

Abstract
The glymphatic system plays a pivotal role in maintaining cerebral homeostasis. Chronic cerebral hypoperfusion, arising from small vessel disease or carotid stenosis, results in cerebrometabolic disturbances ultimately manifesting in white matter injury and cognitive dysfunction. However, whether the glymphatic system serves as a potential therapeutic target for white matter injury and cognitive decline during hypoperfusion remains unknown. Here, we established a mouse model of chronic cerebral hypoperfusion via bilateral common carotid artery stenosis. We found that the hypoperfusion model was associated with significant white matter injury and initial cognitive impairment in conjunction with impaired glymphatic system function. The glymphatic dysfunction was associated with altered cerebral perfusion and loss of aquaporin 4 polarization. Treatment of digoxin rescued changes in glymphatic transport, white matter structure, and cognitive function. Suppression of glymphatic functions by treatment with the AQP4 inhibitor TGN-020 abolished this protective effect of digoxin from hypoperfusion injury. Our research yields new insight into the relationship between hemodynamics, glymphatic transport, white matter injury, and cognitive changes after chronic cerebral hypoperfusion.
Keywords: Chronic cerebral hypoperfusion, Cognitive impairment, Digoxin, Glymphatic system, White matter injury
Introduction
Vascular cognitive impairment (VCI) is a clinical syndrome ranging in severity from mild cognitive impairment to dementia; it arises secondary to cerebrovascular disease [1]. Generally considered to be the second most common subtype of dementia after Alzheimer’s disease, VCI comprises ~20% of dementia cases. With a higher prevalence in low- and middle-income countries, VCI brings a greatly increased risk of death, and imposes a considerable burden on the families of the afflicted and society in general [2]. White matter injury (WMI) induced by chronic cerebral hypoperfusion (CCH) is one of the main underlying pathologies of VCI [3]. Chronic hypoperfusion of white matter brings about a marked reduction in myelin density in VCI patients compared to age-matched controls [4]. The main neuropathological manifestations of WMI are demyelination and inflammation, manifesting in the activation and proliferation of astrocytes and microglia due to a cascade of molecular and cellular changes provoked by ischemic hypoxia [5, 6]. A large prospective multicenter observational study showed an association between periventricular white matter hyperintensity and elevated cerebral amyloid levels that are independent of potential confounding factors, suggesting that WMI is related to impaired clearance of amyloid from the brain parenchyma, which eventually results in dementia [7]. The present lack of effective treatments for VCI caused by CCH motivated our investigation of the pathological relationships between CCH and VCI [8].
The glymphatic system of the brain provides a pathway for the clearance of neurotoxic waste such as soluble amyloid-beta (Aβ) via bulk flow of cerebrospinal fluid (CSF) from para-arterial influx to para-venular efflux of the CSF, together with its various solutes [9, 10]. Aquaporin 4 (AQP4), a water channel with polarized distribution predominantly on astrocytic end-feet surrounding perivascular spaces, facilitates the exchange and mixing between CSF and interstitial fluid. Any loss of the normal polarization of AQP4 expression leads to glymphatic dysfunction [10]. The main driving force for glymphatic flow is cerebral arterial pulsation, and treatments that enhance the pulsation of cerebral arteries thus promote the functioning of the glymphatic system [11]. Previous studies have confirmed that prolonged failure of the glymphatic pathway for perivascular drainage contributes importantly to central nervous system diseases such as Alzheimer’s disease, traumatic brain injury, and stroke, and systemic diseases including diabetes and hypertension, and psychiatric disorders such as depression [12–19]. These insights have inspired efforts to pharmacologically stimulate the glymphatic system with pressor medications such as the β1-adrenergic agonist dobutamine, which increases vessel pulsatility and mean arterial blood pressure [11]. However, such approaches have hitherto proven ineffective in translation to clinical practice, perhaps because hypertension is antagonistic to glymphatic function [17, 20].
Since hemodynamic pulsations are the main drivers of glymphatic transport, we suppose that CCH arising from reduced cerebral blood flow (CBF) manifests in glymphatic impairment, ultimately leading to cognitive dysfunction [20, 21]. The pulsatile driving force provided by cardiac output sustains CBF, and the covariance of these processes may underlie CCH [22, 23]. The cardiac glycoside digoxin has been used since antiquity to stimulate myocardial contractility and thus boost cardiac output [24]. Given this pharmacologic action to raise cardiac output, we hypothesized that digoxin could stimulate glymphatic function and rescue the cognitive impairment arising in the aftermath of CCH by the effect of raising hemodynamics and thus eventually enhancing cerebral vessel pulsatility. In addition, unlike dobutamine, digoxin has parasympathetic effects on the sinoatrial node to reduce heart rate and raises cardiac output without increasing blood pressure, which may also be beneficial in restoring glymphatic function [25].
Here, we successfully established a mouse model of CCH resulting in WMI and cognitive decline, and tested the efficacy of short-term digoxin treatment in this model. For the first time, we showed that glymphatic function was impaired after CCH before notable structural alterations in white matter occurred, and, more importantly, that digoxin treatment ameliorated the disruption of glymphatic transport and rescued the cognitive impairment by restoring hemodynamic function in the brain.
Materials and Methods
Animals
All the experiments were conducted according to the standards recommended by the United States National Institute of Health Guide for the Care and Use of Laboratory Animals, and were approved by the Institutional Animal Care and Use Committee at Tongji Medical College, Huazhong University of Science and Technology. The experimental data were collected from male C57/BL6 mice of SPF grade aged 10 weeks–12 weeks and weighing 20 g–27 g, which were obtained from Hubei SJA Laboratory Animal Co. Ltd., Hubei, China. The mice were housed in a temperature- and humidity-controlled facility with free access to food and water. Efforts were made to minimize the number of experimental animals. Before undergoing any operation, mice were first acclimated in their cages for 7 days to adapt to the new environment and experimental staff, thus reducing interference from environmental stress.
Model Preparation
We used the bilateral common carotid artery stenosis (BCAS) surgical procedure, which is known for its simplicity and robust induction of WMI in the rodent brain. Mice randomly allocated to the sham and experimental groups were anesthetized with isoflurane (1.5%–2%) delivered in medical oxygen by a facemask. Mice in the sham group received bilateral exposure of the carotid arteries, followed by suturing of the exposures, and return to their home cage without further treatment, but mice in the experimental groups underwent the BCAS operation as previously described [26]. In particular, to establish the BCAS model, we used microcoils with internal diameter of 0.18 mm (Sawane Spring Co, Shizuoka, Japan). Mice showing any post-surgical neurological deficiency such as hemiplegia were excluded. A total of 227 mice were used in the experiments, of which six mice were excluded according to our stated criteria.
CBF Measurement
We measured CBF in the different treatment groups using laser speckle flowmetry (Omegazone, Omegawave Inc., Tokyo, Japan) at the presurgical baseline, and at 3 days, 10 days, and 30 days after BCAS. Mice were anesthetized by intraperitoneal (i.p,) injection of 1% pentobarbital, and the scalp was cut along the midline and fully retracted to expose the skull. Skull optical clearing was performed as described previously [27] to clearly expose the cerebrovascular structure. The post-operative CBF values in regions of interest of 6 main vessels were expressed as a percentage of baseline. According to the method described in the article, the ROIs were 0.6 mm away from the raphe [28]. The circular ROIs were preset by the software with the diameter of 50 pixels [15].
Intracisternal Tracer Injection and Drug Administration
The CSF tracer FITC-dextran 40000 Lysine Fixable (FITC-d40000, Invitrogen, USA) was dissolved in artificial CSF at a concentration of 1%. As previously described, a 30-G needle was inserted into the cisterna magna for injection into mice under anesthesia with pentobarbital (1%, i.p) [29, 30]. Then, 10 µL of dissolved CSF tracer was injected at 1 µL/min for 10 min with a syringe pump (Harvard Apparatus, USA). Thirty minutes after the beginning of the injection, the still deeply anesthetized mice were perfused and their brains fixed as above. Brain-wide fluorescent tracer images were captured by a laser zoom-stereo microscope (Nikon, SMZ18, Japan). Then, 100-µm coronal slices were cut and sealed with 50% glycerol for ex vivo measurement of the fluorescence-positive area of the tracer by laser scanning confocal microscopy (Olympus, FV1000, Japan). The regional FITC fluorescent area was quantified by a blinded investigator using ImageJ software (National Institute of Health, Bethesda, MD, USA).
Starting 3 days after the BCAS procedure, digoxin (Sanofi (Hangzhou), China) was administered at a daily dose of 0.5 mg/kg i.p. for 5 days, based on previous study and our preliminary findings [31]. The control group received daily injections of normal saline (NS).
For pharmacological inhibition of AQP4 polarity, TGN-020 (MCE, HY-W008574, China) was used before digoxin treatment, according to a previous report [32]. Mice were treated intraperitoneally with either TGN-020 (250 mg/kg in 20 mL/kg body weight) or vehicle (empty 20% SBE-β-CD, 20 mL/kg).
Endogenous Amyloid-β (Aβ) Measurement by ELISA
To measure glymphatic efflux, mice were sacrificed under anesthesia at 3 days, 10 days, and 30 days after BCAS. Brain tissue was collected promptly, carefully separated into whole brain region, cortex, subcortex and corpus callosum on ice, and the samples were later eventually examined by using a mouse Aβ ELISA kit (CSB-E10787m, Cusabio, China) for assay of brain endogenous Aβ42 concentration (pg/mg brain tissue). The relative Aβ level was expressed as concentration compared to sham for each group.
Immunofluorescence Staining
The mice were sacrificed under anesthesia at 3 days, 10 days, and 30 days after BCAS and transcardially perfused as above. After removal and post-fixation in 4% paraformaldehyde (PFA) overnight at 4 °C, the brains were dehydrated successively in 15%, 20%, and 30% (w/v) sucrose in 4% PFA overnight. Coronal sections were cut at 100 μm and stored temporarily at 4 °C. After blocking in 10% bovine serum albumin and 0.5% Triton X-100 for 1 h at room temperature, the sections were incubated with primary antibodies for 24 h at 4 °C. After rinsing in PBS, sections were incubated with the corresponding secondary antibodies for 1 h at room temperature. The primary antibodies were: rabbit anti-myelin associated glycoprotein (MAG) (1:200; Proteintech, China), mouse anti-glial fibrillary acidic protein (GFAP) (1:200; Sigma, USA), and rabbit anti-AQP4 (1:50; Proteintech, China). The secondary antibodies were: 647-conjugated donkey anti-mouse (1:200; Abcam, USA) and cy3-conjugated goat anti-rabbit (1:300; Jackson Immunoresearch, USA). After washing and mounting, sections were observed under a laser scanning confocal microscope (Olympus, FV1000, Japan).
AQP4 Polarization Analysis
To analyze AQP4 polarization, a confocal microscope (Olympus, FV1000, Japan) was used to acquire immunofluorescence staining images in areas of the cortex (CTX) and corpus callosum (CC). Blinded evaluators analyzed the images with FIJI software (National Institutes of Health, Bethesda, MD, USA): GFAP in the green channel, AQP4 in the red channel, and 4',6-diamidino-2-phenylindole (DAPI) in the blue channel.
Based on the results of previous studies, we identified voids surrounded by zones enriched in DAPI and GFAP as capillaries [18, 33]. Then, a donut-shaped area was drawn within 5 pixels from a capillary, wherein red fluorescence intensity indicated the perivascular expression of AQP4. The fluorescent intensity of a corresponding subregion was recorded as the global AQP4 expression. The ratio of the mean fluorescent intensity of the perivascular AQP4 to its global distribution was calculated as an index of AQP4 polarization. For PVS calculation, 6–10 vessels in cortex and 6–8 vessels in corpus callosum per brain section were evaluated.
Evaluation of Cardiac Function, Blood Pressure, and Heart Rate
In isoflurane-anesthetized mice, cardiac function was recorded with a high-resolution echocardiography machine equipped with a 30-MHz scanning head (VisualSonics Vevo770, VisualSonics, Toronto, Canada) as described previously [34]. Blood pressure and heart rate were recorded using a non-invasive small-animal sphygmomanometer (BP2010A; Softron Biotechnology Ltd., Japan). In this procedure, mice were gently placed head-first in a dark cloth bag, with the tail exposed and warmed by a heater. After the mice were acclimated, we recorded blood pressure and heart rate using a tail cuff. The mean of five repeated measurements was calculated for each mouse.
In-vivo Two-Photon Laser Scanning Microscopy
A 3-mm-diameter craniotomy was drilled in the skull 1.5 mm–2 mm lateral and 1 mm–1.5 mm posterior to bregma, leaving the dura intact. The operated region was covered with artificial CSF and sealed with a glass coverslip. To visualize the vasculature, 100 µL of 1% Texas Red-dextran 70000 Neutral (TR-d70000, Invitrogen, USA) was injected as a bolus through the femoral vein. A Mai Tai laser (Spectral Physics, USA) attached to a confocal scanning system (LSM780, Carl Zeiss, Japan) on an upright microscope (20×/1.0, water, WD 1.8 mm, W Plan-Apochromat 75 mm, DIC sliderSenarmont 75 W PA 20×/1.0 III), was used for imaging. The excitation wavelength was 870 nm. The cerebral vasculature was displayed in a 256 × 256-pixel frame extending from the pial surface to a depth of 150 µm with 2-µm Z-step scanning. To record the velocity and pulsatility of vessels, 4800-ms X-T line scans parallel and orthogonal to the axis of penetrating arteries, respectively, were applied at a 1.6-kHz scan rate. Flow velocity and pulsatility were calculated as previous described [35].
Injection of Human Exogenous Aβ Solution and ELISA
Mice anesthetized with pentobarbital (1%, i.p.) were placed in a stereotaxic frame for head fixation. The body temperature was kept at 37ºC with a heating pad, the scalp was incised and retracted, and a 1-mm-diameter craniotomy was made 1.5 mm lateral and 1 mm anterior to bregma, taking care to avoid hemorrhage, with placement of the needle tip 2.5 mm below the cortical surface. To measure glymphatic efflux, 0.5 µL of a solution containing 10 nmol/L human Aβ42 (Chinapeptides, China) was injected at 0.05 µL/min for 10 min with a syringe pump (Harvard Apparatus, USA) [10, 18, 36]. Mice were sacrificed while still anesthetized 1 h after completion of the injection, and brain tissue was collected for assay of the exogenous human Aβ42 remaining (pg/mg brain tissue), which was determined using a human Aβ ELISA kit (CSB-E10684h, Cusabio, China). The relative Aβ level was expressed as concentration compared to sham for each group.
Luxol Fast Blue (LFB) Staining
The mice were sacrificed while anesthetized at 30 days after BCAS. They were transcardially perfused with ice-cold NS followed by 4% PFA in 0.01 mol/L phosphate-buffered saline (PBS) and the brains harvested. After washing with PBS, the brains were frozen and cut into 20-μm cryostat sections, which were slide-mounted and stored at − 80 °C until use. Thawed slides were post-fixed in 4% PFA, dehydrated in a graded ethanol series (75, 90, and 100%), and then incubated in 0.1% LFB (Servicebio, Wuhan, China) at 60 °C for 10 h. Then the sections were differentiated in Li2CO3 solution for 3 min–5 min, bathed in 70% ethanol to stop differentiation, and sealed with resin. Upon microscopic examination, the severity of white matter lesions was graded as normal (grade 0), disarrangement of nerve fibers (grade 1), formation of marked vacuoles (grade 2), or disappearance of myelinated fibers (grade 3), as described previously [26].
Electron Microscopy
The mice were sacrificed while anesthetized at 30 days after BCAS and perfused intracardially with ice-cold NS. Tissue samples containing the CC were collected and prepared as previously described [35]. Samples were then observed in an electron microscope (Hitachi, HT7700) at 120 kV. G-ratios were calculated as inner axon diameter relative to the total outer diameter.
Morris Water Maze
The Morris water maze test was used to evaluate spatial learning and memory, following conventional methods for mice [37]. In brief, a white circular water maze with a diameter of 150 cm and a depth of 50 cm was filled with water made opaque by addition of powdered milk, and equipped with a 10 cm-diameter white platform hidden 0.5 cm below the water surface. The temperature of the water was 25.0 °C ± 0.5 °C and the test was performed by blinded researchers.
During the first five training days, mice were gently placed into the water with their heads pointed towards the edge of the pool. If they discovered the hidden platform within 60 s, they were allowed to remain in place for an additional 30 s to afford the consolidation of spatial memory cues. On the sixth day, the platform was removed and mice were placed in the quadrant opposed to the pre-existing platform. Their swimming tracks were recorded with a video camera (Hikvision Digital Technology Co., Ltd, China) and analyzed using the Any-Maze Behavioral Tracking System (Stoelting Co. Ltd., USA).
Novel Object Recognition
A novel object recognition test was administered as described in the literature [38]. The test was conducted in a 40 × 40 cm2 open field test box in a double-blind manner. Before each test, the interior of the box was cleaned with double-distilled water and 75% alcohol. First, mice were gently placed into the box without any object and left for a 15-min adaptation period. In a session 24 h later, the mice were left for 5 min in the box, now containing two cubes (objects A and B) of the same color and size. Next, one cube (object B) was replaced with a sphere (object C) of the same size but of a different color prior to testing 24 h later. Mouse behavior was recorded for 5 min using the video camera (Hikvision Digital Technology Co., Ltd.). The video recordings were analyzed using the Any-Maze Behavioral Tracking System (Stoelting Co., Ltd), and the novel object preference ratio was calculated as (time of interest in C/time of interest in (A + C)) %. Mice were excluded for the following reasons: immobility in a corner for > 1 min, crawling on an object, interest in an object for < 2 s, or a preference ratio > 80%, or < 20%.
Study Design and Statistical Analysis
Data were analyzed using GraphPad Prism 8.0 (GraphPad Software Inc., La Jolla, CA, USA). All measurements are presented as the mean ± SD. Data were first tested for normality using the Shapiro-Wilk test. Student’s t-test was applied for comparisons of two groups, and one-way analysis of variance (ANOVA) with Tukey’s post-hoc test for multiple comparisons, when the data were normally distributed. For comparisons of non-normally distributed data, the Mann-Whitney U-test was applied. Pearson’s correlation analysis was used to compare the correlations between CBF, glymphatic function, and AQP4 polarity. P < 0.05 was considered to indicate a statistically significant difference.
Results
The Glymphatic System is Damaged in the Acute Phase but Recovers Gradually After Hypoperfusion
To verify establishment of the BCAS model, laser speckle flowmetry was used to measure the CBF of mice in the sham group, and groups at 3 days, 10 days, and 30 days after the operation. Our results showed that CBF was dramatically reduced to about 40% at 3 days after BCAS, and then recovered gradually with time. At 30 days after BCAS, there was no statistically-significant difference in CBF from the sham group, in accordance with previous reports [26] (Fig. 1A).
Fig. 1.

Glymphatic influx and efflux are impaired after BCAS and recover gradually with time. A Left, representative images of CBF measured by laser speckle flowmetry; right, quantitative analysis of images as in A (n = 5 per group; scale bar, 1 mm). B Representative images of brain-wide CSF influx. Left, models of tracer flux along vessels observed in perivascular spaces; right, brain-wide images of CSF tracer 30 min after intracisternal infusion on the dorsal and ventral sides of the brain at 3 days, 10 days, and 30 days following BCAS (scale bar, 2 mm). C Representative images of CSF tracer influx in sagittal (a1–a4) and coronal (b1–b4) slices. (c1–c4) The perivascular space of penetrating arteries is filled with fluorescent CSF tracer perpendicular (arrowhead) and parallel (arrow) to the slice orientation (red, fluorescent areas with intensity exceeding the preset threshold; scale bars, 2 mm and 500 μm). D Global percentages of fluorescent area in the whole brain, CTX, SCTX, and CC (n = 6 mice per group). E Correlation between global percentage of fluorescent area and CBF. F Relative concentration of endogenous Aβ42 in the whole brain, CTX, SCTX, and CC at 3 days, 10 days, and 30 days following BCAS (n = 6 per group). G Correlation between relative concentration of Aβ42 and CBF. H Relative concentration of human exogenous Aβ42 at 60 min after intra-striatale injection. I Correlation between global percentage of fluorescent areas and clearance ability of human exogenous Aβ42. In A, D, F, and H *P <0.05, **P <0.01, ***P <0.001 vs sham, #P <0.05, ##P <0.01, ###P <0.001 vs 3 days, §P <0.05, §§P <0.01, §§§P <0.001 vs 10 days. PVS, perivascular spaces; ACA, anterior cerebral artery; MCA, middle cerebral artery; PCA, posterior cerebral artery; BA, basilar artery; C. Willis, circle of Willis; CTX, cortex; CC, corpus callosum; SCTX, subcortex; CBF, cerebral blood flow.
Previous studies indicated that the percentage fluorescent area reflects the capacity for glymphatic influx [39]. The fluorescence was evident throughout the telencephalon 30 min after the injection of tracer into the cisterna magna (Fig 1B). The percentage fluorescent area sharply declined to ~25% of baseline at 3 days after BCAS, and gradually rose to ~50% at 10 days post-BCAS. The fluorescent area had recovered completely to baseline levels by 30 days post-injury. Subregional analysis showed similar patterns of the recovery of glymphatic influx in the CTX and CC, whereas in the subcortex (SCTX) the percentage of fluorescent area had already recovered at 10 days after BCAS (Fig 1C, D). Regression analysis of fluorescent area with CBF showed a significantly linear correlation (R2 = 0.94), indicating a strong association between glymphatic influx and CBF level (Fig 1E).
Aβ aggregation in the neocortex has an evident association with the onset of clinical AD. Previous research indicates that ~65% of endogenous Aβ is cleared by the glymphatic system under normal physiological conditions [10]. Given the key neurotoxicity of Aβ42 peptide, we used ELISA to measure the clearance of endogenous Aβ42 from the entire mouse brain. Relative to the sham group, the Aβ42 concentration in the whole brain was 65% higher in the BCAS group at 3 days after the injury but was essentially normal at 30 days following BCAS. The subregional analysis showed similar temporal patterns of Aβ42 clearance in the CTX, SCTX, and CC. An approximately linear relationship was also noted between the relative Aβ42 concentration and CBF (R2 = 0.89) at each time point after BCAS (Fig 1F, G). To further confirm that the clearance of glymphatic system is implicated in the reduced concentration of Aβ42 as the CBF recovers, we injected exogenous human Aβ42 into the corpus callosum, and measured by ELISA the residual of injected peptide in the whole brain region at one hour after injection. Our results showed that the exogenous Aβ42 significantly aggregated at 3 days after BCAS operation and gradually decreased along with time. This result indicated that the clearance of Aβ42 by the glymphatic system played a crucial role in the reduced residual concentration of Aβ (Fig 1H, I). Thus, we found that disruption of glymphatic function relates to the influx and efflux processes following BCAS, which recover over time in parallel with CBF recovery.
De-localization of AQP4 in Astrocytic End-Feet Contributes to Post-BCAS glymphatic Dysfunction
The water channel AQP4 normally has a polarized distribution on the end-feet of perivascular astrocytes, but glymphatic flow fails when the polarized AQP4 distribution is perturbed. Immunofluorescence labeling for AQP4, GFAP, and DAPI showed a sharp decrease in AQP4 polarization on cortical astrocytes at 3 days post-BCAS, which slowly recovered at 10 days and finally normalized at 30 days relative to findings in the sham group. Similarly, the AQP4 depolarization in the CC initially increased but returned to baseline at 10 days after BCAS (Fig 2A–C). Regression analysis showed linear relationships between AQP4 and CBF in the CTX (R2 = 0.79) and CC (R2 = 0.98) (Fig 2D, E). Thus, depolarization of AQP4 in astrocytes occurred in parallel with reduced CBF and glymphatic impairment following BCAS.
Fig. 2.

AQP4 polarity is reduced after BCAS and repolarizes gradually over time. A Left, flow diagram of AQP4-polarization analysis. a1–a4 Representative confocal images of AQP4 immunostaining in coronal sections from sham mice and at 3 days, 10 days and 30 days following BCAS. b1–b4, c1–c4 Representative images of AQP4 depolarization in the CC. d1–d4, e1–e4 Representative images of AQP4 depolarization in the CTX. f1–f4 Representative images of co-localization of AQP4 and GFAP (red, AQP4; green, GFAP; blue, DAPI; scale bars, 200 μm, 50 μm, and 20 μm). B, C Quantitative analysis of AQP4 polarization in the CTX and CC (**P <0.01, ***P <0.001 vs sham; ###P <0.001 vs 3 days; n = 202 in CTX and 69 in CC from 6 mice per group). D, E Regression analysis between the polarity of AQP4 and CBF in the CTX and CC. CTX, cortex; CC, corpus callosum.
Digoxin Narrows Hemodynamic Variation After Hypoperfusion
Digoxin has positive inotropic and negative chronotropic effects in the treatment of myocardial disease [25]; this predicts that it might enhance glymphatic function via effects on hemodynamics. We previously reported a persistent reduction of blood flow velocity and arteriolar pulsatility following BCAS [35]. Thus, we predicted that the hemodynamic changes induced by digoxin would rescue the functional glymphatic deficits in the mouse brain after CCH. Although cardiac ultrasound and hemodynamic monitoring revealed no statistically-significant difference in cardiac output, injection fraction, stroke volume, or blood pressure in mice before and after medication (Fig 3A–H), they showed that heart rate was significantly reduced in the digoxin group post-BCAS (Fig 3I). However, on further analysis, we noted that, compared with vehicle treatment, digoxin reversed the downtrend in stroke volume and restricted the amount of change in cardiac output by >95% after BCAS (Fig 3J, K).
Fig. 3.

Digoxin attenuates downward trends of stroke volume and cardiac output, and reduces heart rate after BCAS. A Time-line of the BCAS operation, digoxin or vehicle medication, and hemodynamic measurements by cardiac ultrasonography and tail-cuff sphygmomanometry before BCAS (baseline), before medication (pre), and after medication (post). B Representative cardiac ultrasonograms before (left) and after (right) digoxin treatment. C–E SV, EF, and CO measured by ultrasonography in vehicle and digoxin-treated mice at baseline, pre-, and post-medication (no statistically-significant difference between groups; n = 14 per group). F–I MBP, SBP, DBP, and HR measured by tail-cuff sphygmomanometer at baseline, pre-, and post-medication (no statistically-significant difference in blood pressure between groups). HR is significantly lower pre-digoxin treatment than pre-treatment (**P <0.01 vs pre. n = 6 per group). J, K Changes in SV and CO between pre- and post-medication (*P <0.05, **P <0.01 vs NS. n = 14 per group). SV, stroke volume; EF, ejection fraction; CO, cardiac output; MBP, mean blood pressure; SBP, systolic blood pressure; DBP, diastolic blood pressure; HR, heart rate.
We then measured the pharmacological effects of digoxin on cerebral vessels. Blood flow velocity and arteriole pulsatility after the intervention were measured using a two-photon microscope. Our results showed that both the flow velocity and the pulsatility of arterioles were decreased after BCAS, but improved after digoxin intervention (Fig 4A–C). The change of CBF was also detected by laser speckle flowmetry, revealing a decline of CBF after BCAS that was rescued by digoxin (Fig 4D).
Fig. 4.

Digoxin increases blood flow velocity and arteriole pulsatility after BCAS. A Left, schematic of in vivo two-photon microscopy of mouse cerebral vessels; right, representative images acquired by two-photon microscopy. B Left, representative images of RBC movement in vessels in sham, vehicle, and digoxin-treated groups at 10 days after BCAS; right, quantitative analysis of data from images as at left (**P <0.01, ***P <0.001 vs sham, #P <0.05 vs NS; n = 12 per group). C Left, representative images of vessel pulsatility, and its calculation as the absolute value of the integral of vascular diameter during recording; right, quantitative analysis of data as at left (***P <0.001 vs sham, #P <0.05 vs NS; n = 12 per group). D Left, representative images of CBF measured by laser speckle flowmetry in the sham, vehicle, and digoxin-treated groups at 10 days after BCAS; right, quantitative results of data as at left (**P <0.01 vs sham, #P <0.05 vs NS; n = 5 per group; scale bar, 1 mm).
Digoxin Promotes Glymphatic Systematic Recovery After Hypoperfusion
We also explored the effects of digoxin on glymphatic influx and efflux after BCAS. After fluorescent tracer delivery into the cisterna magna, global imaging revealed increased tracer penetration to the dorsal and ventral brain surfaces in digoxin-treated mice (Fig 5A). As expected, the ex vivo fluorescence studies at 10 days after BCAS showed enhanced glymphatic influx in the whole brain, CTX, and SCTX, along with an upward tendency in the CC at 30 min in the digoxin-treated group (Fig 5B, C). Thus, we found a remarkable and global facilitation of glymphatic influx by digoxin. To amplify the effect of medical intervention on the clearance of interstitial solutes, we next measured exogenous Aβ42 after intracerebral injection. Similarly, the glymphatic efflux in the whole brain was significantly increased by digoxin treatment (Fig 5D). In brief, digoxin improved glymphatic function and Aβ42 clearance in the aftermath of a hypoperfusion injury.
Fig. 5.
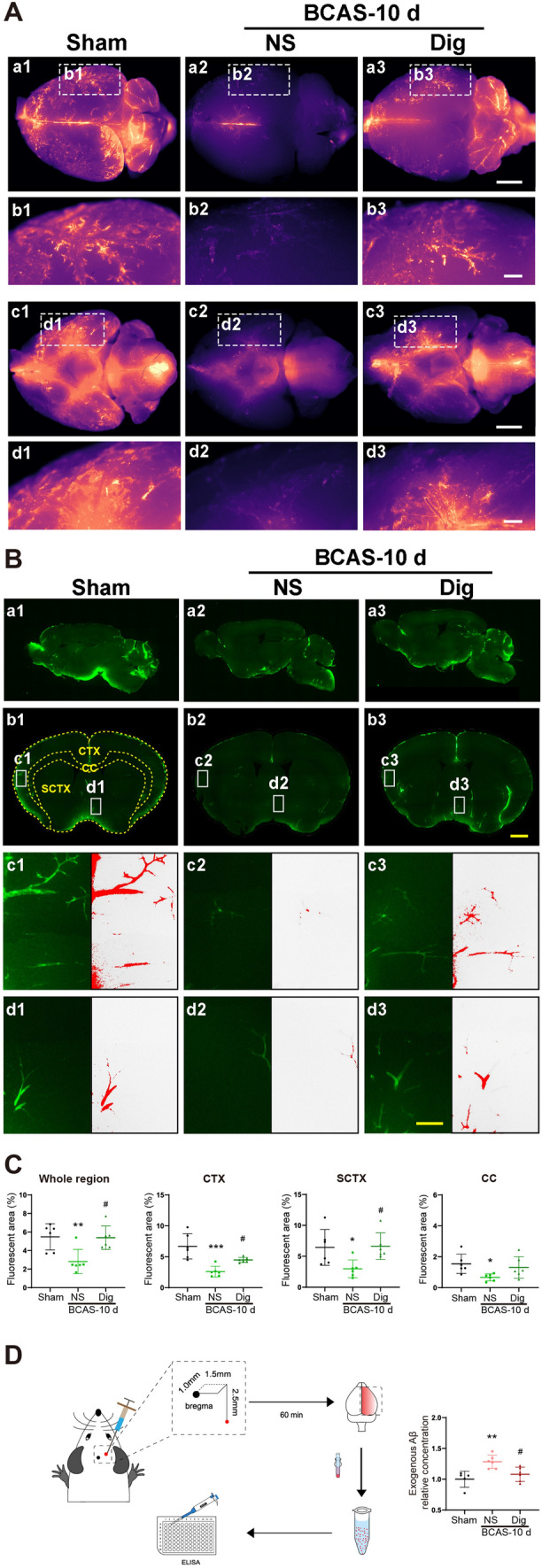
Digoxin ameliorates glymphatic transport after BCAS. A a1–a3, c1–c3 Representative brain-wide images of CSF tracer 30 min after intracisternal infusion on the dorsal and ventral sides in sham, vehicle, and digoxin-treated groups. b1–b3, d1–d3 Enlarged regional views of PVS (scale bars, 2 mm and 500 μm). B a1–a3, b1–b3 Representative sagittal and coronal slice images of CSF tracer influx. c1–c3, d1–d3 Enlarged regional views of PVS (red, fluorescent areas with intensity exceeding the preset threshold; scale bars, 2 mm and 100 μm). C Quantitative analysis of data as in B in the whole brain, CTX, SCTX, and CC (*P <0.05, **P <0.01, ***P <0.001 vs sham, #P <0.05 vs NS; n = 6 per group). D Left, schematic of stereotactic injection of exogenous human Aβ42 and measurement by ELSIA; right, relative concentration of human exogenous Aβ42 60 min after injection in sham, vehicle, and digoxin-treated groups (**P <0.01 vs sham, #P <0.05 vs NS; n = 6 per group). CTX, cortex; SCTX, subcortex; CC, corpus callosum.
Digoxin Accelerates the Process of Repolarization in AQP4 and Attenuates WMI Following CCH
Immunofluorescence findings showed a significant increase in the polarization of AQP4 in astrocyte end-feet in the CTX and CC at 10 days after digoxin treatment, relative to the vehicle control group. Also, as expected, regression analyses between AQP4 polarity and CBF showed linear relations both in the CTX (R2 = 0.74) and CC (R2 = 0.97), suggesting an association with the hemodynamic effects of digoxin on glymphatic function in the hypoperfused brain (Fig 6A–C).
Fig. 6.

Digoxin decreases AQP4 depolarization after BCAS. A Representative confocal images of AQP4 immunostaining in the CC and CTX at 10 days after BCAS in vehicle and digoxin-treated groups. Enlarged views show repolarization of AQP4 after digoxin treatment (red, AQP4; green, GFAP; blue, DAPI; scale bars, 1 mm and 100 μm). B Quantification of AQP4 polarization as in A (**P <0.01, ***P <0.001 vs sham, #P <0.05, ###P <0.001 vs NS; n = 180 in CTX and n =78 in CC from 6 mice per group). C Regression analysis between the polarity of AQP4 and CBF in the CTX and CC. CTX, cortex. CC, corpus callosum.
Previous research has shown that chronic hypoperfusion results in the disruption of white matter integrity [35]. The present findings in the digoxin group did not show any effect of treatment on MAG expression at 10 days after BCAS, whereas its expression was elevated at 30 days relative to that in the vehicle-treated group (Fig 7A, C). Digoxin treatment also significantly reduced the LFB score in the CC, and showed a downward tendency in the corpus striatum at 30 days post-BCAS (Fig 7B, D). In addition, digoxin treatment ameliorated the decrease in the thickness of the myelin sheath in the CC due to CCH, thus confirming protection against WMI (Fig 7E, F).
Fig. 7.

Digoxin ameliorates WMI after BCAS. A Representative confocal images of the CC labeled with MAG in each group at 10 days and 30 days following BCAS. B LFB staining for myelin integrity in the CC, AC, and CPu 30 days after BCAS in each group (scale bar, 100 μm). C Quantitative analysis of the relative expression of MAG as in A (*P <0.05, **P <0.01 vs sham, ##P <0.01 vs NS; n = 10 per group). D Scores from LFB staining as in B (***P <0.001 vs sham, #P <0.05 vs NS; n = 24 from 6 mice per group). E Representative electron microscopy images of axons in the CC 30 days after BCAS in each group (scale bars, 2 µm and 1 µm). F Upper, quantitative analysis of axon diameters and G-ratios; lower, scatter plot of the G-ratios and axon diameters in each group 30 days after BCAS (***P <0.001 vs sham, ###P <0.001 vs NS; n =180 from 6 mice per group). WMI, white matter injury, CC, corpus callosum; CTX, cortex; MAG, myelin-associated glycoprotein; LFB, Luxol fast blue; AC, anterior commissure; CPu, corpus striatum.
Digoxin Rescues Learning and Memory Disturbances Induced by BCAS
Finally, we used the Morris water maze to test the hypothesis that digoxin treatment would rescue the cognitive impairment after BCAS. There were no group differences in escape latency (the time taken to find the platform) on the first day after surgery, nor did the swimming speeds differ, suggesting that all groups had similar motor abilities before any intervention. Vehicle-treated BCAS mice showed a relatively prolonged escape latency on the fourth and fifth days of training sessions, indicating impaired learning (Fig 8A–C). Moreover, on the sixth day (trial day), vehicle-treated mice had poorer performance with respect to platform crossing number, time spent in the platform location, and latency to reach the previous platform location than unlesioned mice, indicating the persistence of spatial memory impairment at 30 days after BCAS. The digoxin-treated mice showed a trend (P = 0.05 at day five) to an improved escape latency on training days four and five, and markedly out-performed the vehicle-treated mice in platform crossings, time spent in the previous platform location, and latency to reach the platform position on the sixth day, suggesting a rescue of spatial learning capacity at 30 days after BCAS (Fig 8D–F).
Fig. 8.

Digoxin rescued cognitive disturbances 30 d after BCAS, and TGN-020 blocked the effect by inhibiting glymphatic transport. A Representative tracks of mice during the training sessions in sham, vehicle, and digoxin-treated groups after BCAS. B Time spent to find the hidden platform each day during training. C Average swimming speed during training in each group. D Numbers of crossings of the zone formerly occupied by the platform on the sixth day in each group. E Time spent in the platform zone on the sixth day in each group. F Mean time to reach the platform zone for the first time on the sixth day in each group. B–F *P <0.05, **P <0.01 vs sham, #P <0.05 vs NS; n = 15 per group. G Percentage of time spent exploring the novel object in each group (** P <0.01 vs sham, #P <0.05 vs NS; n = 10 per group). H Quantitative analysis of CBF measured by laser speckle flowmetry in vehicle and TGN-020 groups at 10 days after BCAS with digoxin treatment. I Quantitative analysis of injected CSF tracers via cisterna magna in the whole brain region, CTX, SCTX, and CC at 10 days in the two groups after BCAS with digoxin treatment, respectively. J White matter injury scored by LFB staining in CC, CPu, and AC at 10 days in the two groups after BCAS with digoxin treatment, respectively. K Quantitative analysis of CSF tracers at 30 days in two groups after BCAS with digoxin treatment, respectively. L White matter injury scored by LFB staining at 30 days in the two groups after BCAS with digoxin treatment, respectively. M The percentage of time mice spend to explore the novel object in the two groups. N, O Representative images of CSF tracer and LFB staining in vehicle and TGN-020 groups at 10 days and 30 days after BCAS with digoxin treatment, respectively. Scale bar, 500μm and 100 μm. H–M (*P <0.05, **P <0.01, ***P <0.001 versus vehicle. n = 6 per group). CTX, cortex; CC, corpus callosum; SCTX, subcortex; CBF, cerebral blood flow; LFB, Luxol fast blue; AC, anterior commissure; CPu, corpus striatum.
We finally used the novel object recognition test to further explore the effects of digoxin on visual learning in BCAS mice. Mice with digoxin treatment showed greater exploration of the novel object at 30 days following BCAS than vehicle-treated mice (Fig 8G), thus confirming that digoxin improved the learning and memory deficits induced by chronic hypoperfusion.
TGN-020 Treatment Blocked Effects of Digoxin on BCAS Mice, Impaired Glymphatic Function, and Worsened Cognitive Competence
TGN-020 is reported to inhibit the expression of AQP4 M23, thus disrupting the polarization of astrocytic AQP4 [40]. Therefore, we applied TGN-020 to suppress the glymphatic transport in order to explore the possible protective mechanism of digoxin after BCAS. Our results showed that CBF was increased after TGN-020 treatment at 10 days after BCAS, which was in accordance with a previous report (Fig 8H) [41]. The effect of digoxin on glymphatic system was obviously blocked at 10 days after BCAS. White matter injury scored by LFB staining showed that there was no statistical difference in CC or anterior commissure between TGN-020 and vehicle groups at 10 days after BCAS, while corpus striatum was significantly impaired in TGN-020 treatment group. At 30 days after BCAS, although there was no statistical significance in glymphatic function between TGN-020 and vehicle groups, both the white matter and the cognitive ability were notably impaired as detected by LFB staining and NOR in the TGN-020 group, respectively (Fig 8I-O). Taken together, the present results demonstrated that TGN-020 treatment elevated CBF but impaired glymphatic system function, which eventually led to white matter injury and cognitive deficits. This indicated that it was the rescued glymphatic system rather than CBF recovery per se that was the dominant factor in ultimately altering BCAS outcomes, and showed that digoxin treatment ameliorated white matter injury and cognitive decline via its potentiation of the glymphatic system.
Discussion
CCH gives rise to WMI, which eventually manifests in cognitive dysfunction, presumably proportional to the extent of white matter damage. We now showed that glymphatic efflux and influx were severely impaired in mice at 3 days after BCAS, followed by a gradual spontaneous recovery in the following weeks. The transient deficit in glymphatic function paralleled the reduction in CBF. Furthermore, treatment with digoxin expedited the recovery of glymphatic function, while rescuing mice from the WMI and cognitive deficits due to CCH. The pathophysiological relationship between reduced CBF and WMI has been a matter of controversy. While some research indicates that white matter hyperintensities arise due to low perfusion [42, 43], other studies have failed to detect such an association [44, 45]. Intriguingly, we saw the onset of impaired glymphatic function at only 3 days after BCAS, far preceding the observable manifestations of WMI. The present results suggested that decreased CBF plays a vital causal role in the triad linking hemodynamics, glymphatic function, and impaired cognition in the murine BCAS model.
Previous research on cognitive deficits induced by CCH has focused on neuroinflammation, blood-brain barrier disruption, or neurovascular unit dysfunction [6]. We now find that the dysfunction of the brain-wide clearance pathway for metabolic wastes (including Aβ42) also contributes to the pathogenesis of WMI and cognitive impairment after CCH. The findings of parallel reductions in glymphatic function and CBF presumably reflect multiple factors. First of all, CCH results in hypoxia and consequent hypercapnic vasodilation [46], which is believed to stunt interstitial solute transport by narrowing the perivascular tunnels where CSF–interstitial fluid exchange occurs [47]. Second, the reduced blood volume causes the collapse of vessels, especially terminal arterioles lacking musculature, thus further compressing the perivascular space. In vivo studies have indicated that the cerebral arterial pulsation acts as a major driving force to promote CSF movement [11, 41]. BCAS-induced hypoperfusion significantly reduces arteriolar pulsatility, thus attenuating glymphatic function. Moreover, hypercapnic vasodilation further constrains the maximum range of vasomotion, which aggravates the restriction of cerebrovascular pulsatility [47]. Previous research has shown that the deep white matter is especially vulnerable to ischemic injury because of its shortage of collateral circulation and low capacity for vascular regulation [48]. Consequently, glymphatic function in deep perivascular compartments is an early casualty of declining local perfusion.
Aβ plaques are one of the canonical neuropathologies of Alzheimer’s disease [49]. Our previous research showed a halving of exogenous Aβ clearance in AQP4-null mice, suggesting that perivascular flow is a key mediator in the removal of soluble Aβ from the brain [10]. Given this background, we hypothesized that impaired Aβ clearance would occur in BCAS-induced CCH. We found that the clearance of endogenous Aβ42 followed a V-shaped function with time, attaining a nadir at 3 days after BCAS, followed by a slow climb towards normal values. Remarkably, recovery of Aβ42 clearance approximately coincided with the restoration of CBF. Interestingly, Thomas et al. showed that perivascular circulation is unaffected by unilateral common carotid artery occlusion, presumably due to rapid accommodation or shunting from the circle of Willis [14]. However, in the present bilateral occlusion model, there was a linear relationship between CBF and glymphatic flux after BCAS, suggesting that decompensated CBF was critical for the pathogenesis of glymphatic failure.
Our study further showed that there was a linear relationship between CBF and AQP4 polarity. It has been proved that the binding between α-syntrophin and the cytoplasmic tail of AQP4 is essential to maintain the polarized expression of AQP4 at the astrocytic membrane, which is sensitive to ischemia [40]. Thus, we suppose that cerebral hypoperfusion can influence glymphatic transport by altering AQP4 polarity.
Digoxin is a cardiotonic glycoside derived from the leaves of Digitalis purpurea, and has long been used for the treatment of heart failure. It increases the force of myocardial contraction and promotes cardiac output by selectively inhibiting the Na+ and K+ pumps in cardiomyocytes [24]. We expected that elevating cardiac output might help to restore the hypoperfusion-induced disturbance of glymphatic transport in the mouse brain [50]. We found that digoxin-treated mice showed less variation of cardiac output and stroke volume than vehicle-treated counterparts. Besides, digoxin also excites the vagus nerve and inhibits the activity of the sinoatrial node, thus slightly reducing the heart rate [24]. We suppose that this phenomenon might prolong the interval for the filling of cerebral arterioles, and this would facilitate substance exchange with the perivascular space. This scenario is consistent with previous findings that glymphatic function is inversely related to heart rate [51].
According to our results, digoxin also has a positive effect on the flow velocity and pulsatility of cerebral vessels, especially penetrating arteries, which are essential for glymphatic transport [10]. This phenomenon is most likely an effect of the hemodynamic changes described above. Our present results that digoxin promoted the restoration of AQP4 polarity and glymphatic function after BCAS reveals that increased cardiac output can indeed override the hypoperfusion in BCAS. By increasing blood volume and vascular pulsation in the brain, digoxin may restore glymphatic function.
CCH leads to cerebral WMI, which is an important factor in the cognitive decline in age-related dementias [52] and hypobaric hypoxia [53]. We confirmed the occurrence of disturbances in visual learning and memory in mice post-BCAS, as seen in previous research [54]. We attribute the rescue of behavioral performance by digoxin to an amelioration of white matter demyelination after CCH upon elevated perfusion.
Interestingly, there are disparate reports in the literature of the association between digoxin treatment and cognitive function. Some reports indicate a potential risk of cognitive decline in patients receiving digoxin treatment [55, 56]. On the other hand, Laudisio et al. reported an improvement in cognitive function among older patients treated with digoxin for heart failure. We speculate that these discrepant results relate to the relatively narrow therapeutic window for digoxin, which can cause fatal arrhythmia, cognitive impairment, or delirium upon excessive dosing [57]. Individual differences may also contribute to negative effects of digoxin. Therefore, in our experiment, we cautiously applied digoxin treatment at a relatively low dose [31] and for only a short term (five days), which proved to ameliorate the reduced hemodynamic indices and cognitive deficits of the hypoperfusion injury.
The translational interpretation of our experiment has limitations due to our exclusive use of young, healthy male mice. In fact, age, obesity, diabetes, and hypertension are important high-risk factors for heart diseases, cognitive impairment, and glymphatic dysfunction. Therefore, further studies shall be needed to investigate the influences of age, sex, and comorbidities in this model. Besides, since Aβ tends to accumulate at a chronic stage following BCAS [58], in terms of clinical translation it would be most meaningful to promote Aβ clearance in a long term after BCAS. Whether digoxin, which we have shown to ameliorate the outcomes by restoring glymphatic function in the short run, would have salutatory effects at the chronic stage of CCH remains unknown. Further studies should be carried to explore the effects of digoxin on clearance capacity for Aβ and BCAS outcomes at a chronic stage after BCAS.
In our experiment, the effect of digoxin was blocked after TGN-020 inhibited the glymphatic function, suggesting that digoxin acted by rescuing the glymphatic disruption. Our result revealed that digoxin improved CBF and enhanced vascular pulsation at the same time, and the latter was closely related to the glymphatic function [11]. However, it remains to be established whether digoxin acted primarily through CBF or arterial pulsation. Resolving these questions calls for further investigations of the separate effects of CBF and vascular pulsation on glymphatic function.
A previous report claimed that persistent reduction of CBF could be a preclinical risk of developing dementia [59]. Conversely, it is well known that attaining improved aerobic fitness increases cardiac output and slows resting heart rate, which resembles the pharmacological effects of digoxin in our study [60]. Previous research has shown that moderate exercise improves glymphatic function in young and aged healthy mice [61, 62]. This suggests that improving cerebral hemodynamics by means of exercise and/or medication could relieve CCH injury by restoring impaired glymphatic function. Besides, a recent population-based cohort study revealed that a high risk of developing dementia is strongly associated with cardiovascular risk factors [63]. This implies the importance of maintaining cardiovascular health and the possibility of protection against VCI by means of cardiovascular interventions. To paraphrase Juvenal’s mens sana in corpore sano, a powerful heart keeps a brain healthy. In conclusion, we show that glymphatic dysfunction contributes to the pathogenesis of cognitive deficits that result from CCH. Persistent reduction in CBF in the CCH model causes glymphatic function impairment that precedes the onset of overt WMI and cognitive impairment, all of which are improved by treatment with digoxin.
Acknowledgments
This work was supported by Grants from the National Natural Science Foundation of China (81873749 and 81801072). We sincerely thank the Optical Bioimaging Core Facility of Wuhan Laboratory for Optoelectronics–Hong Kong University of Science and Technology for support in data acquisition, Dr. P. Liu for helpful discussions during data acquisition, and Prof. Paul Cumming for manuscript review.
Conflict of interests
All authors claim that there are no conflicts of interest.
Footnotes
Jie Cao and Di Yao contributed equally to this work.
Contributor Information
Minghuan Wang, Email: mhwang@tjh.tjmu.edu.cn.
Wei Wang, Email: wwang@vip.126.com.
References
- 1.Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, et al. Vascular contributions to cognitive impairment and dementia: A statement for healthcare professionals from the American heart association/American stroke association. Stroke. 2011;42:2672–2713. doi: 10.1161/STR.0b013e3182299496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O'Brien JT, Thomas A. Vascular dementia. Lancet. 2015;386:1698–1706. doi: 10.1016/S0140-6736(15)00463-8. [DOI] [PubMed] [Google Scholar]
- 3.Dichgans M, Leys D. Vascular cognitive impairment. Circ Res. 2017;120:573–591. doi: 10.1161/CIRCRESAHA.116.308426. [DOI] [PubMed] [Google Scholar]
- 4.Barker R, Ashby EL, Wellington D, Barrow VM, Palmer JC, Kehoe PG, et al. Pathophysiology of white matter perfusion in Alzheimer's disease and vascular dementia. Brain. 2014;137:1524–1532. doi: 10.1093/brain/awu040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen YX, Tian YY, Tian H, Huang QB, Fang YK, Wang W, et al. Tamoxifen promotes white matter recovery and cognitive functions in male mice after chronic hypoperfusion. Neurochem Int 2019, 131: 104566. [DOI] [PubMed]
- 6.Duncombe J, Kitamura A, Hase Y, Ihara M, Kalaria RN, Horsburgh K. Chronic cerebral hypoperfusion: A key mechanism leading to vascular cognitive impairment and dementia. Closing the translational gap between rodent models and human vascular cognitive impairment and dementia. Clin Sci (Lond) 2017, 131: 2451–2468. [DOI] [PubMed]
- 7.Marnane M, Al-Jawadi OO, Mortazavi S, Pogorzelec KJ, Wang BW, Feldman HH, et al. Periventricular hyperintensities are associated with elevated cerebral amyloid. Neurology. 2016;86:535–543. doi: 10.1212/WNL.0000000000002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ben-Ari H, Lifschytz T, Wolf G, Rigbi A, Blumenfeld-Katzir T, Merzel TK, et al. White matter lesions, cerebral inflammation and cognitive function in a mouse model of cerebral hypoperfusion. Brain Res. 2019;1711:193–201. doi: 10.1016/j.brainres.2019.01.017. [DOI] [PubMed] [Google Scholar]
- 9.Iliff JJ, Nedergaard M. Is there a cerebral lymphatic system? Stroke. 2013;44:S93–S95. doi: 10.1161/STROKEAHA.112.678698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid Β. Sci Transl Med 2012, 4: 147ra111. [DOI] [PMC free article] [PubMed]
- 11.Iliff JJ, Wang MH, Zeppenfeld DM, Venkataraman A, Plog BA, Liao YH, et al. Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J Neurosci. 2013;33:18190–18199. doi: 10.1523/JNEUROSCI.1592-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peng WG, Achariyar TM, Li BM, Liao YH, Mestre H, Hitomi E, et al. Suppression of glymphatic fluid transport in a mouse model of Alzheimer's disease. Neurobiol Dis. 2016;93:215–225. doi: 10.1016/j.nbd.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iliff JJ, Chen MJ, Plog BA, Zeppenfeld DM, Soltero M, Yang LJ, et al. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J Neurosci. 2014;34:16180–16193. doi: 10.1523/JNEUROSCI.3020-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaberel T, Gakuba C, Goulay R, Martinez De Lizarrondo S, Hanouz JL, Emery E, et al. Impaired glymphatic perfusion after strokes revealed by contrast-enhanced MRI: A new target for fibrinolysis? Stroke 2014, 45: 3092–3096. [DOI] [PubMed]
- 15.Wang M, Ding F, Deng S, Guo X, Wang W, Iliff JJ, et al. Focal solute trapping and global glymphatic pathway impairment in a murine model of multiple microinfarcts. J Neurosci. 2017;37:2870–2877. doi: 10.1523/JNEUROSCI.2112-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang Q, Zhang L, Ding G, Davoodi-Bojd E, Li Q, Li L, et al. Impairment of the glymphatic system after diabetes. J Cereb Blood Flow Metab. 2017;37:1326–1337. doi: 10.1177/0271678X16654702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mortensen KN, Sanggaard S, Mestre H, Lee H, Kostrikov S, Xavier ALR, et al. Impaired glymphatic transport in spontaneously hypertensive rats. J Neurosci. 2019;39:6365–6377. doi: 10.1523/JNEUROSCI.1974-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu X, Hao J, Yao E, Cao J, Zheng X, Yao D, et al. Polyunsaturated fatty acid supplement alleviates depression-incident cognitive dysfunction by protecting the cerebrovascular and glymphatic systems. Brain Behav Immun. 2020;89:357–370. doi: 10.1016/j.bbi.2020.07.022. [DOI] [PubMed] [Google Scholar]
- 19.Liang S, Lu Y, Li Z, Li S, Chen B, Zhang M, et al. Iron Aggravates the Depressive Phenotype of Stressed Mice by Compromising the Glymphatic System. Neurosci Bull. 2020;36:1542–1546. doi: 10.1007/s12264-020-00539-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mestre H, Tithof J, Du T, Song W, Peng WG, Sweeney AM, et al. Flow of cerebrospinal fluid is driven by arterial pulsations and is reduced in hypertension. Nat Commun. 2018;9:4878. doi: 10.1038/s41467-018-07318-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ogoh S. Relationship between cognitive function and regulation of cerebral blood flow. J Physiol Sci. 2017;67:345–351. doi: 10.1007/s12576-017-0525-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ainslie PN, Brassard P. Why is the neural control of cerebral autoregulation so controversial? F1000Prime Rep 2014, 6: 14. [DOI] [PMC free article] [PubMed]
- 23.Meng LZ, Hou WG, Chui J, Han RQ, Gelb AW. Cardiac output and cerebral blood flow: The integrated regulation of brain perfusion in adult humans. Anesthesiology. 2015;123:1198–1208. doi: 10.1097/ALN.0000000000000872. [DOI] [PubMed] [Google Scholar]
- 24.Ziff OJ, Kotecha D. Digoxin: The good and the bad. Trends in Cardiovascular Medicine. 2016;26:585–595. doi: 10.1016/j.tcm.2016.03.011. [DOI] [PubMed] [Google Scholar]
- 25.Ehle M, Patel C, Giugliano RP. Digoxin: clinical highlights: A review of digoxin and its use in contemporary medicine. Crit Pathw Cardiol. 2011;10:93–98. doi: 10.1097/HPC.0b013e318221e7dd. [DOI] [PubMed] [Google Scholar]
- 26.Shibata M, Ohtani R, Ihara M, Tomimoto H. White matter lesions and glial activation in a novel mouse model of chronic cerebral hypoperfusion. Stroke. 2004;35:2598–2603. doi: 10.1161/01.STR.0000143725.19053.60. [DOI] [PubMed] [Google Scholar]
- 27.Zhang C, Feng W, Zhao YJ, Yu TT, Li PC, Xu TH, et al. A large, switchable optical clearing skull window for cerebrovascular imaging. Theranostics. 2018;8:2696–2708. doi: 10.7150/thno.23686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu S, Li Y, Lu H, Li H, Tong S. Imaging the early cerebral blood flow changes in rat middle cerebral artery occlusion stroke model. Annu Int Conf IEEE Eng Med Biol Soc. 2012;2012:2655–2658. doi: 10.1109/EMBC.2012.6346510. [DOI] [PubMed] [Google Scholar]
- 29.Xavier ALR, Hauglund NL, von Holstein-Rathlou S, Li QL, Sanggaard S, Lou NH, et al. Cannula implantation into the cisterna Magna of rodents. J Vis Exp. 2018;23:57378. doi: 10.3791/57378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Song J, He XZ, Xiong J, Xue R, Ge JH, et al. Quantitative Determination of Glymphatic Flow Using Spectrophotofluorometry. Neurosci Bull. 2020;36:1524–37. doi: 10.1007/s12264-020-00548-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang L, Garrett Injac S, Cui KM, Braun F, Lin Q, Du YC, et al. Systems biology-based drug repositioning identifies digoxin as a potential therapy for groups 3 and 4 medulloblastoma. Sci Transl Med 2018, 10(464). [DOI] [PMC free article] [PubMed]
- 32.Harrison IF, Ismail O, Machhada A, Colgan N, Ohene Y, Nahavandi P, et al. Impaired glymphatic function and clearance of tau in an Alzheimer's disease model. Brain. 2020;143:2576–93. doi: 10.1093/brain/awaa179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeppenfeld DM, Simon M, Haswell JD, D'Abreo D, Murchison C, Quinn JF, et al. Association of perivascular localization of aquaporin-4 with cognition and alzheimer disease in aging brains. JAMA Neurol. 2017;74:91–99. doi: 10.1001/jamaneurol.2016.4370. [DOI] [PubMed] [Google Scholar]
- 34.Toischer K, Hartmann N, Wagner S, Fischer TH, Herting J, Danner BC, et al. Role of late sodium current as a potential arrhythmogenic mechanism in the progression of pressure-induced heart disease. J Mol Cell Cardiol. 2013;61:111–122. doi: 10.1016/j.yjmcc.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang MH, Qin C, Luo X, Wang J, Wang XX, Xie MJ, et al. Astrocytic connexin 43 potentiates myelin injury in ischemic white matter disease. Theranostics. 2019;9:4474–4493. doi: 10.7150/thno.31942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342:373–7. doi: 10.1126/science.1241224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bromley-Brits K, Deng Y, Song WH. Morris water maze test for learning and memory deficits in Alzheimer's disease model mice. J Vis Exp 2011: 2920. [DOI] [PMC free article] [PubMed]
- 38.Leger M, Quiedeville A, Bouet V, Haelewyn B, Boulouard M, Schumann-Bard P, et al. Object recognition test in mice. Nat Protoc. 2013;8:2531–2537. doi: 10.1038/nprot.2013.155. [DOI] [PubMed] [Google Scholar]
- 39.Munk AS, Wang W, Bechet NB, Eltanahy AM, Cheng AX, Sigurdsson B, et al. PDGF-B Is Required for Development of the Glymphatic System. Cell Rep. 2019; 26: 2955-69 e3. [DOI] [PMC free article] [PubMed]
- 40.Vandebroek A, Yasui M. Regulation of AQP4 in the Central Nervous System. Int J Mol Sci. 2020; 21. [DOI] [PMC free article] [PubMed]
- 41.Igarashi H, Tsujita M, Suzuki Y, Kwee IL, Nakada T. Inhibition of aquaporin-4 significantly increases regional cerebral blood flow. Neuroreport. 2013;24:324–8. doi: 10.1097/WNR.0b013e32835fc827. [DOI] [PubMed] [Google Scholar]
- 42.Shi Y, Thrippleton MJ, Makin SD, Marshall I, Geerlings MI, de Craen AJM, et al. Cerebral blood flow in small vessel disease: A systematic review and meta-analysis. J Cereb Blood Flow Metab. 2016;36:1653–1667. doi: 10.1177/0271678X16662891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wong SM, Jansen JFA, Zhang CE, Hoff EI, Staals J, van Oostenbrugge RJ, et al. Blood-brain barrier impairment and hypoperfusion are linked in cerebral small vessel disease. Neurology. 2019;92:e1669–e1677. doi: 10.1212/WNL.0000000000007263. [DOI] [PubMed] [Google Scholar]
- 44.Blair GW, Thrippleton MJ, Shi YL, Hamilton I, Stringer M, Chappell F, et al. Intracranial hemodynamic relationships in patients with cerebral small vessel disease. Neurology. 2020;94:e2258–e2269. doi: 10.1212/WNL.0000000000009483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi Y, Thrippleton MJ, Blair GW, Dickie DA, Marshall I, Hamilton I, et al. Small vessel disease is associated with altered cerebrovascular pulsatility but not resting cerebral blood flow. J Cereb Blood Flow Metab. 2020;40:85–99. doi: 10.1177/0271678X18803956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci. 2011;12:723–738. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goodman JR, Iliff JJ. Vasomotor influences on glymphatic-lymphatic coupling and solute trafficking in the central nervous system. J Cereb Blood Flow Metab. 2020;40:1724–1734. doi: 10.1177/0271678X19874134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petty MA WJ. White matter ischaemia. Brain Res Brain Res Rev. 1999, 31: 58–64. [DOI] [PubMed]
- 49.Insel PS, Hansson O, Mackin RS, Weiner M, Mattsson N, Alzheimer's Disease Neuroimaging Initiative. Amyloid pathology in the progression to mild cognitive impairment. Neurobiol Aging 2018, 64: 76–84. [DOI] [PMC free article] [PubMed]
- 50.Marelli A, Miller SP, Marino BS, Jefferson AL, Newburger JW. Brain in congenital heart disease across the lifespan: The cumulative burden of injury. Circulation. 2016;133:1951–1962. doi: 10.1161/CIRCULATIONAHA.115.019881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lauren M. Hablitz HSV, Qian Sun, Frederik Filip Stæger, Björn Sigurdsson, Kristian N. Mortensen TOL, Maiken Nedergaard. Increased glymphatic influx is correlated with high EEG delta power and low heart rate. Sci. Adv. 2019, 5: eaav5447. [DOI] [PMC free article] [PubMed]
- 52.Hase Y, Horsburgh K, Ihara M, Kalaria RN. White matter degeneration in vascular and other ageing-related dementias. J Neurochem. 2018;144:617–633. doi: 10.1111/jnc.14271. [DOI] [PubMed] [Google Scholar]
- 53.McGuire SA, Ryan MC, Sherman PM, Sladky JH, Rowland LM, Wijtenburg SA, et al. White matter and hypoxic hypobaria in humans. Hum Brain Mapp. 2019;40:3165–3173. doi: 10.1002/hbm.24587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Patel A, Moalem A, Cheng H, Babadjouni RM, Patel K, Hodis DM, et al. Chronic cerebral hypoperfusion induced by bilateral carotid artery stenosis causes selective recognition impairment in adult mice. Neurol Res. 2017;39:910–917. doi: 10.1080/01616412.2017.1355423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huffman JC, Stern TA. Neuropsychiatric consequences of cardiovascular medications. Dialogues Clin Neurosci. 2007;9:29–45. doi: 10.31887/DCNS.2007.9.1/jchuffman. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eisenman DP, McKegney FP. Delirium at therapeutic serum concentrations of digoxin and quinidine. Psychosomatics. 1994;35:91–93. doi: 10.1016/S0033-3182(94)71814-4. [DOI] [PubMed] [Google Scholar]
- 57.Laudisio A, Marzetti E, Pagano F, Cocchi A, Bernabei R, Zuccala G. Digoxin and cognitive performance in patients with heart failure: a cohort, pharmacoepidemiological survey. Drugs Aging. 2009;26:103–112. doi: 10.2165/0002512-200926020-00002. [DOI] [PubMed] [Google Scholar]
- 58.Bannai T, Mano T, Chen X, Ohtomo G, Ohtomo R, Tsuchida T, et al. Chronic cerebral hypoperfusion shifts the equilibrium of amyloid beta oligomers to aggregation-prone species with higher molecular weight. Sci Rep. 2019;9:2827. doi: 10.1038/s41598-019-39494-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mazza M, Marano G, Traversi G, Bria P, Mazza S. Primary cerebral blood flow deficiency and Alzheimer's disease: Shadows and lights. J Alzheimers Dis. 2011;23:375–389. doi: 10.3233/JAD-2010-090700. [DOI] [PubMed] [Google Scholar]
- 60.Egan B, Zierath JR. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013;17:162–184. doi: 10.1016/j.cmet.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 61.He XF, Liu DX, Zhang Q, Liang FY, Dai GY, Zeng JS, et al. Voluntary exercise promotes glymphatic clearance of amyloid beta and reduces the activation of astrocytes and microglia in aged mice. Front Mol Neurosci. 2017;10:144. doi: 10.3389/fnmol.2017.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.von Holstein-Rathlou S, Petersen NC, Nedergaard M. Voluntary running enhances glymphatic influx in awake behaving, young mice. Neurosci Lett. 2018;662:253–258. doi: 10.1016/j.neulet.2017.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liang YJ, Ngandu T, Laatikainen T, Soininen H, Tuomilehto J, Kivipelto M, et al. Cardiovascular health metrics from mid- to late-life and risk of dementia: A population-based cohort study in Finland. PLoS Med. 2020;17:e1003474. doi: 10.1371/journal.pmed.1003474. [DOI] [PMC free article] [PubMed] [Google Scholar]