

Abstract
Chimeric antigen receptor (CAR)-redirected T cell therapy often fails to control tumors in the long term due to selecting cancer cells that downregulated or lost CAR targeted antigen. To reprogram the functional capacities specifically of engineered CAR T cells, we inserted IL12 into the extracellular moiety of a CD28-ζ CAR; both the CAR endodomain and IL12 were functionally active, as indicated by antigen-redirected effector functions and STAT4 phosphorylation, respectively. The IL12-CAR reprogrammed CD8+ T cells toward a so far not recognized natural killer (NK) cell-like signature and a CD94+CD56+CD62Lhigh phenotype closely similar, but not identical, to NK and cytokine induced killer (CIK) cells. In contrast to conventional CAR T cells, IL12-CAR T cells acquired antigen-independent, human leukocyte antigen E (HLA-E) restricted cytotoxic capacities eliminating antigen-negative cancer cells in addition to eliminating cancer cells with CAR cognate antigen. Simultaneous signaling through both the CAR endodomain and IL12 were required for inducing maximal NK-like cytotoxicity; adding IL12 to conventional CAR T cells was not sufficient. Antigen-negative tumors were attacked by IL12-CAR T cells, but not by conventional CAR T cells. Overall, we present a prototype of a new family of CARs that augments tumor recognition and elimination through expanded functional capacities by an appropriate cytokine integrated into the CAR exodomain.
Keywords: CAR T cells, IL12, CIK cells, NK cells, adoptive cell therapy
Graphical abstract
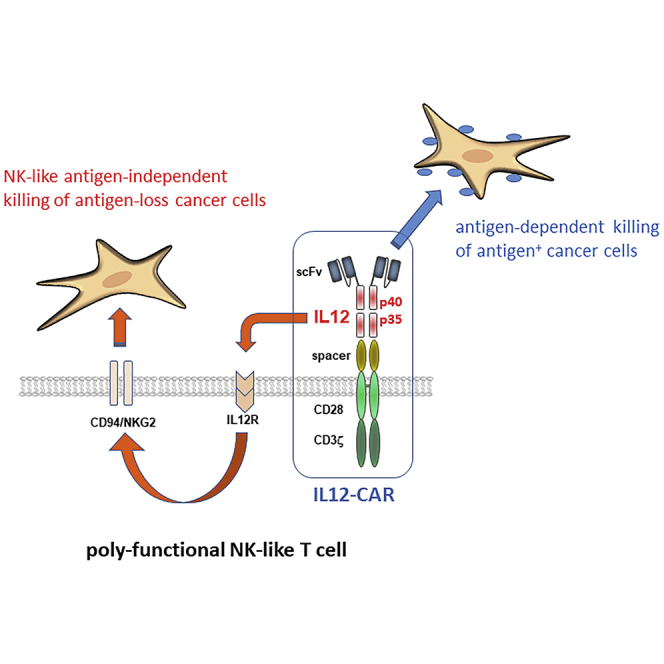
A novel CAR design integrates the IL12 cytokine into the CAR exodomain in order to reprogram T cell function toward poly-functional NK-like cells capable of both CAR-redirected antigen-specific killing and, moreover, IL12 induced antigen-independent killing of those cancer cells that lost CAR cognate antigen.
Introduction
Chimeric antigen receptor (CAR)-engineered T cells showed spectacular efficacy in the treatment of hematologic malignancies by recognizing tumor cells with pre-defined antibody specificity. Downregulating or losing antigen expression, however, is the Achilles heel of CAR-redirected therapy, resulting in tumor relapse by antigen-loss cancer cells as seen by the increasing number of CD19-negative relapses after treatment with CD19 CAR T cells.1 Moreover, primarily antigen-heterogeneous tumors are hard to treat by CAR T cells, as is frequently the case in solid cancers.2 The situation can be addressed by natural killer (NK) cells or cytokine-induced killer (CIK) cells that eliminate those antigen-negative cancer cells invisible to CAR T cells. To attract and activate innate effector cells into the tumor lesion, we and others developed the TRUCK strategy (T cells redirected for antigen-unrestricted cytokine-initiated killing) to deliver transgenic IL12 to the targeted tumor tissue by IL12 secreting CAR T cells.3,4 Thereby IL12 accumulates specifically in the tumor tissue where it fulfills multiple functions, including counteracting suppression by the tumor stroma,5,6 converting Treg cells to pro-inflammatory cells with IFN-γ release,7 and augmenting a Tc17 anti-tumor immune response.8 Accordingly, engineered IL12 release combined with CAR T cell therapy3,9 improved host immunity against tumors.10 Locally deposited IL12 is superior over systemic IL12 release due to far less toxicity, like vascular damage or cytokine release syndrome,11, 12, 13 and due to attracting innate cells to the tumor tissue with fewer systemic effects.
We used IL12 in order to reprogram the functional capacities of the CAR T cell itself without releasing IL12 into the environment. Technically, we inserted a functionally active, single-chain p40-p35 variant of IL12 into the extracellular moiety of the CAR while the CAR remains capable of binding cognate antigen through the scFv and of signaling through the CAR intracellular domains upon antigen engagement. Strikingly, engineering with the IL12-CAR converted cytotoxic T cells toward a CD8+CD56+CD62Lhigh phenotype, downregulated Th2 genes, and upregulated NK and CIK cell-associated genes compared with conventional CAR T cells. Functionally, IL12-CAR T cells gained antigen-independent lytic activities in addition to antigen-dependent cytolysis, making antigen-heterogeneous tumors accessible to an IL12-CAR T cell attack. By concept, IL12-CAR is the first prototype of a hybrid receptor that specifically combines the antigen-redirected activity of a CAR with the T cell maturating activity of a cytokine to tune the T cell function toward eradicating antigen-heterogeneous tumors.
Results
We wanted to combine the activity of IL12 with the antigen-redirected activity of a CAR in one hybrid molecule for display on the T cell surface. Therefore, we inserted the p40-p35 single-chain variant of murine IL12, flanked by a flexible linker, into the CAR exodomain between the scFv and the IgG spacer (Figure 1A); the other CAR domains are of human origin. The CD28 signaling endodomain is modified to prevent lymphocyte-specific protein tyrosine kinase (LCK) binding and thereby finally LCK activation induced IL2 release.14 After transduction, IL12-hybrid CARs were expressed on the T cell surface as detected by flow cytometry (Figure S1A). The transduction efficiency for the hybrid CAR was within the same range as for the conventional CAR without IL12; however, the IL12-CAR levels on the cell surface were lower (Figure S1B).
Figure 1.

T cells with IL12-CAR executed their cytolytic functions in an antigen-dependent fashion
(A–C) (A) Schematic representation of the IL12-CAR. scFv, binding domain; hi, human IgG1 hinge; IL12, (p40-p35) IL12; IgG1-Fc, IgG1 CH2-CH3; CD28, transmembrane and intracellular domains without the lck binding motif; CD3ζ, intracellular domain of CD3ζ. The IL12 domain is of murine origin and binds to the human IL12 receptor; all other CAR domains are of human origin. IL12-CAR T cells with specificity for CEA (B) and Muc1 (C) or non-modified T cells (w/o) for control (0.31–5 × 104 cells/well) were co-cultivated for 48 h in 96 micro-test plates with CEA+ Muc1- LS174T, CEA− Colo320 and Muc1+ MCF7 tumor cells (each 2.5 × 104 cells/well), respectively. Viability of tumor cells was determined by the XTT assay and specific cytotoxicity was calculated. Numbers represent the mean values of technical replicates ±SD. Data from a typical experiment out of three are shown.
To test for CAR-mediated T cell activation, we recorded the cytotoxic activity toward cognate target cells using an anti-Muc1 CAR and an anti-carcinoembryonic antigen (CEA) CAR as examples. As summarized in Figure 1B, T cells with anti-Muc1 CAR and integrated IL12 eliminated Muc1+ MCF7 breast cancer cells. Notably, the weak activity of anti-Muc1 IL12-CAR T cells was improved compared with conventional anti-Muc1 CAR T cells without IL12, which becomes visible at lower effector to target cell ratios. The specific cytolytic activity of the strong anti-CEA CAR T cells against CEA+ LS174T cells was conserved by the IL12-CAR. Data indicate that IL12 integrated into the CAR enhanced the cytotoxic activity of a weak CAR and conserved antigen specificity as defined by the CAR binding domain.
To test for the activity of IL12 when incorporated into the CAR, we determined phospho-STAT4 (pSTAT4) levels15 in engineered T cells. As summarized in Figure 2A, the amount of pSTAT4 was significantly enhanced in IL12-CAR T cells compared with conventional CAR T cells, indicating that IL12 integrated into the CAR exodomain is functional. Noteworthy, increase in pSTAT4 did not require CAR engagement of antigen, indicating constitutively active IL12 in this context.
Figure 2.

IL12 in the CAR exodomain was functional, resulting in differentially expressed proteins in IL12-CAR T cells versus CAR T cells
(A) Flow cytometric analysis of phosphorylated STAT4 (pSTAT4) in conventional and IL12-CAR T cells. Representative pSTAT4 histograms of engineered T cells and mean values ±SD of three donor T cells engineered with the respective CAR are shown. Statistical significance was calculated by Student's t test. ∗p < 0.05. MFI, mean fluorescence intensity. (B) T cells with IL12-CAR and conventional CAR, respectively, were cultivated for 7 days and analyzed for several signature markers by flow cytometry and ELISA. For flow cytometric analysis, gates were set on CAR+ T cells and numbers of CAR T cells with expression of the respective markers were determined. CAR T cells were stained for CD56, CCR1, and CD94 expression. CAR T cells (5 × 104 cells/well) were further stimulated through CD3/CD28 for 72 h with agonistic anti-CD3 and anti-CD28 mAbs (1 μg/mL each), respectively, and recorded for intracellular IL5 and surface TGFβ by flow cytometry. Supernatants were analyzed for IFN-γ by ELISA. CAR T cells were stained for CAR, CD8, CD56, CD62L, and CD94. Data represent mean values ±SD of three to five different donors. Statistical differences were determined by Student's t test.
IL12 signaling is known to enhance IFN-γ secretion by T cells.16 Accordingly, we detected higher IFN-γ levels upon co-incubating anti-CEA IL12-CAR T cells with CEA+ tumor cells compared with CAR T cells without IL12, also indicating functional activity of IL12 (Figures 2B and S2). IFN-γ secretion was also increased in the presence of CEA− Colo320 target cells and in absence of target cells, indicating spontaneous IFN-γ release in an antigen-independent fashion, although at lower levels. Taken together, the IL12-CAR induced signaling through the IL12 receptor pathway resulted in pSTAT4 increase and increase in the pro-inflammatory effector cytokine IFN-γ independently of CAR engagement of antigen. Moreover, IL12 signaling improved CAR-mediated T cell activation through the CD28-CD3ζ endodomain.
We addressed whether the IL12-CAR altered the phenotype of engineered T cells and released functionally relevant factors compared with the CAR without IL12 (Figure 2B). In particular, IL12-CAR T cells upregulated IFN-γ secretion and expression of NK cell-associated receptors like CD56 and CD94, whereas immune repressive proteins like IL5 and TGF-β1 were downregulated compared with conventional CAR T cells. In the IL12-CAR T cell population, the numbers of cells with CD8+CD56+ and CD56+CD94+ phenotype were highly upregulated. IL12-CAR T cells developed predominantly a CD8+CD56+CD62Lhigh phenotype, whereas conventional CD8+CD56+ CAR T cells were rather CD62Llow (Figures 2B and S3).
To address whether the altered phenotype goes along with a global change in gene expression pattern, T cells engineered with the respective CAR were subjected to differential expression analyses. Sorted CAR T cells were stimulated with a CAR-specific anti-idiotypic antibody as surrogate antigen. Differential gene expression analyses revealed 152 genes up- and 108 genes downregulated in IL12-CAR T cells compared with conventional CAR T cells stimulated by antigen under the same conditions (Figure 3). In IL12-CAR T cells, genes associated with cytokine-mediated signaling pathways (Gene Ontology [GO]: 0019221), i.e., Th2 cytokines and immune repression cytokines like TGF-β1, were downregulated. In contrast, genes defining NK cell-mediated immunity (GO: 0002228), cellular defense response (GO: 0006968), and leukocyte chemotaxis (GO: 30595) were highly upregulated. IL-10 was also upregulated as a known result of active IL12 signaling.17
Figure 3.

CAR-stimulated IL12-CAR T cells showed upregulated NK and downregulated Th2-associated genes compared with conventional CAR T cells
(A and B) T cells engineered with IL12-CAR or conventional CAR, both with the same specificity for CEA, were cultured for 7 days and flow sorted. Cells were stimulated for 48 h through their CAR by incubating in microtest-plates coated with the CAR-specific anti-idiotypic antibody BW2064 as surrogate antigen. RNA was extracted and labeled using the Affymetrix WTPLus reagent and probed using the Clariom S Human Array. Raw array data were processed using the Affymetrix Expression Console software. CEL files containing feature intensity values were converted into summarized expression values using RMA, which consists of background adjustment, quantile normalization, and summarization across all chips. (A) Expression data [log(2)] of CAR T cells from three different donors were generated and mean values with differential expression of ≥2.5 times determined. Statistical analysis was performed by ANOVA and values with p < 0.05 were included in further analyses. Differentially expressed genes, i.e., >4-fold and <5-fold, respectively, were depicted. Numbers represent mean values of x-fold differences. (B) Differentially expressed gene sets were subjected to GO analysis. X-fold enrichment (red bars) and x-fold depletion (green bars) of gene sets are depicted. Open bars represent p values of differentially regulated gene sets.
Taken together, IL12 integrated into the IL12-CAR was functional in converting the gene expression profile toward cytotoxic cells that are characterized by high levels of CD56, and of NK cell activating and inhibiting receptors including CD94, Ksp37 (FGFBP2), and granulysin, all indicative of highly cytotoxic T cells.18,19 In contrast, immune-modulating and inhibiting factors like IL5 and TGF-β1 were downregulated. Taken together, gene expression profiling implied a novel phenotype of NK-like T cells induced upon signaling through the IL12-CAR. The CD8+CD56+CD62Lhigh phenotype of IL12-CAR T cells resembled, although was not identical to, CIK cells,20,21 the recently described poly-functional NK cells,22 and anti-microbial cytotoxic T cells.23
CD94/NKG2A was highly upregulated in IL12-CAR T cells compared with conventional CAR T cells (Figure 4A). To test whether IL12-CAR T cells gained poly-functional capacities like NK cells, we assayed for antigen-independent cytotoxicity against a variety of cancer cells. T cells engineered with the anti-CEA IL12-CAR, and for comparison T cells with the conventional anti-CEA CAR without IL12, were co-incubated with CEA+ and CEA− cancer cell lines, healthy fibroblasts, and autologous B cells, respectively (Figure 4B). IL12-CAR T cells with anti-CEA CAR lysed CEA+ cancer cells and also CEA− Daudi, Skov3, and Ovcar3 cancer cells in a CEA antigen-independent fashion. However, the cytotoxicity was restricted to susceptible cancer cells because human healthy fibroblasts and autologous B cells were not lysed. In contrast, the cytotoxic activity of conventional anti-CEA CAR T cells was specifically restricted to CEA+ cancer cells.
Figure 4.

IL12-CAR T cells exhibited HLA-E-restricted NK-like cytotoxicity against tumor cells
(A) T cells were engineered with a conventional CAR or IL12-CAR, each with specificity for CEA. CAR T cells were detected by the anti-IgG1 antibody that binds to the common extracellular spacer, gated and analyzed 4 and 7 days post transduction for CD94 and CD56 expression, respectively. Histograms of a typical experiment are shown. (B) T cells were engineered with anti-CEA and anti-CEA IL12-CAR, respectively, and co-cultivated for 48 h with cancer cell lines, healthy human fibroblasts or autologous B cells at an (E)T ratio of 1:1. (C) Anti-CEA IL12-CAR T cells (2 × 104 T cells/well) were co-cultivated for 48 h with CEA− Skov3 tumor cells (2 × 104/well) that were pretreated for 30 min with the anti-HLA-E antibody 3D12 or an isotype control mAb (each 5 μg/mL). Viability of tumor cells was determined by the XTT assay and specific cytotoxicity was calculated. For recording B cell lysis, cells were labeled with an anti-CD19 mAb and 7-AAD, respectively, and numbers of dead B cells were determined by flow cytometry. Numbers represent mean values of technical replicates ±SD. Representative data of two to four independent experiments are shown. Statistical analyses were performed using Student's t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Since CD94/NKG2A was highly upregulated in IL12-CAR T cells, and not in conventional CAR T cells, we assumed that CD94 is involved in antigen-independent killing. Human leukocyte antigen E (HLA-E) can protect target cells from lysis by CD94/NKG2A-expressing NK cell clones24; we therefore recorded killing of Daudi cells that are deficient in HLA-E25 and sensitive to killing by activated NK cells. As shown in Figure 4B, Daudi cells were eliminated by IL12-CAR T cells but not by conventional CAR T cells. Also, Skov3 ovarian cancer cells were susceptible to IL12-CAR T cell killing, as they were to NK cell killing.26 Since Skov3 cells express some HLA-E on the cell surface,27,28 we asked whether Skov3 killing by IL12-CAR T cells can be further enhanced by blocking CD94/NKG2A binding by the anti-HLA-E antibody 3D12. As shown in Figure 4C, HLA-E blocking substantially improved lysis of CEA-negative Skov3 tumor cells by IL12-CAR T cells, which was not the case for conventional CAR T cells, again indicating a strong NK-like activity of IL12-CAR T cells. NK cell-like cytotoxicity was not restricted to CEA-specific IL12-CAR T cells because IL12-CAR T cells with specificity for Muc-1 also upregulated CD94 and CD56 and also lysed Daudi cells in an antigen-independent fashion (Figure S4). Taken together, we concluded that IL12-CAR T cells acquired antigen-independent, NK-like cytotoxicity toward cancer cells that was mediated, at least in part, by CD94.
We asked whether IL12 within the CAR exodomain acts in cis only on the CAR T cell or also in trans on CAR-negative T cells within the same culture. We propagated IL12-CAR-engineered T cells along with CAR-negative T cells for 5 days to allow potential cross-activation and sorted CAR+ and CAR− T cells to test for their cytotoxicity against CEA+ LS174T and CEA− Daudi tumor cells (Figure 5). Only CD8+ T cells expressing the IL12-CAR exhibited antigen-independent cytotoxicity against Daudi cells; T cells without CAR from the same culture did not, indicating that the IL12-CAR predominantly acts in cis on the CAR T cell itself. CD4+ T cells with IL12-CAR did not induce Daudi killing, moreover indicating that CD8+ T cells and not CD4+ T cells gain the NK-like activity through the IL12-CAR.
Figure 5.

NK-like cytotoxicity of IL12-CAR T cells was mediated by CD8 + CAR + T cells
(A) T cells were engineered with conventional and IL12-CAR, both with specificity for CEA, propagated for 5 days, and sorted into CD4+ and CD8+ cell populations with and without the respective CAR. (B) Sorted T cell subsets were co-cultivated (2 × 104 cells/well) with CEA+ LS174T or CEA− Daudi cells (each 2 × 104 cells/well) for 48 h. Viability of tumor cells was determined by the XTT assay and specific cytotoxicity was calculated. Numbers represent mean values of technical replicates ±SD. Representative data of two independent experiments are shown. Statistical differences were calculated using the Student's t test.
We assumed that repetitive stimulation with antigen differentially affects T cell maturation and target cell recognition dependent on the CAR. T cells were engineered with the conventional anti-CEA CAR and the anti-CEA IL12-CAR, respectively, and propagated in presence of IL2 or the agonistic anti-idiotypic antibody BW2064 as surrogate antigen for the CAR. As summarized in Figure 6A, IL12-CAR T cells rapidly upregulated CD94 followed by a CD56+CD62Lhigh phenotype. Noteworthy, T cells with conventional CAR did not display these changes in phenotype upon repetitive antigen stimulation. Also noteworthy, IL2 stimulation did not increase CD94+ CAR T cells, while stimulation through the IL12-CAR did.
Figure 6.

CAR stimulation induced NK-like differentiation and conserved CAR-specific cytotoxicity of IL12-CAR T cells
(A) T cells were engineered with the IL12-CAR and conventional CAR, respectively, both with the same specificity for CEA. After engraftment (day 0) cells were further stimulated in presence of IL2 (50 U/mL) or the CAR-specific anti-idiotypic antibody BW2064 (2 μg/mL) as surrogate antigen. Cells were analyzed for the indicated markers by flow cytometry. Numbers represent mean values of three to five different donors ±SD. Significant differences were calculated by Student's t test. ∗∗p < 0.01; ∗p < 0.05. (B) CAR T cells were tested for CAR-specific and NK-like cytotoxicity before (day 0) and after CAR-specific stimulation through an anti-idiotypic antibody at day 2 and day 4. CAR T cells were co-cultivated (2 × 104 cells/well) at day 0 or day 7 with CEA+ LS174T or CEA− Skov3 tumor cells (each 2 × 104 cells/well) for 48 h. To remove remaining CAR-specific anti-idiotypic antibody from day 7 cultures, CAR T cells were washed extensively before co-cultivation with target cells. Viability of tumor cells was determined by the XTT assay and specific cytotoxicity was calculated. Numbers represent mean values of technical replicates ±SD. Representative data of two independent experiments are shown. Statistical differences were calculated using Student's t test. n.s., not significant.
Prolonged IL12-CAR stimulation by antigen engagement also affected the cytolytic capacity. Upon stimulation by antigen for 7 days, IL12-CAR T cells still retained their specific cytotoxicity against CEA+ LS174T cells on nearly the same level and without drop in their NK-like cytotoxicity against CEA-negative Skov3 cells (Figure 6B). In contrast, cytotoxicity mediated by the conventional anti-CEA CAR declined upon prolonged antigen or IL12 stimulation, demonstrating beneficial capacities of IL12-CAR T cells.
We addressed whether the antigen-independent cytotoxicity of IL12-CAR T cells toward CEA− cancer cells requires the CAR endodomains. We therefore generated IL12-CAR variants with deleted CD28-CD3ζ or CD3ζ signaling endodomains leaving IL12 in the exodomain (Figure 7A). As summarized in Figure 7B, deletion of the CD3ζ domain reduced antigen-independent killing of CEA− Skov3 cells; deletion of both CD28 and CD3ζ endodomains did not further reduce killing. For comparison, antigen-redirected killing of CEA+ LS174 cells was completely abrogated upon deletion of the CAR CD3ζ endodomain, as expected. IFN-γ release, indicating CD3ζ-mediated T cell activation, was abolished upon deleting the CD3ζ endodomain in both the conventional CAR and the IL12-CAR. We concluded that both IL12 and CD3ζ were required for inducing antigen-independent, NK-like cytotoxicity.
Figure 7.

Conversion to NK-like poly-functional T cells required both IL12 and CD3ζ CAR endodomain
(A) Schematic representation of the IL12-CAR constructs with deletions of the CD3ζ and/or CD28-CD3ζ endodomains. TM, transmembrane domain. (B) CAR T cells were cultured for 4 days, flow sorted, and co-cultured for 48 h in 96-well micro-test plates with CEA+ LS174T or CEA− Skov3 tumor cells (each 2 × 104 cells/well). Viability of tumor cells was determined by the XTT assay and specific cytotoxicity was calculated. IFN-γ in the supernatant was determined by ELISA. Numbers represent mean values of technical replicates ±SD. Statistical differences were determined by Student's t test (∗∗∗p < 0.005; ∗∗p < 0.01; ∗p < 0.05; n.s., not significant). Representative data of two independent experiments are shown. (C) Acquisition of NK-like cytotoxicity by CAR T cells requires both IL12 and CAR signaling. T cells were engineered with the conventional CAR and IL12-CAR, respectively, and co-cultivated for 48 h with CEA− Skov3 or CEA+ LS174T tumor cells (each 2 × 104 cells/well) in presence or absence of the CAR-specific anti-idiotypic antibody BW2064 (4 μg/mL), which serves as a CAR-specific antigen, and added IL12 (5 ng/mL), respectively. Viability of tumor cells was determined by the XTT assay and specific cytotoxicity was calculated. Numbers represent mean values of technical replicates ±SD. Statistical differences were determined by Student's t test. Representative data of two independent experiments are shown. (D) CAR CD3ζ endodomain is required for upregulating CD94 in IL12-CAR T cells. T cells were engineered with the conventional CAR, IL12-CAR, and the CAR variants with truncated endodomains, respectively. After transduction, T cells were cultured for 2 days and analyzed for CAR and CD94 expression by flow cytometry. Numbers represent mean values of seven or eight different donors ±SD. Significant differences were calculated by Student's t test. ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05.
To test whether IL12 is capable of converting conventional CAR T cells toward antigen-independent killing, we added IL12 and BW2064 anti-idiotypic antibody as surrogate CAR antigen to CAR T cells and IL12-CAR T cells, respectively, co-cultured with CEA− Skov3 cells. CAR engagement of antigen in the presence of added IL12 induced cytotoxic activity of conventional CAR T cells against CEA− Skov3 cells in a similar fashion as obtained by the IL12-CAR (Figure 7C). Addition of IL12 or of antigen alone to conventional CAR T cells was not sufficient. Data extend our conclusion that both IL12 and CAR signaling are required and sufficient for inducing NK-like activities. Noteworthy, addition of CAR cognate antigen and addition of IL12 did not further enhance antigen-independent cytotoxicity of IL12-CAR T cells, implying that the IL12-CAR itself without antigen engagement mediated combined IL12 and CD3ζ signaling that was sufficient for acquiring NK-like reactivity.
The IL12-CAR also mediates antigen-redirected killing through target recognition through the scFv domain since adding the anti-idiotypic antibody BW2064, which binds to the CAR scFv-binding domain, specifically blocked lysis of CEA+ LS174T cells; the same data were obtained with the conventional CAR T cells. Addition of IL12 to CAR T cells did not restore the antigen-specific lysis of CEA+ LS174 cells in the presence of the anti-idiotypic antibody. Noteworthy, killing of CEA− Skov3 cells by IL12-CAR T cells was not blocked by the anti-idiotypic antibody, moreover indicating that the NK-like activity of IL12-CAR T cells was not mediated by CAR-mediated target recognition.
In accordance with our conclusion, the IL12-CAR induced strong upregulation of CD94/NKG2 that is far less the case for conventional CAR T cells or IL12-CAR T cells lacking the signaling endodomain (Figure 7D). Data indicate that strong CD94 upregulation also required both IL12 and CD3ζ of the CAR, which is in accordance with our conclusion that both are required to induce antigen-independent killing by IL12-CAR T cells.
We asked whether the antigen-independent cytotoxic activity of IL12-CAR T cells is capable of counteracting progression of CEA− Skov3 tumors. Rag2−/− cγ−/- mice were subcutaneously (s.c.) inoculated with Skov3 cells and established tumors were treated by intravenous (i.v.) injection of T cells with IL12-CAR or conventional CAR, both with specificity for CEA. As summarized in Figure 8, only IL12-CAR T cells reduced Skov3 tumor growth, whereas T cells with conventional anti-CEA CAR and without CAR did not. Data demonstrate the poly-functional NK-like activity of IL12-CAR T cells in eliminating transplanted tumors lacking the CAR cognate antigen, which makes the IL12-CAR superior to a conventional antigen-dependent CAR.
Figure 8.

Anti-CEA IL12-CAR T cells inhibited growth of CEA− Skov3 tumors
(A and B) Groups of male Rag−/− cγ−/− mice (six to nine mice) were subcutaneously injected with Skov3 tumor cells (5 × 106 cells/mouse). One week after tumor inoculation, mice with tumors of similar size were randomized into groups. CAR T cells and T cells without CAR for control were intravenously administered into tumor-bearing mice (each 5 × 106 cells/mouse) and tumor growth was determined by a digital caliper every 2–3 days. (A) AUC for each tumor was calculated. Data represent mean values ±SD of each group expressed in arbitrary units. Statistical differences were determined by the Student's t test. (B) Tumor volumes of treated animals. Numbers represent mean values of each test group. A novel CAR design integrates the IL12 cytokine into the CAR exodomain in order to reprogram T cell function toward poly-functional NK-like cells capable of both CAR-redirected antigen-specific killing and, moreover, IL12-induced antigen-independent killing of those cancer cells that lost CAR cognate antigen.
Discussion
IL12 has the capacity to take the key role in boosting T cell therapy of cancer due to orchestrating the Th1 response and activating cytolytic CD8+ T and NK cells.29 To take advantage of the favorite properties and to combine them with antigen-targeting receptors, we integrated p40-p35 IL12 into the CAR exodomain resulting in a composite IL12 cytokine-CAR that provides both functionally active IL12 and antigen-triggered T cell activation. To our best knowledge, this is the first prototype of a composite CAR that provides a cytokine to modulate T cell function in addition to CAR-driven activation.
Both domains within the CAR are functional since IL12-CAR T cells increased activated pSTAT4 and released enhanced amounts of IFN-γ upon antigen engagement compared with conventional CAR T cells without IL12. Integrated into the CAR, IL12 is constitutively active since phosphorylation of STAT4 increased without antigen engagement; T cell activation is augmented, as indicated by increased IFN-γ release. More strikingly, T cells upregulated genes associated with cytotoxic or NK cell function and downregulated genes associated with a Th2 response and/or a suppressive phenotype. Particularly, the IL12-CAR transformed CD8+ T cells into CD8+CD56+CD62Lhigh cytotoxic T cells with upregulated CD94/NKG2. This is an as-yet unrecognized T cell subset capable of mediating antigen-independent killing. Blocking of HLA-E, the main ligand for CD94/NKG2, enhanced antigen-independent target cell lysis, indicating HLA-E restriction of NK-like cytotoxicity by IL12-CAR T cells. Notably, IL12-CAR T cells with NK-like activity against ovarian cancer also upregulated Ksp37 (FGFBP2); this NK-associated receptor was associated with favorable clinical outcome of ovarian cancer patients.18 Gain of poly-functional NK-like killer capacities was restricted to CD8+ T cells and required both IL12 and CD3ζ of the CAR but did not require antigen engagement. Our conclusion is based on the observation that (1) IL12 integrated into the CAR is required to induce NK-like cytotoxicity since the respective CAR without IL12 does not (Figure 7C). CD3ζ of the CAR is required since deletion of CD3ζ abolishes the effect (Figure 7B). (2) IL12-CAR produced increased levels of pSTAT4 independently of antigen engagement (Figure 3A), indicating spontaneous IL12 signaling in engineered T cells. (3) CD94 along with other NK cell-associated markers was induced in IL12-CAR T cells without engagement of CAR antigen (Figure 4A).
The induced poly-functional capacities of IL12-CAR T cells resemble, but are not identical to, CD8+CD56+ CIK cells that represent terminally differentiated T cells with profound cytotoxic activities.30 CIK cells acquire antigen-independent killing upon repetitive and prolonged stimulation through IFN-γ over weeks accompanied by CD62L downregulation. In contrast, IL12-CAR T cells acquired antigen-independent cytotoxicity within days and maintained high CD62L expression. IL12-CAR T cells are also very similar to the newly described cytotoxic T cells with high granulysin load and anti-microbial activity23 and resemble poly-functional CD3−CD62L+ NK cells,22 which has not yet been reported for T cells. CD3−CD62L+ NK cells have high proliferative capabilities and superior in vivo persistence and tumor-homing capacities. In line with this, CD62L+ NK-T cells, which share some properties with CD62L+ NK cells, also exhibit prolonged persistence and improved anti-tumor activity.31 Our data indicate that the IL12-CAR converts cytotoxic CD8+ T cells toward NK-like cells with beneficial anti-tumor properties within 5 days of re-stimulation.
While the targeting specificity defined by the CAR-binding domain is one of the major advantages for redirected cell therapy, there is also a need for antigen-independent cancer cell elimination, in particular, when targeting tumors with heterogeneous antigen pattern. The shifting antigen load is crucial for the success in CAR therapy since progressively growing tumors with high amplification rates continuously produce antigen-loss cancer cells that are invisible to conventional CAR T cells. To overcome the limitation, CIK cells with their physiologic NK-like activities were engineered with tumor-specific CARs to target antigen-defined tissues and to take advantage of their antigen-independent killing capacities.32, 33, 34 Despite a successful phase I trial with allogeneic donor-derived CAR CIK cells,35 there is currently no broad application in clinical trials. This is likely because the in vitro generation of CIK cells is extremely time consuming, prone to low efficiency in producing CD3+CD56+ cells, and suffers from high variability between donors. Since CIK cells represent terminally differentiated effector cells, their capacity to expand, persist, and release pro-inflammatory cytokines is also poor,30,34,36 which may also account for their poor clinical exploration.
Alternatively, NK cells were engineered with specific CARs37 in order to take advantage of CAR targeting and of antigen-independent killing. A recently reported phase I trial with CD19 CAR NK cells from cord blood demonstrated high efficacy with remarkably low side effects, indicating the feasibility of recruiting of poly-functional effector cells toward specific targets.38 Our data suggest that engineering young peripheral blood T cells with the IL12-CAR represents a more efficient procedure to rapidly obtain poly-functional NK-like T cells with both antigen-directed and antigen-independent anti-tumor cytotoxicity. Tethering IL12 to the CAR, moreover, has the benefit of potentially low toxicity, while systemically delivered IL12 has a very small therapeutic window with the risk of inducing severe cytotoxicity.39 Although a xenogenic mouse model is of limited value for predicting potential toxicity, our results are in line with previous reports on dramatically decreased toxicity of IL12 when attached to the cell membrane.40,41 We therefore do not expect major toxicities due to CAR-bound IL12 in clinical applications. In a more general perspective, the IL12-CAR represents a prototype of a new generation of function-modulating CARs with an integrated cytokine module to shape CAR T cell effector capacities in a specific fashion; other cytokines, in particular single-chain cytokines, may likely be active when inserted into the CAR exodomain to reprogram effector cell functions.
Materials and methods
Cell lines and reagents
HEK293T cells are human embryonic kidney 293 cells that express the SV40 large T antigen.42 The cancer cell lines Colo320 (ATCC CCL 220.1), LS174T (ATCC CL-188), Skov3 (ATCC HTB-79), Ovcar-3 (ATCC HTB-161), MCF7 (ATCC HTB-22), Daudi (ATCC CCL-213), and K562 (ATCC CCL-243) were obtained from ATCC (Rockville, MD). Human dermal fibroblasts were purchased from tebu-bio (Offenbach, Germany). All cell lines were cultured in RPMI 1640 or DMEM medium, 10% (v/v) fetal bovine serum (FBS) (Thermo Fisher Scientific, Erlangen, Germany). Anti-CD3 monoclonal antibody (mAb) OKT3 and anti-CD28 mAb 15 × 108 were purified from OKT3 hybridoma (ATCC CRL 8001) and 15 × 108 hybridoma (kindly provided by Dr. R. van Lier, Red Cross Central Blood Bank, Amsterdam, The Netherlands) supernatants, respectively, by affinity chromatography. The anti-BW431/36 idiotypic antibody BW2064 was described earlier.43 The PE-conjugated F(ab')2 goat anti-human IgG antibody was purchased from Southern Biotechnology (Birmingham, AL). The fluorochrome-conjugated anti-CD3, anti-CD4, and anti-CD8 mAbs were purchased from Miltenyi Biotec (Bergisch Gladbach, Germany). The anti-CD62L and anti-CD56 mAbs were purchased from Immunotools (Friesoythe, Germany). The anti-CD94 and anti-CCR1 mAbs, respectively, were purchased from BioLegend (San Diego, CA). The anti-HLA-E mAb 3D12 was purchased from eBioscience (San Diego, CA). The anti-human TGF-β mAb was purchased from Biozol (IQproducts; Leipzig, Germany), fluorochrome-conjugated isotype controls were from BD Biosciences (San Diego, CA). Matched antibody pairs for capture and detection of human IFN-γ were purchased from BD Biosciences. Recombinant IL-2 was obtained from Endogen (Woburn, MA). Intracellular IL5 was recorded utilizing the anti-IL5 mAb (BioLegend) and BD IntraSure Kit (BD Biosciences) for fixation and permeabilization of CAR T cells. pStat4 was detected by the anti-phospho-STAT4 PerCP-CyTM 5.5 mAb (BD Biosciences)
Preparation of human T cells
Peripheral blood lymphocytes were obtained from healthy donors by Ficoll density centrifugation. T cells were activated by incubation with OKT3 and 15 × 108 mAbs (100 ng/mL each) and IL-2 (500 U/mL) and further propagated in the presence of IL-2 (500 U/mL). Blood was obtained from healthy volunteers through the University Hospital, Department of Transfusion Medicine (ethics approval 21-2224-101).
CARs
Engineering of CARs with the modified CD28-CD3ζ signaling and the modified IgG1-CH2/3 extracellular spacer domains44 and the retroviral modification of T cells were previously described in detail.45, 46, 47 The p40-p35 single-chain variant of the murine IL12 was described earlier.48 To engineer the IL12-CAR, the sequence of p40-p35 IL12 was PCR amplified and flanked on both sides with sequences from the human IgG1 hinge domain incorporating a BamHI and BglII restriction site, respectively. The PCR fragment was digested and incorporated into the BamHI restriction site of the anti-CEA CAR14 between the BW431/26 scFv and the human IgG1 constant domain, respectively. The anti-CEA scFv was substituted by the anti-Muc1 H594 scFv to obtain the IL12 anti-Muc1 CAR.49
Site directed mutagenesis
The IL12-CAR was truncated at the CD3ζ or CD28ζ signaling domains by introducing stop codons at AA positions 1,094 and 1,113 of the IL12-CAR, respectively. Briefly, vector DNA of the IL12-CAR was PCR amplified utilizing the following oligonucleotide primer sets: S-CD28TM-TTCTGGGTGAGGAGTTAAGAGGAGCAGGCTCCT (forward); AS-CD28TM-OL-AGGAGCCTGCTCCTCTTAACTCCTCACCCAGAA (reverse); S-CD28TMIC-TTCGCAGCCTATCGCTGACTGAGAGTGAAGTTCA (forward); AS-CD28TMIC-OL-TGAACTTCACTCTCAGTCAGCGATAGGCTGCGAA (reverse). Amplified DNA was isolated, DpnI digested, and transformed into bacteria. Introduced mutations were confirmed by sequencing.
T cell modification
For engineering with CARs,50 T cells were stimulated and transduced with γ-retrovirus containing supernatants or by co-culture with virus-producing 293T cells as described.51 Retroviruses were produced by 293T cells upon transfection of plasmid DNAs encoding GALV encoding and gag/pol, respectively, and of the plasmid DNA encoding the respective CAR. CAR expression was monitored by flow cytometry using an antibody against the common extracellular IgG1 Fc domain.
Flow cytometry and cell sorting
CAR T cells were stained with fluorochrome-labeled antibodies. IL5-secreting cells were detected by intracellular fluorescence-activated cell sorting (FACS) utilizing the IntraSure fixation and permeabilization kit and the PE-labeled anti-IL5 mAb. Cells were analyzed by a FACSCanto II flow cytometer equipped with the FACSDiva software (BD Biosciences). Fluorochrome-labeled CAR T cells and/or CAR T cell subpopulations were further purified by flow sorting using a FACSAria III cell sorter (BD Biosciences). pSTAT4 was detected by intracellular flow cytometry: Briefly, after labeling with fixable viability dye (eFluor780, Invitrogen), CAR T cells were fixed and permeabilized utilizing the Cytofix/Cytoperm Fixation/Permeabilization Solution Kit (BD Bioscience) according to the manufacturer's recommendations. Cells were intracellularly stained with the PerCP-CyTM 5.5-labeled anti-phospho-STAT4 mAb (BD Biosciences). Gates were set to the live cell population and cells were analyzed on a FACSlyric Flow cytometer equipped with FACSuite software (BD Biosciences). Doublets were discriminated using forward scatter area (FSC-A) versus forward scatter height (FSC-H) gating.
Gene expression analysis
Human peripheral blood T cells were grafted with the respective CAR and cultivated for 7 days. CAR T cells were flow sorted and stimulated through the CAR for 48 h by incubation in microtest-plates coated with the anti-idiotypic mAb BW2064 directed against the scFv of the CAR. RNA from 5 × 106 cells each was extracted, labeled using the Affymetrix WTPLus kit according to manufacturer's guidelines, and probed to the Clariom S Human Array. After staining, arrays were scanned using the Affymetrix Gene-Chip Scanner-3000-7G; quality control analysis was performed using Affymetrix GCOS software. Raw array data were processed using the Affymetrix Transcriptome Analysis Console (TAC) 4.0 software. CEL files containing feature intensity values were converted into summarized expression values using robust multi-array average (RMA), which consists of background adjustment, quantile normalization, and summarization across all chips. Expression data [log(2)] of CAR T cells were generated and x-fold expression of differentially expressed genes determined. Mean values of expression data [log(2)] of CAR T cells from three different donors were determined. Mean values with differential expression of ≥2.5 in mean and ≥2 in individual samples were regarded significant. Statistical analysis was performed by ANOVA and values with p < 0.05 were included in the analysis.
GO analysis
For GO analysis gene sets that were differentially expressed between IL12-CAR T cells and conventional CAR T cells (p < 0.05, fold change cutoff of <−5 or >4) were imported into the GO software tool (http://geneontology.org/).52 Gene sets were uploaded and mapped against the GO Panther database with the human genes reference list (www.pantherdb.org, version 15.0, released 2020.02.14). Significant lists of up- and downregulated genes were statistically compared with the reference list to identify significant over-representation of genes (p < 0.05) within the GO classes for biological process. Classes with significant over-representation were shown after correcting for multiple testing using the Bonferroni correction.
Activation of CAR T cells
CAR T cells (0.125–10 × 104 cells/well) were co-cultivated for 24–48 h in 96-well round-bottom plates with tumor cells (each 2.5–5 × 104 cells/well). Specific cytotoxicity of CAR T cells was monitored by an XTT (2,3-Bis-(2-Methoxy-4-Nitro-5-Sulfophenyl)-2H-Tetrazolium-5-Carboxanilide)-based colorimetric assay53 using the Cell Proliferation Kit II (Roche Diagnostics, Mannheim, Germany). Viability of tumor cells was calculated as mean values of six wells containing only tumor cells subtracted by the mean background level of wells containing medium only. Non-specific formation of formazan due to the presence of T cells was determined from triplicate wells containing T cells in the same number as in the corresponding experimental wells. The number of viable tumor cells in experimental wells was calculated as follows: viability (%) = [OD(experimental wells – corresponding number of T cells)]/[OD(tumor cells without T cells – medium)] × 100. Cytotoxicity (%) was defined as 100 – viability (%). In some experiments, CAR T cells were cultivated in presence of the anti-CEA CAR-specific anti-idiotypic antibody BW2064 (2–4 μg/mL) and/or added IL12 (5 ng/mL) or Skov3 target cells that were pre-incubated for 30 min with anti-HLA mAbs (each 5 μg/mL).
ELISA
IFN-γ in culture supernatants was monitored by ELISA by binding to the solid-phase anti-IFN-γ capture antibody (1 μg/mL) and detection through the biotinylated anti-IFN-γ detection antibody (0.5 μg/mL) (BD Biosciences). The reaction product was visualized by a peroxidase-streptavidin conjugate (1:10,000) and ABTS (Roche Diagnostics).
Suppression of tumor growth by IL12-CAR T cells
Rag2−/− cγ−/− mice (Charles River, Sulzfeld, Germany) (six to nine animals per group) were subcutaneously inoculated with Skov3 ovarian cancer cells (5 × 106 cells/animal). Seven days after tumor inoculation CAR T cells were injected intravenously (5 × 106 cells/animal). T cells without CAR served as control. Tumor volumes were recorded every 2–3 days by digital caliper measurements. Area under the curve (AUC) was determined as described54 and significant differences were determined by the Student's t test. Mouse experiments were done under the approval of the local animal welfare board.
Statistics
Experimental results from independent representative experiments are reported as mean values plus or minus standard deviation (SD). Significance analyses were performed by the two-sided Student's t test using Microsoft Excel and GraphPad Prism, respectively.
Acknowledgments
The authors would like to thank Birgit Hops, Petra Hofmann, Nicole Riet, Danuta Chrobok, Charlotte Schenkel, and Anja Pavlica for excellent technical assistance. This work was supported by grants from the German Federal Ministry of Education and Research through the CD20CAR Time project (Fkz 01EK1507A-C to H.A.) within the funding program Innovations for Individualized Medicine.
Author contributions
A.H., M.B., L.H., G.R., and A.S. conducted experiments. A.H., M.C., and H.A. designed the experiments, interpreted the data, and wrote the manuscript.
Declaration of interests
A.H. and H.A. are inventors of a patent application covering the IL12-CAR design; the paper may affect the value of the patent application. The remaining authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.10.011.
Supplemental information
References
- 1.Majzner R.G., Mackall C.L. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018;8:1219–1226. doi: 10.1158/2159-8290.CD-18-0442. [DOI] [PubMed] [Google Scholar]
- 2.Chen N., Li X., Chintala N.K., Tano Z.E., Adusumilli P.S. Driving CARs on the uneven road of antigen heterogeneity in solid tumors. Curr. Opin. Immunol. 2018;51:103–110. doi: 10.1016/j.coi.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chmielewski M., Kopecky C., Hombach A.A., Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011;71:5697–5706. doi: 10.1158/0008-5472.CAN-11-0103. [DOI] [PubMed] [Google Scholar]
- 4.Heuser C., Diehl V., Abken H., Hombach A. Anti-CD30-IL-12 antibody-cytokine fusion protein that induces IFN-gamma secretion of T cells and NK cell-mediated lysis of Hodgkin’s lymphoma-derived tumor cells. Int. J. Cancer. 2003;106:545–552. doi: 10.1002/ijc.11279. [DOI] [PubMed] [Google Scholar]
- 5.Wang Y., Lin Y.-X., Qiao S.-L., An H.-W., Ma Y., Qiao Z.-Y., Rajapaksha R.P.Y.J., Wang H. Polymeric nanoparticles promote macrophage reversal from M2 to M1 phenotypes in the tumor microenvironment. Biomaterials. 2017;112:153–163. doi: 10.1016/j.biomaterials.2016.09.034. [DOI] [PubMed] [Google Scholar]
- 6.Kunert A., Chmielewski M., Wijers R., Berrevoets C., Abken H., Debets R. Intra-tumoral production of IL18, but not IL12, by TCR-engineered T cells is non-toxic and counteracts immune evasion of solid tumors. Oncoimmunology. 2017;7:e1378842. doi: 10.1080/2162402X.2017.1378842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liaskou E., Patel S.R., Webb G., Bagkou Dimakou D., Akiror S., Krishna M., Mells G., Jones D.E., Bowman S.J., Barone F., et al. Increased sensitivity of Treg cells from patients with PBC to low dose IL-12 drives their differentiation into IFN-γ secreting cells. J. Autoimmun. 2018;94:143–155. doi: 10.1016/j.jaut.2018.07.020. [DOI] [PubMed] [Google Scholar]
- 8.Bowers J.S., Nelson M.H., Kundimi S., Bailey S.R., Huff L.W., Schwartz K.M., Cole D.J., Rubinstein M.P., Paulos C.M. Dendritic cells in irradiated mice trigger the functional plasticity and antitumor activity of adoptively transferred Tc17 cells via IL12 signaling. Clin. Cancer Res. 2015;21:2546–2557. doi: 10.1158/1078-0432.CCR-14-2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koneru M., Purdon T.J., Spriggs D., Koneru S., Brentjens R.J. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology. 2015;4:e994446. doi: 10.4161/2162402X.2014.994446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kueberuwa G., Kalaitsidou M., Cheadle E., Hawkins R.E., Gilham D.E. CD19 CAR T cells expressing IL-12 eradicate lymphoma in fully lymphoreplete mice through induction of host immunity. Mol. Ther. Oncolytics. 2018;8:41–51. doi: 10.1016/j.omto.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sangro B., Mazzolini G., Ruiz J., Herraiz M., Quiroga J., Herrero I., Benito A., Larrache J., Pueyo J., Subtil J.C., et al. Phase I trial of intratumoral injection of an adenovirus encoding interleukin-12 for advanced digestive tumors. J. Clin. Oncol. 2004;22:1389–1397. doi: 10.1200/JCO.2004.04.059. [DOI] [PubMed] [Google Scholar]
- 12.Motzer R.J., Rakhit A., Schwartz L.H., Olencki T., Malone T.M., Sandstrom K., Nadeau R., Parmar H., Bukowski R. Phase I trial of subcutaneous recombinant human interleukin-12 in patients with advanced renal cell carcinoma. Clin. Cancer Res. 1998;4:1183–1191. [PubMed] [Google Scholar]
- 13.Leonard J.P., Sherman M.L., Fisher G.L., Buchanan L.J., Larsen G., Atkins M.B., Sosman J.A., Dutcher J.P., Vogelzang N.J., Ryan J.L. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood. 1997;90:2541–2548. [PubMed] [Google Scholar]
- 14.Kofler D.M., Chmielewski M., Rappl G., Hombach A., Riet T., Schmidt A., Hombach A.A., Wendtner C.-M., Abken H. CD28 costimulation impairs the efficacy of a redirected T-cell antitumor attack in the presence of regulatory T cells which can be overcome by preventing lck activation. Mol. Ther. 2011;19:760–767. doi: 10.1038/mt.2011.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lund R.J., Chen Z., Scheinin J., Lahesmaa R. Early target genes of IL-12 and STAT4 signaling in Th cells. J. Immunol. 2004;172:6775–6782. doi: 10.4049/jimmunol.172.11.6775. [DOI] [PubMed] [Google Scholar]
- 16.Germann T., Gately M.K., Schoenhaut D.S., Lohoff M., Mattner F., Fischer S., Jin S.C., Schmitt E., Rüde E. Interleukin-12/T cell stimulating factor, a cytokine with multiple effects on T helper type 1 (Th1) but not on Th2 cells. Eur. J. Immunol. 1993;23:1762–1770. doi: 10.1002/eji.1830230805. [DOI] [PubMed] [Google Scholar]
- 17.Jeannin P., Delneste Y., Seveso M., Life P., Bonnefoy J.Y. IL-12 synergizes with IL-2 and other stimuli in inducing IL-10 production by human T cells. J. Immunol. 1996;156:3159–3165. [PubMed] [Google Scholar]
- 18.Elgaaen B.V., Haug K.B.F., Wang J., Olstad O.K., Fortunati D., Onsrud M., Staff A.C., Sauer T., Gautvik K.M. POLD2 and KSP37 (FGFBP2) correlate strongly with histology, stage and outcome in ovarian carcinomas. PLoS One. 2010;5:e13837. doi: 10.1371/journal.pone.0013837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaspar A.A., Okada S., Kumar J., Poulain F.R., Drouvalakis K.A., Kelekar A., Hanson D.A., Kluck R.M., Hitoshi Y., Johnson D.E., et al. A distinct pathway of cell-mediated apoptosis initiated by granulysin. J. Immunol. 2001;167:350–356. doi: 10.4049/jimmunol.167.1.350. [DOI] [PubMed] [Google Scholar]
- 20.Schmidt-Wolf I.G., Negrin R.S., Kiem H.P., Blume K.G., Weissman I.L. Use of a SCID mouse/human lymphoma model to evaluate cytokine-induced killer cells with potent antitumor cell activity. J. Exp. Med. 1991;174:139–149. doi: 10.1084/jem.174.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zoll B., Lefterova P., Ebert O., Huhn D., Von Ruecker A., Schmidt-Wolf I.G. Modulation of cell surface markers on NK-like T lymphocytes by using IL-2, IL-7 or IL-12 in vitro stimulation. Cytokine. 2000;12:1385–1390. doi: 10.1006/cyto.2000.0733. [DOI] [PubMed] [Google Scholar]
- 22.Juelke K., Killig M., Luetke-Eversloh M., Parente E., Gruen J., Morandi B., Ferlazzo G., Thiel A., Schmitt-Knosalla I., Romagnani C. CD62L expression identifies a unique subset of polyfunctional CD56dim NK cells. Blood. 2010;116:1299–1307. doi: 10.1182/blood-2009-11-253286. [DOI] [PubMed] [Google Scholar]
- 23.Balin S.J., Pellegrini M., Klechevsky E., Won S.T., Weiss D.I., Choi A.W., Hakimian J., Lu J., Ochoa M.T., Bloom B.R., et al. Human antimicrobial cytotoxic T lymphocytes, defined by NK receptors and antimicrobial proteins, kill intracellular bacteria. Sci. Immunol. 2018;3:eaat7668. doi: 10.1126/sciimmunol.aat7668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Braud V.M., Allan D.S., O’Callaghan C.A., Söderström K., D’Andrea A., Ogg G.S., Lazetic S., Young N.T., Bell J.I., Phillips J.H., et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature. 1998;391:795–799. doi: 10.1038/35869. [DOI] [PubMed] [Google Scholar]
- 25.Marín R., Ruiz-Cabello F., Pedrinaci S., Méndez R., Jiménez P., Geraghty D.E., Garrido F. Analysis of HLA-E expression in human tumors. Immunogenetics. 2003;54:767–775. doi: 10.1007/s00251-002-0526-9. [DOI] [PubMed] [Google Scholar]
- 26.Hoogstad-van Evert J.S., Cany J., van den Brand D., Oudenampsen M., Brock R., Torensma R., Bekkers R.L., Jansen J.H., Massuger L.F., Dolstra H. Umbilical cord blood CD34+ progenitor-derived NK cells efficiently kill ovarian cancer spheroids and intraperitoneal tumors in NOD/SCID/IL2Rgnull mice. Oncoimmunology. 2017;6:e1320630. doi: 10.1080/2162402X.2017.1320630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Enqvist M., Nilsonne G., Hammarfjord O., Wallin R.P.A., Björkström N.K., Björnstedt M., Hjerpe A., Ljunggren H.-G., Dobra K., Malmberg K.-J., et al. Selenite induces posttranscriptional blockade of HLA-E expression and sensitizes tumor cells to CD94/NKG2A-positive NK cells. J. Immunol. 2011;187:3546–3554. doi: 10.4049/jimmunol.1100610. [DOI] [PubMed] [Google Scholar]
- 28.Law K.S., Chen H.-C., Liao S.-K. Non-cytotoxic and sublethal paclitaxel treatment potentiates the sensitivity of cultured ovarian tumor SKOV-3 cells to lysis by lymphokine-activated killer cells. Anticancer Res. 2007;27:841–850. [PubMed] [Google Scholar]
- 29.Xu D., Gu P., Pan P.-Y., Li Q., Sato A.I., Chen S.-H. NK and CD8+ T cell-mediated eradication of poorly immunogenic B16-F10 melanoma by the combined action of IL-12 gene therapy and 4-1BB costimulation. Int. J. Cancer. 2004;109:499–506. doi: 10.1002/ijc.11696. [DOI] [PubMed] [Google Scholar]
- 30.Franceschetti M., Pievani A., Borleri G., Vago L., Fleischhauer K., Golay J., Introna M. Cytokine-induced killer cells are terminally differentiated activated CD8 cytotoxic T-EMRA lymphocytes. Exp. Hematol. 2009;37:616–628.e2. doi: 10.1016/j.exphem.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 31.Tian G., Courtney A.N., Jena B., Heczey A., Liu D., Marinova E., Guo L., Xu X., Torikai H., Mo Q., et al. CD62L+ NKT cells have prolonged persistence and antitumor activity in vivo. J. Clin. Invest. 2016;126:2341–2355. doi: 10.1172/JCI83476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leuci V., Casucci G.M., Grignani G., Rotolo R., Rossotti U., Vigna E., Gammaitoni L., Mesiano G., Fiorino E., Donini C., et al. CD44v6 as innovative sarcoma target for CAR-redirected CIK cells. Oncoimmunology. 2018;7:e1423167. doi: 10.1080/2162402X.2017.1423167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zuo S., Wen Y., Panha H., Dai G., Wang L., Ren X., Fu K. Modification of cytokine-induced killer cells with folate receptor alpha (FRα)-specific chimeric antigen receptors enhances their antitumor immunity toward FRα-positive ovarian cancers. Mol. Immunol. 2017;85:293–304. doi: 10.1016/j.molimm.2017.03.017. [DOI] [PubMed] [Google Scholar]
- 34.Hombach A.A., Rappl G., Abken H. Arming cytokine-induced killer cells with chimeric antigen receptors: CD28 outperforms combined CD28-OX40 “super-stimulation. Mol. Ther. 2013;21:2268–2277. doi: 10.1038/mt.2013.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Magnani C.F., Gaipa G., Lussana F., Belotti D., Gritti G., Napolitano S., Matera G., Cabiati B., Buracchi C., Borleri G., et al. Sleeping Beauty-engineered CAR T cells achieve antileukemic activity without severe toxicities. J. Clin. Invest. 2020;130:6021–6033. doi: 10.1172/JCI138473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schlimper C., Hombach A.A., Abken H., Schmidt-Wolf I.G.H. Improved activation toward primary colorectal cancer cells by antigen-specific targeting autologous cytokine-induced killer cells. Clin. Dev. Immunol. 2012;2012:238924. doi: 10.1155/2012/238924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y., Zhou Y., Huang K.-H., Fang X., Li Y., Wang F., An L., Chen Q., Zhang Y., Shi A., et al. Targeting epidermal growth factor-overexpressing triple-negative breast cancer by natural killer cells expressing a specific chimeric antigen receptor. Cell Prolif. 2020;53:e12858. doi: 10.1111/cpr.12858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu E., Marin D., Banerjee P., Macapinlac H.A., Thompson P., Basar R., Nassif Kerbauy L., Overman B., Thall P., Kaplan M., et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 2020;382:545–553. doi: 10.1056/NEJMoa1910607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lasek W., Zagożdżon R., Jakobisiak M. Interleukin 12: still a promising candidate for tumor immunotherapy? Cancer Immunol. Immunother. 2014;63:419–435. doi: 10.1007/s00262-014-1523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang P., Li X., Wang J., Gao D., Li Y., Li H., Chu Y., Zhang Z., Liu H., Jiang G., et al. Re-designing interleukin-12 to enhance its safety and potential as an anti-tumor immunotherapeutic agent. Nat. Commun. 2017;8:1395. doi: 10.1038/s41467-017-01385-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pan W.-Y., Lo C.-H., Chen C.-C., Wu P.-Y., Roffler S.R., Shyue S.-K., Tao M.-H. Cancer immunotherapy using a membrane-bound interleukin-12 with B7-1 transmembrane and cytoplasmic domains. Mol. Ther. 2012;20:927–937. doi: 10.1038/mt.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Graham F.L., Smiley J., Russell W.C., Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977;36:59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- 43.Hombach A., Koch D., Sircar R., Heuser C., Diehl V., Kruis W., Pohl C., Abken H. A chimeric receptor that selectively targets membrane-bound carcinoembryonic antigen (mCEA) in the presence of soluble CEA. Gene Ther. 1999;6:300–304. doi: 10.1038/sj.gt.3300813. [DOI] [PubMed] [Google Scholar]
- 44.Hombach A., Hombach A.A., Abken H. Adoptive immunotherapy with genetically engineered T cells: modification of the IgG1 Fc “spacer” domain in the extracellular moiety of chimeric antigen receptors avoids “off-target” activation and unintended initiation of an innate immune response. Gene Ther. 2010;17:1206–1213. doi: 10.1038/gt.2010.91. [DOI] [PubMed] [Google Scholar]
- 45.Weijtens M.E., Willemsen R.A., Hart E.H., Bolhuis R.L. A retroviral vector system “STITCH” in combination with an optimized single chain antibody chimeric receptor gene structure allows efficient gene transduction and expression in human T lymphocytes. Gene Ther. 1998;5:1195–1203. doi: 10.1038/sj.gt.3300696. [DOI] [PubMed] [Google Scholar]
- 46.Hombach A., Wieczarkowiecz A., Marquardt T., Heuser C., Usai L., Pohl C., Seliger B., Abken H. Tumor-specific T cell activation by recombinant immunoreceptors: CD3 zeta signaling and CD28 costimulation are simultaneously required for efficient IL-2 secretion and can be integrated into one combined CD28/CD3 zeta signaling receptor molecule. J. Immunol. 2001;167:6123–6131. doi: 10.4049/jimmunol.167.11.6123. [DOI] [PubMed] [Google Scholar]
- 47.Hombach A., Heuser C., Gerken M., Fischer B., Lewalter K., Diehl V., Pohl C., Abken H. T cell activation by recombinant FcepsilonRI gamma-chain immune receptors: an extracellular spacer domain impairs antigen-dependent T cell activation but not antigen recognition. Gene Ther. 2000;7:1067–1075. doi: 10.1038/sj.gt.3301195. [DOI] [PubMed] [Google Scholar]
- 48.Jiang C., Magee D.M., Cox R.A. Construction of a single-chain interleukin-12-expressing retroviral vector and its application in cytokine gene therapy against experimental coccidioidomycosis. Infect. Immun. 1999;67:2996–3001. doi: 10.1128/iai.67.6.2996-3001.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heuser C., Ganser M., Hombach A., Brand H., Denton G., Hanisch F.-G., Abken H. An anti-MUC1-antibody-interleukin-2 fusion protein that activates resting NK cells to lysis of MUC1-positive tumour cells. Br. J. Cancer. 2003;89:1130–1139. doi: 10.1038/sj.bjc.6601267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Golumba-Nagy V., Kuehle J., Abken H. Genetic modification of T cells with chimeric antigen receptors: a laboratory manual. Hum. Gene Ther. Methods. 2017;28:302–309. doi: 10.1089/hgtb.2017.083. [DOI] [PubMed] [Google Scholar]
- 51.Hombach A.A., Görgens A., Chmielewski M., Murke F., Kimpel J., Giebel B., Abken H. Superior therapeutic index in lymphoma therapy: CD30(+) CD34(+) hematopoietic stem cells resist a chimeric antigen receptor T-cell attack. Mol. Ther. 2016;24:1423–1434. doi: 10.1038/mt.2016.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jost L.M., Kirkwood J.M., Whiteside T.L. Improved short- and long-term XTT-based colorimetric cellular cytotoxicity assay for melanoma and other tumor cells. J. Immunol. Methods. 1992;147:153–165. doi: 10.1016/s0022-1759(12)80003-2. [DOI] [PubMed] [Google Scholar]
- 54.Duan F., Simeone S., Wu R., Grady J., Mandoiu I., Srivastava P.K. Area under the curve as a tool to measure kinetics of tumor growth in experimental animals. J. Immunol. Methods. 2012;382:224–228. doi: 10.1016/j.jim.2012.06.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.