

ABSTRACT
Radiotherapy in cancer treatment involves the use of ionizing radiation for cancer cell killing. Although radiotherapy has shown significant improvements on cancer recurrence and mortality, several radiation-induced adverse effects have been documented. Of these adverse effects, radiation-induced cardiovascular disease (CVD) is particularly prominent among patients receiving mediastinal radiotherapy, such as breast cancer and Hodgkin’s lymphoma patients. A number of mechanisms of radiation-induced CVD pathogenesis have been proposed such as endothelial inflammatory activation, premature endothelial senescence, increased ROS and mitochondrial dysfunction. However, current research seems to point to a so-far unexamined and potentially novel involvement of epigenetics in radiation-induced CVD pathogenesis. Firstly, epigenetic mechanisms have been implicated in CVD pathophysiology. In addition, several studies have shown that ionizing radiation can cause epigenetic modifications, especially DNA methylation alterations. As a result, this review aims to provide a summary of the current literature linking DNA methylation to radiation-induced CVD and thereby explore DNA methylation as a possible contributor to radiation-induced CVD pathogenesis.
KEYWORDS: Radiation, Cardiovascular disease, Epigenetics, DNA methylation, Breast cancer
Graphical abstract
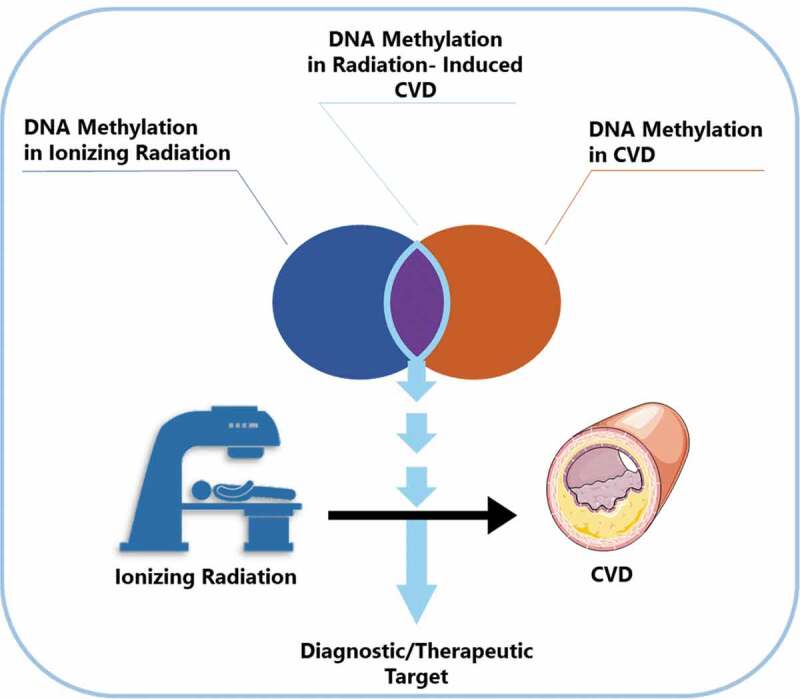
Introduction
Mediastinal radiotherapy is an effective means employed in the treatment of breast cancer, lung cancer, oesophageal cancer, thymoma, and mediastinal lymphoma (Hodgkin’s lymphoma (HL), primary mediastinal B-cell lymphoma (PMBCL), and T-lymphoblastic lymphoma) [1–5]. However, radiotherapy comes with definite side effects such as radiation-induced cardiovascular disease (CVD). In breast cancer, significantly higher rate of major coronary events were observed in irradiated left-sided breast cancer patients over their right-sided breast cancer counterparts [6]. A reason for this difference could be the different mean heart doses after left sided and right sided breast cancer radiotherapy being 3.6 and 1.9 Gy respectively [6–8]. As for Hodgkin’s lymphoma, treatment most commonly employs, in addition to radiotherapy, cardiotoxic chemotherapeutic agents e.g. anthracycline. Consequently, lymphoma survivors have a 5.3–7.3 times increased risk of death from cardiac mortality when compared to the general population [9,10]. The mechanisms underlying radiation-induced CVD are not completely understood. Moreover, a possible contribution of epigenetics mechanisms has recently been proposed.
Previous research has linked altered DNA methylation to the development of many diseases including CVD [11,12]. Conversely, ionizing radiation has been shown to cause DNA methylation alterations both in vitro and in vivo [13–16]. Considering the common occurrence of DNA methylation alterations in CVD and after ionizing radiation exposure, we suggest a previously unaddressed involvement of DNA methylation in radiation-induced CVD.
Radiation in cancer therapy
Radiation therapy employs high doses of ionizing radiation to kill cancer cells and shrink tumours. Radiation exerts its signature lethal cellular effects either directly or indirectly. Indirect effects involve the hydrolysis of water molecules to produce free radicals which can damage cellular compounds, such as DNA [17]. Several factors affect the biological effectiveness of radiation, namely the linear energy transfer (LET), total dose, fractionation rate, and radiosensitivity of the targeted cells or tissues [18]. LET is the energy transferred to the tissue by ionizing radiation (IR) per unit tract length. Simply, more charged, slower moving alpha particles and neutrons have higher LET and subsequently deliver more cell damaging energy to tissues than lesser charged, faster moving X-rays and γ-rays [19].
Despite the beneficial outcomes of cancer radiotherapy, cardiac toxicity occurring due to incidental heart irradiation is an undesirable healthy tissue side effect [7]. Previously, the heart was considered insensitive to radiation doses less than 30 Gy in accordance to the law of Bergonié and Tribondeau [20,21]. According to which, the degree of radiosensitivity of a biological tissue is dependent on its growth rate and degree of differentiation. Consequently, the heart with its non-dividing tissue was dubbed rather non-radiosensitive [21]. However, it was shown that the incidence of excess major coronary events increased by 7.4% per 1 Gy increase in the mean heart dose delivered by breast cancer radiotherapy with no observed safe threshold of dose [6].
Radiation-induced CVD manifestations
Radiation-induced CVD can manifest in different ways: coronary heart disease, pericarditis, cardiomyopathy and valvular heart disease (Figure 1). Due to contemporary radiation-sparing techniques including prone position radiotherapy, image- and dose- guided radiotherapy, intensity modulated radiation therapy (IMRT), stereotactic body radiation therapy (SBRT) and deep inspirational breath hold (DIBH), the incidence of acute pericarditis and cardiomyopathy has been reduced [22–25]. Consequently, coronary heart disease accounts for most of the cardiac mortality attributable to radiotherapy with the first signs of cardiac toxicity typically appearing after 10–15 years of follow-up [26–28]. Risk factors for the development of radiation-induced CVD strongly overlap with conventional risk factors for CVD, such as diabetes, hypertension, obesity, smoking, chronic obstructive pulmonary disease (COPD) and hypercholesterolemia. Additionally, therapy-dependent factors such as the cumulative dose from radiotherapy, chemotherapeutics (e.g. anthracyclins) and regular analgesic use represent additional risk factors [6,29].
Figure 1.
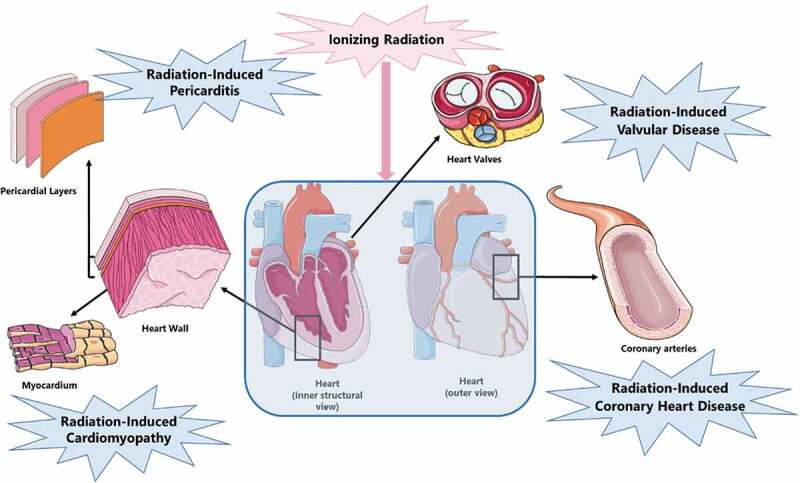
Radiation-induced CVD presentation.
Made with Microsoft Powerpoint using images from Server Medical Art (https://smart.servier.com/).
Radiation-induced coronary heart disease
Radiation-induced coronary heart disease (CHD) -also referred to as myocardial ischaemia- is caused by atherosclerosis of medium and large vessels. The pathogenesis of atherosclerosis involves a complex interplay of lipid accumulation, local inflammation and smooth muscle cell proliferation resulting in the formation of atherosclerotic plaques. Stable atherosclerotic plaques may narrow the lumen and hamper blood flow. However, unstable plaques may rupture and cause total occlusion of blood flow through thrombosis, leading to myocardial infarction. The risk of developing radiation-induced CHD is proportional to dose. This risk also increases within the first 5 years after exposure to radiation and continues for at least 20 years. In addition, radiation-induced CHD occurs at 10% of the tolerance dose of other cardiac tissues (e.g. 36–40 Gy mean heart dose for the pericardium and 40 Gy for the myocardium) [6,30,31].
Interestingly, similar radiation-induced atherosclerotic changes in the carotid artery were observed in Hodgkin’s lymphoma patients who received neck radiotherapy [32,33]. This arterial thickening leads to an increased relative risk for transient ischaemic attack or ischaemic stroke (2 general population risk) [34,35].
Radiation-induced pericarditis
Radiation-induced pericarditis describes a state of acute or delayed onset pericardial inflammation. Acute radiation-induced pericarditis is an early side effect to high radiation doses. The tolerance dose of human pericardium is estimated to be a mean heart dose higher than 36 or 40 Gy or a dose of 50 Gy administered to more than 30% of the heart. Consequently, due to current radiation-sparing techniques, acute radiation-induced pericarditis is considered very rare. On the other hand, chronic radiation-induced pericarditis is one of the most common manifestations of radiation-induced CVD presenting months to years after radiotherapy. However, its risk has also decreased significantly due to the same radiation-sparing techniques. Chronic radiation-induced pericarditis patients develop fibrous thickening of the pericardium even before manifestation of symptoms. This pericardial thickening might progress to chronic constrictive pericarditis which could then lead to intractable heart failure and will require pericardiectomy when it becomes symptomatic [30,36,37].
Radiation-induced cardiomyopathy
Radiation-induced cardiomyopathy most commonly occurs after previous valvular disease or myocardial infarction. The tolerance dose of the human myocardium is approximately 40 Gy. Consequently, pronounced radiation-induced cardiomyopathy occurs at higher doses. The most probable mechanism behind radiation-induced cardiomyopathy is radiation-induced microvasculature damage leading to capillary loss and myocardial hypoxia and cell death. Dead cells are replaced by fibrotic tissue leading to a reduction of myocardial elasticity and distensibility [30,31].
Radiation-induced valvular disease
Radiation-induced valvular disease risk is dose dependent with delayed onset (up to 20 years after radiotherapy). It leads to endocardial fibrosis which starts by thickening and calcification of the valvular endocardium [38,39].
Mechanisms of radiation-induced CVD
Radiation-induced CVD pathophysiology has been extensively described in literature [30,31,40–45]. Inflammatory changes as well as reactive oxygen species (ROS) production appear to be the main causes of the early radiation-induced cardiac tissue damage. On the other hand, persistence of this state of inflammation and oxidative stress results in the delayed radiation-induced tissue damage [44–47].
An early event in radiation-induced cardiac effects is NF-κB activation. NF-κB is a transcription factor implicated in the regulation of immune cell maturation, cell survival, and inflammation signalling pathways [48]. IR activates NF-κB through ROS and double stranded breaks (DSBs) as well as damage-associated molecular patterns (DAMPs) released from stressed or dying cells [42]. DAMP binding to endothelial cells also activates MAPK, and interferon regulatory factor 3 (IRF3) signalling [42,49]. This leads to the expression of various pro-inflammatory cytokines (e.g. Interleukins 1 & 6, interferon-γ (IFN-γ), and Tumour Necrosis Factor (TNF-α)), chemokines (e.g. Monocyte chemoattractant protein-1 or MCP-1), cell adhesion molecules (e.g. Vascular cell adhesion molecule 1 or VCAM-1 and E-Selectin) and matrix metalloproteinases. This ultimately initiates an acute inflammatory state with endothelial activation and inflammatory cell recruitment within minutes of IR exposure. The recruited inflammatory cells (mainly neutrophils) add to the inflammatory state and secrete pro-fibrotic transforming growth factor beta (TGF-β). TGF-β has been shown to initiate myocardial remodelling by inducing cardiac fibroblast differentiation into myofibroblast with increased collagen deposition and eventual fibrosis [50].
In addition to chronic inflammation, chronic oxidative stress is an important pathophysiologic mechanism of radiation-induced CVD. As previously mentioned, IR-induced ROS play a role in the activation of NF-κB. In addition, ROS upregulate several enzymes such as nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase), lipoxygenases (LOXs), nitric oxide synthase (NOS), and cyclooxygenases (COXs). These enzymes contribute to the acute and chronic effects of oxidative stress by inducing production of inflammatory and pro-fibrotic cytokines, prostaglandin production, lipid peroxidation and inhibition of DNA repair [51].
Interestingly, recent studies showed an activation of nucleotide-binding domain and leucine-rich-repeat-containing family pyrin 3 (NLRP3) inflammasome after thoracic irradiation of C57BL/6 mice [52]. Irradiation of THP-1 monocytes led to a similar inflammasome activation resulting in increased expression of interleukin −1β (IL-1β) and IL-18 [52,53]. This is especially important as IL-1 secretion was suggested to participate in the development of radiation-induced CVD. This was further evidenced by amelioration of radiation induced sustained expression of inflammatory mediators in mice after administration of IL-1β blocker directly after irradiation for 2 weeks [54,55].
This state of inflammation and oxidative stress is suggested to initiate a number of pathophysiologic dysfunctions. The first is cell death and premature endothelial senescence [56]. Aside from the direct apoptotic effects of radiation, irradiation of cardiac myocytes stimulates calcium release from the endoplasmic reticulum. This causes mitochondrial calcium overload, mitochondrial membrane swelling and release of apoptotic factors [44]. On the other hand, endothelial cell senescence occurs as a result of activation of Ataxia telangiectasia mutated (ATM)/p53/p21 and protein Kinase B/phosphatidylinositol 3-kinase/mechanistic target of rapamycin (Akt/PI3K/mTOR) pathways [42,56]. In addition, IL-6 induced by NF-κB activation has been previously identified as a critical controller of autocrine senescence [57–59]. Finally, accelerated telomere shortening induced by oxidative stress and mitochondrial dysfunction also contribute to accelerated endothelial senescence [57–60].
The second contributing pathophysiologic dysfunction is impaired endothelium-dependent relaxation of blood vessels. This is mediated mainly by endothelial nitric oxide synthase (eNOS) uncoupling and direct inactivation of NO by superoxide radicals leading to diminished vasodilation [61–64]. Senescent endothelial cells also contribute to decreased NO production due to their lowered eNOS activity [65]. In addition, decreased vasodilating prostacyclin and increased vasoconstrictive endothelin-1 and angiotensin-II levels contribute to chronic vasoconstriction [66,67]. The third contributing pathophysiologic dysfunction is mitochondrial dysfunction. Mitochondrial DNA is especially sensitive to radiation-induced damage due to its limited repair capacity, lack of protective histones, a high exon/intron ratio and its close proximity to the electron transport chain [42]. Furthermore, radiation-induced ROS can cause mutations in mitochondrial DNA as well as damage or alter the expression of proteins required for critical mitochondrial and cellular functions. The damaged mitochondria then generate more ROS which starts a vicious cycle of mitochondrial ROS generation [51,68].
The final contributing pathophysiologic dysfunction is initiation of a pro-coagulative and pro-thrombotic state. Endothelial cell damage with secreted pro-inflammatory cytokines induces the secretion of von Willebrand factor (vWF), platelet-activating factor and tissue factor while reducing thrombomodulin and prostacyclin production [42]. In addition, TGF-β released from irradiated endothelial cells induces fibrogenic activation of vascular smooth muscle cells leading to increased proliferation and migration [69]. This, along with decreased NO availability, leads to increased platelet aggregation and thrombus formation [42]. Recently, the European Society for Medical Oncology (ESMO) proposed aspirin as an antiplatelet as one of the cardioprotective treatments for patients receiving radiotherapy and having a high risk for CVD [70]. However, up to this moment, antiplatelet therapy has not proven to be beneficial in radiation-induced CVD [71–73]. Consequently, non-traditional therapies against CVD may need to be further investigated.
DNA methylation: a possible overlooked mechanism in radiation-induced CVD?
Epigenetic modifications involve different mechanisms, such as histone modifications, changes in DNA methylation and regulation of gene expression through micro-RNAs. Epigenetic modifications induce changes in gene expression that can occur without any changes in the primary DNA sequence. Moreover, epigenetic alterations could be transmitted to future progeny [74]. Alterations in DNA methylation are the most widely researched epigenetic changes and will be the focus of the current review.
DNA methylation
DNA methylation involves methylation of a cytosine base in a CpG dinucleotide to produce 5-methyl cytosine (5-mC). A process that is essential for silencing of transposable elements, genomic imprinting and X-chromosome inactivation [75]. 5-mC represents only ~1% of nucleic acids in the genome [76–78]. This low CpG frequency occurs due to cytosine deamination. Spontaneous cytosine deamination yields uracil. This uracil is then repaired by uracil glycosylase to cytosine. On the other hand, deamination of 5-mC yields thymine which is not corrected by uracil glycosylase leading to a C/T transition. This C/T transition is the most frequent mutation observed in human diseases occuring 10–50 times more frequently than all other base transitions. Consequently, cytosine deamination is the main reason for the observed frequency of CpG dinucleotides being lower than the expected CpG frequency in the absence of deamination [79,80]. The biological significance of this C/T transition is especially apparent when considering arginine deamination. The codon for arginine aminoacid (CGA) can be changed upon oxidative deamination of 5-mC into a stop codon (TGA). The premature appearance of a stop codon leads to the production of truncated and usually inactive proteins [81].
The majority of CpG dinucleotides in the human genome are heavily methylated (~70%) with the rest being unmethylated and mainly part of so called CpG islands (CGIs). CGIs are defined as regions of DNA (>200 bps) that have a GC content of at least 50%, observed CpG frequency of at least 60% of the expected CpG frequency, lack methylation and consequently are not transcriptionally silenced [82]. The expected CpG frequency in the previous definition is the CpG frequency if no cytosine deamination occurs. This high content of CpGs allows for DNA methylation-mediated regulation of gene expression. About 70% of gene promoters are associated with these CGIs and around 50% of these CGIs contain transcription start sites [83]. Hyper- or hyopmethylation of DNA has long been considered to inhibit or activate gene expression, respectively. However, the relationship between the DNA methylation state and gene expression is actually more complex as will be discussed later. Nonpromoter CGIs are CGIs that can be found in inter- and intragenic sequences. These CGIs represent either alternative transcription start sites of protein-coding genes or noncoding RNAs [84,85]. Intragenic or gene body CGIs that are present in actively expressed genes show increased DNA methylation. This can be due to gene body CGI’s ability to block transcription at intragenic promoters, affect intragenic repetitive element activity`and alter mRNA splicing by destabilizing nucleosomes at intron-exon junctions [86]. In general, methylation at CGIs is relatively stable in normal healthy tissue with major methylation pattern alterations associated with various disease states including cancer [87].
Historically, DNA methylation can be classified into two main types: De novo methylation which happens mainly in the developing embryo and maintenance methylation which maintains the methylation patterns from the parent strand in the daughter strand during replication. Consequently, DNA methylation is an epigenetic pattern that is maintained in cellular progeny and is normally stable in non-dividing cells [88,89].
DNA methylation is carried out by the action of DNA methyltransferases or DNMTs. Initiation of DNA methylation in the growing embryo during implantation occurs under the effects of the de novo DNMTs (DNMT-3a and −3b) with the help of DNMT3 like (DNMT3l). Here, the de novo DNMTs transfer methyl groups from S-adenyl methionine (SAM) to cytosines of the unmethylated DNA strand. DNMT3l is a DNMT3 family member which is incapable of individual methyltransferase activity but increases the activity of DNMT-3a and −3b by up to threefold [89]. Following this initial methylation step, the maintenance DNMT (DNMT1) then replicates the methylation pattern in following cell divisions. There, DNMT1 methylates hemimethylated CpG dinucleotides at the replication fork where newly biosynthesized DNA strands are directly methylated (Figure 2) [78,89,90]. This ‘maintenance’ thereby allows inheritance of the methylation patterns of the parent cell. The addition of a methyl group to C5 of a cytosine leads to the production of a stable covalent bond that requires a high degree of energy to be broken. An observation that led to the mistaken belief of the irreversibility of DNA methylation [91,92].
Figure 2.
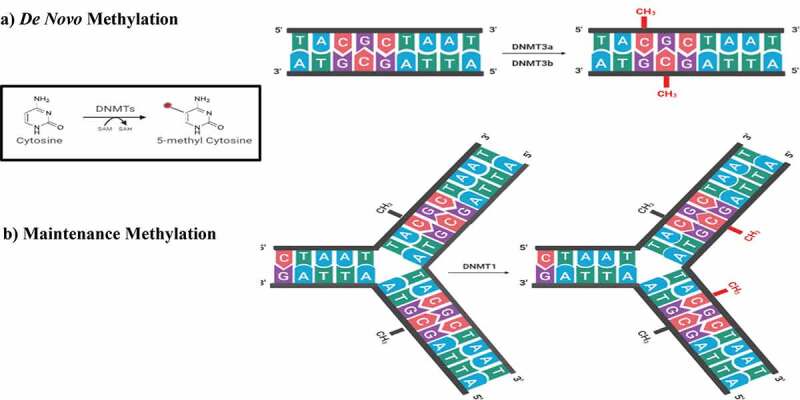
The writers of DNA methylation. DNMTs transfer a methyl group from SAM to the 5th carbon of cytosine. (A) De novo methylation process in the developing embryo by the action of DNMT3a and DNMT3b. (B) Maintenance methylation process during DNA replication to maintain the methylation profile in the daughter cells by the action of DNMT1 present at the replication fork on hemimethylated DNA. DNMT: DNA methyltransferase, SAM: S – adenosyl methionine, SAH: S-adenosyl-L-homocysteine.
Made with Biorender (biorender.com).
While DNA methylation patterns are fairly stable over time, DNA can be demethylated as well. DNA demethylation can occur through passive and active mechanisms. Passive demethylation occurs by inhibition of -or reduction in- DNMT1 levels. This inhibition or reduction of DNMT1 allows newly incorporated cytosine to remain unmethylated thereby causing a replication-dependent dilution of 5-mC [78,93]. Active demethylation involves the modification of the methyl group of 5-mC by enzymatic action and subsequent restoration of the non-methylated cytosine by DNA repair. Active DNA demethylation is mainly mediated by the actions of ten-eleven translocation (TET) enzyme [83].
Does radiation impact DNA methylation and how?
Radiation has been shown in previous studies to cause DNA methylation alterations [15,94–98]. However, the interplay between IR and DNA methylation is highly complex and may be tissue-dependent as well as model and strain-specific [99]. A summary of the literature [13–16,94-96,100–109] addressing the effect of radiation on DNA methylation is provided in Table 1 of Supplementary Materials.
Table 1.
Functional analysis of CVD differentially methylated genes in literature
| PANTHER Pathways | Number of genes | raw P-value | False Discovery Rate (FDR) |
|---|---|---|---|
| JAK/STAT signalling pathway | 3 | 0.0000 | 0.0010 |
| Interferon-gamma signalling pathway | 3 | 0.0001 | 0.0035 |
| PI3 kinase pathway | 3 | 0.0003 | 0.0112 |
| Interleukin signalling pathway | 3 | 0.0012 | 0.0393 |
| PDGF signalling pathway | 3 | 0.0048 | 0.1300 |
| Angiogenesis | 3 | 0.0076 | 0.1770 |
| TGF-beta signalling pathway | 2 | 0.0222 | 0.4040 |
| Inflammation mediated by chemokine and cytokine signalling pathway | 3 | 0.0214 | 0.4380 |
| Androgen/estrogene/progesterone biosynthesis | 1 | 0.0295 | 0.4830 |
| 5-Hydroxytryptamine degredation | 1 | 0.0494 | 0.7360 |
| Adrenaline and noradrenaline biosynthesis | 1 | 0.0667 | 0.7810 |
| Axon guidance mediated by Slit/Robo | 1 | 0.0624 | 0.7870 |
| CCKR signalling map | 2 | 0.0606 | 0.8280 |
| Dopamine receptor mediated signalling pathway | 1 | 0.1250 | 1.0000 |
| p53 pathway | 1 | 0.1840 | 1.0000 |
| Wnt signalling pathway | 2 | 0.1660 | 1.0000 |
| VEGF signalling pathway | 1 | 0.1470 | 1.0000 |
| p53 pathway feedback loops 2 | 1 | 0.1110 | 1.0000 |
| Ras Pathway | 1 | 0.1590 | 1.0000 |
| Oxidative stress response | 1 | 0.1210 | 1.0000 |
| Gonadotropin-releasing hormone receptor pathway | 1 | 0.4170 | 1.0000 |
| Endothelin signalling pathway | 1 | 0.1740 | 1.0000 |
| EGF receptor signalling pathway | 1 | 0.2710 | 1.0000 |
| Cadherin signalling pathway | 1 | 0.3100 | 1.0000 |
| Blood coagulation | 1 | 0.1030 | 1.0000 |
| Apoptosis signalling pathway | 1 | 0.2350 | 1.0000 |
| Alzheimer disease-presenilin pathway | 1 | 0.2500 | 1.0000 |
Research addressing radiation-induced DNA methylation alterations was found to involve different radiation types, doses and sampling times. Subsequently, the task of arriving to a simple conclusion regarding the exact effects of IR on DNA methylation becomes extremely difficult. Some studies address the effects of IR exposure during spaceflight. Subsequently, those employ low doses of high LET protons and heavier high atomic number and energy (HZE) ions such as 56Fe radiation [94,96,107] . On the other hand, studies that address the effects of the medicals application of radiation tend to use different types and doses of radiation in an attempt to mimic the doses regularly received by patients. In addition, even among those studies, different doses, dose intensities and study durations are employed. This is because of the varying aims of these studies which range from the investigation of radiation-induced carcinogenesis, radiation-induced genomic instability and bystander effects [13–15,95,101,102,109,110] to cellular responses to radiation and radiosensitivity [100,103,104,108]. In addition, some studies addressed the difference between the effects of IR on somatic and germinal tissues [16].
Another important and somewhat confounding aspect of the effects of IR on DNA methylation is the common use of in vivo animal models with rather short lifespan compared to humans. This is because the late effects of IR tend to develop in humans several years after the initial radiation. Consequently, investigating these late side effects in animal models requires taking into consideration the equivalent age of the animals [111]. This is indeed difficult as there are wide variations in the developmental durations and phases of these animals versus humans. Consequently, special considerations to the exact developmental stage of the used animal model are necessary to correlate the results to human stage [112].
Despite these difficulties, these studies share trends in DNA methylation alterations in response to IR analysed on both a global and a gene-specific scale. Global DNA methylation offers a representation of the total 5‐mC content in the genome, but lacks gene-specific information. On the other hand, gene-specific methylation focuses on the methylation alterations of specific genes. In Figure 3, we attempt to summarize the studies addressing the effect of radiation on DNA methylation from a global DNA methylation perspective. In Figure 3, we observe a general trend of radiation-induced global hypomethylation which persists for several months in a number of studies employing animal models. This suggests a link between DNA methylation alterations and IR-induced late effects observed years after radiotherapy [14,94,96,110]. Global hypomethylation is mainly evaluated by the methylation of DNA repetitive elements (REs) which account for about 50% of the human genome. Of which, Alu element (Alu) and long interspersed element-1 (LINE-1) are the most abundant human RE sequences. Hypomethylation of these REs will therefore lead to their reactivation, retrotransposition and resultant genomic instability [113]. On the other hand, attempting a similar overview for gene-specific methylation is more difficult. Different studies reported different sets of methylated genes with no clear patterns. Of note, IR was reported to elicit both hyper- and hypo-methylation of individual genes. However, overall radiation-induced gene-specific hypermethylation occurred at a higher frequency than gene-specific hypomethylation.
Figure 3.
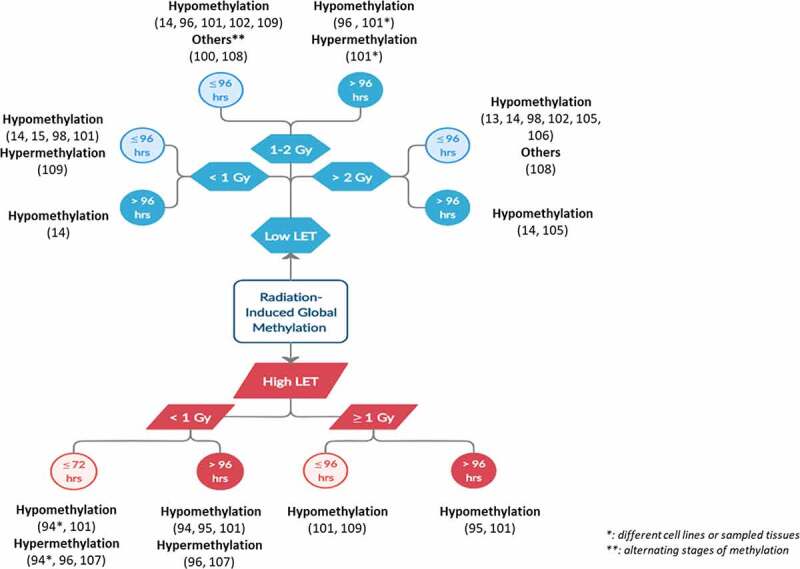
Overview of the shared outcomes of the different studies involving radiation-induced global methylation.
Made with creately (creately.com) and Microsoft Powerpoint.
Within these literature studies, a number of possible mechanisms have been put forward (Figure 4). The first mechanism involves ROS produced by radiation-induced water hydrolysis. These ROS are able to alter DNA methylation patterns through several ways [114] . ROS may act directly on the DNA by oxidizing the guanine base in CGIs forming 8-oxo-20-deoxyguanosine (8-OHdG), which can be repaired by its removal by 8-oxoguanine DNA glycosylase (OGG1) followed by base excision repair (BER). If not repaired, 8-OHdG can prevent the methylation of the adjacent cytosine leading to subsequent hypomethylation. Alternatively, ROS may directly convert 5-mC to 5-hydroxyl mC by interaction of DNA with hydroxyl radicals. ROS may affect DNA methylation indirectly by altering the activity of methylation related enzymes [114]. However, ROS effects on DNMTs seem to be bidirectional. ROS can lead to hypermethylation by upregulating DNMTs or increasing their recruitment by H2O2. ROS can also lead to hypomethylation by reducing the availability of SAM which is an essential cofactor for DNMT activity. Finally, other studies have shown that ROS can lead to DNA hypermethylation due to their nucleophilic action on the 5 position of cytosine molecule leading to its deprotonation and accelerating its reaction with SAM [115,116]. In summary, ROS can lead to hypomethylation by 8-OHdG formation, 5-mC hydroxylation or reducing SAM availability. On the other hand, ROS can also lead to hypermethylation by DNMT upregulation or increased recruitment as well as 5-mC deprotonation. Despite the counter intuitiveness of ROS having opposing effects on DNA methylation, this ‘dual’ effect may contribute to the observed similar opposing effects of radiation on DNA methylation.
Figure 4.
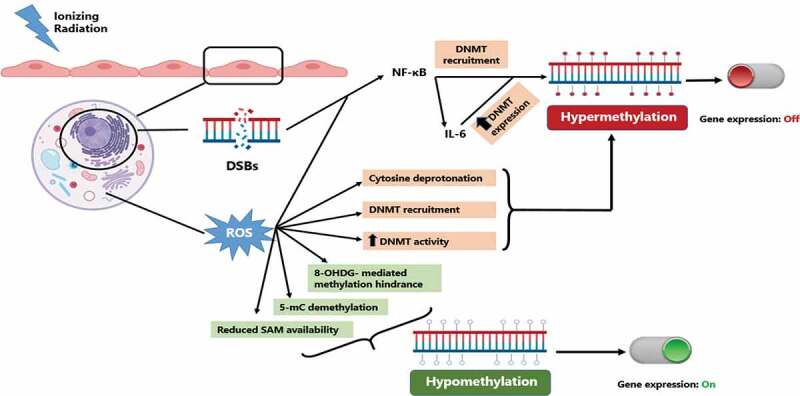
Possible mechanisms of radiation-induced DNA methylation alterations. incident ionizing radiation leads to the production of DSBs in affected cells. these DSBs as well as radiation-induced ROS can lead to NF-κB activation which in turn can lead to DNA hypermethylation and inhibition of gene expression. on the other hand, radiation-induced ROS can cause varying DNA methylation alterations by multiple mechanisms leading to either hypo- and hyper-methylation with differential regulation of gene expression. DSBs: double strand breaks, ROS: reactive oxygen species, 8-OHdG: 8-oxo-20-deoxyguanosine, 5-mC: 5-methyl cytosine, SAM: S-adenosyl methionine, DNMT: DNA methyltransferase.
Made with Biorender (biorender.com) and Microsoft Powerpoint.
Secondly, radiation can also cause DNA hypermethylation by activation of NF-κB [117–120]. NF-κB activation leads to alterations in DNA methylation through two different mechanisms. Firstly, the RelA/p65 subunit of NF-κB can directly recruit DNMT-1 to chromatin [121]. Secondly, NF-κB regulates DNA methylation indirectly by the production of Interleukin-6 (IL-6), which has been shown to also regulate DNMT1 expression leading to increased DNMT1 activity [122–124]. This increased availability/activity of DNMT1 subsequently leads to hypermethylation.
DNA methylation and CVD; possible disease collaborator?
A number of studies have linked DNA methylation alterations to CVD development [125–157]. The focus of these studies is mostly atherosclerosis as it is the underlying cause of most CVDs [158–164]. In most cases, global DNA hypomethylation has been observed in atherosclerosis [165–170]. This state of hypomethylation was detectable in atherosclerosis prone murine aorta even before the development of atherosclerosis [171]. In addition, gene specific hypermethylation has also been observed in atherosclerosis [147,151,156]. Interestingly, focal gene-specific hypermethylation offers higher biological relevance than that of global hypomethylation [162].
We examined the literature concerning differentially methylated genes in CVD (summarized in Table 2 of Supplementary Materials). In most cases, identified differentially methylated genes are associated with key elements in CVD pathogenesis, such as lipid metabolism, inflammation, oxidative stress, atherosclerosis and endothelial cell dysfunction. This consequently points to a contribution of DNA methylation to CVD pathophysiology. However, care should be taken when translating DNA methylation data from these studies to clinically relevant biomarkers. This is in part due to the fact that the effect of DNA methylation on gene expression is of a complex nature. Several studies have pointed out that the state of CpG methylation does not always predict the state of gene expression. Indeed, examination of the DNA methylation of primary human fibroblasts showed that CpG methylation alterations did not always translate to changes in gene expression [172,173]. Furthermore, it was found that better correlation with gene expression was encountered with certain genomic positions (first intron) or associated with chromatin states, particularly those that are representative of active chromatin and transcribed regions [172,174,175]. Another confounder is that many studies investigating differential methylation in CVD are based on a limited, rather low, number of samples which casts doubt on the capacity to extrapolate the results. Therefore, care must be taken when interpreting the results originating from low sample size or those not backed up with gene expression analyses.
The association between altered DNA methylation and CVD is further proven in experimental models of atherosclerosis. Dunn et al. have shown DNMT inhibitor 5-Aza-2′-deoxycytidine (also known as decitabine) to be effective atheroma preventive measures in a ApoE−/- mouse model of atherosclerosis [176]. In addition, decitabine was shown to decrease atherosclerosis development in LDLr−/− mice through decreased macrophage inflammation and suppressed macrophage endoplasmic reticulum stress [177]. This could be explained by decreased methylation and subsequent increased expression of liver X receptor α (LXRα) and peroxisome proliferator-activated receptor γ1 (PPARγ). LXRα and PPARγ are atheroprotective as they regulate lipid metabolism and macrophage inflammation [178–180]. The interested reader is referred to a number of reviews regarding the use of DNA methylation inhibitors as candidates drugs for treating atherosclerosis [181–183].
Functional analysis of differentially methylated genes in CVD
In an attempt to validate the involvement of DNA methylation in CVD pathogenesis, we performed a functional analysis of genes reported in literature to be differentially methylated in CVD (Table 1). This was done by investigating these genes in the pathway analysis functionality of PANTHER (protein analysis through evolutionary relationships) classification system [184,185]. Our PANTHER analysis of differently methylated genes in Table 1 reveals that these genes were connected to a number of pathophysiological mechanisms of CVD, such as inflammation, oxidative stress and endothelial activation. In addition, four pathways showed statistically significant pathway involvement, namely Janus kinase/signal transducers and activators of transcription (JAK/STAT) signalling pathway, interferon-gamma (INF-γ) signalling pathway, phosphoinositide 3 kinase (PI3K) pathway and interleukin signalling pathway. This should come as no surprise as these pathways are associated with cardiac myocyte response to injury and stress as well as cardiovascular inflammation [186–194]. Activation of JAK/STAT signalling contributes to atherosclerosis development by aiding immune cell recruitment and vascular smooth muscle proliferation, hypertrophy, and migration [195]. In addition, STAT1 synergizes with NF-κB in its inflammatory signalling [196]. Conversely, IFN-γ promotes endothelial cell adhesion, immune cell recruitment with a conflicting effect on foam cell formation [197–199]. Also, PI3K catalyzes second messenger phosphatidylinositol (3,4,5)-trisphosphate (PIP3) production. In the heart, four isoforms (PI3Kα, PI3Kβ, PI3Kδ, and PI3Kγ) are differentially expressed in different cell subsets (cardiomyocytes, fibroblasts, endothelial cells and vascular smooth muscle cells). Subsequently, they are associated with varying effects on myocardial contractility, physiological growth and pathobiological remodelling as well as smooth muscle cell and immune cell migration [200–202]. In addition, PI3Ks function as scaffolding proteins in cardiac excitation-contraction coupling and autophagy [201]. Finally, interleukins are a family of cytokines strongly associated with chronic inflammation and atherogenesis. Some interleukins can have proatherogenic effects while fewer interleukins can have atheroprotective effects [197,203–208]. These DNA methylation alterations in pathways associated with atherogenesis suggest a usefulness for DNA methylation based CVD biomarkers. First, Istas et al. found that differentially methylated regions in BRCA1 and CRISP2 genes were reproducibly differentially methylated in independent atherosclerotic human aorta tissue and human carotid plaque samples [156]. In addition, these methylation changes at BRCA1 and CRISP2 genes were consistently associated with subclinical atherosclerosis in an independent sample cohort of middle-aged men. This study provides a concrete example of how gene-specific methylation could be used to monitor the development of atherosclerosis, even before the condition is clinically diagnosable [156]. Second, p16INK4a was found to be differentially methylated by IR while also influencing epicardial adipose tissue development and subsequent CVD risk [209]. This intersection of gene-specific methylation alterations observed after radiation and associated with CVD could offer an untapped source of functional biomarkers.
Conclusions and future directions
DNA methylation is an epigenetic mechanism that has been shown to be implicated in the pathogenesis of many diseases. In healthy individuals, the CpG islands in promoter regions are normally non-methylated thereby allowing their associated genes to be transcriptionally active. For the rest of the genome, CpG dinucleotides are normally methylated. Predictably, alterations in DNA methylation lead to changes in gene expression.
Ionizing radiation has been shown in several studies to cause DNA methylation alterations; most commonly global DNA hypomethylation and gene-specific hypermethylation. However, studies investigating radiation-induced DNA methylation alterations vary in radiation LET and dose, type of investigated model as well as the sampling timeline. These variations make it difficult to answer important questions regarding the possible differences in effect on DNA methylation between high and low LET radiation, as well as if there is a dose threshold that needs to be reached to cause DNA methylation alterations. However, the persistence of radiation-induced DNA methylation alterations after IR exposure suggests the involvement of these DNA methylation alterations in radiation-induced late effects.
On the other hand, several studies reported global hypomethylation in CVD and specifically in atherosclerosis. In addition, hyper- and -less commonly- hypo-methylation of specific genes has also been reported in CVD. Through a gene ontology analysis, we found that the genes that have been reported to be differentially methylated in CVD are associated with pathways related to CVD pathogenesis, such as endothelial activation, oxidative stress and inflammation.
One of the most common adverse effects of mediastinal radiotherapy is radiation-induced CVD, which has the same presentation as atherosclerosis. Radiation-induced DNA damage, oxidative stress and inflammation are considered as major molecular drivers of radiation-induced CVD. Various therapeutic techniques have been employed to reduce the accidental irradiation of the heart during radiotherapy such as prone position radiotherapy, image- and dose- guided radiotherapy, IMRT, SBRT and DIBH. However, despite substantially reducing the dose of the accidental irradiation, radiation-induced cardiac events continue to occur. Furthermore, research is exploring protective agents which interfere with one or more of the identified pathophysiological mechanisms of radiation-induced CVD mainly through reducing chronic inflammation and oxidative stress [44,210,211]. In addition, non-traditional therapies against CVD are worth exploring
The late-onset aspect of radiation-induced CVD constitutes a diagnostic challenge to timely initiation of cardioprotective therapy. Interestingly, radiation-induced DNA methylation alterations are notable immediately after irradiation and often persist long after it. Therefore, DNA methylation alterations in radiation-induced CVD could serve as novel, early biomarkers e.g. p16INK4a.
To the extent of our knowledge, no experimental research has directly investigated the correlation between radiation-induced DNA methylation alterations and resultant CVD. Previous research suggests the possibility of performing targeted DNA methylation [212] and demethylation [213,214]. This would allow studying the effect of prevention/induction of radiation-induced methylation alterations of specific genes. Subsequent effects on gene expression and induction of CVD pathophysiology could then serve as a means to test our theory. It is therefore our recommendation that future studies direct their attention to examining targeted radiation-induced DNA methylation alterations and their involvement in CVD pathogenesis. [194,215–271]
Supplementary Material
Funding Statement
This project (MEDIRAD) has received funding from the Euratom research and training programme 2014-2018 under grant agreement No 755523.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Ratosa I, Ivanetic Pantar M.. Cardiotoxicity of mediastinal radiotherapy. Rep Pract Oncol Radiother. 2019;24:629–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hoppe BS, Bates JE, Mendenhall NP, et al. The meaningless meaning of mean heart dose in mediastinal lymphoma in the modern radiation therapy era. Pract Radiat Oncol. 2020;10(3):e147–54. [DOI] [PubMed] [Google Scholar]
- [3].Van Nimwegen FA, Ntentas G, Darby SC, et al. Risk of heart failure in survivors of Hodgkin lymphoma: effects of cardiac exposure to radiation and anthracyclines. Blood. 2017;129(16):2257–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pohjola‐Sintonen S, Tötterman KJ, Salmo M, et al. Late cardiac effects of mediastinal radiotherapy in patients with Hodgkin’s disease. Cancer. 1987;60(1):31–37. [DOI] [PubMed] [Google Scholar]
- [5].Hahn E, Jiang H, Ng A, et al. Late cardiac toxicity after mediastinal radiation therapy for Hodgkin lymphoma: contributions of coronary artery and whole heart dose-volume variables to risk prediction. Int J Radiat Oncol Biol Phys. 2017. Aug 1;98(5):1116–1123. [DOI] [PubMed] [Google Scholar]
- [6].Darby SC, Ewertz M, McGale P, et al. Risk of ischemic heart disease in women after radiotherapy for breast cancer. N Engl J Med. 2013. Mar 14;368(11):987–998. [DOI] [PubMed] [Google Scholar]
- [7].Drost L, Yee C, Lam H, et al. A systematic review of heart dose in breast radiotherapy. Clin Breast Cancer. 2018. Oct 1;18(5):e819–24. [DOI] [PubMed] [Google Scholar]
- [8].Bogaard VAB, van Den, Ta BDP, Schaaf A, et al. Validation and modification of a prediction model for acute cardiac events in patients with breast cancer treated with radiotherapy based on three-dimensional dose distributions to cardiac substructures. J Clin Oncol. 2017. Apr 10;35(11):1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yu AF, Jones LW.. Modulation of cardiovascular toxicity in Hodgkin lymphoma: potential role and mechanisms of aerobic training. Future Cardiol. 2015;11(4):441–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Boyne DJ, Mickle AT, Brenner DR, et al. Long-term risk of cardiovascular mortality in lymphoma survivors: a systematic review and meta-analysis. Cancer Med. 2018;7(9):4801–4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Muka T, Koromani F, Portilla E, et al. The role of epigenetic modifications in cardiovascular disease: a systematic review. Int J Cardiol. 2016. Jun 1;212:174–183. [DOI] [PubMed] [Google Scholar]
- [12].Zhong J, Agha G, Baccarelli AA. The role of DNA methylation in cardiovascular risk and disease: methodological aspects, study design, and data analysis for epidemiological studies. Circ Res. 2016. Jan 8;118(1):119–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Loree J, Koturbash I, Kutanzi K, et al. Radiation-induced molecular changes in rat mammary tissue: possible implications for radiation-induced carcinogenesis. Int J Radiat Biol. 2006. Jan 3;82(11):805–815. [DOI] [PubMed] [Google Scholar]
- [14].Luzhna L, Ilnytskyy Y, Kovalchuk O. Mobilization of LINE-1 in irradiated mammary gland tissue may potentially contribute to low dose radiation-induced genomic instability. Genes Cancer. 2015;6(1–2):71–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pogribny I, Koturbash I, Tryndyak V, et al. Fractionated low-dose radiation exposure leads to accumulation of DNA damage and profound alterations in DNA and histone methylation in the murine thymus. Mol Cancer Res. 2005. Oct 1;3(10):553–561. [DOI] [PubMed] [Google Scholar]
- [16].Jangiam W, Udomtanakunchai C, Reungpatthanaphong P, et al. Late effects of low-dose radiation on the bone marrow, lung, and testis collected from the same exposed BALB/cJ Mice. Dose-Response. 2018. Oct 19;16(4):155932581881503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Desouky O, Ding N, Zhou G. Targeted and non-targeted effects of ionizing radiation. J Radiat Res Appl Sci. 2015. Apr 1;8(2):247–254. [Google Scholar]
- [18].Baskar R, Dai J, Wenlong N, et al. Biological response of cancer cells to radiation treatment. Front Mol Biosci. 2014;1:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Balaji K, Subramanian B, Yadav P, et al. Radiation therapy for breast cancer: literature review. Med Dosim. 2016;41(3):253–257. [DOI] [PubMed] [Google Scholar]
- [20].Darby SC, Cutter DJ, Boerma M, et al. Radiation-related heart disease: current knowledge and future prospects. Int J Radiat Oncol Biol Phys. 2010. Mar 1;76(3):656–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hayes AW. Principles and methods of toxicology. 5th ed. CRC Press; 2007. [Google Scholar]
- [22].Citrin DE, Mitchell JB. Mechanisms of normal tissue injury from irradiation. Semin Radiat Oncol. 2017;27(4):316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].De Ruysscher D, Niedermann G, Burnet NG, et al. Radiotherapy toxicity. Nat Rev Dis Prim. 2019. Dec 21;5(1):13. [DOI] [PubMed] [Google Scholar]
- [24].Spetz J, Moslehi J, Sarosiek K. Radiation-induced cardiovascular toxicity: mechanisms, prevention, and treatment. Curr Treat Options Cardio Med. 2018. Apr 20;20(4):31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cui S, Li W, Lv X, et al. Folic acid supplementation delays atherosclerotic lesion development by modulating MCP1 and VEGF DNA methylation levels in vivo and in vitro. Int J Mol Sci. 2017. May 5;18(5):990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cuomo JR, Javaheri SP, Sharma GK, et al. How to prevent and manage radiation-induced coronary artery disease. Heart. 2018;104(20):1647–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Armanious MA, Mohammadi H, Khodor S, et al. Cardiovascular effects of radiation therapy. Curr Prob Cancer. 2018;42:433–442. [DOI] [PubMed] [Google Scholar]
- [28].Tapio S. Pathology and biology of radiation-induced cardiac disease. J Radiat Res. 2016. Sep 1;57(5):439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lancellotti P, Nkomo VT, Badano LP, et al. Expert consensus for multi-modality imaging evaluation of cardiovascular complications of radiotherapy in adults: a report from the european association of cardiovascular imaging and the American society of echocardiography. J Am Soc Echocardiogr. 2013;26(9):1013–1032. [DOI] [PubMed] [Google Scholar]
- [30].Cuomo JR, Sharma GK, Conger PD, et al. Novel concepts in radiation-induced cardiovascular disease. World J Cardiol. 2016. Sep 26;8(9):504–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Taunk NK, Haffty BG, Kostis JB, et al. Radiation-induced heart disease: pathologic abnormalities and putative mechanisms. Front Oncol. 2015;5:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dorresteijn LD, Stewart FA, Boogerd W. Stroke as a late treatment effect of Hodgkin’s disease. J Clin Oncol. 2006;24(9):1480. DOI: 10.1200/JCO.2005.04.8538. [DOI] [PubMed] [Google Scholar]
- [33].Bowers DC, McNeil DE, Liu Y, et al. Stroke as a late treatment effect of Hodgkin’s disease: a report from the childhood cancer survivor study. J Clin Oncol. 2005. Sep 20;23(27):6508–6515. [DOI] [PubMed] [Google Scholar]
- [34].Plummer C, Henderson RD, O’Sullivan JD, et al. Ischemic stroke and transient ischemic attack after head and neck radiotherapy: a review. Stroke. 2011;42(9):2410–2418. [DOI] [PubMed] [Google Scholar]
- [35].Dorresteijn LDA, Kappelle AC, Scholz NMJ, et al. Increased carotid wall thickening after radiotherapy on the neck. Eur J Cancer. 2005. May 1;41(7):1026–1030. [DOI] [PubMed] [Google Scholar]
- [36].Yusuf SW, Venkatesulu BP, Mahadevan LS, et al. Radiation-induced cardiovascular disease: a clinical perspective. Front Cardiovasc Med. 2017;26:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Baselet B, Rombouts C, Benotmane AM, et al. Cardiovascular diseases related to ionizing radiation: the risk of low-dose exposure (Review). Int J Mol Med. 2016. Dec 1;38(6):1623–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Heidenreich PA, Kapoor JR. Radiation induced heart disease. Heart. 2008. Nov 25;95(3):252–258. [DOI] [PubMed] [Google Scholar]
- [39].Gujral DM, Lloyd G, Bhattacharyya S. Radiation-induced valvular heart disease. Heart. 2016. Feb 15;102(4):269–276. [DOI] [PubMed] [Google Scholar]
- [40].Halle M, Hall P, Tornvall P. Cardiovascular disease associated with radiotherapy: activation of nuclear factor kappa-B. J Intern Med. 2011. May;269(5):469–477. [DOI] [PubMed] [Google Scholar]
- [41].Sylvester CB, Abe J, Patel ZS, et al. Radiation-induced cardiovascular disease: mechanisms and importance of linear energy transfer. Front Cardiovasc Med. 2018;5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Baselet B, Sonveaux P, Baatout S, et al. Pathological effects of ionizing radiation: endothelial activation and dysfunction. Cell Mol Life Sci. 2019;76:699–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Li M, You L, Xue J, et al. Ionizing radiation-induced cellular senescence in normal, non-transformed cells and the involved DNA damage response: a mini review. Front Pharmacol. 2018;9:522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wang H, Wei J, Zheng Q, et al. Radiation-induced heart disease: a review of classification, mechanism and prevention. Int J Biol Sci. 2019;15(10):2128–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Halle M, Gabrielsen A, Paulsson-Berne G, et al. Sustained inflammation due to nuclear factor-kappa B activation in irradiated human arteries. J Am Coll Cardiol. 2010. Mar 23;55(12):1227–1236. [DOI] [PubMed] [Google Scholar]
- [46].Zhao W, Robbins M. Inflammation and chronic oxidative stress in radiation-induced late normal tissue injury: therapeutic implications. Curr Med Chem. 2008. Dec 30;16(2):130–143. [DOI] [PubMed] [Google Scholar]
- [47].Lee Chuy K, Nahhas O, Dominic P, et al. Cardiovascular complications associated with mediastinal radiation. Curr Treat Options Cardiovasc Med. 2019;21(7):1–20. [DOI] [PubMed] [Google Scholar]
- [48].Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF-κB in the heart: to be or not to NF-κB. Circ Res. 2011;108(9):1122–1132. [DOI] [PubMed] [Google Scholar]
- [49].Land WG. The role of damage-associated molecular patterns (DAMPs) in human diseases part II: dAMPs as diagnostics, prognostics and therapeutics in clinical medicine. Sultan Qaboos Univ Med J. 2015;15(2):e157–70. [PMC free article] [PubMed] [Google Scholar]
- [50].Parichatikanond W, Luangmonkong T, Mangmool S, et al. Therapeutic targets for the treatment of cardiac fibrosis and cancer: focusing on tgf-β Signaling. Front Cardiovasc Med. 2020;7:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wei J, Wang B, Wang H, et al. Radiation-induced normal tissue damage: oxidative stress and epigenetic mechanisms. Oxid Med Cell Longev. 2019;2019(4):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Li X, Gong Y, Li D, et al. Low-dose radiation therapy promotes radiation pneumonitis by activating NLRP3 inflammasome. Int J Radiat Oncol Biol Phys. 2020. Jul 15;107(4):804–814. [DOI] [PubMed] [Google Scholar]
- [53].Huang S, Che J, Chu Q, et al. The role of NLRP3 inflammasome in radiation-induced cardiovascular injury. Front Cell Dev Biol. 2020;8:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mezzaroma E, Mikkelsen RB, Toldo S, et al. Role of interleukin-1 in radiation-induced cardiomyopathy. Mol Med. 2015;21(1):210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Christersdottir T, Pirault J, Gisterå A, et al. Prevention of radiotherapy-induced arterial inflammation by interleukin-1 blockade. Eur Heart J. 2019. Aug 7;40(30):2495–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wang Y, Boerma M, Zhou D. Ionizing radiation-induced endothelial cell senescence and cardiovascular diseases. Radiat Res. 2016. Aug;186(2):153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Liu Y, Bloom SI, Donato AJ. The role of senescence, telomere dysfunction and shelterin in vascular aging. Microcirculation. 2019;26(2):e12487. DOI: 10.1111/micc.12487. Wiley Blackwell [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ayouaz A, Raynaud C, Heride C, et al. Telomeres: hallmarks of radiosensitivity. Biochimie. 2008;90(1):60–72. DOI: 10.1016/j.biochi.2007.09.011. [DOI] [PubMed] [Google Scholar]
- [59].Lustig A, Shterev I, Geyer S, et al. Long term effects of radiation exposure on telomere lengths of leukocytes and its associated biomarkers among atomic-bomb survivors. Oncotarget. 2016;7(26):38989–38998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Minamino T, Komuro I. Role of telomere in endothelial dysfunction in atherosclerosis. Curr Opin Lipidol. 2002;13(5):537–543. [DOI] [PubMed] [Google Scholar]
- [61].Sugihara T, Hattori Y, Yamamoto Y, et al. Preferential impairment of nitric oxide-mediated endothelium-dependent relaxation in human cervical arteries after irradiation. Circulation. 1999. Aug 10;100(6):635–641. [DOI] [PubMed] [Google Scholar]
- [62].Beckman JA, Thakore A, Kalinowski BH, et al. Radiation therapy impairs endothelium-dependent vasodilation in humans. J Am Coll Cardiol. 2001. Mar 1;37(3):761–765. [DOI] [PubMed] [Google Scholar]
- [63].Pathak R, Cheema AK, Boca SM, et al. Modulation of radiation response by the tetrahydrobiopterin pathway. Antioxidants. 2015;4(1):68–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Hong CW, Kim YM, Pyo H, et al. Involvement of inducible nitric oxide synthase in radiation-Induced vascular endothelial damage. J Radiat Res. 2013. Nov;54(6):1036–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Donato AJ, Morgan RG, Walker AE, et al. Cellular and molecular biology of aging endothelial cells. J Mol Cell Cardiol. 2015;89:122–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Estrada-García L, Carrera-Rotllan J, Puig-Parellada P. Effects of oxidative stress and antioxidant treatments on eicosanoid synthesis and lipid peroxidation in long term human umbilical vein endothelial cells culture. Prostaglandins Other Lipid Mediat. 2002;67(1):13–25. [DOI] [PubMed] [Google Scholar]
- [67].Fardid R, Najafi M, Salajegheh A, et al. Radiation-induced non-targeted effect in vivo: evaluation of cyclooygenase-2 and endothelin-1 gene expression in rat heart tissues. J Cancer Res Ther. 2017. Jan 1;13(1):51–55. [DOI] [PubMed] [Google Scholar]
- [68].Azzam EI, Jay-Gerin JP, Pain D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012;327(1–2):48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Milliat F, François A, Isoir M, et al. Influence of endothelial cells on vascular smooth muscle cells phenotype after irradiation: implication in radiation-induced vascular damages. Am J Pathol. 2006;169(4):1484–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Curigliano G, Lenihan D, Fradley M, et al. Management of cardiac disease in cancer patients throughout oncological treatment: ESMO consensus recommendations. Ann Oncol. 2020;31(2):171–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].RON WAKSMAN, PAKALA R, ROY P, et al. Effect of clopidogrel on neointimal formation and inflammation in balloon-denuded and radiated hypercholesterolemic Rabbit iliac arteries. J Interv Cardiol. 2008;21(2):122–128. [DOI] [PubMed] [Google Scholar]
- [72].Hoving S, Heeneman S, Gijbels MJJ, et al. NO-donating aspirin and aspirin partially inhibit age-related atherosclerosis but not radiation-induced atherosclerosis in ApoE null mice. PLoS One. 2010;5(9):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Hoving S, Heeneman S, Gijbels MJJ, et al. Anti-inflammatory and anti-thrombotic intervention strategies using atorvastatin, clopidogrel and knock-down of CD40L do not modify radiation-induced atherosclerosis in ApoE null mice. Radiother Oncol. 2011;101(1):100–108. DOI: 10.1016/j.radonc.2011.09.019. [DOI] [PubMed] [Google Scholar]
- [74].Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010. Oct 13;28(10):1057–1068. [DOI] [PubMed] [Google Scholar]
- [75].Ren W, Gao L, Song J. Structural basis of DNMT1 and DNMT3A-mediated DNA methylation. Genes (Basel). 2018. Dec 11;9(12):620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Lövkvist C, Dodd IB, Sneppen K, et al. DNA methylation in human epigenomes depends on local topology of CpG sites. Nucleic Acids Res. 2016. Jun 20;44(11):5123–5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Walsh CP, Xu GL. Cytosine methylation and DNA repair. In: Current topics in microbiology and immunology. Springer Verlag; 2006. p. 283–315. [DOI] [PubMed] [Google Scholar]
- [78].Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013. Jan;38(1):23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Fryxell KJ, Moon W-J. CpG mutation rates in the human genome are highly dependent on local GC content. Mol Biol Evol. 2005. Mar 1;22(3):650–658. [DOI] [PubMed] [Google Scholar]
- [80].Carlberg C, Molnár F. DNA methylation. In: Human epigenomics. Singapore: Springer Singapore; 2018. p. 57–73. [Google Scholar]
- [81].Romanov GA, Sukhoverov VS. Arginine CGA codons as a source of nonsense mutations: a possible role in multivariant gene expression, control of mRNA quality, and aging. Mol Genet Genomics. 2017. Oct 1;292(5):1013–1026. [DOI] [PubMed] [Google Scholar]
- [82].Strichman-Almashanu LZ, Lee RS, Onyango PO, et al. A genome-wide screen for normally methylated human CpG islands that can identify novel imprinted genes. Genome Res. 2002. Mar 20;12(4):543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Wu H, Zhang Y. Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell. 2014;156(1–2):45–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Sarda S, Das A, Vinson C, et al. Distal CpG islands can serve as alternative promoters to transcribe genes with silenced proximal promoters. Genome Res. 2017. Apr;27(4):553–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011. May 15;25(10):1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Yang X, Han H, De Carvalho DD, et al. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014. Oct;;26(4):577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Luo C, Hajkova P, Ecker JR. Dynamic DNA methylation: in the right place at the right time. Science. 2018;361(6409):1336–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002. Jan 1;16(1):6–21. [DOI] [PubMed] [Google Scholar]
- [89].Feng J, Fan G. Chapter- 4 the role of DNA methylation in the central nervous system and neuropsychiatric disorders. Nov Approaches to Stud Basal Ganglia Relat Neuropsychiatr Disord. 2009;89:67–84. http://www.sciencedirect.com/science/article/pii/S0074774209890041 [DOI] [PubMed] [Google Scholar]
- [90].Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006. Feb 1;31(2):89–97. [DOI] [PubMed] [Google Scholar]
- [91].Ambrosi C, Manzo M, Baubec T. Dynamics and context-dependent roles of DNA methylation. J Mol Biol. 2017;429(10):1459–1475. [DOI] [PubMed] [Google Scholar]
- [92].Unoki M. Recent insights into the mechanisms of De novo and maintenance of DNA methylation in mammals. In: DNA methylation mechanism. IntechOpen; 2019. [Google Scholar]
- [93].Bhutani N, Burns DM, Blau HM. DNA demethylation dynamics. Cell. 2011. Sep 16;146(6):866–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Miousse IR, Shao L, Chang J, et al. Exposure to low-dose 56 Fe-ion radiation induces long-term epigenetic alterations in mouse bone marrow hematopoietic progenitor and stem cells. Radiat Res. 2014;182(1):92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Miousse IR, Chang J, Shao L, et al. Inter-strain differences in LINE-1 DNA methylation in the mouse hematopoietic system in response to exposure to ionizing radiation. Int J Mol Sci. 2017. Jul 4;18(7):1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Kennedy EM, Powell DR, Li Z, et al. Galactic cosmic radiation induces persistent epigenome alterations relevant to human lung cancer. Sci Rep. 2018. Dec 1;8(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Koturbash I, Pogribny I, Kovalchuk O. Stable loss of global DNA methylation in the radiation-target tissue—A possible mechanism contributing to radiation carcinogenesis?. Biochem Biophys Res Commun. 2005. Nov 18;337(2):526–533. [DOI] [PubMed] [Google Scholar]
- [98].Tawa R, Kimura Y KOMURAJ-I, MIYAMURA Y, et al. Effects of X-ray irradiation on genomic DNA methylation levels in mouse tissues. J Radiat Res. 1998. Dec;39(4):271–278. [DOI] [PubMed] [Google Scholar]
- [99].Miousse IR, Kutanzi KR, Koturbash I. Effects of ionizing radiation on DNA methylation: from experimental biology to clinical applications. Int J Radiat Biol. 2017. May;93(5):457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Chaudhry MA, Omaruddin RA. Differential DNA methylation alterations in radiation-sensitive and -resistant cells. DNA Cell Biol. 2012. Jun 1;31(6):908–916. [DOI] [PubMed] [Google Scholar]
- [101].Goetz W, Morgan MNM, Baulch JE. The effect of radiation quality on genomic DNA methylation profiles in irradiated human cell lines. Radiat Res. 2011. May;;175(5):575–587. [DOI] [PubMed] [Google Scholar]
- [102].Kalinich JF, Catravas GN, Snyder SL. The effect of γ radiation on DNA methylation. Radiat Res. 1989. Feb;;117(2):185. [PubMed] [Google Scholar]
- [103].Kuhmann C, Weichenhan D, Rehli M, et al. DNA methylation changes in cells regrowing after fractioned ionizing radiation. Radiother Oncol. 2011. Oct 1;101(1):116–121. [DOI] [PubMed] [Google Scholar]
- [104].Wang J, Zhang Y, Xu K, et al. Genome-wide screen of DNA methylation changes induced by low dose X-ray radiation in mice. PLoS One. 2014;9(3):e90804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Koturbash I, Boyko A, Rodriguez-Juarez R, et al. Role of epigenetic effectors in maintenance of the long-term persistent bystander effect in spleen in vivo. Carcinogenesis. 2007. Aug 1;28(8):1831–1838. [DOI] [PubMed] [Google Scholar]
- [106].Kumar A, Rai PS, Upadhya R, et al. γ-radiation induces cellular sensitivity and aberrant methylation in human tumor cell lines. Int J Radiat Biol. 2011. Nov 7;87(11):1086–1096. [DOI] [PubMed] [Google Scholar]
- [107].Acharya MM, Baddour AAD, Kawashita T, et al. Epigenetic determinants of space radiation-induced cognitive dysfunction. Sci Rep. 2017. Feb 21;7(January):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Antwih DA, Gabbara KM, Lancaster WD, et al. Radiation-induced epigenetic DNA methylation modification of radiation-response pathways. Epigenetics. 2013. Aug 27;8(8):839–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Aypar U, Morgan WF, Baulch JE. Radiation-induced epigenetic alterations after low and high LET irradiations. Mutat Res Mol Mech Mutagen. 2011. Feb 10;707(1–2):24–33. [DOI] [PubMed] [Google Scholar]
- [110].Koturbash I, Miousse IR, Sridharan V, et al. Radiation-induced changes in DNA methylation of repetitive elements in the mouse heart. Mutat Res. 2016;787:43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Sengupta P. The laboratory rat: relating its age with human’s. Int J Prev Med. 2013;4(6):624–630. [PMC free article] [PubMed] [Google Scholar]
- [112].Dutta S, Sengupta P. Men and mice: relating their ages. Life Sci. 2016;152:244–248. [DOI] [PubMed] [Google Scholar]
- [113].Zheng Y, Joyce BT, Liu L, et al. Prediction of genome-wide DNA methylation in repetitive elements. Nucleic Acids Res. 2017. Sep 1;45(15):8697–8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Kietzmann T, Petry A, Shvetsova A, et al. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br J Pharmacol. 2017;174(12):1533–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Yara S, Lavoie J-C LE. Oxidative stress and DNA methylation regulation in the metabolic syndrome. Epigenomics. 2015. Apr;;7(2):283–300. [DOI] [PubMed] [Google Scholar]
- [116].Wu Q, Ni X. ROS-mediated DNA methylation pattern alterations in carcinogenesis. Curr Drug Targets. 2015;16(1):13–19. [DOI] [PubMed] [Google Scholar]
- [117].Lee S-J, Dimtchev A, Lavin MF, et al. A novel ionizing radiation-induced signaling pathway that activates the transcription factor NF-κB. Oncogene. 1998. Oct 9;17(14):1821–1826. [DOI] [PubMed] [Google Scholar]
- [118].Janssens S, Tschopp J. Signals from within: the DNA-damage-induced NF-κB response. Cell Death Differ. 2006. May 13;13(5):773–784. [DOI] [PubMed] [Google Scholar]
- [119].Hei TK, Zhou H, Chai Y, et al. Radiation induced non-targeted response: mechanism and potential clinical implications. Curr Mol Pharmacol. 2011. Jun;;4(2):96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Di Maggio F, Minafra L, Forte G, et al. Portrait of inflammatory response to ionizing radiation treatment. J Inflamm. 2015. Feb 18;12(1):14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Liu Y, Mayo MW, Nagji AS, et al. Phosphorylation of RelA/p65 promotes DNMT-1 recruitment to chromatin and represses transcription of the tumor metastasis suppressor gene BRMS1. Oncogene. 2012. Mar 18;31(9):1143–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Li Y, Deuring J, Peppelenbosch MP, et al. 6-induced DNMT1 activity mediates SOCS3 promoter hypermethylation in ulcerative colitis-related colorectal cancer. Carcinogenesis. 2012. Oct 1;33(10):1889–1896. [DOI] [PubMed] [Google Scholar]
- [123].Brasier AR. The nuclear factor- B-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc Res. 2010. May 1;86(2):211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Metabolic, Endocrine, and Genitourinary Pathobiology . DNA Methyl transferase 1 reduces expression of SRD5A2 in the aging adult prostate. Am J Pathol. 2015;185(3):870–882. http://www.sciencedirect.com/science/article/pii/S0002944014006907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Ghaznavi H, Mahmoodi K, Soltanpour MS. A preliminary study of the association between the ABCA1 gene promoter DNA methylation and coronary artery disease risk. Mol Biol Res Commun. 2018. Jun;7(2):59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Guay S-P, Légaré C, Houde -A-A, et al. Acetylsalicylic acid, aging and coronary artery disease are associated with ABCA1 DNA methylation in men. Clin Epigenetics. 2014. Jul 29;6(1):14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Zhou J, Chen L, Yang X, et al. Preliminary study of the relationship between promoter methylation of the ANGPTL2 gene and coronary heart disease. J Clin Lab Anal. 2018;21:e22702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Nguyen A, Mamarbachi M, Turcot V, et al. Lower methylation of the ANGPTL2 gene in leukocytes from post-acute coronary syndrome patients. PLoS One. 2016. Apr 21;11(4):e0153920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Yamada Y, Horibe H, Oguri M, et al. Identification of novel hyper- or hypomethylated CpG sites and genes associated with atherosclerotic plaque using an epigenome-wide association study. Int J Mol Med. 2018. Feb 2;41(5):2724–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Chen X, Jiang D, Xu L, et al. Elevated methylation of cyclin dependent kinase inhibitor 2B contributes to the risk of coronary heart disease in women. Exp Ther Med. 2018. Nov 2;17(1):205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Zhong J, Chen X, Wu N, et al. Catechol-O-methyltransferase promoter hypomethylation is associated with the risk of coronary heart disease. Exp Ther Med. 2016. Nov 1;12(5):3445–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Miao L, Yin R-X, Zhang Q-H, et al. Integrated DNA methylation and gene expression analysis in the pathogenesis of coronary artery disease. Aging (Albany NY). 2019. Mar 7;11(5):1486–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Niu -P-P, Cao Y, Gong T, et al. Hypermethylation of DDAH2 promoter contributes to the dysfunction of endothelial progenitor cells in coronary artery disease patients. J Transl Med. 2014. Jun 16;12(1):170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Post WS, Goldschmidt-Clermont PJ, Wilhide CC, et al. Methylation of the estrogen receptor gene is associated with aging and atherosclerosis in the cardiovascular system [Internet]. Cardiovasc Res Narnia. 1999. Sep;1(4):985–91. [DOI] [PubMed] [Google Scholar]
- [135].Min J, Weitian Z, Peng C, et al. Correlation between insulin-induced estrogen receptor methylation and atherosclerosis. Cardiovasc Diabetol. 2016. Dec 10;15(1):156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Breitling LP, Salzmann K, Rothenbacher D, et al. F2RL3 methylation, and prognosis in stable coronary heart disease. Eur Heart J. 2012. Nov 1;33(22):2841–2848. [DOI] [PubMed] [Google Scholar]
- [137].Guay S-P, Brisson D, Munger J, et al. ABCA1 gene promoter DNA methylation is associated with HDL particle profile and coronary artery disease in familial hypercholesterolemia. Epigenetics. 2012. May 27;7(5):464–472. [DOI] [PubMed] [Google Scholar]
- [138].Jia L, Zhu L, Wang JZ, et al. Methylation of FOXP3 in regulatory T cells is related to the severity of coronary artery disease. Atherosclerosis. 2013. Jun 1;228(2):346–352. [DOI] [PubMed] [Google Scholar]
- [139].C-X L, Xu R-D, Cao M, et al. FOXP3 demethylation as a means of identifying quantitative defects in regulatory T cells in acute coronary syndrome. Atherosclerosis. 2013. Jul 1;229(1):263–270. [DOI] [PubMed] [Google Scholar]
- [140].Xu LL, Zheng D, Wang L, et al. GCK gene-body hypomethylation is associated with the risk of coronary heart disease. Biomed Res Int. 2014. Feb 17;2014:151723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Fan R WANGW-J, Q-L ZHONG, S-W DUAN, et al. Aberrant methylation of the GCK gene body is associated with the risk of essential hypertension. Mol Med Rep. 2015. Aug 1;12(2):2390–2394. [DOI] [PubMed] [Google Scholar]
- [142].Indumathi B, Katkam SK, Krishna LSR, et al. Dual effect of IL-6-174 G/C polymorphism and promoter methylation in the risk of coronary artery disease among south indians. Indian J Clin Biochem. 2019. Apr 21;34(2):180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Bakshi C, Vijayvergiya R, Aberrant DV. DNA methylation of M1-macrophage genes in coronary artery disease. Sci Rep. 2019. Dec 5;9(1):1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Ghose S, Ghosh S, Tanwar VS, et al. Investigating Coronary Artery Disease methylome through targeted bisulfite sequencing. Gene. 2019. Dec 30;721:144107. [DOI] [PubMed] [Google Scholar]
- [145].Afzali M, Nakhaee A, Tabatabaei SP, et al. Aberrant promoter methylation profile of Niemann-pick type C1 gene in cardiovascular disease. Iran Biomed J. 2013;17(2):77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Jiang D, Zheng D, Wang L, et al. Elevated PLA2G7 gene promoter methylation as a gender-specific marker of aging increases the risk of coronary heart disease in females. Oudejans C, editor. PLoS One. 2013. Mar 28;8(3):e59752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Wei L, Zhao S, Wang G, et al. SMAD7 methylation as a novel marker in atherosclerosis. Biochem Biophys Res Commun. 2018. Feb 5;496(2):700–705. [DOI] [PubMed] [Google Scholar]
- [148].S-C MA, H-P ZHANG, F-Q KONG, et al. Integration of gene expression and DNA methylation profiles provides a molecular subtype for risk assessment in atherosclerosis. Mol Med Rep. 2016. Jun 1;13(6):4791–4799. [DOI] [PubMed] [Google Scholar]
- [149].Nakatochi M, Ichihara S, Yamamoto K, et al. Epigenome-wide association of myocardial infarction with DNA methylation sites at loci related to cardiovascular disease. [DOI] [PMC free article] [PubMed]
- [150].Guay S-P, Légaré C, Brisson D, et al. Epigenetic and genetic variations at the TNNT1 gene locus are associated with HDL-C levels and coronary artery disease. Epigenomics. 2016. Mar 7;8(3):359–371. [DOI] [PubMed] [Google Scholar]
- [151].Huang Y-S, Zhi Y-F, Wang S-R. Hypermethylation of estrogen receptor-α gene in atheromatosis patients and its correlation with homocysteine. Pathophysiology. 2009. Oct 1;16(4):259–265. [DOI] [PubMed] [Google Scholar]
- [152].Akinyemiju T, Do AN, Patki A, et al. Epigenome-wide association study of metabolic syndrome in African-American adults. Clin Epigenetics. 2018. Dec 10;10(1):49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].ÅK H, Mendelson MM, Marioni RE, et al. Epigenetic patterns in blood associated with lipid traits predict incident coronary heart disease events and are enriched for results from genome-wide association studies. Circ Cardiovasc Genet. 2017. Jan;10(1):e001487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Peng P, Wang L, Yang X, et al. A preliminary study of the relationship between promoter methylation of the ABCG1, GALNT2 and HMGCR genes and coronary heart disease. Coleman WB, editor. PLoS One. 2014. Aug 1;9(8):e102265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Pfeiffer L, Wahl S, Pilling LC, et al. DNA methylation of lipid-related genes affects blood lipid levels. Circ Cardiovasc Genet. 2015. Apr;8(2):334–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Istas G, Declerck K, Pudenz M, et al. Identification of differentially methylated BRCA1 and CRISP2 DNA regions as blood surrogate markers for cardiovascular disease. Sci Rep. 2017. Dec 11;7(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Wang P, Shen C, Diao L, et al. Aberrant hypermethylation of aldehyde dehydrogenase 2 promoter upstream sequence in rats with experimental myocardial infarction. Biomed Res Int. 2015. Jan 5;2015:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Glier MB, Green TJ, Devlin AM. Methyl nutrients, DNA methylation, and cardiovascular disease. Mol Nutr Food Res. 2014. Jan;58(1):172–182. [DOI] [PubMed] [Google Scholar]
- [159].Jiang D, Sun M, You L, et al. DNA methylation and hydroxymethylation are associated with the degree of coronary atherosclerosis in elderly patients with coronary heart disease. Life Sci. 2019. May 1;224:241–248. [DOI] [PubMed] [Google Scholar]
- [160].Kim M, Long TI, Arakawa K, et al. Methylation as a biomarker for cardiovascular disease risk. Bader JS, editor. PLoS One. 2010. Mar 15;5(3):e9692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Fernández-Sanlés A, Sayols-Baixeras S, Subirana I, et al. Association between DNA methylation and coronary heart disease or other atherosclerotic events: a systematic review. Atherosclerosis. 2017. Aug 1;263:325–333. [DOI] [PubMed] [Google Scholar]
- [162].Aavik E, Babu M, Ylä-Herttuala S. DNA methylation processes in atheosclerotic plaque. Atherosclerosis. 2019. Feb 1;281:168–179. [DOI] [PubMed] [Google Scholar]
- [163].Hai Z, Zuo W. Aberrant DNA methylation in the pathogenesis of atherosclerosis. Clin Chim Acta. 2016. May 1;456:69–74. [DOI] [PubMed] [Google Scholar]
- [164].Turunen MP, Aavik E, Ylä-Herttuala S. Epigenetics and atherosclerosis. Biochim Biophys Acta Gen Subj. 2009. Sep 1;1790(9):886–891. [DOI] [PubMed] [Google Scholar]
- [165].Hiltunen MO, Turunen MP, Häkkinen TP, et al. DNA hypomethylation and methyltransferase expression in atherosclerotic lesions. Vasc Med. 2002. Feb 3;7(1):5–11. [DOI] [PubMed] [Google Scholar]
- [166].Aavik E, Lumivuori H, Leppanen O, et al. Global DNA methylation analysis of human atherosclerotic plaques reveals extensive genomic hypomethylation and reactivation at imprinted locus 14q32 involving induction of a miRNA cluster. Eur Heart J. 2015. Apr 2;36(16):993–1000. [DOI] [PubMed] [Google Scholar]
- [167].Pogribny IP, Beland FA. DNA hypomethylation in the origin and pathogenesis of human diseases. Cell Mol Life Sci. 2009. Jul 27;66(14):2249–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Zaina SA, Garay-Sevilla ME, Hernández-González MA, et al. Extensive demethylation of normally hypermethylated CpG islands occurs in human atherosclerotic arteries. Int J Mol Med. 2010. Sep 21;26(5):691–700. [DOI] [PubMed] [Google Scholar]
- [169].Jiang Y, Zhang H, Sun T, et al. The comprehensive effects of hyperlipidemia and hyperhomocysteinemia on pathogenesis of atherosclerosis and DNA hypomethylation in Apo E-/- mice. Acta Biochim Biophys Sin (Shanghai). 2012. Oct 1;44(10):866–875. [DOI] [PubMed] [Google Scholar]
- [170].Zaina S, Heyn H, Carmona FJ, et al. DNA methylation map of human atherosclerosis. Circ Cardiovasc Genet. 2014. Oct 1;7(5):692–700. [DOI] [PubMed] [Google Scholar]
- [171].Lund G, Andersson L, Lauria M, et al. DNA methylation polymorphisms precede any histological sign of atherosclerosis in mice lacking apolipoprotein E. J Biol Chem. 2004. Jul 9;279(28):29147–29154. [DOI] [PubMed] [Google Scholar]
- [172].Wagner JR, Busche S, Ge B, et al. The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome Biol. 2014. Feb 20;15(2):R37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Maeder ML, Angstman JF, Richardson ME, et al. Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat Biotechnol. 2013. Dec;31(12):1137–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Lea AJ, Vockley CM, Johnston RA, et al. Genome-wide quantification of the effects of DNA methylation on human gene regulation. Elife. 2018;1:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Anastasiadi D, Esteve-Codina A, Piferrer F. Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigene Chromat. 2018. Jun 29;11(1):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Dunn J, Qiu H, Kim S, et al. Flow-dependent epigenetic DNA methylation regulates endothelial gene expression and atherosclerosis. The Journal of Clinical Investigation. 2014. Jul 1;124(7):3187–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Cao Q, Wang X, Jia L, et al. Inhibiting DNA methylation by 5-Aza-2ⴕ- deoxycytidine ameliorates atherosclerosis through suppressing macrophage inflammation. Endocrinology. 2014. Dec 1;155(12):4925–4938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Calkin AC, Tontonoz P. Liver X receptor signaling pathways and atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30(8):1513–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Grbić E, Peterlin A, Kunej T, et al. PPARγ gene and atherosclerosis: genetic polymorphisms, epigenetics and therapeutic implications. Balk J Med Genet. 2018;21(1):39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Yu J, Qiu Y, Yang J, et al. DNMT1-PPARγ pathway in macrophages regulates chronic inflammation and atherosclerosis development in mice. Sci Rep. 2016;17:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Chistiakov DA, Orekhov AN, Bobryshev YV. Treatment of cardiovascular pathology with epigenetically active agents: focus on natural and synthetic inhibitors of DNA methylation and histone deacetylation. Int J Cardiol. 2017;227:66–82. [DOI] [PubMed] [Google Scholar]
- [182].Nicorescu I, Dallinga GM, de Winther MPJ, et al. Potential epigenetic therapeutics for atherosclerosis treatment. Atherosclerosis. 2019;281:189–197. [DOI] [PubMed] [Google Scholar]
- [183].Schiano C, Vietri MT, Grimaldi V, et al. Epigenetic-related therapeutic challenges in cardiovascular disease. Trends Pharmacol Sci. 2015. Apr 1;36(4):226–235. [DOI] [PubMed] [Google Scholar]
- [184].Thomas PD, Campbell MJ, Kejariwal A, et al. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003. Sep 1;13(9):2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Mi H, Muruganujan A, Huang X, et al. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v14.0). Nat Protoc. 2019. Mar 1;14(3):703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Segiet OA, Piecuch A, Ł M, et al. Role of interleukins in heart failure with reduced ejection fraction. Anatol J Cardiol. 2019;22(6):287–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Abbate A, Toldo S, Marchetti C, et al. Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ Res. 2020. Apr;24(9):1260–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Rose NR. Critical cytokine pathways to cardiac inflammation. J Interferon Cytokine Res. 2011;31(10):705–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Aoyagi T, Matsui T. Phosphoinositide-3 kinase signaling in cardiac hypertrophy and heart failure. Curr Pharm Des. 2011. Jul 11;17(18):1818–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Kitamura Y, Koide M, Akakabe Y, et al. Manipulation of cardiac phosphatidylinositol 3-kinase (PI3K)/Akt signaling by apoptosis regulator through modulating IAP expression (ARIA) regulates cardiomyocyte death during doxorubicin-induced cardiomyopathy. J Biol Chem. 2014. Jan 31;289(5):2788–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Hoyer FF, Nahrendorf M. Interferon-γregulates cardiac myeloid cells in myocardial infarction. Cardiovasc Res. 2019;115:1815–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Ferreira LRP. Interferon-γ and other inflammatory mediators in cardiomyocyte signaling during Chagas disease cardiomyopathy. World J Cardiol. 2014;6(8):782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Wagner MA, Siddiqui MAQ, The JAK. STAT pathway in hypertrophic stress signaling and genomic stress response. JAK-STAT. 2012. Apr;1(2):131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Barry SP, Townsend PA, Latchman DS, et al. Role of the JAK–STAT pathway in myocardial injury. Trends Mol Med. 2007. Feb 1;13(2):82–89. [DOI] [PubMed] [Google Scholar]
- [195].Goswami R. JAK-STAT Signaling in Diseases. 1st ed. Boca Raton: CRC Press; 2020. [Google Scholar]
- [196].Ivashkiv LB. Crosstalk with the Jak-STAT pathway in inflammation. In: Jak-Stat signaling: from basics to disease. Springer-Verlag Wien; 2012. p. 353–70. [Google Scholar]
- [197].Boshuizen MCS, de Winther MPJ. Interferons as essential modulators of atherosclerosis. Arterioscler Thromb Vasc Biol. 2015. Jul 27;35(7):1579–1588. [DOI] [PubMed] [Google Scholar]
- [198].Zhang J, Alcaide P, Liu L, et al. Regulation of endothelial cell adhesion molecule expression by mast cells, macrophages, and neutrophils. PLoS One. 2011;6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Whitman SC, Ravisankar P, Elam H, et al. Exogenous interferon-γ enhances atherosclerosis in apolipoprotein E-/- mice. Am J Pathol. 2000;157(6):1819–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Fougerat A, Gayral S, Malet N, et al. Phosphoinositide 3-kinases and their role in inflammation: potential clinical targets in atherosclerosis?. Clinical Science. 2009;116:791–804. [DOI] [PubMed] [Google Scholar]
- [201].Ghigo A, Li M. Phosphoinositide 3-kinase: friend and foe in cardiovascular disease. Front Pharmacol. 2015. Aug 13;6(Aug):169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Choi KH, Kim JE, Song NR, et al. Phosphoinositide 3-kinase is a novel target of piceatannol for inhibiting PDGF-BB-induced proliferation and migration in human aortic smooth muscle cells. Cardiovasc Res. 2010. Mar 1;85(4):836–844. [DOI] [PubMed] [Google Scholar]
- [203].Buckley ML, Williams JO, Chan Y-H, et al. The interleukin-33-mediated inhibition of expression of two key genes implicated in atherosclerosis in human macrophages requires MAP kinase, phosphoinositide 3-kinase and nuclear factor-κB signaling pathways. Sci Rep. 2019;9(1):11317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Libby P. Interleukin-1 beta as a target for atherosclerosis therapy: biological basis of CANTOS and beyond. J Am Coll Cardiol. 2017;70(18):2278–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Hartman J, Frishman WH. Inflammation and atherosclerosis: A review of the role of interleukin-6 in the development of atherosclerosis and the potential for targeted drug therapy. Cardiology in Review. 2014;22:147–151. [DOI] [PubMed] [Google Scholar]
- [206].Tian Y, Ling X, Chen D, et al. Interleukin‐36 receptor antagonist attenuates atherosclerosis development by inhibiting NLRP3 inflammasome. J Cell Physiol. 2020. Dec 2;235(12):9992–9996. [DOI] [PubMed] [Google Scholar]
- [207].Fatkhullina AR, Peshkova IO, Koltsova EK. The role of cytokines in the development of atherosclerosis. Biochemistry (Moscow). 2016;81:1358–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Von der Thüsen JH, Kuiper J, Van Berkel TJC, et al. Interleukins in atherosclerosis: molecular pathways and therapeutic potential. Pharmacol Rev. 2003;55:133–166. [DOI] [PubMed] [Google Scholar]
- [209].Wouters K, Deleye Y, Hannou SA, et al. The tumour suppressor CDKN2A/p16INK4a regulates adipogenesis and bone marrow-dependent development of perivascular adipose tissue. Diab Vasc Dis Res. 2017. Nov 1;14(6):516–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Musa A, Shabeeb D. Radiation-induced heart diseases: protective effects of natural products. Med (B Aires). 2019. May 9;55(5):126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Kolivand S, Amini P, Saffar H, et al. Selenium-L-methionine modulates radiation injury and Duox1 and Duox2 upregulation in rat’s heart tissues. J Cardiovasc Thorac Res. 2019. Jun 27;11(2):121–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Siddique AN, Nunna S, Rajavelu A, et al. Targeted methylation and gene silencing of VEGF-A in human cells by using a designed Dnmt3a-Dnmt3L single-chain fusion protein with increased DNA methylation activity. J Mol Biol. 2013. Feb 8;425(3):479–491. [DOI] [PubMed] [Google Scholar]
- [213].Xu X, Tao Y, Gao X, et al. A CRISPR-based approach for targeted DNA demethylation. Cell Discov. 2016. May 3;2(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Hanzawa N, Hashimoto K, Yuan X, et al. Targeted DNA demethylation of the Fgf21 promoter by CRISPR/dCas9-mediated epigenome editing. Sci Rep. 2020. Dec 1;10(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Tang C, Oram JF. The cell cholesterol exporter ABCA1 as a protector from cardiovascular disease and diabetes. Biochim Biophys Acta - Mol Cell Biol Lipids. 2009. Jul 1;1791(7):563–572. [DOI] [PubMed] [Google Scholar]
- [216].Rudel LL, Lee RG, Cockman TL. Acyl coenzyme a: cholesterol acyltransferase types 1 and 2: structure and function in atherosclerosis. Curr Opin Lipidol. 2001. Apr;12(2):121–127. [DOI] [PubMed] [Google Scholar]
- [217].Chen C-H, Budas GR, Churchill EN, et al. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008. Sep 12;321(5895):1493–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Gong D, Zhang H, Hu S. Mitochondrial aldehyde dehydrogenase 2 activation and cardioprotection. J Mol Cell Cardiol. 2013. Feb 1;55:58–63. [DOI] [PubMed] [Google Scholar]
- [219].Yang M-Y, Wang Y-B, Han B, et al. Activation of aldehyde dehydrogenase 2 slows down the progression of atherosclerosis via attenuation of ER stress and apoptosis in smooth muscle cells. Acta Pharmacol Sin. 2018. Jan;39(1):48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Kadomatsu T, Endo M, Miyata K, et al. Diverse roles of ANGPTL2 in physiology and pathophysiology. Trends Endocrinol Metab. 2014. May 1;25(5):245–254. [DOI] [PubMed] [Google Scholar]
- [221].van Westerop LLM, Arts-de Jong M, Hoogerbrugge N, et al. Cardiovascular risk of BRCA1/2 mutation carriers: a review. Maturitas. 2016. Sep 1;91:135–139. DOI: 10.1016/j.maturitas.2016.06.012. [DOI] [PubMed] [Google Scholar]
- [222].Chen M, Ni J, Zhang Y, et al. ERAP75 functions as a coactivator to enhance estrogen receptor α transactivation in prostate stromal cells. Prostate. 2008. Sep 1;68(12):1273–1282. [DOI] [PubMed] [Google Scholar]
- [223].Papait R, Kunderfranco P, Stirparo GG, et al. Long noncoding RNA: a new player of heart failure?. J Cardiovasc Transl Res. 2013. Dec 9;6(6):876–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Bastos P, Gomes T, Ribeiro L. Catechol-O-methyltransferase (COMT): an update on Its Role in Cancer, Neurological and Cardiovascular Diseases. Rev Physiol Biochem Pharmacol. 2017;173:1–39. [DOI] [PubMed] [Google Scholar]
- [225].Akhtar S, Gremse F, Kiessling F, et al. CXCL12 promotes the stabilization of atherosclerotic lesions mediated by smooth muscle progenitor cells in Apoe-deficient mice. Arterioscler Thromb Vasc Biol. 2013. Apr;33(4):679–686. [DOI] [PubMed] [Google Scholar]
- [226].Farouk SS, Rader DJ, Reilly MP, et al. CXCL12: a new player in coronary disease identified through human genetics. Trends Cardiovasc Med. 2010. Aug;20(6):204–209. DOI: 10.1016/j.tcm.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].van der Vorst EPC, Döring Y, Weber C. Chemokines and their receptors in Atherosclerosis. J Mol Med. 2015. Sep 15;93(9):963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Zhang J, Liu J, Li Z, et al. Dysfunction of endothelial NO system originated from homocysteine-induced aberrant methylation pattern in promoter region of DDAH2 gene. Chin Med J (Engl). 2007. Dec 5;120(23):2132–2137. [PubMed] [Google Scholar]
- [229].Menazza S, Murphy E. The expanding complexity of estrogen receptor signaling in the cardiovascular system. Circ Res. 2016. Mar 18;118(6):994–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Mahmoodzadeh S, Eder S, Nordmeyer J, et al. Estrogen receptor alpha up-regulation and redistribution in human heart failure. Faseb J. 2006. May 1;20(7):926–934. [DOI] [PubMed] [Google Scholar]
- [231].Deroo BJ, Korach KS. Estrogen receptors and human disease. J Clin Invest. 2006. Mar 1;116(3):561–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Rothenbacher D, Koenig W, Brenner H. Comparison of N-terminal pro–B-natriuretic peptide, C-reactive protein, and creatinine clearance for prognosis in patients with known coronary heart disease. Arch Intern Med. 2006. Dec 11;166(22):2455. [DOI] [PubMed] [Google Scholar]
- [233].Zhang Y, Yang R, Burwinkel B, et al. 3 methylation as a biomarker of current and lifetime smoking exposures. Environ Health Perspect. 2014. Feb;122(2):131–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Waters CE, Saldivar JC, Hosseini SA, et al. FHIT gene product: tumor suppressor and genome "caretaker". Cell Mol Life Sci. 2014. Dec;71(23):4577–4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Di Paola R, Marucci A, Trischitta V. GALNT2 effect on HDL-cholesterol and triglycerides levels in humans: evidence of pleiotropy?. Nutr Metab Cardiovasc Dis. 2017. Apr 1;27(4):281–282. [DOI] [PubMed] [Google Scholar]
- [236].Marucci A, Antonucci A, De Bonis C, et al. GALNT2 as a novel modulator of adipogenesis and adipocyte insulin signaling. Int J Obes. 2019. Apr;30. [DOI] [PubMed] [Google Scholar]
- [237].Webster RJ, Warrington NM, Weedon MN, et al. The association of common genetic variants in the APOA5, LPL and GCK genes with longitudinal changes in metabolic and cardiovascular traits. Diabetologia. 2009. Jan 19;52(1):106–114. [DOI] [PubMed] [Google Scholar]
- [238].Iynedjian PB. Molecular physiology of mammalian glucokinase. Cell Mol Life Sci. 2009. Jan 26;66(1):27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Tracy MR, Dormans JP, Kusumi K. Klippel-feil syndrome. Clin Orthop Relat Res. 2004. Jul;424:183–190. [PubMed] [Google Scholar]
- [240].YAMADA Y, NISHIDA T, HORIBE H, et al. Identification of hypo- and hypermethylated genes related to atherosclerosis by a genome-wide analysis of DNA methylation. Int J Mol Med. 2014. May 1;33(5):1355–1363. [DOI] [PubMed] [Google Scholar]
- [241].Nova-Lampeti E, Aguilera V, Oporto K, et al. Hox genes in adult tissues and their role in endothelial cell differentiation and angiogenesis. In: Endothelial dysfunction - old concepts and new challenges. InTech; 2018. [Google Scholar]
- [242].Gorski DH, Patel CV, Walsh K. Homeobox transcription factor regulation in the cardiovascular system. Trends Cardiovasc Med. 1993. Sep 1;3(5):184–190. [DOI] [PubMed] [Google Scholar]
- [243].Fontes JA, Rose NR, Čiháková D. The varying faces of IL-6: from cardiac protection to cardiac failure. Cytokine. 2015. Jul;74(1):62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].van der Heijden T, Bot I, The KJ. IL-12 cytokine family in cardiovascular diseases. Cytokine. 2019. Oct 1;122:154188. DOI: 10.1016/j.cyto.2017.10.010. [DOI] [PubMed] [Google Scholar]
- [245].Umar S, van der Laarse A. Nitric oxide and nitric oxide synthase isoforms in the normal, hypertrophic, and failing heart. Mol Cell Biochem. 2010. Jan 19;333(1–2):191–201. [DOI] [PubMed] [Google Scholar]
- [246].Shah A. Inducible nitric oxide synthase and cardiovascular disease. Cardiovasc Res. 2000. Jan 1;45(1):148–155. [DOI] [PubMed] [Google Scholar]
- [247].Davies RW, Wells GA, Stewart AFR, et al. A genome-wide association study for coronary artery disease identifies a novel susceptibility locus in the major histocompatibility complex. Circ Cardiovasc Genet. 2012. Apr 1;5(2):217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Missala I, Kassner U, Steinhagen-Thiessen E, et al. Review of the association of lipoprotein(a) and autoimmune diseases and atherosclerosis. Int J Rheumatol. 2012. Dec 5;2012:480784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Clarke R, Peden JF, Hopewell JC, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009. Dec 24;361(26):2518–2528. [DOI] [PubMed] [Google Scholar]
- [250].Rozanski A, Takano APC, Kato PN, et al. M-protein is down-regulated in cardiac hypertrophy driven by thyroid hormone in rats. Mol Endocrinol. 2013. Dec;27(12):2055–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Sequeira V, Nijenkamp LLA, Regan JA, van der Velden J. The physiological role of cardiac cytoskeleton and its alterations in heart failure. Biochim Biophys Acta - Biomembr. 2014. Feb 1;1838(2):700–722. [DOI] [PubMed] [Google Scholar]
- [252].Liu X, Xu H, Xu H, et al. New genetic variants associated with major adverse cardiovascular events in patients with acute coronary syndromes and treated with clopidogrel and aspirin. bioRxiv. 2018;9:411165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012. Apr 1;33(7):829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Yu X-H, Jiang N, Yao P-B, et al. NPC1, intracellular cholesterol trafficking and atherosclerosis. Clin Chim Acta. 2014. Feb 15;429:69–75. [DOI] [PubMed] [Google Scholar]
- [255].Sutton BS, Crosslin DR, Shah SH, et al. Comprehensive genetic analysis of the platelet activating factor acetylhydrolase (PLA2G7) gene and cardiovascular disease in case–control and family datasets. Hum Mol Genet. 2008. May 1;17(9):1318–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Karabina S, Ninio E. Plasma PAFAH/PLA2G7 genetic variability, cardiovascular disease, and clinical trials. Enzymes. 2015;38:145–155. [DOI] [PubMed] [Google Scholar]
- [257].Sorrentino S. The eight human “canonical” ribonucleases: molecular diversity, catalytic properties, and special biological actions of the enzyme proteins. FEBS Lett. 2010. Jun 3;584(11):2194–2200. [DOI] [PubMed] [Google Scholar]
- [258].Koczera P, Martin L, Marx G, et al. The ribonuclease a superfamily in humans: canonical RNases as the buttress of innate immunity. Int J Mol Sci. 2016. Aug 5;17(8):1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].Wei L, Chao N, Gao S, et al. Homocysteine induces vascular inflammatory response via SMAD7 hypermethylation in human umbilical vein smooth muscle cells. Microvasc Res. 2018. Nov 1;120:8–12. [DOI] [PubMed] [Google Scholar]
- [260].Papait R, Greco C, Kunderfranco P, et al. Epigenetics: a new mechanism of regulation of heart failure?. Basic Res Cardiol. 2013. Jul 6;108(4):361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [261].Guo X, Wang X, Wang Y, et al. Variants in the SMARCA4 gene was associated with coronary heart disease susceptibility in Chinese han population. Oncotarget. 2017. Jan 31;8(5):7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Chmielewski S, Piaszyk-Borychowska A, Wesoly J, et al. STAT1 and IRF8 in vascular inflammation and cardiovascular disease: diagnostic and therapeutic potential. Int Rev Immunol. 2016. Sep 2;35(5):434–454. [DOI] [PubMed] [Google Scholar]
- [263].Hammer S, Toenjes M, Lange M, et al. Characterization of TBX20 in human hearts and its regulation by TFAP2. J Cell Biochem. 2008. Jun 1;104(3):1022–1033. [DOI] [PubMed] [Google Scholar]
- [264].Shen T, Zhu Y, Patel J, et al. T-Box20 suppresses oxidized low-density lipoprotein-induced human vascular endothelial cell injury by upregulation of PPAR-γ. Cell Physiol Biochem. 2013;32(5):1137–1150. [DOI] [PubMed] [Google Scholar]
- [265].Johnson JL. Metalloproteinases in atherosclerosis. Eur J Pharmacol. 2017. Dec 5;816:93–106. [DOI] [PubMed] [Google Scholar]
- [266].Guay S-P, Voisin G, Brisson D, et al. Epigenome-wide analysis in familial hypercholesterolemia identified new loci associated with high-density lipoprotein cholesterol concentration. Epigenomics. 2012. Dec;4(6):623–639. [DOI] [PubMed] [Google Scholar]
- [267].Li C, Wang F, Yang Y, et al. Significant association of SNP rs2106261 in the ZFHX3 gene with atrial fibrillation in a Chinese Han GeneID population. Hum Genet. 2011. Mar;129(3):239–246. DOI: 10.1007/s00439-010-0912-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [268].Kao Y-H, Hsu J-C, Chen Y-C, et al. ZFHX3 knockdown increases arrhythmogenesis and dysregulates calcium homeostasis in HL-1 atrial myocytes. Int J Cardiol. 2016. May 1;210:85–92. [DOI] [PubMed] [Google Scholar]
- [269].Xue Y, Shi N, Zhang B, et al. Genetic variant in ZFHX3 geneon 16q22 associated with risk of stroke in Chinese Han population. Int J Clin Exp Pathol. 2016;9:8650–8656. [Google Scholar]
- [270].Liu C, Yao M-D, Li C-P, et al. Silencing of circular RNA-ZNF609 ameliorates vascular endothelial dysfunction. Theranostics. 2017;7(11):2863–2877. DOI: 10.7150/thno.19353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [271].Lin T, Zhang X, Lu Y, et al. Identification of circular RNA related to inflammation-induced lymphangiogenesis by microarray analysis. DNA Cell Biol. 2019. Aug;38(8):887–894. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.