

Abstract
Understanding the characteristics of cancer cells is essential for the development of improved diagnosis and therapeutics. From a gene regulation perspective, the super‐enhancer concept has been introduced to systematically understand the molecular mechanisms underlying the identities of various cell types and has been extended to the analysis of cancer cells and cancer genome alterations. In addition, several characteristic features of super‐enhancers have led to the recognition of the link between gene regulation and biomolecular condensates, which is often mediated by liquid‐liquid phase separation. Several lines of evidence have suggested molecular and biophysical principles and their alterations in cancer cells, which are particularly associated with gene regulation and cell signaling (“ transcriptional” and “signaling” condensates). These findings collectively suggest that the modification of biomolecular condensates represents an important mechanism by which cancer cells acquire various cancer hallmark traits and establish functional innovation for cancer initiation and progression. The condensate model also provides the molecular basis of the vulnerability of cancer cells to transcriptional perturbation and further suggests the possibility of therapeutic targeting of condensates. This review summarizes recent findings regarding the relationships between super‐enhancers and biomolecular condensate models, multiple scenarios of condensate alterations in cancers, and the potential of the condensate model for therapeutic development.
Keywords: biomolecular condensate, cancer, intrinsically disordered region, liquid‐liquid phase separation, super‐enhancer
In this review, we summarize recent findings regarding the relationships between super‐enhancers and the biomolecular condensates, multiple scenarios of condensate alterations in cancer, and the potentials of the condensate model for therapeutic development. Modification of biomolecular condensates may be important mechanisms by which cancer cells acquire various cancer hallmark traits and establish functional innovation for cancer initiation and progression.

1. INTRODUCTION
Malignant transformation is a manifestation of changes in gene expression that promote various cancer hallmark traits, including sustained proliferation, resistance to cell death, and activation of invasion and metastasis. As genomic alterations play an essential role in oncogenesis, understanding alterations in gene expression and genome regulation in cancer cells is essential for the development of improved diagnosis and therapeutics. The identity of various cell types, each of which has essentially the same genome, is determined by cell type‐specific transcriptional programs. Master transcription factors (master TFs) have predominant roles in cell type‐specific transcriptional programs, by activating cell type‐specific enhancers near cell type‐specific genes in many cases. Advances in genome‐wide epigenome analysis, including ChIP‐seq, have unveiled that many of the cell type‐specific enhancers are cooperatively activated by master TFs and Mediator and other transcriptional regulators, and exist as clusters in the genome, leading to the coining of the term “super‐enhancer.” 1 These approaches are also extended to the cancer cells and facilitate the understanding of cancer genome alterations. 2
Several characteristic features of super‐enhancers have led to the recognition of the link between gene regulation and biomolecular condensates, which is often mediated by liquid‐liquid phase separation. 3 In addition, some studies have suggested that some cancer cells bear alterations in the biophysical properties of biomolecular condensates that are particularly associated with cell signaling and gene regulation. These findings collectively suggest that the modification of biomolecular condensates represents the machinery by which cancer cells acquire various cancer hallmark traits and establish functional innovation to facilitate cancer initiation and progression. This review summarizes recent findings regarding the relationships between super‐enhancers and the biomolecular condensate model, multiple scenarios of condensate alterations in cancers, and the potential of the condensate model for therapeutic development. The nuclei include various types of condensates, including heterochromatin condensates; however, this review mainly focuses on transcriptional condensates.
2. SUPER‐ENHANCERS AND CANCER BIOLOGY
Cell type‐specific transcriptional programs are organized by the activation of ~104‐105 cell type‐specific enhancers in one cell type. These active enhancers can be visualized using high‐throughput techniques, including ChIP‐seq, which detects the genome‐wide patterns of histone modifications, such as H3K27ac, and the binding patterns of master TFs and other transcriptional regulators such as the Mediator complex and p300/CBP. In mouse embryonic stem cells (mESCs), reduction in Mediator expression disrupts the expression of ESC‐specific genes and silences master TFs including Oct4. 4 Consistent with this, several hundreds of large clusters of enhancers are densely bound by master TFs and Mediator and are found near genes that play prominent roles in ESC biology. The discovery of these cell type‐specific enhancer clusters led to the proposal of the super‐enhancer concept in 2013. 1 Super‐enhancers are currently defined by the following algorithm: stitching consecutive enhancers marked by Mediator, H3K27ac, or TFs with certain distance cut‐offs (5‐12.5 kb), quantifying the ChIP‐seq signals over the stitched enhancer domains, and then separating super‐enhancers from typical enhancers according to the signal strength with a geometrically defined cut‐off (Figure 1A). H3K27ac ChIP‐seq has been widely used for the convenient identification of super‐enhancers in various cell types, including cancer cells. In typical cases, super‐enhancers are bound by cell type‐specific master TFs and are formed nearby master TFs, thereby indicating a positive feedback loop to establish cell identity and explaining the vulnerability of cell identity for perturbation of master TFs.
FIGURE 1.
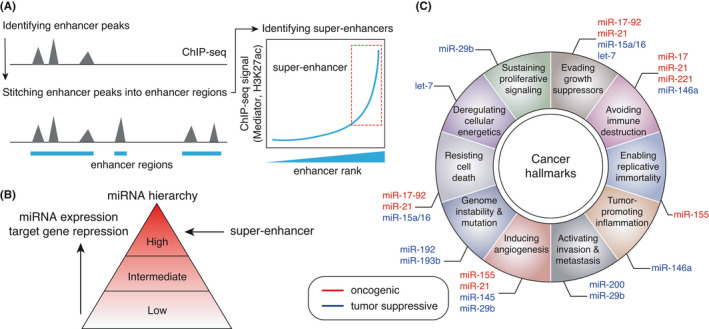
Super‐enhancer, miRNA regulation, and cancer biology. A, Definition of super‐enhancers. B, Impacts of super‐enhancers on miRNA expression and function. Super‐enhancers drive a small subset of cell type‐specific miRNAs that are highly abundant in the relevant cell types and mediate potent target gene repression. C, Cancer hallmark traits and SE‐miRNAs with SE loss or gain in cancer cells. B and C are modified from our previous report 6
Whereas clusters of enhancers can be identified by various approaches and referred to in various terms including “locus control regions” (LCRs), “super‐enhancer” is popularly used to systematically classify the cell type‐specific enhancer regions. 5 We previously reported an integrative meta‐analysis of the connectivity between super‐enhancers and microRNAs (miRNAs) and verified the super‐enhancer concept. 6 In addition to TFs, the expression and function of miRNAs are highly cell type specific. Our integrative analyses of super–enhancer‐associated miRNAs (SE‐RNAs) revealed that the super‐enhancer categorization successfully characterizes a small subset of miRNAs that dominates the miRNA expression pool and target gene repression in various cell types (Figure 1B), and identifies many cell type‐specific miRNAs, whose deletion causes developmental abnormalities in the relevant cell types. 6
Super‐enhancers play multifaceted roles in disease biology. First, disease‐associated genetic variants, detected by Genome‐Wide Association Studies (GWAS), are frequently observed within super‐enhancers of disease‐relevant cell types. 2 Second, in cancer cells, super‐enhancers are frequently generated near various oncogenes and other genes important for various aspects of cancer biology. 2 The tight connection with cancer‐associated genes is also the case for cancer‐associated miRNAs. 6 Systematic analysis of SE‐miRNAs in various cancer cell lines has shown that miRNAs with gain or loss of nearby super‐enhancers tend to have oncogenic or tumor‐suppressive roles, respectively. 6 The target genes of such miRNAs with super‐enhancer alterations are associated with various aspects of cancer hallmark traits (Figure 1C). 6 In addition, we have previously shown that miRNAs with super‐enhancer gain tended to show an association with worse prognosis. 6 These results collectively suggest that alterations in super‐enhancer activity contribute to the phenotypes of cancer cells through the modulation of both cancer‐associated genes and miRNAs. In addition to these scenarios, we recently showed that gain‐of‐function mutation in the seed sequences of miRNA‐140, driven by chondrocyte‐specific super‐enhancers, causes autosomal dominant human skeletal dysplasia (“spondyloepiphyseal dysplasia MIR140 type Nishimura”), extending the roles of super‐enhancers in human diseases. 7 , 8 Although the association between super‐enhancers and long non‐coding RNAs (lncRNAs) in cell identity control has not been explored in depth, certain super–enhancer‐driven lncRNAs (SE‐lncRNAs) have been implicated in cancer pathogenesis. 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 Multifaceted functional crosstalk between super‐enhancers and lncRNAs, including cis and trans gene or super‐enhancer regulation by lncRNAs, has been also suggested. 9 , 13 , 17 , 18
3. SUPER‐ENHANCERS AND CANCER GENOME ALTERATIONS
The abnormal formation of super‐enhancers in cancer cells involves multiple mechanisms. Certain well known examples of genome abnormalities, such as translocation and focal amplification, can be currently viewed as misdirection and misformation of super‐enhancers, respectively. 2 For example, MYC, a well known oncogene, is activated through diverse super–enhancer‐associated mechanisms in various cancer types. In lymphoma and myeloma, chromosomal translocation involving the immunoglobulin locus and MYC locus juxtaposes immunoglobulin super‐enhancers to MYC and induces constitutive expression. 2 A similar scenario, also called as enhancer hijacking, works in solid tumors, including neuroblastomas. 19
In addition, MYC is activated by focal amplification of super‐enhancers located near the 3′ end of the MYC gene in various cancer types, including lung adenocarcinoma, endometrial carcinoma, acute lymphoid leukemia (ALL), and acute myeloid leukemia (AML). 20 , 21 , 22 This is mediated by cell type‐specific amplification of super‐enhancers and chromatin loop re‐wiring. Genomic integration of oncogenic viral DNAs, such as human papillomavirus (HPV) and Epstein‐Barr virus (EBV), can also provide a resource for ectopic (super‐)enhancer formation and peculiar chromatin looping to activate oncogenes. 23 , 24
Non‐coding mutations are also attributable to the dysregulation of super‐enhancers in certain cancer types. In ~5% of T‐cell acute lymphoblastic leukemias (T‐ALL), small insertions (2‐18 base pairs), located ~7.5 kb upstream of the TAL1 transcription start site, create the recognition sites for the MYB oncoprotein, resulting in an ectopic formation of a super‐enhancer through cooperative binding of other TFs and the activation of TAL1 transcription. 25 This provides a representative model to study how non‐coding mutations induce the formation of ectopic super‐enhancers. Although the functional significance of enhancer mutations remains obscure, in contrast with the frequent mutations in the promoters of cancer‐associated genes (eg, TERT), 26 recent studies have gradually expanded knowledge on cancer‐associated mutations in enhancers, including recurrent mutations of PAX5 enhancer regions in chronic lymphocytic leukemia, 27 mutations of ESR1 enhancers in breast cancer, 28 and mutations of FOXA1 enhancers in prostate cancer. 29 Other scenarios for misdirection of super‐enhancer activities include mutations in CTCF binding sites and intrachromosomal deletion. 30 Furthermore, alterations in super‐enhancer programs can be driven by mechanisms other than genomic mutations/structural variations, such as dysregulation of transcriptional factors.
4. SUPER‐ENHANCERS AND THE TRANSCRIPTIONAL CONDENSATE MODEL
Multiple membraneless organelles that form in eukaryotic cells are thought to be important for compartmentalization of various biochemical reactions within cells. 31 As stimulated by a study in 2009 that demonstrated liquid‐liquid phase separation (LLPS) as the driving force of the formation of germline P granules in Caenorhabditis elegans, 32 LLPS has been intensively studied as an emerging regulator of membraneless organelles assembly and various cellular processes, including cellular signaling, gene regulation, and autophagy. Such membraneless compartments are collectively called biomolecular condensates. 31 Although the molecular mechanisms of biomolecular condensate formation remain obscure in many cases and are context dependent, multivalent but individually weak interactions between condensate components, such as protein, RNA, and DNA, are key to determining the phase separation threshold, ie, the solubility limit of the molecule, through modulation of entropy‐driven effects, and promoting biomolecular phase separation. 31 Proteins contribute to multivalent interactions by having multiple modular interaction domains or disordered domains, called “intrinsically disordered regions (IDR)” or “low‐complexity (LC) domains,” which are frequently observed for cellular signaling mediators such as N‐WASP and RNA‐binding and DNA‐binding proteins such as FUS, respectively. RNA and DNA molecules with multiple regions that interact with other DNA and RNA molecules or proteins, also empower condensate formation.
As for gene regulation, multivalent interactions and phase transition capacities of the FET (FUS/EWS/TAF15) RNA‐binding protein family and other RNA‐binding proteins were highlighted in 2012 by identifying their capability to form hydrogels consisting of amyloid‐like fibers in vitro. 33 , 34 Subsequently, it has been shown that hydrogels of LC domains of FET proteins incorporate the repetitive C‐terminal domain (CTD) of RNA polymerase II (RNA Pol II) in a reversible manner sensitive to CTD phosphorylation and that binding between hydrogel polymers and CTD domains correlates with the strength of transcriptional activation by LC domains of FET proteins. 35 The link between multivalent FUS interactions and Pol II CTD recruitment was later extended in the setting of in vitro phase‐separated dynamic FUS droplets (also called as granules), 36 indicating a general property observed beyond protein inclusions and hydrogels. Such in vitro droplet models have been widely used in the condensate field to date. 36
Together with these findings, several functional studies of enhancers have laid the groundwork to connect super‐enhancers and biomolecular condensates. In several studies, but not all, genetic deletion of constituent enhancers within a super‐enhancer has been shown to decrease the activity of the other super‐enhancer constituents and reduce the associated gene expression, which suggests interdependency and high cooperation between super‐enhancer constituents (Figure 2). 37 , 38 We further reported that super‐enhancers can be characterized by a pervasive interaction with the miRNA processing machineries, DGCR8 and Drosha, and that super‐enhancer constituents act cooperatively and facilitate the recruitment of DGCR8 and Drosha and pri‐miRNA processing of cell type‐specific miRNAs. 6 In addition, chromatin contact mapping methods indicate that the clusters of enhancers within super‐enhancers are in close physical contact with one another and with the promoter region of the gene they activate. 39 Based on these results, particularly the multiple layers of cooperation between interacting factors and multiple super‐enhancer constituents, the formation of biomolecular condensates was proposed in 2017 as the operating principle of super‐enhancers (Figure 2). 3 In addition to the previously proposed “transcription factory,” several terms, including “transcriptional condensate,” “transcription hub,” and “MegaTrans complex” have been used to describe such transcription‐associated multimolecular assemblies. 40 , 41 , 42 This condensate model embraces the observation of large clusters of RNA Pol II in living mammalian cells by super‐resolution imaging, 43 and helps, at least partially, to explain the super‐sensitivity of super‐enhancers to transcriptional perturbation, such as inhibition of master TFs and other regulators (Mediator and BRD4), the capacity of an enhancer to simultaneously activate multiple genes in cis and in trans, and the patterns of transcription bursts of strong vs. weak enhancers (Figure 2). 1 , 44 , 45 , 46
FIGURE 2.
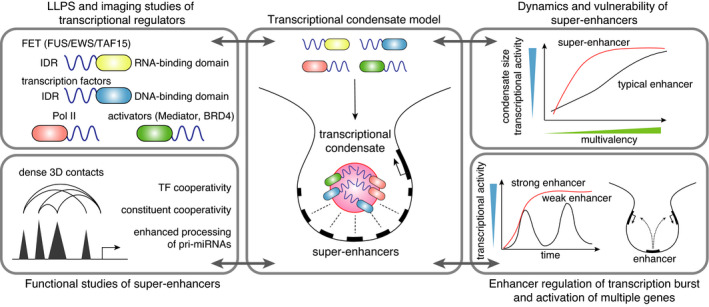
The transcriptional BRD4 condensate model: a link between super‐enhancer and phase separation
Several lines of research, including characterization of LLPS capacity of IDRs in Mediator and BRD4, and imaging of Pol II, Mediator, and BRD4, have provided supporting evidence for this model (Figure 2). 42 , 47 , 48 However, several aspects are currently unclear, including the extent to which LLPS contributes to the in vivo dynamics of super–enhancer‐associated factors, the fraction of super‐enhancers vs. typical enhancers that are associated with condensate formation, and how the mechanisms differ between in vitro and in vivo. Despite these important questions, multivalency appears to be a solid feature of interaction modes of transcriptional regulators, as DNA‐binding and RNA‐binding proteins tend to contain long IDRs relative to other proteins. 49 , 50 , 51 In humans, the large subunit of RNA Pol II contains 52 repeats of a heptapeptide sequence at its CTD, which have LLPS capacity sensitive to CTD phosphorylation, and select interacting partners, such as Mediator and splicing factors, depending on the phosphorylation status. 52 , 53 Mediator and BRD4 have IDRs at the C‐terminus that undergo phase separation in vitro. 47 For TFs, they typically have both DNA‐binding domains (DBD) crucial for genome interactions and activation domains (AD), which are typically disordered and have phase separation capacity. 51 Therefore, the combination of sequence‐specific strong interaction via the DBD and IDR‐mediated multivalent weak interactions is thought to define condensate formation in a context‐dependent manner that is dependent on the modification of components such as phosphorylation of Pol II CTDs. 51 Super‐enhancers facilitate condensate assembly through a high density of TF binding motifs across the local genomic regions. 54
5. ALTERATION IN TRANSCRIPTIONAL CONDENSATES IN CANCER
The pathological significance of altered regulation of biomolecular condensates has recently attracted much attention in parallel with advances in condensate biology. The association with neurodegenerative disorders is well studied, as IDR mutations in FUS in patients with amyotrophic lateral sclerosis (ALS) abrogate the balance between dynamic liquid‐like compartments and solid aggregates and accelerates aberrant phase separation. 55 Furthermore, recent studies have highlighted the link between aberrant condensate dynamics and cancer pathogenesis. In this section, we summarize examples of cancer‐associated alterations in transcription and nuclear condensates (Table 1 and Figure 3). Other condensates related to cytoplasmic cellular signaling and proteostasis are discussed in the following section (Table 1).
TABLE 1.
Summary of target proteins for cancer‐associated alterations in biomolecular condensates
| Protein | Cancer type | Cellular process | Reference |
|---|---|---|---|
| FET fusion | Ewing sarcoma | Transcription | 40, 57, 58 |
| NUP98 fusion | Pediatric AML | Transcription | 59, 60 |
| YAP, TAZ | Breast cancer | Transcription | 61, 62 |
| AKAP95 | Breast cancer | Transcription, RNA splicing | 63 |
| YTHDC1 | AML | RNA modification | 64 |
| ASXL1 | Myeloid neoplasm | Epigenetic regulation | 65 |
| EML4‐ALK | Lung cancer | Cellular signaling | 69 |
| CCDC6‐RET | Lung cancer | Cellular signaling | 69 |
| DnaJB1‐PKAcat | Fibrolamellar hepatocellular carcinoma | Cellular signaling | 70 |
| DACT1 | Breast cancer, prostate cancer | Cellular signaling | 71 |
| SPOP | Solid tumors (prostate cancer) | Protein homeostasis | 72 |
| KEAP1, NRF2 | Multiple cancer types (lung cancer) | Protein homeostasis, metabolism | 73 |
| KRAS | Multiple cancer types (pancreatic cancer) | Stress granule | 74 |
| YB‐1 | Sarcoma | Stress granule | 75 |
| DDX3X | Multiple cancer types (medulloblastoma) | Stress granule | 76 |
FIGURE 3.
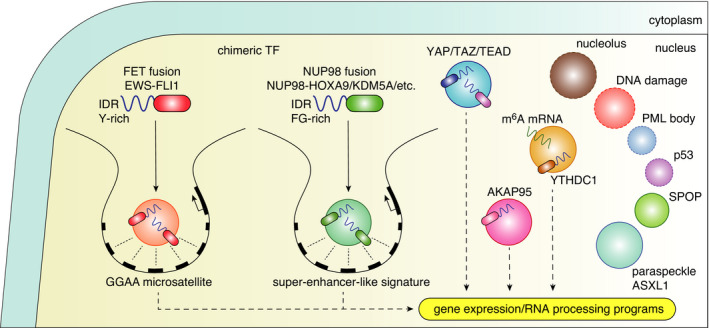
Alterations in transcriptional condensates and other condensates in the nuclei of cancer cells
As DNA‐binding and RNA‐binding proteins frequently contain IDRs, chimeric fusion TFs caused by chromosomal translocation alter the dynamics of condensate formation capacity and transcriptional activity. In the well known FET protein‐related translocation, IDRs of the FET RNA‐binding protein family are fused to a variety of different DBDs of translocation partners, yielding various chimeric TFs, such as EWS‐FLI, EWS‐ERG, and FUS‐ERG, in Ewing sarcoma. 56 EWS‐FLI TFs recruit the BRG1/BRM‐associated factor (BAF) chromatin remodeling complex to the tumor‐specific enhancers in a manner dependent on the tyrosine residues of EWS IDRs and form condensates at the GGAA repeat‐containing microsatellites (Figure 3). 40 , 57 , 58 Analogously, recent studies have shown that NUP98‐involving TF chimeras, recurrently detected in pediatric AML, engage similar mechanisms. 59 , 60 In these cases, phenylalanine and glycine‐rich IDRs of NUP98, which is intrinsically a part of nuclear pore complex, are fused to various proteins involved in transcriptional regulation and epigenetics, including HOXA9, KDM5A, NSD1, DDX10, and PSIP1. Upon fusion, these diverse chimeric proteins form nuclear condensates in an IDR‐dependent manner, interact with various transcriptional regulators, and induce aberrant chromatin looping and leukemogenic gene expression programs (Figure 3). 59 , 60 In both cases, that is, FET and NUP98 fusion, IDRs potentiate target gene activation, possibly through increased chromatin binding and/or retention. 57 , 60 Consistent with the super‐enhancer and condensate model, a high density of DNA‐binding motifs (GGAA repeats for FET chimeras and super‐enhancer‐like signatures for NUP98 chimeras, respectively) not only ensures the specificity of target genes, but also aids in perturbation of their spatiotemporal precise regulation, finally leading to oncogenesis. 58 , 60 Furthermore, given that various NUP98 chimeras share interacting partners and that artificial IDRs can recapitulate the fusion phenotypes, diverse IDRs may have redundant functions to hijack pre‐existing transcriptional machineries, perturb transcriptional programs, and exert neomorphic activities.
Alteration in transcriptional condensates can be induced by dysregulation of the expression levels of transcriptional and RNA processing regulators. YAP and TAZ, which are frequently upregulated in cancers, form condensates and promote gene expression (Figure 3). 61 , 62 The condensates of TAZ were observed as discrete nuclear puncta in breast cancer tissue samples. 62 As splicing factors are well known to form condensates, a recent analysis showed that a regulator of transcription and RNA splicing, AKAP95, which is frequently overexpressed across a large range of human cancers, forms nuclear condensates, supports cancer cell growth, and suppresses oncogene‐induced senescence in an IDR‐dependent manner (Figure 3). 63 In addition, a recent report suggested that YTHDC1, a reader protein of N 6‐methyladenosine (m6A), undergoes LLPS and forms nuclear condensates together with m6A and that nuclear YTHDC1‐m6A condensates are abundant in AML cells relative to normal blood cells and support the cell survival of AML cells by protecting m6A mRNAs including MYC from RNA degradation (Figure 3). 64 Mutations in ASXL1 histone modifier have also been linked to regulation of paraspeckle formation in hematopoietic cells. 65 Current research has mainly focused on TF binding and transcription initiation and a future challenge will be to examine how the condensates are associated with transcription cycles and how they are altered in cancers. Although the functional role of nuclear condensates in cancer cells remains to be elucidated, cancer may be associated with the alteration in various other nuclear condensates. These scenarios include alterations in morphology, size, and number of nucleoli in cancer; alteration in DNA damage‐associated condensates, which involve 53BP1, PARP1, and FET proteins; alteration of promyelocytic leukemia (PML) bodies, whose components can be targeted by PML‐RARA fusions; and phase separation and aggregation properties of wild‐type and mutant p53 (Figure 3). 66 , 67 , 68
6. ALTERATION IN SIGNALING AND OTHER CONDENSATES IN CANCER
Biomolecular condensates also play important roles in cellular signaling cascades. 31 Well known examples include T‐cell receptor signaling clusters and the assembly of nephrin receptors and the downstream effectors, including NCK and N‐WASP. Typically, such signaling condensates are formed at or near the cell membrane and are thought to be important for the full activation of signaling pathways, their spatiotemporal control, and ensuring signaling specificity. Recent studies have highlighted that known cancer‐associated signaling pathways are altered by cancer‐associated mutations. Chimeric kinases, such as EML4‐ALK and CCDC6‐RET in lung cancer, have recently been shown to form membraneless cytoplasmic protein granules, although EML4‐ALK granules are not simple liquid droplets and have solid‐like properties. 69 Importantly, these pathogenic condensates locally concentrate the RAS activating complex GRB2/SOS1 and activate RAS in a lipid membrane‐independent manner (Figure 4). 69 Another study has shown that the type I regulatory subunit of protein kinase A (PKA), RIα, underwent LLPS to form RIα bodies harboring high levels of cAMP and PKA activity, spatially sequester cAMP, and constrain cAMP/PKA signaling for tumor suppression. 70 However, such tumor‐suppressive effects of RIα LLPS are abrogated by loss‐of‐function of RIα and DnaJB1‐PKAcat (the PKA catalytic subunit) fusion oncogene in fibrolamellar hepatocellular carcinoma, leading to increased cell proliferation and anchorage‐independent growth (Figure 4). 70 These studies collectively suggested a link between condensate‐based dysregulation of RAS and cAMP/PKA signaling and cancer. A recent study also reported that TGF‐β‐induced DACT1 forms cytoplasmic condensates to inhibit Wnt signaling and promotes bone metastasis in breast and prostate cancer (Figure 4). 71 Given that many cellular signaling cascades activate specific transcriptional programs, the crosstalk between alterations in “signaling condensates” and “transcriptional condensates” may be a key feature of cancer cells.
FIGURE 4.
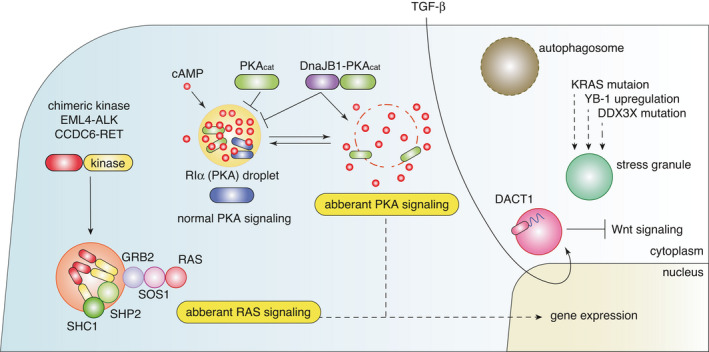
Alterations in signaling condensates and other cytoplasmic condensates in cancer cells
Phase separation is also linked to protein homeostasis (“proteostasis”). In addition to neurodegenerative disorders, cancers appear to be associated with the regulation of condensate‐mediated proteostasis. SPOP, a substrate adaptor of the cullin3‐RING ubiquitin ligase, forms liquid nuclear bodies together with its substrates, such as Death Domain‐Associated Protein (DAXX), and normally suppresses the substrate levels (Figure 3). 72 The tumor suppressor roles of SPOP are perturbed by frequent mutations in solid cancers, particularly in prostate cancer, with increased levels of proto‐oncogenic substrates. 72 The pathogenic effects of SPOP mutations can be partly explained by its interference in LLPS and colocalization with its substrates. 72 In addition, loss‐of‐function mutations in the KEAP1 tumor suppressor gene stabilize the NRF2 transcription factor, leading to alterations in cellular metabolism and adaptation to oxidative stress. 73 A recent study showed that KEAP1 mutants (ANCHOR mutants) form p62‐dependent condensates with NRF2 and probably cause impaired degradation of NRF2. 73
Other condensates potentially associated with cancer biology are stress granules. Although the mechanisms of their alterations, especially alterations in LLPS, remain unclear, several oncogenic events, such as KRAS mutations, YB‐1 upregulation, and DDX3X mutations, are reportedly associated with alterations in stress granules and translation (Figure 4). 74 , 75 , 76 Although the effects on translation may vary and be context dependent, these events appear to enhance stress granule assembly in general. 74 , 75 , 76
7. THERAPEUTIC TARGETING OF BIOMOLECULAR CONDENSATES
Transcription of super–enhancer‐associated genes highly depends on transcriptional regulators such as Mediator and BRD4 and is preferentially downregulated upon transcriptional perturbation by the BET‐bromodomain inhibitor JQ1 and CDK7 inhibitor THZ1 compared with that of typical enhancer‐associated genes. 44 , 77 Therefore, the super‐enhancer and transcriptional condensate models reinforce the rationale of targeting “transcription addiction” and “transcriptional dependency” and partly explain the molecular basis of super‐sensitivity of certain cancer cell types to transcriptional perturbation. 78 However, given that normal cells also utilize super‐enhancer transcriptional programs, the reasons why cancer cells are super‐sensitive to perturbation and the means to target “transcriptional dependency” specifically should be further explored.
A recent study showed that small‐molecule cancer therapeutics, such as cisplatin, mitoxantrone, and tamoxifen, become concentrated in specific protein condensates, independent of the drug target. 79 These findings may be extrapolated to understand the connection between the dynamics of condensates in cancer cells and anti‐cancer drug activities and resistance. 79 In addition, as a condensate‐hardening drug has been recently shown to block the replication of human respiratory syncytial virus in vivo, 80 the pharmaceutical design for direct targeting of condensate dynamics may be promising for cancer therapeutics.
8. CONCLUSION
In the present review, we summarize recent findings regarding the relationships between super‐enhancers and cancer biology, the relationships between super‐enhancers and biomolecular condensate models, and multiple scenarios of condensate alterations in cancers. These findings collectively suggest that the modification of biomolecular condensates represents the machinery by which cancer cells acquire various cancer hallmark traits and establish functional innovation for cancer initiation and progression. From an evolutionary standpoint, multivalent weak interactions can be recognized as a means by which life systems develop new pathways with few and probably less lethal gene mutations. 81 Accumulating evidence in cancer research, particularly that related to transcription and signaling condensates, appears to be consistent with this view. If so, the next important question would be to delineate the shared and context‐dependent molecular grammars of how redundancy and weak cooperation, a key feature of condensates and IDRs, contribute to new phenotypes in cancer, and to construct frameworks for modeling, validating, and therapeutically targeting the functions of altered cancer‐associated condensates. Together with an in‐depth understanding of the biophysical dynamics of biomolecular condensates, understanding the diversity of IDRs in human diseases and cancers may provide further insights into these key issues.
DISCLOSURE
HIS received research grants from the Mochida Memorial Foundation for Medical and Pharmaceutical Research, Sumitomo Foundation, Mitsubishi Foundation, Daiichi Sankyo Foundation of Life Science, Uehara Memorial Foundation, and Takeda Science Foundation. KO declares no conflict of interest.
ACKNOWLEDGMENTS
We thank Dr. Koichi Ogami, Dr. Seiko Yoshino, and Dr. Shintaro Komatsu, and the members of the Suzuki laboratories for their discussions. We apologize to the many researchers whose work has not been cited due to space limitations. This work was supported by a JSPS KAKENHI Home‐Returning Researcher Development Research Grant Number 19K24694 (to HIS), the Mochida Memorial Foundation for Medical and Pharmaceutical Research (to HIS), Grant for Basic Science Research Projects from the Sumitomo Foundation (to HIS), Mitsubishi Foundation (to HIS), Daiichi Sankyo Foundation of Life Science (to HIS), Uehara Memorial Foundation (to HIS), Takeda Science Foundation (to HIS), and Nasu Foundation (to KO).
Suzuki HI, Onimaru K. Biomolecular condensates in cancer biology. Cancer Sci.2022;113:382–391. doi: 10.1111/cas.15232
REFERENCES
- 1. Whyte WA, Orlando DA, Hnisz D, et al. Master transcription factors and mediator establish super‐enhancers at key cell identity genes. Cell. 2013;153:307‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hnisz D, Abraham BJ, Lee TI, et al. Super‐enhancers in the control of cell identity and disease. Cell. 2013;155:934‐947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hnisz D, Shrinivas K, Young RA, Chakraborty AK, Sharp PA. A phase separation model for transcriptional control. Cell. 2017;169:13‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kagey MH, Newman JJ, Bilodeau S, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blobel GA, Higgs DR, Mitchell JA, Notani D, Young RA. Testing the super‐enhancer concept. Nat Rev Genet. 2021;22(12):749‐755. [DOI] [PubMed] [Google Scholar]
- 6. Suzuki HI, Young RA, Sharp PA. Super‐enhancer‐mediated RNA processing revealed by integrative MicroRNA network analysis. Cell. 2017;168:1000‐1014.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grigelioniene G, Suzuki HI, Taylan F, et al. Gain‐of‐function mutation of microRNA‐140 in human skeletal dysplasia. Nat Med. 2019;25:583‐590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Suzuki HI, Spengler RM, Grigelioniene G, Kobayashi T, Sharp PA. Deconvolution of seed and RNA‐binding protein crosstalk in RNAi‐based functional genomics. Nat Genet. 2018;50:657‐661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xiang JF, Yin QF, Chen T, et al. Human colorectal cancer‐specific CCAT1‐L lncRNA regulates long‐range chromatin interactions at the MYC locus. Cell Res. 2014;24:513‐531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu S, Wan L, Yin H, et al. Long noncoding RNA Linc00152 functions as a tumor propellant in Pan‐cancer. Cell Physiol Biochem. 2017;44:2476‐2490. [DOI] [PubMed] [Google Scholar]
- 11. Xie JJ, Jiang YY, Jiang Y, et al. Super‐enhancer‐driven long non‐coding RNA LINC01503, regulated by TP63, is over‐expressed and oncogenic in squamous cell carcinoma. Gastroenterology. 2018;154:2137‐2151.e1. [DOI] [PubMed] [Google Scholar]
- 12. Miano V, Ferrero G, Rosti V, et al. Luminal lncRNAs regulation by ERα‐controlled enhancers in a ligand‐independent manner in breast cancer cells. Int J Mol Sci. 2018;19:593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jiang Y, Jiang YY, Xie JJ, et al. Co‐activation of super‐enhancer‐driven CCAT1 by TP63 and SOX2 promotes squamous cancer progression. Nat Commun. 2018;9:3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Peng L, Jiang B, Yuan X, et al. Super‐enhancer‐associated long noncoding RNA HCCL5 is activated by ZEB1 and promotes the malignancy of hepatocellular carcinoma. Cancer Res. 2019;79:572‐584. [DOI] [PubMed] [Google Scholar]
- 15. Lin X, Spindler TJ, de Souza Fonseca MA, et al. Super‐enhancer‐associated LncRNA UCA1 interacts directly with AMOT to activate YAP target genes in epithelial ovarian cancer. iScience. 2019;17:242‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang X, Zhang R, Wu S, et al. Super‐enhancer LncRNA LINC00162 promotes progression of bladder cancer. iScience. 2020;23:101857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Soibam B. Super‐lncRNAs: identification of lncRNAs that target super‐enhancers via RNA:DNA:DNA triplex formation. RNA. 2017;23:1729‐1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guo ZW, Xie C, Li K, et al. SELER: a database of super‐enhancer‐associated lncRNA‐ directed transcriptional regulation in human cancers. Database. 2019;2019:baz027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zimmerman MW, Liu Y, He S, et al. MYC drives a subset of high‐risk pediatric neuroblastomas and is activated through mechanisms including enhancer hijacking and focal enhancer amplification. Cancer Discov. 2018;8:320‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang X, Choi PS, Francis JM, et al. Identification of focally amplified lineage‐specific super‐enhancers in human epithelial cancers. Nat Genet. 2016;48:176‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Herranz D, Ambesi‐Impiombato A, Palomero T, et al. A NOTCH1‐driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat Med. 2014;20:1130‐1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shi J, Whyte WA, Zepeda‐Mendoza CJ, et al. Role of SWI/SNF in acute leukemia maintenance and enhancer‐mediated Myc regulation. Genes Dev. 2013;27:2648‐2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Warburton A, Redmond CJ, Dooley KE, et al. HPV integration hijacks and multimerizes a cellular enhancer to generate a viral‐cellular super‐enhancer that drives high viral oncogene expression. PLoS Genet. 2018;14:e1007179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Okabe A, Huang KK, Matsusaka K, et al. Cross‐species chromatin interactions drive transcriptional rewiring in Epstein‐Barr virus‐positive gastric adenocarcinoma. Nat Genet. 2020;52:919‐930. [DOI] [PubMed] [Google Scholar]
- 25. Mansour MR, Abraham BJ, Anders L, et al. Oncogene regulation. An oncogenic super‐enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014;346:1373‐1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Elliott K, Larsson E. Non‐coding driver mutations in human cancer. Nat Rev Cancer. 2021;21:500‐509. [DOI] [PubMed] [Google Scholar]
- 27. Puente XS, Beà S, Valdés‐Mas R, et al. Non‐coding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015;526:519‐524. [DOI] [PubMed] [Google Scholar]
- 28. Bailey SD, Desai K, Kron KJ, et al. Noncoding somatic and inherited single‐nucleotide variants converge to promote ESR1 expression in breast cancer. Nat Genet. 2016;48:1260‐1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou S, Hawley JR, Soares F, et al. Noncoding mutations target cis‐regulatory elements of the FOXA1 plexus in prostate cancer. Nat Commun. 2020;11:441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hnisz D, Weintraub AS, Day DS, et al. Activation of proto‐oncogenes by disruption of chromosome neighborhoods. Science. 2016;351:1454‐1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol. 2017;18:285‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brangwynne CP, Eckmann CR, Courson DS, et al. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science. 2009;324:1729‐1732. [DOI] [PubMed] [Google Scholar]
- 33. Kato M, Han TW, Xie S, et al. Cell‐free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012;149:753‐767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Han TW, Kato M, Xie S, et al. Cell‐free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell. 2012;149:768‐779. [DOI] [PubMed] [Google Scholar]
- 35. Kwon I, Kato M, Xiang S, et al. Phosphorylation‐regulated binding of RNA polymerase II to fibrous polymers of low‐complexity domains. Cell. 2013;155:1049‐1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Burke KA, Janke AM, Rhine CL, Fawzi NL. Residue‐by‐residue view of in vitro FUS granules that bind the C‐terminal domain of RNA polymerase II. Mol Cell. 2015;60:231‐241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hnisz D, Schuijers J, Lin CY, et al. Convergence of developmental and oncogenic signaling pathways at transcriptional super‐enhancers. Mol Cell. 2015;58:362‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shin HY, Willi M, HyunYoo K, et al. Hierarchy within the mammary STAT5‐driven Wap super‐enhancer. Nat Genet. 2016;48:904‐911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dowen JM, Fan ZP, Hnisz D, et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell. 2014;159:374‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chong S, Dugast‐Darzacq C, Liu Z, et al. Imaging dynamic and selective low‐complexity domain interactions that control gene transcription. Science. 2018;361:eaar2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu Z, Merkurjev D, Yang F, et al. Enhancer activation requires trans‐recruitment of a mega transcription factor complex. Cell. 2014;159:358‐373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nair SJ, Yang L, Meluzzi D, et al. Phase separation of ligand‐activated enhancers licenses cooperative chromosomal enhancer assembly. Nat Struct Mol Biol. 2019;26:193‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cisse II, Izeddin I, Causse SZ, et al. Real‐time dynamics of RNA polymerase II clustering in live human cells. Science. 2013;341:664‐667. [DOI] [PubMed] [Google Scholar]
- 44. Lovén J, Hoke HA, Lin CY, et al. Selective inhibition of tumor oncogenes by disruption of super‐enhancers. Cell. 2013;153:320‐334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fukaya T, Lim B, Levine M. Enhancer control of transcriptional bursting. Cell. 2016;166:358‐368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lim B, Heist T, Levine M, Fukaya T. Visualization of transvection in living Drosophila embryos. Mol Cell. 2018;70:287‐296.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sabari BR, Dall'Agnese A, Boija A, et al. Coactivator condensation at super‐enhancers links phase separation and gene control. Science. 2018;361:eaar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cho WK, Spille JH, Hecht M, et al. Mediator and RNA polymerase II clusters associate in transcription‐dependent condensates. Science. 2018;361:412‐415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu J, Perumal NB, Oldfield CJ, Su EW, Uversky VN, Dunker AK. Intrinsic disorder in transcription factors. Biochemistry. 2006;45:6873‐6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. van der Lee R, Buljan M, Lang B, et al. Classification of intrinsically disordered regions and proteins. Chem Rev. 2014;114:6589‐6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Boija A, Klein IA, Sabari BR, et al. Transcription factors activate genes through the phase‐separation capacity of their activation domains. Cell. 2018;175:1842‐1855.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Boehning M, Dugast‐Darzacq C, Rankovic M, et al. RNA polymerase II clustering through carboxy‐terminal domain phase separation. Nat Struct Mol Biol. 2018;25:833‐840. [DOI] [PubMed] [Google Scholar]
- 53. Guo YE, Manteiga JC, Henninger JE, et al. Pol II phosphorylation regulates a switch between transcriptional and splicing condensates. Nature. 2019;572:543‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shrinivas K, Sabari BR, Coffey EL, et al. Enhancer features that drive formation of transcriptional condensates. Mol Cell. 2019;75:549‐561.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Patel A, Lee HO, Jawerth L, et al. A liquid‐to‐solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell. 2015;162:1066‐1077. [DOI] [PubMed] [Google Scholar]
- 56. Riggi N, Suvà ML, Stamenkovic I. Ewing's Sarcoma. N Engl J Med. 2021;384:154‐164. [DOI] [PubMed] [Google Scholar]
- 57. Boulay G, Sandoval GJ, Riggi N, et al. Cancer‐specific retargeting of BAF complexes by a prion‐like domain. Cell. 2017;171:163‐178.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zuo L, Zhang G, Massett M, et al. Loci‐specific phase separation of FET fusion oncoproteins promotes gene transcription. Nat Commun. 2021;12:1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Terlecki‐Zaniewicz S, Humer T, Eder T, et al. Biomolecular condensation of NUP98 fusion proteins drives leukemogenic gene expression. Nat Struct Mol Biol. 2021;28:190‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ahn JH, Davis ES, Daugird TA, et al. Phase separation drives aberrant chromatin looping and cancer development. Nature. 2021;595:591‐595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cai D, Feliciano D, Dong P, et al. Phase separation of YAP reorganizes genome topology for long‐term YAP target gene expression. Nat Cell Biol. 2019;21:1578‐1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lu Y, Wu T, Gutman O, et al. Phase separation of TAZ compartmentalizes the transcription machinery to promote gene expression. Nat Cell Biol. 2020;22:453‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li W, Hu J, Shi B, et al. Biophysical properties of AKAP95 protein condensates regulate splicing and tumorigenesis. Nat Cell Biol. 2020;22:960‐972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cheng Y, Xie W, Pickering BF, et al. N(6)‐Methyladenosine on mRNA facilitates a phase‐separated nuclear body that suppresses myeloid leukemic differentiation. Cancer Cell. 2021;39:958‐972.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yamamoto K, Goyama S, Asada S, et al. A histone modifier, ASXL1, interacts with NONO and is involved in paraspeckle formation in hematopoietic cells. Cell Rep. 2021;36:109576. [DOI] [PubMed] [Google Scholar]
- 66. Lafontaine DLJ, Riback JA, Bascetin R, Brangwynne CP. The nucleolus as a multiphase liquid condensate. Nat Rev Mol Cell Biol. 2021;22:165‐182. [DOI] [PubMed] [Google Scholar]
- 67. Jiang S, Fagman JB, Chen C, Alberti S, Liu B. Protein phase separation and its role in tumorigenesis. eLife. 2020;9:e60264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cai D, Liu Z, Lippincott‐Schwartz J. Biomolecular condensates and their links to cancer progression. Trends Biochem Sci. 2021;46:535‐549. [DOI] [PubMed] [Google Scholar]
- 69. Tulpule A, Guan J, Neel DS, et al. Kinase‐mediated RAS signaling via membraneless cytoplasmic protein granules. Cell. 2021;184:2649‐2664.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhang JZ, Lu TW, Stolerman LM, et al. Phase separation of a PKA regulatory subunit controls cAMP compartmentation and oncogenic signaling. Cell. 2020;182:1531‐1544.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Esposito M, Fang C, Cook KC, et al. TGF‐β‐induced DACT1 biomolecular condensates repress Wnt signalling to promote bone metastasis. Nat Cell Biol. 2021;23:257‐267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bouchard JJ, Otero JH, Scott DC, et al. Cancer mutations of the tumor suppressor SPOP disrupt the formation of active, phase‐separated compartments. Mol Cell. 2018;72:19‐36.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Cloer EW, Siesser PF, Cousins EM, et al. p62‐Dependent phase separation of patient‐derived KEAP1 mutations and NRF2. Mol Cell Biol. 2018;38:e00644‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Grabocka E, Bar‐Sagi D. Mutant KRAS enhances tumor cell fitness by upregulating stress granules. Cell. 2016;167:1803‐1813.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Somasekharan SP, El‐Naggar A, Leprivier G, et al. YB‐1 regulates stress granule formation and tumor progression by translationally activating G3BP1. J Cell Biol. 2015;208:913‐929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Valentin‐Vega YA, Wang YD, Parker M, et al. Cancer‐associated DDX3X mutations drive stress granule assembly and impair global translation. Sci Rep. 2016;6:25996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kwiatkowski N, Zhang T, Rahl PB, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511:616‐620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Vervoort SJ, Devlin JR, Kwiatkowski N, Teng M, Gray NS, Johnstone RW. Targeting transcription cycles in cancer. Nat Rev Cancer. 2021. Online ahead of print. [DOI] [PubMed] [Google Scholar]
- 79. Klein IA, Boija A, Afeyan LK, et al. Partitioning of cancer therapeutics in nuclear condensates. Science. 2020;368:1386‐1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Risso‐Ballester J, Galloux M, Cao J, et al. A condensate‐hardening drug blocks RSV replication in vivo. Nature. 2021;595:596‐599. [DOI] [PubMed] [Google Scholar]
- 81. Gao A, Shrinivas K, Lepeudry P, Suzuki HI, Sharp PA, Chakraborty AK. Evolution of weak cooperative interactions for biological specificity. Proc Natl Acad Sci USA. 2018;115:E11053‐E11060. [DOI] [PMC free article] [PubMed] [Google Scholar]