

Abstract
Organelles play important roles in maintaining cellular homeostasis. Organelle stress responses, especially in mitochondria, endoplasmic reticula (ER), and primary cilia, are deeply involved in kidney disease pathophysiology. Mitochondria are the center of energy production in most eukaryotic cells. Renal proximal tubular cells are highly energy demanding and abundant in mitochondria. Mitochondrial dysfunctions in association with energy metabolism alterations produce reactive oxygen species and promote inflammation in proximal tubular cells, resulting in progression of kidney disease. The ER play critical roles in controlling protein quality. Unfolded protein response (UPR) pathways are the adaptive response to ER stress for maintaining protein homeostasis. UPR pathway dysregulation under pathogenic ER stress often occurs in glomerular and tubulointerstitial cells and promotes progression of kidney disease. The primary cilia sense extracellular signals and maintain calcium homeostasis in cells. Dysfunction of the primary cilia in autosomal dominant polycystic kidney disease reduces the calcium concentration in proximal tubular cells, leading to increased cell proliferation and retention of cyst fluid. In recent years, the direct interaction at membrane contact sites has received increased attention in association with the development of imaging technologies. The part of the ER that is directly connected to mitochondria is termed the mitochondria-associated ER membrane (MAM), which regulates calcium homeostasis and phospholipid metabolism in cells. Disruption of MAM integrity collapses cellular homeostasis and leads to diseases such as diabetes and Alzheimer disease. This review summarizes recent research on organelle stress and crosstalk, and their involvement in kidney disease pathophysiology. In addition, potential treatment options that target organelle stress responses are discussed.
Keywords: chronic kidney disease, acute kidney injury, AKI-to-CKD transition, ER stress, lipid metabolism, mitochondria, organelle crosstalk, organelle stress, tubular inflammation, unfolded protein response (UPR)
Introduction
Organelles are confined functional subunits within a cell. Various biochemical reactions that are essential for maintaining the cellular homeostasis, such as energy metabolism and protein quality control, are conducted within organelles. Because organelles are separated from the cytoplasm, each organelle has its own unique role and performs the complex activities of cells. Organelles closely interact with one another at membrane contact sites or via intracellular vesicles. Recent advances in imaging techniques have revealed the dynamic interaction between organelles (1). Organelle stress and crosstalk are deeply involved in the progression of various disorders, including kidney diseases. In this review, we summarize the recent advances in research on organelle stress and crosstalk, in addition to their involvement in the pathophysiology of kidney diseases.
Organelle Damage and Stress Signaling
Mitochondria
Mitochondria are the center of energy production in most eukaryotic cells. Energy substrates such as glucose, amino acids, and fatty acids enter the tricarboxylic acid (TCA) cycle (Figure 1). Glucose is converted into pyruvic acids by an oxygen-independent metabolic pathway called glycolysis. Amino acids are deaminated and converted into some components of the TCA cycle. Fatty acids are broken down to generate acetyl-CoA, which is called β-oxidation. The TCA cycle supplies the electron transport chain with the reduced form of nicotinamide adenine dinucleotide (NADH) and the reduced form of flavin adenine dinucleotide. The electron transport chain consists of a series of electron transporters (complex I–IV) in the inner mitochondrial membrane, and it shuttles electrons from NADH and the reduced form of flavin adenine dinucleotide to molecular oxygen. During this process, protons are pumped from the mitochondrial matrix to the intermembrane space. This proton gradient between the mitochondrial matrix and intermembrane space is used by ATP synthetase to produce energy. These processes are named mitochondrial respiration or oxidative phosphorylation.
Figure 1.
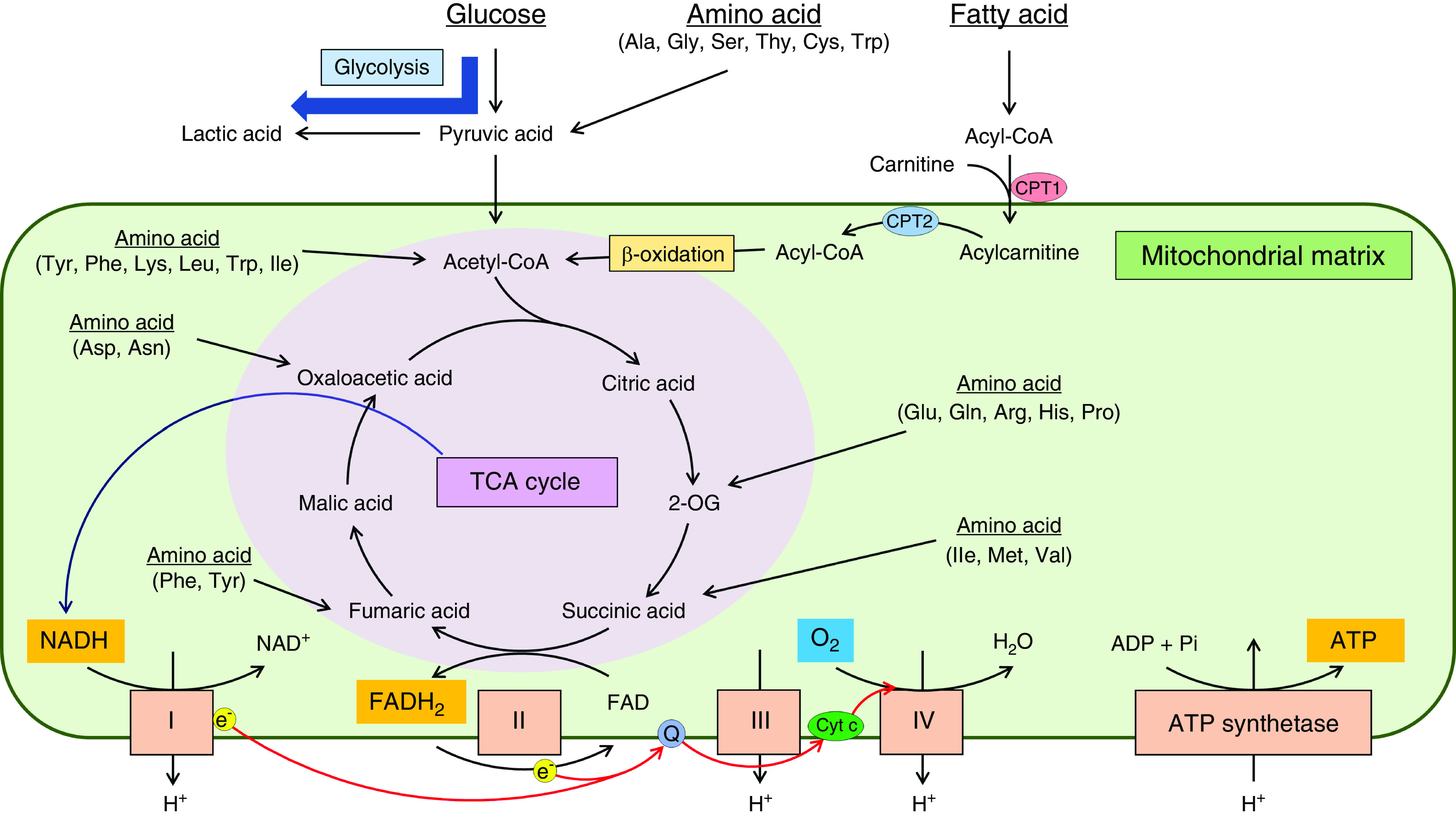
Mitochondria produce energy through oxidative phosphorylation to fuel cellular activity. Glucose, amino acids, and fatty acids enter the tricarboxylic acid (TCA) cycle, which supplies the electron transport chain with the reduced form of NAD (NADH) and the reduced form of flavin adenine dinucleotide (FADH2). The electron transport chain is a series of electron transporters (complex I–IV) in the inner mitochondrial membrane and forms the proton gradient by shuttling electrons from NADH and FADH2 to molecular oxygen (O2) via ubiquinone (Q) and cytochrome c (Cyt c) (the flow of electrons is indicated by red arrows). ATP synthetase produces energy by using the proton gradient between the mitochondrial matrix and intermembrane space. 2-OG, 2-oxoglutarate; Ala, alanine; Arg, arginine; Asn, asparagine; Asp, aspartic acid; CPT1, carnitine palmitoyltransferase I; CPT2, carnitine palmitoyltransferase II; Cys, cysteine; e−, electron; FAD, flavin adenine dinucleotide; Gln, glutamine; Glu, glutamic acid; Gly, glycine; H+, proton; His, histidine; H2O, water; Ile, isoleucine; Leu, leucine; Lys, lysine; Met, methionine; NAD+, oxidized form of NAD; Phe, phenylalanine; Pi, inorganic phosphate; Pro, proline; Ser, serine; Thy, threonine; Trp, tryptophan; Tyr, tyrosine; Val, valine.
In the kidney, proximal tubular cells are abundant in mitochondria, likely because they are highly energy demanding due to the need for reabsorption of glucose and sodium. Thus, mitochondria play crucial roles in maintaining renal function. Peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α), a mitochondrial biogenesis regulator, plays a pivotal role in proximal tubular recovery from AKI by regulating NAD biosynthesis (2). Mitochondrial dysfunction in the proximal tubular cells after AKI results in the progression of CKD (3,4).
Proximal tubular cells predominantly rely on fatty acids as an energy source. Genome-wide transcriptome studies of human kidney samples revealed the lower expression of key enzymes of fatty acid β-oxidation in fibrotic kidneys than in healthy kidneys (Figure 2A) (5). Moreover, restoring fatty acid metabolism by genetic or pharmacologic methods was found to protect mice from tubulointerstitial fibrosis (5). Although it is difficult to discern which occurs first, defective fatty acid β-oxidation or mitochondrial dysfunction, it is apparent that energy production in mitochondria has a key role in CKD progression.
Figure 2.
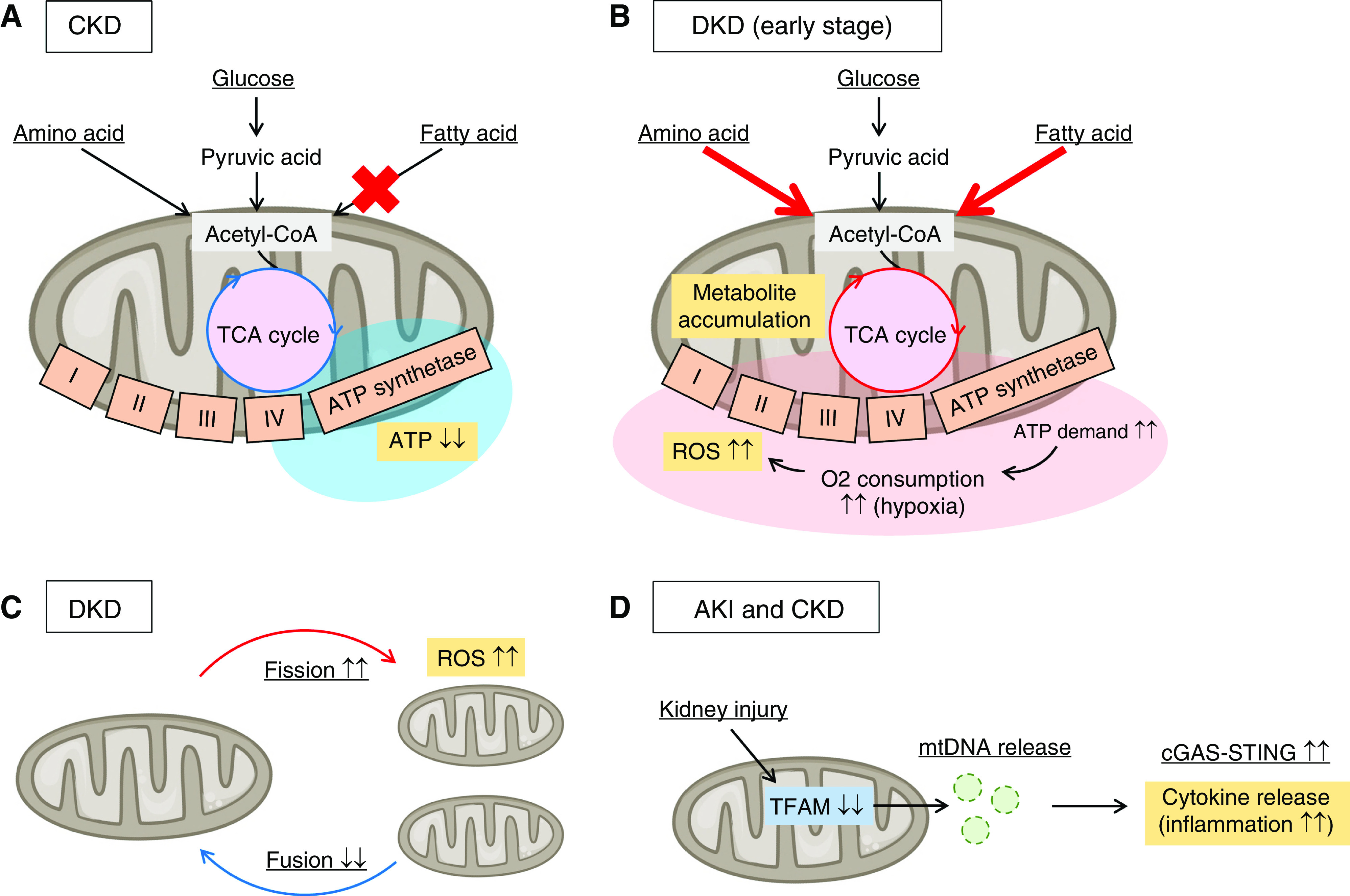
Mitochondrial stress responses are deeply involved in kidney disease progression. (A) Energy metabolism alterations of proximal tubules in CKD are shown. Defective fatty acid β-oxidation and mitochondrial dysfunction are observed in fibrotic kidneys, leading to ATP shortage. (B) Energy metabolism alterations of proximal tubules in the early stage of diabetic kidney disease (DKD) are shown. Mitochondrial respiration (mainly fueled by fatty acids and amino acids) is forcibly activated to meet the energy demand for glucose reabsorption in the hyperglycemic state, which increases oxygen consumption and results in renal hypoxia. Mitochondrial respiration in the hypoxic state produces a large amount of reactive oxygen species (ROS). (C) The imbalance in mitochondrial fission and fusion is observed in DKD. The increase in mitochondrial fission results in ROS overproduction. (D) Mitochondrial defects, including the loss of mitochondrial transcription factor A (TFAM) in proximal tubular cells, induce translocation of mitochondrial DNA (mtDNA) to the cytosol, which activates the innate immune pathway, cGAS-STING. This inflammatory response is observed both in AKI and CKD. O2, oxygen.
Energy metabolism is also altered in the kidneys of patients with diabetic kidney disease (DKD) (Figure 2B). Systemic metabolic disorders such as hyperglycemia and dyslipidemia cause metabolism alterations in renal tissue (6). Glucose and TCA cycle metabolites are accumulated in diabetic renal tissue, which might be related to mitochondrial dysfunction (7,8). These metabolite accumulations may occur because the TCA cycle in mitochondria is forcibly activated to meet the energy demand for glucose reabsorption in the hyperglycemic state (9), which increases oxygen consumption and results in renal hypoxia (10). Mitochondrial respiration in the hypoxic state produces a large amount of reactive oxygen species (ROS), in association with mitochondrial fragmentation (11). The imbalance in mitochondrial fission and fusion is also observed in the diabetic state (Figure 2C); the expression of mitofusin 2, which is essential for mitochondrial fusion, is decreased (12) and the activity of dynamin-related protein 1 (Drp1), a mediator of mitochondrial fission, is conversely increased. Indeed, Drp1 inhibition in podocytes reduces ROS levels in diabetic mouse models (13). Thus, energy metabolism alterations and morphologic alterations of mitochondria are closely related, and result in ROS overproduction in diabetic renal tissue.
Mitochondrial damage directly stimulates innate immune mechanisms and promotes inflammation (Figure 2D). Mitochondrial defects, including the loss of mitochondrial transcription factor A, are observed in the tubular cells of fibrotic kidneys (14). Tubule-specific deletion of mitochondrial transcription factor A induces not only severe metabolic and energy defects, but also translocation of mitochondrial DNA (mtDNA) to the cytosol, which activates the innate immune pathway, cGAS-STING. The cGAS-STING pathway was originally identified as the defensive mechanism against microorganism invasion (15). cGAS senses cytoplasmic double-stranded DNA (dsDNA) and activates STING, which induces many genes related to inflammation (16–21). cGAS senses not only exogenous dsDNA, but also self-dsDNA. Thus, mtDNA translocated to the cytosol after mitochondrial damage is sensed by cGAS and promotes inflammation in the renal tubular cells, which leads to kidney fibrosis (14). This inflammatory mechanism is also observed in the acute phase of cisplatin-induced AKI (22). Thus, mitochondrial dysfunction in association with energy metabolism alterations is closely related to kidney disease progression via ROS production and inflammation.
Endoplasmic Reticulum
Endoplasmic reticula (ER) play critical roles in controlling protein quality (folding and maturation). If cells are exposed to environmental change, the folding process is disturbed and unfolded proteins accumulate in cells, which is called ER stress. Unfolded protein response (UPR) pathways are the cellular response to ER stress for maintaining protein homeostasis (Figure 3) (23,24). UPR pathways are regulated by three sensors in the ER lumen: inositol-requiring enzyme 1 (IRE1), protein kinase R-like ER kinase (PERK), and activating transcription factor 6 (ATF6). These sensors attach to binding Ig protein (BiP/GRP78) and are inactivated in normal conditions. Under ER stress conditions, these ER sensors are separated from BiP/GRP78 and activated. Activated IRE1 induces the splicing of X-box binding protein 1 (XBP1) mRNA (25,26). Activated PERK phosphorylates eukaryotic initiation factor 2α, which promotes the translation of ATF4 (27) and suppresses the translation of other mRNAs to reduce unfolded proteins (28). ATF6 translocates to the Golgi apparatus and is cleaved to form an active fragment (ATF6 p50) (29,30). Spliced XBP1, ATF4, and ATF6 p50 induce the transcription of various UPR target genes such as chaperone, ER-associated protein degradation, and apoptosis-related genes. Note that mitochondria have their own stress response against the accumulation of unfolded protein, which is called mitochondrial UPR (31,32).
Figure 3.

Unfolded protein response (UPR) pathways are the cellular response to endoplasmic reticulum (ER) stress. UPR pathways consist of three sensors: inositol-requiring enzyme 1 (IRE1), protein kinase R-like endoplasmic reticulum kinase (PERK), and activating transcription factor 6 (ATF6), which are activated under ER stress. Activated IRE1 induces the splicing of X-box binding protein 1 (XBP1) mRNA. Activated PERK phosphorylates (P) eukaryotic initiation factor 2α (eIF2α), which promotes the translation of ATF4 and suppresses the translation of other mRNAs to reduce unfolded proteins. ATF6 translocates to the Golgi apparatus and is cleaved to form an active fragment (ATF6 p50). Spliced XBP1, ATF4, and ATF6 p50 induce the transcription of various UPR target genes including chaperone, ER-asociated protein degradation (ERAD), and apoptosis-related genes.
Pathogenic ER stress leads to maladaptive activation of UPR pathways, which causes various diseases such as Parkinson disease (33) and Alzheimer disease (34). The kidneys are also exposed to pathogenic ER stress under oxidative stress, glycative stress, and hypoxia (35). Dysregulation of UPR pathways often occurs in glomerular and tubulointerstitial cells. Pathogenic ER stress induces podocyte injury (36), which is related to the progression of GN (37). Renal tubular cells exposed to high urinary albumin are also affected by ER stress, which leads to tubular apoptosis (38). Pathogenic ER stress is activated after ischemia-reperfusion injury and promotes CKD progression (39). Maladaptive ATF6 activation induced by ischemia-reperfusion injury also suppresses fatty acid β-oxidation, which contributes to tubular apoptosis and subsequent tubulointerstitial fibrosis (40). Moreover, renal erythropoietin (EPO)-producing cells are affected by ER stress. Palmitate, a long-chain saturated fatty acid, induces maladaptive ATF4 activation and deranges EPO production in renal EPO-producing cells (41,42).
The detailed roles of ER stress on kidney disease progression have been clarified by using genetically modified animals. For example, XBP1 is important for maintaining podocytes’ homeostasis. Although podocyte-specific ablation of XBP1 or SEC63, which encodes an ER chaperone protein, does not show glomerular injury up to 1 year of age, podocyte-specific double knockout of these genes demonstrates progressive albuminuria, foot process effacement, and increased glomerular apoptosis (43). The simultaneous inactivation of XBP1 and SEC63 in collecting ducts also induces inflammation and myofibroblast activation, leading to chronic tubulointerstitial kidney injury (44). Moreover, XBP1 plays a critical role in the progression of DKD. Podocyte-specific genetic ablation of XBP1 in mice aggravates DKD pathophysiology. Defective podocyte insulin signaling impairs the nuclear translocation of spliced XBP1, which promotes maladaptive ATF6 activation in DKD (45). Meanwhile, podocyte-specific deletion of IRE1 spontaneously results in foot process effacement and microvillus transformation along with worsening albuminuria with time, which is partly due to reduced autophagy in podocytes (46). In this manner, ER stress is deeply involved in the pathophysiology of kidney diseases.
Primary Cilia
The cilium is a hairlike structure on the cell surface. Nearly all mammalian cells have a single, nonmotile cilium, which is called a primary cilium. Primary cilia sense a wide variety of extracellular signals and transmit them to the interior of cells. Genetic defects of primary cilia cause various diseases, known as ciliopathies (47). Autosomal dominant polycystic kidney disease (ADPKD) is a ciliopathy of the kidney. Polycystin-1 (PC1) and polycystin-2 (PC2), the genes that are mutated in ADPKD, are located in the cilia of the renal proximal tubular cells (48–50). PC1 is a transmembrane mechanosensor receptor, and PC2 is a calcium channel. PC1 regulates cellular calcium influx by physically sensing urinary flow and interacting with PC2, which appropriately maintains the renal tubular diameter. Disruption of cellular calcium homeostasis increases cAMP levels and affects the cell cycle, leading to increased tubular cell proliferation and retention of cyst fluid. In this manner, the signaling pathways activated by the primary cilia play an important role in maintaining the homeostasis of renal tubular environments.
Organelle Crosstalk
Crosstalk between the ER and Mitochondria
Organelles interact with one another to maintain the cellular homeostasis. The direct interaction at membrane contact sites has recently received increased attention in association with the development of imaging technologies. Valm et al. (51) used confocal and lattice light-sheet microscopy (52) and an imaging informatics pipeline to map organelle numbers, volumes, speeds, positions, and dynamic interorganelle contact in live cells, and found that contact between the ER and mitochondria occurs most frequently among organelle interactions. Kakimoto et al. (53) also developed the organelle-targeted, split–green fluorescent protein system to visualize multiple membrane contact sites, including ER-mitochondria contact sites in living cells.
The part of the ER that is directly connected to mitochondria is termed the mitochondria-associated ER membrane (MAM) (Figure 4A) (54,55). Calcium is transported from the ER to mitochondria via the inositol trisphosphate receptor and the voltage-dependent anion channel (56). During the adaptive phase of ER stress, diverse parameters of mitochondrial metabolism are enhanced in association with the increased mitochondrial calcium uptake (57). Although the appropriate increase in calcium concentration activates oxidative phosphorylation in mitochondria, the excessive increase in calcium concentration releases cytochrome c and results in apoptosis. Thus, disruption of MAM integrity collapses cellular homeostasis. For example, disruption of MAM integrity contributes to insulin resistance in the liver (58) and muscles (59). Overexpression of mitofusin 2 (60) and glucose-regulated protein 75 (61), the key molecules for MAM integrity, in hepatocytes prevents palmitate-induced deficiency in insulin signaling. MAM integrity is also involved in progression of kidney disease. Igwebuike et al. (62) have clarified that disruption of MAM integrity, which is also called crossorganelle stress response disruption, occurs at the early stage of gentamicin-induced AKI, which precedes downstream UPR activation and cell death. In contrast, increased connectivity between the ER and mitochondria is observed in patients with familial Alzheimer disease (63). MAMs are critical for phospholipid and cholesterol metabolism, as well as calcium homeostasis; patients with Alzheimer disease exhibit alterations in phospholipid metabolism in the ER and mitochondria, leading to the accumulation of hyperphosphorylated forms of τ proteins in tissues (64). MAMs are also involved in mitochondrial fission. Mitochondrial division occurs at positions at which the ER contact mitochondria and mediate constriction before Drp1 recruitment (65). In this manner, the ER and mitochondria directly interact with one another, which maintains cellular homeostasis.
Figure 4.

Organelles closely interact with one another to maintain cellular functions. (A) The crosstalk between the ER and mitochondria is shown. The part of the ER that is directly connected to mitochondria is termed the mitochondria-associated ER membrane (MAM), which consists of various proteins including inositol trisphosphate receptor (IP3R), voltage-dependent anion channel (VDAC), glucose-regulated protein 75 (GRP75), and mitofusin 2 (Mfn2). MAMs maintain cellular homeostasis. Calcium (Ca2+) is transported from the ER to mitochondria via IP3R and VDAC. The mitochondrial fission also occurs at the MAM’s position. (B) The link between transmembrane and coiled-coil domain family 1 (TMCC1) and coronin 1C at the ER-endosome membrane contact sites defines the timing and position of endosomal fission. (C) The crosstalk between the primary cilia and mitochondria in autosomal dominant polycystic kidney disease (ADPKD) is shown. Polycystin-1 (PC1) and polycystin-2 (PC2) are located in the cilia of the renal proximal tubular cells and regulate cellular calcium homeostasis. The mutations of PC1 and PC2 in ADPKD reduce the cellular calcium concentration. The decrease in calcium concentration suppresses peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α) expression, the master regulator of mitochondrial biogenesis.
Crosstalk between the ER and Endosomes
Endosomes are membrane-bound vesicles that transport a wide range of proteins between the ER and Golgi apparatus, or from the Golgi apparatus to lysosomes inside cells. ER contact defines the timing and position of endosomal fission in a similar manner as mitochondrial fission (Figure 4B). ER membrane contact sites mark the positions at which endosomes undergo fission for cargo sorting (66). The link between transmembrane and coiled-coil domain family 1 (the ER membrane protein) and coronin 1C (the endosome-localized actin regulator) at the ER-endosome membrane contact sites is required for endosomal fission (67).
The crosstalk between the ER and endosomes is also involved in kidney diseases. Mucin 1 kidney disease (MKD) is an autosomal dominant tubulointerstitial kidney disease characterized by gradually progressive tubule-interstitial cyst formation (68). MKD results from a frameshift mutation in the MUC1 gene. The abnormal MUC1 protein (MUC1-fs) is trapped in TMED9 cargo receptor–containing endosomes between the ER and Golgi apparatus, which prevents the unfolded proteins from trafficking through the secretory pathway to the lysosome for degradation (69). Thus, MUC1-fs accumulates in the renal tubular cells, which activates the ATF6 branch of UPR pathways and ultimately leads to tubular injury. A high-content screen was conducted to identify compounds that can remove MUC1-fs. One of the candidate compounds (BRD4780) binds TMED9, releases MUC1-fs, and reroutes it for lysosomal degradation, both in animal models and kidney organoids derived from patients’ induced pluripotent stem cells. This compound is a promising lead for the treatment of kidney diseases induced by organelle stress.
Crosstalk between the Cilia and Mitochondria
Not only primary cilia but also mitochondria are closely involved in the pathophysiology of ADPKD (Figure 4). The mtDNA copy number and PGC-1α expression are reduced in the kidneys of ADPKD model mice, and PGC-1α expression inversely correlates with oxidative stress levels (70). PC1 and PC2, the genes that are mutated in ADPKD, are located in the cilia of the renal proximal tubular cells and regulate cellular calcium homeostasis. The decrease in the cellular calcium concentration suppresses PGC-1α expression via calcineurin, p38 mitogen-activated protein kinase, and nitric oxide synthase deactivation. This finding clearly demonstrates that the crosstalk between the primary cilia and mitochondria is deeply related to the pathophysiology of ADPKD.
Therapeutic Approach Based on Organelle Homeostasis
Although there are no treatments that directly target organelle deficiency to date, certain existing medications indirectly improve organelle homeostasis in the kidney. Filtered glucose in the glomerulus is reabsorbed together with sodium, primarily in the S1/S2 segments of the proximal tubules via sodium-glucose cotransporter 2 (SGLT2). SGLT2 inhibitors were developed to lower blood glucose levels in patients with type 2 diabetes. The Canagliflozin and Renal Events in Diabetes with Established Nephropathy Clinical Evaluation (CREDENCE) trial clearly illustrates that canagliflozin, an SGLT2 inhibitor, improves the renal outcome in patients with type 2 diabetes and CKD (71). SGLT2 inhibition reverses the accumulation of TCA cycle metabolites and relieves oxidative stress (72), which may reduce the mitochondrial burden.
Hypoxia-inducible factor (HIF) prolyl hydroxylase inhibitors (also known as HIF stabilizers) increase endogenous EPO production and serve as novel therapeutic agents against anemia in CKD (73). In addition, HIF induces the metabolic reprogramming from the TCA cycle to glycolysis, which represses oxygen consumption and is critical for the adaptation of cells exposed to hypoxic environments (74,75). Our transcriptome and metabolome analyses of renal tissue in diabetic rat and mouse models have revealed that HIF stabilization counteracts renal energy metabolism alterations and reduces oxidative stress in the early stages of DKD, in association with the improvement in renal pathologic abnormalities (9). This result suggests that HIF stabilization mitigates the mitochondrial burden in diabetic renal tissue and serves as a potential intervention, targeting energy metabolism dysregulation in diabetic kidneys.
The development of novel drugs that directly target organelle stress has been challenging. As indicated in the former section, the mechanism-based strategy with a high-content screen was successful in identifying a promising compound, BRD4780, for the treatment of MKD (69). BRD4780 directly contributes to the degradation of abnormal proteins and reduces ER stress. BRD4780 exhibits no overt toxicity and thus holds significant potential for successful development into a therapeutic agent. Also, therapeutic attempts that target podocyte ER stress have recently been reported. Podocyte ER calcium release channel, type 2 ryanodine receptor (RyR2), undergoes phosphorylation during ER stress. The accelerated podocyte ER calcium efflux due to RyR2 remodeling leads to podocyte injury. Park et al. (76) have identified a chemical compound (K201) and a biotherapeutic protein (mesencephalic astrocyte-derived neurotrophic factor), that can prevent RyR2 remodeling and attenuate podocyte injury in the nephrotic syndrome mouse model. Further attempts, such as these studies, are needed to develop essential treatment options for kidney diseases that directly target organelle stress and crosstalk.
Disclosures
All authors have nothing to disclose.
Funding
This work was supported by the Japan Society for the Promotion of Science Grant-in-Aid for Research Fellow KAKENHI grant 19J11928 (to S. Hasegawa) and Grant-in-Aid for Scientific Research (B) KAKENHI grant 18H02727 (to R. Inagi). R. Inagi also received research funding from Kyowa Kirin Co. Ltd.
Author Contributions
S. Hasegawa wrote the original draft; S. Hasegawa and R. Inagi reviewed and edited the manuscript.
References
- 1.Salvador-Gallego R, Hoyer MJ, Voeltz GK: SnapShot: Functions of endoplasmic reticulum membrane contact sites. Cell 171: 1224–1224.e1, 2017 [DOI] [PubMed] [Google Scholar]
- 2.Tran MT, Zsengeller ZK, Berg AH, Khankin EV, Bhasin MK, Kim W, Clish CB, Stillman IE, Karumanchi SA, Rhee EP, Parikh SM: PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 531: 528–532, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ishimoto Y, Inagi R: Mitochondria: A therapeutic target in acute kidney injury. Nephrol Dial Transplant 31: 1062–1069, 2016 [DOI] [PubMed] [Google Scholar]
- 4.Inoue T, Maekawa H, Inagi R: Organelle crosstalk in the kidney. Kidney Int 95: 1318–1325, 2019 [DOI] [PubMed] [Google Scholar]
- 5.Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, Park AS, Tao J, Sharma K, Pullman J, Bottinger EP, Goldberg IJ, Susztak K: Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med 21: 37–46, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hasegawa S, Jao TM, Inagi R: Dietary metabolites and chronic kidney disease. Nutrients 9: 358, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sas KM, Kayampilly P, Byun J, Nair V, Hinder LM, Hur J, Zhang H, Lin C, Qi NR, Michailidis G, Groop PH, Nelson RG, Darshi M, Sharma K, Schelling JR, Sedor JR, Pop-Busui R, Weinberg JM, Soleimanpour SA, Abcouwer SF, Gardner TW, Burant CF, Feldman EL, Kretzler M, Brosius FC 3rd, Pennathur S: Tissue-specific metabolic reprogramming drives nutrient flux in diabetic complications. JCI Insight 1: e86976, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.You YH, Quach T, Saito R, Pham J, Sharma K: Metabolomics reveals a key role for fumarate in mediating the effects of NADPH oxidase 4 in diabetic kidney disease. J Am Soc Nephrol 27: 466–481, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hasegawa S, Tanaka T, Saito T, Fukui K, Wakashima T, Susaki EA, Ueda HR, Nangaku M: The oral hypoxia-inducible factor prolyl hydroxylase inhibitor enarodustat counteracts alterations in renal energy metabolism in the early stages of diabetic kidney disease. Kidney Int 97: 934–950, 2020 [DOI] [PubMed] [Google Scholar]
- 10.Rosenberger C, Khamaisi M, Abassi Z, Shilo V, Weksler-Zangen S, Goldfarb M, Shina A, Zibertrest F, Eckardt KU, Rosen S, Heyman SN: Adaptation to hypoxia in the diabetic rat kidney. Kidney Int 73: 34–42, 2008 [DOI] [PubMed] [Google Scholar]
- 11.Yu T, Robotham JL, Yoon Y: Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U S A 103: 2653–2658, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zorzano A, Liesa M, Palacín M: Mitochondrial dynamics as a bridge between mitochondrial dysfunction and insulin resistance. Arch Physiol Biochem 115: 1–12, 2009 [DOI] [PubMed] [Google Scholar]
- 13.Ayanga BA, Badal SS, Wang Y, Galvan DL, Chang BH, Schumacker PT, Danesh FR: Dynamin-related protein 1 deficiency improves mitochondrial fitness and protects against progression of diabetic nephropathy. J Am Soc Nephrol 27: 2733–2747, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chung KW, Dhillon P, Huang S, Sheng X, Shrestha R, Qiu C, Kaufman BA, Park J, Pei L, Baur J, Palmer M, Susztak K: Mitochondrial damage and activation of the STING pathway lead to renal inflammation and fibrosis. Cell Metab 30: 784–799.e5, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barber GN: STING: Infection, inflammation and cancer. Nat Rev Immunol 15: 760–770, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishikawa H, Barber GN: STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling [Published correction appears in Nature 456: 274, 2008]. Nature 455: 674–678, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishikawa H, Ma Z, Barber GN: STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461: 788–792, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE: STING is a direct innate immune sensor of cyclic di-GMP. Nature 478: 515–518, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yin Q, Tian Y, Kabaleeswaran V, Jiang X, Tu D, Eck MJ, Chen ZJ, Wu H: Cyclic di-GMP sensing via the innate immune signaling protein STING. Mol Cell 46: 735–745, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abe T, Harashima A, Xia T, Konno H, Konno K, Morales A, Ahn J, Gutman D, Barber GN: STING recognition of cytoplasmic DNA instigates cellular defense. Mol Cell 50: 5–15, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, Serganov AA, Liu Y, Jones RA, Hartmann G, Tuschl T, Patel DJ: Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153: 1094–1107, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maekawa H, Inoue T, Ouchi H, Jao TM, Inoue R, Nishi H, Fujii R, Ishidate F, Tanaka T, Hirokawa N, Nangaku M, Inagi R: Mitochondrial damage causes inflammation via cGAS-STING signaling in acute kidney injury. Cell Rep 29: 1261–1273.e6, 2019 [DOI] [PubMed] [Google Scholar]
- 23.Cox JS, Shamu CE, Walter P: Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73: 1197–1206, 1993 [DOI] [PubMed] [Google Scholar]
- 24.Mori K, Ma W, Gething MJ, Sambrook J: A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 74: 743–756, 1993 [DOI] [PubMed] [Google Scholar]
- 25.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K: XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107: 881–891, 2001 [DOI] [PubMed] [Google Scholar]
- 26.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D: IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA [Published correction appears in Nature 420: 202, 2002]. Nature 415: 92–96, 2002 [DOI] [PubMed] [Google Scholar]
- 27.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D: Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6: 1099–1108, 2000 [DOI] [PubMed] [Google Scholar]
- 28.Harding HP, Zhang Y, Ron D: Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397: 271–274, 1999 [DOI] [PubMed] [Google Scholar]
- 29.Yoshida H, Haze K, Yanagi H, Yura T, Mori K: Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem 273: 33741–33749, 1998 [DOI] [PubMed] [Google Scholar]
- 30.Haze K, Yoshida H, Yanagi H, Yura T, Mori K: Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell 10: 3787–3799, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ: A mitochondrial specific stress response in mammalian cells. EMBO J 21: 4411–4419, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shpilka T, Haynes CM: The mitochondrial UPR: Mechanisms, physiological functions and implications in ageing. Nat Rev Mol Cell Biol 19: 109–120, 2018 [DOI] [PubMed] [Google Scholar]
- 33.Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R: An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell 105: 891–902, 2001 [DOI] [PubMed] [Google Scholar]
- 34.Katayama T, Imaizumi K, Sato N, Miyoshi K, Kudo T, Hitomi J, Morihara T, Yoneda T, Gomi F, Mori Y, Nakano Y, Takeda J, Tsuda T, Itoyama Y, Murayama O, Takashima A, St George-Hyslop P, Takeda M, Tohyama M: Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat Cell Biol 1: 479–485, 1999 [DOI] [PubMed] [Google Scholar]
- 35.Inagi R, Ishimoto Y, Nangaku M: Proteostasis in endoplasmic reticulum--new mechanisms in kidney disease. Nat Rev Nephrol 10: 369–378, 2014 [DOI] [PubMed] [Google Scholar]
- 36.Inagi R, Nangaku M, Onogi H, Ueyama H, Kitao Y, Nakazato K, Ogawa S, Kurokawa K, Couser WG, Miyata T: Involvement of endoplasmic reticulum (ER) stress in podocyte injury induced by excessive protein accumulation. Kidney Int 68: 2639–2650, 2005 [DOI] [PubMed] [Google Scholar]
- 37.Inagi R, Kumagai T, Nishi H, Kawakami T, Miyata T, Fujita T, Nangaku M: Preconditioning with endoplasmic reticulum stress ameliorates mesangioproliferative glomerulonephritis. J Am Soc Nephrol 19: 915–922, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohse T, Inagi R, Tanaka T, Ota T, Miyata T, Kojima I, Ingelfinger JR, Ogawa S, Fujita T, Nangaku M: Albumin induces endoplasmic reticulum stress and apoptosis in renal proximal tubular cells. Kidney Int 70: 1447–1455, 2006 [DOI] [PubMed] [Google Scholar]
- 39.Shu S, Zhu J, Liu Z, Tang C, Cai J, Dong Z: Endoplasmic reticulum stress is activated in post-ischemic kidneys to promote chronic kidney disease. EBioMedicine 37: 269–280, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jao TM, Nangaku M, Wu CH, Sugahara M, Saito H, Maekawa H, Ishimoto Y, Aoe M, Inoue T, Tanaka T, Staels B, Mori K, Inagi R: ATF6α downregulation of PPARα promotes lipotoxicity-induced tubulointerstitial fibrosis. Kidney Int 95: 577–589, 2019 [DOI] [PubMed] [Google Scholar]
- 41.Chiang CK, Nangaku M, Tanaka T, Iwawaki T, Inagi R: Endoplasmic reticulum stress signal impairs erythropoietin production: A role for ATF4. Am J Physiol Cell Physiol 304: C342–C353, 2013 [DOI] [PubMed] [Google Scholar]
- 42.Anusornvongchai T, Nangaku M, Jao TM, Wu CH, Ishimoto Y, Maekawa H, Tanaka T, Shimizu A, Yamamoto M, Suzuki N, Sassa R, Inagi R: Palmitate deranges erythropoietin production via transcription factor ATF4 activation of unfolded protein response. Kidney Int 94: 536–550, 2018 [DOI] [PubMed] [Google Scholar]
- 43.Hassan H, Tian X, Inoue K, Chai N, Liu C, Soda K, Moeckel G, Tufro A, Lee AH, Somlo S, Fedeles S, Ishibe S: Essential role of X-box binding protein-1 during endoplasmic reticulum stress in podocytes. J Am Soc Nephrol 27: 1055–1065, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ishikawa Y, Fedeles S, Marlier A, Zhang C, Gallagher AR, Lee AH, Somlo S: Spliced XBP1 rescues renal interstitial inflammation due to loss of Sec63 in collecting ducts. J Am Soc Nephrol 30: 433–459, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Madhusudhan T, Wang H, Dong W, Ghosh S, Bock F, Thangapandi VR, Ranjan S, Wolter J, Kohli S, Shahzad K, Heidel F, Krueger M, Schwenger V, Moeller MJ, Kalinski T, Reiser J, Chavakis T, Isermann B: Defective podocyte insulin signalling through p85-XBP1 promotes ATF6-dependent maladaptive ER-stress response in diabetic nephropathy. Nat Commun 6: 6496, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaufman DR, Papillon J, Larose L, Iwawaki T, Cybulsky AV: Deletion of inositol-requiring enzyme-1α in podocytes disrupts glomerular capillary integrity and autophagy. Mol Biol Cell 28: 1636–1651, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hildebrandt F, Benzing T, Katsanis N: Ciliopathies. N Engl J Med 364: 1533–1543, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Igarashi P, Somlo S: Genetics and pathogenesis of polycystic kidney disease. J Am Soc Nephrol 13: 2384–2398, 2002 [DOI] [PubMed] [Google Scholar]
- 49.Pazour GJ, San Agustin JT, Follit JA, Rosenbaum JL, Witman GB: Polycystin-2 localizes to kidney cilia and the ciliary level is elevated in orpk mice with polycystic kidney disease. Curr Biol 12: R378–R380, 2002 [DOI] [PubMed] [Google Scholar]
- 50.Yoder BK, Hou X, Guay-Woodford LM: The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol 13: 2508–2516, 2002 [DOI] [PubMed] [Google Scholar]
- 51.Valm AM, Cohen S, Legant WR, Melunis J, Hershberg U, Wait E, Cohen AR, Davidson MW, Betzig E, Lippincott-Schwartz J: Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 546: 162–167, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen BC, Legant WR, Wang K, Shao L, Milkie DE, Davidson MW, Janetopoulos C, Wu XS, Hammer JA 3rd, Liu Z, English BP, Mimori-Kiyosue Y, Romero DP, Ritter AT, Lippincott-Schwartz J, Fritz-Laylin L, Mullins RD, Mitchell DM, Bembenek JN, Reymann AC, Böhme R, Grill SW, Wang JT, Seydoux G, Tulu US, Kiehart DP, Betzig E: Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution. Science 346: 1257998, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kakimoto Y, Tashiro S, Kojima R, Morozumi Y, Endo T, Tamura Y: Visualizing multiple inter-organelle contact sites using the organelle-targeted split-GFP system. Sci Rep 8: 6175, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gatta AT, Levine TP: Piecing together the patchwork of contact sites. Trends Cell Biol 27: 214–229, 2017 [DOI] [PubMed] [Google Scholar]
- 55.Annunziata I, Sano R, d’Azzo A: Mitochondria-associated ER membranes (MAMs) and lysosomal storage diseases. Cell Death Dis 9: 328, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Vliet AR, Verfaillie T, Agostinis P: New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta 1843: 2253–2262, 2014 [DOI] [PubMed] [Google Scholar]
- 57.Bravo R, Vicencio JM, Parra V, Troncoso R, Munoz JP, Bui M, Quiroga C, Rodriguez AE, Verdejo HE, Ferreira J, Iglewski M, Chiong M, Simmen T, Zorzano A, Hill JA, Rothermel BA, Szabadkai G, Lavandero S: Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J Cell Sci 124: 2143–2152, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tubbs E, Theurey P, Vial G, Bendridi N, Bravard A, Chauvin MA, Ji-Cao J, Zoulim F, Bartosch B, Ovize M, Vidal H, Rieusset J: Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 63: 3279–3294, 2014 [DOI] [PubMed] [Google Scholar]
- 59.Tubbs E, Chanon S, Robert M, Bendridi N, Bidaux G, Chauvin MA, Ji-Cao J, Durand C, Gauvrit-Ramette D, Vidal H, Lefai E, Rieusset J: Disruption of mitochondria-associated endoplasmic reticulum membrane (MAM) integrity contributes to muscle insulin resistance in mice and humans. Diabetes 67: 636–650, 2018 [DOI] [PubMed] [Google Scholar]
- 60.de Brito OM, Scorrano L: Mitofusin 2 tethers endoplasmic reticulum to mitochondria [Published correction appears in Nature 513: 266, 2014]. Nature 456: 605–610, 2008 [DOI] [PubMed] [Google Scholar]
- 61.Honrath B, Metz I, Bendridi N, Rieusset J, Culmsee C, Dolga AM: Glucose-regulated protein 75 determines ER-mitochondrial coupling and sensitivity to oxidative stress in neuronal cells. Cell Death Discov 3: 17076, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Igwebuike C, Yaglom J, Huiting L, Feng H, Campbell JD, Wang Z, Havasi A, Pimentel D, Sherman MY, Borkan SC: Cross organelle stress response disruption promotes gentamicin-induced proteotoxicity. Cell Death Dis 11: 217, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hedskog L, Pinho CM, Filadi R, Rönnbäck A, Hertwig L, Wiehager B, Larssen P, Gellhaar S, Sandebring A, Westerlund M, Graff C, Winblad B, Galter D, Behbahani H, Pizzo P, Glaser E, Ankarcrona M: Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc Natl Acad Sci U S A 110: 7916–7921, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schon EA, Area-Gomez E: Mitochondria-associated ER membranes in Alzheimer disease. Mol Cell Neurosci 55: 26–36, 2013 [DOI] [PubMed] [Google Scholar]
- 65.Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK: ER tubules mark sites of mitochondrial division. Science 334: 358–362, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rowland AA, Chitwood PJ, Phillips MJ, Voeltz GK: ER contact sites define the position and timing of endosome fission. Cell 159: 1027–1041, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hoyer MJ, Chitwood PJ, Ebmeier CC, Striepen JF, Qi RZ, Old WM, Voeltz GK: A novel class of ER membrane proteins regulates ER-associated endosome fission. Cell 175: 254–265.e14, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kirby A, Gnirke A, Jaffe DB, Barešová V, Pochet N, Blumenstiel B, Ye C, Aird D, Stevens C, Robinson JT, Cabili MN, Gat-Viks I, Kelliher E, Daza R, DeFelice M, Hůlková H, Sovová J, Vylet’al P, Antignac C, Guttman M, Handsaker RE, Perrin D, Steelman S, Sigurdsson S, Scheinman SJ, Sougnez C, Cibulskis K, Parkin M, Green T, Rossin E, Zody MC, Xavier RJ, Pollak MR, Alper SL, Lindblad-Toh K, Gabriel S, Hart PS, Regev A, Nusbaum C, Kmoch S, Bleyer AJ, Lander ES, Daly MJ: Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nat Genet 45: 299–303, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dvela-Levitt M, Kost-Alimova M, Emani M, Kohnert E, Thompson R, Sidhom EH, Rivadeneira A, Sahakian N, Roignot J, Papagregoriou G, Montesinos MS, Clark AR, McKinney D, Gutierrez J, Roth M, Ronco L, Elonga E, Carter TA, Gnirke A, Melonson M, Hartland K, Wieder N, Hsu JCH, Deltas C, Hughey R, Bleyer AJ, Kmoch S, Zivna M, Baresova V, Kota S, Schlondorff J, Heiman M, Alper SL, Wagner F, Weins A, Golub TR, Lander ES, Greka A: Small molecule targets TMED9 and promotes lysosomal degradation to reverse proteinopathy. Cell 178: 521–535.e23, 2019 [DOI] [PubMed] [Google Scholar]
- 70.Ishimoto Y, Inagi R, Yoshihara D, Kugita M, Nagao S, Shimizu A, Takeda N, Wake M, Honda K, Zhou J, Nangaku M: Mitochondrial abnormality facilitates cyst formation in autosomal dominant polycystic kidney disease. Mol Cell Biol 37: e00337–e00417, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, Edwards R, Agarwal R, Bakris G, Bull S, Cannon CP, Capuano G, Chu PL, de Zeeuw D, Greene T, Levin A, Pollock C, Wheeler DC, Yavin Y, Zhang H, Zinman B, Meininger G, Brenner BM, Mahaffey KW; CREDENCE Trial Investigators : Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med 380: 2295–2306, 2019 [DOI] [PubMed] [Google Scholar]
- 72.Tanaka S, Sugiura Y, Saito H, Sugahara M, Higashijima Y, Yamaguchi J, Inagi R, Suematsu M, Nangaku M, Tanaka T: Sodium-glucose cotransporter 2 inhibition normalizes glucose metabolism and suppresses oxidative stress in the kidneys of diabetic mice. Kidney Int 94: 912–925, 2018 [DOI] [PubMed] [Google Scholar]
- 73.Hasegawa S, Tanaka T, Nangaku M: Hypoxia-inducible factor stabilizers for treating anemia of chronic kidney disease. Curr Opin Nephrol Hypertens 27: 331–338, 2018 [DOI] [PubMed] [Google Scholar]
- 74.Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC: HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 3: 187–197, 2006 [DOI] [PubMed] [Google Scholar]
- 75.Kim JW, Tchernyshyov I, Semenza GL, Dang CV: HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3: 177–185, 2006 [DOI] [PubMed] [Google Scholar]
- 76.Park SJ, Kim Y, Yang SM, Henderson MJ, Yang W, Lindahl M, Urano F, Chen YM: Discovery of endoplasmic reticulum calcium stabilizers to rescue ER-stressed podocytes in nephrotic syndrome. Proc Natl Acad Sci U S A 116: 14154–14163, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]