

SUMMARY
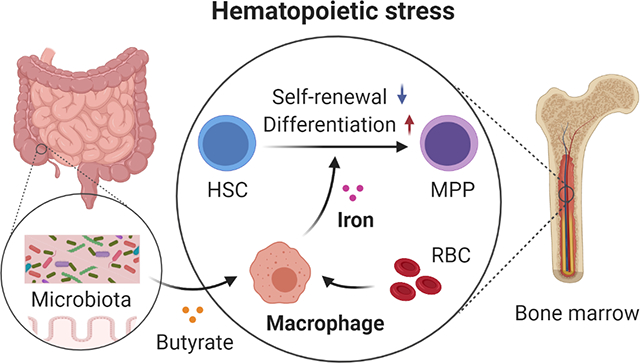
Host microbiota crosstalk is essential for the production and functional modulation of blood cell lineages. Whether, and if so how, the microbiota influences hematopoietic stem cells (HSCs) is unclear. Here, we show that the microbiota regulates HSC self-renewal and differentiation under stress conditions by modulating local iron availability in the bone marrow (BM). In microbiota-depleted mice, HSC self-renewal was enhanced during regeneration, while the commitment toward differentiation was dramatically compromised. Mechanistically, microbiota depletion selectively impaired the recycling of red blood cells (RBCs) by BM macrophages, resulting in reduced local iron levels without affecting systemic iron homeostasis. Limiting iron availability in food (in vivo) or in culture (ex vivo), or by CD169+ macrophage depletion, enhanced HSC self-renewal and expansion. These results reveal an intricate interplay between the microbiota, macrophages and iron, and their essential roles in regulating critical HSC fate decisions under stress.
Keywords: Hematopoietic stem cell, microbiota, macrophage, erythrophagocytosis, iron, self-renewal, fate decision, hematopoietic regeneration
Graphical Abstract
In Brief
Zhang et al. show that the gut microbiota regulates RBC recycling by BM macrophages under stress conditions, which ensures rapid iron access in the BM to control HSC self-renewal and differentiation. Thus, a microbiota-macrophage-iron axis plays an essential role in regulating HSC fate decisions.
INTRODUCTION
Hematopoietic stem cells (HSCs) are largely quiescent and metabolically inactive under steady state (Itkin et al., 2016; Nakamura-Ishizu et al., 2020; Signer et al., 2014; Wilson et al., 2008), but can be activated to rapidly produce progeny in response to inflammation, hematopoietic stress and in diseases (Baldridge et al., 2010; King and Goodell, 2011; Pietras et al., 2015; Takizawa et al., 2017; Yamashita et al., 2020). During hematopoietic recovery following myeloablation (e.g. after anti-cancer chemotherapy), HSCs transiently proliferate and overproduce myeloid-biased multipotent progenitors (MPPs) to support rapid reconstruction of the hematopoietic system (Pietras et al., 2015; Wilson et al., 2008). The proliferation and differentiation of HSCs in response to inflammatory or regenerative stimuli is often associated with a reduction of long-term repopulating activity (Baldridge et al., 2010; Pietras et al., 2015; Takizawa et al., 2017). However, the underlying mechanisms regulating HSC self-renewal and differentiation decisions remain incompletely understood.
Macrophages are myeloid cells that reside in all tissues, where they function as immune sentinels for pathogens, scavengers for apoptotic cells and debris, and coordinators of inflammatory responses and tissue homeostasis (Varol et al., 2015; Wynn et al., 2013). In the bone marrow (BM), macrophages form erythroblastic islands that promote erythropoiesis (Chasis and Mohandas, 2008; Chow et al., 2013; Li et al., 2019; Wei et al., 2019), and regulate HSC egress by interacting with mesenchymal stromal cells (Chow et al., 2011; Christopher et al., 2011; Winkler et al., 2010). Clearance of senescent neutrophils by BM macrophages may reinforce a circadian rhythm in hematopoietic stem and progenitor cells (HSPC) migration (Casanova-Acebes et al., 2013). Additionally, subsets of BM macrophages have been found to closely associate with HSCs and promote their quiescence (Hur et al., 2016; Ludin et al., 2012).
Mammals accommodate a large and diverse community of microorganisms, the microbiota, on mucosal surfaces. The microbiota has a broad influence on host health and diseases, regulating the epithelial, immune, and nervous system through nutrients, microbial components and metabolites (Belkaid and Hand, 2014; Holmes et al., 2012; Zhang and Frenette, 2019). The microbiota has recently emerged as an important regulator of hematopoiesis, with significant contributions to the production of myeloid cells (Balmer et al., 2014; Deshmukh et al., 2014; Josefsdottir et al., 2017; Khosravi et al., 2014; Staffas et al., 2018; Zhang et al., 2015) and MPPs (Lee et al., 2019). However, it remains unclear whether HSC functions can be regulated by the microbiota. Here, we show that the microbiota is critical in regulating HSC regenerative response by controlling iron availability in the BM, and reveal a microbiota-macrophage-iron axis that plays an essential role in regulating HSC self-renewal and differentiation decisions.
RESULTS
Microbiota depletion impairs HSC response during regeneration
To explore the role of microbiota in HSC maintenance, we depleted the microbiota in C57BL/6 mice with broad-spectrum antibiotics (ABX) for 4–6 weeks. In this model, we previously reported significant reductions in total and aged neutrophils in the blood compared to control (Zhang et al., 2015). In the BM, we observed a significant reduction in neutrophils, no differences in short-term (ST) or long-term (LT) repopulating HSCs, but significant reductions in myeloid-biased MPP2 and MPP3 (Figure S1A–C), consistent with other studies (Lee et al., 2019). Thus, the microbiota is dispensable for steady-state HSC maintenance.
We next challenged control and ABX-treated mice with 5-fluorouracil (5FU), a chemotherapy drug that causes severe bone marrow injury followed by rapid regeneration (Lucas et al., 2013). Strikingly, we observed significant delays in the regeneration of most blood lineages, including erythrocytes, myeloid and lymphoid cells, in ABX-treated animals (Figure 1A and Figure S1D). In addition, when mice were challenged with a second dose of 5FU, ABX-treated animals exhibited increased lethality from hematopoietic stress (Figure S1E). Next, we analyzed the BM at day 12 after one dose of 5FU, a time point when HSPCs shut down self-renewal and commit to lineage differentiation (Herault et al., 2017). We found a dramatic reduction in total BM cellularity in ABX-treated animals, with significant reductions of neutrophils, monocytes, B cells and erythroblasts (Figure 1B and Figure S1F). By contrast, the percentage of Lineage− (Lin−) progenitor cells, and the numbers of MPP2 and HSCs were significantly increased in ABX-treated mice (Figure 1C and 1D), suggesting a differentiation blockade. Time-course analyses revealed that HSCs from ABX-treated mice were normally activated by hematopoietic stress to enter cell cycle, but presented significant delays in returning to quiescence (Figure S1G).
Figure 1. Microbiota depletion leads to impaired HSC response in regenerative condition.
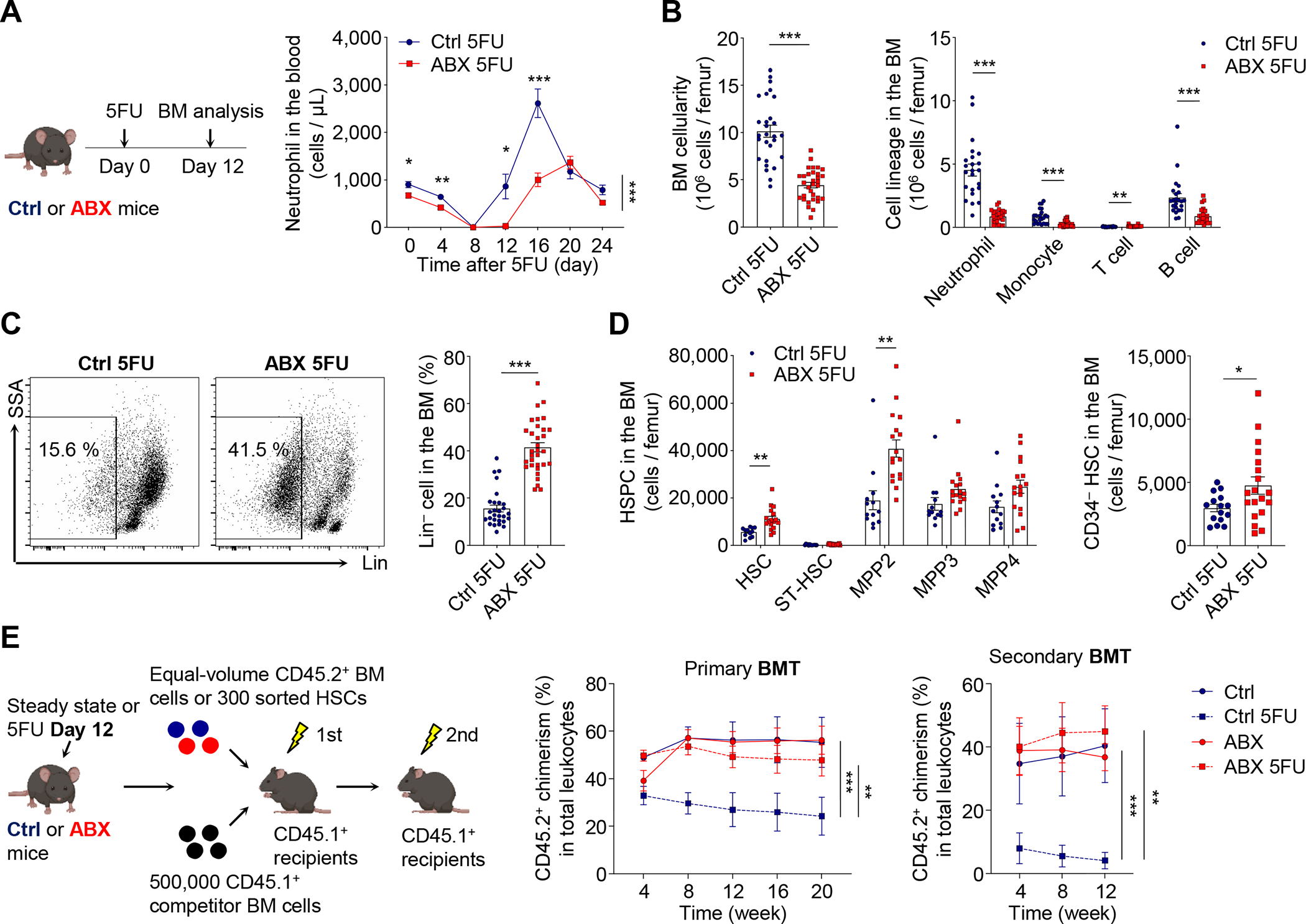
(A) Neutrophils in the blood of control and ABX-treated mice after 5FU challenge (n = 6–27). See also Figure S1D.
(B–D) Total cellularity, cell lineages, Lin− cells, MPPs and HSCs in the BM of control and ABX-treated mice at day 12 after 5FU challenge (n = 12–32). See also Figure S1F–G for erythroblasts and time-course analysis of HSCs.
(E) BMT analysis of control and ABX-treated mice under steady state or at day 12 after 5FU treatment (n = 5–7). See also Figure S1H–I for multi-lineage reconstitution and the HSCT analysis.
* p < 0.05, ** p < 0.01, *** p < 0.001. Error bars, mean ± SEM.
See also Figure S1 and S2 for steady-state and early time-point analyses and the germ-free model.
We further assessed bona fide long-term repopulating HSCs by serial transplantations of BM cells (BMT) from control and ABX-treated mice under steady state or at day 12 after 5FU challenge, into lethally irradiated recipients (Figure 1E). We found that the competitive reconstitution of BM cells from control animals was significantly reduced by 5FU treatment following primary and secondary BMTs (Figure 1E and Figure S1H). Strikingly, ABX treatment completely prevented the reduction in long-term reconstitution during regeneration, without affecting the outcome under steady state (Figure 1E and Figure S1H). These results suggest that ABX treatment enhanced HSC self-renewal, leading to increased numbers of long-term repopulating HSCs during regeneration, while producing a differentiation blockade that dramatically delays BM recovery.
To investigate the effects of ABX treatment on individual HSCs, we performed transplantation assays of sorted HSCs (HSCT) from control or ABX-treated mice under steady state or at day 12 after 5FU challenge (Figure 1E). We found that sorted HSCs from regenerating mice exhibited significant reductions of long-term repopulating activity following HSCT, consistent with a previous report using a sublethal irradiation model (Pietras et al., 2015). In contrast to the BMT results, this reduction was not mitigated by ABX treatment (Figure S1I). In addition, ABX treatment led to an increase of long-term repopulation capacity in phenotypic HSCs under steady state (Figure S1I). Therefore, the ABX-induced increase in long-term reconstitution following BMT was a net result of increased numbers, more active cell cycling, and reduced long-term function of phenotypic HSCs.
To clarify whether the effects of ABX treatment on HSCs were limited to specific time points, we performed BMT and HSCT analyses in control or ABX-treated mice at an earlier time point (day 8) after 5FU challenge. We found no reduction of long-term reconstitution following BMT in 5FU-treated mice at this early time point, while sorted phenotypic HSCs already showed significant reductions of long-term repopulation capacity (Figure S2A). Importantly, ABX treatment led to a significant increase in long-term reconstitution 20 weeks post BMT, but not HSCT (Figure S2A), suggesting increased numbers of long-term repopulating HSCs at this time point.
To rule out the possibility of off-target effects of ABX on hematopoietic recovery, we evaluated HSC response in germ-free (GF) mice. Both ABX-treated and GF mice, have >103-fold reduction of total bacterial load in the intestine, as well as significant dysbiosis compared with control animals (Figure S2B), as reported previously (Zhang et al., 2015). In line with the ABX model, we observed a reduction in neutrophils in the BM, and no defect in long-term repopulating HSCs in GF mice under steady state (Figure S2C–S2F). However, following 5FU challenge, GF mice presented an impaired recovery of total cellularity, neutrophils and B cells in the BM, whereas HSPCs were significantly expanded (Figure S2C and S2D). HSCs from GF mice were actively cycling, and the long-term reconstitution following BMT, but not HSCT, was significantly improved during regeneration (Figure S2E and S2F). These results further suggest that the microbiota regulates HSC self-renewal and differentiation during regeneration.
The microbiota regulates HSC response in different stress conditions
To assess whether a similar response can be observed in another hematopoietic stress condition, we subjected control and ABX-treated mice to a sublethal dose (8 Gy) of total-body irradiation (TBI; Figure 2A). At day 24 post-sublethal TBI, total cellularity, neutrophils, monocytes and B cells were significantly reduced in the BM of ABX-treated mice (Figure 2B and Figure S2G). By contrast, Lin− cells and phenotypic HSCs were significantly expanded and actively cycling (Figure 2C and Figure S2G). To evaluate the effects of TBI and ABX treatment on HSCs, we further performed BMT and HSCT analyses in these animals (Figure 2D). We found that sublethal TBI produced very strong suppression of long-term repopulating activities of BM cells or sorted HSCs from control or ABX-treated mice (Figure 2E). Interestingly, ABX treatment significantly increased long-term reconstitution in both BMT and HSCT experiments (Figure 2E). Taken together, these results suggest that the microbiota regulates HSC fate decisions during different hematopoietic stresses.
Figure 2. The microbiota regulates HSC response in different stress conditions.
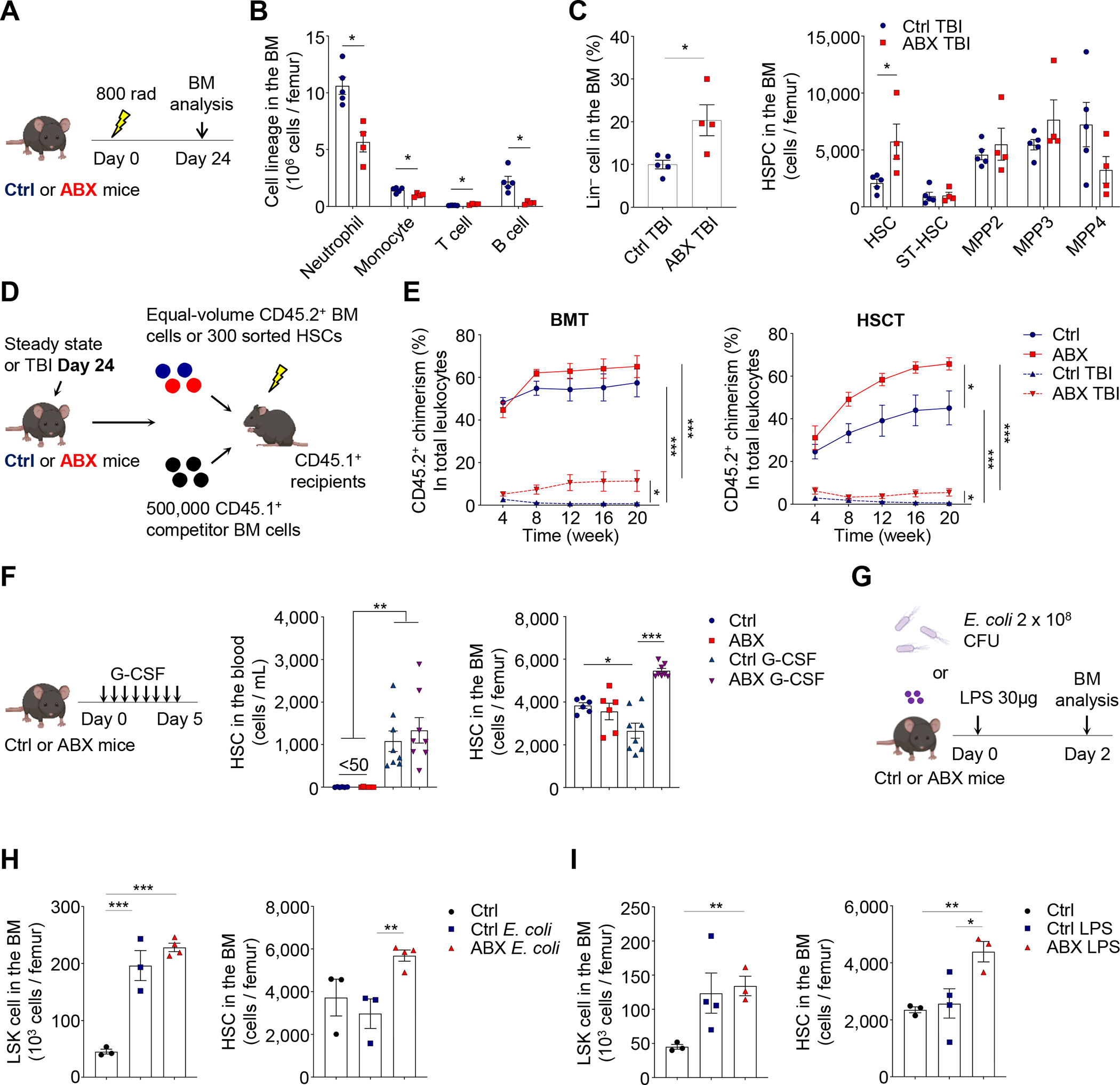
(A–C) Cell lineages, Lin− cells, MPPs and HSCs in the BM of control and ABX-treated mice at day 24 after sublethal TBI (n = 4–5). See also Figure S2G.
(D–E) BMT and HSCT analyses of control and ABX-treated mice under steady state or at day 24 after sublethal TBI (n = 4–9).
(F) HSCs in the blood and BM of control and ABX-treated mice following G-CSF treatment (n = 6–8).
(G–I) LSK cells and HSCs in the BM of control and ABX-treated mice following E. coli infection or LPS challenge (n = 3–4). See also Figure S2H–I.
* p < 0.05, ** p < 0.01, *** p < 0.001. Error bars, mean ± SEM.
See also Figure S3 for niche, HSC localization, and cytokine availability analyses.
We further extended our analyses of the HSC stress response to non-injured inflammatory models. We first performed G-CSF treatment with a procedure that can efficiently mobilize HSCs into the circulation (Gao et al., 2021). We found that ABX treatment led to a significantly increase of HSC numbers in the bone marrow, while not affecting their mobilization to the blood (Figure 2F), consistent with a previous report showing that the germ-free condition mitigated G-CSF-induced HSC suppression in the BM (Schuettpelz et al., 2014). Next, we evaluated the effect of ABX treatment on HSCs in response to systemic E. coli infection (Boettcher et al., 2014). We found that systemic E. coli infection induced robust neutrophil mobilization and striking expansions of HSPCs in both control and ABX-treated mice, but HSCs were significantly expanded only in ABX-treated animals (Figure 2G, 2H and Figure S2H). Further, we analyzed whether ABX treatment produced similar effects on HSCs upon lipopolysaccharide (LPS) challenge (Cai et al., 2018). Consistently, we observed robust neutrophil mobilization and HSPC expansion following LPS challenge in both groups, while HSCs were expanded only in ABX-treated mice (Figure 2G, 2I and Figure S2I). Taken together, these results suggest a broad influence of the microbiota on HSC response in different stress conditions.
Microbiota depletion alters the bone marrow microenvironment
HSCs are maintained in the perivascular niches that can sense and respond to microbial signals (Iwamura et al., 2017; Pinho and Frenette, 2019; Shi et al., 2011). Our findings thus raised the question whether the microbiota mediated the observed effects through the HSC niche. We found that the number of mesenchymal stromal cells (MSCs) and endothelial cells (ECs), two essential niche components (Asada et al., 2017; Ding et al., 2012; Kunisaki et al., 2013; Pinho et al., 2013), were comparable between control and ABX-treated mice following 5FU challenge (Figure S3A). Furthermore, MSCs from ABX-treated mice produced significantly higher levels of niche factors, such as Cxcl12 and Scf (Figure S3B), and immunofluorescence analysis of HSCs with vascular structure revealed a closer association of HSCs in ABX-treated mice with arterioles (Figure S3C and S3D). As hematopoietic and inflammatory cytokines, such as EPO, IL1β, TNF-α and G-CSF, were suggested to play essential roles in hematopoietic regeneration (Bowers et al., 2018; Herault et al., 2017), we evaluated cytokine availability in the BM after 5FU challenge. We observed significantly higher levels of hematopoietic and pro-inflammatory cytokines, including EPO, IL-1β, IL-17 and GM-CSF, but not TNF-α or G-CSF, in ABX-treated mice (Figure S3E, S3F and data not shown). Additionally, levels of the anti-inflammatory cytokine IL-10, but not TGF-β or the glucocorticoid hormone corticosterone, were up-regulated in these animals (Figure S3G). To investigate the in vivo functions of the altered microenvironment, we adoptively transferred Lin− GFP+ cells at day 10 after 5FU challenge, and analyzed homing and differentiation in control and ABX-treated hosts. We found that homing of donor cells to the BM was significantly increased in ABX-treated mice (Figure S3H), consistent with the enhanced Cxcl12 expression by stromal cells (Figure S3B). However, donor cells showed no defects in differentiation, as evidenced by robust myeloid cell production two days after adoptive transfer (Figure S3H). These results suggest that the alterations in the bone marrow microenvironment were secondary effects, rather than the cause of impaired regenerative response.
The microbiota regulates erythrophagocytosis in the bone marrow
During hematopoietic regeneration, we noticed that mice consistently developed an anemia that appeared to exceed that predicted from the normal RBC lifespan (~50 days in mice) (Chow et al., 2013). To investigate this issue, we evaluated RBC half-life using pulse-chase experiments in which Sulfo-NHS-Biotin was injected to label circulating RBCs before 5FU treatment. We found that 5FU treatment indeed accelerated significantly RBC clearance compared to steady-state mice (Figure 3A, left panel). However, the premature RBC clearance was delayed in the early stages of regeneration (until day 8) in ABX-treated animals (Figure 3A, right panel). RBC recycling under steady state occurs through macrophage-mediated erythrophagocytosis mainly in the spleen, but also in the BM and liver (Korolnek and Hamza, 2015). To what extent BM macrophages are involved in emergency RBC recycling of stress conditions remains unclear. BM macrophage numbers were increased in ABX-treated animals under steady state and in the late stage of regeneration (day 12), but were significantly reduced in the early stage (day 5; Figure 3B and S4A). In addition, the expression of genes involved in RBC phagocytosis and degradation (e.g. Lrp1, Hmox1 and Mertk) in BM macrophages was increased following 5FU challenge, but significantly reduced in ABX-treated mice (Figure S4B). Notably, splenic macrophage numbers were significantly reduced under steady state (Zhang et al., 2015), and in the late stage of regeneration (day 12; Figure S4C), suggesting differential regulation of BM and splenic macrophages by the microbiota. These results demonstrate an altered macrophage response during regeneration, and a potential defect in stress-induced erythrophagocytosis in the BM.
Figure 3. The microbiota regulates erythrophagocytosis and iron availability in the BM during regeneration.
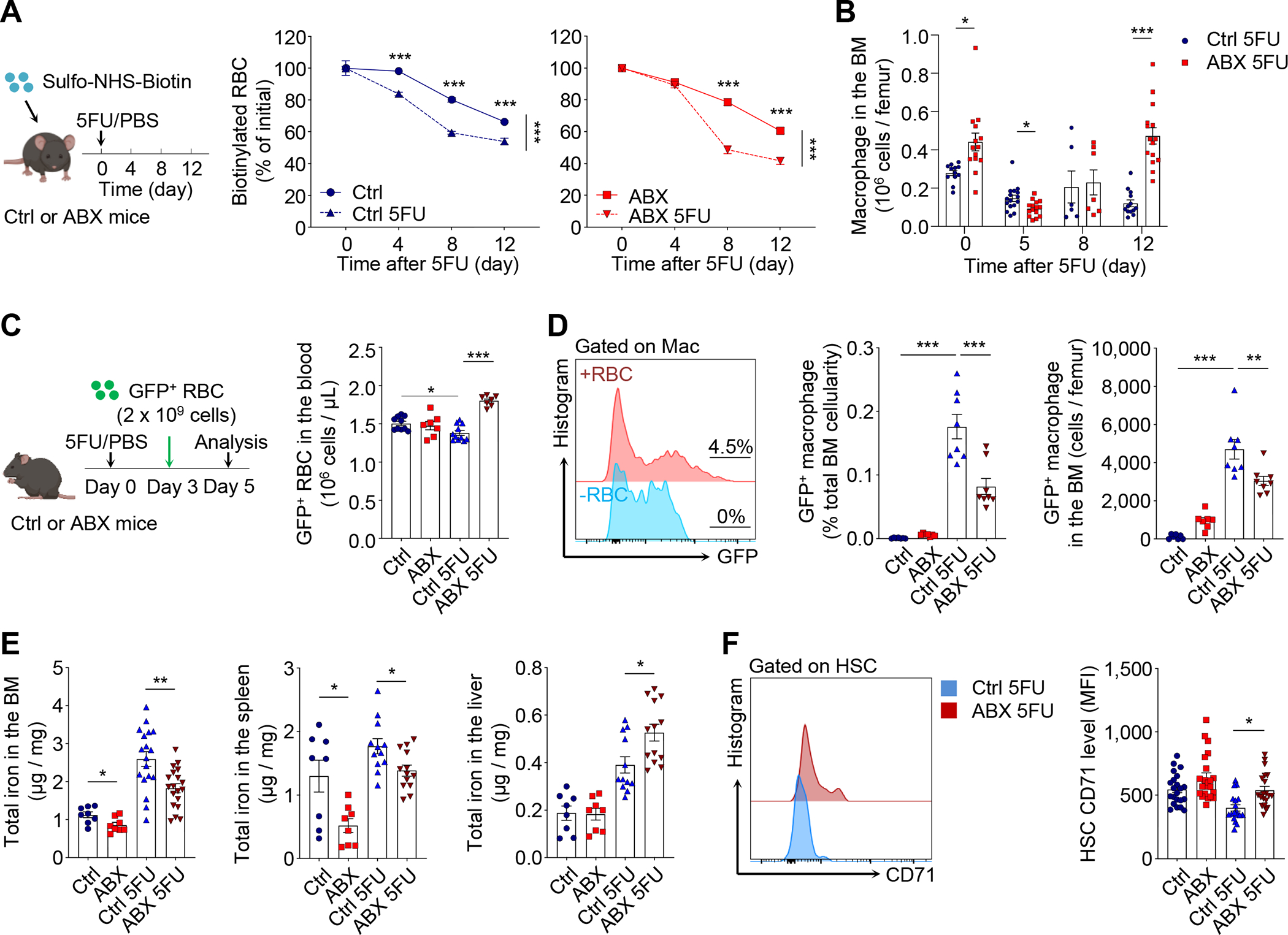
(A) Biotin pulse-chase labelling to evaluate RBC clearance in control and ABX-treated mice after 5FU challenge (n = 4–10).
(B) Time-course analysis of macrophages in the BM of control and ABX-treated mice after 5FU challenge (n = 6–16). See also Figure S4B and S4C for gene expression analyses and splenic macrophages.
(C–D) Adoptive transfer of GFP+ RBCs to evaluate RBC clearance and phagocytosis by macrophages in the BM under steady state or in the early phase of regeneration (day 3–5; n = 7–11). See also Figure S4D–G for other tissues and later time points.
(E) Iron levels in the BM, spleen and liver of control and ABX-treated mice under steady state or at day 12 after 5FU challenge (n= 8–19).
(F) CD71 levels on HSCs in control and ABX-treated mice under steady state or at day 12 after 5FU challenge (n= 18–22). See also Figure S4H–J for germ-free and sublethal irradiation models.
* p < 0.05, ** p < 0.01, *** p < 0.001. Error bars, mean ± SEM.
See also Figure S4K for systemic iron parameters.
To assess the phagocytic activity of macrophages, we adoptively transferred fluorescently labelled RBCs (from UBC-GFP mice) into control and ABX-treated animals, and analyzed the proportion of macrophages that phagocytosed GFP+ RBCs in different tissues two days later (Figure 3C). We found that the numbers of GFP+ RBCs that remained in the circulation were significantly higher in ABX-treated compared to control animals in the early stages (days 3–5) following 5FU challenge, whereas no difference was found at steady state (Figure 3C), suggesting a selective delay of clearance during regeneration. Consistent with the predominant role of the spleen in steady-state RBC recycling (de Back et al., 2014), we observed little phagocytic activity in the BM under steady state (Figure 3D). However, under 5FU-induced stress, robust erythrophagocytosis was triggered in the BM during the early stages (days 3–5) of regeneration, while ABX treatment significantly mitigated the effect (Figure 3D). The stress-induced phagocytic events in the BM were exclusively mediated by macrophages (Figure S4D), and were also robust in the spleen but rarely observed in the liver (Figure S4E). Interestingly, ABX treatment reduced regenerative erythrophagocytosis only in the BM, but not in the spleen or liver (Figure S4E). In addition, active erythrophagocytosis in the BM was limited to the early stages of regeneration and not observed in later stages (days 10–12) (Figure S4F and S4G). These results reveal that the microbiota regulates stress-induced erythrophagocytosis by BM macrophages in the early phase of regeneration.
Acute RBC phagocytosis provides iron to initiate regeneration
The premature sacrifice of RBCs suggested that they may play a key role in the regenerative process. Under steady state, recycling of RBCs by macrophages functions as a major approach to recycle iron for daily erythropoiesis demand (Muckenthaler et al., 2017; Soares and Hamza, 2016). In addition to erythropoiesis, iron is also essential for many fundamental biological processes including energy metabolism, DNA synthesis, epigenetic modification, and signal transduction (Ganz and Nemeth, 2015; Muckenthaler et al., 2017). Emerging data suggest that intracellular iron homeostasis can directly influence HSC maintenance (Chai et al., 2015; Kao et al., 2021; Kao et al., 2018; Muto et al., 2017), which prompted us to hypothesize that stress-induced RBC recycling in the BM may fuel hematopoietic regeneration and regulate HSC self-renewal through iron availability.
To test whether iron availability was altered in microbiota-depleted animals, we analyzed iron levels in the BM, spleen and liver, the three organs involved in stress hematopoiesis. These analyses revealed that the iron levels were significantly reduced in the BM and spleen of ABX-treated animals in both steady-state and regenerative conditions (Figure 3E), while iron levels were not changed under steady state, but increased during regeneration in the liver of these animals (Figure 3E). Since iron is known to be delivered to different cell types through either transferrin, lipocalin or macrophage-mediated pathways, iron can be unevenly distributed within a tissue (Soares and Hamza, 2016; Yang et al., 2002). To assess whether the low iron availability produced by microbiota depletion affected HSCs, we analyzed transferrin receptor 1 (Tfr1, also known as CD71) expression on HSC surface, which correlates negatively with intracellular iron levels (Casey et al., 1988; Tong et al., 2002). We found that CD71 expression was significantly increased on HSCs from ABX-treated mice after 5FU challenge (Figure 3F). Similar reduction of iron levels in the BM and increase of CD71 expression level on HSCs were observed in GF animals following 5FU challenge (Figure S4H). In addition, HSCs from GF mice after 5FU challenge exhibited higher calcein fluorescence, an iron sensor whose fluorescence emission is quenched upon iron binding (Devireddy et al., 2005; Wang et al., 2020), suggesting reduced intracellular iron concentrations (Figure S4I). Consistently, ABX treatment also led to a significant reduction of iron levels in the BM and an increase of CD71 expression level on HSCs in sublethal irradiation-induced hematopoietic stress (Figure S4J).
To investigate whether systemic iron delivery contributes to the observed iron deficiency in the BM, we analyzed hepcidin and serum iron parameters in control and ABX-treated animals. Hepcidin, a key regulator of iron entry into the circulation by inhibiting the iron export channel ferroportin (Ganz and Nemeth, 2015), was significantly less abundant in ABX-treated mice in both steady-state and regenerative conditions (Figure S4K), leading to no change of serum iron and transferrin saturation under steady state but significant increases of both parameters following 5FU challenge (Figure S4K). Thus, microbiota depletion induces iron deficiency in the BM independent of systemic regulation, and local iron levels provided by RBC recycling during hematopoietic stress may fuel HSC differentiation.
Bone marrow macrophages control the iron delivery to HSCs
As macrophages mediated RBC recycling during hematopoietic stress, we sought to understand whether they were required to supply iron to HSCs in regenerative condition. To investigate this issue, we depleted BM macrophages using CD169-DTR mice, which specifically targeted CD169+ macrophages in the BM and several other tissues (Chow et al., 2013; Chow et al., 2011; Gupta et al., 2016; Miyake et al., 2007). Biweekly injections of diphtheria toxin (DT) led to efficient depletion of BM macrophages (by 85%), and a significant delay of RBC clearance after 5FU challenge (Figure 4A and 4B). Strikingly, we observed significant reductions of the iron content in the BM, but not the spleen, liver or serum, of both steady-state and regenerating mice (Figure 4C and Figure S5A), suggesting that CD169+ macrophages selectively regulate local iron availability in the BM. CD169+ macrophage depletion produced no alteration of BM cellularity and HSCs under steady state, but led to increased neutrophil production (Figure 4D and 4E), consistent with previous reports showing enhanced IL-17/G-CSF signaling in mouse models with macrophage deficiencies (Gordy et al., 2011; Hong et al., 2012). Similar to ABX-induced iron deficiency in the BM, CD169+ macrophage depletion led to a significant expansion of Lin− cells and HSCs in regenerative condition, while the differentiation towards downstream lineages was impaired (Figure 4D, 4E and Figure S5B). HSCs from 5FU-treated macrophage-depleted animals exhibited more active cycling, increased CD71 expression and calcein fluorescence, and significant improvement in long-term repopulating activity following BMT, but not HSCT (Figure 4F and Figure S5C–E). If the microbiota regulated HSC responses during regeneration selectively through macrophages, we anticipated that the depletion of macrophages would prevent the altered regeneration induced by ABX treatment. Indeed, we found that the iron reduction, HSC expansion and differentiation blockade induced by ABX treatment were completed abrogated in CD169-DTR animals (Figure 4G, 4H and Figure S5F). These results suggest that BM macrophages are required to supply iron to regenerating HSCs, and to mediate the regulatory signals from the microbiota.
Figure 4. CD169+ BM macrophages control the delivery of iron to HSCs for regeneration.
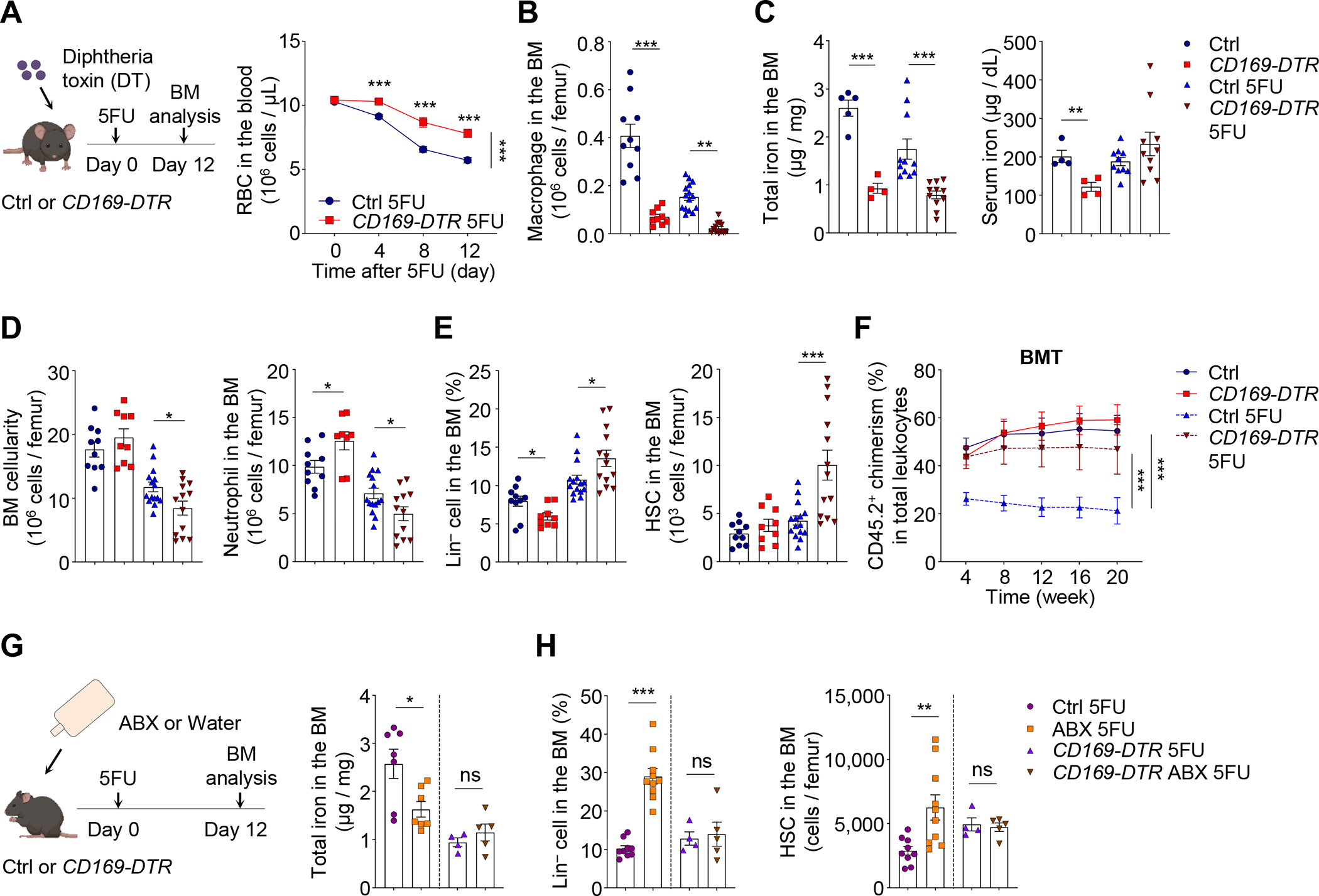
(A) RBCs in the blood of control and CD169-DTR mice after 5FU challenge (n = 9–13).
(B–C) BM macrophages and iron levels in the BM and sera of control and CD169-DTR mice under steady state or at day 12 after 5FU challenge (n = 4–15). See also Figure S5A for iron levels in other tissues.
(D–F) BM cellularity, neutrophils, Lin− cells, HSCs, and long-term reconstitution following BMT in control and CD169-DTR mice under steady state or at day 12 after 5FU challenge (n = 6–15). See also Figure S5B–E.
(G–H) BM iron levels, Lin− cells, and HSCs in control and CD169-DTR mice with or without ABX treatment at day 12 after 5FU challenge (n = 4–10). See also Figure S5F.
* p < 0.05, ** p < 0.01, *** p < 0.001; ns, not significant. Error bars, mean ± SEM.
The microbiota orchestrates regenerative events via short-chain fatty acid
We next sought to understand how the microbiota could regulate emergency iron recycling by BM macrophages to ensure proper HSC response during regeneration. Microbiota-derived pro-inflammatory molecules, such as pathogen-associated molecular patterns (PAMPs), are known to perfuse into the host, modulating the production and functions of hematopoietic cells through toll-like receptor (TLR) pathways (Clarke et al., 2010; Deshmukh et al., 2014; Lee et al., 2019; Zhang et al., 2015). To test this possibility, we analyzed the regenerative process in genetic mouse models with various deficiencies in TLR pathways (Figure 5A). To our surprise, the constitutive deletion of Tlr4, Tlr2, or the adaptor protein Trif, failed to induce any defect in hematopoietic regeneration or HSC response following 5FU challenge (Figure 5B and Figure S5G). In addition, the conditional deletion of the Myd88 adapter in myeloid cells and macrophages (LysM-Cre) or HSCs (Fgd5-CreER) did not reveal any defect in 5FU-induced stress responses (Figure 5B and Figure S5H). Moreover, add-back experiments of PAMP molecules, LPS or single-strand DNA (ssDNA) (Lee et al., 2019; Zhang et al., 2015), did not rescue the differentiation blockade or revert the HSPC expansion in ABX-treated mice following 5-FU challenge (Figure S5I, S5J, and data not shown). These results suggest that TLR/Myd88/TRIF pathways are not relevant for the regulation of hematopoietic regeneration by the microbiota.
Figure 5. The microbiota orchestrates regenerative events in the BM via short-chain fatty acid butyrate.
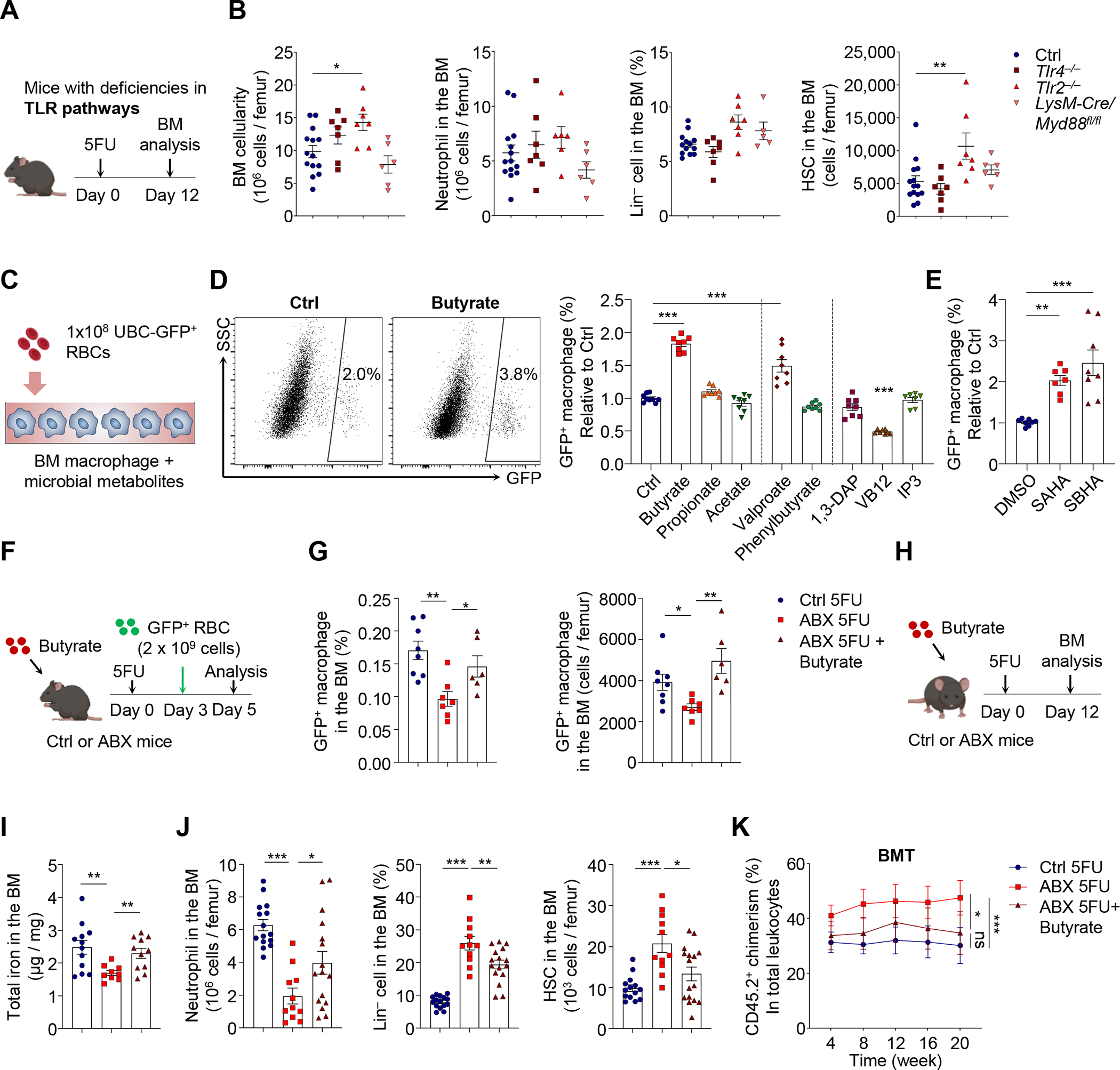
(A–B) BM cellularity, neutrophils, Lin− cells, and HSCs in control, Tlr4−/−, Tlr2 −/− and LysM-Cre/Myd88fl/fl mice at day 12 after 5FU challenge (n = 5–14). See also Figure S5G–J for additional mouse models and the LPS add-back experiment.
(C–E) RBC phagocytosis by bone marrow-derived macrophages ex vivo in the presence of microbial metabolites or HDAC inhibitors (n = 7–9). See also Figure S6A and S6B.
(F–G) RBC phagocytosis in control, ABX-treated and ABX-treated mice supplemented with butyrate, in the early phase of regeneration (day 3–5; n = 6–8). See also Figure S6E for gene expression analyses.
(H–K) BM iron levels, neutrophils, Lin− cells, HSCs, and long-term reconstitution following BMT in control, ABX-treated and ABX-treated mice supplemented with butyrate, at day 12 after 5FU treatment (n = 6–16). See also Figure S6F–H.
* p < 0.05, ** p < 0.01, *** p < 0.001; ns, not significant. Error bars, mean ± SEM.
The microbiota can also regulate host physiological processes via microbial metabolites (Holmes et al., 2012; McCarville et al., 2020). To investigate this possibility, we screened a series of microbial metabolites by co-culturing BM-derived macrophages with GFP+ RBCs in the presence of these molecules (Figure 5C). These candidates included short-chain fatty acids (SCFAs; butyrate, propionate and acetate) (Chang et al., 2014; Schulthess et al., 2019), 1,3-diaminopropane (1,3-DAP) (Das et al., 2020), inositol trisphosphate (IP3) (Wu et al., 2020), and vitamin B12 (VB12) (Koury and Ponka, 2004), which have been reported to influence host cell functions. Interestingly, we found that only the SCFA butyrate significantly enhanced macrophage phagocytosis towards RBCs (Figure 5D). Butyrate mediated this effect via histone deacetylase (HDAC) inhibitory activity, as similar enhancement of erythrophagocytosis was observed with synthetic HDAC inhibitor valproate (targeting Class I), SAHA (targeting Class I and II) and SBHA (targeting Class I, HDAC1 and 3) (Schulthess et al., 2019) (Figure 5D and 5E). Further analyses revealed that butyrate treatment significantly enhanced global histone H3 acetylation (H3ac) levels in cultured BM macrophages, leading to increased expression levels of genes involved in RBC recycling, including Lrp1, Hmox1 and Mertk (Figure S6A and S6B).
SCFAs are mainly produced by the microbiota through anaerobic fermentation of dietary fibers, and depletion of the microbiota has long been known to deplete SCFA species in the intestine (Hoverstad and Midtvedt, 1986; Zarrinpar et al., 2018). To evaluate systemic SCFA levels, we analyzed serum from control or ABX-treated mice with liquid chromatography-mass spectrometry (LC-MS). We found that ABX treatment led to significant reductions of all three major SCFA species, including acetic acid, propionate acid and butyric acid (Figure S6C and S6D). To investigate whether butyrate could modulate macrophage function and HSC regenerative response in vivo, we performed a rescue experiment by adding back butyrate in ABX-treated animals (Figure 5F). We found that butyrate supplementation restored defective BM erythrophagocytosis in the early stages of regeneration, and significantly increased the expression levels of genes involved in RBC recycling in BM macrophages (Figure 5G and Figure S6E). Further, in mice treated during the whole process of regeneration (Figure 5H), butyrate supplementation significantly improved local iron deficiency, BM cellularity, lineage differentiation, and reverted HSC expansion, cell cycling, CD71 expression and calcein fluorescence (Figure 5I, 5J, Figure S6F and S6G). Butyrate supplementation also abrogated the increase of long-term reconstitution following BMT, but not HSCT, in ABX-treated mice after 5FU challenge (Figure 5K and Figure S6H). These results suggest that the microbial metabolite butyrate orchestrates regenerative events in the BM to provide rapid iron access to regenerating HSCs.
Iron availability regulates HSC self-renewal and differentiation decisions
To evaluate further the role of iron in HSC function, we fed mice with formulated diet containing different iron levels (normal 185ppm, low 30ppm, deficient 3ppm) (Kautz et al., 2014) for 3 months. Mice fed with low-iron or iron-deficient diet exhibited increased RBC numbers, reduced hemoglobin levels, mean corpuscular volume (MCV), and lower neutrophil numbers in the circulation, which are consistent with iron deficiency (Figure S7A). These animals showed no significant difference in HSCs or downstream lineages in the BM under steady state, but competitive transplantation experiments revealed significant reductions in long-term repopulating activity following BMT (Figure S7B and S7C). Upon challenge with 5FU, mice fed with low-iron or iron-deficient diet exhibited dramatic expansion of HSCs while regeneration of mature lineages was significantly compromised (Figure 6A, 6B and Figure S7D). Notably, BM macrophages were significantly expanded during regeneration in mice fed with low-iron or iron-deficient diet, while splenic macrophages remained not affected (Figure S7E), showing again differential regulation of macrophages in different organs. HSCs from iron-deficient mice exhibited more active cell cycling, increased CD71 expression and calcein fluorescence (Figure S7F). The expansion of HSCs in these animals correlated with the severity of iron deficiency, leading to dramatic increases of long-term reconstitution following competitive BMT, but not HSCT (Figure 6C and Figure S7G).
Figure 6. Iron availability regulates HSC self-renewal and differentiation during regeneration.
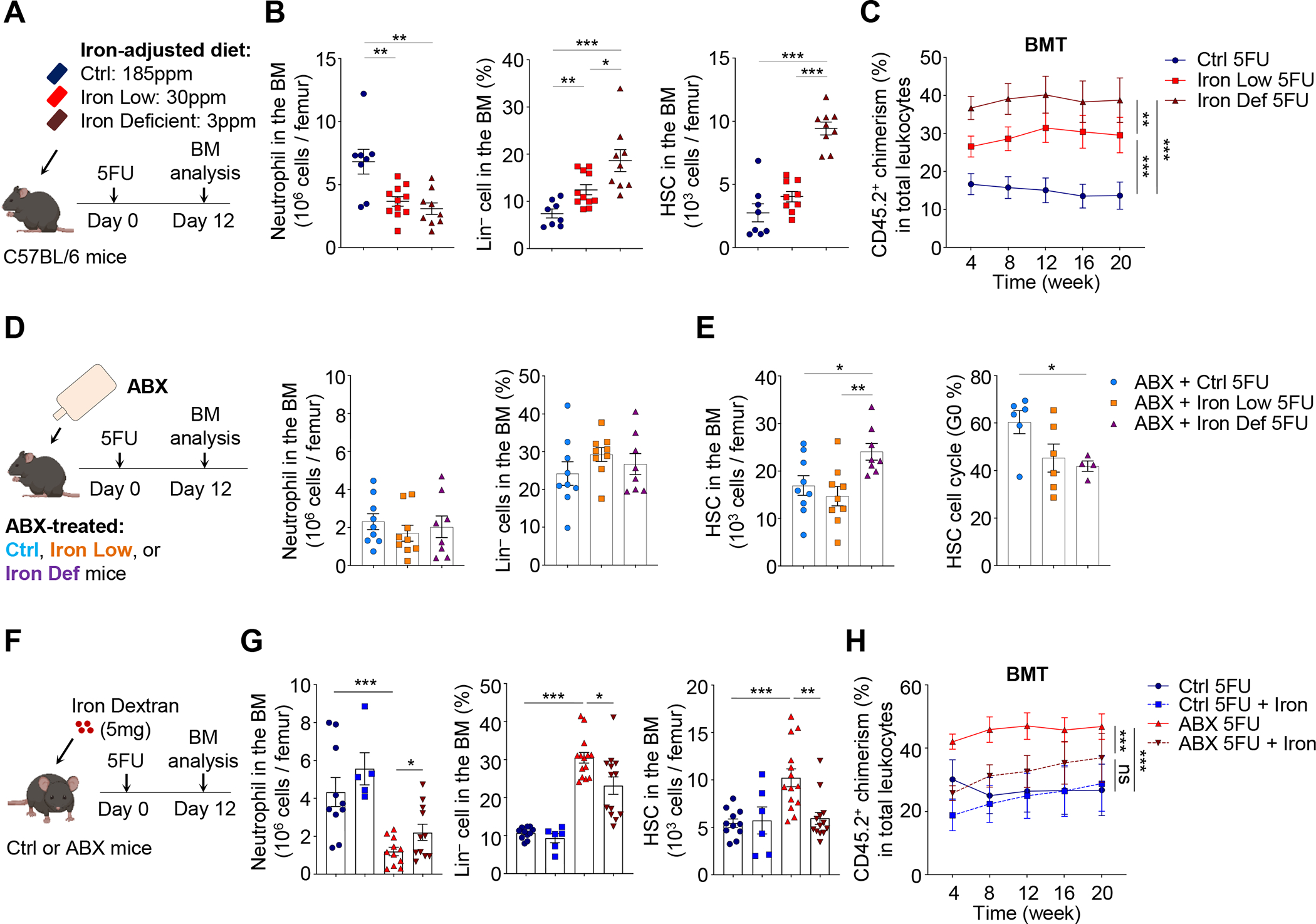
(A–C) Neutrophils, Lin− cells, HSCs, and long-term reconstitution following BMT in mice fed with normal, iron-low or iron-deficient food, at day 12 after 5FU challenge (n= 8–11). See also Figure S7D–G.
(D–E) Neutrophils, Lin− cells, HSCs and HSC cell cycling in the BM of ABX-treated mice fed with normal, iron-low or iron-deficient food, at day 12 after 5FU treatment (n = 4–9).
(F–H) Neutrophils, Lin− cells, HSCs, and long-term reconstitution following BMT in control and ABX-treated mice injected with PBS or iron dextran, at day 12 after 5FU challenge (n = 5–14). See also Figure S7H–L.
* p < 0.05, ** p < 0.01, *** p < 0.001; ns, not significant. Error bars, mean ± SEM.
See also Figure S7 for steady-state analyses of the iron deficiency model.
Since iron deficiency may cause dysbiosis that could complicate interpretation of the results, we induced iron deficiency in ABX-treated mice and challenged them with 5FU to trigger hematopoietic stress (Figure 6D). In these animals, iron deficiency failed to reduce neutrophils or expand Lin− cells, but led to significant expansions, more active cell cycling and increased CD71 expression of HSCs on top of the effects induced by ABX treatment (Figure 6D and 6E, and data not shown). These results suggest that diet-induced iron deficiency could regulate HSC self-renewal and expansion independent of the microbiota.
We next asked whether the restoration of iron levels could rescue ABX-induced HSC dysregulation, and injected ABX-treated animals with iron dextran (Figure 6F). We found that iron add-back significantly rescued local iron deficiency, improved the differentiation blockade and reverted macrophage and HSC expansions (Figure 6G, Figure S7H and S7I). Supplementation of iron during regeneration also shut down HSC cycling, restored CD71 expression on HSCs, and reduced their long-term repopulating activity following BMT, but not HSCT (Figure 6H and Figure S7J–L). Taken together, these results suggest that the microbiota regulates HSC self-renewal and differentiation decisions during regeneration through iron availability in the BM.
Limiting iron availability stimulates HSC self-renewal and expansion ex vivo
Finally, we surmised that if limiting iron availability could promote HSC self-renewal and inhibit differentiation during regeneration, optimization of iron levels would enhance HSC expansion ex vivo. Although extensively studied for decades, robust expansion of transplantable HSCs with long-term repopulating activity has remained a major challenge (Bhatia et al., 1997; Conneally et al., 1997; Kumar and Geiger, 2017; Miller and Eaves, 1997). The titration of hematopoietic cytokines thrombopoietin (TPO) and stem-cell factor (SCF) in serum-free media containing polyvinyl alcohol (PVA) was recently shown to expand functional murine HSCs many folds more than previous approaches (Wilkinson et al., 2019). We noticed that the iron concentrations in the PVA-based culture system was much lower (5.5 mg/L transferrin, binding 138 nM iron) than other published serum-free culture systems (100 – 200 mg/L transferrin, binding 2.5 – 5 μM iron) (Bhatia et al., 1997; Conneally et al., 1997; Miller and Eaves, 1997). It is thus possible that low iron availability might have contributed to the maintenance and expansion of functional HSCs ex vivo. To test this possibility, we titrated the transferrin concentrations (ranging from 5 μg/L to 500 mg/L) in the PVA-based culture system while keeping all other components unchanged. Strikingly, the transferrin levels in the culture system markedly influenced the HSC yield. We found that transferrin concentration at 0.5 mg/L, a level significantly lower than the concentrations used in previous studies (Bhatia et al., 1997; Conneally et al., 1997; Miller and Eaves, 1997; Wilkinson et al., 2019), supported the most robust HSC expansion (Figure 7A and 7B). While higher transferrin concentrations induced HSC differentiation and low yields of phenotypic HSCs (Figure 7B and 7C), transferrin levels lower than 0.5 mg/L triggered survival stress, as evidenced by activation of the cell cycle and ROS production (Figure 7D), and concentrations lower than 5 μg/L did not allow HSC survival (Figure 7A). Transplantation assay revealed that the long-term repopulating activity was significantly higher when HSCs were cultured with a lower concentration of transferrin within the Goldilocks zone (Figure 7E and 7F). To clarify how iron availability in the media influenced intracellular iron levels in HSCs, we analyzed CD71 expression on HSCs cultured with different levels of transferrin. We found that only when transferrin levels decreased from 500 mg/L to 0.5 mg/L, HSCs showed a significant increase of CD71 expression (Figure S7M), with a magnitude (~40%) similar to that observed with ABX treatment in vivo (Figure 3F). However, when transferrin levels decreased further to 0.05 mg/L, HSCs exhibited a 5-fold increase in CD71 expression (Figure S7M). Taken together, these results identify iron levels as a pivotal element regulating HSC self-renewal and differentiation.
Figure 7. Limiting iron availability stimulates HSC self-renewal and expansion ex vivo.

(A–B) Total cell and CD150+ HSC numbers in the PVA-based culture system with defined transferrin levels (n = 7–8).
(C–D) Lin− cell ratio, cell cycling and ROS production of HSCs in the PVA-based culture system with defined transferrin levels (n = 4–8).
(E) Transplantation analysis of HSCs expanded with 500 mg/L or 0.5 mg/L transferrin (n = 4–5).
(F) Model figure showing a Goldilocks zone for HSC ex vivo expansion, with higher or lower iron levels impairing HSC self-renewal.
* p < 0.05, ** p < 0.01, *** p < 0.001. Error bars, mean ± SEM.
DISCUSSION
Here, we have uncovered a long-ranged function of the microbiota in regulating iron availability in the HSC niche during regeneration. Microbiota-derived SCFAs promote emergency erythrophagocytosis by BM macrophages to distribute iron that fuels hematopoietic regeneration. In microbiota- or CD169+ macrophage-depleted mice, emergency erythrophagocytosis is impaired, thereby limiting local iron availability to HSPCs for differentiation. Our results indicate that low iron availability enhance HSC self-renewal and promote their expansion in vitro and in vivo. Thus, a microbiota-macrophage-iron axis plays an essential role in regulating HSC fate decisions to accommodate hematopoietic stress.
Local regulation of iron availability
Systemic iron homeostasis is regulated by hepcidin, which inhibits and induces degradation of ferroportin on enterocytes, macrophages and hepatocytes, thus shutting down iron efflux into the circulation (Muckenthaler et al., 2017). Hepcidin is produced in the liver in response to systemic iron levels, and can be induced by the inflammatory cytokine IL-6, and suppressed by the erythropoietic hormone erythroferrone (Kautz et al., 2014). We found that hepcidin levels were significantly reduced in ABX-treated mice, leading to no change under steady-state but increased serum iron level during regeneration (Figure S4K). The reduced hepcidin levels may reflect lower IL-6 levels (Meisel et al., 2018), or increased erythropoietic demand in these animals. The increased serum iron level in ABX-treated mice during regeneration may also result from increased intestinal iron absorption (Das et al., 2020). Despite the increased serum iron level, we observed significant local iron deficiency in the BM (Figure 3E). Our results thus uncover a mechanism for rapid iron access independently from traditional routes via hepcidin- or intestinal epithelium-mediated systemic regulation. Instead, iron supply in the BM under stress conditions is largely controlled by emergency RBC recycling mechanisms through macrophages. Interestingly, erythrophagocytosis and shortened lifespan of RBCs also contribute to the pathogenesis of anemia induced by inflammation or chronic diseases (Ganz, 2019; Libregts et al., 2011; Mitlyng et al., 2006; Zoller et al., 2011). However, the reason for this phenomenon had remained unclear. Our data provide a possible explanation in which under stress conditions, the organism borrows on the RBC capital of a rich accessible source of iron to jump-start hematopoiesis to meet the emergency demand. This paradigm may represent a strategy that has evolved from immune defense against pathogens. During an infection, hepcidin production is induced by inflammatory cytokines to shut down iron efflux into the plasma, thus limiting iron availability to invading pathogens (Ganz and Nemeth, 2015), while active RBC recycling is triggered in the BM to provide rapid local iron access for burst production of immune cells.
Broad macrophage functions in iron allocation
Macrophages form erythroblastic islands that promote the maturation of erythroid precursors, and also destroy senescent RBC to recycle iron for daily tissue demand (Chasis and Mohandas, 2008; Muckenthaler et al., 2017). Under steady state, splenic red pulp macrophages are the major cell population that scrutinizes circulating RBCs and removes senescent ones from the circulation (de Back et al., 2014). BM macrophages are also equipped with the machinery for RBC scavenging and iron recycling, and have been reported to deliver iron locally via ferritin secretion or ferroportin-mediated export (Delaby et al., 2005; Knutson et al., 2005; Leimberg et al., 2008; Li et al., 2019). When damaged RBCs accumulate in hemolytic conditions, the liver contributes significantly to erythrocyte disposal by recruiting iron-loaded monocytes, which differentiate into a transient iron-recycling macrophage population (Theurl et al., 2016). Our results are in line with previous reports that the spleen takes the major responsibility of steady-state RBC recycling. However, our results indicate that the situation changes during regenerative stress where robust erythrophagocytosis occurs in both BM and spleen, while the liver is not meaningfully involved (Figure S4E). The relative contribution of the three organs in erythrocyte disposal is thus dynamic and responsive to different physiological conditions. By depletion of CD169+ macrophages, we have shown that iron levels are specifically reduced in the BM, but not in the spleen or liver (Figure 4C and Figure S5A). This may result from a compensation by CD169− macrophage populations, or by other iron-loaded cell types, such as hepatocytes. We have also identified the microbial metabolite SCFA butyrate as the microbiota-derived signal that regulates the erythrophagocytic function of BM macrophages (Figure 5 and Figure S6). These findings support other studies suggesting that butyrate can imprint antimicrobial programs in intestinal macrophages (Chang et al., 2014; Schulthess et al., 2019). Notably, different directions of macrophage polarization have been proposed to associate with these functions; antimicrobial defense is enhanced with polarization towards inflammation whereas heme-iron metabolism is involved in polarization towards tissue repair and regeneration (Cairo et al., 2011). It is thus possible that macrophages in different tissues possess distinct transcriptional and epigenetic programs that may yield tissue-specific responses to butyrate-induced HDAC inhibition. Our results extend the iron recycling functions of macrophages beyond erythropoiesis, to the HSC niche by directly regulating HSC self-renewal and differentiation decisions under stress.
Harnessing iron for HSC expansion and cancer contraction
HSCs are the basis of BM transplantation, the only cure for many hematological diseases (Notta et al., 2011; Osawa et al., 1996). Our findings that limiting iron availability improved the expansion of functional HSCs ex vivo thus have broad implications in current HSC expansion protocols (Figure 7). In hematological malignancies, leukemic and pre-leukemic cells exhibit enhanced iron metabolism, which is essential for maintaining their rapid growth rate (Torti et al., 2018). In addition, excessive iron triggers the production of reactive oxygen species (ROS), induces DNA damage and impairs the functions of healthy stem cells (Chai et al., 2015; Torti et al., 2018). Iron overload has been correlated with poor prognosis in leukemias and myelodysplastic syndrome (MDS) (Gattermann, 2018; Wang et al., 2019). Iron chelators have been found to have potent antitumor activity (Callens et al., 2010; Roth et al., 2012; Whitnall et al., 2006), and is under intense investigations in clinical trials for leukemias or MDS. Interestingly, the microbiota has been recently described to play a role in the development of pre-leukemic myeloproliferation in Tet2−/− mice (Meisel et al., 2018). It would thus be interesting to define further whether the interplay between the microbiota, macrophage and iron also plays a role in these diseases.
Regulation of HSC fate decisions under stress by the microbiota
Previous studies have suggested that the microbiota regulates myeloid cell production (Balmer et al., 2014; Deshmukh et al., 2014; Khosravi et al., 2014). In addition, high-fat diet induces a myeloid bias in hematopoiesis which can be transmitted by fecal transplantation (Luo et al., 2015). The microbiota also regulates the production of MPPs, but is dispensable for the maintenance of HSCs under steady state (Iwamura et al., 2017; Lee et al., 2019; Zhang et al., 2015). It is notable that other studies have reported a reduction of phenotypic HSCs in ABX-treated animals, although the long-term reconstitution following BMT was not affected (Josefsdottir et al., 2017). Here we analyzed both phenotypic HSCs and their long-term repopulating activity in the ABX and GF models, and identified an essential role for the microbiota in regulating HSC response in different stress conditions (Figure 1, 2, Figure S1 and S2). Although our results speak to the function of microbiota at the whole-community level, it would be of interest to explore in future studies whether certain commensals are specifically involved in hematopoietic stress, as described in inflammatory diseases (Bloom et al., 2011). The promotion of lineage differentiation by the microbiota, which is critical for the recovery after BM transplantation and chemotherapy, calls for attention to the use of antibiotics and the impact of iron deficiency in this vulnerable patient population, and the possibility of manipulating the microbiota to hasten hematopoietic recovery.
HSCs are dormant under steady state, which protects them from genotoxic insult and ensures their long-term repopulating activity (Wilson et al., 2008). The commitment of HSCs to differentiation may involve a reprogramming of transcriptional, epigenetic and metabolic programs (Cabezas-Wallscheid et al., 2017; Cabezas-Wallscheid et al., 2014; Herault et al., 2017; Laurenti and Gottgens, 2018). The quiescence and activation of HSCs are known to be regulated by a complex network of transcription factors, signaling pathways, and their interactions with the microenvironment (Calvi and Link, 2015; Hoggatt et al., 2016; Pinho and Frenette, 2019; Rossi et al., 2012). The present studies add a layer to this complexity by revealing an essential role for iron distribution in the niche, which is controlled by the microbiota and macrophages, in regulating HSC self-renewal and differentiation decisions under stress conditions. Our results demonstrate the mechanisms underlying iron-mediated regulation of HSCs in vivo, and bring forward the prospect that targeting iron regulatory pathways may have significant impact on stem cell-based hematological diseases.
Limitations of the study
It remains to be determined whether the microbiota regulates HSC response with the same macrophage-iron mechanism in different stress conditions. Further, our analyses of BM macrophages relied on the common macrophage marker F4/80. Characterization of the heterogeneity and functional sub-populations is required to further understand this population. Finally, it also needs to be explored how macrophages allocate iron to different cell types in the BM, and how intracellular iron levels influence the metabolic and transcriptional programs underlying HSC self-renewal and differentiation.
STAR METHODS
KEY RESOURCES TABLE
Key resources table is submitted as a separate file.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Mouse Ly-6G/Ly-6C (Gr-1) eFluor 450 | eBioscience | Cat# 48-5931-82, RRID: AB_1548797 |
| Anti-Mouse CD115 APC | eBioscience | Cat# 17-1152-82, RRID: AB_1210789 |
| Anti-Mouse CD3e PerCP-Cy5.5 | eBioscience | Cat# 45-0031-82, RRID: AB_1107000 |
| Anti-Mouse B220 APC-eFluor 780 | eBioscience | Cat# 47-0452-82, RRID: AB_1518810 |
| Anti-Mouse CD11b PE-Cy7 | eBioscience | Cat# 25-0112-82, RRID: AB_469588 |
| Anti-Mouse CD11b PE | eBioscience | Cat# 12-0112-83, RRID: AB_2734870 |
| Anti-Mouse Ter119 PE | eBioscience | Cat# 12-5921-83, RRID: AB_466043 |
| Anti-Mouse Ter119 APC-eFluor 780 | eBioscience | Cat# 47-5921-82, RRID: AB_1548786 |
| Anti-Mouse Ter119 PerCp-Cy5.5 | eBioscience | Cat# 45-5921-82, RRID: AB_925765 |
| Anti-Mouse CD45.1 PE-Cy7 | eBioscience | Cat# 25-0453-82, RRID: AB_469629 |
| Anti-Mouse CD45 PerCp-Cy5.5 | eBioscience | Cat# 45-0451-82, RRID: AB_1107002 |
| Anti-Mouse Ly-6A/E (Sca-1) FITC | eBioscience | Cat# 11-5981-82, RRID: AB_465333 |
| Anti-Mouse Ly-6A/E (Sca-1) PE-Cy7 | eBioscience | Cat# 25-5981-82, RRID: AB_469669 |
| Anti-Mouse Ly-6A/E (Sca-1) APC | eBioscience | Cat# 17-5981-83, RRID: AB_469488 |
| Anti-Mouse CD135 (Flt3) PerCP-eFluor 710 | eBioscience | Cat# 46-1351-82, RRID: AB_10733393 |
| Anti-Mouse CD48 PerCP-eFluor 710 | eBioscience | Cat# 46-0481-82, RRID: AB_10853483 |
| Anti-Mouse CD48 PE-Cy7 | eBioscience | Cat# 25-0481-80, RRID: AB_1724087 |
| Anti-Mouse CD48 Biotin | eBioscience | Cat# 13-0481-85, RRID: AB_466471 |
| Anti-Mouse CD34 eFluor 660 | eBioscience | Cat# 50-0341-80, RRID: AB_10609352 |
| Anti-Mouse CD71 (Tfr1) FITC | eBioscience | Cat# 11-0711-85, RRID: AB_465125 |
| Anti-Mouse CD71 (Tfr1) PE | eBioscience | Cat# 12-0711-83, RRID: AB_465741 |
| Anti-Mouse Ki67 eFluor 660 | eBioscience | Cat# 50-5698-82, RRID: AB_2574235 |
| Anti-Mouse CD51 PE | eBioscience | Cat# 12-0512-83, RRID: AB_465705 |
| Anti-Mouse CD140a (PDGFRA) APC | eBioscience | Cat# 17-1401-81, RRID: AB_529482 |
| Anti-Mouse CD41 Biotin | eBioscience | Cat# 13-0411-82, RRID: AB_763484 |
| Biotin Mouse Lineage Panel | BD Biosciences | Cat# 559971, RRID: AB_10053179 |
| Anti-Mouse CD45.2 FITC | BioLegend | Cat# 109806, RRID: AB_313443 |
| Anti-Mouse CD117 (c-Kit) Brilliant Violet 421 | BioLegend | Cat# 105828, RRID: AB_11204256 |
| Anti-Mouse CD117 (c-Kit) PE-Cy7 | BioLegend | Cat# 105814, RRID: AB_313223 |
| Anti-Mouse F4/80 PE | BioLegend | Cat# 123110, RRID: AB_893486 |
| Anti-Mouse CD150 (SLAM) PE | BioLegend | Cat# 115904, RRID: AB_313683 |
| Anti-Mouse CD48 Alexa Fluor 647 | BioLegend | Cat# 103416, RRID: AB_571987 |
| Anti-Mouse CD16/32 APC-Cy7 | BioLegend | Cat# 101328, RRID: AB_2104158 |
| Anti-Mouse CD44 PE-Cy7 | BioLegend | Cat# 103030, RRID: AB_830787 |
| Anti-Mouse CD31 Alexa Fluor 647 | BioLegend | Cat# 102516, RRID: AB_2161029 |
| Anti-Mouse CD31 PE-Cy7 | BioLegend | Cat# 102524, RRID: AB_2572182 |
| Anti-Mouse CD144 Alexa Fluor 647 | BioLegend | Cat# 138006, RRID: AB_10569114 |
| Anti-Mouse/Human Histone H3ac (pan-acetyl) | Active Motif | Cat# 61637, RRID: AB_2793714 |
| Bacterial and virus strains | ||
| E. coli HST08 | Takara Bio | Cat# 636766 |
| Chemicals, peptides, and recombinant proteins | ||
| Ampicillin | Sigma-Aldrich | Cat# A0166 |
| Neomycin | Sigma-Aldrich | Cat# N6386 |
| Metronidazole | Sigma-Aldrich | Cat# M1547 |
| Vancomycin | Pharmacy of Jack D. Weiler Hospital | N/A |
| Lipopolysaccharides (LPS) | Sigma-Aldrich | Cat# L2630 |
| E. coli ssDNA / LyoVec | Invivogen | Cat# tlrl-ssec |
| 5-Fluorouracil (5FU) | Sigma-Aldrich | Cat# F6627 |
| Tamoxifen | Sigma-Aldrich | Cat# T5648 |
| G-CSF (NEUPOGEN/Filgrastim, 300 μg/ml) | Pharmacy of Jack D. Weiler Hospital | N/A |
| Collagenase Type IV | Gibco | Cat# 17104019 |
| Dispase II | Gibco | Cat# 17105041 |
| DNase I | Sigma-Aldrich | Cat# DN25 |
| Percoll Density Gradient Media | Sigma-Aldrich | Cat# GE17-0891-01 |
| Foxp3 / Transcription Factor Staining Buffer Set | eBioscience | Cat# 00-5523-00 |
| Calcein, AM | Invitrogen | Cat# C3099 |
| Dihydroethidium | Invitrogen | Cat# D11347 |
| EZ-link Sulfo-NHS-Biotin | Thermo Scientific | Cat# 21217 |
| Lympholyte-M Cell Separation Media | Cedarlane | Cat# CL5030 |
| Mouse M-CSF | ProSpec | Cat# CYT-439 |
| Sodium butyrate | Sigma-Aldrich | Cat# 303410 |
| Sodium propionate | Sigma-Aldrich | Cat# P1880 |
| Sodium acetate | Sigma-Aldrich | Cat# S2889 |
| Valproic acid sodium salt | Sigma-Aldrich | Cat# P4543 |
| Sodium phenylbutyrate | Sigma-Aldrich | Cat# SML0309 |
| Vitamin B12 | Sigma-Aldrich | Cat# V6629 |
| 1,3-Diaminopropane | Sigma-Aldrich | Cat# D23602 |
| D-myo-Inositol 1,4,5-Trisphosphate | Sigma-Aldrich | Cat# 407137 |
| Vorinostat (SAHA) | Sigma-Aldrich | Cat# SML0061 |
| Suberohydroxamic acid (SBHA) | Sigma-Aldrich | Cat# 390585 |
| Iron-Dextran | Sigma-Aldrich | Cat# D8517 |
| Diphtheria toxin | Sigma-Aldrich | Cat# D0564 |
| Iron-adjusted diet | Teklad/Envigo | N/A |
| Insulin, human recombinant | Gibco | Cat# 12585014 |
| Sodium selenite | Sigma-Aldrich | Cat# 214485 |
| Ethanolamine | Sigma-Aldrich | Cat# 411000 |
| Poly-vinyl alcohol | Sigma-Aldrich | Cat# P8136 |
| Animal-Free Recombinant Murine TPO | PeproTech | Cat# AF-315-14 |
| Animal-Free Recombinant Murine SCF | PeproTech | Cat# AF-250-03 |
| Human holo-Transferrin | Sigma-Aldrich | Cat# T0665 |
| Human Fibronectin Coated 96-well Microplates | R&D Systems | Cat# CWP001 |
| Critical commercial assays | ||
| RNA to cDNA EcoDry Premix system | Clontech | Cat# 639546 |
| Dynabeads mRNA DIRECT Purification Kit | Invitrogen | Cat# 61012 |
| FastStart Universal SYBR Green Master Mix | Roche | Cat# 4913914001 |
| QIAamp Fast DNA Stool Mini Kit | Qiagen | Cat# 51604 |
| Mouse IL-17A ELISA | Invitrogen | Cat# BMS6001, RRID: AB_2575601 |
| Mouse IL-1beta ELISA | Invitrogen | Cat# BMS6002, RRID: AB_2575603 |
| Mouse GM-CSF ELISA | Invitrogen | Cat# BMS612, RRID: AB_2575678 |
| Mouse IL-10 ELISA | Invitrogen | Cat# BMS614, RRID: AB_2575685 |
| Mouse TGF-beta1 ELISA | Invitrogen | Cat# BMS608/4TWO, RRID: AB_2575669 |
| Mouse Erythropoietin/EPO ELISA | R&D Systems | Cat# MEP00B |
| Mouse G-CSF ELISA | R&D Systems | Cat# MCS00 |
| Mouse TNF-alpha ELISA | R&D Systems | Cat# MTA00B |
| Corticosterone ELISA | Abcam | Cat# ab108821, RRID: AB_2889904 |
| Hepcidin Murine-Compete ELISA Kit | Intrinsic LifeSciences | Cat# HMC-001 |
| LD Columns for MACS | Miltenyi Biotec | Cat# 130-042-901 |
| Iron/TIBC Reagent Set | Pointe Scientific | Cat# I750460 |
| Experimental models: Organisms/strains | ||
| Mouse: Cxcl12gfp (Cxcl12-GFP) | Gift from T. Nagasawa (Osaka University, Japan) | N/A |
| Mouse: CD169DTR (CD169-DTR) | Gift from M. Merad (Icahn School of Medicine at Mount Sinai, US) | N/A |
| Mouse: C57BL/6-Tg(UBC-GFP)30Scha/J (UBC-GFP) | The Jackson Laboratory | RRID: IMSR_JAX:004353 |
| Mouse: B6.B10ScN-Tlr4lps-del/JthJ (Tir4−/−) | The Jackson Laboratory | RRID: IMSR_JAX:007227 |
| Mouse: B6.129-Tlr2tm1Kir/J (Tlr2−/−) | The Jackson Laboratory | RRID: IMSR_JAX:004650 |
| Mouse: C57BL/6J-Ticam1Lps2/J (Trif−/−) | The Jackson Laboratory | RRID: IMSR_JAX:005037 |
| Mouse: B6.129P2-Lyz2tm1(cre)Ifo/J (LysM-Cre) | The Jackson Laboratory | RRID: IMSR_JAX:004781 |
| Mouse: B6.129P2(SJL)-Myd88tm1Defr/J (Myd88fl/fl) | The Jackson Laboratory | RRID: IMSR_JAX:008888 |
| Mouse: C57BL/6N-Fgd5tm3(cre/ERT2)Djr/J (Fgd5-CreER) | The Jackson Laboratory | RRID: IMSR_JAX:027789 |
| Mouse: C57BL/6J (CD45.2) | The Jackson Laboratory | RRID: IMSR_JAX:000664 |
| Mouse: B6.SJL-Pprca Pepcb/BoyJ (CD45.1) | The Jackson Laboratory | RRID: IMSR_JAX:002014 |
| Mouse: Germ-free B6NTac (C57BL/6) | Taconic | N/A |
| Oligonucleotides | ||
| Primers for Actb, See Table S1 | Eurofins Genomics | N/A |
| Primers for Lrp1, See Table S1 | Eurofins Genomics | N/A |
| Primers for Hmox1, See Table S1 | Eurofins Genomics | N/A |
| Primers for Mertk, See Table S1 | Eurofins Genomics | N/A |
| Primers for Pan-bacteria, See Table S2 | Eurofins Genomics | N/A |
| Primers for Bacteroidetes, See Table S2 | Eurofins Genomics | N/A |
| Primers for Firmicutes, See Table S2 | Eurofins Genomics | N/A |
| Primers for γ-Proteobacteria, See Table S2 | Eurofins Genomics | N/A |
| Primers for Actinobacteria, See Table S2 | Eurofins Genomics | N/A |
| Software and algorithms | ||
| SlideBook | Intelligent Imaging Innovations | RRID:SCR_014300 |
| FlowJo | FlowJo LLC | RRID:SCR_008520 |
| BD FACSDiva Software | BD Biosciences | RRID:SCR_001456 |
| GraphPad Prism | GraphPad Software | RRID:SCR_002798 |
| Biorender | Biorender | RRID:SCR_018361 |
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dachuan Zhang (dachuan.zhang@einsteinmed.org).
Materials availability
Mouse lines and reagents generated in this study will be provided by the lead contact upon request.
Data and code availability
There are no standardized datatypes reported in this paper.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse models
Cxcl12-GFP and CD169-DTR mice have been previously described (Asada et al., 2017; Chow et al., 2013; Chow et al., 2011). C57BL/6-Tg(UBC-GFP)30Scha/J (UBC-GFP), B6.B10ScN-Tlr4lps-del/JthJ (Tlr4−/−), B6.129-Tlr2tm1Kir/J (Tlr2−/−), C57BL/6J-Ticam1Lps2/J (Trif−/−), B6.129P2-Lyz2tm1(cre)Ifo/J (LysM-Cre), B6.129P2(SJL)-Myd88tm1Defr/J (Myd88fl/fl), C57BL/6N-Fgd5tm3(cre/ERT2)Djr/J (Fgd5-CreER), C57BL/6J (CD45.2) and B6.SJL-Ptprca Pepcb/BoyJ (CD45.1) mice were purchased from The Jackson Laboratory. Germ-free C57BL/6 mice were purchased from Taconic, and maintained in sterile isolators in the Gnotobiotic Facility at Albert Einstein College of Medicine. Six to eight-week-old mice of both genders were used for experiments. All mice were housed in specific pathogen-free conditions and fed with autoclaved food and water. Experimental procedures performed on mice were approved by the Institutional Animal Care and Use Committee of Albert Einstein College of Medicine.
METHOD DETAILS
Antibiotic treatment
Wild-type or CD169-DTR mice were treated with Ampicillin (1g/L), Neomycin (1g/L), Metronidazole (1g/L) and Vancomycin (1g/L) in drinking water for 4 – 6 weeks. Mice were gavaged with 500 μl antibiotic solution before the treatment started, which effectively prevented dehydration induced by the treatment in the first two weeks (Hill et al., 2010). Antibiotics were purchased from Sigma or Jack D. Weiler Hospital of Albert Einstein College of Medicine. Drinking water containing antibiotics was changed once a week. For microbial product add-back experiment, ABX-treated or wild-type mice were fed with 1 mg LPS (0111:B4, Sigma) or 20 μg ssDNA (E. coli ssDNA/LyoVec, Invivogen) by intragastric gavage every three days starting two days before 5-FU treatment. For butyrate add-back experiment, ABX-treated mice were injected subcutaneously with 500 mg/kg butyrate twice a day starting two days before 5-FU treatment.
In vivo treatments
For the induction of hematopoietic stress, mice were injected with one dose of 200 mg/kg body weight 5FU. For the evaluation of susceptibility, mice were challenged with a second dose of 5FU at day 7 following the first injection (Takizawa et al., 2017), and survival time, defined as the time from the first injection until death, was recorded. For the sublethal irradiation model, mice were subjected to 8 Gy irradiation, split into two doses with 3 hours apart as described (Hooper et al., 2009), and analyzed 24 days after irradiation. For induction of Cre recombination, Fgd5-CreER/Myd88fl/fl mice were injected intraperitoneally with 2 mg tamoxifen (Sigma) dissolved in corn oil (Sigma) every other day for 3 weeks. For G-CSF treatment, G-CSF was administered subcutaneously at a dose of 125 μg/kg twice a day for 8 doses, and mice were analyzed 3h after the final morning dose. G-CSF (NEUPOGEN/Filgrastim, 300 μg/ml) was purchased from Jack D. Weiler Hospital of Albert Einstein College of Medicine. For systemic E. coli infection, control or ABX-treated mice (ABX treatment was stopped 5 days before bacteria injection) were injected intravenously with 2 × 108 CFU E. coli, and analyzed two days later. For LPS challenge, mice were injected intravenously with 30 μg LPS (0111:B4, Sigma), and analyzed two days later.
Blood counts
Blood was collected from the retro-orbital plexus with 5 μl 0.5M EDTA, and analyzed on the ADVIA 120 hematology system (SIEMENS).
Cell preparation
Single cell suspension was prepared as described previously (Chow et al., 2013; Pinho et al., 2013). Briefly, BM cells were gently flushed out from bones, and homogenized by pipetting. Spleens were mashed through a 40 μm filter. Liver cells were mechanically diced and digested with 1 mg/ml Type IV collagenase (Gibco), 2 mg/ml Dispase (Gibco) and 1 mg/ml DNAse (Sigma) for one hour. The liver suspension was then homogenized through a 19G syringe needle, filtrated, and pelleted with a 30% Percoll gradient (GE Healthcare). Single cell suspension from blood, the BM, spleen or liver was then subjected to staining with or without RBC lysis.
Flow cytometry and cell sorting
Cells were surface-stained in PEB buffer (PBS containing 0.5% BSA and 2mM EDTA) for 30–60 min on ice. Multiparametric flow cytometric analyses were performed on a LSRII equipped with FACS Diva 6.1 software (BD Biosciences) and analyzed with FlowJo software (Tree Star). Dead cells were excluded by FSC, SSC and 4’,6-diamino-2-phenylindole (DAPI, Sigma) staining. Cell sorting experiments were performed on Aria Cell Sorter (BD Biosciences) or MoFlo Astrios (Beckman Coulter). For the analysis of CD71, calcein fluorescence and ROS production, EDTA was omitted from the buffer to prevent interference. Neutrophils were gated by Gr-1hi CD115lo SSChi; T cells, B cells and monocytes were gated by CD3+ CD11b−, B220+ CD11b− and CD115hi CD11b+, respectively; Hematopoietic progenitor and stem cells were identified by lineage cocktail (Gr-1, CD11b, CD3, B220 and Ter119), Sca-1, c-Kit, CD135, CD150, CD48, CD34 and CD16/32; macrophages were identified by Gr-1lo CD115lo F4/80+ SSClo; erythroblasts were identified by CD71 and Ter119; mesenchymal stem cells and endothelial cells were identified by CD45− CD31− CD51+ CD140a+ and CD45− CD31+ Sca-1+, as described previously (Chow et al., 2013; Pietras et al., 2015; Pinho et al., 2013; Wilson et al., 2008; Xu et al., 2018). Cell cycle was analyzed with Ki67 and Hoechst 33342 (Sigma). Fluorophore-conjugated, biotinylated, or primary antibodies against mouse Gr-1 (RB6–8C5), CD115 (AFS98), CD3 (145–2C11), B220 (RA3–6B2), CD11b (M1/70), Ter119 (Ter-119), CD45.1 (A20), CD45 (30-F11), Sca-1 (D7), CD135 (A2F10), CD48 (HM48–1), CD34 (RAM34), CD71 (R17217), Ki67 (SolA15), CD51 (RMV-7), and CD140a (APA5) were from eBioscience. Biotin Mouse Lineage Panel was from BD Pharmingen. Antibodies against CD45.2 (104), c-Kit (2B8), F4/80 (BM8), CD150 (TC15–12F12.2), CD48 (HM48–1), CD16/32 (93), CD44 (IM7), and CD31 (MEC13.3) were from BioLegend. Antibody against H3ac was from Active Motif. For intracellular staining, cells were fixed and permeabilized with Foxp3 / Transcription Factor Staining Buffer Set from eBioscience. For calcein staining, surface-stained cells were incubated with 5 nM Calcein AM (Invitrogen) for 5 minutes at 37 °C. For ROS detection, surface-stained cells were incubated with 7 μM Dihydroethidium (DHE) for 20 minutes at 37 °C.
Bone marrow and HSC transplantations
To account for the dynamic changes of total BM cellularity during regeneration, the competitive BM transplantation assays were performed by transplanting 5 × 105 CD45.1+ competitor cells with equal volumes of BM cells (matching the volume of competitor cells; usually ~3% of total cells from a femur) from control or ABX-treated mice (CD45.2+) with or without 5FU treatment, into lethally irradiated CD45.1+ recipients (12 Gy in two split doses), as reported previously (Asada et al., 2017; Bruns et al., 2014; Kunisaki et al., 2013). Irradiation was performed using a Cesium Mark 1 irradiator (JL Shepherd & Associates). Chimeric mice were analyzed for 20 weeks after transplantation, followed by secondary transplantation of 3 × 106 BM cells from primary recipients into lethally irradiated CD45.1+ recipients. HSCTs were performed by transplanting 300 sorted phenotypic HSCs (CD45.2+) with 5 × 105 CD45.1+ competitor cells, into lethally irradiated CD45.1+ recipients. For the HSC ex vivo culture experiment, transplantation assays were performed by transplanting 1/50 of the HSCs expanded with 500 mg/L or 0.5 mg/L transferrin (CD45.2+), plus 1 × 106 competitive CD45.1+ BM cells, into lethally irradiated CD45.1+ recipients. Transplantation assays were also performed in other models as described in figure legends.
Quantitative real-time PCR (Q-PCR)
Messenger RNA extraction from 2000 sorted macrophages using Dynabeads mRNA DIRECT™ Kit (Life technologies) and reverse transcription using RNA to cDNA EcoDry Premix (Clontech) were preformed according to the manufacturer’s protocols. Q-PCR was performed with FastStart Universal SYBR Green Master Mix (Roche) on QuantStudio 6 Flex Real-Time PCR Systems (Applied Biosystems). The PCR protocol started with one cycle at 95 °C (10 min) and continued with 40 cycles at 95 °C (15 s) and 60 °C (1 min). Expression of Actb was used as a standard. The average threshold cycle number (Ct) for each tested gene was used to quantify the relative expression of each gene: 2[Ct(standard) − Ct(gene)]. Primers were listed in the Key Resources Table and Table S1.
16S rDNA quantification
Stool pellets were collected and total DNA was extracted using the QIAamp Fast DNA Stool Mini Kit (Qiagen), followed by real-time q-PCR using FastStart Universal SYBR Green Master Mix (Roche). Absolute copy numbers were calculated based on standard curves derived from a serial dilution of plasmids containing bacterial DNA sequence from each phylum. NCBI Reference Sequences of Escherichia coli, Enterococcus faecalis, Bacteroides fragilis, and Bifidobacterium adolescentis were used to prepare standards. Primers were used as previously described (Bacchetti De Gregoris et al., 2011; Guo et al., 2008; Hartman et al., 2009), and listed in the Key Resources Table and Table S2.
Whole-mount immunofluorescence imaging
Whole-mount tissue preparation, HSC immunofluorescence staining and imaging analysis of the sternum in control and ABX-treated Cxcl12-GFP mice following 5FU challenge were performed as described previously (Kunisaki et al., 2013; Pinho et al., 2018). Briefly, the vasculature of was stained by i.v. injection of Alexa Fluor 647 anti-CD144 (BV13, Biolegend) and Alexa Fluor 647 anti-CD31, and after 20 min mice were perfused with 4% PFA followed by cold PBS. Sternum bones were carefully harvested, cleaned and transected with a surgical blade into individual pieces. Each piece was bisected sagittally to expose the marrow cavity. Fragments were further fixed with 4% PFA for 20 minutes, blocked and permeabilized in PBS with 20% normal goat serum and 0.5% Triton X-100, and stained with antibodies (biotin anti-Lineage panel cocktail, biotin anti-CD41, biotin anti-CD48 and PE anti-CD150) for 3 days. The tissues were then incubated with streptavidin eFluor 450 for 2 hours. Images were acquired using a ZEISS AXIO examiner D1 microscope (Zeiss) with a confocal scanner unit, CSUX1CU (Yokogawa), and reconstructed in 3D with SlideBook software (Intelligent Imaging Innovations). HSCs were identified as CD150+ Lin− CD48− by immunofluorescence. For each HSC, the Euclidean distance to the closest sinusoid or arteriole was measured in order to generate distribution maps.
ELISA
Cytokine levels in plasma and bone marrow extracellular fluid (BMEF) from control and ABX-treated mice under steady state or 12 days after 5FU treatment were measured using ELISA kits (IL-17, IL-1β, GM-CSF, IL-10 and TGF-β kits from Invitrogen; EPO, G-CSF and TNF-α kits from R&D systems; Corticosterone kit from Abcam; Hepcidin kit from Intrinsic LifeSciences) according to the manufacturer’s instructions.
Adoptive transfer and pulse-chase labelling
For progenitor homing and differentiation assay, Lin− cells were enriched by immunomagnetic depletion using biotin anti-Lineage panel antibodies, streptavidin magnetic beads and LD Columns (Miltenyi Biotec), and >90% purity was confirmed by flow cytometry. 6 × 105 Lin− cells from UBC-GFP mice were transferred into control or ABX-treated mice at Day 10 after 5FU challenge via retro-orbital injections. Mice were analyzed two hours or two days after the transfer. For RBC clearance assay, control and ABX-treated mice were injected with 100 mg/kg EZ-Link™ Sulfo-NHS-Biotin (Thermo Scientific) one day before chasing started.
RBC phagocytic assay
For in vivo RBC phagocytosis assay, RBCs were harvested from 5FU-stressed UBC-GFP mice using Lympholyte-M Cell Separation Media (Cedarlane). BSA was avoided in the RBC isolation process to prevent interference. GFP+ RBCs were transferred into control and ABX-treated mice at indicated time points, and phagocytic activities in the BM, spleen and liver were analyzed two days later. For in vitro phagocytosis assay, BM-derived macrophages were prepared by plating total BM cells in complete medium containing RPMI1640 with Glutamine (Corning), 10% FBS, 1% P/S, 10mM HEPES and 10 ng/mL M-CSF (Prospec) in non-treated Petri dishes. Non-adherent cells were washed away on day 6, and macrophages were harvested with Trypsin/EDTA solution (Corning) on day 7. Macrophages were then dispensed into 6-well plates and co-cultured with GFP+ RBCs for 18 hours. Microbial metabolites and HDAC inhibitors were added to the co-culture medium at the concentration of 1 mM with the following exceptions: VB12 (100 μM), IP3 (10 μM), SAHA (1 μM), and SBHA (10 μM). Macrophages were harvested with cell scraper (Falcon) for flow cytometry analysis. Microbial metabolites and HDAC inhibitors were referred to as below: 1,3-DAP, 1,3-diaminopropane; VB12, vitamin B12; IP3, inositol trisphosphate; SAHA (Vorinostat), suberoylanilide hydroxamic acid; SBHA, suberoyl bis-hydroxamic acid.
Tissue iron quantification
Quantification of tissue iron stores were performed as described previously (Li et al., 2017). Briefly, iron was extracted from tissues using acid solution containing hydrochloride acid and trichloroacetic acid, and then assayed with chromogen reagent containing bathophenanthroline sulfonate and thioglycolic acid. Tissues were dried and weight before acid treatment. For the analyses of bone marrow, at least two tibias were flushed with water to collect total bone marrow, or with PBS followed by RBC lysis to collect bone marrow leukocytes. For quantification of serum iron parameters, a ferrozine-based Iron/TIBC Reagent Set (Pointe Scientific) was used. The absorption was measured at 540 nm for bathophenanthroline sulfonate, or at 560nm for ferrozine by Absorbance Microplate Reader (BioTek).
Iron manipulation in vivo
For the induction of iron deficiency, mice were fed with iron-adjusted diet containing 185ppm (normal), 30ppm (low-iron), or 3ppm iron (iron-deficient; Teklad) for three months before steady-state analyses or 5FU challenge. To exclude the influence of diet-induced iron deficiency on the microbiome, mice fed with different iron-adjusted diet were treated with ABX for one month to deplete the microbiota. For the add-back of iron during regeneration, ABX-treated mice were injected subcutaneously with 5mg iron dextran (Sigma) or PBS after 5FU challenge.
Macrophage depletion
For the depletion of CD169+ macrophages, wild-type and CD169-DTR mice were injected subcutaneously with 10μg/kg body weight diphtheria toxin (Sigma) twice a week, starting one week before 5FU. Notably, female macrophage-depleted mice tended to develop increased bone formation after 5FU challenge, which made the collection of BM cells difficult in a small fraction of these mice.
LC-MS analysis of SCFA
SCFA levels were measured by the Proteomics and Metabolomics Shared Resource at Duke University, using a UPLC-MS/MS method as described previously (Han et al., 2015). In brief, this method utilized Acquity UPLC columns coupled to Xevo TQ-S triple quadrupole mass spectrometry (Waters) to perform quantitative multiplexed analysis of SCFAs.
Serum-free culture system
HSCs were cultured using a PVA-based albumin-free culture system as described in a previous report (Wilkinson et al., 2019). Briefly, HSCs were cultured in media composed of F12 medium (Gibco), 10 mg/L insulin (Gibco), 6.7 μg/L sodium selenite (Sigma), 2 mg/L ethanolamine (Sigma), 1% P/S and L-Glutamine (Gibco), 10 mM HEPES (Gibco), 0.1% poly-vinyl alcohol (PVA, Sigma), 100 ng/ml murine TPO (PeproTech), 10 ng/ml murine SCF (PeproTech), and human holo-transferrin (Sigma) with defined concentrations ranging from 5 μg/L to 500 mg/L. Fifty sorted Lin− Sca-1+ c-Kit+ CD48− CD150+ CD34− HSCs were used as input, and cultured in human fibronectin coated 96-well microplates (R&D systems) for 18–21 days. Complete medium change was initiated 5 days after culture started, and performed every 2–3 days. Cultured cells were split 1:3 into new plates at ~90% confluency, and harvested for analyses and transplantation when they reach confluency for the second time.
QUANTIFICATION AND STATISTICAL ANALYSIS
No statistical methods were used to predetermine the sample size. Sample sizes were determined based on similar models of hematopoiesis in previous studies from our laboratory (Asada et al., 2017; Bowers et al., 2018; Kunisaki et al., 2013; Lucas et al., 2013; Pinho et al., 2013; Pinho et al., 2018; Xu et al., 2018). Key experiments (ABX treatment models, iron manipulation experiments, and iron assays) were performed blind to group allocations, and validated by two investigators independently. Unpaired two-sided Student’s t-tests were used to compare two groups. One-way ANOVA, two-way ANOVA, or multiple t-test followed by Holm-Sidak multiple comparison test were used for multiple-group comparisons. Log-rank test was used to compare survival curves. Statistical analyses were performed using Graph Pad Prism 8 software and the results were presented in figures and figure legends.
Supplementary Material
Highlights.
Microbiota depletion impairs HSC response under stress conditions
BM macrophages recycle RBCs to provide iron to HSCs for regeneration
The microbiota regulates BM macrophage functions via short-chain fatty acid
Local iron availability controls HSC self-renewal and differentiation decisions
ACKNOWLEDGEMENTS
We are deeply saddened by the passing of Dr. Paul S. Frenette on July 26th, 2021. We will forever be indebted to him for his superb mentorship. His rigorous and creative thinking, out-of-the box approach, and unsurpassed enthusiasm for science dramatically influenced the co-authors and laid the foundation for this manuscript. He will be deeply missed by all, and his research and approach to science will live on through his trainees.
We are grateful to C. Prophete, G. Amatuni, and J. Kazmi for expert technical assistance; D. Sun for assistance in cell sorting; L. Dubois, W. Thompson, M. Foster and A. Moseley for assistance in LC-MS analysis. We also thank U. Steidl for helpful discussion and advice. This work was supported by R01 grants from the National Institutes of Health (HL157948, HL069438, DK056638, HL116340 to P.S.F.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCE
- Asada N, Kunisaki Y, Pierce H, Wang Z, Fernandez NF, Birbrair A, Ma’ayan A, and Frenette PS (2017). Differential cytokine contributions of perivascular haematopoietic stem cell niches. Nat Cell Biol 19, 214–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacchetti De Gregoris T, Aldred N, Clare AS, and Burgess JG (2011). Improvement of phylum- and class-specific primers for real-time PCR quantification of bacterial taxa. J Microbiol Methods 86, 351–356. [DOI] [PubMed] [Google Scholar]
- Baldridge MT, King KY, Boles NC, Weksberg DC, and Goodell MA (2010). Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature 465, 793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmer ML, Schurch CM, Saito Y, Geuking MB, Li H, Cuenca M, Kovtonyuk LV, McCoy KD, Hapfelmeier S, Ochsenbein AF, et al. (2014). Microbiota-derived compounds drive steady-state granulopoiesis via MyD88/TICAM signaling. J Immunol 193, 5273–5283. [DOI] [PubMed] [Google Scholar]
- Belkaid Y, and Hand TW (2014). Role of the microbiota in immunity and inflammation. Cell 157, 121–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia M, Bonnet D, Kapp U, Wang JC, Murdoch B, and Dick JE (1997). Quantitative analysis reveals expansion of human hematopoietic repopulating cells after short-term ex vivo culture. J Exp Med 186, 619–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom SM, Bijanki VN, Nava GM, Sun L, Malvin NP, Donermeyer DL, Dunne WM Jr., Allen PM, and Stappenbeck TS (2011). Commensal Bacteroides species induce colitis in host-genotype-specific fashion in a mouse model of inflammatory bowel disease. Cell Host Microbe 9, 390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettcher S, Gerosa RC, Radpour R, Bauer J, Ampenberger F, Heikenwalder M, Kopf M, and Manz MG (2014). Endothelial cells translate pathogen signals into G-CSF-driven emergency granulopoiesis. Blood 124, 1393–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers E, Slaughter A, Frenette PS, Kuick R, Pello OM, and Lucas D (2018). Granulocyte-derived TNFalpha promotes vascular and hematopoietic regeneration in the bone marrow. Nat Med 24, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns I, Lucas D, Pinho S, Ahmed J, Lambert MP, Kunisaki Y, Scheiermann C, Schiff L, Poncz M, Bergman A, et al. (2014). Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat Med 20, 1315–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabezas-Wallscheid N, Buettner F, Sommerkamp P, Klimmeck D, Ladel L, Thalheimer FB, Pastor-Flores D, Roma LP, Renders S, Zeisberger P, et al. (2017). Vitamin A-Retinoic Acid Signaling Regulates Hematopoietic Stem Cell Dormancy. Cell 169, 807–823 e819. [DOI] [PubMed] [Google Scholar]
- Cabezas-Wallscheid N, Klimmeck D, Hansson J, Lipka DB, Reyes A, Wang Q, Weichenhan D, Lier A, von Paleske L, Renders S, et al. (2014). Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell 15, 507–522. [DOI] [PubMed] [Google Scholar]
- Cai Z, Kotzin JJ, Ramdas B, Chen S, Nelanuthala S, Palam LR, Pandey R, Mali RS, Liu Y, Kelley MR, et al. (2018). Inhibition of Inflammatory Signaling in Tet2 Mutant Preleukemic Cells Mitigates Stress-Induced Abnormalities and Clonal Hematopoiesis. Cell Stem Cell 23, 833–849 e835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairo G, Recalcati S, Mantovani A, and Locati M (2011). Iron trafficking and metabolism in macrophages: contribution to the polarized phenotype. Trends Immunol 32, 241–247. [DOI] [PubMed] [Google Scholar]
- Callens C, Coulon S, Naudin J, Radford-Weiss I, Boissel N, Raffoux E, Wang PH, Agarwal S, Tamouza H, Paubelle E, et al. (2010). Targeting iron homeostasis induces cellular differentiation and synergizes with differentiating agents in acute myeloid leukemia. J Exp Med 207, 731–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvi LM, and Link DC (2015). The hematopoietic stem cell niche in homeostasis and disease. Blood 126, 2443–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova-Acebes M, Pitaval C, Weiss LA, Nombela-Arrieta C, Chevre R,N,AG, Kunisaki Y, Zhang D, van Rooijen N, Silberstein LE, et al. (2013). Rhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell 153, 1025–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey JL, Hentze MW, Koeller DM, Caughman SW, Rouault TA, Klausner RD, and Harford JB (1988). Iron-responsive elements: regulatory RNA sequences that control mRNA levels and translation. Science 240, 924–928. [DOI] [PubMed] [Google Scholar]
- Chai X, Li D, Cao X, Zhang Y, Mu J, Lu W, Xiao X, Li C, Meng J, Chen J, et al. (2015). ROS-mediated iron overload injures the hematopoiesis of bone marrow by damaging hematopoietic stem/progenitor cells in mice. Sci Rep 5, 10181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang PV, Hao L, Offermanns S, and Medzhitov R (2014). The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci U S A 111, 2247–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chasis JA, and Mohandas N (2008). Erythroblastic islands: niches for erythropoiesis. Blood 112, 470–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow A, Huggins M, Ahmed J, Hashimoto D, Lucas D, Kunisaki Y, Pinho S, Leboeuf M, Noizat C, van Rooijen N, et al. (2013). CD169(+) macrophages provide a niche promoting erythropoiesis under homeostasis and stress. Nat Med 19, 429–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow A, Lucas D, Hidalgo A, Mendez-Ferrer S, Hashimoto D, Scheiermann C, Battista M, Leboeuf M, Prophete C, van Rooijen N, et al. (2011). Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med 208, 261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopher MJ, Rao M, Liu F, Woloszynek JR, and Link DC (2011). Expression of the G-CSF receptor in monocytic cells is sufficient to mediate hematopoietic progenitor mobilization by G-CSF in mice. J Exp Med 208, 251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, and Weiser JN (2010). Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med 16, 228–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conneally E, Cashman J, Petzer A, and Eaves C (1997). Expansion in vitro of transplantable human cord blood stem cells demonstrated using a quantitative assay of their lympho-myeloid repopulating activity in nonobese diabetic-scid/scid mice. Proc Natl Acad Sci U S A 94, 9836–9841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das NK, Schwartz AJ, Barthel G, Inohara N, Liu Q, Sankar A, Hill DR, Ma X, Lamberg O, Schnizlein MK, et al. (2020). Microbial Metabolite Signaling Is Required for Systemic Iron Homeostasis. Cell Metab 31, 115–130 e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Back DZ, Kostova EB, van Kraaij M, van den Berg TK, and van Bruggen R (2014). Of macrophages and red blood cells; a complex love story. Front Physiol 5, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaby C, Pilard N, Hetet G, Driss F, Grandchamp B, Beaumont C, and Canonne-Hergaux F (2005). A physiological model to study iron recycling in macrophages. Exp Cell Res 310, 43–53. [DOI] [PubMed] [Google Scholar]
- Deshmukh HS, Liu Y, Menkiti OR, Mei J, Dai N, O’Leary CE, Oliver PM, Kolls JK, Weiser JN, and Worthen GS (2014). The microbiota regulates neutrophil homeostasis and host resistance to Escherichia coli K1 sepsis in neonatal mice. Nat Med 20, 524–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devireddy LR, Gazin C, Zhu X, and Green MR (2005). A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell 123, 1293–1305. [DOI] [PubMed] [Google Scholar]
- Ding L, Saunders TL, Enikolopov G, and Morrison SJ (2012). Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481, 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganz T (2019). Anemia of Inflammation. N Engl J Med 381, 1148–1157. [DOI] [PubMed] [Google Scholar]
- Ganz T, and Nemeth E (2015). Iron homeostasis in host defence and inflammation. Nat Rev Immunol 15, 500–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Zhang D, Xu C, Li H, Caron KM, and Frenette PS (2021). Nociceptive nerves regulate haematopoietic stem cell mobilization. Nature 589, 591–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattermann N (2018). Iron overload in myelodysplastic syndromes (MDS). Int J Hematol 107, 55–63. [DOI] [PubMed] [Google Scholar]
- Gordy C, Pua H, Sempowski GD, and He YW (2011). Regulation of steady-state neutrophil homeostasis by macrophages. Blood 117, 618–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Xia X, Tang R, Zhou J, Zhao H, and Wang K (2008). Development of a real-time PCR method for Firmicutes and Bacteroidetes in faeces and its application to quantify intestinal population of obese and lean pigs. Lett Appl Microbiol 47, 367–373. [DOI] [PubMed] [Google Scholar]
- Gupta P, Lai SM, Sheng J, Tetlak P, Balachander A, Claser C, Renia L, Karjalainen K, and Ruedl C (2016). Tissue-Resident CD169(+) Macrophages Form a Crucial Front Line against Plasmodium Infection. Cell Rep 16, 1749–1761. [DOI] [PubMed] [Google Scholar]
- Han J, Lin K, Sequeira C, and Borchers CH (2015). An isotope-labeled chemical derivatization method for the quantitation of short-chain fatty acids in human feces by liquid chromatography-tandem mass spectrometry. Anal Chim Acta 854, 86–94. [DOI] [PubMed] [Google Scholar]
- Hartman AL, Lough DM, Barupal DK, Fiehn O, Fishbein T, Zasloff M, and Eisen JA (2009). Human gut microbiome adopts an alternative state following small bowel transplantation. Proc Natl Acad Sci U S A 106, 17187–17192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herault A, Binnewies M, Leong S, Calero-Nieto FJ, Zhang SY, Kang YA, Wang X, Pietras EM, Chu SH, Barry-Holson K, et al. (2017). Myeloid progenitor cluster formation drives emergency and leukaemic myelopoiesis. Nature 544, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DA, Hoffmann C, Abt MC, Du Y, Kobuley D, Kirn TJ, Bushman FD, and Artis D (2010). Metagenomic analyses reveal antibiotic-induced temporal and spatial changes in intestinal microbiota with associated alterations in immune cell homeostasis. Mucosal Immunol 3, 148–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoggatt J, Kfoury Y, and Scadden DT (2016). Hematopoietic Stem Cell Niche in Health and Disease. Annu Rev Pathol 11, 555–581. [DOI] [PubMed] [Google Scholar]
- Holmes E, Li JV, Marchesi JR, and Nicholson JK (2012). Gut microbiota composition and activity in relation to host metabolic phenotype and disease risk. Cell Metab 16, 559–564. [DOI] [PubMed] [Google Scholar]
- Hong C, Kidani Y,N,AG, Phung T, Ito A, Rong X, Ericson K, Mikkola H, Beaven SW, Miller LS, et al. (2012). Coordinate regulation of neutrophil homeostasis by liver X receptors in mice. J Clin Invest 122, 337–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper AT, Butler JM, Nolan DJ, Kranz A, Iida K, Kobayashi M, Kopp HG, Shido K, Petit I, Yanger K, et al. (2009). Engraftment and reconstitution of hematopoiesis is dependent on VEGFR2-mediated regeneration of sinusoidal endothelial cells. Cell Stem Cell 4, 263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoverstad T, and Midtvedt T (1986). Short-chain fatty acids in germfree mice and rats. J Nutr 116, 1772–1776. [DOI] [PubMed] [Google Scholar]
- Hur J, Choi JI, Lee H, Nham P, Kim TW, Chae CW, Yun JY, Kang JA, Kang J, Lee SE, et al. (2016). CD82/KAI1 Maintains the Dormancy of Long-Term Hematopoietic Stem Cells through Interaction with DARC-Expressing Macrophages. Cell Stem Cell 18, 508–521. [DOI] [PubMed] [Google Scholar]
- Itkin T, Gur-Cohen S, Spencer JA, Schajnovitz A, Ramasamy SK, Kusumbe AP, Ledergor G, Jung Y, Milo I, Poulos MG, et al. (2016). Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature 532, 323–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamura C, Bouladoux N, Belkaid Y, Sher A, and Jankovic D (2017). Sensing of the microbiota by NOD1 in mesenchymal stromal cells regulates murine hematopoiesis. Blood 129, 171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefsdottir KS, Baldridge MT, Kadmon CS, and King KY (2017). Antibiotics impair murine hematopoiesis by depleting the intestinal microbiota. Blood 129, 729–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao Y-R, Chen J, Kumari R, Tatiparthy M, Ma Y, Aivalioti MM, Zintiridou A, Thiruthuvanathan V, Reisz JA, Stranski S, et al. (2021). Cytoplasmic labile iron accumulates in aging stem cells perturbing a key rheostat for identity control. bioRxiv. [Google Scholar]
- Kao YR, Chen J, Narayanagari SR, Todorova TI, Aivalioti MM, Ferreira M, Ramos PM, Pallaud C, Mantzaris I, Shastri A, et al. (2018). Thrombopoietin receptor-independent stimulation of hematopoietic stem cells by eltrombopag. Sci Transl Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, and Ganz T (2014). Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet 46, 678–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosravi A, Yanez A, Price JG, Chow A, Merad M, Goodridge HS, and Mazmanian SK (2014). Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe 15, 374–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King KY, and Goodell MA (2011). Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nat Rev Immunol 11, 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson MD, Oukka M, Koss LM, Aydemir F, and Wessling-Resnick M (2005). Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin 1 overexpression and down-regulated by hepcidin. Proc Natl Acad Sci U S A 102, 1324–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korolnek T, and Hamza I (2015). Macrophages and iron trafficking at the birth and death of red cells. Blood 125, 2893–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koury MJ, and Ponka P (2004). New insights into erythropoiesis: the roles of folate, vitamin B12, and iron. Annu Rev Nutr 24, 105–131. [DOI] [PubMed] [Google Scholar]
- Kumar S, and Geiger H (2017). HSC Niche Biology and HSC Expansion Ex Vivo. Trends Mol Med 23, 799–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, Mizoguchi T, Wei Q, Lucas D, Ito K, et al. (2013). Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 502, 637–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurenti E, and Gottgens B (2018). From haematopoietic stem cells to complex differentiation landscapes. Nature 553, 418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kim H, You G, Kim YM, Lee S, Le VH, Kwon O, Im SH, Kim YM, Kim KS, et al. (2019). Bone marrow CX3CR1+ mononuclear cells relay a systemic microbiota signal to control hematopoietic progenitors in mice. Blood 134, 1312–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leimberg MJ, Prus E, Konijn AM, and Fibach E (2008). Macrophages function as a ferritin iron source for cultured human erythroid precursors. J Cell Biochem 103, 1211–1218. [DOI] [PubMed] [Google Scholar]
- Li H, Choesang T, Bao W, Chen H, Feola M, Garcia-Santos D, Li J, Sun S, Follenzi A, Pham P, et al. (2017). Decreasing TfR1 expression reverses anemia and hepcidin suppression in beta-thalassemic mice. Blood 129, 1514–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Wang Y, Zhao H, Zhang H, Xu Y, Wang S, Guo X, Huang Y, Zhang S, Han Y, et al. (2019). Identification and transcriptome analysis of erythroblastic island macrophages. Blood 134, 480–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libregts SF, Gutierrez L, de Bruin AM, Wensveen FM, Papadopoulos P, van Ijcken W, Ozgur Z, Philipsen S, and Nolte MA (2011). Chronic IFN-gamma production in mice induces anemia by reducing erythrocyte life span and inhibiting erythropoiesis through an IRF-1/PU.1 axis. Blood 118, 2578–2588. [DOI] [PubMed] [Google Scholar]
- Lucas D, Scheiermann C, Chow A, Kunisaki Y, Bruns I, Barrick C, Tessarollo L, and Frenette PS (2013). Chemotherapy-induced bone marrow nerve injury impairs hematopoietic regeneration. Nat Med 19, 695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludin A, Itkin T, Gur-Cohen S, Mildner A, Shezen E, Golan K, Kollet O, Kalinkovich A, Porat Z, D’Uva G, et al. (2012). Monocytes-macrophages that express alpha-smooth muscle actin preserve primitive hematopoietic cells in the bone marrow. Nat Immunol 13, 1072–1082. [DOI] [PubMed] [Google Scholar]
- Luo Y, Chen GL, Hannemann N, Ipseiz N, Kronke G, Bauerle T, Munos L, Wirtz S, Schett G, and Bozec A (2015). Microbiota from Obese Mice Regulate Hematopoietic Stem Cell Differentiation by Altering the Bone Niche. Cell Metab 22, 886–894. [DOI] [PubMed] [Google Scholar]
- McCarville JL, Chen GY, Cuevas VD, Troha K, and Ayres JS (2020). Microbiota Metabolites in Health and Disease. Annu Rev Immunol 38, 147–170. [DOI] [PubMed] [Google Scholar]
- Meisel M, Hinterleitner R, Pacis A, Chen L, Earley ZM, Mayassi T, Pierre JF, Ernest JD, Galipeau HJ, Thuille N, et al. (2018). Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature 557, 580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CL, and Eaves CJ (1997). Expansion in vitro of adult murine hematopoietic stem cells with transplantable lympho-myeloid reconstituting ability. Proc Natl Acad Sci U S A 94, 13648–13653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitlyng BL, Singh JA, Furne JK, Ruddy J, and Levitt MD (2006). Use of breath carbon monoxide measurements to assess erythrocyte survival in subjects with chronic diseases. Am J Hematol 81, 432–438. [DOI] [PubMed] [Google Scholar]
- Miyake Y, Asano K, Kaise H, Uemura M, Nakayama M, and Tanaka M (2007). Critical role of macrophages in the marginal zone in the suppression of immune responses to apoptotic cell-associated antigens. J Clin Invest 117, 2268–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muckenthaler MU, Rivella S, Hentze MW, and Galy B (2017). A Red Carpet for Iron Metabolism. Cell 168, 344–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto Y, Nishiyama M, Nita A, Moroishi T, and Nakayama KI (2017). Essential role of FBXL5-mediated cellular iron homeostasis in maintenance of hematopoietic stem cells. Nat Commun 8, 16114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura-Ishizu A, Ito K, and Suda T (2020). Hematopoietic Stem Cell Metabolism during Development and Aging. Dev Cell 54, 239–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notta F, Doulatov S, Laurenti E, Poeppl A, Jurisica I, and Dick JE (2011). Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science 333, 218–221. [DOI] [PubMed] [Google Scholar]
- Osawa M, Hanada K, Hamada H, and Nakauchi H (1996). Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science 273, 242–245. [DOI] [PubMed] [Google Scholar]
- Pietras EM, Reynaud D, Kang YA, Carlin D, Calero-Nieto FJ, Leavitt AD, Stuart JM, Gottgens B, and Passegue E (2015). Functionally Distinct Subsets of Lineage-Biased Multipotent Progenitors Control Blood Production in Normal and Regenerative Conditions. Cell Stem Cell 17, 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho S, and Frenette PS (2019). Haematopoietic stem cell activity and interactions with the niche. Nat Rev Mol Cell Biol 20, 303–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho S, Lacombe J, Hanoun M, Mizoguchi T, Bruns I, Kunisaki Y, and Frenette PS (2013). PDGFRalpha and CD51 mark human nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J Exp Med 210, 1351–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho S, Marchand T, Yang E, Wei Q, Nerlov C, and Frenette PS (2018). Lineage-Biased Hematopoietic Stem Cells Are Regulated by Distinct Niches. Dev Cell 44, 634–641 e634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi L, Lin KK, Boles NC, Yang L, King KY, Jeong M, Mayle A, and Goodell MA (2012). Less is more: unveiling the functional core of hematopoietic stem cells through knockout mice. Cell Stem Cell 11, 302–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth M, Will B, Simkin G, Narayanagari S, Barreyro L, Bartholdy B, Tamari R, Mitsiades CS, Verma A, and Steidl U (2012). Eltrombopag inhibits the proliferation of leukemia cells via reduction of intracellular iron and induction of differentiation. Blood 120, 386–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettpelz LG, Borgerding JN, Christopher MJ, Gopalan PK, Romine MP, Herman AC, Woloszynek JR, Greenbaum AM, and Link DC (2014). G-CSF regulates hematopoietic stem cell activity, in part, through activation of Toll-like receptor signaling. Leukemia 28, 1851–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulthess J, Pandey S, Capitani M, Rue-Albrecht KC, Arnold I, Franchini F, Chomka A, Ilott NE, Johnston DGW, Pires E, et al. (2019). The Short Chain Fatty Acid Butyrate Imprints an Antimicrobial Program in Macrophages. Immunity 50, 432–445 e437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C, Jia T, Mendez-Ferrer S, Hohl TM, Serbina NV, Lipuma L, Leiner I, Li MO, Frenette PS, and Pamer EG (2011). Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating toll-like receptor ligands. Immunity 34, 590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signer RA, Magee JA, Salic A, and Morrison SJ (2014). Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 509, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares MP, and Hamza I (2016). Macrophages and Iron Metabolism. Immunity 44, 492–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staffas A, Burgos da Silva M, Slingerland AE, Lazrak A, Bare CJ, Holman CD, Docampo MD, Shono Y, Durham B, Pickard AJ, et al. (2018). Nutritional Support from the Intestinal Microbiota Improves Hematopoietic Reconstitution after Bone Marrow Transplantation in Mice. Cell Host Microbe 23, 447–457 e444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa H, Fritsch K, Kovtonyuk LV, Saito Y, Yakkala C, Jacobs K, Ahuja AK, Lopes M, Hausmann A, Hardt WD, et al. (2017). Pathogen-Induced TLR4-TRIF Innate Immune Signaling in Hematopoietic Stem Cells Promotes Proliferation but Reduces Competitive Fitness. Cell Stem Cell 21, 225–240 e225. [DOI] [PubMed] [Google Scholar]
- Theurl I, Hilgendorf I, Nairz M, Tymoszuk P, Haschka D, Asshoff M, He S, Gerhardt LM, Holderried TA, Seifert M, et al. (2016). On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat Med 22, 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong X, Kawabata H, and Koeffler HP (2002). Iron deficiency can upregulate expression of transferrin receptor at both the mRNA and protein level. Br J Haematol 116, 458–464. [PubMed] [Google Scholar]
- Torti SV, Manz DH, Paul BT, Blanchette-Farra N, and Torti FM (2018). Iron and Cancer. Annu Rev Nutr 38, 97–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varol C, Mildner A, and Jung S (2015). Macrophages: development and tissue specialization. Annu Rev Immunol 33, 643–675. [DOI] [PubMed] [Google Scholar]
- Wang F, Lv H, Zhao B, Zhou L, Wang S, Luo J, Liu J, and Shang P (2019). Iron and leukemia: new insights for future treatments. J Exp Clin Cancer Res 38, 406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, He X, Wu Q, Jiang L, Chen L, Yu Y, Zhang P, Huang X, Wang J, Ju Z, et al. (2020). Transferrin receptor 1-mediated iron uptake plays an essential role in hematopoiesis. Haematologica 105, 2071–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Q, Boulais PE, Zhang D, Pinho S, Tanaka M, and Frenette PS (2019). Maea expressed by macrophages, but not erythroblasts, maintains postnatal murine bone marrow erythroblastic islands. Blood 133, 1222–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitnall M, Howard J, Ponka P, and Richardson DR (2006). A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc Natl Acad Sci U S A 103, 14901–14906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson AC, Ishida R, Kikuchi M, Sudo K, Morita M, Crisostomo RV, Yamamoto R, Loh KM, Nakamura Y, Watanabe M, et al. (2019). Long-term ex vivo haematopoietic-stem-cell expansion allows nonconditioned transplantation. Nature 571, 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, et al. (2008). Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 135, 1118–1129. [DOI] [PubMed] [Google Scholar]
- Winkler IG, Sims NA, Pettit AR, Barbier V, Nowlan B, Helwani F, Poulton IJ, van Rooijen N, Alexander KA, Raggatt LJ, et al. (2010). Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood 116, 4815–4828. [DOI] [PubMed] [Google Scholar]
- Wu SE, Hashimoto-Hill S, Woo V, Eshleman EM, Whitt J, Engleman L, Karns R, Denson LA, Haslam DB, and Alenghat T (2020). Microbiota-derived metabolite promotes HDAC3 activity in the gut. Nature 586, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, Chawla A, and Pollard JW (2013). Macrophage biology in development, homeostasis and disease. Nature 496, 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Gao X, Wei Q, Nakahara F, Zimmerman SE, Mar J, and Frenette PS (2018). Stem cell factor is selectively secreted by arterial endothelial cells in bone marrow. Nat Commun 9, 2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Dellorusso PV, Olson OC, and Passegue E (2020). Dysregulated haematopoietic stem cell behaviour in myeloid leukaemogenesis. Nat Rev Cancer 20, 365–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Goetz D, Li JY, Wang W, Mori K, Setlik D, Du T, Erdjument-Bromage H, Tempst P, Strong R, et al. (2002). An iron delivery pathway mediated by a lipocalin. Mol Cell 10, 1045–1056. [DOI] [PubMed] [Google Scholar]
- Zarrinpar A, Chaix A, Xu ZZ, Chang MW, Marotz CA, Saghatelian A, Knight R, and Panda S (2018). Antibiotic-induced microbiome depletion alters metabolic homeostasis by affecting gut signaling and colonic metabolism. Nat Commun 9, 2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Chen G, Manwani D, Mortha A, Xu C, Faith JJ, Burk RD, Kunisaki Y, Jang JE, Scheiermann C, et al. (2015). Neutrophil ageing is regulated by the microbiome. Nature 525, 528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, and Frenette PS (2019). Cross talk between neutrophils and the microbiota. Blood 133, 2168–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoller EE, Lykens JE, Terrell CE, Aliberti J, Filipovich AH, Henson PM, and Jordan MB (2011). Hemophagocytosis causes a consumptive anemia of inflammation. J Exp Med 208, 1203–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
There are no standardized datatypes reported in this paper.