

Abstract
Cell death plays an important role during pathogen infections. Here, we report that interferon-γ (IFNγ) sensitizes macrophages to Toll-like receptor (TLR)-induced death that requires macrophage-intrinsic death ligands and caspase-8 enzymatic activity, which trigger the mitochondrial apoptotic effectors, BAX and BAK. The pro-apoptotic caspase-8 substrate BID was dispensable for BAX and BAK activation. Instead, caspase-8 reduced pro-survival BCL-2 transcription and increased inducible nitric oxide synthase (iNOS), thus facilitating BAX and BAK signaling. IFNγ-primed, TLR-induced macrophage killing required iNOS, which licensed apoptotic caspase-8 activity and reduced the BAX and BAK inhibitors, A1 and MCL-1. The deletion of iNOS or caspase-8 limited SARS-CoV-2-induced disease in mice, while caspase-8 caused lethality independent of iNOS in a model of hemophagocytic lymphohistiocytosis. These findings reveal that iNOS selectively licenses programmed cell death, which may explain how nitric oxide impacts disease severity in SARS-CoV-2 infection and other iNOS-associated inflammatory conditions.
Keywords: Toll-like receptor, interferon, TNF, apoptosis, BAX and BAK, caspase-8, iNOS, SARS-CoV-2, COVID-19, hemophagocytic lymphohistiocytosis
Graphical abstract
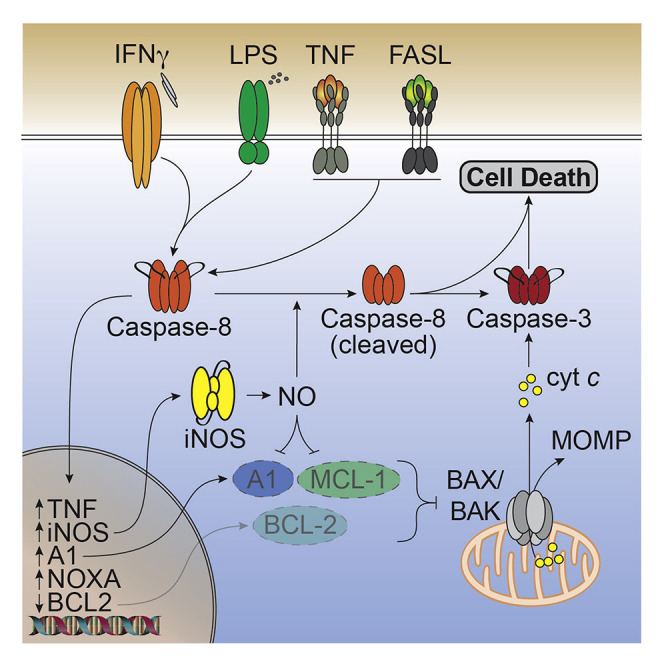
IFNγ modulates TLR signaling in macrophages, but the mechanism behind how their combined actions enact cell death is unclear. Simpson and colleagues demonstrate that both inducible nitric oxide synthase (iNOS) and caspase-8 promote mitochondrial-driven cell death during IFNγ and TLR signaling and increase SARS-CoV-2 disease severity.
Introduction
Macrophages are front-line sentinels that are essential for the rapid host response against invading pathogens (Ginhoux et al., 2016; Wynn et al., 2013). Through the Toll-like receptor (TLR) family, macrophages detect pathogen molecules to coordinate an appropriate cellular response that includes the production of inflammatory cytokines and chemokines that initiate immune cell infiltration and activation at sites of infection (Takeuchi and Akira, 2010). T lymphocytes and natural killer cells act in concert with macrophages by, in part, producing the pro-inflammatory cytokine, interferon-gamma (IFNγ) (Ivashkiv, 2018). IFNγ has a profound impact on the consequences of TLR activation by augmenting macrophage anti-pathogen signaling (Schroder et al., 2006), and their combined actions orchestrate the innate immune response (Hu et al., 2008; Hu and Ivashkiv, 2009; Su et al., 2015).
TLR signaling can also induce programmed cell death (Salaun et al., 2007) that contextually can either confer protection from pathogens by eliminating infected cells (Chow et al., 2016; Doerflinger et al., 2020; Jorgensen et al., 2017; Orzalli et al., 2021; Speir et al., 2016; Suzuki et al., 2018), or increase immunogenic molecules being released from cells, which in some circumstances can cause pathological inflammation and tissue damage (Wallach et al., 2014). Indeed, mammalian genetic variants that increase cell death signaling cause severe inflammation (Alehashemi and Goldbach-Mansky, 2020; Hildebrand et al., 2020; Lalaoui et al., 2020; Newton et al., 2019a; Rickard et al., 2014a; Taraborrelli et al., 2018). Even apoptotic cell death mediated by the death-receptor-activated caspase-8, or through the mitochondrial apoptotic effectors BAX and BAK (BAX/BAK), which have typically been considered immunogenically silent modalities, can result in the activation of inflammatory cytokines (Bossaller et al., 2012; Chauhan et al., 2018; Feltham et al., 2017; Gurung et al., 2014; Taraborrelli et al., 2018; Vince et al., 2018; Vince et al., 2012). Therefore, cell death signaling is specifically primed in macrophages that recognize pathogens and is heavily regulated to tailor the innate immune response.
IFNγ can also prime programmed cell death signaling. IFNγ sensitizes fibroblasts, hepatocytes, and keratinocytes to necroptotic cell death that is dependent on the pseudokinase, mixed lineage kinase domain-like (MLKL) (Günther et al., 2016; Lauffer et al., 2018; Thapa et al., 2011; Weir et al., 2021). Similarly, IFNγ may promote caspase-1-mediated cell killing (Chin et al., 1997) or death receptor-induced apoptosis in some cancer-derived cell lines (Takeda et al., 2002; Tanzer et al., 2017; Xu et al., 1998). Patients suffering from the pathogen-associated hyper-inflammatory condition, hemophagocytic lymphohistiocytosis (HLH), can benefit from IFNγ blockade (Locatelli et al., 2020), while certain pathogens including Mycobacterium tuberculosis and Legionella pneumophilia have been reported to inhibit IFNγ receptor signaling (Fortune et al., 2004; Kincaid and Ernst, 2003; Yang et al., 2020). While IFNγ may contribute to the death of infected cells (Herbst et al., 2011; Janssen et al., 2002; Niedelman et al., 2013; Sedger et al., 1999), how IFNγ might invoke immune cell death and the consequences thereof remain incompletely understood.
In this study, we sought to understand how IFNγ combines with pathogen-derived TLR ligands to alter macrophage fate. We reveal that IFNγ and TLR activation triggered macrophage death involving the death-receptor-induced caspase-8 and BAX/BAK-driven mitochondrial apoptotic processes, which were both licensed by iNOS via its enzymatic product nitric oxide (NO). Caspase-8 promoted BAX/BAK activation independent of the caspase-8 substrate BID by, in part, transcriptionally regulating BCL-2 and iNOS, the latter of which reduced the amount of the pro-survival proteins, MCL-1 and A1. In mice, the deletion of Nos2 or Casp8 reduced SARS-CoV-2 infection-driven weight loss without impacting peak viral burdens, thereby implicating an iNOS-caspase-8 cell death axis in damaging host immune responses. These findings identify multiple targets of iNOS that explain how nitric oxide sculpts specific programmed cell death responses following the cellular sensing of host-derived cytokines and pathogen ligands.
Results
Sustained activation of macrophages by IFNγ and TLR signaling triggers cell death
To examine how IFNγ signaling might alter macrophage fate during pathogen infections, we primed wild-type (WT) bone-marrow-derived macrophages (BMDMs) with IFNγ followed by treatment with the TLR4 agonist lipopolysaccharide (LPS), the TLR1 and TLR2 heterodimer agonist Pam3CysSK4 (P3C), or the TLR3 agonist polyinosinic:polycytidylic acid (PolyI:C). Only IFNγ primed cells underwent significant TLR-induced macrophage death (Figure 1 A), which was not recapitulated when cells were primed with IFNβ (Figure 1B). IFNγ-primed BMDMs stimulated with LPS (hereafter termed IFNγ/LPS) exhibited apoptotic body formation and were often observed to be phagocytosed by neighboring cells (Figure S1A and Videos S1, S2, S3, and S4) and, as expected, presented a typical “activated” phenotype with increased Nos2, Il1b, Tnf, and CD86, and reduced Ym1 expression (Figure S1B). Unstimulated BMDMs (F4/80hi:CD11bhi:CD206hi) became MHCIIhi in the presence of IFNγ (Figures S1B–S1D), and IFNγ/LPS treatment caused an increase in both inflammatory TNF and HMGB1 release (Figure S1E and S1F). Priming cells with LPS prior to IFNγ stimulation did not cause cell death, while IFNγ/LPS co-stimulation only resulted in macrophage death by 48 h (Figure S1G).
Figure 1.

IFNγ primes macrophages for TLR-induced cell death
(A and B) Wild-type (WT) bone-marrow-derived macrophages (BMDMs) were treated with (A) IFNγ (50 ng/mL) or (B) IFNβ (1,000 U/mL, n = 5) overnight, then with either LPS (50 ng/mL, n = 5), Pam3CSK4 (P3C, 500 ng/mL, n = 6), or PolyI:C (10 μg/mL, n = 6) for 24 h. Cell death was assessed by propidium iodide (PI) exclusion as measured by flow cytometry.
(C) WT or Tnf−/−Faslgld/gldTrail−/− BMDMs were treated with IFNγ (50 ng/mL) overnight then with LPS (50 ng/mL) (left), or P3C (500 ng/mL) (right) for 24 or 48 h ± TNF (100 ng/mL). Cell death was assessed by PI exclusion as measured by flow cytometry (n = 5).
(D) Immunoblot of WT BMDMs treated with IFNγ (50 ng/mL) overnight then with LPS (50 ng/mL) for 8, 12, or 24 h. Treatment with the BCL-2, BCL-XL, and BCL-W inhibitor, ABT-737 (737, 1 μM) and cycloheximide (CHX, 10 μg/mL) ± Q-VD-OPh (QVD, 20 μM) for 4 h was used as a control (n = 3).
(E) Immunoblot of Tnf−/−Faslgld/gldTrail−/− BMDMs that were treated with IFNγ (50 ng/mL) overnight then with LPS (50 ng/mL) for 12, 24, or 48 h (n = 2).
Data represent the mean value ± SD, or a representative immunoblot, from n independent experiments. p > 0.05 (n.s.), p ≤ 0.001 (∗∗∗), p ≤ 0.0001 (∗∗∗∗). See also Figures S1 and S2 and Videos S1, S2, S3, and S4.
WT BMDMs were stimulated with IFNγ (50 ng/mL) and imaged live every 20 min for 24 h. Composite video of brightfield (gray) and PI stain (red) is shown. Magnification is set at 20x, scale bar (white): 100 μm. Related to Figure 1.
WT BMDMs were primed with IFNγ (50 ng/mL) overnight and then stimulated with LPS (50 ng/mL) and imaged live every 20 min for 24 h. Imaging began at LPS time 0 h (e.g., after IFNγ priming). Composite video of brightfield (gray) and PI stain (red) is shown. Magnification is set at 20x, scale bar (white): 100 μm. Related to Figure 1.
WT BMDMs were stimulated with ABT-737 (1 μM) plus cycloheximide (CHX, 10 μg/mL) to trigger BAX/BAK dependent apoptosis and imaged live every 20 min for 24 h. Composite video of brightfield (gray) and PI stain (red) is shown. Magnification is set at 20x, scale bar (white): 100 μm. Related to Figure 1.
WT BMDMs were primed with IFNγ (50 ng/mL) overnight and then stimulated with LPS (50 ng/mL) and imaged live every 15 min for 24 h. Imaging began at LPS time 0 h (after IFNγ priming). Composite video of brightfield (gray) and PI stain (red) is shown. Magnification is set at 63x, scale bar (white): 10 μm. Related to Figures 1 and S1A.
We next tested whether death ligands might contribute to IFNγ and TLR-induced macrophage death. Co-deletion of TNF, TRAIL, and FASL signaling (Tnf −/− Fasl gld/gld Trail −/−) conveyed significant protection to IFNγ/LPS or IFNγ priming followed by P3C (IFNγ/P3C)-mediated killing that could be restored by the addition of exogenous TNF (Figure 1C). Tnfr1, Tnfr2, or Tnf deletion alone did not prevent IFNγ/LPS- or IFNγ/P3C-induced cell death (Figure S2A). However, Tnfr1, Tnfr2, or Tnf deletion reduced IFNγ/LPS-induced BMDM death when experiments were performed in the absence of macrophage colony stimulating factor (M-CSF) containing L929 cell-conditioned medium (Figure S2A). Concurrently, increased mRNA expression of the death ligand Fasl—but not Fas, Tnfsf10 (TRAIL), nor Tnfrsf10b (DR5)—was only observed when IFNγ/LPS-stimulated BMDMs were cultured in L929 cell-conditioned medium (Figure S2B). These data indicate that IFNγ priming sensitizes BMDMs to TLR-activation-induced cell death that is driven in part by the death ligand TNF and via expression of Fasl.
To further discern the mode of IFNγ/LPS-induced macrophage killing, we assessed relevant markers of cell death activation by immunoblot. IFNγ/LPS stimulation triggered the activation-associated processing of caspase-8, caspase-9, and caspase-3 that were all reduced in Tnf −/− Fasl gld/gld Trail −/− macrophages (Figures 1D and 1E). We also observed cleavage of pyroptotic Gasdermin D (GSDMD) into its pore-forming p30 fragment and the inactive p43 fragment (Figure 1D) (Kayagaki et al., 2015; Shi et al., 2015; Taabazuing et al., 2017). Although caspase-8 cleavage and cell death is often associated with loss of the cellular or X-linked inhibitor of apoptosis proteins (cIAP1, cIAP2, and XIAP) (Lawlor et al., 2017; Lawlor et al., 2015), reduced cIAP1 and XIAP were only observed upon or after caspase processing, indicating that their loss was likely a consequence of macrophage killing, rather than a cause (Figure 1D).
Efficient IFNγ/LPS-induced cell death requires caspase-8 and the apoptotic effectors BAX/BAK
Our cell death and immunoblot data suggested that TNF- and FASL-mediated activation of caspases should contribute to IFNγ/LPS-induced macrophage death. We therefore generated BMDMs from Casp8 −/− Mlkl −/− and Mlkl −/− control mice to interrogate the function of the critical death receptor initiator caspase, caspase-8. Casp8 −/− Mlkl −/− BMDMs (but not Mlkl −/− BMDMs) were largely protected against IFNγ/LPS-induced killing (Figure 2 A) and displayed reduced caspase-9 and caspase-3 cleavage, resembling the co-deletion of TNF, FASL, and TRAIL signaling (Figure 2B). In agreement with previous work (He et al., 2011; Lawlor et al., 2015; Najjar et al., 2016), Casp8 −/− Mlkl −/− BMDMs were resistant to cell death induced by LPS and the SMAC-mimetic compound, compound A (Cp. A) (Vince et al., 2007), as well as necroptosis triggered by LPS and the broad-spectrum caspase inhibitor Z-VAD-fmk (Z-VAD), while Mlkl −/− BMDMs were only resistant against LPS- and Z-VAD-induced necroptosis (Figure 2A).
Figure 2.

Both extrinsic caspase-8 and the mitochondrial apoptosis effector proteins BAX/BAK contribute to IFNγ/LPS-induced macrophage killing
(A) WT, Mlkl−/−, or Casp8−/−Mlkl−/− BMDMs were treated with IFNγ (50 ng/mL) overnight then with LPS (50 ng/mL) for 24 or 48 h. Treatment with LPS and Compound A (Cp. A, 1 μM) (extrinsic apoptosis) or LPS and Z-VAD-fmk (Z-VAD, 20 μM) (necroptosis) for 24 h were used as controls. Cell death was assessed by PI exclusion as measured by flow cytometry (n = 4).
(B) Immunoblot analysis of WT, Mlkl−/−, or Casp8−/−Mlkl−/− BMDMs that were treated with IFNγ (50 ng/mL) overnight then with LPS (50 ng/mL) for 8, 12 or 24 h. WT BMDMs were treated with LPS and Cp. A for 12 h as a control (n = 3).
(C) WT and Bax−/−Bak−/− BMDMs were treated with IFNγ (50 ng/mL) overnight then with LPS (50 ng/mL) for 24 or 48 h. ABT-737 (1 μM) plus cycloheximide (CHX, 10 μg/mL) treatment for 6 h was used as a control. Cell death was assessed by PI exclusion as measured by flow cytometry (n = 4).
(D) Immunoblot of WT and Bax−/−Bak−/− BMDMs that were treated with IFNγ (50 ng/mL) overnight then with LPS (50 ng/mL) for 12, 24, or 48 h (n = 3).
Data represent the mean value ± SD, or a representative immunoblot, from n independent experiments. p > 0.05 (n.s.), p ≤ 0.05 (∗), p ≤ 0.01 (∗∗), p ≤ 0.001 (∗∗∗), p ≤ 0.0001 (∗∗∗∗). See also Figure S3.
The p30 fragment of GSDMD, which causes pyroptosis, was also reduced in the absence of caspase-8 (Figure 2B). However, GSDMD was not required for IFNγ/LPS-induced macrophage cell death (Figure S3A), neither were the inflammatory caspases, caspase-1 and 11, NLRP3, nor the IFN-inducible guanylate-binding proteins (GBP2, GBP4, GBP8, GBP9, and GBP5) that expose pathogens to the cell death machinery (Figures S3B–S3E). Moreover, neither ferroptosis inhibition with Ferrostatin-1 or Deferiprone, nor the combined deletion of pyroptotic effectors Gsdmd and Gsdme, had any impact on IFNγ/LPS-induced macrophage death (Figures S3F–S3G). Similarly, the deletion of cell-lysis-associated ninjurin-1 (Ninj1) protein in immortalized BMDMs (iBMDMs) did not abrogate IFNγ/LPS-killing, despite blocking LDH release into the cell supernatant (Figures S3H–S3J) (Kayagaki et al., 2021).
Caspase-9 cleavage, which is associated with BAX- and BAK-mediated mitochondrial apoptosis, was also observed in IFNγ/LPS stimulated BMDMs (Figures 1D and 1E and 2B). We therefore generated Bak −/− Bax −/− BMDMs to definitively test the contribution of mitochondrial apoptosis to IFNγ/LPS killing. These cells were protected against mitochondrial apoptosis induced by the BH3-mimetic ABT-737 (van Delft et al., 2006) and CHX (Vince et al., 2018) and were significantly resistant to IFNγ/LPS-induced macrophage death at 24 h, with some protection provided up to 48 h post-LPS treatment (Figure 2C). This was associated with reduced processing of caspase-9 and delayed processing of caspase-3, while caspase-8 processing remained prevalent even after 48 h of IFNγ/LPS stimulation (Figure 2D). Overall, this genetic analysis shows that IFNγ/LPS treatment elicits a specific caspase-8- and BAX/BAK-dependent cell death that does not require MLKL, GSDMD, GSDME, caspase-1, caspase-11, ferroptosis, nor NINJ1.
BAX/BAK activation from IFNγ/LPS stimulation is driven by caspase-8, but not the caspase-8 substrate BID
Casp8−/−Mlkl−/− BMDMs exhibited reduced caspase-9 cleavage and a more sustained resistance to IFNγ/LPS-induced cell death than Bak −/− Bax −/− BMDMs, suggesting that caspase-8 contributes to the activation of BAX/BAK. Cytochrome c release from the inter-mitochondrial space into the cytosol is a hallmark of BAX/BAK-mediated apoptosis (Czabotar et al., 2014). We therefore assessed the amount of mitochondrial cytochrome c in WT, Mlkl −/− and Casp8 −/− Mlkl −/− BMDMs to understand if caspase-8 promoted BAX/BAK activation during IFNγ/LPS killing. As expected, the chemical activation of BAX/BAK with ABT-737 and CHX (Vince et al., 2018) caused a significant reduction in cytochrome c staining (Figure 3 A). WT and Mlkl −/− BMDMs stimulated with IFNγ/LPS lost cytochrome c in a time-dependent manner, while Casp8 −/− Mlkl −/− BMDMs retained cytochrome c (Figure 3A), indicating that caspase-8 promotes both BAX/BAK-dependent and -independent macrophage death induced by IFNγ/LPS signaling.
Figure 3.

Caspase-8 triggers mitochondrial cytochrome c loss and cell death through a BID-independent mechanism following IFNγ/LPS treatment
(A) WT, Mlkl−/−, or Casp8−/−Mlkl−/− BMDMs were treated with IFNγ (50 ng/mL) overnight then with LPS (50 ng/mL) for 12, 24, 36, or 48 h. Treatment with ABT-737 (1 μM) and cycloheximide (CHX, 10 μg/mL) for 5 h was used as a positive control for BAX/BAK activation (see Figure 2C). Cytochrome c retention was measured by intracellular cytochrome c staining and flow cytometric analysis (n = 3).
(B) WT and Bid−/− BMDMs were treated with IFNγ (50 ng/mL) overnight then with LPS (50 ng/mL) for 24 or 48 h. Cell death was assessed by PI exclusion as measured by flow cytometry (n = 6).
(C) Immunoblot of WT and Bid−/− BMDMs that were treated with IFNγ (50 ng/mL) overnight then with LPS (50 ng/mL) for 8 or 16 h. WT BMDMs treated with ABT-737 (1 μM) plus cycloheximide (CHX, 10 μg/mL) for 4 h were used as a positive control (n = 3).
(D) WT or Bid−/− BMDMs were treated with IFNγ (50 ng/mL) overnight then with LPS (50 ng/mL) for 16 or 24 h. Treatment with ABT-737 (1 μM) and cycloheximide (CHX, 10 μg/mL) for 6 h was used as a positive control for BAX/BAK activation. Cytochrome c retention was measured by intracellular cytochrome c staining and flow cytometric analysis (n = 3).
(E) Immunoblot analysis of WT BMDMs that had been primed with IFNγ (50 ng/mL) overnight then stimulated with LPS (50 ng/mL) for 8, 12, or 24 h (n = 3).
Data represent the mean value ± SD, or a representative immunoblot, from n independent experiments. p > 0.05 (n.s.), p ≤ 0.05 (∗), p ≤ 0.001 (∗∗∗), p ≤ 0.0001 (∗∗∗∗).
Caspase-8 cleaves the BH3-only protein BID, which can subsequently trigger activation of BAX/BAK (Czabotar et al., 2014; Li et al., 1998). However, Bid −/− BMDMs died at a comparable rate to WT macrophages upon IFNγ/LPS treatment (Figure 3B). Consistent with this finding, although IFNγ/LPS resulted in the cleavage of BID, loss of BID did not reduce caspase-3 or caspase-9 processing, nor cytochrome c loss from the mitochondria (Figures 3C and 3D). Therefore, caspase-8 can trigger the activation of BAX/BAK and the mitochondrial apoptotic pathway independently of BID.
MCL-1 and BCL-XL are both crucial for limiting BAX/BAK activation in macrophages (Vince et al., 2018); however, IFNγ/LPS treatment only reduced MCL-1 and not BCL-XL (Figure 3E). MCL-1 loss alone is not sufficient to kill BMDMs (Vince et al., 2018). Therefore BID-independent BAX/BAK activation by IFNγ/LPS likely involves MCL-1 loss and another level of caspase-8-mediated regulation.
IFNγ/LPS triggers caspase-8-mediated transcriptional programming in macrophages to increase pro-apoptotic NOXA and reduce pro-survival BCL-2
We reasoned that caspase-8 might promote BAX/BAK activation by regulating genes that control BAX/BAK activity, particularly as caspase-8 has been reported to alter macrophage gene transcription (Allam et al., 2014; DeLaney et al., 2019; Gitlin et al., 2020; Philip et al., 2016). We therefore performed 3′ mRNA-sequencing, comparing IFNγ/LPS treated Mlkl −/− and Casp8 −/− Mlkl −/− BMDMs. Loss of caspase-8 resulted in significant changes in gene expression upon treatment with IFNγ/LPS (Figures 4 A–4C; S4A and S4B). Gene ontology (GO) analyses revealed several processes that were affected in the absence of caspase-8, including the inflammatory response, cytokine production, and leukocyte migration (Figure S4C).
Figure 4.

Caspase-8 is required for transcriptional programming of macrophages upon stimulation with IFNγ/LPS, resulting in elevated NOXA and reduced BCL-2 transcripts
(A–D) Mlkl−/− and Casp8−/−Mlkl−/− BMDMs were treated with IFNγ (50 ng/mL) overnight then with LPS (50 ng/mL) for 7 h followed by RNA isolation and 3′ mRNA-sequencing. (A) Multidimensional scaling (MDS) plot and (B) differentially expressed genes (DEGs) that are up- or down-regulated in Casp8−/−Mlkl−/− BMDMs in comparison to Mlkl−/− cells are shown. The effect of Casp8 deletion on genes associated with cell death was assessed by (C) a volcano plot and (D) a heatmap plot of DEGs involved in distinct cell death signaling pathways (see Table S1) that are enriched in Mlkl−/− BMDMs versus Casp8−/−Mlkl−/− BMDMs . Adjusted p ≤ 0.05 and cut-off values logFC ≥ 1 or logFC ≤ −1 (n = 3).
(E) Immunoblot of WT, Mlkl−/−, or Casp8−/−Mlkl−/− BMDMs that were treated with IFNγ (50 ng/mL) overnight then with LPS (50 ng/mL) for 2, 4, 6, or 8 h (n = 2).
(F) WT or Casp8−/−Mlkl−/− BMDMs were treated with IFNγ (50 ng/mL) overnight then with LPS (50 ng/mL) for 4, 8, or 16 h. Bcl2 expression was assessed by quantitative PCR (qPCR) using Hprt as a housekeeping gene. Baseline Bcl2 expression (dCT) is shown (right) as a control (n = 3).
(G) Casp8+/+ (WT [circles] or Ripk3−/− [triangles]) or Casp8−/− (Casp8−/−Mlkl−/− [circles] or Casp8−/−Ripk3−/− [triangles]) BMDMs were treated with IFNγ (50 ng/mL) overnight then with LPS (50 ng/mL) ± the BCL-2 inhibitor, ABT-199 (1 μM), or DMSO for 16 or 24 h. Treatments with LPS and Compound A (Cp. A 1 μM) (extrinsic apoptosis) or ABT-737 (1 μM) plus cycloheximide (CHX, 10 μg/mL) (intrinsic apoptosis) were used as controls. Cell death was assessed by PI exclusion as measured by flow cytometry (n = 4).
(H) WT BMDMs were treated with the MCL-1 inhibitor S63845 (10 μM) ± ABT-199 (1 μM) for 12 h. Cell death was assessed by PI exclusion as measured by flow cytometry (n = 3).
Data represent the mean value ± SD, or a representative immunoblot, from n independent experiments. p > 0.05 (n.s.), p ≤ 0.05 (∗), p ≤ 0.01 (∗∗), p ≤ 0.001 (∗∗∗). See also Figures S4 and S5 and Tables S1–S3.
Comparing IFNγ/LPS-treated Casp8 −/− Mlkl −/− to Mlkl −/− control cells, gene set enrichment analysis (GSEA) showed that genes associated with IFNα and IFNγ responses were increased in the absence of caspase-8, and genes associated with inflammatory responses, glycolysis pathways, and apoptosis were decreased in the absence of caspase-8 (Figure S4D). Further prediction analyses performed using the transcriptional regulatory relationships unraveled by sentence-based text mining (TRRUST) database (Han et al., 2018) suggested that caspase-8 promotes the expression of genes regulated by the transcription factors JUN, NF-κB1, FOXO3, RELA, and c-REL, and limits the expression of genes regulated by ETS2, ETV2, JUNB, ESR1, and ETV4 (Figures S5A and S5B). These changes were associated with increased expression of the macrophage-related Lyz1 and Lyz2 genes and reduced inflammatory Il6 gene expression and TNF secretion in caspase-8 deficient cells (Figures S5C and S5D). While c-REL promotes the expression of a subset of caspase-8-dependent genes (DeLaney et al., 2019), Rel −/− BMDMs exhibited equivalent cell death responses to WT BMDMs upon treatment with IFNγ/LPS (Figure S5E). These data show how upon sensing of IFNγ/LPS caspase-8 regulates gene transcription to promote an inflammatory and metabolically active macrophage phenotype.
We next compared the significantly enriched caspase-8-regulated genes to a boutique list of genes associated with apoptosis, pyroptosis, and necroptosis (Figures 4C and 4D; Table S1) and generated a heatmap of cell death-associated genes derived from apoptosis-related GO pathways enriched in our RNA-seq dataset (Figure S5F; Tables S2 and S3). Several of these changes at the mRNA level, including A1 and IL-1β, were validated at the protein level by immunoblot analysis (Figure 4E) or by qPCR in the case of Bcl2 (Figure 4F). Of the cell-death-associated genes, increased expression of the pro-apoptotic BCL-2 family member Pmaip1 (NOXA) and decreased expression of pro-survival Bcl2 (BCL-2) in the presence of caspase-8 correlated with BAX/BAK activation and apoptosis in WT macrophages treated with IFNγ/LPS (Figures 4C and 4D).
NOXA can promote proteasome-dependent degradation of MCL-1 (Willis et al., 2005); however, NOXA-deficient (Pmaip1 −/−) BMDMs died after IFNγ/LPS stimulation at a rate comparable to WT BMDMs (Figure S5G). Moreover, MCL-1 loss occurred as efficiently in Casp8 −/− Mlkl −/− BMDMs as in WT or Mlkl −/− BMDMs (Figure 4E). In contrast, the clinically approved BCL-2 inhibitor venetoclax (ABT-199) (Souers et al., 2013) was able to partially restore IFNγ/LPS-induced killing of BMDMs that was prevented in the absence of caspase-8 (Figure 4G). This suggests that BCL-2 restrains BAX/BAK to prevent apoptosis of BMDMs exposed to IFNγ/LPS, and that caspase-8-mediated transcriptional silencing of Bcl2 contributes to IFNγ/LPS-induced killing of these cells. In agreement, in the absence of IFNγ/LPS treatment, combined BCL-2 and MCL-1 inhibition was sufficient to cause some macrophage death (Figure 4H). Therefore, IFNγ/LPS likely results in caspase-8-mediated transcriptional suppression of Bcl2 that combines with MCL-1 depletion to facilitate BAX/BAK activation and subsequent apoptotic cell death.
Caspase-8 catalytic activity promotes apoptosis of BMDMs induced by treatment with IFNγ/LPS
Caspase-8 controls both cell death and gene transcription via a scaffolding role and/or its enzymatic activity (DeLaney et al., 2019; Fritsch et al., 2019; Gitlin et al., 2020; Kang et al., 2015; Newton et al., 2019b). To test if caspase-8 catalytic activity was required for IFNγ/LPS-induced cell killing, we stimulated BMDMs from enzymatically inactive caspase-8 (Casp8 C362S/C362S Ripk3 −/−) or control (WT, Ripk3 −/− and Casp8 −/− Ripk3 −/−) animals (Fritsch et al., 2019). Comparable with Mlkl −/− and Casp8 −/− Mlkl −/− BMDMs, Ripk3 −/−, Casp8 −/− Ripk3 −/−, and Casp8 C362S/C362S Ripk3 −/− macrophages were resistant to necroptosis (LPS and caspase inhibition with IDN-6556 (IDN)), while Casp8 −/− Ripk3 −/− and Casp8 C362S/C362S Ripk3 −/− were also resistant to extrinsic apoptosis (LPS and IAP inhibition with Cp. A) (Figure 5 A). All genotypes were sensitive to mitochondrial BAX/BAK-driven apoptosis (ABT-737 and CHX) (Figure 5A). Enzymatic inactivation of caspase-8 provided protection from IFNγ/LPS-induced macrophage death and mitochondrial cytochrome c release to almost the same extent as Casp8 deletion (Figures 5A and 5B). IFNγ/LPS stimulated Casp8 −/− Ripk3 −/− BMDMs also displayed reduced caspase-9 p17 and caspase-3 p17 fragments compared to control cells, while Casp8 C362S/C362S Ripk3 −/− BMDMs had an intermediate effect (Figure 5C). The residual caspase-9 and −3 processing in Casp8 C362S/C362S Ripk3 −/− BMDMs treated with IFNγ/LPS likely caused the small amount of processed caspase-8C362S/C362S observed (Figure 5C) (Woo et al., 1999). Akin to the Casp8 −/− Mlkl −/− macrophages (Figure 4E), both Casp8 −/− Ripk3 −/− and Casp8 C362S/C362S Ripk3 −/− BMDMs also displayed reduced IFNγ/LPS-mediated induction of A1 and IL-1β compared to control cells (Figure 5D). Therefore, caspase-8 catalytic activity is required for IFNγ/LPS-mediated expression of BAX/BAK antagonists, activation of BAX/BAK, and apoptotic cell death.
Figure 5.

Caspase-8 enzymatic activity is required for IFNγ/LPS-induced killing of macrophages
(A and B) WT, Ripk3−/−, Casp8−/−Ripk3−/−, or Casp8C362S/C362SRipk3−/− (Casp8CS/CSRipk3−/−) BMDMs were primed with IFNγ (50 ng/mL) overnight then stimulated with LPS (50 ng/mL) for 24 or 48 h. Treatment with LPS and Compound A (Cp. A, 1 μM) for 24 h (extrinsic apoptosis), LPS and IDN-6556 (IDN, 5 μM) for 24 h (necroptosis), and ABT-737 (1 μM) and cycloheximide (CHX, 10 μg/mL) for 6 h (intrinsic apoptosis) were used as controls. (A) Cell death and (B) cytochrome c retention was measured by PI exclusion as measured by flow cytometry or intracellular cytochrome c staining and flow cytometric analysis (n = 3).
(C and D) Immunoblot of WT, Ripk3−/−, Casp8−/−Ripk3−/−, or Casp8CS/CSRipk3−/− BMDMs primed with IFNγ (50 ng/mL) overnight and stimulated with LPS (50 ng/mL) for (C) 16 and 24 h, or (D) 2–8 h. Ponceau stain is provided as a loading control (n = 2).
(E) WT or Mlkl−/− BMDMs were treated with IFNγ (50 ng/mL) overnight then with LPS (50 ng/mL) ± IDN (5 μM) or DMSO for 16, 24, or 48 h. LPS and IDN or IFNγ and IDN treatment for 24 h (necroptosis) was used as a control. Cell death was assessed by PI exclusion as measured by flow cytometry (n = 3).
(F) Immunoblot of WT or Mlkl−/− BMDMs that were treated with IFNγ (50 ng/mL) overnight then with LPS (50 ng/mL) ± IDN (5 μM) or DMSO for 8 or 16 h (n = 2).
Data represent the mean value ± SD, or a representative immunoblot, from n independent experiments. p > 0.05 (n.s.), p ≤ 0.01 (∗∗), p ≤ 0.0001 (∗∗∗∗). See also Figure S6.
To test if the inhibition of caspase-dependent apoptosis could mimic genetic caspase-8 loss, we stimulated BMDMs generated with IFNγ/LPS in the presence of the broad-spectrum caspase inhibitors, IDN-6556 (IDN) (Figure 5E) or Z-VAD (Figure S6A). The inhibition of caspase-8 triggers MLKL-dependent necroptosis upon LPS stimulation (He et al., 2011; Najjar et al., 2016), therefore, Mlkl −/− BMDMs were also examined. As described previously (Figure 2), Mlkl −/− BMDMs died comparably to WT BMDMs upon IFNγ/LPS stimulation and were resistant to necroptosis triggered by LPS with either Z-VAD or IDN (Figures 5E and S6A). Similarly, Z-VAD and IDN both exacerbated IFNγ/LPS-induced killing in WT BMDMs (Figures 5 and S6A). In the presence of a caspase inhibitor, Mlkl −/− BMDMs were resistant to the necroptosis induced by caspase-inhibition in WT, IFNγ/LPS stimulated BMDMs, but failed to further protect BMDMs from cell death caused by IFNγ/LPS stimulation (Figures 5E and S6A). Moreover, IFNγ/LPS-induced cytochrome c release from the mitochondria was still observed in MLKL-deficient macrophages in the presence of IDN, albeit slightly reduced (Figure S6B).
Caspase inhibition limited IFNγ/LPS-mediated caspase-9, but not caspase-8, processing and reduced the expression of IL-1β, but had no impact on MCL-1 loss (Figure 5F), as expected based on our genetic data (Figure 4E) and previous studies (Brumatti et al., 2016; Jaco et al., 2017; Lalaoui et al., 2020). In contrast to Casp8 C362S/C362S Ripk3 −/− BMDMs, apoptosis, necroptosis, and pyroptosis deficient (Casp1 −/− Casp3 −/− Casp7 −/− Casp9 −/− Bid −/− Mlkl −/− Gsdmd −/− gene targeted) iBMDMs (Doerflinger et al., 2020) still died upon treatment with IFNγ/LPS, even in the presence of IDN to inhibit the remaining caspase activity (Figures S6C and S6D). Therefore, caspase-8 activity is required to activate BAX/BAK during IFNγ/LPS signaling, and this activity is not efficiently blocked by broad-spectrum caspase inhibitors. As reported previously (Ekert et al., 2004; Marsden et al., 2004; Marsden et al., 2002), these data suggest that BAX/BAK-mediated irreversible damage to mitochondria causes cell death even when downstream caspase function is eliminated.
iNOS-generated nitric oxide licenses IFNγ/LPS-induced killing of macrophages by caspase-8 and BAX/BAK
Our RNA-sequencing data identified genes associated with nitric oxide (NO) production, including inducible nitric oxide synthase (iNOS), as being downregulated upon Casp8 deletion (Figure S6E). iNOS has been linked to cellular toxicity (Dubey et al., 2016; Kiang et al., 2008; Li et al., 2019; Murphy, 1999; Okada et al., 1998; Oyadomari et al., 2001; Seminara et al., 2007; Sennlaub et al., 2002; Snyder et al., 2009; Taylor et al., 2003), and further analysis confirmed that optimal protein expression of iNOS required caspase-8 activity (Figure S6F). We therefore explored the possibility that iNOS may contribute to IFNγ/LPS-induced cell death.
IFNγ/LPS treatment induced robust iNOS expression and this correlated with increased concentrations of nitrite (NO2 −) in the cell supernatant (Figure 6 A), a product of NO oxidization. Despite a delay in protein expression, the final concentration of NO2 − detected in cell supernatants was comparable between IFNγ/LPS-treated WT and Casp8 −/− Mlkl −/− BMDMs (Figure S6G). However, IFNγ alone could induce significant NO2 − in Casp8 −/− Mlkl −/− BMDMs, but not control cells (Figure S6G). Therefore, we normalized the death-associated NO2 − production (i.e., post-treatment with LPS) to the IFNγ-primed baseline. In this analysis, Casp8 −/− Mlkl −/− BMDMs displayed both delayed kinetics and reduced maximal NO2 − concentration attributable to LPS stimulation of IFNγ primed cells (Figure S6H). In contrast, Bak −/− Bax −/− and Tnf −/− Fasl gld/gld Trail −/− macrophages treated with IFNγ and/or LPS generated similar amounts of NO2 − to WT cells (Figure S6I), thereby placing NO production upstream of BAX/BAK activation and cell death.
Figure 6.

IFNγ/LPS-induced iNOS sensitizes macrophages to caspase-8 and BAX/BAK-mediated death
(A) WT BMDMs were primed with IFNγ (50 ng/mL) overnight then stimulated with LPS (50 ng/mL) for 2–24 h. Nitrite (NO2−) production and iNOS expression were measured by the Griess assay and immunoblot (n = 3).
(B and C) WT BMDMs were primed with IFNγ (50 ng/mL) overnight then stimulated with LPS (50 ng/mL) ± the iNOS inhibitor 1400W (10 μM) or DMSO for 24 h. (B) Nitrite (NO2−) production and cell death were measured by the Griess assay and (C) PI exclusion as measured by flow cytometry (n = 4).
(D) WT BMDMs were primed with IFNγ (50 ng/mL) overnight then stimulated with LPS (50 ng/mL) ± 1400W (10 μM) for 24 h. The nitric oxide donor SNAP (200 μM) or DMSO were provided 8 h post-treatment with LPS. Cell death was assessed by PI exclusion as measured by flow cytometry (n = 5).
(E) WT BMDMs were treated with 1400W (10 μM) ± IFNγ (50 ng/mL) overnight then stimulated with LPS (50 ng/mL) or TNF (100 ng/mL) for 24 h. SNAP (200 μM) or DMSO were added to cells 8 h post-treatment with LPS or TNF. Cell death was assessed by PI exclusion as measured by flow cytometry (n = 4).
(F, G, and H) WT or Nos2−/− BMDMs were primed with IFNγ (50 ng/mL) overnight then stimulated with LPS (50 ng/mL) or Pam-3-CSK4 (P3C, 500 ng/mL) for 16, 24, or 48 h. ABT-737 (1 μM) and cycloheximide (CHX, 10 μg/mL) treatment for (G) 6 h or (H) 2 h was used as a positive control. (F) Cell death and (G) cytochrome c retention was assessed by PI exclusion as measured by flow cytometry or intracellular cytochrome c staining and flow cytometric analysis (n = 6). (H) Cell death pathway activation was assessed by immunoblot of cell supernatants (S/N) and cell lysates (n = 2).
Data represent the mean value ± SD, or a representative immunoblot, from n independent experiments. p ≤ 0.05 (∗), p ≤ 0.001 (∗∗∗), p ≤ 0.0001 (∗∗∗∗). See also Figure S6 and S7.
The iNOS-specific inhibitor, 1400W (Garvey et al., 1997), reduced NO2 − production and almost completely protected macrophages from IFNγ/LPS-induced killing (Figure 6B and 6C). Similarly, inhibition of iNOS with 1400W effectively prevented IFNγ/LPS-induced killing of Casp1 −/− Casp3 −/− Casp7 −/− Casp9 −/− Bid −/− Mlkl −/− Gsdmd −/− gene-targeted iBMDMs (Figure S6D). NO restoration with S-Nitroso-N-acetyl-DL-penicillamine (SNAP), in a manner that mimics the kinetics of endogenous NO production, re-sensitized iNOS-inhibited BMDMs to IFNγ/LPS-induced cell death, without exhibiting toxicity by itself (Figure 6D, S7A, and S7B). SNAP treatment also moderately sensitized iNOS-inhibited macrophages to TNF or LPS-induced killing although, in the case of LPS, this was further enhanced when BMDMs were primed with IFNγ (Figure 6E).
We next generated BMDMs from iNOS-deficient (Nos2 −/−) mice. Nos2 −/− BMDMs did not produce NO2 − upon stimulation with IFNγ/LPS or IFNγ/P3C (Figure S7C) and were protected from IFNγ/LPS- or IFNγ/P3C-induced cell death (Figure 6F) in addition to mitochondrial cytochrome c release (Figure 6G), akin to iNOS-inhibited cells (Figure S7D). IFNγ/LPS-treated Nos2 −/− BMDMs or iNOS-inhibited WT macrophages displayed reduced processing of caspase-8, caspase-9, caspase-3 and GSDMD, and decreased HMGB1 release into the cellular supernatant (Figures 6H and S7E). iNOS inhibition or Nos2 deletion also prevented the IFNγ/LPS-induced reduction in MCL-1, and stabilized A1 protein (Figures 6H and S7E). These data demonstrate a crucial role for iNOS in reducing MCL-1 and A1 upon IFNγ/LPS stimulation, which in combination with caspase-8-mediated suppression of Bcl2, will sensitize macrophages to BAX/BAK activation and mitochondrial apoptosis (Figure S7F).
Both iNOS and caspase-8 contribute to SARS-CoV-2 disease severity, but only caspase-8 causes hemophagocytic lymphohistiocytosis (HLH) lethality
Recently, Karki et al. described a caspase-8 and iNOS-mediated macrophage death pathway resulting from co-treatment with IFNγ and TNF (Karki et al., 2021). This study implicated this cell death in murine SARS-CoV-2 infection and hemophagocytic lymphohistiocytosis (HLH) disease models as IFNγ and TNF neutralizing antibodies confer significant protection from mortality (Karki et al., 2021). However, whether iNOS, caspase-8, or cell death alter disease severity in models of SARS-CoV-2 infection or HLH has not been examined using relevant gene targeted mice.
HLH-like disease is induced in mice by treatment with polyI:C for 24 h followed by low-dose LPS injection (Wang et al., 2019). In agreement with the idea that death ligands such as TNF contribute to HLH pathology (Karki et al., 2021), Tnf −/− Fasl gld/gld Trail −/− mice were protected from disease (decreased core body temperature) when compared to WT animals (Figure 7 A). Casp8 −/− Ripk3 −/−, but not Ripk3 −/−, mice were also protected from the HLH-induced decreases in core body temperature (Figure 7A). In contrast to death ligand or caspase-8 deficiency, Nos2 −/− mice behaved like WT mice, rapidly dropping their core body temperature and reaching the ethical endpoint requiring humane euthanasia (Figure 7B).
Figure 7.

iNOS and caspase-8 influence the host response to SARS-CoV-2, but only caspase-8 impacts hemophagocytic lymphohistiocytosis (HLH) disease severity
(A and B) Rectal temperatures of (A) wild-type (WT, n = 12), Ripk3−/− (n = 6), Casp8−/−Ripk3−/− (n = 10), and Tnf−/−Faslgld/gldTrail−/− (n = 6) mice, or (B) WT (n = 14) and Nos2−/− (n = 12) mice injected with PolyI:C (10 mg/kg) for 24 h followed by LPS (5 mg/kg) to induce HLH-like disease.
(C) Cleaved caspase-3 immunohistochemistry of endpoint small intestine sections taken from mice treated as described (A and B). Each image represents a separate mouse. Positive cells are indicated with red arrows. Scale bar, 100 μm.
(D and E) Endpoint plasma TNF, IL-1β and IL-6 concentrations of mice treated as described in (D) A and (E) B.
(F and G) (F) TCID50 infectious units per lung and (G) percentage weight loss of WT (n = 22), Nos2+/− (n = 18) or Nos2−/− (n = 20) mice infected with 1.5 × 107 TCID50 infectious units of SARS-CoV-2 for three days.
(H and I) (H) TCID50 infectious units per lung and (I) percentage weight loss of WT (n = 21), Mlkl−/− (n = 23), Casp8+/−Mlkl−/− (n = 17), or Casp8−/−Mlkl−/− (n = 15) mice infected with 1.5 × 107 TCID50 infectious units of SARS-CoV-2 for three days.
Data represent the mean value ± SEM pooled from at least 2 independent experimental cohorts of mice. p > 0.05 (n.s.), p ≤ 0.05 (∗), p ≤ 0.01 (∗∗), p ≤ 0.001 (∗∗∗), p ≤ 0.0001 (∗∗∗∗).
Immunohistochemical staining for cleaved caspase-3 revealed prominent cell death in the small intestinal mucosal layer of WT, Ripk3 −/−, and Nos2 −/− mice, but not in Casp8 −/− Ripk3 −/− or Tnf −/− Fasl gld/gld Trail −/− mice, highlighting cell death as a feature of this model associated with poorer disease outcomes (Figure 7C). Endpoint plasma TNF, but not IL-6, concentrations correlated with treatment outcome and were reduced in Casp8 −/− Ripk3 −/− and Tnf −/− Fasl gld/gld Trail −/− animals compared to WT, Ripk3 −/− and Nos2 −/− mice (Figures 7D and 7E). Therefore, although ligands for death receptors and caspase-8-driven cell death likely contribute to murine HLH disease severity, this cell death does not require iNOS.
Next, we used the N501Y+D614G strain of SARS-CoV-2, which can infect C57BL/6 mice and causes a non-lethal disease, to assess the contribution of iNOS and caspase-8 signaling to viral replication and disease severity. Homozygous, but not heterozygous, deletion of Nos2 caused a minor, yet significant, reduction in lung viral titers at three days post-infection (the time point representing peak viral loads) (Figure 7F). SARS-CoV-2 infection also triggered a 5–10% loss in total body weight in WT mice within 3 days, while heterozygous or homozygous deletion of Nos2 largely ameliorated this response (Figure 7G).
SARS-CoV-2 infected WT, Casp8 −/− Mlkl −/−, Casp8 +/− Mlkl −/−, and Mlkl −/− mice all exhibited similar peak viral loads, showing that neither caspase-8- nor MLKL-dependent death impact viral burdens (Figure 7H). However, identical to Nos2 −/− mice, Casp8 −/− Mlkl −/−, but not Mlkl −/− mice, almost completely prevented SARS-CoV-2-induced weight loss (Figure 7I). These data indicate that iNOS and caspase-8 both drive a damaging host response in this non-lethal SARS-CoV-2 model and is consistent with the positive relationship between Nos2 expression and COVID-19 disease severity reported in patients (Karki et al., 2021).
Discussion
We have performed a detailed genetic and biochemical analysis delineating how IFNγ priming sensitizes macrophages to TLR-induced killing. Our genetic experiments define a requirement for iNOS generated NO in the licensing of apoptotic caspase-8 and caspase-8-driven BAX/BAK activity, the latter of which occurs independent of the caspase-8 substrate, BID. The removal of MCL-1 and BCL-XL induces the activation of BAX/BAK and apoptotic death in macrophages (Vince et al., 2018), yet in the context of IFNγ and TLR signaling, we show that iNOS activity reduces A1 and MCL-1 stability, while caspase-8 represses inducible Bcl2 expression. The combined reduction of these pro-survival BCL-2 family members likely promotes BAX/BAK triggering. However, we cannot rule out a role for additional cell death regulators that contribute to BAX/BAK activation and apoptosis, particularly as BCL-2 inhibition only partly restored IFNγ/LPS killing in caspase-8 deficient cells (where IFNγ/LPS-mediated reductions in MCL-1 and A1 still occur). In this regard, BID cleavage was observed during IFNγ/LPS killing, and even though BID was dispensable for IFNγ/LPS-induced activation of BAX/BAK, caspase-8 processing of BID may nevertheless act in concert with the combined reduction in MCL-1, A1, and BCL-2 to unleash the mitochondrial apoptotic pathway.
IFNγ sensitizes macrophages to TNF-induced cell death (Karki et al., 2021), yet our data show that the cell death signaling that occurs during pathogenic insult is likely to be more complex. The presence of M-CSF (i.e., L929-medium) overcame the requirement of TNF during IFNγ/TLR cell death signaling, likely via M-CSF-driven expression of Fasl. Moreover, while IFNγ and TNF (IFNγ/TNF) killing was reported to require GSDME (Karki et al., 2021), IFNγ/LPS can cause cell death in the combined absence of both GSDMD and GSDME. It has also been suggested that mitochondrial apoptosis does not contribute to IFNγ/TNF killing, as macrophages still die in the absence of the key apoptosome component, APAF1 (Karki et al., 2021). However, APAF1 deficiency only delays, not prevents, BAX/BAK-dependent cell death (Ekert et al., 2004; Marsden et al., 2002). In the context of IFNγ/LPS-induced macrophage killing, our genetic data clearly demonstrate that BAX/BAK-mediated cell death is the primary avenue of cell death, which can be diverted to alternative cell death streams should BAX/BAK activation be compromised. Ultimately, it is possible that IFNγ/TNF-driven cell death responses do not require BAX/BAK and are therefore different to those elicited by IFNγ/TLR signaling, although this possibility remains to be experimentally tested.
The mutation of the catalytic cysteine of caspase-8 mimicked caspase-8 deficiency to a large degree, and in addition to preventing IFNγ/LPS-induced cell death, the loss of caspase-8 catalytic activity reduced the expression of genes (i.e., IL-1β and A1) that were also reduced upon Casp8 deletion. Caspase-8 can cleave and inactivate NEDD4 binding protein 1 (N4BP1) to facilitate TLR-induced transcriptional responses, which accounts for approximately half of the LPS-induced transcriptional effects mediated by caspase-8 (Gitlin et al., 2020). Whether caspase-8-mediated transcriptional changes in Bcl2a1, iNOS, Pmaip1, and Bcl2, or other cell death regulators identified in our analysis, are controlled via N4BP1 cleavage and/or the nucleation of a caspase-8 transcriptional scaffolding complex (Henry and Martin, 2017; Kreuz et al., 2004; Varfolomeev et al., 2005), in response to IFNγ/LPS treatment, remains to be determined.
Our in vivo genetic studies indicate that a death receptor caspase-8 signaling axis has a significant role in HLH-like disease, but neither the cell death pathology nor disease-induced temperature loss was altered by Nos2 deletion. Both iNOS and caspase-8 are required for IFNγ/TNF- (Karki et al., 2021) and IFNγ/LPS-induced death of BMDMs. Therefore, our findings provide genetic evidence that questions the proposal that iNOS drives caspase-8-dependent cell death and disease severity in this HLH model (Karki et al., 2021).
Contrary to our findings in HLH, the loss of both caspase-8 and iNOS improved the impact of SARS-CoV-2 infection on the host. This response occurred independent of changes in peak viral loads, indicating that the protection from SARS-CoV-2-induced weight loss upon iNOS or Casp8 deletion is not caused by altered viral clearance. Caspase-8 activity and associated inflammation occurs in SARS-CoV-2 infected cells and humanized ACE-2 mice (Li et al., 2020). Similarly, NOS2 expression positively correlates with disease severity in humans (Karki et al., 2021). Our genetic experiments documenting an important role for both iNOS and caspase-8 in a murine model of SARS-CoV-2-driven disease now show that they are not simply markers of infection, but that their signaling causes a damaging host response that likely reflects increased cell death. Additional genetic experiments to evaluate the contribution of BAX/BAK and the role of iNOS and caspase-8 in lethal SARS-CoV-2 infection models, will be of interest to pursue.
Even though the key actions of NO that license the programmed cell death we have described remain to be determined, both iNOS deficiency and iNOS variants have been linked to human diseases outside of SARS-CoV-2 infection, including cell death-associated infections and inflammatory conditions (Dhillon et al., 2014; Drutman et al., 2020; Hague et al., 2004; de Jesus Trovoada et al., 2014). Moreover, NO produced by endothelial NOS or neuronal NOS could potentially replace iNOS-derived NO in select cell types and settings, resulting in programmed cell death in other disease contexts. Therefore, the genetic characterization of the selective IFNγ and TLR-mediated cell death pathway licensed by NO points to potential therapeutic strategies for targeting aberrant iNOS and/or NO production in relevant cell death- and inflammation-associated conditions.
Limitations of study
Our study relied on the use of mice to delineate the molecular determinants responsible for causing IFNγ and TLR-mediated cell death. Future efforts to validate this pathway in relevant primary human cells is important as substantial differences exist in the transcriptional regulation of murine and human iNOS and there is no consensus on human NOS2 gene induction (Gross et al., 2014; Schneemann et al., 1993; Young et al., 2018). The ethical endpoint of our HLH model was when mice reached a body core temperature of 30°C, meaning disease lethality was not directly assessed. In addition, although our findings establish both iNOS and caspase-8 as contributing to SARS-CoV-2- disease severity, our study does not document the cell types dying following SARS-CoV-2 infection, nor which are protected from death when iNOS and caspase-8 are deleted. Finally, the actions and/or substrates of NO that license IFNγ and TLR-mediated apoptotic caspase-8 and BAX/BAK activity remain to be defined, although the reported nitrosylation of FAS, cFLIP, and IAPs (Leon-Bollotte et al., 2011; Romagny et al., 2018; Talbott et al., 2014; Wu et al., 2015) may contribute. Further experiments to address this aspect will be important for identifying targets that might be manipulated for therapeutic benefit in iNOS-driven diseases.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-cleaved caspase-3 | R&D Systems | Cat#: AF835; RRID: AB_2243952 |
| Rat anti-F4/80 (FITC) | eBioscience | Cat#: 11-4801-81; RRID: AB_2735037; Clone: BM8 |
| Rat anti-MHCII (AF700) | eBioscience | Cat#: 56-5321-82; RRID: AB_494009; Clone: M5/114.15.2 |
| Rat anti-CD206 (APC) | eBioscience | Cat#: 47-2061-82; RRID: AB_2802285; Clone: MR6F3 |
| Rat anti-CD11b (PE-Cy7) | eBioscience | Cat#: 25-0112-82; RRID: AB_469588; Clone: M1/70 |
| Human anti-cytochrome c (APC) | Miltenyl Biotec | Cat#: 130-111-180; RRID: AB_2651489; Clone: REA702 |
| Rat anti-A1 | The laboratory of Marco Herold, (Lang et al., 2014) | Clone: 6D6 |
| Mouse anti-β-actin | Sigma | Cat#: A1978; RRID: AB_476692; Clone: AC-15 |
| Rabbit anti-BCL-XL | Cell Signaling Technology | Cat#: 2764; RRID: AB_2228008; Clone: 54H6 |
| Rat anti-BID | The laboratory of Andreas Strasser, (Kaufmann et al., 2007) | Clone: 2D1 |
| Mouse anti-caspase-1 | Adipogen | Cat#: AG-20B-0042-C100; RRID: AB_2755041; Clone: Casper-1 |
| Rabbit anti-cleaved caspase-3 | Cell Signaling Technology | Cat#: 9661; RRID: AB_2341188 |
| Rabbit anti-caspase-3 | Cell Signaling Technology | Cat#: 9665; RRID: AB_2069872; Clone: 8G10 |
| Rabbit anti-cleaved caspase-8 (Asp387) | Cell Signaling Technology | Cat#: 9429; RRID: AB_2068300 |
| Rabbit anti-cleaved caspase-8 (Asp387) | Cell Signaling Technology | Cat#: 8592; RRID: AB_10891784; Clone: D5B2 |
| Rat anti-caspase-8 | Enzo Life Sciences | Cat#: ALX-804-448-C100; RRID: AB_2050953; Clone: 3B10 |
| Rabbit anti-cleaved caspase-9 | Cell Signaling Technology | Cat#: 9509; RRID: AB_2073476 |
| Mouse anti-caspase-9 | Cell Signaling Technology | Cat#: 9508; RRID: AB_2068620 |
| Mouse anti-cIAP1 | Enzo Life Sciences | Cat#: ALX-803-335; RRID: AB_2227905; Clone: 1E1-1-10 |
| Rabbit anti-GSDMD | Abcam | Cat#: ab209845; RRID: AB_2783550; Clone: EPR19828 |
| Rabbit anti-HMGB1 (HRP) | Abcam | Cat#: ab195012; Clone: EPR3507 |
| Goat anti-IL-1β | R&D Systems | Cat#: AF-401-NA; RRID: AB_416684 |
| Mouse anti-iNOS | BD Transduction | Cat#: 610328; RRID: AB_397718 |
| Rabbit anti-MCL-1 | Cell Signaling Technology | Cat#: 5453; RRID: AB_10694494 |
| Rat anti-MLKL | Merck Millipore, (Murphy et al., 2013) | Cat#: MABC604; RRID: AB_2820284; Clone: 3H1 |
| Rabbit anti-NINJ1 | Invitrogen | Cat#: PA5-95755; RRID: AB_2807557 |
| Mouse anti-XIAP | MBL | Cat#: M044-3; RRID: AB_592998 |
| Peroxidase-AffiniPure Rabbit anti-Goat IgG (H+L) | Jackson ImmunoResearch Labs | Cat#: 305-035-003; RRID: AB_2339400 |
| Peroxidase-AffiniPure Goat anti-Mouse IgG (H+L) | Jackson ImmunoResearch Labs | Cat#: 115-035-003; RRID: AB_10015289 |
| Peroxidase-AffiniPure Goat anti-Rabbit IgG (H+L) | Jackson ImmunoResearch Labs | Cat#: 111-035-003; RRID: AB_2313567 |
| Peroxidase-AffiniPure Goat Anti-Rat IgG (H+L) | Jackson ImmunoResearch Labs | Cat#: 112-035-003; RRID: AB_2338128 |
| Bacterial and virus strains | ||
| SARS-CoV-2 (hCoV-19/Australia/VIC2089/2020) N501Y+D614G strain | The laboratory of Marc Pellegrini | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant mouse IFNγ | R&D | Cat#: 485-MI |
| Recombinant mouse IFNβ | PBL Assay Science | Cat#: 12405-1 |
| Ultrapure LPS from E. coli O111:B4 | InvivoGen | Cat#: tlrl-3pelps |
| Pam-3-CSK4 | InvivoGen | Cat#: tlrl-pms |
| Poly(I:C) LMW | InvivoGen | Cat#: tlrl-picw |
| Recombinant human FLAG-TNF | The laboratory of Rebecca Feltham | N/A, generated in-house |
| Z-VAD-fmk | R&D Systems | Cat#: FMK001 |
| IDN-6556 | TetraLogic Pharmaceuticals | N/A |
| 1400W | Abcam | Cat#: ab120165 |
| ABT-199 | Active Biochem | Cat#: A-1231 |
| S63845 | Active Biochem | Cat#: A-6044 |
| Deferiprone | Sigma-Aldrich | Cat#: Y0001976 |
| Ferrostatin-1 | Sigma-Aldrich | Cat#: SML0583 |
| Nigericin | Sigma-Aldrich | Cat#: N7143 |
| SNAP | Sigma-Aldrich | Cat#: N3398 |
| Compound A (Cp.A, 12911) | TetraLogic Pharmaceuticals | N/A |
| ABT-263 | Active Biochem | Cat#: A-1001 |
| ABT-737 | Active Biochem | Cat#: A-1002 |
| cycloheximide | Sigma-Aldrich | Cat#: C7698 |
| RSL3 | Sigma-Aldrich | Cat#: SML2234 |
| QVD-OPh | MP Biomedicals | Cat#: 03OPH109 |
| Propidium iodide | Sigma-Aldrich | Cat#: P4170 |
| Poly(I:C) HMW | InvivoGen | Cat#: tlrl-pic |
| Ultrapure LPS from E. coli 055:B5 | InvivoGen | Cat#: tlrl-pb5lps |
| Lipofectamine 2000 Transfection Reagent | Thermo Fisher | Cat#: 11668027 |
| Puromycin | InvivoGen | Cat#: ant-pr-1 |
| Sulfanilamide | Sigma-Aldrich | Cat#: S9251 |
| N-(1-napthyl)ethylenediamine dihydrochloride | Sigma-Aldrich | Cat#: 222488 |
| Critical commercial assays | ||
| TNF ELISA | ThermoFisher | Cat#: 88-7324-88 |
| IL-1β ELISA | R&D Systems | Cat#: DY401 |
| IL-6 ELISA | ThermoFisher | Cat#: 88-7064-88 |
| Griess reagent system | Promega | Cat#: G2930 |
| ISOLATE II RNA Mini Kit | Meridian Bioscience | Cat#: BIO-52073 |
| Cytotoxicity Detection Kit (LDH) | Roche | Cat#: 11644793001 |
| 4–12% Bis-Tris NuPAGE gel system | ThermoFisher | Cat#: NP0321BOX |
| Superscript III Reverse Transcriptase Kit | ThermoFisher | Cat#: 18080093 |
| eBioscienceTM IC Fixation Buffer | ThermoFisher | Cat#: 00-8222-49 |
| eBioscienceTM Permeabilization buffer | ThermoFisher | Cat#: 00-8333-56 |
| Maxima SYBR Green qPCR Master Mix (2X), with separate ROX vial | ThermoFisher | Cat#: K0251 |
| QuantSeq 3′mRNA-Seq Library Prep Kit | Lexogen | N/A |
| SuperSignal™ West Atto Ultimate Sensitivity Substrate | ThermoFisher | Cat#: A38556 |
| Clarity Western ECL Substrate | Bio-Rad | Cat#: 1705061 |
| Deposited data | ||
| Raw RNA-sequencing data | This paper | GEO: GSE161179 |
| Experimental models: Cell lines | ||
| Human: HEK293T | The laboratory of James Vince | N/A |
| Mouse: Wildtype immortalized BMDM (iBMDM) | This paper | N/A, generated in-house |
| Mouse: Mlkl−/− iBMDM | This paper | N/A, generated in-house |
| Mouse: Mlkl−/−Ninj1−/− iBMDM (sgRNA1) | This paper | N/A, generated in-house |
| Mouse: Mlkl−/−Ninj1−/− iBMDM (sgRNA2) | This paper | N/A, generated in-house |
| Mouse: Casp1−/−Casp3−/−Casp7−/−Casp9−/−Bid−/−Mlkl−/−Gsdmd−/−iBMDM | The laboratory of Marco Herold, (Doerflinger et al., 2020) | N/A |
| Human: Vero cells | The laboratory of Marc Pellegrini | Clone: CCL81 |
| Experimental models: Organisms/strains | ||
| Mouse: Wildtype: C57BL/6J | In-house | JAX stock #000664 |
| Mouse: Tnf−/−FasLgld/gldTrail−/−: C57BL/6 | In-house, (Ebert et al., 2020) | N/A |
| Mouse: Tnf−/−: C57BL/6-Tnf−/−/J | In-house, (Körner et al., 1997) | N/A |
| Mouse: Tnfr1−/−: C57BL/6-Tnfrsf1atm1Imx/J | In-house, (Peschon et al., 1998) | JAX stock #003242 |
| Mouse: Tnfr2−/−: B6.129S2-Tnfrsf1btm1Mwm/J | In-house, (Erickson et al., 1994) | JAX stock #002620 |
| Mouse: Mlkl−/−: C57BL/6-Mlkl−/−/J | In-house, (Murphy et al., 2013) | N/A |
| Mouse: Casp8−/−Mlkl−/−: B6.129P2-Casp8tm1RazMlkl−/−/J | In-house, (Alvarez-Diaz et al., 2016) | N/A |
| Mouse: Baxfl/flBak−/−: C57BL/6-Baxfl/flBak−/− | (Takeuchi et al., 2005) | N/A |
| Mouse: Bax−/−Bak−/−: C57BL/6-Baxfl/flBak−/−Vav-Cre | In-house | N/A |
| Mouse: Wildtype: C57BL/6-Ly5.1: B6.SJL-Ptprca Pepcb/BoyJ | In-house, (Yang et al., 2002) | JAX stock #002014 |
| Mouse: Gsdmd−/−: C57BL/6N-Gsdmd−/−/J | In-house, (Kayagaki et al., 2015) | JAX stock #032410 |
| Mouse: Gsdme−/−: C57BL/6N-Gsdmeem1Fsha/J | A kind gift from Genentech, (Kayagaki et al., 2021) | JAX stock #032411 |
| Mouse: Gsdmd−/−Gsdme−/−: C57BL/6N-Gsdmd−/−Gsdmeem1Fsha/J | In house, this paper | N/A |
| Mouse: Casp1−/−Casp11−/−: NOD.129S2(B6)-Casp1tm1SeshCasp4del/LtJ | In-house, (Li et al., 1995) | JAX stock # 004947 |
| Mouse: Nlrp3−/−: C57BL/6-Nlrp3tm1Tsc/J | In-house, (Martinon et al., 2006) | MGI ID: 3721141 |
| Mouse: Rel−/−: C57BL/6-Rel−/−/J | In-house, (Köntgen et al., 1995) | N/A |
| Mouse: Bid−/−: C57BL/6-Bid−/−/J | In-house, (Kaufmann et al., 2007) | N/A |
| Mouse: Pmaip1−/−: C57BL/6-Pmaip1tm1Ast/J | In-house, (Villunger et al., 2003) | JAX stock #011068 |
| Mouse: Nos2−/−: B6.129P2-Nos2tm1Lau/J | In-house, (Laubach et al., 1995) | JAX stock #002609 |
| Mouse: Ripk3−/−: C57BL/6N-Ripk3−/−/J | In-house, (Newton et al., 2004) | N/A |
| Mouse: Casp8−/−Ripk3−/−: C57BL/6N-Ripk3−/−B6.129-Casp8tm1Hed/J | In-house, (Rickard et al., 2014b) | N/A |
| Mouse: Wildtype: C57BL/6N | From the laboratory of Hamid Kashkir | N/A |
| Mouse: Ripk3−/−: C57BL/6N-Ripk3−/− | From the laboratory of Hamid Kashkir, (Newton et al., 2004) | N/A |
| Mouse: Casp8−/−Ripk3−/−: C57BL/6N-Ripk3−/−B6.129-Casp8tm1Hed | From the laboratory of Hamid Kashkir, (Fritsch et al., 2019) | N/A |
| Mouse: Casp8C632S/ C632SRipk3−/−: C57BL/6N | From the laboratory of Hamid Kashkir, (Fritsch et al., 2019) | N/A |
| Mouse: Gbp2−/− | Australian National University, Laboratories of Si Ming Man and Gaetan Burgio | N/A |
| Mouse: Gbp5−/− | Australian National University, Laboratories of Si Ming Man and Gaetan Burgio | N/A |
| Mouse: Gbp4−/−Gbp8−/−Gbp9−/− | Australian National University, Laboratories of Si Ming Man and Gaetan Burgio | N/A |
| Oligonucleotides | ||
| See Table S4 | N/A | |
| Recombinant DNA | ||
| Plasmid LV04 with Ninj1 gRNA: 5′ TCCGCAGCGCTCTTCTTGTTGG | (Metzakopian et al., 2017) | N/A |
| Plasmid LV04 with Ninj1 gRNA: 5′ ACCACAAGGGGCACGAAGAAGG | (Metzakopian et al., 2017) | N/A |
| Plasmid FUGW-pFU-Cas9-FLAG-mCherry | Addgene | Cat# 70182 |
| Plasmid pMDLg/pRRE (pMDL) | Addgene | Cat# 12251 |
| Plasmid pMD2.G (VSVg) | Addgene | Cat# 12259 |
| Plasmid pRSV-REV | Addgene | Cat# 12253 |
| Software and algorithms | ||
| Software: WEASEL (Version 2.7) | Frank Battye | https://frankbattye.com.au/Weasel/ |
| Software: GraphPad PRISM (Version 8.4.3) | GraphPad | Graphpad.com |
| Software: ImageJ (Version 2.1.0/1.53k) | (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| Software: Image Lab (Version 6.1.0) | Bio-Rad | https://www.bio-rad.com/en-au/product/image-lab-software?ID=KRE6P5E8Z#fragment-6 |
| Software: Adobe Illustrator 2021 (Version 25.0) | Adobe | http://www.adobe.com/Illustrator |
| Software: LAS-X | Leica Microsystems | https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
| Software: CaseCenter | 3D HisTech | https://www.3dhistech.com/solutions/casecenter/ |
| Cutadapt (v1.9) | (Martin, 2011) | https://journal.embnet.org/index.php/embnetjournal/article/view/200 |
| HISAT2 | (Kim et al., 2019) | https://idp.nature.com/authorize?response_type=cookie&client_id=grover&redirect_uri=https%3A%2F%2Fwww.nature.com%2Farticles%2Fs41587-019-0201-4 |
| FeatureCounts, Rsubread package (version 1.34.7) | (Liao et al., 2014; 2019) | https://www.ncbi.nlm.nih.gov/pubmed/24227677 |
| limma version 3.40.6 | (Law et al., 2014) | https://genomebiology.biomedcentral.com/articles/10.1186/gb-2014-15-2-r29 |
| GSEA2-2.2.2 | (Liberzon et al., 2015; Subramanian et al., 2005) | https://www.gsea-msigdb.org/gsea/index.jsp |
| Metascape | (Zhou et al., 2019) | https://genomebiology.biomedcentral.com/articles/10.1186/gb-2014-15-2-r29 |
| ggplot2 (version 3.2.1) | (Wickham, 2009) | https://link.springer.com/book/10.1007/978-0-387-98141-3 |
| pheatmap (version 1.0.12) | Package “pheatmap,” Version 1.0.12 (2018) | https://rdrr.io/cran/pheatmap/ |
| Other | ||
| Bullet Blender | Next Advance Inc | N/A |
| Inhalation Exposure System | Glas-Col | N/A |
| 24-well non-treated tissue culture plates | Falcon | Cat#: 351147 |
| μ-Slide 8 Well Glass Bottom | Ibidi | Cat#: 80827 |
| LSR II flow cytometer | Becton Dickinson | N/A (discontinued) |
| Immoblon-E polyvinyl difluoride membranes | Merck Millipore | Cat#: IEVH85R |
| ChemiDoc Imaging System | Bio-Rad | Cat#: 17001401 |
| Leica SP8 microscope | Leica Microsystems | N/A |
| ViiA 7 Real-Time PCR System | Applied Biosystems | Cat#: 4453545 |
| 4200 Tapestation system | Agilent | Cat#: G2991BA |
| NextSeq 500 | Illumina | N/A |
| VersaMaxTM Tunable Microplate Reader | Molecular Devices | N/A |
| Pierce™ Centrifuge Columns | ThermoFisher | Cat#: 89868 |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, James E. Vince (vince@wehi.edu.au).
Materials availability
All unique reagents generated in this study are available from the lead contact with a completed materials transfer agreement.
Experimental model and subject details
Mice
Bax−/−Bak−/−, Gsdmd−/− (Kayagaki et al., 2015), Gsdme −/− (kindly provided by Genentech (Kayagaki et al., 2021)), Gsdmd −/− Gsdme −/− (bred in-house,), Mlkl −/− (Murphy et al., 2013), Casp8 −/− Mlkl −/− (Alvarez-Diaz et al., 2016), Casp1 −/− Casp11 −/− (Li et al., 1995), Nlrp3 −/− (Martinon et al., 2006), Rel −/− (Köntgen et al., 1995), Pmaip1 −/− (Villunger et al., 2003), Nos2 −/− (JAX stock #002609; (Laubach et al., 1995)), Ripk3 −/− (Newton et al., 2004), Casp8 −/− Ripk3 −/− (Rickard et al., 2014b), Bid −/− (Kaufmann et al., 2007), Tnf −/− (Körner et al., 1997), Tnfr1 −/− (Peschon et al., 1998), Tnfr2 −/− (Erickson et al., 1994) and Tnf −/−FasLgld/gld Trail −/− (Ebert et al., 2020) mice were all backcrossed and maintained in-house on a C57BL/6J background under specific pathogen-free conditions at the Walter and Eliza Hall Institute of Medical Research (WEHI), Australia. Animal rooms were maintained at approximately 21°C ± 3°C at 40–70% humidity with a timed 14/10 h light dark cycle. All procedures were approved by the WEHI Animal Ethics Committee (Australia). Wild-type (WT) C57BL/6J mice and all gene-targeted animals were bred at WEHI and/or obtained from WEHI animal supplies (Kew, Australia). None of the mice used in our experiments had been previously used for other procedures. The animals presented with a healthy status and were selected independently of their gender for generating bone marrow-derived macrophages. Female and male mice were at least 6-weeks old at the time of experimentation. Bax flox/flox Bak −/− mice (Takeuchi et al., 2005) were crossed with Vav-Cre transgenic mice (or compound gene deleted mice used) to generate mice lacking both BAX and BAK in the hematopoietic system and are referred to as Bax −/− Bak −/− mice. To expand the numbers of Bax −/− Bak −/− mice available for derivation of macrophages, bone marrow was harvested from Bax −/− Bak −/− and WT C57BL/6-Ly5.2 donor mice. C57BL/6-Ly5.1 recipient mice (Yang et al., 2002) were lethally irradiated (2 × 550 Rad, 4 h apart), then injected intravenously with 3-5x106 bone marrow cells and allowed to reconstitute for 8 weeks. Reconstitution efficiency was assessed using staining for Ly5.1 and Ly5.2 surface markers and flow cytometry.
Casp8C632S/C632SRipk3−/− (Fritsch et al., 2019) animals, maintained on a C57BL/6N background, were housed in the animal care facility of the University of Cologne under standard pathogen-free conditions with a 12 h light/dark schedule and provided with food and water ad libitum. Studies with these mice were performed after approval by relevant government authorities (LANUV, NRW, Germany) in accordance with the German animal protection law, using wild-type, Ripk3 −/− and Casp8 −/− Ripk3 −/− mice maintained on a C57BL/6N background as controls.
The Gbp2 −/−, Gbp5 −/− and Gbp4 −/− Gbp8 −/− Gbp9 −/− animals were housed under specific pathogen-free conditions. All the procedures related to the Gbp2 −/−, Gbp5 −/− and Gbp4 −/− Gbp8 −/− Gbp9 −/− mice were approved by the Australian National University Animal Experimentation Committee (AEEC protocol A2017/44) according to the National Health and Medical Research Council (NHMRC) Australian code of practice. Gbp2 −/− and Gbp4 −/− Gbp8 −/− Gbp9 −/− mice were generated using CRISPR-Cas9-mediated gene editing technology as previously described (Jiang et al., 2019; O’Brien et al., 2019). The mouse genomic sequences were obtained from Ensembl (Ensembl.org). Cas9 protein and the single guide RNA (sgRNA) were purchased from IDT with the following sequences: For Gbp2, sgRNA1: 5′-GTGTGTGCCTCACCCCAAGA AGG-3′, sgRNA2: 5′-GACGATTCCGCTAACTTTGT GGG-3′ and sgRNA3: 5′-TCGTTGCTCAGACTTGCTGG GGG-3′ respectively targeting exons 3, 5 and 8. For Gbp4;Gbp8;Gbp9, sgRNA1: 5′-GGTGGAGGCGGGGTATGGTG AGG-3′ and sgRNA2: 5′-CATGGAGAGTGGAATTTGAG AGG-3′ targeting upstream of the Gbp4 gene, and sgRNA3: 5′-AGCTCACTGCTTCTCCATAC TGG-3′ and sgRNA4: 5′-TTCATCTCTTGTAAGATGGG AGG-3′ targeting downstream of the Gbp8 gene. The nucleases were delivered into the pronucleus of the fertilized zygotes with the following concentrations: Cas9 protein (50 ng/μl) was co-injected with a mixture of sgRNA (2.5 ng/μl). After the micro-injection of the zygotes were incubated overnight at 37°C under 5% CO2 and two-cell stage embryos were surgically transferred into the uterus of the pseudo-pregnant CFW/Crl mice. DNA was extracted from the ear punches of the mice using a crude DNA extraction protocol and PCR amplification. The PCR products were then purified with a PCR Clean-Up System (Promega) kit according to the manufacturer’s instructions. The Sanger sequencing was performed in the Biomolecular Resource facilities at the Australian National University. The Sanger sequencing identified an 8,770 bp deletion between exon 3 and exon 8 of Gbp2 and 127 kb deletion from the intergenic region upstream of Gbp4 to the intergenic region downstream of Gbp8.
Bone marrow-derived macrophages (BMDMs)
Bone marrow-derived macrophages were prepared from bone marrow cells harvested from femoral, tibial, and pelvic bones. Cells were cultured on 15-cm non-treated dishes for 6 days (37°C, 10% CO2) in 25 mL Dulbecco’s modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS, Sigma), 50 U/mL penicillin and 50 μg/mL streptomycin and supplemented with 15%–20% L929 cell-conditioned medium. An additional 10 mL culture medium was added on day 3. Unless otherwise specified in the figure legends, differentiated BMDMs were harvested and replated in sterile 24-well non-treated tissue culture plates at 6x105 cells/well in a final volume of 600 μL DMEM/FCS supplemented with 20% L929 conditioned medium. BMDMs that were to be stimulated with compound A were plated at 4x105 cells/well in a final volume of 600 μL DMEM/FCS supplemented with 20% L929 conditioned medium. For imaging experiments, 2x105 BMDMs were seeded into μ-Slide 8 Well Glass Bottom (Ibidi, 80827) in 200 μL DMEM/FCS supplemented with 20% L929 conditioned medium.
Immortalized BMDMs (iBMDMs)
Low-passage iBMDMs were cultured in DMEM containing 10% fetal bovine serum (FBS, Sigma), 50 U/mL penicillin and 50 μg/mL streptomycin and incubated at 37°C, 10% CO2. Mlkl −/− iBMDMs used to generate Ninj1 −/− cells were generated as described (De Nardo et al., 2018). Casp1 −/− Casp3 −/− Casp7 −/− Casp9 −/− Bid −/− Mlkl −/− Gsdmd −/− gene targeted immortalized BMDMs (iBMDMs) were a kind gift from the Herold laboratory (Doerflinger et al., 2020). For experiments, iBMDMs were plated in sterile 24-well treated plates at 2x105 cells/well.
Method details
SARS-CoV-2 Infection
All procedures involving animals and live SARS-CoV-2 were conducted in an OGTR-approved Physical Containment Level 3 (PC-3) facility at the Walter and Eliza Hall Institute of Medical Research (Cert-3621) and were approved by The Walter and Eliza Hall Institute of Medical Research Animal Ethics Committee (2020.016). SARS-CoV-2 infection of mice was performed using an inhalation exposure system (Glas-Col, LLC). Briefly, caged animals were placed in compartmented mesh baskets within the sealed 141 L chamber of a Glas-Col Inhalation Exposure System and exposed to 1.5 × 107 TCID50 infectious units of venturi-nebulised SARS-CoV-2 N501Y+D614G strain virus for 30 min. SARS-CoV-2 N501Y+D614G strain virus was generated from hCoV-19/Australia/VIC2089/2020 that had been by serial passaged in mice (the laboratory of Marc Pellegrini). Mice of mixed sexes used for experimentation were 6 – 10 weeks of age. Mice were weighed prior to infection and at the experimental endpoint to determine the percentage weight loss for each infected mouse.
Median Tissue Culture Infectious Dose (TCID50) assay
Animals were humanely euthanized and lungs removed and homogenized in a Bullet Blender (Next Advance Inc) in 1 mL DME media (ThermoFisher) containing steel homogenization beads (Next Advance Inc). Samples were clarified by centrifugation at 10,000 rcf for 5 min. SARS-CoV-2 lung TCID50 was determined by plating 1:7 serially diluted lung tissue homogenate onto confluent layers of Vero cells (clone CCL81) in DME media (ThermoFisher) containing 0.5 μg/mL trypsin-TPCK (ThermoFisher) in replicates of six on 96-well plates. Plates were incubated at 37°C supplied with 5% CO2 for four days before measuring cytopathic effect under light microscope. The TCID50 calculation was performed using the Spearman Karber method (Hierholzer and Killington, 1996).
PolyI:C and LPS induced murine HLH
Hemophagocytic Lymphohistiocytosis (HLH) was induced by sequential challenge with polyI:C and LPS as previously described (Wang et al., 2019). Mice were injected intraperitoneally with high molecular weight polyI:C (InvivoGen, tlrl-pic) at 10 mg/kg body weight reconstituted in PBS. 24 h post polyI:C injection, mice were injected intraperitoneally with LPS (InvivoGen, tlrl-pb5lps) at 5 mg/kg body weight reconstituted in PBS. Rectal temperature monitoring was then performed every hour until the ethical experimental endpoint, which was defined by a temperature reading of 30°C or below. All mice were humanely euthanized once one mouse reached the ethical experimental endpoint at which point samples were taken for histological and plasma cytokine analysis. Mice of mixed sexes were age-matched for each experiment and were used between 8-15 weeks of age. The results presented are pooled from data of at least two experimental cohorts per genotype performed on separate occasions.
Histology and Immunohistochemistry (IHC)
Small intestines were fixed in 10% neutral buffered formalin, paraffin embedded, and sectioned for routine histology. IHC sections were stained with anti-cleaved caspase-3 antibodies (R&D, AF835) and a HRP-conjugated rabbit secondary antibody. Slides were scanned using a 3D Histech Brightfield Scan x20 and images were taken using CaseCenter software at 20x magnification. Scale bars = 100 μm.
CRISPR/Cas9 genome editing
Ninj1−/− immortalized BMDMs were generated based on a CRISPR/Cas9 protocol described previously (Baker and Masters, 2018). pFU-Cas9-mCherry plasmid DNA was transiently transfected into HEK293T cells alongside pMDL (packaging), RSV-REV (packaging) and VSVg (envelope) using Lipofectamine 2000 diluted in OptiMEM (Thermo Fisher Scientific) to generate lentiviral particles in DMEM. The cell culture supernatant was collected 48 h later and filtered through a 0.45 μm filter prior to cell transduction. Lentiviral transduction was performed by replacing normal cell culture medium with DMEM containing lentivirus particles for 24 h. Transduced, Cas9-mCherry positive cells were selected using flow cytometry. Lentiviral particles harboring Ninj1 targeting (exon-13 #1: 5′ TCCGCAGCGCTCTTCTTGTTGG, exon-13 #2: 5′ ACCACAAGGGGCACGAAGAAGG) sgRNAs cloned into the LV04 (Metzakopian et al., 2017) plasmid were generated and transduced into Cas9 positive, Mlkl −/− iBMDMs using the method described above. Non-transduced control and transduced cells were selected in puromycin (2 μg/mL) until all control cells had died to obtain polyclonal Ninj1 −/− cell populations. Gene disruption was confirmed by immunoblot analysis of the targeted protein and functional analysis.
Cell stimulation
Unless otherwise stated in figure legends, after BMDMs or iBMDMs had adhered to the plate, cells were primed where indicated with IFNγ (50 ng/mL, recombinant mouse, R&D; 485-MI), IFNβ (1000 U/mL, PBL Assay Science; 12405-1) or treated with vehicle overnight (16-24 h). Subsequently, LPS (50 ng/mL, InvivoGen; tlrl-3pelps), Pam-3-CSK4 (500 ng/mL, InvivoGen; tlrl-pms), PolyI:C (10 μg/mL, InvivoGen; tlrl-picw), FLAG-TNF (100 ng/mL recombinant human, in-house) or vehicle (control) were added for up to 48 h. Where multiple time points were used for analysis, TLR stimulations were performed in a reverse time-course fashion so that all cells were harvested at the same time. Single treatment controls were added for the longest time point measured. Inhibitors: Z-VAD-fmk (20 μM, Z-VAD-FMK, R&D Systems; FMK001), IDN-6556 (5 μM, kindly gifted by TetraLogics Pharmaceuticals) and 1400W (10 μM, Abcam; ab120165), ABT-199 (1 μM, Active Biochem; A-1231), S63845 (10 μM, Active Biochem; A-6044), were added at the same time as TLR stimulation. Deferiprone (DFP, 150 μM, Sigma; Y0001976) or Ferrostatin-1 (Fer-1, 2 μM, Sigma; SML0583) were added 15 min prior to TLR stimulation. Nigericin (10 μM, Sigma; N7143) and SNAP (100 – 200 μM, Sigma; N3398) stimulation was performed as described in the relevant figure legends. LPS and compound A (1 μM, Cp.A, TetraLogic Pharmaceuticals), LPS and Z-VAD-fmk or LPS and IDN-6556 stimulations were performed for 24 h for flow cytometric analysis and 12 h for immunoblot analysis. Treatments with ABT-263 (1 μM, Active Biochem; A-1001), ABT-737 (1 μM, Active Biochem; A-6044) or cycloheximide (10 μg/mL, Sigma; C7698) were conducted for 6 h for flow cytometric analysis or 2–4 h for immunoblot analysis, unless otherwise stated in the figure legends. DFP and Fer-1 were added 15 min before the addition of RSL3 (500 nM, Sigma; SML2234) for 24 h. QVD-Oph (20 μM, MP Biomedicals; 03OPH109) was added 15 min before treatment with ABT-737. For RNA-sequencing BMDMs were primed with IFNγ (50 ng/mL) for 16 h, followed by stimulation with LPS for 7 h.
Flow cytometry
To evaluate cell viability, cells were detached from plates using fresh EDTA (5 mM) in PBS, pooled with cell supernatants containing propidium iodide (PI, 10 μg/mL) and recorded using an LSR II flow cytometer (Becton Dickinson, NJ). PI exclusion analysis for each sample was performed with 10,000 single cell, non-debris events using a FSC-A versus PI FACS plot. For cell surface marker expression analysis, stimulated BMDMs were harvested as described above and incubated with the following antibodies: F4/80-FITC (1:200, eBioscience; clone BM8), MHCII-AF700 (1:200, eBiocience; M5/114.15.2), CD206-APC (1:200, eBioscience; MR6F3) and CD11b-PE-Cy7 (1:400, eBioscience; M1/70) for 30 min on ice. Cells were washed once in PBS prior to analysis by flow cytometry. Analysis of cell surface marker expression was performed on 10,000 single cell, PI negative, gated events. Flow cytometry data were analyzed using WEASEL version 2.7 software (Frank Battye).
Cytochrome c retention assay
Mitochondrial retention of cytochrome c was examined based on a protocol previously described (Waterhouse and Trapani, 2003). Briefly, cells were permeabilized in 0.025% digitonin (w/v) in MELB (20 mM HEPES pH 7.5, 250 mM sucrose, 1 mM EDTA, 50 mM KCl, 2.5 mM MgCl2) for 10 min on ice. Pelleted cell fractions were obtained by centrifugation (18,000 rcf, 5 min) and fixed in eBioscience IC Fixation Buffer (ThermoFisher Scientific) for 30 min on ice. Fixed cells were washed twice in eBioscience Permeabilization buffer (ThermoFisher Scientific) and then incubated in eBioscience Permeabilization buffer containing an antibody against cytochrome c conjugated to APC (Miltenyl Biotec; 130-111-180, 1:100) for 1 h on ice. Cells were then washed twice in eBioscience Permeabilization buffer and analyzed on an LSR II flow cytometer (Becton Dickinson, NJ). Unstained permeabilized cells were used as controls for flow cytometry analysis.
Immunoblots
Cells or cell-free supernatants were lysed in SDS (2%) lysis buffer with β-mercaptoethanol (143 mM). Cell lysates were centrifuged through a low protein binding polypropylene column (Pierce) to shred DNA. Lysates were separated on 4%–12% gradient gels (Invitrogen), then proteins were transferred onto nitrocellulose (Amersham) or Immoblon-E polyvinyl difluoride membranes (Merck Millipore; IEVH85R). Ponceau staining was performed routinely to evaluate protein loading accuracy. Membranes were blocked with skim milk (5%, Devondale) in TBS containing 0.1% Tween 20 (TBS-T) for 20 min. Membranes were probed with primary antibodies overnight at 4°C (all diluted in 5% BSA TBS-T with 0.04% azide at 1:1000 unless stated otherwise): A1 (in-house (Lang et al., 2014)), β-actin (Sigma; A-1798), BCL-XL (Cell Signaling; 2764), BID (2D1; in-house (Kaufmann et al., 2007)), caspase-1 (Adipogen; AG-20B-0042-C100), cleaved caspase-3 (Cell Signaling; 9661), cleaved caspase-8 Asp387 (Cell Signaling; 9429, 8592), cleaved caspase-9 (Cell Signaling; 9509), pro-caspase-3 (Cell Signaling; 9665), pro-caspase-9 (Cell Signaling; 9508), pro-caspase-8 (in-house, 3B10), cIAP1 (Alexis Biochemicals; ALX-803-355), GSDMD (Abcam; ab209845), HMGB1 (Abcam, ab195012), IL-1β (R&D; AF-401-NA), iNOS (BD Transduction; 610328), MCL-1 (Cell Signaling; 5453), MLKL (in-house (Murphy et al., 2013); 3H1), NINJ1 (Invitrogen, PA5-95755) and XIAP (MBL; M044-3). Relevant horseradish peroxidase-conjugated secondary antibodies (Jaxon laboratories) were diluted 1:10,000 in 5% skimmed milk in TBS-T applied for 1 h at room temperature. Membranes were subjected to 3 × 5 min washes in TBS-T between antibody incubations and 4 × 5 min washes after secondary antibody incubation. Membranes were developed using ECL (Millipore, Bio-Rad) using the ChemiDoc Touch Imaging System (Bio-Rad) and Image Lab software.
Quantitative Polymerase Chain Reaction (qPCR)
Total RNA was extracted from stimulated cells using the ISOLATE II RNA Mini Kit (Meridian Bioscience). 1 μg RNA was used to generate cDNA with the SuperScript III Reverse Transcriptase (Invitrogen). 40 ng cDNA samples, or nuclease free water controls were prepared in duplicate for gene expression using the Maxima SYBR Green qPCR Master Mix (2X), with separate ROX vial (ThermoFisher; K0251). Samples were assayed on the ViiA 7 Real-Time PCR System (Applied Biosystems) with the following protocol (1 cycle (hold stage): 95.0°C for 10 min; 42 cycles (PCR stage): 95.0°C for 15 s, 51.3°C for 25 s, 72.0°C for 19 s; continuous (melt curve stage): 95.0°C for 15 s, 51.3°C for 1 min, 95.0°C for 19 s). Relative gene expression was normalized to an internal housekeeping reference gene (Hprt or 18S) and displayed as the fold-change compared to an unstimulated control sample, as previously described (Livak and Schmittgen, 2001). Where different groups (genotype, medium conditions) are compared side-by-side, gene expression of one group is displayed as the fold-change compared to an unstimulated control sample of that group, to ensure intrinsic differences in untreated conditions are accounted for. Where differences between untreated conditions of different groups are shown, the data are displayed as the dCt (i.e., the Ct value of the gene of interest normalized to Hprt or 18S). Primer sequences used to assay gene expression are as follows: mm18S (For: 5′ GTAACCCGTTGAACCCCATT, Rev: 5′ CCATCCAATCGGTAGTAGCG), mmBcl2 (For: 5′ CCTGTGGATGACTGAGTACCTG, Rev: 5′ AGCCAGGAGAAATCAAACAGAGG), mmCd86 (For: 5′ TCAGTGATCGCCAACTTCAG, Rev: 5′ TTAGGTTTCGGGTGACCTTG), mmFasl (For: 5′ GAAGGAACTGGCAGAACTCCGT, Rev: 5′ GCCACACTCCTCGGCTCTTTTT), mmFas (For: 5′ CTGCGATTCTCCTGGCTGTGAA, Rev: 5′ CAACAACCATAGGCGATTTCTGG), mmHprt (For: 5′ TGAAGTACTCATTATAGTCAAGGGCA, Rev: 5′ CTGGTGAAAAGGACCTCTCG), mmIl10 (For: 5′ GGTTGCCCAGCCTTATCGGA, Rev: 5′ ACCTGCTCCACTGCCTTGCT), mmIl1b (For: 5; AGTTGACGGACCCCAAAAG, Rev: 5′ AGCTGGATGCTCTCATCAGG), mmNos2 (For: 5′ GCCACCAACAATGGCAACA, Rev: 5′ CGTACCGGATGAGCTGTGAATT), mmTnfrsf10b (For: 5′ TGTGTCGATGCAAACCAGGCAC, Rev: 5′ GCCGTTTTGGAGACACACTTCC), and mmTnfsf10 (For: 5′ GGAAGACCTCAGAAAGTGGCAG, Rev: 5′ TTTCCGAGAGGACTCCCAGGAT), mmYm1 (For: 5′ GGGCATACCTTTATCCTGAG, Rev: 5′ CCACTGAAGTCATCCATGTC). Specificity of each primer set was confirmed by the observation of a single peak in the melt curve graph of each qPCR run.
RNA sequencing preparation and analysis
Total RNA was extracted using the ISOLATE II RNA Mini Kit (Meridian Bioscience). The extracted RNA was analyzed on the Agilent 4200 Tapestation prior to library preparation. High quality RNA with RINe values greater than 9 was used for downstream application. 3′ mRNA-sequencing libraries were prepared using 100 ng of total RNA using the QuantSeq 3′mRNA-Seq Library Prep (Lexogen) according to the manufacturer’s instructions and sequenced on the NextSeq 500 (Illumina). The single-end 75 bp were demultiplexed using Casavav1.8.2 and Cutadapt (v1.9) was used for read trimming (Martin, 2011). The trimmed reads were subsequently mapped to the mouse genome (mm10) using HISAT2 (Kim et al., 2019). FeatureCounts from Rsubread package (version 1.34.7) was used for read counting after which genes with less than 2 counts per million reads (CPM) in at least 3 samples were excluded from downstream analysis (Liao et al., 2014; 2019). Count data were normalized using the trimmed mean of M values (TMM) method and differential gene expression analysis was performed using the limma-voom pipeline (limma version 3.40.6) (Law et al., 2014; Liao et al., 2014; Robinson and Oshlack, 2010). Comparisons between IFNγ/LPS-treated Mlkl −/− versus Casp8 −/− Mlkl −/− samples were made. GSEA 2.2.2 was used for Gene set enrichment analysis (GSEA) (Liberzon et al., 2015; Subramanian et al., 2005). Gene ontology (GO) and TRRUST analysis was performed using Metascape (Zhou et al., 2019). ggplot2 (version 3.2.1) (Wickham, 2009) was used to plot the volcano plots and pheatmap (version 1.0.12) (https://rdrr.io/cran/pheatmap/) was used to generate heatmaps. A list of genes associated with iNOS and arginine metabolism was obtained from Young et al. (2018). The datasets generated during this study are available at GEO: GSE161179.
Griess assay
Nitrite (NO2 −) in cell supernatants was measured based on the Griess reagent system (Promega). Cell supernatants were assayed in duplicate and a sodium nitrite standard curve ranging from 100 μM – 1.56 μM in DMEM was assayed in triplicate. 50 μl sulfanilamide (1% w/v, Sigma) in phosphoric acid (5% v/v, Sigma) was added to 50 μl cell supernatants for 5 min followed by the addition of 50 μl N-1-napthylethylenediamine dihydrochloride (0.1% w/v, Sigma) in water. Samples were incubated for 5 min in the dark and absorbance was measured on a VersaMaxTM Tunable Microplate Reader (Molecular Devices, CA). Replicates were averaged and corrected for background absorbance. Data values were interpolated from the background corrected sodium nitrite standard.
Cytokine analysis
Plasma concentrations of TNF, IL-1β and IL-6 were measured by TNF (Invitrogen, 88-7324-88), IL-1β (R&D, DY401) and IL-6 (Invitrogen, 88-7064-88) ELISA according to the manufacturer’s instructions. TNF in the supernatants from stimulated cells was measured by TNF ELISA according to the manufacturer’s instructions.
Imaging
BMDMs were stimulated as described in the relevant figure legends with PI (1 μg/mL and imaged on a Leica SP8 microscope (LAS-X software) set at 3 – 4 images per hour for 24 h. Matched time-lapsed brightfield and fluorescent (PI) images were merged and stitched into videos using ImageJ (Version 2.1.0/1.53k) (Schneider et al., 2012).
Lactate Dehydrogenase (LDH) assay
Supernatants from stimulated iBMDMs were harvested and analyzed in duplicate for LDH measurements according to the manufacturer’s instruction (Roche, 11644793001).
Quantification and statistical analysis
Each data point from graphs of BMDM or mouse experiments represents an independent biological replicate (i.e., different mouse). All graphs of iBMDM experiments display data points from experiments performed on the same cell line. Replicates from In vitro experiments were acquired either on different days or by different researchers with separate reagents and are displayed as the mean ± standard deviation (SD). In vivo data are pooled from at least two cohorts and are displayed as the mean ± standard error of the mean (SEM). Statistical comparisons of treatments between paired samples of the same genotype (i.e., treatment A versus treatment B) were performed using a parametric, paired t test. Statistical comparisons of different genotypic groups receiving the same treatment were performed using a two-way ANOVA, with a Bonferroni (two genotypes) or Tukey (three or more genotypes) post hoc correction for multiple comparisons. Paired t tests and two-way ANOVA analyses assumed Gaussian distribution and equal standard deviations between experimental and control groups. All graphical data were prepared and analyzed in GraphPad PRISM (Version 8.4.3). In all analyses significance was defined at 0.05, with p > 0.05 (n.s.), p ≤ 0.05 (∗), p ≤ 0.01 (∗∗), p ≤ 0.001 (∗∗∗), p ≤ 0.0001 (∗∗∗∗).
Acknowledgments
We thank Associate Professor G. Dewson for valuable experimental advice and critical reading of the manuscript and Dr. P. Bouillet for kind donation of Pmaip1 −/− and Bid −/− mice. We gratefully acknowledge grant support from the National Health and Medical Research Council (NHMRC) of Australia (project grants: 1145788 to J.E.V., K.E.L.; 1101405 to J.E.V.; 1162765 to K.E.L.; 1165591 to E.D.H.; 1143105 to M.J.H. and A.S.; 1183848 to T.N; 1137989 to J.R.G; ideas grants: 1183070 to J.E.V.; 1181089 to K.E.L.; 1182649 to J.R.G; investigator grants: 1194144 to H.A.; 1175011 to M.P.; 1107149 & 1195038 to J.S.; program grant 101671 to A.S.), the German Research Foundation (SFB1403, project no. 414786233 to H.K.), fellowships (1141466 to J.E.V.; 1020363 to A.S.; 1144014 to S.A.C.; 1159488 to E.D.H.), and the Leukemia and Lymphoma Society (LLS SCOR 7015-18 to A.S., M.J.H., and J.S.). K.E.L. and T.F are Australian Research Council (ARC) Future Fellows (FT190100266 to K.E.L and FT170100313 to T.N.). A.R. is supported by the Co-Funded Monash Graduate Scholarship (CF-MGS) from Monash University. M.J.H. is an NHMRC Senior Research Fellow (1156095). G.B. is funded by the National Collaborative Research Infrastructure Strategy (NCRIS) via Phenomics Australia. D.S.S. is supported by a philanthropic PhD scholarship from the Walter and Eliza Hall Institute of Medical Research. R.F. is supported by the Galbraith Family Charitable Trust. This work was also supported by operational infrastructure grants through the Australian Government Independent Research Institute Infrastructure Support Scheme and the Victorian State Government Operational Infrastructure Support Scheme, Australia.
Author contributions
The project was conceived by D.S.S., J.E.V., and R.F.; the experiments were designed by D.S.S., J.E.V., and R.F.; and the manuscript was written by D.S.S., J.E.V., and R.F. Experiments were performed by D.S.S., J.P., A.W., I.Y.K., M.F., M.R., J.P.C., K.C.D., M.S., T.M.D., S.H., L.M., M.D., H.A., M.D., Y.D., S.A.C., H.T., A.R., S.H.C., K.E.L., R.F., and J.E.V. Expert advice, essential mice, and reagents were provided by A.S.H., R.S.N., T.N., S.E.N., G.B., S.M.M., J.R.G., M.J.H., E.D.H., A.S., J.S., M.P., and H.K. All authors assisted with data interpretation and manuscript editing.
Declaration of interests
The authors declare that D.S.S., J.P., A.W., I.Y.K., M.R., J.P.C., K.C.D., S.H., H.A., M.D., Y.D., L.M., M.D., A.S.H., S.E.N., J.R.G., M.J.H., E.D.H., A.S., J.S., M.P., R.F., and J.E.V. are employees or former employees of the Walter and Eliza Hall Medical Institute, which receives milestone payments from Genentech and AbbVie for the development of ABT-199 for cancer therapy. J.E.V. sits on the advisory board of Avammune Therapeutics.
Inclusion and diversity
One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. One or more of the authors of this paper self-identifies as living with a disability.
Published: February 8, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.immuni.2022.01.003.
Supplemental information
Data and code availability
-
•
RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. All raw data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Alehashemi S., Goldbach-Mansky R. Human Autoinflammatory Diseases Mediated by NLRP3-, Pyrin-, NLRP1-, and NLRC4-Inflammasome Dysregulation Updates on Diagnosis, Treatment, and the Respective Roles of IL-1 and IL-18. Front. Immunol. 2020;11:1840. doi: 10.3389/fimmu.2020.01840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allam R., Lawlor K.E., Yu E.C., Mildenhall A.L., Moujalled D.M., Lewis R.S., Ke F., Mason K.D., White M.J., Stacey K.J., et al. Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. EMBO Rep. 2014;15:982–990. doi: 10.15252/embr.201438463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Diaz S., Dillon C.P., Lalaoui N., Tanzer M.C., Rodriguez D.A., Lin A., Lebois M., Hakem R., Josefsson E.C., O’Reilly L.A., et al. The Pseudokinase MLKL and the Kinase RIPK3 Have Distinct Roles in Autoimmune Disease Caused by Loss of Death-Receptor-Induced Apoptosis. Immunity. 2016;45:513–526. doi: 10.1016/j.immuni.2016.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker P.J., Masters S.L. In: Innate Immune Activation: Methods and Protocols. De Nardo D., De Nardo C.M., editors. Springer; 2018. Generation of Genetic Knockouts in Myeloid Cell Lines Using a Lentiviral CRISPR/Cas9 System; pp. 41–55. [DOI] [PubMed] [Google Scholar]
- Bossaller L., Chiang P.I., Schmidt-Lauber C., Ganesan S., Kaiser W.J., Rathinam V.A., Mocarski E.S., Subramanian D., Green D.R., Silverman N., et al. Cutting edge: FAS (CD95) mediates noncanonical IL-1β and IL-18 maturation via caspase-8 in an RIP3-independent manner. J. Immunol. 2012;189:5508–5512. doi: 10.4049/jimmunol.1202121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumatti G., Ma C., Lalaoui N., Nguyen N.Y., Navarro M., Tanzer M.C., Richmond J., Ghisi M., Salmon J.M., Silke N., et al. The caspase-8 inhibitor emricasan combines with the SMAC mimetic birinapant to induce necroptosis and treat acute myeloid leukemia. Sci. Transl. Med. 2016;8:339ra69. doi: 10.1126/scitranslmed.aad3099. [DOI] [PubMed] [Google Scholar]
- Chauhan D., Bartok E., Gaidt M.M., Bock F.J., Herrmann J., Seeger J.M., Broz P., Beckmann R., Kashkar H., Tait S.W.G., et al. BAX/BAK-Induced Apoptosis Results in Caspase-8-Dependent IL-1beta Maturation in Macrophages. Cell Rep. 2018;25:2354–2368. doi: 10.1016/j.celrep.2018.10.087. e2355. [DOI] [PubMed] [Google Scholar]
- Chin Y.E., Kitagawa M., Kuida K., Flavell R.A., Fu X.Y. Activation of the STAT signaling pathway can cause expression of caspase 1 and apoptosis. Mol. Cell. Biol. 1997;17:5328–5337. doi: 10.1128/mcb.17.9.5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow S.H., Deo P., Naderer T. Macrophage cell death in microbial infections. Cell. Microbiol. 2016;18:466–474. doi: 10.1111/cmi.12573. [DOI] [PubMed] [Google Scholar]
- Czabotar P.E., Lessene G., Strasser A., Adams J.M. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- de Jesus Trovoada M., Martins M., Ben Mansour R., Sambo Mdo.R., Fernandes A.B., Antunes Gonçalves L., Borja A., Moya R., Almeida P., Costa J., et al. NOS2 variants reveal a dual genetic control of nitric oxide levels, susceptibility to Plasmodium infection, and cerebral malaria. Infect. Immun. 2014;82:1287–1295. doi: 10.1128/IAI.01070-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nardo D., Kalvakolanu D.V., Latz E. In: Macrophages: Methods and Protocols. Rousselet G., editor. Springer; 2018. Immortalization of Murine Bone Marrow-Derived Macrophages; pp. 35–49. [DOI] [PubMed] [Google Scholar]
- DeLaney A.A., Berry C.T., Christian D.A., Hart A., Bjanes E., Wynosky-Dolfi M.A., Li X., Tummers B., Udalova I.A., Chen Y.H., et al. Caspase-8 promotes c-Rel-dependent inflammatory cytokine expression and resistance against Toxoplasma gondii. Proc. Natl. Acad. Sci. USA. 2019;116:11926–11935. doi: 10.1073/pnas.1820529116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon S.S., Mastropaolo L.A., Murchie R., Griffiths C., Thöni C., Elkadri A., Xu W., Mack A., Walters T., Guo C., et al. Higher activity of the inducible nitric oxide synthase contributes to very early onset inflammatory bowel disease. Clin. Transl. Gastroenterol. 2014;5:e46–e47. doi: 10.1038/ctg.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerflinger M., Deng Y., Whitney P., Salvamoser R., Engel S., Kueh A.J., Tai L., Bachem A., Gressier E., Geoghegan N.D., et al. Flexible Usage and Interconnectivity of Diverse Cell Death Pathways Protect against Intracellular Infection. Immunity. 2020;53:533–547.e7. doi: 10.1016/j.immuni.2020.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drutman S.B., Mansouri D., Mahdaviani S.A., Neehus A.-L., Hum D., Bryk R., Hernandez N., Belkaya S., Rapaport F., Bigio B., et al. Fatal Cytomegalovirus Infection in an Adult with Inherited NOS2 Deficiency. N. Engl. J. Med. 2020;382:437–445. doi: 10.1056/NEJMoa1910640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey M., Nagarkoti S., Awasthi D., Singh A.K., Chandra T., Kumaravelu J., Barthwal M.K., Dikshit M. Nitric oxide-mediated apoptosis of neutrophils through caspase-8 and caspase-3-dependent mechanism. Cell Death Dis. 2016;7:e2348. doi: 10.1038/cddis.2016.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert G., Lopaticki S., O’Neill M.T., Steel R.W.J., Doerflinger M., Rajasekaran P., Yang A.S.P., Erickson S., Ioannidis L., Arandjelovic P., et al. Targeting the Extrinsic Pathway of Hepatocyte Apoptosis Promotes Clearance of Plasmodium Liver Infection. Cell Rep. 2020;30:4343–4354. doi: 10.1016/j.celrep.2020.03.032. e4344. [DOI] [PubMed] [Google Scholar]
- Ekert P.G., Read S.H., Silke J., Marsden V.S., Kaufmann H., Hawkins C.J., Gerl R., Kumar S., Vaux D.L. Apaf-1 and caspase-9 accelerate apoptosis, but do not determine whether factor-deprived or drug-treated cells die. J. Cell Biol. 2004;165:835–842. doi: 10.1083/jcb.200312031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson S.L., de Sauvage F.J., Kikly K., Carver-Moore K., Pitts-Meek S., Gillett N., Sheehan K.C., Schreiber R.D., Goeddel D.V., Moore M.W. Decreased sensitivity to tumour-necrosis factor but normal T-cell development in TNF receptor-2-deficient mice. Nature. 1994;372:560–563. doi: 10.1038/372560a0. [DOI] [PubMed] [Google Scholar]
- Feltham R., Vince J.E., Lawlor K.E. Caspase-8: not so silently deadly. Clin. Transl. Immunology. 2017;6:e124. doi: 10.1038/cti.2016.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortune S.M., Solache A., Jaeger A., Hill P.J., Belisle J.T., Bloom B.R., Rubin E.J., Ernst J.D. Mycobacterium tuberculosis inhibits macrophage responses to IFN-gamma through myeloid differentiation factor 88-dependent and -independent mechanisms. J. Immunol. 2004;172:6272–6280. doi: 10.4049/jimmunol.172.10.6272. [DOI] [PubMed] [Google Scholar]
- Fritsch M., Günther S.D., Schwarzer R., Albert M.C., Schorn F., Werthenbach J.P., Schiffmann L.M., Stair N., Stocks H., Seeger J.M., et al. Caspase-8 is the molecular switch for apoptosis, necroptosis, and pyroptosis. Nature. 2019;575:683–687. doi: 10.1038/s41586-019-1770-6. [DOI] [PubMed] [Google Scholar]
- Garvey E.P., Oplinger J.A., Furfine E.S., Kiff R.J., Laszlo F., Whittle B.J., Knowles R.G. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J. Biol. Chem. 1997;272:4959–4963. doi: 10.1074/jbc.272.8.4959. [DOI] [PubMed] [Google Scholar]
- Ginhoux F., Schultze J.L., Murray P.J., Ochando J., Biswas S.K. New insights into the multidimensional concept of macrophage ontogeny, activation, and function. Nat. Immunol. 2016;17:34–40. doi: 10.1038/ni.3324. [DOI] [PubMed] [Google Scholar]
- Gitlin A.D., Heger K., Schubert A.F., Reja R., Yan D., Pham V.C., Suto E., Zhang J., Kwon Y.C., Freund E.C., et al. Integration of innate immune signalling by caspase-8 cleavage of N4BP1. Nature. 2020;587:275–280. doi: 10.1038/s41586-020-2796-5. [DOI] [PubMed] [Google Scholar]
- Gross T.J., Kremens K., Powers L.S., Brink B., Knutson T., Domann F.E., Philibert R.A., Milhem M.M., Monick M.M. Epigenetic silencing of the human NOS2 gene: rethinking the role of nitric oxide in human macrophage inflammatory responses. J. Immunol. 2014;192:2326–2338. doi: 10.4049/jimmunol.1301758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günther C., He G.W., Kremer A.E., Murphy J.M., Petrie E.J., Amann K., Vandenabeele P., Linkermann A., Poremba C., Schleicher U., et al. The pseudokinase MLKL mediates programmed hepatocellular necrosis independently of RIPK3 during hepatitis. J. Clin. Invest. 2016;126:4346–4360. doi: 10.1172/JCI87545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurung P., Anand P.K., Malireddi R.K., Vande Walle L., Van Opdenbosch N., Dillon C.P., Weinlich R., Green D.R., Lamkanfi M., Kanneganti T.D. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 2014;192:1835–1846. doi: 10.4049/jimmunol.1302839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hague S., Peuralinna T., Eerola J., Hellström O., Tienari P.J., Singleton A.B. Confirmation of the protective effect of iNOS in an independent cohort of Parkinson disease. Neurology. 2004;62:635–636. doi: 10.1212/01.wnl.0000110191.38152.29. [DOI] [PubMed] [Google Scholar]
- Han H., Cho J.W., Lee S., Yun A., Kim H., Bae D., Yang S., Kim C.Y., Lee M., Kim E., et al. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Research. 2018;46(D1):D380–D386. doi: 10.1093/nar/gkx1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S., Liang Y., Shao F., Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl. Acad. Sci. USA. 2011;108:20054–20059. doi: 10.1073/pnas.1116302108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry C.M., Martin S.J. Caspase-8 Acts in a Non-enzymatic Role as a Scaffold for Assembly of a Pro-inflammatory “FADDosome” Complex upon TRAIL Stimulation. Mol Cell. 2017;65:715–729 e715. doi: 10.1016/j.molcel.2017.01.022. [DOI] [PubMed] [Google Scholar]
- Herbst S., Schaible U.E., Schneider B.E. Interferon gamma activated macrophages kill mycobacteria by nitric oxide induced apoptosis. PLoS ONE. 2011;6:e19105. doi: 10.1371/journal.pone.0019105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hierholzer J.C., Killington R.A. In: Virology Methods Manual. 1st. Kangro H., Mahy B., editors. Academic Press; 1996. Virus isolation and quantitation; pp. 25–46. [Google Scholar]
- Hildebrand J.M., Kauppi M., Majewski I.J., Liu Z., Cox A.J., Miyake S., Petrie E.J., Silk M.A., Li Z., Tanzer M.C., et al. A missense mutation in the MLKL brace region promotes lethal neonatal inflammation and hematopoietic dysfunction. Nat. Commun. 2020;11:3150. doi: 10.1038/s41467-020-16819-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X., Ivashkiv L.B. Cross-regulation of signaling pathways by interferon-gamma: Implications for immune responses and autoimmune diseases. Immunity. 2009;31:539–550. doi: 10.1016/j.immuni.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X., Chakravarty S.D., Ivashkiv L.B. Regulation of interferon and Toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunol. Rev. 2008;226:41–56. doi: 10.1111/j.1600-065X.2008.00707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivashkiv L.B. IFNγ: signalling, epigenetics, and roles in immunity, metabolism, disease, and cancer immunotherapy. Nat. Rev. Immunol. 2018;18:545–558. doi: 10.1038/s41577-018-0029-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaco I., Annibaldi A., Lalaoui N., Wilson R., Tenev T., Laurien L., Kim C., Jamal K., Wicky John S., Liccardi G., et al. MK2 Phosphorylates RIPK1 to Prevent TNF-Induced Cell Death. Mol Cell. 2017;66:698–710 e695. doi: 10.1016/j.molcel.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen R., Van Wengen A., Verhard E., De Boer T., Zomerdijk T., Ottenhoff T.H., Van Dissel J.T. Divergent role for TNF-alpha in IFN-gamma-induced killing of Toxoplasma gondii and Salmonella typhimurium contributes to selective susceptibility of patients with partial IFN-gamma receptor 1 deficiency. J. Immunol. 2002;169:3900–3907. doi: 10.4049/jimmunol.169.7.3900. [DOI] [PubMed] [Google Scholar]
- Jiang S.H., Athanasopoulos V., Ellyard J.I., Chuah A., Cappello J., Cook A., Prabhu S.B., Cardenas J., Gu J., Stanley M., et al. Functional rare and low frequency variants in BLK and BANK1 contribute to human lupus. Nat. Commun. 2019;10:2201. doi: 10.1038/s41467-019-10242-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen I., Rayamajhi M., Miao E.A. Programmed cell death as a defence against infection. Nat. Rev. Immunol. 2017;17:151–164. doi: 10.1038/nri.2016.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S., Fernandes-Alnemri T., Rogers C., Mayes L., Wang Y., Dillon C., Roback L., Kaiser W., Oberst A., Sagara J., et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat. Commun. 2015;6:7515. doi: 10.1038/ncomms8515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karki R., Sharma B.R., Tuladhar S., Williams E.P., Zalduondo L., Samir P., Zheng M., Sundaram B., Banoth B., Malireddi R.K.S., et al. Synergism of TNF-alpha and IFN-gamma Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell. 2021;184:149–168 e117. doi: 10.1016/j.cell.2020.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann T., Tai L., Ekert P.G., Huang D.C., Norris F., Lindemann R.K., Johnstone R.W., Dixit V.M., Strasser A. The BH3-only protein bid is dispensable for DNA damage- and replicative stress-induced apoptosis or cell-cycle arrest. Cell. 2007;129:423–433. doi: 10.1016/j.cell.2007.03.017. [DOI] [PubMed] [Google Scholar]
- Kayagaki N., Stowe I.B., Lee B.L., O’Rourke K., Anderson K., Warming S., Cuellar T., Haley B., Roose-Girma M., Phung Q.T., et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- Kayagaki N., Kornfeld O.S., Lee B.L., Stowe I.B., O’Rourke K., Li Q., Sandoval W., Yan D., Kang J., Xu M., et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature. 2021;591:131–136. doi: 10.1038/s41586-021-03218-7. [DOI] [PubMed] [Google Scholar]
- Kiang J.G., Krishnan S., Lu X., Li Y. Inhibition of inducible nitric-oxide synthase protects human T cells from hypoxia-induced apoptosis. Mol. Pharmacol. 2008;73:738–747. doi: 10.1124/mol.107.041079. [DOI] [PubMed] [Google Scholar]
- Kim D., Paggi J.M., Park C., Bennett C., Salzberg S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019;37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kincaid E.Z., Ernst J.D. Mycobacterium tuberculosis exerts gene-selective inhibition of transcriptional responses to IFN-gamma without inhibiting STAT1 function. J. Immunol. 2003;171:2042–2049. doi: 10.4049/jimmunol.171.4.2042. [DOI] [PubMed] [Google Scholar]
- Köntgen F., Grumont R.J., Strasser A., Metcalf D., Li R., Tarlinton D., Gerondakis S. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995;9:1965–1977. doi: 10.1101/gad.9.16.1965. [DOI] [PubMed] [Google Scholar]
- Körner H., Cook M., Riminton D.S., Lemckert F.A., Hoek R.M., Ledermann B., Köntgen F., Fazekas de St Groth B., Sedgwick J.D. Distinct roles for lymphotoxin-alpha and tumor necrosis factor in organogenesis and spatial organization of lymphoid tissue. Eur. J. Immunol. 1997;27:2600–2609. doi: 10.1002/eji.1830271020. [DOI] [PubMed] [Google Scholar]
- Kreuz S., Siegmund D., Rumpf J.J., Samel D., Leverkus M., Janssen O., Häcker G., Dittrich-Breiholz O., Kracht M., Scheurich P., Wajant H. NFkappaB activation by Fas is mediated through FADD, caspase-8, and RIP and is inhibited by FLIP. J. Cell Biol. 2004;166:369–380. doi: 10.1083/jcb.200401036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalaoui N., Boyden S.E., Oda H., Wood G.M., Stone D.L., Chau D., Liu L., Stoffels M., Kratina T., Lawlor K.E., et al. Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature. 2020;577:103–108. doi: 10.1038/s41586-019-1828-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang M.J., Brennan M.S., O’Reilly L.A., Ottina E., Czabotar P.E., Whitlock E., Fairlie W.D., Tai L., Strasser A., Herold M.J. Characterisation of a novel A1-specific monoclonal antibody. Cell Death Dis. 2014;5:e1553. doi: 10.1038/cddis.2014.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laubach V.E., Shesely E.G., Smithies O., Sherman P.A. Mice lacking inducible nitric oxide synthase are not resistant to lipopolysaccharide-induced death. Proc. Natl. Acad. Sci. USA. 1995;92:10688–10692. doi: 10.1073/pnas.92.23.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffer F., Jargosch M., Krause L., Garzorz-Stark N., Franz R., Roenneberg S., Böhner A., Mueller N.S., Theis F.J., Schmidt-Weber C.B., et al. Type I Immune Response Induces Keratinocyte Necroptosis and Is Associated with Interface Dermatitis. J. Invest. Dermatol. 2018;138:1785–1794. doi: 10.1016/j.jid.2018.02.034. [DOI] [PubMed] [Google Scholar]
- Law C.W., Chen Y., Shi W., Smyth G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor K.E., Khan N., Mildenhall A., Gerlic M., Croker B.A., D’Cruz A.A., Hall C., Kaur Spall S., Anderton H., Masters S.L., et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 2015;6:6282. doi: 10.1038/ncomms7282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor K.E., Feltham R., Yabal M., Conos S.A., Chen K.W., Ziehe S., Graß C., Zhan Y., Nguyen T.A., Hall C., et al. XIAP Loss Triggers RIPK3- and Caspase-8-Driven IL-1β Activation and Cell Death as a Consequence of TLR-MyD88-Induced cIAP1-TRAF2 Degradation. Cell Rep. 2017;20:668–682. doi: 10.1016/j.celrep.2017.06.073. [DOI] [PubMed] [Google Scholar]
- Leon-Bollotte L., Subramaniam S., Cauvard O., Colas S.P., Paul C., Godard C., Ruiz A.M., Legembre P., Jeannin J.F., Bettaieb A. S-Nitrosylation of the Death Receptor Fas Promotes Fas Ligand–Mediated Apoptosis in Cancer Cells. YGAST. 2011;140:2009–2018. doi: 10.1053/j.gastro.2011.02.053. e2004. [DOI] [PubMed] [Google Scholar]
- Li P., Allen H., Banerjee S., Franklin S., Herzog L., Johnston C., McDowell J., Paskind M., Rodman L., Salfeld J., et al. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell. 1995;80:401–411. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- Li H., Zhu H., Xu C.J., Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/S0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- Li X., Shang B., Li Y.N., Shi Y., Shao C. IFNγ and TNFα synergistically induce apoptosis of mesenchymal stem/stromal cells via the induction of nitric oxide. Stem Cell Res. Ther. 2019;10:18. doi: 10.1186/s13287-018-1102-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Zhang Y., Guan Z., Li H., Ye M., Chen X., Shen J., Zhou Y., Shi Z.L., Zhou P., Peng K. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal Transduct. Target. Ther. 2020;5:235. doi: 10.1038/s41392-020-00334-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019;47:e47. doi: 10.1093/nar/gkz114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Locatelli F., Jordan M.B., Allen C., Cesaro S., Rizzari C., Rao A., Degar B., Garrington T.P., Sevilla J., Putti M.C., et al. Emapalumab in Children with Primary Hemophagocytic Lymphohistiocytosis. N. Engl. J. Med. 2020;382:1811–1822. doi: 10.1056/NEJMoa1911326. [DOI] [PubMed] [Google Scholar]
- Marsden V.S., O’Connor L., O’Reilly L.A., Silke J., Metcalf D., Ekert P.G., Huang D.C., Cecconi F., Kuida K., Tomaselli K.J., et al. Apoptosis initiated by Bcl-2-regulated caspase activation independently of the cytochrome c/Apaf-1/caspase-9 apoptosome. Nature. 2002;419:634–637. doi: 10.1038/nature01101. [DOI] [PubMed] [Google Scholar]
- Marsden V.S., Ekert P.G., Van Delft M., Vaux D.L., Adams J.M., Strasser A. Bcl-2-regulated apoptosis and cytochrome c release can occur independently of both caspase-2 and caspase-9. J. Cell Biol. 2004;165:775–780. doi: 10.1083/jcb.200312030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:3. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- Martinon F., Pétrilli V., Mayor A., Tardivel A., Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- Metzakopian E., Strong A., Iyer V., Hodgkins A., Tzelepis K., Antunes L., Friedrich M.J., Kang Q., Davidson T., Lamberth J., et al. Enhancing the genome editing toolbox: genome wide CRISPR arrayed libraries. Sci. Rep. 2017;7:2244. doi: 10.1038/s41598-017-01766-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M.P. Nitric oxide and cell death. Biochim. Biophys. Acta. 1999;1411:401–414. doi: 10.1016/s0005-2728(99)00029-8. [DOI] [PubMed] [Google Scholar]
- Murphy J.M., Czabotar P.E., Hildebrand J.M., Lucet I.S., Zhang J.G., Alvarez-Diaz S., Lewis R., Lalaoui N., Metcalf D., Webb A.I., et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39:443–453. doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- Najjar M., Saleh D., Zelic M., Nogusa S., Shah S., Tai A., Finger J.N., Polykratis A., Gough P.J., Bertin J., et al. RIPK1 and RIPK3 Kinases Promote Cell-Death-Independent Inflammation by Toll-like Receptor 4. Immunity. 2016;45:46–59. doi: 10.1016/j.immuni.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K., Sun X., Dixit V.M. Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol. Cell. Biol. 2004;24:1464–1469. doi: 10.1128/MCB.24.4.1464-1469.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K., Wickliffe K.E., Dugger D.L., Maltzman A., Roose-Girma M., Dohse M., Kőműves L., Webster J.D., Dixit V.M. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature. 2019;574:428–431. doi: 10.1038/s41586-019-1548-x. [DOI] [PubMed] [Google Scholar]
- Newton K., Wickliffe K.E., Maltzman A., Dugger D.L., Reja R., Zhang Y., Roose-Girma M., Modrusan Z., Sagolla M.S., Webster J.D., Dixit V.M. Activity of caspase-8 determines plasticity between cell death pathways. Nature. 2019;575:679–682. doi: 10.1038/s41586-019-1752-8. [DOI] [PubMed] [Google Scholar]
- Niedelman W., Sprokholt J.K., Clough B., Frickel E.M., Saeij J.P. Cell death of gamma interferon-stimulated human fibroblasts upon Toxoplasma gondii infection induces early parasite egress and limits parasite replication. Infect. Immun. 2013;81:4341–4349. doi: 10.1128/IAI.00416-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien A.R., Wilson L.O.W., Burgio G., Bauer D.C. Unlocking HDR-mediated nucleotide editing by identifying high-efficiency target sites using machine learning. Sci. Rep. 2019;9:2788. doi: 10.1038/s41598-019-39142-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada S., Zhang H., Hatano M., Tokuhisa T. A physiologic role of Bcl-xL induced in activated macrophages. J. Immunol. 1998;160:2590–2596. [PubMed] [Google Scholar]
- Orzalli M.H., Prochera A., Payne L., Smith A., Garlick J.A., Kagan J.C. Virus-mediated inactivation of anti-apoptotic Bcl-2 family members promotes Gasdermin-E-dependent pyroptosis in barrier epithelial cells. Immunity. 2021;54:1447–1462. doi: 10.1016/j.immuni.2021.04.012. e1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari S., Takeda K., Takiguchi M., Gotoh T., Matsumoto M., Wada I., Akira S., Araki E., Mori M. Nitric oxide-induced apoptosis in pancreatic beta cells is mediated by the endoplasmic reticulum stress pathway. Proc. Natl. Acad. Sci. USA. 2001;98:10845–10850. doi: 10.1073/pnas.191207498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschon J.J., Torrance D.S., Stocking K.L., Glaccum M.B., Otten C., Willis C.R., Charrier K., Morrissey P.J., Ware C.B., Mohler K.M. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J. Immunol. 1998;160:943–952. [PubMed] [Google Scholar]
- Philip N.H., DeLaney A., Peterson L.W., Santos-Marrero M., Grier J.T., Sun Y., Wynosky-Dolfi M.A., Zwack E.E., Hu B., Olsen T.M., et al. Activity of Uncleaved Caspase-8 Controls Anti-bacterial Immune Defense and TLR-Induced Cytokine Production Independent of Cell Death. PLoS Pathog. 2016;12:e1005910. doi: 10.1371/journal.ppat.1005910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickard J.A., Anderton H., Etemadi N., Nachbur U., Darding M., Peltzer N., Lalaoui N., Lawlor K.E., Vanyai H., Hall C., et al. TNFR1-dependent cell death drives inflammation in Sharpin-deficient mice. eLife. 2014;3 doi: 10.7554/eLife.03464. 10.7554/eLife.03464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickard J.A., O’Donnell J.A., Evans J.M., Lalaoui N., Poh A.R., Rogers T., Vince J.E., Lawlor K.E., Ninnis R.L., Anderton H., et al. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell. 2014;157:1175–1188. doi: 10.1016/j.cell.2014.04.019. [DOI] [PubMed] [Google Scholar]
- Robinson M.D., Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagny S., Bouaouiche S., Lucchi G., Ducoroy P., Bertoldo J.B., Terenzi H., Bettaieb A., Plenchette S. S-Nitrosylation of cIAP1 Switches Cancer Cell Fate from TNFα/TNFR1-Mediated Cell Survival to Cell Death. Cancer Res. 2018;78:1948–1957. doi: 10.1158/0008-5472.CAN-17-2078. [DOI] [PubMed] [Google Scholar]
- Salaun B., Romero P., Lebecque S. Toll-like receptors’ two-edged sword: when immunity meets apoptosis. Eur. J. Immunol. 2007;37:3311–3318. doi: 10.1002/eji.200737744. [DOI] [PubMed] [Google Scholar]
- Schneemann M., Schoedon G., Hofer S., Blau N., Guerrero L., Schaffner A. Nitric oxide synthase is not a constituent of the antimicrobial armature of human mononuclear phagocytes. J. Infect. Dis. 1993;167:1358–1363. doi: 10.1093/infdis/167.6.1358. [DOI] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K., Sweet M.J., Hume D.A. Signal integration between IFNgamma and TLR signalling pathways in macrophages. Immunobiology. 2006;211:511–524. doi: 10.1016/j.imbio.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Sedger L.M., Shows D.M., Blanton R.A., Peschon J.J., Goodwin R.G., Cosman D., Wiley S.R. IFN-gamma mediates a novel antiviral activity through dynamic modulation of TRAIL and TRAIL receptor expression. J. Immunol. 1999;163:920–926. [PubMed] [Google Scholar]
- Seminara A.R., Ruvolo P.P., Murad F. LPS/IFNgamma-induced RAW 264.7 apoptosis is regulated by both nitric oxide-dependent and -independent pathways involving JNK and the Bcl-2 family. Cell Cycle. 2007;6:1772–1778. doi: 10.4161/cc.6.14.4438. [DOI] [PubMed] [Google Scholar]
- Sennlaub F., Courtois Y., Goureau O. Inducible nitric oxide synthase mediates retinal apoptosis in ischemic proliferative retinopathy. J Neurosci. 2002;22:3987–3993. doi: 10.1523/jneurosci.22-10-03987.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J., Zhao Y., Wang K., Shi X., Wang Y., Huang H., Zhuang Y., Cai T., Wang F., Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- Snyder C.M., Shroff E.H., Liu J., Chandel N.S. Nitric oxide induces cell death by regulating anti-apoptotic BCL-2 family members. PLoS ONE. 2009;4:e7059. doi: 10.1371/journal.pone.0007059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souers A.J., Leverson J.D., Boghaert E.R., Ackler S.L., Catron N.D., Chen J., Dayton B.D., Ding H., Enschede S.H., Fairbrother W.J., et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- Speir M., Lawlor K.E., Glaser S.P., Abraham G., Chow S., Vogrin A., Schulze K.E., Schuelein R., O’Reilly L.A., Mason K., et al. Eliminating Legionella by inhibiting BCL-XL to induce macrophage apoptosis. Nat. Microbiol. 2016;1:15034. doi: 10.1038/nmicrobiol.2015.34. [DOI] [PubMed] [Google Scholar]
- Su X., Yu Y., Zhong Y., Giannopoulou E.G., Hu X., Liu H., Cross J.R., Rätsch G., Rice C.M., Ivashkiv L.B. Interferon-γ regulates cellular metabolism and mRNA translation to potentiate macrophage activation. Nat. Immunol. 2015;16:838–849. doi: 10.1038/ni.3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T., Okamoto T., Katoh H., Sugiyama Y., Kusakabe S., Tokunaga M., Hirano J., Miyata Y., Fukuhara T., Ikawa M., et al. Infection with flaviviruses requires BCLXL for cell survival. PLoS Pathog. 2018;14:e1007299. doi: 10.1371/journal.ppat.1007299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taabazuing C.Y., Okondo M.C., Bachovchin D.A. Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages. Cell Chem. Biol. 2017;24:507–514.e4. doi: 10.1016/j.chembiol.2017.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K., Smyth M.J., Cretney E., Hayakawa Y., Kayagaki N., Yagita H., Okumura K. Critical role for tumor necrosis factor-related apoptosis-inducing ligand in immune surveillance against tumor development. J. Exp. Med. 2002;195:161–169. doi: 10.1084/jem.20011171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O., Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- Takeuchi O., Fisher J., Suh H., Harada H., Malynn B.A., Korsmeyer S.J. Essential role of BAX,BAK in B cell homeostasis and prevention of autoimmune disease. Proc. Natl. Acad. Sci. USA. 2005;102:11272–11277. doi: 10.1073/pnas.0504783102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbott S.J., Luanpitpong S., Stehlik C., Azad N., Iyer A.K.V., Wang L., Rojanasakul Y. S-nitrosylation of FLICE inhibitory protein determines its interaction with RIP1 and activation of NF-κB. Cell Cycle. 2014;13:1948–1957. doi: 10.4161/cc.28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzer M.C., Khan N., Rickard J.A., Etemadi N., Lalaoui N., Spall S.K., Hildebrand J.M., Segal D., Miasari M., Chau D., et al. Combination of IAP antagonist and IFNγ activates novel caspase-10- and RIPK1-dependent cell death pathways. Cell Death Differ. 2017;24:481–491. doi: 10.1038/cdd.2016.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taraborrelli L., Peltzer N., Montinaro A., Kupka S., Rieser E., Hartwig T., Sarr A., Darding M., Draber P., Haas T.L., et al. LUBAC prevents lethal dermatitis by inhibiting cell death induced by TNF, TRAIL and CD95L. Nat. Commun. 2018;9:3910. doi: 10.1038/s41467-018-06155-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor E.L., Megson I.L., Haslett C., Rossi A.G. Nitric oxide: a key regulator of myeloid inflammatory cell apoptosis. Cell Death Differ. 2003;10:418–430. doi: 10.1038/sj.cdd.4401152. [DOI] [PubMed] [Google Scholar]
- Thapa R.J., Basagoudanavar S.H., Nogusa S., Irrinki K., Mallilankaraman K., Slifker M.J., Beg A.A., Madesh M., Balachandran S. NF-kappaB protects cells from gamma interferon-induced RIP1-dependent necroptosis. Mol. Cell. Biol. 2011;31:2934–2946. doi: 10.1128/mcb.05445-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Delft M.F., Wei A.H., Mason K.D., Vandenberg C.J., Chen L., Czabotar P.E., Willis S.N., Scott C.L., Day C.L., Cory S., et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varfolomeev E., Maecker H., Sharp D., Lawrence D., Renz M., Vucic D., Ashkenazi A. Molecular determinants of kinase pathway activation by Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand. J. Biol. Chem. 2005;280:40599–40608. doi: 10.1074/jbc.M509560200. [DOI] [PubMed] [Google Scholar]
- Villunger A., Michalak E.M., Coultas L., Müllauer F., Böck G., Ausserlechner M.J., Adams J.M., Strasser A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- Vince J.E., Wong W.W., Khan N., Feltham R., Chau D., Ahmed A.U., Benetatos C.A., Chunduru S.K., Condon S.M., McKinlay M., et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell. 2007;131:682–693. doi: 10.1016/j.cell.2007.10.037. [DOI] [PubMed] [Google Scholar]
- Vince J.E., Wong W.W., Gentle I., Lawlor K.E., Allam R., O’Reilly L., Mason K., Gross O., Ma S., Guarda G., et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity. 2012;36:215–227. doi: 10.1016/j.immuni.2012.01.012. [DOI] [PubMed] [Google Scholar]
- Vince J.E., De Nardo D., Gao W., Vince A.J., Hall C., McArthur K., Simpson D., Vijayaraj S., Lindqvist L.M., Bouillet P., et al. The Mitochondrial Apoptotic Effectors BAX/BAK Activate Caspase-3 and -7 to Trigger NLRP3 Inflammasome and Caspase-8 Driven IL-1beta Activation. Cell Rep. 2018;25:2339–2353. doi: 10.1016/j.celrep.2018.10.103. e2334. [DOI] [PubMed] [Google Scholar]
- Wallach D., Kang T.B., Kovalenko A. Concepts of tissue injury and cell death in inflammation: a historical perspective. Nat. Rev. Immunol. 2014;14:51–59. doi: 10.1038/nri3561. [DOI] [PubMed] [Google Scholar]
- Wang A., Pope S.D., Weinstein J.S., Yu S., Zhang C., Booth C.J., Medzhitov R. Specific sequences of infectious challenge lead to secondary hemophagocytic lymphohistiocytosis-like disease in mice. Proc. Natl. Acad. Sci. USA. 2019;116:2200–2209. doi: 10.1073/pnas.1820704116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse N.J., Trapani J.A. A new quantitative assay for cytochrome c release in apoptotic cells. Cell Death Differ. 2003;10:853–855. doi: 10.1038/sj.cdd.4401263. [DOI] [PubMed] [Google Scholar]
- Weir A., Hughes S., Rashidi M., Hildebrand J.M., Vince J.E. Necroptotic movers and shakers: cell types, inflammatory drivers and diseases. Curr. Opin. Immunol. 2021;68:83–97. doi: 10.1016/j.coi.2020.09.008. [DOI] [PubMed] [Google Scholar]
- Wickham H. Springer; 2009. ggplot2. [DOI] [Google Scholar]
- Willis S.N., Chen L., Dewson G., Wei A., Naik E., Fletcher J.I., Adams J.M., Huang D.C. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–1305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo M., Hakem A., Elia A.J., Hakem R., Duncan G.S., Patterson B.J., Mak T.W. In vivo evidence that caspase-3 is required for Fas-mediated apoptosis of hepatocytes. J. Immunol. 1999;163:4909–4916. [PubMed] [Google Scholar]
- Wu W., Wan O.W., Chung K.K.K. S-nitrosylation of XIAP at Cys 213 of BIR2 domain impairs XIAP’s anti-caspase 3 activity and anti-apoptotic function. Apoptosis. 2015;20:491–499. doi: 10.1038/35065125. [DOI] [PubMed] [Google Scholar]
- Wynn T.A., Chawla A., Pollard J.W. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., Fu X.-Y., Plate J., Chong A.S.F. IFN-γ induces cell growth inhibition by Fas-mediated apoptosis: requirement of STAT1 protein for up-regulation of Fas and FasL expression. Cancer Res. 1998;58:2832–2837. [PubMed] [Google Scholar]
- Yang G., Hisha H., Cui Y., Fan T., Jin T., Li Q., Lian Z., Hosaka N., Li Y., Ikehara S. A new assay method for late CFU-S formation and long-term reconstituting activity using a small number of pluripotent hemopoietic stem cells. Stem Cells. 2002;20:241–248. doi: 10.1634/stemcells.20-3-241. [DOI] [PubMed] [Google Scholar]
- Yang C., McDermot D.S., Pasricha S., Brown A.S., Bedoui S., Lenz L.L., van Driel I.R., Hartland E.L. IFNγ receptor down-regulation facilitates Legionella survival in alveolar macrophages. J. Leukoc. Biol. 2020;107:273–284. doi: 10.1002/JLB.4MA1019-152R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young R., Bush S., Lefevre L., McCulloch M., Lisowski Z., Muriuki C., Waddell L., Sauter K., Pridans C., Clark E., Hume D.A. Species-Specific Transcriptional Regulation of Genes Involved in Nitric Oxide Production and Arginine Metabolism in Macrophages. ImmunoHorizons. 2018:1–12. doi: 10.4049/immunohorizons.1700073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A.H., Tanaseichuk O., Benner C., Chanda S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019;10:1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
WT BMDMs were stimulated with IFNγ (50 ng/mL) and imaged live every 20 min for 24 h. Composite video of brightfield (gray) and PI stain (red) is shown. Magnification is set at 20x, scale bar (white): 100 μm. Related to Figure 1.
WT BMDMs were primed with IFNγ (50 ng/mL) overnight and then stimulated with LPS (50 ng/mL) and imaged live every 20 min for 24 h. Imaging began at LPS time 0 h (e.g., after IFNγ priming). Composite video of brightfield (gray) and PI stain (red) is shown. Magnification is set at 20x, scale bar (white): 100 μm. Related to Figure 1.
WT BMDMs were stimulated with ABT-737 (1 μM) plus cycloheximide (CHX, 10 μg/mL) to trigger BAX/BAK dependent apoptosis and imaged live every 20 min for 24 h. Composite video of brightfield (gray) and PI stain (red) is shown. Magnification is set at 20x, scale bar (white): 100 μm. Related to Figure 1.
WT BMDMs were primed with IFNγ (50 ng/mL) overnight and then stimulated with LPS (50 ng/mL) and imaged live every 15 min for 24 h. Imaging began at LPS time 0 h (after IFNγ priming). Composite video of brightfield (gray) and PI stain (red) is shown. Magnification is set at 63x, scale bar (white): 10 μm. Related to Figures 1 and S1A.
Data Availability Statement
-
•
RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. All raw data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.