

Abstract
Macrophages constitute a major component in human hepatocellular carcinoma (HCC) and perform various functions to facilitate disease progression. Reprogramming or reconstituting the tumor surveillance phenotypes of macrophages represents an attractive immunotherapeutic strategy in cancer treatments. The current study identified CD169 as a potential target for macrophage repolarization since it signified a population of macrophages positively correlated with an activated immune signature and better prognosis of patients with HCC. In vitro experiments revealed that a low dose of type I interferon (IFN) could effectively reprogram human monocyte-derived macrophages to upregulate CD169 expression, and such induced CD169+ macrophages exhibited significantly enhanced phagocytotic and CD8+ T cell-activating capacities compared to controls. A low dose of IFNα also inhibited hepatoma growth in mice in vivo, presumably through polarizing the CD169+ macrophage population and enhancing CD8+ T cell activities. Notably, IFNα also induced substantial PD-L1 expression on macrophages in vivo, and thus blockade of PD-L1 could further increase the anti-tumor efficacy of IFNα in the treatment of HCC. We propose a low dose of IFNα in combination with a PD-L1 blocking agent as a potential anti-tumor therapeutic strategy via its effects on macrophage polarization.
Keywords: CD169, Siglec-1, macrophage polarization, hepatocellular carcinoma, interferon, PD-L1, immunotherapy
Graphical abstract
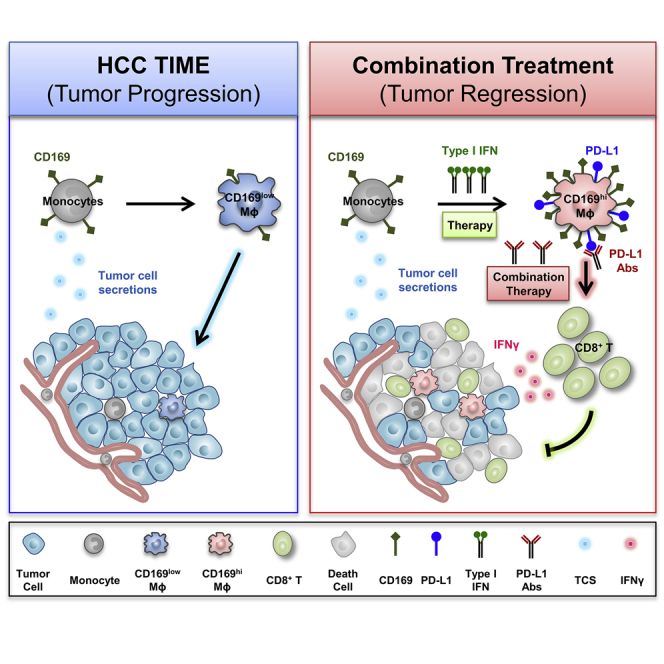
Liao et al. report that a low dosage of type I IFNs could induce anti-tumor CD169+ macrophage polarization both in vitro and in vivo and increase the activation of cytotoxic T cells. Furthermore, blockage of PD-L1 might attenuate the inhibitory effects which induced by IFNα therapy and enhance anti-tumor immunity.
Introduction
Macrophages are highly plastic and diverse immune populations that play important roles in almost every aspect of tumor progression.1, 2, 3 From the very first immune surveillance role to the findings that macrophages in most solid tumors function as enablers rather than gate keepers, from the highly generalized M2 definition to the acknowledgment that tumor-infiltrating macrophages comprise different subsets with regard to their distinct distribution, phenotypes, and functions, and from the monotonous pro-tumor effects to the discovery that at least some macrophages with anti-tumor capacity still exist within tumor microenvironments, our knowledge about tumor-associated macrophages (TAMs) have evolved through time, as have the relentless efforts of trying to target these cells.
Targeting TAMs has proven to be an attractive therapeutic strategy, but also an uphill battle due to several obstacles.4, 5, 6 Substantial tissue-infiltrating macrophages are derived from monocytes recruited from peripheral blood, and thus organ-selective and sustained depletion of this population is difficult to achieve, let alone risking losing the already scarce subsets with anti-tumor capacities. For efforts aiming to reprogram the pro-tumor macrophages, there are just too many subsets, with not enough applicable, tumor-specific markers to differentiate them from their normal tissue-resident counterparts, risking systemic side effects. Given the relatively small amounts of infiltrating macrophages with anti-tumor potentials, it is tempting to hypothesize that by somehow enlarging these not so diverse subsets, they can ultimately outweigh the pro-tumor wing and “reconstitute” or tilt the balance toward a tumor-hostile immune environment.
CD169 (Siglec-1/sialoadhesin) belongs to the sialic acid-binding immunoglobulin (Ig)-type lectins (Siglecs) family and is predominantly expressed on sinusoidal macrophages and activated monocytes that capture antigens and evoke adaptive immunity.7,8 Our previous study suggested a positive role played by CD169+ macrophages in prohibiting hepatocellular carcinoma (HCC) progression.9 Based on that, the current results further reveal a positive association between CD169+ macrophage infiltration and inflammatory immune signatures within human HCC tumor tissues. Type I interferon (IFN) was identified as an effective stimulator to polarize the development of CD169+ macrophages with T cell-activating and tumor-inhibiting potentials both in vitro and in vivo, and PD-L1 blocking antibody could further enhance the anti-tumor effects exhibited by a low dose of IFNα.
Results
The level of CD169+ macrophages is downregulated in HCC tumors and negatively correlated with disease progression
We performed gene-level analysis of SIGLEC1 (CD169 gene) in 50 samples from The Cancer Genome Atlas liver hepatocellular carcinoma (TCGA-LIHC) dataset10 and found that SIGLEC1 was significantly decreased in tumor tissue (1.102 ± 0.139 FPKM [fragments per kilobase of transcript per million mapped reads]) compared with paired non-tumor liver tissues (2.283 ± 0.222 FPKM) (Figure S1A). The downregulation of SIGLEC1 in tumor tissues was confirmed in an independent external cohort (GEO: GSE14520) of patients with pathologically confirmed HCC (Figure S1B, n = 234).
To validate the expression of CD169 in macrophages in tumors in situ, we double stained sections of HCC samples with anti-CD68 (maker for macrophages) and anti-CD169 antibody. As shown in Figures 1A–1C, while CD169 signals were selectively expressed by CD68+ cells in both tissue regions, they were significantly lower in tumor than in adjacent normal liver tissues (n = 8). Consistently, by analyzing tissue microarray (TMA) data of a total of 488 paired samples (488 HCC and 488 adjacent normal liver) stained with anti-CD169 antibody, we found that levels of CD169+ cells were significantly downregulated in tumor (188.8 ± 10.11 cells/mm2) compared with paired non-tumor liver tissues (730.2 ± 13.43 cells/mm2) (Figures 1D and S2). These patients were then divided into two groups according to their median value of CD169 expression in tumor or non-tumor tissues. As shown in Figures 1E and 1F, patients with high CD169+ cell density in tumor tissues exhibited better overall survival (OS) and lower recurrence than did those with low CD169+ cell density, while no correlation was found between the density of CD169+ cells in non-tumor liver tissues and patient OS or recurrence (Figure S3). Univariate and multivariate regression analyses demonstrated that the level of CD169+ macrophage infiltration in tumor tissue was an independent prognostic factor for HCC progression (Table S2).
Figure 1.

Level of CD169+ macrophages is downregulated in HCC tumors and negatively correlated with disease progression
(A) Immunofluorescence staining shows DAPI (blue), CD68 (green), and CD169 (red) expression and localization in liver hepatocellular carcinoma (HCC) tissue. Scale bars, 50 μm. (B and C) The density of CD169+ cells (B) and the ratio of CD169+/CD68+ cells (C) in paired non-tumor (N) and tumor (T) HCC tissues; n = 8 (8 HCC and 8 adjacent normal liver tissues). (D) IHC staining shows CD169+ cell densities of 488 paired non-tumor and tumor regions in HCC tissue specimen. (E and F) 488 HCC patients were divided into two groups according to the median of the CD169+ cell densities in tumor regions. The cumulative overall survival (E) and recurrence (F) were calculated using the Kaplan-Meier method. Wilcoxon matched-pairs signed rank test (B–D) and log-rank test (E and F): ∗∗p < 0.01, ∗∗∗∗p < 0.0001.
CD169 indicates an inflammatory immune signature in HCC tumors
Using Gene Ontology (GO) data, we identified 935 genes correlated with CD169 expression in HCC tissues (n = 365, R ≥ 0.4, p < 0.05) (Figure 2A). Interestingly, among the top 30 enrichment GO terms, the signatures of antigen-presenting cell (APC), T cell, and B cell activation were enriched intensively, suggesting that CD169 might be a beneficial marker indicating inflammatory immune response within HCC tumors (Figure 2A). Supporting this finding, gene set enrichment analysis (GSEA) showed that genes indicating an inflammatory response signature (HALLMARK_INFLAMMATORY_RESPONSE) were dominantly enriched in SIGLEC1high HCC tumors (Figure 2B). We further performed a genome-wide data analysis using data from the Gene Expression Omnibus database. As shown in Figure 2C and Table S3, SIGLEC1high tumors exhibited a positive correlation with an inflamed cancer gene profile indicating T cell accumulation11 and a negative correlation with gene profiles indicating tumor immune dysfunction and exclusion (TIDE),12 T cell exhaustion,13,14 regulatory T cell (Treg) accumulation,15 immune checkpoint blockade (ICB) resistance,16 myeloid-derived suppressor cell (MDSC) accumulation,17 M2 phenotypes of TAMs,18 and cancer-associated fibroblast (CAF) accumulation.19
Figure 2.

CD169 indicates an inflammatory immune signature in HCC tumors
(A) Top 30 biological process (GO terms) enriched in 935 genes correlated with SIGLEC1 expression in HCC samples from TCGA dataset (Spearman’s r > 0.4, p < 0.05). (B and C) GSEA of inflammatory response signature (B) and inflamed-cancer signature (C) in 183 SIGLEC1hi and 182 SIGLEC1low counterparts from TCGA dataset. The enrichment scores, nominal (NOM) p values and false discovery rate (FDR) q values were calculated by GSEA with weighted enrichment statistics and the ratio of classes for the metric as input parameters. (D and E) Correlations between SIGLEC1 and CD8A gene were calculated in 365 patients of HCC samples from TCGA dataset (D) and the GEO: GSE14520 dataset (E). (F) Correlations between CD169+ cell and CD8+ cell density were calculated in 488 HCC tumor tissue specimens. (G and H) 488 patients were divided into four groups according to the median of the CD169+ cell and CD8+ cell density in tumor regions. The cumulative overall survival and recurrence were calculated using the Kaplan-Meier method and then analyzed with a log-rank test. p values and r values were calculated based on the analysis of Pearson’s correlation (D–F).
Given that CD169 mainly functions as a co-stimulator to elicit T cell responses, we analyzed correlations between levels of CD169 expression and those of CD8+ T cells in HCC. Using TCGA-LIHC and GEO: GSE14520 RNA sequencing (RNA-seq) data, we found that RNA levels of SIGLEC1 were positively correlated with those of CD8A (CD8 gene) in HCC tumor tissues (Figures 2D and 2E), and such correlation was confirmed at the protein level by analyzing TMA data of 488 paired samples (488 HCC and 488 adjacent normal liver) stained with anti-CD169 and anti-CD8 antibodies (Figure 2F). Furthermore, when HCC patients were divided into four groups according to their levels of CD169+ and CD8+ cell infiltration in tumor tissues, patients in the CD8hiCD169hi group (5-year OS rate, 72.23%; recurrence rate, 54.29%) exhibited the best prognosis when compared to those in the CD8lowCD169low group (5-year OS rate, 41.97%; recurrence rate, 69.29%), CD8lowCD169hi group (5-year OS rate, 62.53%; recurrence rate, 68.43%), and CD8hiCD169low group (5-year OS rate, 51.85%; recurrence rate, 72.51%) (Figures 2G and 2H).
We then tried to evaluate mechanisms underlying the downregulation of CD169 on TAMs of HCC. To that end, CD14+ monocytes were purified from the peripheral blood of healthy donors and treated with SK hepatoma tumor cell supernatants (TCSs). As shown in Figure 3, levels of CD169 expression were significantly downregulated on TCS-treated monocytes compared to control.
Figure 3.

Hepatoma cell supernatants induce CD169 downregulation on macrophages
(A and B) Monocyte enrichment by using CD14+ magnetic beads and cultured with medium or SK TCS for 0–3 days. The expression of HLA-DR and CD169 (A) and the ratio of CD169+ cells in CD14+ cells (B) were determined by flow cytometry; n = 6 or 7. ∗p < 0.05, ∗∗p < 0.01; multiple t test (B).
The above results suggested that CD169 represented a strong marker to predict an inflammatory microenvironment of human HCC and that tumor-derived soluble factors could reduce its expression in tumor tissues.
Type I IFN could induce CD169 upregulation on tumor-exposed macrophages
Given the repressive role of CD169 in HCC disease progression, repolarizing a CD169+ macrophage population might represent a possible anti-tumor therapeutic strategy. Interestingly, in in vitro experiments, when total peripheral blood mononuclear cells (PBMCs), instead of purified CD14+ monocytes, were subjected to the treatment of QGY, HepG2, or SK TCS, monocytes did not reduce their CD169 levels. Instead, these cells upregulated the expression of CD169 compared to their counterparts treated with medium, THP-1, or normal liver cell line 7702-derived supernatants (Figures 4A and 4B). The upregulation of CD169 expression on monocytes was time-dependent when total PBMCs were treated with SK TCS (Figure 4C). These results indicated that when enough CD14− leukocytes were present in the culture system, they might antagonize the effects of TCS and induce the expression of CD169 on monocytes.
Figure 4.

Type I IFN could induce CD169 upregulation on tumor-exposed macrophages
(A and B) PBMCs purified by the adherent treatment method were cultured with the indicated TCS for 2 days. Flow cytometry patterns and gate frequencies in percentages (A) and statistical results (B) are shown; n = 16. (C) PBMCs enriched by using the adherent treatment method were cultured with medium or SK TCS for 0–3 days, after which the ratio of CD169+ cells in CD14+ cells was determined by flow cytometry; n = 6 or 7. (D) Supernatants from PBMCs pretreated with medium or SK TCS for 48 h were collected (designated as PSN and SK-PSN, respectively). Thereafter, CD14+ monocytes were isolated with magnetic bead sorting and cultured with medium, SK TCS, PSN, or SK-PSN for 48 h; n = 4 or 5. (E) CD14+ monocytes were exposure to different fluid components for 2 days, after which the CD169+ cell ratio was determined by flow cytometry; n = 6. The exosome and supernatant components of SK-PSN were separated by ultracentrifugation; the proteins, lipids, and small molecules in SK-PSN were removed by high-temperature treatment (100°C, 30 min), fumed silica, and DCC, respectively. (F) CD14+ monocytes were cultured with medium, SK TCS, or 10 pg/mL IFNα2/IFNβ/IFNγ, respectively. Two days later, the ratio of CD169+ cells in CD14+ cells was determined by flow cytometry; n = 5. (G) CD14+ monocytes were cultured in the presence or absence of 5 μg/mL anti-IFNAR2 or 1 μg/mL anti-IFNγ in various conditions. Two days later, the ratio of CD169+ cells in CD14+ cells was determined by flow cytometry; n = 4. Data represent mean ± SEM. ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; NS, not significant by multiple t test (C) and an unpaired Student’s t test (B and E–G).
To examine mechanisms underlying the upregulation of CD169 expression on TCS-exposed monocytes, we collected supernatants from PBMCs pretreated with medium or SK TCS for 48 h (designated as PSN and SK-PSN, respectively; Figure S4) and treated purified CD14+ monocytes with medium, SK TCS, PSN, or SK-PSN for 48 h. As shown in Figure 4D, while SK TCS reduced CD169 expression on monocytes compared to control, SK-PSN could counter its effects and markedly upregulate levels of CD169 expression on these cells in a dose-dependent manner. In contrast, PSN exhibited no significant effect on monocyte expression of CD169. While depletion of lipid with fumed silica or small molecules with dextran-coated charcoal (DCC) in SK-PSN did not impact its effects on inducing CD169 expression by monocytes, denaturation of protein components by heating SK-PSN could significantly attenuate its CD169-inducing effects (Figure 4E). Furthermore, exosomes derived from SK-PSN could not mimic effects of their source on CD169 expression, indicating that some soluble protein components might be responsible for the induction of CD169 on monocytes (Figure 4E).
To figure out the origins of these active protein components, CD4+ T cells (CD14−CD19−CD4+CD8−), CD8+ T cells (CD14−CD19−CD4−CD8+), and non-T cells (CD14−CD19−CD4−CD8−) were sorted out from healthy PBMCs and cocultured with CD14+ monocytes at various ratios under the exposure to SK TCS. As shown in Figure S5A, all three groups of these cells could mimic effects of SK-PSN to upregulate CD169 expression on monocytes. In addition, supernatants from PBMCs or CD3+ T cells activated by anti-CD3/CD28 antibodies could also mimic effects of SK-PSN in inducing monocyte expression of CD169 (Figures S5B and S5C).
Given that activated leukocytes could produce large amounts of IFN, and GSEA data analysis showed enrichments of both IFNα and IFNγ signatures in CD169hi HCC patients (Figure S6), we hypothesized that IFNs may be involved in mediating the CD169-inducing effects of SK-PSN. As shown in Figure 4F, while IFNγ showed no significant effects on CD169 expression, a low dose of IFNα2 (10 pg/mL) and IFNβ (10 pg/mL) could both induce significant expression of CD169 on tumor-exposed purified monocytes compared to controls. Accordingly, type I IFN receptor neutralization antibody could attenuate the CD169-inducing effects of SK PSN on monocytes, whereas type II IFN neutralization antibody exhibited no such effects compared to control groups (Figure 4G). These results indicated that a low dose of type I IFN could counter the effects of tumor supernatants and polarize a CD169+ macrophage population.
IFNα-induced CD169+ macrophages enhanced phagocytotic and autologous T cell-activating abilities in vitro
To examine the possible functions of type I IFN-induced CD169+ macrophages, we evaluated the phagocytotic and T cell-activating capacities of these cells. As shown in Figures 5A and 5D, CD14+-purified monocytes were treated with or without SK-PSN (designated as CD169low and CD169hi groups, respectively) for 48 h. CD169hi monocytes showed significantly enhanced phagocytotic capacity to both mitomycin C (MMC)-induced apoptotic SK hepatoma cells and fluorescein isothiocyanate-labeled dextran (dextran-FITC) molecules compared to the CD169low group. We then treated CD14+ monocytes with or without IFNα in the presence of SK TCS for 2 days, and then cocultured these cells with autologous CD3+ T cells for another 5 days. As shown in Figure 5E, levels of IFNγ production were significantly increased in the IFNα-treated group compared to controls, and levels of CD8+ T cell proliferation were also markedly upregulated after coculturing with IFNα-polarized macrophages in comparison to those of CD8+ T cells cocultured with control macrophages (Figures 5F and 5H). These data implicated that IFNα could polarize a CD169+ macrophage population with enhanced phagocytotic and autologous T cell-activating abilities in vitro.
Figure 5.

IFNα-induced CD169+ macrophages enhanced phagocytotic and autologous T cell-activating abilities in vitro
(A–D) CD14+ monocytes were precultured with PSN or SK-PSN in the presence of SK TCS for 2 days, and CD169low or CD169hi monocytes were collected for further phagocytosis assays. (A) Monocytes (5 × 105 cells) were cocultured with mitomycin C-treated and CFSE-labeled SK tumor cells in the indicated ratios for 20 h. Phagocytosis was measured by flow cytometry, and the percentage of CFSE+ monocytes is shown; n = 5. (B–D) Monocytes (5 × 105 cells) were incubated with 1 mg/mL dextran-FITC molecules for 60 min at 0°C (to measure non-specific adherence) or at 37°C (to measure phagocytosis), and the ratio of FITC+ cells and the mean fluorescence intensity (MFI) of FITC on CD14+ cells were measured by flow cytometry; n = 5. (E–H) CD14+ monocytes were incubated with or without 10 pg/mL IFNα2 in the presence of SK TCS. Two days later, pretreated monocytes were cocultured with CFSE-labeled autologous CD3+ T cells for 5 days (monocyte/T cell ratio of 1:2); n = 6. (E) IFNγ in the supernatant was detected by ELISA. (F–H) CD8+ T cell proliferation was detected by flow cytometry. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; paired Student’s t test (A, C–E, G, and H).
IFNα induces CD169 upregulation on macrophages and inhibits tumor progression in vivo
We went on to evaluate the possible anti-tumor effects of type I IFN-induced CD169+ macrophages in mice in vivo. A hepatoma-bearing mouse model was established by subcutaneously (s.c.) inoculating mouse Hepa1-6 cells into the right flanks of immunocompetent C57BL/6 mice for 8 days. Thereafter, 10 μg of IFNα-IgG or PBS (no treatment control [NTC]) was administered intravenously (i.v.), and the immune status of the tumor microenvironment was analyzed by flow cytometry after another 3 days. t-Distributed stochastic neighbor embedding (tSNE) analysis on a panel including 11 phenotypic markers (FSC, SSC, CD45, 7-aminoactinomycin D [7-AAD], CD11b, Ly6G, Ly6C, major histocompatibility complex class II [MHC class II], F4/80, PD-L1, and CD169) was performed, and the distribution of each cluster was color-coded and shown in Figures 6A and S7. According to the expression of MHC class II, PD-L1, and CD169, the F4/80hiLy6Cint cell population could be further divided into two clusters, that is, an activated cluster (F4/80hiLy6Cint-act) and a non-activated cluster (F4/80hiLy6Cint-non-act) (Figure S8). Our analysis found that the levels of CD3+, CD4+, CD8+ T cells and CD11b+ Ly6G+ neutrophil/MDSC infiltration in tumor tissues were not impacted by the treatment of IFNα-IgG (Figure 6B). While the ratio of F4/80hiLy6Cint-act cells in total CD45+ cells did not change after the treatment of IFNα-IgG, the ratio of F4/80hiLy6Cint-non-act cells was significantly decreased, and the ratios of F4/80intLy6Chi and F4/80lowLy6Clow populations were increased in tumor tissues after the exposure to IFNα-IgG (Figure 6C). While CD169 was predominantly expressed by the F4/80hiLy6Cint-act cluster, its expression was significantly upregulated after the treatment of IFNα-IgG (Figures 6D and 6E). The increased tumor infiltration of CD169+ TAMs after the IFNα-IgG treatment was confirmed through immunohistochemistry (IHC) staining (Figure S9). Notably, IFNα-IgG could markedly reduce tumor volumes as well as tumor weights in mice in vivo (Figures 6F and 6G).
Figure 6.

IFNα induces CD169 upregulation on macrophages and inhibits tumor progression in vivo
(A–G) WT C57BL/6 mice were injected subcutaneously with 1 × 106 Hepa1-6 cells. Then, 10 μg of IFNα-IgG or PBS (no treatment control [NTC]) was administered intravenously on day 8, and tumor tissues were collected on day 11; n = 10. (A–E) Immunocytes were isolated from freshly dissociated tumor specimens, and they were analyzed by multiparametric flow cytometry analysis. Represented FACS data were analyzed using the tSNE algorithm (A), and statistical analysis was conducted as shown in (B)–(E). (F) Tumor volume. (G) Tumor weight. Data represent mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001; NS, not significant; multiple t test (B–E) and unpaired Student’s t test (F and G).
Blockage of PD-L1 augments the anti-tumor efficacy of IFNα in vivo
Interestingly, while IFNα did not increase PD-L1—it actually slightly decreased PD-L1—expression on macrophages in in vitro experiments, it did significantly upregulate levels of PD-L1 expression on different macrophage populations in mice in vivo (Figures 7A–7C). The upregulated PD-L1 might indicated a negative feedback response after the activation of immune components in tumor microenvironments and suggested a possible combination therapeutic effects of IFNα and PD-L1 blocking agents. To confirm this hypothesis, we treated mice with established Hepa1-6 tumors with PBS (NTC), IFNα-IgG, anti-PD-L1 blocking antibody, or IFNα-IgG in combination with anti-PD-L1 blocking antibody. As shown in Figures 7D–7G, a combination of IFNα-IgG and anti-PD-L1 blocking antibody induced significantly more infiltration of IFNγ+ CD8+ T cells in tumor tissues and it also exhibited much more effective function in terms of inhibiting tumor growth in vivo compared to either treatment alone.
Figure 7.

Blockage of PD-L1 augments the anti-tumor efficacy of IFNα in vivo
(A and B) CD14+ monocytes were cultured with medium or 10 pg/mL IFNα2, SK TCS, or SK TCS plus 10 pg/mL IFNα2, respectively. Two days later, the ratio of CD169+ cells in CD14+ cells (A) and the expression of PD-L1 on CD14+ cells (B) were determined by flow cytometry; n = 11. (C) Wild-type (WT) C57BL/6 mice were injected subcutaneously with 1 × 106 Hepa1-6 cells. Then, 10 μg of IFNα-IgG or PBS (NTC) was administered intravenously on day 8, and tumoral tissues were collected on day 11; n = 10. Multiparametric flow cytometry analysis was used on of live immunocytes that were isolated from freshly dissociated tumor specimens, and PD-L1 expression levels on various myeloid cells were analyzed. (D–G) WT C57BL/6 mice were injected subcutaneously with 1 × 106 Hepa1-6 cells. Then, 10 μg of IFNα-IgG was administered intravenously on days 10, 12 and 14. Meanwhile, 100 μg of anti-PD-L1 was administered intraperitoneally on days 10 and 14. Spleen tissues were collected on day 21; n = 9 or 10. (D and E) IFNγ-expressing CD8+ T cells were detected from tumor-bearing mice spleens. (F) Tumor size (in cm). (G) Tumor growth curve. Data represent mean ± SEM of two independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; NS, not significant; paired Student’s t test (A and B), multiple t test (C), unpaired Student’s t test (E), and two-way ANOVA (G).
Discussion
Discoveries of immune checkpoint molecules (PD-1/PD-L1 and CTLA4) and the success of their targeting in the treatment of various cancers have invigorated studies in the field of T cell (re)activation. Yet the limited efficacy of current immune checkpoint blockade therapy warrants the search for new immune-based anti-tumor strategies.20,21 In the present study, we revealed that a low dose of IFNα in combination with PD-L1 blocking agent could polarize a CD169+ macrophage population with anti-tumor capacity, thus representing a potential novel therapeutic strategy for the treatment of HCC.
Levels of CD169 expression were very high on macrophages infiltrating the non-tumor liver tissues of HCC patients compared to those derived from PBMCs. Reasons for such particular high expression were not clear, but might possibly be attributed to the organ characteristics22 and virus-related/unrelated basal inflammatory conditions23,24—which might include low levels of IFNα—within non-tumor liver tissue. The fact that CD169 mainly functions as a costimulatory signal9,25 and would not activate T cells without a first antigen-presenting signal might explain why its high expression on non-tumor macrophages did not pose a threat to normal liver tissues, and it also provides a rationale for the design of polarizing a CD169+ tumor-infiltrating macrophage population as a therapeutic strategy since this strategy might exhibit very few, if any, side effects given the already high levels of CD169 expression on non-tumor macrophages.
Compared to those in paired non-tumor liver tissues, levels of CD169 expression were significantly lower on macrophages in tumor tissues of HCC. With hepatoma supernatants only partially reduced CD169 expression on PBMC-derived monocytes/macrophages, other regulating mechanisms might exist that possibly include some antagonizing signals derived from stroma cells, or not enough supporting signals, such as IFNα, derived from activated lymphocytes within tumor microenvironments. Definite mechanisms underlining the low levels of CD169 expression on TAMs require further exploration and validation.
The conclusion that type I IFN could polarize a CD169+ macrophage population with anti-tumor potentials was reached through three sets of experiments. First, a low dose of type I IFN could induce the upregulation of CD169 expression on monocyte-derived normal/tumor-supernatant-exposed macrophages, while type I IFN blocking antibody could significantly antagonize activated lymphocyte/supernatant-induced CD169 expression on macrophages. Second, IFNα-induced CD169+ macrophages exhibited significantly enhanced phagocytotic and CD8+ T cell-activating capacities in comparison to control groups in vitro. Third, a low dose of IFNα could increase levels of CD169+ macrophage infiltration in tumor tissues, stimulate IFNγ production in CD8+ T cells, and inhibit hepatoma growth in mice in vivo. We hypothesized that the observed functions of IFNα might rest on its effects in switching the balance between pro-tumor and anti-tumor macrophage subpopulations within tumor microenvironments, without effectively impacting the pro-tumor subsets essentially.
Different from immune checkpoint blockage strategies aiming to release effective T cells from the inhibitory grip of PD-1/PD-L1 or CTLA4,26, 27, 28, 29 IFNα tends to activate/restimulate CD8+ T cells indirectly by polarizing CD169+ macrophages within tumors. Interestingly, while IFNα induced mainly activated phenotypes in macrophages in vitro, it could induce substantial levels of PD-L1 expression on macrophages in mice in vivo, which might have resulted from the activated T cell-derived feedback signals such as IFNγ within in vivo tumor microenvironments. Accordingly, PD-L1 blocking antibody could significantly enhance the anti-tumor efficacy of IFNα, presumably through the synergistic or compensatory effects between these two agents on macrophages. It is therefore tempting to hypothesize that the proposed therapeutic strategy of IFNα in combination with PD-L1 blocking antibody would have reverberated effects beyond the induced CD169+ macrophages since it might also impact other PD-L1-positive resident macrophage populations, thus further shifting the balance of macrophage subsets toward an anti-tumor-dominant feature within tumor microenvironments. Moreover, given that activated lymphocytes could produce significant amounts of IFNα, a therapeutic strategy aiming to increase the permeabilization and accumulation of activated T cells in tumor tissues might also inhibit disease progression partially via polarizing the CD169+ macrophage population within tumor microenvironments.
The present study did not provide direct evidence that IFNα-polarized CD169+ macrophages enhanced their phagocytotic and T cell-activating capacity via the CD169 molecule itself in vitro, nor did it prove that IFNα inhibits tumor progression via CD169 or activated CD8+ T cells in vivo, although a low dose of IFNα did not impact the survival of cancer cells themselves in in vitro cultures. Meanwhile, IFNα treatment induced a decrease of non-activated macrophages, while it increased the percentages of F4/80intLy6Chi and F4/80lowLy6Clow populations in CD11b+Ly6G− cells. Population conversions might account for such ratio shifts. However, this hypothesis requires further evidence using methods such as single-cell sequencing to map the fates of each cell population. Nevertheless, given its potential in activating macrophages and thereafter CD8+ T cells and rebalancing the pro-tumor versus anti-tumor components within tissue microenvironments, the combination of IFNα and PD-L1 blocking agent might represent an applicable therapeutic strategy in the treatment of HCC.
Materials and methods
Patients and specimens
TCGA clinical data were downloaded from the data portal of Genomic Data Commons (GDC; https://portal.gdc.cancer.gov/), and 365 HCC samples with available prognostic information and whole mRNA expression data, of which 50 have paired HCC and adjacent normal liver information, were enrolled in this study.10 In addition, the GEO: GSE14520 raw data are available at the NCBI’s GEO database, and 234 HCC samples were enrolled in this study. Patients were divided into two groups according to the median value.
A total of 488 patients with pathologically confirmed HCC who underwent surgery at the Sun Yat-sen University Cancer Center (SYSUCC) from January 5, 2006 to September 17, 2010 were included in this study. This study was approved by the Ethics Committee of SYSUCC (GZR2019-081). The patients provided informed consent for participation in the present study. The patients did not receive any immunotherapy or neoadjuvant therapy before the operation. The clinical data of the patients were extracted from their electronic medical records and are listed in Table S1.
The clinical stage of tumors was determined according to the tumor-node-metastasis (TNM) classification system of the American Joint Committee on Cancer (AJCC; 2018-01-01, 8th edition). The follow-up period ended in July 14, 2014, and the median survival time was 63 months (range, 1–121 months). Recurrence was diagnosed pathologically via surgical biopsy and/or radiologically via computed tomography or positron emission tomography. OS was defined as the length of time between surgery and death or the last follow-up examination.
TMA construction and IHC
Formalin-fixed, paraffin-embedded (FFPE) tissue specimens were used to construct TMAs as previously described.30 Briefly, hematoxylin and eosin (H&E)-stained slides were reviewed by pathologists who were blinded to patients’ clinical characteristics and outcomes. TMAs were constructed using two separate 1.0-mm tissue cores taken from both regions of adjacent non-tumor liver (≥2 cm from the edge of the tumor) and tumor (a total of four punches for each specimen). TMAs containing the tissue cores were then cut into 5-μm sections for IHC staining.
The slides were incubated with a rabbit monoclonal primary antibody against CD169 (AF5197, 1:200, R&D Systems, USA) or CD8 (ZA-0508, 1:200, Zsbio Commerce Store, China) or a rat monoclonal primary antibody against mouse CD169 (3D6.112, 1:500, BioLegend, USA) at 4°C overnight. Immunostaining was performed using horseradish peroxidase (HRP)-conjugated anti-rabbit and anti-mouse antibodies from Dako EnVision systems (DakoCytomation, Glostrup, Denmark) or an anti-rat antibodies kit (PV-9004, 1:200, Zsbio Commerce Store, China) and was developed with peroxidase and 3,3′-diaminobenzidine tetrahydrochloride. All sections were counterstained with Mayer’s hematoxylin and mounted in non-aqueous mounting medium.
IHC image analysis was performed as previously described.3 TMA slides were scanned at ×20 magnification and evaluated by computerized image analysis system built with a Vectra 2.0 automated quantitative pathology imaging system and InForm Cell Analysis 2.2 (PerkinElmer). Images were processed using Nuance image analysis software (PerkinElmer) for unmixing of diaminobenzidine (DAB) and hematoxylin signals and to establish a spectral library. To segment different tissue regions, a pathologist manually drew the training regions of tumor and non-tumor areas to train the machine-learning tissue segmentation, which was later applied to all tissue images. Following tissue segmentation, images were subject to color deconvolution using the established spectral library, and cell numbers were estimated with the counting object module of InForm Cell Analysis 2.2 (PerkinElmer). The results of the automated measurements were verified and compared with the counting results obtained by a pathologist.
Immunofluorescence staining
Multiplex immunofluorescence staining was obtained using a tyramide signal amplification (TSA) fluorescence kit (Panovue, Beijing, China). Briefly, re-hydrated FFPE sections were incubated at 4°C overnight with mouse anti-human CD68 (M0876, Dako, Carpinteria, CA USA) or sheep anti-human CD169 antibodies (AF5197, R&D Systems, Minneapolis, MN, USA), followed by HRP-conjugated secondary antibody incubation and TSA. The slides were microwave heat treated after each TSA operation. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) in the TSA fluorescence kit after all of the antigens had been labeled.
To obtain multispectral images, the stained slides were scanned using a Zeiss LSM880 microscope (Zeiss, Germany) and analyzed using ZEN 2011 software (Zeiss, Germany). Two independent observers blinded to the outcome counted and analyzed single- or double-positive cells in each of five representative fields at ×400 magnification (0.125 mm2 per field).
Cell culture and TCS preparation
All cells, including the mouse hepatoma cell line Hepa1-6, the human monocytic cell line THP-1, the normal (HL-7702) cell line, and tumor (SK, QGY and HepG2) human hepatocytes, were obtained from the American Type Culture Collection (ATCC). All cells were tested for mycoplasma contamination using the single-step polymerase chain reaction (PCR) method and cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin (Sigma-Aldrich), and 100 μg/mL streptomycin (Sigma-Aldrich) in a humidified 5% CO2 incubator at 37°C. TCSs were prepared by plating 5 × 106 tumor cells in 10 mL of complete medium in 10-cm dishes for 24 h, after which the medium was changed to DMEM, which contained 10% human AB serum. The cells were cultured for 48 h in this medium, after which supernatants were collected, centrifuged, and stored in aliquots at −80°C.
Isolation and culture of monocytes
Blood samples were obtained from healthy donors attending the Guangzhou Blood Center. PBMCs were isolated from buffy coats derived from the blood of healthy donors by Ficoll density gradient as described previously.31
For PBMC isolation by the adherent treatment method, cells in DMEM alone were plated at 5 × 106/well in 24-well plates for 1.5 h, washed, and then cultured in DMEM containing 10% human AB serum for 16 h to remove residual lymphocytes. These cells (in addition to CD14+ monocytes) included approximately 30% other leukocytes (e.g., T cells, B cells) and were cultured with medium or 15% SK TCS for 48 h, and supernatants were collected as PBMC condition supernatants (PSN) or SK TCS-treated PBMC condition supernatants (SK-PSN).
For the CD14+ monocyte purification by magnetic beads method, PBMCs were incubated with CD14 magnetic beads (130-050-201, Miltenyi Biotec, Germany) at 4°C for 20 min. After washing with bead buffer, the cell suspension of labeled cells was loaded onto a MACS column, which was placed in the magnetic field pf MACS separator. The magnetically labeled CD14+ monocytes were retained within the column and were washed three times to remove the unlabeled cells. After removing the column from the separator, labeled CD14+ monocytes were flushed out and resuspended with DMEM, and CD14+ cell purity was greater than 95%. The cells in DMEM alone were plated at 5 × 105/well in 24-well plates for 20–30 min and then cultured in DMEM containing 10% human AB serum for 8 h to remove residual nonadherent cells. Thereafter, the monocytes in DMEM containing AB serum were cultured in the presence of SK TCS (15%), PSN (50%), SK-PSN (50%), or medium alone for 2 days.
Depletion of proteins, lipids, or small molecules from the SK-PSN
For protein removal, the SK-PSN was incubated at 100°C for 30 min to inactivate the protein and then dropped into an ice bath immediately to cool down. Precipitates were removed by centrifugation at 2,000 × g for 15 min. For lipid removal, the SK-PSN was treated with fumed silica. Briefly, fumed silica and SK-PSN were mixed (20 mg/mL) and incubated at 37°C on a shaker for 2 h. The mixture was centrifuged at 2,000 × g for 15 min, and the precipitates were removed. For small molecule removal, the SK-PSN was treated with DCC. In short, DCC was added into SK-PSN to a final concentration of 0.05 mg/mL (weight per volume [w/v]) and incubated at 4°C overnight. The DCC was removed by centrifugation at 2,000 × g for 15 min at 4°C, and filtration of the supernatants was accomplished with a 0.22-μm filter. All supernatants were collected for further experiments.
Flow cytometry
Human leukocytes were stained with the following reagents to identify the expression of surface markers: PD-L1-phycoerythrin (PE) and CD8-Alexa Fluor (AF)750 (eBioscience, San Diego, CA, USA); CD4-Brilliant Violet (BV)421 (BioLegend, San Diego, CA, USA); CD14-AF700, histocompatibility leukocyte antigen (HLA)-DR-BV421, PD-L1-PE, and 7-AAD (BD Pharmingen, San Diego, CA, USA); and CD8-PE (Beckman Coulter, Fullerton, CA, USA).
Mouse leukocytes were stained with the following reagents to identify the expression of surface markers: CD45-BV570, CD11b-AF700, CD3-PE, CD4-PC7, PD-1-allophycocyanin (APC), Ly6C-BV421, F4/80-APC, CD274-BV605, and CD45-AF700 (BioLegend, San Diego, CA USA); CD169-PE and CD8α-eFluor 450 (eBioscience, San Diego, CA, USA); CD39-peridinin-chlorophyll-protein (PerCP)-eFluor 710, MHC class II-FITC, and a Live/Dead fixable blue dead cell stain kit (Invitrogen, Carlsbad, CA, USA); and Ly6G-PE-CF594, CD11b-PC7, and 7-AAD (BD Pharmingen, San Diego, CA, USA). Moreover, to measure intracellular perforin and IFNγ, leukocytes were stimulated at 37°C for 5 h with brefeldin A (BFA; Invitrogen, Carlsbad, CA, USA), stained for surface markers, fixed, permeabilized with IntraPre reagent (Beckman Coulter, Fullerton, CA, USA), and then stained with IFNγ-AF488 (eBioscience, San Diego, CA, USA) antibodies.
Stained cells were analyzed on a CytoFLEX S flow cytometer (Beckman Coulter). Data were analyzed using FlowJo v10 software (Tree Star).
To more carefully characterize the phenotype of myeloid cells within tumor tissues, a set of markers that relate to myeloid cell subtype and function were discriminated by flow cytometry. We performed a tSNE analysis on a panel including 11 phenotypic markers (FSC, SSC, CD45, 7-AAD, CD11b, Ly6G, Ly6C, MHC class II, F4/80, PD-L1, and CD169). By gating on cells positive for each region of tumors and then overlaying those events on top of the tSNE plots, we can see the contribution of each of the markers to the overall structure of the population.
Phagocytosis
Endocytic uptake of a fluid-phase antigen was measured by using dextran-FITC (60–76 kDa; FD70S-100MG, Sigma-Aldrich, USA) as described previously.32 Briefly, CD169low or CD169hi monocytes were incubated in DMEM containing 10% FBS plus 1 mg/mL dextran-FITC for 60 min at 0°C (to measure non-specific adherence) or at 37°C (to measure energy-dependent uptake). After washing with ice-cold PBS three times, the cells were digested by 10% EDTA, and collected for flow cytometry detection. For apoptotic the tumor cell phagocytosis assay, SK tumor cells were preincubated in 5 μM carboxyfluorescein succinimidyl ester (CFSE) for 10 min at 37°C. After washing twice with DMEM containing 10% FBS, the cells were incubated with 100 μg/mL mitomycin C for 10 min at 37°C. After washing twice with DMEM containing 10% FBS, the cells were collected and cocultured with 5 × 106 cells/well CD169low or CD169hi monocytes in 24-well plates for 24 h at the indicated ratio. The phagocytosis efficiency was determined as the percentage of CD14+ cells containing CFSE-derived green fluorescence.
T cell proliferation assay
CD14+ monocytes were enriched by magnetic beads, and autologous flow-through cells were cultured with RPMI 1640 medium with 10% AB serum. In addition, these CD14+ monocytes were purified further using flow cytometry, and then 1–2 × 105 cells/well in 96-round well plates were stimulated with medium, 15% SK TCS, 100 pg/mL IFNα, or 15% SK TCS plus 100 pg/mL IFNα, respectively. Two days later, monocytes were collected, and the expression of CD169 on CD14+ cells was measured by flow cytometry. Meanwhile, autologous CD3+ T cells were sorted by flow cytometry and labeled with 1 μM CFSE. Moreover, CD169low or CD169hi monocytes were cocultured with CFSE-labeled autologous CD3+ T cells (monocytes/T cell ratio of 1:2) in RPMI 1640 medium with 10% AB serum, 5 μg/mL anti-CD3 (coating; 16-0038-38, eBioscience, USA), and 40 U/mL recombinant interleukin (IL)-2 (14-8029, eBioscience, USA). Five days later, IFNγ in the supernatant was detected by ELISA, and cells were collected for proliferation detection by flow cytometry. The division index, which is the average number of cell divisions that a cell in the original population has undergone, was calculated using FlowJo v10 software (Tree Star).
ELISA
The concentrations of IFNγ in monocyte and T cell coculture supernatants were detected using ELISA kits according to the manufacturer’s instructions (88-7316, eBioscience, USA).
Mice and tumor treatments
Wild-type male C57BL/6 mice were purchased from Guangdong Medical Laboratory Animal Center (Guangzhou, China). All mice were maintained under specific pathogen-free conditions and were used between 6 and 8 weeks of age in accordance with the experimental animal guidelines set by the Institutional Animal Care and Use Committee of Sun Yat-sen University Cancer Center (L102042017110F).
A total of 1 × 106 Hepa1-6 tumor cells were s.c. transplanted into the flanks of the mice. Tumor volumes were measured along three orthogonal axes (a, b, and c) and calculated as tumor volume = abc/2. Then, 10 μg of IFNα-IgG (a single-chain variable fragment with a human IgG1 Fc in the C-terminal region and murine IFNα4a inserted into the N-terminal region, which exhibits a longer half-life, lower effective dose, fewer side effects, and better stability in comparison to murine IFNα as described previously33,34) or PBS (NTC) was administered i.v. on day 8, and tumor tissues were collected on day 11. For combination therapeutic assays, 10 μg of IFNα-IgG was administered i.v. on days 10, 12, and 14. Meanwhile, 100 μg of anti-PD-L1 was administered intraperitoneally (i.p.) on days 10 and 14. Tumor and spleen tissues were collected on day 21.
Isolation of leukocytes from tissues
Fresh tumor-infiltrating leukocytes and splenocytes were obtained as follows. In short, fresh tissue specimens were cut into small pieces and digested in RPMI 1640 supplemented with 50 μg/mL collagenase IV (C-5138, Sigma-Aldrich, USA), 50 μg/mL DNase I (DN25, Sigma-Aldrich, USA), and 10% FBS (11011-8611, Sijiqing Biological, China) at 37°C for 40 min. Dissociated cells were filtered through a 70-μm mesh and separated by low speed (70 × g) centrifugation to remove parenchymal cells. The nonparenchymal cells were washed and resuspended in medium supplemented with 2% heat-inactivated FBS for fluorescence-activated cell sorting (FACS) analysis.
Statistical analyses
Data were analyzed using Prism 8.0 software (GraphPad) and are presented as the mean ± SEM. A Kolmogorov-Smirnov test was used to analyze normality, and the significance of differences between groups was determined by the Wilcoxon matched-pairs signed rank test. OS was defined as the time from resection to death by any cause. Recurrence was defined as the time from the initiation of treatment to the new event by independent radiological assessment. The Kaplan-Meier method and log-rank test were used to estimate survival. Univariate and multivariate analyses were performed using the Cox proportional hazards model. A Student’s t test was applied for comparing means of two groups, and ANOVA was used for multiple comparisons. The associations between parametric, nonparametric, and stratified variables were evaluated using Pearson’s, Spearman’s, and the χ2 tests (or a Fisher’s exact test when appropriate), respectively. p values less than 0.05 were consider significant.
Acknowledgments
This work was supported by project grants from the National Natural Science Foundation of China (31900651 to J.L. and 81773054 to Y.W.) and the Guangdong Basic and Applied Basic Research Foundation (2021A1515012620 to J.L.). The authors wish to thank Lingyan Zhu and Yunfeng Chen for help with flow cytometry and cell sorting (School of Life Science, Sun Yat-sen University). IFNα-IgG were kindly provided by Yang-Xin Fu (Institute of Biophysics, Chinese Academy of Sciences).
Author contributions
Conception and design, J.L., Y.W., and L.Z.; development of methodology, J.L., C.W., and J.X.; acquisition of the data, J.L., D.-N.Z., J.-Z.L., and Q.-M.H.; analysis and interpretation of the data, J.L. and Y.W.; administrative, technical, or material support, W.-P.W., C.W., J.X., and C.-X.H.; writing, reviewing, and revising the manuscript, J.L., Y.W., and L.Z.; study supervision, W.-P.W. and L.Z.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.09.021.
Contributor Information
Wei-Ping Wen, Email: wenwp@mail.sysu.edu.cn.
Yan Wu, Email: wuyan32@mail.sysu.edu.cn.
Supplemental information
References
- 1.Mantovani A., Marchesi F., Malesci A., Laghi L., Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017;14:399–416. doi: 10.1038/nrclinonc.2016.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeNardo D.G., Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019;19:369–382. doi: 10.1038/s41577-019-0127-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu C., Lin J., Weng Y., Zeng D.N., Xu J., Luo S., Xu L., Liu M., Hua Q., Liu C.Q., et al. Myeloid signature reveals immune contexture and predicts the prognosis of hepatocellular carcinoma. J. Clin. Invest. 2020;130:4679–4693. doi: 10.1172/JCI135048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cassetta L., Pollard J.W. Targeting macrophages: therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018;17:887–904. doi: 10.1038/nrd.2018.169. [DOI] [PubMed] [Google Scholar]
- 5.Feng M., Jiang W., Kim B.Y.S., Zhang C.C., Fu Y.X., Weissman I.L. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer. 2019;19:568–586. doi: 10.1038/s41568-019-0183-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xia Y., Rao L., Yao H., Wang Z., Ning P., Chen X. Engineering macrophages for cancer immunotherapy and drug delivery. Adv. Mater. 2020;32:e2002054. doi: 10.1002/adma.202002054. [DOI] [PubMed] [Google Scholar]
- 7.Fraschilla I., Pillai S. Viewing Siglecs through the lens of tumor immunology. Immunol. Rev. 2017;276:178–191. doi: 10.1111/imr.12526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duan S., Paulson J.C. Siglecs as immune cell checkpoints in disease. Annu. Rev. Immunol. 2020;38:365–395. doi: 10.1146/annurev-immunol-102419-035900. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y., Li J.Q., Jiang Z.Z., Li L., Wu Y., Zheng L. CD169 identifies an anti-tumour macrophage subpopulation in human hepatocellular carcinoma. J. Pathol. 2016;239:231–241. doi: 10.1002/path.4720. [DOI] [PubMed] [Google Scholar]
- 10.Cancer Genome Atlas Research Network. Electronic address: wheeler@bcm.edu. Cancer Genome Atlas Research Network Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169:1327–1341.e23. doi: 10.1016/j.cell.2017.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou P., Shaffer D.R., Alvarez Arias D.A., Nakazaki Y., Pos W., Torres A.J., Cremasco V., Dougan S.K., Cowley G.S., Elpek K., et al. In vivo discovery of immunotherapy targets in the tumour microenvironment. Nature. 2014;506:52–57. doi: 10.1038/nature12988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang P., Gu S., Pan D., Fu J., Sahu A., Hu X., Li Z., Traugh N., Bu X., Li B., et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018;24:1550–1558. doi: 10.1038/s41591-018-0136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giordano M., Henin C., Maurizio J., Imbratta C., Bourdely P., Buferne M., Baitsch L., Vanhille L., Sieweke M.H., Speiser D.E., et al. Molecular profiling of CD8 T cells in autochthonous melanoma identifies Maf as driver of exhaustion. EMBO J. 2015;34:2042–2058. doi: 10.15252/embj.201490786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Philip M., Fairchild L., Sun L., Horste E.L., Camara S., Shakiba M., Scott A.C., Viale A., Lauer P., Merghoub T., et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature. 2017;545:452–456. doi: 10.1038/nature22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wakamatsu E., Mathis D., Benoist C. Convergent and divergent effects of costimulatory molecules in conventional and regulatory CD4+ T cells. Proc. Natl. Acad. Sci. USA. 2013;110:1023–1028. doi: 10.1073/pnas.1220688110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Twyman-Saint Victor C., Rech A.J., Maity A., Rengan R., Pauken K.E., Stelekati E., Benci J.L., Xu B., Dada H., Odorizzi P.M., et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520:373–377. doi: 10.1038/nature14292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yaddanapudi K., Rendon B.E., Lamont G., Kim E.J., Al Rayyan N., Richie J., Albeituni S., Waigel S., Wise A., Mitchell R.A. MIF is necessary for late-stage melanoma patient MDSC immune suppression and differentiation. Cancer Immunol. Res. 2016;4:101–112. doi: 10.1158/2326-6066.CIR-15-0070-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beyer M., Mallmann M.R., Xue J., Staratschek-Jox A., Vorholt D., Krebs W., Sommer D., Sander J., Mertens C., Nino-Castro A., et al. High-resolution transcriptome of human macrophages. PLoS ONE. 2012;7:e45466. doi: 10.1371/journal.pone.0045466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calon A., Espinet E., Palomo-Ponce S., Tauriello D.V., Iglesias M., Céspedes M.V., Sevillano M., Nadal C., Jung P., Zhang X.H., et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell. 2012;22:571–584. doi: 10.1016/j.ccr.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kubli, S.P., Berger, T., Araujo, D.V., Siu, L.L., and Mak, T.W. (2021). Beyond immune checkpoint blockade: Emerging immunological strategies. Nat. Rev. Drug Discov..Published online March 8, 2021. 10.1038/s41573-021-00155-y. [DOI] [PubMed]
- 21.Meric-Bernstam F., Larkin J., Tabernero J., Bonini C. Enhancing anti-tumour efficacy with immunotherapy combinations. Lancet. 2021;397:1010–1022. doi: 10.1016/S0140-6736(20)32598-8. [DOI] [PubMed] [Google Scholar]
- 22.Behnke K., Zhuang Y., Xu H.C., Sundaram B., Reich M., Shinde P.V., Huang J., Modares N.F., Tumanov A.V., Polz R., et al. B cell-mediated maintenance of cluster of differentiation 169-positive cells is critical for liver regeneration. Hepatology. 2018;68:2348–2361. doi: 10.1002/hep.30088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Llovet J.M., Kelley R.K., Villanueva A., Singal A.G., Pikarsky E., Roayaie S., Lencioni R., Koike K., Zucman-Rossi J., Finn R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers. 2021;7:6. doi: 10.1038/s41572-020-00240-3. [DOI] [PubMed] [Google Scholar]
- 24.York M.R., Nagai T., Mangini A.J., Lemaire R., van Seventer J.M., Lafyatis R. A macrophage marker, Siglec-1, is increased on circulating monocytes in patients with systemic sclerosis and induced by type I interferons and Toll-like receptor agonists. Arthritis Rheum. 2007;56:1010–1020. doi: 10.1002/art.22382. [DOI] [PubMed] [Google Scholar]
- 25.Liao J., Zeng D.N., Li J.Z., Hua Q.M., Xiao Z., He C., Mao K., Zhu L.Y., Chu Y., Wen W.P., et al. Targeting adenosinergic pathway enhances the anti-tumor efficacy of sorafenib in hepatocellular carcinoma. Hepatol. Int. 2020;14:80–95. doi: 10.1007/s12072-019-10003-2. [DOI] [PubMed] [Google Scholar]
- 26.Finn R.S., Ryoo B.Y., Merle P., Kudo M., Bouattour M., Lim H.Y., Breder V., Edeline J., Chao Y., Ogasawara S., et al. KEYNOTE-240 investigators Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: A randomized, double-blind, phase III trial. J. Clin. Oncol. 2020;38:193–202. doi: 10.1200/JCO.19.01307. [DOI] [PubMed] [Google Scholar]
- 27.Finn R.S., Qin S., Ikeda M., Galle P.R., Ducreux M., Kim T.Y., Kudo M., Breder V., Merle P., Kaseb A.O., et al. IMbrave150 Investigators Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N. Engl. J. Med. 2020;382:1894–1905. doi: 10.1056/NEJMoa1915745. [DOI] [PubMed] [Google Scholar]
- 28.Yau T., Kang Y.K., Kim T.Y., El-Khoueiry A.B., Santoro A., Sangro B., Melero I., Kudo M., Hou M.M., Matilla A., et al. Efficacy and safety of nivolumab plus ipilimumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib: The CheckMate 040 randomized clinical trial. JAMA Oncol. 2020;6:e204564. doi: 10.1001/jamaoncol.2020.4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zou W., Wolchok J.D., Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016;8:328rv4. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu J., Ding T., He Q., Yu X.J., Wu W.C., Jia W.H., Yun J.P., Zhang Y., Shi M., Shao C.K., et al. An in situ molecular signature to predict early recurrence in hepatitis B virus-related hepatocellular carcinoma. J. Hepatol. 2012;57:313–321. doi: 10.1016/j.jhep.2012.03.027. [DOI] [PubMed] [Google Scholar]
- 31.Kuang D.M., Wu Y., Chen N., Cheng J., Zhuang S.M., Zheng L. Tumor-derived hyaluronan induces formation of immunosuppressive macrophages through transient early activation of monocytes. Blood. 2007;110:587–595. doi: 10.1182/blood-2007-01-068031. [DOI] [PubMed] [Google Scholar]
- 32.Moghaddami M., Mayrhofer G., Cleland L.G. MHC class II compartment, endocytosis and phagocytic activity of macrophages and putative dendritic cells isolated from normal tissues rich in synovium. Int. Immunol. 2005;17:1117–1130. doi: 10.1093/intimm/dxh291. [DOI] [PubMed] [Google Scholar]
- 33.Liao J., Luan Y., Ren Z., Liu X., Xue D., Xu H., Sun Z., Yang K., Peng H., Fu Y.X. Converting lymphoma cells into potent antigen-presenting cells for interferon-induced tumor regression. Cancer Immunol. Res. 2017;5:560–570. doi: 10.1158/2326-6066.CIR-16-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xuan C., Steward K.K., Timmerman J.M., Morrison S.L. Targeted delivery of interferon-alpha via fusion to anti-CD20 results in potent antitumor activity against B-cell lymphoma. Blood. 2010;115:2864–2871. doi: 10.1182/blood-2009-10-250555. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.