

Abstract
There are more amino acid permutations within a 40-residue sequence than atoms on Earth. This vast chemical search space hinders the use of human learning to design functional polymers. Here we show how machine learning enables de novo design of abiotic nuclear-targeting miniproteins to traffic antisense oligomers to the nucleus of cells. We combined high-throughput experimentation with a directed evolution-inspired deep learning approach in which the molecular structures of natural and unnatural residues are represented as topological fingerprints. The model is able to predict activities beyond the training dataset, and simultaneously deciphers and visualizes sequence-activity predictions. The predicted miniproteins, termed “Mach”, reach 10 kDa average mass, are more effective than any previously known variant in cells, and can also deliver proteins into the cytosol. The Mach miniproteins are nontoxic and efficiently deliver antisense cargo in mice. These results demonstrate that deep learning can decipher design principles to generate highly active biomolecules that are unlikely to be discovered by empirical approaches.
Introduction
The vast chemical search space hinders design of functional macromolecules by empirical approaches alone.1 It is hypothesized that machine learning can enable interpolation in high-dimensional search spaces by bridging the gaps between experimental training data points.2,3 Recent works have shown promise using a variety of input representations and quantitative activity prediction for design of new antimicrobial peptides and antibody CDR3 loops.4–6 For cell-penetrating peptides (CPPs), similar strategies involving binary classifiers have been used to optimize activity.7–10 We sought to further address this challenge by using a large standardized dataset and an advanced input representation combined with deep learning to simultaneously design new functional miniproteins and quantitatively predict their activity.
Successful design of functional polymers can have considerable implications for medicine. For example, anticancer miniproteins have been shown to access intracellular targets.11,12 Similarly, CPPs are short (5-20 residue) sequences that can enhance intracellular delivery of biomolecules, such as oligonucleotides and proteins, that otherwise cannot efficiently cross the cell membrane.13–17 While promising, variation in experimental design has resulted in inconsistent and sometimes contradictory datasets. For example, penetratin has different efficacy as a CPP depending on the assay and the cargo.18 These inconsistent results preclude the development of sequence-activity relationships and complicate the use of machine learning models to design analogs de novo.19–21
We overcome these challenges by de novo design of abiotic miniproteins that deliver an active cargo, antisense phosphorodiamidate morpholino oligomer (PMO), to the nucleus of cells. The miniproteins described here are distinct in that they have a defined function (PMO delivery) and are significantly longer (30-80 residues) than CPPs (5-20 residues). While PMO has recently been approved for the treatment of Duchenne muscular dystrophy, a major challenge remains with their poor cellular permeability.13–17,22,23 High doses of PMO of up to 50 mg/kg are required for in vivo efficacy.24 It has been shown that nuclear delivery can be improved by attaching PMO to CPPs, and the first clinical success of this strategy has been demonstrated just this year.25,26 Development of advanced, novel sequences for antisense delivery would rapidly accelerate the development of these gene therapies.
Here we report a deep learning-based design strategy with predictive power fueled by robust input data containing unnatural residues and structures. Our framework includes generation of starting sequences, a predictor to predict the activity of a sequence, and an optimizer to improve the activity of the sequence. A library containing 600 unique antisense-miniprotein conjugates was constructed using linear combinations of three peptides, or “modules” (Fig. 1a). A quantitative activity readout was achieved using an in vitro assay in which nuclear delivery of PMO results in enhanced green fluorescent protein (EGFP) fluorescence (Fig. 1b–c). Residues were encoded as fingerprints to provide chemical structure information, labeled with corresponding activity data, and used to train a predictor neural network (Fig. 1d). A “CPP thesaurus” dataset was used to train a generator neural network to produce novel sequences that are “CPP-like” to be used as seeds for optimization. These novel sequences were then optimized in the predictor-optimizer loop to increase predicted activity while minimizing similarity to the library, and minimizing length and arginine content to mitigate toxicity.27 The output is hundreds of de novo designed sequences with a broad spectrum of predicted activity (Fig. 1e).
Fig. 1. Machine learning model based on directed evolution predicts highly active abiotic miniproteins for macromolecule delivery.
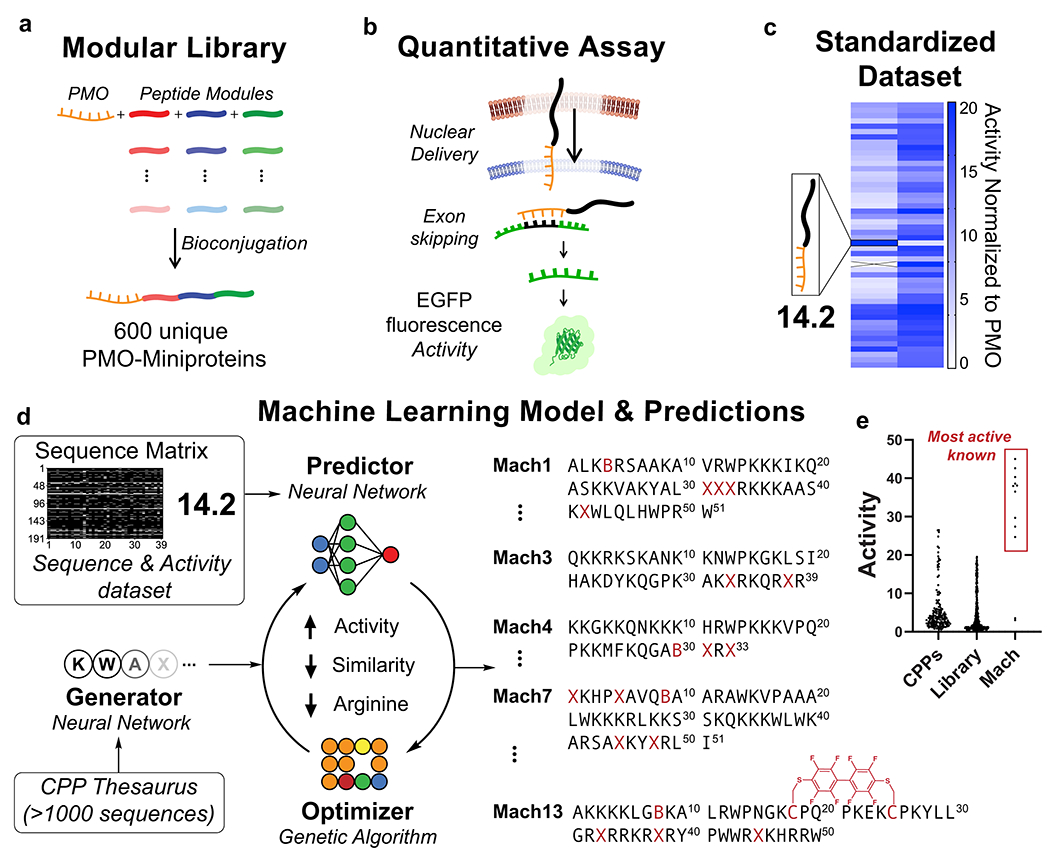
(a) A 600-membered library of PMO-miniprotein conjugates was synthesized using linear combinations of abiotic peptide modules. (b) A standardized in vitro activity assay tests for nuclear delivery using a quantitative fluorescence readout. (c) Members of the modular library exhibit a broad spectrum of activities. Each bit corresponds to a PMO-peptide in the library and its corresponding activity. (d) Sequences are encoded into a fingerprint matrix, labeled with experimental activity, and used to train a machine learning model. The model designs novel sequences in a loop based on directed evolution. X = aminohexanoic acid, B = β-alanine, C = cysteine macrocycles linked through decafluorobiphenyl. (e) Normalized activity from peptides designed in this work (Mach) compared to peptides in the modular library and known CPPs tested using the same assay.
The model is also interpretable: we can visualize the decision-making process and identify structure-activity relationships that are consistent with empirical observations. From these predictions, we discovered best-in-class abiotic “Mach” (Machine Learning) nuclear-targeting miniproteins that improve PMO delivery by 50-fold and are effective in animals. Mach miniproteins are nontoxic and noninflammatory, and are able to deliver macromolecules other than PMO to the cytosol. Our approach has the potential to be extended to the design of peptides with other functions, although further work is required in these directions.
Results
Assembly of a Standardized Dataset
Recently, we demonstrated that linear combinations of known CPP sequences into chimeric miniproteins can synergistically improve delivery of PMO compared to each CPP alone.28 We hypothesized that expanding this approach to a larger, more diverse library of linear combinations of CPPs would access a wide range of sequences and activities. We designed a synthetic method to assemble this library via bioconjugation of peptide “modules” into hundreds of novel PMO-miniproteins. Our rationale is that such a library would enable a broad sequence diversity and spectrum of activities and would be ideal to train machine learning models (Fig. 1).
Our synthesis strategy employs four modules: one for PMO and three for distinct pools of peptide sequences containing diverse structure and function, including nuclear-targeting peptides and peptides containing unnatural residues and cysteine-linked macrocycles (Supplementary Table 11).29 The constructs were synthesized in a series of bioconjugation reactions that are chemoselective and irreversible, yielding products of sufficient crude purity for direct testing in vitro (Supplementary Fig. 18). The resulting library contained 600 miniproteins, composed of combinations of 57 total peptides.
The resulting dataset was broad in terms of both peptide sequences and range of activity, quantified by a high-throughput nuclear-targeting assay.19 The activity-based assay that is used to acquire the training data provides a direct, quantitative readout of the activity characteristic we want to enhance—specifically nuclear delivery. In this assay, HeLa cells stably transfected with an EGFP gene interrupted by a mutated intron of β-globin (IVS2-654) produce a non-fluorescent EGFP protein. Successful delivery of PMO IVS2-654 to the nucleus results in corrective splicing and EGFP synthesis. The amount of PMO delivered to the nucleus is therefore correlated with EGFP fluorescence, quantified by flow cytometry. Activity is reported as mean fluorescence intensity (MFI) relative to PMO alone (Fig.1c, e, Supplementary Information). The most active construct improved PMO delivery by nearly 20-fold, while the median activity was 3-fold.
Developing the Deep Learning Model
Inspired by directed evolution, we leveraged fingerprint sequence representations to develop a machine learning-based generator-predictor-optimizer triad. In this framework, the generator produces novel cell-penetrating sequences, the predictor quantitatively estimates the activity for a given sequence, and the optimizer evolves towards the most optimal miniprotein sequence.
The standardized dataset of activity-labeled sequences from the modular library allowed for development and training of a quantitative regressor algorithm. This approach enabled us to overcome the limitations of other efforts in the computational CPP literature which employed binary classifiers of active versus inactive sequences.2,3,8,30–32 These previous predictors were mostly trained using physicochemical descriptors, with datasets obtained from non-standardized experiments and containing only natural residues.7,10,33,34 Inclusion of chemically diverse unnatural moieties using this strategy is challenging because such physicochemical descriptors may not be readily available. The ability to predict the effect of unnatural residues expands the chemical search space, and may lead to enhanced macromolecule delivery.35 One-hot residue encodings can be extended to represent unnatural residues in the training data. We were interested, however, in encoding the molecular structure of each residue. Therefore, to predict activity of de novo-designed nuclear-targeting abiotic miniproteins, we evaluated a topological representation based on stacking traditional cheminformatics fingerprints for each residue along the sequence.36 This representation extends the approach of using one-hot encodings for quantitative structure activity relationship predictors in the peptide literature,4,5 provides chemical structure information for unnatural residues, and may leverage weight sharing across structurally similar residues. Combined with quantitative experimental readouts, this polymer representation allows us to access the diverse pool of unnatural residues and structures and quantitatively predict delivery activity.
Peptide sequences are represented as matrices comprised of residue fingerprints in the columns, padded with zeros until each sequence matrix is the same length. Individual residue fingerprints are bit-vectors based on the molecular graph of the whole monomer, including backbone and side-chain. We used 2048-bit ECFP6 fingerprints generated by RDKit (Fig. 2a, SM Appendix 3) but other structural descriptors may be used.37 For analysis and visualization of fingerprints, we removed all indices which are inactive across all residues, resulting in a condensed 191-bit fingerprint (SM Appendix 3). Each bit in the vector corresponds to a substructure, and is active/inactive depending on the presence/absence of the particular substructure. Representing residues as chemical structures, rather than discrete choices, eases the use of both natural and unnatural residues and leverages chemical similarity between residues. The fingerprints are then compiled into a row matrix to encode the amide backbone of the peptide sequence (Fig. 2b). This representation method is also more effective than typical one-hot encodings at using inherent chemistry to predict novel sequences and is able to predict activities of sequences containing a new residue not in the training set (Supplementary Table 8, Supplentary Information Section 2.5).
Fig. 2. Machine learning-based generator-predictor-optimizer loop predicts nuclear-targeting abiotic miniproteins.
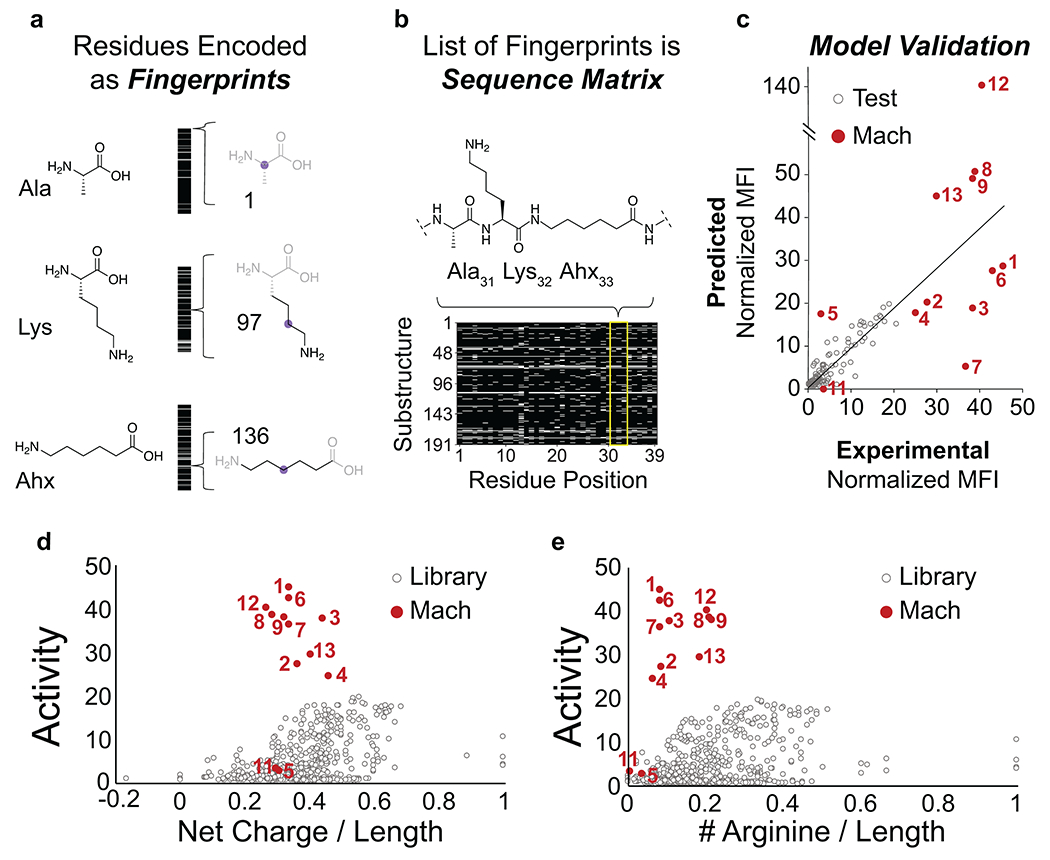
(a) Each amino acid residue is represented as a unique fingerprint, constructed as a bit-vector encoding for the presence or absence of 191 possible substructures in the residue. (b) Sequences are represented as residue fingerprints stacked in a row matrix. (c) Performance of machine learning model, comparing the predicted and experimental activity values for the holdout test set and novel Mach sequences. Twelve predicted “Mach” peptides were synthesized and tested in the same activity assay (red) and compared to the modular library (grey) in relation to (d) relative charge and (e) arginine content.
The predictor neural network quantitatively estimated normalized MFI for a given sequence. Pairs of sequence representations and corresponding experimental activities were used to train a convolutional neural network (CNN). The training dataset consisted of PMO-miniproteins from the modular library as well as other conjugates previously tested in the same assay.7 A randomly-selected 20% of the dataset was saved for validation of the predictive accuracy of the algorithm. The root mean squared error (RMSE) on the validation set was 0.4 of the standard deviation of the training data. The prediction relative error was found to be 11% as long as the predicted activity fell within the range of training values (normalized activity of 0.32-19.5) (Fig. 2c). Tests were conducted against other model architectures, using both fingerprint and one-hot encodings in both regression and classification tasks (Supplementary Tables 1–5; Supplementary Figs. 2–3; Supplementary Information Sections 2.1, 2.2). We also explicitly tested whether reported models (hosted as webservers) were able to predict activity of the Mach miniproteins accurately (Supplementary Table 6). We observed that most of these models were limited by the range of the training data, and that only the CNN-FP model was able to extrapolate in the codomain and generate predicted activity values (validated by experimental activity values) that were greater than any in the training set. This ability to extrapolate, however, came at a cost in average accuracy because of the increased statistical noise of extrapolated predictions. Models based on the topological representations added only minimal increase in performance over one-hot encodings on the validation dataset, and performed similarly or worse on the Mach dataset, due to outliers with extreme predicted activity values (Supplementary Table 2). However, a CNN model using one-hot encodings, despite its lowest overall average error, was not able to extrapolate in the codomain space, unlike when using fingerprint representations. To investigate the role of outliers that impact model performance, we used model ensembling and found the ensembled CNN one-hot model is superior for the validation dataset, whereas the ensembled CNN-FP model is superior for the Mach dataset, likely due to its ability to extrapolate in the codomain (Supplementary Table 3). Further efforts should be focused on how to accurately predict activity values that reach beyond that of the training set. We investigated the CNN model’s ability to extrapolate from the training dataset and found that experimental activity above a threshold of ~8 is necessary to accurately predict peptides with activity beyond that of the training dataset (Supplementary Fig. 4, Supplementary Table 7). Finally, inclusion of unnatural amino acids was required for high-activity predictions, as predicting canonical sequences using the same model resulted in a significant drop in predicted activity (Supplementary Fig. 6).
We developed a generator based on a recurrent neural network (RNN) that captured the ontology of CPPs and generated “CPP-like” starter sequences. We trained the generator using a nested long short-term memory (LSTM) neural network architecture, which is better able to capture long-range correlations in sequence data.38 We trained the algorithm using a “CPP thesaurus,” a collection of sequences from both our modular library and the literature.39 Because the model is learning sequence grammar and has no role in activity predictions, no quantitative labels are necessary and we can use a large dataset of available sequences. Other strategies for generating seed sequences also resulted in predicted peptides with high predicted activity. For example, starting with the top 50 performers from the PMO-CPP library resulted in the highest predicted activity values. However, we confirmed that the generator approach led to predicted sequences that better met our three criteria simultaneously: high predicted activity, low similarity, and low Arg content, compared to other methods of generating seed sequences (Supplementary Fig. 5, Supplementary Table 9). It is possible for the other methods of seed selection and optimization to also produce optimal peptide sequences, but experimental validation is required to adequately compare these methods.
The optimizer completed the loop based on directed evolution. Sequences from the generator were randomly mutated and evaluated against an objective function, which maximized activity as predicted by the CNN model, and minimized length, arginine content, and similarity to the library while retaining water solubility estimated with net charge of the sequence (Supplementary Table 10). After 1000 iterations over each sequence, the model delivered hundreds of unique sequences with a wide range of predicted activity values. Along with highly active sequences, we predicted inactive sequences as negative control. By directing the evolution of the optimizer in the opposite direction, i.e., minimizing MFI, but keeping other constraints the same, we were able to generate an inactive sequence (Mach11) that appeared similar in amino acid composition to the active predictions. After synthesis, the Mach11 conjugate displayed low experimental activity, demonstrating the robustness of the model in predicting the activity of a unique sequence (Fig. 2c).
Interpreting the predictor model
We interpreted the predictor CNN by visualizing the residue substructures that are important in its decision-making process. This type of visualization was a longstanding attribution challenge that was recently addressed for image classification and more recently with small molecule design.40–42 We developed an analogous tool to correlate the input sequence representation with predicted activity. This process generated bit-wise positive and negative activation values for each chemical substructure in the sequence. Bits with higher activation indicated the features that most strongly influence the final activity prediction.
As an example, for the predicted Mach3 sequence the two C-terminal aminohexanoic acid (Ahx) residues were the most positively activated (Fig. 3a), followed by arginine (Arg). The alkyl backbone in Ahx was the most activated substructure (Fig. 3b). A similar trend was observed for active sequences and substructures in the training dataset (Supplementary Fig. 7–8).
Fig. 3. Interpretation of predictor CNN unveils activated substructures.
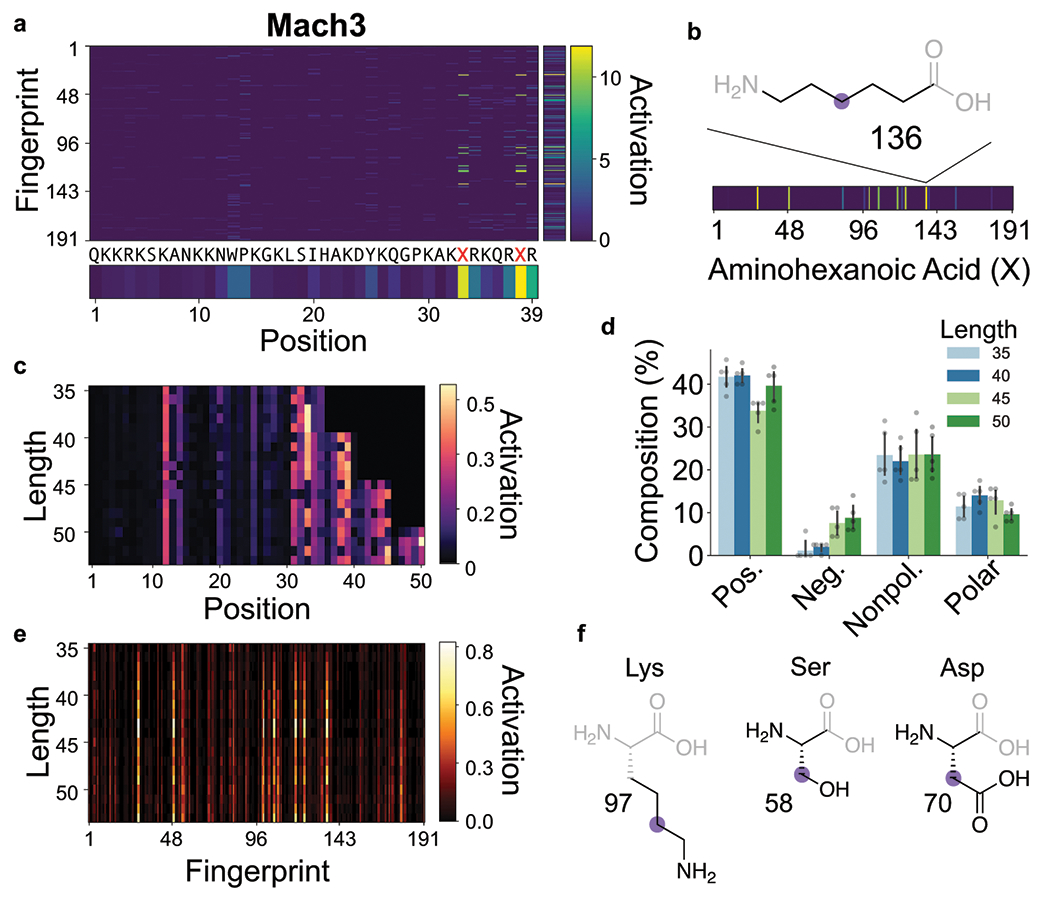
(a) CNN positive activation gradient map was calculated for input sequence representation of Mach3. The averaged activation values over fingerprint indices and residue positions are shown. Fingerprint index represents a corresponding substructure. (b) The activation gradient map of aminohexanoic acid in Mach3 indicates the activated substructures of this residue. The alkyl backbone substructure (136) is shown. (c) Gradient maps of predicted sequences with lengths 35, 40, 45 and 50 are shown relative to residue position. (d) Percent composition of each type of residue (positive, negative, nonpolar, and polar) relative to predicted sequences with lengths 35, 40, 45 and 50 is shown. Each bar represents group mean ± SD, n = 5. (e) Gradient maps of predicted sequences with lengths 35, 40, 45 and 50 are shown relative to substructure fingerprints. (f) Several residues and substructures that are consistently activated across all sequence lengths are shown, including the amine side chain of Lys, polar side chain of Ser, and the carboxylic acid side chain of Asp.
We used this visualization approach to better understand how the trained model designed sequences. We chose five random sequences of different lengths, seeded them in the predictor-optimizer loop to maximize activity contingent upon other design constraints, and visualized the activations for the best predictions. Again, a higher activation can be seen for C-terminal residues (Fig. 3c), most likely due to the attachment of PMO to the N-terminus. We also observed that the general composition of charged and hydrophobic residues remained unchanged across different sequence lengths (Fig. 3d). Particular residue fingerprints were activated irrespective of the sequence length, such as the side chains of Lys, Ser, and Asp (Fig. 3e–f). Consistent with earlier observations, a strong preference for polar and charged side chains as well as for Ahx was evident. We investigated whether the attribution feature is useful toward post-hoc mutations to Mach miniproteins, and found a significant boost in activity when mutating Ahx (6-carbon chain) to aminoundecanoic acid (11-carbon chain) in Mach3 (Supplementary Fig. 16).
Mach miniproteins enhance PMO delivery
We synthesized and characterized twelve candidates from hundreds of miniproteins predicted by the model, selecting diverse sequences and predicted activities. Mach1, 2 and 6 were selected because they had high predicted activity among 50-mer sequences. Mach3 was selected as a mid-length peptide (39 residues), Mach4 was selected as a shorter sequence (33 residues) with only two Arg residues, and Mach5 was selected because it was predicted to have moderate activity while having the lowest net charge (10.5). Mach7 was initially designed to be a negative control—where the sequence of Mach1 was rearranged until the model predicted the lowest activity. Mach8 and 9 were selected from a list of much longer miniproteins (around 80 residues) and Mach12 and 13 were selected from sequences that contained Cys-linked macrocycles. Finally, Mach11 was selected from a list of sequences for which the activity was optimized in the negative direction, to show that the algorithm could predict peptides of similar length, charge, and amino acid composition, but with no PMO delivery activity. Each candidate was synthesized using automated fast-flow solid-phase peptide synthesis, and when applicable, the two cysteine residues were connected with decafluorobiphenyl as previously reported (Supplementary Fig. 1).43,19 Conjugation of azido-Mach to PMO IVS2-654 was achieved in the same manner as in the library. The final PMO-Mach constructs are described in Supplementary Table 12.
Nearly all sequences predicted to have activity greater than 20-fold did indeed surpass the highest performing modular library construct, with the exception of Mach5. Because the model is extrapolating outside the range of the training data, the predicted and experimental activity of PMO-Mach constructs shows greater % error than the test dataset (Fig. 2c). The PMO-Mach constructs were first tested for PMO delivery in the HeLa 654 assay as was done with the library (Supplementary Fig. 20).
Physicochemical properties of validated predictions show little correlation with PMO activity. We compared the activities of Mach constructs to the training library in relation to various physicochemical properties (Fig. 2d–e). While library constructs clearly show an increase in activity with an increase in arginine content relative to length, and net charge relative to length, there is no obvious correlation between activity of Mach constructs and these same properties. In addition, truncated versions of PMO-Mach constructs do not retain the activity of the parent sequences (Supplementary Fig. 17). These observations suggest that the model is taking advantage of sequence-activity relationships that go beyond sequence length and charge.
Several PMO-Mach constructs have greater potency than previously characterized PMO-CPPs, while remaining nontoxic. This type of macromolecular delivery is a historic challenge, often suffering from either membrane toxicity or endosomal entrapment. We first verified that PMO-Mach constructs enter cells via energy-dependent uptake using a panel of chemical endocytosis inhibitors and the HeLa 654 assay (Supplementary Fig. 13). We then performed dose-response experiments to characterize activity in the EGFP assay and toxicity in a lactate dehydrogenase (LDH) release assay. PMO-Mach2, 3, 4, and 7 each had an EC50 value near 1 μM and were nontoxic at the concentrations tested, as determined by viability staining with propidium iodide (PI) and an LDH release assay (Fig. 4a–c, Supplementary Fig. 21–22). Extending toxicity tests to higher concentrations in renal cells showed that no toxicity was observed at the highest concentration needed for maximum PMO activity in HeLa 654 cells (Supplementary Fig. 23). We compared these results to a previously well-performing CPP for PMO delivery, Bpep-Bpep.28 This peptide has similar activity, but is composed of mostly Arg residues and exhibits cytotoxicity above 10 μM (Supplementary Fig. 22). This contrast between Mach peptides and Bpep-Bpep indicates that there is no apparent direct connection between toxicity and cargo delivery efficacy. PMO-Mach constructs have high activity, low arginine content, and a wide therapeutic window, highlighting their suitability for cytosolic and nuclear delivery.
Fig. 4. Mach miniproteins are highly active in vitro and in vivo and deliver other biomacromolecules into the cytosol.
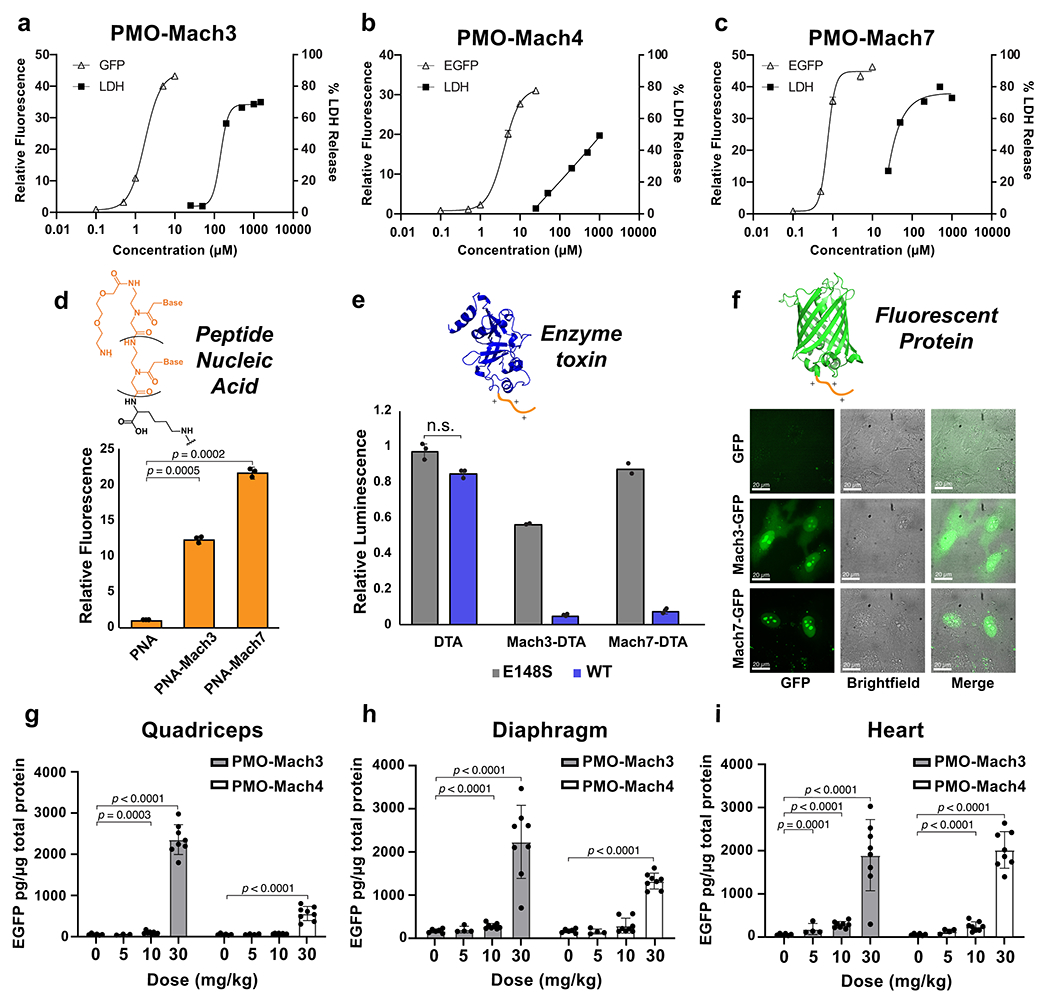
(a-c) Shown are dose-response curves corresponding to activity in the EGFP assay and toxicity in the LDH assay for PMO-Mach3, 4, and 7. Activity is shown as fluorescence intensity relative to unconjugated PMO at 5 μM, and toxicity is shown as LDH release relative to a lysis control. (d) The relative fluorescence of Mach conjugated to PNA 654 are compared to PNA alone, as determined by EGFP assay. (e) Comparison of the toxicity of wild-type and inactive mutant DTA and DTA(E148S) alone or conjugated to Mach3 or Mach7. Delivery of the active toxin to the cytosol results in toxicity as measured by luminescence. (f) Confocal micrographs displaying green fluorescence produced by EGFP, Mach3-EGFP, or Mach7-EGFP in HeLa cells after 3 h incubation at 10 μM. (g-i) EGFP synthesis in EGFP transgene mice after treatment with PMO-Mach. Dose-response EGFP protein level in (g) quadriceps (h) diaphragm and (i) heart. (a-c) Points show mean ± SD, for EGFP assay n = 3, and mean for LDH assay, n = 2 distinct samples. EGFP assay experiments were repeated at different concentration ranges with similar results, reported in Supplementary Figure 21 (d) Each bar represents group mean ± SD, n = 3 distinct samples, p-values calculated from student’s two-tailed t-test (PNA-Mach3 p = 0.0005, PNA-Mach7 p = 0.0002. (e) Each bar represents group mean ± SD, n = 3, except for Mach3-DTA(E148S) and Mach7-DTA(E148S) which show mean, n = 2 distinct samples. Full concentration curves are reported in Supplementary Figure 25. (f) Scale bar shows 10 μm. This experiment was conducted twice independently with similar results. (g-i) Saline (n = 6), Mach3 and Mach4 at 5 mg/kg (n = 4), all other n = 8 mice. Each bar represents group mean ± SD, p-values calculated from two-tailed Mann-Whitney U test. (Quadriceps: PMO-Mach3 10 mg/kg p = 0.0003, PMO-Mach3 30 mg/kg p < 0.0001, PMO-Mach4 30 mg/kg p < 0.0001; Diaphragm: PMO-Mach3 10 mg/kg p < 0.0001, PMO-Mach3 30 mg/kg p <0.0001, PMO-Mach4 30 mg/kg p < 0.0001; Heart: PMO-Mach3 5 mg/kg p = 0.0001, PMO-Mach3 10 mg/kg p < 0.0001, PMO-Mach3 30 mg/kg p < 0.0001, PMO-Mach4 10 mg/kg p < 0.0001, PMO-Mach4 30 mg/kg p < 0.0001.)
Mach miniproteins deliver other biomacromolecules
Mach miniproteins are versatile in that they can deliver other large biomolecules to the cytosol. Peptide nucleic acid (PNA), is a class of synthetic antisense oligonucleotides that has the same mechanism of action as PMO but has a highly flexible backbone structure.44 We tested for delivery of a PNA variant of PMO 654 that is compatible with the EGFP assay. Each of the four Mach miniproteins tested was able to significantly enhance PNA delivery (Fig. 4d, Supplementary Fig. 24).
In addition to antisense oligonucleotides, Mach peptides can also deliver charged proteins, such as Diphtheria toxin A (DTA). DTA is a 21 kDa anionic protein segment containing the catalytic domain of the toxin but lacking the portions that endow cell entry.45 Delivery of this enzyme can be monitored using a cell proliferation assay as it inhibits protein synthesis in the cytosol. We found that Mach-DTA constructs were delivered into the cell cytosol significantly more efficiently than protein alone, and that covalent linkage was required for delivery (Fig. 4e, Supplementary Fig. 25). Furthermore, we confirmed that toxicity is due to the cytosolic delivery of active DTA by comparing the wild-type constructs to those containing DTA(E148S), a mutant with 300-fold lower activity that the wild-type.46 As expected, the mutant DTA conjugates lead to significantly reduced toxicity.
Conjugation to Mach miniproteins also improves the delivery of EGFP, a fluorescent protein commonly used as a reporter. Confocal micrographs of HeLa cells displayed diffuse green fluorescence in the cytosol and intense fluorescence in the nucleus after incubation with Mach-EGFP (Fig. 4f). This observation is in contrast with the EGFP alone condition, in which no diffuse fluorescence was observed in either location, indicating reduced uptake.
PMO-Mach restore protein synthesis in mice
After verifying Mach miniproteins’ propensity for in vitro macromolecule delivery, we looked towards in vivo antisense applications. In vitro tests with human macrophages suggested that the constructs are not inflammatory and therefore may be safe to evaluate in animals (Supplementary Fig. 15). Existing predictive models also suggest that Mach sequences would not be T cell epitopes (Supplementary Fig. 12).
Lastly, we demonstrated that PMO-Mach constructs safely correct protein synthesis in animals. Transgenic mice containing the same EGFP IVS2-654 gene as used in cell assays were given a single intravenous injection of varying doses of PMO-Mach3 or PMO-Mach4 and evaluated after 7 days. Both constructs exhibited a dose-dependent increase in EGFP expression in quadriceps, diaphragm, and heart (Fig. 4g–i). PMO delivery to the heart is a critical but challenging objective. Here we observe similar levels of protein synthesis in both skeletal and cardiac tissue. In addition, there were no significant changes in the level of renal function biomarkers 7-days post-treatment (Supplementary Fig. 26). These findings indicate that Mach miniproteins may be safe delivery materials for PMO to muscle tissue.
Discussion
We demonstrate a method to efficiently sample the vast chemical search space of functional peptides using machine learning and standardized experimentation. Our model was applied to the design of abiotic miniproteins that can deliver an antisense PMO to the nucleus with greater efficiency than any previously known polypeptide-based variant. Importantly, the new constructs are effective in animals and are non-toxic up to a dose of 30 mg/kg. These miniproteins are versatile intracellular carriers, delivering other classes of biomolecules to the nucleus and cytosol, including antisense PNA, fluorescent protein, and enzymes. The core strengths of our model lie in: (1) standardized quantitative activity data, (2) the model’s ability to extrapolate beyond the training set and (3) a visual attribution tool to interpret the decision-making process of the model.
A critical factor in building a robust machine learning model was the training dataset; the 600-member library was synthesized by combining peptide modules, and tested in a standardized assay that provides quantitative activity information. Synthesis and testing of the modular PMO-CPP library produced a broad spectrum of sequence and activity data with which we trained the model. By representing peptide sequences as topological fingerprints rather than categorical choices or descriptors such as molecular weight, charge, and hydrophobicity, the model has access to inherent structural information and can be used on monomers not encountered in the training data. The standardized activity values allowed us to use a quantitative regressor, rather than an active/inactive classifier, and thus design sequences with a broad spectrum of activity predictions. While we have previously tested CPPs designed by other machine learning methods, we found that they were not able to deliver PMO.7 The CNN model using fingerprints was able to extrapolate predicted activity beyond that of the training dataset, while models using other frameworks and representations were not. While the other models and methods to generate seed sequences may be able to produce sequences with high experimental activity, the ability to predict that high activity is critical for the informed selection of predicted sequences to validate. Since our goal is to discover unique peptides with higher activity than any previously known, a model able to predict values outside the range of the training data is required, thus necessitating the use of CNN with fingerprint representations.
The interpretability of the model is an additional advantage. By overlaying the output of the predictor with the sequence matrix of a given peptide, we can visualize the activated residues and substructures important for the decision-making process. Several observations from the interpretations match our current understanding of CPP motifs, such as the benefit of cationic residues. The model also identified Ahx as an important residue, one which has only been investigated in the context of endosomal escape in Arg-rich sequences.47 This tool allowed for post-hoc analysis to validate empirical hypotheses and enhance the activity of Mach3 by mutating aminohexanoic acid to aminoundecanoic acid, an amino acid not present in the training dataset.
In addition to PMO, Mach peptides deliver other antisense oligonucleotides as well as functional proteins into the cell cytosol. Delivery of EGFP reveals diffuse green fluorescence in the cytosol and clear accumulation of EGFP to the nucleus. We believe that Mach peptides may contain nuclear localization sequences (NLS), which have been described previously and are typically lysine-rich.48 However, nuclear localization is not solely due to the cationic charge, as shown by a previous study.49 The model likely selected for such NLS sequences because the activity used in the training was acquired from a nuclear delivery-based assay.
PMO-Mach conjugates effected a dose-dependent increase in protein synthesis in all three examined mouse muscle tissues including heart after a single intravenous injection. The mouse model used contains the same transgene as the in vitro assay, and the Mach sequences recapitulated the in vitro results in vivo, indicating that the model implemented here could be applied to data obtained from animal experiments. If a sequence-activity training set were generated from data obtained in animals, then this model may be applicable further downstream in the drug design pipeline. A greater challenge remains toward in vivo delivery to target tissues. In Duchenne muscular dystrophy, PMO must access the nucleus of muscle cells to have therapeutic effect. Targeting to cardiac tissue is a primary concern given that the leading cause of death from this disease is heart failure. Our animal model confirmed localization of the PMO-Mach constructs to the heart, suggesting a potential solution to the tissue targeting challenge.
In conclusion, this strategy illustrates how deep learning can be applied to de novo design of functional abiotic miniproteins. The Mach miniproteins are the most effective PMO delivery constructs developed to date and are effective in animals. Our machine learning framework could potentially be repurposed to discover sequence-optimized peptides with other desired activities, solely requiring a standardized high-quality input dataset. We envision that this strategy will enable the rapid future design of de novo functional peptides with impact on chemical, biological and material sciences.
Methods
Peptide synthesis:
All peptides and miniproteins were synthesized by automated solid-phase peptide synthesis as previously described.43,50 If the predicted sequence contained a cysteine macrocycle, we subsequently utilized SNAr chemistry to link the two cysteine residues with decafluorobiphenyl before additional purification. PMO was acquired from Sarepta and functionalized with a DBCO acid handle. A complete description of the synthesis, purification, and characterization protocols can be found in the supplementary information.
Mach miniproteins were conjugated to PMO via strain-promoted azide-alkyne cycloaddition. PMO-DBCO (5 mM in water) was stoichiometrically combined with azide-peptide (5 mM in water) and incubated at room temperature until reaction completed (between 2 and 12 hours), monitored by LCMS. The reaction was purified using reversed-phase HPLC (Agilent Zorbax SB C3 column: 21.2 x 100 mm, 5 μm) and a linear gradient from 2 to 60% B (solvent A: 100 mM ammonium acetate in water pH 7.2; solvent B: acetonitrile) over 58 min (1% B / min). Pure fractions were pooled as determined by LCMS and lyophilized.
50 nmol of PNA 654 (O-GCTATTACCTTAACCCAG-Lys(DBCO)) was purchased from PNABio. PNA-DBCO (1 mM in water) was stoichiometrically combined with azide-peptide (1 mM in water) and incubated at 4 °C for 12 hours. The product was then used in cell assays without purification. Conversion was checked by LCMS.
Library synthesis:
The library was synthesized in a combinatorial fashion and analyzed by LCMS 51. The 600-member library was synthesized using 50 peptide members in module 4 (SM).
Reaction 1:
PMO-DBCO was dissolved in water to 10 mM concentration (determined by UV-Vis). The module 2 peptides were dissolved in water containing 0.1% TFA at 10 mM concentration (determined gravimetrically; the molecular weight was calculated to include 0.5 trifluoroacetate counter ions per lysine, arginine, and histidine residue). In a microcentrifuge tube, 50 μL each of PMO-DBCO solution and module 2 peptide solution were mixed and incubated for 1 h. The product was analyzed by LC-MS and dried by lyophilization. Lastly, the product was resuspended in 100 μL of DMSO to provide a 5 mM solution and stored at −20 °C.
Reaction 2:
Stock solutions were prepared by dissolving module 3 peptides and module 4 peptides in water at 10 mM concentration (determined gravimetrically). For each reaction, 4 μL of module 3 peptide was mixed with 4 μL of module 4 peptide in a PCR tube. Separately, the copper bromide solution was prepared by mixing 1 mL of degassed DMSO with 2.8 mg copper (I) bromide under N2 to afford a 20 mM solution. Under ambient conditions, 4 μL of the CuBr solution was added to the mixture of module peptides 3 and 4. The reaction was capped and the reaction was allowed to proceed for 2 hours; the small amount of O2 present during reaction setup does not substantially impede reaction progress. After 2 hours, 2 μL of a 100 mM solution of Na2HPO4 was added. The PCR tube was then sonicated, vortexed, and centrifuged. To remove the solvent, the PCR tube was centrifuged under vacuum using a Savant SPD121P Speed-Vac set at 35 °C for 2 hours. Lastly, the product was resuspended in 16 μL of DMSO to provide a 5 mM solution and stored at −80 °C. The product was analyzed by LCMS.
Reaction 3:
The final modular construct was synthesized through the combination of module 1-2 and module 3-4. First, 1.6 μL of reaction 2 was added to a 384-well plate. Separately, 30 μL of reaction 1 was mixed with 15 μL of TCEP solution (100 mM TCEP·HCl in 50/50 water/DMSO containing 400 mM NaOH) and 75 μL DMSO. Then, 1.6 μL of the reaction 1 solution was added to reaction 2 in the 384 well plate. Each individual reaction ultimately contained 0.4 μL of reaction 1 (at 5 mM in DMSO), 1.6 μL of reaction 2 (at 5 mM in DMSO), 0.2 μL TCEP solution (at 100 mM in water/DMSO), and 1 μL DMSO. Excess reaction 2 was used to force the reaction to go to completion; the presence of copper hinders the efficiency of this conjugation. Reaction 1 was used as a limiting reagent to avoid excess PMO, which is the active component for the cell culture assays. The reaction was allowed to proceed for 2 hours, and then the plate was stored at −80 °C. The reaction was analyzed by LCMS.
EGFP assay:
HeLa 654 cells obtained from the University of North Carolina Tissue Culture Core facility were maintained in MEM supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) penicillin-streptomycin at 37 °C and 5% CO2. 18 h prior to treatment, the cells were plated at a density of 5,000 cells per well in a 96-well plate in MEM supplemented with 10% FBS and 1% penicillin-streptomycin.
For testing of the library, on the day of the experiment, the 384 well plate containing the crude reaction mixtures in DMSO was diluted to 100 μM by the addition of 16.8 μL of PBS to the 3.2 μL reaction mixture. Then, each construct was diluted to 5 μM in MEM supplemented with 10% FBS and 1% penicillin-streptomycin. For individual peptide testing, PMO-peptides were dissolved in PBS without Ca2+ or Mg2+ at a concentration of 1 mM (determined by UV) before being diluted in MEM. Cells were incubated at the designated concentrations for 22 h at 37 °C and 5% CO2. Next, the treatment media was removed, and the cells were washed once before being incubated with 0.25% Trypsin-EDTA for 15 min at 37 °C and 5% CO2. Lifted cells were transferred to a V-bottom 96-well plate and washed once with PBS, before being resuspended in PBS containing 2% FBS and 2 μg/mL propidium iodide (PI). Flow cytometry analysis was carried out on a BD LSRII flow cytometer at the Koch Institute. Gates were applied to the data to ensure that cells that were positive for propidium iodide or had forward/side scatter readings that were sufficiently different from the main cell population were excluded. Each sample was capped at 5,000 gated events (SM).
Analysis was conducted using Graphpad Prism 7 and FlowJo. For each sample, the mean fluorescence intensity (MFI) and the number of gated cells was measured. To report activity, triplicate MFI values were averaged and normalized to the PMO alone condition.
Inverse Design Model:
Generator – Recurrent Neural Network.
The generator is a data-driven tool to generate new peptide sequences that follow the ontology of cell penetrating peptides to seed the optimization from likely starting points, and is based on recurrent neural network (RNN) - Nested LSTM architecture.38 It was trained using one-hot encoding representations of the amino acids to predict the next amino acid in the sequence, from the preceding sequence. The inputs were size 5 to 50 amino acids, left-padding with zeros and representing termination with a unique token. The training dataset comprised of 1,150 sequences and a total of 19,800 sequence-next character pairs, including the non-modular sequences used in the creation of the library and sequences from CPPSite2.0.39 The training was performed using 80% of this dataset, and validated using the remaining 20%. A validation accuracy of 76% was obtained in the training. For the model, multiple combinations of LSTM and Nested LSTM layers were tried with different cell sizes.38 The final model was chosen after the optimization of hyperparameters. All hyperparameters were optimized using SigOpt.52
Predictor – Convolutional Neural Network.
The predictor, based on convolutional neural network (CNN), estimates the normalized fluorescence intensity from PMO delivery by a given peptide sequence, as measured in the HeLa 654 assay. The model was trained on a row matrix of residue fingerprints. The row matrix of 2048-bit vectors (vector of 0s and 1s) represents the arrangement of the residues along the backbone of the peptide chain. This representation is analogous to 1D image with 2048 color channels. Fingerprints have radius 3 and were generated using RDKit.53 By combining the CPP library from this work as well as the collection of CPPs from previous work, we compiled 640 PMO-peptides sequences for training.7
We used fingerprints and one-hot encodings to train non-CNN models such as those based on support vector regression, Gaussian process regression, kernel ridge regression, k-nearest neighbors regression and XGBoost regression.
Optimizer.
The optimization was done using genetic algorithm (GA), where single residue mutations involved insertion, deletion and swapping, and multi-residue mutation was done using hybridization. For hybridization, the sequence length and position to be hybridized, and the hybridized sequence (from the list of all CPPs) were all chosen randomly. In the case of hybridization mutation, the selection and replacement of motifs was done at random without conservation of the sequence length. For the case of mutations with cysteine macrocycles, explicit conditions were built in to keep the number and position of cysteine residues separate in the case of a single through-space covalent bond or bicycle. A constrained hybridization condition conserving the sequence length was also set-up for specific optimization tasks. In the case of cysteine macrocycles, different fingerprints were used to denote the residues. The GA was used the following objective function, starting from LSTM-generated sequences and taking 1000 evolution steps:
Set up of Generator-Predictor-Optimizer Loop:
The generator was primed with a 5-long random sequence from the training dataset and sampled until a termination character is produced. The randomly sampled sequences were then set-up for optimization. The directed evolution of the generated sequences was carried using the predictor-optimizer feedback loop. Each sequence was mutated by the optimizer. Post mutations, the normalized fluorescence values for the new sequence was predicted by the predictor and the optimization parameters (similarity, % arginine, length, net charge) were calculated. The objective function (equation with optimization parameters) was evaluated for both the old and mutated sequences. If the value for the mutated sequence was higher for the mutated than the older, then the old sequence was replaced by the mutated sequence. 1000 such optimization rounds were conducted for each sequence. The output was hundreds of sequences with varying predicted activity.
Toxicity assays:
Cytotoxicity assays were performed in both HeLa 654 cells and human RPTEC (Human Renal Proximal Tubule Epithelial cells, TH-1, ECH001, Kerafast). RPTEC were maintained in high glucose DMEM supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) penicillin-streptomycin at 37 °C and 5% CO2. Treatment of RPTEC was performed as with the HeLa 654 cells. After treatment, supernatant was transferred to a new 96-well plate. To each well of the 96-well plate containing supernatant, described above, was added CytoTox 96 Reagent (Promega). The plate was shielded from light and incubated at room temperature for 30 minutes. Equal volume of Stop Solution was added to each well, mixed, and the absorbance of each well was measured at 490 nm. The blank measurement was subtracted from each measurement, and % LDH release was calculated as % cytotoxicity = 100 × Experimental LDH Release (OD490) / Maximum LDH Release (OD490).
Synthesis and testing of Mach-DTA:
Mach-LPSTGG peptides were synthesized and purified by standard protocol as described. G5-DTA (50 μM) was incubated with either Mach3-LPSTGG (250 μM) or Mach7-LPSTGG (750 μM) and SrtA* (2.5 μM) for 90 minutes at 4 °C in SrtA buffer (10 mM CaCl2, 50 mM Tris, 150 mM NaCl, pH 7.5). The reaction was monitored by LCMS and gel electrophoresis. After 90 minutes, Mach-DTA conjugate was isolated using HiLoad 26/600 Superdex 200 prep grade size exclusion chromatography column (GE Healthcare, UK) in 20 mM Tris, 150 mM NaCl, pH 7.5 buffer. Fractions containing the pure product as determined by LCMS and gel electrophoresis were concentrated using a centrifugal filter unit (10K, Millipore).
To test for DTA delivery to the cytosol, HeLa cells were plated at 5,000 cells/well in a 96-well plate the day before the experiment. Wild-type and mutant constructs of G5-DTA, Mach3-DTA, and Mach7-DTA, as well as Mach3-LPSTGG and Mach7-LPSTGG were prepared at varying concentrations in complete media and transferred to the plate. Cell proliferation was measured after 48 h using the CellTiter-Glo assay.
Synthesis and testing of Mach-EGFP:
G5-EGFP (60 μM) was incubated with either Mach3-LPSTGG (1000 μM) or Mach7-LPSTGG (1000 μM) and SrtA* (5 μM) in SrtA buffer (10 mM CaCl2, 50 mM Tris, 150 mM NaCl, pH 7.5) for 90 minutes at room temperature under exclusion of light. The reaction was monitored by LCMS and gel electrophoresis. After 90 minutes, Mach-eGFP conjugate was isolated using cation exchange chromatography using HiTrap SP HP cation exchange chromatography column (GE Healthcare, UK) in (0–100 %B over 20 CV) where A: 50 mM NaCl, 20 mM Tris, pH 7.5 buffer and B: 1 M NaCl, 20 mM Tris, pH 12 buffer. Fractions containing the pure product as determined by LCMS and gel electrophoresis were immediately desalted and concentrated using a centrifugal filter unit (10K, Millipore).
To visualize delivery of EGFP into cells, HeLa were plated at 5,000 cells/well in a coverslip glass-bottomed 96-well plate the day before the experiment. Mach3-EGFP, Mach7-EGFP, or EGFP were added to each well at 10 μM and incubated at 37 °C and 5% CO2 for 3 h. Treatment media was replaced with fresh media 1 h before being imaged in the W.M. Keck microscopy facility on an RPI Spinning Disk Confocal microscope on brightfield and GFP setting (488 nm, 150 mW OPSL excitation laser, 525/50 nm emission).
In vivo studies
EGFP-654 transgenic mice (FVB/NJ mice transformed with CX-EGFP-654 plasmid) obtained from Dr. Ryszard Kole’s lab54 ubiquitously express EGFP-654 transgene throughout body under chicken β-actin promoter. Identical to the HeLa 654 cell line, a mutated nucleotide 654 at intron 2 of human β-globin gene interrupts EGFP-654 coding sequence and prevents proper translation of EGFP protein. The antisense activity of PMO blocks aberrant splicing and resulted in EGFP expression, the same as in the HeLa 654 assay. In this study, 6- to 8- week-old male EGFP-654 mice bred at Charles River Laboratory were shipped to the vivarium at Sarepta Therapeutics (Cambridge, MA). These mice were group housed with ad libitum access to food and water. All animal protocols were approved by and conducted in accordance with the Institutional Animal Care and Use Committee (IACUC) of Sarepta Therapeutics.
After 3 days of acclimation, mice were randomized into groups to receive a single i.v. tail vein injection of either saline or PMO-peptide (PMO-Mach3 or PMO-Mach4) at the indicated doses; 5, 10 and 30 mg/kg. 7-days after the injection, the mice were euthanized for serum and tissue sample collection. Quadriceps, diaphragm, heart were rapidly dissected, snap-frozen in liquid nitrogen and stored at −80 °C until analysis.
Serum from all groups were collected 7 days post-injection and tested for kidney injury markers using a Vet Axcel Clinical Chemistry System (Alfa Wassermann Diagnostic Technologies, LLC.) Specifically, serum BUN, creatinine, and cystatin C levels were measured using ACE® Creatinine Reagent (Alfa Wassermann, Cat# SA1012), ACE® Blood Urea Nitrogen Reagent (Alfa Wassermann, Cat# SA2024) and Diazyme Cystatin C immunoassay (Diazyme Laboratories, Cat# DX133C-K), respectively, per manufacturer’s recommendation.
20-25 mg of mouse tissue was homogenized in RIPA buffer (Thermo Fisher, Cat# 89900) with protease inhibitor cocktail (Roche, 04693124001) using a Fast Prep 24-5G instrument (MP Biomedical). Homogenates were centrifuged at 12,000 g for 10 min at 4 °C. The resultant supernatant lysates were quantified by Pierce BCA Protein Assay Kit (Thermo Fisher, Cat# 23225) and saved for EGFP expression measurement. Specifically, 80 μg of lysates were aliquoted in each well in a black-wall clear-bottom 96-well microplate (Corning). EGFP fluorescent intensity of each sample was measured in duplicates using a SpectraMAx i3x microplate reader (Molecular devices) by default setting. The average EGFP fluorescent intensity of each sample was then plotted against a standard curve constructed by recombinant EGFP protein (Origen, Cat#TP790050) to quantify EGFP protein level per μg protein lysate.
Supplementary Material
Acknowledgments:
We thank Dr. Alexander R. Loftis and Jacob Rodriguez for assistance with recombinant protein expression, Dr. Coralie Backlund for assistance with immunoassays, Wendy C. Salmon at the W. M. Keck Microscopy Facility at the Whitehead Institute for help with imaging, the Swanson Biotechnology Center Flow Cytometry Facility at the Koch Institute for the use of their flow cytometers, and Bryan Mastis and Sam Foley for help with in vivo studies. We also thank Zi-Ning Choo for igniting our interest in machine learning. This research was funded by Sarepta Therapeutics, by the MIT-SenseTime Alliance on Artificial Intelligence, and by an award from the Abdul Latif Jameel Clinic for Machine Learning in Health (J-Clinic). C.K.S. (4000057398) acknowledges the National Science Foundation Graduate Research Fellowship (NSF Grant No. 1122374) for research support.
Competing interests:
B.L.P. is a co-founder of Amide Technologies and Resolute Bio. Both companies focus on the development of protein and peptide therapeutics. The following authors are inventors on patents and patent applications related to the technology described: J.M.W., C.M.F. and B.L.P are co-inventors on patents WO 2020028254A1 (February 6, 2020), WO2019178479A1 (September 19, 2019), WO2019079386A1 (April 25, 2019), and WO2019079367A1 (April 24, 2019), describing trimeric peptides for antisense delivery, chimeric peptides for antisense delivery, cell-penetrating peptides for antisense delivery, and bicyclic peptide oligonucleotide conjugates, respectively. A.M., K.B., C.L.W., and J.A.W. are employees of Sarepta Therapeutics, and Sarepta Therapeutics provided a portion of the funding for the work.
Footnotes
Code availability: All code used for model training and analysis is available at https://github.com/learningmatter-mit/peptimizer, and archived in Zenodo repository at https://zenodo.org/record/4815385#.YK_VCjZKhhE. Tutorial Jupyter notebooks are also in the repository, and demo Google Colab notebooks can be found at github.com/pikulsomesh/tutorials.
Data availability:
The main data supporting the findings of the current study are available within the paper and its Supplementary Information. The Supplementary Information provides additional methods information, supplementary figures and data. Supplementary Table 1 includes sequences and activity of the modular library. Source Data for Figs 1–4 are provided with the paper. Data used for training of the model has been made available at https://github.com/learningmatter-mit/peptimizer, and archived in Zenodo repository.55
References
- 1.Exploring chemical space: Can AI take us where no human has gone before? Chemical & Engineering News https://cen.acs.org/physical-chemistry/computational-chemistry/Exploring-chemical-space-AI-take/98/i13.
- 2.Zhavoronkov A et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. doi: 10.1038/s41587-019-0224-x. [DOI] [PubMed] [Google Scholar]
- 3.Stokes JM et al. A deep learning approach to antibiotic discovery. Cell 180, 688–702 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spänig S & Heider D Encodings and models for antimicrobial peptide classification for multi-resistant pathogens. BioData Min. 12, 7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Witten J & Witten Z Deep learning regression model for antimicrobial peptide design. bioRxiv 692681 (2019) doi: 10.1101/692681. [DOI] [Google Scholar]
- 6.Liu G et al. Antibody complementarity determining region design using high-capacity machine learning. Bioinformatics 36, 2126–2133 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wolfe JM et al. Machine Learning To Predict Cell-Penetrating Peptides for Antisense Delivery. ACS Cent. Sci 4, 512–520 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Su R, Hu J, Zou Q, Manavalan B & Wei L Empirical comparison and analysis of web-based cell-penetrating peptide prediction tools. Brief Bioinform (2019). [DOI] [PubMed] [Google Scholar]
- 9.Sanders WS, Johnston CI, Bridges SM, Burgess SC & Willeford KO Prediction of cell penetrating peptides by support vector machines. PLoS Comput. Biol 7, e1002101 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manavalan B, Subramaniyam S, Shin TH, Kim MO & Lee G Machine-Learning-Based Prediction of Cell-Penetrating Peptides and Their Uptake Efficiency with Improved Accuracy. J. Proteome Res 17, 2715–2726 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Crook ZR, Nairn NW & Olson JM Miniproteins as a Powerful Modality in Drug Development. Trends Biochem. Sci 45, 332–346 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beaulieu M-E et al. Intrinsic cell-penetrating activity propels Omomyc from proof of concept to viable anti-MYC therapy. Sci. Transl. Med 11, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Juliano RL The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 44, 6518–6548 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slastnikova TA, Ulasov AV, Rosenkranz AA & Sobolev AS Targeted intracellular delivery of antibodies: The state of the art. Front. Pharmacol 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miersch S & Sidhu SS Intracellular targeting with engineered proteins. F1000Research 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trenevska I, Li D & Banham AH Therapeutic antibodies against intracellular tumor antigens. Front. Immunol 8, 1001 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu A, Tang R, Hardie J, Farkas ME & Rotello VM Promises and pitfalls of intracellular delivery of proteins. Bioconjug. Chem 25, 1602–1608 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Illien F et al. Quantitative fluorescence spectroscopy and flow cytometry analyses of cell-penetrating peptides internalization pathways: optimization, pitfalls, comparison with mass spectrometry quantification. Sci. Rep 6, 36938 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolfe JM et al. Perfluoroaryl Bicyclic Cell-Penetrating Peptides for Delivery of Antisense Oligonucleotides. Angew. Chem 130, 4846–4849 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Betts C et al. Pip6-PMO, a new generation of peptide-oligonucleotide conjugates with improved cardiac exon skipping activity for DMD treatment. Mol. Ther.-Nucleic Acids 1, e38 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boisguérin P et al. Delivery of therapeutic oligonucleotides with cell penetrating peptides. Adv. Drug Deliv. Rev 87, 52–67 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chery J RNA therapeutics: RNAi and antisense mechanisms and clinical applications. Postdoc J. 4, 35–50 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mendell JR et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol 74, 637–647 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Moulton J & Jiang S Gene knockdowns in adult animals: PPMOs and vivo-morpholinos. Molecules 14, 1304–1323 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McClorey G & Banerjee S Cell-Penetrating Peptides to Enhance Delivery of Oligonucleotide-Based Therapeutics. Biomedicines 6, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inc ST Sarepta Therapeutics Announces Positive Clinical Results from MOMENTUM, a Phase 2 Clinical Trial of SRP-5051 in Patients with Duchenne Muscular Dystrophy Amenable to Skipping Exon 51. GlobeNewswire News Room http://www.globenewswire.com/news-release/2020/12/07/2140613/0/en/Sarepta-Therapeutics-Announces-Positive-Clinical-Results-from-MOMENTUM-a-Phase-2-Clinical-Trial-of-SRP-5051-in-Patients-with-Duchenne-Muscular-Dystrophy-Amenable-to-Skipping-Exon-5.html (2020).
- 27.Cardozo AK et al. Cell-permeable peptides induce dose- and length-dependent cytotoxic effects. Biochim. Biophys. Acta BBA - Biomembr 1768, 2222–2234 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Fadzen CM et al. Chimeras of Cell-Penetrating Peptides Demonstrate Synergistic Improvement in Antisense Efficacy. Biochemistry 58, 3980–3989 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Wolfe J Peptide Conjugation to Enhance Oligonucleotide Delivery. (2018). [Google Scholar]
- 30.Wei L, Tang J & Zou Q SkipCPP-Pred: an improved and promising sequence-based predictor for predicting cell-penetrating peptides. BMC Genomics 18, 742 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pandey P, Patel V, George NV & Mallajosyula SS KELM-CPPpred: Kernel extreme learning machine based prediction model for cell-penetrating peptides. J. Proteome Res 17, 3214–3222 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Chen B et al. Predicting HLA class II antigen presentation through integrated deep learning. Nat. Biotechnol 37, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee EY, Wong GCL & Ferguson AL Machine learning-enabled discovery and design of membrane-active peptides. Bioorg. Med. Chem 26, 2708–2718 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.A Dobchev D et al. Prediction of cell-penetrating peptides using artificial neural networks. Curr. Comput. Aided Drug Des 6, 79–89 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Jearawiriyapaisarn N et al. Sustained dystrophin expression induced by peptide-conjugated morpholino oligomers in the muscles of mdx mice. Mol. Ther 16, 1624–1629 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morgan HL The Generation of a Unique Machine Description for Chemical Structures-A Technique Developed at Chemical Abstracts Service. J. Chem. Doc 5, 107–113 (1965). [Google Scholar]
- 37.Rogers D & Hahn M Extended-Connectivity Fingerprints. J. Chem. Inf. Model 50, 742–754 (2010). [DOI] [PubMed] [Google Scholar]
- 38.Moniz JRA & Krueger D Nested LSTMs. arXiv:1801.10308 (2018). [Google Scholar]
- 39.Agrawal P et al. CPPsite 2.0: a repository of experimentally validated cell-penetrating peptides. Nucleic Acids Res. 44, D1098–D1103 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Selvaraju RR et al. Grad-CAM: Visual Explanations from Deep Networks via Gradient-Based Localization. Proc. IEEE Int. Conf. Comput. Vis 618–626 (2017) doi: 10.1109/ICCV.2017.74. [DOI] [Google Scholar]
- 41.McCloskey K, Taly A, Monti F, Brenner MP & Colwell LJ Using attribution to decode binding mechanism in neural network models for chemistry. Proc. Natl. Acad. Sci 116, 11624–11629 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanchez-Lengeling B et al. Machine Learning for Scent: Learning Generalizable Perceptual Representations of Small Molecules. ArXiv191010685 Phys. Stat (2019). [Google Scholar]
- 43.Hartrampf N et al. Synthesis of proteins by automated flow chemistry. Science 368, 980–987 (2020). [DOI] [PubMed] [Google Scholar]
- 44.Hanvey JC et al. Antisense and antigene properties of peptide nucleic acids. Science 258, 1481–1485 (1992). [DOI] [PubMed] [Google Scholar]
- 45.Choe S et al. The crystal structure of diphtheria toxin. Nature 357, 216 (1992). [DOI] [PubMed] [Google Scholar]
- 46.Wilson BA, Reich KA, Weinstein BR & Collier RJ Active-site mutations of diphtheria toxin: effects of replacing glutamic acid-148 with aspartic acid, glutamine, or serine. Biochemistry 29, 8643–8651 (1990). [DOI] [PubMed] [Google Scholar]
- 47.Abes S et al. Vectorization of morpholino oligomers by the (R-Ahx-R)4 peptide allows efficient splicing correction in the absence of endosomolytic agents. J. Controlled Release 116, 304–313 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Cerrato CP, Künnapuu K & Langel Ü Cell-penetrating peptides with intracellular organelle targeting. Expert Opin. Drug Deliv 14, 245–255 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Nischan N et al. Covalent attachment of cyclic TAT peptides to GFP results in protein delivery into live cells with immediate bioavailability. Angew. Chem. Int. Ed 54, 1950–1953 (2015). [DOI] [PubMed] [Google Scholar]
- 50.Mijalis AJ et al. A fully automated flow-based approach for accelerated peptide synthesis. Nat. Chem. Biol 13, 464 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Wolfe JM Peptide Conjugation to Enhance Oligonucleotide Delivery. (Massachusetts Institute of Technology, 2018). [Google Scholar]
- 52.Clark S & Hayes P SigOpt WebPage. SigOpt Web page (2019). [Google Scholar]
- 53.Landrum G RDKit: Open-source cheminformatics. RDKit Open-Source Cheminformatics (2006). [Google Scholar]
- 54.Sazani P et al. Systemically delivered antisense oligomers upregulate gene expression in mouse tissues. Nat. Biotechnol 20, 1228–1233 (2002). [DOI] [PubMed] [Google Scholar]
- 55.Mohapatra Somesh. learningmatter-mit/peptimizer: Initial Release. (Zenodo, 2021). doi: 10.5281/zenodo.4815385 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The main data supporting the findings of the current study are available within the paper and its Supplementary Information. The Supplementary Information provides additional methods information, supplementary figures and data. Supplementary Table 1 includes sequences and activity of the modular library. Source Data for Figs 1–4 are provided with the paper. Data used for training of the model has been made available at https://github.com/learningmatter-mit/peptimizer, and archived in Zenodo repository.55