

Abstract
Iontophoresis is an electrical current-based, non-invasive drug delivery technology, which is particularly suitable for intraocular drug delivery. Current ocular iontophoresis devices use low current intensities that significantly limit macromolecule and nanoparticle (NP) delivery efficiency. Increasing current intensity leads to ocular tissue damage due to Joule heating and electrochemical (EC) reactions. Here, we describe a hydrogel ionic circuit (HIC)-based iontophoresis device for high-efficiency intraocular macromolecule and NP delivery. Our HIC-based device is capable of minimizing Joule heating, effectively buffering EC reaction-generated pH changes, and absorbing electrode overpotential-induced heating. As a result, our device allows safe application of high current intensities (~87 mA cm−2, more than 10 times higher than current ocular iontophoresis devices) to the eye with minimal ocular cell death and tissue damage. Our high-intensity iontophoresis significantly enhances intraocular macromolecule and NP delivery to both anterior and posterior segments by up to 300 times compared to conventional iontophoresis. Therapeutically effective concentrations of bevacizumab and dexamethasone are delivered to target tissue compartments within 10–20 min of iontophoresis application. Our studies highlight the significant safety enhancement enabled by a HIC-based device design and the potential of our device to deliver therapeutic doses of macromolecule and NP ophthalmic drugs within a clinically relevant time frame.
Keywords: Iontophoresis, Ocular drug delivery, Ionic circuit, Hydrogel electrode, Aqueous two-phase systems
Graphical Abstract
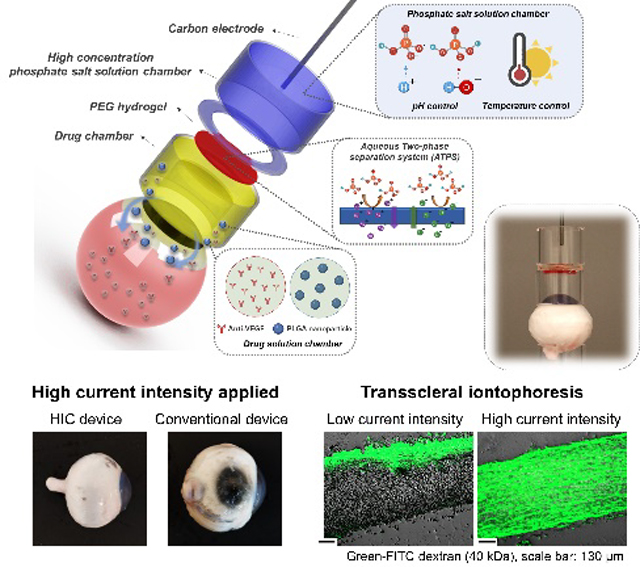
A hydrogel ionic circuit (HIC) based iontophoresis device is developed for highly effective intraocular delivery of macromolecules and nanoparticle drugs. The HIC-based device allows safe application of high current intensities (~87 mA cm−2) to the ocular tissue. Moreover, therapeutically effective concentrations of bevacizumab and dexamethasone (nanoparticle formula) can be delivered to target tissues within 10–20 min of iontophoresis application.
1. Introduction
Macromolecular and NP ophthalmic drugs have seen increasing utility in ocular disease treatment. Monoclonal antibodies against human vascular endothelial growth factor (anti-VEGF, e.g. ranibizumab and bevacizumab), have been successfully used in treating retinal and corneal vascular diseases[1, 2]. NP ophthalmic formula provides sustained drug release and the ability to target specific tissues through surface modification[3]. Several NP ophthalmic drugs have been FDA-approved for treating dry eye syndrome and for photodynamic therapy[4].
Despite the promising therapeutic efficacy of macromolecular and NP ophthalmic drugs, their intraocular delivery presents a significant challenge. The large sizes of macromolecular and NP drugs slow their permeation in ocular tissues compared to small-molecule drugs[5]. When eye drops are used, only a small fraction (typically <1%) of the applied macromolecular and NP drugs is delivered into the eye even with multiple doses per day[6]. Although drug-eluting contact lenses effectively increase drug residence time, they do not increase drug permeation rate. A ranibizumab-eluting contact lens failed to deliver a therapeutic concentration of ranibizumab into the posterior segment despite extended wear for several days[7]. Systemically administered macromolecules and NPs need to overcome the blood-aqueous/retinal barriers, and are subject to liver modification and kidney clearance[1], leading to a low bioavailability of less than 0.1%[8]. Due to the challenges of topical and systemic routes, intraocular injection remains the most effective method for delivering macromolecules and NPs. However, the injection procedure carries a risk of potentially blinding complications, including endophthalmitis, retinal detachment, and cataract[9]. Needle phobia can cause anxiety and reduce patient compliance[10]. Moreover, intraocular injection needs to be performed by a specialist. The uneven distribution of specialists between rural and urban areas[11] and between developing and developed countries[12] leads to a disparate treatment provision. There is a critical need for a next-generation intraocular macromolecule and NP delivery technology that is safe, non-invasive, highly efficacious, and allows easy operation by patients or caregivers without special trainings.
Iontophoresis is a constant (DC) electrical current-based, non-invasive drug delivery technology. It can be easily applied using portable or wearable devices[13]. The utility of iontophoresis in ocular drug delivery has been explored[14]. The basic design of an ocular iontophoretic device consists of a continuous DC power source and two electrodes: the drug-loading or donor electrode (i.e. iontophoresis applicator) and the counter electrode. The drug-loading electrode is filled with drug solution and placed on the eye. The counter electrode is placed at a distal site on the body surface, such as the forehead and the neck, to close the circuit. At present, Ocuphor™ (IOMED Inc., Salt Lake City, Utah, USA), Eyegate II (EyeGate Pharmaceuticals Inc., Waltham, MA, USA), and Visulex (Aciont Inc., Salt Lake City, Utah, USA) are the major iontophoresis devices that are being developed for ocular drug delivery applications[15]. The OcuPhor™ Delivery System has been tested for transscleral iontophoretic delivery of amikacin using a current intensity up to 4 mA and a duration of 20 minutes[16]. The drug-loading electrode is a silicon shell that contains a silver-silver chloride conductive component and a hydrogel sponge as a drug reservoir. The drug-loading electrode is placed on the sclera under the lower eyelid and the counter electrode is placed on the back or the neck[16]. Eyegate II Delivery System has been used for transscleral delivery of small-molecule ophthalmic drugs (e.g. dexamethasone phosphate). The cup-shaped drug-loading electrode consists of an annular silicone probe with a 0.5 cm2 contact area and a 13 mm inner diameter[17]. The drug-loading electrode is placed on the eye, and the counter electrode is placed on the forehead. Eyegate II uses a current intensity up to 3.5 mA and an iontophoresis duration of 4 min[18]. The Visulex™ Delivery System has a cup-shaped drug-loading electrode. It has been tested for transscleral delivery of macromolecules (e.g. immunoglobulin G and bevacizumab) using a current intensity of 3 mA (1.4 mA cm−2) and a duration of 20 minutes[19].
Although effective in delivering small molecules[20], current ocular iontophoresis has low efficiency when delivering macromolecules and NPs due to their larger sizes. Pescina et al. reported that an anodal iontophoresis at 3.8 mA cm−2 achieved a transscleral bevacizumab permeation rate of 37.01 μg cm−2 per hour. To achieve the dose delivered by intravitreal injection (IVI), which is 600 μg[21], 10–20 hours of iontophoresis application is required. Jae Hwan Jung et al. reported that a 0.7 mA iontophoresis only enhanced the penetration of 20 nm NPs into rabbit eyes by 34% compared to passive diffusion[22].
Increasing current intensity can increase iontophoretic drug delivery efficiency[23]. However, applying high current intensities presents a significant challenge for current ocular iontophoresis devices. All current devices conduct electron currents, which have to be converted to ion currents at the device/tissue interface. For DC current, this conversion requires electrochemical (EC) reactions[24]. These EC reactions can induce significant pH changes (due to water electrolysis[25]) and local heating (due to electrode overpotential[26]) that can damage ocular tissues when the current intensity is high[27]. Therefore, current ocular iontophoresis studies typically use current intensities that do not exceed 7.5 mA cm−2 [13]. Zhang et al. reported significant ocular damage when 15 mA cm−2 was used[28].
A potential solution to this issue is the newly developed ionically conductive materials. They conduct ion currents, so when they are used to construct the iontophoresis device, EC reaction-based current conversion does not happen at the device/tissue interface. These materials include hydrogels containing high-concentration NaCl/LiCl[29], ionic liquid hydrogels[30], and polyelectrolyte hydrogels[31]. However, NaCl/LiCl-containing hydrogels are not stable in aqueous tissue environments due to ion diffusion[32]. Polyelectrolytes and ionic liquids have low conductivity (in general <20 mS cm−1)[31, 33], which can induce significant Joule heating to damage tissue when current intensity is high. Ionic liquids also have cyto- and tissue-toxicity[34]. Therefore, these materials are not suitable for ocular iontophoresis at high current intensities.
To address the issues of current ionic conductors, we developed a novel ion current-conducting circuit device, dubbed hydrogel ionic circuit (HIC)[35]. In HIC, fluidic channels filled with saturated phosphate salt solutions with a conductivity up to 51.31 mS cm−1 conduct ion currents with low Joule heating. Importantly, these phosphate salt solution channels are encapsulated in a polyethylene glycol (PEG) hydrogel matrix. The PEG hydrogel formed an aqueous two-phase separation (ATPS) with the phosphate salt solutions, which prevented phosphate ions from diffusing into the PEG hydrogel or the surrounding environment while still permitting the transmission of ion current. This allowed our HIC device to remain stable in an aqueous tissue environment and to route ion currents to biological tissues without inducing EC reactions at the device/tissue interface. We therefore hypothesize that an HIC-based ocular iontophoresis system can safely apply high-intensity ion currents to enhance drug delivery efficiency without causing tissue damage.
In this paper, we developed an HIC-based ocular iontophoresis device for high-efficiency intraocular delivery of macromolecules and NPs. Our device exhibited long-term stability in aqueous environments. The high-concentration phosphate salt solutions in our device had minimal impact on the ocular tissue directly interfacing with our device. The optimal device design for minimizing temperature and pH changes during high-intensity current application was determined through mathematical, simulation and experimental methods. The safety of our optimal device design was verified using in vitro ocular cell cultures and rabbit whole eyes. Importantly, the ability to safely apply high-intensity currents allowed our HIC-based device to significantly enhance the intraocular delivery of macromolecules and NPs, compared to conventional iontophoresis devices. Dextran (40 kDa), bevacizumab and a NP formula of dexamethasone (DEX) were used as model drugs in our drug delivery tests. Therapeutically effective concentrations of bevacizumab and DEX were delivered into target ocular tissues within 10–20 min.
2. Concept and design of HIC-based ocular iontophoresis device
Figure 1 showed the working mechanism and design of our HIC-based ocular iontophoresis device. During iontophoresis, the electron current from a DC current source was converted to ion current at the current source/HIC interface (at the carbon electrode) through EC reactions. The high-concentration phosphate salt solutions in our HIC device served three important roles: 1) they effectively buffered the pH changes induced by EC reactions due to their higher concentration of HPO42−/H2PO4− ions than physiological phosphate-buffered saline (PBS); 2) they absorbed the heat generated by the overpotential of EC reactions; and 3) they conducted ion currents with low Joule heating due to their high conductivities. The ion currents were then transmitted to the drug chamber through a PEG hydrogel membrane to induce iontophoretic drug flux. The PEG hydrogel formed an ATPS with the phosphate salt solutions. As shown in Figure 1, the HIC-based drug-loading device was attached to the front of the eye, and the counter device was attached to the back of the eye in our ex vivo drug delivery experiments to complete the circuit. In future in vivo studies, the counter device can be placed at a distal site on the body surface (e.g. forehead, neck, ear, etc.).
Figure 1.
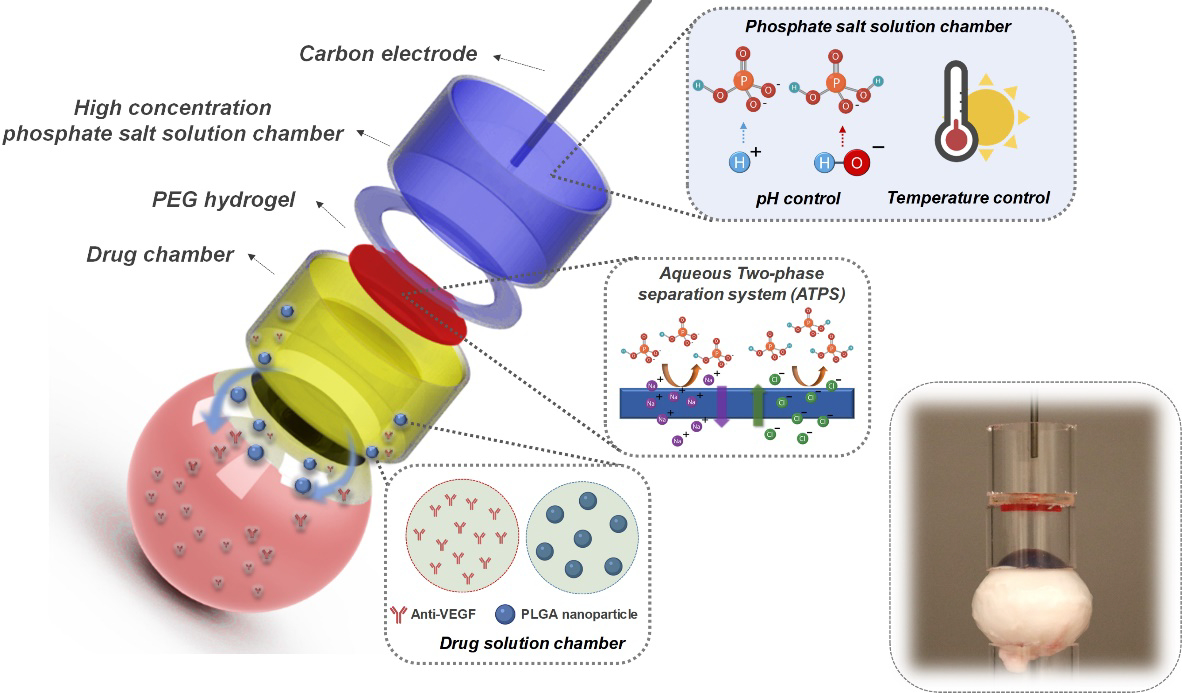
Schematic of the hydrogel ionic circuit (HIC)-based high intensity iontophoresis device for intraocular delivery of macromolecules and NPs. The inset shows a digital camera image of the actual device. The drug-loading device was attached to the front of the eye. The counter device was attached to the back of the eye.
2.1. Minimization of EC reaction-induced pH changes
The Faradaic charge transfer between electron current-conducting electrodes and ion current-conducting media decomposes water molecules and generates pH changes. Although non-polarizable electrodes (e.g. Ag/AgCl) can transfer charges through reactions between electrode material and soluble ions without decomposing water[36], the electrode material (e.g. AgCl) may be quickly depleted during high-intensity DC current application, after which water decomposition can still happen[37]. In this section, we estimated the pH changes induced by high-intensity current application in our device and the amount of HPO42−/H2PO4− ions that was required to buffer these changes to maintain a safe ocular pH of 6.5 to 8.5[38]. We assumed that all electron transferred at the current source/HIC interface were used to decompose water molecules[39]. The amount of HPO42−/H2PO4− ions required to buffer these changes was then calculated using the Henderson-Hasselbalch equation: pH = pKa + log (Cs/Cas), where pKa was the negative logarithm of the acid dissociation constant for the phosphate salt, and Cs and Cas were molarities of the weak acid and its conjugate base (Supporting Information). Using a 100 mA, 15 min iontophoresis as an example, a total of 5.62 × 1020 hydrogen ions and hydroxide ions would be generated at the anode and cathode interfaces, respectively. 0.9 mmol disodium phosphate and sodium dihydrogen phosphate would be required to react with these hydrogen/hydroxide ions to maintain an ocular surface pH between 6.5 and 8.5. This method was used to calculate the volume of anode and cathode phosphate salt solutions in our ex vivo safety experiments.
2.2. Minimization of overpotential and Joule heating induced temperature increase
Two sources of heat were involved in our HIC-based iontophoresis device: the electrode overpotential-induced heat at the current source/HIC interface (Supporting Information) and the Joule heating. In this section, we estimated the heat generation during iontophoresis using a computer-aided finite-element analysis (FEA) (Supporting Information). Our FEA results highlighted two important functions of the high-concentration phosphate salt solutions in our device in temperature control: reducing Joule heating due to their high conductivity and absorbing overpotential-induced heat. Our goal was to keep the ocular surface temperature below 43°C to minimize thermal damage to ocular tissues[40].
The FEA results (Figure S1A, Supporting Information) showed the temperature distribution in our device and the eye immediately after a 100 mA, 15 min iontophoresis. With the phosphate salt solution in the device, limited Joule heating was induced and the temperature on the ocular surface was kept below 43°C. In contrast, when the phosphate salt solution was replaced with 1X PBS, higher Joule heat was generated and the ocular surface temperature increased to 47.84°C (Figure S1B, Supporting Information). These results indicated that the high-concentration phosphate salt solutions in our device effectively reduced Joule heating and prevented overheating on ocular surface. Also noted in Figure S1A (Supporting Information) was that the overpotential-induced heat was concentrated around the carbon electrode and did not affect the eye. This indicated that the volume of the phosphate salt solution used in the FEA was sufficient to absorb overpotential-induced heat. In conventional ocular iontophoresis devices, the metal/carbon electrodes were in direct contact with the drug solution[19], which typically had a volume of less than 1 mL[41]. This small liquid volume in conventional devices was not able to absorb the overpotential-induced heat and the ocular surface temperature increased to 52.76°C (Figure S1C, Supporting Information). It should be noted our FEA used a simplified model that did not account for secondary effects of EC reactions, such as the gas bubble generation and the fluid flow within phosphate salt solutions. These effects may lead to higher Joule heating or faster heat transfer to the ocular surface. Therefore, the volume of the phosphate salt solution required to maintain a safe ocular surface temperature was experimentally determined in our ex vivo safety studies.
3. Safety evaluation of HIC-based ocular iontophoresis device
3.1. The stability of phosphate/PEG ATPS
In our device, the ATPS served a critical role to minimize the out-diffusion of high-concentration phosphate salt ions, so they did not affect the drug solution or the ocular tissues. In this section, we examined the long-term stability of the phosphate-PEG hydrogel ATPS in an aqueous tissue environment. Our PEG hydrogel was prepared by photocrosslinking of a precursor solution containing 10% w/w poly(ethylene glycol) dimethacrylate (PEGDMA, molecular weight (MW): 8000) and 5% w/w poly(ethylene glycol) diacrylate (PEGDA, MW: 700). This PEG formula exceeded the minimal PEG concentration for the formation of phosphate-PEG ATPS[42] (Figure S2–S4, Supporting Information) and provided sufficient mechanical strength to support the weight of phosphate salt solution in our device (Figure S5, Supporting Information). Saturated Na2HPO4 solution (0.6 M, pH = 9.0) was used in the anode device to buffer the hydrogen ions generated by anode reactions (Figure S6A, Supporting Information). To buffer the hydroxide ions generated by cathode reactions, a NaH2PO4 solution was required. However, high-concentration NaH2PO4 solution had a pH that was much lower than 6.5 (Figure S6B, Supporting Information), which might cause ocular tissue damage. Therefore, a mixture solution containing 0.6 M NaH2PO4 and 0.48 M Na2HPO4 was used in our cathode device, which had a pH of 6.4.
To evaluate ATPS stability, we first immersed our PEG hydrogel in anode and cathode phosphate salt solutions and measured its conductivity over a 2-week period. The PEG hydrogel maintained a conductivity that was 6–12 times lower than the phosphate salt solutions with less than 5% fluctuation during the 2-week period (Figure 2A–B), indicating a stable ATPS. We next showed that once the ATPS was formed, the high-concentration phosphate salt solutions in our device had minimal impact on the surrounding environment. We immersed our HIC-based anode and cathode devices in 1X PBS for 1 hour, and monitored the conductivity and pH of the phosphate salt solutions in our device and the PBS. The conductivity of the phosphate salt solutions in our device and the PBS changed less than 6% (Figure 2C–D). The pH of the phosphate salt solutions in our device had less than 1% change (Figure 2E). The pH of PBS had more profound changes, but they remained between 6.5 and 8.5 (Figure 2F). We tried to replace the phosphate salt solutions in our device with 3 M NaCl, which did not form ATPS with PEG hydrogel, and repeated our experiment. After immersing our device in 1X PBS for 1 hour, the conductivity of PBS increased by over 90% (Figure 2C gray column), and the conductivity of NaCl solution in the device decreased by over 10% (Figure 2D gray line), indicating the critical role of ATPS. Finally, we demonstrated that our device had minimal impact on cell viability due to ATPS (Figure 2G). After immersing our device in in vitro cultures of corneal epithelium (Human Pr. HCEC) and endothelium cells (BCE C/D-1b), retinal pigmented epithelium cells (RPE-19), and choroid/retina endothelium cells (RF/6A) for 1 hour, the viability of all four cell types was minimally affected.
Figure 2.

ATPS between PEG hydrogel and high-concentration phosphate salt solutions. (A) The electrical conductivity of PEG hydrogel compared to that of high-concentration phosphate salt solutions after soaking PEG hydrogel in anode and cathode phosphate salt solutions for 24 h. (B) Long-term stability of the electrical conductivity of PEG hydrogel when soaked in anode and cathode phosphate salt solutions for two weeks. (C) The change of the electrical conductivity of the PBS after immersing our HIC-based anode and cathode devices loaded with phosphate salt solutions and our HIC device loaded with 3M NaCl in PBS for 60 min. (D) The change of the electrical conductivity of the phosphate salt solutions loaded in our HIC-based anode (blue line) and cathode (green line) devices when the devices were immersed in PBS for 60 min. The gray line showed the change of the electrical conductivity of 3M NaCl loaded in our HIC device when the device was immersed in PBS for 60 min. (E-F) pH changes of the high-concentration phosphate salt solutions loaded in our HIC-based device and the PBS when immersing our device in PBS for 60 min. (G) Cell viabilities (normalized to untreated control) of four types of ocular cells after immersing HIC-based device in their cell culture media for 1 h (scale bar: 200 μm).
3.2. The ex vivo safety of high-intensity ion current application
In this section, we evaluated the safety of high-intensity ion current application by our HIC-based devices. Our HIC-based anode or cathode device under investigation was amounted on the equator of an isolated porcine eye (Figure 3A). The drug chamber was filled with 1X PBS. A conventional ocular iontophoresis device constructed by directly inserting a carbon electrode in the drug chamber was tested as a comparison (Figure 3B). A high current of 100 mA (65 mA cm−2, 8.6 times higher than typical ocular iontophoresis current density) was applied for 15 min. During the test, the average peak temperature on the surface of the eye treated by our device remained below 43°C (Figure 3C). In contrast, the conventional iontophoresis device increased ocular surface temperature up to 63 ± 5 °C on the anode side and 48 ± 4 °C on the cathode side (Figure 3C). The cumulative equivalent exposure minutes at 43°C (CEM43) calculated from the temperature-versus-time data has been used as a reliable determinant of thermal damage to biological tissues[40, 43]. Dewhirst et al. reported that the threshold CEM43 for inducing significant thermal damage in retina and cornea was 40 minutes[43]. Using the temperature-versus-time data (Figure S7, Supporting Information), we calculated the average CEM43 induced by our HIC-based iontophoresis anode and cathode devices in our ex vivo safety test, which were 2.04 and 0.04 minutes, respectively. The conventional iontophoresis anode and cathode devices had an average CEM43 of 7.75 × 108 and 166.73 minutes. The CEM43 induced by our HIC-based devices was lower than the threshold for significant thermal damage, while the CEM43 induced by conventional iontophoresis devices was considerably higher than the threshold. These results showed that our HIC-based device was capable of maintaining thermal safety during high-intensity iontophoresis. These results showed that our HIC-based device was capable of reducing temperature increase during high-intensity iontophoresis. We also measured the pH in the drug chamber, on the ocular surface, and in the vitreous of the eye immediately after current application (Figure 3D–F). When HIC-based device was used, the pH at these three locations all remained between 6.5 and 8.5. The conventional iontophoresis device, however, changed the pH at the three locations to below 3 on the anode side and above 11.5 on the cathode side. There results demonstrated the ability of our HIC-based device to maintain a safe pH in drug chamber and ocular tissue during high-intensity iontophoresis. Figure 3G showed photographs of the porcine eyes taken after high-intensity current application at 100 mA for 15 min. The conventional iontophoresis device induced severe ocular tissue damage especially at the device contact area due to chemical/thermal burns, which did not happen to the eye treated with HIC-based device.
Figure 3.
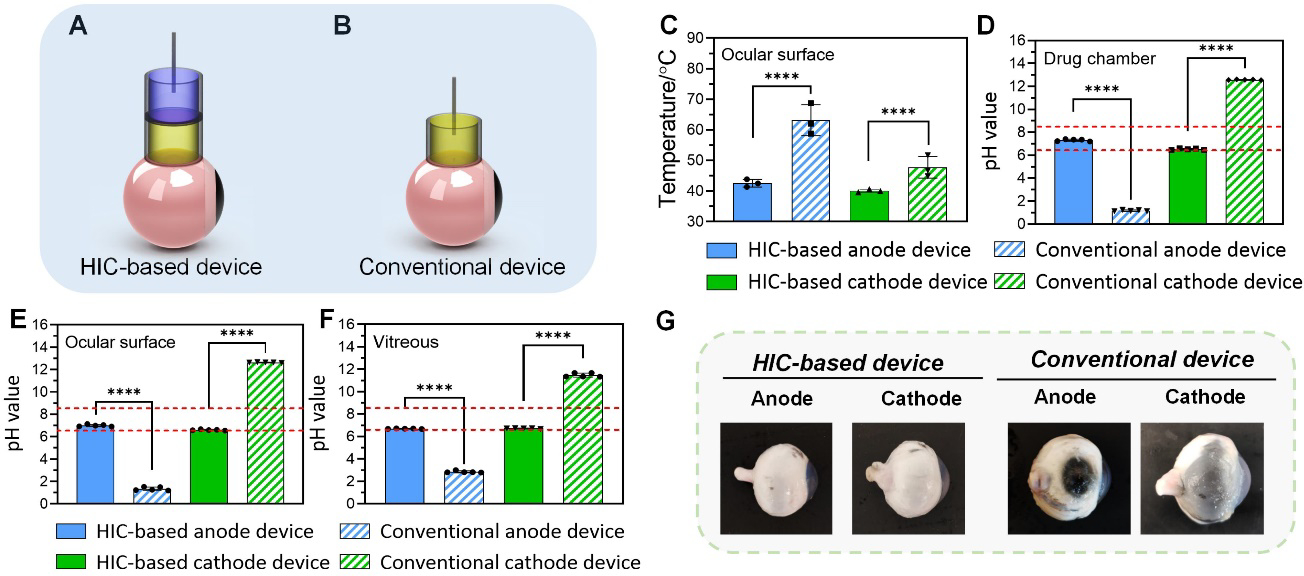
Ex vivo safety test of high-intensity ion current application. (A-B) Schematic of the ex vivo safety test setup. (A) HIC-based device. (B) Conventional ocular iontophoresis device, where carbon electrode was directly inserted in drug solution (1X PBS). (C) The peak temperature on the surface of the eye treated by a 100 mA current for 15 min applied by our HIC-based device and a conventional iontophoresis device. (D-F) pH changes of the PBS in the drug chamber (D), on the ocular surface (E) and in the vitreous (F) after 100 mA current application for 15 min by our HIC-based device and a conventional iontophoresis device. (G) Photographs of porcine eyeballs after the application of 100 mA current for 15 min by our HIC-based device and a conventional iontophoresis device.
4. Enhanced macromolecule delivery to the ocular tissue compartments
4.1. Enhancement of drug permeation by high-intensity iontophoresis
To determine whether the high-intensity ion current safely applied by our HIC-based device could enhance the iontophoretic intraocular drug delivery, we performed an in vitro transscleral (porcine) drug delivery study using a Franz-type cell setup (Figure S8, Supporting Information) and dextran (fluorescein isothiocyanate (FITC)-labeled, MW: 40 kDa) as a model drug. Dextran-40 kDa is a suitable surrogate to ophthalmic anti-VEGF for iontophoresis studies due to their similar hydrodynamic diameters and net charge[44, 45]. We tested several different current intensities from 0 mA (i.e. passive diffusion) to 100 mA. The amount of dextran permeated into the receipt chamber was measured at 3, 6, 9, 12, and 15 min using a fluorescent plate reader. The increase of dextran in the receipt chamber had an initial lag phase of 3 min, where minimal dextran was detected in the receipt chamber no matter the current intensity applied (Figure 4A). Such a lag phase was well-documented in the literature and was attributed to the time required by drug molecules to penetrate sclera[44]. After the lag phase, the amount of dextran permeated increased approximately linearly with time and the current intensity applied. At 100 mA, the total amount of dextran transported through sclera at 15 min was 1,077 ± 62 μg. This was close to or higher than the amount of ophthalmic anti-VEGF administered in IVI (1.25 mg for bevacizumab and 0.5 mg for ranibizumab[46]). This indicated the potential of our device to deliver a therapeutic dose of anti-VEGF within a short period of time. The enhancement of drug permeation was further illustrated in Figure 4B–C, where the permeation coefficient (Pc) of dextran at different current intensities was shown. The Pc was significantly increased at higher current intensities, and had a linear relationship (R2 = 0.9688) with the applied current intensity between 40 and 100 mA. When using 100 mA current intensity, the transscleral Pc of dextran 40kDa was increased over 350 times compared to the conventional iontophoresis using 4.5 mA (7.5 mA cm−2) current intensity.
Figure 4.
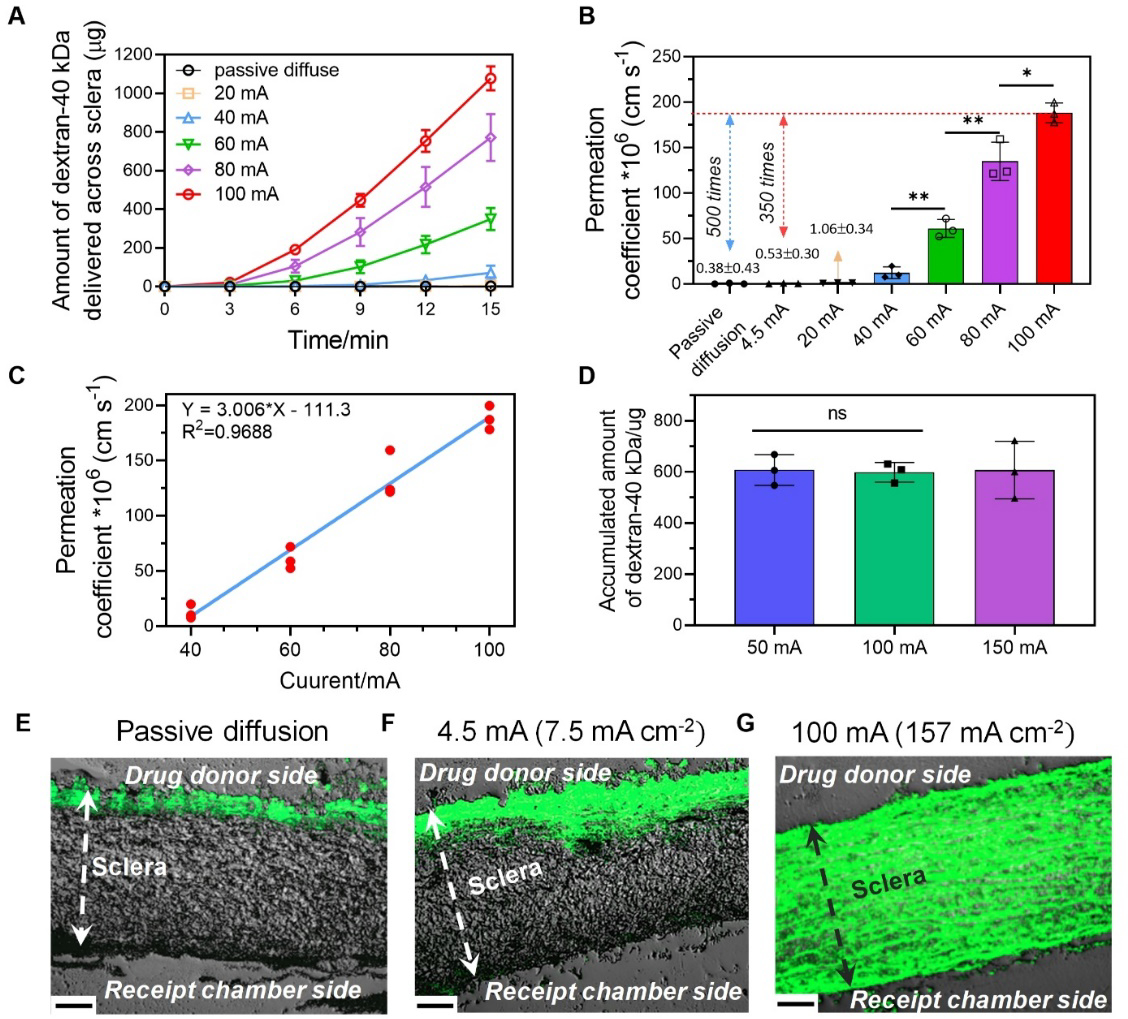
Transscleral delivery of dextran-40 kDa using HIC-based iontophoresis device. (A) Accumulated amount of dextran-40 kDa collected from the receipt chamber of the Franz-type cell at different current intensities and time points. (B-C) The transscleral permeation coefficient of dextran-40 kDa as a function of the applied current. (D) Accumulated amount of dextran-40 kDa collected from the receipt chamber when using different current intensities and iontophoresis durations while keeping the total charge applied the same charge. (E-G) Representative fluorescent images of the cryo-sectioned porcine sclera after transscleral iontophoresis under different conditions: (E) Passive diffusion for 15 min; (F) Conventional iontophoresis at 4.5 mA (7.5 mA cm−2) for 15 min; and (G) High-intensity ion current iontophoresis at 100 mA (157 mA cm−2) for 15 min. Scale bars in (E-G) were 130 μm.
Since the iontophoretic drug flux should be proportional to current intensity, the total amount of drug permeated (flux × time) should be proportional to the total amount of electrical charge applied (current intensity × time). We verified this by varying the current intensity and iontophoresis duration, while keeping the total charge unchanged. As can be seen in Figure 4D and Figure S9 (Supporting Information), the three iontophoresis protocols tested (50 mA-30 min, 100 mA-15 min and 150 mA-10 min) all applied 1,500 coulombs total charge and transferred similar amount of dextran to the receipt chamber. This result provided a practical means to reduce iontophoresis duration by increasing current intensity, while achieving the same drug dose. Figure 4E–G showed the fluorescent dextran distribution in sclera after passive diffusion, low-intensity iontophoresis at 7.5 mA cm−2, and high-intensity iontophoresis at 100 mA (157 mA cm−2) for 15 min. When passive diffusion (Figure 4E) or low-intensity iontophoresis (Figure 4F) was applied, dextran was only present near the upper boundary of the sclera. However, with high-intensity iontophoresis (Figure 4G), dextran penetrated the entire thickness of the sclera. These images provided direct proof that high-intensity iontophoresis enhanced drug permeation in ocular tissues.
4.2. Macromolecules delivery to the posterior segment by high-intensity iontophoresis
Our Franz cell study demonstrated the significant enhancement of transscleral macromolecule delivery enabled by our high-intensity iontophoresis. Here, we took a step further to determine the efficacy of our device in enhancing the macromolecule delivery to the posterior segment using excised rabbit whole eyes. Dextran-40kDa and bevacizumab were used as model drugs. Our HIC-based anode was loaded with drug solutions and was attached to the front of the eye (Figure S10, Supporting Information). Our HIC-based cathode device served as the counter device and was attached to the back of the eye. The cornea was covered by a plastic film, so drugs could only entered the posterior segment at pars plana. We first tested the delivery of dextran-40 kDa. High-intensity iontophoresis at 100 mA (87 mA cm−2) was applied for 20 min, and the amount of dextran delivered into the vitreous was measured every 5 min (Figure 5A). Compared to passive diffusion and conventional iontophoresis at 7.5 mA cm−2, our high-intensity iontophoresis increased the amount of dextran delivered into the vitreous by 217 and 122 times, respectively, at 20 min. Figure 5B showed the dextran concentration distribution in different ocular tissue segments at 20 min. A significant concentration of dextran presented in all tissue segments from conjunctive/sclera to vitreous. The concentration distribution resembled a diffusion profile, which was common for iontophoretic drug delivery. In contrast, for conventional iontophoresis and passive diffusion, the concentration of dextran was considerably lower in all tissue segments (Figure S11A–B, Supporting Information). In light of our promising results from the dextran experiment, we next sought to determine whether high-intensity iontophoresis could deliver a therapeutic concentration of bevacizumab into the posterior segment. 25 mg mL−1 bevacizumab was loaded in the drug chamber and 100 mA (87 mA cm−2) iontophoresis was applied for 20 min. At the end of the iontophoresis, 692 ± 119 μg bevacizumab was delivered into the vitreous (Figure 5C), which was close to the amount delivered by IVI (787 ± 52 μg). Similar to dextran, a diffusion-like concentration gradient of bevacizumab was established from conjunctiva to vitreous (Figure 5D).
Figure 5.
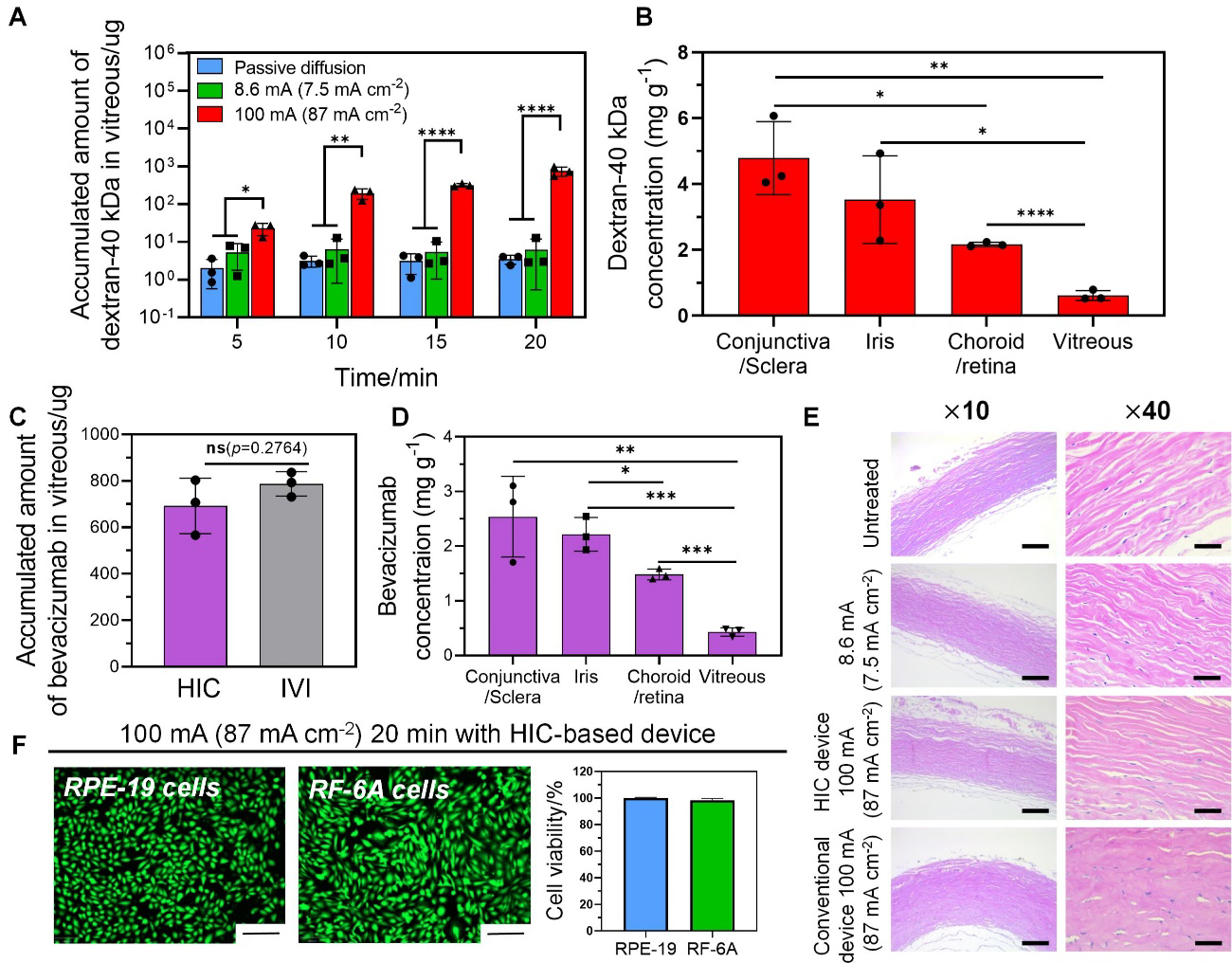
Drug delivery of dextran-40 kDa and bevacizumab to the posterior segments of excised rabbit eyeballs by high-intensity iontophoresis applied by our HIC-based device. (A) Accumulated amount of dextran-40 kDa in vitreous after iontophoresis (100 mA (87 mA cm−2)) for 5 min, 10 min, 15 min, and 20 min, respectively. (B) Accumulated concentration of dextran in various ocular tissue segments after high-intensity iontophoresis at 100 mA (87 mA cm−2) for 20 min. (C) Accumulated amount of bevacizumab in the vitreous after high-intensity iontophoresis (100 mA (87 mA cm−2), 20 min), and an intravitreal injection of bevacizumab (1.25 mg 50 μL−1). (D) Accumulated concentration of bevacizumab in various ocular tissue segments after high-intensity iontophoresis at 100 mA (87 mA cm−2) for 20 min. (E) Hematoxylin and Eosin (H&E) staining images for histological analysis of the sclera after iontophoresis under different test conditions (no treatment, low-intensity iontophoresis at 10 mA (7.5 mA cm−2) for 20 min applied by our HIC-based device, high-intensity iontophoresis at 100 mA (87 mA cm−2) for 20 min applied by our HIC-based device and a conventional device) (scale bars: ×10–100μm, ×40–25 μm). (F) The viability of retinal pigmented epithelium cell (RPE-19) and choroid/retina endothelial cell (RF/6A) (normalized to untreated control) after treated by 100 mA (87 mA cm−2) ion current for 20 min applied by our HIC-based device (scale bar: 200 μm).
Using freshly excised rabbit whole eyes, we showed that the 100 mA (87 mA cm−2) 20 min iontophoresis applied by our device did not induce appreciable scleral tissue damage compared to the untreated control (Figure 5E). However, in the sclera sample treated by a conventional iontophoresis device applying the same current intensity (Figure 5E), fluid accumulation and swelling of the collagen bundles were observed. As a result, the cleft spaces between collagen bundles were missing and the collagen bundles had a more homogenized appearance compared to the untreated control. We also showed that high-intensity current at 100 mA (87 mA cm−2) applied by our HIC-based device for 20 min had minimal impact on the viability of in vitro cultured ARPE-19 cells and RF/6A cells (Figure 5F).
Compared to previous transscleral bevacizumab iontophoresis studies that used low-intensity currents[47], our device achieved over 100-time enhancement of bevacizumab permeation rate. This reduced the time required to deliver a therapeutic dose of bevacizumab from more than 10 hours[47] to 20 min. Another marked difference between our high-intensity iontophoresis and conventional iontophoresis was a shortened lag phase from 30 min[47] to less than 5 min. Our high-intensity iontophoresis device also increased the drug penetration into deeper ocular tissues. A 2020 study examined the distribution of human immunoglobulin G (IgG, as a surrogate of bevacizumab) in rabbit eyes after low-intensity iontophoresis at 3.6 mA cm−2 for 20 min[41]. Although a high total amount of IgG (727.1 μg) was delivered into the eye, the majority of the IgG was found in conjunctiva/sclera. Only 5.8% (42.1 μg) and 1.0% (7.2 μg) of the delivered drug reached retina/choroid and vitreous, respectively. The high drug amount delivered to conjunctive/sclera would not support a sustained drug concentration in retina and vitreous, because the choroidal blood flow presented a significant barrier to drug permeation[48]. So a frequent dosing regimen would be necessary. In contrast, our high-intensity iontophoresis device delivered a therapeutic dose of bevacizumab into the vitreous within 20 min that was comparable to IVI (Figure 5C). Therefore, a monthly dosing regimen similar to IVI could be used. Both frozen and freshly excised, unfrozen rabbit whole eyes were used in our posterior segment macromolecule delivery studies. The freezing procedure at −80°C was shown to have little impact on drug permeation[49] (Figure S12, Supporting Information).
4.3. Intracorneal macromolecule delivery by high-intensity iontophoresis
In light of our promising posterior segment macromolecule delivery results, here we sought to determine the utility of the high-intensity iontophoresis in delivering macromolecules to the anterior segment. We chose to study intracorneal delivery because anti-VEGF has been routinely administered into cornea through invasive subconjunctival or intrastromal injections to treat corneal neovascularization[50]. We first tested the intracorneal delivery of dextran-40 kDa on excised rabbit whole eyes using high-intensity iontophoresis at 100 mA (77 mA cm−2) applied by our HIC-based device. The iontophoresis was applied for 10 min, and the corneal tissues were examined under fluorescent microscope to visualize the distribution of the dextran. The total amount of dextran in cornea was also measured (Figure 6A). Our high-intensity iontophoresis significantly improved dextran delivery efficiency compared to passive diffusion and conventional iontophoresis at 7.5 mA cm−2. The amount of dextran delivered by high-intensity iontophoresis at 10 min was 1876 ± 264 μg, while only 30 ± 17 μg and 115 ± 31 μg were delivered into cornea by passive diffusion and conventional iontophoresis, respectively. Figure 6B showed the distribution of dextran within the cornea after high-intensity iontophoresis, conventional iontophoresis or passive diffusion for 10 min. Dextran was not visible in the cornea after passive diffusion due to the barrier function of the corneal epithelium[51]. Conventional iontophoresis at 7.5 mA cm−2 delivered more dextran into the corneal, but they mainly accumulated near the surface of the cornea. When high-intensity iontophoresis was applied, dextran penetrated the entire thickness of cornea with visible dextran in the stromal and endothelial layers at as early as 4 min. The significant enhancement of intracorneal dextran delivery by our high-intensity iontophoresis indicated its potential to deliver a therapeutic dose of anti-VEGF to cornea within a short period of time. Currently, 1.25 mg bevacizumab is injected through intrastromal route to treat corneal neovascularization[52]. We tested the intracorneal bevacizumab delivery by high-intensity iontophoresis at 100 mA (77 mA cm−2). Our results showed that 1170 ± 332 μg bevacizumab was delivered into cornea after 10 min iontophoresis application (Figure 6C), resulting in a bevacizumab concentration of 4,680.88 μg g−1. It was reported that intraocular bevacizumab has a half-life of 4.3 to 5.6 days[53]. The minimal bevacizumab concentration to completely block VEGF-induced endothelial cell growth was 500 ng mL−1[54]. Therefore, the bevacizumab concentration delivered by our device was able to support sustained anti-VEGF effect for 56.7 to 73.9 days.
Figure 6.
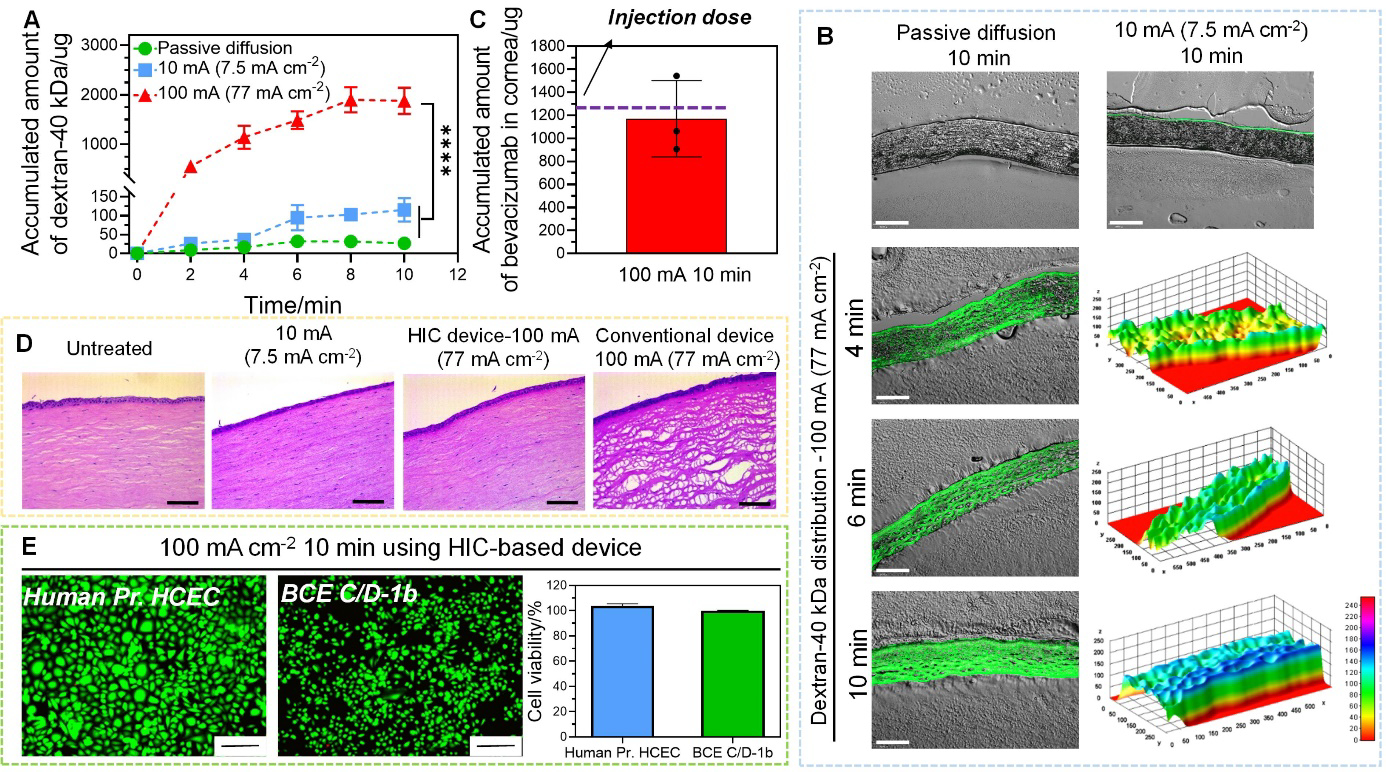
Intracorneal delivery of dextran-40 kDa and bevacizumab by high-intensity iontophoresis using HIC-based device. (A) Accumulated amount of dextran-40 kDa extracted from cornea after passive diffusion, low-intensity iontophoresis at 10 mA (7.5 mA cm−2), and high-intensity iontophoresis at 100 mA (77 mA cm−2) applied for 2 min, 4 min, 6 min, 8 min, and 10 min. (B) Fluorescent images of cryo-sectioned rabbit corneas after intracorneal delivery of dextran-40 kDa under different conditions (passive diffusion, low-intensity iontophoresis at 10 mA (7.5 mA cm−2), and high-intensity iontophoresis at 100 mA (77 mA cm−2)). The fluorescent intensity was converted to a 3D stack using surface plotting from ImageJ software for cornea samples treated with high-intensity iontophoresis (scale bar: 200 μm). (C) Accumulated amount of bevacizumab extracted from cornea after high-intensity iontophoresis at 100 mA (77 mA cm−2) applied for 10 min. (D) H&E images for histological analysis of the cornea after iontophoresis under different test conditions (no treatment, low-intensity iontophoresis at 10 mA (7.5 mA cm−2) applied by our HIC-based device for 10 min, high-intensity iontophoresis at 100 mA (77 mA cm−2) applied by our HIC-based device and a conventional iontophoresis device for 10 min) (scale bar: 25 μm). (E) The viability of corneal epithelial (Human Pr. HCEC) and endothelial cell (BCE C/D-1b) (normalized to untreated control) after treated by 100 mA (77 mA cm−2) current for 10 min by our HIC-based device (scale bar: 200 μm).
Using freshly excised rabbit whole eyes, we showed that when our device was used to apply the high current intensity of 100 mA (77 mA cm−2) 10 min, the cornea remained structurally intact (Figure 6D). However, when conventional iontophoresis device was used, the corneal tissue showed marked vacuolization with prominent disruption of the stromal lamellae (Figure 6D). Besides, high-intensity iontophoresis applied by our device did not induce significant changes in the viability of in vitro cultured Human Pr. HCEC and BCE C/D-1b cells compared to the untreated control (Figure 6E).
5. Intraocular delivery of NPs by high-intensity iontophoresis
In this section, we aimed to determine the efficacy of our high-intensity iontophoresis in enhancing the intraocular delivery of NPs. Poly(lactic-co-glycolic acid) (PLGA) NP was used as a model drug carrier, because PLGA NPs allowed sustained drug release and were widely used in ophthalmic drug delivery. A chitosan (CS) coating was used to introduce positive charges to the NP to enhance iontophoretic efficiency. Both FITC and DEX were loaded in PLGA NPs as our model drugs and NP delivery tests were performed on isolated rabbit eyeballs. FITC allowed easy quantification of NP delivery efficiency. DEX is commonly used to treat corneal inflammation and neovascularization[55]. Our CS-FITC-PLGA NPs had a diameter of 247 ± 9 nm and a zeta potential of 20 ± 2 mV. Our CS-DEX-PLGA NPs had a diameter of 264 ± 2 nm and a zeta potential of 21 ± 1 mV (Figure S13A–D, Supporting Information). We first evaluated the high-intensity iontophoretic delivery of CS-FITC-PLGA NP into cornea and sclera at 100 mA. The corneal and scleral tissues were collected at different time points (4, 7, and 10 min for intracorneal delivery; 5, 10, and 15 min for intrascleral delivery), and the fluorescent intensity in those tissues was measured. Our high-intensity iontophoresis significantly enhanced the delivery of CS-FITC-PLGA NP into both cornea (Figure 7A) and sclera (Figure 7B), compared to passive diffusion and conventional iontophoresis at 7.5 mA cm−2. Next, we evaluated the intracorneal and intrascleral delivery of CS-DEX-PLGA NPs for 10 and 15 min, respectively. Similar to CS-FITC-PLGA NPs, high-intensity iontophoresis significantly enhanced the permeation rate of CS-DEX-PLGA NPs into cornea (Figure 7C) and sclera (Figure 7D), compared to passive diffusion and conventional iontophoresis at 7.5 mA cm−2. After 10 min high-intensity iontophoresis, the concentration of DEX in cornea reached 61 ± 8 μg g−1, while only 1 ± 1 μg g−1 and 7 ± 2 μg g−1 of DEX were achieved for passive diffusion and conventional iontophoresis. In sclera, the concentration of DEX reached 113 ± 29 μg g−1, while passive diffusion and conventional iontophoresis only delivered 1 ± 0.2 μg g−1 and 6 ± 3 μg g−1 DEX, respectively. The corneal and scleral concentrations of DEX delivered by our high-intensity iontophoresis were more than 10 times higher than the DEX concentrations achieved by a conventional method that combined subconjunctival and intravenous injections in rabbit, which were 5 ± 1 μg g−1 in cornea and 11 ± 3 μg g−1 in sclera[56]. These results indicated that the CS-DEX-PLGA NPs delivered by our high-intensity iontophoresis had a potential to provide sustained therapeutic concentration of DEX in ocular tissues. Our NP delivery results were also 20 to 150 times better than other non-invasive intraocular NP delivery methods. Xu et al. reported that a topical application of DEX-loaded nanomicelles in live rabbit eyes resulted in a corneal and scleral DEX concentration of 2 and 0.4 μg g−1, respectively[57]. Besides topical administration, suprachoroidal injection was used to deliver a Laponite-based DEX NP formula into the posterior segment, which resulted a scleral DEX concentration of 283.53 ng mL−1[58]. The sustained release characteristics of PLGA NP could be easily tailored by changing the composition of the material[59], and a one-month sustained release has been demonstrated[60]. This showed a great potential of our high-intensity iontophoretic DEX NP delivery technology to support long-term, sustained anti-inflammatory effect within the eye.
Figure 7.
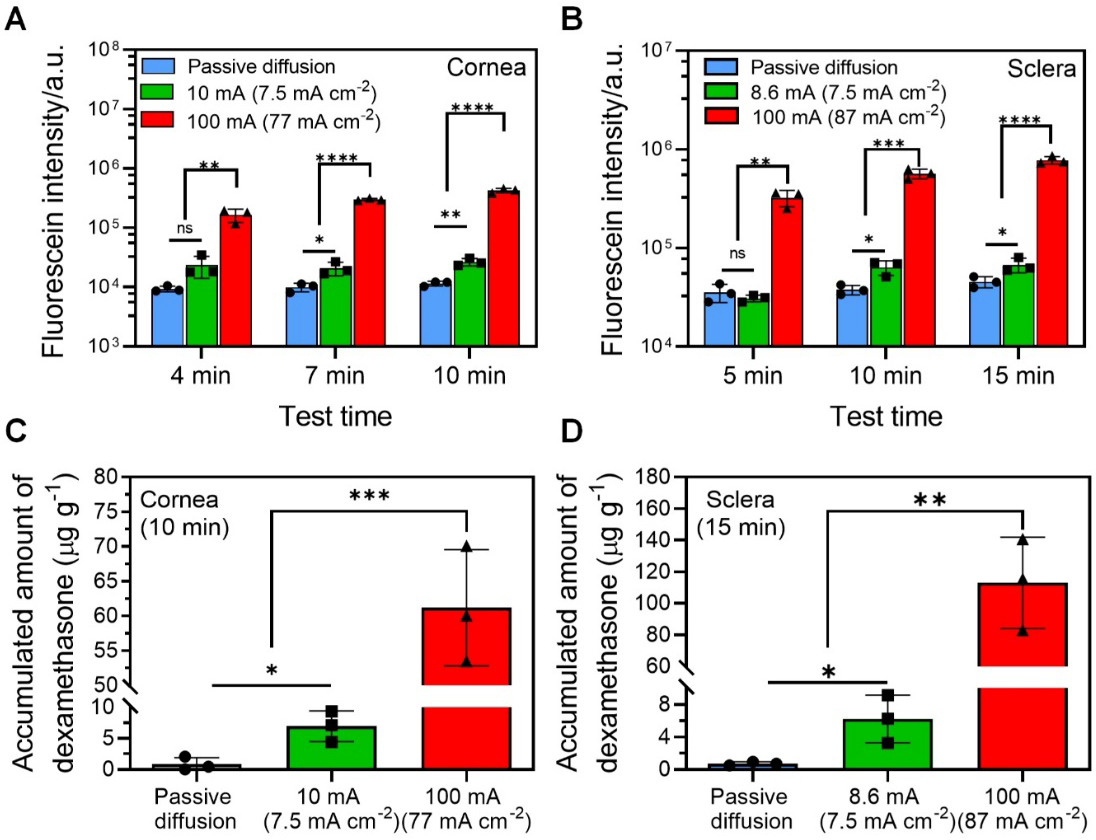
Intraocular delivery of NPs by high-intensity iontophoresis using HIC-based device. (A) Intracorneal delivery of chitosan (CS)-FITC-PLGA NPs by using different delivery methods (passive diffusion, conventional low-intensity iontophoresis at 10 mA (7.5 mA cm−2), and high-intensity iontophoresis at 100 mA (77 mA cm−2) for 4 min, 7 min, and 10 min). (B) Intrascleral delivery of CS-FITC-PLGA NPs by using different delivery methods (passive diffusion, conventional low-intensity iontophoresis at 8.6 mA (7.5 mA cm−2), and high-intensity iontophoresis at 100 mA (87 mA cm−2) for 5 min, 10 min, and 15 min). (C) Accumulated concentration of DEX in cornea using different delivery conditions (passive diffusion, conventional low-intensity iontophoresis at 10 mA (7.5 mA cm−2), and high-intensity iontophoresis at 100 mA (77 mA cm−2) for 10 min). (D) Accumulated concentration of DEX in sclera using different delivery conditions (passive diffusion, conventional low-intensity iontophoresis at 8.6 mA (7.5 mA cm−2), and high-intensity iontophoresis at 100 mA (87 mA cm−2) for 15 min).
6. Limitations and outlook
This study presents a critical proof-of-concept for the ex vivo safety and macromolecule/NP delivery efficiency of our HIC-based, high-intensity ocular iontophoresis device. The current study has several limitations. Future research needs to be conducted to further evaluate our technology before its clinical translation.
Although our device can deliver therapeutic concentrations of bevacizumab and dexamethasone in a NP formula to various ocular tissues, the iontophoresis duration required to achieve these results is still long (up to 20 minutes). A short iontophoresis duration is critical for high patient compliance, as the most cited reason for discontinuation of iontophoresis drug delivery by patients is lack of time[61]. Clinical trials of iontophoretic ocular drug delivery typically used a short duration of less than 5 minutes[23]. To further reduce the iontophoresis duration, a higher drug loading concentration can be used to increase the drug flux based on our results (Figure S14 in Supporting Information). Further increasing the iontophoretic current intensity can also increase the drug flux and reduce the iontophoresis duration (Figure 4D and Figure S9 in supporting Information). Due to the relatively low net charge and large size of macromolecule and NP ophthalmic drugs, it is reasonable to assume that the dominating mechanism of their iontophoresis is electroosmosis. Electroosmosis is an electric field-induced solvent flow, which can transport low-charge and neutral substances. It has been reported that the electroosmotic flow rate is sensitive to the ion strength of the solvent. Lower ion strength increases electroosmotic flow rate[15, 19, 62]. This phenomenon can be exploited to increase the iontophoretic drug delivery efficiency and reduce the iontophoresis duration.
Due to the high-intensity nature of our iontophoresis technology, it will be critical to determine its in vivo safety, particularly the impact of high-intensity ion current on the survival and function of retina. It will also be important to characterize the in vivo pharmacokinetics of the drug delivered by our technology. Clinically relevant animal models of corneal and choroidal neovascularization and uveitis can be used to evaluate the therapeutic efficacy of bevacizumab and dexamethasone delivered by our technology.
7. Conclusion
In this paper, we described an HIC-based, high-intensity ocular iontophoresis device. We demonstrated the safety of our device by showing that minimal pH/temperature changes on ocular surface, cell death, and ocular tissue damage were induced by high-intensity iontophoresis applied by our device. We also demonstrated the significant enhancement of intraocular macromolecule and NP delivery enabled by our high-intensity iontophoresis. The significantly enhanced drug delivery efficiency, safety and non-invasiveness of our HIC-based, high-intensity ocular iontophoresis device will enhance patient compliance to intraocular macromolecule and NP administrations. Our device can be easily operated by caregivers without special medical trainings, which will particularly benefit rural or home-bound patients and patients in developing countries who have limited access to ophthalmologists.
8. Materials and Methods
8.1. Materials
PEGDMA was purchased from Polysciences (Warrington, PA, USA). PEGDA, Irgacure 2959, benzophenone, FITC labeled dextran-40 kDa, PLGA, dexamethasone, chitosan were purchased from Sigma-Aldrich (St. Louis, MO, USA). FITC-PLGA was purchased from Nanosoft Biotechnology (Lewisville, NC, USA). Acrylic sheets and very-high-bond (VHB) foam tape were purchased from Mcmaster-Carr (Robbinsville, NJ, USA). Bevacizumab solution (Avastin) was purchased from Genentech, Inc (San Francisco, CA, USA). Anti-bevacizumab antibodies (HCA 182 and HCA184P), HISPEC Assay Diluent (BUF049) were purchased from Bio-Rad (Raleigh, NC, USA). The standard ABTS ELISA development kit was purchased from Peprotech (Rochy Hill, NJ, USA). Retinal pigmented epithelium cells (RPE-19, ATCC CRL-2302), choroidal/retina endothelial cells (RF/6A, ATCC CRL-1780), primary corneal epithelial cells (Human Pr. HCEC, ATCC PCS-700–010), and corneal endothelial cells (BCE C/D-1b, ATCC CRL-2048) were purchased from ATCC (Manassas, Virginia, USA).
8.2. Fabrication and characterization of HIC-based device
The composition of PEG hydrogel precursor solution was 10% w/v PEGDMA, 5% v/v PEGDA and 1% w/v irgacure 2959 in deionized water. The PEG hydrogel was fabricated by ultraviolet (UV) photo-crosslinking. The viscoelastic behaviors of these hydrogels were investigated by using a parallel plate rheometer (HR-2, TA instruments, New Castle, DE). By performing an angular frequency (0.1 to 100 rad s−1) on the hydrogel samples, the shear storage modulus (G’) and shear loss modulus (G”) of different samples were measured. To evaluate the stability of phosphate/PEG phase separation, PEG hydrogel discs (diameter (D): 10 mm, thickness (T): 2 mm) were soaked in various phosphate salt solutions and their conductivity was tested before soaking and at day 1, 3, 5, 8, 13 and 15. The pH and conductivity of phosphate salt solutions were measured by a pH meter (Accumet AE150 instrument; Fisher Scientific, Waltham, MA) and a Five Easy Plus conductivity meter (Mettler Toledo, Columbus, OH), respectively. The resistivity of the hydrogel discs were obtained using a BK Precision 894 LCR meter and the conductivity of hydrogels were calculated from the following equation:
| (1) |
, where ρ (Ω cm−1) and σ (mS cm−1) represent the resistivity and conductivity of hydrogels respectively.
Our devices were designed using Adobe Illustrator 2019. All devices were fabricated using acrylic plastic and laser micromachining (Trotec Speedy 300, Trotec., MI, USA, cut-through mode). For the HIC-based device, a layer of 0.55 mm thick VHB tape was attached to the bottom surface of the phosphate salt solution chamber. The VHB tape (concentric shape, OD: 14 mm, ID: 7 mm) was then treated with 10% w/v benzophenone in ethanol for 5 min. PEG hydrogels (D: 14 mm, T: 0.4 mm) were slightly exposed under UV, and then placed on the VHB tape and over-exposed under UV to form a good bonding between the PEG hydrogels and the VHB tape[63]. In order to increase the conductivity of the PEG hydrogel, it was soaked in anode phosphate salt solution or cathode phosphate salt solution for 24 h before use. To verify the effect of two-phase separation, the phosphate salt solution chambers of the HIC-based device were filled with anode phosphate salt solution (3 mL), cathode phosphate salt solution (3 mL), or 3 M sodium chloride (3 mL, 3 mol L−1). The HIC-based device were then immersed in PBS solution (5 mL) for 15 min, 30 min, 45 min, and 60 min, respectively. At each time point, the pH and conductivity of the phosphate salt solutions and PBS were measured. Since the HIC-based device was in direct contact with ocular tissues, the biocompatibility of the HIC-based device was also evaluated. The phosphate salt solution chambers of the HIC-based device were filled with anode or cathode phosphate salt solution (3 mL), then the HIC-based device were immersed in cell culture wells (6-well plate) with complete growth media (5 mL) for 60 min after cells were seeded and cultured overnight. After test, Live/Dead viability kit was used to simultaneously visualize live and dead cells and the fluorophore images were obtained from a fluorescent microscope (DMI 6000 B, Leica, Bannockburn, IL, USA). Four types of cell lines were used: ARPE-19, RF/6A, Human Pr. HCEC and BCE C/D-1b. The cell culture procedures were followed with the standard protocols.
8.3. In vitro safety test of high-intensity ion current application
Cells were seeded in 8 mm × 8 mm areas in cell culture dishes defined by a PDMS stencil, and were cultured overnight. The PDMS stencils were removed the next day and the test device was assembled to the cell culture dish (Figure S15, Supporting Information). The test device had a rectangular fluidic chamber so the current density experienced by the cells can be precisely defined. complete growth media (16 mL) was added in the test device. Then 87 mA cm−2 DC current intensity was conducted on ARPE-19 and RF/6A cell lines for 20 min and 77 mA cm−2 current intensity was applied on Human Pr. HCEC and BCS C/D-1b cells for 10 min. After test, LIVE/DEAD stain was used to visualize live and dead cells.
8.4. Ex vivo safety test of high-intensity ion current application
Frozen-preserved porcine whole eyes were used for ex vivo safety test of high-intensity ion current application after thaw to room temperature in PBS. The test system demonstrated in Figure 3A were put on a thermal plate set at 37 °C to maintain a physiological ocular temperature. The dimension of the PEG hydrogel in the HIC-based device under investigation was set as diameter = 24 mm, thickness = 0.2 mm. Before running the test, 1X PBS (1.5 mL) and phosphate salt solution at room temperature were added into drug chamber and phosphate salt solution chamber of HIC-based device, respectively. The HIC-based counter device had a much larger drug chamber filled with 1X PBS (around 20 mL) aiming to minimize the pH/temperature impact of the counter device, so that only the pH/temperature changes induced by the device under investigation were picked up. Then 100 mA DC current was applied for 15 min continuously on the eyeball surface. The temperature of device/tissue interface was monitored by a K-type thermocouple (Digi-Sense™ Standard Precalibrated Thermocouple, Vernon Hills, IL, USA). After the test, the pH of drug solution, device/tissue interface, and vitreous were measured using a flat pH probe (Sensorex Corporation, Stanton, CA). The test setup with carbon electrode directly inserted into the drug chamber was used as the conventional control.
8.5. Transscleral dextran-40 kDa delivery by high-intensity iontophoresis using a Franz-type cell setup
The transscleral iontophoresis tests of dextran were performed in Franz-type cells using sclera (dissected from frozen porcine eyeball, which was preserved in −80 °C, purchased from Pel-Freez LLC, AR, USA) as the barrier. Previous study demonstrated that the freezing procedure did not modify the permeability toward dextran-40 kDa[64]. The test schematic was shown in Figure S8 (Supporting Information). The receipt chamber contained PBS (11 mL). The drug chamber was filled with FITC-dextran (40 kDa) solution (500 μL). The phosphate salt solution chambers were filled with anode or cathode phosphate salt solution (2 mL). The PEG hydrogels of the anode and cathode HIC-based device were immersed in drug solution and receipt solution, respectively. To evaluate the effect of current on the dextran delivery efficacy, 20 mA, 40 mA, 60 mA, 80 mA and 100 mA DC currents were applied for up to 15 min, respectively, and compared with passive diffusion (0 mA). To explore the effect of charge on the dextran delivery efficacy, 50 mA-30 min, 100 mA-15 min, and 150 mA-10 min iontophoresis conditions were applied. At predetermined time intervals, the receipt solutions were sampled to determine the amount of drug permeated through the sclera using a Biotek Synergy H1 hybrid multi-mode microplate reader. The transscleral permeation coefficient (Pc, cm s−1) was calculated as
| (2) |
, where J is the transscleral flux (μg (cm2·h)−1), C is the concentration of the drug solution (μg mL−1)
8.6. Posterior and anterior segment delivery of dextran-40 kDa in whole eyes by high-intensity iontophoresis
The freshly excised or frozen rabbit eyeballs were used for dextran distribution in posterior eye segment tests, which were obtained form Pel-freez. All the rabbit eyeballs for the tests were intact with conjunctiva attached. Our preliminary test results showed that the freezing procedure did not modify the drug delivery efficiency toward dextran-40 kDa (Figure S12, Supporting Information). For evaluating the effect of iontophoresis time on the dextran delivery efficacy to the posterior eye segment, frozen rabbit eyes were used after thaw to room temperature in PBS. A Glad ClingWrap plastic film was attached to the cornea with cyanoacrylate tissue glue to prevent drug solution from being transported into cornea. Therefore, the drug solution was only in contact with sclera near pars plana, and the contact area was 1.15 cm2 (Figure 10). 25 mg mL−1 dextran solution (2 mL) was filled in the drug chamber of HIC-based anode device. For the HIC-based counter device, PBS (12 mL) and cathode phosphate salt solution (10 mL) were added to minimize the pH/temperature impact of the HIC-based counter device. After soaking in anode phosphate salt solution for 24 hours, the PEG hydrogels of anode HIC-based device were immersed in drug solution. 100 mA (87 mA cm−2) DC current was applied for 5 min, 10 min, 15 min and 20 min, respectively. For iontophoresis of dextran to the anterior segment, the test setup is the same to the test of dextran iontophoresis to the posterior eye segments, except the drug chamber (filled with 25 mg mL−1 dextran solution (2 mL)) was applied on the cornea with a contact area of 1.3 cm2. 100 mA DC current (77 mA cm−2) was conducted for 4, 6, 8, and 10 min, respectively. Passive diffusion and conventional iontophoresis (7.5 mA cm−2) were also tested using same test device for same time points as the passive control and conventional iontophoresis control. After each test, the drug solutions were removed and the drug donors were thrice washed with sodium chloride (0.9%). Then the eyeballs were preserved in −80 °C for 30 min to stop the diffusion of permeant and then stored in −20 °C fridge overnight. For iontophoresis of dextran to the posterior eye segments, iris, choroid/retina, vitreous, and conjunctiva/sclera were collected and weighted. For the dextran iontophoresis to the anterior eye segment, only cornea was collected and weighted. All tissues were mixed with PBS (10 mL) and homogenized using a hand homogenizer (Fisher Scientific, Hampton, NH, USA). Each dextran extraction process was repeated twice. The supernatant was separated from the tissue matrix by using a centrifuge (Eppendorf centrifuge 5810R) at 4000 rpm for 20 min and analyzed using a microplate reader.
8.7. Cryo-section and fluorescent microscopy
The ocular tissues were cryo-sectioned in order to evaluate the dextran distribution in different tissues. Briefly, the rabbit cornea was dissected from the rabbit eyeball after tests and dehydrated in acetone for 2.5 h. The sample was then air dried for 30 min, embedded in tissue freezing medium (OCT), and sectioned at −20 °C to obtain 10 μm cornea sections. For porcine sclera cryo-section, the sclera was embedded in OCT and sectioned at 10 μm. Fluorescein imaging was performed with the fluorescent microscope.
8.8. Hematoxylin and Eosin (H&E) staining of ocular tissues
Ocular tissues were collected after iontophoresis and fixed in 4% PBS-buffered paraformaldehyde at room temperature for 24 h. The samples were then dehydrated using a graded series of ethanol (70%, 90%, 95%, and 100%), embedded in paraffin and sectioned at 4 μm thickness. H&E staining was performed based on the standard protocol. Samples were imaged using an optical microscope (Leica DMi1; Leica Microsystems, Germany).
8.9. Posterior and anterior segment delivery of bevacizumab in whole eyes and Enzyme-Linked Immunosorbent Assay (ELISA) for bevacizumab
The test setup of bevacizumab iontophoresis to the posterior segment and anterior segment of eyes was same to the ex vivo iontophoresis test of dextran-40 kDa (Figure S10, Supporting Information). 25 mg mL−1 of bevacizumab solution was used for posterior eye segment iontophoresis test, and 10 mg mL−1 bevacizumab diluted by PBS containing 0.05% (w/v) bovine serum albumin (BSA) was used for anterior segment iontophoresis. After tests, cornea in anterior segment iontophoresis test and posterior segment tissues (iris, choroidal/retina, vitreous, conjunctiva/sclera) in posterior segment iontophoresis test were dissected from eyeball and immersed in bevacizumab extraction solvent (10 mL, PBS containing 1% w/v BSA). The mixture of tissue and extraction solvent was rotated 24 h at 4 °C, and the supernatant was collected after centrifuge at 4000 rpm for 20 min. In order to completely extract bevacizumab from ocular tissues, the process repeated three times. The amount of bevacizumab was then quantitatively measured using the following ELISA protocol. A 96-well microtiter plate was coated with anti-bevacizumab capture antibody HCA182 (100 μL, 1 μg mL−1 in PBS) with required number of wells. After incubated overnight at 4 °C, the microtiter plate was washed five times with PBST (PBS with 0.05% Tween-20). It was then blocked by adding 5% BSA in PBST (100 μL) to each well and incubated for 1 h at room temperature, and then washed five times with PBST. For the standard curve, a dilution series of bevacizumab in BUF049 in duplicate were prepared in a range of 0–200 ng mL−1. Diluted standards (100 μL) and test samples (100 μL) were added to the wells. Then the microtiter plate was incubated for 1 h at room temperature. After washed with PBST five times, HRP conjugated detection antibody HCA184P (100 μL, 4 μg mL−1 in BUF049) were added in each well and incubated for another 1 h at room temperature and prevented from light. ABTS (100 μL) was added to each well and the microtiter plate was incubated for 30 min at room temperature and protected from light for color development. The absorbance was determined using a microplate reader at 405 nm, with the subtraction of the reference absorbance at 650 nm.
8.10. Fabrication of CS-FITC-PLGA NPs and CS-DEX-PLGA NPs
CS-FITC-PLGA NPs were prepared using a method adapted from published literature with modifications[65]. Briefly, 5 mg mL−1 of FITC-PLGA (MW: 5 kDa) was dissolved in acetone. Solution (1 mL) was added dropwise into CS solution (3 mL, 0.05% w/v, 1% acetic acid in deionized water). The mixture was then stirred in open air (speed: 460 rpm) at room temperature for 6 hours until the acetone was completely evaporated. The NPs were isolated with 10 kDa MWCO Amicon Ultra-4 Centrifugal Filters (Millipore, CA, USA). The NPs were then washed twice by deionized water. After the last centrifuge, the CS-FITC-PLGA NPs were suspended with PBS and stored at 4 °C for further use. The particle size distribution and zeta potential of CS-FITC-PLGA NPs were determined with dynamic light scattering and electrophoretic light scattering (Zetasizer Nano ZS), respectively.
CS-DEX-PLGA NPs were prepared using a method adapted from published literature with modifications[55]. Briefly, PLGA (20 mg, MW: 24–38 kDa) and DEX (2 mg) were dissolved in dichloromethane (800 μL) and acetone (200 μL), respectively. They were then mixed together and drop-wised to deionized water containing 1% acetic acid (5 mL), which also contains 0.5 mg mL−1 CS and 5 mg mL−1 Poly(vinyl alcohol) (PVA, MW: 13–23 kDa). After sonication for 3 min (20% set amplitude, ice-water bath), the mixture was magnetic stirred (speed: 400 rpm) overnight at room temperature. NP suspensions were then isolated from the free DEX and PVA with 50 kDa MWCO Amicon Ultra-4 Centrifugal Filters (Millipore, CA, USA). The NPs were washed twice by deionized water. After the last centrifuge, the CS-DEX-PLGA NPs was suspended with PBS and stored at 4 °C for further use. The particle size distribution and zeta potential of NPs were determined with dynamic light scattering and electrophoretic light scattering, respectively.
8.11. Intraocular delivery of CS-FITC-PLGA NPs and CS-DEX-PLGA NPs by high-intensity iontophoresis
The iontophoretic delivery of CS-FITC-PLGA NPs and CS-DEX-PLGA NPs into the anterior and posterior segments were evaluated using the test setup described in Figure S10 (Supporting Information). Briefly, NP suspensions (2 mL, collected from NP fabrication process) were loaded in the drug chamber. 100 mA current intensity was used for both intracorneal and intrascleral delivery. For intracorneal delivery, the resulting current density was 77 mA cm−2, which was applied for 4 min, 7 min, and 10 min, respectively. For intrascleral delivery, the resulting current density was 87 mA cm−2, which was applied at pars plana for 5 min, 10 min, and 15 min, respectively. After iontophoresis, the ocular surface contacted with NP suspensions were triply washed with sodium chloride (0.9%). To quantify CS-FITC-PLGA NP delivery efficiency, the ocular tissues (cornea and sclera) were mixed with PBS (1 mL) and homogenized using a high-speed beads mill homogeniser (Fisher Scientific, Hampton, NH, USA). The supernatant was collected after centrifuge (BT604 mini centrifuge, BT labsystems, St Louis, MO, USA) at 10,000 rpm for 10 min. This extraction process of FITC was repeated three times. Then the fluorescein intensity of the supernatant was analyzed using a microplate reader. To quantify CS-DEX-PLGA NP delivery efficiency, ocular tissues were cut into small pieces and were immersed in deionized water containing 2% acetic acid (10 mL) and stirred at room temperature overnight to dissolve the CS. After centrifugation (5500 rpm; 30 min), the precipitant was collected and dissolved in acetonitrile (5 mL). After water-bath sonication (10 min), the supernatant was collected and evaporated completely. Samples were re-dissolved in chloroform:methanol mixture (1:4, v/v), and the supernatant was collected after centrifugation (10,000 rpm, 10 min). Then the supernatant was evaporated completely and the samples were re-dissolved in acetonitrile (1 ml). The DEX concentration was measured by using a UV-Vis spectrometry at 241 nm and compared to the calibration curve of DEX prepared in acetonitrile (Figure S16, Supporting Information). The intraocular passive diffusion and low intensity iontophoresis of CS-FITC-PLGA NPs and CS-DEX-PLGA NPs were also tested at the same time points as passive diffusion control and conventional iontophoresis controls.
8.12. Statistical Analysis
Unpaired student’s t-tests were used for determining the statistical difference between samples (GraphPad Software, SanDiego, CA, USA). The data in the figures were marked by * for p < 0.05, ** for p < 0.01, *** for p < 0.005, and **** for p < 0.001. All statistics were performed using at least 3 replicates.
Supplementary Material
Acknowledgements
This research was supported by Departmental Start-Up Fund provided by the Holland Regenerative Medicine Program at the University of Nebraska Medical Center. This research was also supported by the National Institute of General Medical Sciences, U54 GM115458, which funds the Great Plains IDeA-CTR Network. The content in this publication was solely the responsibility of the authors and did not necessarily represent the official views of the NIH. The authors acknowledge Dr. Meghal Gagrani for the instruction of ocular tissue dissections; Dr. Wen Xue for the instruction of ELISA assay; Dr. Bo Liu for the instruction of nanoparticle fabrication; Dr. Cuifen Wang for the paraffin section and H&E staining of ocular tissues; Dr. Mike Punsoni and Dr. Dominick J. Dimaio for the interpretation of ocular tissue damage from H&E staining images.
Contributor Information
Fan Zhao, Mary & Dick Holland Regenerative Medicine Program, University of Nebraska Medical Center, Omaha, NE, 68198, USA; Department of Surgery, University of Nebraska Medical Center, Omaha, NE, USA.
Shan Fan, Ophthalmology and Visual Sciences, University of Nebraska Medical Center, Omaha, NE, 68198, USA.
Deepta Ghate, Truhlsen Eye Institute, University of Nebraska Medical Center, Omaha, NE, 68198, USA.
Svetlana Romanova, Department of Pharmaceutical Sciences, University of Nebraska Medical Center, Omaha, NE, 68198, USA.
Siwei Zhao, Mary & Dick Holland Regenerative Medicine Program, University of Nebraska Medical Center, Omaha, NE, 68198, USA; Department of Surgery, University of Nebraska Medical Center, Omaha, NE, USA.
Data availability
All relevant data are included in the main manuscript and the Supporting Information. Additional data are available from the corresponding authors upon reasonable request.
Reference
- [1].Kim YC, Chiang B, Wu X, Prausnitz MR, J Control Release 2014, 190, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Gross JG, Glassman AR, Liu D, Sun JK, Antoszyk AN, Baker CW, Bressler NM, Elman MJ, Ferris FL 3rd, Gardner TW, Jampol LM, Martin DF, Melia M, Stockdale CR, Beck RW, Diabetic N Retinopathy Clinical Research, JAMA Ophthalmol 2018, 136, 1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Meza-Rios A, Navarro-Partida J, Armendariz-Borunda J, Santos A, Ophthalmol Ther 2020, 9, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gorantla S, Rapalli VK, Waghule T, Singh PP, Dubey SK, Saha RN, Singhvi G, Rsc Adv 2020, 10, 27835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gaudana R, Ananthula HK, Parenky A, Mitra AK, The AAPS journal 2010, 12, 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Nomoto H, Shiraga F, Kuno N, Kimura E, Fujii S, Shinomiya K, Nugent AK, Hirooka K, Baba T, Invest Ophthalmol Vis Sci 2009, 50, 4807; [DOI] [PubMed] [Google Scholar]; Civiale C, Licciardi M, Cavallaro G, Giammona G, Mazzone MG, Int J Pharm 2009, 378, 177. [DOI] [PubMed] [Google Scholar]
- [7].Schultz C, Breaux J, Schentag J, Morck D, Clin Exp Optom 2011, 94, 212. [DOI] [PubMed] [Google Scholar]
- [8].Furrer E, Berdugo M, Stella C, Behar-Cohen F, Gurny R, Feige U, Lichtlen P, Urech DM, Invest Ophthalmol Vis Sci 2009, 50, 771; [DOI] [PubMed] [Google Scholar]; Sakai T, Ishihara T, Higaki M, Akiyama G, Tsuneoka H, Invest Ophthalmol Vis Sci 2011, 52, 1516. [DOI] [PubMed] [Google Scholar]
- [9].Jager RD, Aiello LP, Patel SC, Cunningham ET Jr., Retina 2004, 24, 676. [DOI] [PubMed] [Google Scholar]
- [10].Polat O, Inan S, Ozcan S, Dogan M, Kusbeci T, Yavas GF, Inan UU, Turk J Ophthalmol 2017, 47, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Finger RP, Xie J, Fotis K, Parikh S, Cummins R, Mitchell P, Guymer RH, Clin Exp Ophthalmol 2017, 45, 143. [DOI] [PubMed] [Google Scholar]
- [12].Sommer A, Taylor HR, Ravilla TD, West S, Lietman TM, Keenan JD, Chiang MF, Robin AL, Mills RP, S. Council of the American Ophthalmological, JAMA Ophthalmol 2014, 132, 640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Eljarrat-Binstock E, Domb AJ, Journal of Controlled Release 2006, 110, 479. [DOI] [PubMed] [Google Scholar]
- [14].Zhao S, Mehta AS, Zhao M, Cell Mol Life Sci 2020, 77, 2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Soni V, Pandey V, Tiwari R, Asati S, Tekade RK, in Basic Fundamentals of Drug Delivery, Elsevier, 2019, 473. [Google Scholar]
- [16].Vollmer DL, Szlek MA, Kolb K, Lloyd LB, Parkinson TM, Journal of ocular pharmacology and therapeutics 2002, 18, 549; [DOI] [PubMed] [Google Scholar]; Myles ME, Neumann DM, Hill JM, Advanced drug delivery reviews 2005, 57, 2063. [DOI] [PubMed] [Google Scholar]
- [17].Behar-Cohen F, El Aouni A, Gautier S, David G, Davis J, Chapon P, Parel J, Experimental eye research 2002, 74, 51; [DOI] [PubMed] [Google Scholar]; Kusama S, Sato K, Matsui Y, Kimura N, Abe H, Yoshida S, Nishizawa M, Nature communications 2021, 12, 1; [DOI] [PubMed] [Google Scholar]; Huang D, Chen Y-S, Rupenthal ID, Advanced drug delivery reviews 2018, 126, 96. [DOI] [PubMed] [Google Scholar]
- [18].Perez VL, Wirostko B, Korenfeld M, From S, Raizman M, Journal of Ocular Pharmacology and Therapeutics 2020, 36, 75. [DOI] [PubMed] [Google Scholar]
- [19].Molokhia S, Papangkorn K, Butler C, Higuchi JW, Brar B, Ambati B, Li SK, Higuchi WI, Journal of Ocular Pharmacology and Therapeutics 2020, 36, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Behar-Cohen FF, El Aouni A, Gautier S, David G, Davis J, Chapon P, Parel JM, Exp Eye Res 2002, 74, 51; [DOI] [PubMed] [Google Scholar]; Kiran Vaka SR, Sammeta SM, Day LB, Murthy SN, Current eye research 2008, 33, 661. [DOI] [PubMed] [Google Scholar]
- [21].Sinapis CI, Routsias JG, Sinapis AI, Sinapis DI, Agrogiannis GD, Pantopoulou A, Theocharis SE, Baltatzis S, Patsouris E, Perrea D, Clinical Ophthalmology (Auckland, NZ) 2011, 5, 697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Jung JH, Chiang B, Grossniklaus HE, Prausnitz MR, J Control Release 2018, 277, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gratieri T, Santer V, Kalia YN, Expert opinion on drug delivery 2017, 14, 1091. [DOI] [PubMed] [Google Scholar]
- [24].Biesheuvel PM, Dykstra JE, in arXiv e-prints, 2018, arXiv:1809.02930. [Google Scholar]
- [25].Millet P, in Hydrogen Production: Electrolysis, (Ed: Godula-Jopek A), Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany: 2015, 36. [Google Scholar]
- [26].Abe I, in Energy Carriers And Conversion Systems - Volume 1, (Ed: Ohta T), EOLSS Publishers Co. Ltd., Oxford, United Kingdom: 2009, 146. [Google Scholar]
- [27].Stecker MM, Patterson T, Netherton BL, Am J Electroneurodiagnostic Technol 2006, 46, 315. [PubMed] [Google Scholar]
- [28].Zhang Y, Chen Y, Qi Y, Huang D, Yanga M, Yu X, Hu Y, Li Z, Sensors and Actuators B: Chemical 2016, 237, 1007. [Google Scholar]
- [29].Keplinger C, Sun JY, Foo CC, Rothemund P, Whitesides GM, Suo Z, Science 2013, 341, 984; [DOI] [PubMed] [Google Scholar]; Zhou Y, Wan C, Yang Y, Yang H, Wang S, Dai Z, Ji K, Jiang H, Chen X, Long Y, Advanced Functional Materials 2019, 29, 1806220. [DOI] [PubMed] [Google Scholar]
- [30].Zhou T, Gao X, Dong B, Sun N, Zheng L, Journal of Materials Chemistry A 2016, 4, 1112. [Google Scholar]
- [31].Lee CJ, Wu H, Hu Y, Young M, Wang H, Lynch D, Xu F, Cong H, Cheng G, ACS Appl Mater Interfaces 2018, 10, 5845. [DOI] [PubMed] [Google Scholar]
- [32].Yuk H, Lu B, Zhao X, Chem Soc Rev 2019, 48, 1642. [DOI] [PubMed] [Google Scholar]
- [33].Liu Z, Zhang Y, Yang T, Liu Y, Zhou W, Wang Z, Liu Y, Kong T, Journal of Materials Chemistry C 2020, 8, 2320; [Google Scholar]; Ren Y, Guo J, Liu Z, Sun Z, Wu Y, Liu L, Yan F, Sci Adv 2019, 5, eaax0648.31467977 [Google Scholar]
- [34].Flieger J, Flieger M, Int J Mol Sci 2020, 21.;; [DOI] [PMC free article] [PubMed] [Google Scholar]; Leitch AC, Abdelghany TM, Probert PM, Dunn MP, Meyer SK, Palmer JM, Cooke MP, Blake LI, Morse K, Rosenmai AK, Oskarsson A, Bates L, Figueiredo RS, Ibrahim I, Wilson C, Abdelkader NF, Jones DE, Blain PG, Wright MC, Food Chem Toxicol 2020, 136, 111069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhao S, Tseng P, Grasman J, Wang Y, Li W, Napier B, Yavuz B, Chen Y, Howell L, Rincon J, Omenetto FG, Kaplan DL, Adv Mater 2018, 30, e1800598. [DOI] [PubMed] [Google Scholar]
- [36].Sieg A, Wascotte V, Journal of drug targeting 2009, 17, 690. [DOI] [PubMed] [Google Scholar]
- [37].Chopra P, Hao J, Li SK, International journal of pharmaceutics 2010, 388, 107; [DOI] [PMC free article] [PubMed] [Google Scholar]; Liu H, Liu C, Jiang L, Liu J, Yang Q, Guo Z, Cai X, Electroanalysis: An International Journal Devoted to Fundamental and Practical Aspects of Electroanalysis 2008, 20, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gonnering R, Edelhauser HF, Van Horn DL, Durant W, Investigative ophthalmology & visual science 1979, 18, 373. [PubMed] [Google Scholar]
- [39].Abe I, Energy carriers and conversion systems 2009, 1. [Google Scholar]
- [40].Yarmolenko PS, Moon EJ, Landon C, Manzoor A, Hochman DW, Viglianti BL, Dewhirst MW, International Journal of Hyperthermia 2011, 27, 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Molokhia S, Papangkorn K, Butler C, Higuchi JW, Brar B, Ambati B, Li SK, Higuchi WI, J Ocul Pharmacol Ther 2020, 36, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hey MJ, Jackson DP, Yan H, Polymer 2005, 46, 2567. [Google Scholar]
- [43].Dewhirst MW, Viglianti B, Lora-Michiels M, Hanson M, Hoopes P, International journal of hyperthermia 2003, 19, 267. [DOI] [PubMed] [Google Scholar]
- [44].Nicoli S, Ferrari G, Quarta M, Macaluso C, Santi P, European journal of pharmaceutical sciences 2009, 36, 486; [DOI] [PubMed] [Google Scholar]; Pescina S, Ferrari G, Govoni P, Macaluso C, Padula C, Santi P, Nicoli S, Journal of Pharmacy and Pharmacology 2010, 62, 1189. [DOI] [PubMed] [Google Scholar]
- [45].Wen H, Hao J, Li SK, Journal of pharmaceutical sciences 2013, 102, 892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bakri SJ, Snyder MR, Reid JM, Pulido JS, Singh RJ, Ophthalmology 2007, 114, 855; [DOI] [PubMed] [Google Scholar]; Bakri SJ, Snyder MR, Reid JM, Pulido JS, Ezzat MK, Singh RJ, Ophthalmology 2007, 114, 2179. [DOI] [PubMed] [Google Scholar]
- [47].Pescina S, Ferrari G, Govoni P, Macaluso C, Padula C, Santi P, Nicoli S, J Pharm Pharmacol 2010, 62, 1189. [DOI] [PubMed] [Google Scholar]
- [48].Gaudana R, Ananthula HK, Parenky A, Mitra AK, AAPS J 2010, 12, 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Pescina S, Padula C, Santi P, Nicoli S, Eur J Pharm Sci 2011, 42, 503. [DOI] [PubMed] [Google Scholar]
- [50].Than A, Liu C, Chang H, Duong PK, Cheung CMG, Xu C, Wang X, Chen P, Nature communications 2018, 9, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chen H, Journal of drug targeting 2015, 23, 597. [DOI] [PubMed] [Google Scholar]
- [52].Ucgul RK, Celebi S, Yilmaz NS, Bukan N, Ucgul AY, Eye 2021, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Stewart MW, Retina 2007, 27, 1196. [DOI] [PubMed] [Google Scholar]
- [54].Wang Y, Fei D, Vanderlaan M, Song A, Angiogenesis 2004, 7, 335. [DOI] [PubMed] [Google Scholar]
- [55].Wang B, Tang Y, Oh Y, Lamb NW, Xia S, Ding Z, Chen B, Suarez MJ, Meng T, Kulkarni V, Nanomedicine: Nanotechnology, Biology and Medicine 2019, 17, 119; [DOI] [PMC free article] [PubMed] [Google Scholar]; Soiberman U, Kambhampati SP, Wu T, Mishra MK, Oh Y, Sharma R, Wang J, Al Towerki AE, Yiu S, Stark WJ, Biomaterials 2017, 125, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Huang X, Duan Y, Zhao L, Liu S, Qin D, Zhang F, Lin D, Journal of Drug Delivery Science and Technology 2020, 57, 101639. [Google Scholar]
- [57].Xu X, Sun L, Zhou L, Cheng Y, Cao F, Carbohydrate polymers 2020, 227, 115356. [DOI] [PubMed] [Google Scholar]
- [58].Prieto E, Cardiel MJ, Vispe E, Idoipe M, Garcia-Martin E, Fraile JM, Polo V, Mayoral JA, Pablo LE, Rodrigo MJ, Biomedical Materials 2020, 15, 065021. [DOI] [PubMed] [Google Scholar]
- [59].Kobashi H, Ciolino JB, Cornea 2018, 37 Suppl 1, S94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Ciolino JB, Stefanescu CF, Ross AE, Salvador-Culla B, Cortez P, Ford EM, Wymbs KA, Sprague SL, Mascoop DR, Rudina SS, Trauger SA, Cade F, Kohane DS, Biomaterials 2014, 35, 432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Özcan D, Güleç AT, Journal of cutaneous medicine and surgery 2014, 18, 109. [DOI] [PubMed] [Google Scholar]
- [62].Li S, Cao W, Hui YS, Wen W, Nanoscale research letters 2014, 9, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Yuk H, Zhang T, Parada GA, Liu X, Zhao X, Nature communications 2016, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Pescina S, Padula C, Santi P, Nicoli S, European journal of pharmaceutical sciences 2011, 42, 503. [DOI] [PubMed] [Google Scholar]
- [65].Kang M, Hong J, Jung M, Kwon SP, Song SY, Kim HY, Lee JR, Kang S, Han J, Koo JH, Advanced Materials 2020, 32, 2003368; [DOI] [PubMed] [Google Scholar]; Takeuchi I, Takeshita T, Suzuki T, Makino K, Colloids and Surfaces B: Biointerfaces 2017, 160, 520. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data are included in the main manuscript and the Supporting Information. Additional data are available from the corresponding authors upon reasonable request.