

Summary
Triggering receptor expressed on myeloid cells-2 (TREM2) is a cell surface receptor on macrophages and microglia that senses and responds to disease associated signals to regulate the phenotype of these innate immune cells. The TREM2 signaling pathway has been implicated in a variety of diseases ranging from neurodegeneration in the central nervous system to metabolic disease in the periphery. We report here that TREM2 is a thyroid hormone regulated gene and its expression in macrophages and microglia is stimulated by thyroid hormone and synthetic thyroid hormone agonists (thyromimetics). Our findings report the endocrine regulation of TREM2 by thyroid hormone, and provide a unique opportunity to drug the TREM2 signaling pathway with orally active small molecule therapeutic agents.
eTOC blurb:
In this article, Ferrara et al. show that an important immune system signaling hub, TREM2, is transcriptionally regulated by thyroid hormone. This renders TREM2 druggable by small molecule synthetic thyroid hormone derivatives which impart beneficial phagocytic and anti-inflammatory properties in relevant cell types and disease model mice.
Graphical Abstract
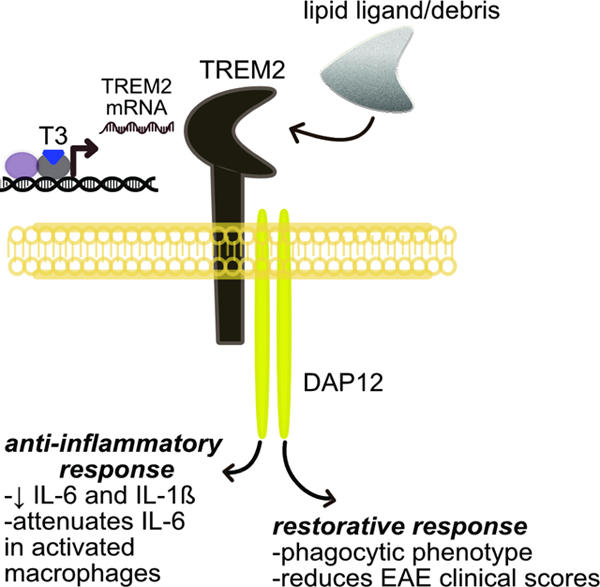
Introduction
Thyroid hormone (TH) provides essential regulation of many critical processes in vertebrate biology (Yen, 2001). Thyroxine (T4) is the predominant form of TH produced in and secreted from the thyroid gland, but its deiodinated metabolite 3,5,3’-triiodothyronine (T3) is the active form of TH that binds thyroid hormone receptors (TR) with high affinity. TRs bind to regulatory DNA sequences called thyroid hormone response elements (TRE) in the promoter regions of TH regulated genes, and T3 binding to TR in the cell nucleus activates TR to either stimulate or suppress transcription of these genes. Through such regulation of target genes TH action plays an important developmental role in the central nervous system (CNS) and periphery, as well as regulation of metabolism and homeostasis in many organs and cell types in the periphery. It is known that TH exerts effects on the immune system, in particular the innate immune cells, such as TR-expressing tissue-resident macrophages and microglia in the CNS; however, the mechanistic basis of TH-dependent effects on innate immunity is not well understood (De Vito et al., 2011).
Triggering receptor expressed on myeloid cells (TREM2) has emerged recently as a major regulator of the innate immune response and an important new therapeutic target connected to a number of diseases in the CNS and periphery (Deczkowska et al., 2020). Expressed as a cell surface protein on macrophages and microglia, activation of TREM2 initiates a signal transduction cascade that triggers a switch in these cells away from a pro-inflammatory phenotype to an anti-inflammatory, phagocytic, restorative phenotype. Homozygous loss of function mutations in TREM2, or DAP12, a molecule that interacts with TREM2 to facilitate TREM2 signaling, causes Nasu-Hakola disease, a rare inborn error resulting in premature dementia, loss of myelin, and bone abnormalities (Xing et al., 2015). In addition, heterozygous TREM2 missense variants have been shown to be risk factors for common neurodegenerative diseases such as Alzheimer’s disease (AD), frontotemporal dementia, Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) (Deczkowska et al., 2020; Krasemann et al., 2017; Ulrich and Holtzman, 2016). In the periphery,TREM2 expressed on circulating monocytes and tissue-resident macrophages has been implicated to play a beneficial role in controlling/resolving obesity/excess adiposity (Jaitin et al., 2019), non-alcoholic steatohepatitis (NASH) (Xiong et al., 2019), and hepatocellular carcinoma (Tang et al., 2019). In most of these diseases it appears that activation of the TREM2 pathway by increasing surface expression of TREM2 on macrophages and/or microglia and/or activating TREM2 with an agonist ligand is likely to be therapeutically beneficial. One of the main obstacles to targeting TREM2 for therapeutic benefit is that it falls into the category of “undruggable” or at least difficult to drug targets. This results from the nature of the ligands that bind to and activate TREM2. The general consensus is that TREM2 does not bind to a single, discrete ligand, but instead binds and is activated by various proteins, lipids, and other molecular debris arising from damaged cells (Kober and Brett, 2017).
Here we report the discovery that TREM2 is a positively regulated thyroid hormone target gene. TREM2 expression and signaling through the TREM2 pathway is increased both in vitro and in vivo by treatment with T3, or the peripheral and CNS-penetrating thyromimetic agents sobetirome and Sob-AM2, respectively. This discovery effectively renders TREM2 “druggable” by systemically dosed small molecules with sufficient drug-like properties for targeting the TREM2 pathway in either the CNS or periphery.
Results
Our interest in TREM2 was initiated from reports that microglia and macrophages increased phagocytic capacity upon treatment with the retinoid X receptor (RXR) agonist bexarotene (Natrajan et al., 2015), and treatment of TREM2-expressing myeloid lineage cells with other nuclear receptor agonists including those for PPAR and LXR, also stimulated phagocytosis (Savage et al., 2015). Based on comprehensive genome-wide studies in mice, it has been reported that the Trem2 gene is regulated by an RXR-dependent enhancer (Daniel et al., 2014). Because a major biological function of RXR is to form heterodimers at positively-regulated response elements with thyroid hormone receptor (TR) and other members of the nuclear receptor family including PPAR and LXR, we examined this enhancer from murine macrophages. We found the following sequence embedded in the putative RXR enhancer of Trem2—AGGGAG-GTTA-AGGTCA (Fig.1). This sequence resembles a DR4 TRE with two hexanucleotide binding sites for TR and RXR (AGGGAG and AGGTCA) separated by a four-nucleotide spacer (GTTA). The AGGGAG half-site is contained in a vitamin D receptor (VDR) response element that binds VDR and RXR as a heterodimer and is likely the RXR binding half-site.(Liu et al., 1996) The highly similar sequence AGGGAC, differing by one nucleotide is a half-site found in the DR4 TRE of rat growth hormone.(Koenig et al., 1987) The AGGTCA half-site is found in many naturally occurring DR4 TRE’s,(Harbers et al., 1996; Wu et al., 2001) and the sequence TA-AGGTCA that exists in the murine Trem2 promoter has been shown to be a preferred TR-binding half-site.(Katz and Koenig, 1993) This putative DR4 TRE in the TREM2 promoter would be expected to make TREM2 a positively regulated thyroid hormone target gene which should increase TREM2 expression upon binding thyroid hormone agonists to TR (Fig. 1a).
Figure 1: Thyroid hormone response element found in promoter of TREM2.

RXR-TR heterodimer associated with a DR4-TRE located in the promoter region of TREM2 (a, PDB ID: 2NLL and 4ZO1) and endogenous and synthetic TR ligands used in this study (b).
We verified that TREM2 expression is indeed positively regulated by thyroid hormone in both murine (Fig. 2a) and human (Fig. 2b) microglia, murine macrophages (Fig. 2c), and in mouse brain homogenate (Fig. 2d). Sobetirome is a synthetic T3 agonist drug that has a larger therapeutic index (TI) than T3, and Sob-AM2 is a prodrug of sobetirome that greatly facilitates delivery of sobetirome to the CNS from a systemic dose (Fig. 1b) (Meinig et al., 2017; Meinig et al., 2019; Scanlan, 2010). We examined the ability of these agents to drive TREM2 expression and found that sobetirome stimulates TREM2 expression in mouse and human microglia (Fig. 2a,b), and hypothyroid mice treated with Sob-AM2 (i.p.) had increased Trem2 expressed in brain compared to hypothyroid control (Fig. 2d). To verify the involvement of TR in the observed TREM2 regulation by TH, the TR antagonist NH-3 (Fig. 1b) was employed in combination with T3 (Nguyen et al., 2002). NH-3 was not only found to block T3 induction of Trem2 expression in murine microglia, but to suppress Trem2 expression significantly below the basal level observed in vehicle treated cells (Fig. 2a). As mentioned above, activation of the TREM2 signaling pathway induces both a pro-phagocytic and an anti-inflammatory response in monocytes and macrophages. We found that expression of the phagocytic marker Cd68 was increased in microglia by treatment with either T3 or sobetirome, and the T3 effect was blocked by the TR antagonist NH-3 (Fig. 2e). In addition, expression of the pro-inflammatory cytokine interleukin-1β (IL-1β) was significantly decreased by T3 and sobetirome in mouse primary microglia (Fig. 2f).
Figure 2: RT-qPCR demonstrating that T3 and thyromimetics regulate TREM2 expression in vitro and in vivo.

TR agonists T3 (10 nM) and sobetirome (1 μM) upregulate Trem2 in mouse (a) and human (b) primary microglia, (c) mouse macrophage cell line RAW 264.7, (d) hypothyroid mouse whole brain extract (3.05 μmol/kg T3 and 30.5 μmol/kg Sob-AM2), and alter the transcript levels of TREM2 pathway-connected genes by upregulating (e) Cd68 and downregulating (f) Il1b in mouse primary microglia (n = 3–5 as denoted). In contrast, TR antagonist NH-3 (2 μM NH-3 with 10 nM T3) downregulated Trem2 (a) and Cd68 (e) in mouse primary microglia. Statistical significance was determined by a 2-tailed, unpaired t test for comparisons between vehicle and group then were plotted together. Asterisks represent significant difference from vehicle unless otherwise noted (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001). All graphs show mean ± SEM.
We next examined thyroid hormone regulated gene expression of the pro-inflammatory cytokine interleukin-6 (IL-6) to test the breadth of anti-inflammatory effects that would be expected by thyromimetic stimulation of the TREM2 pathway. We tested whether IL-6 expression stimulated from an inflammatory challenge in macrophages could be suppressed by thyromimetic stimulation of TREM2 expression. (Fig. 3). While not specifically used in the context of studying TREM2 biology, it has been shown that macrophages incubated with coronavirus (SARS-CoV and SARS-CoV-2) spike S1 protein induce the production of pro-inflammatory cytokines including IL-6 (chen et al., 2020; Dosch et al., 2009; Theobald et al., 2020; Wang et al., 2007). RAW 264.7 cells stimulated with SARS-CoV-2 spike S1 protein induced a surge in Il6 expression compared to unstimulated cells, and this response was significantly attenuated upon treatment with T3 (Fig. 3a). These results were reproduced in an IL-6 ELISA using mouse primary lung macrophages under the exact same experimental conditions (Fig. 3e). T3 treatment also significantly suppressed Il6 (Fig. 3b) and Il1b expression (Fig. 3c) in mouse primary lung macrophages stimulated with S1 protein. Concomitant with suppression of these pro-inflammatory cytokines, we observed stimulation of Trem2 expression by T3 in murine primary lung macrophages (Fig. 3d).
Figure 3: Anti-inflammatory effects of thyroid hormone treatment in macrophages after inflammatory challenge by the SARS-CoV-2 S1 protein.

RT-qPCR of mouse RAW264.7 macrophage cells (a) and mouse primary lung macrophages (b-d) stimulated with 10 μg/mL SARS-CoV-2 S1 protein with and without treatment with 10 nM T3 (n = 5–7 as denoted). T3 treatment suppresses pro-inflammatory cytokine expression (Il6 and Il1b, a-c) and upregulates Trem2 expression following inflammatory stimulation with the S1 protein (d). T3 suppression of IL-6 was validated by ELISA from the same experiment as in b-d (e). Statistical significance was determined by a 2-tailed, unpaired t test for comparisons between vehicle and group then were plotted together or using one-way ANOVA (P < 0.05). Asterisks represent significant difference from vehicle unless otherwise noted (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001). All graphs show mean ± SEM.
The finding that TH agonists upregulate both TREM2 and CD68 expression predicts that phagocytosis will be stimulated by these drugs. This prompted us to directly examine phagocytosis, which is an established consequence of TREM2 signaling pathway activation in myeloid lineage cells. In order to ascertain the phagocytic response upon drug treatment, mouse primary microglia were cultured on glass coverslips and treated with drugs in the presence of fluorescent beads used as a phagocytosis substrate that can be monitored and quantified by fluorescence microscopy (Kleinberger et al., 2014; Lian et al., 2016). Cellular bead uptake was evaluated via 3D volume visualization down orthogonal axes to confirm complete entrapment as shown in Fig. 4g. Microscopy of cells stained for CD11b microglial expression showed that T3 and sobetirome treatment significantly increased phagocytosis compared to vehicle control as judged by the number of beads engulfed by the microglia (Fig. 4a-c’). Conversely, the T3 antagonist NH-3 treatment was found to suppress phagocytic uptake of beads compared to vehicle-treated control (Fig. 4d,d’). Quantification of these microscopy data demonstrate that TH agonists stimulate, while TH antagonists block, phagocytosis in microglia (Fig. 4h). These results corroborate existing literature reporting augmented phagocytosis upon T3 treatment in macrophages (Perrotta et al., 2014).
Figure 4: T3 and sobetirome stimulate and NH-3 blocks phagocytosis by microglia.

C57BL/6 mouse primary microglia cells in culture were treated with DMSO vehicle (a + a’), 10 nM T3 (b + b’), 1 μM sobetirome (c + c’), or 2 μM NH-3 (d + d’) for 24 h before 2 μm diameter fluorescent beads (~100/cell) were introduced 2 h before cells were fixed and stained for Cd11b (green). (e) Elongated morphology of an unactivated/ramified microglia cell. (f) Morphology of an activated microglia cell upon treatment with sobetirome (see supporting information). (g) Three-dimensional view of beads inside the uppermost cell in the sobetirome treatment group (image generated in Imaris). (h) Quantification of phagocytosed beads per treatment group. Scale bars are 20 μm (a-d), 10–20 μm (a’-d’), and 8–10 μm (e-g) as noted. Statistical significance was determined by a 2-tailed, unpaired t test for comparisons between vehicle and group then were plotted together. Asterisks represent significant difference from vehicle unless otherwise noted (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001). All graphs show mean ± SEM.
We have shown previously that CNS-penetrating thyromimetics stimulate myelin repair in different murine models of demyelination (Hartley et al., 2019). One such model is experimental autoimmune encephalomyelitis (EAE), which is a demyelination model stimulated by an autoimmune attack that has parallels to the human disease multiple sclerosis. The autoimmune insult in EAE produces demyelination and axonal degeneration, particularly in the lumbar spinal cord region in mice. We have shown previously that in mice with EAE, T3, sobetirome, and Sob-AM2 treatment improves clinical disease scores, and reduces demyelination and axonal degeneration within the spinal cord (Chaudhary et al., 2020). Thus, the question arises as to whether this results in part from thyromimetic stimulation of the TREM2 signaling pathway in microglia and macrophages to produce an anti-inflammatory phenotype, and induce phagocytosis of myelin debris, which is known to be a prerequisite to myelin repair (Lloyd and Miron, 2019). EAE was induced in C57Bl/6 mice within a 21-day treatment regimen, where mice were administered once daily injections of vehicle, T3, or Sob-AM2 starting 7 days after immunization before disease onset and continuing until day 21 when euthanasia and tissue collection occurred. Spinal cord sections co-stained with DAPI and TREM2 antibodies in the dorsal white matter of the lumbar section of spinal cords were analyzed for TREM2 content (Fig. 5a-c, see also Supplemental Fig. S1). Compared to vehicle, TREM2 protein expression was increased approximately four-fold and three-fold by T3 and Sob-AM2 treatment, respectively (Fig. 5d). Immunohistochemical analysis of this same spinal cord region showed that the population of CD11b positive cells, which correspond to microglia and/or macrophages, were not statistically different between Sob-AM2 treated mice and vehicle (see Supplemental Fig. S2) (Chaudhary et al., 2020). This indicates that the increase in TREM2 staining was not a result of increased myeloid cells in the spinal cord lesions. This increase in TREM2 protein expression observed by immunohistochemical analysis of spinal cord sections was confirmed by ELISA of spinal cord homogenate from unprocessed/unstained samples of the same groups (Fig. 5e). Improvement in EAE clinical scores (Fig. 5f) was observed in the mice that received T3 or Sob-AM2 compared to control, which correlates with augmented TREM2 expression observed in stained spinal cord sections.
Figure 5: Treatment of EAE mice with T3 and Sob-AM2 upregulates TREM2 expression in diseased spinal cord regions.

(a-c) EAE mice were treated with vehicle, T3 (0.4 mg/kg), or Sob-AM2 (1 mg/kg) i.p. for 15 days (day 7–21). (a-c) Representative immunofluorescence images showing TREM2 protein expression within the dorsal white matter in the lumbar section of spinal cord. Scale bars: 100 μm. (d) Cells expressing TREM2 (white) within the dorsal white matter were quantified. T3 and Sob-AM2 increased TREM2 expression four-fold and three-fold compared to vehicle, respectively. Data represents images from vehicle (n = 18), T3 (n = 12), and Sob-AM2 (n = 12). Two sections were used from each mouse. Significance was determined using one-way ANOVA (P < 0.05). (e) ELISA measurement of TREM2 in spinal cord extracts from the same cohorts of EAE mice (n = 3) used in a-d. (f) Daily EAE clinical disease scores from vehicle (n = 18), T3 (0.4 mg/kg, n = 15), and Sob-AM2 (1 mg/kg, n = 28) treated cohorts. Statistical significance was determined by a two-tailed, unpaired t test comparing vehicle and treatment group per day with each t test performed independently. Asterisks represent significant difference from vehicle unless otherwise noted. (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001). All graphs show mean ± SEM. See also Supplemental Figures S1 and S2.
Discussion
TREM2 has emerged as a central node in the innate immune response governing whether myeloid lineage macrophages and microglia have a pro-inflammatory or anti-inflammatory, restorative, healing phenotype in response to pathological insults. Here we report the discovery that TREM2 is positively regulated by thyroid hormone. The promoter region of the TREM2 gene contains a sequence that resembles a DR4-TRE which presents binding sites for the RXR-TR heterodimer. RXR binding was shown previously to this TREM2 enchancer, and it remains to be shown that TR is also capable of binding. However, this putative TRE would be expected to stimulate TREM2 expression in response to thyroid hormone agonists which is what we observed. We found that TREM2 expression was increased in both microglia and macrophages by addition of either T3 or the synthetic T3 analog sobetirome, and this T3 agonist induction of TREM2 was blocked by the T3 antagonist NH-3. We also found that the pro-inflammatory cytokines IL-1β and IL-6 which are downregulated by induction of the TREM2 pathway were downregulated by T3 and sobetirome in microglia and macrophages that had been stimulated with the pro-inflammatory SARS-CoV-2 spike protein. Activation of the TREM2 pathway induces phagocytosis in these myeloid cells and T3 and sobetirome were found to induce phagocytosis in microglia whereas the T3 antagonist NH-3 blocked phagocytosis. Finally using the murine EAE model we showed that demyelinated regions of the spinal cord from mice treated with Sob-AM2, a CNS-penetrating prodrug of sobetirome, contained more TREM2 positive cells than control mice. Importantly, mice treated with Sob-AM2 had reduced clinical impairment, demyelination and axonal degeneration, suggesting that thyromimetic stimulation of TREM2 could result in protection in the inflammatory disease EAE. On the basis of these results, we report that TREM2, and by extension the TREM2 pathway, is subject to endocrine regulation by thyroid hormone.
It has become increasingly apparent that thyroid hormone action plays a role in modulating innate immunity (Montesinos and Pellizas, 2019). Control of intracellular T3 levels in macrophages has been shown to be critical for these cells to respond appropriately to inflammatory signals (Anne et al., 2017; van der Spek et al., 2018). In addition, thyroid status dictates the degree to which rats respond to an inflammatory insult in which hypothyroidism induces increased pro-inflammatory cytokine production while hyperthyroidism inhibits this response (Rittenhouse and Redei, 1997). Administration of T3 blocks NLRP3 inflammasome activation, which depends upon NF-kB activation and results in IL-1β production and release, in different models of liver injury (Dong et al., 2018; Vargas and Videla, 2017). In a model of kidney injury, deletion of the TRα isoforms result in increased injury, and isolated macrophages lacking TRα produce excessive levels of IL-1β compared to WT control (Furuya et al., 2017). In addition to these examples of driving the transition to an anti-inflammatory phenotype in macrophages, TH has also been shown to promote phagocytosis in macrophages (Perrotta et al., 2014), a response that we recapitulated here in microglia treated with T3. Collectively, these myeloid cell observations from both in vitro and in vivo experiments can be explained by thyroid hormone activation of the TREM2 pathway.
The sequelae of Nasu-Hakola disease provides genetic evidence that functional TREM2 and its signaling pathway in myeloid cells is essential for a normal, disease-free human lifespan (Xing et al., 2015). Over the past few years it has become increasingly apparent that the biology of the TREM2 signaling pathway is more involved in responding to pathological conditions as opposed to homeostatic physiology (Deczkowska et al., 2020). For example, TREM2 is known to play a major role in a disparate collection of diseases including neurodegenerative diseases (Konishi and Kiyama, 2018; Krasemann et al., 2017), metabolic diseases (Jaitin et al., 2019; Xiong et al., 2019), infectious diseases (Chen et al., 2013; Wu et al., 2015), cancer (Donatelli et al., 2014; Tang et al., 2019; Yao et al., 2016; Zhang et al., 2018), and stroke (Gervois and Lambrichts, 2019). Heterozygous missense mutations in TREM2 are associated with strong increased risk of developing late onset Alzheimer’s disease (AD) (Guerreiro et al., 2012; Jonsson et al., 2013), and increased cerebrospinal fluid (CSF) levels of the soluble extracellular domain of TREM2 (sTREM2) has been shown to correlate with reduced disease severity in AD (Ewers et al., 2019). This suggests that increased expression of TREM2 and/or increased signaling through the TREM2 pathway in microglia may be therapeutically beneficial in AD. It has also been known for some time that decreased thyroid hormone action in the CNS is detrimental while increased thyroid hormone action in the CNS correlates with clinical benefit in AD and other diseases of cognitive impairment (Accorroni et al., 2017; Choi et al., 2017; Davis et al., 2008; Hogervorst et al., 2008; Johansson et al., 2013; Juárez-Cedillo et al., 2017; Sampaolo et al., 2005). Our finding that TREM2 is a thyroid hormone regulated gene whose expression is increased by T3 acting on TR in microglia suggests a mechanistic connection between elevated levels of T3 in the CNS and clinical benefit in AD and related disorders. The beneficial effect of increased T3 in the brain may in part relate to increased stimulation of TREM2 expression in microglia and increased activity through the TREM2 pathway.
Another neurodegenerative disease that TREM2 appears associated with is multiple sclerosis (MS), an autoimmune disease that causes inflammatory demyelination and degeneration of axons in the CNS. Similar to AD, sTREM2 levels in the CSF are elevated in both relapsing remitting and primary progressive MS patients compared to CSF from patients with non-inflammatory neurological disease (Piccio et al., 2008). In addition, TREM2-expressing myeloid cells have been detected in CSF and in demyelinating lesions upon autopsy from MS subjects (Piccio et al., 2008). Macrophages and microglia release proinflammatory cytokines and probably contribute to demyelination and axonal degeneration in MS. In addition, microglia and macrophages play an important role in clearing myelin debris in demyelinating lesions that occur in MS via phagocytosis, which is a prerequisite to forming new myelin from mature oligodendrocytes (Lloyd and Miron, 2019). We have shown previously that the thyroid hormone receptor agonists sobetirome and Sob-AM2 promote remyelination in murine gliatoxin and genetic models of demyelination, and have further shown that like thyroid hormone, these agents stimulate oligodendrogenesis in vitro and in vivo (Hartley et al., 2019). Our findings presented here that sobetirome induces TREM2 expression through TR activation in myeloid cells which stimulates phagocytosis in microglia constitutes a second beneficial mechanism of action in addition to oligodendrogenesis for thyroid hormone agonists as myelin repair agents.
This leads to the question of how the TREM2 pathway can be best engaged by drugs for therapeutic benefit. One approach would be to develop TREM2 agonists or antagonists that engage TREM2 directly to activate or block downstream signaling. A problem with this approach is that endogenous ligands for TREM2 appear to be a heterogenous collection of molecules associated with cell damage as opposed to a discrete metabolite or protein which is more amenable to drug discovery approaches. Some success has been reported with anti-TREM2 antibodies that act as agonists upon TREM2 binding (Cignarella et al., 2020; Piccio et al., 2007), but the challenge with biologic agents such as antibodies is their intrinsic limits on distribution to different tissues and compartments affected by disease. For example, distribution from blood to the CNS is difficult with antibodies, and many of the diseases that would benefit from TREM2 engagement are localized in the CNS. Our findings that TREM2 is a thyroid hormone regulated gene and that TREM2 expression and the TREM2 pathway can be activated or blocked by T3 agonists or antagonists, respectively opens the door to a therapeutic approach based on orally active small molecules. A number of studies have demonstrated that increased expression of TREM2 in microglia and/or macrophages alone serves to activate the TREM2 signaling pathway (Ewers et al., 2019; Jiang et al., 2014; Lee et al., 2018; Takahashi et al., 2007), and this is consistent with our observations that T3 agonists increase TREM2 expression and activate the TREM2 pathway, whereas the T3 antagonist NH-3 blocks TREM2 expression and downstream signaling such as that involved in phagocytosis. The T3 agonist sobetirome would be representative of a drug candidate designed to stimulate the TREM2 pathway in the periphery for therapeutic benefit. Sobetirome therapy would be indicated, for example, for diseases of the liver that may benefit from activation of TREM2 such as NASH (Xiong et al., 2019), immunemediated damage following liver injury (Perugorria et al., 2019), and hepatocellular carcinoma (Tang et al., 2019). Alternatively, for peripheral diseases that may benefit from temporary blockade of the TREM2 pathway such as early in respiratory viral infection (Wu et al., 2015) and certain cancers in which a robust immune response would be beneficial (Yao et al., 2016; Zhang et al., 2018), the T3 antagonist NH-3 could be employed. However, during later stages of viral infection when cytokine storm can present, as can occur in COVID-19 critical illness, activation of the TREM2 pathway by sobetirome to drive macrophages to transition to an anti-inflammatory, resolving phenotype would be more appropriate for therapeutic benefit (Mehta et al., 2020; Merad and Martin, 2020). The CNS-penetrating sobetirome prodrug Sob-AM2 would be the appropriate agent for AD, MS, and other neurodegenerative diseases for which therapeutic benefit would potentially come from TREM2 activation in the CNS while minimizing TREM2 activation and other thyromimetic activity in the periphery. And finally, should there be an application for blocking TREM2 activation in the CNS, the N-methyl amide prodrug of antagonist NH-3 provides enhanced distribution of NH-3 to the CNS while minimizing T3 antagonism in the periphery (Ferrara and Scanlan, 2020).
In conclusion, we have found that TREM2 expression is regulated by thyroid hormone, and the presence of a putative DR4 TRE in the TREM2 promoter region suggests that this regulation is a direct result of T3 agonist binding to TR associated with the TREM2 promoter. Regardless of mechanism, our findings suggest that TREM2 and its signaling pathway in macrophages and microglia are subject to regulation by a major endocrine system. Endogenous regulation of TREM2 at the level of gene expression is likely to have important physiological and pathophysiological ramifications for TREM2-mediated innate immunity. This finding also represents a path toward developing small molecule therapeutics that either activate or suppress the TREM2 signaling pathway selectively in the CNS or periphery. The ability to “drug” TREM2 and its pathway with small molecule agents possessing good, drug-like properties would be an important medical advance for the diverse collection of diseases that intersect with TREM2 biology.
Significance
The TREM2 pathway has recently emerged as a signaling hub within the innate immune system that responds to disease associated damage, and is connected to a variety of peripheral and neurological diseases. We report here that TREM2 is a thyroid hormone regulated gene and its expression in macrophages and microglia is stimulated by the endogenous thyroid hormone T3 as well as the synthetic thyroid hormone agonist sobetirome. The synthetic thyroid hormone antagonist NH-3 suppresses TREM2 expression and blocks T3 induction of expression indicating that thyroid hormone receptors are involved in the observed TREM2 regulation. T3 and sobetirome suppress pro-inflammatory cytokine production from myeloid cells and induce phagocytic behavior in microglia, both of which are phenotypes that result from activation of the TREM2 pathway. In murine experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis, treatment with Sob-AM2, a CNS-penetrating sobetirome prodrug, results in an improvement in clinical disease score, reduced damage to myelin, and increased Trem2 expression in disease lesion resident microglia. Here we report the endocrine regulation of TREM2 and the TREM2 pathway which should serve to advance the understanding of TREM2 biology. In addition, this discovery effectively renders the TREM2 pathway druggable by clinical stage orally active small molecule thyromimetics with excellent drug-like properties which may open the door to new medications for neurological and peripheral diseases that currently lack effective therapeutic options.
STAR Methods
Resource availability
Lead contact:
Requests or information regarding resources and reagent should be addressed to Prof. Thomas S. Scanlan (scanlant@ohsu.edu).
Materials availability:
This study did not generate any unique reagents. Chemical compounds were synthesized using commercially available reagents. Primary cells, cell lines, antibodies, and other reagents were purchased from commercial vendors described in the key resource table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mouse monoclonal anti-CD111b | AbD Serotec | Cat# MCA711, RRID:AB_321292 |
| donkey anti-mouse secondary antibody Alexa Fluor 488 | Thermo Fisher | Cat# A-21202, RRID:AB_141607 |
| rat monoclonal anti-TREM2 | EMD Millipore | Cat# MABN2320 |
| goat anti-rat secondary antibody Alexa Fluor 647 | Thermo Fisher | Cat# A-21247, RRID:AB_141778 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 3,3′,5-Triiodo-L-thyronine | Sigma-Aldrich | Cat# T2877 |
| sobetirome | Scanlan laboratory; (Placzek et al., 2016) | N/A |
| Sob-AM2 | Scanlan laboratory; (Meinig et al., 2017) | N/A |
| NH-3 | Scanlan laboratory; (Placzek and Scanlan, 2015) | N/A |
| SARS-CoV-2 S1 protein | ACROBiosystems | Cat# S1N-C52H3 |
| 2 micron fluorescent latex beads | Sigma-Aldrich | Cat# L0280 |
| MOG35–55 peptide (amino acid sequence MEVGWYRSPFSRVV HLYRNGK) | PolyPeptide Laboratories | N/A |
| Heat killed Mycobacterium tuberculosis H37 Ra | Difco | Cat# 231141 |
| Pertussis toxin | List Biological Laboratories, Inc | Cat# 181 |
| complete Freund’s adjuvant | BD | Cat# 263810 |
| microglia media | ScienCell | Cat# 1901 |
| microglia media | Celprogen | Cat# M37089-01S |
| Critical Commercial Assays | ||
| mouse TREM2 ELISA kit | Reddot Biotech Inc. | Cat# RD-TREM2-Mu |
| mouse IL-6 ELISA kit | R&D Systems | Cat# M6000B |
| Experimental Models: Cell Lines | ||
| mouse primary microglia | ScienCell | Cat# M1900-57 |
| human primary microglia | Celprogen | Cat# 37089-01 |
| RAW 264.7 | ATCC | Cat# TIB-71 |
| mouse primary lung macrophages | CellBiologics | Cat# C57-2313F |
| Experimental Models: Organisms/Strains | ||
| Mouse: Wild-type: C57BL/6J | The Jackson Laboratory | JAX: 000664 |
| Oligonucleotides: See Supplemental Information Table S1 | ||
| Software and Algorithms | ||
| Prism 8 | GraphPad |
https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| ZEN 2 | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| IMARIS | Bitplane | https://imaris.oxinst.com/ |
Data and code availability:
All data reported in this paper will be shared by the lead contact upon request. All datasets generated or analyzed during this study are included in this article. No code was generated in this study. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
Cell lines:
Human primary microglia, mouse primary microglia, mouse primary lung macrophages, and RAW 264.7 cells were purchased from commercial sources and cultured as described in the Method details.
Animals:
Female wild-type C57BL/6 mice, aged 8-to-10 weeks, were purchased from Jackson Laboratory (Bar Harbor, ME) and housed in climate-controlled rooms with a 12-hour light/12-hour dark cycle with ad libitum access to food and water. Mice were made hypothyroid by receiving 0.1% (w/v) methimazole and 0.2% (w/v) potassium perchlorate (Sigma-Aldrich) in drinking water for 2 weeks (Hackenmueller et al., 2012). All experiments were approved by the IACUC committee at the VA Portland Health Care System (VAPORHCS) or OHSU.
Method details
Reagents.
T3 was purchased and used as received from Sigma-Aldrich. Sobetirome, Sob-AM2, and NH-3 were prepared as described in the literature (see below). (Meinig et al., 2017; Placzek et al., 2016; Placzek and Scanlan, 2015). Vehicle and drug stocks for cell culture experiments were prepared in dimethyl sulfoxide (DMSO). For in vivo experiments, all drugs were prepared at concentrations suitable for an i.p. injection of 150 μL per 26-g mouse. T3 drug stocks were prepared in 8 mM NaOH in saline and sobetirome and Sob-AM2 drug stocks were prepared in 50% DMSO in saline solutions. Vehicle stock solutions of 50% DMSO in saline (vehicle for sobetirome and Sob-AM2) or 8 mM NaOH in saline (T3 vehicle) were prepared and administered within the appropriate experiments. Drug concentrations are described in the figure legends for individual experiments. SARS-CoV-2 S1 protein was purchased from ACROBiosystems (#S1N-C52H3) and used as received. Information on other specific reagents is listed below within the Methods description for that particular experiment.
Sob-AM2, 2-(4-(4-Hydroxy-3-isopropylbenzyl)-3,5-dimethylphenoxy)-N-methylacetamide
Sobetirome (155 mg, 0.47 mmol) was dissolved in MeOH (3 mL) in a sealed tube. Sulfuric acid (1 drop) was added, and the reaction was sealed and then heated to 65 °C for 1 h while stirring. The reaction was allowed to come to room temperature. TLC analysis (1:30 MeOH/DCM) showed complete conversion to the intermediate methyl ester. To the intermediate reaction mixture a solution of 40% methyl amine in water (610 μL, 7.05 mmol, 15 equiv)was added. The reaction was resealed and, again, heated to 65 °C for 1 h. The reaction flask was allowed to return to room temperature, and the contents were added to 0.5 N NaOH (20 mL) in a separtory funnel and subsequently extracted with DCM (3 × 100 mL). The organic layers were combined, dried with Na2SO4, and concentrated. Purification by flash chromatography (0–6% MeOH in DCM) gave the product as a white solid (144 mg, 90%). Purity: 97% (HPLC). 1H NMR (400 MHz, CD3CN): 6.98 (br, 1 H), 6.89 (s, 1 H), 6.68 (s, 2 H), 6.62 (d, 1 H, J = 8.6 Hz), 6.54 (dd, J = 8.4, 2.4 Hz, 1 H), 4.37 (s, 2 H), 3.87 (s, 2 H), 3.16 (septet, J = 6.9 Hz, 1 H), 2.75 (d, J = 4.9 Hz, 3 H), 2.20 (s, 6 H), 1.12 (d, J = 6.9 Hz, 6 H). 13C NMR (101 MHz, CDCl3) δ 169.4, 155.1, 151.0, 138.9, 134.3, 131.7, 131.2, 126.1, 125.2, 115.1, 114.0, 67.2, 33.7, 27.1, 25.8, 22.6, 20.5. HRMS (ESI) m/z [M + Na+] C21H27NO3Na+ requires 364.1883, found 364.1890.
NH-3, {4-[4-Hydroxy-3-isopropyl-5-(4-nitrophenylethynyl)-benzyl]-3,5-dimethylphenoxy}acetic acid
Formation of the protected sobetirome skeleton via Grignard coupling {Placzek, 2016 #98} was followed by sequential 5’ outer ring iodination and Sonogashira coupling to afford tert-butyl 2-(4-(3-isopropyl-4-(methoxymethoxy)-5-((4-nitrophenyl)ethynyl)benzyl)-3,5-dimethylphenoxy)acetate.{Placzek, 2015 #29} To a solution of tert-butyl 2-(4-(3-isopropyl-4-(methoxymethoxy)-5-((4-nitrophenyl)ethynyl)benzyl)-3,5-dimethylphenoxy)acetate (201 mg, 0.351 mmol) and MeOH (5 mL) was added 2 M NaOH (4.06 mL, 8.11 mmol). The reaction mixture was allowed to stir at room temperature for 2 h and then concentrated to remove the methanol. The mixture was then acidified with 20 mL of 1 M HCl and the resulting aqueous layer was extracted three times with DCM. The combined organic fractions were washed with brine, dried with Mg2SO4, and concentrated under reduced pressure. The crude mixture was then dissolved in 20 mL of THF and 6 mL of H2O. Conc. HCl (2.0 mL) was added and the solution was stirred at room temperature for 48 h. The reaction mixture was then diluted with H2O and the resulting aqueous layer was extracted three times with DCM. The combined organic fractions were washed with brine, dried with Mg2SO4, and concentrated under reduced pressure. Purification of the residue with flash chromatography (silica, 0%–4% MeOH/DCM+1% AcOH) yielded NH-3 (102 mg, 61%). 1H NMR (400 MHz, CDCl3): δ 8.20 (d, J=8.7 Hz, 2H), 7.64 (d, J=8.7 Hz, 2H), 7.02 (d, J=2 Hz, 1H), 6.70 (d, J=2 Hz, 1H), 6.66 (s, 2H), 5.30 (br, 1H), 4.69 (s, 2H), 3.90 (s, 2H), 3.26 (sept, J=6.8 Hz, 1H), 2.21 (s, 6H), 1.20 (d, J=6.8 Hz, 6H).
Cell culture.
Mouse (ScienCell, #M1900–57) and human primary (Celprogen, #37089–01) microglia cells were purchased from and cultured according to the manufacturer’s protocol in proprietary microglia media (ScienCell, #1901) containing 5% FBS, 1% microglia growth supplement, and 1% antibiotic solution and proprietary microglia media (Celprogen, #M37089–01S), respectively. Primary cells were plated and used within 72 h after plating at the passage from which they were isolated. RAW 264.7 cells were purchased from ATCC (ATCC® TIB-71™) and cultured according to the manufacturer’s protocol in DMEM containing 10% FBS. Mouse primary lung macrophages were purchased from CellBiologics (#C57–2313F) and cultured according to the manufacturer’s protocol in DMEM containing 10% FBS. All cells were incubated at 37°C in the presence of 5% CO2. For experiments, cells were plated in either 6, 12, or 24 well plates at ~2 × 105 cells per well.
RT-qPCR.
All cell cultures were serum-starved for 24 h before drug treatment, then treated with either DMSO vehicle, 10 nM T3, 1 μM sobetirome, or 2 μM NH-3 in the presence of 10 nM T3 for 24 h before RNA extraction. For mouse whole brain data, male C57Bl/6 mice (8–10 weeks old) were made hypothyroid according to standard procedure.(Hackenmueller et al., 2012) Mouse cohorts were administered DMSO vehicle, 3.05 μmol/kg T3, or 30.5 μmol/kg Sob-AM2 and euthanized 6 h post-injection. Brains were immediately stored in 10-fold excess RNALater (Thermo Fisher) for RNA preservation. RNA was purified using either the RNeasy Mini Kit (Qiagen) or the PureLink RNA Mini kit with TRIzol extractions (Life Technologies) and quantified using a NanoDrop. PCR reactions were run on 1 μg of RNA per sample to afford cDNA using the QuantiTect Reverse Transcription kit (Qiagen). RT-qPCR was performed on an Applied Biosciences 7500 Real-Time PCR system following the QuantiTect SYBR Green PCR kit protocols (Qiagen) using Cyclophilin A (Ppia) and Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as housekeeping genes for mouse and human samples, respectively. Samples were run with technical duplicates and results were analyzed using the ΔΔCt relative quantification method (Pfaffl, 2001).
Mouse primers:
f-Ppia 5′-AGGGTGGTGACTTTACACGC-3′, r-Ppia 5′-CTTGCCATCCAGCCATTCAG-3′; f-Trem2 5’-GACCTCTCCACCAGTTTCTCC-3’, r-Trem2 5’-TACATGACACCCTCAAGGACTG-3’; f-Cd68 5’-TTCTGCTGTGGAAATGCAAG-3’, r-Cd68 5’-GAGAAACATGGCCCGAAGT-3’; f-Il1b 5′-TCCAGGATGAGGACATGAGCAC-3′, r-Il1b 5′-GAACGTCACACACCAGCAGGTTA-3′, f-Il6 5’-GTTGCCTTCTTGGGACTGATG-3’, r-Il6 5’-CATACAATCAGAATTGCCATTGC-3’ (Ferrara et al., 2018; Humbert-Claude et al., 2016; Jiang et al., 2014; Sun et al., 2019; Zhao et al., 2017).
Human primers:
f-GAPDH 5’-CAGGAGGCATTGCTGATGAT-3’, r-GAPDH 5’-GAAGGCTGGGGCTCATTT-3’; f-TREM2 5’-ACAGAAGCCAGGGACACATC-3’, r-TREM2 5’-CCTCCCATCATCTTCCTTCA-3’ (Min et al., 2018; Rai et al., 2016).
Phagocytosis assay and immunocytochemistry.
Mouse primary microglia cells were plated onto poly-L-lysine coated glass coverslips at ~5 × 104 cells per coverslip seated within a 12 well plate (3 coverslips per group, 4 groups total). Cells were treated with DMSO vehicle, 10 nM T3, 1 μM sobetirome, or 2 μM NH-3 for 24 h before the addition of 3 μL of a fluorescent latex bead suspension (L0280, Sigma) in complete DMEM in a ~100:1 bead-to-cell ratio for 2 h before the end of the experiment. Each well was then stripped of media, washed with PBS, and cells were fixed with 4% paraformaldehyde in PBS for 10 min at room temperature. The cells were then permeabilized with PBS containing 0.05% saponin (saponin was included in all subsequent incubations and washes) and stained for microglial marker CD11b using monoclonal rat anti-mouse CD11b (1:200 dilution, AbD Serotec, #MCA711) in conjunction with Alexa Fluor 488-conjugated donkey anti-mouse secondary antibody (1:400 dilution,Thermo Fisher, A21202). Coverslips were then washed and mounted with ProLong® Gold antifade reagent (Life Technologies). Two fields per coverslip were imaged for a total of six fields of microglial cells per group imaged with a Zeiss ApoTome.2 at 20x magnification collecting z-stack images to verify the entrapment of fluorescent beads within the CD11b-stained cell field. Images were acquired and processed with ZEN 2 (blue edition) version 3.1 software (Zeiss), ImageJ/FIJI, and IMARIS software (Bitplane). Blue fluorescent beads were colorized to red during image post-processing in IMARIS for ease of visualization.(Schneider et al., 2012)
EAE experiment, histology, and immunofluorescence.
Immunization of female C57BL/6 mice (The Jackson Laboratory, Bar Harbor, ME, ages 8–10 weeks) with 200 μg of myelin oligodendrocyte glycoprotein (MOG)35–55 (PolyPeptide Laboratories, San Diego, CA) in complete Freund’s adjuvant containing 400 μg of Mycobacterium tuberculosis per mouse (subcutaneous injection of 0.2ml volume), followed by pertussis toxin (List Biological labs Inc), was administered via intraperitoneal (i.p.) injection at day 0 (75ng per mouse) and day 2 (200ng per mouse) after immunization. All mice were scored for clinical signs of EAE daily using a 9-point scale and received one-time daily i.p. injections of vehicle (50% DMSO or 8mM NaOH, both in saline) or drug (T3 0.4 mg/kg or Sob-AM2 1 mg/kg) starting at day 7 through euthanasia on day 21 post-immunization. Each group contained 6–8 mice and the experiment was repeated 3 times. At 21 days post-immunization, mice were euthanized with carbon dioxide and spinal columns were removed. Columns were immersed in 4% paraformaldehyde (PFA) for 24–48 hours, then spinal cords were extracted and fixed in a microwave for 1 hour. Free-floating 40 μm lateral sections were collected from the lumbar region using a vibratome, then stored in PBS at 4°C. Tissues were permeabilized with 0.05% Triton X-100 in PBS for 30 minutes, washed in PBS, then blocked with 5% donkey serum in PBS for 3 hours. Sections were incubated in primary monoclonal anti-TREM2 antibody (EMD Millipore, MABN2320, 1:250) in blocking buffer overnight at 4°C. Tissues were washed in PBS, then incubated in Alexa Fluor 647-conjugated goat anti-rat secondary antibody (Thermo Fisher, A21247, 1:200) and DAPI (1:50000) in PBS overnight at 4°C. Sections were washed and mounted on slides using ProLong® Gold antifade reagent (Life Technologies) and imaged using Zeiss 780 laser scanning confocal microscope at 20x. Cells expressing TREM2 were quantified within the region of dorsal white matter. Images were acquired and processed with ZEN 2 (blue edition) version 3.1 software (Zeiss), ImageJ/FIJI, and IMARIS software (Bitplane). TREM2 was colorized white in ImageJ/FIJI during post-processing for ease of visualization. Quantification of TREM2 concentration via ELISA was performed using a TREM2 ELISA kit (Reddot Biotech Inc). following the manufacturer’s instructions.
Quantification and statistical analysis
Statistical significance was determined using 1-way ANOVA with Dunnett’s post-test or two-tailed, unpaired Student’s t tests between two groups and then plotted together graphically as denoted in each figure legend (P < 0.05). For in vivo experiments, there were no differences in effects between the different vehicles (8 mM NaOH in saline or 50% DMSO in saline), so data from the different vehicles were combined into a single vehicle control group. Replicates for each experiment were as stated in the specific figure legend and in the corresponding methods. Sample sizes for animal experiments were informed by previous literature accounts or from preliminary data to minimize total animal numbers as appropriate. Analysis was carried out in GraphPad Prism 8 without further modifications. The ROUT method was used to identify and eliminate outliers. Significance level was set to *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001. All graphs show mean ± SEM.
Supplementary Material
Highlights:
TREM2 is regulated by thyroid hormone (T3) and thyromimetics
T3 and thyromimetics produce anti-inflammatory effects in microglia and macrophages
T3 and thyromimetics induce phagocytosis in microglia
EAE mice treated with T3 or thyromimetic present with reduced clinical scores
Acknowledgments
This research was supported by NIH grants DK52798 (T.S.S.) and GM133804 (B.A.N.), and the National Multiple Sclerosis Society grants RG 5199A4 and RG-1607-25053 to D.B., RG 5106A1/1 and RG-2001-35775 to B.E., the Race to Erase MS to D.B., the OHSU Laura Fund for Innovation in Multiple Sclerosis to D.B. and T.S.S. We would like to thank the Advanced Light Microscopy Core (supported by NIH P30 NS061800) at OHSU for technical assistance.
Footnotes
Declaration of interests
The authors declare the following competing financial interest(s): S.J.F. and T.S.S. are inventors of licensed patent applications claiming central nervous system-penetrating prodrugs of nuclear receptor modulators and their uses, including drugs acting on the thyroid hormone receptors. T.S.S., D.B., and B.E. are co-founders of Autobahn Therapeutics, and T.S.S. is a Senior Advisor to Autobahn Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Accorroni A, Giorgi FS, Donzelli R, Lorenzini L, Prontera C, Saba A, Vergallo A, Tognoni G, Siciliano G, Baldacci F, et al. (2017). Thyroid hormone levels in the cerebrospinal fluid correlate with disease severity in euthyroid patients with Alzheimer’s disease. Endocrine 55, 981–984. [DOI] [PubMed] [Google Scholar]
- Anne H.v.d.S., Eric F, and Anita B. (2017). Thyroid hormone metabolism in innate immune cells. Journal of Endocrinology 232, R67–R81. [DOI] [PubMed] [Google Scholar]
- Chaudhary P, Marracci GH, Calkins E, Pocius E, Bensen AL, Scanlan TS, Emery B, and Bourdette DN (2020). Thyroid hormone and thyromimetics inhibit myelin and axonal degeneration and oligodendrocyte loss in EAE. bioRxiv, 2020.2012.2020.423638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Zhang K, Jin Y, Zhu T, Cheng B, Shu Q, and Fang X. (2013). Triggering receptor expressed on myeloid cells-2 protects against polymicrobial sepsis by enhancing bacterial clearance. American journal of respiratory and critical care medicine 188, 201–212. [DOI] [PubMed] [Google Scholar]
- Chen y., Feng Z, Diao B, Wang, Wang G, Wang C, Tan Y, Liu L, Wang C, Liu Y, et al. (2020). The Novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Directly Decimates Human Spleens and Lymph Nodes. medRxiv, 2020.2003.2027.20045427. [Google Scholar]
- Choi HJ, Byun MS, Yi D, Sohn BK, Lee JH, Lee JY, Kim YK, and Lee DY (2017). Associations of thyroid hormone serum levels with in-vivo Alzheimer’s disease pathologies. Alzheimer’s research & therapy 9, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cignarella F, Filipello F, Bollman B, Cantoni C, Locca A, Mikesell R, Manis M, Ibrahim A, Deng L, Benitez BA, et al. (2020). TREM2 activation on microglia promotes myelin debris clearance and remyelination in a model of multiple sclerosis. Acta Neuropathologica 140, 513–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel B, Nagy G, Hah N, Horvath A, Czimmerer Z, Poliska S, Gyuris T, Keirsse J, Gysemans C, Van Ginderachter JA, et al. (2014). The active enhancer network operated by liganded RXR supports angiogenic activity in macrophages. Genes & Development 28, 1562–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JD, Podolanczuk A, Donahue JE, Stopa E, Hennessey JV, Luo L-G, Lim Y-P, and Stern RA (2008). Thyroid hormone levels in the prefrontal cortex of post-mortem brains of Alzheimer’s disease patients. Curr Aging Sci 1, 175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vito P, Incerpi S, Pedersen JZ, Luly P, Davis FB, and Davis PJ (2011). Thyroid Hormones as Modulators of Immune Activities at the Cellular Level. Thyroid 21, 879–890. [DOI] [PubMed] [Google Scholar]
- Deczkowska A, Weiner A, and Amit I. (2020). The Physiology, Pathology, and Potential Therapeutic Applications of the TREM2 Signaling Pathway. Cell 181, 1207–1217. [DOI] [PubMed] [Google Scholar]
- Donatelli SS, Zhou JM, Gilvary DL, Eksioglu EA, Chen X, Cress WD, Haura EB, Schabath MB, Coppola D, Wei S, et al. (2014). TGF-β-inducible microRNA-183 silences tumor-associated natural killer cells. Proceedings of the National Academy of Sciences of the United States of America 111, 4203–4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Yang H, Li C, Liu Q, Bai Q, and Zhang Z. (2018). Triiodothyronine alleviates alcoholic liver disease injury through the negative regulation of the NLRP3 signaling pathway. Exp Ther Med 16, 1866–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosch SF, Mahajan SD, and Collins AR (2009). SARS coronavirus spike protein-induced innate immune response occurs via activation of the NF-κB pathway in human monocyte macrophages in vitro. Virus Research 142, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewers M, Franzmeier N, Suárez-Calvet M, Morenas-Rodriguez E, Caballero MAA, Kleinberger G, Piccio L, Cruchaga C, Deming Y, Dichgans M, et al. (2019). Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer’s disease. Science translational medicine 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara SJ, Bourdette D, and Scanlan TS (2018). Hypothalamic-Pituitary-Thyroid Axis Perturbations in Male Mice by CNS-Penetrating Thyromimetics. Endocrinology 159, 2733–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara SJ, and Scanlan TS (2020). A CNS-Targeting Prodrug Strategy for Nuclear Receptor Modulators. Journal of Medicinal Chemistry 63, 9742–9751. [DOI] [PubMed] [Google Scholar]
- Furuya F, Ishii T, Tamura S, Takahashi K, Kobayashi H, Ichijo M, Takizawa S, Kaneshige M, Suzuki-Inoue K, and Kitamura K. (2017). The ligand-bound thyroid hormone receptor in macrophages ameliorates kidney injury via inhibition of nuclear factor-κB activities. Scientific Reports 7, 43960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervois P, and Lambrichts I. (2019). The Emerging Role of Triggering Receptor Expressed on Myeloid Cells 2 as a Target for Immunomodulation in Ischemic Stroke. Frontiers in immunology 10, 1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JSK, Younkin S, et al. (2012). TREM2 Variants in Alzheimer’s Disease. New England Journal of Medicine 368, 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackenmueller SA, Marchini M, Saba A, Zucchi R, and Scanlan TS (2012). Biosynthesis of 3-Iodothyronamine (T1AM) Is Dependent on the Sodium-Iodide Symporter and Thyroperoxidase but Does Not Involve Extrathyroidal Metabolism of T4. Endocrinology 153, 5659–5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbers M, Wahlstrom GM, and Vennstrom B. (1996). Transactivation by the thyroid hormone receptor is dependent on the spacer sequence in hormone response elements containing directly repeated half-sites. Nucleic Acid Res 24, 2252–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley MD, Banerji T, Tagge IJ, Kirkemo LL, Chaudhary P, Calkins E, Galipeau D, Shokat MD, DeBell MJ, Van Leuven S, et al. (2019). Myelin repair stimulated by CNS-selective thyroid hormone action. JCI Insight 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogervorst E, Huppert F, Matthews FE, and Brayne C. (2008). Thyroid function and cognitive decline in the MRC Cognitive Function and Ageing Study. Psychoneuroendocrinology 33, 1013–1022. [DOI] [PubMed] [Google Scholar]
- Humbert-Claude M, Duc D, Dwir D, Thieren L, Sandström von Tobel J, Begka C, Legueux F, Velin D, Maillard MH, Do KQ, et al. (2016). Tollip, an early regulator of the acute inflammatory response in the substantia nigra. Journal of Neuroinflammation 13, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, Lundgren P, Bleriot C, Liu Z, Deczkowska A, et al. (2019). Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell 178, 686–698.e614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Tan L, Zhu X-C, Zhang Q-Q, Cao L, Tan M-S, Gu L-Z, Wang H-F, Ding Z-Z, Zhang Y-D, et al. (2014). Upregulation of TREM2 Ameliorates Neuropathology and Rescues Spatial Cognitive Impairment in a Transgenic Mouse Model of Alzheimer’s Disease. Neuropsychopharmacology 39, 2949–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson P, Almqvist EG, Johansson JO, Mattsson N, Hansson O, Wallin A, Blennow K, Zetterberg H, and Svensson J. (2013). Reduced cerebrospinal fluid level of thyroxine in patients with Alzheimer’s disease. Psychoneuroendocrinology 38, 1058–1066. [DOI] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, et al. (2013). Variant of TREM2 associated with the risk of Alzheimer’s disease. The New England journal of medicine 368, 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juárez-Cedillo T, Basurto-Acevedo L, Vega-García S, Sánchez-Rodríguez Martha A, Retana-Ugalde R, Juárez-Cedillo E, Gonzalez-Melendez Roberto C, and Escobedo-de-la-Peña J. (2017). Prevalence of thyroid dysfunction and its impact on cognition in older mexican adults: (SADEM study). Journal of endocrinological investigation 40, 945–952. [DOI] [PubMed] [Google Scholar]
- Katz RW, and Koenig RJ (1993). Nonbiased identification of DNA sequences that bind thyroid hormone receptor alpha 1 with high affinity. J Biol Chem 268, 19392–19397. [PubMed] [Google Scholar]
- Kleinberger G, Yamanishi Y, Suárez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, et al. (2014). TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Science translational medicine 6, 243ra286. [DOI] [PubMed] [Google Scholar]
- Kober DL, and Brett TJ (2017). TREM2-Ligand Interactions in Health and Disease. Journal of Molecular Biology 429, 1607–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig RJ, Brent G, Warne RL, Larsen PR, and Moore DD (1987). Thyroid hormone receptor binds to a site in the rat growth hormone promoter required for induction by thyroid hormone. Proc Natl Acad Sci 84, 5670–5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi H, and Kiyama H. (2018). Microglial TREM2/DAP12 Signaling: A Double-Edged Sword in Neural Diseases. Frontiers in Cellular Neuroscience 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O’Loughlin E, Xu Y, Fanek Z, et al. (2017). The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47, 566–581.e569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CYD, Daggett A, Gu X, Jiang LL, Langfelder P, Li X, Wang N, Zhao Y, Park CS, Cooper Y, et al. (2018). Elevated TREM2 Gene Dosage Reprograms Microglia Responsivity and Ameliorates Pathological Phenotypes in Alzheimer’s Disease Models. Neuron 97, 1032–1048.e1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian H, Roy E, and Zheng H. (2016). Microglial Phagocytosis Assay. Bio-protocol 6, e1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Lee M-H, Cohen M, Bommakanti M, and Freedman LP (1996). Transcriptional activation of the Cdk inhibitor p21 by vitamin D3 leads to the induced differentiation of the myelomonocytic cell line U937. Genes Dev 10, 142–153. [DOI] [PubMed] [Google Scholar]
- Lloyd AF, and Miron VE (2019). The pro-remyelination properties of microglia in the central nervous system. Nature Reviews Neurology 15, 447–458. [DOI] [PubMed] [Google Scholar]
- Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, and Manson JJ (2020). COVID-19: consider cytokine storm syndromes and immunosuppression. The Lancet 395, 1033–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinig JM, Ferrara SJ, Banerji T, Banerji T, Sanford-Crane HS, Bourdette D, and Scanlan TS (2017). Targeting Fatty-Acid Amide Hydrolase with Prodrugs for CNS-Selective Therapy. ACS Chemical Neuroscience 8, 2468–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinig JM, Ferrara SJ, Banerji T, Banerji T, Sanford-Crane HS, Bourdette D, and Scanlan TS (2019). Structure–Activity Relationships of Central Nervous System Penetration by Fatty Acid Amide Hydrolase (FAAH)-Targeted Thyromimetic Prodrugs. ACS Medicinal Chemistry Letters 10, 111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merad M, and Martin JC (2020). Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nature Reviews Immunology 20, 355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min Z, Tang Y, Hu X-T, Zhu B-L, Ma Y-L, Zha J-S, Deng X-J, Yan Z, and Chen G-J (2018). Cosmosiin Increases ADAM10 Expression via Mechanisms Involving 5’UTR and PI3 K Signaling. Frontiers in Molecular Neuroscience 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montesinos M.d.M., and Pellizas CG (2019). Thyroid Hormone Action on Innate Immunity. Frontiers in Endocrinology 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natrajan MS, de la Fuente AG, Crawford AH, Linehan E, Nuñez V, Johnson KR, Wu T, Fitzgerald DC, Ricote M, Bielekova B, et al. (2015). Retinoid X receptor activation reverses age-related deficiencies in myelin debris phagocytosis and remyelination. Brain 138, 3581–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen N-H, Apriletti JW, Cunha Lima ST, Webb P, Baxter JD, and Scanlan TS (2002). Rational Design and Synthesis of a Novel Thyroid Hormone Antagonist That Blocks Coactivator Recruitment. Journal of Medicinal Chemistry 45, 3310–3320. [DOI] [PubMed] [Google Scholar]
- Perrotta C, Buldorini M, Assi E, Cazzato D, De Palma C, Clementi E, and Cervia D. (2014). The Thyroid Hormone Triiodothyronine Controls Macrophage Maturation and Functions: Protective Role during Inflammation. The American Journal of Pathology 184, 230–247. [DOI] [PubMed] [Google Scholar]
- Perugorria MJ, Esparza-Baquer A, Oakley F, Labiano I, Korosec A, Jais A, Mann J, Tiniakos D, Santos-Laso A, Arbelaiz A, et al. (2019). Non-parenchymal TREM-2 protects the liver from immunemediated hepatocellular damage. Gut 68, 533–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW (2001). A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Research 29, e45-e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccio L, Buonsanti C, Cella M, Tassi I, Schmidt RE, Fenoglio C, Rinker J 2nd, Naismith RT, Panina-Bordignon P, Passini N, et al. (2008). Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain 131, 3081–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccio L, Buonsanti C, Mariani M, Cella M, Gilfillan S, Cross AH, Colonna M, and Panina-Bordignon P. (2007). Blockade of TREM-2 exacerbates experimental autoimmune encephalomyelitis. European journal of immunology 37, 1290–1301. [DOI] [PubMed] [Google Scholar]
- Placzek AT, Ferrara SJ, Hartley MD, Sanford-Crane HS, Meinig JM, and Scanlan TS (2016). Sobetirome prodrug esters with enhanced blood–brain barrier permeability. Bioorganic & Medicinal Chemistry 24, 5842–5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Placzek AT, and Scanlan TS (2015). New synthetic routes to thyroid hormone analogs: d6-sobetirome, 3H-sobetirome, and the antagonist NH-3. Tetrahedron 71, 5946–5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai V, Rao VH, Shao Z, and Agrawal DK (2016). Dendritic Cells Expressing Triggering Receptor Expressed on Myeloid Cells-1 Correlate with Plaque Stability in Symptomatic and Asymptomatic Patients with Carotid Stenosis. PLOS ONE 11, e0154802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittenhouse PA, and Redei E. (1997). Thyroxine Administration Prevents Streptococcal Cell Wall-Induced Inflammatory Responses*. Endocrinology 138, 1434–1439. [DOI] [PubMed] [Google Scholar]
- Sampaolo S, Campos-Barros A, Mazziotti G, Carlomagno S, Sannino V, Amato G, Carella C, and Di Iorio G. (2005). Increased Cerebrospinal Fluid Levels of 3,3′,5′-Triiodothyronine in Patients with Alzheimer’s Disease. The Journal of Clinical Endocrinology & Metabolism 90, 198–202. [DOI] [PubMed] [Google Scholar]
- Savage JC, Jay T, Goduni E, Quigley C, Mariani MM, Malm T, Ransohoff RM, Lamb BT, and Landreth GE (2015). Nuclear Receptors License Phagocytosis by Trem2+ Myeloid Cells in Mouse Models of Alzheimer’s Disease. The Journal of Neuroscience 35, 6532–6543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlan TS (2010). Sobetirome: a case history of bench-to-clinic drug discovery and development. Heart Failure Reviews 15, 177–182. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nature Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Ma J, Li D, Li P, Zhou X, Li Y, He Z, Qin L, Liang L, and Luo X. (2019). Interleukin-10 inhibits interleukin-1β production and inflammasome activation of microglia in epileptic seizures. Journal of Neuroinflammation 16, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Prinz M, Stagi M, Chechneva O, and Neumann H. (2007). TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLoS medicine 4, e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Lv B, Yang B, Chen Y, Yuan F, Ma L, Chen S, Zhang S, and Xia J. (2019). TREM2 acts as a tumor suppressor in hepatocellular carcinoma by targeting the PI3K/Akt/β-catenin pathway. Oncogenesis 8, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theobald S, Simonis A, Kreer C, Zehner M, Fischer J, Albert M-C, Malin J, Gräb J, Winter S, Silva U.S.d., et al. (2020). The SARS-CoV-2 spike protein primes inflammasome-mediated interleukin-1-beta secretion in COVID-19 patient-derived macrophages (Research Square). [Google Scholar]
- Ulrich JD, and Holtzman DM (2016). TREM2 Function in Alzheimer’s Disease and Neurodegeneration. ACS Chemical Neuroscience 7, 420–427. [DOI] [PubMed] [Google Scholar]
- van der Spek AH, Surovtseva OV, Jim KK, van Oudenaren A, Brouwer MC, Vandenbroucke-Grauls CMJE, Leenen PJM, van de Beek D, Hernandez A, Fliers E, et al. (2018). Regulation of Intracellular Triiodothyronine Is Essential for Optimal Macrophage Function. Endocrinology 159, 2241–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas R, and Videla LA (2017). Thyroid hormone suppresses ischemia-reperfusion-induced liver NLRP3 inflammasome activation: Role of AMP-activated protein kinase. Immunology Letters 184, 92–97. [DOI] [PubMed] [Google Scholar]
- Wang W, Ye L, Ye L, Li B, Gao B, Zeng Y, Kong L, Fang X, Zheng H, Wu Z, et al. (2007). Upregulation of IL-6 and TNF-α induced by SARS-coronavirus spike protein in murine macrophages via NF-κB pathway. Virus Research 128, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K, Byers DE, Jin X, Agapov E, Alexander-Brett J, Patel AC, Cella M, Gilfilan S, Colonna M, Kober DL, et al. (2015). TREM-2 promotes macrophage survival and lung disease after respiratory viral infection. Journal of Experimental Medicine 212, 681–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Xu B, and Koenig RJ (2001). Thyroid hormone response element sequence and the recruitment of retinoid X receptors for thyroid hormone responsiveness. J Biol Chem 276, 3929–3936. [DOI] [PubMed] [Google Scholar]
- Xing J, Titus AR, and Humphrey MB (2015). The TREM2-DAP12 signaling pathway in Nasu-Hakola disease: a molecular genetics perspective. Res Rep Biochem 5, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, Zhao X-Y, Ji Y, Li C, Guo L, et al. (2019). Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Molecular Cell 75, 644–660.e645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Li H, Chen J, Xu W, Yang G, Bao Z, Xia D, Lu G, Hu S, and Zhou J. (2016). TREM-2 serves as a negative immune regulator through Syk pathway in an IL-10 dependent manner in lung cancer. Oncotarget 7, 29620–29634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen PM (2001). Physiological and Molecular Basis of Thyroid Hormone Action. Physiological Reviews 81, 1097–1142. [DOI] [PubMed] [Google Scholar]
- Zhang X, Wang W, Li P, Wang X, and Ni K. (2018). High TREM2 expression correlates with poor prognosis in gastric cancer. Human pathology 72, 91–99. [DOI] [PubMed] [Google Scholar]
- Zhao H, Liu A, Shen L, Xu C, Zhu Z, Yang J, Han X, Bao F, and Yang W. (2017). Isoforskolin downregulates proinflammatory responses induced by Borrelia burgdorferi basic membrane protein A. Exp Ther Med 14, 5974–5980. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request. All datasets generated or analyzed during this study are included in this article. No code was generated in this study. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.