

ABSTRACT
The oncogenic MUC1-C protein drives dedifferentiation of castrate resistant prostate cancer (CRPC) cells in association with chromatin remodeling. The present work demonstrates that MUC1-C is necessary for expression of IFNGR1 and activation of the type II interferon-gamma (IFN-γ) pathway. We show that MUC1-C→ARID1A/BAF signaling induces IFNGR1 transcription and that MUC1-C-induced activation of the NuRD complex suppresses FBXW7 in stabilizing the IFNGR1 protein. MUC1-C and NuRD were also necessary for expression of the downstream STAT1 and IRF1 transcription factors. We further demonstrate that MUC1-C and PBRM1/PBAF are necessary for IRF1-induced expression of (i) IDO1, WARS and PTGES, which metabolically suppress the immune tumor microenvironment (TME), and (ii) the ISG15 and SERPINB9 inhibitors of T cell function. Of translational relevance, we show that MUC1 associates with expression of IFNGR1, STAT1 and IRF1, as well as the downstream IDO1, WARS, PTGES, ISG15 and SERPINB9 immunosuppressive effectors in CRPC tumors. Analyses of scRNA-seq data further demonstrate that MUC1 correlates with cancer stem cell (CSC) and IFN gene signatures across CRPC cells. Consistent with these results, MUC1 associates with immune cell-depleted “cold” CRPC TMEs. These findings demonstrate that MUC1-C integrates chronic activation of the type II IFN-γ pathway and induction of chromatin remodeling complexes in linking the CSC state with immune evasion.
KEYWORDS: MUC1-C, CRPC, IFN-gamma, PBRM1, immunosuppression
Introduction
Castration resistant prostate cancer (CRPC) is effectively treated with agents that target the androgen receptor (AR) pathway.1–4 However, resistance to this therapy inevitably develops, often from progression to more aggressive disease with neuroendocrine (NE) features.1–4 Subsequent treatment options are then limited, emphasizing an unmet need for other therapeutic strategies. Immunotherapy represents an attractive approach for treating advanced PC,5 although success with immune checkpoint inhibitors (ICIs) has been limited in comparison to other types of cancers.6 Evaluation of the anti-PD1 antibody pembrolizumab in docetaxel-resistant metastatic CRPC (mCRPC) demonstrated modest anti-tumor activity and improvement in overall survival.7 In a retrospective analysis, pembrolizumab was more effective in the setting of microsatellite instability high (MSI-H) mCRPCs, suggesting that mutational burden may be a contributing factor for response.8 Other studies have identified the immunosuppressive PC tumor microenvironment (TME) as a critical factor in the lack of response to immunotherapy.9 Given the complexities of immunosuppressive TMEs,10 no single mechanism has been attributable to ICI resistance. However, inherently “cold tumors” characterized by depletion or dysfunction of immune effector cells are likely a significant contributing factor.9,11 The infiltration of regulatory T cells (T-regs), myeloid-derived suppressor cells (MDSCs) and tumor associated macrophages (TAMs) also play a role.9 In addition, intrinsic PC tumor cell production of immunosuppressive factors, such as IDO1, IL-10 and TGF-beta, contributes to immune effector cell-depleted PC TMEs that preclude responsiveness to ICIs.9 These pleiotropic mechanisms challenge the identification of strategies to circumvent CRPC immune evasion.
The MUC1 gene emerged in mammals to provide protection of epithelial niches from loss of homeostasis.12 MUC1 encodes an N-terminal (MUC1-N) subunit that contributes to a physical mucous barrier and a transmembrane C-terminal (MUC1-C) subunit that is activated by stress.12 MUC1-C contributes to inflammatory, proliferative and remodeling responses associated with the wound healing response.12 However, prolonged MUC1-C activation in association with chronic inflammation promotes oncogenesis by driving activation of the epithelial mesenchymal transition (EMT) and epigenetic reprogramming in cancer cells.12,13 Upregulation of MUC1 in PC associates with aggressive disease and poor patient outcomes.14–18 In PC cells, MUC1-C suppresses the AR axis and activates MYC signaling pathways that drive lineage plasticity in neuroendocrine prostate cancer (NEPC) progression.19 In this capacity, MUC1-C induces the Yamanaka pluripotency factors19 that function as pioneer TFs in the reprogramming of embryonic stem cells (ESCs).20 MUC1-C integrates MYC-induced pluripotency factor expression with activation of E2F1 and induction of the embryonic stem cell esBAF and PBAF chromatin remodeling complexes in driving changes in chromatin accessibility and PC dedifferentiation.21–23 Increasing evidence has linked cell fate specification and the cancer stem cell (CSC) state to immune evasion. In this respect, lineage plasticity in cancer represents a major challenge responsible for immune evasion and poor clinical outcomes.24–30 These findings have collectively invoked the possibility that MUC1-C-induced lineage plasticity and dedifferentiation in CRPC could contribute to an immunosuppressive TME. In support of this notion, the present work demonstrates that MUC1-C promotes “cold” immune effector cell-depleted CRPC TMEs by chronic activation of tumor intrinsic inflammatory and immunosuppressive pathways.
Materials and methods
Cell culture
Human DU-145 cells (ATCC) were cultured in RPMI1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum (FBS; GEMINI Bio-Products, West Sacramento, CA, USA), 100 μg/ml streptomycin and 100 U/ml penicillin. LNCaP-AI cells were grown in phenol red-free RPMI1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) containing 10% charcoal-stripped FBS (Millipore Sigma, Burlington, MA, USA).19 Human NCI-H660 NEPC cells (ATCC) were cultured in RPMI1640 medium with 5% FBS, 10 nM β-estradiol (Millipore Sigma), 10 nM hydrocortisone, 1% insulin-transferrin-selenium (Thermo Fisher Scientific) and 2 mM L-glutamine (Thermo Fisher Scientific). Cells were treated with 10 ng/ml human recombinant IFN-γ (Stemcell Technologies, Vancouver, BC, Canada). Authentication of the cells was performed by short tandem repeat (STR) analysis every 4 months. Cells were monitored for mycoplasma contamination using the MycoAlert Mycoplasma Detection Kit (Lonza, Rockland, ME, USA) every 3 months.
Tetracycline-inducible gene silencing
MUC1shRNA (MISSION shRNA TRCN0000122938; Sigma), MTA1shRNA (MISSION shRNA TRCN0000230496), MBD3shRNA (MISSION shRNA TRCN0000274441), PBRM1shRNA (MISSION shRNA TRCN0000235890), JUNshRNA (sc-29223-SH, Santa Cruz Biotechnologies), ARID1AshRNA (MISSION shRNA TRCN0000059092) was inserted into the pLKO-puro vector. Guide RNA (#1: GATCGTCAGGTTATATCGAG; #2: TGAACTGTGTCTCCACGTCG) targeting MUC1 exon 4 were inserted into the lentiCRISPR v2 (Plasmid 52961; Addgene).22 The viral vectors were produced in 293 T cells. Cells transduced with the vectors were selected for growth in 2 μg/ml puromycin. For tet-inducible vectors, cells were treated with 0.1% DMSO as the vehicle control or 500 ng/ml doxycycline (DOX; Millipore Sigma).
Quantitative reverse-transcription PCR (qRT-PCR)
Total cellular RNA was isolated using Trizol reagent (Thermo Fisher Scientific). cDNAs were synthesized and amplified as described.19 Primers used for qRT-PCR are listed in Supplementary Table S1.
Immunoblot analysis
Whole cell lysates were prepared in RIPA buffer containing protease inhibitor cocktail (Thermo Fisher Scientific, Waltham, MA, USA). Immunoblotting was performed with anti-MUC1-C (16564, 1:1000 dilution; Cell Signaling Technology (CST), Danvers, MA, USA), anti-STAT1 (9172S, 1:1000 dilution; CST), anti-IRF1 (#8478S, 1:1000 dilution; CST), anti-IFNGR1 (34808, 1:1000; CST), anti-GAPDH (#5174S, 1:1000 dilution; CST) and anti-β-actin (A5441; 1:100000 dilution; Sigma, St. Louis, MO, USA), anti-FBXW7 (ab109617, 1:1000; abcam, Cambridge, MA, USA), anti-MTA1 (5647, 1:1000; CST), anti-MBD3 (14540, 1:1000; CST), anti-IDO1 (86630, 1:1000; CST), anti-WARS (GTX110223, 40037, 1:1000; Gene Tex), anti-PTGES (ab180589, 1:1000; abcam), anti-PBRM1(A301-591A, 1:10000; Bethyl Laboratories, Montgomery, TX, USA), anti-ARID1A (12354, 1:500; CST), anti-ISG15 (sc166755, 1:2000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-SERPINB9 (PA5-51038, 1:2000; Invitrogen, Waltham, MA, USA).
Coimmunoprecipitation studies
Nuclear lysates from DU-145 cells were precipitated with a control IgG or anti-MUC1-C. Input and precipitated proteins were immunoblotted with anti-JUN (9165, 1:1000; CST).
RNA-seq analysis
Total RNA from cells cultured in triplicates was isolated using Trizol reagent (Invitrogen). TruSeq Stranded mRNA (Illumina, San Diego, CA, USA) was used for library preparation. Raw sequencing reads were aligned to the human genome (GRCh38.74) using STAR. Raw feature counts were normalized and differential expression analysis using DESeq2. Differential expression rank order was utilized for subsequent Gene Set Enrichment Analysis (GSEA), performed using the fgsea (v1.8.0) package in R. Gene sets queried included those from the Hallmark Gene Sets available through the Molecular Signatures Database (MSigDB).
Chromatin immunoprecipitation (ChIP)
ChIP was performed on cells crosslinked with 1% formaldehyde for 5 min at 37°C, quenched with 2 M glycine and washed with PBS, and then sonicated in a Covaris E220 sonicator to generate 300–600 bp DNA fragments. Immunoprecipitation was performed using a control IgG (Santa-Cruz Biotechnology) and antibodies against MUC1-C (D5K91; CST), JUN (32137; CST), ARID1A (12354; CST), EP300 (D2X6N; CST), H3K27ac (ab4729; Abcam), H3K4me1 (ab8895; Abcam) and H3K4me3 (ab8580; Abcam). Precipitated DNAs were detected by PCR using primers listed in Supplemental Table 1. Quantitation was performed on immunoprecipitated DNA using SYBR-green and the CFX384 real-time PCR machine (Bio-Rad, USA). Data are reported as fold enrichment relative to IgG.
ATAC-seq
ATAC-seq libraries were generated from three biologically independent replicates per condition. Library preparation and quality control were performed as described.23,31 Peak calling for all libraries was performed using MACS2.
Statistical analysis
Each experiment was performed at least three times. Data are expressed as the mean ± SD. The unpaired Mann-Whitney U test was used to determine differences between means of groups. A p-value of <0.05 denoted by an asterisk (*) was considered statistically significant.
Analysis of publicly available cohort data
TCGA-PRAD, SU2C-CRPC and Beltran cohort expression and clinical annotations were obtained from the Genomic Data Commons (GDC) data portal and processed via TCGAbiolinks package in R using TCGAWorkflow guided practices.32 Normalized expression and clinical annotations were obtained directly from cBioPortal. Differential expression associated with MUC1 expression (MUC1-high = top quartile; MUC1-low = bottom quartile) within each respective cohort was determined by TCGAbiolinks/edgeR or limma.33 Gene set enrichment analysis of differential expression was assessed using the clusterProfiler package. Queried gene sets derived from the Hallmark, Canonical pathways, and GO Biological Processes Ontology collections were retrieved from the MSigDB. Cell type enrichment within each sample was estimated from bulk expression via xCell34 and TIP35 analyses.
Analysis of CRPC scRNA-seq dataset
Processed scRNA-seq data comprising cells captured from 14 metastatic CRPC samples36 were obtained directly from the Broad Institute Single Cell Portal (https://singlecell.broadinstitute.org/single_cell). Normalized expression (transcripts per kilobase million (TPM)) and previously determined cell annotation were utilized. Tumor cells (n = 836) were isolated from prior annotations, and tumor cell gene expression was imputed using MAGIC,37 implemented via the Rmagic package in R. CRPC cell imputed expression was re-analyzed via Seurat38 for variable feature selection, dimensionality reduction (PCA), and uniform manifold approximation and projection (UMAP) low-dimensional representation. Single-cell pathway enrichment was performed by AUCell,39 using select HALLMARK or curated pathways representing AR signaling (AR, FKBP5, TMPRSS2, KLK3, NKX3-1, STEAP4, PMEPA1, PLPP1, PART1, ALDH1A3, DPP4) or prostate CSC (KLF4, NANOG, POU5F1, MYC, SOX2, CD44, PSCA, PROM1, EZH2, ABCG2, ALDH1A1, TGM2, KIT, ARID1A) signatures. Associations between MUC1 expression and select genes or signatures within CRPC cells were examined by Pearson correlation analysis.
Results
MUC1 associates with chronic activation of the IFN-γ inflammatory response pathway in prostate carcinomas
MUC1-C is aberrantly expressed in CRPC and has been linked to CRPC progression.19,21,22 Analysis of the TCGA-PRAD dataset derived from 333 primary PCs40 demonstrated that MUC1-high PCs significantly associate with activation of the HALLMARK INTERFERON GAMMA RESPONSE gene signature (Figure 1a). Similar results were obtained from analysis of 266 metastatic CRPCs in the SU2C-CRPC dataset4 (Figure 1b), suggesting that MUC1 may functionally contribute to activation of the type II IFN pathway. Further analysis of the TCGA-PRAD dataset showed that MUC1-high PCs have significantly increased levels of IFNGR1 and IFNGR2 expression as compared to that in MUC1-low tumors (Figure 1c). Stimulation of the IFNGR1 receptor complex by IFN-γ activates the downstream STAT1 and IRF1 transcription factors (TFs) that drive IFN response genes (ISGs) and chronic inflammatory responses in cancer cells.41–43 Notably, MUC1-high PCs were significantly associated with upregulation of STAT1 and IRF1 expression (Figure 1d). Moreover, we found that MUC1 significantly associates with the HALLMARK INFLAMMATORY RESPONSE gene signature in the TCGA-PRAD (Figure 1e) and SU2C-CRPC (Figure 1f) datasets, in support of activating the IFN-γ pathway and promoting chronic inflammation in PC cells.
Figure 1.
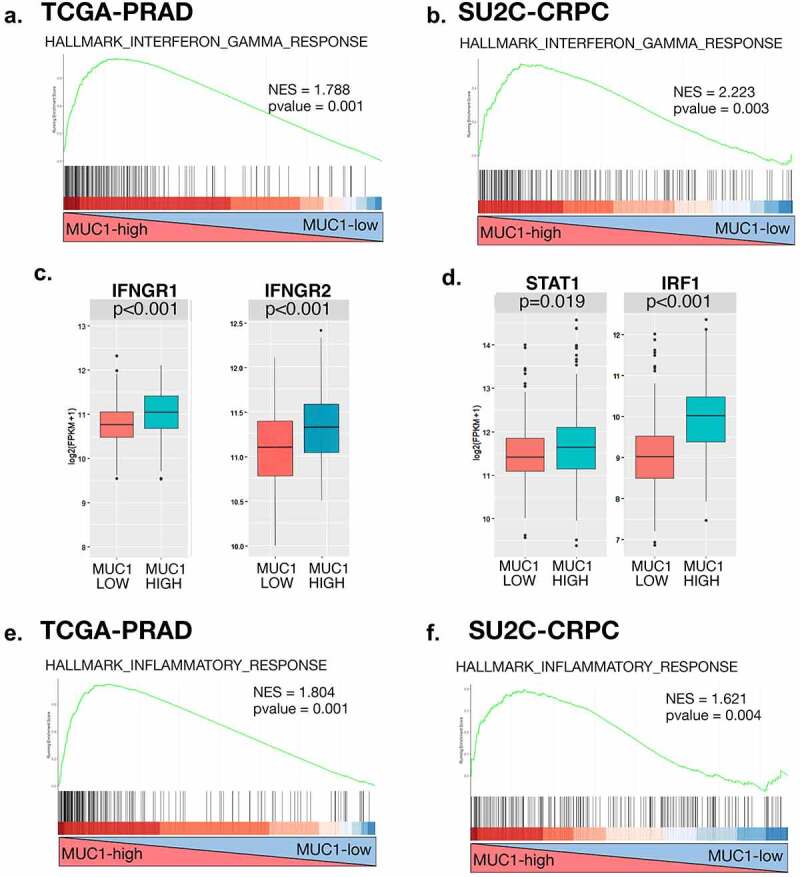
Expression of MUC1 in PC tumors associates with chronic activation of the type II IFNG pathway. (a and b). Enrichment plots for the HALLMARK INTERFERON GAMMA RESPONSE pathway, comparing MUC1-high to MUC1-low PC tumors in the TCGA-PRAD (a). and SU2C-CRPC (b). cohorts. (c and d). Normalized expression data for the TCGA-PRAD cohort were downloaded from cBioPortal, and median expression used to group samples into MUC1-high and MUC1-low groups. Expression of IFNGR1 and IFNGR2 (c). and downstream STAT1 and IRF1 (d). genes was assessed in MUC1-high and MUC1-low groups using a Wilcoxon rank-sum test. Boxplots represent the 1st quartile, median and 3rd quartile of each distribution. Whiskers extend to the maximum and minimum values up to 1.5*interquartile range (IQR). (e and f). Enrichment plots for the HALLMARK INFLAMMATORY RESPONSE pathway, comparing MUC1-high to MUC1-low PC tumors in the TCGA-PRAD (e) and SU2C-CRPC (f) cohorts.
MUC1-C activates the IFNGR1 distal enhancer-like sequence
In extending these observations linking MUC1-C to regulation of the IFN-γ pathway, we silenced MUC1-C in DU-145 CRPC cells with a tet-inducible MUC1shRNA and found that MUC1-C is necessary for expression of IFNGR1 transcripts (Figure 2a) and protein (Figure 2b). Stable MUC1-C silencing with single guide RNAs (sgRNAs) confirmed that MUC1-C is necessary for IFNGR1 expression (Figure 2c). In addition, similar results were obtained in (i) LNCaP-AI cells that were derived from androgen-dependent LNCaP cells selected for growth under androgen-independent (AI) conditions and overexpress MUC1-C19(Supplemental Figs. S1a and S1b), (ii) PC3 CRPC cells19(Supplemental Fig. S1c) and (iii) NCI-H660 NEPC cells (Supplemental Fig. S1d). In investigating the mechanism responsible for MUC1-C-induced IFNGR1 expression, we assessed the effects of silencing MUC1-C on the IFNGR1 gene at a distal enhancer-like signature (dELS) that includes a putative AP-1 binding motif (Figure 2d). Along these lines, nuclear MUC1-C forms complexes with JUN/AP-1 (Supplemental Fig. S1e)23 and we detected occupancy of MUC1-C and JUN on the IFNGR1 dELS region (Figure 2d). Consistent with JUN-mediated recruitment of the BAF chromatin remodeling complex,44 we also detected occupancy of ARID1A (Figure 2d). Of significance for MUC1-C-mediated IFNGR1 activation, silencing MUC1-C decreased JUN and ARID1A occupancy (Figure 2e). Moreover, silencing JUN and ARID1A suppressed IFNGR1 expression (Supplemental Figs. S1f-S1i). Silencing MUC1-C also decreased H3K4me3 levels (Figure 2f) and chromatin accessibility of the dELS (Figure 2g), indicating that MUC1-C activates IFNGR1 by a mechanism involving JUN, ARID1A and remodeling of chromatin.
Figure 2.
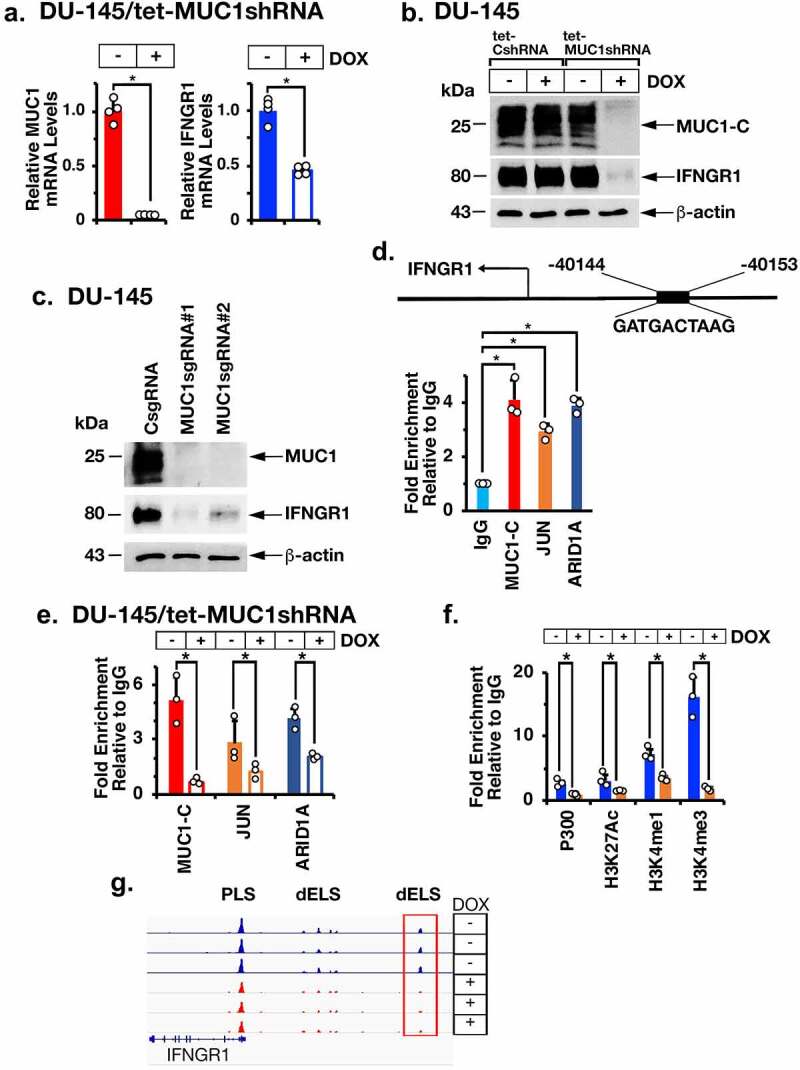
The MUC1-C→ARID1A/BAF pathway is necessary for activation of IFNGR1 expression. (a). DU-145/tet-MUC1shRNA cells treated with vehicle or DOX for 7 days were analyzed for MUC1-C and IFNGR1 mRNA levels by qRT-PCR using primers listed in Supplemental Table S1. The results (mean ± SD of 4 determinations) are expressed as relative mRNA levels compared to that obtained for vehicle-treated cells (assigned a value of 1). (b). Lysates from DU-145/tet-CshRNA and DU-145/tet-MUC1shRNA treated with vehicle or DOX for 7 days were immunoblotted with antibodies against the indicated proteins. (c). Lysates from DU-145 expressing a CsgRNA, MUC1sgRNA#1 or MUC1sgRNA#2 were immunoblotted with antibodies against the indicated proteins. (d). Schema of the IFNGR1 with highlighting of a JUN/AP-1 binding site in the dELS. Soluble chromatin from DU-145 cells was precipitated with a control IgG, anti-MUC1-C, anti-JUN and anti-ARID1A. (e). Soluble chromatin from DU-145/tet-MUC1shRNA cells treated with vehicle or DOX was precipitated with a control IgG, anti-MUC1-C, anti-JUN and anti-ARID1A. (f). Soluble chromatin from DU-145/tet-MUC1shRNA cells treated with vehicle or DOX was precipitated with a control IgG, anti-EP300, anti-H3K27ac, anti-H3K27me1 and anti-H3K4me3. The DNA samples were amplified by qPCR with primers for the IFNGR1 dELS region. The results (mean ± SD of 3 determinations) are expressed as fold enrichment relative to that obtained with the IgG control (assigned a value of 1). (g and h). Genome browser snapshots of ATAC-seq data from the IFNGR1 dELS in DU-145/tet-MUC1shRNA cells treated with vehicle or DOX for 7 days (g). Chromatin was analyzed for accessibility by nuclease digestion (h). The results (mean ± SD of 3 determinations) are expressed as % untreated chromatin.
MUC1-C suppresses FBXW7 to promote IFNGR1 expression
The FBXW7 ubiquitin E3 ligase induces proteasomal IFNGR1 degradation and thereby downregulation of the IFN-γ signaling pathway.45 In determining whether MUC1-C also contributes to regulation of IFNGR1 through this pathway, we found that silencing MUC1-C results in the induction of FBXW7 mRNA and protein (Figure 3a–b; Supplemental Fig. S2a-S2c). In a gain-of-function study, we also found that overexpression of MUC1-C suppresses FBXW7 and upregulates IFNGR1 expression (Supplemental Fig. S2d). MUC1-C represses gene expression by activation of the nucleosome remodeling and deacetylation (NuRD) complex that consists in part of MTA1 and MBD3.46 Silencing MUC1-C resulted in suppression of MTA1 and MBD3 expression in DU-145 and LNCaP-AI cells (Figure 3c; Supplemental Fig. S2e). Moreover, silencing MTA1 or MBD3 was associated with induction of FBXW7 and downregulation of IFNGR1 levels (Figure 3d–e; Supplemental Fig. S2f). In extending these results, suppression of IFNGR1 levels by silencing MUC1-C (Figure 3f), MTA1 (Figure 3g) or MBD3 (Figure 3h) was abrogated by treatment with the proteosome inhibitor MG-132, confirming the involvement of MUC1-C and the NuRD complex in regulating IFNGR1 stability. Taken together, these results indicate that MUC1-C drives IFNGR1 expression by transcriptional and posttranscriptional mechanisms.
Figure 3.
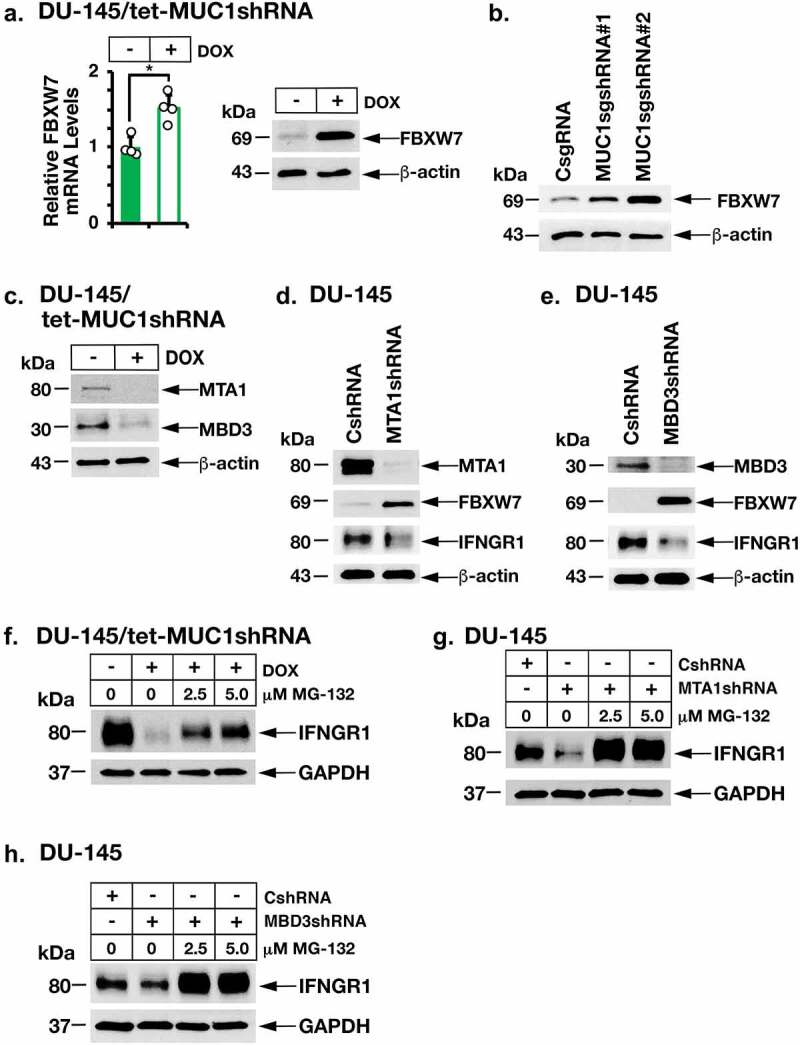
MUC1-C→NuRD signaling upregulates IFNGR1 by repressing FBXW7 expression. (a). DU-145/tet-MUC1shRNA cells treated with vehicle or DOX for 7 days were analyzed for FBXW7 mRNA levels (left). The results (mean ± SD of 4 determinations) are expressed as relative mRNA levels compared to that obtained for vehicle-treated cells (assigned a value of 1). Lysates were immunoblotted with antibodies against the indicated proteins (right). (b). Lysates from DU-145 cells expressing a CsgRNA, MUC1sgRNA#1 or MUC1sgRNA#2 were immunoblotted with antibodies against the indicated proteins. (c). Lysates from DU-145/tet-MUC1shRNA treated with vehicle or DOX for 7 days were immunoblotted with antibodies against the indicated proteins. (d and e). Lysates from DU-145 expressing a CshRNA, MTA1shRNA (d) or MBD3shRNA (e). were immunoblotted with antibodies against the indicated proteins. (f). DU-145/tet-MUC1shRNA cells were treated with vehicle or DOX for 7 days in the presence of the indicated concentrations of MG-132. Lysates were immunoblotted with antibodies against the indicated proteins. (g and h). DU-145 cells expressing a CshRNA, MTA1shRNA (g) or MBD3shRNA (h) were treated with the indicated concentrations of MG-132. Lysates were immunoblotted with antibodies against the indicated proteins.
MUC1-C induces STAT1 and IRF1 expression
Stimulation of IFNGR1 by IFN-γ activates the STAT1→IRF1 pathway.47 Consistent with MUC1-C-mediated induction of IFNGR1, we found that silencing MUC1-C decreases STAT1 and IRF1 transcripts (Figure 4a; Supplemental Fig. S3a) and protein (Figure 4b–c; Supplemental Figs. S3b and S3c). In addition, MUC1-C was necessary for induction of STAT1 and IRF1 expression in the response to IFN-γ stimulation (Figure 4d). As found for IFNGR1, silencing MTA1 or MBD3 suppressed STAT1 and IRF1 expression (Figure 4e). In addition, silencing MUC1-C decreased chromatin accessibility of the STAT1 (Figure 4f) and IRF1 (Figure 4g) genes, in support of a MUC1-C-driven pathway that drives IFNGR1, STAT1 and IRF1.
Figure 4.
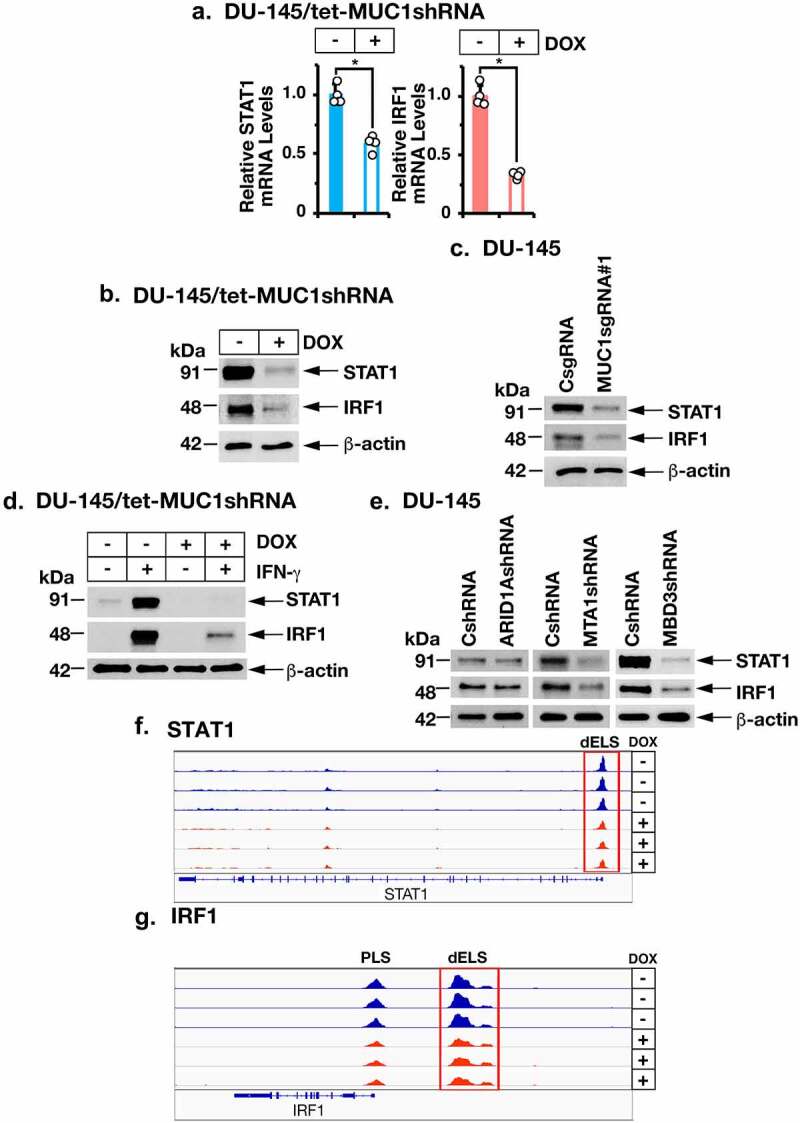
MUC1-C and NuRD drive STAT1 and IRF1 expression. (a and b). DU-145/tet-MUC1shRNA cells treated with vehicle or DOX for 7 days were analyzed for the indicated mRNA levels (a). The results (mean ± SD of 4 determinations) are expressed as relative mRNA levels compared to that obtained for vehicle-treated cells (assigned a value of 1). Lysates were immunoblotted with antibodies against the indicated proteins (b). (c). Lysates from DU-145 cells expressing a CsgRNA or MUC1sgRNA#1 were immunoblotted with antibodies against the indicated proteins. (d). DU-145/tet-MUC1shRNA cells treated with vehicle or DOX for 7 days were stimulated with 10 ng/ml IFN-γ for 24 hours. Lysates were immunoblotted with antibodies against the indicated proteins. (e). Lysates from DU-145 expressing a CshRNA, ARID1AshRNA, MTA1shRNA or MBD3shRNA were immunoblotted with antibodies against the indicated proteins. (f and g). Genome browser snapshots of ATAC-seq data from the STAT1 (f) and IRF1 (g) genes in DU-145/tet-MUC1shRNA cells treated with vehicle or DOX for 7 days.
MUC1-C and PBRM1 are necessary for induction of the metabolic IDO1, WARS and PTGES immunosuppressive factors
The finding that MUC1-C induces IRF1 invoked the possibility that MUC1-C is necessary for activation of downstream effectors in the type II IFN pathway. To identify type II IFN genes that are regulated by MUC1-C, RNA-seq was performed on DU-145/tet-MUC1shRNA cells treated with vehicle or DOX and then stimulated with IFN-γ. Analysis of the datasets demonstrated that MUC1-C drives IFN-γ-induced activation and repression of the HALLMARK INTERFERON GAMMA RESPONSE gene signature (Figure 5a; Supplemental Fig. S4a) and other enriched pathways that include the HALLMARK INTERFERON ALPHA RESPONSE (Supplemental Figs. S4b and S4c). Specifically, we found that MUC1-C is necessary for IFN-γ-induced STAT1 and IRF1 expression (Figure 5b). As determined by Epigenetic Landscape in Silico deletion Analysis (LISA),48 MUC1-induced genes were enriched for STAT1 and IRF1 regulation (Supplemental Fig. S4d), indicating that MUC1-C drives the IFN-γ response by activating STAT1→IRF1 signaling. Consistent with this notion, we found that MUC1-C is necessary for expression of immunosuppressive indoleamine-2,3-dioxygenase-1 (IDO1), tryptophanyl-tRNA synthetase (WARS, WRS) and PTGES effectors (Figure 5b). IRF1 induces (i) IDO1, which reduces tryptophan (Trp) levels in the TME that are necessary for T cell proliferation and function,49 (ii) WARS that is associated with IDO1 expression and protects cancer cells from Trp depletion,50,51 and (iii) PTGES, which is required for the synthesis of PGE2, an inhibitor of T cell function.52 Analysis of the TCGA-PRAD and SU2C-CRPC datasets further demonstrated that MUC1-high PCs significantly associate with upregulation of IDO1 (Figure 5c; Supplemental Fig. S5a), as well as IDO2 and tryptophan-2,3-dioxygenase (TDO2), which also degrade Trp (Supplemental Figs. S5a and S5b). In addition, MUC1 was significantly associated with WARS and PTGES expression (Figure 5c; Supplemental Fig. S5c). In support of these observations, we confirmed that MUC1-C is necessary for constitutive (Figure 5d–e; Supplemental Figs. S6a-S6d) and IFN-γ-induced (Figure 5f; Supplemental Figs. S6e and S6f) expression of IDO1, WARS and PTGES transcripts and protein. The polybromo-associated BAF (PBAF) chromatin remodeling complex, which includes BRG1, PBRM1/BAF180, ARID2/BAF200 and BRD7, is activated by the MUC1-C→E2F1 signaling pathway in CRPC cells.22 PBRM1 has been associated with conferring resistance to T cell-mediated killing of tumor cells.53–55 Of interest in that regard, we found that PBRM1 is dispensable for constitutive STAT1 and IRF1 expression (Figure 5g). However, and as found for MUC1-C, PBRM1 was necessary for expression of IDO1, WARS and PTGES (Figure 5g). PBRM1 was also necessary for IFN-γ-stimulated upregulation IDO1, WARS and PTGES (Figure 5h), indicating that the MUC1-C→PBRM1 pathway functions in promoting induction of these IRF1 target genes.
Figure 5.
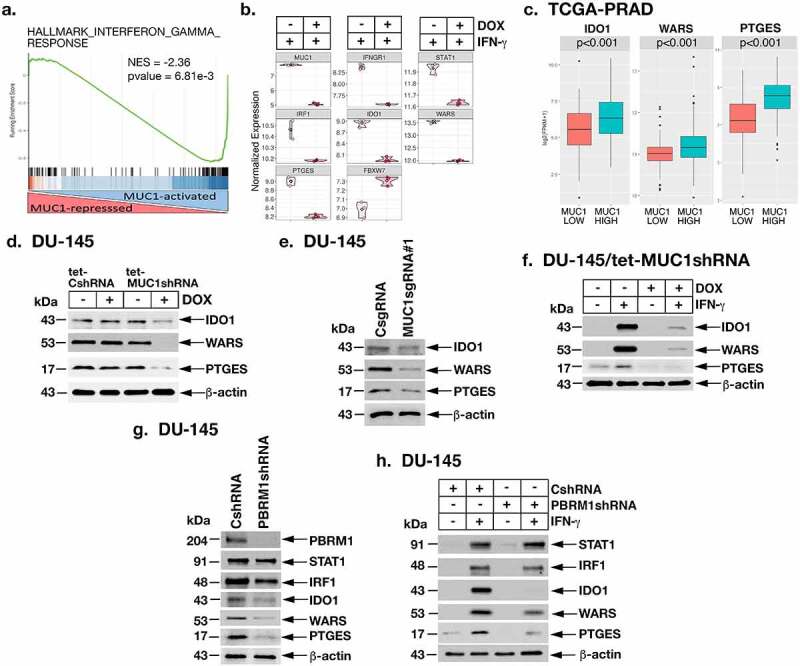
MUC1-C→PBRM1/PBAF pathway induces IDO1, WARS and PTGES. (a and b). RNA-seq was performed in triplicates on DU-145/tet-MUC1shRNA cells treated with vehicle or DOX for 7 days and then stimulated with 10 ng/ml IFN-γ for 24 hours. The datasets were analyzed by GSEA using the HALLMARK INTERFERON GAMMA RESPONSE gene signature comparing DOX-treated with vehicle-treated cells (a). Analysis of the indicated genes in vehicle- and DOX-treated cells showing significant differences in mRNA levels (b). (c). Expression of IDO1, WARS and PTGES in the TCGA-PRAD cohort was assessed in MUC1-high and MUC1-low groups using a Wilcoxon rank-sum test. (d). Lysates from DU-145/tet-CshRNA and DU-145/tet-MUC1shRNA treated with vehicle or DOX for 7 days were immunoblotted with antibodies against the indicated proteins. (e). Lysates from DU-145 expressing a CsgRNA or MUC1sgRNA#1 were immunoblotted with antibodies against the indicated proteins. (f). DU-145/tet-MUC1shRNA cells treated with vehicle or DOX for 7 days were stimulated with 10 ng/ml IFN-γ for 24 hours. Lysates were immunoblotted with antibodies against the indicated proteins. (g). Lysates from DU-145/CshRNA and DU-145/PBRM1shRNA cells were immunoblotted with antibodies against the indicated proteins. (h). Lysates from DU-145/CshRNA and DU-145/PBRM1shRNA cells stimulated with 10 ng/ml IFN-γ for 24 hours were immunoblotted with antibodies against the indicated proteins.
MUC1-C→PBRM1 signaling induces the ISG15 and SERPINB9 inhibitors of CTL function
Analysis of the IFN-γ-stimulated DU-145 cell dataset further demonstrated that MUC1-C is necessary for induction of (i) ISG15, which encodes a ubiquitin-like protein involved in innate immunity that suppresses CTL function in the TME,56–58 and (ii) SERPINB9, a member of the serpin family encoding a granzyme B inhibitor that confers resistance to CTL killing.59,60 Analysis of the TCGA-PRAD dataset demonstrated that MUC1 is associated with ISG15 and SERPINB9 expression (Figure 6a). Studies in DU-145 and LNCaP-AI cells further showed that silencing MUC1-C downregulates ISG15 and SERPINB9 mRNA (Supplemental Fig. S7a) and protein (Figure 6b–c) levels. We also found that PBRM1 is necessary for ISG15 and SERPINB9 expression (Figure 6d; Supplemental Fig. S7b), supporting involvement of the MUC1-C→PBRM1 pathway. Consistent with these results, MUC1-C (Figure 6e–f; Supplemental Fig. S7c) and PBRM1 (Figure 6f; Supplemental Fig. S7d) were necessary for IFN-γ-induced expression of these suppressive effectors of CTL function.
Figure 6.
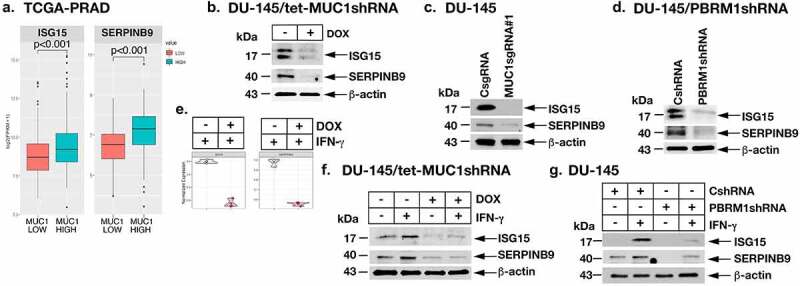
MUC1-C and PBRM1/PBAF drive ISG15 and SERPINB9 expression. (a). Expression of ISG15 and SERPINB9 in the TCGA-PRAD cohort was assessed in MUC1-high and MUC1-low groups using a Wilcoxon rank-sum test. (b). Lysates from DU-145/tet-MUC1shRNA treated with vehicle or DOX for 7 days were immunoblotted with antibodies against the indicated proteins. (c). Lysates from DU-145/CsgRNA or DU-145/MUC1sgRNA#1 were immunoblotted with antibodies against the indicated proteins. (d). Lysates from DU-145/CshRNA and DU-145/PBRM1shRNA cells were immunoblotted with antibodies against the indicated proteins. (e and f). DU-145/tet-MUC1shRNA cells treated with vehicle or DOX for 7 days were stimulated with 10 ng/ml IFN-γ for 24 hours. Analysis of the indicated genes in vehicle- and DOX-treated cells showing significant differences in mRNA levels (e). Lysates were immunoblotted with antibodies against the indicated proteins (f). (g). Lysates from DU-145/CshRNA and DU-145/PBRM1shRNA cells stimulated with 10 ng/ml IFN-γ for 24 hours were immunoblotted with antibodies against the indicated proteins.
MUC1 associates with suppression of the CRPC tumor immune microenvironment
In support of MUC1-C involvement in promoting suppression of the PC TME, analysis of the TCGA-PRAD dataset demonstrated that MUC1 associates with enrichment of REACTOME INTERLEUKIN 10 SIGNALING and GO RESPONSE TO TRANSFORMING GROWTH FACTOR BETA pathways (Figure 7a). GSEA confirmed that MUC1-high tumors significantly associate with activation the IL-10 (Supplemental Fig. S8a) and TGFB1 (Supplemental Fig. S8b) gene signatures, which function as negative regulators of the immune TME.9,61 We also found that MUC1-high tumors associate with expression of CCL5 (Supplemental Fig. S8c), an inflammatory chemokine that recruits TAMs, MDSCs and T-regs into the TME and inhibits CTL activity.62 Consistent with these findings, MUC1 was significantly associated with negative regulation of (i) T cell and NK cell mediated immunity, (ii) T cell proliferation, and (iii) B cell activation (Figure 7b). In extending these analyses to the Beltran cohort (67 CRPC/NEPC samples),63 tumors were stratified by MUC1-high and MUC1-low expression. Hierarchical clustering based on cell type estimation (xCell)34 demonstrated that MUC1-high clusters associate with decreased estimates of immune cell infiltration (Figure 7c). Further analysis by immune cell type demonstrated that MUC1-high tumors significantly associate with decreases in CD4+ memory T cells, Th2 cells, B cells and M2 macrophages, among others, as well as the ImmuneScore (Figure 7d; Supplemental Fig. S8d).
Figure 7.
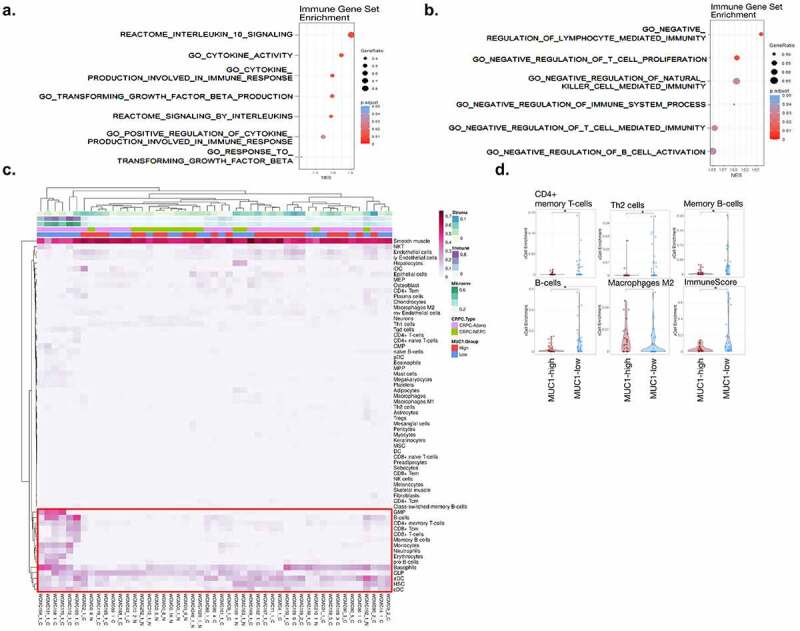
Association of MUC1-high expressing CRPC tumors with immune cell depletion. (a and b). Association of MUC1 expression with the indicated REACTOME and GO immune gene datasets in the TCGA-PRAD cohort. (c). Heatmap depicting cell type enrichment analysis in MUC1-high and MUC1-low tumors from the Beltran cohort. (d). Select cell-type enrichments determined between MUC1-high and MUC1-low tumors from the Beltran cohort. The asterisk represents significant difference (Wilcox rank-sum test, p < .05) between groups.
MUC1 associates with intratumoral CSC and IFN signatures in CRPCs
To examine MUC1 as it relates to tumor cell heterogeneity and the CSC state, we analyzed a scRNA-seq dataset containing 836 CRPC cells obtained from 14 patients with metastatic disease to assess the distribution of MUC1 expression36 (Figure 8a–b; Supplemental Fig. S9a). This dataset comprises CRPC cells of both adenocarcinoma and small cell morphology, thus spanning an array of CRPC transcriptional phenotypes. Single-cell pathway enrichment was performed to examine the cellular distributions of AR, CSC and IFN signaling pathways (Figure 8c, Fig. S9b-c). The CSC signature was strongly enriched in CRPC cells of small cell phenotype, as well as subsets of adenocarcinoma cells, which was inversely correlated with AR signaling (Figure 8c). Notably, IFN and inflammatory signatures were significantly and positively associated with CSC enrichment. Similarly, MUC1 expression significantly associated with CSC and IFN signatures across CRPC cells, typified by significant associations with CSC-related (SOX2, NANOG, KIT, EZH2, KL4, ARID1A) and to a lesser extent IFN-related (IRF1, STAT1, IFNGR1, IFNGR2) genes (Figure 8d). MUC1 was inversely correlated with AR and AR target genes (KLK3, TMPRSS2, NKX3-1, DPP4) (Figure 8d). These results suggest that MUC1, as well as IFN, signatures, reside in CRPC cells with CSC characteristics rather than being homogeneously distributed across all CRPC cells.
Figure 8.
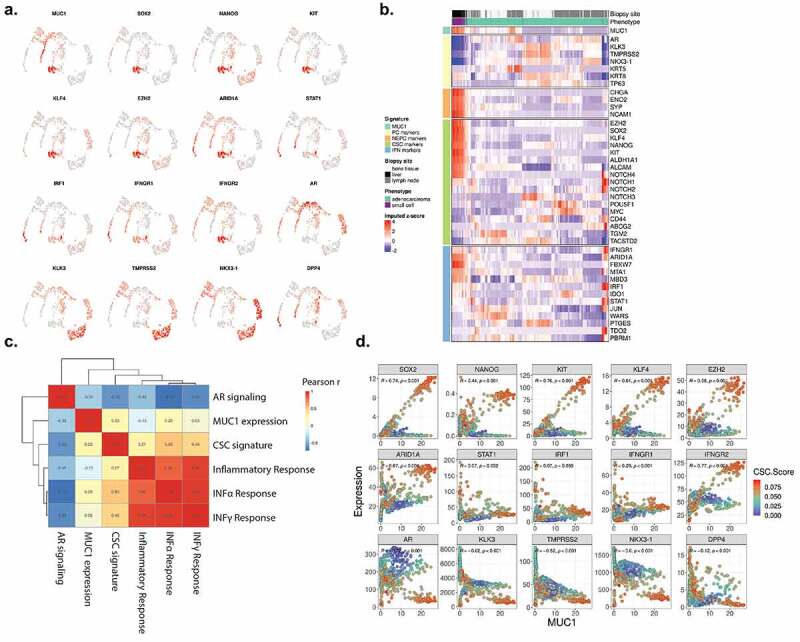
MUC1 associates with CSC and IFN signatures in individual CRPC cells. (a). Imputed expression of MUC1 and select genes associated with CSC (SOX2, NANOG, KIT, KLF4, EZH2, ARID1A), interferon signaling (STAT1, IRF1, IFNGR1, IFNGR2), and androgen signaling (AR, KLK3, TMPRSS2, NKX3-1, DPP4). (b). Heatmap depicting CRPC cell expression of candidate genes associated with AR signaling, NEPC, CSC and interferon signaling. (c). Single-cell enrichment was performed for curated AR and CSC signatures and select HALLMARK pathways. Correlation of enrichment scores amongst pathways and MUC1 expression is shown. (d). Correlation analysis of candidate gene expression with MUC1 expression across CRPC cells. CSC signature enrichment across cells is displayed as blue to red scale.
Discussion
The type II IFN-γ pathway plays opposing roles in tumor immune surveillance and evasion that are dependent on context of both the tumor cell and TME.64,65 Intrinsic activation of the IFN-γ pathway in cancer cells contributes to chronic inflammation, immune evasion and progression.64,65 Despite this importance, little is known about induction of the IFNGR1 gene, which is essential for activating this pathway.56 Analysis of the TCGA-PRAD and SU2C-CRPC RNA-seq datasets revealed that MUC1 associates with IFNGR1, as well as IFNGR2, expression. Moreover, we found that MUC1-high PC tumors significantly associate with activation of the HALLMARK INTERFERON GAMMA RESPONSE pathway. These findings invoked the possibility that MUC1-C is necessary for induction of the IFNGR1 gene and the IFN-γ pathway (Figure 9). To address this notion, we studied CRPC and NEPC cells and found that MUC1-C is necessary for expression of IFNGR1 transcripts. MUC1-C→E2F1 signaling induces ARID1A and other components of the esBAF chromatin remodeling complex in NEPC progression.21 ARID1A mutations, which drive cancer development, limit chromatin accessibility and expression of IFN-responsive genes.66 MUC1-C forms complexes with JUN and ARID1A in promoting chromatin accessibility of NOTCH1 and other stemness-associated genes in CRPC cells.23 In the present work, we found that MUC1-C, JUN and ARID1A form a complex on the IFNGR1 dELS region that induces chromatin accessibility, H3K4 trimethylation and IFNGR1 expression. These findings provided direct evidence for a MUC1-C-induced pathway that integrates induction of ARID1A/BAF with intrinsic activation of the CSC state23 and IFN-γ pathway in CRPC progression (Figure 9).
Figure 9.
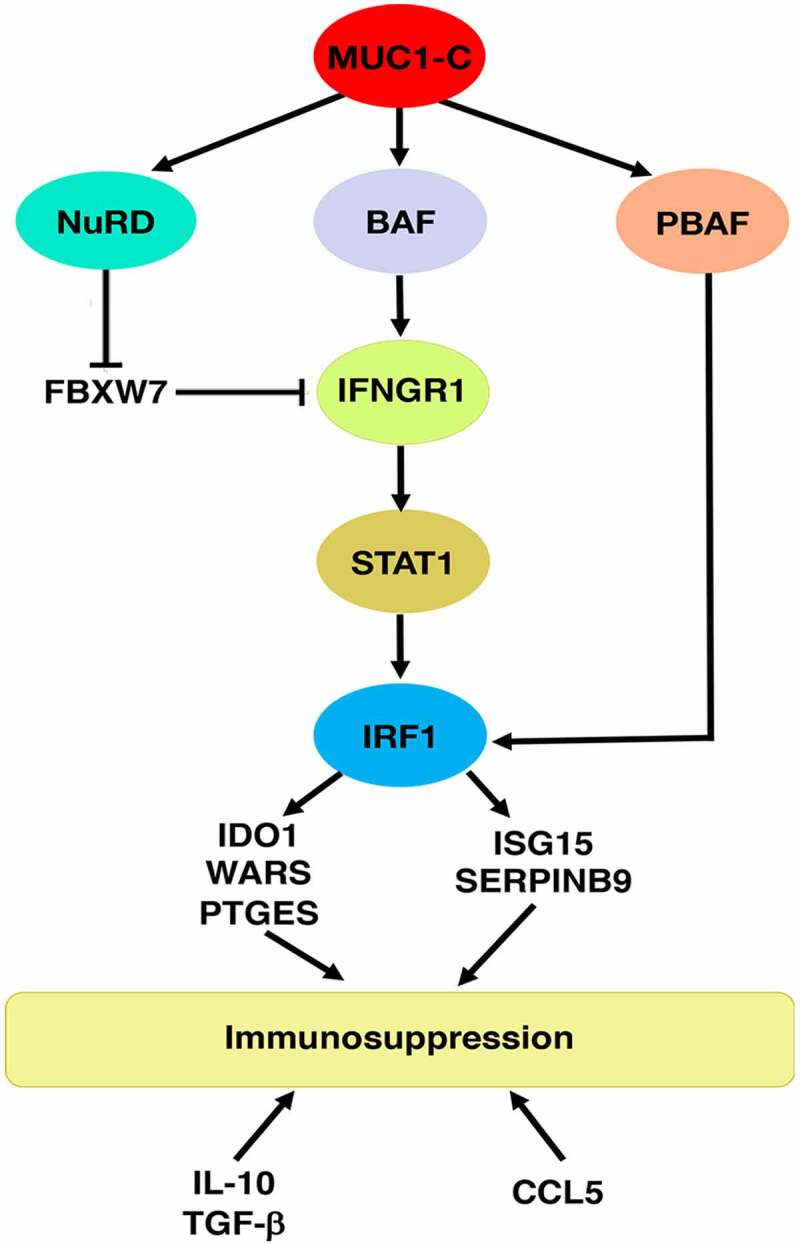
Schema depicting MUC1-C-induced chronic activation of the type II IFNG pathway, chromatin remodeling complexes and immunosuppression. MUC1-C drives expression of the BAF, NuRD and PBAF complexes. MUC1-C activates IFNGR1 by forming a complex with JUN and recruiting ARID1A/BAF to a dELS, which increases chromatin accessibility, H3K4 trimethylation and IFNGR1 expression. MUC1-C also stabilizes IFNGR1 by NuRD-mediated repression of FBXW7, an effector of IFNGR1 degradation. In turn, MUC1-C contributes to upregulation of STAT1, as well as IRF1, which interacts with PBRM1/PBAF in inducing expression of (i) IDO1, WARS and PTGES that metabolically suppress the immune TME, and (ii) the ISG15 and SERPINB9 inhibitors of T cell function. MUC1-C-high PC tumors also associate with increased expression of the immunosuppressive IL-10 and TGFB1 cytokines and the CCL5 chemokine. Consistent with these results, MUC1-C drives negative regulation and depletion of immune effectors in the PC TME.
While performing these experiments, we recognized that MUC1-C is also playing a role in the posttranscriptional regulation of IFNGR1 expression. In this regard, other work had demonstrated that the FBXW7 ubiquitin ligase promotes degradation of the IFNGR1 protein.45 Those studies showed that the ELF5 TF induced FBXW7 expression and in turn IFNGR1 destabilization.45 There is no known link between MUC1-C and the regulation of FBXW7. MUC1-C activates the NuRD complex, which includes MTA1 and MBD3, in suppressing gene expression46 and has been linked to the regulation of FBXW7 transcription.67 We found that MUC1-C downregulates FBXW7 by a mechanism involving MTA1 and MBD3. MUC1-C, MTA1 and MBD3 were necessary for suppression of FBXW7 and, as a result, stabilization of the IFNGR1 protein (Figure 9). Taken together with the effects of MUC1-C on IFNGR1 transcription, these findings uncovered another previously unrecognized MUC1-C-driven pathway that increases IFNGR1 expression by a posttranscriptional mechanism. Stimulation of IFNGR1 by IFN-γ activates the downstream STAT1 and IRF1 effectors of the type II IFN pathway. Consistent with MUC1-C-induced upregulation of IFNGR1, we found that MUC1 is associated with STAT1 and IRF1 expression in CRPC/NEPC tumors (Figure 9). In addition, silencing MUC1-C, MTA1 and MBD3 in CRPC cells decreased chromatin accessibility and expression of STAT1 and IRF1, indicating that MUC1-C is necessary for activation of the IFNGR1→STAT1/IRF1 pathway (Figure 9).
IRF1 is an essential regulator of downstream effectors, such as IDO1,49 WARS50,51 and PTGES,52 that promote immunosuppression of the TME by metabolically inhibiting T cell functions. IRF1 also drives ISG1556–58 and SERPINB959,60 that confer resistance of cancer cells to CTL killing. Consistent with the demonstration that MUC1-C is necessary for induction of IRF1, we found that silencing MUC1-C in CRPC/NEPC cells suppresses expression of these immunosuppressive effectors. In addition, MUC1-high PC tumors significantly associated with upregulation of IDO1, WARS, PTGES, ISG15 and SERPNB9. As reported for ARID1A/BAF,21 MUC1-C→E2F1 signaling activates the PBAF chromatin remodeling complex, integrating the ARID1A/BAF and PBRM1/PBAF pathways in CRPC cells.22 Unexpectedly, we found that PBRM1 is necessary for induction of IDO1, WARS, PTGES, ISG15 and SERPINB9 (Figure 9). PBRM1 has been associated with conferring resistance to T cell-mediated killing of melanoma cells.53 In addition, PBRM1 deficiency has been associated with (i) clinical benefit to ICI treatment,54 and, in contrast, (ii) a less immunogenic TME and ICI resistance.55 Our findings that the MUC1-C→E2F1→PBRM1/PBAF signaling induces effectors that inhibit CTL functions support involvement of this pathway in immune evasion (Figure 9).
MUC1-C drives lineage plasticity and dedifferentiation in the progression of CRPC and NEPC cells.19 MUC1-C-induced activation of the BAF and NuRD complexes has been linked to dedifferentiation and progression of CSCs.21,46 MUC1-C also activates the PBRM1/PBAF complex in regulating redox balance and lineage plasticity in CSCs.22 These and the present findings lend support to a model in which MUC1-C integrates activation of (i) the BAF, NuRD and PBAF complexes, (ii) IFNGR1 and the immunosuppressive IFN-γ pathway, and (iii) dedifferentiation and CSC progression (Figure 9). In this regard, cancer cell stemness is intimately associated with resistance to treatment with immunotherapeutic agents, albeit by mechanisms that remain unclear.24–27,68 Our findings in individual CRPC cells with a small cell phenotype demonstrate that MUC1-C associates with CSC and IFN gene signatures. These results suggest that MUC1 is selectively expressed in CRPC cells with CSC characteristics and chronic inflammatory signaling, linking stemness with immune evasion. In further support of the notion that MUC1-C integrates the CSC state with immune suppression, MUC1-C drives dedifferentiation in triple-negative breast cancer (TNBC) cells23,46 and contributes to suppression of the TNBC immune TME.69
Of potential translational relevance, antibodies and vaccines have been developed against the MUC1-N subunit with a particular emphasis on targeting the VNTR region.70 Clinical trials of these agents have demonstrated the induction of immune responses, but not effective anti-tumor activity.70 CAR T cells directed against the MUC1-N VNTR are now being evaluated in the clinic [Tmmunity Therapeutics; NCT04025216]. MUC1-N is shed from the cancer cell surface and circulates at increased levels in cancer patients, posing potential obstacles for directing anti-MUC1-N CAR T cells to tumors.12 In contrast, MUC1-C is not shed from the surface of cancer cells and the MUC1-C extracellular domain thus represents another potential target for CAR T cells.12 Nonetheless, treating solid tumors with CAR T cells has had limited success to date71 and, based on the present results and those in TNBCs,69 the effects of MUC1-C on suppression of the immune TME could represent a significant challenge for this field. In this regard, targeting the MUC1-C extracellular domain with antibody-drug conjugates72 and the MUC1-C intracellular domain with the GO-203 inhibitor12 provide alternative approaches for killing MUC1-C-expressing tumor cells and reversing the associated immunosuppressive TME73,74 that could be used in combination with other immunotherapeutics.
Supplementary Material
Funding Statement
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under grant numbers [CA97098, CA166480 and CA233084] awarded to D. Kufe and [CA232979] awarded to S. Liu.
Author contributions
Conceptualization, MH, AF, MDL, DK; Methodology, MH, AF, NY, YM; Investigation, MH, AF, NY, YM; Bioinformatics Analysis, AF, HGW, QH, TL, MDL, SL; Writing-Original Draft, D.K.; Writing-Review and Editing, MH, MDL, DK; Funding Acquisition, MO, SL, KKW, DK.
Disclosure statement
DK has equity interests in Genus Oncology, Reata Pharmaceuticals, and HillstreamBioPharma, and is a paid consultant to Reata and CanBas.
Data availability
The accession numbers for the RNA-seq data are GEO Submission GSE139335 and GSE184896.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Puca L, Vlachostergios PJ, Beltran H.. Neuroendocrine differentiation in prostate cancer: emerging biology, models, and therapies. Cold Spring Harb Perspect Med. 2019;9(2):cshperspect.a030593. doi: 10.1101/cshperspect.a030593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davies AH, Beltran H, Zoubeidi A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat Rev Urol. 2018;15(5):271–15. doi: 10.1038/nrurol.2018.22. [DOI] [PubMed] [Google Scholar]
- 3.Aggarwal R, Huang J, Alumkal JJ, Zhang L, Feng FY, Thomas GV, Weinstein AS, Friedl V, Zhang C, Witte ON, et al. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: a multi-institutional prospective study. J Clin Oncol. 2018;36(24):2492–2503. doi: 10.1200/JCO.2017.77.6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, Cieslik M, Benelli M, Robinson D, Van Allen EM, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci USA. 2019;116(23):11428–11436. doi: 10.1073/pnas.1902651116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cha HR, Lee JH, Ponnazhagan S. Revisiting immunotherapy: a focus on prostate cancer. Cancer Res. 2020;80(8):1615–1623. doi: 10.1158/0008-5472.CAN-19-2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nicholson LT, Fong L. Immune checkpoint inhibition in prostate cancer. Trends Cancer. 2020;6(3):174–177. doi: 10.1016/j.trecan.2020.01.003. [DOI] [PubMed] [Google Scholar]
- 7.Antonarakis ES, Piulats JM, Gross-Goupil M, Goh J, Ojamaa K, Hoimes CJ, Vaishampayan U, Berger R, Sezer A, Alanko T, et al. Pembrolizumab for treatment-refractory metastatic castration-resistant prostate cancer: multicohort, open-label Phase II KEYNOTE-199 study. J Clin Oncol. 2020;38(5):395–405. doi: 10.1200/JCO.19.01638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barata P, Agarwal N, Nussenzveig R, Gerendash B, Jaeger E, Hatton W, Ledet E, Lewis B, Layton J, Babiker H, et al. Clinical activity of pembrolizumab in metastatic prostate cancer with microsatellite instability high (MSI-H) detected by circulating tumor DNA. J Immunother Cancer. 2020;8(2):e001065. doi: 10.1136/jitc-2020-001065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stultz J, Fong L. How to turn up the heat on the cold immune microenvironment of metastatic prostate cancer. Prostate Cancer Prostatic Dis. 2021;24(3):697–717. doi: 10.1038/s41391-021-00340-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spranger S, Gajewski TF. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat Rev Cancer. 2018;18(3):139–147. doi: 10.1038/nrc.2017.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao SG, Lehrer J, Chang SL, Das R, Erho N, Liu Y, Sjöström M, Den RB, Freedland SJ, Klein EA, et al. The immune landscape of prostate cancer and nomination of PD-L2 as a potential therapeutic target. J Natl Cancer Inst. 2019;111(3):301–310. doi: 10.1093/jnci/djy141. [DOI] [PubMed] [Google Scholar]
- 12.Kufe D. MUC1-C in chronic inflammation and carcinogenesis; emergence as a target for cancer treatment. Carcinogenesis. 2020;41(9):1173–1183. doi: 10.1093/carcin/bgaa082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li W, Zhang N, Jin C, Long MD, Rajabi H, Yasumizu Y, Fushimi A, Yamashita N, Hagiwara M, Zheng R, et al. MUC1-C drives stemness in progression of colitis to colorectal cancer. JCI Insight. 2020;5(12):137112. doi: 10.1172/jci.insight.137112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lapointe J, Li C, Higgins JP, van de Rijn M, Bair E, Montgomery K, Ferrari M, Egevad L, Rayford W, Bergerheim U, et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc Natl Acad Sci USA. 2004;101(3):811–816. doi: 10.1073/pnas.0304146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andrén O, Fall K, Andersson SO, Rubin MA, Bismar TA, Karlsson M, Johansson J-E, Mucci LA. MUC-1 gene is associated with prostate cancer death: a 20-year follow-up of a population-based study in Sweden. Br J Cancer. 2007;97(6):730–734. doi: 10.1038/sj.bjc.6603944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eminaga O, Wei W, Hawley SJ, Auman H, Newcomb LF, Simko J, Hurtado-Coll A, Troyer DA, Carroll PR, Gleave ME, et al. MUC1 expression by immunohistochemistry is associated with adverse pathologic features in prostate cancer: a multi-institutional study. PLoS One. 2016;11(11):e0165236. doi: 10.1371/journal.pone.0165236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Genitsch V, Zlobec I, Thalmann GN, Fleischmann A. MUC1 is upregulated in advanced prostate cancer and is an independent prognostic factor. Prostate Cancer Prostatic Dis. 2016;19(3):242–247. doi: 10.1038/pcan.2016.11. [DOI] [PubMed] [Google Scholar]
- 18.Lin X, Gu Y, Kapoor A, Wei F, Aziz T, Ojo D, Jiang Y, Bonert M, Shayegan B, Yang H, et al. Overexpression of MUC1 and genomic alterations in its network associate with prostate cancer progression. Neoplasia. 2017;19(11):857–867. doi: 10.1016/j.neo.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yasumizu Y, Rajabi H, Jin C, Hata T, Pitroda S, Long MD, Hagiwara M, Li W, Hu Q, Liu S, et al. MUC1-C drives lineage plasticity in progression to neuroendocrine prostate cancer. Nat Commun. 2020;11(1):338. doi: 10.1038/s41467-019-14219-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takahashi K, Yamanaka S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat Rev Mol Cell Biol. 2016;17(3):183–193. doi: 10.1038/nrm.2016.8. [DOI] [PubMed] [Google Scholar]
- 21.Hagiwara M, Yasumizu Y, Yamashita N, Rajabi H, Fushimi A, Long MD, Li W, Bhattacharya A, Ahmad R, Oya M, et al. MUC1-C activates the BAF (mSWI/SNF) complex in prostate cancer stem cells. Cancer Res. 2021;81(4):1111–1122. doi: 10.1158/0008-5472.CAN-20-2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hagiwara M, Fushimi A, Yamashita N, Battacharya A, Rajabi H, Long M, Yasumizu Y, Oya M, Liu S, Kufe D. MUC1-C activates the PBAF chromatin remodeling complex in integrating redox balance with progression of human prostate cancer stem cells. Oncogene. 2021;40(30):4920–4940. doi: 10.1038/s41388-021-01899-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhattacharya B, Fushimi A, Yamashita N, Hagiwara M, Morimoto Y, Rajabi H, Long MD, Abdulla M, Ahmad R, Street K, et al. MUC1-C dictates JUN and BAF-mediated chromatin remodeling at enhancer signatures in cancer stem cells. Mol Cancer Res. In Press, 2022;molcanres.MCR-21-0672-E.2021. doi: 10.1158/1541-7786.MCR-21-0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith BA, Balanis NG, Nanjundiah A, Sheu KM, Tsai BL, Zhang Q, Park JW, Thompson M, Huang J, Witte ON, et al. A human adult stem cell signature marks aggressive variants across epithelial cancers. Cell Rep. 2018;24(12):3353–66.e5. doi: 10.1016/j.celrep.2018.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Angelis ML, Francescangeli F, La Torre F, Zeuner A. Stem cell plasticity and dormancy in the development of cancer therapy resistance. Front Oncol. 2019;9:626. doi: 10.3389/fonc.2019.00626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miranda A, Hamilton PT, Zhang AW, Pattnaik S, Becht E, Mezheyeuski A, Bruun J, Micke P, de Reynies A, Nelson BH, et al. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc Natl Acad Sci USA. 2019;116(18):9020–9029. doi: 10.1073/pnas.1818210116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malta TM, Sokolov A, Gentles AJ, Burzykowski T, Poisson L, Weinstein JN, Kamińska B, Huelsken J, Omberg L, Gevaert O, et al. Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell. 2018;173(2):338–54 e15. doi: 10.1016/j.cell.2018.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hepburn AC, Steele RE, Veeratterapillay R, Wilson L, Kounatidou EE, Barnard A, Berry P, Cassidy JR, Moad M, El-Sherif A, et al. The induction of core pluripotency master regulators in cancers defines poor clinical outcomes and treatment resistance. Oncogene. 2019;38(22):4412–4424. doi: 10.1038/s41388-019-0712-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miao Y, Yang H, Levorse J, Yuan S, Polak L, Sribour M, Singh B, Rosenblum MD, Fuchs E. Adaptive immune resistance emerges from tumor-initiating stem cells. Cell. 2019;177(5):1172–86 e14. doi: 10.1016/j.cell.2019.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quintanal-Villalonga A, Chan JM, Yu HA, Pe’er D, Sawyers CL, Sen T, Rudin CM. Lineage plasticity in cancer: a shared pathway of therapeutic resistance. Nat Rev Clin Oncol. 2020;17(6):360–371. doi: 10.1038/s41571-020-0340-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol. 2015;109(1):1–9. doi: 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mounir M, Lucchetta M, Silva TC, Olsen C, Bontempi G, Chen X, Noushmehr H, Colaprico A, Papaleo E. New functionalities in the TCGAbiolinks package for the study and integration of cancer data from GDC and GTEx. PLoS Comput Biol. 2019;15(3):e1006701. doi: 10.1371/journal.pcbi.1006701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18(1):220. doi: 10.1186/s13059-017-1349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu L, Deng C, Pang B, Zhang X, Liu W, Liao G, Yuan H, Cheng P, Li F, Long Z, et al. TIP: a web server for resolving tumor immunophenotype profiling. Cancer Res. 2018;78(23):6575–6580. doi: 10.1158/0008-5472.CAN-18-0689. [DOI] [PubMed] [Google Scholar]
- 36.He MX, Cuoco MS, Crowdis J, Bosma-Moody A, Zhang Z, Bi K, Kanodia A, Su M-J, Ku S-Y, Garcia MM, et al. Transcriptional mediators of treatment resistance in lethal prostate cancer. Nat Med. 2021;27(3):426–433. doi: 10.1038/s41591-021-01244-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Dijk D, Sharma R, Nainys J, Yim K, Kathail P, Carr AJ, Burdziak C, Moon KR, Chaffer CL, Pattabiraman D, et al. Recovering gene interactions from single-cell data using data diffusion. Cell. 2018;174(3):716–29 e27. doi: 10.1016/j.cell.2018.05.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol. 2015;33(5):495–502. doi: 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aibar S, Gonzalez-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine J-C, Geurts P, Aerts J, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods. 2017;14(11):1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cancer Genome Atlas Research Network, The molecular taxonomy of primary prostate cancer. Cell. 2015;163(4):1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheon H, Holvey-Bates EG, Schoggins JW, Forster S, Hertzog P, Imanaka N, Rice CM, Jackson MW, Junk DJ, Stark GR, et al. IFNbeta-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. 2013;32(20):2751–2763. doi: 10.1038/emboj.2013.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheon H, Borden EC, Stark GR. Interferons and their stimulated genes in the tumor microenvironment. Semin Oncol. 2014;41(2):156–173. doi: 10.1053/j.seminoncol.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stark GR, Cheon H, Wang Y. Responses to cytokines and interferons that depend upon JAKs and STATs. Cold Spring Harb Perspect Biol. 2018;10(1):a028555. doi: 10.1101/cshperspect.a028555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vierbuchen T, Ling E, Cowley CJ, Couch CH, Wang X, Harmin DA, Roberts CWM, Greenberg ME. AP-1 transcription factors and the BAF complex mediate signal-dependent enhancer selection. Mol Cell. 2017;68(6):1067–82 e12. doi: 10.1016/j.molcel.2017.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Singh S, Kumar S, Srivastava RK, Nandi A, Thacker G, Murali H, Kim S, Baldeon M, Tobias J, Blanco MA, et al. Loss of ELF5-FBXW7 stabilizes IFNGR1 to promote the growth and metastasis of triple-negative breast cancer through interferon-gamma signalling. Nat Cell Biol. 2020;22(5):591–602. doi: 10.1038/s41556-020-0495-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hata T, Rajabi H, Takahashi H, Yasumizu Y, Li W, Jin C, Long MD, Hu Q, Liu S, Fushimi A, et al. MUC1-C activates the NuRD complex to drive dedifferentiation of triple-negative breast cancer cells. Cancer Res. 2019;79(22):5711–5722. doi: 10.1158/0008-5472.CAN-19-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murtas D, Maric D, De Giorgi V, Reinboth J, Worschech A, Fetsch P, Filie A, Ascierto ML, Bedognetti D, Liu Q, et al. IRF-1 responsiveness to IFN-gamma predicts different cancer immune phenotypes. Br J Cancer. 2013;109(1):76–82. doi: 10.1038/bjc.2013.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qin Q, Fan J, Zheng R, Wan C, Mei S, Wu Q, Sun H, Brown M, Zhang J, Meyer CA, et al. Lisa: inferring transcriptional regulators through integrative modeling of public chromatin accessibility and ChIP-seq data. Genome Biol. 2020;21(1):32. doi: 10.1186/s13059-020-1934-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prendergast GC, Malachowski WP, DuHadaway JB, Muller AJ. Discovery of IDO1 inhibitors: from bench to bedside. Cancer Res. 2017;77(24):6795–6811. doi: 10.1158/0008-5472.CAN-17-2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adam I, Dewi DL, Mooiweer J, Sadik A, Mohapatra SR, Berdel B, Keil M, Sonner JK, Thedieck K, Rose AJ, et al. Upregulation of tryptophanyl-tRNA synthethase adapts human cancer cells to nutritional stress caused by tryptophan degradation. Oncoimmunology. 2018;7(12):e1486353. doi: 10.1080/2162402X.2018.1486353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ahn YH, Oh SC, Zhou S, Kim TD. Tryptophanyl-tRNA synthetase as a potential therapeutic target. Int J Mol Sci. 2021;22(9):4523. doi: 10.3390/ijms22094523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu AA, Drake V, Huang HS, Chiu S, Zheng L. Reprogramming the tumor microenvironment: tumor-induced immunosuppressive factors paralyze T cells. Oncoimmunology. 2015;4(7):e1016700. doi: 10.1080/2162402X.2015.1016700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, Tsoucas D, Qiu X, Lim K, Rao P, et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science. 2018;359(6377):770–775. doi: 10.1126/science.aao1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miao D, Margolis CA, Gao W, Voss MH, Li W, Martini DJ, Norton C, Bossé D, Wankowicz SM, Cullen D, et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science. 2018;359(6377):801–806. doi: 10.1126/science.aan5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu XD, Kong W, Peterson CB, McGrail DJ, Hoang A, Zhang X, Lam T, Pilie PG, Zhu H, Beckermann KE, et al. PBRM1 loss defines a nonimmunogenic tumor phenotype associated with checkpoint inhibitor resistance in renal carcinoma. Nat Commun. 2020;11(1):2135. doi: 10.1038/s41467-020-15959-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Michalska A, Blaszczyk K, Wesoly J, Bluyssen HAR. A positive feedback amplifier circuit that regulates interferon (IFN)-stimulated gene expression and controls type I and type II IFN responses. Front Immunol. 2018;9:1135. doi: 10.3389/fimmu.2018.01135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sandy Z, Da Costa IC, Schmidt CK. More than meets the ISG15: emerging roles in the DNA damage response and beyond. Biomolecules. 2020;10(11):1557. doi: 10.3390/biom10111557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen RH, Xiao ZW, Yan XQ, Han P, Liang FY, Wang JY, Yu S-T, Zhang T-Z, Chen S-Q, Zhong Q, et al. Tumor cell-secreted ISG15 promotes tumor cell migration and immune suppression by inducing the macrophage M2-like phenotype. Front Immunol. 2020;11:594775. doi: 10.3389/fimmu.2020.594775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24(10):1550–1558. doi: 10.1038/s41591-018-0136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiang L, Wang YJ, Zhao J, Uehara M, Hou Q, Kasinath V, Ichimura T, Banouni N, Dai L, Li X, et al. Direct tumor killing and immunotherapy through anti-SerpinB9 therapy. Cell. 2020;183(5):1219–33 e18. doi: 10.1016/j.cell.2020.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol. 2011;29(1):71–109. doi: 10.1146/annurev-immunol-031210-101312. [DOI] [PubMed] [Google Scholar]
- 62.Aldinucci D, Borghese C, Casagrande N. The CCL5/CCR5 axis in cancer progression. Cancers (Basel). 2020;12(7):1765. doi: 10.3390/cancers12071765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi BVSK, Varambally S, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;22(3):298–305. doi: 10.1038/nm.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mojic M, Takeda K, Hayakawa Y. The dark side of IFN-gamma: its role in promoting cancer immunoevasion. Int J Mol Sci. 2017;19(1):89. doi: 10.3390/ijms19010089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Castro F, Cardoso AP, Goncalves RM, Serre K, Oliveira MJ. Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front Immunol. 2018;9:847. doi: 10.3389/fimmu.2018.00847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li J, Wang W, Zhang Y, Cieslik M, Guo J, Tan M, Green MD, Wang W, Lin H, Li W, et al. Epigenetic driver mutations in ARID1A shape cancer immune phenotype and immunotherapy. J Clin Invest. 2020;130(5):2712–2726. doi: 10.1172/JCI134402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao S, Choi M, Overton JD, Bellone S, Roque DM, Cocco E, Guzzo F, English DP, Varughese J, Gasparrini S, et al. Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc Natl Acad Sci U S A. 2013;110(8):2916–2921. doi: 10.1073/pnas.1222577110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mohiuddin IS, Wei SJ, Kang MH. Role of OCT4 in cancer stem-like cells and chemotherapy resistance. Biochim Biophys Acta Mol Basis Dis. 2019;1866 (4) :165432. doi: 10.1016/j.bbadis.2019.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yamashita N, Long M, Fushimi A, Yamamoto M, Hata T, Hagiwara M, Bhattacharya A, Hu Q, Wong K-K, Liu S, et al. MUC1-C integrates activation of the IFN-gamma pathway with suppression of the tumor immune microenvironment in triple-negative breast cancer. JImmunother Cancer. 2021;9(1):e002115. doi: 10.1136/jitc-2020-002115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Taylor-Papadimitriou J, Burchell JM, Graham R, Beatson R. Latest developments in MUC1 immunotherapy. Biochem Soc Trans. 2018;46(3):659–668. doi: 10.1042/BST20170400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miao L, Zhang Z, Ren Z, Li Y. Reactions related to CAR-T cell therapy. Front Immunol. 2021;12:663201. doi: 10.3389/fimmu.2021.663201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Panchamoorthy G, Jin C, Raina D, Bharti A, Yamamoto M, Adeebge D, Zhao Q, Bronson R, Jiang S, Li L, et al. Targeting the human MUC1-C oncoprotein with an antibody-drug conjugate. JCI Insight. 2018;3(12):e99880. doi: 10.1172/jci.insight.99880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bouillez A, Adeegbe D, Jin C, Hu X, Tagde A, Alam M, Rajabi H, Wong K-K, Kufe D. MUC1-C promotes the suppressive immune microenvironment in non-small cell lung cancer. Oncoimmunology. 2017;6(9):e1338998. doi: 10.1080/2162402X.2017.1338998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Maeda T, Hiraki M, Jin C, Rajabi H, Tagde A, Alam M, Bouillez A, Hu X, Suzuki Y, Miyo M, et al. MUC1-C induces PD-L1 and immune evasion in triple-negative breast cancer. Cancer Res. 2018;78(1):205–215. doi: 10.1158/0008-5472.CAN-17-1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the RNA-seq data are GEO Submission GSE139335 and GSE184896.