

Abstract
The serine/threonine kinase AKT functions as a critical node of the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (m-TOR) signaling pathway. Aberrant activation and overexpression of AKT are strongly correlated with numerous human cancers. To date, only two AKT degraders with no structure–activity relationship (SAR) results have been reported. Through extensive SAR studies on various linkers, E3 ligase ligands, and AKT binding moieties, we identified two novel and potent AKT proteolysis targeting chimera (PROTAC) degraders: von Hippel–Lindau (VHL)-recruiting degrader 13 (MS98) and cereblon (CRBN)-recruiting degrader 25 (MS170). These two compounds selectively induced robust AKT protein degradation, inhibited downstream signaling, and suppressed cancer cell proliferation. Moreover, these two degraders exhibited good plasma exposure levels in mice through intraperitoneal injection. Overall, our comprehensive SAR studies led to the discovery of degraders 13 and 25, which are potentially useful chemical tools to investigate biological and pathogenic functions of AKT in vitro and in vivo.
INTRODUCTION
The serine/threonine kinase AKT, also known as protein kinase B (PKB), is encoded by three closely related genes in humans: AKT1 (PKB-α), AKT2 (PKB-β), and AKT3 (PKB-γ).1–3 AKT is an important component of the PI3K/AKT/ mammalian target of rapamycin (m-TOR) signaling pathway and regulates fundamental cellular and physiological processes, such as cell growth and survival, apoptosis, transcription, migration, and protein synthesis.4–7 PI3K/AKT/m-TOR signaling is one of the most frequently dysregulated pathway in the initiation and propagation of human cancers.8–10 Hyperactivation or overexpression of AKT is associated with a variety of human malignancies, including breast, prostate, lung, colon, brain, pancreatic, and ovarian cancer; gastric carcinoma; and melanoma.9,11,12 Consequently, AKT has been recognized as an oncogenic therapeutic target for several decades.12–14
Several highly potent adenosine triphosphate (ATP)-competitive AKT inhibitors (such as GSK690693,15 GSK2110183,16,17 GSK2141795,16 GDC-0068,18 and AZD536319) and allosteric inhibitors (such as MK-220620,21 and ARQ-09222) are currently being investigated in clinics for oncology indications (Figure 1).3,14 ATP-competitive AKT inhibitors, such as GDC-0068, paradoxically lead to the elevated hyperphosphorylation of AKT at Thr308 and Ser473 that stabilizes AKT active conformation.18,23 Moreover, the clinical efficacy of these ATP-competitive inhibitors is compromised by lacking selectivity over closely related AGC kinase family members.24 Allosteric inhibitors, such as MK-2206 and ARQ092, possess a high degree of selectivity for AKT1/2/3 and inhibit the phosphorylation of two regulatory sites: Thr308 and Ser473. However, these allosteric inhibitors did not achieve sufficient antitumor activities in clinical studies.25,26 Recently, several covalent allosteric AKT inhibitors have been developed by irreversibly targeting AKT at Cys296.27–29 For example, borussertib exerts potent tumor regression in KRAS-mutant patient-derived xenografts when it is combined with the MEK inhibitor trametinib, but further investigation is needed to explore its therapeutic potential. Moreover, kinase-independent functions of AKT can also promote cancer cell survival.7,30 Therefore, an alternative therapeutic strategy is highly desirable for the treatment of AKT-associated human cancers.
Figure 1.
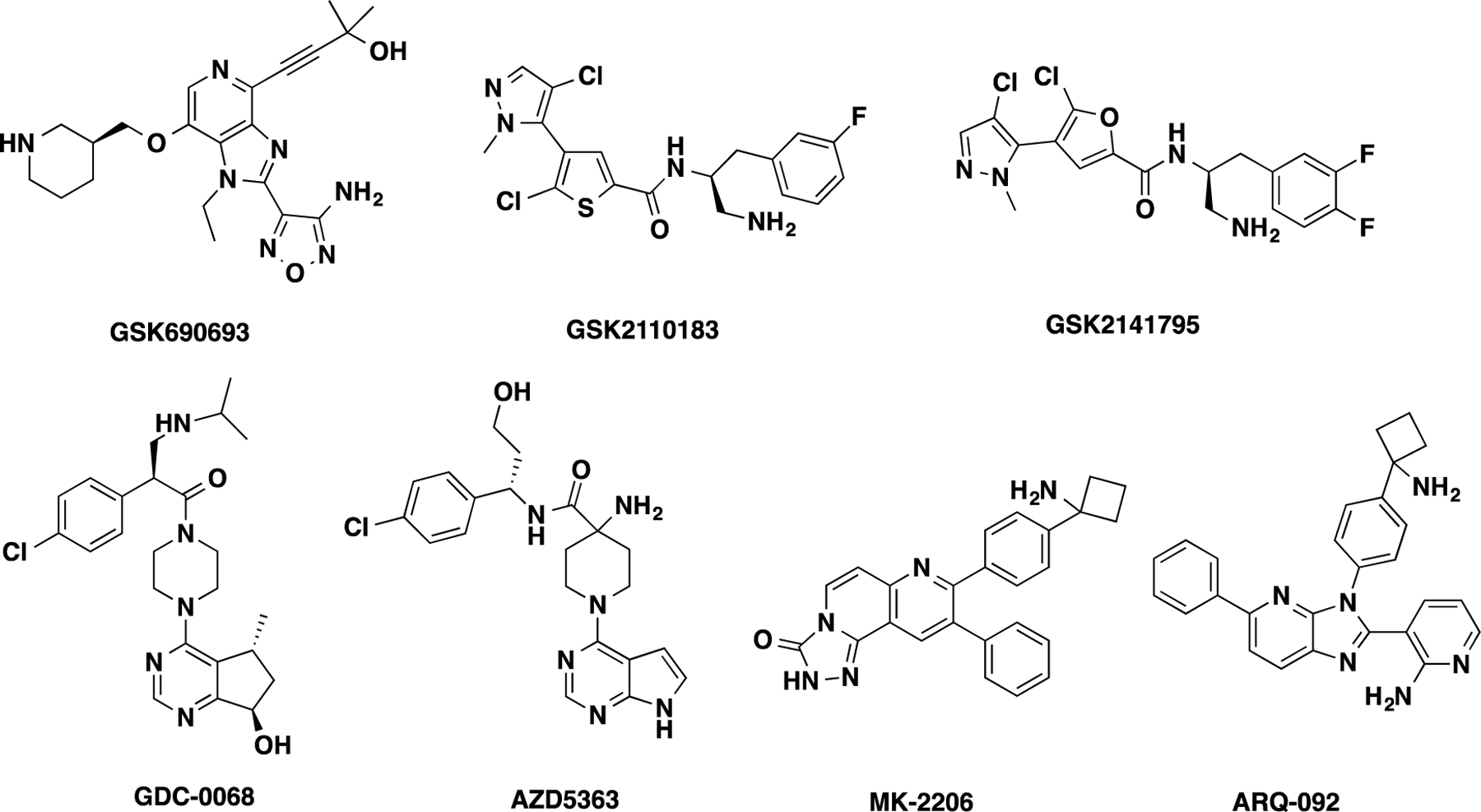
Representative ATP-competitive and allosteric inhibitors of AKT.
Targeted protein degradation using the proteolysis targeting chimera (PROTAC) technology has gained substantial attention from both academic institutions and pharmaceutical industry since 2015. This technology has been utilized to degrade a wide range of oncological and clinically relevant targets.31–35 PROTACs are heterobifunctional small molecules that engage target proteins and E3 ubiquitin ligases, inducing polyubiquitination and subsequent degradation of the target proteins at the proteasome.36–39 PROTACs are able to temporally eliminate both catalytic and noncatalytic (such as scaffolding) functions of the targeted enzymes. Because functional binders (inhibitors or activators) of the targeted protein are not required for developing effective PROTACs, this technology has the potential to target a broad spectrum of previously inaccessible proteome.40–42 Moreover, isoform-selective PROTACs can be achieved using the binders that are not isoform-selective.43–46 Overall, these features highlight a few potential advantages of PROTACs over conventional small-molecule inhibitors.
Two AKT PROTAC degraders have been reported to date.47,48 You et al. reported a potent and selective GDC-0068-based cereblon (CRBN)-recruiting AKT degrader, INY-03-041, which induced sustained AKT degradation with prolonged inhibition of downstream signaling.47 We very recently reported MS21, a potent and selective AZD5363-based von Hippel–Lindau (VHL)-recruiting AKT degrader, which was efficacious in vivo.48 However, no structure–activity relationship (SAR) studies have been reported to date. Herein, we report extensive SAR studies for developing AKT PROTACs by exploring various linkers, E3 ligase ligands, and AKT binding moieties. Through these studies, we discovered multiple series of AKT degraders derived from two AKT inhibitors, GDC-0068 and GSK690693, by recruiting either VHL or CRBN E3 ligase. We further characterized two GDC-0068-based degraders: 13 (MS98, which recruits VHL) and 25 (MS170, which recruits CRBN). Both degraders promoted selective AKT protein degradation, reduced downstream signaling, and inhibited cancer cell proliferation in a concentration- and time-dependent manner. In addition, both degraders are bioavailable in mice and could be used for in vivo efficacy studies. Overall, we present multiple promising AKT degraders, which are potentially useful for investigating pathophysiological functions of AKT.
RESULTS AND DISCUSSION
Design, Synthesis, and Evaluation of GDC-0068-Based VHL-Recruiting AKT Degraders.
GDC-0068 is a highly potent and selective ATP-competitive pan-AKT kinase inhibitor.18 As illustrated in Figure 2A, the cocrystal structure of AKT1 in the complex with GDC-0068 (PDB: 4EKL) revealed that the isopropyl group was solvent-exposed, which offered a suitable exit vector for attaching a linker. In addition, the N–H of the isopropylamino group interacts with Glu 234 and Glu 278 side chains through hydrogen bonds (Figure 2B). On the basis of these observations, we designed two precursors (1 and 2) by replacing the isopropyl moiety with propionic acid and ethylamine groups to serve as bridge groups for linker installation (Figure 2C).
Figure 2.
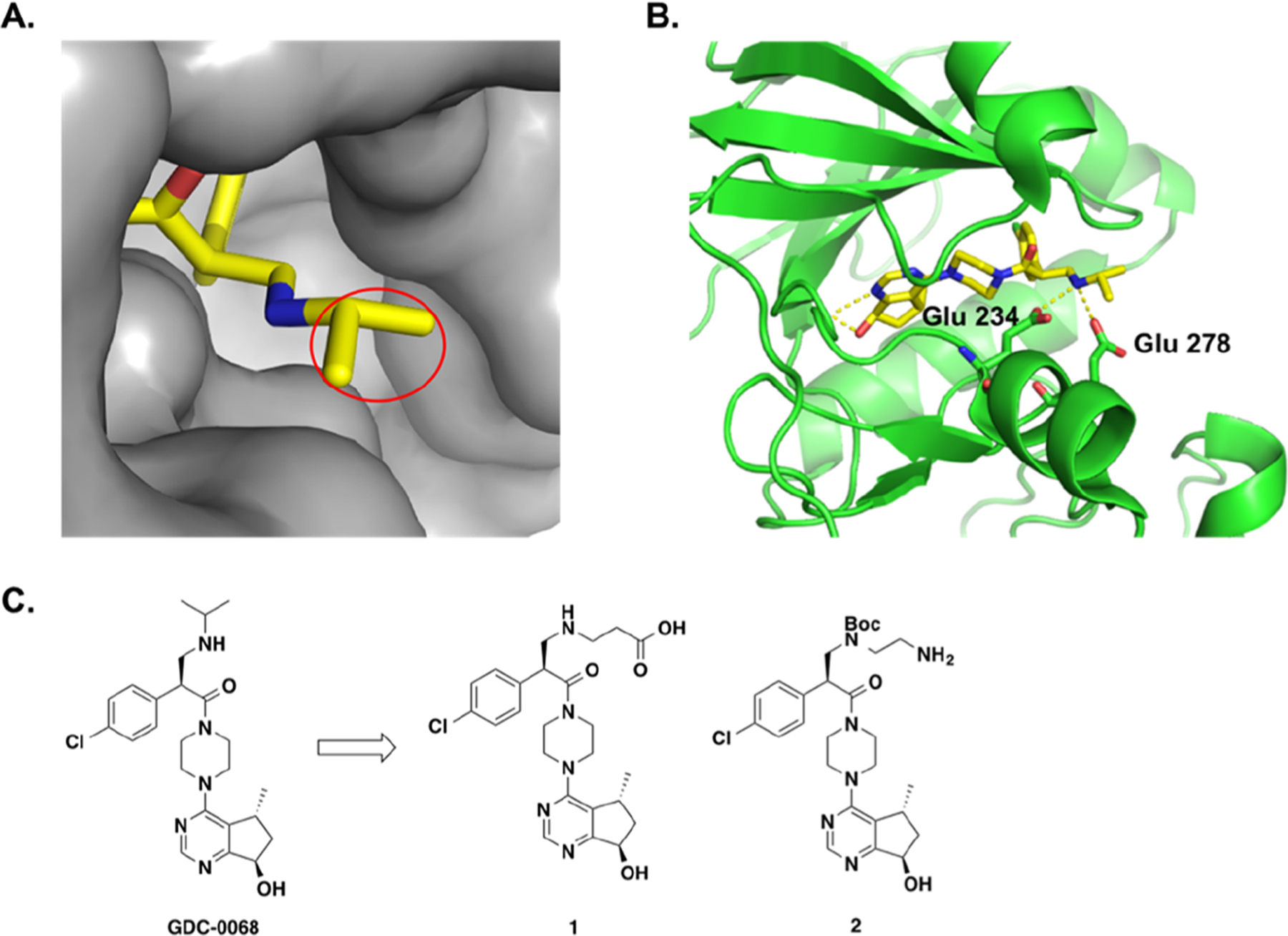
Schematic design of GDC-0068-based AKT putative degraders. (A) The crystal structure of AKT1 in complex with GDC-0068 (PDB: 4EKL) indicates that the isopropyl group is solvent-exposed. (B) Key hydrogen bonding interactions are indicated by yellow dotted lines. (C) Design of precursors for AKT degraders.
It is known that linker types, physicochemical properties, and lengths play critical roles in the successful development of PROTAC degraders.49,50 Therefore, we designed and synthesized a series of putative VHL-recruiting AKT degraders (3–11) using either a polyethylene glycol (PEG) or an alkylene linker with different linker lengths (Figure 3A). Their effects on reducing total AKT (T-AKT) and phosphorylated AKT (P-AKT) protein levels were assessed by western blot (WB) analysis in BT474 cells (a PIK3CAK111N mutant and HER2 positive breast cancer cell line)51 after 24 h treatment with 1, 5, and 10 μM compound concentrations (Figure 4). We also evaluated their effects on inhibiting the phosphorylation of two downstream targets: PRAS40 (P-PRAS40) and S6 (P-S6). In this assay, DMSO, GDC-0068, VHL ligand (VHL-1), and CRBN ligand pomalidomide (POM) were used as controls. As expected, GDC-0068, VHL-1, and POM had no detectable effect on the T-AKT protein level. Consistent with earlier reports,18,23 GDC-0068 treatment effectively inhibited downstream signaling but also resulted in AKT hyperphosphorylation. Compound 3, bearing the shortest PEG linker (one PEG unit), induced significant T-AKT and P-AKT protein degradation at concentrations of 5 and 10 μM. However, its inhibition potency against the downstream signaling, P-PRAS40 and P-S6, was less than GDC-0068. Compounds 4 and 5, bearing longer PEG linkers, were less effective in AKT degradation. Compound 6, with a butylene linker, concentration-dependently reduced the protein levels of T-AKT and P-AKT and exhibited a similar inhibitory effect as GDC-0068 against P-S6. Increasing the alkylene linker length (compounds 7–11) resulted in more potent AKT degradation, although an obvious ″hook effect″ was observed for more potent degraders (8–11) at higher concentrations. For example, compound 11, bearing the longest linker (decylene), exhibited near-complete degradation of T-AKT and P-AKT at 1 μM. However, at 5 and 10 μM, a ″hook effect″ was observed. Moreover, compound 11 also inhibited P-PRAS40 and P-S6. However, it was less effective than the parent inhibitor GDC-0068 in inhibiting P-PRAS40 and P-S6 at the same concentration (1 μM).
Figure 3.
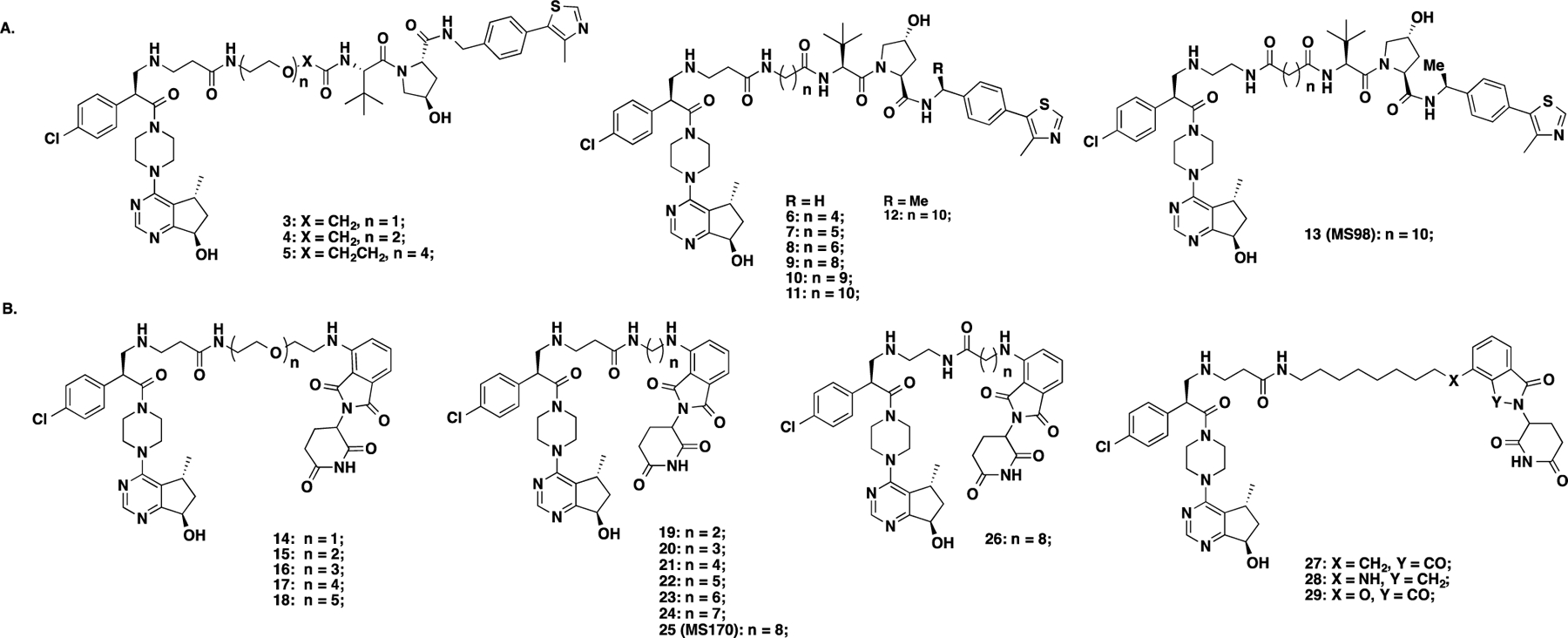
Chemical structures of GDC-0068-based VHL-recruiting (A) and CRBN-recruiting (B) AKT putative degraders.
Figure 4.

Effects of GDC-0068-based VHL-recruiting compounds on degrading AKT and inhibiting downstream signaling. BT474 cells were treated with GDC-0068 (1 μM); VHL-1 (1 μM); pomalidomide (POM, 1 μM); compounds 3–11 at 1, 5, and 10 μM; or compounds 12–13 at 1, 3, and 10 μM for 24 h. The cell lysates were analyzed by western blotting to examine the protein levels of total AKT (T-AKT) and phosphorylated AKT at serine 473 (P-AKT (S473)), and downstream signaling inhibition of P-PRAS40 (T246) and P-S6 (S240/244). β-Actin was used as the loading control.
It has been reported that the VHL ligand (S,R,S)-AHPC-Me (VHL-2), which adds a benzylic methyl group on VHL-1, could increase the binding affinity to the VHL E3 ligase and lead to more effective PROTACs.52–54 Therefore, we replaced VHL-1 in compound 11 with VHL-255,56 and obtained compound 12. In addition, we designed an analog of 12, compound 13 with a reversed amide group (Figure 3A). Both 12 and 13 displayed remarkable effects on degrading T-AKT and P-AKT, and more profound inhibition of P-PRAS40 and P-S6 than 11 (Figure 4). Because 13 exhibited slightly better T-AKT and P-AKT degradation effects and less ″hook effect″ than 12, we selected compound 13 for further biological characterization.
Design, Synthesis, and Evaluation of GDC-0068-Based CRBN-Recruiting AKT Degraders.
In addition to exploring VHL-recruiting degraders, we also designed CRBN-recruiting degraders 14–25 by conjugating GDC-0068 to pomalidomide through various PEG and alkylene linkers (Figure 3B). Similar to the VHL-recruiting degraders, the effects of these putative CRBN-recruiting degraders on degrading T-AKT and P-AKT and inhibiting downstream signaling were evaluated in BT474 cells (Figure 5). Among the five compounds with PEG linkers, compounds 15–18 (with two to four PEG unit linkers) were effective in reducing T-AKT and P-AKT protein levels at 1 μM, while compound 14 (with one PEG unit linker) was not very effective at all concentrations tested. However, compounds 15 to 18 were less effective than GDC-0068 in inhibiting P-PRAS40 and P-S6 (Figure 5). Among the compounds with alkylene linkers, compounds with ethylene (19), propylene (20), butylene (21), and pentylene (22) linkers were marginally effective in inducing AKT degradation at 1–10 μM. On the other hand, compounds with relatively longer linkers, such as 23 (hexylene), 24, (heptylene) and 25 (octylene), were highly effective in reducing T-AKT and P-AKT protein levels. Among these three compounds, 25 was the most effective AKT degrader without a ″hook effect″ and was also the most effective in inhibiting the downstream signaling, P-PRAS40 and P-S6.
Figure 5.

Effects of GDC-0068-based CRBN-recruiting compounds on degrading AKT and inhibiting downstream signaling. BT474 cells were treated with GDC-0068 (1 μM), VHL-1 (1 μM), POM (1 μM), or indicated compounds at 1, 5, and 10 μM for 24 h. The cell lysates were analyzed by western blotting to examine the protein levels of T-AKT, P-AKT (S473), P-PRAS40, (T246) and P-S6 (S240/244). β-Actin was used as the loading control.
We further designed four degraders, 26–29, which have the same linker length as 25 but different linker types or CRBN binders (Figure 3B). While compound 26, which has a reverse amide linker, did not reduce the P-AKT level, it partially degraded T-AKT (Figure 5). Surprisingly, compound 26 inhibited P-PRAS40 at 1 μM as effectively as GDC-0068. It is unclear what the major contributor to this strong downstream signaling inhibition is. One possibility is that the partial T-AKT degradation may contribute to the observed inhibition of P-PRAS40. This warrants further investigation. In addition, removing one of the two carbonyl oxygen atoms in the phthalimide moiety (28) completely abolished AKT degradation activity. On the contrary, changing the NH-group at the linker attachment site of POM to a methylene group (27) or oxygen atom (29) maintained the excellent AKT degradation and downstream signaling inhibition effects. Overall, compounds 25, 27, and 29 were very effective in inducing T-AKT and P-AKT degradation (>95% degradation at 1 μM) and inhibiting P-PRAS40 and P-S6. To rank these three compounds, we compared their cell growth inhibition effect using a colony formation assay with BT474 cells. From this study, compound 25 exhibited slightly better potency than 27 and much better potency than 29 in inhibiting the colony formation of these cells (Figure S1). We therefore selected compound 25 for further characterization.
Design, Synthesis, and Evaluation of GSK690693-Based AKT Degraders.
To explore other AKT binding moieties, we next designed and synthesized a set of putative AKT degraders (31–60) with a modified AKT binding moiety based on GSK69069315 (Figure 6). Analysis of the cocrystal structure of GSK690693-AKT2 (PDB: 3D0E, Figure S2) and the reported SAR results of the amine-containing side chain moiety revealed that the piperidinyl ring is solvent-exposed.15 Based on synthesis considerations, intermediate 30 was designed as a degrader precursor by replacing the piperidinyl group with a propylamino group, which was coupled with a butyric acid bridge group (Figure S2). This modified AKT binding moiety was then linked to VHL-1 or POM via various linkers (Figure 6).
Figure 6.
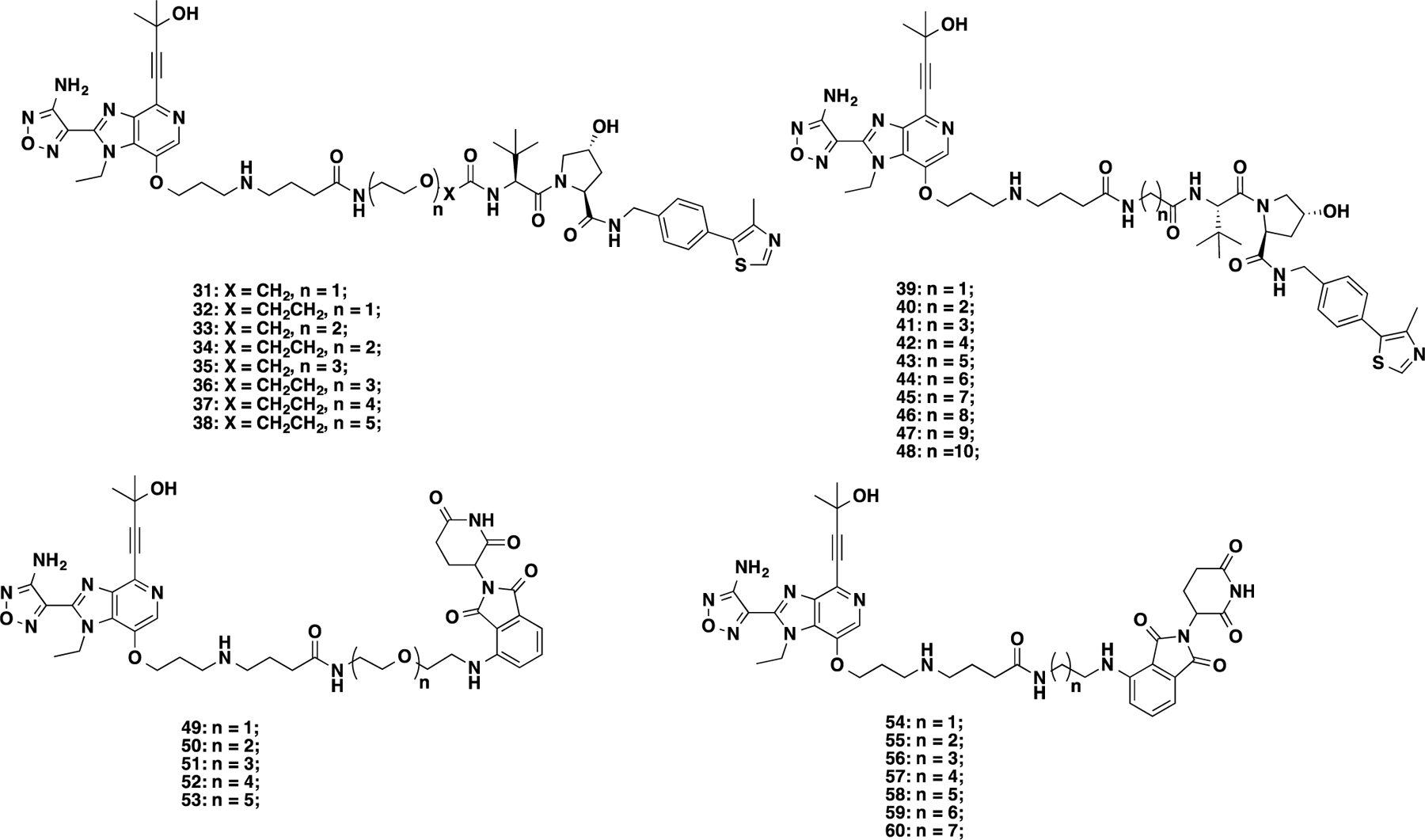
Chemical structures of GSK690693-based AKT degraders.
The effects of putative degraders 31–60 on degrading T-AKT and P-AKT and inhibiting P-PRAS40 and P-S6 were assessed in PC3 cells (a PETN loss prostate cancer cell line)57 treated with 1 μM of the test compound for 24 h (Figure 7). Western blotting analysis showed that compound 48 from the VHL series and compound 60 from the CRBN series were the most effective in degrading T-AKT and P-AKT. Compared to GDC-0068-based degraders with alkylene linkers, a similar SAR trend on the linker length was observed for GSK690693-based degraders with alkylene linkers. That is, compounds 48 and 60 have the longest alkylene linker in VHL- and CRBN-recruiting series, respectively. On the other hand, GSK690693-based VHL-recruiting degraders with a PEG linker, 31–38, were completely ineffective in degrading T-AKT and P-AKT, while CRBN-recruiting degraders with a PEG linker, 49–53, were marginally effective or ineffective. Interestingly, degraders 48 and 60 displayed a similar effect as GSK690693 on suppressing P-S6 but were less effective than GSK690693 in inhibiting P-PRAS40. Mainly due to this, we did not select degraders 48 and 60 for further characterization in this study. Nevertheless, compounds 48 and 60 are interesting AKT degraders, which could be useful for the research community to study AKT functions.
Figure 7.
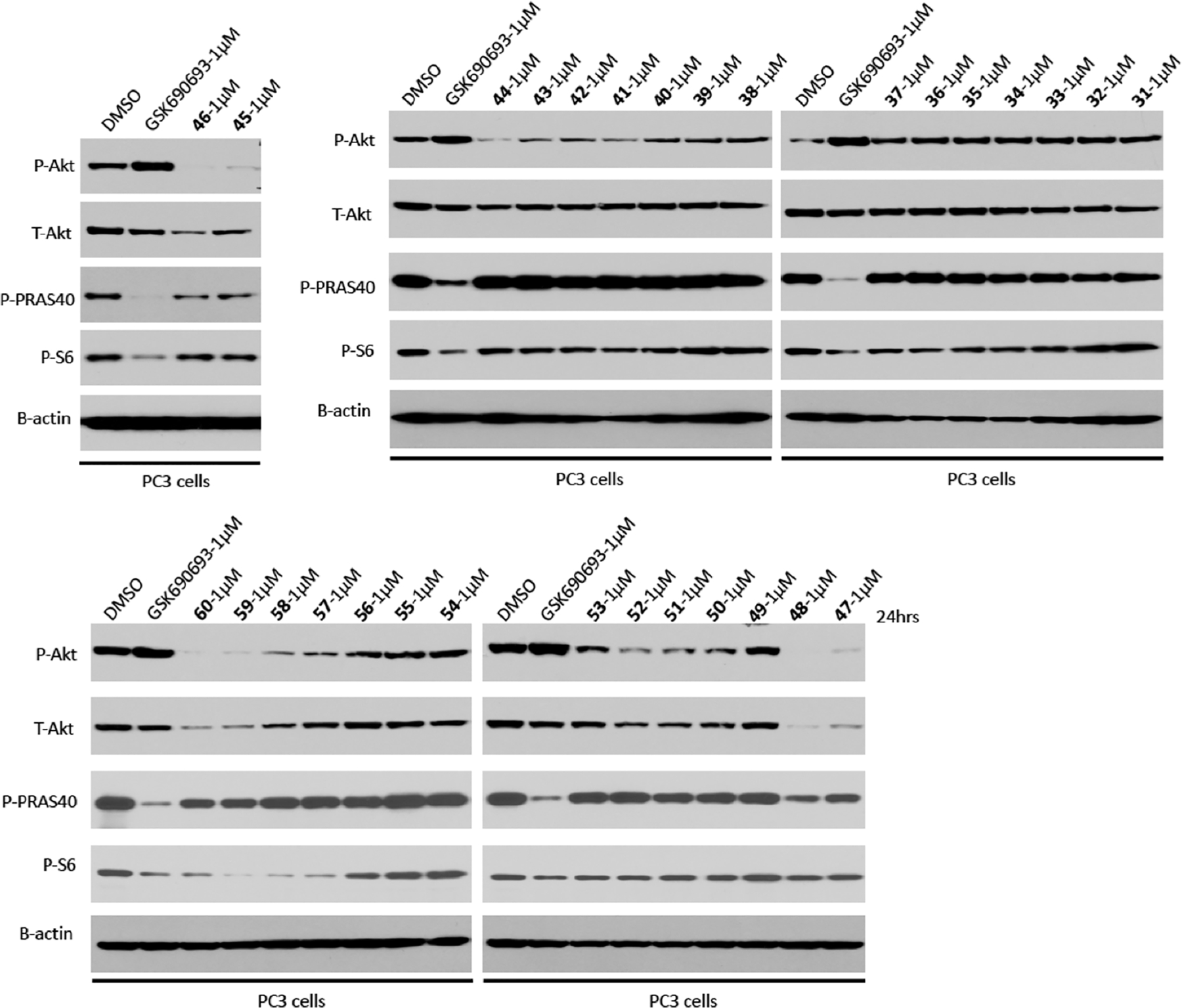
Effects of GSK690693-based AKT degraders on degrading AKT and inhibiting downstream signaling. PC3 cells were treated with GSK690693 or indicated compounds at 1 μM for 24 h. The cell lysates were analyzed by western blotting to examine the protein levels of T-AKT, P-AKT (S473), P-PRAS40 (T246), and P-S6 (S240/244). β-Actin was used as the loading control.
After extensively exploring a variety of linkers, E3 ligase ligands, and AKT binding moieties and identifying multiple highly effective AKT degraders, we next sought to further characterize AKT degraders 13 and 25 in a battery of biochemical and cellular assays. To help elucidate the mechanism of action of AKT degradation and characterize cellular effects, we developed two negative control compounds, 61 (MS98N) and 62 (MS170N), for degraders 13 and 25, respectively (Figure 8). Compound 61 was designed to abolish VHL engagement by incorporating a diastereoisomer of VHL-2.52 Similarly, compound 62 was designed to abolish CRBN engagement by incorporating a methyl group at the glutarimide moiety.58 In addition, we also used the previously reported AKT degrader INY-03–041 as a positive control (Figure 8).
Figure 8.
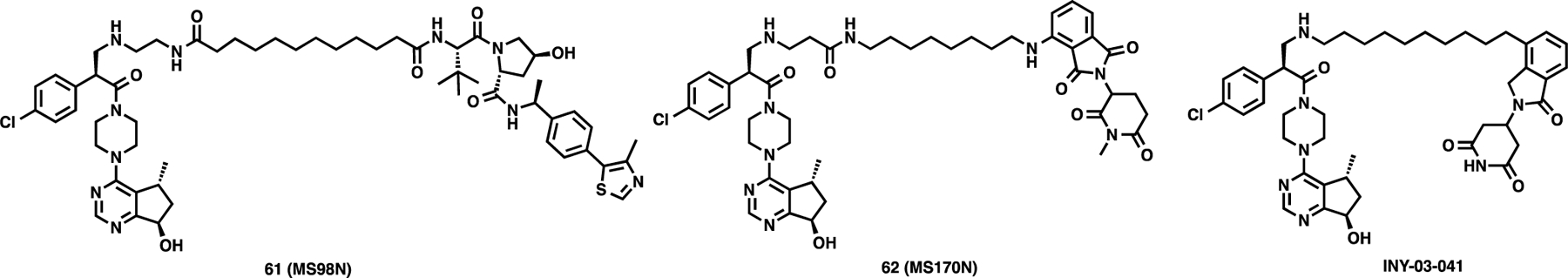
Chemical structures of negative control compounds 61 and 62 and the previously reported AKT degrader INY-03–041.
Binding Affinities of Compounds 13, 25, 61, and 62 to Three AKT Isoforms.
Using a competitive binding assay, we assessed binding affinities of the lead degraders 13 and 25 and their negative controls 61 and 62 to AKT1/2/3 in comparison to the parental inhibitor GDC-0068 (Figure 9). In this assay, inhibitor GDC-0068 displayed very high binding affinities to AKT1 (Kd = 0.64 ± 2.6 nM) and AKT3 (Kd = 2.5 ± 1.4 nM) and lower binding affinity to AKT2 (Kd = 35 ± 2.4 nM). Compared to GDC-0068, degraders 13 and 25 and their corresponding negative control compounds 61 and 62 exhibited 2- to 6-fold lower binding affinities to AKT1 (13: Kd = 4.0 ± 4.6 nM, 25: Kd = 1.3 ± 1.6 nM, 61: Kd = 1.9 ± 2.7 nM, and 62: Kd = 1.8 ± 2.6 nM). As for AKT2, all tested compounds (13: Kd = 140 ± 4.5 nM, 25: Kd = 77 ± 6.9 nM, 61: Kd = 84 ± 3.1 nM, and 62: Kd = 74 ± 4.2 nM) showed 2- to 4-fold decreased binding affinities compared to GDC-0068. Similarly, binding affinities of degraders 13 (Kd = 8.1 ± 2.7 nM), 25 (Kd = 6.5 ± 3.1 nM), and negative controls 61 (Kd = 4.2 ± 2.6 nM) and 62 (Kd = 12 ± 2.4 nM) to AKT3 decreased by 2- to 5-fold compared with GDC-0068. Overall, while these degraders and negative controls displayed 2- to 6-fold lower binding affinities to AKT isomers compared with the parent inhibitor, all of these compounds still retained high binding affinities for all three AKT isoforms, indicating that the attachment of linkers and E3 ligase ligands at the solvent-exposed moiety of GDC-0068 is tolerated.
Figure 9.
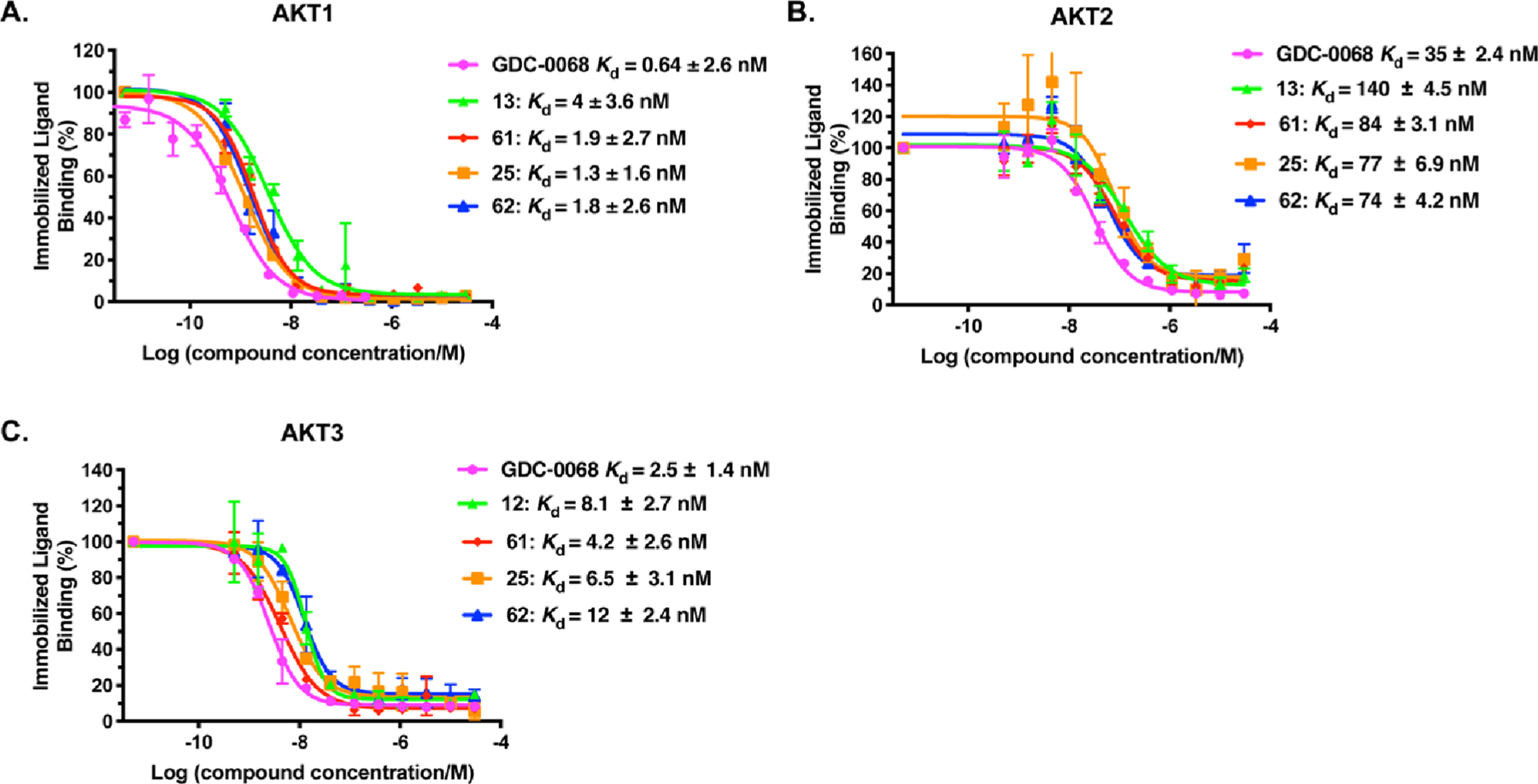
GDC-0068, 13, 61, 25, and 62 bind AKT1 (A), AKT2 (B), and AKT3 (C) with high affinities. The Kd determinations were performed in a competitive binding assay in duplicate. Error bars represent ± SEM from duplicated experiments.
Compounds 13 and 25 Concentration- and Time-Dependently Induced AKT Degradation through the Ubiquitin-Proteasome System (UPS).
We next determined the potency of degraders 13 and 25 in reducing AKT protein levels in BT474 cells (Figure 10A–D). We found that compounds 13 and 25 concentration-dependently depleted cellular T-AKT with DC50 values of 78 ± 64 and 32 ± 18 nM, respectively. Compound 13 induced T-AKT degradation substantially at 100 nM and achieved near-complete depletion of T-AKT at 1 μM. Compound 25 exhibited a slightly higher potency than compound 13 with a significant decrease of the AKT protein level at 30 nM and complete degradation of T-AKT at 300 nM. No ″hook effect″ was observed for both compounds at concentrations up to 10 μM. As anticipated, negative control compounds 61 and 62 did not degrade T-AKT (Figure 10,FE). In addition, we found that the previously reported CRBN-recruiting AKT degrader INY-03–041 was slightly more potent than degrader 25 (Figure 10G). However, a very significant ″hook effect″ was observed for INY-03–041 at high concentrations. In addition, we assessed the effect of 13, 25, and INY-03–041 on degrading AKT in PC3 and MDA-MB-468 cells and found that both 13 and 25 effectively induced AKT degradation in a concentration-dependent manner (Figure S3). Interestingly, 13 exhibited similar potency as INY-03–041, while 25 was less potent than 13 and INY-03–041 in PC3 cells (Figure S3A–C). In addition, while both 13 and 25 were less potent than INY-03–041 in MDA-MB-468 cells (Figure S3D–F), INY-03–041 displayed an obvious ″hook effect″ at higher concentrations in MDA-MB-468 cells (Figure S3F), similar to what was observed in BT474 cells (Figure 10G). We also confirmed that the negative controls 61 and 62 had no effect on the AKT protein levels in MDA-MB-468 cells (Figure S3G–H).
Figure 10.
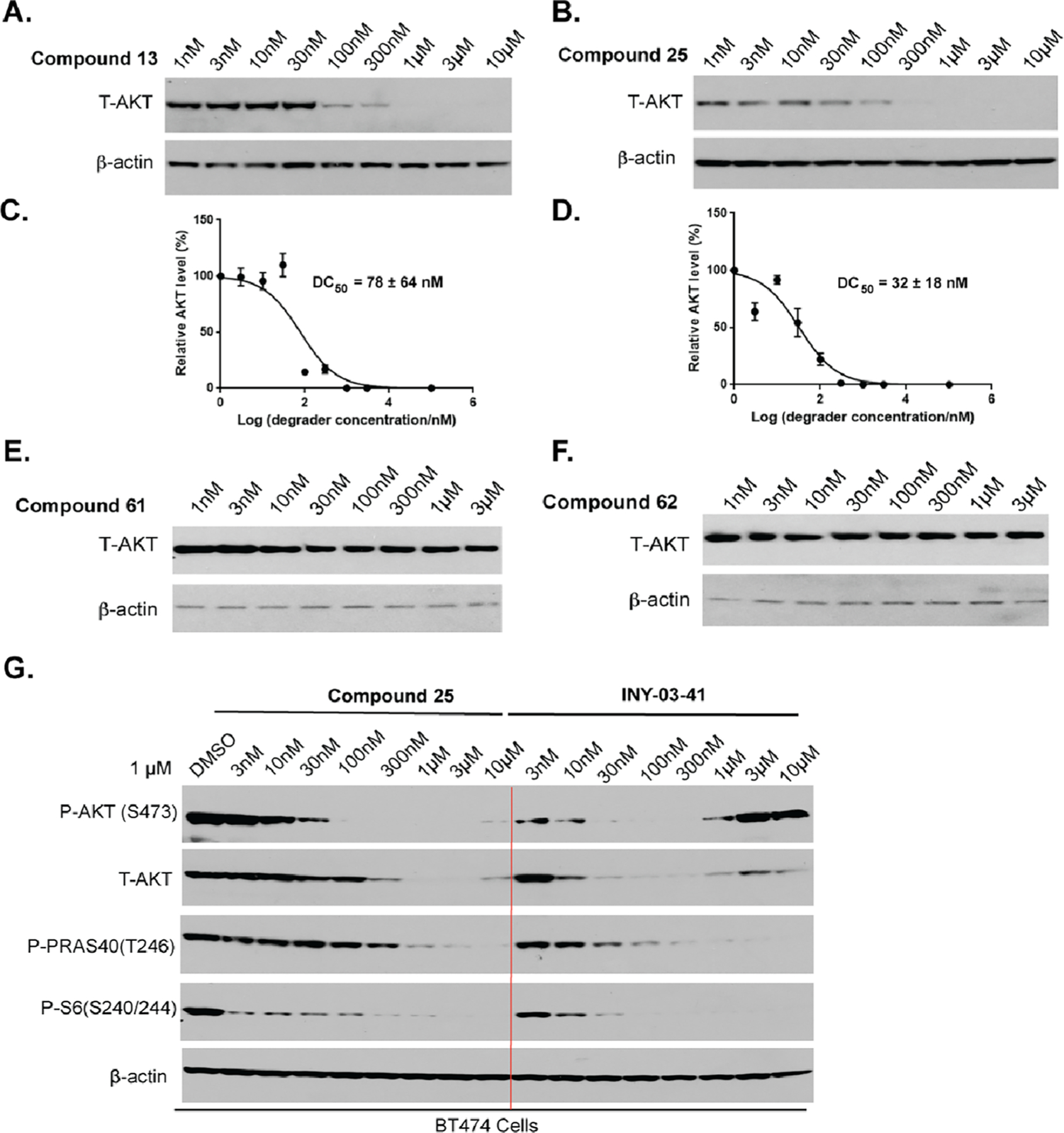
Compounds 13 and 25, but not 61 and 62, potently induced AKT degradation in BT474 cells. BT474 cells were treated with 13 (A, C), 25 (B, D), 61 (E), or 62 (F) at indicated concentrations for 24 h. (G) Effects of CRBN-recruiting AKT degraders 25 and INY-03–041 on AKT degradation and downstream signaling inhibition in BT474 cells treated with the test compound at indicated concentrations.
We also investigated the kinetics of AKT degradation and downstream signaling inhibition induced by compounds 13 and 25 in BT474 cells, with GDC-0068 as a control (Figure 11). As expected, GDC-0068 had no effect on the T-AKT protein level and increased the P-AKT protein level. On the other hand, compound 13 (at 1 μM) induced rapid T-AKT and P-AKT degradation (Figure 11A). Obvious degradation occurred at 4 h, and near-complete degradation was achieved at 8 h. Similarly, compound 25 degraded T-AKT and P-AKT in a time-dependent manner (Figure 11B). Significant P-AKT and T-AKT degradation occurred as early as 4 and 8 h, respectively. Both compounds maintained T-AKT and P-AKT degradation for at least 24 h. In addition, compounds 13 and 25 inhibited the downstream signaling, albeit the inhibition induced by 13 and 25 was not as rapid as that induced by GDC-0068. Taken together, these data indicate that 13 and 25 can induce rapid and sustained AKT degradation.
Figure 11.
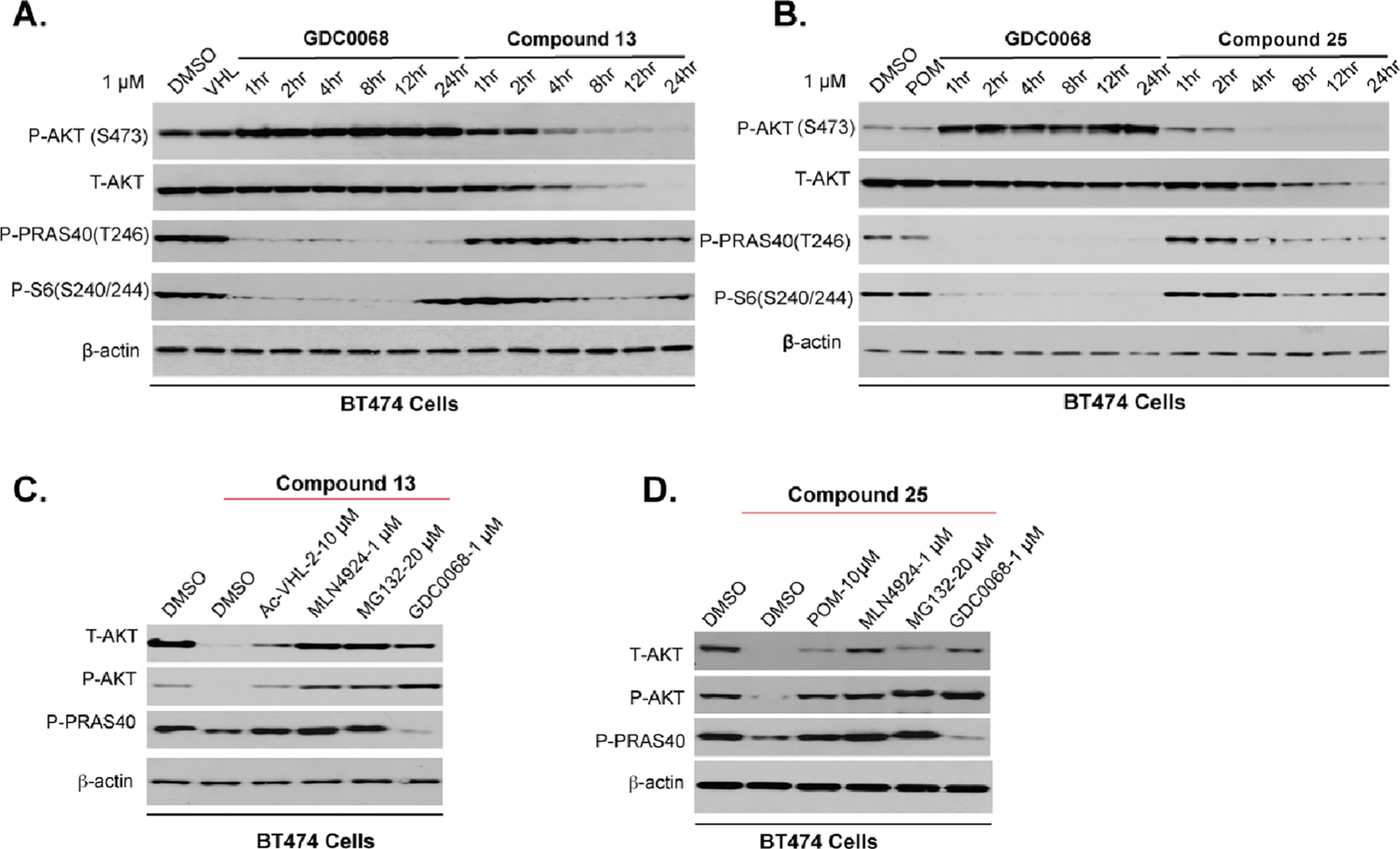
Compounds 13 and 25 induced AKT degradation and inhibited downstream signaling in a time-, E3 ligase- and proteasome-dependent manner in BT474 cells. BT474 cells were treated with 1 μM of compound 13 (A) or compound 25 (B) for the indicated time, with GDC-0068 as a control. Cell lysates were collected at the indicated time point for western blotting. MOA studies of 13 (C) or 25 (D) induced AKT degradation and downstream signaling inhibition. BT474 cells were pretreated with DMSO or the indicated compound (Ac-VHL-2 (10 μM), POM (10 μM), MLN4924 (1 μM), MG132 (10 μM), or GDC-0068 (1 μM)) for 2 h and subsequently treated with 13 or 25 for an additional 22 h. Levels of the indicated proteins were accessed by western blotting. β-Actin was used as the loading control.
To demonstrate that the AKT downregulation induced by 13 and 25 was through the UPS system, we next performed a set of competition experiments (Figure 11C,D). In case of the VHL-recruiting degrader 13 (Figure 11C), pretreatment with an excess of acetyl-capped VHL-2 (Ac-VHL-2) partially rescued AKT degradation and P-PRAS40 inhibition, suggesting that VHL binding is required for 13-induced AKT degradation and downstream signaling inhibition. Pretreatment with the NEDD8-activating enzyme (NAE) inhibitor MLN492459 or proteasome inhibitor MG132 completely prevented 13-mediated degradation of T-AKT and P-AKT and inhibition of P-PRAS40, indicating that the cullin-ring ubiquitin ligase complexes and proteasome are necessary for the observed AKT degradation and downstream signaling inhibition. These results also suggest that compound 13’s AKT degradation activity, instead of its AKT inhibition activity, is the primary contributor to compound 13’s downstream signaling inhibition effect. Additionally, pretreatment with GDC-0068 also blocked the AKT degradation induced by 13. Similarly, pretreatment with the CRBN ligand POM, MLN4924, MG132, or GDC-0068 rescued 25-induced AKT degradation (Figure 11D). Pretreatment with POM, MLN4924, or MG132 also blocked 25-mediated downstream signaling inhibition. Collectively, these results, together with that negative controls 61 and 62 were unable to degrade AKT, support that the AKT degradation induced by 13 and 25 is mediated by the UPS and requires the engagement of AKT and the corresponding E3 ligase (VHL or CRBN).
Compounds 13 and 25 Exhibited Remarkable Selectivity for AKT in Unbiased Global Proteomic Studies.
To assess whether the expression levels of other proteins are affected by AKT degraders 13 and 25, we conducted unbiased quantitative tandem mass tag (TMT) labeling mass spectrometry (MS)-based proteomic studies. The samples for the proteomic studies were prepared by the treatment of BT474 cells with 13, 25, 61, or 62 at 1 μM for 24 h. Immunoblot analysis confirmed that AKT1, AKT2, and T-AKT protein levels were greatly depleted by degraders 13 and 25, but not the negative controls 61 and 62, in these samples (Figure S4). We did not detect AKT3 in these samples, which is consistent with the previous report that the expression of AKT3 in BT474 cells is too low to be detected.60 This TMT-labeled proteomic approach enabled quantification of over 8000 proteins (Figure 12). Compared to the negative control 61, compound 13 significantly and selectively downregulated the abundance of AKT1 and AKT2, out of over 8000 quantified proteins, demonstrating that degrader 13 is highly selective for AKT1 and AKT2 (Figure 12A). Similarly, compared to the negative control 62, CRBN-recruiting compound 25 significantly reduced the protein levels of AKT1 and AKT2, as well as 3-hydroxy-3-methylglutaryl-CoA synthase 1 (HMGCS1) and the CRBN neo-substrate ZFP91, again suggesting that 25 is a selective degrader of AKT1 and AKT2 (Figure 12B). We did not detect AKT3 in these proteomic studies, in agreement with our WB analysis results (Figure S4). Overall, the results from our MS-based proteomic studies suggest that 13 and 25 are highly selective AKT degraders.
Figure 12.
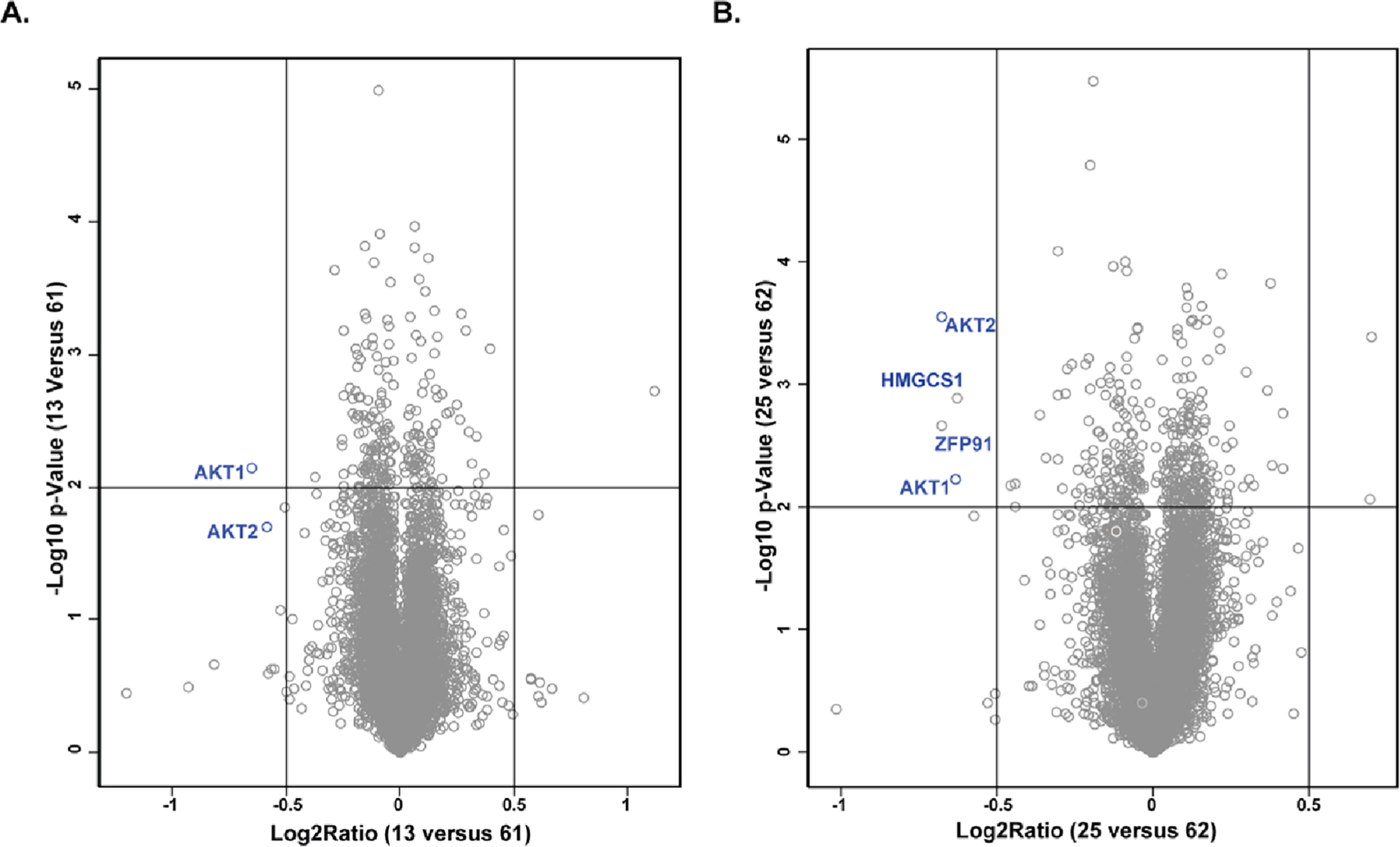
Compounds 13 (A) and 25 (B) selectively degraded AKT1 and AKT2 in unbiased global MS-based proteomic studies. BT474 cells were treated with 13, 61, 25, or 62 at 1 μM for 24 h before they were collected for mass spectrometry analysis. The log2 fold change is shown on the x axis and the negative log10 p value is shown on the y axis for two independent biological replicates of each treatment.
Compounds 13 and 25 Effectively Inhibited the Proliferation in Multiple Human Cancer Cell Lines.
We first evaluated the antiproliferative activities of 13 and 25 in BT474 cells, with GDC-0068, 61, 62, and INY-03–041 as controls (Figure 13A). Both VHL-recruiting degrader 13 (GI50 = 1.3 ± 0.3 μM) and CRBN-recruiting degrader 25 (GI50 = 0.7 ± 0.2 μM) effectively inhibited the proliferation in BT474 cells. Their potencies were slightly weaker than those of GDC-0068 (GI50 = 0.3 ± 0.03 μM) and INY-03–041 (GI50 = 0.4 ± 0.2 μM). Of note, 13 was 2- to 3-fold more potent than its control compound, 61 (GI50 = 3.1 ± 0.4 μM), and 25 showed 5-fold improved potency over its control compound, 62 (GI50 = 3.3 ± 0.8 μM), suggesting that the antiproliferation activities of 13 and 25 are partially due to their AKT degradation effect. We next examined their antiproliferation activities in two additional cancer cell lines: PC3 and MDA-MB-468 (a triple-negative breast cell line). Both 13 and 25 effectively inhibited the growth in these two cancer cell lines, with comparable potencies to GDC-0068 and INY-03–041 (Figure 13B,C). Next, we evaluated the effect of these compounds on cell apoptosis in MDA-MB-468 cells and found that neither the AKT inhibitor GDC-0068 nor degraders 13 and 25 induced more than 3% apoptosis (Figure S5). Therefore, these AKT degraders inhibited the growth of MDA-MB-468 cells mainly through an antiproliferative effect. Collectively, these results indicate that AKT degraders 13 and 25 can suppress the growth in multiple human cancer cell lines.
Figure 13.
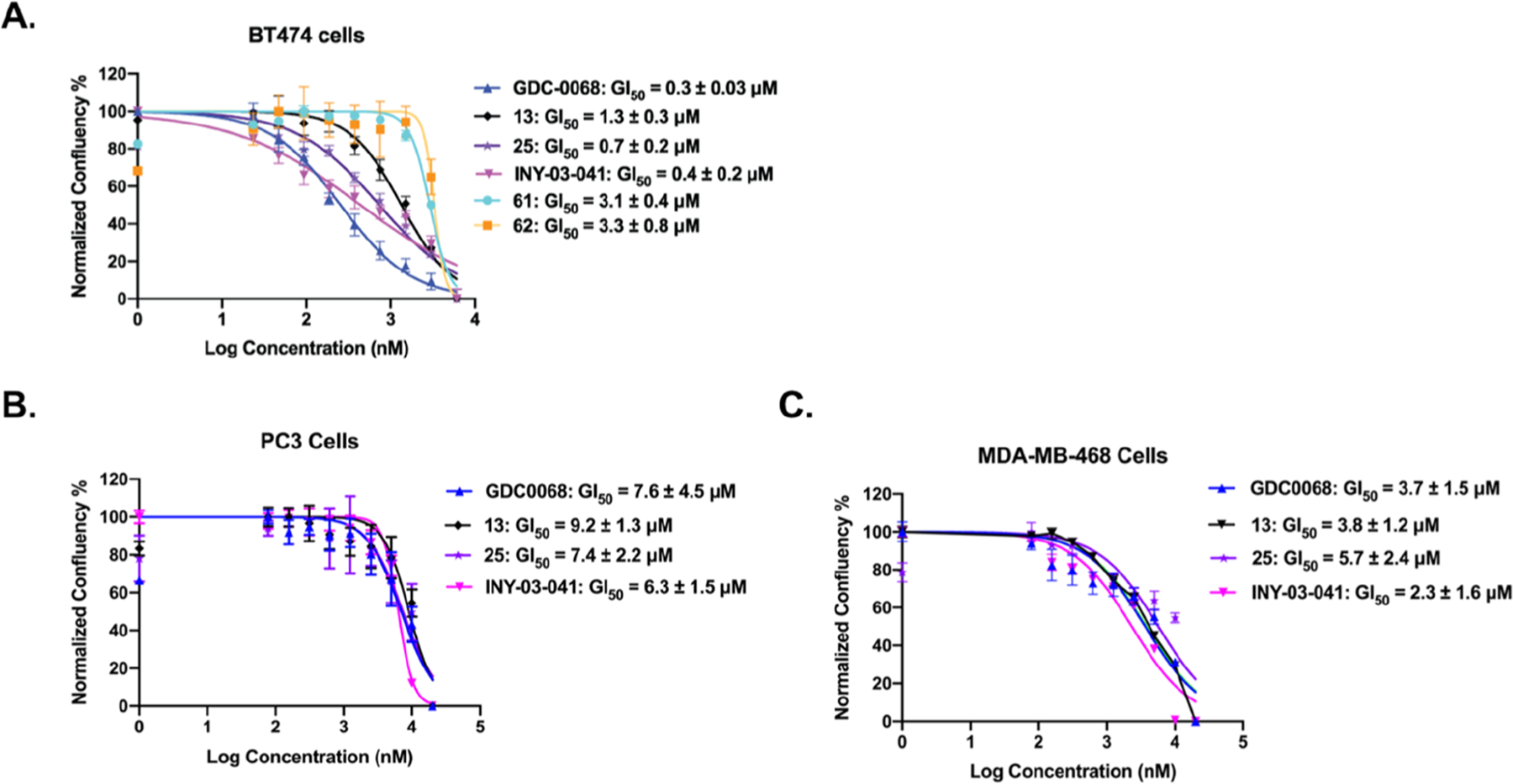
AKT degraders 13 and 25 effectively inhibited the proliferation in multiple cancer cell lines. BT474 (A), PC3 (B), and MDA-MB-468 (C) cells were treated with the indicated compound at indicated concentrations for 5 days. Cell growth was determined using calculations derived from phase-contrast images in IncuCyte, and error bars indicate the standard errors from three independent experiments.
Compounds 13 and 25 Were Bioavailable in Mice.
We next examined in vivo mouse pharmacokinetic (PK) properties of compounds 13 and 25 following a single intraperitoneal (IP) injection of each compound at a dose of 50 mg/kg. The maximum plasma concentration (Cmax) of 13 reached approximately 3.5 μM at 2 h, and the plasma concentrations remained above 3 μM over 8 h (Figure 14A). Similarly, compound 25 was also bioavailable in mice via IP injection (Figure 14B). Although the Cmax (1.4 μM at 2 h) of 25 was about 2.5-fold less than that of 13, its exposure levels in plasma were still good. The sufficient in vivo mouse PK properties of 13 and 25 make them suitable for in vivo efficacy studies. The in vivo PK properties of INY-03–041 have not been reported.47 It is also worth noting that both 13 and 25 were well tolerated in the mouse PK studies. No clinical signs were observed in the test mice.
Figure 14.
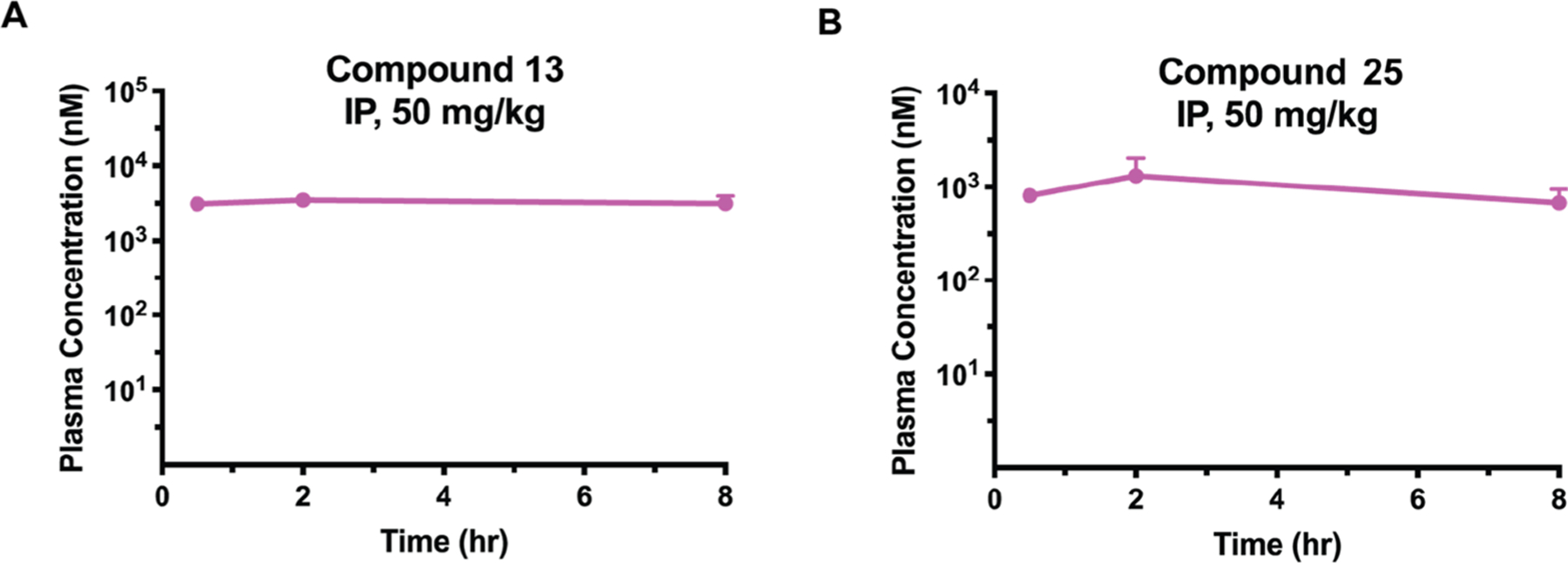
AKT degraders 13 and 25 were bioavailable in mouse PK studies. Plasma concentrations of 13 (A) and 25 (B) following a single 50 mg/kg IP injection over 8 h in male Swiss albino mice. Experiments were carried out in biological triplicates, with points representing mean concentrations ± SEM.
Synthesis.
Synthetic routes for intermediates 1 and 2 are outlined in Scheme 1. The amide coupling reaction between known compounds 63 and 64 and subsequent deprotection of the Boc group under acidic conditions yielded intermediate 65.18 The nucleophilic substitution of commercially available ethyl 3-bromopropanoate (66) with 65 followed by the hydrolysis of the ethyl ester afforded the carboxylic acid precursor 1. The amine intermediate 2 was prepared in a similar manner from benzyl (2-iodoethyl)carbamate (67) and 65 through nucleophilic substitution, Boc protection, and Cbz deprotection.
Scheme 1. Synthesis of Intermediates 1 and 2a.
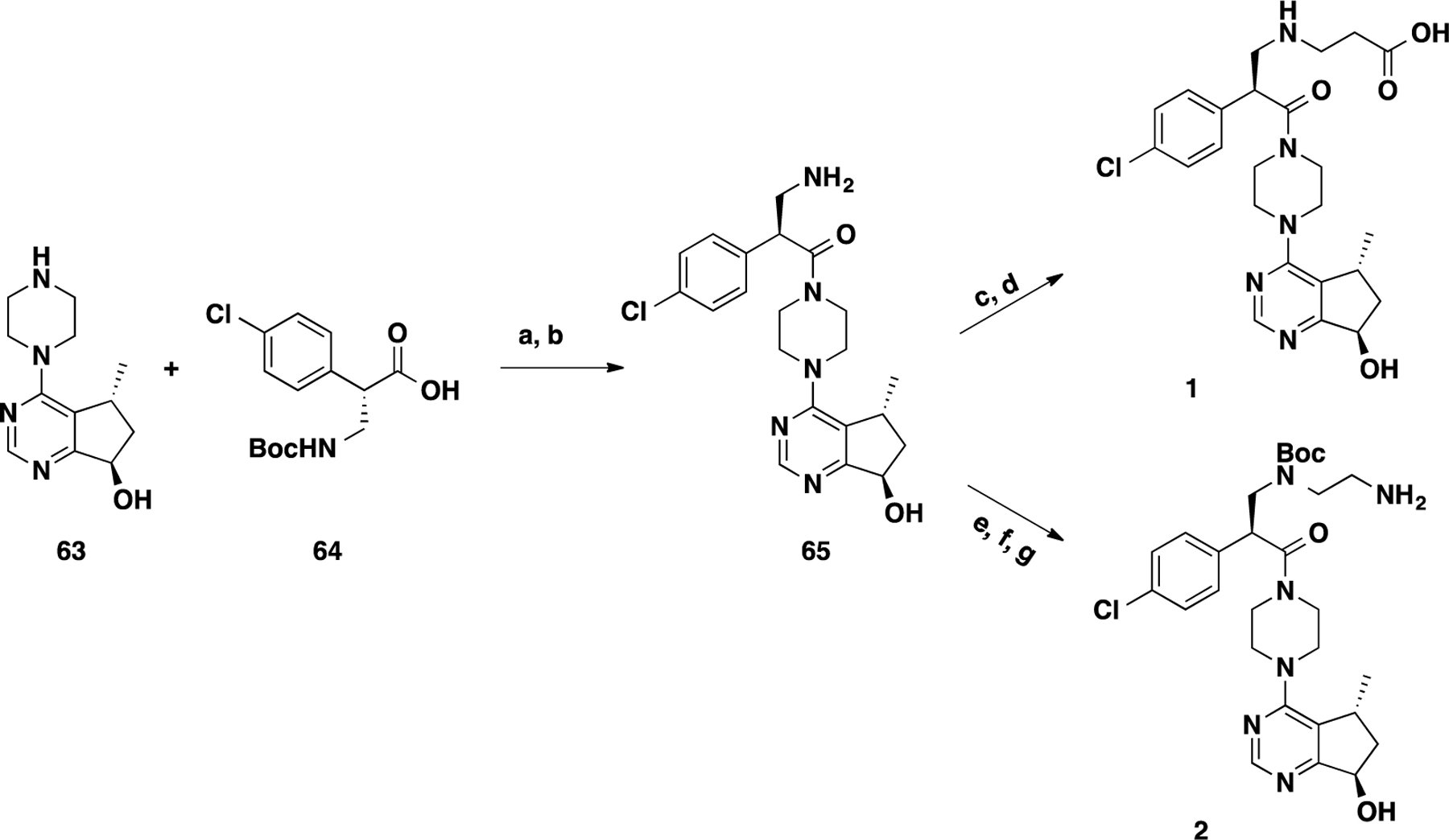
aReaction conditions: (a) 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI), 1-hydroxy-7-azabenzo-triazole (HOAt), N-methylmorpholine (NMM), DMSO, rt, 12 h; (b) TFA/DCM, rt, 30 min; (c) ethyl 3-bromopropanoate (66), K2CO3, DMF, 80 °C, 12 h; (d) LiOH, THF, H2O, rt, 12 h; (e) benzyl (2-iodoethyl)carbamate (67), K2CO3, CH3CN, 80 °C, 8 h; (f) Boc2O, Et3N, DCM, rt, 2 h; and (g) H2, Pd/C, MeOH, rt, 1 h.
Compounds 3–11 were prepared by amide coupling reactions between linker-attached VHL ligands 68–7661 and intermediate 1 (Scheme 2A). The synthesis of 12 and 13 is outlined in Scheme 2B. Briefly, the amide coupling reaction between the VHL-2 ligand (77) and Boc protected amino acid 78 followed by Boc deprotection afforded amine intermediate 79, which was coupled with intermediate 1 to provide 12. Similarly, the amide coupling between 77 and diacid 80 provided acid intermediate 81, which was converted to 13 by an amide coupling reaction followed by Boc deprotection.
Scheme 2. Synthesis of GDC-0068-Based VHL-Recruiting Compounds 3–11 (A) and 12 and 13 (B)a.
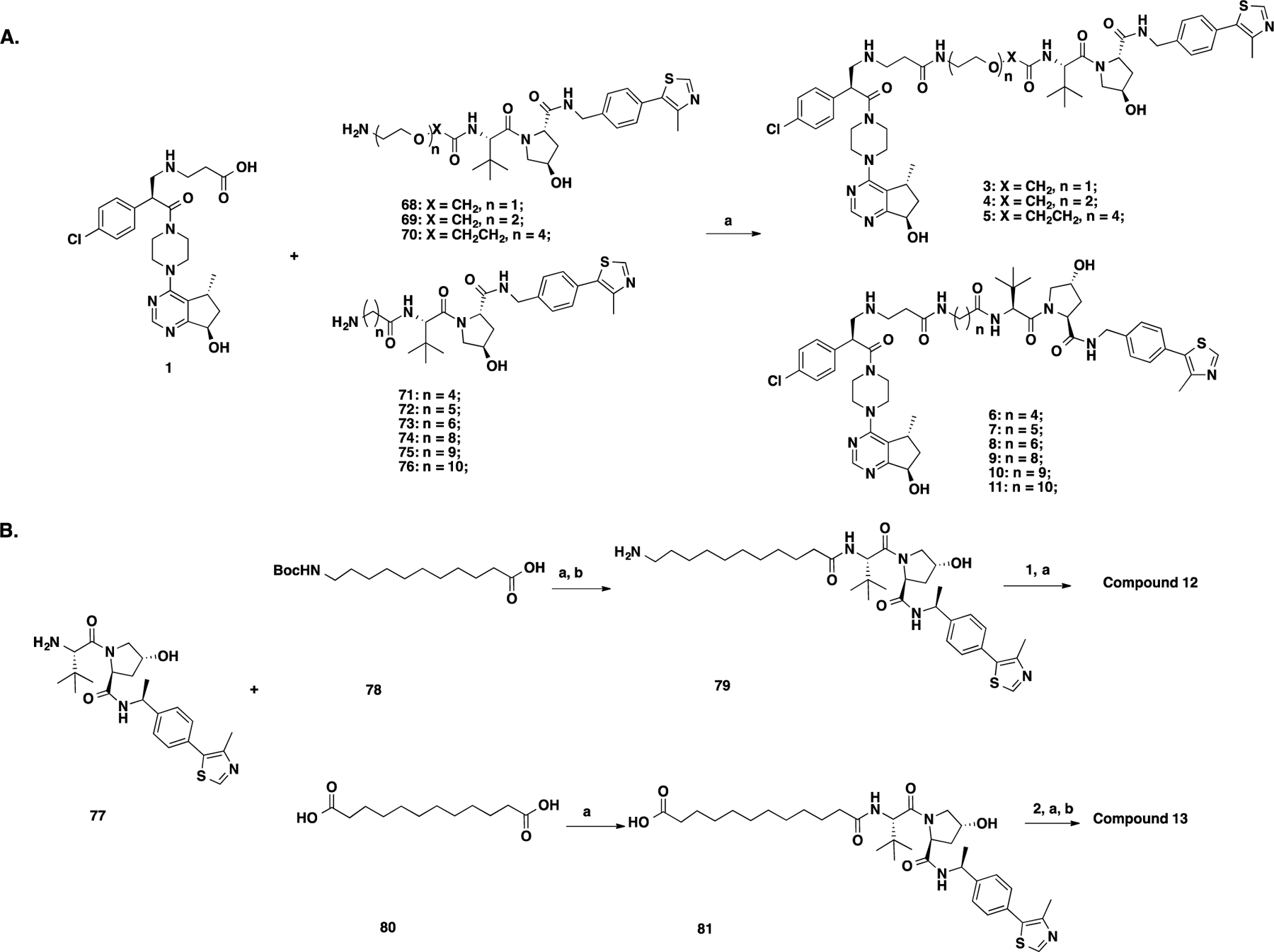
aReaction conditions: (a) EDCI, HOAt, NMM, DMSO, rt, 12 h and (b) TFA/DCM, rt, 30 min.
Synthetic routes for compounds 14–29 are included in Scheme 3. Compounds 14–25 were synthesized using the amide coupling reactions between intermediate 1 and different linker-attached pomalidomide analogs 82–9361 (Scheme 3A). Similarly, compound 26 was synthesized through the amide coupling reaction between intermediate 2 and 94 (Scheme 3B). The synthesis of compound 27 commenced with the conversion of oct-7-yn-1-ol (95) to its corresponding oct-7-yn-1-yl 4-methylbenzenesulfonate (96), which was subjected to the substitution with sodium cyanide to generate non-8-ynenitrile (97). The Sonogashira coupling reaction between 97 and commercially available 4-bromo-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione followed by the hydrogenation reaction to reduce alkynyl and cyanide groups furnished intermediate 99. Amide coupling between 99 and intermediate 1 yielded compound 27 (Scheme 3C). Compounds 28 and 29 were prepared from tert-butyl (8-iodoctyl)carbamate (100) and 101 or 102 through a sequence of nucleophilic substitution, Boc-deprotection, and amide coupling reaction with intermediate 1 (Scheme 3D).61
Scheme 3. Synthesis of GDC-0068-Based CRBN-Recruiting Compounds 14–25 (A), 26 (B), 27 (C), and 28 and 29 (D)a.
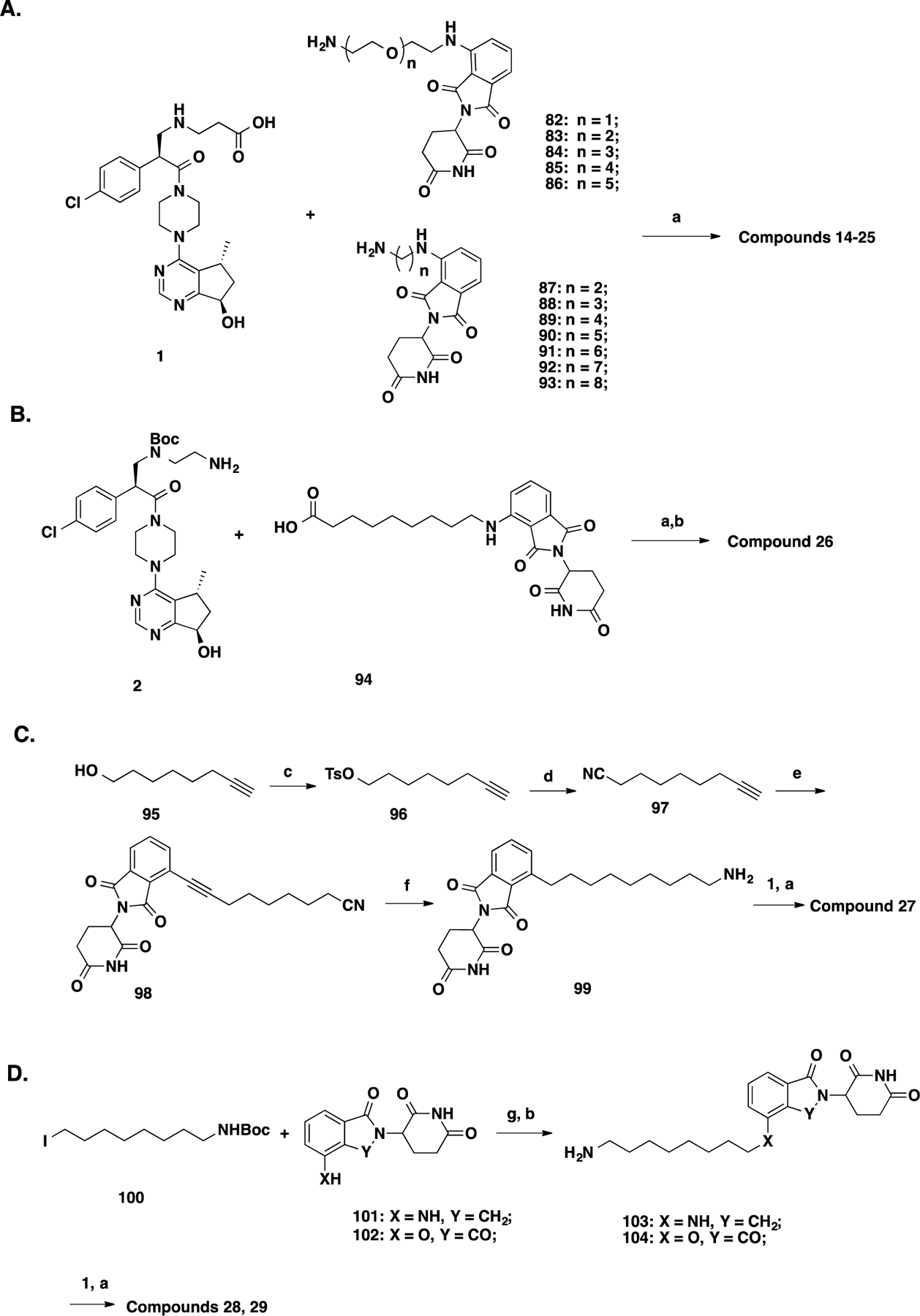
aReaction conditions: (a) EDCI, HOAt, NMM, DMSO, rt, 12 h; (b) TFA/DCM, rt, 30 min; (c) 4-toluenesulfonyl chloride, NEt3, DCM, 0 °C to rt, 4 h; (d) NaCN, NaI, DMF, 50 °C, 8 h; (e) 4-bromo-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione, Pd(PPh3)2Cl2, CuI, NEt3, DMF, 70 °C, 2 h; (f) Raney Ni, H2, MeOH/NH3.H2O (3/1), rt, 1 h; and (g) NaHCO3, KI, DMF, 60 °C, overnight.
GSK690693-based degraders 31–60 were prepared following the synthetic route outlined in Scheme 4. Reductive amination between 10515 and methyl 4-oxobutanoate followed by Boc-protection afforded intermediate 106. The subsequent Sonogashira coupling of 106 and 2-methylbut-3-yn-2-ol yielded 107, which was converted to compound 30 through an ester hydrolysis reaction. Amide coupling reactions between intermediate 30 and linker-attached E3 ligase ligands 68–76, 82–93, and 108–116 followed by Boc deprotection afforded the designed bifunctional compounds 31–60.
Scheme 4. Synthesis of Compounds 31–60a.
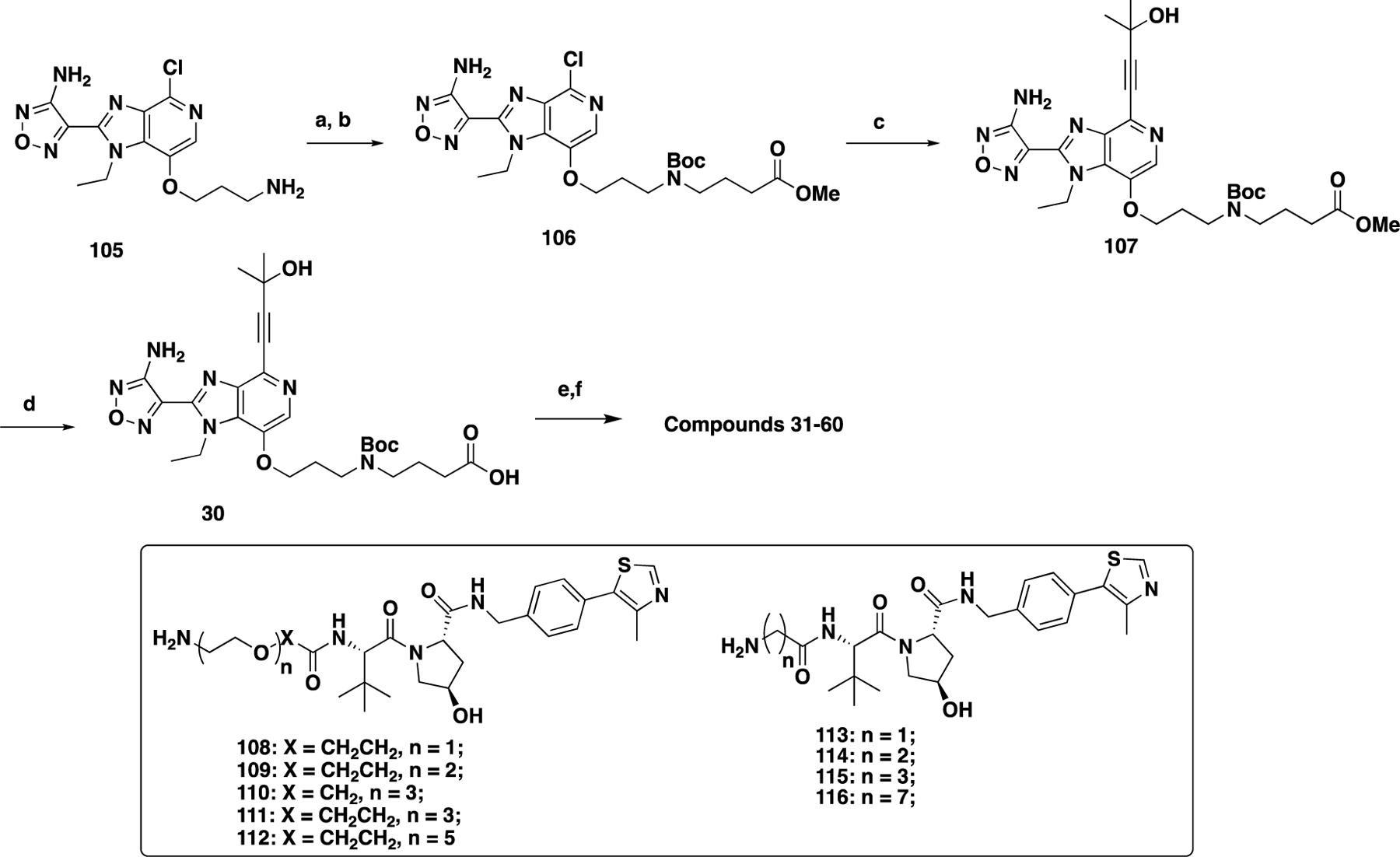
aReaction conditions: (a) methyl 4-oxobutanoate, NaBH(OAc)3, DCM, rt; (b) Boc2O, Et3N, DCM, rt; (c) 2-methylbut-3-yn-2-ol, Pd(PPh3)4, Zn powder, DBU, NaI, DMSO, 80 °C; (d) NaOH (3 N), MeOH, 60 °C; (e) 68–76, 108–116, and 82–93, EDCI, HOAt, NMM, DMSO, rt, 12 h; and (f) TAF/DCM, rt, 30 min.
Synthetic routes for compounds 61 and 62 are outlined in Scheme 5. Intermediate 118 was prepared by amide coupling between intermediate 117 and commercially available dodecanedioic acid (80). Amide coupling between intermediate 2 and 118 followed by Boc removal provided 61. The nucleophilic aromatic substitution (SNAr) of intermediate 119 with tert-butyl(8-aminoctyl)carbamate (120) and the subsequent Boc-deprotecting reaction afforded intermediate 121.61 Amide coupling of 121 and intermediate 1 provided compound 62.
Scheme 5. Synthesis of Negative Control Compounds 61 and 62a.
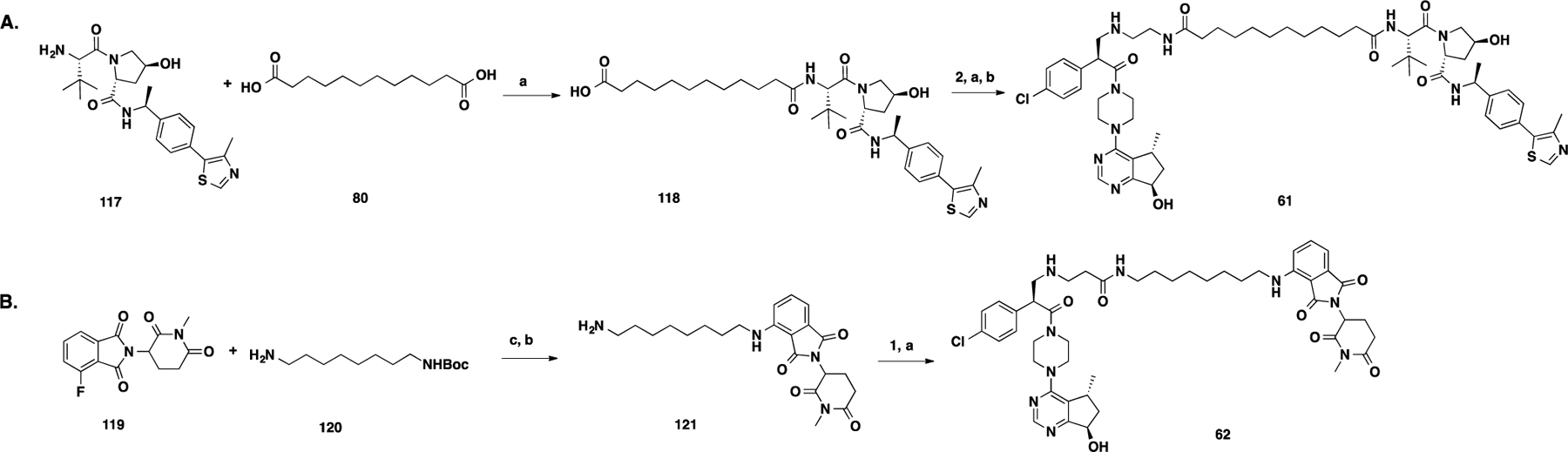
aReaction conditions: (a) EDCI, HOAt, NMM, DMSO, rt, 12 h; (b) TFA/DCM, rt, 30 min; and (c) NMP, DIPEA, MW, 100 °C, 1 h.
CONCLUSIONS
To date, only two AKT PROTAC degraders and no SAR studies have been reported. We conducted comprehensive SAR studies by the design, synthesis, and evaluation of a large set of novel compounds to explore various AKT binders, linkers, and E3 ligase ligands. Through these extensive SAR studies, we discovered multiple promising AKT PROTAC degraders derived from AKT inhibitors GDC-0068 and GSK690693, including compounds 13, 25, 48, and 60. Among them, the GDC-0068-based VHL-recruiting compound 13 and CRBN-recruiting compound 25 were further characterized in biochemical, cellular, proteomic, and PK studies. We also developed the corresponding negative controls of 13 and 25, compounds 61 and 62, which maintained good binding affinity to AKT but were unable to bind VHL and CRBN, respectively. We show that 13 and 25, but not 61 and 62, concentration- and time-dependently reduced the protein levels of T-AKT and P-AKT and inhibited the downstream signaling such as P-PRAS40 and P-S6 in BT474 cells. Through a series of rescue experiments, we demonstrate that the AKT degradation and downstream signaling inhibition induced by 13 and 25 were mediated by the UPS and required the engagement with AKT and the corresponding E3 ligase (VHL or CRBN). Using unbiased quantitative MS-based global proteomic studies, we show that both 13 and 25 are highly selective AKT degraders. Moreover, compounds 13 and 25 effectively suppressed the proliferation in multiple human cancer cell lines. Lastly, both 13 and 25 were bioavailable in mouse PK studies via IP injection and could be suitable for in vivo efficacy studies. Overall, our comprehensive SAR studies resulted in the discovery of multiple novel, potent ,and selective AKT PROTAC degraders.
EXPERIMENTAL SECTION
Chemistry General Procedures.
All commercially available chemical reagents were used directly in syntheses without further purification. Microwave-heated reactions were performed with a Discover SP microwave system with an Explorer 12 Hybrid Autosampler by CEM (Buckingham, UK). A Teledyne ISCO CombiFlash Rf+ instrument and HP C18 RediSep Rf reverse phase columns equipped with UV detector were used to conduct flash chromatography. All final compounds for biological evaluation were purified with preparative high-performance liquid chromatography (HPLC) on an Agilent Prep 1200 series with the UV detector set to 254 or 220 nm with a flow rate of 40 mL/min at room temperature. Crude samples were injected into a Phenomenex Luna 750 × 30 mm, 5 μm C18 column, with the gradient program set to 10% of methanol or acetonitrile (B) in H2O containing 0.1% TFA (A) progressing from 10 to 100% of methanol or acetonitrile (B). The purity of all compounds for biological activity was >95% as assessed by HPLC-HRMS. All HPLC spectra was obtained by using an Agilent 1200 series system with a DAD detector and a 2.1 × 150 mm Zorbax 300SB-C18 5 μm column for chromatography. Samples (0.5 μL) were injected onto a C18 column at room temperature with the flow rate of 0.4 mL/min. Chromatography was performed with the solvent as follows: water containing 0.1% formic acid was designated as solvent A, while acetonitrile containing 0.1% formic acid was designated as solvent B. The linear gradient was set such that 1% B was used from 0 to 1 min, 1–99% B from 1 to 4 min, and 99% B from 4 to 8 min. High-resolution mass spectra (HRMS) data were acquired in positive ion mode using an Agilent G1969A API-TOF with an electrospray ionization (ESI) source. All compounds were also characterized using either a Bruker (Billerica, MA) DRX Nuclear Magnetic Resonance (NMR) spectrometer (400, 500, and 600 MHz, 1H NMR) or a Bruker DXI 800 MHz spectrometer (800 MHz 1H NMR, 201 MHz 13C NMR). Chemical shifts for all compounds are reported in units of parts per million (ppm, δ) relative to residual solvent peaks. 1H NMR data are reported in the following format: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, and m = multiplet), coupling constant, and integration.
Synthetic route, procedures, and characterization of compounds 68–76, 82–93, and 108–116 are depicted in the Supporting Information.
3-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)piperazin-1-yl)-3-oxopropyl)amino)propanoic Acid (1).
To a solution of intermediate 6518 (360 mg, 0.86 mmol) in DMF (10 mL) was added potassium carbonate (358 mg, 2.6 mmol, 3 equiv). The resulting suspension was stirred at 80 °C for 15 min before ethyl 3-bromopropanoate (66, 310 mg, 1.72 mmol, 2 equiv) was added. After the reaction was stirred overnight, water was added and the mixture was extracted with ethyl acetate (3 × 10 mL), dried over Na2SO4, filtered, and evaporated. The resulting mixture was purified by flash chromatography (DCM/ MeOH = 10:1) to afford the product ethyl 3-(((S)-2-(4-chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)piperazin-1-yl)-3-oxopropyl)amino)-propanoate (257 mg, yield 58%). ESI [M + H]+ (m/z): 416.2. The obtained intermediate was dissolved in THF/H2O (1:1). To the solution was added lithium hydroxide (24 mg, 1 mmol). After stirring overnight at room temperature, the reaction mixture was concentrated and the residue was purified by reverse phase column (10–100% methanol/0.1% TFA in H2O) to afford intermediate 1 as a white solid in TFA salt form (238 mg, yield 97%). 1H NMR (600 MHz, CD3OD) δ 8.57 (d, J = 4.5 Hz, 1H), 7.47 (dt, J = 8.7, 2.3 Hz, 2H), 7.36 (dd, J = 8.4, 6.0 Hz, 2H), 5.28 (t, J = 7.9 Hz, 1H), 4.50 (ddd, J = 9.7, 6.6, 4.1 Hz, 1H), 4.37 (td, J = 7.9, 4.6 Hz, 1H), 4.24–4.10 (m, 1H), 4.09–4.01 (m, 1H), 3.95–3.81 (m, 4H), 3.65 (dd, J = 12.9, 8.9 Hz, 3H), 3.52–3.38 (m, 2H), 3.21 (dd, J = 12.8, 4.6 Hz, 1H), 2.78 (t, J = 6.4 Hz, 2H), 2.28 (dd, J = 12.9, 7.4 Hz, 1H), 2.17 (ddt, J = 12.6, 8.3, 4.1 Hz, 1H), 1.19 (dd, J = 21.1, 7.0 Hz, 3H). HRMS (m/z) for C24H31ClN5O + [M + H]+: calculated 488.2059, found 488.2057.
tert-Butyl (2-aminoethyl)((S)-2-(4-chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)-piperazin-1-yl)-3-oxopropyl)carbamate (2).
To a solution of intermediate 65 (467 mg, 1.12 mmol, 1.1 equiv) in CH3CN was added potassium carbonate (706 mg, 5 mmol, 5 equiv). After the resulting suspension was stirred at 80 °C for 15 min, benzyl (2-iodoethyl)carbamate (67, 305 mg, 1.0 mmol) was added. After the reaction was stirred at 80 °C for 8 h, the reaction mixture was filtered, and the filtrate was concentrated. The resulting residue was purified by preparative HPLC to afford the desired intermediate as a white solid (201 mg, yield 30%). The white solid (201 mg, 0.34 mmol) was dissolved in DCM (5 mL). To the resulting solution were added triethylamine (92 μL, 0.68 mmol, 2 equiv) and di-tert-butyl dicarbonate (89 mg, 0.4 mmol, 1.2 equiv). After the reaction was stirred at room temperature for 2 h, the solvent was removed under reduced pressure. The resulting residue was purified by flash chromatography (MeOH/DCM = 1:9). The product was obtained as a white solid (160 mg, yield 68%). This product was dissolved in methanol (6 mL) followed by the addition of 10% palladium on carbon (16 mg, 10% of weight). After stirring under H2 for 4 h, the mixture was filtered, and the filtrate was concentrated. The resulting mixture was purified by preparative HPLC to afford the title compound 2 as a white solid in TFA salt form (91 mg, yield 71%). 1H NMR (600 MHz, CD3OD) δ 8.58 (d, J = 3.7 Hz, 1H), 7.44–7.35 (m, 2H), 7.32 (s, 2H), 5.31 (t, J = 8.0 Hz, 1H), 4.32 (s, 1H), 4.21 (s, 1H), 3.98 (d, J = 8.2 Hz, 1H), 3.85 (s, 2H), 3.79–3.65 (m, 6H), 3.57 (d, J = 13.4 Hz, 2H), 3.51–3.41 (m, 2H), 3.03 (s, 1H), 2.32–2.25 (m, 1H), 2.21–2.16 (m, 1H), 1.41 (s, 9H), 1.17 (d, J = 7.0 Hz, 3H). HRMS (m/z) for C28H40ClN6O4+ [M + H]+: calculated 559.2794, found 559.2782.
(2S,4R)-1-((2S,15S)-2-(tert-Butyl)-15-(4-chlorophenyl)-16-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]-pyrimidin-4-yl)piperazin-1-yl)-4,10,16-trioxo-6-oxa-3,9,13-triaza-hexadecanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)-pyrrolidine-2-carboxamide (3).
To a solution of intermediate 1 (12 mg, 0.02 mmol) in DMSO (1 mL) were added linker 68 (11.3 mg, 0.02 mmol, 1.0 equiv), EDCI (1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide, 5.8 mg, 0.03 mmol, 1.5 equiv), HOAt (1-hydroxy-7-azabenzo-triazole, 4.1 mg, 0.03 mmol, 1.5 equiv), and NMM (N-methylmorpholine, 6.1 mg, 0.06 mmol, 3.0 equiv). After stirring overnight at room temperature, the reaction mixture was purified by preparative HPLC (10–100% methanol/0.1% TFA in H O) to afford title compound 3 as a white solid in TFA salt form (18.2 mg, yield 91%). 1H NMR (600 MHz, CD3OD) δ 8.97 (s, 1H), 8.58 (s, 1H), 7.54–7.39 (m, 6H), 7.39–7.30 (m, 2H), 5.31 (t, J = 8.0 Hz, 1H), 4.71 (s, 1H), 4.61–4.45 (m, 4H), 4.42–4.36 (m, 1H), 4.17 (s, 1H), 4.08–4.03 (m, 1H), 3.99–3.76 (m, 7H), 3.71–3.54 (m, 7H), 3.47–3.36 (m, 3H), 3.28–3.23 (m, 1H), 2.80–2.64 (m, 3H), 2.48 (s, 3H), 2.33–2.22 (m, 2H), 2.17 (dt, J = 12.7, 8.2 Hz, 1H), 2.09 (ddd, J = 13.4, 9.4, 4.4 Hz, 1H), 1.17 (d, J = 6.9 Hz, 3H), 1.04 (s, 9H). HRMS (m/z) for C50H66ClN10O8S+ [M + H]+: calculated 1001.4469, found 1001.4472.
(2S,4R)-1-((2S,18S)-2-(tert-Butyl)-18-(4-chlorophenyl)-19-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]-pyrimidin-4-yl)piperazin-1-yl)-4,13,19-trioxo-6,9-dioxa-3,12,16-tri-azanonadecanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)-pyrrolidine-2-carboxamide (4).
Compound 4 was synthesized following the standard procedure for preparing compound 3 from intermediate 1 (12 mg, 0.02 mmol) and 69 (12.3 mg, 0.02 mmol, 1.0 equiv). Compound 4 was obtained as a white solid in TFA salt form (9.4 mg, yield 45%). 1H NMR (600 MHz, CD3OD) δ 8.95 (s, 1H), 8.57 (s, 1H), 7.53–7.37 (m, 6H), 7.34 (dd, J = 8.5, 2.1 Hz, 2H), 5.31 (t, J = 7.9 Hz, 1H), 4.75 (s, 1H), 4.63–4.45 (m, 4H), 4.40 (d, J = 15.5 Hz, 1H), 4.17 (s, 1H), 4.03 (d, J = 2.0 Hz, 2H), 3.96–3.79 (m, 6H), 3.74–3.47 (m, 13H), 3.39 (t, J = 9.5 Hz, 1H), 3.28–3.22 (m, 3H), 2.68 (t, J = 6.3 Hz, 1H), 2.47 (s, 3H), 2.32–2.24 (m, 2H), 2.17 (dt, J = 12.7, 8.1 Hz, 1H), 2.08 (ddd, J = 13.5, 9.6, 4.3 Hz, 1H), 1.17 (dd, J = 7.0, 2.0 Hz, 3H), 1.04 (s, 9H). HRMS (m/z) for C52H70ClN10O9S+ [M + H]+: calculated 1045.4731, found 1045.4738.
(2S,4R)-1-((2S,25S)-2-(tert-Butyl)-25-(4-chlorophenyl)-26-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]-pyrimidin-4-yl)piperazin-1-yl)-4,20,26-trioxo-7,10,13,16-tetraoxa-3,19,23-triazahexacosanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (5).
Compound 5 was synthesized following the standard procedure for preparing compound 3 from intermediate 1 (12 mg, 0.02 mmol) and 70 (14.3 mg, 0.02 mmol, 1.0 equiv). Compound 5 was obtained as a white solid in TFA salt form (11.2 mg, yield 48%). 1H NMR (600 MHz, CD3OD) δ 8.95 (s, 1H), 8.58 (s, 1H), 7.54–7.39 (m, 6H), 7.39–7.28 (m, 2H), 5.31 (t, J = 8.0 Hz, 1H), 4.65 (s, 1H), 4.60–4.47 (m, 4H), 4.36 (d, J = 15.5 Hz, 1H), 4.18 (s, 1H), 3.98–3.77 (m, 6H), 3.76–3.57 (m, 19H), 3.53 (t, J = 5.4 Hz, 2H), 3.44–3.33 (m, 4H), 3.29–3.23 (m, 2H), 2.67 (t, J = 6.3 Hz, 2H), 2.58 (ddd, J = 15.0, 7.5, 5.2 Hz, 1H), 2.48 (s, 3H), 2.33–2.26 (m, 1H), 2.25–2.15 (m, 2H), 2.12–2.04 (m, 1H), 1.17 (d, J = 6.9 Hz, 3H), 1.04 (s, 9H). HRMS (m/z) for C57H80ClN10O11S+ [M + H]+: calculated 1147.5412, found 1147.5434.
(2S,4R)-1-((S)-2-(5-(3-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)-piperazin-1-yl)-3-oxopropyl)amino)propanamido)pentanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)-benzyl)pyrrolidine-2-carboxamide (6).
Compound 6 was synthesized following the standard procedure for preparing compound 3 from intermediate 1 (12 mg, 0.02 mmol) and 71 (11.3 mg, 0.02 mmol, 1.0 equiv). Compound 6 was obtained as a white solid in TFA salt form (7.6 mg, yield 38%). 1H NMR (600 MHz, CD3OD) δ 8.93 (s, 1H), 8.57 (s, 1H), 7.51–7.39 (m, 6H), 7.39–7.32 (m, 2H), 5.31 (t, J = 7.9 Hz, 1H), 4.62 (s, 1H), 4.59–4.48 (m, 4H), 4.36 (d, J = 15.4 Hz, 1H), 4.18 (s, 1H), 3.96–3.86 (m, 4H), 3.81 (dt, J = 10.9, 6.1 Hz, 2H), 3.70–3.60 (m, 5H), 3.40 (t, J = 8.9 Hz, 1H), 3.27 (dd, J = 12.6, 3.8 Hz, 2H), 3.22–3.16 (m, 2H), 2.67–2.63 (m, 2H), 2.47 (s, 3H), 2.33–2.26 (m, 3H), 2.23–2.16 (m, 2H), 2.08 (ddd, J = 13.3, 9.1, 4.5 Hz, 1H), 1.65–1.57 (m, 2H), 1.51 (t, J = 7.4 Hz, 2H), 1.17 (d, J = 7.3 Hz, 3H), 1.03 (s, 9H). HRMS (m/z) for C51H68ClN10O7S+ [M +H]+: calculated 999.4676, found 999.4678.
(2S,4R)-1-((S)-2-(6-(3-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)-piperazin-1-yl)-3-oxopropyl)amino)propanamido)hexanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)-benzyl)pyrrolidine-2-carboxamide (7).
Compound 7 was synthesized following the standard procedure for preparing compound 3 from intermediate 1 (12 mg, 0.02 mmol) and 72 (11.6 mg, 0.02 mmol, 1.0 equiv). Compound 7 was obtained as a white solid in TFA salt form (12.7 mg, yield 63%). 1H NMR (600 MHz, CD3OD) δ 8.97 (s, 1H), 8.58 (d, J = 4.4 Hz, 1H), 7.54–7.38 (m, 6H), 7.38–7.32 (m, 2H), 5.31 (t, J = 8.0 Hz, 1H), 4.63 (s, 1H), 4.60–4.47 (m, 4H), 4.37 (d, J = 15.4 Hz, 1H), 4.18 (s, 1H), 3.97–3.78 (m, 6H), 3.72–3.54 (m, 5H), 3.40 (dd, J = 10.8, 7.1 Hz, 1H), 3.29–3.24 (m, 2H), 3.18 (t, J = 7.1 Hz, 2H), 2.67–2.61 (m, 2H), 2.48 (s, 3H), 2.34–2.14 (m, 5H), 2.08 (ddd, J = 13.3, 9.2, 4.5 Hz, 1H), 1.66–1.57 (m, 2H), 1.50 (q, J = 7.3 Hz, 2H), 1.38–1.30 (m, 2H), 1.17 (d, J = 7.1 Hz, 3H), 1.03 (s, 9H). HRMS (m/z) for C52H70ClN10O7S+ [M + H]+: calculated 1013.4833, found 1013.4847.
(2S,4R)-1-((S)-2-(7-(3-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)-piperazin-1-yl)-3-oxopropyl)amino)propanamido)heptanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)-benzyl)pyrrolidine-2-carboxamide (8).
Compound 8 was synthesized following the standard procedure for preparing compound 3 from intermediate 1 (12 mg, 0.02 mmol) and 73 (11.9 mg, 0.02 mmol, 1.0 equiv). Compound 8 was obtained as a white solid in TFA salt form (8.6 mg, yield 42%). 1H NMR (600 MHz, CD3OD) δ 8.95 (s, 1H), 8.58 (s, 1H), 7.54–7.38 (m, 6H), 7.36 (t, J = 6.6 Hz, 2H), 5.38–5.26 (m, 1H), 4.64 (s, 1H), 4.61–4.48 (m, 4H), 4.37 (d, J = 15.6 Hz, 1H), 4.18 (s, 1H), 4.02–3.81 (m, 6H), 3.73–3.53 (m, 5H), 3.41 (s, 1H), 3.27 (d, J = 6.0 Hz, 2H), 3.23–3.10 (m, 2H), 2.72–2.57 (m, 2H), 2.48 (s, 3H), 2.33–2.17 (m, 5H), 2.10 (s, 1H), 1.66–1.58 (m, 2H), 1.53–1.46 (m, 2H), 1.39–1.31 (m, 4H), 1.17 (d, J = 7.5 Hz, 3H), 1.03 (s, 9H). HRMS (m/z) for C53H72ClN10O7S+ [M + H]+: calculated 1027.4989, found 1027.4983.
(2S,4R)-1-((S)-2-(9-(3-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)-piperazin-1-yl)-3-oxopropyl)amino)propanamido)nonanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)-benzyl)pyrrolidine-2-carboxamide (9).
Compound 9 was synthesized following the standard procedure for preparing compound 3 from intermediate 1 (12 mg, 0.02 mmol) and 74 (12.4 mg, 0.02 mmol, 1.0 equiv). Compound 9 was obtained as a white solid in TFA salt form (9.9 mg, yield 47%). 1H NMR (600 MHz, CD3OD) δ 8.96 (s, 1H), 8.58 (d, J = 3.7 Hz, 1H), 7.55–7.39 (m, 6H), 7.39–7.32 (m, 2H), 5.31 (t, J = 8.0 Hz, 1H), 4.63 (s, 1H), 4.60–4.48 (m, 4H), 4.36 (d, J = 15.5 Hz, 1H), 4.18 (s, 1H), 3.97–3.78 (m, 6H), 3.73–3.60 (m, 5H), 3.40 (t, J = 8.9 Hz, 1H), 3.29–3.23 (m, 2H), 3.17 (dd, J = 7.8, 6.3 Hz, 2H), 2.67–2.61 (m, 2H), 2.48 (s, 3H), 2.34–2.14 (m, 5H), 2.08 (s, 1H), 1.59 (d, J = 7.0 Hz, 2H), 1.48 (d, J = 7.1 Hz, 2H), 1.35–1.29 (m, 8H), 1.18 (d, J = 7.0 Hz, 3H), 1.03 (s, 9H). HRMS (m/z) for C55H76ClN10O7S+ [M + H]+: calculated 1055.5302, found 1055.5303.
(2S,4R)-1-((S)-2-(10-(3-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)-piperazin-1-yl)-3-oxopropyl)amino)propanamido)decanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)-benzyl)pyrrolidine-2-carboxamide (10).
Compound 10 was synthesized following the standard procedure for preparing compound 3 from intermediate 1 (12 mg, 0.02 mmol) and 75 (12.7 mg, 0.02 mmol, 1.0 equiv). Compound 10 was obtained as a white solid in TFA salt form (2.8 mg, yield 13%). 1H NMR (600 MHz, CD3OD) δ 8.93 (s, 1H), 8.58 (s, 1H), 7.54–7.40 (m, 6H), 7.36 (d, J = 8.5 Hz, 2H), 5.31 (d, J = 8.2 Hz, 1H), 4.63 (s, 1H), 4.61–4.43 (m, 4H), 4.36 (d, J = 15.3 Hz, 1H), 4.19 (s, 1H), 4.03–3.78 (m, 6H), 3.76–3.56 (m, 5H), 3.49–3.39 (m, 1H), 3.17 (d, J = 7.7 Hz, 2H), 3.03–2.95 (m, 1H), 2.91–2.83 (m, 1H), 2.65 (d, J = 8.9 Hz, 2H), 2.47 (s, 3H), 2.36–2.14 (m, 5H), 2.13–2.06 (m, 1H), 1.69–1.56 (m, 2H), 1.56–1.46 (m, 2H), 1.41–1.25 (m, 10H), 1.18 (d, J = 7.0 Hz, 3H), 1.04 (s, 9H). HRMS (m/z) for C56H78ClN10O7S+ [M + H]+: calculated 1069.5459, found 1069.5464.
(2S,4R)-1-((S)-2-(11-(3-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)-piperazin-1-yl)-3-oxopropyl)amino) propanam ido)-undecanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methyl-thiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (11).
Compound 11 was synthesized following the standard procedure for preparing compound 3 from intermediate 1 (12 mg, 0.02 mmol) and 76 (13.0 mg, 0.02 mmol, 1.0 equiv). Compound 11 was obtained as a white solid in TFA salt form (11.3 mg, yield 52%). 1H NMR (600 MHz, CD3OD) δ 8.96 (s, 1H), 8.58 (d, J = 3.9 Hz, 1H), 7.53–7.38 (m, 6H), 7.38–7.28 (m, 2H), 5.31 (t, J = 8.0 Hz, 1H), 4.63 (s, 1H), 4.60–4.44 (m, 4H), 4.36 (d, J = 15.5 Hz, 1H), 4.18 (s, 1H), 3.97–3.78 (m, 6H), 3.72–3.59 (m, 5H), 3.45–3.36 (m, 1H), 3.29–3.24 (m, 2H), 3.17 (t, J = 7.1 Hz, 2H), 2.67–2.62 (m, 2H), 2.48 (s, 3H), 2.34–2.16 (m, 5H), 2.12–2.04 (m, 1H), 1.60 (dt, J = 15.2, 7.4 Hz, 2H), 1.49 (t, J = 7.0 Hz, 2H), 1.36–1.26 (m, 12H), 1.18 (d, J = 6.9 Hz, 3H), 1.03 (s, 9H). HRMS (m/z) for C57H80ClN10O7S+ [M + H]+: calculated 1083.5615, found 1083.5637.
(2S,4R)-1-((S)-2-(11-(3-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)-piperazin-1-yl)-3-oxopropyl)amino) propanam ido)-undecanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (12).
Compound 12 was synthesized following the standard procedure for preparing compound 3 from intermediate 1 (6.01 mg, 0.01 mmol) and 79 (7.4 mg, 0.01 mmol, 1.0 equiv). Compound 12 was obtained as a white solid in TFA salt form (6.5 mg, yield 59%). 1H NMR (600 MHz, CD3OD) δ 8.87 (s, 1H), 8.52 (s, 1H), 7.43 (dt, J = 15.8, 8.5 Hz, 6H), 7.37 (dd, J = 8.6, 6.8 Hz, 2H), 5.19 (t, J = 7.5 Hz, 1H), 5.03–4.98 (m, 1H), 4.62 (s, 1H), 4.59–4.50 (m, 2H), 4.43 (s, 1H), 4.13–4.03 (m, 1H), 4.02–3.90 (m, 1H), 3.91–3.70 (m, 7H), 3.62 (t, J = 11.0 Hz, 4H), 3.59–3.51 (m, 1H), 3.43–3.37 (m, 1H), 3.26 (dd, 1H), 3.17 (t, J = 7.2 Hz, 2H), 2.65 (t, J = 6.0 Hz, 2H), 2.47 (s, 3H), 2.32–2.27 (m, 1H), 2.26–2.14 (m, 3H), 1.98–1.90 (m, 1H), 1.50 (d, J = 7.0 Hz, 3H), 1.31 (s, 16H), 1.15 (d, J = 7.0 Hz, 3H), 1.04 (s, 9H). HRMS (m/z) for C58H82ClN10O7S+ [M + H]+: calculated 1097.5772, found 1097.57567.
N1-(2-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)piperazin-1-yl)-3-oxopropyl)amino)ethyl)-N12-((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)dodecanediamide (13).
Compound 13 was synthesized following the standard amide coupling procedure for preparing compound 3 from intermediate 2 (11.2 mg, 0.01 mmol) and 81 (13.1 mg, 0.01 mmol, 1.0 equiv). The resulting product was dissolved in TFA (1 mL) and DCM (1 mL). The mixture was stirred at room temperature for 30 min. The solvent was removed, and the mixture was purified by preparative HPLC. Compound 13 was obtained as a white solid in TFA salt form (17.4 mg, yield 79%). 1H NMR (600 MHz, CD3OD) δ 8.98 (s, 1H), 8.58 (s, 1H), 7.49–7.41 (m, 6H), 7.36 (dd, J = 8.5, 1.8 Hz, 2H), 5.31 (t, J = 8.0 Hz, 1H), 5.00 (q, J = 7.0 Hz, 1H), 4.62 (s, 1H), 4.59–4.50 (m, 2H), 4.46–4.42 (m, 1H), 4.24–4.14 (m, 1H), 4.13–3.99 (m, 1H), 3.99–3.90 (m, 2H), 3.90–3.78 (m, 3H), 3.75 (dd, J = 11.0, 4.0 Hz, 1H), 3.73–3.59 (m, 4H), 3.51–3.46 (m, 2H), 3.46–3.36 (m, 1H), 3.35–3.27 (m, 2H), 3.19 (t, J = 5.7 Hz, 2H), 2.48 (s, 3H), 2.36–2.14 (m, 5H), 2.00–1.91 (m, 1H), 1.67–1.53 (m, 4H), 1.50 (d, J = 7.0 Hz, 3H), 1.39–1.24 (m, 12H), 1.24–1.12 (m, 3H), 1.04 (s, 9H). 13C NMR (151 MHz, CD3OD) δ 176.70, 174.64, 171.83, 170.92, 169.36, 159.67, 151.49, 149.25, 147.64, 144.28, 134.38, 133.49, 131.98, 130.11, 129.62 (2C), 129.50 (2C), 129.10 (2C), 126.22 (2C), 126.06, 121.02, 70.78, 69.56, 67.64, 59.19, 57.60, 56.57, 50.23, 48.74, 48.64, 48.17, 45.49, 45.42, 44.99, 44.39, 41.40, 37.39, 36.43, 36.19, 35.74, 35.44, 35.27, 35.11, 29.16, 29.14, 29.04, 28.94, 28.93, 25.66 (3C), 25.30, 20.98, 19.10, 14.36. tR = 4.07 min; HRMS (m/z) for C58H82ClN10O7S+ [M + H]+: calculated 1097.5772, found 1097.5756.
3-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)piperazin-1-yl)-3-oxopropyl)amino)-N-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethyl)propenamide (14).
Compound 14 was synthesized following the standard procedure for preparing compound 3 from intermediate 1 (6.0 mg, 0.01 mmol) and 82 (4.8 mg, 0.01 mmol, 1.0 equiv). Compound 14 was obtained as a yellow solid in TFA salt form (2.1 mg, yield 25%). 1H NMR (600 MHz, CD3OD) δ 8.56 (s, 1H), 7.57 (t, J = 8.4 Hz, 1H), 7.43 (d, J = 6.5 Hz, 2H), 7.34 (d, J = 8.2 Hz, 2H), 7.16–6.96 (m, 2H), 5.29 (t, J = 7.8 Hz, 1H), 5.07 (d, J = 12.1 Hz, 1H), 4.49 (s, 1H), 4.22–3.93 (m, 3H), 3.92–3.75 (m, 4H), 3.71 (s, 2H), 3.67–3.53 (m, 5H), 3.50 (s, 2H), 3.46–3.33 (m, 3H), 3.27–3.14 (m, 2H), 2.87 (t, J = 14.8 Hz, 1H), 2.79–2.62 (m, 4H), 2.31–2.22 (m, 1H), 2.22–2.05 (m, 2H), 1.16 (d, J = 6.9 Hz, 3H). HRMS (m/z) for C41H49ClN9O8+ [M + H]+: calculated 830.3387, found 830.3385.
3-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)piperazin-1-yl)-3-oxopropyl)amino)-N-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)ethyl)propenamide (15).
Compound 15 was synthesized following the standard procedure for preparing compound 3 from intermediate 1 (6.0 mg, 0.01 mmol) and 83 (5.0 mg, 0.01 mmol, 1.0 equiv). Compound 15 was obtained as a yellow solid in TFA salt form (5.8 mg, yield 64%). 1H NMR (600 MHz, CD3OD) δ 8.57 (s, 1H), 7.59–7.53 (m, 1H), 7.45 (dd, J = 8.5, 1.8 Hz, 2H), 7.39–7.29 (m, 2H), 7.13–7.03 (m, 2H), 5.30 (t, J = 7.9 Hz, 1H), 5.07 (dd, J = 12.7, 5.5 Hz, 1H), 4.58–4.45 (m, 1H), 4.20–3.97 (m, 2H), 3.96–3.77 (m, 4H), 3.74–3.70 (m, 2H), 3.71–3.58 (m, 8H), 3.56 (t, J = 5.5 Hz, 2H), 3.51 (t, J = 5.2 Hz, 2H), 3.43–3.33 (m, 3H), 3.29–3.22 (m, 2H), 2.88 (ddd, J = 17.7, 13.8, 5.3 Hz, 1H), 2.80–2.67 (m, 2H), 2.67–2.60 (m, 2H), 2.28 (dd, J = 12.8, 7.5 Hz, 1H), 2.22–2.08 (m, 2H), 1.23–1.12 (m, 3H). HRMS (m/z) for C43H53ClN9O9+ [M + H]+: calculated 874.3649, found 874.3650.
3-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)piperazin-1-yl)-3-oxopropyl)amino)-N-(2-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy) ethoxy) ethoxy)ethyl)-propenamide (16).
Compound 16 was synthesized following the standard procedure for preparing compound 3 from intermediate 1 (6.1 mg, 0.01 mmol) and 84 (5.4 mg, 0.01 mmol, 1.0 equiv). Compound 16 was obtained as a yellow solid in TFA salt form (4.7 mg, yield 50%). 1H NMR (600 MHz, CD3OD) δ 8.57 (s, 1H), 7.56 (ddd, J = 8.5, 7.0, 1.3 Hz, 1H), 7.49–7.41 (m, 2H), 7.39–7.29 (m, 2H), 7.15–6.99 (m, 2H), 5.30 (t, J = 7.9 Hz, 1H), 5.07 (dd, J = 12.9, 5.4 Hz, 1H), 4.56–4.48 (m, 1H), 4.16 (s, 1H), 4.05 (s, 1H), 3.95–3.78 (m, 4H), 3.72 (t, J = 5.1 Hz, 2H), 3.70–3.55 (m, 12H), 3.56–3.48 (m, 4H), 3.43–3.37 (m, 1H), 3.35 (q, J = 3.9, 2.6 Hz, 2H), 3.29–3.20 (m, 2H), 2.92–2.83 (m, 1H), 2.80–2.68 (m, 2H), 2.68–2.61 (m, 2H), 2.29 (dd, J = 12.8, 7.5 Hz, 1H), 2.21–2.09 (m, 2H), 1.17 (d, J = 7.0 Hz, 3H). HRMS (m/z) for C45H57ClN9O10+ [M + H]+: calculated 918.3911, found 918.3916.
3-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)piperazin-1-yl)-3-oxopropyl)amino)-N-(14-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-3,6,9,12-tetraoxatetradecyl)propenamide (17).
Compound 17 was synthesized following the standard procedure for preparing compound 3 from intermediate 1 (9.9 mg, 0.0165 mmol) and 85 (9.3 mg, 0.0165 mmol, 1.0 equiv). Compound 17 was obtained as a yellow solid in TFA salt form (3.5 mg, yield 22%). 1H NMR (600 MHz, CD3OD) δ 8.57 (s, 1H), 7.60–7.52 (m, 1H), 7.49–7.41 (m, 1H), 7.37–7.24 (m, 3H), 7.10 (d, J = 8.6 Hz, 1H), 7.06 (d, J = 7.1 Hz, 1H), 5.29 (s, 1H), 5.13–5.00 (m, 1H), 4.58–4.42 (m, 1H), 4.16 (s, 1H), 4.03 (s, 1H), 3.95–3.74 (m, 4H), 3.72 (t, J = 5.2 Hz, 2H), 3.70–3.54 (m, 14H), 3.56–3.50 (m, 4H), 3.44–3.32 (m, 4H), 3.30–3.22 (m, 3H), 2.93–2.80 (m, 1H), 2.79–2.68 (m, 2H), 2.68–2.59 (m, 2H), 2.34–2.25 (m, 1H), 2.22–2.08 (m, 2H), 1.17 (d, J = 7.0 Hz, 3H). HRMS (m/z) for C47H61ClN9O11+ [M + H]+: calculated 962.4174, found 962.4167.
3-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)piperazin-1-yl)-3-oxopropyl)amino)-N-(17-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-3,6,9,12,15-pentaoxaheptadecyl)-propenamide (18).
Compound 18 was synthesized following the standard procedure for preparing compound 3 from intermediate 1 (9.9 mg, 0.0165 mmol) and 86 (10.0 mg, 0.0165 mmol, 1.0 equiv). Compound 18 was obtained as a yellow solid in TFA salt form (5.0 mg, yield 30%). 1H NMR (600 MHz, CD3OD) δ 8.58 (d, J = 3.6 Hz, 1H), 7.56 (dd, J = 8.6, 7.0 Hz, 1H), 7.49–7.42 (m, 1H), 7.39–7.22 (m, 3H), 7.10 (d, J = 8.5 Hz, 1H), 7.05 (d, J = 7.1 Hz, 1H), 5.30 (t, J = 8.0 Hz, 1H), 5.10–5.05 (m, 1H), 4.56–4.44 (m, 1H), 4.16 (s, 1H), 4.11–3.97 (m, 1H), 3.96–3.77 (m, 4H), 3.72 (t, J = 5.2 Hz, 2H), 3.70–3.55 (m, 18H), 3.56–3.49 (m, 4H), 3.43–3.32 (m, 4H), 3.30–3.22 (m, 3H), 2.94–2.82 (m, 1H), 2.79–2.69 (m, 2H), 2.67 (dd, J = 10.2, 4.0 Hz, 2H), 2.34–2.23 (m, 1H), 2.22–2.06 (m, 2H), 1.17 (d, J = 7.0 Hz, 3H). HRMS (m/z) for C49H65ClN9O12+ [M + H]+: calculated 1006.4436, found 1006.4449.
3-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)piperazin-1-yl)-3-oxopropyl)amino)-N-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethyl)propenamide (19).
Compound 19 was synthesized following the standard procedure for preparing compound 3 from intermediate 2 (6.1 mg, 0.01 mmol) and 87 (4.3 mg, 0.01 mmol, 1.0 equiv). Compound 19 was obtained as a yellow solid in TFA salt form (5.8 mg, yield 74%). 1H NMR (600 MHz, CD3OD) δ 8.55 (s, 1H), 7.62–7.54 (m, 1H), 7.42–7.37 (m, 4H), 7.13 (d, J = 8.6 Hz, 1H), 7.05 (dd, J = 7.0, 2.3 Hz, 1H), 5.26 (t, J = 7.7 Hz, 1H), 5.10–5.06 (m, 1H), 4.57 (d, J = 9.1 Hz, 1H), 4.06 (s, 2H), 3.85 (t, J = 14.7 Hz, 3H), 3.76–3.56 (m, 4H), 3.56–3.40 (m, 4H), 3.39–3.32 (m, 2H), 3.29–3.24 (m, 2H), 2.92–2.79 (m, 1H), 2.73 (t, J = 18.1 Hz, 2H), 2.66 (t, J = 5.6 Hz, 2H), 2.25 (t, J = 10.2 Hz, 1H), 2.20–2.08 (m, 2H), 1.14 (dd, J = 7.0, 1.7 Hz, 3H). HRMS (m/z) for C39H45ClN9O + [M + H]+: calculated 786.3125, found 786.3130.
3-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)piperazin-1-yl)-3-oxopropyl)amino)-N-(3-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)propyl)propenamide (20).
Compound 20 was synthesized following the standard procedure for preparing compound 14 from intermediate 1 (6.1 mg, 0.01 mmol) and 88 (4.4 mg, 0.01 mmol, 1.0 equiv). Compound 20 was obtained as a yellow solid in TFA salt form (7.8 mg, yield 97%). 1H NMR (600 MHz, CD3OD) δ 8.51 (s, 1H), 7.57–7.53 (m, 1H), 7.44 (d, J = 8.3 Hz, 2H), 7.35 (d, J = 8.2 Hz, 2H), 7.08–7.02 (m, 2H), 5.20 (t, J = 7.6 Hz, 1H), 5.13–5.02 (m, 1H), 4.61–4.48 (m, 1H), 4.11–3.89 (m, 2H), 3.90–3.67 (m, 4H), 3.68–3.46 (m, 4H), 3.41 (t, J = 6.5 Hz, 2H), 3.39–3.31 (m, 3H), 3.28–3.21 (m, 2H), 2.91–2.81 (m, 1H), 2.81–2.55 (m, 4H), 2.28–2.02 (m, 3H), 1.85 (t, J = 6.6 Hz, 2H), 1.11 (d, J = 7.0 Hz, 3H). HRMS (m/z) for C40H47ClN9O7+ [M + H]+: calculated 800.3281, found 800.3284.
3-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)piperazin-1-yl)-3-oxopropyl)amino)-N-(4-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)butyl)propenamide (21).
Compound 21 was synthesized following the standard procedure for preparing compound 3 from intermediate 1 (6.1 mg, 0.01 mmol) and 89 (4.6 mg, 0.01 mmol, 1.0 equiv). Compound 21 was obtained as a yellow solid in TFA salt form (5.7 mg, yield 70%). 1H NMR (600 MHz, CD3OD) δ 8.53 (s, 1H), 7.56 (t, J = 7.8 Hz, 1H), 7.44 (d, J = 8.4 Hz, 2H), 7.35 (dd, J = 8.4, 4.1 Hz, 2H), 7.05 (t, J = 9.5 Hz, 2H), 5.23 (s, 1H), 5.10–5.02 (m, 1H), 4.52 (s, 1H), 4.07 (d, J = 11.6 Hz, 1H), 4.03–3.89 (m, 1H), 3.89–3.71 (m, 4H), 3.71–3.50 (m, 4H), 3.42–3.33 (m, 3H), 3.26 (d, J = 15.5 Hz, 4H), 2.91–2.80 (m, 1H), 2.80–2.56 (m, 4H), 2.30–2.02 (m, 3H), 1.79–1.55 (m, 4H), 1.14 (d, J = 6.8 Hz, 3H). HRMS (m/z) for C41H49ClN9O7+ [M + H]+: calculated 814.3438, found 814.3443.
3-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)piperazin-1-yl)-3-oxopropyl)amino)-N-(5-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)pentyl)propenamide (22).
Compound 22 was synthesized following the standard procedure for preparing compound 3 from intermediate 1 (6.1 mg, 0.01 mmol) and 90 (4.7 mg, 0.01 mmol, 1.0 equiv). Compound 22 was obtained as a yellow solid in TFA salt form (4.7 mg, yield 57%). 1H NMR (600 MHz, CD3OD) δ 8.54 (s, 1H), 7.55 (t, J = 7.8 Hz, 1H), 7.44 (d, J = 8.1 Hz, 2H), 7.35 (dd, J = 8.4, 2.8 Hz, 2H), 7.04 (t, J = 7.8 Hz, 2H), 5.25 (s, 1H), 5.13–5.00 (m, 1H), 4.58–4.45 (m, 1H), 4.16–3.91 (m, 2H), 3.91–3.72 (m, 4H), 3.70–3.54 (m, 4H), 3.38–3.31 (m, 4H), 3.28–3.17 (m, 3H), 2.91–2.82 (m, 1H), 2.79–2.68 (m, 2H), 2.65 (d, J = 6.2 Hz, 2H), 2.28–2.21 (m, 1H), 2.21–2.14 (m, 1H), 2.13–2.07 (m, 1H), 1.73–1.65 (m, 2H), 1.57 (t, J = 7.5 Hz, 2H), 1.46 (d, J = 7.7 Hz, 2H), 1.22–1.07 (m, 3H). HRMS (m/z) for C42H51ClN9O7 + [M + H]+: calculated 828.3594, found 828.3597.
3-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)piperazin-1-yl)-3-oxopropyl)amino)-N-(6-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)hexyl)propenamide (23).
Compound 23 was synthesized following the standard procedure for preparing compound 3 from intermediate 1 (6.1 mg, 0.01 mmol) and 91 (4.9 mg, 0.01 mmol, 1.0 equiv). Compound 23 was obtained as a yellow solid in TFA salt form (4.7 mg, yield 57%). 1H NMR (600 MHz, CD3OD) δ 8.55 (s, 1H), 7.55 (dd, J = 8.6, 7.1 Hz, 1H), 7.45 (d, J = 8.0 Hz, 2H), 7.39–7.27 (m, 2H), 7.04 (t, J = 7.2 Hz, 2H), 5.28 (t, J = 7.8 Hz, 1H), 5.05 (dd, J = 12.8, 5.5 Hz, 1H), 4.56–4.48 (m, 1H), 4.14 (s, 1H), 4.09–3.97 (m, 1H), 3.92–3.73 (m, 4H), 3.70–3.56 (m, 4H), 3.42–3.32 (m, 4H), 3.28–3.23 (m, 1H), 3.19 (t, J = 7.0 Hz, 2H), 2.90–2.81 (m, 1H), 2.77–2.68 (m, 2H), 2.68–2.60 (m, 2H), 2.27 (dd, J = 12.9, 7.4 Hz, 1H), 2.21–2.13 (m, 1H), 2.13–2.06 (m, 1H), 1.71–1.65 (m, 2H), 1.58–1.51 (m, J = 7.2 Hz, 2H), 1.49–1.43 (m, 2H), 1.40 (d, J = 7.2 Hz, 2H), 1.16 (d, J = 7.0 Hz, 3H). HRMS (m/z) for C43H53ClN9O7+ [M + H]+: calculated 842.3751, found 842.3758.
3-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)piperazin-1-yl)-3-oxopropyl)amino)-N-(7-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)heptyl)propenamide (24).
Compound 24 was synthesized following the standard procedure for preparing compound 3 from intermediate 1 (6.1 mg, 0.01 mmol) and 92 (5.2 mg, 0.01 mmol, 1.0 equiv). Compound 24 was obtained as a yellow solid in TFA salt form (8.8 mg, yield 98%). 1H NMR (600 MHz, CD3OD) δ 8.57 (d, J = 4.4 Hz, 1H), 7.55 (dd, J = 8.6, 7.1 Hz, 1H), 7.45 (d, J = 8.4 Hz, 2H), 7.39–7.29 (m, 2H), 7.04 (dd, J = 7.8, 6.1 Hz, 2H), 5.30 (t, J = 7.9 Hz, 1H), 5.05 (dd, J = 12.8, 5.5 Hz, 1H), 4.54–4.50 (m, 1H), 4.16 (s, 1H), 4.04 (d, J = 22.0 Hz, 1H), 3.97–3.76 (m, 4H), 3.71–3.58 (m, 4H), 3.40 (t, J = 8.9 Hz, 1H), 3.34 (d, J = 6.9 Hz, 2H), 3.29–3.25 (m, 2H), 3.18 (t, J = 6.8 Hz, 2H), 2.86 (ddd, J = 17.6, 14.0, 5.4 Hz, 1H), 2.79–2.67 (m, 2H), 2.63 (t, J = 6.3 Hz, 2H), 2.28 (dd, J = 12.7, 7.5 Hz, 1H), 2.21–2.15 (m, 1H), 2.14–2.08 (m, 1H), 1.69–1.63 (m, 2H), 1.54–1.50 (m, 2H), 1.47–1.30 (m, 6H), 1.17 (d, J = 6.9 Hz, 3H). HRMS (m/z) for C44H55ClN9O7+ [M + H]+: calculated 856.3907, found 856.3912.
3-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)piperazin-1-yl)-3-oxopropyl)amino)-N-(8-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)octyl)propenamide (25).
Compound 25 was synthesized following the standard procedure for preparing compound 3 from intermediate 1 (6.1 mg, 0.01 mmol) and 93 (5.3 mg, 0.01 mmol, 1.0 equiv). Compound 25 was obtained as a yellow solid in TFA salt form (2.8 mg, yield 32%). 1H NMR (600 MHz, CD3OD) δ 8.56 (s, 1H), 7.55 (dd, J = 8.6, 7.0 Hz, 1H), 7.45 (d, J = 8.2 Hz, 2H), 7.41–7.29 (m, 2H), 7.04 (dd, J = 7.8, 3.6 Hz, 2H), 5.28 (t, J = 7.8 Hz, 1H), 5.05 (dd, J = 12.8, 5.5 Hz, 1H), 4.52 (dd, J = 9.3, 4.1 Hz, 1H), 4.15 (s, 1H), 4.10–3.97 (m, 1H), 3.94–3.74 (m, 4H), 3.74–3.50 (m, 4H), 3.44–3.32 (m, 4H), 3.28–3.24 (m, 1H), 3.17 (t, J = 7.2 Hz, 2H), 2.90–2.81 (m, 1H), 2.78–2.67 (m, 2H), 2.63 (t, J = 6.2 Hz, 2H), 2.27 (dd, J = 12.9, 7.4 Hz, 1H), 2.21–2.14 (m, 1H), 2.14–2.06 (m, 1H), 1.71–1.61 (m, 2H), 1.53–1.25 (m, 10H), 1.16 (d, J = 7.0 Hz, 3H). 13C NMR (201 MHz, CD3OD) δ 173.30, 171.00, 170.35, 169.44, 169.35, 167.92, 161.30, 159.61, 148.93, 146.92, 135.90, 134.30, 133.61, 132.49, 129.58 (2C), 129.47 (2C), 120.97, 116.66, 110.38, 109.54, 70.68, 50.20, 48.80, 45.53, 45.42, 44.96, 44.44, 44.36, 41.98, 41.45, 41.33, 39.06, 36.48, 30.84, 29.62, 28.82 (2C), 28.77 (2C), 26.40 (2C), 22.41, 19.15. tR = 4.03 min; HRMS (m/z) for C45H57ClN9O7+ [M + H]+: calculated 870.4064, found 870.4078.
N-(2-(((S)-2-(4-chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)piperazin-1-yl)-3-oxopropyl)amino)ethyl)-9-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)nonanamide (26).
Compound 26 was synthesized following the standard procedure for preparing compound 13 from intermediate 2 (19.2 mg, 0.034 mmol) and 9-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)nonanoic acid (94, 14.7 mg, 0.034 mmol, 1.0 equiv). Compound 26 was obtained as a yellow solid in TFA salt form (22.1 mg, yield 74%). 1H NMR (600 MHz, CD3OD) δ 8.57 (d, J = 3.9 Hz, 1H), 7.58–7.32 (m, 5H), 7.12–6.90 (m, 2H), 5.30 (t, J = 7.9 Hz, 1H), 5.06 (ddd, J = 12.9, 5.6, 3.4 Hz, 1H), 4.64–4.52 (m, 1H), 4.24–3.31 (m, 15H), 3.24–3.15 (m, 2H), 2.94–2.79 (m, 2H), 2.76–2.57 (m, 1H), 2.34–2.02 (m, 5H), 1.71–1.49 (m, 4H), 1.41–1.27 (m, 8H), 1.16 (d, J = 7.0 Hz, 3H). HRMS (m/z) for C45H57ClN9O7+ [M + H]+: calculated 870.4064, found 870.4056.
3-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)piperazin-1-yl)-3-oxopropyl)amino)-N-(9-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)nonyl)propenamide (27).
Compound 27 was synthesized following the standard procedure for preparing compound 3 from intermediate 1 (9.7 mg, 0.02 mmol) and 4-(9-aminononyl)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (99, 8 mg, 0.025 mmol, 1.0 equiv). Compound 27 was obtained as a white solid in TFA salt form (11.2 mg, yield 64%). 1H NMR (600 MHz, CD3OD) δ 8.57 (s, 1H), 7.74–7.68 (m, 2H), 7.65–7.59 (m, 1H), 7.47–7.42 (m, 2H), 7.36 (dd, J = 8.4, 1.2 Hz, 2H), 5.31 (t, J = 8.0 Hz, 1H), 5.17–5.10 (m, 1H), 4.54 (dd, J = 9.4, 4.1 Hz, 1H), 3.97–3.75 (m, 3H), 3.71–3.59 (m, 3H), 3.47–3.23 (m, 6H), 3.16 (t, J = 7.2 Hz, 2H), 3.12–3.06 (m, 2H), 2.94–2.84 (m, 1H), 2.80–2.69 (m, 2H), 2.67–2.62 (m, 2H), 2.32–2.25 (m, 1H), 2.21–2.11 (m, 2H), 1.69–1.62 (m, 2H), 1.48 (t, J = 7.1 Hz, 2H), 1.36 (s, 5H), 1.30 (d, J = 3.4 Hz, 5H), 1.17 (d, J = 7.0 Hz, 3H). HRMS (m/z) for C46H57ClN9O7+ [M + H]+: calculated 869.4112, found 869.4123.
3-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)piperazin-1-yl)-3-oxopropyl)amino)-N-(8-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)amino)octyl)propenamide (28).
Compound 28 was synthesized following the standard procedure for preparing compound 3 from intermediate 1 (12.2 mg, 0.025 mmol) and 3-(4-((8-aminooctyl)amino)-1-oxoisoindolin-2-yl)piperidine-2,6-dione (103, 9.7 mg, 0.025 mmol, 1.0 equiv). Compound 28 was obtained as a yellow solid in TFA salt form (8.8 mg, yield 41%). 1H NMR (600 MHz, CD3OD) δ 8.57 (s, 1H), 7.51–7.42 (m, 2H), 7.38–7.33 (m, 2H), 7.27 (ddd, J = 8.9, 6.9, 1.3 Hz, 1H), 7.21–7.12 (m, 1H), 7.00–6.90 (m, 1H), 5.30 (t, J = 7.9 Hz, 1H), 5.13 (dd, J = 13.4, 5.1 Hz, 1H), 4.57–4.48 (m, 1H), 4.36 (dd, J = 16.7, 1.1 Hz, 1H), 4.27 (d, J = 16.7 Hz, 1H), 4.19–4.12 (m, 1H), 3.97–3.46 (m, 8H), 3.44–3.23 (m, 6H), 3.21–3.10 (m, 2H), 3.02–2.81 (m, 2H), 2.68–2.59 (m, 2H), 2.51–2.43 (m, 1H), 2.32–2.24 (m, 1H), 2.21–2.10 (m, 2H), 1.56–1.43 (m, 4H), 1.39–1.24 (m, 8H), 1.24–1.08 (m, 3H). HRMS (m/z) for C45H59ClN9O6+ [M + H]+: calculated 856.4271, found 856.4264.
3-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)piperazin-1-yl)-3-oxopropyl)amino)-N-(8-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)octyl)propenamide (29).
Compound 29 was synthesized following the standard procedure for preparing compound 3 from intermediate 1 (12.2 mg, 0.025 mmol) and 4-((8-aminooctyl)oxy)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (104, 10 mg, 0.025 mmol, 1.0 equiv). Compound 29 was obtained as a white solid in TFA salt form (10.2 mg, yield 47%). 1H NMR (600 MHz, CD3OD) δ 8.57 (d, J = 2.2 Hz, 1H), 7.81–7.74 (m, 1H), 7.49–7.38 (m, 4H), 7.38–7.33 (m, 2H), 5.30 (t, J = 8.0 Hz, 1H), 5.10 (dd, J = 12.8, 5.5 Hz, 1H), 4.61–4.49 (m, 1H), 4.25–4.13 (m, 3H), 3.98–3.76 (m, 3H), 3.73–3.58 (m, 4H), 3.43–3.36 (m, 1H), 3.35–3.28 (m, 2H), 3.21–3.16 (m, 2H), 2.96–2.82 (m, 1H), 2.79–2.67 (m, 2H), 2.64 (t, J = 6.2 Hz, 2H), 2.31–2.24 (m, 1H), 2.23–2.03 (m, 2H), 1.86–1.81 (m, 2H), 1.59–1.45 (m, 4H), 1.46–1.33 (m, 8H), 1.17 (d, J = 7.0 Hz, 3H). HRMS (m/z) for C45H56ClN8O8+ [M + H]+: calculated 871.3904, found 871.3938.
4-((3-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]pyridin-7-yl)oxy)propyl)-(tert-butoxycarbonyl)amino)butanoic acid (30).
The intermediate 105 was synthesized following a known procedure.15 To the suspension of intermediate 105 (119 mg × 3, 0.5 mmol) and methyl-4-oxobutanonate (58 mg × 3, 0.5 mmol) in 5 mL of DCM was added sodium triacetoxyborohydride (211 mg × 3, 1.0 mmol) in three portions. Once the reaction mixture became a clear solution, a saturated NaHCO3 solution was added. The aqueous phase was extracted with DCM (10 mL × 3), dried over Na2SO4, filtered, and concentrated. The resulting residue was dissolved in DCM (15 mL). To this solution were added di-tert-butyl decarbonate (272 mg, 1.25 mmol) and triethylamine (188 mg, 1.86 mmol). The reaction was stirred for 30 min before the reaction mixture was concentrated. The residue was purified by silica gel column (hexane/EA = 1:3) to afford intermediate 106 as a yellow solid (178 mg, yield 53%). ESI m/z 538.8 [M + H]+. To the solution of intermediate 106 (178 mg, 0.34 mmol) in DMSO (5 mL) were added 2-methylbut-3-yn-2-ol (371 μL, 4.1 mmol), zinc powder (68 mg, 1.02 mmol), NaI (16 mg, 0.11 mmol), DBU (153 μL,1.02 mmol), and triethylamine (207 μL, 1.02 mmol). The reaction was degassed for 5 min, before the catalyst Pd(PPh3)4 (40 mg, 10 mol %) was added. The reaction was purged with nitrogen, sealed, and heated to 80 °C for 1 h. Saturated NH4Cl solution was added, and the aqueous layer was extracted with ethyl acetate, dried over Na2SO4, filtered, and concentrated under reduced pressure to afford the intermediate 107, which was used in the next step without further purification. The intermediate 107 (146 mg, 0.25 mmol) was dissolved in methanol (5 mL). To the resulting solution was added NaOH (0.5 mL, 3 N). The reaction was heated at 60 °C for 1 h before the reaction mixture was concentrated. The residue was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford title compound 30 as a white solid in TFA salt form (128.3 mg, yield 90%). 1H NMR (500 MHz, DMSO-d6) δ 8.15 (s, 1H), 7.03 (s, 2H), 4.93–4.74 (m, 2H), 4.40–4.26 (m, 2H), 3.39 (t, J = 7.1 Hz, 2H), 3.22 (t, J = 7.2 Hz, 2H), 2.21 (t, J = 7.2 Hz, 2H), 2.13–2.02 (m, 2H), 1.82–1.69 (m, 2H), 1.47 (t, J = 7.0 Hz, 3H), 1.41–1.30 (m, 15H). HRMS (m/z) for C27H38N7O7+ [M + H]+: calculated 572.2827, found 572.2838.
(2S,4R)-1-((S)-17-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]pyridin-7-yl)-oxy)-2-(tert-butyl)-4,10-dioxo-6-oxa-3,9,14-triazaheptadecanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (31).
To a solution of intermediate 30 (9.1 mg, 0.016 mmol) in DMSO (1 mL) were added 68 (9.1 mg, 0.016 mmol, 1.0 equiv), EDCI (4.6 mg, 0.024 mmol, 1.5 equiv), HOAt (3.3 mg, 0.024 mmol, 1.5 equiv), and NMM (4.8 mg, 0.048 mmol, 3.0 equiv). After stirring overnight at room temperature, the resulting mixture was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford the desired product. After this product was dissolved in DCM (1 mL), the reaction mixture was treated with TFA (1 mL) for 30 min. After the solvent was evaporated, the residue was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford compound 31 as a white solid in TFA salt form (6.1 mg, yield 39%). 1H NMR (500 MHz, CD3OD) δ 8.97 (d, J = 5.7 Hz, 1H), 8.23 (s, 1H), 7.51–7.34 (m, 4H), 5.05 (q, J = 7.0 Hz, 2H), 4.74–4.65 (m, 1H), 4.64–4.28 (m, 6H), 4.13–3.93 (m, 2H), 3.93–3.77 (m, 2H), 3.68–3.49 (m, 2H), 3.43–3.35 (m, 4H), 3.24–3.07 (m, 2H), 2.58–2.33 (m, 7H), 2.27 (dd, J = 13.2, 7.6 Hz, 1H), 2.10 (ddd, J = 13.5, 9.4, 4.4 Hz, 1H), 2.00 (h, J = 7.2 Hz, 2H), 1.69 (s, 6H), 1.58 (t, J = 7.0 Hz, 3H), 1.05 (s, 9H). HRMS (m/z) for C48H65N12O9S+ [M + H]+: calculated 985.4713, found 985.4728.
(2S,4R)-1-((S)-18-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]pyridin-7-yl)-oxy)-2-(tert-butyl)-4,11-dioxo-7-oxa-3,10,15-triazaoctadecanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (32).
Compound 32 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 108 (12.4 mg, 0.016 mmol, 1.0 equiv). Compound 32 was obtained as a white solid in TFA salt form (11.8 mg, yield 74%). 1H NMR (500 MHz, CD3OD) δ 8.97 (s, 1H), 8.24 (s, 1H), 7.55–7.32 (m, 4H), 5.05 (q, J = 7.1 Hz, 2H), 4.71–4.63 (m, 1H), 4.64–4.48 (m, 5H), 4.41 (d, J = 15.4 Hz, 1H), 3.96–3.80 (m, 2H), 3.74–3.68 (m, 2H), 3.50 (t, J = 5.4 Hz, 2H), 3.38–3.33 (m, 4H), 3.16 (t, J = 7.0 Hz, 2H), 2.56–2.36 (m, 9H), 2.29–2.24 (m, 1H), 2.13–2.08 (m, 1H), 2.03–1.95 (m, 2H), 1.69 (s, 6H), 1.58 (t, J = 7.1 Hz, 3H), 1.05 (s, 9H). HRMS (m/z) for C49H67N12O9S+ [M + H]+: calculated 999.4869, found 999.4875.
(2S,4R)-1-((S)-20-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]pyridin-7-yl)-oxy)-2-(tert-butyl)-4,13-dioxo-6,9-dioxa-3,12,17-triazaicosanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (33).
Compound 33 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 69 (9.8 mg, 0.016 mmol, 1.0 equiv). Compound 33 was obtained as a white solid in TFA salt form (10.6 mg, yield 64%). 1H NMR (500 MHz, CD3OD) δ 9.01 (d, J = 2.9 Hz, 1H), 8.26 (d, J = 3.0 Hz, 1H), 7.53–7.44 (m, 4H), 5.06 (q, J = 7.0 Hz, 2H), 4.95–4.71 (m, 1H), 4.64–4.31 (m, 6H), 4.03–4.01 (m, 2H), 3.94–3.78 (m, 2H), 3.80–3.58 (m, 4H), 3.56–3.48 (m, 2H), 3.46–3.26 (m, 4H), 3.22–3.08 (m, 2H), 2.59–2.35 (m, 7H), 2.36–2.23 (m, 1H), 2.19–2.06 (m, 1H), 2.03–1.93 (m, 2H), 1.70 (s, 6H), 1.59 (t, J = 7.0 Hz, 3H), 1.06 (s, 9H). HRMS (m/z) for C50H69N12O10S+ [M + H]+: calculated 1029.4975, found 1029.4983.
(2S,4R)-1-((S)-21-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]pyridin-7-yl)-oxy)-2-(tert-butyl)-4,14-dioxo-7,10-dioxa-3,13,18-triazahenicosanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (34).
Compound 34 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 109 (13.1 mg, 0.016 mmol, 1.0 equiv). Compound 34 was obtained as a white solid in TFA salt form (6.4 mg, yield 38%). 1H NMR (500 MHz, CD3OD) δ 9.01 (s, 1H), 8.26 (s, 1H), 7.54–7.36 (m, 4H), 5.07 (q, J = 7.1 Hz, 2H), 4.67 (d, J = 6.2 Hz, 1H), 4.64–4.47 (m, 5H), 4.39 (d, J = 15.4 Hz, 1H), 3.91 (d, J = 11.0 Hz, 1H), 3.82 (dd, J = 11.0, 3.9 Hz, 1H), 3.80–3.69 (m, 2H), 3.66–3.56 (m, 4H), 3.50 (t, J = 5.4 Hz, 2H), 3.36–3.31 (m, 4H), 3.17 (t, J = 7.0 Hz, 2H), 2.59 (ddd, J = 15.0, 7.3, 5.2 Hz, 1H), 2.55–2.34 (m, 8H), 2.28–2.20 (m, 1H), 2.10 (ddd, J = 13.3, 9.1, 4.5 Hz, 1H), 2.03–1.96 (m, 2H), 1.70 (s, 6H), 1.59 (t, J = 7.1 Hz, 3H), 1.05 (s, 9H). HRMS (m/z) for C51H71N12O10S+ [M + H]+: calculated 1043.5131, found 1043.5145.
(2S,4R)-1-((S)-23-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]pyridin-7-yl)-oxy)-2-(tert-butyl)-4,16-dioxo-6,9,12-trioxa-3,15,20-triazatricosanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (35).
Compound 35 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 110 (13.5 mg, 0.016 mmol, 1.0 equiv). Compound 35 was obtained as a white solid in TFA salt form (8.1 mg, yield 47%). 1H NMR (500 MHz, CD3OD) δ 9.00 (s, 1H), 8.25 (s, 1H), 7.49–7.41 (m, 4H), 5.06 (q, J = 6.9 Hz, 2H), 4.75–4.64 (m, 1H), 4.64–4.45 (m, 5H), 4.48–4.30 (m, 1H), 4.15–3.97 (m, 2H), 3.93–3.78 (m, 2H), 3.78–3.57 (m, 8H), 3.49 (t, J = 5.4 Hz, 2H), 3.34–3.24 (m, 4H), 3.17 (d, J = 7.1, 2.4 Hz, 2H), 2.54–2.33 (m, 7H), 2.33–2.21 (m, 1H), 2.17–2.05 (m, 1H), 2.04–1.92 (m, 2H), 1.75–1.63 (m, 6H), 1.59 (t, J = 7.1 Hz, 3H), 1.06 (s, 9H). HRMS (m/z) for C52H73N12O11S+ [M + H]+: calculated 1073.5237, found 1073.5231.
(2S,4R)-1-((S)-24-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]pyridin-7-yl)-oxy)-2-(tert-butyl)-4,17-dioxo-7,10,13-trioxa-3,16,21-triazatetracosanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (36).
Compound 36 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 111 (13.8 mg, 0.016 mmol, 1.0 equiv). Compound 36 was obtained as a white solid in TFA salt form (10.7 mg, yield 61%). 1H NMR (500 MHz, CD3OD) δ 9.02 (s, 1H), 8.27 (s, 1H), 7.59–7.33 (m, 4H), 5.07 (q, J = 7.2 Hz, 2H), 4.73–4.62 (m, 1H), 4.62–4.47 (m, 5H), 4.39 (dd, J = 15.6, 2.6 Hz, 1H), 3.94–3.87 (m, 1H), 3.87–3.66 (m, 3H), 3.70–3.54 (m, 8H), 3.50 (t, J = 5.4 Hz, 2H), 3.37–3.30 (m, 4H), 3.17 (t, J = 7.0 Hz, 2H), 2.60 (ddd, J = 15.0, 7.5, 5.2 Hz, 1H), 2.52–2.30 (m, 8H), 2.27–2.21 (m, 1H), 2.10 (ddd, J = 13.3, 9.2, 4.4 Hz, 1H), 2.04–1.97 (m, 2H), 1.70 (s, 6H), 1.60 (t, J = 7.1 Hz, 3H), 1.06 (s, 9H). HRMS (m/z) for C53H75N12O11S+ [M + H]+: calculated 1087.5393, found 1087.5383.
(2S,4R)-1-((S)-27-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]pyridin-7-yl)-oxy)-2-(tert-butyl)-4,20-dioxo-7,10,13,16-tetraoxa-3,19,24-triazaheptacosanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)-pyrrolidine-2-carboxamide (37).
Compound 37 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 70 (11.4 mg, 0.016 mmol, 1.0 equiv). Compound 37 was obtained as a white solid in TFA salt form (12.6 mg, yield 70%). 1H NMR (500 MHz, CD3OD) δ 9.02 (s, 1H), 8.27 (s, 1H), 7.56–7.33 (m, 4H), 5.07 (q, J = 7.1 Hz, 2H), 4.65 (s, 1H), 4.62–4.46 (m, 5H), 4.38 (d, J = 15.5 Hz, 1H), 3.90 (d, J = 11.0 Hz, 1H), 3.82 (dd, J = 11.0, 3.9 Hz, 1H), 3.77–3.71 (m, 2H), 3.67–3.55 (m, 12H), 3.51 (t, J = 5.4 Hz, 2H), 3.40–3.32 (m, 4H), 3.17 (t, J = 7.1 Hz, 2H), 2.60 (ddd, J = 15.0, 7.3, 5.3 Hz, 1H), 2.55–2.29 (m, 8H), 2.26–2.22 (m, 1H), 2.10 (ddd, J = 13.3, 9.1, 4.5 Hz, 1H), 2.03–1.98 (m, 2H), 1.70 (s, 6H), 1.60 (t, J = 7.1 Hz, 3H), 1.05 (s, 9H). HRMS (m/z) for C55H79N12O12S+ [M + H]+: calculated 1131.5656, found 1131.5653.
(2S,4R)-1-((S)-30-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]pyridin-7-yl)-oxy)-2-(tert-butyl)-4,23-dioxo-7,10,13,16,19-pentaoxa-3,22,27-triazatriacontanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)-pyrrolidine-2-carboxamide (38).
Compound 38 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 112 (15.2 mg, 0.016 mmol, 1.0 equiv). Compound 38 was obtained as a white solid in TFA salt form (9.4 mg, yield 50%). 1H NMR (500 MHz, CD3OD) δ 8.99 (s, 1H), 8.25 (s, 1H), 7.53–7.36 (m, 4H), 5.07 (q, J = 7.1 Hz, 2H), 4.66 (s, 1H), 4.61–4.47 (m, 5H), 4.38 (d, J = 15.5 Hz, 1H), 3.90 (d, J = 11.1 Hz, 1H), 3.86–3.70 (m, 3H), 3.66–3.56 (m, 16H), 3.50 (t, J = 5.4 Hz, 2H), 3.36–3.28 (m, 4H), 3.18 (t, J = 7.0 Hz, 2H), 2.60 (ddd, J = 14.8, 7.4, 5.1 Hz, 1H), 2.52–2.32 (m, 8H), 2.27–2.21 (m, 1H), 2.15–2.02 (m, 1H), 1.99 (q, J = 6.8 Hz, 2H), 1.69 (s, 6H), 1.59 (t, J = 7.1 Hz, 3H), 1.06 (s, 9H). HRMS (m/z) for C57H83N12O13S+ [M + H]+: calculated 1175.5918, found 1175.5911.
(2S,4R)-1-((S)-2-(2-(4-((3-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]-pyridin-7-yl)oxy)propyl)amino)butanamido)acetamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)-pyrrolidine-2-carboxamide (39).
Compound 39 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 113 (11.4 mg, 0.016 mmol, 1.0 equiv). Compound 39 was obtained as a white solid in TFA salt form (9.3 mg, yield 62%). 1H NMR (500 MHz, CD3OD) δ 9.01 (d, J = 6.5 Hz, 1H), 8.25 (d, J = 6.2 Hz, 1H), 7.56–7.39 (m, 4H), 5.06 (dd, J = 12.7, 5.7 Hz, 2H), 4.83–4.31 (m, 7H), 4.08–3.78 (m, 4H), 3.46–3.35 (m, 2H), 3.21 (q, J = 7.2, 5.9 Hz, 2H), 2.80–2.29 (m, 7H), 2.33–2.18 (m, 1H), 2.18–1.90 (m, 3H), 1.70 (s, 6H), 1.64–1.51 (m, 3H), 1.06 (d, J = 10.6 Hz, 9H). HRMS (m/z) for C46H61N12O8S+ [M + H]+: calculated 941.4451, found 941.4458.
(2S,4R)-1-((S)-2-(3-(4-((3-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]-pyridin-7-yl)oxy)propyl)amino)butanamido)propanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)-pyrrolidine-2-carboxamide (40).
Compound 40 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 114 (11.7 mg, 0.016 mmol, 1.0 equiv). Compound 40 was obtained as a white solid in TFA salt form (11.3 mg, yield 72%).1H NMR (500 MHz, CD3OD) δ 9.02 (s, 1H), 8.26 (s, 1H), 7.51–7.28 (m, 4H), 5.07 (q, J = 7.0 Hz, 2H), 4.62 (s, 1H), 4.59–4.47 (m, 5H), 4.40 (d, J = 15.5 Hz, 1H), 3.95 (d, J = 11.0 Hz, 1H), 3.82 (dd, J = 11.0, 3.9 Hz, 1H), 3.47–3.33 (m, 4H), 3.17 (t, J = 7.2 Hz, 2H), 2.57–2.33 (m, 9H), 2.26 (dd, J = 13.3, 7.6 Hz, 1H), 2.11 (ddd, J = 13.4, 9.3, 4.3 Hz, 1H), 2.02–1.96 (p, J = 6.8 Hz, 2H), 1.70 (s, 6H), 1.60 (t, J = 7.1 Hz, 3H), 1.05 (s, 9H). HRMS (m/z) for C47H63N12O8S+ [M + H]+: calculated 955.4607, found 955.4613.
(2S,4R)-1-((S)-2-(4-(4-((3-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]-pyridin-7-yl)oxy)propyl)amino)butanamido)butanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)-pyrrolidine-2-carboxamide (41).
Compound 41 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 115 (11.9 mg, 0.016 mmol, 1.0 equiv). Compound 41 was obtained as a white solid in TFA salt form (9.3 mg, yield 60%). 1H NMR (500 MHz, CD3OD) δ 9.02 (s, 1H), 8.27 (s, 1H), 7.58–7.34 (m, 4H), 5.07 (q, J = 7.3, 6.3 Hz, 2H), 4.75–4.48 (m, 6H), 4.40 (dd, J = 15.5, 4.9 Hz, 1H), 3.96–3.77 (m, 2H), 3.40–3.32 (m, 4H), 3.18 (q, J = 7.2, 6.4 Hz, 2H), 2.63–2.19 (m, 10H), 2.11 (ddd, J = 13.3, 9.2, 4.6 Hz, 1H), 2.00 (p, J = 6.6 Hz, 2H), 1.85–1.64 (m, 8H), 1.60 (t, J = 7.0 Hz, 3H), 1.06 (d, J = 4.6 Hz, 9H). HRMS (m/z) for C48H65N12O8S+ [M + H]+: calculated 969.4764, found 969.4754.
(2S,4R)-1-((S)-2-(5-(4-((3-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]-pyridin-7-yl)oxy)propyl)amino)butanamido)pentanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)-pyrrolidine-2-carboxamide (42).
Compound 42 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 71 (9.1 mg, 0.016 mmol, 1.0 equiv). Compound 42 was obtained as a white solid in TFA salt form (6.7 mg, yield 43%). 1H NMR (500 MHz, CD3OD) δ 8.98 (s, 1H), 8.24 (s, 1H), 7.52–7.37 (m, 4H), 5.06 (q, J = 7.1 Hz, 2H), 4.63 (s, 1H), 4.58–4.51 (m, 5H), 4.39 (d, J = 15.4 Hz, 1H), 3.91 (d, J = 11.0 Hz, 1H), 3.82 (dd, J = 11.0, 4.0 Hz, 1H), 3.41–3.26 (m, 2H), 3.22–3.04 (m, 4H), 2.51–2.35 (m, 7H), 2.35–2.18 (m, 3H), 2.17–2.05 (m, 1H), 2.03–1.96 (m, J = 6.9 Hz, 2H), 1.69 (s, 6H), 1.65–1.40 (m, 7H), 1.04 (s, 9H). HRMS (m/z) for C49H67N12O8S+ [M + H]+: calculated 983.4920, found 983.4927.
(2S,4R)-1-((S)-2-(6-(4-((3-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]-pyridin-7-yl)oxy)propyl)amino)butanamido)hexanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)-pyrrolidine-2-carboxamide (43).
Compound 43 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 72 (9.3 mg, 0.016 mmol, 1.0 equiv). Compound 43 was obtained as a white solid in TFA salt form (6.7 mg, yield 42%). 1H NMR (500 MHz, CD3OD) δ 8.97 (s, 1H), 8.23 (s, 1H), 7.54–7.35 (m, 4H), 5.06 (q, J = 7.0 Hz, 2H), 4.64 (s, 1H), 4.61–4.47 (m, 5H), 4.38 (d, J = 15.5 Hz, 1H), 3.92 (d, J = 11.0 Hz, 1H), 3.83 (dd, J = 10.9, 3.9 Hz, 1H), 3.39–3.32 (m, 2H), 3.17 (t, J = 7.0 Hz, 2H), 3.08 (t, J = 7.1 Hz, 2H), 2.50 (s, 3H), 2.48–2.35 (m, 4H), 2.34–2.20 (m, 3H), 2.11 (ddd, J = 13.3, 9.1, 4.5 Hz, 1H), 2.03–1.96 (m, 2H), 1.69 (s, 6H), 1.65–1.54 (m, 5H), 1.49–1.45 (m, J = 7.3 Hz, 2H), 1.39–1.25 (m, 2H), 1.05 (s, 9H). HRMS (m/z) for C50H69N12O8S+ [M + H]+: calculated 997.5077, found 997.5079.
(2S,4R)-1-((S)-2-(7-(4-((3-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]-pyridin-7-yl)oxy)propyl)amino)butanamido)heptanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)-pyrrolidine-2-carboxamide (44).
Compound 44 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 73 (9.5 mg, 0.016 mmol, 1.0 equiv). Compound 44 was obtained as a white solid in TFA salt form (13.9 mg, yield 86%). 1H NMR (500 MHz, CD3OD) δ 9.11–8.85 (m, 1H), 8.25 (s, 1H), 7.58–7.32 (m, 4H), 5.09–5.06 (m, J = 9.3, 4.8 Hz, 2H), 4.72–4.48 (m, 6H), 4.39 (dd, J = 15.4, 3.3 Hz, 1H), 3.92 (d, J = 10.7 Hz, 1H), 3.83 (d, J = 10.9, 3.6 Hz, 1H), 3.33–3.27 (m, 2H), 3.22–3.14 (m, 2H), 3.11–2.95 (m, 2H), 2.57–2.17 (m, 10H), 2.15–2.09 (m, 1H), 2.04–1.97 (m, 2H), 1.73–1.51 (m, 9H), 1.51–1.41 (m, 2H), 1.41–1.23 (m, 6H), 1.05 (s, 9H). HRMS (m/z) for C51H71N12O8S+ [M + H]+: calculated 1011.5233, found 1011.5239.
(2S,4R)-1-((S)-2-(8-(4-((3-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]-pyridin-7-yl)oxy)propyl)amino)butanamido)octanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)-pyrrolidine-2-carboxamide (45).
Compound 45 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 116 (12.8 mg, 0.016 mmol, 1.0 equiv). Compound 45 was obtained as a white solid in TFA salt form (8.6 mg, yield 52%). 1H NMR (500 MHz, CD3OD) δ 8.97 (s, 1H), 8.22 (s, 1H), 7.57–7.19 (m, 4H), 5.14–4.97 (m, 2H), 4.67–4.43 (m, 6H), 4.38 (dd, J = 15.5, 2.6 Hz, 1H), 3.92 (d, J = 11.0 Hz, 1H), 3.82 (dd, J = 11.0, 3.9 Hz, 1H), 3.32 (d, J = 2.2 Hz, 2H), 3.19–3.15 (m, 2H), 3.10–3.06 (m, 2H), 2.54–2.35 (m, 8H), 2.34–2.18 (m, 2H), 2.13–2.06 (m, 1H), 2.04–1.93 (m, 2H), 1.77–1.52 (m, 9H), 1.48–1.40 (m, J = 7.2 Hz, 2H), 1.39–1.12 (m, 8H), 1.05 (s, 9H). HRMS (m/z) for C52H73N12O8S+ [M + H]+: calculated 1025.5390, found 1025.5387.
(2S,4R)-1-((S)-2-(9-(4-((3-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]-pyridin-7-yl)oxy)propyl)amino)butanamido)nonanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)-pyrrolidine-2-carboxamide (46).
Compound 46 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 74 (10 mg, 0.016 mmol, 1.0 equiv). Compound 46 was obtained as a white solid in TFA salt form (7 mg, yield 42%). 1H NMR (500 MHz, CD3OD) δ 8.95 (s, 1H), 8.21 (s, 1H), 7.54–7.31 (m, 4H), 5.06 (q, J = 7.1 Hz, 2H), 4.65 (s, 1H), 4.62–4.48 (m, 5H), 4.38 (d, J = 15.5 Hz, 1H), 3.92 (d, J = 11.0 Hz, 1H), 3.83 (dd, J = 11.0, 3.9 Hz, 1H), 3.37–3.29 (m, J = 2.8 Hz, 2H), 3.17 (t, J = 6.9 Hz, 2H), 3.05 (t, J = 7.1 Hz, 2H), 2.52–2.37 (m, 7H), 2.34–2.19 (m, 3H), 2.15–2.04 (m, 1H), 2.03–1.95 (m, 2H), 1.69 (s, 6H), 1.65–1.54 (m, 3H), 1.48–1.42 (m, 2H), 1.30 (d, J = 13.7 Hz, 10H), 1.05 (s, 9H). HRMS (m/z) for C53H75N12O8S+ [M + H]+: calculated 1039.5546, found 1039.5534.
(2S,4R)-1-((S)-2-(10-(4-((3-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]-pyridin-7-yl)oxy)propyl)amino)butanamido)decanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)-pyrrolidine-2-carboxamide (47).
Compound 47 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 75 (13.2 mg, 0.016 mmol, 1.0 equiv). Compound 47 was obtained as a white solid in TFA salt form (3.9 mg, yield 23%). 1H NMR (500 MHz, CD3OD) δ 8.92 (d, J = 4.5 Hz, 1H), 8.17 (d, J = 5.8 Hz, 1H), 7.62–7.29 (m, 4H), 5.15–5.00 (m, 2H), 4.65 (d, J = 2.5 Hz, 1H), 4.63–4.46 (m, 5H), 4.42–4.29 (m, 1H), 3.92 (d, J = 11.0 Hz, 1H), 3.83 (dd, J = 11.0, 3.9 Hz, 1H), 3.35–3.29 (m, 2H), 3.17 (dd, J = 7.8, 5.9 Hz, 2H), 3.08–3.03 (m, J = 7.3, 3.5 Hz, 2H), 2.58–2.18 (m, 10H), 2.11 (ddd, J = 13.2, 9.0, 4.5 Hz, 1H), 2.04–1.89 (m, 2H), 1.76–1.50 (m, 9H), 1.42 (t, J = 7.0 Hz, 2H), 1.38–1.12 (m, 12H), 1.05 (s, 9H). HRMS (m/z) for C54H77N12O8S+ [M + H]+: calculated 1053.5703, found 1053.5711.
(2S,4R)-1-((S)-2-(11-(4-((3-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]-pyridin-7-yl)oxy)propyl)amino)butanamido)undecanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)-pyrrolidine-2-carboxamide (48).
Compound 48 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 76 (10.4 mg, 0.016 mmol, 1.0 equiv). Compound 48 was obtained as a white solid in TFA salt form (4.8 mg, yield 26%). 1H NMR (500 MHz, CD3OD) δ 8.96 (d, J = 2.6 Hz, 1H), 8.22 (d, J = 2.5 Hz, 1H), 7.58–7.32 (m, 4H), 5.13–5.00 (m, 2H), 4.65 (s, 1H), 4.63–4.50 (m, 5H), 4.37 (dd, J = 15.4, 2.8 Hz, 1H), 3.92 (d, J = 11.0 Hz, 1H), 3.83 (dd, J = 11.0, 3.9 Hz, 1H), 3.33–3.30 (m, 2H), 3.21–3.14 (m, 2H), 3.06 (dd, J = 9.4, 4.5 Hz, 2H), 2.54–2.38 (m, 7H), 2.36–2.20 (m, 3H), 2.11 (ddd, J = 13.3, 9.1, 4.5 Hz, 1H), 2.06–1.91 (m, 2H), 1.75–1.57 (m, 9H), 1.43 (t, J = 7.1 Hz, 2H), 1.39–1.22 (m, 14H), 1.05 (s, 9H). HRMS (m/z) for C55H79N12O8S+ [M + H]+: calculated 1067.5859, found 1067.5853.
4-((3-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]pyridin-7-yl)oxy)propyl)-amino)-N-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethyl)butanamide (49).
Compound 49 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 82 (7.6 mg, 0.016 mmol, 1.0 equiv). Compound 49 was obtained as a yellow solid in TFA salt form (9.6 mg, yield 26%). 1H NMR (500 MHz, CD3OD) δ 8.17 (s, 1H), 7.50 (dd, J = 8.6, 7.1 Hz, 1H), 7.01 (t, J = 8.2 Hz, 2H), 5.06 (dd, J = 12.7, 5.5 Hz, 1H), 4.99 (q, J = 7.1 Hz, 2H), 4.50 (t, J = 5.8 Hz, 2H), 3.66 (t, J = 5.1 Hz, 2H), 3.52 (t, J = 5.3 Hz, 2H), 3.44 (t, J = 5.1 Hz, 2H), 3.40–3.26 (m, 4H), 3.14 (t, J = 6.8 Hz, 2H), 2.88 (ddd, J = 17.3, 13.8, 5.3 Hz, 1H), 2.81–2.63 (m, 2H), 2.48 (t, J = 6.4 Hz, 2H), 2.39–2.35 (m, 2H), 2.17–2.10 (m, 1H), 2.01–1.94 (m, 2H), 1.69 (s, 6H), 1.53 (t, J = 7.1 Hz, 3H). HRMS (m/z) for C39H48N11O9+ [M + H]+: calculated 814.3631, found 814.3611.
4-((3-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]pyridin-7-yl)oxy)propyl)-amino)-N-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)ethyl)butanamide (50).
Compound 50 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 83 (8.3 mg, 0.016 mmol, 1.0 equiv). Compound 50 was obtained as a yellow solid in TFA salt form (7.8 mg, yield 57%). 1H NMR (500 MHz, CD3OD) δ 8.16 (s, 1H), 7.53 (dd, J = 8.5, 7.1 Hz, 1H), 7.05 (dd, J = 15.6, 7.8 Hz, 2H), 5.09–4.97 (m, 3H), 4.51 (t, J = 5.9 Hz, 2H), 3.73 (t, J = 5.2 Hz, 2H), 3.70–3.60 (m, 4H), 3.55–3.46 (m, 4H), 3.34–3.24 (m, 4H), 3.14 (t, J = 7.0 Hz, 2H), 2.87 (ddd, J = 17.6, 14.0, 5.3 Hz, 1H), 2.80–2.64 (m, 2H), 2.46–2.34 (m, 4H), 2.17–2.11 (m, 1H), 1.99–1.95 (m, 2H), 1.69 (s, 6H), 1.56 (t, J = 7.1 Hz, 3H). HRMS (m/z) for C41H52N11O10+ [M + H]+: calculated 858.3893, found 858.3879.
4-((3-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]pyridin-7-yl)oxy)propyl)-amino)-N-(2-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)ethoxy)ethyl)butanamide (51).
Compound 51 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 84 (9 mg, 0.016 mmol, 1.0 equiv). Compound 51 was obtained as a yellow solid in TFA salt form (11.3 mg, yield 78%). 1H NMR (500 MHz, CD3OD) δ 8.23 (s, 1H), 7.51 (dd, J = 8.6, 7.1 Hz, 1H), 7.02 (dd, J = 19.2, 7.8 Hz, 2H), 5.09–4.99 (m, 3H), 4.52 (t, J = 5.9 Hz, 2H), 3.73 (t, J = 5.2 Hz, 2H), 3.68 (s, 4H), 3.69–3.63 (m, 2H), 3.66–3.55 (m, 2H), 3.49 (m, 4H), 3.37–3.25 (m, 4H), 3.15 (t, J = 7.0 Hz, 2H), 2.88 (ddd, J = 17.6, 14.0, 5.3 Hz, 1H), 2.82–2.65 (m, 2H), 2.47–2.38 (m, 4H), 2.16–2.10 (m, 1H), 2.01–1.96 (m, 2H), 1.70 (s, 6H), 1.57 (t, J = 7.1 Hz, 3H). HRMS (m/z) for C43H56N11O11+ [M + H]+: calculated 902.4155, found 902.4134.
4-((3-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]pyridin-7-yl)oxy)propyl)-amino)-N-(14-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-3,6,9,12-tetraoxatetradecyl)butanamide (52).
Compound 52 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 85 (9 mg, 0.016 mmol, 1.0 equiv). Compound 52 was obtained as a yellow solid in TFA salt form (5.2 mg, yield 34%). 1H NMR (500 MHz, CD3OD) δ 8.19 (s, 1H), 7.52 (dd, J = 8.6, 7.1 Hz, 1H), 7.04 (dd, J = 19.6, 7.8 Hz, 2H), 5.09–4.98 (m, 3H), 4.51 (t, J = 5.8 Hz, 2H), 3.73 (t, J = 5.2 Hz, 2H), 3.70–3.60 (m, 10H), 3.58 (dd, J = 6.1, 3.4 Hz, 2H), 3.49 (t, J = 5.3 Hz, 4H), 3.34–3.25 (m, 4H), 3.16 (t, J = 7.0 Hz, 2H), 2.88 (ddd, J = 17.5, 14.0, 5.3 Hz, 1H), 2.81–2.65 (m, 2H), 2.46 (t, J = 6.5 Hz, 2H), 2.41–2.37 (m, 2H), 2.17–2.10 (m, 1H), 2.01–1.96 (m, 2H), 1.69 (s, 6H), 1.57 (t, J = 7.1 Hz, 3H). HRMS (m/z) for C45H60N11O12+ [M + H]+: calculated 946.4417, found 946.4412.
4-((3-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]pyridin-7-yl)oxy)propyl)-amino)-N-(17-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-3,6,9,12,15-pentaoxaheptadecyl)butanamide (53).
Compound 53 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 86 (9.6 mg, 0.016 mmol, 1.0 equiv). Compound 53 was obtained as a yellow solid in TFA salt form (12.8 mg, yield 81%). 1H NMR (500 MHz, CD3OD) δ 8.24 (d, J = 3.6 Hz, 1H), 7.60–7.45 (m, 1H), 7.07–7.01 (m, 2H), 5.07–5.03 (m, 3H), 4.54 (q, J = 5.8, 5.0 Hz, 2H), 3.82–3.56 (m, 18H), 3.49 (q, J = 5.3 Hz, 4H), 3.37–3.27 (m, 4H), 3.23–3.15 (m, 2H), 2.93–2.82 (m, 1H), 2.83–2.72 (m, 2H), 2.47–2.37 (m, 4H), 2.15–2.10 (m, 1H), 2.01–1.97 (m, 2H), 1.70 (d, J = 3.7 Hz, 6H), 1.62–1.53 (m, 3H). HRMS (m/z) for C47H64N11O13+ [M + H]+: calculated 990.4680, found 990.4668.
4-((3-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]pyridin-7-yl)oxy)propyl)-amino)-N-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)-amino)ethyl)butanamide (54).
Compound 54 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 87 (7 mg, 0.016 mmol, 1.0 equiv). Compound 54 was obtained as a yellow solid in TFA salt form (12.1 mg, yield 98%). 1H NMR (500 MHz, CD3OD) δ 8.26 (d, J = 13.2 Hz, 1H), 7.55–7.44 (m, 1H), 7.02 (dd, J = 14.2, 7.8 Hz, 2H), 5.09–4.98 (m, 3H), 4.62–4.51 (m, 2H), 3.36–3.32 (m, 6H), 3.15 (t, J = 6.6 Hz, 2H), 2.87 (ddd, J = 17.2, 13.8, 5.3 Hz, 1H), 2.81–2.62 (m, 2H), 2.51–2.43 (m, 2H), 2.43–2.37 (m, 2H), 2.18–2.12 (m, 1H), 2.01–1.94 (m, 2H), 1.69 (s, 6H), 1.64–1.52 (m, 3H). HRMS (m/z) for C37H44N11O8+ [M + H]+: calculated 770.3369, found 770.3356.
4-((3-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]pyridin-7-yl)oxy)propyl)-amino)-N-(3-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)-amino)propyl)butanamide (55).
Compound 55 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 88 (7.1 mg, 0.016 mmol, 1.0 equiv). Compound 55 was obtained as a yellow solid in TFA salt form (11.7 mg, yield 93%). 1H NMR (500 MHz, CD3OD) δ 8.25 (s, 1H), 7.51 (dd, J = 8.6, 7.1 Hz, 1H), 6.99 (dd, J = 7.8, 4.6 Hz, 2H), 5.08–4.98 (m, 3H), 4.54 (t, J = 5.8 Hz, 2H), 3.39–3.29 (m, 4H), 3.31–3.22 (m, 2H), 3.18 (t, J = 6.6 Hz, 2H), 2.87 (ddd, J = 17.5, 14.0, 5.4 Hz, 1H), 2.80–2.64 (m, 2H), 2.51 (t, J = 6.4 Hz, 2H), 2.45–2.38 (m, 2H), 2.16–2.08 (m, 1H), 2.00 (q, J = 6.5 Hz, 2H), 1.77 (q, J = 6.4 Hz, 2H), 1.70 (s, 6H), 1.56 (t, J = 7.1 Hz, 3H). HRMS (m/z) for C38H46N11O8+ [M + H]+: calculated 784.3525, found 784.3534.
4-((3-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]pyridin-7-yl)oxy)propyl)-amino)-N-(4-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)-amino)butyl)butanamide (56).
Compound 56 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 89 (7.3 mg, 0.016 mmol, 1.0 equiv). Compound 56 was obtained as a yellow solid in TFA salt form (9.6 mg, yield 75%). 1H NMR (500 MHz, CD3OD) δ 8.25 (s, 1H), 7.51 (t, J = 7.8 Hz, 1H), 7.03–6.96 (m, 2H), 5.04 (q, J = 7.1, 6.3 Hz, 3H), 4.54 (t, J = 5.8 Hz, 2H), 3.47–3.01 (m, 8H), 2.93–2.62 (m, 3H), 2.59–2.31 (m, 4H), 2.18–2.07 (m, 1H), 2.01–1.95 (m, J = 6.4 Hz, 2H), 1.82–1.45 (m, 13H). HRMS (m/z) for C39H48N11O8+ [M + H]+: calculated 798.3682, found 798.3667.
4-((3-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]pyridin-7-yl)oxy)propyl)-amino)-N-(5-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)-amino)pentyl)butanamide (57).
Compound 57 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 90 (7.6 mg, 0.016 mmol, 1.0 equiv). Compound 57 was obtained as a yellow solid in TFA salt form (10.6 mg, yield 82%). 1H NMR (500 MHz, CD3OD) δ 8.18 (s, 1H), 7.49 (dd, J = 8.5, 7.1 Hz, 1H), 6.98 (dd, J = 7.8, 6.4 Hz, 2H), 5.08–4.96 (m, 3H), 4.51 (t, J = 5.8 Hz, 2H), 3.34–3.23 (m, 4H), 3.20–3.07 (m, 4H), 2.88 (ddd, J = 17.0, 13.7, 5.2 Hz, 1H), 2.81–2.64 (m, 2H), 2.47 (t, J = 6.5 Hz, 2H), 2.41–2.36 (m, 2H), 2.18–2.08 (m, 1H), 2.01–1.95 (m, J = 6.7 Hz, 2H), 1.69 (s, 6H), 1.69–1.60 (m, 2H), 1.58–1.47 (m, 5H), 1.44–1.38 (m, 2H). HRMS (m/z) for C40H50N11O8+ [M + H]+: calculated 812.3838, found 812.3854.
4-((3-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]pyridin-7-yl)oxy)propyl)-amino)-N-(6-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)-amino)hexyl)butanamide (58).
Compound 58 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 91 (6.5 mg, 0.016 mmol, 1.0 equiv). Compound 58 was obtained as a yellow solid in TFA salt form (11.9 mg, yield 90%). 1H NMR (500 MHz, CD3OD) δ 8.24 (s, 1H), 7.60–7.48 (m, 1H), 7.15–6.86 (m, 2H), 5.07–5.01 (m, 3H), 4.54 (q, J = 5.8, 4.9 Hz, 2H), 3.34–3.16 (m, 8H), 2.98–2.67 (m, 3H), 2.43–2.35 (m, 4H), 2.19–2.10 (m, 1H), 2.01–1.97 (m, 2H), 1.83–1.15 (m, 17H). HRMS (m/z) for C41H52N11O8+ [M + H]+: calculated 826.3995, found 826.3987.
4-((3-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]pyridin-7-yl)oxy)propyl)-amino)-N-(7-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)-amino)heptyl)butanamide (59).
Compound 59 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 92 (8 mg, 0.016 mmol, 1.0 equiv). Compound 59 was obtained as a yellow solid in TFA salt form (12.5 mg, yield 93%). 1H NMR (500 MHz, CD3OD) δ 8.22 (s, 1H), 7.53 (ddd, J = 8.5, 7.2, 2.0 Hz, 1H), 7.05–7.02 (m, 2H), 5.09–5.00 (m, 3H), 4.53 (t, J = 5.9 Hz, 2H), 3.39–3.27 (m, 4H), 3.16 (t, J = 6.9 Hz, 2H), 3.09 (t, J = 7.1 Hz, 2H), 2.93–2.82 (m, 1H), 2.81–2.66 (m, 2H), 2.46–2.39 (m, 4H), 2.18–2.11 (m, 1H), 2.01–1.96 (m, 2H), 1.72–1.62 (m, 8H), 1.58 (t, J = 7.1 Hz, 3H), 1.49–1.29 (m, 8H). HRMS (m/z) for C42H54N11O8+ [M + H]+: calculated 840.4151, found 840.4157.
4-((3-((2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-4-(3-hydroxy-3-methylbut-1-yn-1-yl)-1H-imidazo[4,5-c]pyridin-7-yl)oxy)propyl)-amino)-N-(8-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)-amino)octyl)butanamide (60).
Compound 60 was synthesized following the standard procedure for preparing compound 31 from intermediate 30 (9.1 mg, 0.016 mmol) and 93 (8.2 mg, 0.016 mmol, 1.0 equiv). Compound 60 was obtained as a yellow solid in TFA salt form (12.6 mg, yield 98%). 1H NMR (500 MHz, CD3OD) δ 8.25 (d, J = 2.8 Hz, 1H), 7.54 (ddd, J = 8.5, 7.1, 2.3 Hz, 1H), 7.02 (dd, J = 7.8, 3.0 Hz, 2H), 5.10–5.01 (m, 3H), 4.54 (t, J = 5.8 Hz, 2H), 3.34–3.27 (m, 4H), 3.17 (td, J = 7.3, 2.6 Hz, 2H), 3.09–3.07 (m, 2H), 2.92–2.82 (m, 1H), 2.81–2.66 (m, 2H), 2.48–2.36 (m, 4H), 2.18–2.10 (m, 1H), 2.03–1.92 (m, 2H), 1.75–1.62 (m, 8H), 1.65–1.55 (m, 3H), 1.49–1.25 (m, 10H). HRMS (m/z) for C43H56N11O8+ [M + H]+: calculated 854.4308, found 854.4313.
N1-(2-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)piperazin-1-yl)-3-oxopropyl)amino)ethyl)-N12-((S)-1-((2R,4S)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)dodecanediamide (61).
Compound 61 was synthesized following the standard procedure for preparing compound 13 from intermediate 2 (48.1 mg, 0.073 mmol) and 12-(((S)-1-((2R,4S)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)-phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-12-oxododecanoic acid (118, 40.9 mg, 0.073 mmol, 1.0 equiv). Compound 61 was obtained as a white solid in TFA salt form (32.2 mg, yield 40%). 1H NMR (800 MHz, CD3OD) δ 8.98 (s, 1H), 8.59 (s, 1H), 7.54 (d, J = 7.9 Hz, 2H), 7.48 (d, J = 8.1 Hz, 4H), 7.39 (dd, J = 8.8, 3.1 Hz, 2H), 5.34 (t, J = 7.9 Hz, 1H), 5.05 (q, J = 6.9 Hz, 1H), 4.61–4.53 (m, 2H), 4.51 (s, 1H), 4.49–4.44 (m, 1H), 4.24–4.15 (m, 1H), 4.14–4.02 (m, 1H), 4.00–3.91 (m, 3H), 3.91–3.80 (m, 1H), 3.77–3.63 (m, 5H), 3.55–3.48 (m, 2H), 3.48–3.40 (m, 1H), 3.22 (t, J = 5.9 Hz, 2H), 2.52 (s, 3H), 2.35–2.27 (m, 2H), 2.27–2.16 (m, 4H), 2.16–2.06 (m, 1H), 1.65–1.57 (m, 4H), 1.48 (d, J = 7.1 Hz, 3H), 1.37–1.26 (m, 14H), 1.20 (d, J = 7.0 Hz, 3H), 1.09 (s, 9H). 13C NMR (201 MHz, CD3OD) δ 176.58, 175.02, 172.10, 170.87, 169.37, 160.62, 159.54, 151.66, 148.41, 147.26, 144.19, 134.36, 133.53, 132.35, 129.78, 129.60 (2C), 129.50 (2C), 129.02 (2C), 126.45 (2C), 120.93, 70.55, 69.12, 59.36, 58.17, 55.49, 50.23, 48.54 (2C), 45.55, 45.49, 45.00, 44.44, 44.33, 41.40 (2C), 37.58, 36.58, 35.70, 35.46, 35.18, 34.29, 29.14, 29.03, 29.00, 28.94 (2C), 25.66 (3C), 25.51, 25.29, 21.20, 19.17, 14.35. tR = 3.90 min; HRMS (m/z) for C58H82ClN10O7S+ [M + H]+: calculated 1097.5772, found 1097.5772.
3-(((S)-2-(4-Chlorophenyl)-3-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)piperazin-1-yl)-3-oxopropyl)amino)-N-(8-((2-(1-methyl-2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)octyl)propenamide (62).
Compound 113 was synthesized following the standard procedure for preparing compound 3 from intermediate 1 (19.8 mg, 0.04 mmol) and 4-((8-aminooctyl)amino)-2-(1-methyl-2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (121, 16.8 mg, 0.04 mmol, 1.0 equiv). Compound 62 was obtained as a yellow solid in TFA salt form (15.5 mg, yield 44%). 1H NMR (800 MHz, CD3OD) δ 8.60 (s, 1H), 7.58 (t, J = 7.8 Hz, 1H), 7.48 (d, J = 7.9 Hz, 2H), 7.39 (d, J = 8.0 Hz, 2H), 7.07 (dd, J = 7.9, 5.1 Hz, 2H), 5.34 (t, J = 8.0 Hz, 1H), 5.11 (dd, J = 13.0, 5.6 Hz, 1H), 4.56 (dd, J = 9.6, 4.1 Hz, 1H), 4.19 (d, J = 10.8 Hz, 1H), 3.99–3.80 (m, 4H), 3.75–3.61 (m, 4H), 3.48–3.40 (m, 1H), 3.38–3.26 (m, 8H), 3.17 (s, 3H), 2.95–2.87 (m, 2H), 2.77–2.61 (m, 3H), 2.32 (dd, J = 12.6, 7.4 Hz, 1H), 2.25–2.16 (m, 1H), 2.15–2.08 (m, 1H), 1.74–1.63 (m, 2H), 1.56–1.50 (m, 2H), 1.50–1.44 (m, 2H), 1.44–1.35 (m, 5H), 1.20 (d, J = 6.9 Hz, 3H). 13C NMR (201 MHz, CD3OD) δ 172.28, 171.00, 170.11, 169.48, 169.36, 167.93, 160.71, 159.59, 148.46, 146.96, 135.89, 134.38, 133.53, 132.53, 129.62 (2C), 129.46 (2C), 120.94, 116.63, 110.38, 109.58, 70.56, 50.18, 49.46, 45.58, 45.46, 44.96 (2C), 44.45, 44.32, 41.99, 41.43 (2C), 39.04, 36.58, 31.10, 29.54, 28.85, 28.82 (2C), 26.42 (2C), 25.95, 21.66, 19.15. tR = 4.08 min; HRMS (m/z) for C46H59ClN9O7+ [M + H]+: calculated 884.4220, found 884.4215.
(2S,4R)-1-((S)-2-(11-Aminoundecanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)-pyrrolidine-2-carboxamide (79).
To a solution of (2S,4R)-1-((S)-2-amino-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide53 (77, 22.2 mg, 0.05 mmol) in DMSO (1 mL) were added commercially available 11-((tert-butoxycarbonyl)amino)undecanoic acid (78, 15.1 mg, 0.05 mmol, 1.5 equiv), EDCI (14.4 mg, 0.075 mmol, 1.5 equiv), HOAt (10.2 mg, 0.075 mmol, 1.5 equiv), and NMM (15.2 mg, 0.15 mmol, 3.0 equiv). After stirring overnight at room temperature, the reaction mixture was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford the desired intermediate, which was dissolved in TFA/DCM (1 mL/1 mL). The mixture was stirring for 30 min and concentrated. The residue was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford the title compound 79 as a white solid (21.8 mg, yield 53%). 1H NMR (400 MHz, CD3OD) δ 8.97–8.80 (m, 1H), 7.48–7.42 (m, 4H), 5.02 (d, J = 6.9 Hz, 1H), 4.64 (s, 1H), 4.58 (t, J = 8.3 Hz, 1H), 4.45 (s, 1H), 3.92–3.71 (m, 2H), 2.96–2.83 (m, 2H), 2.49 (d, J = 7.6 Hz, 3H), 2.34–2.13 (m, 2H), 1.62 (d, J = 16.1 Hz, 4H), 1.52 (d, J = 7.8, 2.9 Hz, 3H), 1.35 (s, 14H), 1.05 (s, 9H). HRMS (m/z) for C34H54N5O4S+ [M + H]+: calculated 628.3891, found 628.3896.
12-(((S)-1-((2S,4R)-4-Hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)-phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-12-oxododecanoic acid (81).
Compound 81 was synthesized following the standard procedure for preparing compound 79 from intermediate 77 (58.4 mg, 0.13 mmol) and dodecanedioic acid (80, 36 mg, 0.16 mmol, 1.2 equiv). Compound 81 was obtained as a white solid (39.4 mg, yield 46%). 1H NMR (600 MHz, CD3OD) δ 9.09 (s, 1H), 7.44 (q, J = 8.0 Hz, 4H), 5.00 (q, J = 7.0 Hz, 1H), 4.62 (s, 1H), 4.57 (t, J = 8.5 Hz, 1H), 4.43 (s, 1H), 3.88 (d, J = 11.0 Hz, 1H), 3.74 (dd, J = 11.0, 3.9 Hz, 1H), 2.50 (s, 3H), 2.35–2.22 (m, J = 6.9 Hz, 4H), 2.19 (dd, J = 13.4, 8.4 Hz, 1H), 1.94 (ddd, J = 13.5, 9.0, 4.5 Hz, 1H), 1.62–1.55 (m, 4H), 1.50 (d, J = 7.0 Hz, 3H), 1.34–1.29 (m, 14H), 1.04 (s, 9H). HRMS (m/z) for C35H53N4O6S+ [M + H]+: calculated 657.3680, found 657.3667.
4-(9-Aminononyl)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (99).
To a solution of oct-7-yn-1-ol (95, 1.0 g, 7.94 mmol) in DCM (20 mL) were added NEt3 (3.2 mL, 23.8 mmol, 3 equiv) and 4-toluenesulfonyl chloride (2.3 g, 11.9 mmol, 1.5 equiv). The reaction was stirring at room temperature for 2 h, and solvent was removed. The residue was purified by flash chromatography (hexane/EA = 8:1), and oct-7-yn-1-yl 4-methylbenzenesulfonate (96, 1.86 g, yield 84%) was obtained. To a solution of 4-methylbenzenesulfonate (96, 280 mg, 1 mmol) in DMF (5 mL) were added sodium cyanide (250 mg, 5 mmol, 5.0 equiv) and sodium iodide (15 mg, 0.1 mmol, 0.1 equiv). The reaction was heated at 60 °C for 3 h, and water was added, extracted with EtOAc (15 mL × 3), washed with brine, and dried over Na2SO4. The solvent was removed, and the residue was purified by flash chromatography (hexane/EA = 9:1). The non-8-ynenitrile (97, 126 mg) was obtained in 93% yield. To a solution of commercially available 4-bromo-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (33.7 mg, 0.1 mmol) and non-8-ynenitrile (97, 27 mg, 0.2 mmol, 2 equiv) in DMA (1.5 mL) were added tetrakis(triphenylphosphine)-palladium (0) (2.3 mg, 2 mol %), copper iodide (1 mg, 4 mol %), and triethylamine (22 mg, 0.3 mmol, 3.0 equiv). The reaction was heated at 100 °C for 12 h, cooled to rt, and filtered through Celite. The filtrate was evaporated, and the mixture was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford the corresponding product (31.4 mg, yield 80%). This intermediate was dissolved in MeOH, and Raney nickel (5 mg) was added. After the reaction was stirred at room temperature under hydrogen for 1 h, it was filtered and concentrated. The resulting residue was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford the title product 99 as a white solid (29.8 mg, yield 94%). 1H NMR (600 MHz, CD3OD) δ 7.72–7.70 (m, 2H), 7.62 (dd, J = 5.8, 2.9 Hz, 1H), 5.12 (dd, J = 12.6, 5.5 Hz, 1H), 3.09 (dd, J = 8.8, 6.8 Hz, 2H), 2.92–2.87 (m, 2H), 2.79–2.69 (m, 2H), 2.18–2.05 (m, 1H), 1.69–1.59 (m, 5H), 1.41–1.32 (m, 10H). HRMS (m/z) for C22H30N3O4+ [M + H]+: calculated 400.2231, found 400.2257.
3-(4-((8-Aminooctyl)amino)-1-oxoisoindolin-2-yl)piperidine-2,6-dione (103).
To a solution of 3-(4-amino-1-oxoisoindolin-2-yl)-piperidine-2,6-dione (101, 52 mg, 0.2 mmol) in DMF (2 mL) were added tert-butyl (8-iodooctyl)carbamate (100, 85.2 mg, 0.24 mmol, 1.2 equiv) and NaHCO3 (40 mg, 0.46 mmol, 2.3 equiv). After the reaction was stirred at 60 °C overnight, water was added to quench the reaction. The reaction mixture was extracted with EtOAc (5 mL × 3), dried over Na2SO4, filtered, and concentrated. The residue was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford the desired intermediate as a yellow solid (68.8 mg, yield 71%). This intermediate (68.8 mg, 0.14 mmol) was dissolved in TFA/DCM (1/1 mL). After the reaction solution was stirred at room temperature for 30 min, the solvent was evaporated and the resulting residue was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford the title compound 103 as a yellow solid (56.4 mg, yield 99%). 1H NMR (600 MHz, CD3OD) δ 7.42 (d, J = 3.7 Hz, 2H), 7.21 (dd, J = 5.9, 3.0 Hz, 1H), 5.14 (dd, J = 13.5, 5.0 Hz, 1H), 4.52–4.29 (m, 2H), 3.83–3.65 (m, 2H), 2.95–2.90 (m, 2H), 2.56–2.35 (m, 1H), 2.24–2.09 (m, 1H), 1.73–1.58 (m, 2H), 1.58–1.46 (m, 2H), 1.43–1.26 (m, 10H). HRMS (m/z) for C H N O + [M + H]+: calculated 387.2391, found 387.2376.
4-((8-Aminooctyl)oxy)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (104).
Compound 104 was synthesized following the standard procedure for preparing compound 103 from 2-(2,6-dioxopiperidin-3-yl)-4-hydroxyisoindoline-1,3-dione (102, 55 mg, 0.2 mmol) and tert-butyl (8-iodooctyl)carbamate (100, 85.2 mg, 0.24 mmol, 1.2 equiv). Compound 104 was obtained as a white solid (28.2 mg, 70% yield for two steps). 1H NMR (600 MHz, CD3OD) δ 7.75 (t, J = 7.9 Hz, 1H), 7.41 (dd, J = 8.4, 3.9 Hz, 2H), 5.09 (dd, J = 12.9, 5.4 Hz, 1H), 4.21 (t, J = 6.3 Hz, 2H), 2.98–2.81 (m, 3H), 2.81–2.62 (m, 2H), 2.12 (dd, J = 12.8, 6.1 Hz, 1H), 1.89–1.82 (m, 2H), 1.64 (q, J = 7.5 Hz, 2H), 1.54 (q, J = 7.4 Hz, 2H), 1.48–1.31 (m, 6H). HRMS (m/z) for C21H28N3O5+ [M + H]+: calculated 402.2023, found 402.2034.
12-(((S)-1-((2R,4S)-4-Hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)-phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-12-oxododecanoic acid (118).
To the solution of (2R,4S)-1-((S)-2-amino-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide55 (117, 22.0 mg, 0.05 mmol) in DMSO (1 mL) were added dodecanedioic acid (80, 17.3 mg, 0.075 mmol, 1.5 equiv), EDCI (14.4 mg, 0.075 mmol, 1.5 equiv), HOAt (10.2 mg, 0.075 mmol, 1.5 equiv), and NMM (15.2 mg, 0.15 mmol, 3.0 equiv). After stirring overnight at room temperature, the reaction mixture was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford the title compound 118 as a white solid (12.4 mg, yield 38%). 1H NMR (600 MHz, CD3OD) δ 9.07 (s, 1H), 7.53 (d, J = 8.2 Hz, 2H), 7.50–7.38 (m, 2H), 5.02 (q, J = 7.0 Hz, 1H), 4.55 (dd, J = 8.3, 6.4 Hz, 1H), 4.48 (s, 1H), 4.46–4.41 (m, 1H), 3.94 (dd, J = 10.8, 4.9 Hz, 1H), 3.68 (dd, J = 10.8, 3.5 Hz, 1H), 2.51 (s, 3H), 2.33–2.22 (m, 3H), 2.22–2.15 (m, 2H), 2.13–2.05 (m, 1H), 1.61–1.53 (m, 4H), 1.45 (d, J = 7.1 Hz, 3H), 1.33–1.19 (m, 12H), 1.06 (s, 9H). HRMS (m/z) for C35H53N4O6S+ [M + H]+: calculated 657.3680, found 657.3693.
4-((8-Aminooctyl)amino)-2-(1-methyl-2,6-dioxopiperidin-3-yl)-isoindoline-1,3-dione (121).
To a solution of 4-fluoro-2-(1-methyl-2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione61 (119, 58 mg, 0.2 mmol) in 1-methyl-2-pyrrolidinone (1 mL) were added commercially available tert-butyl (8-aminooctyl)carbamate (120, 49 mg, 0.2 mmol) and N,N-diisopropylethylamine (77 mg, 0.6 mmol, 3 equiv). After stirring at 100 °C under microwave for 1 h, the reaction mixture was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford the desired intermediate, which was dissolved in TFA/DCM (1/1 mL). The mixture was stirred for 30 min at room temperature before it was concentrated. The resulting residue was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford title compound 121 as a yellow solid (66.8 mg, yield 81%). 1H NMR (600 MHz, CD3OD) δ 7.52 (dd, J = 8.5, 7.1 Hz, 1H), 7.06–6.91 (m, 2H), 5.06 (dd, J = 13.0, 5.4 Hz, 1H), 3.12 (s, 3H), 2.94–2.81 (m, 4H), 2.74–2.56 (m, 1H), 2.14–1.99 (m, 1H), 1.64 (q, J = 7.3 Hz, 4H), 1.50–1.28 (m, 10H). HRMS (m/z) for C22H31N4O4+ [M + H]+: calculated 415.2340, found 415.2345.
Cell Culture.
All cell lines were purchased from ATCC and authenticated. Cell lines were regularly tested in the lab for mycoplasma. All cells were cultured at 37 °C and 5% CO2. Cell lines were cultured in 1× DMEM (Corning, 10–013-CV) with 10% fetal bovine serum (Atlanta Biologicals S11150) and 1× penicillin/ streptomycin. Cells were split using 0.05 or 0.25% trypsin (Corning 25–051-Cl or 25–053-Cl, respectively) before they reached full confluence, and media were changed every 3–4 days.
Western Blotting Analysis.
Total cell lysates were collected with an immunoprecipitation (IP) lysis buffer (20 mM Tris [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerolphosphate, 1 mM sodium orthovanadate, 1 mM PMSF, and protease inhibitor cocktail). Whole cell lysates were obtained by sonication followed by centrifugation, and protein concentration was measured by a BCA Protein Assay Kit (Pierce). Cell lysates were boiled with 2× sample buffer (125 mM Tris–HCl at pH 6.8, 10% β-ME, 2% SDS, 20% glycerol, 0.05% bromophenol blue, and 8 M urea). Protein lysates were loaded into 4–12% Bis–Tris gels and resolved by electrophoresis. Samples were then blotted on a PVDF membrane (Millipore IPVH00010) using the wet transfer technique (Invitrogen). Membranes were blocked in 5% milk–TBST for 1 h, washed in TBST for 10 min, and incubated in primary antibody in 5% milk–TBST or 5% BSA–TBST at 4 °C for 16 h. Membranes were rinsed (3 × 6 min) in TBST, incubated in horseradish peroxidase-conjugated secondary antibodies in 5% milk–TBST for 1 h, and rinsed again in TBST (3 × 6 min). Membranes were visualized using the chemiluminescence system (Thermo 34080, 37075) on an autoradiography film (Denville E3018). Primary antibodies were as follows: β-actin (Sigma A5316), p-AKT (Ser473 CST-9271), total AKT (CST-9272), p-S6 (Ser240/244, CST-5364), and p-PRAS40 (Thr246, CST-2997). Secondary antibodies were as follows: mouse (Thermo 31432) and rabbit (Thermo 31460).
Cell Colony Formation Assay.
Cells were cultured for 14 days in the presence of different compounds at indicated concentrations. Media with compound were replenished every 2 days. At the end of the experiment, media were aspirated and viable cells were stained with 0.5% crystal violet dye. Quantification analysis was performed as previously described.62
AKT Binding Assay.
Binding affinities of GDC0068, 13, 25, 61, and 62 to three AKT isoforms (AKT1, AKT2 and AKT3) were determined by DiscoverX using the KINOMEscan assay, a competition binding assay that quantitatively measures the ability of a compound to compete with an immobilized, active-site directed ligand. The assay was performed by combining three components: DNA-tagged AKT, immobilized ligand, and a test compound. The ability of the test compound to compete with the immobilized ligand was measured by quantitative PCR of the DNA tag. The Kd values were determined by using an 11-point 3-fold compound dilution (the top concentration of 10 μM for GDC-0068 and 30 μM for the other four compounds) with three DMSO control points in duplicates.
Cell Proliferation and Apoptosis Assays.
Experiments were carried out in 96-well plates in triplicates (Corning 720089). A total of 1–3 × 103 cells per well were grown in the presence of indicated compounds, and cells were then monitored for 5 days using the IncuCyte live cell imaging system (Essen BioScience, Ann Arbor, MI, USA), which was placed in a cell culture incubator operated at 37 °C and 5% CO2. Cell confluence was determined using calculations derived from phase-contrast images readings on an IncuCyte ZOOM (Essen Biosciences) on live cells over time. For the measurement of cell apoptosis, DRAQ7 (Cell Signaling # 7406) at 1.5 μM was included in the medium and apoptotic red counts were measured in an IncuCyte FLR automated incubator microscope.
TMT-Based Global Proteomic Profiling Sample Preparation.
Trypsin was purchased from Promega. All chemicals were HPLC-grade unless specifically indicated. The TMT11plex Isobaric Label Reagent was purchased from Thermofisher (cat. # A34808). The cell pellets were resuspended in 8 M urea and 50 mM Tris–HCl (pH 8.0), reduced with dithiothreitol (5 mM final) for 30 min at room temperature, and alkylated with iodoacetamide (15 mM final) for 45 min in the dark at room temperature. Samples were diluted fourfold with 25 mM Tris–HCl (pH 8.0) and 1 mM CaCl2 and digested with trypsin at a 1:100 (w/w, trypsin/protein) ratio overnight at room temperature. Peptides were desalted on homemade C18 stage tips, and 100 μg of each peptide sample was labeled with the TMT reagent following the manufacturer’s instruction. The mixture of labeled peptides was desalted and fractionated into 24 fractions in 10 mM trimethylammonium bicardonate (TMAB) buffer containing 5–40% acetonitrile.
Mass Spectrometry Analysis.
Dried peptides were dissolved in 0.1% formic acid and 2% acetonitrile. For global profiling samples, peptide concentration was measured with a Pierce Quantitative Colorimetric Peptide Assay (Thermofisher). A total of 0.5 μg of each fraction were analyzed on a Q-Exactive HF-X coupled with an Easy nanoLC 1200 (Thermo Fisher Scientific, San Jose, CA). Peptides were loaded onto a nanoEase MZ HSS T3 Column (100 Å, 1.8 μm, 75 μm × 150 mm, Waters). Analytical separation of all peptides was achieved with a 100 min gradient. A linear gradient of 5 to 10% buffer B over 5 min, 10 to 31% buffer B over 70 min, and 31 to 75% buffer B over 15 min was executed at a 300 nL/min flow rate followed by a ramp to 100% B in 1 min and 9 min wash with 100% B, where buffer A was aqueous 0.1% formic acid and buffer B was 80% acetonitrile and 0.1% formic acid. LC–MS experiments were also carried out in a data-dependent mode with full MS (externally calibrated to a mass accuracy of <5 ppm and a resolution of 120,000 for TMT-labeled samples at m/z 200) followed by high energy collision-activated dissociation-MS/MS with a resolution of 30,000 for TMT-labeled global samples at m/z 200. High energy collision-activated dissociation-MS/MS was used to dissociate peptides at a normalized collision energy of 32 eV (for TMT-labeled sample) in the presence of nitrogen bath gas atoms. Dynamic exclusion was 45 or 20 s.
Raw Proteomics Data Processing and Analysis.
Mass spectra were processed, and peptide identification was performed using the MaxQuant software version 1.6.10.43 (Max Planck Institute, Germany). All protein database searches were performed against the UniProt human protein sequence database (UP000005640). A false discovery rate (FDR) for both peptide-spectrum match (PSM) and protein assignment was set at 1%. Search parameters included up to two missed cleavages at Lys/Arg on the sequence, oxidation of methionine, and protein N-terminal acetylation as a dynamic modification. Carbamidomethylation of cysteine residues was considered as a static modification. Peptide identifications are reported by filtering of reverse and contaminant entries and assigning to their leading razor protein. The TMT reporter intensity found in MaxQuant was for quantitation. Data processing and statistical analysis were performed on Perseus (Version 1.6.10.50). Protein quantitation was performed using the TMT reporter intensity found in MaxQuant, and a two-sample t test on two biological replicates was used with a p value of 1% to report statistically significant protein abundance fold-changes.
Mouse Pharmacokinetic Study.
Compounds 13 and 25 (in their HCl salt form) were dissolved in 5% DMA, 25% Solutol HS-15, and 70% normal saline as formulation. Three male Swiss albino mice were administered intraperitoneally with a solution formulation of each compound at a 50 mg/kg dose. The formulation vehicle was 5% v/v NMP, 5% v/v Solutol HS-15, and 90% v/v normal saline. Blood samples (approximately 60 μL) were collected from the three test mice at 0.5, 2, and 8 h. Plasma was harvested by centrifugation of the blood and stored at −70 ± 10 °C until analysis. Plasma samples were quantified by the fit-for-purpose LC–MS/MS method. Compound concentrations in plasma at each time point are the average values from three test mice. Error bars represent ± SEM. Experiments involving mice were performed according to the Institutional Animal Care and Use Committee (IACUC)-approved protocol.
Supplementary Material
ACKNOWLEDGMENTS
J.J. acknowledges the support by an endowed professorship by the Icahn School of Medicine at Mount Sinai. This research was supported in part by the grant R35CA220491 (to R.P.) from the National Cancer Institute (NCI) at the National Institutes of Health (NIH). R.P. and J.J. were also supported by the P30CA196521 grant from the NCI at the NIH. J.X. is supported by a T32 postdoc fellow training grant (T32CA078207) and Leo and Julia Forchheimer Foundation Postdoc Fellowship. This work utilized the NMR Spectrometer Systems at Mount Sinai acquired with funding from National Institutes of Health SIG grants 1S10OD025132 and 1S10OD028504.
ABBREVIATIONS USED
- ATP
adenosine triphosphate
- Boc
tert-butyloxycarbonyl
- Cbz
carboxybenzyl
- CRBN
cereblon
- CRLs
cullin-ring ubiquitin ligases
- EDCI
1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide
- HOAt
1-hydroxy-7-azabenzo-triazole
- IMiDs
immunomodulatory drugs
- MOA
mechanism of actions
- MS
mass spectrometry
- m-TOR
mammalian target of rapamycin
- NAE
NEDD8-activating enzyme
- NMM
N-methylmorpholine
- P-AKT
phosphorylated AKT
- PEG
polyethylene glycol
- PI3K
phosphatidylinositol 3-kinase
- PK
pharmacokinetic
- POM
pomalidomide
- PROTACs
proteolysis targeting chimeras
- SAR
structure–activity relationships
- VHL
von Hippel–Lindau
- T-AKT
total AKT
- TMT
tandem mass tag
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.1c01476
The authors declare the following competing financial interest(s): J.J., R.P., J.L. J.X. and X.Y. are inventors of a patent application filed by the Icahn School of Medicine at Mount Sinai. J.J. is a cofounder, scientific advisory board member and equity shareholder in Cullgen Inc. and a consultant for Cullgen Inc., EpiCypher Inc., and Accent Therapeutics Inc. R.P. is a shareholder and advisor of Therapten Bioscience, Inc. The Jin laboratory received research funds from Celgene Corporation, Levo Therapeutics, and Cullgen, Inc.
Contributor Information
Xufen Yu, Mount Sinai Center for Therapeutics Discovery, Department of Pharmacological Sciences, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States; Department of Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Jia Xu, Department of Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Ling Xie, Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Li Wang, Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Yudao Shen, Mount Sinai Center for Therapeutics Discovery, Department of Pharmacological Sciences, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States; Department of Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Kaitlyn M. Cahuzac, Department of Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States
Xian Chen, Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Jing Liu, Mount Sinai Center for Therapeutics Discovery, Department of Pharmacological Sciences, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States; Department of Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Ramon E. Parsons, Department of Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States
Jian Jin, Mount Sinai Center for Therapeutics Discovery, Department of Pharmacological Sciences, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States; Department of Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
REFERENCES
- (1).Scheid MP; Woodgett JR PKB/AKT: functional insights from genetic models. Nat. Rev. Mol. Cell Biol 2001, 2, 760–768. [DOI] [PubMed] [Google Scholar]
- (2).Toker A Achieving specificity in Akt signaling in cancer. Adv. Biol. Regul 2012, 52, 78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Nitulescu GM; Van De Venter M; Nitulescu G; Ungurianu A; Juzenas P; Peng Q; Olaru OT; Gradinaru D; Tsatsakis A; Tsoukalas D; Spandidos DA; Margina D The Akt pathway in oncology therapy and beyond (Review). Int. J. Oncol 2018, 53, 2319–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Vivanco I; Sawyers CL The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [DOI] [PubMed] [Google Scholar]
- (5).Cheng JQ; Lindsley CW; Cheng GZ; Yang H; Nicosia SV The Akt/PKB pathway: molecular target for cancer drug discovery. Oncogene 2005, 24, 7482–7492. [DOI] [PubMed] [Google Scholar]
- (6).Manning BD; Cantley LC AKT/PKB signaling: navigating downstream. Cell 2007, 129, 1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Manning BD; Toker A AKT/PKB signaling: navigating the network. Cell 2017, 169, 381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Polivka J Jr.; Janku F Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol. Ther 2014, 142, 164–175. [DOI] [PubMed] [Google Scholar]
- (9).Mayer IA; Arteaga CL The PI3K/AKT Pathway as a target for cancer treatment. Annu. Rev. Med 2016, 67, 11–28. [DOI] [PubMed] [Google Scholar]
- (10).Fruman DA; Chiu H; Hopkins BD; Bagrodia S; Cantley LC; Abraham RT The PI3K pathway in human disease. Cell 2017, 170, 605–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Tokunaga E; Oki E; Egashira A; Sadanaga N; Morita M; Kakeji Y; Maehara Y Deregulation of the Akt pathway in human cancer. Curr. Cancer Drug Targets 2008, 8, 27–36. [DOI] [PubMed] [Google Scholar]
- (12).Roy N; Bordoloi D; Monisha J; Padmavathi G; Kotoky J; Golla R; Kunnumakkara A Specific Targeting of Akt Kinase Isoforms: Taking the precise path for prevention and treatment of cancer. Curr. Drug Targets 2017, 18, 421–435. [DOI] [PubMed] [Google Scholar]
- (13).Nitulescu GM; Margina D; Juzenas P; Peng Q; Olaru OT; Saloustros E; Fenga C; Spandidos D; Libra M; Tsatsakis AM Akt inhibitors in cancer treatment: the long journey from drug discovery to clinical use (Review). Int. J. Oncol 2016, 48, 869–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Shariati M; Meric-Bernstam F Targeting AKT for cancer therapy. Expert Opin. Invest. Drugs 2019, 28, 977–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Heerding DA; Rhodes N; Leber JD; Clark TJ; Keenan RM; Lafrance LV; Li M; Safonov IG; Takata DT; Venslavsky JW; Yamashita DS; Choudhry AE; Copeland RA; Lai Z; Schaber MD; Tummino PJ; Strum SL; Wood ER; Duckett DR; Eberwein D; Knick VB; Lansing TJ; McConnell RT; Zhang S; Minthorn EA; Concha NO; Warren GL; Kumar R Identification of 4-(2-(4-amino-1,2,5-oxadiazol-3-yl)-1-ethyl-7-{[(3S)-3-piperidinylmethyl]oxy}−1H-imidazo[4,5-c]pyridin-4-yl)-2-methyl-3-butyn-2-ol (GSK690693), a novel inhibitor of AKT kinase. J. Med. Chem 2008, 51, 5663–5679. [DOI] [PubMed] [Google Scholar]
- (16).Dumble M; Crouthamel MC; Zhang SY; Schaber M; Levy D; Robell K; Liu Q; Figueroa DJ; Minthorn EA; Seefeld MA; Rouse MB; Rabindran SK; Heerding DA; Kumar R Discovery of novel AKT inhibitors with enhanced anti-tumor effects in combination with the MEK inhibitor. PLoS One 2014, 9, No. e100880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Spencer A; Yoon S-S; Harrison SJ; Morris S; Smith D; Freedman SJ; Brigandi R; Oliff A; Opalinska JB; Chen C Novel AKT inhibitor GSK2110183 shows favorable safety, pharmacokinetics, and clinical activity in multiple myeloma. preliminary results from a phase I first-time-in-human study. Blood 2011, 118, 1856–1856. [Google Scholar]
- (18).Blake JF; Xu R; Bencsik JR; Xiao D; Kallan NC; Schlachter S; Mitchell IS; Spencer KL; Banka AL; Wallace EM; Gloor SL; Martinson M; Woessner RD; Vigers GPA; Brandhuber BJ; Liang J; Safina BS; Li J; Zhang B; Chabot C; Do S; Lee L; Oeh J; Sampath D; Lee BB; Lin K; Liederer BM; Skelton NJ Discovery and preclinical pharmacology of a selective ATP-competitive Akt inhibitor (GDC-0068) for the treatment of human tumors. J. Med. Chem 2012, 55, 8110–8127. [DOI] [PubMed] [Google Scholar]
- (19).Addie M; Ballard P; Buttar D; Crafter C; Currie G; Davies BR; Debreczeni J; Dry H; Dudley P; Greenwood R; Johnson PD; Kettle JG; Lane C; Lamont G; Leach A; Luke RWA; Morris J; Ogilvie D; Page K; Pass M; Pearson S; Ruston L Discovery of 4-amino-N-[(1S)-1-(4-chlorophenyl)-3-hydroxypropyl]-1-(7H-pyrrolo[2,3-d]pyrimidin –4-yl)piperidine-4-carboxamide (AZD5363), an orally bioavailable, potent inhibitor of Akt kinases. J. Med. Chem 2013, 56, 2059–2073. [DOI] [PubMed] [Google Scholar]
- (20).Hirai H; Sootome H; Nakatsuru Y; Miyama K; Taguchi S; Tsujioka K; Ueno Y; Hatch H; Majumder PK; Pan BS; Kotani H MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther 2010, 9, 1956–1967. [DOI] [PubMed] [Google Scholar]
- (21).Zhao YY; Tian Y; Zhang J; Xu F; Yang YP; Huang Y; Zhao HY; Zhang JW; Xue C; Lam MH; Yan L; Hu ZH; Dinglin XX; Zhang L Effects of an oral allosteric AKT inhibitor (MK-2206) on human nasopharyngeal cancer in vitro and in vivo. Drug Des. Devel. Ther 2014, 8, 1827–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lapierre JM; Eathiraj S; Vensel D; Liu Y; Bull CO; Cornell-Kennon S; Iimura S; Kelleher EW; Kizer DE; Koerner S; Makhija S; Matsuda A; Moussa M; Namdev N; Savage RE; Szwaya J; Volckova E; Westlund N; Wu H; Schwartz B Discovery of 3-(3-(4-(1-aminocyclobutyl)phenyl)-5-phenyl-3H-imidazo[4,5-b]pyridin-2-yl)pyridin –2-amine (ARQ 092): an orally bioavailable, selective, and potent allosteric AKT inhibitor. J. Med. Chem 2016, 59, 6455–6469. [DOI] [PubMed] [Google Scholar]
- (23).Okuzumi T; Fiedler D; Zhang C; Gray DC; Aizenstein B; Hoffman R; Shokat KM Inhibitor hijacking of Akt activation. Nat. Chem. Biol 2009, 5, 484–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Huck BR; Mochalkin I Recent progress towards clinically relevant ATP-competitive Akt inhibitors. Bioorg. Med. Chem. Lett 2017, 27, 2838–2848. [DOI] [PubMed] [Google Scholar]
- (25).Do K; Speranza G; Bishop R; Khin S; Rubinstein L; Kinders RJ; Datiles M; Eugeni M; Lam MH; Doyle LA; Doroshow JH; Kummar S Biomarker-driven phase 2 study of MK-2206 and selumetinib (AZD6244, ARRY-142886) in patients with colorectal cancer. Invest. New Drugs 2015, 33, 720–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Keppler-Noreuil KM; Sapp JC; Lindhurst MJ; Darling TN; Burton-Akright J; Bagheri M; Dombi E; Gruber A; Jarosinski PF; Martin S; Nathan N; Paul SM; Savage RE; Wolters PL; Schwartz B; Widemann BC; Biesecker LG Pharmacodynamic study of miransertib in individuals with proteus syndrome. Am. J. Hum. Genet 2019, 104, 484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Weisner J; Landel I; Reintjes C; Uhlenbrock N; Trajkovic-Arsic M; Dienstbier N; Hardick J; Ladigan S; Lindemann M; Smith S; Quambusch L; Scheinpflug R; Depta L; Gontla R; Unger A; Müller H; Baumann M; Schultz-Fademrecht C; Günther G; Maghnouj A; Müller MP; Pohl M; Teschendorf C; Wolters H; Viebahn R; Tannapfel A; Uhl W; Hengstler JG; Hahn SA; Siveke JT; Rauh D Preclinical efficacy of covalent-allosteric AKT inhibitor Borussertib in combination with Trametinib in KRAS-mutant pancreatic and colorectal cancer. Cancer Res 2019, 79, 2367–2378. [DOI] [PubMed] [Google Scholar]
- (28).Uhlenbrock N; Smith S; Weisner J; Landel I; Lindemann M; Le TA; Hardick J; Gontla R; Scheinpflug R; Czodrowski P; Janning P; Depta L; Quambusch L; Müller MP; Engels B; Rauh D Structural and chemical insights into the covalent-allosteric inhibition of the protein kinase Akt. Chem. Sci 2019, 10, 3573–3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Quambusch L; Landel I; Depta L; Weisner J; Uhlenbrock N; Muller MP; Glanemann F; Althoff K; Siveke JT; Rauh D Covalent-Allosteric Inhibitors to Achieve Akt Isoform-Selectivity. Angew. Chem., Int. Ed. Engl 2019, 58, 18823–18829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Vivanco I; Chen ZC; Tanos B; Oldrini B; Hsieh WY; Yannuzzi N; Campos C; Mellinghoff IK A kinase-independent function of AKT promotes cancer cell survival. Elife 2014, 3, No. e03751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Burslem GM; Crews CM Small-molecule modulation of protein homeostasis. Chem. Rev 2017, 117, 11269–11301. [DOI] [PubMed] [Google Scholar]
- (32).Lai AC; Crews CM Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discov 2017, 16, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Schapira M; Calabrese MF; Bullock AN; Crews CM Targeted protein degradation: expanding the toolbox. Nat. Rev. Drug Discov 2019, 18, 949–963. [DOI] [PubMed] [Google Scholar]
- (34).Dale B; Cheng M; Park K-S; Kaniskan HÜ; Xiong Y; Jin J Advancing targeted protein degradation for cancer therapy. Nat. Rev. Cancer 2021, 21, 638–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Schneider M; Radoux CJ; Hercules A; Ochoa D; Dunham I; Zalmas LP; Hessler G; Ruf S; Shanmugasundaram V; Hann MM; Thomas PJ; Queisser MA; Benowitz AB; Brown K; Leach AR The PROTACtable genome. Nat. Rev. Drug Discov 2021, 20, 789–797. [DOI] [PubMed] [Google Scholar]
- (36).Sakamoto KM; Kim KB; Kumagai A; Mercurio F; Crews CM; Deshaies RJ Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Winter GE; Buckley DL; Paulk J; Roberts JM; Souza A; Dhe-Paganon S; Bradner JE DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Deshaies RJ Protein degradation: Prime time for PROTACs. Nat. Chem. Biol 2015, 11, 634–635. [DOI] [PubMed] [Google Scholar]
- (39).Salami J; Crews CM Waste disposal-an attractive strategy for cancer therapy. Science 2017, 355, 1163–1167. [DOI] [PubMed] [Google Scholar]
- (40).Gechijian LN; Buckley DL; Lawlor MA; Reyes JM; Paulk J; Ott CJ; Winter GE; Erb MA; Scott TG; Xu M; Seo HS; Dhe-Paganon S; Kwiatkowski NP; Perry JA; Qi J; Gray NS; Bradner JE Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nat. Chem. Biol 2018, 14, 405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Bassi ZI; Fillmore MC; Miah AH; Chapman TD; Maller C; Roberts EJ; Davis LC; Lewis DE; Galwey NW; Waddington KE; Parravicini V; Macmillan-Jones AL; Gongora C; Humphreys PG; Churcher I; Prinjha RK; Tough DF Modulating PCAF/GCN5 Immune cell function through a PROTAC approach. ACS Chem. Biol 2018, 13, 2862–2867. [DOI] [PubMed] [Google Scholar]
- (42).Silva MC; Ferguson FM; Cai Q; Donovan KA; Nandi G; Patnaik D; Zhang T; Huang HT; Lucente DE; Dickerson BC; Mitchison TJ; Fischer ES; Gray NS; Haggarty SJ Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. Elife 2019, 8, No. e45457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Jiang B; Wang ES; Donovan KA; Liang Y; Fischer ES; Zhang T; Gray NS Development of Dual and Selective Degraders of Cyclin-Dependent Kinases 4 and 6. Angew. Chem. Int. Ed. Engl 2019, 58, 6321–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Tovell H; Testa A; Zhou H; Shpiro N; Crafter C; Ciulli A; Alessi DR Design and characterization of SGK3-PROTAC1, an isoform specific SGK3 kinase PROTAC degrader. ACS Chem. Biol 2019, 14, 2024–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Khan S; Zhang X; Lv D; Zhang Q; He Y; Zhang P; Liu X; Thummuri D; Yuan Y; Wiegand JS; Pei J; Zhang W; Sharma A; McCurdy CR; Kuruvilla VM; Baran N; Ferrando AA; Kim YM; Rogojina A; Houghton PJ; Huang G; Hromas R; Konopleva M; Zheng G; Zhou D A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat. Med 2019, 25, 1938–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Bai L; Zhou H; Xu R; Zhao Y; Chinnaswamy K; McEachern D; Chen J; Yang C-Y; Liu Z; Wang M; Liu L; Jiang H; Wen B; Kumar P; Meagher JL; Sun D; Stuckey JA; Wang S A potent and selective small-molecule degrader of STAT3 achieves complete tumor regression in vivo. Cancer Cell 2019, 36, 498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).You I; Erickson EC; Donovan KA; Eleuteri NA; Fischer ES; Gray NS; Toker A Discovery of an AKT degrader with prolonged inhibition of downstream signaling. Cell Chem. Biol 2020, 27, 66–73.e7. e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Xu J; Yu X; Martin TC; Bansal A; Cheung K; Lubin A; Stratikopoulos E; Cahuzac KM; Wang L; Xie L; Zhou R; Shen Y; Wu X; Yao S; Qiao R; Poulikakos PI; Chen X; Liu J; Jin J; Parsons R AKT degradation selectively inhibits the growth of PI3K/ PTEN pathway mutant cancers with wild-type KRAS and BRAF by destabilizing Aurora kinase B. Cancer Discov 2021, DOI: 10.1158/2159-8290.CD-20-0815. [DOI] [PMC free article] [PubMed]
- (49).Bondeson DP; Smith BE; Burslem GM; Buhimschi AD; Hines J; Jaime-Figueroa S; Wang J; Hamman BD; Ishchenko A; Crews CM Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem. Biol 2018, 25, 78–87.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Smith BE; Wang SL; Jaime-Figueroa S; Harbin A; Wang J; Hamman BD; Crews CM Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun 2019, 10, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Dai X; Cheng H; Bai Z; Li J Breast cancer cell line classification and its relevance with breast tumor subtyping. J. Cancer 2017, 8, 3131–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Raina K; Lu J; Qian Y; Altieri M; Gordon D; Rossi AMK; Wang J; Chen X; Dong H; Siu K; Winkler JD; Crew AP; Crews CM; Coleman KG PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 7124–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Hu J; Hu B; Wang M; Xu F; Miao B; Yang CY; Wang M; Liu Z; Hayes DF; Chinnaswamy K; Delproposto J; Stuckey J; Wang S Discovery of ERD-308 as a highly potent proteolysis targeting chimera (PROTAC) degrader of estrogen receptor (ER). J. Med. Chem 2019, 62, 1420–1442. [DOI] [PubMed] [Google Scholar]
- (54).Han X; Wang C; Qin C; Xiang W; Fernandez-Salas E; Yang CY; Wang M; Zhao L; Xu T; Chinnaswamy K; Delproposto J; Stuckey J; Wang S Discovery of ARD-69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of androgen receptor (AR) for the treatment of prostate cancer. J. Med. Chem 2019, 62, 941–964. [DOI] [PubMed] [Google Scholar]
- (55).Wei J; Hu J; Wang L; Xie L; Jin MS; Chen X; Liu J; Jin J Discovery of a first-in-class mitogen-activated protein kinase kinase 1/2 degrader. J. Med. Chem 2019, 62, 10897–10911. [DOI] [PubMed] [Google Scholar]
- (56).Shen Y; Gao G; Yu X; Kim H; Wang L; Xie L; Schwarz M; Chen X; Guccione E; Liu J; Bedford MT; Jin J Discovery of first-in-class protein arginine methyltransferase 5 (PRMT5) degraders. J. Med. Chem 2020, 63, 9977–9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Chetram MA; Odero-Marah V; Hinton CV Loss of PTEN permits CXCR4-mediated tumorigenesis through ERK1/2 in prostate cancer cells. Mol. Cancer Res 2011, 9, 90–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Fischer ES; Böhm K; Lydeard JR; Yang H; Stadler MB; Cavadini S; Nagel J; Serluca F; Acker V; Lingaraju GM; Tichkule RB; Schebesta M; Forrester WC; Schirle M; Hassiepen U; Ottl J; Hild M; Beckwith REJ; Harper JW; Jenkins JL; Thomä NH Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512, 49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Soucy TA; Smith PG; Milhollen MA; Berger AJ; Gavin JM; Adhikari S; Brownell JE; Burke KE; Cardin DP; Critchley S; Cullis CA; Doucette A; Garnsey JJ; Gaulin JL; Gershman RE; Lublinsky AR; McDonald A; Mizutani H; Narayanan U; Olhava EJ; Peluso S; Rezaei M; Sintchak MD; Talreja T; Thomas MP; Traore T; Vyskocil S; Weatherhead GS; Yu J; Zhang J; Dick LR; Claiborne CF; Rolfe M; Bolen JB; Langston SP An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [DOI] [PubMed] [Google Scholar]
- (60).Iacovides DC; Johnson AB; Wang N; Boddapati S; Korkola J; Gray JW Identification and quantification of AKT isoforms and phosphoforms in breast cancer using a novel nanofluidic immunoassay. Mol. Cell. Proteomics 2013, 12, 3210–3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Cheng M; Yu X; Lu K; Xie L; Wang L; Meng F; Han X; Chen X; Liu J; Xiong Y; Jin J Discovery of potent and selective epidermal growth factor receptor (EGFR) bifunctional small-molecule degraders. J. Med. Chem 2020, 63, 1216–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Guzman C; Bagga M; Kaur A; Westermarck J; Abankwa D ColonyArea: an ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS One 2014, 9, No. e92444. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.