

Abstract
Extremotolerant organisms from all domains of life produce protective intrinsically disordered proteins (IDPs) in response to desiccation stress. In vitro, many of these IDPs protect enzymes from dehydration stress better than U.S. Food and Drug Administration‐approved excipients. However, as with most excipients, their protective mechanism is poorly understood. Here, we apply thermogravimetric analysis, differential scanning calorimetry, and liquid‐observed vapor exchange (LOVE) NMR to study the protection of two model globular proteins (the B1 domain of staphylococcal protein G [GB1] and chymotrypsin inhibitor 2 [CI2]) by two desiccation‐tolerance proteins (CAHS D from tardigrades and PvLEA4 from an anhydrobiotic midge), as well as by disordered and globular protein controls. We find that all protein samples retain similar amounts of water and possess similar glass transition temperatures, suggesting that neither enhanced water retention nor vitrification is responsible for protection. LOVE NMR reveals that IDPs protect against dehydration‐induced unfolding better than the globular protein control, generally protect the same regions of GB1 and CI2, and protect GB1 better than CI2. These observations suggest that electrostatic interactions, charge patterning, and expanded conformations are key to protection. Further application of LOVE NMR to additional client proteins and protectants will deepen our understanding of dehydration protection, enabling the streamlined production of dehydrated proteins for expanded use in the medical, biotechnology, and chemical industries.
Keywords: amide proton exchange, desiccation, nuclear magnetic resonance, tardigrades
1. INTRODUCTION
Protein‐based therapeutics (i.e., biologics) such as insulin, vaccines, and antibodies are among the most precise and effective drugs on the market. Yet, the relatively short shelf‐lives of proteins in aqueous solution, along with the high costs of refrigerated transport and storage, hinder their widespread use. 1 , 2 , 3 To increase their stability and mitigate challenges associated with the so‐called “cold chain,” many biologics and industrial enzymes are dehydrated. However, given the key role of water in protein structure and function, many proteins cannot withstand dehydration, irreversibly unfolding and aggregating during the process. 4 , 5 , 6
To protect proteins from dehydration‐induced damage, additives called excipients are added before drying. 4 , 6 , 7 , 8 Yet, despite decades of research, our understanding of the mechanisms of protein dehydration protection is limited, in part because we were unable to study the effects of dehydration on protein structure at high resolution. 9 A great deal of information on dry protein structure comes from mass spectrometry of proteins in the gas phase. Such efforts show an increased importance of hydrogen bonds and charge–charge interactions, which, together with the missing hydrophobic effect, leave proteins in an ensemble of collapsed, nonnative states. 10 Nevertheless, although a valuable tool, 11 mass spectrometry provides peptide level—rather than residue level—information, leaving an incomplete picture of dehydrated protein structure.
In practice, our inadequate understanding of how dehydration impacts protein structure, and how excipients mitigate its effects, means that formulation must be approached empirically for each protein, a time‐ and resource‐intensive process that often fails. 4 , 6 , 8 , 12 Gaining a deeper understanding of dehydration protection and how it relates to client protein properties would enable the prediction of effective excipient formulations for dehydrated biologics, in turn, making these life‐saving drugs more affordable and accessible. 13
To this end, we developed liquid‐observed vapor exchange (LOVE) NMR, a solution NMR technique enabling the study of dehydrated protein structure and protection at the residue level. 14 , 15 Based on the well‐established principle that amide protons are less likely to exchange with deuterons from the environment if involved in intra‐ or intermolecular H‐bonds, 16 , 17 , 18 LOVE NMR uses solution NMR to quantify the extent of hydrogen–deuterium exchange between D2O vapor and the amide protons of a dried protein. Following correction for solution back‐exchange, 15 the percent of amide‐proton signal remaining after vapor exchange reflects the fraction of the dry protein population for which a given residue is involved in an inter‐ or intramolecular H‐bond, that is, the “%Protected.” 14 Thus, LOVE NMR can determine where, and to what degree, excipients interact with dehydrated proteins and/or prevent dehydration‐induced unfolding. Such information can be used to elucidate mechanisms of protection and explain why a given excipient will work for some client proteins but not others.
Here, we apply LOVE NMR to study the dehydration protection of two model proteins (Figure 1a)—the B1 domain of staphylococcal protein G (GB1) and chymotrypsin inhibitor 2 (CI2)—by two classes of desiccation‐tolerance proteins—the cytosolic abundant heat soluble (CAHS) proteins and late embryogenesis abundant (LEA) proteins 19 , 20 —and two controls—bovine serum albumin (BSA) and gelatin. CAHS proteins comprise a family of intrinsically disordered proteins (IDPs) unique to tardigrades, microscopic animals well known for their ability to survive extreme stresses in their dehydrated “anhydrobiotic” form. 20 , 21 , 22 CAHS proteins are necessary for tardigrades to survive desiccation and sufficient to protect heterologously expressing cells and enzymes from desiccation damage. 3 , 23 LEA proteins, which are better studied, are IDPs implicated in the desiccation tolerance of many plants and animals and are thought to protect against dehydration‐induced aggregation by acting as “molecular shields.” 24 , 25 , 26 , 27 , 28
FIGURE 1.
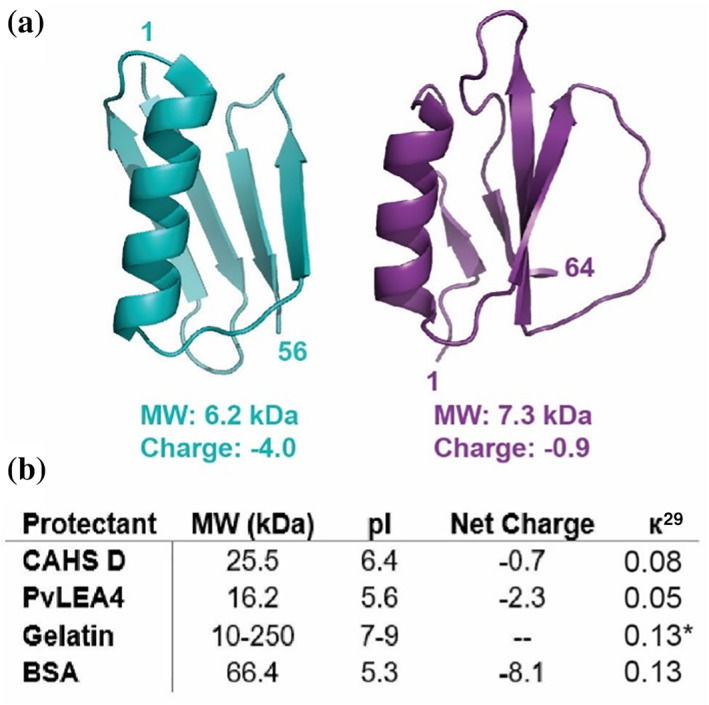
Proteins used in this study. (a) Structures of client proteins GB1 (left, PDB 2QMT) and CI2 (right, PDB 2CI2). (b) Properties of protectant proteins. Net charge is calculated at pH 6.5. *к calculated for porcine collagen α‐1 chain preprotein
Although CAHS and LEA proteins lack homology (Figure S2), they share several properties, including a relatively low molecular weight, a high degree of disorder, and extensive charge patterning (represented by a near‐zero к value 29 [Figure 1b]). Unlike LEA proteins, however, CAHS proteins are thought to form reversible, concentration‐dependent hydrogels. 30 To investigate the importance of disorder and gelation in dehydration protection by desiccation‐tolerance proteins, we, therefore, included the globular protein BSA and the disordered, gelling protein mixture, gelatin, as controls.
2. RESULTS
2.1. Water content and glass transition temperature
To determine if differences in protection are related to a cosolute's ability to retain water and/or form glasses, we performed thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) on GB1 samples lyophilized alone or in the presence of 5 g/L CAHS D, PvLEA4, gelatin, or BSA (Figure 2). Immediately after 24 hr of drying, samples possess 7–8% water by mass, which represents less than one layer of surface water on GB1 (Table S1). The glass transition temperatures (T g) are also similar, ranging from 54°C to 56°C (Figure 2b; Figure S3). After exposure to 75% relative humidity for 24 hr at room temperature, all samples absorb similar amounts of water, increasing the total water content to ~14%, 14 , 31 which still comprises less than a single layer of water, and, thus, does not substantially affect T g (Table S1 and Figure S3). 32 As expected, the added water in the vapor‐exposed sample decreases the denaturation temperature (Figure S4). 31 , 33
FIGURE 2.
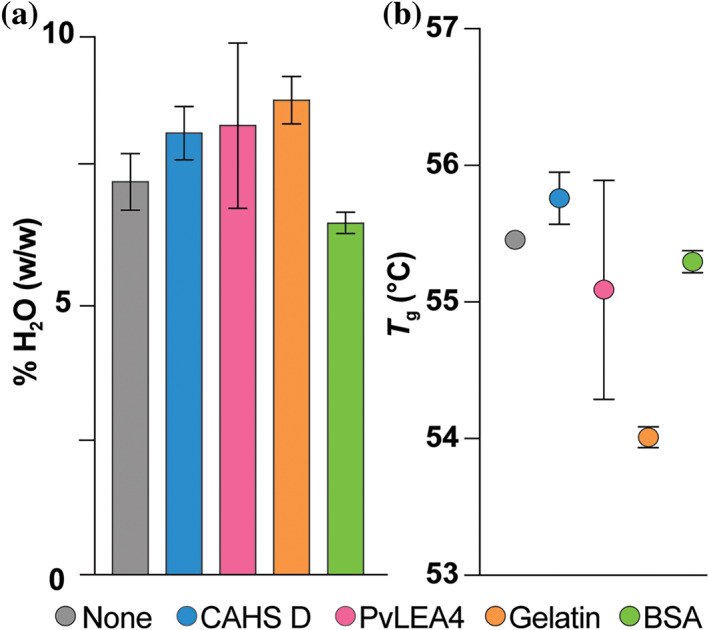
Percent H2O by mass (a) and glass transition temperature (T g) (b) of lyophilized GB1 with indicated protectants. Samples (650 μl) comprising 500 μM (~3 g/L) GB1 alone or with 5 g/L protectant were lyophilized for 24 hr, and immediately analyzed. Uncertainties in %H2O are the standard deviation from three independent measurements for GB1 with CAHS D, four independent measurements for GB1 with PvLEA4, and the range of two independent measurements for other data shown, including T g
2.2. Residue‐level protection
To compare where and how well each protein protects clients from dehydration‐induced unfolding, we performed LOVE NMR on GB1 and CI2 alone and in the presence of CAHS D, PvLEA4, gelatin, or BSA. As with the TGA and DSC experiments, LOVE NMR experiments were performed on lyophilized samples that, before drying, comprised 500 μM (~3 g/L) client protein alone or with 5 g/L protectant in a total volume of 650 μL. %Protected values (Tables S2 and S3) of the proteins dried in buffer alone 15 were subtracted from those dried in the presence of cosolute to give the change in protection (Δ%Protected).
Drying in the presence of 5 g/L CAHS D, PvLEA4, or gelatin increases the average %Protected value of GB1 by 30 ± 20%, 20 ± 20%, and 20 ± 10%, respectively, where the uncertainty is the standard deviation from the mean (Figure 3a,b). By contrast, drying in 5 g/L BSA brings minimal additional protection, increasing the average %Protected by 2 ± 7% (Figure 3c). Testing the null hypothesis using binomial analysis, 35 the probability that the protection arises randomly is <10−5 for CAHS D, LEA, and gelatin, but >0.05 for BSA (Table S4).
FIGURE 3.
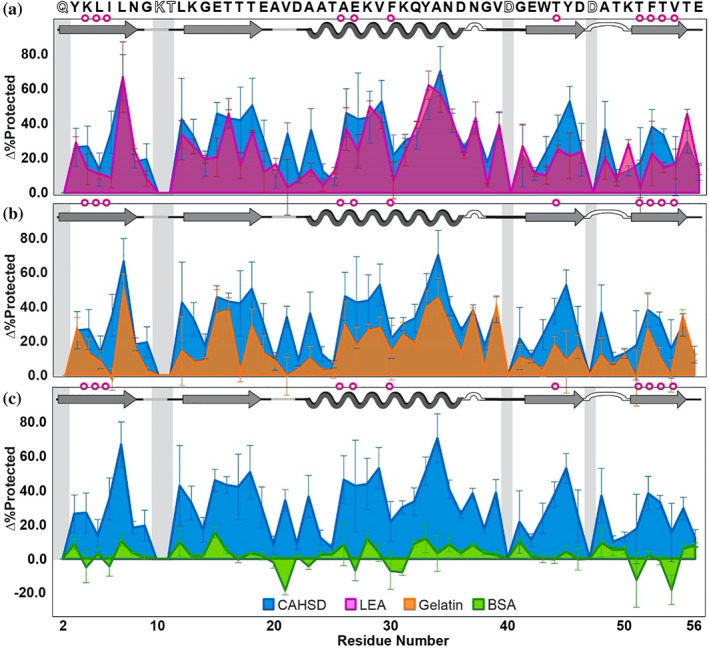
Change in GB1 dry‐state protection due to freeze‐drying in CAHS D versus (a) PvLEA4, (b) gelatin, and (c) BSA. Δ%Protected = %Protected+cosolute − %Protected−cosolute. Primary structure of GB1 (PDB 2QMT) is shown at the top. Secondary structure is shown atop each panel. Magenta circles indicate solution global‐unfolding residues. 34 Gray boxes indicate missing data from rapid back exchange (open letters in primary structure). Error bars represent standard deviations from the mean propagated from triplicate analysis
CAHS D, PvLEA4, and gelatin give similar profiles for GB1 (Figure 3a,b), with those of PvLEA and CAHS D appearing most similar. CAHS D consistently outperforms the other protein cosolutes at residues I6, V21, A23, T44, Y45, and T51, all but two of which (V21 and Y45) are, or immediately neighbor, global‐unfolding residues. 34 However, CAHS D is outperformed by PvLEA4 at residues T25, K50, and T55.
CAHS D, PvLEA4, and gelatin also protect CI2 (Figure 4), but the average Δ%Protected values, 7 ± 7%, 3 ± 3%, and 6 ± 4%, respectively, are smaller than those for GB1. Like GB1, CI2 is least protected by BSA, with a 2 ± 4% increase; unlike GB1, however, only gelatin exhibits a probability <0.05 that the observed protection trends arise from random data (Table S4). Although all protein cosolutes offer a similar level of protection for most regions of CI2, inspection of the protection profiles suggests that with this client protein, CAHS D behaves most like gelatin, especially in the α‐helix and C‐terminal β‐sheets (Figure 4b). This result contrasts with the protection profiles of GB1, which show that CAHS D protects most like PvLEA4.
FIGURE 4.
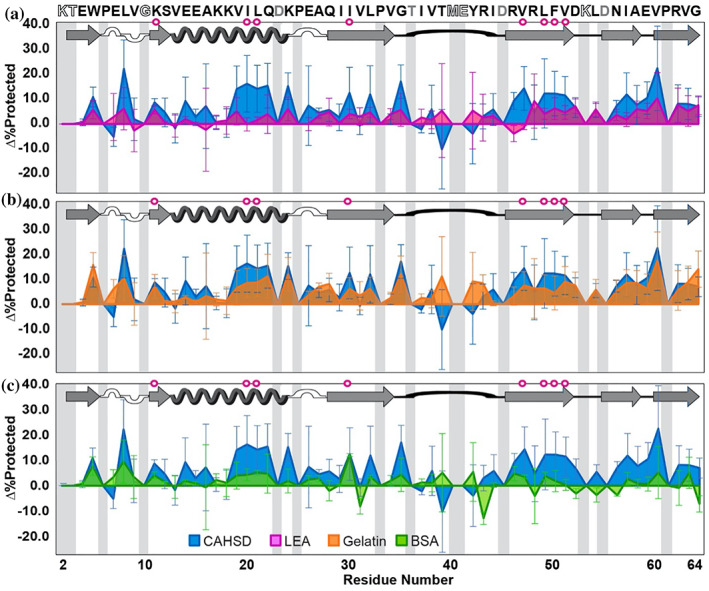
Change in CI2 dry‐state protection due to freeze‐drying in CAHS D versus (a) PvLEA4, (b) gelatin, and (c) BSA. Δ%Protected = %Protected+cosolute − %Protected−cosolute. Primary structure of CI2 (PDB 2CI2) is shown at the top. Secondary structure is shown atop each panel. Magenta circles indicate solution global‐unfolding residues. Gray boxes indicate missing data from rapid back exchange (open letters in primary structure) and/or overlapping crosspeaks (gray letters in primary structure). Error bars represent standard deviations from the mean propagated from triplicate analysis
3. DISCUSSION
The observation that CAHS D and PvLEA4 both protect CI2 less well than GB1 shows that dehydration protection is client protein‐dependent. Moreover, the observation that the disordered proteins generally protect the same regions of GB1 suggests they operate via a similar mechanism.
There are three main models of cosolute‐mediated dehydration protection, which, though distinct, are not mutually exclusive: the vitrification, water replacement, and molecular shield hypotheses. The vitrification hypothesis poses that cosolutes protect by enveloping the client protein in a glassy matrix, thereby inhibiting large motions such as global unfolding. This idea is supported by studies showing positive correlations between T g and long‐term stability in the dehydrated state. 36 The water‐replacement hypothesis posits that cosolutes protect proteins from dehydration‐induced unfolding by replacing water‐mediated H‐bonds. 12 , 37 This model is supported by the observation that the protective ability of certain sugars correlates with their ability to maintain the position of the amide II band, which is sensitive to changes in secondary structure and hydrogen bonding with water. 38 Finally, the molecular shield hypothesis envisions protectants forming barriers between partially unfolded client proteins, thus preventing irreversible aggregation. 27 , 39 Support for this mechanism arises from the observation that an LEA protein inhibits aggregation but fails to prevent intramolecular changes upon drying. 40
The observation that CAHS D generally performs better than PvLEA4 despite having a lower protectant‐to‐client mole ratio (~2:5 vs. 3:5) suggests that most interactions between the desiccation‐protective proteins and client proteins are non‐specific. It is unlikely, however, that dehydration protection arises from enhanced water retention or the properties of the glassy matrix, because the dried mixtures protect to different degrees yet possess nearly the same water content and T g (Figure 2). The latter observation suggests, in turn, that vitrification is not the main source of protection.
The difference between the average Δ%Protected experienced by GB1 and CI2 may provide a window into the mechanism of dehydration‐protective proteins. Although both client proteins are similarly sized, are similarly stable in solution, 41 , 42 and possess a 4β + 1α architecture, GB1 possesses several‐fold more net charge than CI2 under the conditions used here (Figure 1a), suggesting that electrostatic interactions are important for protection. However, given that both client proteins are negatively charged and that BSA has the most negative charge of all the protectants (Figure 1b), it is unlikely that repulsive electrostatic interactions, which stabilize proteins in solution, 43 are responsible for protection. Perhaps, electrostatic interactions enable client proteins to be sequestered into the matrix of protectants, a phenomenon observed with other dehydration‐protective proteins. 44
Fascinatingly, the residue‐level pattern of GB1 protection by CAHS D resembles that of GB1 chemical‐shift perturbations caused by the ionic liquid 1‐butyl‐3‐methylimidazolium bromide (Figure S5, p < .05), that is, residues that see larger ionic‐liquid‐induced shifts in solution tend to be more protected by CAHS D in the dry state. 45 This correlation suggests that proximal interactions with alternating charges (e.g., intermolecular salt bridges or charge‐assisted H‐bonds) may prevent dehydration‐induced unfolding—an idea consistent with simulations and mass spectrometry experiments that suggest salt bridges and ionic H‐bonds stabilize proteins in the gas phase. 46 , 47 A pronounced role for intermolecular charge–charge interactions in dehydration protection is also consistent with the observation that most dehydration protective proteins are disordered, 48 , 49 , 50 and that many disordered proteins, including those used in this study, possess a high degree of charge patterning (low к value, Figure 1b). 29 The apparent importance of charge patterning and non‐specific intermolecular interactions aligns well with both the water‐replacement and molecular shield hypotheses.
Although all the disordered proteins protect better than BSA, residue‐level differences in their protection profiles point to subtle differences in protective mechanism. For instance, CAHS D prevents dehydration‐induced exposure of global‐unfolding residues better than PvLEA4, 35 , 51 signifying that CAHS D is better at preventing global unfolding. In addition, CAHS D appears to behave most like gelatin in the protection of CI2. Together, these observations suggest that gelation of CAHS D supplements the dehydration protection afforded by electrostatics to further prevent global unfolding, perhaps by confining the client protein in a gel matrix. 52 Finally, the observation that PvLEA4 protects the polar and/or charged GB1 residues T25, K50, and T55 better than CAHS D, yet generally performs worse than CAHS D in protecting the more neutral client protein CI2, suggests that electrostatic interactions are more important to the protective mechanism of PvLEA4.
4. CONCLUSIONS
Our residue‐level study of dehydration protection of two client proteins by two IDPs involved in animal desiccation tolerance confirm that protection is client‐protein dependent and that desiccation‐tolerance proteins generally protect better than those unrelated to dehydration stress response. Comparing protection trends of CAHS D and PvLEA4 to those of BSA and gelatin suggest that disorder and charge patterning are integral to dehydration protection, supporting both the molecular shield and water‐replacement hypotheses. Application of LOVE NMR to other client proteins and protectants will further advance our understanding of dehydration protection, knowledge that can, in turn, be used to streamline the production, storage, and shipping of protein products, making these valuable and lifesaving products less expensive and more accessible.
5. MATERIALS AND METHODS
5.1. Materials
Ampicillin, kanamycin sulfate, bovine serum albumin (Sigma Aldrich), trisodium citrate (Agros Organics), citric acid monohydrate, and HEPES (Thermo Fisher) were used without further purification. H2O with a resistivity of >17 MΩ cm−1 was used to prepare buffers. Unflavored porcine gelatin (Knox) was dialyzed against H2O to remove small molecules (ThermoScientific Snakeskin™ dialysis tubing, 3,500 Da molecular weight cutoff), and lyophilized before resuspension in 1.5 mM HEPES to a final concentration of 5 g/L. pH values are direct readings, uncorrected for the deuterium isotope effect. 53 For LOVE NMR experiments, the relative humidity is 75 ± 5% (Fisherbrand TraceableGO™ Bluetooth datalogging digital hygrometer). 14
5.2. Expression and purification of client proteins
The pET11a plasmid (Novagen) containing the gene for the T2Q variant of the immunoglobulin G binding domain of streptococcal G was provided by Leonard D. Spicer's laboratory at Duke University (Durham, North Carolina). This variant, which we call GB1, was chosen, because the mutation prevents N‐terminal deamidation. 54 The pET28a plasmid (Novagen) containing the gene for truncated wild‐type barley chymotrypsin inhibitor 2 from (CI2) was provided by Dr. Andrew Lee's laboratory at UNC‐Chapel Hill. The first 19 residues of full length CI2 are excluded, because they are disordered, and, therefore, exchange too quickly for observation via NMR. Residue 20 of full‐length CI2 is referred as Residue 1.
15N‐enriched GB1 and CI2 were expressed in Agilent BL21 Gold (DE3) Escherichia coli in minimal media and purified as described. 55 , 56 The concentration of purified protein was determined from the absorbance at 280 nm (A280) (Nanodrop One, Thermo Fisher) using an extinction coefficient of 9,530 M−1 cm−1 for GB1 and 7,040 M−1 cm−1 for CI2. 57 Purity was confirmed by Q‐TOF mass spectrometry (ThermoScientific, Q Exactive HF‐X) in the UNC Mass Spectrometry Chemical Research and Teaching Core Laboratory. Purified protein was exchanged into H2O by dialysis (ThermoScientific Snakeskin™ dialysis tubing, 3,500 Da molecular weight cutoff), and divided into aliquots, such that resuspension in 650 μL gives a protein concentration of 500 μM. Aliquots were flash‐frozen, lyophilized, and stored at −20°C.
5.3. Expression and purification of protectant proteins
pET28b plasmids encoding the genes for CAHS D or PvLEA4, both fused to a N‐terminal hexahistadine (His‐) tag and a TEV protease cleavage site, were ordered from Gene Universal Inc. Vectors were transformed into Agilent BL21 Gold (DE3) E. coli as described. 23 A single colony was used to inoculate 100 ml of Lennox Luria broth (Fisher, 10 g/L tryptone, 5 g/L yeast extract, 5 g/L NaCl) supplemented with the antibiotic kanamycin to a final concentration of 60 μg/ml. The culture was shaken at 37°C overnight (New Brunswick Scientific I26 incubator, 225 rpm); 10 ml of the overnight culture was used to inoculate 1 L of kanamycin‐supplemented LB; 1 L cultures were shaken at 37°C until they reached an optical density at 600 nm of 0.6–0.8, at which point protein expression was induced by adding isopropyl β‐D‐1‐thiogalactopyranoside (1 mM final concentration); 3 hr after induction, cells were harvested via centrifugation at 4,000g. The cell pellet from each culture was resuspended in 10 ml of 20 mM Tris, pH 7.5, and stored at −20°C.
On the day of purification, frozen cell slurries from 3 L of culture were thawed at room temperature and lysed via heat shock at 95°C for 15 min. Lysates were clarified by centrifugation at 15,000g for 45 min, passed through a 0.45 μm filter, and purified via affinity chromatography on a Ni‐NTA column (5 ml GE HisTrap HP) using a GE AKTA Start FPLC.
To prevent proteins from gelling on the column, filtered lysates were mixed with an equal volume of 3 M urea containing 10 mM imidazole, 500 mM NaCl, and 20 mM sodium phosphate, pH 7.4 (HUA). To prevent carbamylation by NH₄OCN, 58 urea solutions were deionized with 5 g/L Dowex® MB Mixed Ion Exchange Resin (Sigma). The resin was removed with a 0.22 μm filter (Corning Inc. 431161, Corning, NY).
Affinity chromatography was performed at room temperature in HUA. After sample loading, the Ni‐NTA column was washed with three column volumes of HUA followed by a 29‐column‐volume gradient of 10–150 mM imidazole for CAHS D and 10–100 mM imidazole for PvLEA4. SDS‐PAGE (Bio‐Rad 4–20% Criterion™ TGX™ Gels) was used to identify fractions containing purified, His‐tagged protein.
To remove the His‐tag, the fractions were pooled, dialyzed at room temperature against 50 mM Tris (pH 8.1) containing 500 μM EDTA and 1 mM DTT for 4 hr (ThermoScientific Snakeskin™ dialysis tubing, 3,500 Da molecular weight cutoff), and then incubated with 1 mg TEV protease at room temperature with gentle shaking for 16–24 hr. After digestion, the His‐tag and protease were removed via incubation with loose Ni‐NTA resin for 12–16 hr.
To remove the remaining buffer salts, the solution was subjected to six additional rounds of dialysis (>4 hr each) at room temperature against deionized H2O (ThermoScientific Snakeskin™ dialysis tubing, 3,500 Da molecular weight cutoff). Finally, the solution was heat‐shocked to resolubilize protein that precipitated during dialysis, passed through a 0.45 μm filter, flash‐frozen, lyophilized, and stored at −20°C.
5.4. Thermogravimetric analysis
Aliquots of purified, lyophilized, 15N‐enriched GB1 were resuspended in 650 μl of 1.5‐mM HEPES pH 6.5 with or without 5 g/L protectant to a final client protein concentration of 500 μM, flash‐frozen, and lyophilized (LABCONCO FreeZone 1 Liter Benchtop Freeze Dry System) for 24 hr. T 0 samples were analyzed immediately, while T 24 samples were analyzed after 24 hr in a chamber with a controlled relative humidity of 75 ± 5% created as described above. Samples weighing ~0.5–1.5 mg were loaded into a TA Instruments model 550 thermogravimetric analyzer on an open Pt pan and heated from 25°C to 170°C at a rate of 5°C/min under a N2(g) sample purge of 60 ml/min and a balance purge of 40 ml/min. The well‐defined mass loss ending around 130°C was used to quantify the contents of H2O and D2O (Figure S6). 59 , 60 Thermograms were analyzed using Trios V5.1.0.56403 software.
5.5. Differential scanning calorimetry
Aliquots of purified, lyophilized, 15N‐enriched GB1 were resuspended in 650 μl of 1.5‐mM HEPES pH 6.5 with or without 5 g/L protectant to a final client protein concentration of 500 μM, flash‐frozen, and lyophilized as described above. T 0 samples were analyzed immediately, while T 24 samples were analyzed after 24 hr in a chamber with a controlled relative humidity of 75 ± 5% created as described above. Samples weighing ~1.0–2.5 mg were sealed in Tzero Hermetic Aluminum pans and loaded into a TA Instruments model 250 differential scanning calorimeter equipped with a TA Instruments Refrigerated Cooling System 90. An identical, empty pan was used as a reference. The sample cell was under 50 ml/min N2(g) purge. To eliminate differing thermal history effects on the reversible glass transition, samples were heated at 7.5°C/min to ~5°C above their glass transition temperature, and then cooled to 10°C, where they remained for 1 min. 61 , 62 , 63 , 64 Samples were again heated at 7.5°C/min to about 15°C above their denaturation temperature, and from this scan both glass transition temperature and denaturation temperature were measured. Samples were cooled to 10°C, and once more heated at 7.5°C/min to confirm the irreversibility of denaturation, as expected in protein samples with this water content. 64 Thermograms were analyzed using Trios V5.1.0.56403 software, with the midpoint of the endothermal shift in the baseline on the thermogram reported as the glass transition temperature (T g), 61 , 62 , 63 , 65 and the minimum of the denaturation endothermic peak reported as the denaturation temperature (T m) (Figure S6). 31 , 66 , 67
5.6. NMR
Unless noted, experiments were performed in triplicate on a Bruker Avance III HD spectrometer with a cryogenic QCI probe at a 1H Larmor frequency of 600 MHz. For LOVE NMR, sensitivity‐enhanced 15N–1H heteronuclear single‐quantum coherence (HSQC) spectra were acquired in ~10 min (128 increments in the 15N dimension, 4 scans per increment) with sweep widths of 3,041 Hz in the 15N dimension and 8,418 Hz in the 1H dimension. Spectra were processed with NMRPipe. 68 Crosspeaks were integrated using NMRViewJ. 69 Backbone resonances of GB1 and CI2 at pH 4.5, 4°C were assigned using isotopically enriched protein expressed in minimal media containing 13C‐enriched D‐glucose and 15N‐enriched NH4Cl (Cambridge Isotope Labs) as the sole sources of carbon and nitrogen, respectively, and then purified as described. 55 , 56 HNCACB spectra were acquired with 10% sampling in the indirect dimensions using a Poisson gap scheduling scheme. 70 , 71 Spectra were processed using NMRpipe and reconstructed with the SMILE algorithm. 72 Assigned spectra are shown in Figure S8.
5.7. Liquid observed vapor exchange NMR
For each experiment, two identical aliquots of purified, lyophilized, 15N‐enriched protein were resuspended in 650 μl of 1.5‐mM HEPES pH 6.5 with or without 5 g/L protectant to a final client protein concentration of 500 μM, flash‐frozen, and lyophilized (LABCONCO FreeZone 1 Liter Benchtop Freeze Dry System) for 24 hr. Following lyophilization, one sample, designated T 0, was immediately resuspended in 650 μl of cold quench buffer (100 mM citrate buffer, pH 4.5, 90% H2O/10% D2O) and transferred to an NMR spectrometer for spectrum acquisition at 4°C. The second sample, designated T 24, was placed, with the cap open, in a chamber with a controlled relative humidity of ~75%, prepared as described. 14 After 24 hr, the T 24 sample was resuspended in 650 μl of cold quench buffer and a spectrum obtained using the same parameters that were used for the T 0 sample. To enable back‐exchange correction, the T 24 sample was left in the spectrometer at 4°C for ~12 hr, during which time an additional 10–12 spectra were acquired. The time between resuspension and initiation of the first T 24 spectrum acquisition was 10 min, ~8 min of which were spent at 4°C. To ensure differences in cross peak volume arise solely from vapor exchange and not from concentration differences, after spectra were acquired, client protein concentration of the T 0 and T 24 samples was determined via A280, as described above, and used to normalize crosspeak volumes as described below. Spectra were processed with NMRPipe, 68 crosspeaks were integrated using the ellipse volume integration tool in NMRViewJ, 69 and %Protected for each condition was determined as described below.
5.8. Identifying residues that back‐exchange completely before spectrum acquisition
To differentiate residues that are highly protected from back‐exchange from those that back‐exchange completely before the first T 24 spectrum was acquired, a sample of 15N‐enriched protein was resuspended to 500 μM in 1.5 mM HEPES, lyophilized for 24 hr, resuspended in cold D2O‐based quench buffer (100 mM citrate, pH 4.5 >98% D2O), and immediately transferred to a spectrometer at 4°C for acquisition of a 15N–1H HSQC spectrum using the same acquisition parameters used for the T 0 and T 24 samples. Resonances present in H2O but not in D2O are presumed to back‐exchange completely before the first T 24 spectrum is acquired. Given that an exchange rate cannot be estimated for these residues, we cannot approximate the pre‐back‐exchange signal, and therefore, these residues are omitted from the dataset (see figure captions for lists). We call these resonances “quench labeled.”
5.9. Estimating V T24 *, the peak volume of T 24 sample pre‐back‐exchange
For non‐quench‐labeled T 24 resonances that exhibited a ≤5% increase in peak volume over ~12 hr, back‐exchange during the time between resuspension and full acquisition of an HSQC spectrum (20 min here) is assumed to be negligible. Therefore, for these residues, V T24 * is presumed to be equivalent to the peak volume obtained from integrating the initial T 24 spectrum, V T24 .
For T 24 resonances that exhibited a >5% increase in peak volume over ~12 hr, peak volumes from the 10–12 spectra acquired serially for the T 24 sample were plotted as a function of time and fit, using the nonlinear least‐squares algorithm in Matlab, to the three‐parameter equation V(t) = A(1 − e −Bt) + C, where t is the time between protein resuspension and signal acquisition, V(t) is the peak volume at time t, A is the maximum possible change in peak volume, B is the observed rate constant, and C is a constant equivalent to the initial peak volume before back‐exchange. For these residues, the fitted value for C was used to estimate the pre‐back‐exchange peak volume, V T24 *.
The estimated deadtime was 20 min (based on the 10 min between resuspension and initiation of spectrum acquisition plus 10 min for spectrum acquisition). In practice, this estimation means timepoints are shifted by 20 min; for example, the first spectrum acquired for T 24 corresponds to the 20‐min timepoint.
Any small differences in GB1 concentration between the T 0 and T 24 samples (due to, e.g., incomplete resuspension) were determined post‐experiment via the absorbance at 280 nm, as described above. The A 280 values were used to normalize the calculated V T24* to V T0 for all experiments except for those with BSA, because all protectants besides BSA had negligible absorption at 280 nm. BSA samples were assumed to resuspend completely.
5.10. Calculating average %Protected and Δ%Protected from vapor exchange
To determine the average percent of the dried protein population for which a given amide proton is protected from vapor exchange, the average, concentration‐normalized value of V T24* (obtained as described above) was divided by the average peak volume of the corresponding crosspeak in the non‐vapor‐exchanged protein sample (T 0) and multiplied by 100%, that is,
Average Δ%Protected, that is, the percent change in client protein dry state protection due to drying in the presence of a protectant, was calculated as:
Uncertainties were obtained using triplicate analysis and standard propagation of error.
CONFLICT OF INTEREST
The authors declare no competing interest.
AUTHOR CONTRIBUTIONS
Candice Crilly: Conceptualization (lead); data curation (equal); formal analysis (equal); investigation (equal); methodology (lead); validation (equal); visualization (equal); writing – original draft (lead); writing – review and editing (lead). Julia Brom: Conceptualization (supporting); data curation (equal); formal analysis (equal); investigation (equal); methodology (supporting); writing – original draft (equal); writing – review and editing (equal). Owen Warmuth: Formal analysis (supporting); investigation (supporting); validation (supporting); visualization (supporting). Harrison Esterly: Data curation (supporting); investigation (supporting); methodology (supporting); resources (supporting). Gary Joseph Pielak: Conceptualization (lead); funding acquisition (lead); project administration (lead); writing – original draft (equal); writing – review and editing (equal).
Supporting information
Appendix S1: Supporting information
ACKNOWLEDGMENTS
This research was supported by NIH grant R01GM127291 to Gary J. Pielak and an NIH training grant (T32GM008570) to Candice J. Crilly. The authors thank Stu Parnham from the UNC Biomolecular NMR Core and Brandie Ehrmann from the UNC Chemistry Mass Spectrometry Core Laboratory for equipment maintenance and advice. The authors also thank Dr. Krisztina Varga for sharing chemical shift perturbation data, the Pielak lab for helpful discussion, and Elizabeth Pielak for comments on the manuscript.
Crilly CJ, Brom JA, Warmuth O, Esterly HJ, Pielak GJ. Protection by desiccation‐tolerance proteins probed at the residue level. Protein Science. 2022;31:396–406. 10.1002/pro.4231
Candice J. Crilly and Julia A. Brom contributed equally to the study.
Funding information National Institutes of Health, Grant/Award Numbers: R01GM127291, T32GM008570
REFERENCES
- 1. Morrow T, Felcone LH. Defining the difference: What makes biologics unique. Biotechnol Healthc. 2004;1:24–29. [PMC free article] [PubMed] [Google Scholar]
- 2. Frokjaer S, Otzen DE. Protein drug stability: A formulation challenge. Nat Rev Drug Discov. 2005;4:298–306. [DOI] [PubMed] [Google Scholar]
- 3. Piszkiewicz S, Gunn KH, Warmuth O, et al. Protecting activity of desiccated enzymes. Protein Sci. 2019;28:941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang W. Lyophilization and development of solid protein pharmaceuticals. Int J Pharm. 2000;203:1–60. [DOI] [PubMed] [Google Scholar]
- 5. Walters RH, Bhatnagar B, Tchessalov S, Izutsu KI, Tsumoto K, Ohtake S. Next generation drying technologies for pharmaceutical applications. J Pharm Sci. 2014;103:2673–2695. [DOI] [PubMed] [Google Scholar]
- 6. Bjelosevic M, Zvonar Pobirk A, Planinsek O, Ahlin Grabnar P. Excipients in freeze‐dried biopharmaceuticals: Contributions toward formulation stability and lyophilisation cycle optimisation. Int J Pharm. 2020;576:1–12. [DOI] [PubMed] [Google Scholar]
- 7. Ohtake S, Kita Y, Arakawa T. Interactions of formulation excipients with proteins in solution and in the dried state. Adv Drug Deliv Rev. 2011;63:1053–1073. [DOI] [PubMed] [Google Scholar]
- 8. Piszkiewicz S, Pielak GJ. Protecting enzymes from stress‐induced inactivation. Biochemistry. 2019;58:3825–3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moorthy BS, Iyer LK, Topp EM. Characterizing protein structure, dynamics and conformation in lyophilized solids. Curr Pharm Des. 2015;21:5845–5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Breuker K, McLafferty FW. Stepwise evolution of protein native structure with electrospray into the gas phase, 10−12 to 102 s. Proc Natl Acad Sci U S A. 2008;105:18145–18152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hodge EA, Benhaim MA, Lee KK. Bridging protein structure, dynamics, and function using hydrogen/deuterium‐exchange mass spectrometry. Protein Sci. 2020;29:843–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mensink MA, Frijlink HW, van der Voort MK, Hinrichs WL. How sugars protect proteins in the solid state and during drying (review): Mechanisms of stabilization in relation to stress conditions. Eur J Pharm Biopharm. 2017;114:288–295. [DOI] [PubMed] [Google Scholar]
- 13. Hill AB, Kilgore C, McGlynn M, Jones CH. Improving global vaccine accessibility. Curr Opin Biotechnol. 2016;42:67–73. [DOI] [PubMed] [Google Scholar]
- 14. Crilly CJ, Brom JA, Kowalewski ME, Piszkiewicz S, Pielak GJ. Dried protein structure revealed at the residue level by liquid‐observed vapor exchange NMR. Biochemistry. 2021;60:152–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Crilly C, Eicher JE, Warmuth O, Atkin JM, Pielak GJ. Water's variable role in protein stability uncovered by liquid‐observed vapor exchange NMR. Biochemistry. 2021;60:3041–3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Englander SW, Kallenbach NR. Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q Rev Biophys. 1983;16:521–655. [DOI] [PubMed] [Google Scholar]
- 17. Desai UR, Osterhout JJ, Klibanov AM. Protein structure in the lyophilized state: A hydrogen isotope exchange/NMR study with bovine pancreatic trypsin inhibitor. J Am Chem Soc. 1994;116:9420–9422. [Google Scholar]
- 18. Percy AJ, Rey M, Burns KM, Schriemer DC. Probing protein interactions with hydrogen/deuterium exchange and mass spectrometry‐a review. Anal Chim Acta. 2012;721:7–21. [DOI] [PubMed] [Google Scholar]
- 19. Li S, Chakraborty N, Borcar A, Menze MA, Toner M, Hand SC. Late embryogenesis abundant proteins protect human hepatoma cells during acute desiccation. Proc Natl Acad Sci U S A. 2012;109:20859–20864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yamaguchi A, Tanaka S, Yamaguchi S, et al. Two novel heat‐soluble protein families abundantly expressed in an anhydrobiotic tardigrade. PLoS One. 2012;7:e44209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rebecchi L, Guidetti R, Borsari S, Altiero T, Bertolani R. Dynamics of long‐term anhydrobiotic survival of lichen‐dwelling tardigrades. Hydrobiologia. 2006;558:23–30. [Google Scholar]
- 22. Jonsson KI, Rabbow E, Schill RO, Harms‐Ringdahl M, Rettberg P. Tardigrades survive exposure to space in low Earth orbit. Curr Biol. 2008;18:R729–R731. [DOI] [PubMed] [Google Scholar]
- 23. Boothby TC, Tapia H, Brozena AH, et al. Tardigrades use intrinsically disordered proteins to survive desiccation. Mol Cell. 2017;65(6):975–984.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wise MJ, Tunnacliffe A. Popp the question: What do LEA proteins do? Trends Plant Sci. 2004;9:13–17. [DOI] [PubMed] [Google Scholar]
- 25. Battaglia M, Olvera‐Carrillo Y, Garciarrubio A, Campos F, Covarrubias AA. The enigmatic LEA proteins and other hydrophilins. Plant Physiol. 2008;148:6–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hand SC, Menze MA, Toner M, Boswell L, Moore D. LEA proteins during water stress: Not just for plants anymore. Annu Rev Physiol. 2011;73:115–134. [DOI] [PubMed] [Google Scholar]
- 27. Hatanaka R, Hagiwara‐Komoda Y, Furuki T, et al. An abundant LEA protein in the anhydrobiotic midge, PvLEA4, acts as a molecular shield by limiting growth of aggregating protein particles. Insect Biochem Mol Biol. 2013;43:1055–1067. [DOI] [PubMed] [Google Scholar]
- 28. Kamilari M, Jorgensen A, Schiott M, Mobjerg N. Comparative transcriptomics suggest unique molecular adaptations within tardigrade lineages. BMC Genomics. 2019;20:607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Das RK, Pappu RV. Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc Natl Acad Sci U S A. 2013;110:13392–13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yagi‐Utsumi M, Aoki K, Watanabe H, et al. Desiccation‐induced fibrous condensation of CAHS protein from an anhydrobiotic tardigrade bioRxiv: 2021.2006.2022.449423. [DOI] [PMC free article] [PubMed]
- 31. Bell LN, Hageman MJ, Muraoka LM. Thermally induced denaturation of lyophilized bovine somatotropin and lysozyme as impacted by moisture and excipients. J Pharm Sci. 1995;84:707–712. [DOI] [PubMed] [Google Scholar]
- 32. Towns JK. Moisture content in proteins: Its effects and measurement. J Chromatogr A. 1995;705:115–127. [DOI] [PubMed] [Google Scholar]
- 33. Tsereteli GI, Belopolskaya TB, Grunina NA, Vaveliouk OL. Calorimetric study of the glass transition process in humid proteins and DNA. J Therm Anal Calorim. 2000;62:89–99. [Google Scholar]
- 34. Orban J, Alexander P, Bryan P, Khare D. Assessment of stability differences in the protein G B1 and B2 domains from hydrogen‐deuterium exchange: Comparison with calorimetric data. Biochemistry. 1995;34:15291–15300. [DOI] [PubMed] [Google Scholar]
- 35. Taylor J. Propogation of uncertainties. In: McGuire A, editor. Introduction to error analysis, the study of uncertainties in physical measurements. 2nd ed. Melville, NY: University Science Books, 1997; p. 45–92. [Google Scholar]
- 36. Grasmeijer N, Stankovic M, de Waard H, Frijlink HW, Hinrichs WLJ. Unraveling protein stabilization mechanisms: Vitrification and water replacement in a glass transition temperature controlled system. Biochim Biophys Acta Proteins Proteomics. 2013;1834:763–769. [DOI] [PubMed] [Google Scholar]
- 37. Crowe JH, Clegg JS, Crowe LM. Anhydrobiosis: The water replacement hypothesis. In: Reid DS, editor. The properties of water in foods isopow 6. Boston, MA: Springer, 1998; p. 440–455. [Google Scholar]
- 38. Carpenter JF, Crowe JH. An infrared spectroscopic study of the interactions of carbohydrates with dried proteins. Biochemistry. 1989;28:3916–3922. [DOI] [PubMed] [Google Scholar]
- 39. Tunnacliffe A, Wise MJ. The continuing conundrum of the LEA proteins. Naturwissenschaften. 2007;94:791–812. [DOI] [PubMed] [Google Scholar]
- 40. Chakrabortee S, Tripathi R, Watson M, et al. Intrinsically disordered proteins as molecular shields. Mol Biosyst. 2012;8:210–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gronenborn AM, Filpula DR, Essig NZ, et al. A novel, highly stable fold of the immunoglobulin binding domain of streptococcal protein G. Science. 1991;253:657–661. [DOI] [PubMed] [Google Scholar]
- 42. Roesler KR, Rao AG. Conformation and stability of barley chymotrypsin inhibitor‐2 (ci‐2) mutants containing multiple lysine substitutions. Protein Eng Des Sel. 1999;12:967–973. [DOI] [PubMed] [Google Scholar]
- 43. Cohen RD, Pielak GJ. A cell is more than the sum of its (dilute) parts: A brief history of quinary structure. Protein Sci. 2017;26:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Belott C, Janis B, Menze MA. Liquid‐liquid phase separation promotes animal desiccation tolerance. Proc Natl Acad Sci U S A. 2020;117:27676–27684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Warner L, Gjersing E, Follett SE, Elliott KW, Dzyuba SV, Varga K. The effects of high concentrations of ionic liquid on GB1 protein structure and dynamics probed by high‐resolution magic‐angle‐spinning NMR spectroscopy. Biochem Biophys Rep. 2016;8:75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Konermann L. Molecular dynamics simulations on gas‐phase proteins with mobile protons: Inclusion of all‐atom charge solvation. J Phys Chem B. 2017;121:8102–8112. [DOI] [PubMed] [Google Scholar]
- 47. Breuker K, Brüschweiler S, Tollinger M. Electrostatic stabilization of a native protein structure in the gas phase. Angew Chem Int Ed. 2011;50:873–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Graether SP, Boddington KF. Disorder and function: A review of the dehydrin protein family. Front Plant Sci. 2014;5:576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Das RK, Ruff KM, Pappu RV. Relating sequence encoded information to form and function of intrinsically disordered proteins. Curr Opin Struct Biol. 2015;32:102–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Boothby TC, Pielak GJ. Intrinsically disordered proteins and desiccation tolerance: Elucidating functional and mechanistic underpinnings of anhydrobiosis. Bioessays. 2017;39:1700119. [DOI] [PubMed] [Google Scholar]
- 51. Itzhaki LS, Neira JL, Fersht AR. Hydrogen exchange in chymotrypsin inhibitor 2 probed by denaturants and temperature. J Mol Biol. 1997;270:89–98. [DOI] [PubMed] [Google Scholar]
- 52. Eggers DK, Valentine JS. Molecular confinement influences protein structure and enhances thermal protein stability. Protein Sci. 2001;10:250–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Glasoe PK, Long FA. Use of glass electrodes to measure acidities in deuterium oxide. J Phys Chem. 1960;64:188–190. [Google Scholar]
- 54. Smith CK, Withka JM, Regan L. A thermodynamic scale for the beta‐sheet forming tendencies of the amino acids. Biochemistry. 1994;33:5510–5517. [DOI] [PubMed] [Google Scholar]
- 55. Charlton LM, Barnes CO, Li C, Orans J, Young GB, Pielak GJ. Residue‐level interrogation of macromolecular crowding effects on protein stability. J Am Chem Soc. 2008;130:6826–6830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Monteith WB, Pielak GJ. Residue level quantification of protein stability in living cells. Proc Natl Acad Sci U S A. 2014;111:11335–11340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. [DOI] [PubMed] [Google Scholar]
- 58. Warner R. The kinetics of the hydrolysis of urea and of arginine. J Biol Chem. 1942;142:705–723. [Google Scholar]
- 59. Joan C. May EG, Wheeler RM, and Westy Jerry (1982) Determination of residual moisture in freeze‐dried viral vaccines: Karl fischer, gravimetric and thermogravimetric methodologies. J Biol Stand 10 249–259. [DOI] [PubMed] [Google Scholar]
- 60. May JC, Wheeler RM, Grim E. The gravimetric method for the determination of residual moisture in freeze‐dried biological products. Cryobiology. 1989;26:277–284. [DOI] [PubMed] [Google Scholar]
- 61. Aguilera JM, Levi G, Karel M. Effect of water content on the glass transition and caking of fish protein hydrolyzates. Biotechnol Prog. 1993;9:651–654. [Google Scholar]
- 62. Rouilly A, Orliac O, Silvestre F, Rigal L. DSC study on the thermal properties of sunflower proteins according to their water content. Polymer. 2001;42:10111–10117. [Google Scholar]
- 63. Kalichevsky MT, Jaroszkiewicz EM, Blanshard JMV. Glass transition of gluten. 1: Gluten and gluten‐sugar mixtures. Int J Biol Macromol. 1992;14:257–266. [DOI] [PubMed] [Google Scholar]
- 64. Sochava IV, Smirnova OI. Heat capacity of hydrated and dehydrated globular proteins. Denaturation increment of heat capacity. Food Hydrocoll. 1993;6:513–524. [PubMed] [Google Scholar]
- 65. Duddu SP, Monte PRD. Effect of glass transition temperature on the stability of lyophilized formulations containing a chimeric therapeutic monoclonal antibody. Pharm Res. 1997;14:591–595. [DOI] [PubMed] [Google Scholar]
- 66. Medina‐Vivanco M, Sobral PJA, Sereno AM, Hubinger MD. Denaturation and the glass transition temperatures of myofibrillar proteins from osmotically dehydrated tilapia: Effect of sodium chloride and sucrose. Int J Food Prop. 2007;10:791–805. [Google Scholar]
- 67. Samouillan V, Delaunay F, Dandurand J, et al. The use of thermal techniques for the characterization and selection of natural biomaterials. J Func Biomater. 2011;2:230–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on unix pipes. J Biomol NMR. 1995;6:277–293. [DOI] [PubMed] [Google Scholar]
- 69. Johnson BA, Blevins RA. NMR view: A computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603–614. [DOI] [PubMed] [Google Scholar]
- 70. Miljenović TM, Jia X, Mobli M. Nonuniform sampling in biomolecular NMR. In: Webb GA, editor. Modern magnetic resonance. 2nd ed. Cham, Switzerland: Springer International Publishing, 2017; p. 1–21. [Google Scholar]
- 71. Hyberts SG, Takeuchi K, Wagner G. Poisson‐gap sampling and forward maximum entropy reconstruction for enhancing the resolution and sensitivity of protein NMR data. J Am Chem Soc. 2010;132:2145–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ying J, Delaglio F, Torchia DA, Bax A. Sparse multidimensional iterative lineshape‐enhanced (smile) reconstruction of both non‐uniformly sampled and conventional NMR data. J Biomol NMR. 2017;68:101–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting information