

Abstract
The SARS coronavirus 2 (SARS CoV-2) causes Coronavirus Disease (COVID-19), is an emerging viral infection. SARS CoV-2 infects target cells by attaching to Angiotensin-Converting Enzyme (ACE2). SARS CoV-2 could cause cardiac damage in patients with severe COVID-19, as ACE2 is expressed in cardiac cells, including cardiomyocytes, pericytes, and fibroblasts, and coronavirus could directly infect these cells. Cardiovascular disorders are the most frequent comorbidity found in COVID-19 patients. Immune cells such as monocytes, macrophages, and T cells may produce inflammatory cytokines and chemokines that contribute to COVID-19 pathogenesis if their functions are uncontrolled. This causes a cytokine storm in COVID-19 patients, which has been associated with cardiac damage. Tregs are a subset of immune cells that regulate immune and inflammatory responses. Tregs suppress inflammation and improve cardiovascular function through a variety of mechanisms. This is an exciting research area to explore the cellular, molecular, and immunological mechanisms related to reducing risks of cardiovascular complications in severe COVID-19. This review evaluated whether Tregs can affect COVID-19-related cardiovascular complications, as well as the mechanisms through which Tregs act.
Keywords: SARS CoV-2, COVID-19, Treatment, Tregs, Cardiovascular complications
Graphical abstract
1. Introduction
The SARS coronavirus 2 (SARS CoV-2) infects target cells by attaching to the angiotensin-converting enzyme-2 (ACE2). Coronavirus disease (COVID-19) is caused by the SARS coronavirus 2 (SARS CoV-2) [1]. This viral disease, which has a mortality rate of 2.2%, causes various symptoms such as nausea, cough, acute respiratory distress syndrome (ARDS), severe pneumonia, cardiovascular complications, and organ dysfunction [1], [2].
COVID-19 has so far appeared in four waves [3]. The latest one has been caused by the new Omicron variant [4]. Previous waves were related to the Ancestral, Beta, and Delta variants [4]. In comparison to previous variants, the Omicron variant had a lower rate of hospital admissions and a lower severity of COVID-19 [5], [6]. The mortality rate of Omicron variant has been reduced significantly from the original 2.2 [7], [8]. The high levels of previous infection and vaccination coverage are likely responsible for the changing clinical presentation of SARS-CoV-2 Omicron infection [9]. However, due to ability of COVID-19 to produce repeat infection, a critical concern has been whether humans will experience reinfections with this pathogen, which might enable COVID-19 to become endemic [10].
Tregs, which comprise 5–15% of the CD4+ T cells in the peripheral blood [11], play a significant role in maintaining self-tolerance and suppressing autoimmunity [11], [12], [13], [14]. Tregs express several cell surface molecules, including CD25, CD45RA, CD45RO, CD62L, CD127, CD103, cytotoxic T-lymphocyte antigen-4 (CTLA-4, or CD152), CCR6, HLA-DR, CD39, CD95, ICOS, CD147, glucocorticoid-induced TNF receptor family-related gene (GITR), and programmed cell death 1 (PD-1) that enable them to be isolated and characterized [15]. Furthermore, FOXP3 is a Treg-specific transcription factor that plays a significant role in Treg differentiation, development, and function [16]. In addition, IL-2 as a T-cell growth factor has contributed to developing and promoting natural Treg activity [17].
Tregs are divided into two major groups: those originating from the thymus (nTregs) and those induced in the periphery (iTregs). Immature CD4+ T cells with a high self-antigen affinity differentiate into natural Tregs during the T cell growth process (nTreg) in the thymus [18]. nTregs are recognized by the expression of CD4, CD25, and FOXP3 markers [18].
Inducible Tregs (iTregs) are Tregs derived from virgin CD4+ T cells in the presence of TGF-β in the peripheral tissues that respond to exogenous or self-antigens [19], [20], [21]. iTregs and nTregs are different regarding their epigenetic status and stability [22]. In addition, iTregs are characterized by their cytokine profile [19]. In humans, a variety of iTreg subsets have been recognized, including CD8+ Treg, Th3, Tr1, and natural killer Treg (NK Treg) [22]. These iTreg subsets suppressed immune responses, while they differ in cell surface markers and formation sites [22].
Through direct cell interaction and the release of anti-inflammatory cytokines, both natural and iTregs regulated the proliferation and activities of innate immune cells (dendritic cells and macrophages). They also suppressed self-reactive lymphocytes, including Th1, Th2, Th17, and B cells [23]. IL-10 and TGF-β are multifunctional cytokines secreted by different immune cells, including Tregs (primarily Th1 and Th3) and Th2 cells [24]. IL-10 producing nTregs contributes to eliminating pathogens in viral, fungal and, bacterial infections [25], [26]. TGF-β has been implicated in maintaining natural Tregs in the thymus and inducing iTregs differentiation [27]. TGF-β was found to play a role in Th17 cells differentiation [28], [29], [30]. Tregs release IL-35, which inhibits T-cell proliferation by binding to the IL-12R2 receptor [31].
Aside from nTregs FOXP3-positive T cells could be polarized from FOXP3-negative T cells in the presence of TGF-β [32]. It was also shown that activation of Tregs producing high amounts of TGF-β with the addition of IL-6 induces CD4+CD25+Foxp3− T cells to differentiate into IL-17-producing cells in the absence of other cells [33]. Hence, activated Tregs themselves differentiate into IL-17-producing cells in the presence of a source of IL-6. However, cytokines may enhance the proliferative response and potentiate their FOXP3 expression and suppressive activities. One of these cytokines is interferon β (IFNβ) used for multiple sclerosis therapy [34], [35]. One of the mechanisms of cytokine actions is induction of the proliferation of CD4+CD25+Foxp3− regulatory T cells through up-regulation of GITRL on dendritic cells [35]. From the view of this point, CD4+CD25+Foxp3− T cells have dual effects on the course of an immune response that includes the immune status in COVID-19 infection [32], [33].
Recent research suggests that Tregs phenotype and function may be unstable in an inflammatory environment, with unanticipated plasticity toward Th1 and Th17 cells in autoimmune diseases and viral infections [20], [36]. Tregs may lose their regulatory function and even show a pro-inflammatory activity [20]. Explanations for Treg plasticity include epigenetic and posttranslational modifications [36]. It was found that Tregs converted into Th1-like cells producing IFN-γ, co-expressing FOXP3 and Tbet (the main transcription factor of Th1 cells) with the upregulation of CXCR3 other classical Th1 markers in vivo [20]. The presence of IFN-γ producing cells was also observed in FOXP3 expressing cells after PMA/Ionomycin stimulation or after prolonged in vitro expansion of FOXP3+ Tregs [20]. Th1-like Tregs are associated with several autoimmune diseases in humans, including T1D (type-1 diabetes) and multiple sclerosis (MS) [20]. Additionally, it was demonstrated that Treg-Th17 conversion occurred in the presence of IL-1β and on epigenetic modifications resulting in the up-regulation of ROR-γ (the specific transcription factor of Th17 cells) expression [20], [37], [38]. Th17-like conversion in vivo was recently proposed for tumor-infiltrating Tregs isolated from human ovarian tumors [39]. Interestingly, according to the findings, Tregs produced in the presence of vitamin C + RA established a more stable population when exposed to an inflammatory environment in vitro or in vivo [36], suggesting a possible strategy for reducing Treg plasticity in inflammatory conditions like in COVID-19 infection.
According to our knowledge, very little research has been done to investigate or hypothesize the effects of Tregs on cardiovascular complications in severe COVID-19 infection. Therefore, this review aimed to seek whether the cardiovascular complications caused by COVID-19 infection could be affected by Tregs as well as the mechanisms by which Tregs act.
2. COVID-19
COVID-19 is a respiratory viral infection generated by the SARS coronavirus (SARS CoV-2), first found in Wuhan, China, in December 2019 [40]. The World Health Organization (WHO) announced the new coronavirus is a worldwide outbreak on March 11, 2020 [41].
SARS CoV-2 is a coronaviridae virus with a single-stranded RNA genome [42]. This virus has a genome that is about 30,000 nucleotides (27–32 kb) and encodes structural and accessory proteins [42]. SARS-CoV preserves 79% and 50% of its genetic sequence with MERS and SARS-CoV-1, two other coronaviridae viruses, respectively, and attaches to ACE2 as the receptor for cell infection [43]. According to a large body of evidence, COVID-19 infection causes multi-organ dysfunction in the lung, heart, brain, large intestine, kidneys, and spleen compared to other coronaviruses that are only concerned with respiratory infections express the ACE2 receptor [43], [44], [45], [46], [47], [48].
In the normal viral clearance process, COVID-19 recruits and activates T-helper 1 cells (Th1 CD4+ cells) at the site of inflammation, which can eliminate infected cells and prevent the virus from spreading and replicating [48]. Neutralizing antibodies could then block viral attachments to cells, and macrophages would then phagocytize the neutralized viruses as well as apoptotic cells [48]. The viral load rises during the first week of infection and gradually decreases over the next few days. SARS CoV-2 antibodies start to rise 10 days after infection, and most patients become seroconverted within the first twenty days [49].
3. Immunopathology of COVID-19
It has been shown that alternations in the proportions of immune cells have been associated with progression, severity, and death in most severe COVID-19 patients [50], [51], [52], [53]. The total neutrophils are increased while total lymphocytes are reduced, increasing the neutrophil-lymphocyte ratio (NLR) in these patients [50], [51]. Patients with severe COVID-19 also tend to have a lower frequency of basophils, monocytes, and eosinophils [54]. Moreover, increased neutrophils and decreased lymphocytes have been shown to correlate with the severity of COVID-19 infection [55].
Lymphopenia in COVID-19 patients has been found in several studies [56], [57], [58], [59], [60]. In severe COVID-19 patients, lymphocytes were less than 5% within two weeks of disease onset [56], [57], [58], [59], [60]. Despite the decrease in T cell numbers, their functions were normal [61] or even hyper-activated, as evidenced by the high proportion of HLA-DR (a marker of TCD4+ cell activation) and CD38 (a marker of TCD8+ cell activation) double-positive populations [62], [63].
It was found that B and T lymphocytes and NK cells were significantly diminished in severe COVID-19 patients [64], [65], [66]. However, T-cell frequency (both T CD4+ and TCD8+ cells) are more impaired than other immune cells [61], SARS-CoV-2 affects the proportion of T helper CD4+ subpopulations including, Th1, Th17, Th2 cells, and Tregs, in COVID-19 patients in a different manner [67], [68].
During COVID-19 infection, both anti-inflammatory (Th2) and pro-inflammatory (Th1) responses are activated, resulting in an increase in several cytokines (IFN-γ, IL-6, IL-1, TNF-α, IL-12, IL-10, and IL-2) in severe COVID-19 infection [67], [69]. SARS CoV and MERS CoV usually caused Th1 immunity, contributing to excess secretion of inflammatory cytokines (IFN-γ, IL-12, TNF-α, and IL-1), related to significant pulmonary complications a high death rate [70], [71].
In patients experiencing severe COVID-19, the percentage of Tregs decreased dramatically while the percentage of Th17 cells (as the inflammatory cells) increased [61], [72], resulting in a decline in the Treg/Th17 cell ratio [62], [73]. The reduced Treg/Th17 ratio is associated with the unregulated systemic inflammation in acute lung damage, like acute respiratory distress syndrome [74], [75]. Reduced Treg numbers in severe COVID-19 cases reflect inadequate modulation of pro-inflammatory immune reactions, which could exacerbate inflammatory reactions and tissue damage [76].
Inflammation has already been identified as the main contributor to the pathogenesis of severe COVID-19 [77]. Excess pro-inflammatory cytokine production has been shown in COVID-19 cases due to elevated Th17 cell activity [62], [68]. IL-17A and CXCLs chemokines attract myeloid cells like neutrophils to the infection site and activate matrix metalloproteinase. This leads to the recruitment of more inflammatory cells like Th1 and Th17 cells and the excessive secretion of inflammatory cytokines, which intensifies uncontrolled systemic inflammation [68]. This is known as a “cytokine storm,” resulting in tissue damage and viral sepsis, both of which have fatal consequences [68]. Other symptoms of severe COVID-19 include acute respiratory distress syndrome and respiratory and cardiac failure [68].
Indeed, augmented levels of circulating TNF-α, IL-6, and IL-1 (the major pro-inflammatory cytokines) cause naive CD4+ T cells to differentiate into Th17 cells while inhibiting Tregs, resulting in a Treg/Th17 ratio imbalance [22]. Inducing tissue factor expression on mononuclear cells could lead to coagulation activation and thrombin production, resulting in disseminated intravascular coagulation (DIC) and, eventually, pulmonary embolism [22]. In addition, it may have a role in the rapid decline in pulmonary oxygen exchange shown in COVID-19 patients [22]. TNF-α and IL-6 levels in the blood have been proposed as determinants of disease severity [22].
4. Cardiac damage in COVID-19
Cardiovascular diseases are the most frequent comorbidity detected in COVID-19 patients [78], [79], [80]. The mortality risk in COVID-19 patients with cardiovascular diseases is more significant than in COVID-19 patients with other disorders such as diabetes mellitus and chronic pulmonary disease [74], [75]. In COVID-19 patients, increased levels of several cardiac injury biomarkers were found, including cardiac sensitivity troponin I (hs-TnI), N-terminal pro-B-type natriuretic peptide (NT-proBNP), and C-reactive protein (CRP) [12], [15]. COVID-19-induced heart inflammation resulted in various clinical outcomes, including right ventricle and cardiac amyloidosis, concentric left ventricular hypertrophy with a dilated left ventricle, and severe hypokinetic arrhythmias (ranging from tachycardia and bradycardia to asystole) [12].
SARS CoV-2 can affect cardiac tissue either directly or indirectly [22] (Fig. 1 ). The expression of ACE2 by cardiac cells such as pericytes, cardiomyocytes, vascular smooth muscle cells, and fibroblasts could enable SARS CoV-2 to infect these cells directly [22], [81]. Inflammatory cytokines and chemokines including IL-2, IL-6, TNF-α, IL-1, monocyte chemoattractant protein 1 (MCP-1), and macrophage inflammatory protein 1- (MIP-1), which are the main contributors of cytokine storm, are implicated in the cardiac injury indirectly. [11], [13], [14], [16], [18], [19], [22], [82], [83], [84], [85]. This indicates that the immune system plays a part in cardiovascular complications in COVID-19 patients as infiltration of monocytes and T cells observed in autopsies [11], [13], [14], [16], [18], [19], [22], [62], [82], [83], [84], [85], [86]. Mononuclear infiltrates are associated with areas of cardiomyocyte necrosis, which determines myocarditis in COVID-19 patients according to the Dallas criteria [87], [88]. Elevated levels of inflammatory cytokines and chemokines may lead to myocarditis, heart failure, hypertension, coronary arterial diseases (CAD), myocardial infarction (MI), and cardiac arrhythmias [24], [25], [26]. In addition, hypoxemia, metabolic disturbances, systemic inflammation, or myocarditis may all cause arrhythmias in patients whit COVID-19 [89], [90], [91]. These patients can experience acute coronary syndromes due to the elevated thrombotic proclivity, as shown by increased D-dimer levels, although the incidence of such cases is unclear [89], [90], [91]. Heart failure is another risk associated with coagulation defects in COVID-19 patients [27].
Fig. 1.

Proposed mechanisms of cardiac injury.
Cardiac tissue may be influenced by SARSCov2 infection (or Covid19) directly or indirectly. ACE2 expression by cardiac cells could directly infect these cells by a coronavirus. Indirectly, inflammatory cytokines that are the leading cause of cytokine storms are implicated in cardiac damage.
5. Tregs impairment in COVID-19
According to current evidence, the frequency of peripheral Tregs is significantly reduced in patients with severe COVID-19 infection compared to patients with moderate disease [61], [72], [92]. It is important to note that Treg depletion in mice infected with murine coronavirus resulted in a rise in acute encephalitis mortality, demonstrating the protective role of Tregs during acute COVID-19 infection [93]. Obesity is also a risk factor for COVID-19. Evidence from obese subjects and relevant animal models showed that the percentage of Tregs in the blood and visceral adipose tissues is low, indicating a higher state of inflammation in obese individuals [94].
The following are possible explanations for the reduction of Tregs in severe COVID-19:
IL-2 serves as a growth factor for Tregs by increasing the expression of FOXP3 (the master transcription factor of Tregs) [95]. As observed in the bronchoalveolar lavage of patients with severe COVID-19, decreased IL-2 levels may lead to Tregs apoptosis and decreased FOXP3 gene expression [95]. Furthermore, inflammatory conditions in severe COVID-19 patients probably improve proteolytic cleavage of cell surface CD25 (IL-2R), resulting in increased levels of soluble CD25 [61], [72]. Soluble CD25 may potentially interact with IL-2 signaling and bioavailability, contributing to rising Treg apoptosis (Fig. 1) [61], [72].
One of the reasons for the decrease in the percentage of Tregs in peripheral blood is the possibility of Tregs migrating to the lungs to regulate adverse inflammatory responses in patients with COVID-19 [96]. As noted, Tregs can suppress inflammation [23], leading to the regulation of the activity of macrophage, Th1, and Th17 cells that are contributed to cytokine storm occurrence during viral infection [23]. As previously mentioned (Section 3), the cause of cytokine storm could be due to uncontrolled inflammation and inappropriate immune responses leading to severe lung damage which is the leading cause of morbidity and mortality in COVID-19 [97]. In this light, it has been proposed that Treg therapy may be one option for treating patients with severe COVID-19 [97], [98]. However, the dosage of Tregs and complementary therapies for SARS-CoV-2 infection must be approached [98]. A study demonstrated that three transfusions of allogeneic cord blood Tregs substantially reduced the levels of the major cytokines that contributed to the cytokine storm, including IL-12, IL-6, TNF-α and, IFN-γ [98]. In addition, after transfusion of Tregs, the levels of IL-8 and MC-1 (two well-known chemokines in lung injury) also drastically were diminished [98].
6. The role of Tregs in cardiac homeostasis and various cardiovascular complications
Tregs are contributed to cardiac, immune tolerance, and the breakdown of immune tolerance to self-antigens in the heart may cause cardiac inflammation [99]. According to recent research, Tregs are essential for vascular and cardiovascular function [100]. PDL-1 expression in the heart cells as a ligand of PD-1 (one of the functional surface markers of Tregs) may support that point [99]. Tregs in the heart have higher proliferative rates than Tregs in blood and lymphoid tissue, suggesting that local renewal is particularly important for Tregs expansion in the heart, even in the absence of cardiac injury [101]. In addition, tolerogenic DCs were detected in the heart tissue that primed antigen for Treg cell activation [102]. The development of an inflammatory network, which involves inflammatory cell aggregation and the production of inflammatory cytokines, can affect the progression of cardiovascular diseases [103]. As a result, suppressing inflammatory responses is a potential candidate for preventing and treating myocardial infarction, atherosclerosis, myocarditis, heart failure, and hypertension [103]. Clinical trials indicated that the frequency and function of circulating Tregs were lower in patients with chronic heart disease relative to healthy individuals [104], [105]. Tregs can suppress the pro-inflammatory cells and the production of pro-inflammatory cytokines, both of which are associated with cardiovascular complications [103]. Tregs dysfunction may lead to uncontrolled inflammatory responses of Th1 and Th17 cells and, consequently, myocardial infarction and heart failure [106].
Myocarditis is an inflammatory cardiac disease caused by various infectious agents and autoimmune diseases [107], [108], [109]. Viral infections could trigger myocarditis by inducing immediate cytotoxic reactions, post-viral inappropriate immune responses, and autoimmunity [103]. Tregs protected against myocarditis in animal models infected with Coxsackievirus B3 (CVB3), hepatitis C, and herpes simplex virus by minimizing viral-induced immunopathology and suppressing tissue injury due to viral-induced immunological responses [110].
Infiltration of different inflammatory cells including, neutrophils, monocytes, and lymphocytes (particularly Th1 and Th17 cells) in the myocardium, increased the severity of myocarditis [103]. Tregs have been reported to regulate inflammatory cells activation, which limits the anti-viral immune response and prevents myocarditis progression [103], [111]. It was discovered that adoptive transfer of Tregs reduced viral load and immune cell infiltration in the heart, in the pancreas and, in the spleen, which was associated to decrease expression of the coxsackie-adenovirus receptor (CAR), less activation of p38 MAP kinase, and increased Akt activation [112], [113]. These alternations were caused by TGF-β, which triggered a paracrine positive feedback loop and converted naive CD4+ T cells into regulatory CD4+ T cells [111]. So, inhibition of p38 MAP kinase is an effective strategy in treating viral diseases [114]. The P38 Mitogen-Activated Protein (MAP) Kinase is one of the kinases involved in the inflammatory response [115], [116]. The phosphoinositide 3 kinases (PI3K)-Akt axis improves to differentiate helper T-cell subsets [117]. The phosphorylation of a variety of downstream effector molecules by AKT is involved in cell growth, metabolism, and survival [117]. AKT enhanced FOXP3 gene expression in Tregs, resulting in Tregs with a stable phenotype and functions [117].
Both Th1 and Th17 cells have been found to play a role in the initiation and progression of myocarditis [118], [119], [120]. Tregs produce IL-10, which reduces the severity of CVB3-induced viral myocarditis by suppressing the release of Th17-related cytokines (IL-17A and IL-6) [119]. In addition, IL-10 inhibited the immune response by lowering MHC II complexes and B7 family co-stimulatory molecules on antigen-presenting cell surface, including dendritic cells and macrophages [111], [119].
Treg impairments were detected in CAD patients due to the decrease in Treg frequency and downregulation of FOXP3, IL-10, and TGF-β gene expressions and higher IFN-γ and hsCRP levels [121]. Increased apoptosis induced by inflammation and oxidative stress was also a cause of Treg impairment [121]. In the angiotensin-II-induced hypertension model, Treg deficiency was attributed to increased pro-inflammatory factors like IFN-γ and IL-17A [122]. These cytokines play a part in the synthesis and degradation of vasoconstrictors and vasodilators and the expression of Angiotensin-II, which contributed to artery inflammation and high blood pressure [123]. It was shown that the transfusion of Tregs reduced heart hypertrophy, fibrosis, and arrhythmia in the angiotensin-II-induced hypertension model [124], [125], [126].
Myocardial infarction (MI) happens when blood flow to a part of the heart is inhibited or interrupted, resulting in injury to the cardiac muscle [127]. A decreased percentage of circulating Tregs has been associated with a higher risk of heart failure hospitalization, inversely correlated to IL-6 levels [127]. Transfusion of Tregs improved infarct size and left ventricular dilation in a mouse model of myocardial infarction [128]. Inflammatory myeloid cells such as neutrophils, monocytes, and T cells increased in the infracted myocardium in this model [128]. It was found that Tregs may affect myeloid cell infiltration by modulating the expression of chemokines, which is involved in homing of inflammatory cells [129]. TNF-α and IFN-γ secretion by infiltrated cells has been indicated to induce M1 macrophage polarization [130], [131]. M1 macrophage potentially has adverse effects on inflamed myocardium by releasing pro-inflammatory cytokines (IL-1, IL-6, IL-12, and TNF-α), chemokines (MCP-1, CXCL1-3, CXCL5, and CXCL8-10), high levels of inducible nitric oxide synthase (iNOS), and reactive oxygen species (ROS) which all attribute to enhance the inflammatory responses in inflamed myocardium [130], [131]. Interestingly, Tregs improved M2-like monocyte differentiation post-MI by producing IL-10, TGFβ, and IL-13 in-vivo and in-vitro [132]. As a result, cardiac healing after MI was improved [133], [134]. Activated M2-like macrophages released anti-inflammatory cytokines such as IL-10, IL-13, and TGF-β that are essential in wound healing, tissue remodeling, and angiogenesis [135], [136]. Monocytic cells released osteopontin in response to TGF-β and IL-10 [132]. In the healing myocardium, osteopontin significantly impacts collagen production and matrix assembly [137].
7. The potential of Treg therapy in reducing cardiovascular complications in COVID19 patients
7.1. The probable role of Tregs in alleviating cardiovascular complications in COVID-19
At a glance at previous sections, however, a variety of immune responses have been identified in COVID-19 infection [138], the immunological alternations primarily trending to an anti-inflammatory state (Th2 and Tregs) neutralized COVID-19 inflammatory reactions (cytokine storm) [134]. Current studies showed that the frequency of peripheral Tregs fundamentally diminished in patients with severe COVID-19 infection [61], [72], [92]. Tregs impairment can lead to cytokine storm since Tregs regulate inflammatory responses [61], [72], [92]. Cardiovascular complications in COVID19 patients are caused by a hyper-inflammation state in the cytokine storm, which causes elevated myocardial oxygen consumption, endothelial dysfunction, and suppressed cardiac activity [140], [141]. Additionally, the autopsy revealed increased mononuclear cell infiltration through the myocardium in COVID-19 patients with cardiovascular complications, indicating the increased inflammatory responses in the heart tissue [62].
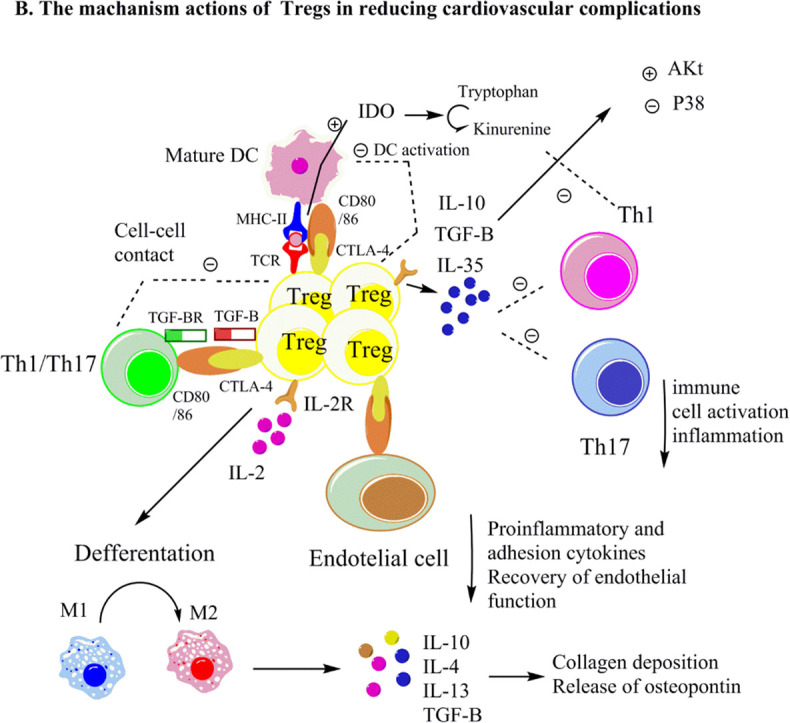
Here, we propose that Tregs may decrease the severity of cardiovascular complications in COVID-19 patients. Tregs, control immune responses especially inflammatory responses of Th17 and Th1 cells [142], [143]. This could be due to the interaction of surface markers of Tregs including, PD-L1, CD25, and CTLA-4, with the ligands on the target cells or/and the secretion of TGF-β and IL-10 (as the tolerogenic cytokines) that stimulate apoptosis and inhibit the cytotoxicity of Th1 and Th17 cells [144], [145], [146] (Fig. 2 ). As noted, Th1 and Th17 cells could exacerbate inflammatory conditions in cardiovascular diseases through secreting inflammatory cytokines, including IFN-γ, IL-17A, and IL-6, which may play a role in cytokine storm and the pathogenesis of severe COVID-19. Furthermore, these cytokines regulated the synthesis and destruction of vasoconstrictors and vasodilators, leading to increased blood pressure [123]. Therefore, Tregs may control inflammatory reactions and blood pressure, reducing the severity of cardiovascular complications associated with COVID-19 infection.
Fig. 2.

A- Tregs impairment in severe COVID-19. B- Proposed mechanisms of actions of Tregs in alleviating cardiovascular complications in COVID-19.
Tregs regulate Th17 and Th1 cell inflammatory activation by molecular interactions (like CD25 and CTLA-4 with ligands on target cells), TGF-β, and IL-10 secretion (as the tolerogenic cytokines). Tregs may enhance cardiac wound healing via activating M2 macrophages, reducing p38 MAP kinase, and increasing Akt activation. Notably, IL-10 and TGF-β could accelerate extracellular matrix deposition and cardiac healing by enhancing collagen deposition and triggering osteopontin release from monocytes and macrophages.
In another aspect, Tregs are critical in controlling endothelium-dependent relaxation in coronary arterioles and arterial blood pressure [100]. Endothelial cells regulate vascular tone and aortic stiffness (one reason for high blood pressure) by releasing relaxing factors like prostacyclin and nitric oxide [131], [147]. Of note, SARS-CoV-2 can infect vascular endothelial cells [148], [149]. Emerging evidence indicates that endothelial dysfunction and arterial hypertension are the critical characteristics of COVID-19 infection [150], [151], [152]. This includes the involvement of vascular endothelium in leukocyte attraction, which leads to cytokine secretion and tissue damage, both of which are key elements in ARDS, and cardiovascular complications [150], [151], [152].
It was discovered that transferring Tregs into hypertensive mice lowered arterial blood pressure and boosted endothelium-dependent relaxation in coronary arterioles by minimizing inflammatory cytokines and macrophage infiltration [151], [152]. Mechanistically, it may result from releasing IL-10, TGF-β, and IL-35 in a paracrine-dependent manner [153]. In addition, these cytokines may reduce oxidative stress by inactivating NOX, which controls endothelium-dependent relaxation [152]. As a result, it is reasonable to hypothesize that Tregs could reduce the severity of COVID-19 infection by increasing endothelial function and lowering high blood pressure, both of which are induced by COVID-19 infection.
Tregs have been found to reduce viral load, limit antiviral immune responses, and prevent myocarditis by regulating inflammatory responses [103], [111]. Tregs also have been shown to modulate Th1 and Th17 cells by releasing IL-10, suggesting that Tregs may be able to prevent myocarditis in COVID-19 patients [118], [119], [120]. Mechanistically, Tregs suppressed p38 MAP kinase activation and increased Akt activation (Fig. 2) by secreting TGF-β [112], [113]. As a result, it's not unexpected that TGF-β is involved in neutralizing adverse immunological responses; Akt activation, p38 inhibition, and immune control by Tregs would all be anticipated. This likely resulted in a reduced viral load and immune infiltration in the case of COVID-19 infection.
In COVID-19 patients, myocardial infarction has been associated with heart failure [154]. Tregs may enhance wound healing in COVID-19 patients with myocardial infarction by influencing macrophage differentiation. Tregs may promote M2-like monocyte differentiation after myocardial infraction by secreting TGF-β, IL-13, and IL-10 in fractioned myocardium in vivo and in vitro [132], [133], [134]. TGF- also promoted collagen deposition by myofibroblasts and accelerated the formation of scar tissue [155]. In addition, TGF-β, and IL-10, as previously discussed, can cause monocytes and macrophages to produce osteopontin [132].
Osteopontin is a glycoprotein with various activities, including cell adhesion and migration, and it contributes to matrix assembly and wound healing following myocardial infarction [132], [137]. Therefore, it's intriguing to suggest that Tregs may have a role in accelerating extracellular matrix deposition and cardiac healing in patients with severe COVID-19 by increasing collagen and osteopontin levels.
7.2. Therapeutic opportunities for Tregs in cardiovascular complications associated with severe COVID-19
Tregs, as noted previously, may reduce the severity of cardiovascular complications in COVID-19 patients. Therapeutic interventions for enhancing tolerance based on the adoptive transfer of Tregs are an effective strategy to treat Treg-mediated diseases, including autoimmunity, spontaneous abortion and, tissue transplantation [156], [157]. This includes isolating in vivo differentiated Tregs, expanding Tregs ex vivo, or generating iTreg cells in vitro and subsequent transfer into the body [156], [157]. This strategy could be used to treat cardiovascular complications caused by severe COVID-19 infection. To utilize Treg immunotherapy, meticulously designed clinical trials and precise Tregs expansion planning need to be employed, using the newest scientific technologies in Treg biology in COVID-19 treatment. In addition, probable adverse effects of artificially reinforcing Tregs, including diminished immune surveillance against tumors, need to be considered. Another issue that needs attention is the appropriate dose and subsets of Tregs [157]. The critical challenges for utilizing Treg therapy in treating cardiovascular complications in severe COVID-19 are the diagnosis of Treg cell deficiency and determining the appropriate time of adoptive transfer of Tregs. To our knowledge, the investigation of Treg cell deficiency in patients with severe COVID-19 was not the primary endpoint of any of the studies. The establishment of a standardized concept of minimum necessary Treg markers will be a good step.
8. Concluding remarks and future directions
Tregs are the key regulators of immune responses. Multiple pathways for controlling immune reactions have been proposed, emphasizing the anti-inflammatory properties of Tregs. Uncontrolled inflammation has been demonstrated in the pathogenesis of COVID-19, which may be related to a reduction in the frequency of Tregs and functions in severe COVID-19 infection. As a result, COVID-19 patients suffer from severe lung injury and cardiovascular complications, the leading causes of morbidity and mortality.
Tregs cell therapy is widely used to treat various autoimmune and inflammatory diseases in animal models, and some clinical trials are going on. Rigorously designed clinical trials and detailed Tregs manufacturing planning should be considered for evaluation in clinical trials to evaluate their effectiveness in improving clinical outcomes and reducing cardiovascular compilations of patients with severe COVID-19. In this light, we propose that the adoptive transfer of autologous Tregs may be one option for treating patients with severe COVID-19 with cardiovascular compilations. However, the dosage of Tregs and complementary therapies for SARS-CoV-2 infection must be approached. Furthermore, the probable adverse effects of artificially reinforcing Tregs, including diminished immune surveillance against tumors, need to be taken into account. As we proposed in previous research, adoptive transfer of Tregs could be an important clinical approach for disorders with inflammatory roots, such as spontaneous abortion. According to the current evidence, we suggest that controlling inflammatory responses and improving endothelial and atrial function, lowering high blood pressure, lowering the viral load and limiting adverse antiviral immune responses, as well as improving cardiac healing are the possible beneficial effects of adoptive transfer of Tregs in alleviating cardiovascular complications including, myocarditis, CAD, hypertension, myocardial infarction and cardiac failure in COVID-19 patients. According to the new findings, we also propose for adoptive transfer of Tregs, expansion of autologous Tregs in the presence of vitamin C + RA established a population that was more stable when exposed to an inflammatory, suggesting a possible strategy for reducing Treg plasticity in inflammatory conditions like in COVID-19 infection.
Mechanisms of actions of transferred Tregs may be mediated at the molecular and cellular levels. Tregs regulate the function of pro-inflammatory cells such as Th17 and Th1 cells and mononuclear cell infiltration through molecular interactions (PD-L1, CD25, and CTLA-4 with ligands on target cells), and the secretion of TGF-β and IL-10 (as the tolerogenic cytokines). Adoptive transfer of Tregs may enhance cardiac wound healing via activating M2-like, reducing p38 MAP kinase, and increasing Akt activation. Additionally, by enhancing collagen deposition and inducing osteopontin release from monocytes and macrophages, IL-10 and TGF-β may accelerate extracellular matrix deposition and cardiac healing.
Nutrients with immune-modulatory properties that boost Tregs differentiation, proliferation, and functions like vitamin D, vitamin A, niacin and, short-chain fatty could be the natural solution in this scenario. As we indicated, VitD3 could enhance the frequency and functions of Tregs (FOXP3 and GITR) [143], [158], [159], [160]. Statin drugs, which are known to have immunomodulatory activities and induce Tregs [[161], [162]], have also been shown to be associated with reduced COVID-19 outcomes including mortality [163], [164], [165]. Furthermore, in future studies, the role of antiviral drugs such as valproic acid, which has been shown to have beneficial effects on Tregs, can be investigated in reducing cardiovascular risks in COVID-19.
In conclusion, the physiological advantages of Tregs and Treg adoptive transfer are exciting research and clinical fields to investigate the cellular, molecular, and immunological mechanisms that contribute to the treatment of cardiovascular complications in severe COVID-19. Therefore, potential clinical trials that could pave the way in reducing cardiovascular complications in patients with severe COVID-19 are recommended.
Funding source
All sources of funding should also be acknowledged, and you should declare any involvement of study sponsors in the study design; collection, analysis and interpretation of data; the writing of the manuscript; the decision to submit the manuscript for publication. If the study sponsors had no such involvement, this should be stated.
CRediT authorship contribution statement
Nafiseh Saghafi - participate with the conception, study design, drafting the manuscript.
Seyed Abdolrahim Rezaee - involved with the study design, critical revision, editing and approval of the final draft of the study.
Amir Abbas Momtazi-Borojeni - contributed with the study design, critical review, editing and the support of the final draft.
Fataneh Tavasolian - contributed with the conception, study design, critical revision, editing and the final approval of the manuscript.
Elham Abdollahi - contributed with the study design, critical review, editing and the support of the final draft.
Thozhukat Sathyapalan - contributed with the conception, study design, critical revision, editing and the final approval of the manuscript.
Amirhossein Sahebkar - contributed with the conception, study design, critical revision, editing and the final approval of the manuscript.
Declaration of competing interest
A conflicting interest exists when professional judgment concerning a primary interest (such as patient's welfare or the validity of research) may be influenced by a secondary interest (such as financial gain or personal rivalry). It may arise for the authors when they have financial interest that may influence their interpretation of their results or those of others. Examples of potential conflicts of interest include employment, consultancies, stock ownership, honoraria, paid expert testimony, patent applications/registrations, and grants or other funding.
References
- 1.Fried J., Ramasubbu K., Bhatt R., Topkara V., Clerkin K., Horn E., et al. The variety of cardiovascular presentations of COVID-19. Circulation. 2020;141(23):1930–1936. doi: 10.1161/CIRCULATIONAHA.120.047164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choi H.M., Moon S.Y., Yang H.I., Kim K.S. Understanding viral infection mechanisms and patient symptoms for the development of COVID-19 therapeutics. Int. J. Mol. Sci. 2021;22(4):1737. doi: 10.3390/ijms22041737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duong D. What’s important to know about the new COVID-19 variants? Can. Med. Assoc. J. 2021;193(4):E141–E142. doi: 10.1503/cmaj.1095915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abdool Karim S.S., de Oliveira T., Loots G. Appropriate names for COVID-19 variants. Science. 2021;371(6535):1215. doi: 10.1126/science.abh0836. [DOI] [PubMed] [Google Scholar]
- 5.Maslo C., Friedland R., Toubkin M., Laubscher A., Akaloo T., Kama B. Characteristics and outcomes of hospitalized patients in South Africa during the COVID-19 omicron wave compared with previous waves. JAMA. 2021 doi: 10.1001/jama.2021.24868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Christie B. British Medical Journal Publishing Group; 2021. Covid-19: Early Studies Give Hope Omicron is Milder Than Other Variants. [DOI] [PubMed] [Google Scholar]
- 7.Christensen P.A., Olsen R.J., Long S.W., Snehal R., Davis J.J., Saavedra M.O. medRxiv; 2022. Early Signals of Significantly Increased Vaccine Breakthrough, Decreased Hospitalization Rates, and Less Severe Disease in Patients With COVID-19 Caused by the Omicron Variant of SARS-CoV-2 in Houston, Texas. 2021.12. 30.21268560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sagar M., Reifler K., Rossi M., Miller N.S., Sinha P., White L.F., et al. Recent endemic coronavirus infection is associated with less-severe COVID-19. J. Clin. Invest. 2021;131(1) doi: 10.1172/JCI143380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abdullah F., Myers J., Basu D., Tintinger G., Ueckermann V., Mathebula M., et al. Decreased severity of disease during the first global omicron variant covid-19 outbreak in a large hospital in tshwane, South Africa. Int. J. Infect. Dis. 2021;116:38–42. doi: 10.1016/j.ijid.2021.12.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Veldhoen M., Simas J.P. Endemic SARS-CoV-2 will maintain post-pandemic immunity. Nat. Rev. Immunol. 2021;21(3):131–132. doi: 10.1038/s41577-020-00493-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang J., Zheng Y., Gou X., Pu K., Chen Z., Guo Q., et al. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: a systematic review and meta-analysis. Int. J. Infect. Dis. 2020;94:91–95. doi: 10.1016/j.ijid.2020.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fried J.A., Ramasubbu K., Bhatt R., Topkara V.K., Clerkin K.J., Horn E., et al. The variety of cardiovascular presentations of COVID-19. Circulation. 2020;141(23):1930–1936. doi: 10.1161/CIRCULATIONAHA.120.047164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richardson S., Hirsch J.S., Narasimhan M., Crawford J.M., McGinn T., Davidson K.W., et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. JAMA. 2020;323(20):2052–2059. doi: 10.1001/jama.2020.6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo T., Fan Y., Chen M., Wu X., Zhang L., He T., et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19) JAMA Cardiol. 2020;5(7):811–818. doi: 10.1001/jamacardio.2020.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lippi G., Lavie C.J., Sanchis-Gomar F. Cardiac troponin I in patients with coronavirus disease 2019 (COVID-19): evidence from a meta-analysis. Prog. Cardiovasc. Dis. 2020;63(3):390–391. doi: 10.1016/j.pcad.2020.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhatla A., Mayer M.M., Adusumalli S., Hyman M.C., Oh E., Tierney A., et al. COVID-19 and cardiac arrhythmias. Heart Rhythm. 2020;17(9):1439–1444. doi: 10.1016/j.hrthm.2020.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zaim S., Chong J.H., Sankaranarayanan V., Harky A. COVID-19 and multi-organ response. Curr. Probl. Cardiol. 2020;100618 doi: 10.1016/j.cpcardiol.2020.100618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheung E.W., Zachariah P., Gorelik M., Boneparth A., Kernie S.G., Orange J.S., et al. Multisystem inflammatory syndrome related to COVID-19 in previously healthy children and adolescents in New York City. JAMA. 2020;324(3):294–296. doi: 10.1001/jama.2020.10374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sala S., Peretto G., Gramegna M., Palmisano A., Villatore A., Vignale D., et al. Acute myocarditis presenting as a reverse tako-tsubo syndrome in a patient with SARS-CoV-2 respiratory infection. Eur. Heart J. 2020;41(19):1861–1862. doi: 10.1093/eurheartj/ehaa286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kleinewietfeld M., Hafler D.A. The plasticity of human Treg and Th17 cells and its role in autoimmunity. Semin. Immunol. 2013;25(4):305–312. doi: 10.1016/j.smim.2013.10.009. Elsevier. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bach J.F. Regulatory T cells under scrutiny. Nat. Rev. Immunol. 2003;3(3):189–198. doi: 10.1038/nri1026. [DOI] [PubMed] [Google Scholar]
- 22.Chen L., Li X., Chen M., Feng Y., Xiong C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020;116(6):1097–1100. doi: 10.1093/cvr/cvaa078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Christoffersson G., von Herrath M. Regulatory immune mechanisms beyond regulatory T cells. Trends Immunol. 2019;40(6):482–491. doi: 10.1016/j.it.2019.04.005. [DOI] [PubMed] [Google Scholar]
- 24.Prabhu S.D., Frangogiannis N.G. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ. Res. 2016;119(1):91–112. doi: 10.1161/CIRCRESAHA.116.303577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pinto J.M., Boyden P.A. Electrical remodeling in ischemia and infarction. Cardiovasc. Res. 1999;42(2):284–297. doi: 10.1016/s0008-6363(99)00013-9. [DOI] [PubMed] [Google Scholar]
- 26.Bruins P., Velthuis Ht, Yazdanbakhsh A.P., Jansen P.G., van Hardevelt F.W., de Beaumont E.M., et al. Activation of the complement system during and after cardiopulmonary bypass surgery: postsurgery activation involves C-reactive protein and is associated with postoperative arrhythmia. Circulation. 1997;96(10):3542–3548. doi: 10.1161/01.cir.96.10.3542. [DOI] [PubMed] [Google Scholar]
- 27.Ker J. Inflammation, immunity and infection in atherothrombosis. Med. Chron. 2019;2019(3):33. [Google Scholar]
- 28.Veldhoen M., Hocking R.J., Atkins C.J., Locksley R.M., Stockinger B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24(2):179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 29.Konkel J.E., Zhang D., Zanvit P., Chia C., Zangarle-Murray T., Jin W., et al. Transforming growth factor-β signaling in regulatory T cells controls T helper-17 cells and tissue-specific immune responses. Immunity. 2017;46(4):660–674. doi: 10.1016/j.immuni.2017.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fujimoto Y., Kuramoto N., Yoneyama M., Azuma Y.-T. Interleukin-19 as an immunoregulatory cytokine. Curr. Mol. Pharmacol. 2021;14(2):191–199. doi: 10.2174/1874467213666200424151528. [DOI] [PubMed] [Google Scholar]
- 31.Collison L.W., Delgoffe G.M., Guy C.S., Vignali K.M., Chaturvedi V., Fairweather D., et al. The composition and signaling of the IL-35 receptor are unconventional. Nat. Immunol. 2012;13(3):290. doi: 10.1038/ni.2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karin N., Wildbaum G. The role of chemokines in adjusting the balance between CD4+ effector T cell subsets and FOXp3-negative regulatory T cells. Int. Immunopharmacol. 2015;28(2):829–835. doi: 10.1016/j.intimp.2015.03.037. [DOI] [PubMed] [Google Scholar]
- 33.Xu L., Kitani A., Fuss I., Strober W. Cutting edge: regulatory T cells induce CD4+ CD25− Foxp3− T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-β. J. Immunol. 2007;178(11):6725–6729. doi: 10.4049/jimmunol.178.11.6725. [DOI] [PubMed] [Google Scholar]
- 34.Axtell R.C., Steinman L. Type 1 interferons cool the inflamed brain. Immunity. 2008;28(5):600–602. doi: 10.1016/j.immuni.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 35.Kappos L., Freedman M.S., Polman C.H., Edan G., Hartung H.-P., Miller D.H., et al. Long-term effect of early treatment with interferon beta-1b after a first clinical event suggestive of multiple sclerosis: 5-year active treatment extension of the phase 3 BENEFIT trial. Lancet Neurol. 2009;8(11):987–997. doi: 10.1016/S1474-4422(09)70237-6. [DOI] [PubMed] [Google Scholar]
- 36.Bhela S., Varanasi S.K., Jaggi U., Sloan S.S., Rajasagi N.K., Rouse B.T. The plasticity and stability of regulatory T cells during viral-induced inflammatory lesions. J. Immunol. 2017;199(4):1342–1352. doi: 10.4049/jimmunol.1700520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ivanova E.A., Orekhov A.N. T helper lymphocyte subsets and plasticity in autoimmunity and cancer: an overview. Biomed. Res. Int. 2015;2015 doi: 10.1155/2015/327470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li N., Saghafi N., Ghaneifar Z., Rezaee S.A., Rafatpanah H., Abdollahi E. Evaluation of the effects of 1, 25VitD3 on inflammatory responses and IL-25 expression. Front. Genet. 2021;12 doi: 10.3389/fgene.2021.779494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Janikashvili N., Chikovani T., Audia S., Bonnotte B., Larmonier N. Hindawi; 2015. T Lymphocyte Plasticity in Autoimmunity and Cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ienca M., Vayena E. On the responsible use of digital data to tackle the COVID-19 pandemic. Nat. Med. 2020;26(4):463–464. doi: 10.1038/s41591-020-0832-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cucinotta D., Vanelli M. WHO declares COVID-19 a pandemic. Acta Biomed. Ateneo Parmense. 2020;91(1):157. doi: 10.23750/abm.v91i1.9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Helmy Y.A., Fawzy M., Elaswad A., Sobieh A., Kenney S.P., Shehata A.A. The COVID-19 pandemic: a comprehensive review of taxonomy, genetics, epidemiology, diagnosis, treatment, and control. J. Clin. Med. 2020;9(4):1225. doi: 10.3390/jcm9041225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bouaziz J., Even M., Isnard-Bogillot F., Vesale E., Nikpayam M., Mihalache A. COVID-19 in pregnancy: what do we really know? F1000Research. 2020;9(362):362. Elsevier. [Google Scholar]
- 44.Zhang Y., Geng X., Tan Y., Li Q., Xu C., Xu J., et al. New understanding of the damage of SARS-CoV-2 infection outside the respiratory system. Biomed. Pharmacother. 2020;110195 doi: 10.1016/j.biopha.2020.110195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu H., Zhong L., Deng J., Peng J., Dan H., Zeng X., et al. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 2020;12(1):1–5. doi: 10.1038/s41368-020-0074-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Athari S.Z., Mohajeri D., Nourazar M.A., Doustar Y. Updates on coronavirus (COVID-19) and kidney. J. Nephropathology. 2020;9(4) [Google Scholar]
- 47.Chen C., Zhou Y., Wang D.W. SARS-CoV-2: a potential novel etiology of fulminant myocarditis. Herz. 2020;45(3):230–232. doi: 10.1007/s00059-020-04909-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Unudurthi S.D., Luthra P., Bose R.J., McCarthy J., Kontaridis M.I. Cardiac inflammation in COVID-19: lessons from heart failure. Life Sci. 2020;118482 doi: 10.1016/j.lfs.2020.118482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Q.-J., Yao Y.-Z., Song J.-S., Wang Q., Xu L.-Y., Bao Z.-J., et al. Kinetic changes in virology, specific antibody response and imaging during the clinical course of COVID-19: a descriptive study. BMC Infect. Dis. 2020;20(1):1–11. doi: 10.1186/s12879-020-05549-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu X., Zhang R., He G. Hematological findings in coronavirus disease 2019: indications of progression of disease. Ann. Hematol. 2020;99:1421–1428. doi: 10.1007/s00277-020-04103-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bastug A., Bodur H., Erdogan S., Gokcinar D., Kazancioglu S., Kosovali B.D., et al. Clinical and laboratory features of COVID-19: predictors of severe prognosis. Int. Immunopharmacol. 2020;88 doi: 10.1016/j.intimp.2020.106950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tavasolian F., Hatam G.R., Mosawi S.H., Saadi M.I., Abdollahi E., Jamialahmadi T., Sathyapalan T., Sahebkar A. The immune response and effectiveness of COVID-19 therapies. Adv. Exp. Med. Biol. 2021;1321:115–126. doi: 10.1007/978-3-030-59261-5_10. [DOI] [PubMed] [Google Scholar]
- 53.Ghale-Noie Z.N., Salmaninejad A., Bergquist R., Mollazadeh S., Hoseini B., Sahebkar A. Genetic aspects and immune responses in Covid-19: important organ involvement. Adv. Exp. Med. Biol. 2021;1327:3–22. doi: 10.1007/978-3-030-71697-4_1. [DOI] [PubMed] [Google Scholar]
- 54.Schett G., Sticherling M., Neurath M.F. COVID-19: risk for cytokine targeting in chronic inflammatory diseases? Nat. Rev. Immunol. 2020;20(5):271–272. doi: 10.1038/s41577-020-0312-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kong M., Zhang H., Cao X., Mao X., Lu Z. Higher level of neutrophil-to-lymphocyte is associated with severe COVID-19. Epidemiol. Infect. 2020;148 doi: 10.1017/S0950268820001557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Varchetta S., Mele D., Oliviero B., Mantovani S., Ludovisi S., Cerino A., et al. Unique immunological profile in patients with COVID-19. Cell. Mol. Immunol. 2020;1–9 doi: 10.1038/s41423-020-00557-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yan W., Chen D., Bigambo F.M., Wei H., Wang X., Xia Y. Differences of blood cells, lymphocyte subsets and cytokines in COVID-19 patients with different clinical stages: a network meta-analysis. BMC Infect. Dis. 2021;21(1):156. doi: 10.1186/s12879-021-05847-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feng X., Li S., Sun Q., Zhu J., Chen B., Xiong M., et al. Immune-inflammatory parameters in COVID-19 cases: a systematic review and meta-analysis. Front. Med. 2020;7:301. doi: 10.3389/fmed.2020.00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen R., Sang L., Jiang M., Yang Z., Jia N., Fu W., et al. Longitudinal hematologic and immunologic variations associated with the progression of COVID-19 patients in China. J. Allergy Clin. Immunol. 2020;146(1):89–100. doi: 10.1016/j.jaci.2020.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carissimo G., Xu W., Kwok I., Abdad M.Y., Chan Y.-H., Fong S.-W., et al. Whole blood immunophenotyping uncovers immature neutrophil-to-VD2 T-cell ratio as an early marker for severe COVID-19. Nat. Commun. 2020;11(1):1–12. doi: 10.1038/s41467-020-19080-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qin C., Zhou L., Hu Z., Zhang S., Yang S., Tao Y. Dysregulation of immune response in patients with coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020;71(15):762–768. doi: 10.1093/cid/ciaa248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu Z., Shi L., Wang Y., Zhang J., Huang L., Zhang C., et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020;8(4):420–422. doi: 10.1016/S2213-2600(20)30076-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Libby P., Simon D.I. Inflammation and thrombosis: the clot thickens. J. Am. Heart Assoc. 2001;103(13):1718–1720. doi: 10.1161/01.cir.103.13.1718. [DOI] [PubMed] [Google Scholar]
- 64.Liu B., Han J., Cheng X., Yu L., Zhang L., Wang W., et al. Reduced numbers of T cells and B cells correlates with persistent SARS-CoV-2 presence in non-severe COVID-19 patients. Sci. Rep. 2020;10(1):1–9. doi: 10.1038/s41598-020-73955-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van Eeden C., Khan L., Osman M.S., Cohen Tervaert J.W. Natural killer cell dysfunction and its role in COVID-19. Int. J. Mol. Sci. 2020;21(17):6351. doi: 10.3390/ijms21176351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maucourant C., Filipovic I., Ponzetta A., Aleman S., Cornillet M., Hertwig L., et al. Natural killer cell immunotypes related to COVID-19 disease severity. Sci. Immunol. 2020;5(50) doi: 10.1126/sciimmunol.abd6832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dashraath P., Wong J.L.J., Lim M.X.K., Lim L.M., Li S., Biswas A., et al. Coronavirus disease 2019 (COVID-19) pandemic and pregnancy. Am. J. Obstet. Gynecol. 2020;222(6):521–531. doi: 10.1016/j.ajog.2020.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Muyayalo K.P., Huang D.H., Zhao S.J., Xie T., Mor G., Liao A.H. COVID-19 and Treg/Th17 imbalance: potential relationship to pregnancy outcomes. Am. J. Reprod. Immunol. 2020;84(5) doi: 10.1111/aji.13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wong S.F., Chow K.M., Leung T.N., Ng W.F., Ng T.K., Shek C.C., et al. Pregnancy and perinatal outcomes of women with severe acute respiratory syndrome. Am. J. Obstet. Gynecol. 2004;191(1):292–297. doi: 10.1016/j.ajog.2003.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mahallawi W.H., Khabour O.F., Zhang Q., Makhdoum H.M., Suliman B.A. MERS-CoV infection in humans is associated with a pro-inflammatory Th1 and Th17 cytokine profile. Cytokine. 2018;104:8–13. doi: 10.1016/j.cyto.2018.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang F., Hou H., Luo Y., Tang G., Wu S., Huang M., et al. The laboratory tests and host immunity of COVID-19 patients with different severity of illness. JCI Insight. 2020;5(10) doi: 10.1172/jci.insight.137799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu D., Yang X.O. TH17 responses in cytokine storm of COVID-19: an emerging target of JAK2 inhibitor fedratinib. J. Microbiol. Immunol. Infect. 2020;53(3):368–370. doi: 10.1016/j.jmii.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chan J., Ng C., Chan Y., Mok T., Lee S., Chu S., et al. Short term outcome and risk factors for adverse clinical outcomes in adults with severe acute respiratory syndrome (SARS) Thorax. 2003;58(8):686–689. doi: 10.1136/thorax.58.8.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Booth C.M., Matukas L.M., Tomlinson G.A., Rachlis A.R., Rose D.B., Dwosh H.A., et al. Clinical features and short-term outcomes of 144 patients with SARS in the greater Toronto area. JAMA. 2003;289(21):2801–2809. doi: 10.1001/jama.289.21.JOC30885. [DOI] [PubMed] [Google Scholar]
- 76.Cao X. COVID-19: immunopathology and its implications for therapy. Nat. Rev. Immunol. 2020;20(5):269–270. doi: 10.1038/s41577-020-0308-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jafarzadeh A., Chauhan P., Saha B., Jafarzadeh S., Nemati M. Contribution of monocytes and macrophages to the local tissue inflammation and cytokine storm in COVID-19: lessons from SARS and MERS, and potential therapeutic interventions. Life Sci. 2020;118102 doi: 10.1016/j.lfs.2020.118102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tajbakhsh A., Gheibi Hayat S.M., Taghizadeh H., Akbari A., inabadi, M., Savardashtaki, A., Johnston, T.P., Sahebkar, A. COVID-19 and cardiac injury: clinical manifestations, biomarkers, mechanisms, diagnosis, treatment, and follow up. Expert Rev. Anti-Infect. Ther. 2021;19(3):345–357. doi: 10.1080/14787210.2020.1822737. [DOI] [PubMed] [Google Scholar]
- 79.Lewek J., Jatczak-Pawlik I., Maciejewski M., Jankowski P., Banach M. COVID-19 and cardiovascular complications - Preliminary results of the LATE-COVID study. Arch. Med. Sci. 2021;17(3):818–822. doi: 10.5114/aoms/134211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moayed M.S., Rahimi-Bashar F., Vahedian-Azimi A., Sathyapalan T., Guest P.C., Jamialahmadi T., Sahebkar A. Cardiac Injury in COVID-19: a systematic review. Adv. Exp. Med. Biol. 2021;1321:325–333. doi: 10.1007/978-3-030-59261-5_29. [DOI] [PubMed] [Google Scholar]
- 81.Litvinukova M., Talavera-Lopez C., Maatz H., Reichart D., Worth C.L., Lindberg E.L. BioRxiv; 2020. Cells and Gene Expression Programs in the Adult Human Heart. [Google Scholar]
- 82.Shi S., Qin M., Shen B., Cai Y., Liu T., Yang F. Association of cardiac injury with mortality in hospitalized patients with COVID-19 in Wuhan, China. JAMA Cardiol. 2020;5(7):802–810. doi: 10.1001/jamacardio.2020.0950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Doyen D., Moceri P., Ducreux D., Dellamonica J. Myocarditis in a patient with COVID-19: a cause of raised troponin and ECG changes. Lancet. 2020;395(10235):1516. doi: 10.1016/S0140-6736(20)30912-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Inciardi R.M., Lupi L., Zaccone G., Italia L., Raffo M., Tomasoni D., et al. Cardiac involvement in a patient with coronavirus disease 2019 (COVID-19) JAMA Cardiol. 2020;5(7):819–824. doi: 10.1001/jamacardio.2020.1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ruan Q., Yang K., Wang W., Jiang L., Song J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020;46(5):846–848. doi: 10.1007/s00134-020-05991-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yao X., Li T., He Z., Ping Y., Liu H., Yu S. A pathological report of three COVID-19 cases by minimally invasive autopsies. Zhonghua Bing Li Xue Za Zhi. 2020;49 doi: 10.3760/cma.j.cn112151-20200312-00193. E009-E. [DOI] [PubMed] [Google Scholar]
- 87.Aretz H.T. Myocarditis: the Dallas criteria. Hum. Pathol. 1987;18(6):619–624. doi: 10.1016/s0046-8177(87)80363-5. [DOI] [PubMed] [Google Scholar]
- 88.Fung G., Luo H., Qiu Y., Yang D., McManus B. Myocarditis. Circ. Res. 2016;118(3):496–514. doi: 10.1161/CIRCRESAHA.115.306573. [DOI] [PubMed] [Google Scholar]
- 89.Wang D., Hu B., Hu C., Zhu F., Liu X., Zhang J., et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. JAMA. 2020;323(11):1061–1069. doi: 10.1001/jama.2020.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhou F., Yu T., Du R., Fan G., Liu Y., Liu Z., et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yang X., Yu Y., Xu J., Shu H., Liu H., Wu Y., et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir. Med. 2020;8(5):475–481. doi: 10.1016/S2213-2600(20)30079-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang W., Su B., Pang L., Qiao L., Feng Y., Ouyang Y., et al. High-dimensional immune profiling by mass cytometry revealed immunosuppression and dysfunction of immunity in COVID-19 patients. Cell. Mol. Immunol. 2020;17(6):650–652. doi: 10.1038/s41423-020-0447-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Anghelina D., Zhao J., Trandem K., Perlman S. Role of regulatory T cells in coronavirus-induced acute encephalitis. Virology. 2009;385(2):358–367. doi: 10.1016/j.virol.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Feuerer M., Herrero L., Cipolletta D., Naaz A., Wong J., Nayer A., et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 2009;15(8):930–939. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kalfaoglu B., Almeida-Santos J., Tye C.A., Satou Y., Ono M. T-cell hyperactivation and paralysis in severe COVID-19 infection revealed by single-cell analysis. Front. Immunol. 2020;11:2605. doi: 10.3389/fimmu.2020.589380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stephen-Victor E., Das M., Karnam A., Pitard B., Gautier J.-F., Bayry J. Potential of regulatory T-cell-based therapies in the management of severe COVID-19. Eur. Respir. J. 2020;56(3) doi: 10.1183/13993003.02182-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Soy M., Keser G., Atagündüz P., Tabak F., Atagündüz I., Kayhan S. Cytokine storm in COVID-19: pathogenesis and overview of anti-inflammatory agents used in treatment. Clin. Rheumatol. 2020;39:2085–2094. doi: 10.1007/s10067-020-05190-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gladstone D.E., Kim B.S., Mooney K., Karaba A.H., D'Alessio F.R. Regulatory T cells for treating patients with covid-19 and acute respiratory distress syndrome: two case reports. Ann. Intern. Med. 2020;173(10):852–853. doi: 10.7326/L20-0681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sattler S., Fairchild P., Watt F.M., Rosenthal N., Harding S.E. The adaptive immune response to cardiac injury—the true roadblock to effective regenerative therapies? NPJ Regen. Med. 2017;2(1):1–5. doi: 10.1038/s41536-017-0022-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yamashita T., Sasaki N., Kasahara K., Hirata K.-i. Anti-inflammatory and immune-modulatory therapies for preventing atherosclerotic cardiovascular disease. J. Cardiol. 2015;66(1):1–8. doi: 10.1016/j.jjcc.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 101.Bansal S.S., Ismahil M.A., Goel M., Zhou G., Rokosh G., Hamid T., et al. Dysfunctional and proinflammatory regulatory T-lymphocytes are essential for adverse cardiac remodeling in ischemic cardiomyopathy. Circulation. 2019;139(2):206–221. doi: 10.1161/CIRCULATIONAHA.118.036065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.de Souza Santos E., de Aragão-França L.S., Meira C.S., Cerqueira J.V., Vasconcelos J.F., Nonaka C.K.V., et al. Tolerogenic dendritic cells reduce cardiac inflammation and fibrosis in chronic chagas disease. Front. Immunol. 2020;11 doi: 10.3389/fimmu.2020.00488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Meng X., Yang J., Dong M., Zhang K., Tu E., Gao Q., et al. Regulatory T cells in cardiovascular diseases. Nat. Rev. Cardiol. 2016;13(3):167–179. doi: 10.1038/nrcardio.2015.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Li N., Bian H., Zhang J., Li X., Ji X., Zhang Y. The Th17/Treg imbalance exists in patients with heart failure with normal ejection fraction and heart failure with reduced ejection fraction. Clin. Chim. Acta. 2010;411(23–24):1963–1968. doi: 10.1016/j.cca.2010.08.013. [DOI] [PubMed] [Google Scholar]
- 105.Tang T.-T., Ding Y.-J., Liao Y.-H., Yu X., Xiao H., Xie J.-J., et al. Defective circulating CD4+ CD25+ Foxp3+ CD127low regulatory T-cells in patients with chronic heart failure. Cell. Physiol. Biochem. 2010;25(4–5):451–458. doi: 10.1159/000303050. [DOI] [PubMed] [Google Scholar]
- 106.Bansal S.S., Ismahil M.A., Goel M., Patel B., Hamid T., Rokosh G., et al. Activated T lymphocytes are essential drivers of pathological remodeling in ischemic heart failure. Circ. Heart Fail. 2017;10(3) doi: 10.1161/CIRCHEARTFAILURE.116.003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Greulich S., Seitz A., Müller K.A., Grün S., Ong P., Ebadi N., et al. Predictors of mortality in patients with biopsy-proven viral myocarditis: 10-year outcome data. J. Am. Heart Assoc. 2020;9(16) doi: 10.1161/JAHA.119.015351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Watanabe K., Sukumaran V., Veeraveedu P.T., Thandavarayan R.A., Gurusamy N., Ma M. Regulation of inflammation and myocardial fibrosis in experimental autoimmune myocarditis. Inflamm. Allergy Drug Targets. 2011;10(3):218–225. doi: 10.2174/187152811795564091. [DOI] [PubMed] [Google Scholar]
- 109.Luetkens J.A., Isaak A., Zimmer S., Nattermann J., Sprinkart A.M., Boesecke C., et al. Diffuse myocardial inflammation in COVID-19 associated myocarditis detected by multiparametric cardiac magnetic resonance imaging. Circ. Cardiovasc. Imaging. 2020;13(5) doi: 10.1161/CIRCIMAGING.120.010897. [DOI] [PubMed] [Google Scholar]
- 110.Rouse B.T., Sarangi P.P., Suvas S. Regulatory T cells in virus infections. Immunol. Rev. 2006;212(1):272–286. doi: 10.1111/j.0105-2896.2006.00412.x. [DOI] [PubMed] [Google Scholar]
- 111.Vdovenko D., Eriksson U. Regulatory role of CD4+ T cells in myocarditis. J Immunol Res. 2018;2018 doi: 10.1155/2018/4396351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Huber S.A., Feldman A.M., Sartini D. Coxsackievirus B3 induces T regulatory cells, which inhibit cardiomyopathy in tumor necrosis factor-α transgenic mice. Circ. Res. 2006;99(10):1109–1116. doi: 10.1161/01.RES.0000249405.13536.49. [DOI] [PubMed] [Google Scholar]
- 113.Pappritz K., Savvatis K., Lindner D., Westermann D., Melzig M., Schultheiss H., et al. Administration of regulatory T cells ameliorates myocardial inflammation in experimental myocarditis. Eur. Heart J. 2013;34(suppl_1) [Google Scholar]
- 114.Adler H.S., Kubsch S., Graulich E., Ludwig S., Knop J., Steinbrink K. Activation of MAP kinase p38 is critical for the cell-cycle–controlled suppressor function of regulatory T cells. Blood. 2007;109(10):4351–4359. doi: 10.1182/blood-2006-09-047563. [DOI] [PubMed] [Google Scholar]
- 115.Amir M., Somakala K., Ali S. p38 MAP kinase inhibitors as anti inflammatory agents. Mini-Rev. Med. Chem. 2013;13(14):2082–2096. doi: 10.2174/13895575113136660098. [DOI] [PubMed] [Google Scholar]
- 116.Shi Y., Fukuoka M., Li G., Liu Y., Chen M., Konviser M., et al. Regulatory T cells protect mice against coxsackievirus-induced myocarditis through the transforming growth factor beta-coxsackie-adenovirus receptor pathway. Circulation. 2010;121(24):2624–2634. doi: 10.1161/CIRCULATIONAHA.109.893248. [DOI] [PubMed] [Google Scholar]
- 117.Pompura S.L., Dominguez-Villar M. The PI3K/AKT signaling pathway in regulatory T-cell development, stability, and function. J. Leukoc. Biol. 2018;103(6):1065–1076. doi: 10.1002/JLB.2MIR0817-349R. [DOI] [PubMed] [Google Scholar]
- 118.Yuan J., Yu M., Lin Q.-W., Cao A.-L., Yu X., Dong J.-H., et al. Th17 cells contribute to viral replication in coxsackievirus B3-induced acute viral myocarditis. J. Immunol. 2010;185(7):4004–4010. doi: 10.4049/jimmunol.1001718. [DOI] [PubMed] [Google Scholar]
- 119.Nindl V., Maier R., Ratering D., De Giuli R., Züst R., Thiel V., et al. Cooperation of T h1 and T h17 cells determines transition from autoimmune myocarditis to dilated cardiomyopathy. Eur. J. Immunol. 2012;42(9):2311–2321. doi: 10.1002/eji.201142209. [DOI] [PubMed] [Google Scholar]
- 120.Wei H., Lin C.-K., Lu S.-J., Wen Y.-X., Yuan S., Liu Y.-L. CD11b is involved in coxsackievirus B3-induced viral myocarditis in mice by inducing Th17 cells. Open Life Sci. 2020;15(1):1024–1032. doi: 10.1515/biol-2020-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hasib L., Lundberg A., Zachrisson H., Ernerudh J., Jonasson L. Functional and homeostatic defects of regulatory T cells in patients with coronary artery disease. J. Intern. Med. 2016;279(1):63–77. doi: 10.1111/joim.12398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mian M.O.R., Barhoumi T., Briet M., Paradis P., Schiffrin E.L. Deficiency of T-regulatory cells exaggerates angiotensin II-induced microvascular injury by enhancing immune responses. J. Hypertens. 2016;34(1):97–108. doi: 10.1097/HJH.0000000000000761. [DOI] [PubMed] [Google Scholar]
- 123.Nguyen H., Chiasson V.L., Chatterjee P., Kopriva S.E., Young K.J., Mitchell B.M. Interleukin-17 causes rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc. Res. 2013;97(4):696–704. doi: 10.1093/cvr/cvs422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Barhoumi T., Kasal D.A., Li M.W., Shbat L., Laurant P., Neves M.F., et al. T regulatory lymphocytes prevent angiotensin ii–induced hypertension and vascular injury. Hypertension. 2011;57(3):469–476. doi: 10.1161/HYPERTENSIONAHA.110.162941. [DOI] [PubMed] [Google Scholar]
- 125.Sharir R., Semo J., Shimoni S., Ben-Mordechai T., Landa-Rouben N., Maysel-Auslender S., et al. Experimental myocardial infarction induces altered regulatory T cell hemostasis, and adoptive transfer attenuates subsequent remodeling. PLoS One. 2014;9(12) doi: 10.1371/journal.pone.0113653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tang T.-T., Yuan J., Zhu Z.-F., Zhang W.-C., Xiao H., Xia N., et al. Regulatory T cells ameliorate cardiac remodeling after myocardial infarction. Basic Res. Cardiol. 2012;107(1):1–17. doi: 10.1007/s00395-011-0232-6. [DOI] [PubMed] [Google Scholar]
- 127.Okamoto N., Noma T., Ishihara Y., Miyauchi Y., Takabatake W., Oomizu S., et al. Prognostic value of circulating regulatory T cells for worsening heart failure in heart failure patients with reduced ejection fraction. Int. Heart J. 2014;55(3):271–277. doi: 10.1536/ihj.13-343. [DOI] [PubMed] [Google Scholar]
- 128.Carrillo-Salinas F.J., Ngwenyama N., Anastasiou M., Kaur K., Alcaide P. Heart inflammation: immune cell roles and roads to the heart. Am. J. Pathol. 2019;189(8):1482–1494. doi: 10.1016/j.ajpath.2019.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kaikita K., Hayasaki T., Okuma T., Kuziel W.A., Ogawa H., Takeya M. Targeted deletion of CC chemokine receptor 2 attenuates left ventricular remodeling after experimental myocardial infarction. Am. J. Pathol. 2004;165(2):439–447. doi: 10.1016/S0002-9440(10)63309-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chistiakov D.A., Bobryshev Y.V., Nikiforov N.G., Elizova N.V., Sobenin I.A., Orekhov A.N. Macrophage phenotypic plasticity in atherosclerosis: the associated features and the peculiarities of the expression of inflammatory genes. Int. J. Cardiol. 2015;184:436–445. doi: 10.1016/j.ijcard.2015.03.055. [DOI] [PubMed] [Google Scholar]
- 131.Sica A., Mantovani AJTJoci. Macrophage Plasticity and Polarization: In Vivo Veritas. 122(3) 2012. pp. 787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Weirather J., Hofmann U.D., Beyersdorf N., Ramos G.C., Vogel B., Frey A., et al. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ. Res. 2014;115(1):55–67. doi: 10.1161/CIRCRESAHA.115.303895. [DOI] [PubMed] [Google Scholar]
- 133.Xia N., Lu Y., Gu M., Li N., Liu M., Jiao J., et al. A unique population of regulatory T cells in heart potentiates cardiac protection from myocardial infarction. Circulation. 2020;142(20):1956–1973. doi: 10.1161/CIRCULATIONAHA.120.046789. [DOI] [PubMed] [Google Scholar]
- 134.Harel-Adar T., Mordechai T.B., Amsalem Y., Feinberg M.S., Leor J., Cohen S. Modulation of cardiac macrophages by phosphatidylserine-presenting liposomes improves infarct repair. Proc. Natl. Acad. Sci. 2011;108(5):1827–1832. doi: 10.1073/pnas.1015623108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Shapouri-Moghaddam A., Mohammadian S., Vazini H., Taghadosi M., Esmaeili S.A., Mardani F., et al. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018;233(9):6425–6440. doi: 10.1002/jcp.26429. [DOI] [PubMed] [Google Scholar]
- 136.Momtazi-Borojeni A.A., Abdollahi E., Nikfar B., Chaichian S., Ekhlasi-Hundrieser M. Curcumin as a potential modulator of M1 and M2 macrophages: new insights in atherosclerosis therapy. Heart Fail. Rev. 2019;24(3):399–409. doi: 10.1007/s10741-018-09764-z. [DOI] [PubMed] [Google Scholar]
- 137.Singh M., Foster C.R., Dalal S., Singh K. Osteopontin: role in extracellular matrix deposition and myocardial remodeling post-MI. J. Mol. Cell. Cardiol. 2010;48(3):538–543. doi: 10.1016/j.yjmcc.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Brodin P. Immune determinants of COVID-19 disease presentation and severity. Nat. Med. 2021;27(1):28–33. doi: 10.1038/s41591-020-01202-8. [DOI] [PubMed] [Google Scholar]
- 139.Berhan Y. What immunological and hormonal protective factors lower the risk of COVID-19 related deaths in pregnant women? J. Reprod. Immunol. 2020;103180 doi: 10.1016/j.jri.2020.103180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tay M.Z., Poh C.M., Rénia L., MacAry P.A., Ng L.F. The trinity of COVID-19: immunity, inflammation and intervention. Nat. Rev. Immunol. 2020;20(6):363–374. doi: 10.1038/s41577-020-0311-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Moore J.B., June C.H. Cytokine release syndrome in severe COVID-19. Science. 2020;368(6490):473–474. doi: 10.1126/science.abb8925. [DOI] [PubMed] [Google Scholar]
- 142.Jørgensen N., Persson G., Hviid T.V.F. The tolerogenic function of regulatory T cells in pregnancy and cancer. Front. Immunol. 2019;10:911. doi: 10.3389/fimmu.2019.00911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Abdollahi E., Rezaee S.A., Saghafi N., Rastin M., Clifton V., Sahebkar A., et al. Evaluation of the effects of 1, 25 vitamin D3 on regulatory T cells and T helper 17 cells in vitamin D-deficient women with unexplained recurrent pregnancy loss. Curr. Mol. Pharmacol. 2020;13(4):306–317. doi: 10.2174/1874467213666200303130153. [DOI] [PubMed] [Google Scholar]
- 144.Mjösberg J., Berg G., Jenmalm M.C., Ernerudh J. FOXP3+ regulatory T cells and T helper 1, T helper 2, and T helper 17 cells in human early pregnancy decidua. Biol. Reprod. 2010;82(4):698–705. doi: 10.1095/biolreprod.109.081208. [DOI] [PubMed] [Google Scholar]
- 145.Robertson S.A., Care A.S., Moldenhauer L.M. Regulatory T cells in embryo implantation and the immune response to pregnancy. J. Clin. Invest. 2018;128(10):4224–4235. doi: 10.1172/JCI122182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Powell R.M. University of Birmingham; 2018. Novel T Cell Function and Specificity at the Human Maternal-fetal Interface. [Google Scholar]
- 147.Leloup A.J., Van Hove C.E., De Moudt S., De Keulenaer G.W., Fransen P. Ex vivo aortic stiffness in mice with different eNOS activity. Am. J. Phys. Heart Circ. Phys. 2020;318(5):H1233–H1244. doi: 10.1152/ajpheart.00737.2019. [DOI] [PubMed] [Google Scholar]
- 148.Varga Z., Flammer A.J., Steiger P., Haberecker M., Andermatt R., Zinkernagel A.S., et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395(10234):1417–1418. doi: 10.1016/S0140-6736(20)30937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Colmenero I., Santonja C., Alonso-Riaño M., Noguera-Morel L., Hernández-Martín A., Andina D., et al. SARS-CoV-2 endothelial infection causes COVID-19 chilblains: histopathological, immunohistochemical and ultrastructural study of seven paediatric cases. Br. J. Dermatol. 2020;183(4):729–737. doi: 10.1111/bjd.19327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Evans P.C., Rainger G.E., Mason J.C., Guzik T.J., Osto E., Stamataki Z., et al. Endothelial dysfunction in COVID-19: a position paper of the ESC working Group for Atherosclerosis and Vascular Biology, and the ESC Council of basic cardiovascular science. Cardiovasc. Res. 2020;116(14):2177–2184. doi: 10.1093/cvr/cvaa230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Matrougui K., Kassan M., Choi S., Nair D., Gonzalez-Villalobos R.A., Chentoufi A.A., et al. Natural regulatory T cells control coronary arteriolar endothelial dysfunction in hypertensive mice. Am. J. Pathol. 2011;178(1):434–441. doi: 10.1016/j.ajpath.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kassan M., Galan M., Partyka M., Trebak M., Matrougui K. Interleukin-10 released by CD4+ CD25+ natural regulatory T cells improves microvascular endothelial function through inhibition of NADPH oxidase activity in hypertensive mice. Arterioscler. Thromb. Vasc. Biol. 2011;31(11):2534–2542. doi: 10.1161/ATVBAHA.111.233262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kassan M., Wecker A., Kadowitz P., Trebak M., Matrougui K. CD4+ CD25+ Foxp3 regulatory T cells and vascular dysfunction in hypertension. J. Hypertens. 2013;31(10):1939. doi: 10.1097/HJH.0b013e328362feb7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Everaert B.R., Muylle J., Bartholomeus Twickler T. Emerging cardiological issues during the COVID-19 pandemic. Eur. J. Clin. Investig. 2020;50(7) doi: 10.1111/eci.13270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Darby I.A., Laverdet B., Bonté F., Desmoulière A. Fibroblasts and myofibroblasts in wound healing. Clin. Cosmet. Investig. Dermatol. 2014;7:301. doi: 10.2147/CCID.S50046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Duffy S.S., Keating B.A., Moalem-Taylor G. Adoptive transfer of regulatory T cells as a promising immunotherapy for the treatment of multiple sclerosis. Front. Neurosci. 2019;13:1107. doi: 10.3389/fnins.2019.01107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mohammadi S., Abdollahi E., Nezamnia M., Esmaeili S.-A., Tavasolian F., Sathyapalan T., et al. Adoptive transfer of tregs: a novel strategy for cell-based immunotherapy in spontaneous abortion: lessons from experimental models. Int. Immunopharmacol. 2021;90 doi: 10.1016/j.intimp.2020.107195. [DOI] [PubMed] [Google Scholar]
- 158.Abdollahi E., Saghafi N., Rezaee S.A.R., Rastin M., Jarahi L., Clifton V., et al. Evaluation of 1, 25 (OH) 2D3 effects on FOXP3, ROR-γt, GITR, and CTLA-4 gene expression in PBMCs of vitamin D-deficient women with unexplained recurrent pregnancy loss. Iran. Biomed. J. 2020;24(5):290. doi: 10.29252/ibj.24.5.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lavi Arab F., Rastin M., Faraji F., Zamani Taghizadeh Rabe S., Tabasi N., Khazaee M., et al. Assessment of 1, 25-dihydroxyvitamin D3 effects on treg cells in a mouse model of systemic lupus erythematosus. Immunopharmacol. Immunotoxicol. 2015;37(1):12–18. doi: 10.3109/08923973.2014.968255. [DOI] [PubMed] [Google Scholar]
- 160.Ghoryani M., Sahebari M., Mahmoudi M., Abdollahi N., Reihani H., Rabe S.Z.T., et al. Immunomodulatory vitamin D effects on regulatory T-cells and cytokines in an in vitro study on patients with systemic lupus erythematosus. Food Agric. Immunol. 2016;27(3):377–387. [Google Scholar]
- 161.Shahbaz S.K., Sadeghi M., Koushki K., Penson P.E., Sahebkar A. Regulatory T cells: possible mediators for the anti-inflammatory action of statins. Pharmacol. Res. 2019;149 doi: 10.1016/j.phrs.2019.104469. [DOI] [PubMed] [Google Scholar]
- 162.Ma X., Liu S., Li T., Yuan H. Intensive statin treatment ameliorate the Th17/Treg functional imbalance in patients with non-ST elevation acute coronary syndrome underwent percutaneous coronary intervention. Clin. Cardiol. 2020;43(4):379–385. doi: 10.1002/clc.23326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Vahedian-Azimi A., Mohammadi S.M., Beni F.H., Banach M., Guest P.C., Jamialahmadi T., Sahebkar A. Improved COVID-19 ICU admission and mortality outcomes following treatment with statins: a systematic review and meta-analysis. Arch. Med. Sci. 2021;17(3):579–595. doi: 10.5114/aoms/132950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Vahedian-Azimi A., Rahimibashar F., Najafi A., Kidde J., Shahriary A., Shojaei S., Pourhoseingholi M.A., Jamialahmadi T., Sahebkar A. Association of in-hospital use of statins, aspirin, and renin-angiotensin-aldosterone inhibitors with mortality and ICU admission due to COVID-19. Adv. Exp. Med. Biol. 2021;1327:205–214. doi: 10.1007/978-3-030-71697-4_17. [DOI] [PubMed] [Google Scholar]
- 165.Vahedian-Azimi A., Mohammadi S.M., Banach M., Beni F.H., Guest P.C., Al-Rasadi K., Jamialahmadi T., Sahebkar A. Improved COVID-19 Outcomes following Statin Therapy: An Updated Systematic Review and Meta-analysis. Biomed. Res. Int. 2021;2021 doi: 10.1155/2021/1901772. [DOI] [PMC free article] [PubMed] [Google Scholar]