W, Ying H. Glucose metabolism in pancreatic cancer. Cancers. 2019;11(10):1460. doi: 10.3390/cancers11101460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luan W, Wang Y, Chen X, Shi Y, Wang J, Zhang J, Qian J, Li R, Tao T, Wei W, et al. PKM2 promotes glucose metabolism and cell growth in gliomas through a mechanism involving a let-7a/c-Myc/hnRNPA1 feedback loop. Oncotarget. 2015;6(15):13006. doi: 10.18632/oncotarget.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gupta A, Ajith A, Singh S, Panday RK, Samaiya A, Shukla S. PAK2–c-Myc–PKM2 axis plays an essential role in head and neck oncogenesis via regulating Warburg effect. Cell Death Dis. 2018;9(8):1–15. doi: 10.1038/s41419-018-0887-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiang S, Gu H, Jin L, Thorne RF, Zhang XD, Wu M. LncRNA IDH1-AS1 links the functions of c-Myc and HIF1α via IDH1 to regulate the Warburg effect. Proc Natl Acad Sci. 2018;115(7):E1465–E74. doi: 10.1073/pnas.1711257115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu X, Zhou Y, Peng J, Xie B, Shou Q, Wang J. Silencing c-Myc enhances the antitumor activity of Bufalin by suppressing the HIF-1α/SDF-1/CXCR4 pathway in pancreatic cancer cells. Front Pharmacol. 2020;11:495. doi: 10.3389/fphar.2020.00495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He T-L, Zhang Y-J, Jiang H, X-h L, Zhu H, Zheng K-L. The c-Myc–LDHA axis positively regulates aerobic glycolysis and promotes tumor progression in pancreatic cancer. Med Oncol. 2015;32(7):187. doi: 10.1007/s12032-015-0633-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qiu H, Jackson AL, Kilgore JE, Zhong Y, Chan LL-Y, Gehrig PA, Zhou C, Bae-Jump VL. JQ1 suppresses tumor growth through downregulating LDHA in ovarian cancer. Oncotarget. 2015;6(9):6915. doi: 10.18632/oncotarget.3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rong Y, Wu W, Ni X, Kuang T, Jin D, Wang D, Lou W. Lactate dehydrogenase A is overexpressed in pancreatic cancer and promotes the growth of pancreatic cancer cells. Tumor Biol. 2013;34(3):1523–1530. doi: 10.1007/s13277-013-0679-1. [DOI] [PubMed] [Google Scholar]
- 25.Mohammad GH, Olde Damink S, Malago M, Dhar DK, Pereira SP, Muders M. Pyruvate kinase M2 and lactate dehydrogenase A are overexpressed in pancreatic cancer and correlate with poor outcome. PLoS One. 2016;11(3):e0151635. doi: 10.1371/journal.pone.0151635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maftouh M, Avan A, Sciarrillo R, Granchi C, Leon L, Rani R, Funel N, Smid K, Honeywell R, Boggi U, et al. Synergistic interaction of novel lactate dehydrogenase inhibitors with gemcitabine against pancreatic cancer cells in hypoxia. Br J Cancer. 2014;110(1):172–182. doi: 10.1038/bjc.2013.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.James AD, Richardson DA, Oh I-W, Sritangos P, Attard T, Barrett L, Bruce JIE. Cutting off the fuel supply to calcium pumps in pancreatic cancer cells: role of pyruvate kinase-M2 (PKM2). Br J Cancer. 2020;122(2):266–278. doi: 10.1038/s41416-019-0675-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim DJ, Park YS, Kang MG, You Y-M, Jung Y, Koo H, Kim J-A, Kim M-J, Hong S-M, Lee KB, et al. Pyruvate kinase isoenzyme M2 is a therapeutic target of gemcitabine-resistant pancreatic cancer cells. Exp Cell Res. 2015;336(1):119–129. doi: 10.1016/j.yexcr.2015.05.017. [DOI] [PubMed] [Google Scholar]
- 29.Shen L, O’Shea JM, Kaadige MR, Cunha S, Wilde BR, Cohen AL, Welm AL, Ayer DE. Metabolic reprogramming in triple-negative breast cancer through Myc suppression of TXNIP. Proc Natl Acad Sci. 2015;112(17):5425–5430. doi: 10.1073/pnas.1501555112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu G, Zhou L, Liu H, Shan Y, Zhang X. MicroRNA-224 promotes pancreatic cancer cell proliferation and migration by targeting the TXNIP-mediated HIF1α pathway. Cell Physiol Biochem. 2018;48(4):1735–1746. doi: 10.1159/000492309. [DOI] [PubMed] [Google Scholar]
- 31.Liang Y, Wang H, Chen B, Mao Q, Xia W, Zhang T, Song X, Zhang Z, Xu L, Dong G, et al. circDCUN1D4 suppresses tumor metastasis and glycolysis in lung adenocarcinoma by stabilizing TXNIP expression. Mol Ther Nucleic Acids. 2021;23:355–368. doi: 10.1016/j.omtn.2020.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao S, Chen M, Wei W, Zhang X, Zhang M, Yao Y, Lv Y, Ling T, Wang L, Zou X, et al. Crosstalk of mTOR/PKM2 and STAT3/c‐Myc signaling pathways regulate the energy metabolism and acidic microenvironment of gastric cancer. J Cell Biochem. 2019;120(2):1193–1202. doi: 10.1002/jcb.26915. [DOI] [PubMed] [Google Scholar]
- 33.Wang J, Ye C, Chen C, Xiong H, Xie B, Zhou J, Chen Y, Zheng S, Wang L. Glucose transporter GLUT1 expression and clinical outcome in solid tumors: a systematic review and meta-analysis. Oncotarget. 2017;8(10):16875. doi: 10.18632/oncotarget.15171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ahuja P, Zhao P, Angelis E, Ruan H, Korge P, Olson A, Wang Y, Jin ES, Jeffrey FM, Portman M, et al. Myc controls transcriptional regulation of cardiac metabolism and mitochondrial biogenesis in response to pathological stress in mice. J Clin Invest. 2010;120(5):1494–1505. doi: 10.1172/JCI38331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dadson K, Hauck L, Hao Z, Grothe D, Rao V, Mak TW, Billia F. The E3 ligase Mule protects the heart against oxidative stress and mitochondrial dysfunction through Myc-dependent inactivation of Pgc-1α and Pink1. Sci Rep. 2017;7(1):1–14. doi: 10.1038/srep41490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu C-L, Yang P-S, Wang T-Y, Huang S-Y, Kuo Y-H, Cheng S-P. PGC1α downregulation and glycolytic phenotype in thyroid cancer. J Cancer. 2019;10(16):3819. doi: 10.7150/jca.30018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xing F, Luan Y, Cai J, Wu S, Mai J, Gu J, Zhang H, Li K, Lin Y, Xiao X, et al. The anti-Warburg effect elicited by the cAMP-PGC1α pathway drives differentiation of glioblastoma cells into astrocytes. Cell Rep. 2017;18(2):468–481. doi: 10.1016/j.celrep.2016.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ji S, Qin Y, Liang C, Huang R, Shi S, Liu J, Jin K, Liang D, Xu W, Zhang B, et al. FBW7 (F-box and WD repeat domain-containing 7) negatively regulates glucose metabolism by targeting the c-Myc/TXNIP (thioredoxin-binding protein) axis in pancreatic cancer. Clin Cancer Res. 2016;22(15):3950–3960. doi: 10.1158/1078-0432.CCR-15-2380. [DOI] [PubMed] [Google Scholar]
- 39.Miller DM, Thomas SD, Islam A, Muench D, Sedoris K. c-Myc and cancer metabolism. Clin Cancer Res. 2012;18(20):5546–53. doi: 10.1158/1078-0432.CCR-12-0977 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sato K, Hikita H, Myojin Y, Fukumoto K, Murai K, Sakane S, Tamura T, Yamai T, Nozaki Y, Yoshioka T, et al. Hyperglycemia enhances pancreatic cancer progression accompanied by elevations in phosphorylated STAT3 and Myc levels. Plos One. 2020;15(7):e0235573. doi: 10.1371/journal.pone.0235573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao P, Tchernyshyov I, Chang T-C, Lee Y-S, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458(7239):762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. 2010;35(8):427–433. doi: 10.1016/j.tibs.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bott AJ, Peng I-C, Fan Y, Faubert B, Zhao L, Li J, Neidler S, Sun Y, Jaber N, Krokowski D, et al. Oncogenic Myc induces expression of glutamine synthetase through promoter demethylation. Cell Metab. 2015;22(6):1068–1077. doi: 10.1016/j.cmet.2015.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roux C, Riganti C, Borgogno SF, Curto R, Curcio C, Catanzaro V, Digilio G, Padovan S, Puccinelli MP, Isabello M, et al. Endogenous glutamine decrease is associated with pancreatic cancer progression. Oncotarget. 2017;8(56):95361. doi: 10.18632/oncotarget.20545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol Elsevier. 2002;29(6Q):15–18. doi: 10.1053/sonc.2002.37263. [DOI] [PubMed] [Google Scholar]
- 46.Huang X, Sun J, Chen G, Niu C, Wang Y, Zhao C, Sun J, Huang H, Huang S, Liang Y, et al. Resveratrol promotes diabetic wound healing via SIRT1-FOXO1-c-Myc signaling pathway-mediated angiogenesis. Front Pharmacol. 2019;10:421. doi: 10.3389/fphar.2019.00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baudino TA, McKay C, Pendeville-Samain H, Nilsson JA, Maclean KH, White EL, Davis AC, Ihle JN, Cleveland JL. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 2002;16(19):2530–2543. doi: 10.1101/gad.1024602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smits M, Wurdinger T, van Het Hof B, Drexhage JA, Geerts D, Wesseling P, Noske DP, Vandertop WP, Vries HE, Reijerkerk A, et al. Myc‐associated zinc finger protein (MAZ) is regulated by miR‐125b and mediates VEGF‐induced angiogenesis in glioblastoma. FASEB J. 2012;26(6):2639–2647. doi: 10.1096/fj.11-202820. [DOI] [PubMed] [Google Scholar]
- 49.Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT, et al. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet. 2006;38(9):1060–1065. doi: 10.1038/ng1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen C, Cai S, Wang G, Cao X, Yang X, Luo X, Feng Y, Hu J. c-Myc enhances colon cancer cell-mediated angiogenesis through the regulation of HIF-1α. Biochem Biophys Res Commun. 2013;430(2):505–511. doi: 10.1016/j.bbrc.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 51.Arany Z, Foo S-Y, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J, Rangwala SM, et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1α. Nature. 2008;451(7181):1008–1012. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- 52.Zhang Z, Liu M, Hu Q, Xu W, Liu W, Sun Q, Ye Z, Fan G, Xu X, Yu X, et al. 2019. FGFBP1, a downstream target of the FBW7/c-Myc axis, promotes cell proliferation and migration in pancreatic cancer. Am J Cancer Res. 9(12):2650. [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu H-Y, Bai W-D, Liu J-Q, Zheng Z, Guan H, Zhou Q, Piazza V, Gemmi M. Up-regulation of FGFBP1 signaling contributes to miR-146a-induced angiogenesis in human umbilical vein endothelial cells. Sci Rep. 2016;6(1):1–11. doi: 10.1038/s41598-016-0001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao B, Liu L, Mao J, Zhang Z, Wang Q, Li Q. PIM1 mediates epithelial-mesenchymal transition by targeting Smads and c-Myc in the nucleus and potentiates clear-cell renal-cell carcinoma oncogenesis. Cell Death Dis. 2018;9(3):1–14. doi: 10.1038/s41419-018-0348-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial–mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu M, Zhu H, Yang S, Wang Z, Bai J, Xu N. c‐Myc suppressed E‐cadherin through miR‐9 at the post‐transcriptional level. Cell Biol Int. 2013;37(3):197–202. doi: 10.1002/cbin.10039. [DOI] [PubMed] [Google Scholar]
- 57.Jiang L, Meng W, Yu G, Yin C, Wang Z, Liao L, Meng F. MicroRNA-144 targets APP to regulate AML1/ETO+ leukemia cell migration via the p-ERK/c-Myc/MMP-2 pathway. Oncol Lett. 2019;18(2):2034–2042. doi: 10.3892/ol.2019.10477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu Y, Zhou S, Shi J, Zhang X, Shentu L, Chen Z, Zhou L. c-Myc transactivates GP73 and promotes metastasis of hepatocellular carcinoma cells through GP73-mediated MMP-7 trafficking in a mildly hypoxic microenvironment. Oncogenesis. 2019;8(10):1–11. doi: 10.1038/s41389-019-0166-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Z, Li Y, Ahmad A, Banerjee S, Azmi AS, Kong D, Sarkar FH. Pancreatic cancer: understanding and overcoming chemoresistance. Nat Rev Gastroenterol Hepatol. 2011;8(1):27. doi: 10.1038/nrgastro.2010.188. [DOI] [PubMed] [Google Scholar]
- 60.Neoptolemos JP, Springfeld C, Hackert T, Büchler MW. 2021. The evolution of adjuvant trials in pancreatic cancer. In: Textbook of pancreatic cancer. Springer; p. 743–761. doi: 10.1007/978-3-030-53786-9_48. [DOI] [Google Scholar]
- 61.Parasido EM, Avetian GS, Brody J, Winter J, Londin E, Pishvaian M, Glasgow, E, Byers, S, Narla, G, Albanese, C . Targeting c-MYC and MAPK pathway to overcome pancreatic cancer drug resistance. AACR. doi: 10.1158/1538-7445.AM2019-1283; .2019 [DOI] [Google Scholar]
- 62.Kim N, Kang M-J, Lee SH, Son JH, Lee JE, Paik WH, Ryu JK, Kim Y-T. Fisetin enhances the cytotoxicity of gemcitabine by down-regulating ERK-MYC in MiaPaca-2 human pancreatic cancer cells. Anticancer Res. 2018;38(6):3527–3533. doi: 10.21873/anticanres.12624. [DOI] [PubMed] [Google Scholar]
- 63.Kunnumakkara AB, Guha S, Krishnan S, Diagaradjane P, Gelovani J, Aggarwal BB. Curcumin potentiates antitumor activity of gemcitabine in an orthotopic model of pancreatic cancer through suppression of proliferation, angiogenesis, and inhibition of nuclear factor-κB–regulated gene products. Cancer Res. 2007;67(8):3853–3861. doi: 10.1158/0008-5472.CAN-06-4257. [DOI] [PubMed] [Google Scholar]
- 64.Zhang C, Huang L, Xiong J, Xie L, Ying S, Jia Y, Yao Y, Song X, Zeng Z, Yuan J, et al. Isoalantolactone inhibits pancreatic cancer proliferation by regulation of PI3K and Wnt signal pathway. Plos One. 2021;16(3):e0247752. doi: 10.1371/journal.pone.0247752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Collins MA, Bednar F, Zhang Y, Brisset J-C, Galbán S, Galbán CJ, Rakshit S, Flannagan KS, Adsay NV, Pasca Di Magliano M, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012;122(2):639–653. doi: 10.1172/JCI59227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stellas D, Szabolcs M, Koul S, Li Z, Polyzos A, Anagnostopoulos C, Cournia Z, Tamvakopoulos C, Klinakis A, Efstratiadis A, et al. Therapeutic effects of an anti-Myc drug on mouse pancreatic cancer. JNCI J Nat Cancer Inst. 2014;106(12). doi: 10.1093/jnci/dju320. [DOI] [PubMed] [Google Scholar]
- 67.Blake DR, Vaseva AV, Hodge RG, Kline MP, Gilbert TS, Tyagi V, Huang D, Whiten GC, Larson JE, Wang X, et al. Application of a MYC degradation screen identifies sensitivity to CDK9 inhibitors in KRAS-mutant pancreatic cancer. Sci Signal. 2019;12(590). doi: 10.1126/scisignal.aav7259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eser S, Reiff N, Messer M, Seidler B, Gottschalk K, Dobler M, Hieber M, Arbeiter A, Klein S, Kong B, et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell. 2013;23(3):406–420. doi: 10.1016/j.ccr.2013.01.023. [DOI] [PubMed] [Google Scholar]
- 69.Qu D, Weygant N, Yao J, Chandrakesan P, Berry WL, May R, Pitts K, Husain S, Lightfoot S, Li M, et al. Overexpression of DCLK1-AL increases tumor cell invasion, drug resistance, and KRAS activation and can be targeted to inhibit tumorigenesis in pancreatic cancer. J Oncol. 2019;2019:1–11. doi: 10.1155/2019/6402925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morris JP, Wang SC, Hebrok M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat Rev Cancer. 2010;10(10):683–695. doi: 10.1038/nrc2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sharma N, Nanta R, Sharma J, Gunewardena S, Singh KP, Shankar S, Srivastava RK. PI3K/AKT/mTOR and sonic hedgehog pathways cooperate together to inhibit human pancreatic cancer stem cell characteristics and tumor growth. Oncotarget. 2015;6(31):32039. doi: 10.18632/oncotarget.5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhong Z, Sepramaniam S, Chew XH, Wood K, Lee MA, Madan B, Virshup DM. PORCN inhibition synergizes with PI3K/mTOR inhibition in Wnt-addicted cancers. Oncogene. 2019;38(40):6662–6677. doi: 10.1038/s41388-019-0908-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Asano T, Yao Y, Zhu J, Li D, Abbruzzese JL, Reddy SA. The PI 3-kinase/Akt signaling pathway is activated due to aberrant Pten expression and targets transcription factors NF-κ B and c-Myc in pancreatic cancer cells. Oncogene. 2004;23(53):8571–8580. doi: 10.1038/sj.onc.1207902. [DOI] [PubMed] [Google Scholar]
- 74.Allen-Petersen BL, Risom T, Feng Z, Wang Z, Jenny ZP, Thoma MC, Pelz KR, Morton JP, Sansom OJ, Lopez CD, et al. Activation of PP2A and inhibition of mTOR synergistically reduce MYC signaling and decrease tumor growth in pancreatic ductal adenocarcinoma. Cancer Res. 2019;79(1):209–219. doi: 10.1158/0008-5472.CAN-18-0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Terracciano F, Capone A, Montori A, Rinzivillo M, Partelli S, Panzuto F, Pilozzi E, Arcidiacono PG, Sette C, Capurso G, et al. MYC upregulation confers resistance to everolimus and establishes vulnerability to cyclin-dependent kinase inhibitors in pancreatic neuroendocrine neoplasm cells. Neuroendocrinology. 2020;111(8):739–751. doi: 10.1159/000509865. [DOI] [PubMed] [Google Scholar]
- 76.Pourdehnad M, Truitt ML, Siddiqi IN, Ducker GS, Shokat KM, Ruggero D. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc Natl Acad Sci. 2013;110(29):11988–11993. doi: 10.1073/pnas.1310230110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ravitz MJ, Chen L, Lynch M, Schmidt EV. c-myc repression of TSC2 contributes to control of translation initiation and Myc-induced transformation. Cancer Res. 2007;67(23):11209–11217. doi: 10.1158/0008-5472.CAN-06-4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Csibi A, Lee G, Yoon S-O, Tong H, Ilter D, Elia I, Fendt S-M, Roberts T, Blenis J. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr Biol. 2014;24(19):2274–2280. doi: 10.1016/j.cub.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chang TM, Chu PY, Hung WC, Shan YS, Lin HY, Huang KW, Chang JS, Chen L-T, Tsai H-J. c‐Myc promotes lymphatic metastasis of pancreatic neuroendocrine tumor through VEGFC upregulation. Cancer Sci. 2021;112(1):243. doi: 10.1111/cas.14717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chang T-M, Shan Y-S, Chu P-Y, Jiang SS, Hung W-C, Chen Y-L, Tu H-C, Lin H-Y, Tsai H-J, Chen L-T, et al. The regulatory role of aberrant phosphatase and tensin homologue and liver kinase B1 on AKT/mTOR/c-Myc axis in pancreatic neuroendocrine tumors. Oncotarget. 2017;8(58):98068. doi: 10.18632/oncotarget.20956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ma J, Sawai H, Ochi N, Matsuo Y, Xu D, Yasuda A, Takahashi H, Wakasugi T, Takeyama H. PTEN regulate angiogenesis through PI3K/Akt/VEGF signaling pathway in human pancreatic cancer cells. Mol Cell Biochem. 2009;331(1–2):161–171. doi: 10.1007/s11010-009-0154-x. [DOI] [PubMed] [Google Scholar]
- 82.Vogt PK. PI 3-kinase, mTOR, protein synthesis and cancer. Trends Mol Med. 2001;7(11):482–484. doi: 10.1016/S1471-4914(01)02161-X. [DOI] [PubMed] [Google Scholar]
- 83.Cui J, Jiang W, Wang S, Wang L, Xie K. Role of Wnt/β-catenin signaling in drug resistance of pancreatic cancer. Curr Pharm Des. 2012;18(17):2464–2471. doi: 10.2174/13816128112092464. [DOI] [PubMed] [Google Scholar]
- 84.Sun Y, Zhu Q, Zhou M, Yang W, Shi H, Shan Y, Zhang Q, Yu F. Restoration of miRNA-148a in pancreatic cancer reduces invasion and metastasis by inhibiting the Wnt/β-catenin signaling pathway via downregulating maternally expressed gene-3. Exp Ther Med. 2019;17(1):639–648. doi: 10.3892/etm.2018.7026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Madan B, Harmston N, Nallan G, Montoya A, Faull P, Petretto E, Virshup DM. Temporal dynamics of Wnt-dependent transcriptome reveal an oncogenic Wnt/MYC/ribosome axis. J Clin Invest. 2019;128(12):5620–5633. doi: 10.1172/JCI122383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lemieux E, Cagnol S, Beaudry K, Carrier J, Rivard N. Oncogenic KRAS signalling promotes the Wnt/β-catenin pathway through LRP6 in colorectal cancer. Oncogene. 2015;34(38):4914–4927. doi: 10.1038/onc.2014.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yu L, Li X, Li H, Chen H, Liu H. Rab11a sustains GSK3β/Wnt/β-catenin signaling to enhance cancer progression in pancreatic cancer. Tumor Biol. 2016;37(10):13821–13829. doi: 10.1007/s13277-016-5172-1. [DOI] [PubMed] [Google Scholar]
- 88.Li Y-J, Wei Z-M, Meng Y-X, Ji X-R. β-catenin up-regulates the expression of cyclinD1, c-myc and MMP-7 in human pancreatic cancer: relationships with carcinogenesis and metastasis. World J Gastroenterol WJG. 2005;11(14):2117. doi: 10.3748/wjg.v11.i14.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Garg B, Giri B, Majumder K, Dudeja V, Banerjee S, Saluja A. Modulation of post-translational modifications in β-catenin and LRP6 inhibits Wnt signaling pathway in pancreatic cancer. Cancer Lett. 2017;388:64–72. doi: 10.1016/j.canlet.2016.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhan T, Zhu Q, Han Z, Tan J, Liu M, Liu W, Chen W, Chen X, Chen X, Deng J, et al. miR-455-3p functions as a tumor suppressor by restraining Wnt/β-catenin signaling via TAZ in pancreatic cancer. Cancer Manag Res. 2020;12:1483. doi: 10.2147/CMAR.S235794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hao Y-H, Lafita-Navarro MC, Zacharias L, Borenstein-Auerbach N, Kim M, Barnes S, Kim J, Shay J, DeBerardinis RJ, Conacci-Sorrell M, et al. Induction of LEF1 by MYC activates the WNT pathway and maintains cell proliferation. Cell Commun Signaling. 2019;17(1):1–16. doi: 10.1186/s12964-019-0444-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zacarías-Fluck MF, Jauset T, Martínez-Martín S, Kaur J, Casacuberta-Serra S, Massó-Vallés D, Serrano Del Pozo E, Martín-Fernández G, González-Larreategui Í, López-Estévez S, et al. The Wnt signaling receptor Fzd9 is essential for Myc-driven tumorigenesis in pancreatic islets. Life Sci Alliance. 2021;4(5):e201900490. doi: 10.26508/lsa.201900490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hayes TK, Neel NF, Hu C, Gautam P, Chenard M, Long B, Aziz M, Kassner M, Bryant KL, Pierobon M, et al. Long-term ERK inhibition in KRAS-mutant pancreatic cancer is associated with MYC degradation and senescence-like growth suppression. Cancer Cell. 2016;29(1):75–89. doi: 10.1016/j.ccell.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhai S, Xu Z, Xie J, Zhang J, Wang X, Peng C, Li H, Chen H, Shen B, Deng X, et al. Epigenetic silencing of LncRNA LINC00261 promotes c-myc-mediated aerobic glycolysis by regulating miR-222-3p/HIPK2/ERK axis and sequestering IGF2BP1. Oncogene. 2021;40(2):277–291. doi: 10.1038/s41388-020-01525-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, Gunda V, Pierobon M, Waters AM, George SD, et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med. 2019;25(4):628–640. doi: 10.1038/s41591-019-0368-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sheng W, Chen C, Dong M, Wang G, Zhou J, Song H, Li Y, Zhang J, Ding S. Calreticulin promotes EGF-induced EMT in pancreatic cancer cells via Integrin/EGFR-ERK/MAPK signaling pathway. Cell Death Dis. 2017;8(10):e3147–e. doi: 10.1038/cddis.2017.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sheng W, Shi X, Lin Y, Tang J, Jia C, Cao R, et al. Musashi2 promotes EGF-induced EMT in pancreatic cancer via ZEB1-ERK/MAPK signaling. J Exp Clin Cancer Res. 2020;39:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tanaka T, Kaida T, Yokoi K, Ishii S, Nishizawa N, Kawamata H, Katoh H, Sato T, Nakamura T, Watanabe M, et al. Critical relevance of genomic gains of PRL-3/EGFR/c-myc pathway genes in liver metastasis of colorectal cancer. Oncol Lett. 2019;17(1):1257–1266. doi: 10.3892/ol.2018.9728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Diersch S, Wirth M, Schneeweis C, Jörs S, Geisler F, Siveke JT, Rad R, Schmid RM, Saur D, Rustgi AK, et al. Kras G12D induces EGFR-MYC cross signaling in murine primary pancreatic ductal epithelial cells. Oncogene. 2016;35(29):3880–3886. doi: 10.1038/onc.2015.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhong H, Sanchez C, Spitrzer D, Plambeck-Suess S, Gibbs J, Hawkins WG, Denardo D, Gao F, Pufahl RA, Lockhart AC, et al. Synergistic effects of concurrent blockade of PI3K and MEK pathways in pancreatic cancer preclinical models. PloS One. 2013;8(10):e77243. doi: 10.1371/journal.pone.0077243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zou Z, Chang H, Li H, Wang S. Induction of reactive oxygen species: an emerging approach for cancer therapy. Apoptosis. 2017;22(11):1321–1335. doi: 10.1007/s10495-017-1424-9. [DOI] [PubMed] [Google Scholar]
- 102.Suzuki S, Okada M, Shibuya K, Seino M, Sato A, Takeda H, Seino S, Yoshioka T, Kitanaka C. JNK suppression of chemotherapeutic agents-induced ROS confers chemoresistance on pancreatic cancer stem cells. Oncotarget. 2015;6(1):458. doi: 10.18632/oncotarget.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. ….
- 104.Rodríguez-Enríquez S, Pacheco-Velázquez SC, Marín-Hernández Á, Gallardo-Pérez JC, Robledo-Cadena DX, Hernández-Reséndiz I, García-García JD, Belmont-Díaz J, López-Marure R, Hernández-Esquivel L, et al. Resveratrol inhibits cancer cell proliferation by impairing oxidative phosphorylation and inducing oxidative stress. Toxicol Appl Pharmacol. 2019;370:65–77. doi: 10.1016/j.taap.2019.03.008. [DOI] [PubMed] [Google Scholar]
- 105.Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, Barneda D, Sellers K, Campos-Olivas R, Graña O, et al. MYC/PGC-1α balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. 2015;22(4):590–605. doi: 10.1016/j.cmet.2015.08.015. [DOI] [PubMed] [Google Scholar]
- 106.Liang C, Shi S, Liu M, Qin Y, Meng Q, Hua J, Ji S, Zhang Y, Yang J, Xu J, et al. PIN1 maintains redox balance via the c-Myc/NRF2 axis to counteract Kras-induced mitochondrial respiratory injury in pancreatic cancer cells. Cancer Res. 2019;79(1):133–145. doi: 10.1158/0008-5472.CAN-18-1968. [DOI] [PubMed] [Google Scholar]
- 107.Ding L, Cao J, Lin W, Chen H, Xiong X, Ao H, Yu M, Lin J, Cui Q. The roles of cyclin-dependent kinases in cell-cycle progression and therapeutic strategies in human breast cancer. Int J Mol Sci. 2020;21(6):1960. doi: 10.3390/ijms21061960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tarrado‐Castellarnau M, de Atauri P, Tarragó‐Celada J, Perarnau J, Yuneva M, Thomson TM, Cascante M. De novo MYC addiction as an adaptive response of cancer cells to CDK4/6 inhibition. Mol Syst Biol. 2017;13(10):940. doi: 10.15252/msb.20167321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Du S, Wang H, Cai J, Ren R, Zhang W, Wei W, Shen X. Apolipoprotein E2 modulates cell cycle function to promote proliferation in pancreatic cancer cells via regulation of the c-Myc–p21Waf1 signalling pathway. Biochem Cell Biol. 2020;98(2):191–202. doi: 10.1139/bcb-2018-0230. [DOI] [PubMed] [Google Scholar]
- 110.Fisher RP. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J Cell Sci. 2005;118(22):5171–5180. doi: 10.1242/jcs.02718. [DOI] [PubMed] [Google Scholar]
- 111.Peterlin BM, Price DH. Controlling the elongation phase of transcription with P-TEFb. Mol Cell. 2006;23(3):297–305. doi: 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 112.Morrison AH, Byrne KT, Vonderheide RH. Immunotherapy and prevention of pancreatic cancer. Trends Cancer. 2018;4(6):418–428. doi: 10.1016/j.trecan.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liang M-Q, Yu F-Q, Chen C. C-Myc regulates PD-L1 expression in esophageal squamous cell carcinoma. Am J Transl Res. 2020;12(2):379. [PMC free article] [PubMed] [Google Scholar]
- 114.Wang J, Jia Y, Zhao S, Zhang X, Wang X, Han X, Wang Y, Ma M, Shi J, Liu L, et al. BIN1 reverses PD-L1-mediated immune escape by inactivating the c-MYC and EGFR/MAPK signaling pathways in non-small cell lung cancer. Oncogene. 2017;36(45):6235–6243. doi: 10.1038/onc.2017.217. [DOI] [PubMed] [Google Scholar]
- 115.Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, Iyer AK. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol. 2017;8:561. doi: 10.3389/fphar.2017.00561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Han H, Jain AD, Truica MI, Izquierdo-Ferrer J, Anker JF, Lysy B, Sagar V, Luan Y, Chalmers ZR, Unno K, et al. Small-molecule MYC inhibitors suppress tumor growth and enhance immunotherapy. Cancer Cell. 2019;36(5):483–97. e15. doi: 10.1016/j.ccell.2019.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pan Y, Fei Q, Xiong P, Yang J, Zhang Z, Lin X, Pan M, Lu F, Huang H. Synergistic inhibition of pancreatic cancer with anti-PD-L1 and c-Myc inhibitor JQ1. Oncoimmunology. 2019;8(5):e1581529. doi: 10.1080/2162402X.2019.1581529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fan P, Ma J, Jin X. Far upstream element-binding protein 1 is up-regulated in pancreatic cancer and modulates immune response by increasing programmed death ligand 1. Biochem Biophys Res Commun. 2018;505(3):830–836. doi: 10.1016/j.bbrc.2018.10.009. [DOI] [PubMed] [Google Scholar]
- 119.Wang R-Q, Geng J, Sheng W-J, Liu X-J, Jiang M, Zhen Y-S. The ionophore antibiotic gramicidin A inhibits pancreatic cancer stem cells associated with CD47 down-regulation. Cancer Cell Int. 2019;19:1–13. doi: 10.1186/s12935-018-0719-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ren S, Cai Y, Hu S, Liu J, Zhao Y, Ding M, Chen X, Zhan L, Zhou X, Wang X, et al. Berberine exerts anti-tumor activity in diffuse large B-cell lymphoma by modulating c-myc/CD47 axis. Biochem Pharmacol. 2021;188:114576. doi: 10.1016/j.bcp.2021.114576. [DOI] [PubMed] [Google Scholar]
- 121.Michaels AD, Newhook TE, Adair SJ, Morioka S, Goudreau BJ, Nagdas S, Mullen MG, Persily JB, Bullock TNJ, Slingluff CL, et al. CD47 blockade as an adjuvant immunotherapy for resectable pancreatic cancer. Clin Cancer Res. 2018;24(6):1415–1425. doi: 10.1158/1078-0432.CCR-17-2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pan Y, Lu F, Fei Q, Yu X, Xiong P, Yu X, Dang Y, Hou Z, Lin W, Lin X, et al. Single-cell RNA sequencing reveals compartmental remodeling of tumor-infiltrating immune cells induced by anti-CD47 targeting in pancreatic cancer. J Hematol Oncol. 2019;12(1):1–18. doi: 10.1186/s13045-019-0822-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Golubovskaya V, Berahovich R, Zhou H, Xu S, Harto H, Li L, Chao -C-C, Mao MM, Wu L. CD47-CAR-T cells effectively kill target cancer cells and block pancreatic tumor growth. Cancers. 2017;9(12):139. doi: 10.3390/cancers9100139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Saravia J, Zeng H, Dhungana Y, Blanco DB, Nguyen T-LM, Chapman NM, Wang Y, Kanneganti A, Liu S, Raynor JL, et al. Homeostasis and transitional activation of regulatory T cells require c-Myc. Sci Adv. 2020;6(1):eaaw6443. doi: 10.1126/sciadv.aaw6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Martinez-Bosch N, Vinaixa J, Navarro P. Immune evasion in pancreatic cancer: from mechanisms to therapy. Cancers. 2018;10(1):6. doi: 10.3390/cancers10010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chen H, Shan G. The physiological function of long-noncoding RNAs. Non-coding RNA Res. 2020;5(4):178–184. doi: 10.1016/j.ncrna.2020.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liu S, Zheng Y, Zhang Y, Zhang J, Xie F, Guo S, Gu J, Yang J, Zheng P, Lai J, et al. Methylation-mediated LINC00261 suppresses pancreatic cancer progression by epigenetically inhibiting c-Myc transcription. Theranostics. 2020;10(23):10634. doi: 10.7150/thno.44278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yu Q, Zhou X, Xia Q, Shen J, Yan J, Zhu J, Li X, Shu M. Long non-coding RNA CCAT1 that can be activated by c-Myc promotes pancreatic cancer cell proliferation and migration. Am J Transl Res. 2016;8(12):5444. [PMC free article] [PubMed] [Google Scholar]
- 129.Li L, Chen H, Gao Y, Wang Y-W, Zhang G-Q, Pan S-H, Ji L, Kong R, Wang G, Jia Y-H, et al. Long noncoding RNA MALAT1 promotes aggressive pancreatic cancer proliferation and metastasis via the stimulation of autophagy. Mol Cancer Ther. 2016;15(9):2232–2243. doi: 10.1158/1535-7163.MCT-16-0008. [DOI] [PubMed] [Google Scholar]
- 130.Peng W-X, He R-Z, Zhang Z, Yang L, Mo -Y-Y. LINC00346 promotes pancreatic cancer progression through the CTCF-mediated Myc transcription. Oncogene. 2019;38(41):6770–6780. doi: 10.1038/s41388-019-0918-z. [DOI] [PubMed] [Google Scholar]
- 131.He J, Li F, Zhou Y, Hou X, Liu S, Li X, Zhang Y, Jing X, Yang L. LncRNA XLOC_006390 promotes pancreatic carcinogenesis and glutamate metabolism by stabilizing c-Myc. Cancer Lett. 2020;469:419–428. doi: 10.1016/j.canlet.2019.11.021. [DOI] [PubMed] [Google Scholar]
- 132.Ou Z-L, Luo Z, Lu Y-B. Long non-coding RNA HULC as a diagnostic and prognostic marker of pancreatic cancer. World J Gastroenterol. 2019;25(46):6728. doi: 10.3748/wjg.v25.i46.6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Olivero CE, Martínez-Terroba E, Zimmer J, Liao C, Tesfaye E, Hooshdaran N, Schofield JA, Bendor J, Fang D, Simon MD, et al. p53 activates the long noncoding RNA Pvt1b to inhibit Myc and suppress tumorigenesis. Mol Cell. 2020;77(4):761–74. e8. doi: 10.1016/j.molcel.2019.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Xiong G, Liu C, Yang G, Feng M, Xu J, Zhao F, You L, Zhou L, Zheng L, Hu Y, et al. Long noncoding RNA GSTM3TV2 upregulates LAT2 and OLR1 by competitively sponging let-7 to promote gemcitabine resistance in pancreatic cancer. J Hematol Oncol. 2019;12(1):1–18. doi: 10.1186/s13045-019-0777-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fatma H, Siddique HR. Role of long non-coding RNAs and MYC interaction in cancer metastasis: a possible target for therapeutic intervention. Toxicol Appl Pharmacol. 2020;399:115056. doi: 10.1016/j.taap.2020.115056. [DOI] [PubMed] [Google Scholar]
- 136.Shen S, Yao T, Xu Y, Zhang D, Fan S, Ma J. CircECE1 activates energy metabolism in osteosarcoma by stabilizing c-Myc. Mol Cancer. 2020;19(1):1–17. doi: 10.1186/s12943-020-01269-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lu TX, Rothenberg ME. MicroRNA. J Allergy Clin Immunol. 2018;141(4):1202–1207. doi: 10.1016/j.jaci.2017.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dey S, Kwon JJ, Liu S, Hodge GA, Taleb S, Zimmers TA, Wan J, Kota J. miR-29a is repressed by MYC in pancreatic cancer and its restoration drives tumor-suppressive effects via Downregulation of LOXL2. Mol Cancer Res. 2020;18(2):311–323. doi: 10.1158/1541-7786.MCR-19-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Azmi AS, Li Y, Muqbil I, Aboukameel A, Senapedis W, Baloglu E, Landesman Y, Shacham S, Kauffman MG, Philip PA, et al. Exportin 1 (XPO1) inhibition leads to restoration of tumor suppressor miR-145 and consequent suppression of pancreatic cancer cell proliferation and migration. Oncotarget. 2017;8(47):82144. doi: 10.18632/oncotarget.19285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Liu Y, Li X, Zhu S, Zhang J, Yang M, Qin Q, Deng S-C, Wang B, Tian K, Liu L, et al. Ectopic expression of miR-494 inhibited the proliferation, invasion and chemoresistance of pancreatic cancer by regulating SIRT1 and c-Myc. Gene Ther. 2015;22(9):729–738. doi: 10.1038/gt.2015.39. [DOI] [PubMed] [Google Scholar]
- 141.Gibori H, Eliyahu S, Krivitsky A, Ben-Shushan D, Epshtein Y, Tiram G, Blau R, Ofek P, Lee JS, Ruppin E, et al. Amphiphilic nanocarrier-induced modulation of PLK1 and miR-34a leads to improved therapeutic response in pancreatic cancer. Nat Commun. 2018;9(1):1–18. doi: 10.1038/s41467-017-02283-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chang T-C, Zeitels LR, Hwang H-W, Chivukula RR, Wentzel EA, Dews M, Jung J, Gao P, Dang CV, Beer MA, et al. Lin-28B transactivation is necessary for Myc-mediated let-7 repression and proliferation. Proc Natl Acad Sci. 2009;106(9):3384–3389. doi: 10.1073/pnas.0808300106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang Y, Li J, Guo S, Ouyang Y, Yin L, Liu S, Zhao Z, Yang J, Huang W, Qin H, et al. Lin28B facilitates the progression and metastasis of pancreatic ductal adenocarcinoma. Oncotarget. 2017;8(36):60414. doi: 10.18632/oncotarget.19578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sureban SM, May R, Weygant N, Qu D, Chandrakesan P, Bannerman-Menson E, Ali N, Pantazis P, Westphalen CB, Wang TC, et al. XMD8-92 inhibits pancreatic tumor xenograft growth via a DCLK1-dependent mechanism. Cancer Lett. 2014;351(1):151–161. doi: 10.1016/j.canlet.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 145.Sureban SM, May R, Lightfoot SA, Hoskins AB, Lerner M, Brackett DJ, Postier RG, Ramanujam R, Mohammed A, Rao CV, et al. DCAMKL-1 regulates epithelial–mesenchymal transition in human pancreatic cells through a miR-200a–dependent mechanism. Cancer Res. 2011;71(6):2328–2338. doi: 10.1158/0008-5472.CAN-10-2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Blackwood EM, Eisenman RN. Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science. 1991;251(4998):1211–1217. doi: 10.1126/science.2006410. [DOI] [PubMed] [Google Scholar]
- 147.Jeong K-C, Kim K-T, Seo -H-H, Shin S-P, Ahn K-O, Ji M-J, Park WS, Kim I-H, Lee S-J, Seo HK, et al. Intravesical instillation of c-MYC inhibitor KSI-3716 suppresses orthotopic bladder tumor growth. J Urol. 2014;191(2):510–518. doi: 10.1016/j.juro.2013.07.019. [DOI] [PubMed] [Google Scholar]
- 148.Jeong K-C, Ahn K-O, Yang C-H. Small-molecule inhibitors of c-Myc transcriptional factor suppress proliferation and induce apoptosis of promyelocytic leukemia cell via cell cycle arrest. Mol Biosyst. 2010;6(8):1503–1509. doi: 10.1039/c002534h. [DOI] [PubMed] [Google Scholar]
- 149.Choi SH, Mahankali M, Lee SJ, Hull M, Petrassi HM, Chatterjee AK, Schultz PG, Jones KA, Shen W. Targeted disruption of Myc–Max oncoprotein complex by a small molecule. ACS Chem Biol. 2017;12(11):2715–2719. doi: 10.1021/acschembio.7b00799. [DOI] [PubMed] [Google Scholar]
- 150.Sheikh‐Zeineddini N, Bashash D, Safaroghli‐Azar A, Riyahi N, Shabestari RM, Janzamin E, Safa M. Suppression of c‐Myc using 10058‐F4 exerts caspase‐3‐dependent apoptosis and intensifies the antileukemic effect of vincristine in pre‐B acute lymphoblastic leukemia cells. J Cell Biochem. 2019;120(8):14004–14016. doi: 10.1002/jcb.28675. [DOI] [PubMed] [Google Scholar]
- 151.Lv M, Wang Y, Wu W, Yang S, Zhu H, Hu B, Chen Y, Shi C, Zhang Y, Mu Q, et al. C-Myc inhibitor 10058-F4 increases the efficacy of dexamethasone on acute lymphoblastic leukaemia cells. Mol Med Rep. 2018;18(1):421–428. doi: 10.3892/mmr.2018.8935. [DOI] [PubMed] [Google Scholar]
- 152.Sayyadi M, Safaroghli-Azar A, Pourbagheri-Sigaroodi A, Abolghasemi H, Anoushirvani AA, Bashash D. c-Myc inhibition using 10058-F4 increased the sensitivity of acute promyelocytic leukemia cells to arsenic trioxide via blunting PI3K/NF-κB axis. Arch Med Res. 2020;51(7):636–644. doi: 10.1016/j.arcmed.2020.06.002. [DOI] [PubMed] [Google Scholar]
- 153.Zhang M, Fan H-Y, Li S-C. Inhibition of c-Myc by 10058-F4 induces growth arrest and chemosensitivity in pancreatic ductal adenocarcinoma. Biomed Pharmacother. 2015;73:123–128. doi: 10.1016/j.biopha.2015.05.019. [DOI] [PubMed] [Google Scholar]
- 154.Wang C, Zhang J, Yin J, Gan Y, Xu S, Gu Y, Yang Y, Fan C, Lu S, Peng X, et al. Alternative approaches to target Myc for cancer treatment. Signal Transduction Targeted Ther. 2021;6(1):1–14. doi: 10.1038/s41392-020-00451-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wang C, Fang H, Zhang J, Gu Y. Targeting “undruggable” c-Myc protein by synthetic lethality. Front Med. 2021;15(1):1–10. doi: 10.1007/s11684-020-0741-5. [DOI] [PubMed] [Google Scholar]
- 156.Morgado-Pascual JL, Rayego-Mateos S, Tejedor L, Suarez-Alvarez B, Ruiz-Ortega M. Bromodomain and extraterminal proteins as novel epigenetic targets for renal diseases. Front Pharmacol. 2019;10:1315. doi: 10.3389/fphar.2019.01315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Coudé -M-M, Braun T, Berrou J, Dupont M, Bertrand S, Masse A, Raffoux E, Itzykson R, Delord M, Riveiro ME, et al. BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget. 2015;6(19):17698. doi: 10.18632/oncotarget.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal R, Qi J, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, Sims RJ. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci. 2011;108(40):16669–16674. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Jin X, Fang R, Fan P, Zeng L, Zhang B, Lu X, Liu T. PES1 promotes BET inhibitors resistance and cells proliferation through increasing c-Myc expression in pancreatic cancer. J Exp Clin Cancer Res. 2019;38(1):1–16. doi: 10.1186/s13046-019-1466-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kumar K, Raza SS, Knab LM, Chow CR, Kwok B, Bentrem DJ, Popovic R, Ebine K, Licht JD, Munshi HG, et al. GLI2-dependent c-MYC upregulation mediates resistance of pancreatic cancer cells to the BET bromodomain inhibitor JQ1. Sci Rep. 2015;5(1):1–9. doi: 10.1038/srep09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wang B, Fan P, Zhao J, Wu H, Jin X, Wu H. FBP1 loss contributes to BET inhibitors resistance by undermining c-Myc expression in pancreatic ductal adenocarcinoma. J Exp Clin Cancer Res. 2018;37(1):1–12. doi: 10.1186/s13046-018-0888-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Xie F, Huang M, Lin X, Liu C, Liu Z, Meng F, Wang C, Huang Q. The BET inhibitor I-BET762 inhibits pancreatic ductal adenocarcinoma cell proliferation and enhances the therapeutic effect of gemcitabine. Sci Rep. 2018;8(1):1–10. doi: 10.1038/s41598-018-26496-0. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 164.Jauset T, Massó-Vallés D, Martínez-Martín S, Beaulieu M-E, Foradada L, Fiorentino FP, Yokota J, Haendler B, Siegel S, Whitfield JR, et al. BET inhibition is an effective approach against KRAS-driven PDAC and NSCLC. Oncotarget. 2018;9(27):18734. doi: 10.18632/oncotarget.24648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Arnold HK, Sears RC. Protein phosphatase 2A regulatory subunit B56α associates with c-Myc and negatively regulates c-Myc accumulation. Mol Cell Biol. 2006;26(7):2832–2844. doi: 10.1128/MCB.26.7.2832-2844.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Liu L, Eisenman RN. Regulation of c-Myc protein abundance by a protein phosphatase 2A–glycogen synthase kinase 3β–negative feedback pathway. Genes Cancer. 2012;3(1):23–36. doi: 10.1177/1947601912448067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Farrell AS, Allen-Petersen B, Daniel CJ, Wang X, Wang Z, Rodriguez S, Impey S, Oddo J, Vitek MP, Lopez C, et al. Targeting inhibitors of the tumor suppressor PP2A for the treatment of pancreatic cancer. Mol Cancer Res. 2014;12(6):924–939. doi: 10.1158/1541-7786.MCR-13-0542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Farrington CC, Yuan E, Mazhar S, Izadmehr S, Hurst L, Allen-Petersen BL, Janghorban M, Chung E, Wolczanski G, Galsky M, et al. Protein phosphatase 2A activation as a therapeutic strategy for managing MYC-driven cancers. J Biol Chem. 2020;295(3):757–770. doi: 10.1016/S0021-9258(17)49933-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lambrecht C, Libbrecht L, Sagaert X, Pauwels P, Hoorne Y, Crowther J, Louis JV, Sents W, Sablina A, Janssens V, et al. Loss of protein phosphatase 2A regulatory subunit B56δ promotes spontaneous tumorigenesis in vivo. Oncogene. 2018;37(4):544–552. doi: 10.1038/onc.2017.350. [DOI] [PubMed] [Google Scholar]
- 170.Yang JY, Huo YM, Yang MW, Shen Y, Liu DJ, Fu XL, Tao L-Y, He R-Z, Zhang J-F, Hua R, et al. SF3B1 mutation in pancreatic cancer contributes to aerobic glycolysis and tumor growth through a PP2A–c‐Myc axis. Mol Oncol. 2021;15(11):3076–3090. doi: 10.1002/1878-0261.12970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Young Lim SHS D, Lee M-H, Malakhova M, Kurinov I, Wu Q, Xu J, Xu J, Jiang Y, Dong Z, Liu K, et al. A natural small molecule, catechol, induces c-Myc degradation by directly targeting ERK2 in lung cancer. Oncotarget. 2016;7(23):35001. doi: 10.18632/oncotarget.9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Fonseca BD, Smith EM, Yelle N, Alain T, Bushell M, Pause A. The ever-evolving role of mTOR in translation. Semin Cell Dev Biol Elsevier. 2014;36:102–112. doi: 10.1016/j.semcdb.2014.09.014. [DOI] [PubMed] [Google Scholar]
- 173.Alzahrani AS. 2019. PI3K/Akt/mTOR inhibitors in cancer: at the bench and bedside. In: Seminars in cancer biology. Elsevier; p. 125–132. [DOI] [PubMed] [Google Scholar]
- 174.Li Y, Seto E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med. 2016;6(10):a026831. doi: 10.1101/cshperspect.a026831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Edderkaoui M, Chheda C, Soufi B, Zayou F, Hu RW, Ramanujan VK, Pan X, Boros LG, Tajbakhsh J, Madhav A, et al. An inhibitor of GSK3B and HDACs kills pancreatic cancer cells and slows pancreatic tumor growth and metastasis in mice. Gastroenterology. 2018;155(6):1985–98. e5. doi: 10.1053/j.gastro.2018.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lee HS, Park SB, Kim SA, Kwon SK, Cha H, Ro S, Takumi T, Hashimoto R, Otani H, Pazour GJ, et al. A novel HDAC inhibitor, CG200745, inhibits pancreatic cancer cell growth and overcomes gemcitabine resistance. Sci Rep. 2017;7(1):1–9. doi: 10.1038/s41598-016-0028-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Liu T, Tee AE, Porro A, Smith SA, Dwarte T, Liu PY, Iraci N, Sekyere E, Haber M, Norris MD, et al. Activation of tissue transglutaminase transcription by histone deacetylase inhibition as a therapeutic approach for Myc oncogenesis. Proc Natl Acad Sci. 2007;104(47):18682–18687. doi: 10.1073/pnas.0705524104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Marshall GM, Gherardi S, Xu N, Neiron Z, Trahair T, Scarlett C, Chang DK, Liu PY, Jankowski K, Iraci N, et al. Transcriptional upregulation of histone deacetylase 2 promotes Myc-induced oncogenic effects. Oncogene. 2010;29(44):5957–5968. doi: 10.1038/onc.2010.332. [DOI] [PubMed] [Google Scholar]
- 179.Zhu C, Chen Q, Xie Z, Ai J, Tong L, Ding J, Geng M. The role of histone deacetylase 7 (HDAC7) in cancer cell proliferation: regulation on c-Myc. J Mol Med. 2011;89(3):279–289. doi: 10.1007/s00109-010-0701-7. [DOI] [PubMed] [Google Scholar]
- 180.X-h F, Zhang X, Yang H, X-w X, Z-l H, Yan J, Zheng X-L, Wei -R-R, Zhang Z-Q, Tang S-R, et al. CUDC-907 displays potent antitumor activity against human pancreatic adenocarcinoma in vitro and in vivo through inhibition of HDAC6 to downregulate c-Myc expression. Acta Pharmacol Sin. 2019;40(5):677–688. doi: 10.1038/s41401-018-0108-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kretzner L, Scuto A, Dino PM, Kowolik CM, Wu J, Ventura P, Jove R, Forman SJ, Yen Y, Kirschbaum MH, et al. Combining histone deacetylase inhibitor vorinostat with Aurora kinase inhibitors enhances lymphoma cell killing with repression of c-Myc, hTERT, and microRNA levels. Cancer Res. 2011;71(11):3912–3920. doi: 10.1158/0008-5472.CAN-10-2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Seo S-K, Jin H-O, Woo S-H, Kim Y-S, An S, Lee J-H, Hong S-I, Lee K-H, Choe T-B, Park I-C, et al. Histone deacetylase inhibitors sensitize human non-small cell lung cancer cells to ionizing radiation through acetyl p53-mediated c-myc down-regulation. J Thorac Oncol. 2011;6(8):1313–1319. doi: 10.1097/JTO.0b013e318220caff. [DOI] [PubMed] [Google Scholar]
- 183.Liu P, Xu N, Malyukova A, Scarlett C, Sun Y, Zhang X, Ling D, Su S-P, Nelson C, Chang DK, et al. The histone deacetylase SIRT2 stabilizes Myc oncoproteins. Cell Death Differ. 2013;20(3):503–514. doi: 10.1038/cdd.2012.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Buchholz M, Schatz A, Wagner M, Michl P, Linhart T, Adler G, Gress TM, Ellenrieder V. Overexpression of c‐myc in pancreatic cancer caused by ectopic activation of NFATc1 and the Ca2+/calcineurin signaling pathway. EMBO J. 2006;25(15):3714–3724. doi: 10.1038/sj.emboj.7601246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Köenig A, Linhart T, Schlengemann K, Reutlinger K, Wegele J, Adler G, Singh G, Hofmann L, Kunsch S, Büch T, et al. NFAT-induced histone acetylation relay switch promotes c-Myc-dependent growth in pancreatic cancer cells. Gastroenterology. 2010;138(3):1189–99. e2. doi: 10.1053/j.gastro.2009.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ding L, Billadeau DD. Glycogen synthase kinase-3β: a novel therapeutic target for pancreatic cancer. Expert Opin Ther Targets. 2020;24(5):417–426. doi: 10.1080/14728222.2020.1743681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Schild C, Wirth M, Reichert M, Schmid RM, Saur D, Schneider G. PI3K signaling maintains c‐myc expression to regulate transcription of E2F1 in pancreatic cancer cells. Mol Carcinog Published Cooperation Univ Texas MD Anderson Cancer Center. 2009;48(12):1149–1158. doi: 10.1002/mc.20569. [DOI] [PubMed] [Google Scholar]
- 188.Bang D, Wilson W, Ryan M, Yeh JJ, Baldwin AS. GSK-3α promotes oncogenic KRAS function in pancreatic cancer via TAK1–TAB stabilization and regulation of noncanonical NF-κB. Cancer Discov. 2013;3(6):690–703. doi: 10.1158/2159-8290.CD-12-0541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Wan B-S, Cheng M, Zhang L. Insulin-like growth factor 2 mRNA-binding protein 1 promotes cell proliferation via activation of AKT and is directly targeted by microRNA-494 in pancreatic cancer. World J Gastroenterol. 2019;25(40):6063. doi: 10.3748/wjg.v25.i40.6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Mahapatra L, Andruska N, Mao C, Le J, Shapiro DJ. A novel IMP1 inhibitor, BTYNB, targets c-Myc and inhibits melanoma and ovarian cancer cell proliferation. Transl Oncol. 2017;10(5):818–827. doi: 10.1016/j.tranon.2017.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Lambrianidou A, Sereti E, Soupsana K, Komini C, Dimas K, Trangas T. Trangas T. mTORC2 deploys the mRNA binding protein IGF2BP1 to regulate c-MYC expression and promote cell survival. Cell Signal. 2021;80:109912. doi: 10.1016/j.cellsig.2020.109912. [DOI] [PubMed] [Google Scholar]
- 192.Mackedenski S, Wang C, Li W-M, Lee CH. Characterizing the interaction between insulin-like growth factor 2 mRNA-binding protein 1 (IMP1) and KRAS expression. Biochem J. 2018;475(17):2749–2767. doi: 10.1042/BCJ20180575. [DOI] [PubMed] [Google Scholar]
- 193.Struntz NB, Chen A, Deutzmann A, Wilson RM, Stefan E, Evans HL, Ramirez MA, Liang T, Caballero F, Wildschut MHE, et al. Stabilization of the Max homodimer with a small molecule attenuates Myc-driven transcription. Cell Chem Biol. 2019;26(5):711–23. e14. doi: 10.1016/j.chembiol.2019.02.009. [DOI] [PubMed] [Google Scholar]
- 194.Hall Z, Wilson CH, Burkhart DL, Ashmore T, Evan GI, Griffin JL. Myc linked to dysregulation of cholesterol transport and storage in nonsmall cell lung cancer. J Lipid Res. 2020;61(11):1390–1399. doi: 10.1194/jlr.RA120000899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Yang G, Zhao F, Qiu J, Yueze L, Taiping Z, Zhao Y. OLR1 Promotes Invasion and Metastasis of Pancreatic Cancer by Regulating the Expression of HMGA2 through c-Myc. J Am Coll Surg. 2019;229(4):e148–e9. doi: 10.1016/j.jamcollsurg.2019.08.1133. [DOI] [Google Scholar]
- 196.Yang G, Xiong G, Feng M, Zhao F, Qiu J, Liu Y, Cao Z, Wang H, Yang J, You L, et al. OLR1 promotes pancreatic cancer metastasis via increased c-Myc expression and transcription of HMGA2. Molecular Cancer Research. 2020;18(5):685–697. doi: 10.1158/1541-7786.MCR-19-0718. [DOI] [PubMed] [Google Scholar]
- 197.Zhang S, Mo Q, Wang X. Oncological role of HMGA2. Int J Oncol. 2019;55(4):775–788. doi: 10.3892/ijo.2019.4856. [DOI] [PubMed] [Google Scholar]
- 198.Hristov AC, Cope L, Reyes MD, Singh M, Iacobuzio-Donahue C, Maitra A, et al. HMGA2 protein expression correlates with lymph node metastasis and increased tumor grade in pancreatic ductal adenocarcinoma. Mod Pathol. 2009;22(1):43–49. doi: 10.1038/modpathol.2008.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Zhao S, Gao X, Zang S, Li Y, Feng X, Yuan X. MicroRNA-383-5p acts as a prognostic marker and inhibitor of cell proliferation in lung adenocarcinoma by cancerous inhibitor of protein phosphatase 2A. Oncol Lett. 2017;14(3):3573–3579. doi: 10.3892/ol.2017.6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Shams R, Aghdaei HA, Behmanesh A, Sadeghi A, Zali M, Salari S, Padrón JM. MicroRNAs targeting MYC expression: trace of hope for pancreatic cancer therapy. a systematic review. Cancer Manag Res. 2020;12:2393. doi: 10.2147/CMAR.S245872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.González V, Hurley LH. The c- MYC NHE III 1: function and regulation. Annu Rev Pharmacol Toxicol. 2010;50(1):111–129. doi: 10.1146/annurev.pharmtox.48.113006.094649. [DOI] [PubMed] [Google Scholar]
- 202.Boxer LM, Dang CV. Translocations involving c-myc and c-myc function. Oncogene. 2001;20(40):5595–5610. doi: 10.1038/sj.onc.1204595. [DOI] [PubMed] [Google Scholar]
- 203.Davis AC, Wims M, Spotts GD, Hann SR, Bradley A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 1993;7(4):671–682. doi: 10.1101/gad.7.4.671. [DOI] [PubMed] [Google Scholar]
- 204.Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, Karnezis AN, Swigart LB, Nasi S, Evan GI, et al. Modelling Myc inhibition as a cancer therapy. Nature. 2008;455(7213):679–683. doi: 10.1038/nature07260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.He C, Jiang H, Geng S, Sheng H, Shen X, Zhang X, Zhu S, Chen X, Yang C, Gao H, et al. 2014. Expression and prognostic value of c-Myc and Fas (CD95/APO1) in patients with pancreatic cancer. Int J Clin Exp Pathol. 7(2):742. [PMC free article] [PubMed] [Google Scholar]
- 206.Mirza Z, Karim S. Nanoparticles-based drug delivery and gene therapy for breast cancer: recent advancements and future challenges. Semin Cancer Biol Elsevier. 2021;69:226–237. doi: 10.1016/j.semcancer.2019.10.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.