

Abstract
The state of somatic energy stores in metazoans is communicated to the brain, which regulates key aspects of behaviour, growth, nutrient partitioning and development1. The central melanocortin system acts through melanocortin 4 receptor (MC4R) to control appetite, food intake and energy expenditure2. Here we present evidence that MC3R regulates the timing of sexual maturation, the rate of linear growth and the accrual of lean mass, which are all energy-sensitive processes. We found that humans who carry loss-of-function mutations in MC3R, including a rare homozygote individual, have a later onset of puberty. Consistent with previous findings in mice, they also had reduced linear growth, lean mass and circulating levels of IGF1. Mice lacking Mc3r had delayed sexual maturation and an insensitivity of reproductive cycle length to nutritional perturbation. The expression of Mc3r is enriched in hypothalamic neurons that control reproduction and growth, and expression increases during postnatal development in a manner that is consistent with a role in the regulation of sexual maturation. These findings suggest a bifurcating model of nutrient sensing by the central melanocortin pathway with signalling through MC4R controlling the acquisition and retention of calories, whereas signalling through MC3R primarily regulates the disposition of calories into growth, lean mass and the timing of sexual maturation.
Pro-opiomelanocortin (POMC), which encodes several melanocortin peptides, is expressed in neurons of the hypothalamic arcuate nucleus2, which are activated by key hormonal signals of caloric balance, leptin3 and insulin4. These hormones also negatively regulate the activity of neurons that release the melanocortin receptor antagonist, agouti-related peptide (AgRP)5. The actions of α-melanocyte-stimulating hormone (α-MSH) and β-MSH on MC4R are necessary for the normal control of food intake and energy expenditure6,7. Humans (and mice) who lack MC4R are obese and hyperphagic and have reduced basal energy expenditure6,8–12. However, they have normal or even accelerated early linear growth and no retardation of pubertal development11, both of which are impaired by caloric deprivation13 or leptin deficiency14,15. This suggests either that POMC-derived peptides are not responsible for transmitting nutritional signals to those particular downstream processes or that a different melanocortin receptor is involved. MC3R is the only other melanocortin receptor that is predominantly expressed in the brain16,17. Mice lacking Mc3r have been reported to have a normal reproductive development, fertility and no change in food intake, but develop an altered body composition with a high ratio of fat-to-lean mass and impaired linear growth18–20. Human genome-wide association studies (GWAS) have identified common variants in the vicinity of MC3R, which are associated with both adult height21 and age at menarche22. While rare functionally compromised heterozygous variants in MC3R have been reported in humans, no consistent phenotype has been reported23, although associations with height24 and obesity25–27 have been suggested. We set out to establish the role of MC3R in human physiology by seeking naturally occurring mutations that resulted in functional impairment of the receptor, and by studying the relationship with relevant human phenotypes. We identify a strong and previously unreported effect of MC3R loss-of-function (LoF) mutations on pubertal timing in humans, and provide evidence for the conservation of this pathway in mice. Consistent with phenotypes previously described in mice that are deficient in MC3R, we report that human MC3R deficiency is also associated with reduced childhood growth, adult height and lean mass.
Heterozygous MC3R phenotypes
Using whole-exome sequence data from approximately 200,000 UK Biobank (UKBB) participants, we found that 0.82% of individuals carried at least one rare (minor allele frequency (MAF) of less than 0.2%), predicted deleterious variant in MC3R (Supplementary Table 1). We undertook aggregated gene burden tests focused on traits relevant to growth, body composition and pubertal timing. The 812 female MC3R rare mutation carriers had a 4.7-month delay in age at menarche compared with non-carriers (beta = 0.39 years, P = 6.4 × 10−12), an effect size that is approximately three times larger than the most significantly associated common variant in the genome (the LIN28B locus)22. The gene burden score for MC3R was also associated with delayed voice breaking in men, shorter adult and childhood stature, lower sitting height, lower circulating levels of IGF1, lower total body lean mass, and lower appendicular lean mass (ALM)-to-body mass index (BMI) (ALM/BMI) ratio, an established measure of sarcopenia28,29 (Supplementary Table 2).
To determine whether there was a quantitative relationship between the degree of functional impact of individual non-synonymous mutations and phenotypic outcomes, we selected three missense mutations that were sufficiently common in the full approximately 500,000 UKBB sample to allow robust testing of association with phenotypes (MAF ≥ 0.05% using array genotypes). We identified p.F45S and p.R220S with MAFs of 0.06% and 0.19%, respectively (Extended Data Fig. 1). The third variant, p.V44I (MAF = 10.09%; Extended Data Fig. 1), is in strong linkage disequilibrium (r2 = 0.97) with a previously identified GWAS signal for age at menarche (rs3746619 located in the 5′ untranslated region)22. We measured the ability of these mutants to generate cAMP in human embryonic kidney (HEK293) cells in vitro, upon stimulation by [Nle4, d-Phe7]-α-MSH (NDP-MSH). p.F45S exhibited severely impaired signalling compared with wild-type (WT) MC3R, p.R220S showed partial LoF and p.V44I was indistinguishable from the WT (Fig. 1a–d, Supplementary Table 3).
Fig. 1 |. Non-synonymous variants of MC3R and association with phenotypes.
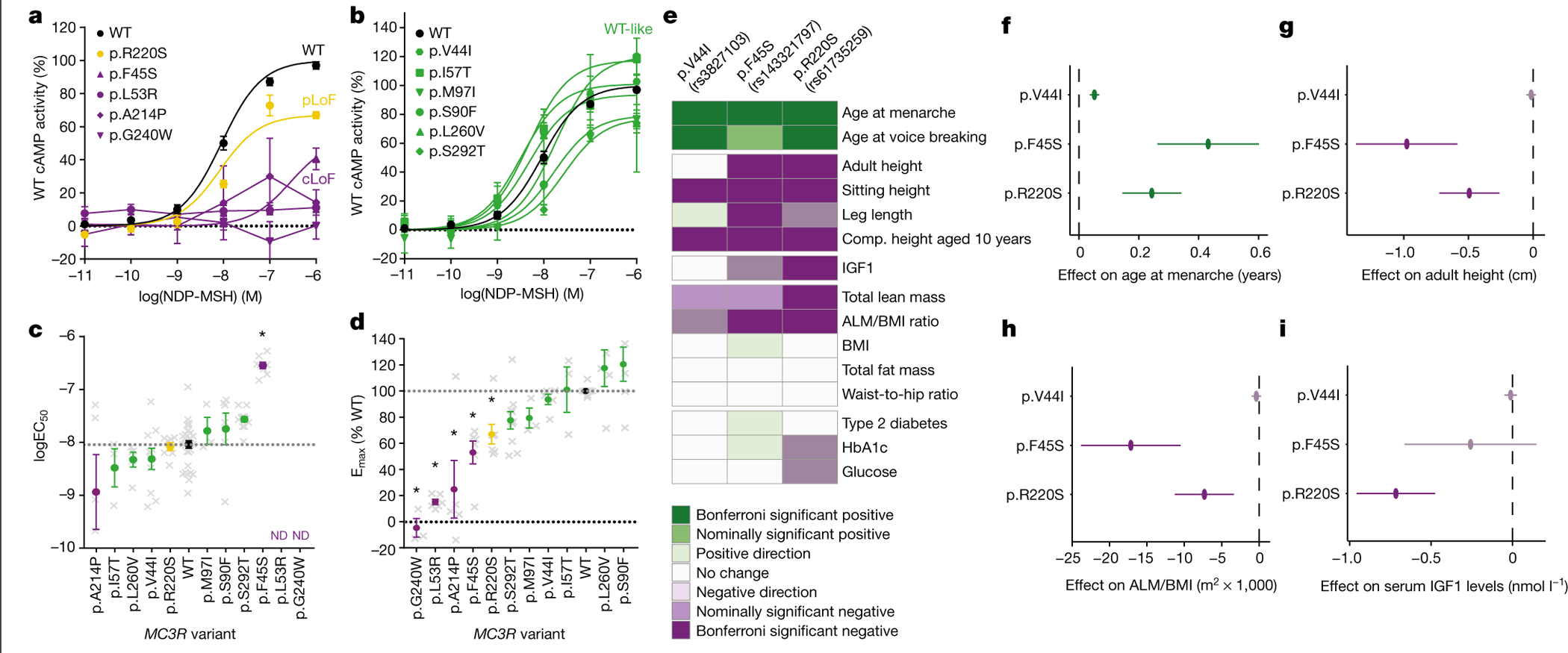
a, b, Dose-dependent cAMP accumulation activity of MC3R mutants stimulated by NDP-MSH. MC3R mutants are grouped by their functional classification: LoF (a) and WT-like (b). Data are normalized to the percentage of WT (black). Mean ± s.e.m. is shown. Dotted line shows 0% WT cAMP activity; N and P values are listed in Supplementary Table 3. cLoF, complete LoF; pLoF, partial LoF. c, d, logEC50 (c) and Emax (d) values of MC3R mutants. Mean ± s.e.m. is shown. Grey crosses show individual data points; grey dotted line shows WT response; black dotted line shows 0% WT activity. Bonferroni *P < 0.05 using one-way ANOVA; N and P values are listed in Supplementary Table 3. e, Heatmap showing the phenotypic association of MC3R variants in the UKBB. Bonferroni threshold = 0.0025. f–i, The effect of UKBB MC3R variants on the age at menarche (f), adult height (g), the ALM/BMI ratio (h), and plasma levels of IGF1 (in nmol l−1) (i). Beta ± 95% CI is shown. Dashed line indicates an effect size of 0; colour indicates level of significance (key in e); MAF and P values are listed in Supplementary Table 2.
While all three variants were individually and jointly associated with delay of pubertal onset in both women and men (Fig. 1e, Supplementary Tables 2, 4), individuals heterozygous for the rarer p.F45S and p.R220S variants, which result in a more substantial disruption of cAMP signalling, had a greater delay in pubertal onset, with female carriers of the p.F45S mutation having a 5.16-month delay (Fig. 1f). These variants were also associated with reduced growth, as indicated by shorter total and sitting height in adults (Fig. 1e, g, Supplementary Table 2), and shorter relative childhood height at 10 years of age (Fig. 1e, Supplementary Table 2). The much more common p.V44I variant was also significantly associated with age at puberty and height, albeit with a substantially smaller effect size (Fig. 1e–g). Although this variant exhibits no significant difference from WT in the cAMP assay, we hypothesize that the large number of carriers (approximately 50,000) allowed us to discern a phenotypic impact of a reduction in signalling resulting from this mutation that is not discernible in a heterologous overexpression system (approximately 94% of WT; Fig. 1b, d). Alternatively, the effect may be explained by its linkage disequilibrium with the 5′ untranslated region variant (or other non-coding variants) that could affect the expression of MC3R. Individuals who are carriers of these three variants also had lower total body lean mass and a reduced ALM/BMI ratio (Fig. 1e, h, Supplementary Table 2). Some heterogeneity existed between individual variant associations—notably, associations with childhood height were more consistent than with adult height (Fig. 1e, Supplementary Table 2) and p.R220S was also associated with lower circulating levels of IGF1 (Fig. 1e, i, Supplementary Table 2). Of note, no variant showed any association with BMI, waist-to-hip ratio, fat mass, type 2 diabetes, circulating levels of HbA1c or random glucose (Fig. 1e, Supplementary Table 2). Phenome-wide association analyses across publicly available GWAS summary statistics in the UKBB and additional cohorts demonstrated that pubertal onset and height had the strongest associations (Extended Data Fig. 2), with no other traits reaching significance after multiple test correction.
To study the impact of MC3R LoF throughout development, we studied 5,993 unrelated participants from the Avon Longitudinal Study of Parents and Children (ALSPAC)30. Using a pooled amplicon next-generation sequencing approach31, we identified seven rare, non-synonymous variants in MC3R that were predicted to be deleterious in silico by SIFT and Polyphen2 (Extended Data Fig. 1, Supplementary Table 3) and found three variants, p.F45S, p.L53R and p.A214P, which all exhibited complete LoF in generating cAMP (Fig. 1a–d, Supplementary Table 3). We then used Sanger sequencing to identify a total of six heterozygous carriers of any one of the three LoF mutations and performed an aggregated burden test on anthropometric trajectories and pubertal timing. We found that despite the small sample size (N = 6), MC3R LoF mutations were associated with lower height throughout childhood, adolescence and early adulthood, with a trend towards lower lean mass and lower weight (Extended Data Fig. 3, Supplementary Table 5, also see Supplementary Information, Supplementary Tables 14, 15). No effect on pubertal onset was discernible in this small group (Supplementary Table 6).
To explore the effect of MC3R variants on the plasma proteome and metabolome, we used data from the Fenland study32 and the European Prospective Investigation of Cancer (EPIC)-Norfolk33,34, respectively. We identified IGFBP1, a liver-derived protein that is known to be suppressed by growth hormone35, as the most strongly associated target (Supplementary Table 7). The two most strongly associated metabolites with MC3R p.F45S, pipecolate (beta = 1.1, s.e. = 0.33, P = 9.6 × 10−4) and 4-hydroxyphenylpyruvate (beta = 0.96, s.e. = 0.31, P = 0.0025), are metabolites of lysine and tyrosine, respectively, and probably reflect increased proteolysis (Supplementary Table 8). These associations, while potentially illuminating, did not reach stringent, multiple test-corrected thresholds (see Methods).
MC3R LoF homozygous phenotype
In the exome data from participants in the Genes & Health study, in whom 18.8% report parental relatedness36, we found two rare, homozygous non-synonymous mutations, p.M97I and p.G240W (Extended Data Fig. 1), each in one participant. While p.M97I signalled normally, the p.G240W mutant receptor was completely unresponsive (Fig. 1a–d, Supplementary Table 3).
The participant carrying p.G240W was invited for phenotypic assessment under ethically approved recall protocols and gave their informed consent for publication of results. The participant is a man of Bangladeshi origin, in his early 40s whose parents are second cousins. The mutation is in an 8.3-Mb genomic region of homozygosity, consistent with consanguineous inheritance.
The participant reported a history of significantly delayed puberty, starting in his early 20s, after which he subsequently fathered children. He was of markedly short stature, −2.95 s.d. of the mean by WHO reference37. His sitting height ratio was below the normal range for South Asian individuals and had reduced circulating levels of IGF1 (Supplementary Table 9). In contrast to the finding in heterozygote individuals, he has been overweight/obese since early childhood and currently has a BMI of 40.4 kg/m2 (Supplementary Table 9), accompanied by type 2 diabetes and hypertension, both well-controlled. Inspection of his exome sequence for all known monogenic obesity genes did not reveal any pathogenic mutations.
Whole-body dual-energy X-ray absorptiometry scanning (Fig. 2a) revealed a high percentage of body fat at 48.5% (Fig. 2a–c), but a low total lean mass for his level of BMI (Fig. 2d). His ALM/BMI ratio, an index of sarcopenia, was below normal (Fig. 2e, Supplementary Table 9).
Fig. 2 |. Characteristics of an individual who is homozygous for the MC3R p.G240W mutation.
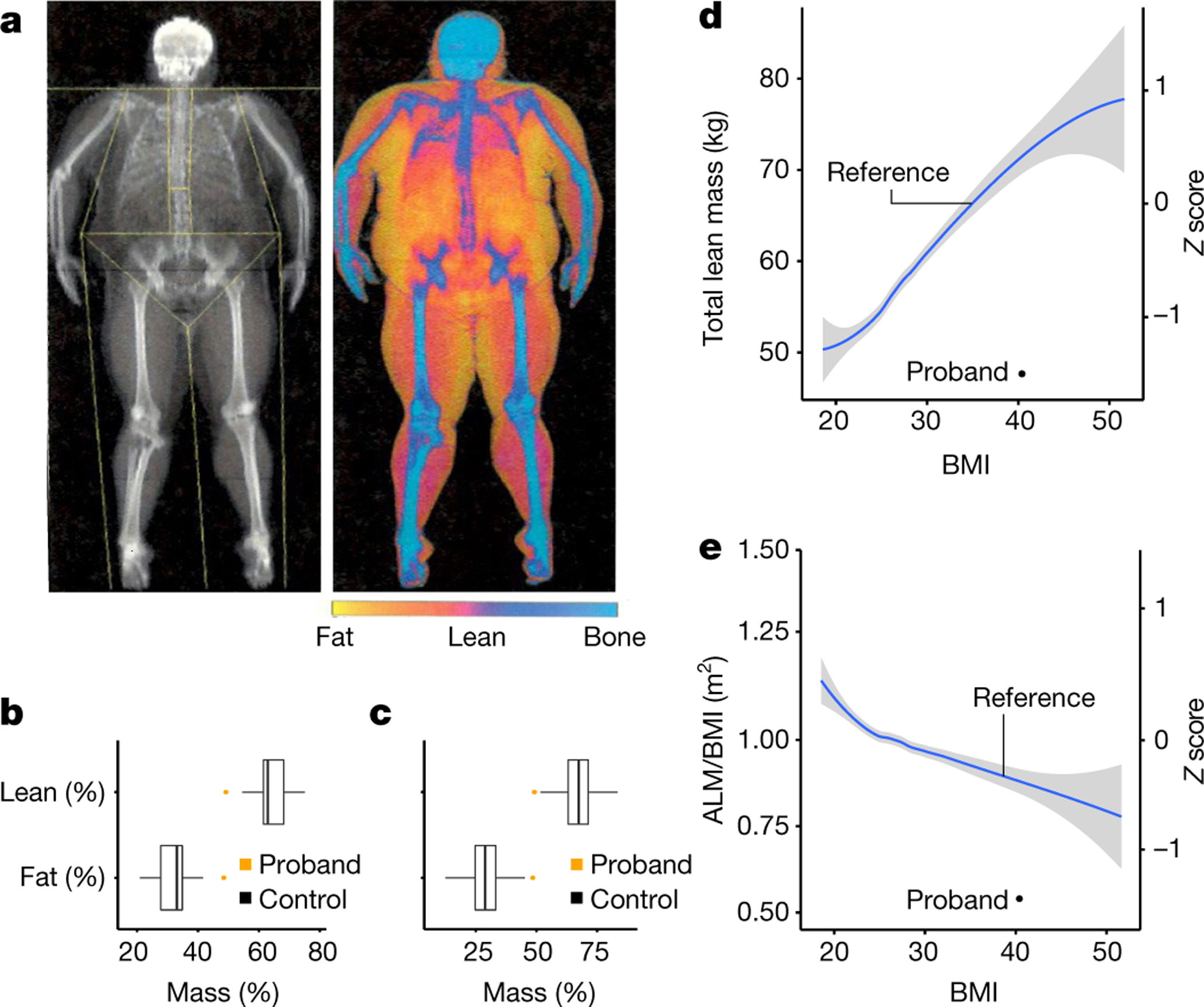
a, Whole-body dual-energy X-ray absorptiometry image of the MC3R p.G240W proband. b, c, Boxplots of the percentage of fat and lean mass in proband (orange) compared with South Asian men (N = 36) (b) and age-matched European men aged 44–54 years (N = 417) (c) in the UKBB. The centre line indicates the median, the box is the interquartile range, and the whiskers indicate 1.5 times the interquartile range. d, e, The total lean mass (black circle) (d) and the ALM/BMI ratio (e) of the proband compared with men from the UKBB with a BMI from 18 to 52 (the mean is indicated in blue and the grey shading indicates the 95% CI; N = 2,356).
Conserved role of MC3R in mice
Male mice lacking Mc3r had a 2-day delay in the onset of sexual maturation compared with WT littermates (Fig. 3a), with female mice showing a similar trend (Fig. 3b). In mature female Mc3r-null mice, the length of the oestrous cycle was significantly prolonged (Fig. 3c, d). To establish whether the known effect of acute caloric deficiency on cycle length required MC3R, WT and MC3R-deficient mice were subjected to an overnight fast. In WT mice, this resulted in a more than twofold prolongation of oestrous cycle length. In the absence of Mc3r, the effect of fasting on cycle length was abolished (Fig. 3c, d, Extended Data Fig. 4a, b).
Fig. 3 |. The role of MC3R in sexual maturation and regulation of the oestrous cycle.
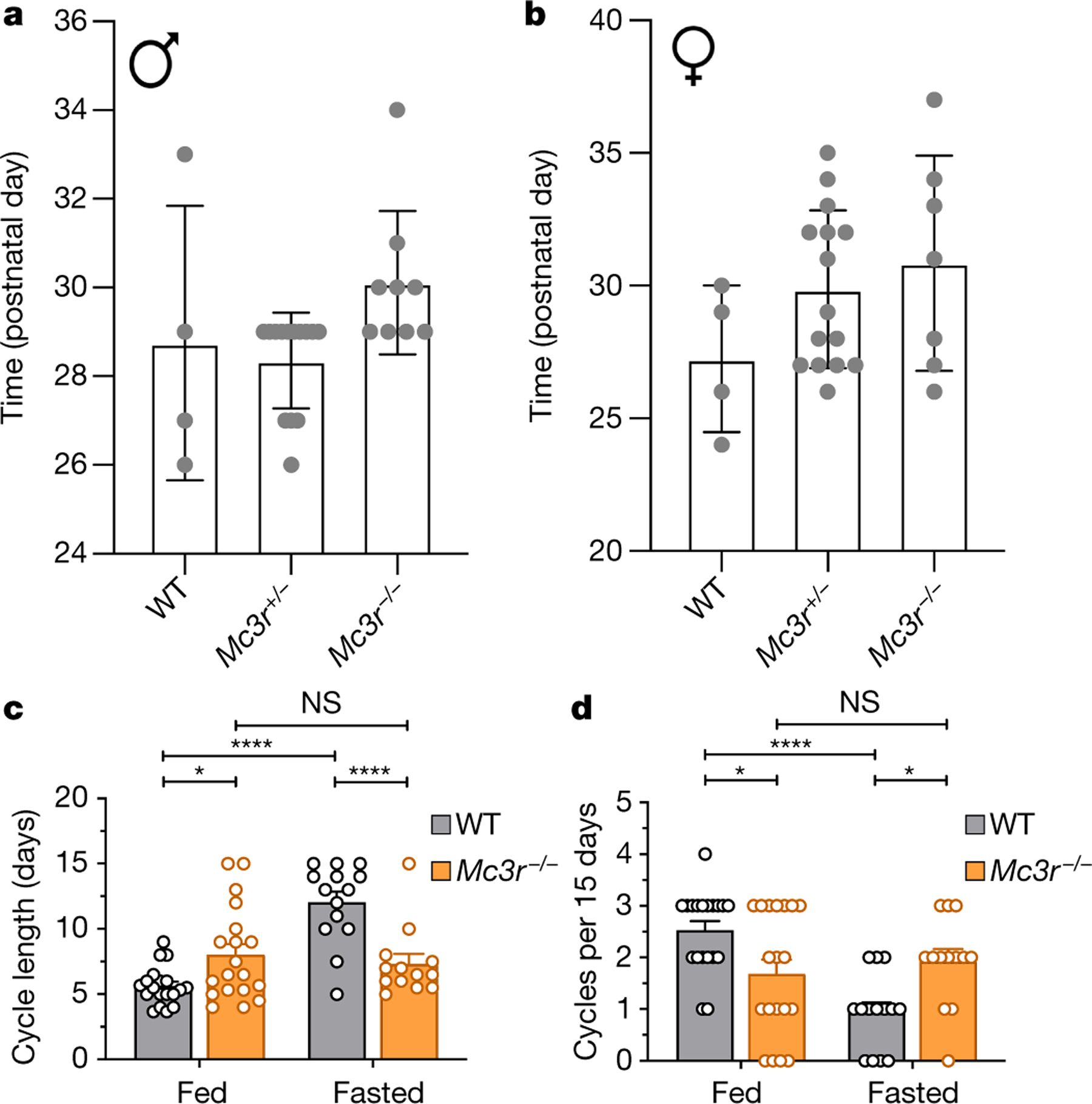
a, b, Day of pubertal maturation as measured by the preputial separation in male mice (a) (N values: 4 (WT), 14 (for Mc3r+/−) and 9 (Mc3r−/−); Kruskal–Wallis test, P = 0.015), and first oestrous cycle in female mice (b) (N values: 4 (WT), 15 (Mc3r+/−) and 7 (Mc3r−/−); Kruskal–Wallis test, P = 0.280). Mean ± s.e.m. is shown. c, Quantification of the length of the oestrous cycle in WT (N values: 19 (fasted) and 38 (fed)) and Mc3r−/− mice (N values: 14 (fasted) and 19 (fed)) in either ad libitum fed and fasted conditions. Mean ± s.e.m. is shown (two-way ANOVA with Bonferroni post-hoc; NS, not significant; *P ≤ 0.05; ****P ≤ 0.0001). d, Number of oestrous cycles per 15 days in WT and Mc3r−/− mice in ad libitum fed and fasted conditions. Mean ± s.e.m. is shown (two-way ANOVA with Bonferroni post-hoc; *P ≤ 0.05; ****P ≤ 0.0001).
Mc3r expression in the hypothalamus
Using a single-cell RNA sequencing dataset of the arcuate nucleus38, a study39 recently reported that Mc3r expression was significantly enriched in neurons expressing kisspeptin, neurokinin B and dynorphin (so-called KNDy neurons) and in growth hormone-releasing hormone (GHRH) neurons. We undertook an expanded analysis including three additional studies40–42 (Extended Data Fig. 5a, Supplementary Table 10, gene markers in Supplementary Table 11), increasing the number of neurons interrogated to 18,427; 1,166 of which expressed Mc3r (Fig. 4a, Extended Data Fig. 5b, gene markers in Supplementary Table 12). This analysis strengthened evidence for co-expression of Mc3r in KNDy neurons (controlling reproduction) and GHRH neurons (controlling growth) (Fig. 4b). Using single-molecule in situ fluorescent hybridization, we validated the co-expression of Mc3r + Tac2 (Fig. 4c, d), Mc3r + Kiss1 (Extended Data Fig. 5c–e) and Mc3r + Ghrh (Fig. 4c, d) in the arcuate nucleus. Leptin regulates the activity of both KNDy43 and GHRH neurons44,45; we therefore assessed the expression of the leptin receptor gene (Lepr), Mc3r and Mc4r in the KNDy and GHRH neurons from the full dataset of 18,427 cells. Both clusters expressed more Mc3r than Lepr and Mc4r (Extended Data Fig. 6a–c). We also established that the expression of MC3R in KNDy and GHRH neurons is conserved in humans by single-molecule in situ fluorescent hybridization (Extended Data Fig. 7a, b). Finally, we studied female mice at postnatal day 16 (P16; infantile), P28 (juvenile) and P48 (sexual maturation), and found that Mc3r mRNA was detected in 40–60% of Kiss1-expressing KNDy neurons in the arcuate nucleus with no change in proportion with age (Fig. 4e). By contrast, in the Kiss1 neurons of the anteroventral periventricular nucleus, which is necessary for the pre-ovulatory gonadotropin-releasing hormone (GnRH) surge46, there was a significant increase in the number of Kiss1 + Mc3r co-expressing cells from P28 to P48 (Fig. 4f, Extended Data Fig. 8a–c).
Fig. 4 |. Mc3r expression in the mouse hypothalamus.
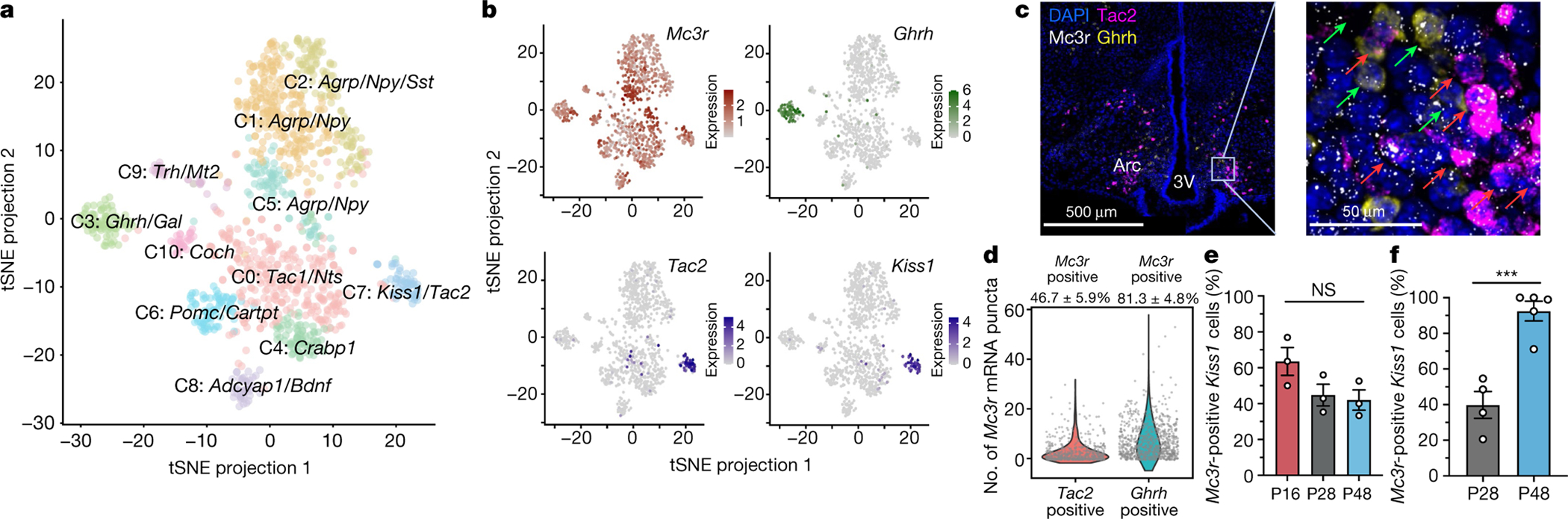
a, Single-cell RNA sequencing of 1,166 Mc3r-expressing neurons reveal 11 distinct clusters (C0–C10). A tSNE plot is shown. b, tSNE plots showing normalized expression of Mc3r (dark red), Ghrh (dark green), Tac2 and Kiss1 (dark blue). c, Single-molecule in situ fluorescent hybridization showing colocalization of Mc3r (white) with Tac2 (magenta) and Ghrh (yellow) in a representative mediobasal hypothalamus (N = 6 mice). Selected Mc3r + Tac2 positive and Mc3r + Ghrh double-positive neurons are indicated by red and green arrows, respectively. Arc, arcuate nucleus; 3V, third ventricle. d, Quantification of Mc3r mRNA expression in Tac2 and Ghrh neurons. Co-expression percentage ± s.e.m. is shown at the top (N = 6 mice). e, The number of Mc3r + Kiss1 co-expressing neurons in the arcuate nucleus at P16, P28 and P48 (one-way ANOVA with Tukey’s post-hoc, Mean ± s.e.m. is shown; NS, P > 0.05; N = 3 mice for all age groups). f, There was an increase in the number of Mc3r + Kiss1 co-expressing neurons with age in the anteroventral periventricular nucleus (two-tailed Student’s unpaired t-test, Mean ± s.e.m. is shown; ***P < 0.001; N = 4 mice for both P28 and P48).
Summary and conclusions
Caloric deprivation is associated with reduced linear growth and delay in the onset of puberty13, whereas over-nourished children tend to grow more rapidly and enter puberty earlier47. Increased macronutrient availability is thought to underpin the progressive increase in height and decrease at age of onset of puberty that has occurred globally over the past century or more48. Leptin and insulin provide signals of nutritional sufficiency to hypothalamic neurons that express melanocortin agonists and antagonists. While these act on MC4R to control food intake and energy expenditure, no such clarity has existed regarding the link between nutritional status and the control of linear growth or the onset of puberty.
The robust association between MC3R LoF mutations and pubertal delay found in our study indicates a role for MC3R in the control of the human reproductive axis. The striking insensitivity of MC3R-deficient mice to the reproductive impact of a period of fasting and the evidence that these mice have delayed sexual maturation indicate conservation of this biology across species. MC3R-deficient mice have been previously reported to be reproductively unimpaired, but those studies did not subject the mice to fasting and may have failed to detect a subtle delay in the timing of sexual maturation18,19. Women who are obese with homozygous mutations that disrupt POMC do not initiate pubertal development49. When treated with setmelanotide, an agonist with ten times selectivity for MC4R over MC3R, they lose weight but remain hypogonadal49.
The effects of MC3R on the reproductive axis may involve direct action on GnRH neurons50. We provide evidence that Mc3r expression is enriched in KNDy neurons in the arcuate nucleus, a site where inhibition of kisspeptin neurons has been shown to impair gonadotropic responses to melanocortins51. Mc3r expression was also high in kisspeptin neurons in the anteroventral periventricular nucleus, which is known to be important for the pre-ovulatory surge of gonadotropins46. In the latter population, Mc3r expression increased with postnatal development in a manner consistent with a role in the timing of sexual maturation.
Consistent with reports of reduced femoral length in mice lacking Mc3r19, we found that humans defective in MC3R signalling have reduced linear growth, correlating with the severity of receptor dysfunction. MC3R status also appears to influence the accrual of lean mass in humans, mirroring previous reports in mice of a low lean-to-fat tissue ratio18,19. The involvement of the growth hormone–IGF1 axis in this phenotype seems likely as, consistent with previous findings in Mc3r-null mice19, the levels of IGF1 were reduced in human mutation carriers. In humans and mice, subpopulations of GHRH neurons express MC3R.
The effect of MC3R deficiency on height is disproportionate, with greater impact on trunk than leg length. We hypothesize that this occurs because a state of relative growth hormone deficiency throughout childhood and adolescence is partially offset by a longer period of limb growth due to the later onset of puberty, which delays epiphyseal fusion, permitting an extended period of long bone growth.
Consistent with what has been described in MC3R-deficient mice18,19, humans with impaired MC3R signalling have shown evidence for reduced lean mass. Growth hormone is known to influence body composition52 and is a candidate for this effect, but we cannot exclude additional MC3R-dependent pathways. In that regard, the association of MC3R dysfunction with raised circulating levels of breakdown products of amino acid metabolism is notable.
Whether mutations in MC3R predispose to human obesity is unclear23. While Mc3r-null mice have a high ratio of fat-to-lean mass, they are not markedly obese, and heterozygous mice have no alterations in their weight or body composition18,19. Consistent with this, heterozygous human carriers of LoF mutations do not have elevated fat mass. By contrast, our homozygous null proband has been obese since early childhood, with no evidence for mutations in known obesity genes. MC3R is expressed in both POMC and AgRP neurons and could influence their function in controlling energy balance39. Resolution of this question will require the identification of additional humans who are homozygous for LoF MC3R mutations.
We have described a new clinical syndrome of MC3R deficiency. Analysis of the MC3R gene should become part of the routine genetic analysis of patients with delayed puberty, short stature and low levels of IGF1. Our data suggest the potential utility of MC3R agonists in some patients with delayed puberty and/or short stature and also potentially in sarcopenia, a condition in which low lean mass, including muscle, contributes to disability in various chronic disorders53.
In summary, across the animal kingdom, nutritional status is a critical determinant of linear growth and the timing of reproductive maturity54. MC3R appears to have an important role in linking signals of caloric sufficiency that act through POMC-expressing neurons to the control of growth and reproduction. This provides a plausible mechanistic basis for the global secular trends towards taller human height and earlier onset of puberty that have accompanied higher levels of caloric availability48.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-021-04088-9.
Methods
In vitro cAMP accumulation assay
HEK293 cells were obtained from laboratory stock and maintained with Dulbecco’s modified eagle medium high glucose (Invitrogen), supplemented with 10% FBS (Invitrogen), 1% Glutamax (Invitrogen), 100 U/ml penicillin and 100 mg/ml streptomycin (Sigma-Aldrich). HEK293 cells were kept in 37 °C humidified air with 5% CO2. The cell line tested negative for mycoplasma contamination, it was not a commonly misidentified cell line and not authenticated.
Site-directed mutagenesis on WT human N-FLAG-MC3R pcDNA3.1(+) was performed using Agilent QuikChange Lightning kit (Santa Clara) to generate all MC3R variants for cAMP activity measurement.
Plasmid (10 ng) carrying WT MC3R and variants were transfected into HEK293 cells using Lipofectamine 3000 (Invitrogen) 48 h before starting the cAMP assay. An increasing dose of NDP-MSH (Bachem) from 10−13 to 10−5 M was administered the following day for 2 h in PBS (Sigma-Aldrich) before measurement of the intracellular cAMP concentration using a luminescence-based HitHunter cAMP Assay for Small Molecules (DiscoverX 90– 0075SM25, Eurofins DiscoverX) and the Tecan Infinite M1000 Pro microplate reader. The cAMP standard curve was measured for each experiment following the standard manufacturer’s protocol and was used to transform luminescence values to cAMP concentrations for downstream analyses.
The baseline and maximal cAMP concentrations were normalized to MC3R WT from the same experiment, and a three-point sigmoidal dose–response curve was fitted to each individual replicate to determine the average relative maximal efficacy (Emax) and the log half-maximal effective concentration (logEC50). The average relative Emax and logEC50 values were used for LoF determination. The logEC50 was not used for mutants that exhibited no response. One-way analysis of variance (ANOVA) was used to compare the Emax and logEC50 of each MC3R mutation to the MC3R WT response. All calculations were performed with GraphPad Prism 7.
The LoF classifications were defined as follows: complete LoF (cLoF): Emax ≤ 25% WT or EC50 ≥ 50× WT; partial LoF (pLoF): 25% WT < Emax ≤ 75% WT or 5× WT ≤ EC50 < 50× WT; WT-like: 125% WT > Emax > 75% WT or 0.2× WT < EC50 < 5× WT
UKBB phenotype association
Cohort information.
The UKBB is a large and prospective study of approximately 500,000 participants aged 40–69 years, recruited between 2006 and 2010 (ref. 55). All analyses conducted using the UKBB Resource were done under application numbers 32974 and 44448.
Phenotype measurements.
We considered candidate anthropometric, puberty timing and metabolic traits. The following specific filters were used: age at menarche was filtered to correspond to the ReproGen consortium definition22 for analyses conducted with genotyping array and imputed data and the full cohort (approximately 100,000 female participants) was used for the whole-exome sequencing (WES) data. Type 2 diabetes was identified on the basis of probable diabetes56 plus any mention of code E11 in hospital episode statistics (HES, main or secondary) or death (underlying or contributory cause); body composition variables (total lean mass and appendicular lean mass) were derived from prediction equations based on demographic, anthropometric and bioelectrical impedance values57. Waist-to-hip ratio was adjusted for BMI and the residuals from this were rank-based inverse normally transformed.
UKBB WES data processing and quality control.
The VCF and PLINK files for the WES data of 200,643 UKBB participants, made available in October 2020, were downloaded and used for the analysis. The data processing and quality control were performed as previously described58. The quality control filters used were: QUAL (variant-site-level quality score); and AQ (variant-site-level allele quality score) between 20 and 99. We also defined a heterozygous genotype call as imbalanced if the allelic balance was 0.25 or less or 0.8 or more and excluded it from the analysis.
UKBB WES variant annotation.
We annotated the MC3R variants using the Ensembl variant effect predictor (VEP)59 tool release 99 based on human genome build GRCh38. The CADD v1.660 VEP plugin was used to provide prediction scores for deleteriousness.
WES gene burden tests.
We selected all rare alleles (MAF of less than 0.2%) in MC3R, which were annotated as ‘high’ or ‘moderate’ impact by VEP, excluding those that were annotated as benign by PolyPhen2. Gene burden scores were created by collapsing variants above to define a binary call denoting whether an individual carries none versus one or more rare, predicted damaging alleles in MC3R. The reported effect estimates represent the trait difference between MC3R mutation carriers and non-carriers. These dummy variables were then transformed into BGEN genotype call format for association testing using BOLT-LMM61. Only common, autosomal variants that passed the quality control and were present on both genotyping array types in the UKBB were included in the genetic relationship matrix (GRM). Genotyping array type, age at baseline and the first ten genetically derived principal components were included as covariates. Samples were excluded from analysis if they failed UKBB quality control, were of non-European ancestry or if the participant withdrew consent from the study.
Selection of variants from the UKBB.
To identify directly genotyped variants covered on the UKBB Axiom array (Affymetrix), we extracted genotype counts in the coding region of MC3R available in UKBB using plink v1.9 (ref.62). Genotyping quality was assessed using plink v1.9 and cluster plots of raw genotype intensity data. Variants that have a MAF of more than 0.05% were taken forward in analysis (Supplementary Table 3). VEP (v99)59 and CADD (v1.6)60 were used to annotate the extracted variants and assess their predicted deleteriousness.
Genotype measurements.
Genotypes included: imputed genotype data were used for two variants—rs3827103 and rs61735259—to maximize sample size (info score of more than 0.96). Directly genotyped data was used for rs143321797 due to its low MAF (0.06%), and genotype cluster plots were manually inspected to ensure genotype reliability63. Furthermore, genotype concordance for non-reference carriers was examined across WES, genotyping array and imputation for rs143321797 and rs61735259 (Supplementary Table 13).
Statistical analyses.
Individual variant associations with outcomes were assessed under additive genetic models. For the individual variants, associations were tested using mixed linear models implemented in BOLT-LMM61, which allow the inclusion of related individuals. Phenome-wide analyses were performed in up to 451,301 individuals. The variant-based models were performed, adjusted for age, sex (where appropriate) and the first ten genetic principal components as provided by the UKBB64, with two outcomes additionally controlling for height where this is stated.
Phenome-wide association study.
A phenome-wide association study was conducted using publicly available GWAS summary statistics from five different repositories: GWAS of 633 ICD10-coded disease phenotypes from the UKBB provided by the Neale laboratory (http://www.nealelab.is/uk-biobank) where data were systematically coded using an algorithm-based approach to determine the most appropriate analysis65, Open Targets Genetics66, Open GWAS IEU67, Global Biobank Engine68 and Phenoscanner69. Summary statistics were extracted for the three coding MC3R variants rs3827103, rs143321797 and rs61735259.
We considered studies with more than 5,000 individuals and excluded binary traits where there were less than 0.1% of cases in the cohort. We manually pruned the list of phenotypes to retain only non-redundant traits by choosing the largest available study covering all variants in cases where a phenotype available was included in multiple datasets.
We used the grs.summary() function from R package gtx (v0.0.8; http://cran.r-project.org/web/packages/gtx), which enables multiple single-nucleotide polymorphism (SNP) genetic risk score analysis using single SNP summary statistics, across 478 traits for which summary statistics for all three variants were available. Weights for each variant’s CADD Phred score (v1.6) were used in the analysis: rs143321797-C = 26.2; rs6173525-A = 23; rs3827103-A = 19. We used a Bonferroni significance threshold to control for multiple testing (P < 1.046 × 10−4).
ALSPAC
Cohort information.
The ALSPAC is a prospective birth cohort from the southwest of England established to study environmental and genetic characteristics that influence health, development and growth of children and their parents30. Full details of the cohort and study design have been described previously and are available at http://www.alspac.bris.ac.uk.
Ethical approval for the study was obtained from the ALSPAC Ethics and Law Committee and the Local Research Ethics Committees. Written informed consent was obtained from mothers at recruitment, from the main carers (usually the mothers) for assessments on the children from ages 7 to 16 years and, from age 16 years onwards, the children gave ritten informed consent at all assessments. Consent for biological samples was collected in accordance with the Human Tissue Act (2004) and informed consent for the use of data collected via questionnaires and clinics was obtained from participants following the recommendations of the ALSPAC Ethics and Law Committee at the time.
Measurements.
Weight and length of each participant were measured at birth and at 4, 8, 12 and 18 months. Weight (to the nearest 50 g) and height (to the nearest millimetre) were measured from 25 months to 24 years. For weight, the participant was encouraged to pass urine and undress to their underclothes. For height, children were positioned with their feet flat and heels together, standing straight so that their heels and shoulders came into contact with the vertical backboard. Equipment used for each measurement were comparable (for example, Fereday 100 kg combined scale, Soenhle scale, Seca scale and Tanita Body Fat Analyser for weight, and Harpenden Neonatometer or Stadiometer, Kiddimetre and Leicester measure for height). Growth trajectories were carried out using linear spline multilevel modelling of weight and height from birth to when participants were 10 years of age. Any missing clinic values were replaced with age-specific predicted values from growth trajectories70.
Fat and lean masses (in kilograms) were measured when participants were a mean age of 10, 12, 14, 15, 18 and 24 years using the Lunar prodigy narrow fan beam densitometer dual-energy X-ray absorptiometry (DEXA) scanner.
Puberty onset was defined by age at menarche in girls and age at peak height velocity (APV) in boys. Age at menarche was assessed from up to nine annual postal questionnaires relating to pubertal development completed by the participants from the age of 8 to 17 years. Each questionnaire asked whether menstrual periods had started and, if so, at what age. Earlier questionnaires were completed by the study mothers on their daughter’s behalf and, from about age 15 years, the questionnaires were completed by the study child. The first-reported age at menarche was used. APV was estimated using superimposition by translation and rotation (SITAR) growth curve analysis, using height measurements taken between ages 5 and 20 years71.
Full details of all measures used in this study are available on the online dictionary: http://www.bristol.ac.uk/alspac/researchers/our-data/.
Pooled high-throughput sequencing of MC3R in ALSPAC.
The pooled sequencing workflow of MC3R was conducted as previously described31. Briefly 20 ng DNA samples representing 5,993 unrelated individuals used in analyses were randomly combined into pools of 50 at the Medical Research Council Biorepository Unit. Pooled DNA (10 ng) was used for MC3R single-exon PCR with Q5 Hot Start High-Fidelity DNA Polymerase (NEB) and MC3R exon primers −331 bp upstream (5′-TGGAACAGCAAAGTTCTCCCT-3′) and +61 bp downstream (5′-CCTCACGTGGATGGAAAGTC-3′) of the protein-coding region, yielding a PCR product of 1,375 bp. The PCR product was purified using Agencourt Ampure XP beads (Beckman Coulter), and quantified with the QuantiFluor dsDNA system (Promega) using the Tecan Infinite M1000 Pro plate reader. Purified PCR product (1 ng) was used to construct the sequencing libraries with the Nextera XT Library Preparation Kit and Nextera XT Index V2 barcodes (Illumina) according to manufacturer’s instruction. Ampure Xp beads were used to purify the libraries, which were then quantified using the Kapa Library Quantification Kit (Roche) and the Quantstudio 7 Flex Real Time PCR instrument (Thermo Fisher Scientific). All libraries were combined at 10 nM for paired-end sequencing at 150 bp (PE150) on the Illumina HiSeq4000 instrument at the CRUK Cambridge Institute Genomics Core. An even coverage was achieved, with a mean per-pool per-base sequencing depth at 45,490 ± 436-fold (s.e.m., data not shown), throughout the protein-coding region of MC3R.
High-throughput sequencing bioinformatics.
BWA MEM (0.7.12)72 was used to align the sequence reads to the human GRCh38 (hg38) genome. PCR de-duplication was performed using Picard 1.127 (https://broadinstitute.github.io/picard/). GATK 3.8 (https://gatk.broadinstitute.org/) was used to perform insertion and deletion (indel) realignment and base quality score recalibration according to GATK Best Practices. The variants were called by mpileup2snp and mpileup2indel function from Varscan 2.4.2 (ref. 73) with the following parameters: variant coverage of 100X or more, ‘Strand Filter’ = ‘ON’, variant allele frequency (VAF) of 0.6% or more and P < 0.05.
ALSPAC MC3R LoF variant selection.
Using high-throughput sequencing, we initially identified 20 non-synonymous variants in MC3R (data not shown). Seven variants were predicted to be deleterious by SIFT, Polyphen-2 and CADD v1.6 (Supplementary Table 3) and were taken forward for functional characterization for their cAMP activity. We identified three complete LoF (cLoF) variants p.F45S, p.L53R and p.A214P. Subsequently, we went back to the original ALSPAC DNA samples and validated six heterozygous carriages: four p.F45S, one p.L53R and one p.A214P via traditional Sanger sequencing described below.
Sanger sequencing for variant validation and carrier identification.
Original DNA samples from participants were validated by Sanger sequencing. The MC3R coding region was first amplified, using GoTaq Green (Promega) Master Mix with 10 ng DNA per 10 μl PCR and MC3R exon primers (as above). MC3R PCR cycling conditions were as follows: one cycle of 95 °C for 5 min; 35 cycles of 95 °C for 30 s, 60 °C for 30 s and 72 °C for 2 min and one cycle of 72 °C for 5 min.
Unincorporated primers and dNTPs were removed using 20 units of exonuclease I (Exo; NEB) and 1 unit of shrimp alkaline phosphatase (SAP; NEB) at 37 °C for 20 min and then 80 °C for 15 min. Of the Exo/SAP product, 1 μl was used in the Sanger sequencing reactions with 0.5 μl of BigDye Terminator v3.1, 2 μl 5x sequencing buffer, 0.5 μM sequencing primer and up to 10 μl using nuclease-free water. The Sanger sequencing cycling conditions were 24 cycles of 95 °C for 10 s, 50 °C for 5 s, and 60 °C for 4 min.
Sanger sequencing reactions were purified using the AxyPrep MAG PCR Clean-Up Kit (Axygen, Corning Inc.) according to the manufacturer’s instructions. Purified sequencing products were resuspended in 30 μl nuclease-free water and analysed on a 3730 DNA Analyzer (Thermo Fisher). The data were analysed on Sequencer 4.8 Build 3767 (Gene Codes Corporation).
Associations between MC3R and anthropometric traits and puberty onset.
Of the 5,993 individuals sequenced, five individuals had missing identifier information for linkage with the wider ALSPAC dataset and 214 individuals were duplicated; therefore, these exclusions left 5,774 participants in the sequenced set. After merging in all of the required clinic and questionnaire data from the ALSPAC cohort and excluding related individuals (details on these exclusions are available74), 5,724 remained in the sequenced set for all analyses, 5,717 of which had complete information on sex, comprising the final sample for analyses. We grouped the six MC3R mutation carriers of three identified MC3R cLoF mutations into the ‘MC3R mutations’ group.
The associations of the MC3R LoF with BMI, height, weight, lean mass, and fat mass at different ages, and age at puberty onset were assessed using linear regression. All analyses and estimates, except for age at puberty onset, were adjusted for age and sex.
The Fenland study
Cohort information.
The Fenland study is a population-based cohort of 12,435 participants born between 1950 and 1975 who underwent detailed phenotyping at the baseline visit between 2005 and 2015, which has previously described in detail75. The study was approved by the Cambridge Local Research Ethics Committee (ref. 04/Q0108/19) and all participants provided written informed consent. In brief, the participants were recruited from general practice surgeries in the Cambridgeshire region in the UK. Individuals were not enrolled in the cohort if they were clinically diagnosed with diabetes mellitus, a terminal illness or psychotic disorder, were unable to walk unaided, or were pregnant or lactating.
Measurements.
Proteomics profiling has previously been described76,77. Proteomics profiling was performed on fasted EDTA samples collected at baseline by SomaLogic Inc. using DNA aptamer-based technology. Relative protein abundances of 4,775 human protein targets were evaluated by 4,979 aptamers (SomaLogic V4).
Statistical analyses.
Fenland participants (N = 10,708) had both phenotypes and genetic data after excluding ancestry outliers and related individuals. Association analyses for variants of interest were performed as previously described77. Briefly, within the three genotyping subsets, aptamer abundances were transformed to follow a normal distribution using rank-based inverse normal transformation, and were then adjusted for age, sex, sample collection site and first ten genetic principal components. The residuals were then used as input for the genetic association analyses using an additive model with BGENIE (v1.3)64. The results for the three genotyping arrays were combined in a fixed-effects meta-analysis in METAL (v 2011–03-25)78.
We first prioritized a total of 14 proteins from the insulin-like growth factor family of proteins targeted by 15 aptamers at a rigorous Bonferroni significance threshold (P < 0.0033). We further considered all proteins targeted by the platform at a lenient multiple testing threshold of P < 1 × 10−4.
The EPIC-Norfolk study
Cohort information.
The EPIC-Norfolk study is a prospective cohort of 25,639 individuals aged between 40 and 79 years and living in the county of Norfolk in the UK at recruitment. The study was approved by the Norfolk Research Ethics Committee (REC 500 ref. 98CN01) and all participants gave their written consent before entering the study79.
Measurements.
Genotyping, imputation and untargeted metabolite profiling of baseline non-fasted serum samples from 9,712 unrelated European individuals in the EPIC-Norfolk cohort was performed using the Discovery HD4 platform (Metabolon, Inc.), as previously described32,34.
Statistical analysis.
Linear regression models adjusted for age, sex, time of blood sample, time of fasting and the first ten genetic principal components were run for each MC3R variant and metabolite pair in R (v3.6.0). A total of 656 metabolites with a known chemical identity were included in the analysis. Statistical significance was considered at a Bonferroni significance threshold of P < 7.6 × 10−5.
Genes & Health
Cohort information.
Genes & Health is an ongoing community-based population study comprising (at 31 August 2021) 48,960 British Bangladeshi individuals and British Pakistani individuals36. Genes & Health operates under approval from the National Research Ethics Committee (London and Southeast), and the Health Research Authority (reference 14/LO/124); Queen Mary University of London is the Sponsor. Genes & Health incorporates stage 1 (health record access and saliva DNA collection) on all volunteers and stage 2 (focused recall studies) procedures on selected volunteers, including recall-by-genotype. Exome sequencing has been performed on all volunteers reporting parental relatedness (N = 5,236) and genotyping (Illumina GSAv3EA+MD chip) on all volunteers. Informed consent is taken at both stage 1 and stage 2, and allows analysis of health and genetic data and publication of results.
Identification of MC3R variants in Genes & Health.
Non-synonymous variants for MC3R were identified from public exome data available on the Genes & Health website (https://genesandhealth.org); summary data were downloaded in September 2019. The exome sequencing of Genes & Health is described in ref. 80.
Genes & Health clinical recall and measurements.
The Genes & Health proband was recruited and recalled to the study under stage 2 procedures described above. Clinical assessment was performed using standard operating protocols designed for metabolic phenotyping in the Genes & Health study, and were performed by qualified medical staff and a bilingual research assistant. All measurements were taken wearing light clothing and with footwear removed, and after voiding urine and a 10-h fast. Height was measured in centimetres (to the nearest 0.5 cm) using a stadiometer, with feet spaced slightly apart with the back of heels and buttocks touching the stadiometer and facing straight ahead. Weight was measured (to the nearest 0.1 kg) using a Tanita TBF-300 scales and body composition analyser. Blood pressure was measured (to the nearest 1 mm/Hg) using a GE Carescape V100 automated blood pressure monitor.
Whole-body DEXA scanning (Hologic, Horizon W, S/N 100091, Auto Whole Body protocol), was performed as part of routine clinical care within the NHS, 1 month after the research clinical assessment. Height (155.0 cm) and weight (96.96 kg) were remeasured at the time of scanning and were consistent with the research assessment (height 155.0 cm and weight 97.8 kg). The DEXA-derived values were used to compute all DEXA-based measurements, including lean and fat mass. We calculated sitting height and sitting height ratio of the skeletal views from the DEXA scan. Anatomical landmarks were used to calculate the sitting height (upper border of the skull to the superior border of the greater trochanter), and the standing height (upper border of the head to the base of the calcaneum, proportioned to clinical height measurement).
Venepuncture was undertaken after a 10 h overnight fast, using a Vacutainer system. Blood plasma was separated from lithium heparin tubes, collected and stored on ice, for insulin, c-peptide, leptin and adiponectin assays. Blood serum was obtained using serum separator tubes for lipid and bone profile, liver and renal function, follicle-stimulating and luteinizing hormone, testosterone, thyroid function tests, sex hormone-binding globulin, cortisol (collected at room temperature), and IGF1 (collected on ice). Adrenocorticotropic hormone was assayed from plasma collected using an EDTA tube on ice. Full blood count and HbA1c were assayed from EDTA whole blood, and plasma glucose from a fluoride oxalate tube. All samples were assayed at the University of Cambridge Core Biochemistry Assay Laboratory.
Genes & Health proband comparison to the UKBB.
The Genes & Health proband was compared to men who had DEXA imaging data available in the UKBB. This cohort was further stratified by self-reported ethnicity (field 21,000) into European men, the majority of the cohort, (N = 2,367; 2,356 with both BMI and DEXA measures) and South Asian men (N = 36) to allow matched assessment of the proband with individuals of the same ethnic background. South Asian ethnicity was defined as individuals who reported to be of Indian, Bangladeshi and Pakistani ethnicity. Total lean and total fat percentage were compared with age-matched men of European ethnicity to account for age effect. These included men within a 10-year span closest to that of the proband, aged 44–54 years at the second study visit when DEXA images were obtained (N = 417).
Individuals with missing data were removed from the comparison. The percentages of lean and fat mass were calculated using DEXA total lean and fat mass variables and total mass as defined by the DEXA measurements. BMI at the second health check was used to allow comparison across different BMI ranges, to match the study visit when DEXA images were obtained. Appendicular lean mass was calculated using the sum of lean mass from legs and arms in kilograms, divided by BMI. Z scores of these measures were calculated to aid cross-trait comparison within these subgroups of interest.
Laboratory animals
The mouse strains used in the reproductive function of MC3R included C57BL/6J (the Jackson laboratory) and Mc3r knockouts (bred in-house at the University of Michigan). Male and female mice were group-housed at 20–24 °C with a 12-h light/12-h dark cycle and provided ad libitum access to food. The experiments were previously approved by the University of Michigan and Vanderbilt University Institutional Animal Care and Use offices (Institutional Animal Care and Use Committee).
Mouse studies performed in Cambridge were in accordance with UK Home Office Legislation regulated under the Animals (Scientific Procedures) Act 1986 Amendment, Regulations 2012, following ethical review by the University of Cambridge Animal Welfare and Ethical Review Body (AWERB). For the adult in situ hybridization experiments, three adult male + three female (Tac2 + Ghrh + Mc3r), and one female + two males (Kiss1 + Tac2 + Mc3r) C57BL/6J mice at 6–8 weeks were housed in individually ventilated cages in controlled temperature (20_24 °C) facilities with a 12-h light/dark cycle (lights on 06:00–18:00) and ad libitum access to food and water in the animal facility at the Anne McLaren Building, University of Cambridge.
Human post-mortem tissue
An anonymized human hypothalamic tissue sample was provided by the Cambridge Brain Bank from a female donor aged 95 at the time of death. The donor gave informed written consent for the use of tissue for research, and samples obtained were used in accordance with the Research Ethics Committee Approval number 10/H0308/56.
Assessment of puberty onset and fertility
Puberty onset in WT, Mc3r+/− and Mc3r−/− was determined by daily examination for preputial separation in male mice. First oestrous in female mice was identified by daily vaginal smears. To visualize first oestrous, the vaginal cells were flushed by introducing 100 μl of sterile saline using a sterile transfer pipette. The saline was slowly released into the vagina and drawn back into the tip; this was repeated four to five times in the same sterile pipette and the cell suspension was then transferred into a 24-well plate. The fluid was then mounted onto a glass slide and the smear was viewed on an inverted compound light microscope.
For the fasting study, animals were randomized and were either fasted or left ad libitum fed overnight before the assessment of their oestrous cycle progression.
The researchers were blinded to the genotype/treatment for the experiments. Power calculation was performed, and the N is shown in the corresponding figure legends.
Single-molecule fluorescent in situ hybridization
For the Mc3r expression in adult mice, animals were euthanized with a lethal administration of sodium pentobarbital (50 mg/kg) intraperitoneally and were perfused with 10% formalin in PBS. The brains were excised after the perfusion and further fixed in 10% formalin for 24 h at 4 °C. The following day, the brains were immersed in 25% sucrose and ProClin 300 (1:2,000; Sigma) in PBS solution and kept at 4 °C. After 24 h, the brains were embedded in optimal cutting temperature (OCT) compound and frozen in Novec 7000 (Sigma) and dry ice, followed by storage at −80 °C until use.
Cryosections (16 μm) containing the hypothalamus were prepared on a Leica CM1950 cryostat (Wetzlar) at −12 °C. For single-molecule fluorescent in situ hybridization, sections were baked at 65 °C for 1 h and fixed in 4% PFA solution at 4 °C for 15 min. Slides were then washed and dehydrated in PBS and ethanol gradients from 50% to 100% for a total of 30 min. Slides were air dried.
For the human single-molecule fluorescent in situ hybridization, a fresh tissue block of human hypothalamus was fixed in 10% neutral-buffered formalin at room temperature for 24 h, transferred to 70% ethanol, and processed into paraffin. Sections (6 μm) were cut and mounted onto Superfrost Plus slides (Thermo Fisher) in an RNase-free environment, and then dried overnight at 37 °C. Sections containing the mediobasal hypothalamus were deparaffinized and rehydrated.
Multiplex single-molecule fluorescent in situ hybridization was performed as previously described81 on a Leica Bond RX automated stainer, using RNAScope Multiplex Fluorescent V2 reagents (Advanced Cell Diagnostics (ACD)). Slides underwent heat-induced epitope retrieval with Epitope Retrieval Solution 2 (Leica) at 95 °C for 5 min. Slides were then incubated in RNAScope Protease III reagent at 42 °C for 15 min, before being treated with RNAScope hydrogen peroxide for 10 min at room temperature to inactivate endogenous peroxidases. Double-Z mRNA probes for mouse Ghrh (Mm-Ghrh-C2), Tac2 (Mm-Tac2-C3), Kiss1 (Mm-Kiss1-C4), Mc3r (Mm-Mc3r), and human MC3R (Hs-MC3R), GHRH (Hs-GHRH-C2) and KISS1 (Hs-KISS1-C3) were designed by ACD for RNAScope on Leica Automated Systems. Slides were incubated in RNAScope 2.5 LS probes for 2 h at room temperature. DNA amplification trees were built through consecutive incubations in AMP1 (preamplifier), AMP2 (background reduction) and AMP3 (amplifier) reagents for 15–30 min each at 42 °C. Slides were washed in LS Rinse buffer between incubations. After amplification, probe channels were detected sequentially via horseradish peroxidase–TSA labelling. To develop the C1–C3 probe signals, samples were incubated in channel-specific horseradish peroxidase reagents for 30 min, TSA fluorophores for 30 min and horseradish peroxidase-blocking reagent for 15 min at 42 °C. The probes in C1, C2 and C3 channels were labelled using Opal 520 (Akoya Biosciences), Opal 570 (Akoya) and Opal 650 (Akoya) fluorophores (diluted 1:500), respectively. Samples were then incubated in DAPI (0.25 μg/ml; Sigma-Aldrich) for 20 min at room temperature to mark cell nuclei. Slides were mounted using approximately 90 μl of Prolong Diamond Antifade (Thermo Fisher) and standard coverslips (24 × 50 mm2; Thermo Fisher). Slides were dried at room temperature for 24 h before storage at 4 °C. Images were acquired using a Perkin Elmer CLS Operetta high-content screening confocal microscope using ×5 and ×40 objectives with Harmony software version 4.9. Randomization and blinding were not relevant as these were observational for in situ studies with no sample groups. No previous power calculation was performed.
For the study of Mc3r expression in the hypothalamic arcuate nucleus and anteroventral periventricular nucleus in female mice from a prepubertal to a postpubertal state, the animals were randomized and brains were harvested at P16, P28 and P48. No previous power calculation was performed; N is shown in the corresponding figure legends. The animals were anaesthetized with tribromoethanol and perfused transcardially with saline followed by fixative (4% paraformaldehyde in borate buffer, pH 9.5). Brains were post-fixed in a solution of 20% sucrose in fixative and cryoprotected in 20% sucrose in 0.2 M potassium PBS (KPBS). Four series of 20-μm-thick frozen sections were collected using a sliding microtome. Sections containing the arcuate nucleus or anteroventral periventricular nucleus were mounted onto SuperFrost Plus slides (Thermo Fisher), and in situ hybridization was performed according to the RNAscope fluorescence multiplex kit user manual for fixed-frozen tissue (ACD) using the RNAscope probes Mm-Mc3r-C and Mm-Kiss1-O1-C3. Images of the arcuate nucleus and anteroventral periventricular nucleus of each animal were obtained using a laser scanning confocal microscope (Zeiss LSM 800). Confocal image stacks were collected through the z axis at a frequency of 0.8 μm using a ×20 objective (NA 0.8). The researcher was blinded to the age of the animals for this experiment.
Imaging analysis
For the adult mouse and human study, data from Harmony (v4.9) were converted into OME TIFF pyramidal format. Individual imaging fields were collapsed along the z axis into maximum projections and subsequently flatfield corrected. Microscope registered coordinates were then used to tile mosaics of all imaging fields in the dataset. OME TIFF files were then read into QuPath v0.2.3 (ref.82) for analysis. Hypothalamic regions were annotated in QuPath, within which StarDist83 was used for nuclear segmentation using the pre-trained ‘dsb2018_heavy_augment’ machine learning model with the default settings. Segmented nuclei were expanded by 2.5 μm to estimate the cell boundary. Cells were classified as Ghrh or Tac2 positive based on median channel intensity within the nuclear region, and the subcellular detection algorithm was use to count the number of Mc3r spots within each cell. The data were exported into .csv format for downstream analysis.
For the developmental study in mice, three-dimensional representations of labelled cells were digitally rendered using Imaris software (version 9.2.0, Bitplane). To determine overall Mc3r mRNA abundance, a region of interest (ROI) was placed around either the arcuate nucleus or anteroventral periventricular nucleus, and the total density of Mc3r labelling was quantified using the spots function. The total numbers of labelled Kiss1 neurons in the arcuate nucleus, Kiss1 in the anteroventral periventricular nucleus, as well as numbers of these neuronal populations that co-express Mc3r, were counted manually in each image stack, aided by Imaris software (Bitplane, v9.3). Only neurons with labelling that was three times that of background were considered positively labelled for Mc3r mRNA. Background for each section was determined by placing ten cell-sized ROIs in user-defined areas, where Mc3r labelling appeared to be lacking, and averaging the number of spots counted in each background ROI.
Single-cell RNA sequencing data analysis
Raw sequence reads from published murine hypothalamic single-cell studies were obtained from Gene Expression Omnibus (GEO accessions GSE93374, GSE87544, GSE92707 and GSE74672; https://www.ncbi.nlm.nih.gov/geo/). Experimental details for the datasets are listed in Supplementary Table 10.
For the drop-seq experiments GSE93374 and GSE87544, the 3′ adaptor of the biological read was first trimmed with Cutadapt 1.16 using ‘AAAAAA’, and the trimmed read was subsequently mapped with RNA STAR 2.7.5b84 to the mouse GRCm38 genome. Read 1, which contained the cell barcode (12 nt) and the unique molecular identifer (UMI; 8 nt), was first split using fastxtrimmer (http://hannonlab.cshl.edu/fastx_toolkit/) and then Fgbio 1.1.0 (http://fulcrumgenomics.github.io/fgbio/) was used to attach information back onto the mapped data generated from read 2. Gene-level UMI count was performed using drop-seq tools 2.3.0 (https://github.com/broadinstitute/Drop-seq/) with a modified gene model from Ensembl V100, where the predicted gene Gm28040 was removed to recover reads for Kiss1. For the smart-seq2 experiments GSE92707 and GSE74672, reads were mapped to the mouse GRCm38 genome and gene-level expression was counted using STAR 2.5.0a with Ensembl V100 gene model.
Gene-level counts from all four datasets were processed separatedly using Seurat v3.285: count data were normalized and scaled using the default options. Variable gene expression was determined using the ‘VST’ selection method and cell clustering was performed using the shared nearest neighbours (SNN) algorithm using the default parameters. Clusters with high Snap25 and Syt1 expression were considered neuronal and were extracted for subsequent integration analysis. Pre-integration, cells with detectable Olig1 in each of the datasets were removed. For GSE93374, we detected contaminating red blood cells and they were removed using the expression of Hba-a1, Hba-a2, Hbb-bs and Hbb-bt. For GSE74672, cells from animals treated with paraformal dehyde were also removed from the downstream analysis.
The integration of the four neuronal datasets was performed using the Seurat v3 (ref.85) standard integration workflow. Briefly, the raw count datasets were renormalized and variable features were determined by ‘mvp’, followed by the use of canonical correlation analysis and mutual nearest neighbours algorithm with ‘ndims = 50’ and ‘k.filter = 150’ to integrate the four datasets into a single 18,427 neuron superset. The integrated data were rescaled, 30 principal components were recalculated via principal component analysis and used for t-distributed stochastic neighbour embedding (tSNE) and SNN clustering analysis with ‘resolution = 1’ to generate the 28 final clusters. Characteristic gene markers for each cluster were determined using the non-parametric Wilcoxon rank-sum test. The marker list is available in Supplementary Table 11.
For the Mc3r subset, the cells were selected by their expression of Mc3r (raw count ≥ 1). Similar to above, the subset was reclustered using 25 principal components and a SNN resolution of 1. Characteristic gene markers for each cluster were determined using the non-parametric Wilcoxon rank-sum test. The marker list is available in Supplementary Table 12.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
All data used in the genetic association analyses are available from the UKBB upon application (https://www.ukbiobank.ac.uk). Data from the Fenland cohort can be requested by bona fide researchers for specified scientific purposes via the study website (https://www.mrc-epid.cam.ac.uk/research/studies/fenland/information-for-researchers/). Data will either be shared through an institutional data sharing agreement or arrangements will be made for analyses to be conducted remotely without the necessity for data transfer. The EPIC-Norfolk data can be requested by bona fide researchers for specified scientific purposes via the study website (https://www.mrc-epid.cam.ac.uk/research/studies/epic-norfolk/). Data will either be shared through an institutional data sharing agreement or arrangements will be made for analyses to be conducted remotely without the need for data transfer. ALSPAC data are available through a system of managed open access. Full details of the cohort and study design have been previously described and are available at http://www.alspac.bris.ac.uk. Please note that the study website contains details of all the data that are available through a fully searchable data dictionary and variable search tool (http://www.bristol.ac.uk/alspac/researchers/our-data/). Data for this project were accessed under the project number B2891. The application steps for ALSPAC data access are as follows: (1) please read the ALSPAC access policy, which describes the process of accessing the data in detail and outlines the costs associated with doing so. (2) You may also find it useful to browse the fully searchable research proposals database, which lists all research projects that have been approved since April 2011. (3) Please submit your research proposal for consideration by the ALSPAC Executive Committee. You will receive a response within 10 working days to advise you whether your proposal has been approved. If you have any questions about accessing data, please email alspac-data@ ext-link bristol.ac.uk. For Genes & Health, data are available via http://www.genesandhealth.org/. Publicly available GWAS datasets utilized in the phenome-wide association study analyses are available from the Neale laboratory (http://www.nealelab.is/uk-biobank), Open Targets Genetics (https://genetics.opentargets.org/), Global Biobank Engine (https://biobankengine.stanford.edu/), Open GWAS IEU (https://gwas.mrcieu.ac.uk/) and Phenoscanner (http://www.phenoscanner.medschl.cam.ac.uk/). Mouse single-cell RNA sequencing data are available from Gene Expression Omnibus (GEO) accessions GSE93374, GSE87544, GSE92707 and GSE74672.
Code availability
Programming scripts were written to assist in the execution of publicly available functions and computer programs in our compute environment. For access to these scripts, readers may contact the corresponding author.
Extended Data
Extended Data Fig. 1 |. Functionally characterised MC3R mutations.
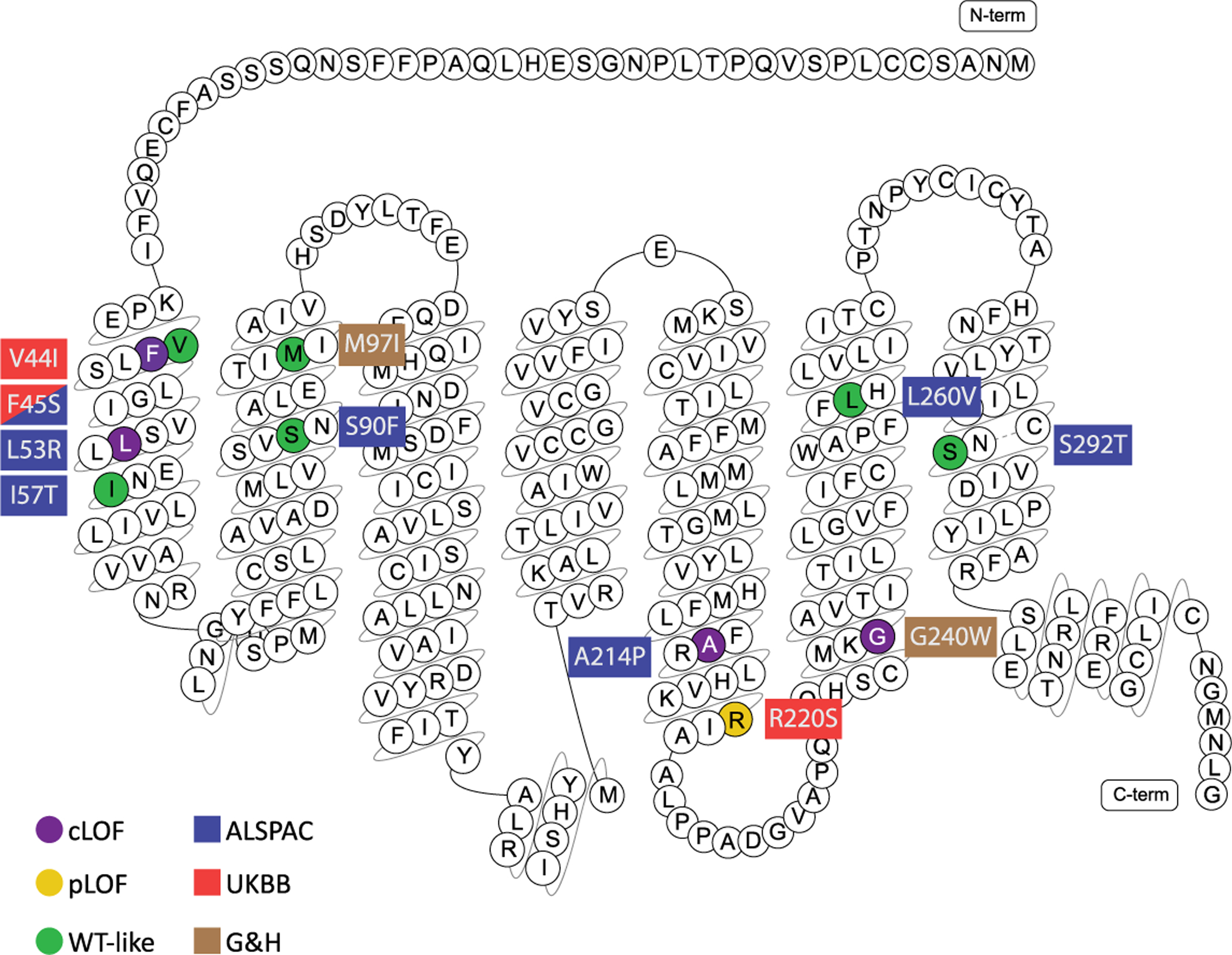
Complete, partial loss-of-function (LoF) and wild-type like mutations are marked in purple, dark yellow and green respectively. Coloured rectangles indicate cohort(s) in which mutations were identified: Red = UK Biobank (UKBB); Blue = Avon Longitudinal Study of Parents & Children (ALSPAC); Light Brown = Genes & Health (G&H).
Extended Data Fig. 2 |. PheWAS analysis of MC3R genetic risk score.
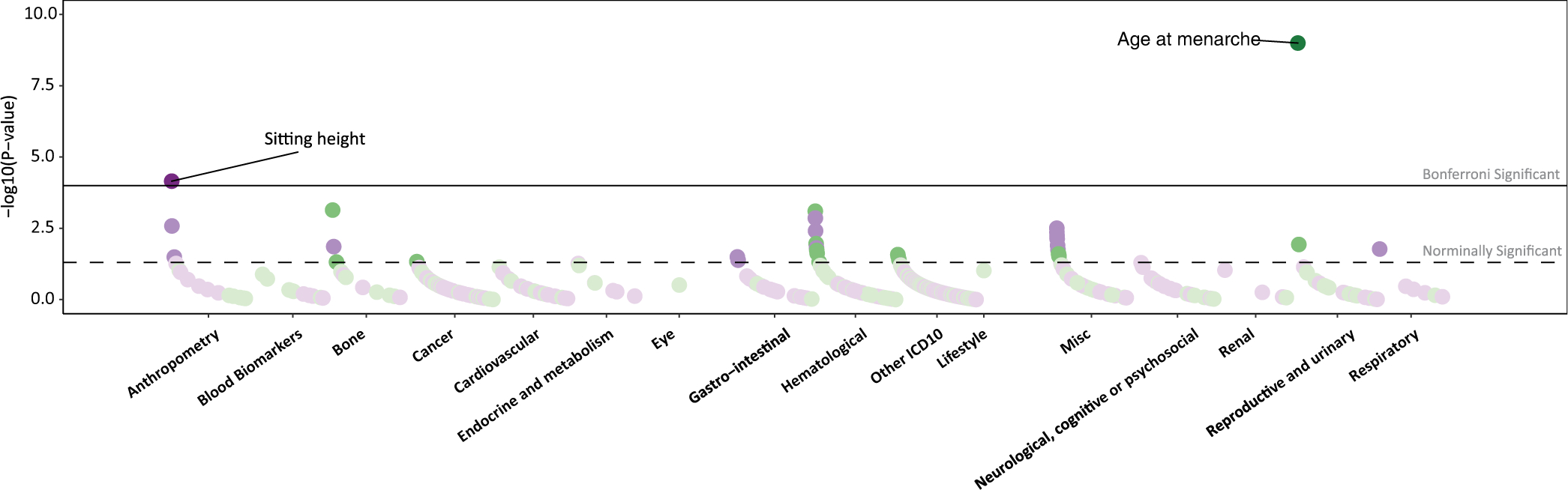
A CADD-weighted MC3R genetic risk score was constructed (see Methods) and used to conduct a phenome-wide analysis (pheWAS) with publicly available summary statistics. Solid black line indicates Bonferroni multiple-testing threshold of p < 1.046e-4, dashed line indicates nominal significance threshold p < 0.05.
Extended Data Fig. 3 |. Effect of MC3R Loss-of-Function mutations on height (cm) across time.
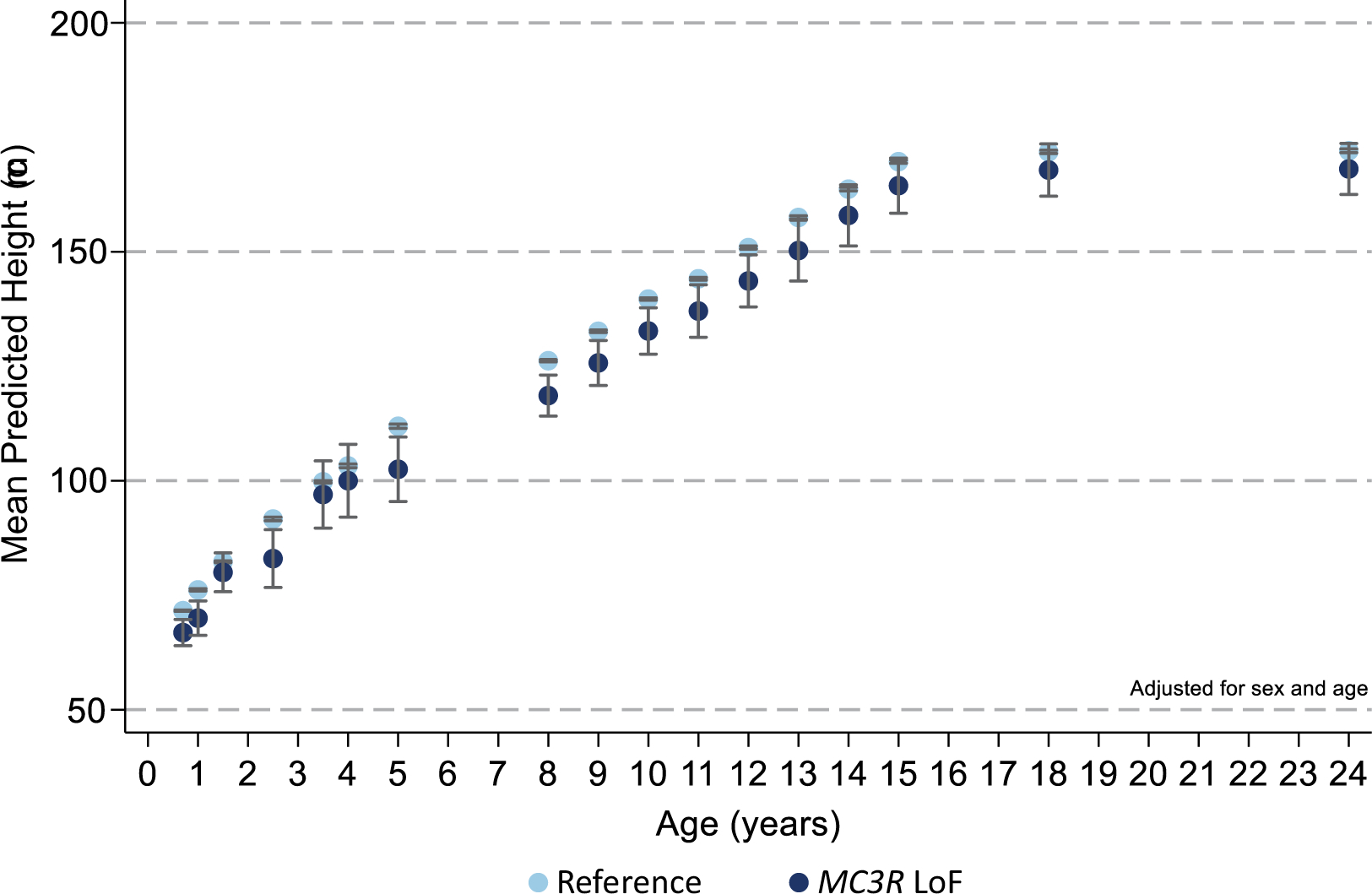
Carriers of MC3R LoF mutations (dark blue) had lower height throughout early life course compared to the reference group (light blue) after adjusting for sex and age. Figures only show results where the mutation group was represented by at least one individual at all time points between birth and 24 years. Mean ± 95% CI shown, N and p-values are listed in Supplementary Table 4.
Extended Data Fig. 4 |. MC3R is essential for normal cycle length and for fasting-induced suppression of the reproductive axis.
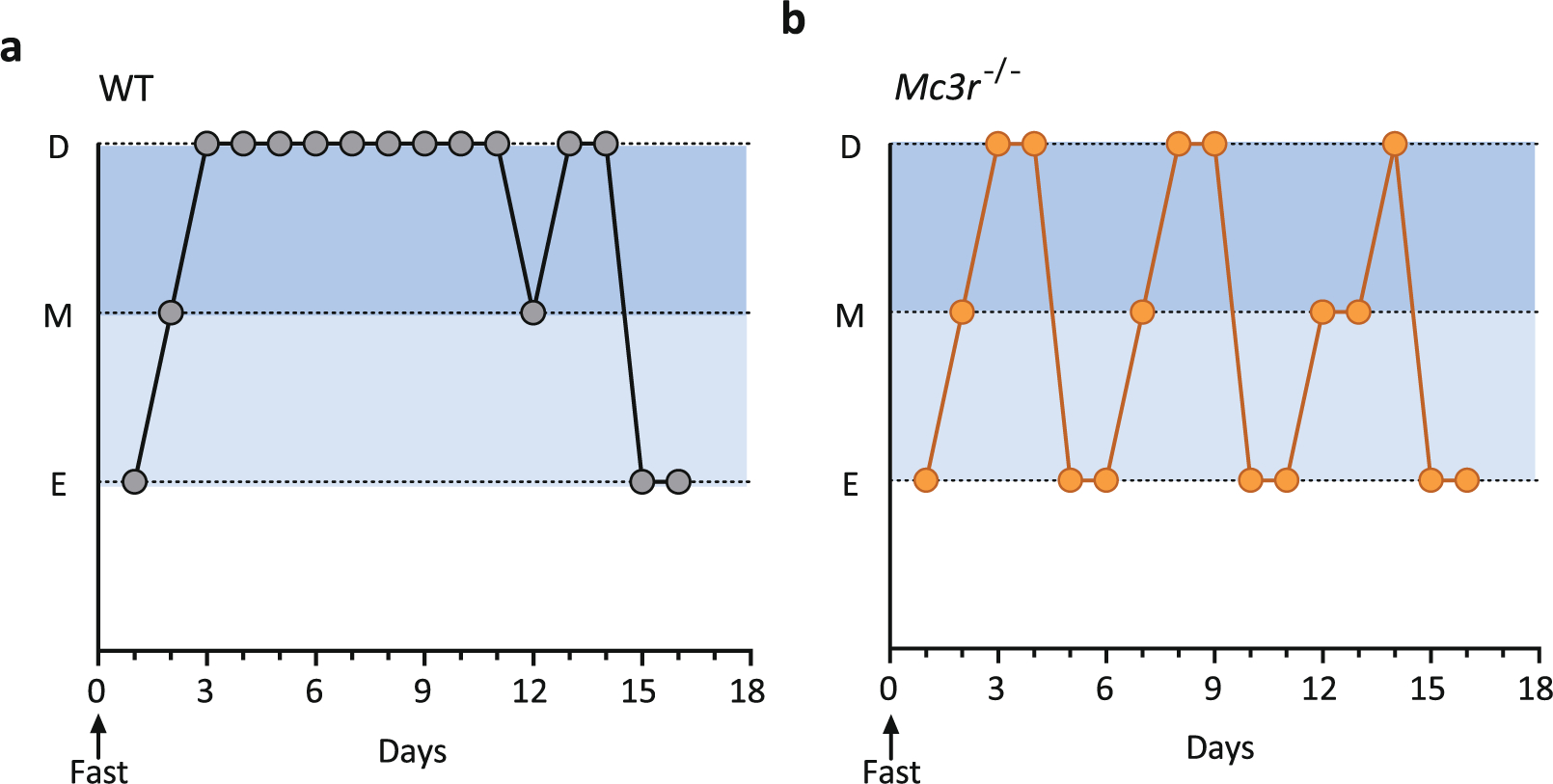
a, b, Representative traces of progression through the oestrous cycle in WT (a) and Mc3r−/− (b) mice following an overnight fast. D = Dioestrous; M = Metoestrous; E = Oestrous.
Extended Data Fig. 5 |. Mc3r is expressed in several cell populations in the mouse hypothalamus.
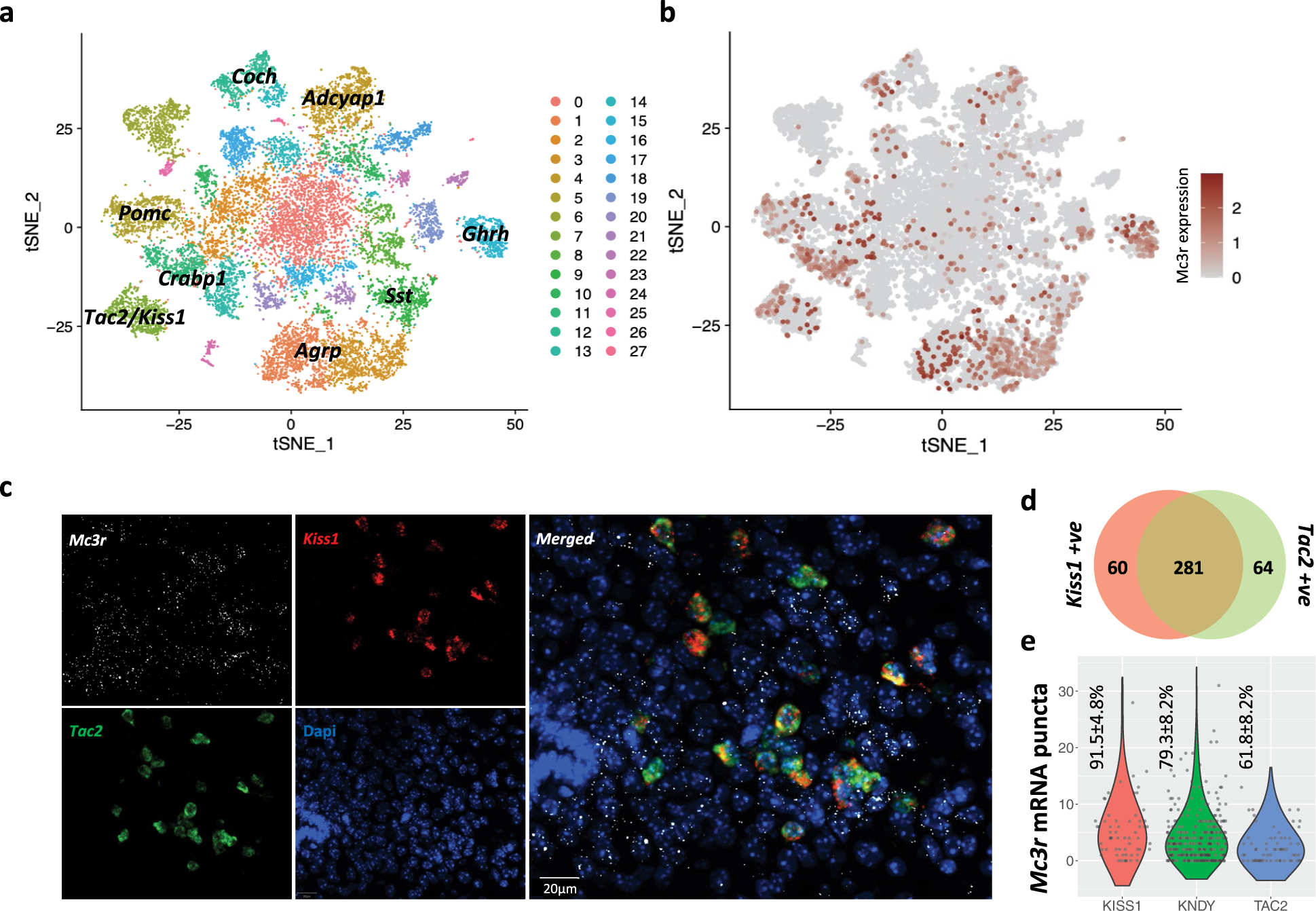
a, T-SNE plot showing 28 neuronal clusters (0–27) of the mouse hypothalamus from a combined dataset consisting of 18,427 neurons from 4 published studies. b, Mc3r is expressed in several neuronal populations (log2 normalised expression in dark red). c, Multiplexed smFISH showing the co-expression of Mc3r (white) Kiss1 (red) and Tac2 (green) in the arcuate nucleus. (Representative example shown, n = 3 mice, scale bar = 20μm). d, Venn diagram showing the number of cells expressing Kiss1 (left, red), Tac2 (right, green), or both (KNDy, centre). e, Violin plots showing the number of Mc3r mRNA punta in Kiss1 only, KNDy, and Tac2 only cells. Mean percentages of cells ± SEM with detected Mc3r are shown, data collected from 3 mice.
Extended Data Fig. 6 |. Expression of Mc3r and Lepr in KNDy and GHRH neurons.
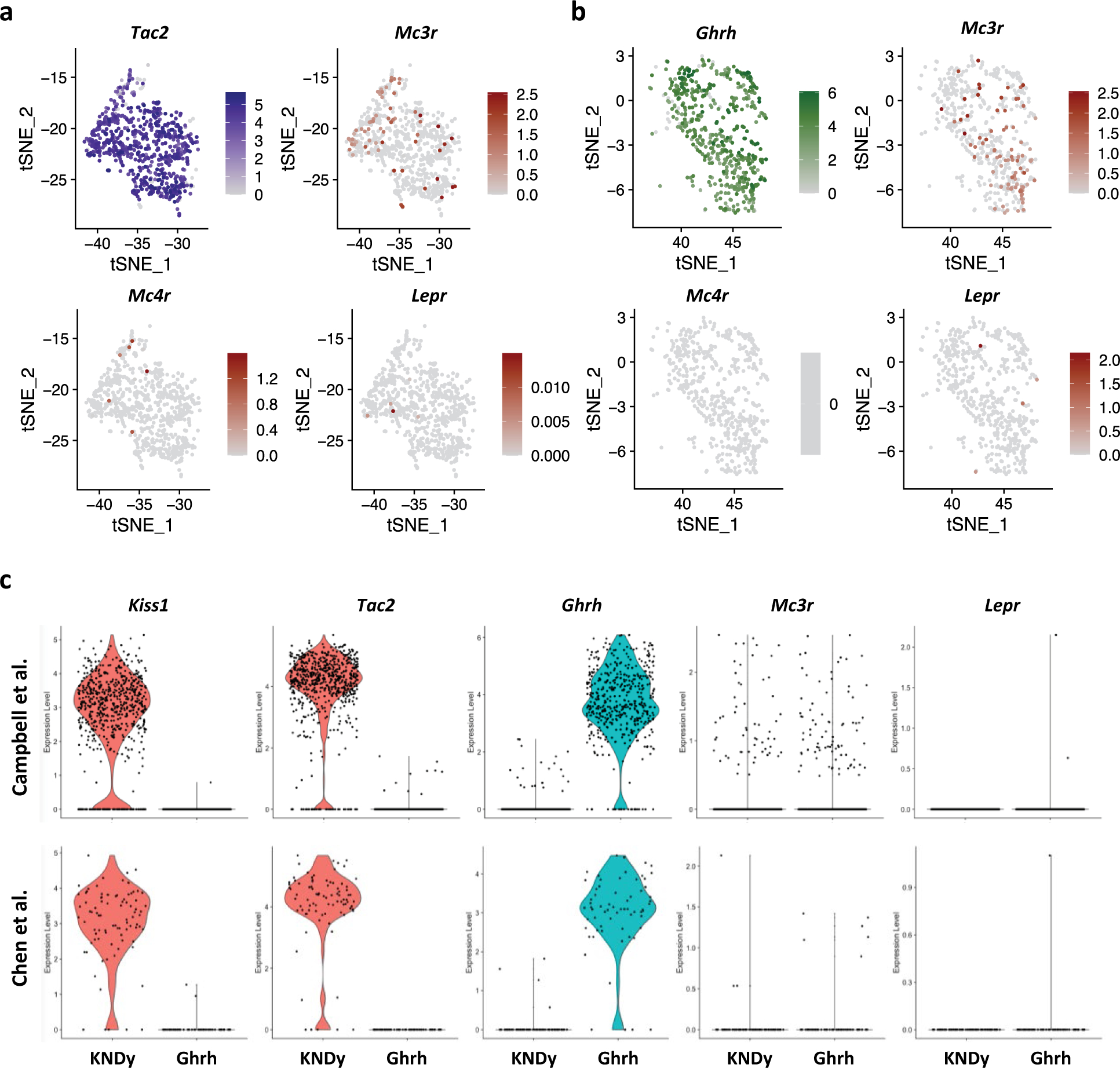
a, b, Mc3r expression is more prominent compared to Mc4r and Lepr in Tac2 (KNDy) (cluster 7, blue) (a) and GHRH neurons (cluster 15, green) (b). c, Violin plots showing expression of Kiss1, Tac2, Ghrh, Mc3r and Lepr in KNDy and Ghrh neurons in the Campbell38 and the Chen42 dataset separately. The Lam40 and Romanov41 datasets are not shown due to low cell count (<10).
Extended Data Fig. 7 |. Human smFISH showing the co-expression of MC3R, KISS1, and GHRH in the human hypothalamic arcuate nucleus.
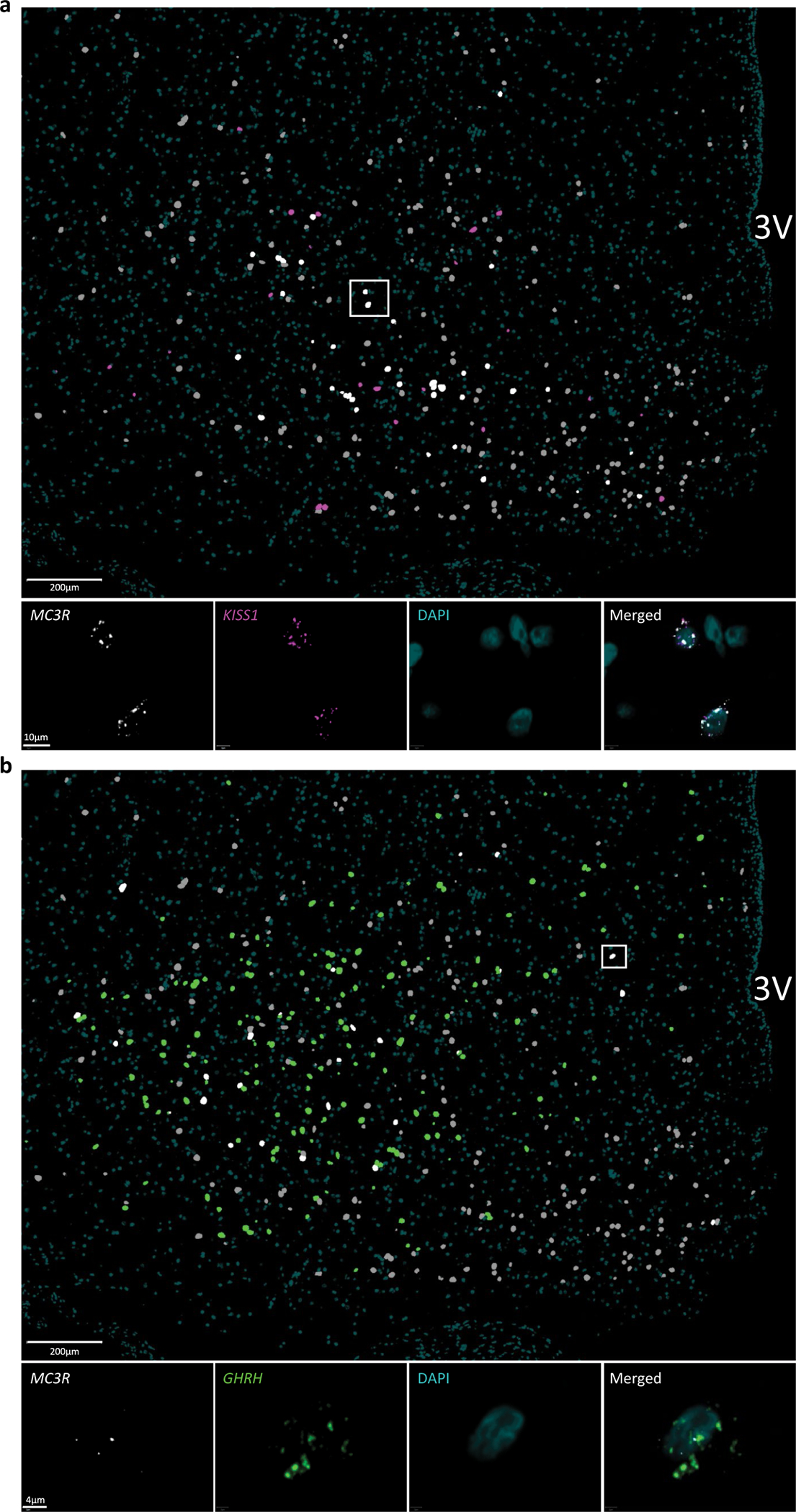
a, Annotated overview MC3R and KISS1 co-expression: MC3R = grey, KISS = magenta and MC3R+KISS1 = white (scale bar = 200μm). High-powered micrograph (squared area) below shows the staining of MC3R (white) and Kiss1 (magenta) mRNA punta in 2 representative cells (teal = DAPI, scale bar = 10μm). N = 2 slides. b, Annotated overview of MC3R and GHRH co-expression: MC3R = grey, GHRH = green and MC3R+KISS1 = white (scale bar = 200μm). High-powered micrograph (squared area) below shows the staining of MC3R and GHRH mRNA punta in a representative cell (teal = DAPI, scale bar = 4μm). N = 2 slides.
Extended Data Fig. 8 |. Mc3r expression in kisspeptin neurons in the mouse hypothalamus at P16, P28 and P48.
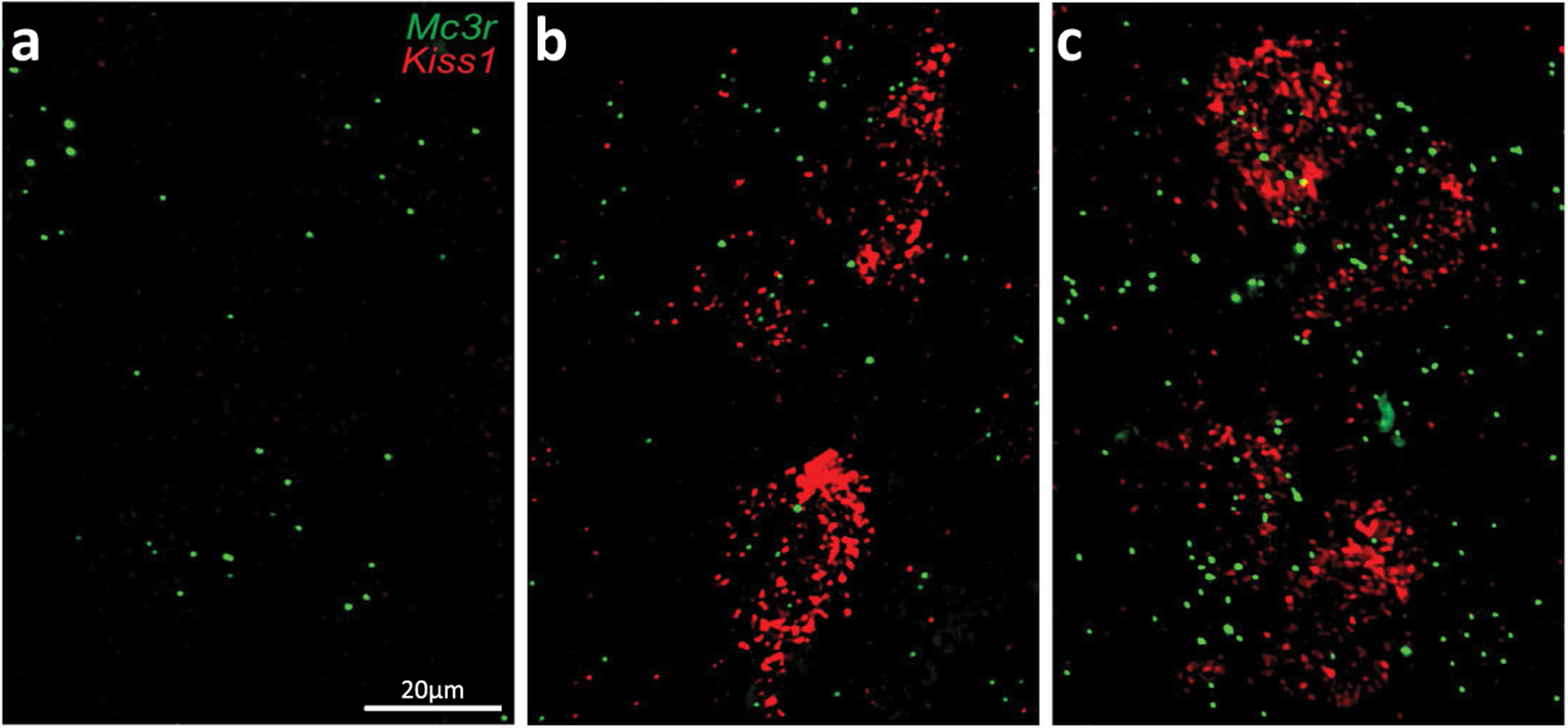
a–c, Representative smFISH showing the co-expression of Mc3r and Kiss1 in the anteroventral periventricular nucleus (AVPV) at P16 (a); P28 (b) and P48 (c) (N = 3 mice for all age groups): Mc3r = green, Kiss1 = red (scale bar = 20μm).
Supplementary Material
Acknowledgements
MC3R genetic analysis, next-generation sequencing and functional analysis were supported by the UK Medical Research Council (MRC) Metabolic Diseases Unit (MC_UU_00014/1), Wellcome (WT 095515/Z/11/Z) and the National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre. K.R., K.D., D.R., I.C., A.P.C. and G.S.Y. are supported by the MRC Metabolic Disease Unit (MC_UU_00014/1). S.O. is supported by a Wellcome Investigator award (WT 095515/Z/11/Z) and the NIHR Cambridge Biomedical Research Centre. B.Y.H.L. is supported by a Biotechnology and Biological Sciences Research Council (BBSRC) Project Grant (BB/S017593/1). A.W. and S.B. hold PhD studentships supported by Wellcome. J.A.T. is supported by an NIHR Clinical Lectureship (CL-2019-14-504). A.M. holds a PhD studentship supported jointly by the University of Cambridge Experimental Medicine Training Initiative programme in partnership with AstraZeneca (EMI-AZ). G.K.C.D. is supported by the BBSRC Doctoral Training Programme. Next-generation sequencing was performed via Wellcome–MRC IMS Genomics and transcriptomics core facility supported by the MRC (MC_UU_00014/5) and the Wellcome (208363/Z/17/Z) and the Cancer Research UK Cambridge Institute Genomics Core. The histology core is supported by the MRC (MC_UU_00014/5). We thank P. Barker and K. Burling of the Cambridge NIHR Biomedical Research Centre Clinical Biochemistry Assay Laboratory for their assistance with biochemical assays. The EPIC-Norfolk study (https://doi.org/10.22025/2019.10.105.00004) has received funding from the MRC (MR/N003284/1 and MC-UU_12015/1) and Cancer Research UK (C864/A14136). The genetics work in the EPIC-Norfolk study was funded by the MRC (MC_PC_13048). Metabolite measurements in the EPIC-Norfolk study were supported by the MRC Cambridge Initiative in Metabolic Science (MR/L00002/1) and the Innovative Medicines Initiative Joint Undertaking under EMIF grant agreement no. 115372. We are grateful to all of the participants who have been part of the project and to the many members of the study teams at the University of Cambridge who have enabled this research. The Fenland study (https://doi.org/10.22025/2017.10.101.00001) is funded by the MRC (MC_UU_12015/1). We are grateful to all of the volunteers and to the general practitioners and practice staff for assistance with recruitment. We thank the Fenland study investigators, Fenland study co-ordination team and the Epidemiology Field, Data and Laboratory teams. We further acknowledge support for genomics and metabolomics from the MRC (MC_PC_13046). Proteomic measurements were supported and governed by a collaboration agreement between the University of Cambridge and Somalogic. F.R.D., N.J.W., K.K.O., C.L. and J.R.B.P. are funded by the MRC (MC_UU_12015/1, MC_UU_12015/2, MC_UU_00006/1 and MC_UU_00006/2). N.J.W. is an NIHR Senior Investigator. We are grateful for funding to the BIA prediction equations, supported by the NIHR Biomedical Research Centre Cambridge (IS-BRC-1215-20014). The NIHR Cambridge Biomedical Research Centre is a partnership between Cambridge University Hospitals NHS Foundation Trust and the University of Cambridge, funded by the NIHR. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care. We thank A. Mesut Erzurumluoglu, L. Wittemans, E. Wheeler, I. Stewart, M. Pietzner, M. Koprulu, E. De Lucia Rolfe, R. Powell and N. Kerrison for providing help with and access to GWAS meta-analysis summary statistics for body composition measures and biomarkers in the UKBB, metabolomics measures in the EPIC-Norfolk study, proteomics measures in the MRC Fenland study, as well as help with genotype quality control in the Fenland study and the UKBB. The MRC, Wellcome (217065/Z/19/Z) and the University of Bristol provide core support for ALSPAC. Genome-wide association data were generated by sample logistics and genotyping facilities at Wellcome Sanger Institute and LabCorp (Laboratory Corporation of America) using support from 23andMe. A.G.S. was supported by the study of ‘Dynamic longitudinal exposome trajectories in cardiovascular and metabolic non-communicable diseases’ (H2020-SC1-2019-Single-Stage-RTD, project ID 874739). K.W. was supported by the Elizabeth Blackwell Institute for Health Research, University of Bristol and the Wellcome Institutional Strategic Support Fund (204813/Z/16/Z). N.T. is a Wellcome Trust Investigator (202802/Z/16/Z), is the principal investigator of the ALSPAC (MRC & WT 217065/Z/19/Z), is supported by the University of Bristol NIHR Biomedical Research Centre (BRC-1215-2001), the MRC Integrative Epidemiology Unit (MC_UU_00011) and works within the Cancer Research UK Integrative Cancer Epidemiology Programme (C18281/A19169). We are extremely grateful to all of the families who took part in the ALSPAC study, the midwives for their help in recruiting them, and the whole ALSPAC team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists and nurses. The Rowitch laboratory receives funding from Wellcome and the ERC Advanced Grant (REP-789054-1). Genes & Health is/has recently been core funded by Wellcome (WT102627 and WT210561), the MRC (M009017), Higher Education Funding Council for England Catalyst, Barts Charity (845/1796), Health Data Research UK (for London substantive site), and research delivery support from the NHS NIHR Clinical Research Network (North Thames). Additional funding for recall was provided by a pump priming award to S.F. (SCA/PP/12/19) from the Diabetes Research and Wellness Foundation. E.G.B. and X.D. are supported by the Wellcome (208987/Z/17/Z) and Barts Charity (project grant to E.G.B.). We thank Social Action for Health, Centre of The Cell, members of our Community Advisory Group, and staff who have recruited and collected data from volunteers; the NIHR National Biosample Centre (UK Biocentre), the Social Genetic and Developmental Psychiatry Centre (King’s College London), Wellcome Sanger Institute, and Broad Institute for sample processing, genotyping, sequencing and variant annotation; Barts Health NHS Trust, NHS Clinical Commissioning Groups (Hackney, Waltham Forest, Tower Hamlets and Newham), East London NHS Foundation Trust, Bradford Teaching Hospitals NHS Foundation Trust, and Public Health England (especially D. Wyllie) for GDPR-compliant data sharing; and most of all, we thank all of the volunteers participating in Genes & Health. R.D.C. receives funding from US National Institutes of Health (NIH) grants DK070332 and DK126715. P.S. is funded by NIH F32HD095620 and K99DK127065. R.B.S. receives funding from the NIH (DK106476). M.N.B. is funded by the NIH (F32DK123879). This research has been conducted using data from UK Biobank, a major biomedical database (https://www.ukbiobank.ac.uk), application numbers 32974 and 44448.
Competing interests S.O. has undertaken remunerated consultancy work for Pfizer, AstraZeneca, GSK and ERX Pharmaceuticals. D.A.v.H. has an unrestricted research grant from Alnylam Pharmaceuticals. P.S. and R.D.C. hold equity in Courage Therapeutics Inc. and are inventors of intellectual property optioned to Courage Therapeutics Inc. R.D.C. chairs the Scientific Advisory Board at Courage Therapeutics Inc. All remaining authors declare no competing interests.
Footnotes
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-021-04088-9.
Peer review information Nature thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Reprints and permissions information is available at http://www.nature.com/reprints.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Friedman JM The function of leptin in nutrition, weight, and physiology. Nutr. Rev. 60, S1–S14; discussion S68–S84, S85–S87 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Cone RD Anatomy and regulation of the central melanocortin system. Nat. Neurosci. 8, 571–578 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Cowley MA et al. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 411, 480–484 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Hill JW et al. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab. 11, 286–297 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Varela L & Horvath TL Leptin and insulin pathways in POMC and AgRP neurons that modulate energy balance and glucose homeostasis. EMBO Rep. 13, 1079–1086 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen AS et al. Role of the melanocortin-4 receptor in metabolic rate and food intake in mice. Transgenic Res. 9, 145–154 (2000). [DOI] [PubMed] [Google Scholar]
- 7.Fan W, Boston BA, Kesterson RA, Hruby VJ & Cone RD Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature 385, 165–168 (1997). [DOI] [PubMed] [Google Scholar]
- 8.Vaisse C, Clement K, Guy-Grand B & Froguel P A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat. Genet. 20, 113–114 (1998). [DOI] [PubMed] [Google Scholar]
- 9.Yeo GS et al. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat. Genet. 20, 111–112 (1998). [DOI] [PubMed] [Google Scholar]
- 10.Huszar D et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 88, 131–141 (1997). [DOI] [PubMed] [Google Scholar]
- 11.Farooqi IS et al. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N. Engl. J. Med. 348, 1085–1095 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Krakoff J et al. Lower metabolic rate in individuals heterozygous for either a frameshift or a functional missense MC4R variant. Diabetes 57, 3267–3272 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown PI & Brasel J in The Malnourished Child Nestlé Nutrition Workshop Series (eds Lewinter-Suskind L & Suskind RM) 213–228 (Nestlé Nutrition Institute and Vevey/Raven Press, 1990). [Google Scholar]
- 14.Clement K et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 392, 398–401 (1998). [DOI] [PubMed] [Google Scholar]
- 15.Strobel A, Issad T, Camoin L, Ozata M & Strosberg AD A leptin missense mutation associated with hypogonadism and morbid obesity. Nat. Genet. 18, 213–215 (1998). [DOI] [PubMed] [Google Scholar]
- 16.Roselli-Rehfuss L et al. Identification of a receptor for gamma melanotropin and other proopiomelanocortin peptides in the hypothalamus and limbic system. Proc. Natl Acad. Sci. USA 90, 8856–8860 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gantz I et al. Molecular cloning of a novel melanocortin receptor. J. Biol. Chem. 268, 8246–8250 (1993). [PubMed] [Google Scholar]
- 18.Butler AA et al. A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology 141, 3518–3521 (2000). [DOI] [PubMed] [Google Scholar]
- 19.Chen AS et al. Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nat. Genet. 26, 97–102 (2000). [DOI] [PubMed] [Google Scholar]
- 20.Renquist BJ et al. Melanocortin-3 receptor regulates the normal fasting response. Proc. Natl Acad. Sci. USA 109, E1489–E1498 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wood AR et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet. 46, 1173–1186 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Day FR et al. Genomic analyses identify hundreds of variants associated with age at menarche and support a role for puberty timing in cancer risk. Nat. Genet. 49, 834–841 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Demidowich AP, Jun JY & Yanovski JA Polymorphisms and mutations in the melanocortin-3 receptor and their relation to human obesity. Biochim. Biophys. Acta Mol. Basis Dis 1863, 2468–2476 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marouli E et al. Rare and low-frequency coding variants alter human adult height. Nature 542, 186–190 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mencarelli M et al. Rare melanocortin-3 receptor mutations with in vitro functional consequences are associated with human obesity. Hum. Mol. Genet. 20, 392–399 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Zegers D et al. Identification of three novel genetic variants in the melanocortin-3 receptor of obese children. Obesity (Silver Spring) 19, 152–159 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Lee YS, Poh LK & Loke KY A novel melanocortin 3 receptor gene (MC3R) mutation associated with severe obesity. J. Clin. Endocrinol. Metab. 87, 1423–1426 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Studenski SA et al. The FNIH Sarcopenia Project: rationale, study description, conference recommendations, and final estimates. J. Gerontol. A Biol. Sci. Med. Sci. 69, 547–558 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim TN et al. Comparisons of three different methods for defining sarcopenia: an aspect of cardiometabolic risk. Sci. Rep. 7, 6491 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boyd A et al. Cohort profile: the ‘children of the 90s’-the index offspring of the Avon Longitudinal Study of Parents and Children. Int. J. Epidemiol. 42, 111–127 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wade KH et al. Loss-of-function mutations in the melanocortin 4 receptor in a UK birth cohort. Nat. Med. 27, 1088–1096 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lotta LA et al. A cross-platform approach identifies genetic regulators of human metabolism and health. Nat. Genet. 53, 54–64 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khaw KT et al. Combined impact of health behaviours and mortality in men and women: the EPIC-Norfolk prospective population study. PLoS Med. 5, e12 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pietzner M et al. Plasma metabolites to profile pathways in noncommunicable disease multimorbidity. Nat. Med. 27, 471–479 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tapanainen J et al. Short and long term effects of growth hormone on circulating levels of insulin-like growth factor-I (IGF-I), IGF-binding protein-1, and insulin: a placebo-controlled study. J. Clin. Endocrinol. Metab. 73, 71–74 (1991). [DOI] [PubMed] [Google Scholar]
- 36.Finer S et al. Cohort profile: East London Genes & Health (ELGH), a community-based population genomics and health study in British Bangladeshi and British Pakistani people. Int. J. Epidemiol. 49, 20–21i (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Onis M et al. Development of a WHO growth reference for school-aged children and adolescents. Bull. World Health Organ. 85, 660–667 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Campbell JN et al. A molecular census of arcuate hypothalamus and median eminence cell types. Nat. Neurosci. 20, 484–496 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sweeney P et al. The melanocortin-3 receptor is a pharmacological target for the regulation of anorexia. Sci. Transl. Med. 13, eabd6434 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lam BYH et al. Heterogeneity of hypothalamic pro-opiomelanocortin-expressing neurons revealed by single-cell RNA sequencing. Mol. Metab. 6, 383–392 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Romanov RA et al. Molecular interrogation of hypothalamic organization reveals distinct dopamine neuronal subtypes. Nat. Neurosci. 20, 176–188 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen R, Wu X, Jiang L & Zhang Y Single-cell RNA-seq reveals hypothalamic cell diversity. Cell Rep. 18, 3227–3241 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Backholer K et al. Kisspeptin cells in the ewe brain respond to leptin and communicate with neuropeptide Y and proopiomelanocortin cells. Endocrinology 151, 2233–2243 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Cocchi D, De Gennaro Colonna V, Bagnasco M, Bonacci D & Muller EE Leptin regulates GH secretion in the rat by acting on GHRH and somatostatinergic functions. J. Endocrinol. 162, 95–99 (1999). [DOI] [PubMed] [Google Scholar]
- 45.Tannenbaum GS, Gurd W & Lapointe M Leptin is a potent stimulator of spontaneous pulsatile growth hormone (GH) secretion and the GH response to GH-releasing hormone. Endocrinology 139, 3871–3875 (1998). [DOI] [PubMed] [Google Scholar]
- 46.Wang L & Moenter SM Differential roles of hypothalamic AVPV and arcuate kisspeptin neurons in estradiol feedback regulation of female reproduction. Neuroendocrinology 110, 172–184 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dunger DB, Ahmed ML & Ong KK Effects of obesity on growth and puberty. Best Pract. Res. Clin. Endocrinol. Metab. 19, 375–390 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Hauspie RC, Vercauteren M & Susanne C Secular changes in growth and maturation: an update. Acta Paediatr. Suppl. 423, 20–27 (1997). [DOI] [PubMed] [Google Scholar]
- 49.Kuhnen P et al. Proopiomelanocortin deficiency treated with a melanocortin-4 receptor agonist. N. Engl. J. Med. 375, 240–246 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Roa J & Herbison AE Direct regulation of GnRH neuron excitability by arcuate nucleus POMC and NPY neuron neuropeptides in female mice. Endocrinology 153, 5587–5599 (2012). [DOI] [PubMed] [Google Scholar]
- 51.Manfredi-Lozano M et al. Defining a novel leptin–melanocortin–kisspeptin pathway involved in the metabolic control of puberty. Mol. Metab 5, 844–857 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salomon F, Cuneo RC, Hesp R & Sonksen PH The effects of treatment with recombinant human growth hormone on body composition and metabolism in adults with growth hormone deficiency. N. Engl. J. Med. 321, 1797–1803 (1989). [DOI] [PubMed] [Google Scholar]
- 53.Doherty TJ Invited review: aging and sarcopenia. J. Appl. Physiol. 95, 1717–1727 (2003). [DOI] [PubMed] [Google Scholar]
- 54.McCance RA & Widdowson EM The determinants of growth and form. Proc. R. Soc. Lond. B Biol. Sci. 185, 1–17 (1974). [DOI] [PubMed] [Google Scholar]
- 55.Sudlow C et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eastwood SV et al. Algorithms for the capture and adjudication of prevalent and incident diabetes in UK Biobank. PLoS ONE 11, e0162388 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Powell RM et al. Development and validation of total and regional body composition prediction equations from anthropometry and single frequency segmental bioelectrical impedance with DEXA. Preprint at medRxiv 10.1101/2020.12.16.20248330 (2020). [DOI] [Google Scholar]
- 58.Zhao Y et al. GIGYF1 loss of function is associated with clonal mosaicism and adverse metabolic health. Nat. Commun. 12, 4178 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McLaren W et al. The Ensembl variant effect predictor. Genome Biol. 17, 122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rentzsch P, Witten D, Cooper GM, Shendure J & Kircher M CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Loh PR et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat. Genet. 47, 284–290 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chang CC et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Van Hout CV et al. Exome sequencing and characterization of 49,960 individuals in the UK Biobank. Nature 586, 749–756 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bycroft C et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Millard LAC, Davies NM, Gaunt TR, Davey Smith G & Tilling K Software application profile: PHESANT: a tool for performing automated phenome scans in UK Biobank. Int. J. Epidemiol. 47, 29–35 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ghoussaini M et al. Open Targets Genetics: systematic identification of trait-associated genes using large-scale genetics and functional genomics. Nucleic Acids Res. 49, D1311–D1320 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Elsworth B et al. The MRC IEU OpenGWAS data infrastructure. Preprint at bioRxiv 10.1101/2020.08.10.244293 (2020). [DOI] [Google Scholar]
- 68.McInnes G et al. Global Biobank Engine: enabling genotype–phenotype browsing for biobank summary statistics. Bioinformatics 35, 2495–2497 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kamat MA et al. PhenoScanner V2: an expanded tool for searching human genotype–phenotype associations. Bioinformatics 35, 4851–4853 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Howe LD et al. Changes in ponderal index and body mass index across childhood and their associations with fat mass and cardiovascular risk factors at age 15. PLoS ONE 5, e15186 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Frysz M, Howe LD, Tobias JH & Paternoster L Using SITAR (superimposition by translation and rotation) to estimate age at peak height velocity in Avon Longitudinal Study of Parents and Children. Wellcome Open Res. 3, 90 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li H Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at arXiv https://export.arxiv.org/abs/1303.3997 (2013). [Google Scholar]
- 73.Koboldt DC et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wade KH et al. Loss-of-function mutations in the melanocortin 4 receptor in a UK birth cohort. Nat. Med. 27, 1088–1096 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lindsay T et al. Descriptive epidemiology of physical activity energy expenditure in UK adults (the Fenland study). Int. J. Behav. Nutr. Phys. Act. 16, 126 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Williams SA et al. Plasma protein patterns as comprehensive indicators of health. Nat. Med. 25, 1851–1857 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pietzner M et al. Genetic architecture of host proteins involved in SARS-CoV-2 infection. Nat. Commun. 11, 6397 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Willer CJ, Li Y & Abecasis GR METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Day N et al. EPIC-Norfolk: study design and characteristics of the cohort. European Prospective Investigation of Cancer. Br. J. Cancer 80 95–103 (1999). [PubMed] [Google Scholar]
- 80.Narasimhan VM et al. Health and population effects of rare gene knockouts in adult humans with related parents. Science 352, 474–477 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bayraktar OA et al. Astrocyte layers in the mammalian cerebral cortex revealed by a single-cell in situ transcriptomic map. Nat. Neurosci. 23, 500–509 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bankhead P et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 7, 16878 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schmidt U, Weigert M, Broaddus C & Myers G Cell Detection with Star-Convex Polygons in MICCAI 2018265–273 (Springer Nature; Switzerland, 2018) [Google Scholar]
- 84.Widmann J et al. RNASTAR: an RNA structural alignment repository that provides insight into the evolution of natural and artificial RNAs. RNA 18, 1319–1327 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stuart T et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data used in the genetic association analyses are available from the UKBB upon application (https://www.ukbiobank.ac.uk). Data from the Fenland cohort can be requested by bona fide researchers for specified scientific purposes via the study website (https://www.mrc-epid.cam.ac.uk/research/studies/fenland/information-for-researchers/). Data will either be shared through an institutional data sharing agreement or arrangements will be made for analyses to be conducted remotely without the necessity for data transfer. The EPIC-Norfolk data can be requested by bona fide researchers for specified scientific purposes via the study website (https://www.mrc-epid.cam.ac.uk/research/studies/epic-norfolk/). Data will either be shared through an institutional data sharing agreement or arrangements will be made for analyses to be conducted remotely without the need for data transfer. ALSPAC data are available through a system of managed open access. Full details of the cohort and study design have been previously described and are available at http://www.alspac.bris.ac.uk. Please note that the study website contains details of all the data that are available through a fully searchable data dictionary and variable search tool (http://www.bristol.ac.uk/alspac/researchers/our-data/). Data for this project were accessed under the project number B2891. The application steps for ALSPAC data access are as follows: (1) please read the ALSPAC access policy, which describes the process of accessing the data in detail and outlines the costs associated with doing so. (2) You may also find it useful to browse the fully searchable research proposals database, which lists all research projects that have been approved since April 2011. (3) Please submit your research proposal for consideration by the ALSPAC Executive Committee. You will receive a response within 10 working days to advise you whether your proposal has been approved. If you have any questions about accessing data, please email alspac-data@ ext-link bristol.ac.uk. For Genes & Health, data are available via http://www.genesandhealth.org/. Publicly available GWAS datasets utilized in the phenome-wide association study analyses are available from the Neale laboratory (http://www.nealelab.is/uk-biobank), Open Targets Genetics (https://genetics.opentargets.org/), Global Biobank Engine (https://biobankengine.stanford.edu/), Open GWAS IEU (https://gwas.mrcieu.ac.uk/) and Phenoscanner (http://www.phenoscanner.medschl.cam.ac.uk/). Mouse single-cell RNA sequencing data are available from Gene Expression Omnibus (GEO) accessions GSE93374, GSE87544, GSE92707 and GSE74672.