

ABSTRACT
Staphylococcus aureus is an opportunistic pathogen that causes a wide range of infections and food poisoning in humans with antibiotic resistance, specifically to methicillin, compounding the problem. Bacteriophages (phages) provide an alternative treatment strategy, but these only infect a limited number of circulating strains and may quickly become ineffective due to bacterial resistance. To overcome these obstacles, engineered phages have been proposed, but new methods are needed for the efficient transformation of large DNA molecules into S. aureus to “boot-up” (i.e., rescue) infectious phages. We presented a new, efficient, and reproducible DNA transformation method, NEST (non-electroporation Staphylococcus transformation), for S. aureus to boot-up purified phage genomic DNA (at least 150 kb in length) and whole yeast-assembled synthetic phage genomes. This method was a powerful new tool for the transformation of DNA in S. aureus and will enable the rapid development of engineered therapeutic phages and phage cocktails against Gram-positive pathogens.
IMPORTANCE The continued emergence of antibiotic-resistant bacterial pathogens has heightened the urgency for alternative antibacterial strategies. Phages provide an alternative treatment strategy but are difficult to optimize. Synthetic biology approaches have been successfully used to construct and rescue genomes of model phages but only in a limited number of highly transformable host species. In this study, we used a new, reproducible, and efficient transformation method to reconstitute a functional nonmodel Siphophage from a constructed synthetic genome. This method will facilitate the engineering of Staphylococcus and Enterococcus phages for therapeutic applications and the engineering of Staphylococcus strains by enabling transformation of higher molecular weight DNA to introduce more complex modifications.
KEYWORDS: bacteriophage assembly, bacteriophage genetics, phage engineering, synthetic biology, transformation
INTRODUCTION
Infections caused by Staphylococcus aureus are difficult to treat due to an increased rate of antibiotic resistance and the bacterium’s ability to quickly adapt to changing conditions (1, 2). Of particular concern are infections caused by methicillin-resistant S. aureus (MRSA), which account for over 300,000 hospitalizations and over 10,000 deaths in the US (3). The use of bacteriophage (phage), viruses that infect bacteria as an alternative therapy to treat antibiotic-resistant bacterial pathogens has gained a clinical resurgence (4, 5). Although these recent reports note success in treating otherwise terminal infections, there is still the problem of finding the right phage that infects the strain of interest. To overcome this obstacle, the engineering of phages using synthetic genomics methods has been demonstrated (6–8). For this approach to be successful, transformation and boot-up (i.e., rescue) of the synthetic or recombinant genome (i.e., packaging of the genome into an infectious virion) in S. aureus must be efficient. Several methods to transform Staphylococci have been developed, including electroporation (9–15), calcium-dependent (16–21), partial cell wall removal (22), protoplast fusion (23–27), and “natural” competence (28–32), but these are inefficient and require large amounts of purified DNA (e.g., 3 to 24 μg) and are prohibitive to the transformation of large (i.e., >100 kb) phage genomes.
For model phages of model organisms, boot-up of recombinant or modified phage genomic DNA can occur by in vivo packaging following the transformation of highly competent cells or by in vitro packaging systems. Transformation by electroporation or chemical competence works well for phages of easily transformable Gram-negative bacteria (e.g., Escherichia coli and Klebsiella pneumoniae) (33–36) and for the Gram-positive bacterium Mycobacterium smegmatis (37, 38). However, for phages of other more difficult to transform bacteria (e.g., S. aureus and Enterococcus faecalis), transformation represents a major bottleneck for phage genome engineering (6). The use of in vitro transcription-translation systems for phage boot-up is even more limited, having only been successful with E. coli phages (39–44).
Since the 1960s, phages of Bacillus subtilis were shown to boot-up (i.e., transfect) using methods that take advantage of natural competence in B. subtilis (45, 46). The mechanism of natural competence in B. subtilis has been well studied (47–49), and the conditions and genetics needed to bolster the expression of competence genes for efficient transformation in B. subtilis have also been worked out (50, 51). However, in S. aureus, despite having homologs to conserved components for natural competence (28), the conditions and gene regulation needed to induce high-level expression of natural competence are unknown (29, 30). Low-level transfection using 10 μg of phage 44AHJD DNA was observed from S. aureus cells treated with lysostaphin (22), and transfection was observed using a high concentration of ϕ80α DNA (i.e., 10 μg/mL) only on calcium-treated S. aureus cells lysogenized with ϕ11 (16, 18).
The production of L-form Listeria monocytogenes has been reported to enable boot-up of Listeria and Bacillus phages from synthetic DNA and “reactivation” (i.e., transfection) of S. aureus phage K from virion-derived DNA (52). Our attempts to generate L-forms of L. monocytogenes EDGe or S. aureus RN4220 capable of transforming either plasmid DNA or “reactivation” of phage K DNA isolated from virions were unsuccessful presumably because multiple mutations are required to make stable competent L-forms. Because L-forms have deficient cell walls and can resemble protoplasts (53), enzymatic treatment with lysozyme and β-lactam antibiotics can produce L-form bacteria without the need for selection of complex mutants that produce L-forms (54).
We described a new method that utilizes enzymatic treatment with ampicillin and lysozyme, NEST (non-electroporation Staphylococcus transformation), for the transformation of plasmid and phage DNA (at least 150 kb) into S. aureus. The method was faster, easier, more robust, and more reproducible than producing permanent L-form cells as well as more efficient and less expensive than electroporation. Lastly, we showed that this method enabled reproducible boot-up of synthetic S. aureus phages and cross-genus boot-up of E. faecalis phages by S. aureus cells for use in bioengineering.
RESULTS
NEST versus electroporation.
The efficiency of the NEST method was compared with a widely used electroporation method to transform plasmid DNA into restriction-deficient S. aureus RN4220 (Fig. 1) (10). The effect of increased DNA concentration on the efficiency of transformation was tested for plasmids differing in size and selectable markers (Table 1). For NEST, S. aureus cells were grown in hypertonic HI-sucrose media to late log phase growth followed by treatment with ampicillin and lysozyme. NEST demonstrated a 10- to 128-fold, 56- to 618-fold, and 27- to 68-fold increase in transformation efficiency compared to the electroporation method for pCM28 (5.6 kb), pCAS9counter (9.5 kb), and pGF35 (9.0 kb), respectively (Fig. 2). A linear relationship was observed between the amount of plasmid DNA and transformation efficiency for both plasmids. NEST was able to transform S. aureus with as little as 10 ng of E. coli-purified pCAS9counter, which did not transform by electroporation with this amount of DNA.
FIG 1.
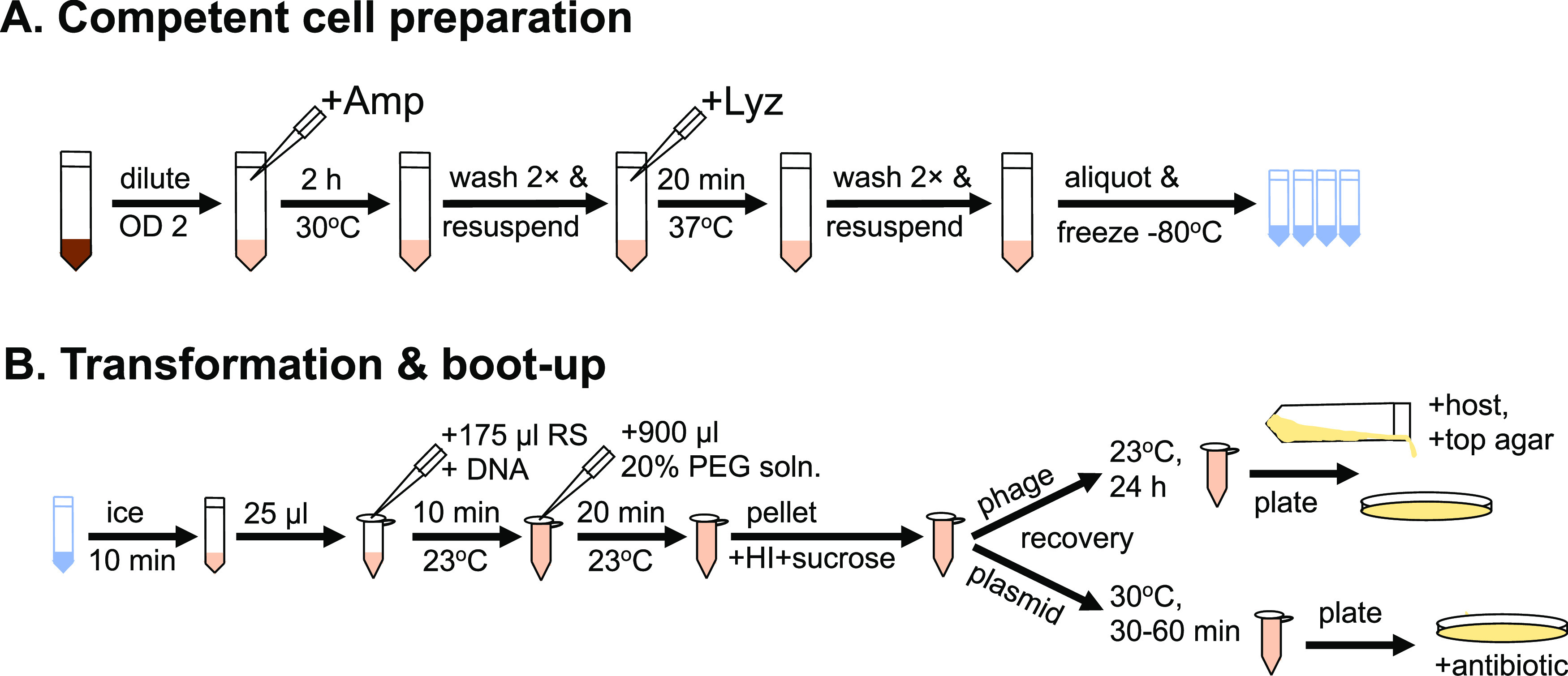
Overview of NEST. (A) The basic steps needed to make stocks of S. aureus competent cells by NEST. (B) Frozen competent cells are thawed, incubated with either plasmid or phage DNA and PEG before plating as indicated. See Material and Methods for details.
TABLE 1.
Plasmids used in this study
| Plasmids | Origin of replication (S. aureus, E. coli) | Selective marker | Size (bp) | Reference |
|---|---|---|---|---|
| pCM28 | pC194, ColE1 | Chloramphenicol (12.5 μg/mL) in S. aureus, Ampicillin (50 μg/mL) in E. coli | 5594 | (77, 85) |
| pCAS9counter | E194ts, ColE1 | Erythromycin (10 μg/mL) in S. aureus, Ampicillin (50 μg/mL) in E. coli | 9533 | (79) |
| pGF35 (pMSP3535-GFP) | pAMβ1, ColE1 | Erythromycin (10 μg/mL) in S. aureus, Erythromycin (300 μg/mL) in E. coli | 9016 | (78) |
FIG 2.

Comparison of NEST versus electroporation transformation methods. The effects of DNA amount and size on the efficiency of transformation between NEST and electroporation were determined using different amounts (10 ng, 100 ng, 500 ng, and 1,000 ng) of plasmids pCM28, pCAS9counter, and pFG25 purified from E. coli DH10B. For the NEST method, plasmids were transformed using a 20% PEG solution to the S. aureus RN4220 competent cells prepared using HI seed media. For electroporation, the electrocompetent cells prepared with B2 media were electroporated (2.3 kV, 100 Ω, 25 μF) with the indicated plasmids. Cells were plated on NYE media plates with 12.5 μg/mL chloramphenicol for pCM28 and 10 μg/mL erythromycin for pCAS9counter and pGF35 and incubated at 30°C for 24 to 48 h. Each data point represents the mean from three independent experiments (except for pGF35 electroporation, which had two independent experiments), and the error bars indicate standard error.
NEST does not depend on polyethylene glycol (PEG).
PEG-mediated DNA uptake has been widely used in bacteria and yeast transformations (55). Previously, it was noted that PEG is indispensable for the attachment of the DNA around intact cells and spheroplasts in yeast and increases the transformation efficiency (56) and to transform DNA by fusion of S. aureus protoplasts (23–27). To test whether PEG is required for NEST, we performed transformation experiments of pCM28 in RN4220 competent cells and calculated the transformation efficiency. NEST without PEG produced 1.33 × 104 ± 2.00 × 102 transformants per 108 colony forming unit (CFU) while NEST with PEG produced 2.24 × 104 ± 5.37 × 103 transformants, which was an ∼2-fold increase in transformation efficiency compared with the same competent cells without PEG.
NEST of other S. aureus strains.
Because many clinically relevant S. aureus strains are refractory to DNA transformation by standard methods, due to clonal complex-specific restriction-modification (R-M) systems (57, 58), we tested the ability of NEST to transform plasmid DNA into methicillin-susceptible S. aureus (MSSA) and MRSA isolates. MRSA S. aureus strains JE2 (CC8, (59)), Mu50 (CC5) and MW2 (CC1) and MSSA S. aureus strains MRSN 7983 (CC8) and HER1049 (CC25 (60)) were transformed with S. aureus-grown plasmid DNA using the NEST method and the transformation efficiency was determined (Fig. S1). For all strains tested, NEST had a greater transformation efficiency than electroporation by as much as 3 logs. For NEST, MW2 (CC1) and HER1049 (CC25) had the lowest transformation efficiency (∼2 logs lower than CC8 strains) but ∼2 logs better than electroporation. There was no difference between MRSA and MSSA strains. Electroporation performed the worst on Mu50 (CC5), MW2 (CC1), and HER1049 (CC25).
Long-term stability of NEST competent cells.
An important utility of competent cell preparation is long-term stability and reliability without the need to prepare fresh cells for each experiment. We performed NEST on competent cells stored at −80°C after 1 year using pCM28, and there was no significant reduction in transformation efficiency at lower DNA concentrations of DNA and less than a log difference at 0.5 and 1 μg of DNA (Fig. S2).
Transfection of S. aureus phage genomic DNA.
Satisfying the need for an efficient transformation procedure for S. aureus, we next tested whether NEST was capable of transfecting (i.e., transforming and booting-up) virion-purified phage DNA. We used gDNA of Siphophage SA75 and Myophage K whose genome sizes are 43 kb and 148 kb, respectively. Purified phage gDNA was mixed with NEST competent S. aureus RN4220 cells and PEG. The sample was incubated at room temperature (RT) for 24 h to allow for DNA uptake, phage gene expression, and packaging. This workflow is illustrated in Fig. 3A For phage K transfection, we mixed S. aureus MRSN 7983 as an indicator host strain with the transfection supernatant, whereas SA75 phage plaque formation was observed on S. aureus RN4220 as its indicator host strain. We were able to observe plaque formation within 24 to 36 h for both SA75 and phage K (Fig. 3B). SA75 produced more plaque-forming units (PFU) than phage K. The total time frame for the complete experiment from the preparation of competent cells, transformation, and boot-up was less than 4 days. Similar to plasmid transformation, we found a linear correlation between phage gDNA quantity and phage plaque production for both SA75 and phage K (Fig. 3C). With as little as 100 ng of gDNA, NEST produced ∼100 PFU per 108 competent cells.
FIG 3.
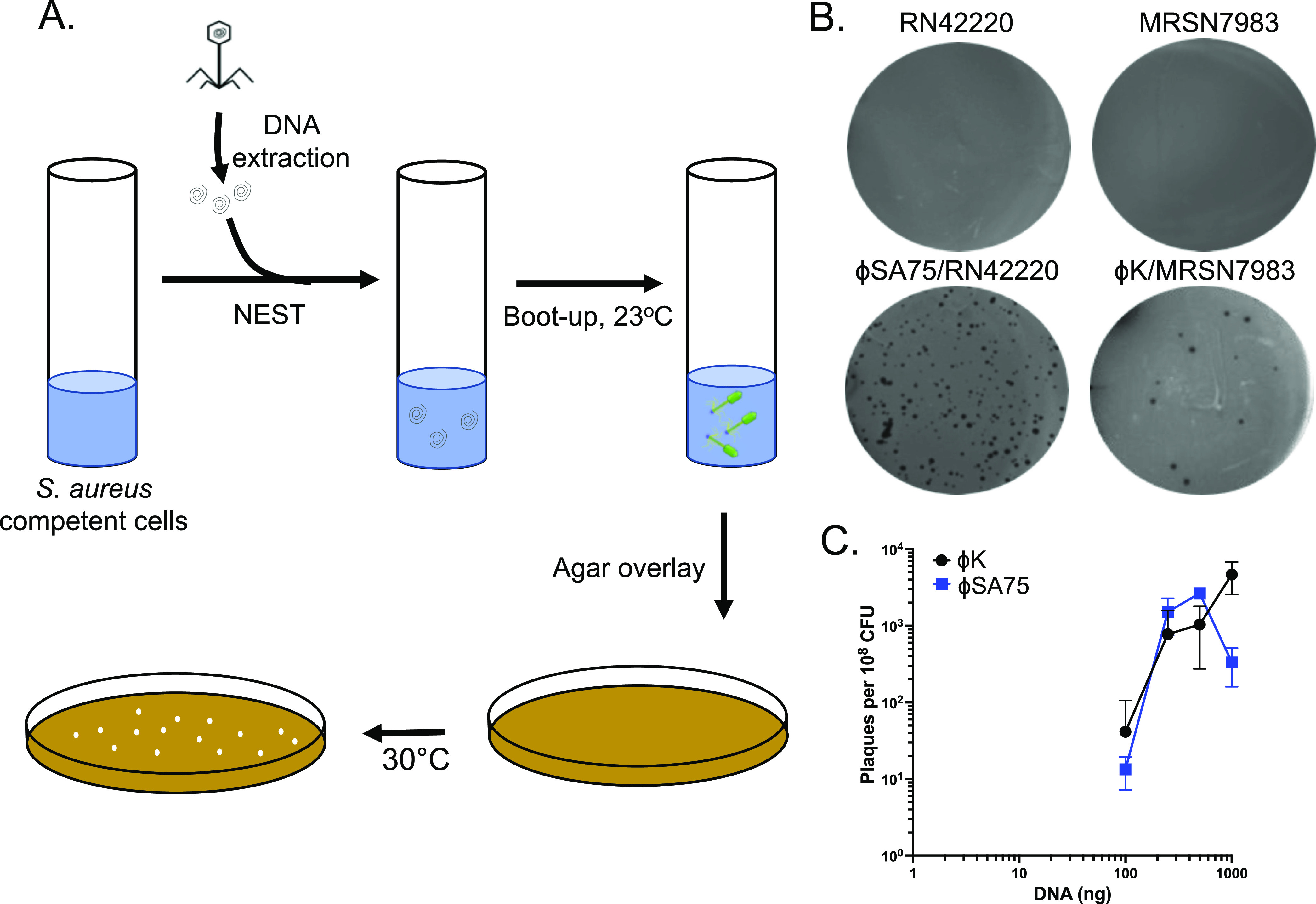
Transfection of S. aureus phage genomic DNA using NEST. (A) Workflow of the boot-up of S. aureus phage genomic DNA using NEST method. Phenol-chloroform extracted gDNA was transformed into S. aureus competent cells using 20% PEG solution. After a brief incubation, PEG solution was removed using centrifugation, and pellets were dissolved in HI seed media. Samples were incubated at RT for 24 h followed by the top agar overlay method and plates were incubated at 30°C for 24 to 48 h to visualize the plaques. (B) phage SA75 and phage K were booted-up using 100 ng of gDNA and between 1.13 × 108 to 6.13 × 108 CFU of S. aureus competent cells prepared by the NEST method. S. aureus MRSN 7983 was used as an indicator strain for phage K. (C) Efficiency of boot-up was determined by calculating the plaques per 108 CFU using different amounts of gDNA of phage SA75 and phage K. Each data point represents the mean from three independent experiments, and the error bars indicate standard error.
Boot-up of yeast-assembled S. aureus phage genomes.
To further evaluate the efficiency of the NEST method in phage engineering, we tested the ability of NEST to transform and boot-up a synthetic phage genome. A synthetic SA75 phage genome was constructed as a single fragment (SA75YC) as well as four overlapping fragments assembled into a complete genome (SA75YA) by transformation-associated recombination (TAR) cloning (Fig. 4 and Fig. S3). Because the packaged phage genome is terminally redundant, a terminal repeat fragment was generated using PCR and added to make identical direct repeats at both ends of the genome (Fig. S3). The synthetic genomes with the terminal repeat sequences were assembled in yeast and transformed into E. coli to obtain a higher yield of the cloned circular phage genome. SA75YC and SA75YA boot-up efficiency were 5.5-fold and 8-fold times higher than the SA75gDNA, respectively (Fig. 4).
FIG 4.
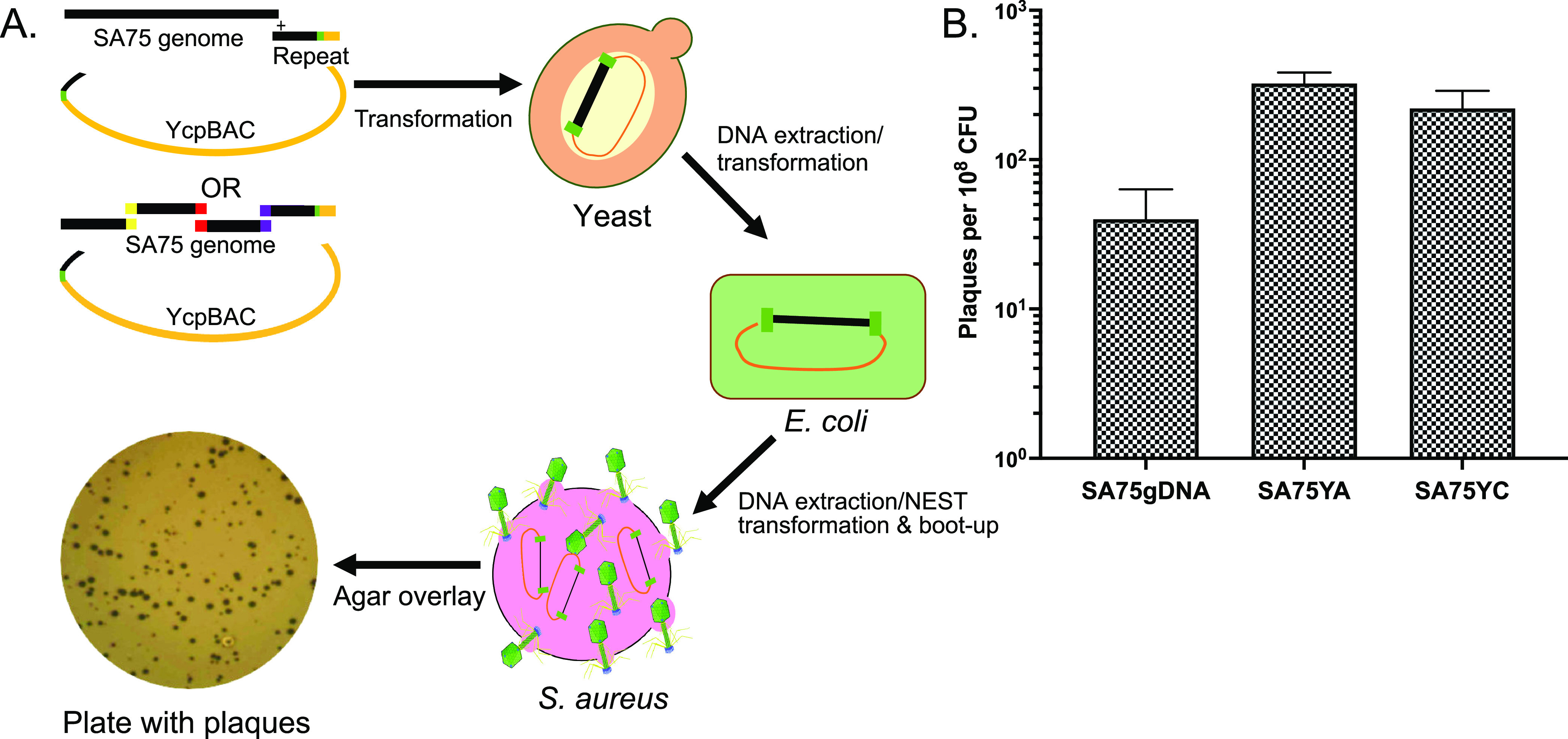
Boot-up of yeast-assembled synthetic S. aureus phage genomes using NEST. (A) The boot-up of either the whole yeast cloned (YC) or yeast assembled (YA) genomes of phage SA75 is depicted. To make SA75YC, cloned SA75 DNA, a PCR-generated repeat fragment, and a linear Ycp/BAC vector harboring terminal homology to the phage DNA and unique restriction site (ISce-I) were transformed into S. cerevisiae (yeast). Similarly, to make SA75YA, TAR-cloned fragments are transformed into yeast with the linear Ycp/BAC vector. DNA from positive clones was transformed into E. coli DH10B cells to produce high concentration plasmid stocks. These plasmids were further transfected/transformed into S. aureus competent cells using the NEST method, incubated at RT for 24 h, and plated using the agar overlay method to observe plaques. (B) To determine the efficiency of boot-up of SA75 DNA isolated from packaged genomes and synthetic genomes, 100 ng of DNA was incubated with 1.13 × 108 CFU of NEST competent cells for 30 min before plating. The efficiency of boot-up was computed by calculating the number of plaques formed per 108 CFU. Each data point represents the mean from three independent experiments, and the error bars indicate standard error.
Cross genus transfection of phages using NEST.
To further explore the utility of the NEST method to rescue other Gram-positive phages, we transfected gDNA of E. faecalis phages vB_EfaS_Ef5.1 (41.1 kb), vB_EfaS_Ef5.2 (41.4 kb), vB_EfaS_Ef5.3 (39.1 kb), vB_EfaS_Ef5.4 (40.6 kb), and vB_EfaS_Ef6.4 (41.1 kb) (61) in S. aureus competent cells. We were able to efficiently transfect/boot-up all the above potentially therapeutic phages and infect their original host strains E. faecalis AH5 or AH6 (61) (Fig. 5).
FIG 5.

Cross-genus transfection of E. faecalis phage genomic DNA using NEST. Appropriate amount (100 to 200 ng) of E. faecalis phage ɸ5.1, ɸ5.2, ɸ5.3, ɸ5.4 and ɸ6.4 gDNA was transformed into S. aureus cells (∼9 × 108 CFU) using the NEST method and incubated for 24 h at 23°C. (A) Transformants were mixed with the corresponding indicator strain (E. faecalis AH5 for ɸ5.1, ɸ5.2, ɸ5.3, and ɸ5.4, or AH6 for ɸ6.4) and plated on BHI media using the top agar overlay method and incubated at 30°C for 24 to 48 h to observe the plaques. The control plate (Cntl) was a lawn of S. aureus RN4220 cells mixed with E. faecalis AH5 lacking phage DNA. (B) The efficiency of boot-up was determined by calculating the number of plaques per 108 CFU of NEST competent cells of each E. faecalis phage and SA75 for comparison. Each data point represents the mean from three independent experiments, and the error bars indicate standard error.
DISCUSSION
The transformation of purified phage virion gDNA and subsequent packaging is called transfection. While methods were devised in the early 1970s to transfect S. aureus, they were limited to certain laboratory strains lysogenic for ϕ11 (18). Other methods of S. aureus transformation required the use of expensive and dose-sensitive lysostaphin to make protoplasts (23) and PEG for the fusion of the protoplasts (23–27). The most widely used method of S. aureus transformation has been electroporation, with the Schenk and Laddaga method being highly cited in the literature (10). We attempted transfection of phage SA75 DNA by electroporation with no success. This was likely due to the increased size of the phage genomic DNA (∼40 to 150 kb DNA) compared to the ∼5.5 kb plasmid (62). We next attempted to transfect SA75 DNA into Listeria monocytogenes EDGe L-forms following a procedure described previously (52, 63–65) but were again unsuccessful. Cells producing the characteristic “fried egg” appearance were made but were never competent, even for plasmid DNA. In addition to Listeria, we attempted to make S. aureus L-forms using a similar approach with no successful transformation of plasmid or transfection of phage DNA. We suspected that the formation of stable L-form bacteria requires multiple mutations that are difficult to obtain. Because L-forms are deficient in cell walls and can resemble protoplasts (53), enzymatic treatment with lysozyme and β-lactam antibiotics can produce L-form bacteria without the need for selection of complex mutants that produce L-forms (54). We adapted the use of ampicillin, lysozyme, and sucrose that can make efficient electrocompetent Listeria (66) but omitted the electroporation step. The new method of transformation, called NEST, allows for robust and reproducible booting-up of engineered phage of Gram-positive bacteria for therapeutic, sanitation, and diagnostic purposes.
The NEST method showed a 10- to 618-fold increase in transformation efficiency compared with the electroporation method for plasmids with sizes of 5.5 and 9.5 kb. NEST was better at transforming the larger plasmid and could transform both clinically derived MSSA and MRSA isolates, further increasing the utility of this method. However, the efficient transformation of these non-laboratory strains has traditionally been passaging of plasmids through RN4220 before the transformation of clinical isolates to provide CC8-specific methylation patterns or more recently, the use of IMXXB E. coli strains that mimic methylation patterns of CC1, CC8, and CC30 S. aureus isolates (67).
There are several examples of bacteria capable of importing foreign DNA via an active transport mechanism (68). However, unlike Bacillus subtilis, conditions for high-efficiency natural transformation of S. aureus have remained elusive (29, 30). While we do not know the mechanism of transformation by NEST, we do know that none of the buffers used in NEST have enough Ca2+ (e.g., 0.1 M (19)) to enable the transformation of S. aureus. Additionally, the requirement for a ϕ11 prophage to enable transformation (18) is not met because S. aureus strain RN4220 has been cured of prophages (and validated by searching the genome sequence using Phage_Finder (69)). Lastly, NEST functions without the addition of PEG. Although NEST uses PEG in the standard protocol, when omitted, we still get more transformants than with electroporation (Fig. S1). PEG is also not required for phage boot-up (data not shown).
As with any method of transformation, the efficiency of NEST is at the mercy of restriction systems that are present in the recipient S. aureus strains. However, NEST seems to be able to overcome some restriction pressure in the tested strains. The observed patterns of transformation efficiency can be explained with respect to the known CC-specific restriction systems (58). CC8 strains carry one restriction system in common with both CC5 (i.e., CC5-1, ATC[N]5CCT) and CC1 (i.e., CC1-2, CCAY[N]6TGT) strains. RN4220, a CC8 strain, should methylate these recognition sites if present on the plasmid of interest. In our case, pCM28 had 1 CC5-1 site and 2 CC1-2 sites based on the plasmid DNA sequence (GenBank accession number MN956986). In addition to these recognition sites, CC5 also encodes CC5-2 (CCAY[N]6GTA) and CC1 also encodes CC1-1 (CCAY[N]5TTAA). The plasmid pCM28 does not have the CC5-2 recognition sequence, but has 1 predicted CC1-1 site, making it vulnerable to restriction by the CC1-1 restriction system. From these known recognition site patterns, we predict that pCM28 would be protected from restriction in Mu50 (CC5) with CC5-1 methylated and CC5-2 missing. However, the efficiency of transformation is not as good as for RN4220 or JE2 (both CC8 strains) but is equivalent to MRSN 7983 (CC8). Perhaps this may be due to differences in cell preparation or other factors that influence competence by NEST. Curiously, the electroporation transformation efficiency of Mu50 was much worse compared to CC8 strains. The patterns of known restriction recognition sites do not explain this difference, which suggests that there is another explanation for the observed differences by electroporation. For MW2 (CC1) NEST transformation, we did observe a 2-log reduction in transformation efficiency compared to CC8 strains, which may be explained by the presence of the CC1-1 recognition site on pCM28.
The restriction deficient and methylation competent RN4220 has historically been used as an intermediate strain for plasmid transfer and phage transduction assays (70). Among other mutations in RN4220 (71), two restriction factors, HsdR from a type I restriction system and SauUSI from a type IV system, were inactivated via chemical mutagenesis (70, 72–74). Although IMXXB E. coli strains are available that remove cytosine methylation recognized by type IV RM systems of S. aureus and methylate adenine residues recognized by type I RM systems of specific S. aureus clonal complexes (67), we needed the booted-up progeny phages to incorporate these protective measures rather than the input DNA undergoing boot-up. In other words, the methylation or removal of methylation must occur in the boot-up host strain, RN4220, for the resulting phages to be protected from restriction in the target host strains. NEST seems to be limited to S. aureus, perhaps further suggesting that it may function through a S. aureus-specific DNA uptake pathway or that we have issues with restriction systems different from the S. aureus.
The NEST method of transforming plasmid DNA and transfecting phage DNA has several advantages over existing methods, such as being (i) capable of transforming relatively large DNA (>10 Kbp) into S. aureus (the largest tested was ∼150 Kbp), (ii) less expensive than electroporation (i.e., no cuvettes, electroporator) and lysostaphin-based protoplasting methods, (iii) faster than methods using permanent L-form cells, (iv) stable at −80°C for at least 1 year, and (v) reproducible. We were able to efficiently transfect morphologically diverse Myoviridae phage K and Siphoviridae SA75 with genome sizes of 148 kb and 43 kb, respectively. However, we observed increased PFU in a dose-dependent manner of DNA addition only for phage K (Fig. 3C). While we do not fully understand the reason why SA75 boots-up less than phage K when using 1,000 ng of DNA, we hypothesize that it may have to do with differences in phage development because phage K is a virulent phage while SA75 is a temperate phage. Perhaps as more temperate phage genomes enter a cell (i.e., high MOI), they form lysogens (75) or pseudolysogens. However, we have not observed the lysogenization of RN4220 by SA75.
NEST also enabled boot-up of the synthetic SA75 TAR-cloned phage genome but was unable to boot-up a cloned phage K genome. Several yeast-cloned phage K fragments and the complete genome were unable to propagate in E. coli. We were also unable to isolate pure cloned phage K from yeast that was free from yeast chromosomal DNA. The copy number of cloned phage K in yeast may also be contributing to low yield because the YCpBAC contains a yeast centromeric origin of replication rather than the 2 μm origin or replication. Because phage-purified phage K DNA can be booted-up in S. aureus, this suggests that the inability of cloned phage K to be rescued may be due to issues with toxicity in E. coli and with purification of large DNA from yeast than with size. In addition to booting-up the native and synthetic Staphylococcus phages, the NEST method demonstrated the efficient cross-genus boot-up of Enterococcus phages.
MATERIALS AND METHODS
Bacterial strains, bacteriophage, and plasmids.
S. aureus RN4220 (ST8/CC8, NR-45946) and S. epidermidis SK135 (HM-118) were obtained from BEI Resources. S. aureus strains Mu50 (ST5/CC5) and MW2 (ST1/CC1) were obtained from the JCVI PFGRC strain collection. S. aureus bacteriophage SA75 was obtained from Sangryeol Ryu, Seoul National University, Seoul, South Korea (76). E. faecalis bacteriophage DNA and host strains AH5 and AH6 were obtained from Bernd Schnabl, University of California-San Diego (UCSD), USA (61). S. aureus HER1049 (ST25/CC25) was obtained from Félix d'Hérelle Reference Center for Bacterial Virus (QC, Canada). S. aureus MRSN 7983 (ST8/CC8), pCM28 (77), and phage K were obtained from Mikeljon Nikolich, WRAIR, USA. Plasmid pGF35, pMSP3535 (Addgene plasmid number 46886) (78) with GFP inserted, was provided by Roy Stevens, Kornberg School of Dentistry, Temple University, USA. pCAS9counter was a gift from Steve Salipante (Addgene plasmid number 107192) (79). For S. aureus plasmid selection, we used chloramphenicol (12.5 μg/mL) or erythromycin (10 μg/mL) and for E. coli propagation of plasmids, ampicillin (50 μg/mL) or erythromycin (300 μg/mL) was used.
Isolation of plasmid and phage DNA.
Phage genomic DNA was purified as previously described with a few modifications (76). In brief, 500 μL of phage lysate (titer ∼109 PFU/mL) was treated at 37°C for 1 h with 125U of Benzonase (Sigma), 10 U of rDNase-I (Invitrogen), and 10 μL of RNase cocktail solution (Invitrogen) to remove residual S. aureus host DNA and RNA. Benzonase was deactivated by treating the sample with 50 μL of 0.5 M EDTA and 50 μL 0.5 M EGTA at 70°C for 10 min. Proteinase K (New England BioLabs Inc., Ipswich, MA) and SDS were added to final concentrations of 50 g/mL and 0.5% SDS was added and incubated at 56°C for 1 h followed by phenol-chloroform DNA purification. DNA purity was confirmed by restriction digestion and agarose gel electrophoresis.
Plasmids propagated in E. coli DH10B were extracted using the PureLink HiPure Plasmid Midiprep kit (Invitrogen, Vilnius, Lithuania) following the manufacturer’s recommended procedure. Plasmids from S. aureus were also extracted with the same kit with minor modifications. In brief, S. aureus was cultured in 100 mL heart infusion (HI) broth and grown until the optical density at 600 nm (OD600) was between 1.9 and 2.0. Cells were pelleted by centrifugation at 2,429 × g for 10 min at room temperature (23°C) and resuspended in 4 mL of Buffer R3 with 200 μg of Lysostaphin (Sigma-Aldrich, MO, USA) and 40 μg of lysozyme (Sigma-Aldrich, MO, USA) and incubated at 37°C for 1h. Subsequent procedures were followed as described in the manufacturer's recommended procedure.
Electroporation procedure.
S. aureus cells were made electrocompetent as described previously (10). In brief, 4 mL of an overnight-grown culture of S. aureus strain RN4220 grown in B2 broth was diluted 1/25 into to 96 mL of the same broth and incubated at 37°C with constant aeration (180 RPM) until they reached mid-log-phase of growth (OD600 ∼0.5). Cells were harvested by centrifugation at 8000 × g at room temperature (23°C) for 20 min. Cells were washed three times with an equal volume of sterile deionized water, followed by two washes with 1/5 and 1/10 volumes of 10% glycerol solution. Cells resuspended in a 10% glycerol solution were incubated at room temperature (23°C) for 15 min. Cells were centrifuged again, and the pellet was resuspended in 3.2 mL of 10% glycerol solution and aliquoted into 70 μL and frozen at −80°C.
For electroporation, 70 μL of electrocompetent cells were mixed with 10 to 1000 ng of plasmid DNA and 60 μL of this cell-DNA mix (∼9 × 108 CFU) was transferred to 0.1 cm gap electroporation cuvette (Bio-Rad, CA). The cuvette was placed into a Gene Pulser Xcell (Bio-Rad, CA) and electroporated at 2.3 kV, 100 Ω resistance with 25 μF capacitance. The cells were resuspended in 390 μL of B2 broth transferred to 15 mL tubes and incubated at 30°C for 1 h. The cells were plated on NYE media plates (80) with 12.5 μg/mL chloramphenicol for pCM28 or 10 μg/mL erythromycin for pCAS9counter and pGF35 and incubated at 30°C for 24 to 48 h.
NEST competent cell preparation.
This procedure was inspired by a method to electroporate Listeria monocytogenes (66) but modified with four key differences, including the use of HI media (Bacto Heart Infusion broth BD-238400) instead of BHI media, growth temperature (30°C instead of 37°C), cells grown to saturation rather than early log phase, and elimination of the need for electroporation. A single colony of S. aureus strain RN4220 was grown in a 50 mL conical tube containing 5 mL of HI at 37°C with aeration (225 RPM) for 8 h. This culture was subcultured in a 2 L Erlenmeyer flask containing 500 mL of HI seed media (autoclaved HI broth, pH 7.4 supplemented with 500 mM sucrose [Sigma-Aldrich S9378] and 0.2 μm filtered) at 1:1000 dilution and incubated with aeration (225 RPM) at 30°C overnight. The concentration of cells was then adjusted to an OD600 of 2.0 with fresh HI seed media. Ampicillin (Sigma-Aldrich A9618) was added to a final concentration of 10 μg/mL and incubated with aeration at 30°C for another 2 h before being chilled on ice for 10 min and harvested by centrifugation (5,500 × g, 4°C, 15 min). The cell pellet was washed in 100 mL of ice-cold washing buffer (1 mM HEPES [Sigma-Aldrich H8651], 500 mM sucrose, pH 7.0) without resuspending and centrifuged for another 10 min. This cell pellet was resuspended in 20 mL of wash buffer, transferred to a 50 mL conical tube and washed with an equal volume of ice-cold wash buffer, centrifuged again for 10 min, and gently resuspended with incremental addition of wash buffer to a final volume of 50 mL. Before the addition of lysozyme, the cells were mixed by gentle inversion five to six times. Lysozyme (Sigma-Aldrich L6876) was added to a final concentration of 10 μg/mL (47,080 units/mg) and incubated without shaking at 37°C for 20 min. To remove the lysozyme, cells were pelleted by centrifugation (3,500 × g, 4°C, 10 min) and washed with ice-cold wash buffer twice. The cells were resuspended in 2.5 mL of ice-cold resuspension solution (1 mM HEPES, 500 mM sucrose, 10% glycerol [Fisher BP-229-1], pH 7.0) using wide-bore pipette tips and frozen at −80°C in 50 μL aliquots. All subsequent manipulations of the cells were performed with wide-bore pipette tips.
NEST transformation of plasmid DNA.
A 50 μL of an aliquot of frozen competent cells was thawed on ice for 10 min. Half of the volume was transferred to a new tube and 175 μL of ice-cold resuspension solution was added. This equates to ∼9 × 108 CFU per transformation. Plasmid DNA (10 to 1000 ng) was added to the cells, mixed using wide-bore pipette tips and incubated at room temperature (i.e., 23°C) for 10 min followed by the addition of 900 μL of 20% PEG solution (20% PEG 8,000, 1 mM HEPES, 500 mM sucrose, 10% glycerol, pH 7.0). The tube was gently inverted eight to 10 times to resuspend the cells and incubated at room temperature for 20 min. Cells were pelleted in a microcentrifuge at 2,300 × g at room temperature for 5 min. The supernatant was discarded, and residual PEG was removed by pipette following quick centrifugation. The cell pellet was carefully resuspended in 1 mL of HI seed medium and incubated statically at 30°C for 30 to 60 min. The cells were plated on HI media plates with 12.5 μg/mL chloramphenicol for pCM28 or 10 μg/mL erythromycin for pCAS9counter and pGF35 and incubated at 30°C for 24 to 48 h.
Transformation efficiency.
Transformation efficiency was calculated by first determining the number of transformants (CFU for plasmids and PFU for phages) per volume plated and then multiplying by the total volume in the transformation mixture (1 mL) and any dilution factor used. This number was divided by the number of viable bacterial cells used in the transformation mixture to get the number of transformants per cell. This number was multiplied by 108 to get the number of transformants per 108 cells.
Construction of a synthetic phage SA75 genome.
Cloning of phage SA75 was performed using TAR cloning in yeast (81, 82). Oligonucleotide primers (Table S1) were designed to generate overlapping fragments to assemble the SA75 genome (i.e., SA75YA) and clone SA75 in one piece (i.e., SA75YC) into yeast using methods previously described (83, 84). PCR-positive clones were transformed into E. coli DH10B to obtain a high yield of purified plasmid DNA. Cloned phage genomes were further validated by digestion with I-SceI, which is a rare cutter that excises the cloned phage genome, and other restriction enzymes unique to the phage genome. Terminal direct repeats of 500 bp were added to the ends of the phage genome using a similar TAR cloning procedure. The final assembled genome was confirmed by PCR, restriction digests, Sanger sequencing of select regions, and Illumina sequencing.
Boot-up S. aureus and E. faecalis phages using S. aureus competent cells.
Phage DNA was transformed into S. aureus RN4220 competent cells as described above for plasmid DNA, except that following the addition of HI seed media for recovery, the cells were incubated at room temperature (i.e., 23°C) instead of 30°C for 30 min to 24 h as noted in the figure legends. The number of cells that are used in the transformation mix varied from 1.13 × 108 CFU to 9 × 108 CFU as noted in the figure legends. For S. aureus phage SA75 boot-up, we removed 200 μL of the transformation mix and plated using the top agar overlay method. For boot-up of E. faecalis phages,100 to 200 ng of DNA was used, and the transformation mix was centrifuged at 4,500 × g at room temperature for 10 min to separate released phage from the S. aureus RN4220 boot-up cells. The booted-up phages were propagated by mixing 100 μL of the supernatant with 100 μL of log-phase cultured phage propagation host strain (i.e., E. faecalis AH5 or AH6) and plated on BHI media using the top agar overlay method. The plates were incubated at 30°C for 24 to 36 h for plaque formation.
Data availability.
Newly determined plasmid sequence data have been deposited in GenBank under accession number MN956986, and sequences for the synthetic yeast-assembled SA75 phage genome have been deposited in GenBank under accession number OL638402 as well as illustrated in Fig. S3.
ACKNOWLEDGMENTS
In the conduct of research utilizing recombinant DNA, the investigators adhered to NIH guidelines for research involving recombinant DNA molecules. We thank Mikeljon Nikolich for supplying the plasmid pCM28, phage K, and S. aureus MRSN 7983. We thank Roy Stevens for providing Enterococcus faecalis JH2-2 and pGF35 plasmid. We also thank Bernd Schnabl for providing the E. faecalis bacteriophages used in this study. Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the Department of Defense.
N.A.G., R.D., R.B., A.T. were responsible for plasmid transformation experiments. R.D. was responsible for the cloning of phage genomes, generation of figures and tables. N.A.G. and R.D. were responsible for phage boot-up. N.A.G., R.D., L.M.O., A.T., S.V., and D.E.F. were responsible for the analysis and interpretation of data. R.D., L.M.O., S.V., and D.E.F. were responsible for the drafting of the manuscript; S.V. and D.E.F. were responsible for the study concept and design and editing of the manuscript. D.E.F. was responsible for study supervision.
We declare no conflict of interest.
Footnotes
Supplemental material is available online only.
Contributor Information
Derrick E. Fouts, Email: dfouts@jcvi.org.
Martha Vives, Unversidad de los Andes.
REFERENCES
- 1.Weems JJ, Jr. 2001. The many faces of Staphylococcus aureus infection. Recognizing and managing its life-threatening manifestations. Postgrad Med 110:24. 24–6, 29–31, 35–6. 10.3810/pgm.2001.10.1042. [DOI] [PubMed] [Google Scholar]
- 2.Tong SYC, Davis JS, Eichenberger E, Holland TL, Fowler VG, Jr.. 2015. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev 28:603–661. 10.1128/CMR.00134-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Centers for Disease Control and Prevention (U. S.). Antibiotic resistance threats in the United States, 2019. https://www.cdc.gov/DrugResistance/Biggest-Threats.html.
- 4.Schooley RT, Biswas B, Gill JJ, Hernandez-Morales A, Lancaster J, Lessor L, Barr JJ, Reed SL, Rohwer F, Benler S, Segall AM, Taplitz R, Smith DM, Kerr K, Kumaraswamy M, Nizet V, Lin L, McCauley MD, Strathdee SA, Benson CA, Pope RK, Leroux BM, Picel AC, Mateczun AJ, Cilwa KE, Regeimbal JM, Estrella LA, Wolfe DM, Henry MS, Quinones J, Salka S, Bishop-Lilly KA, Young R, Hamilton T. 2017. Development and use of personalized bacteriophage-based therapeutic cocktails to treat a patient with a disseminated resistant Acinetobacter baumannii infection. Antimicrob Agents Chemother 61:e00954-17. 10.1128/AAC.00954-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dedrick RM, Guerrero-Bustamante CA, Garlena RA, Russell DA, Ford K, Harris K, Gilmour KC, Soothill J, Jacobs-Sera D, Schooley RT, Hatfull GF, Spencer H. 2019. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat Med 25:730–733. 10.1038/s41591-019-0437-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pires DP, Cleto S, Sillankorva S, Azeredo J, Lu TK. 2016. Genetically engineered phages: a review of advances over the last decade. Microbiol Mol Biol Rev 80:523–543. 10.1128/MMBR.00069-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kilcher S, Loessner MJ. 2019. Engineering bacteriophages as versatile biologics. Trends Microbiol 27:355–367. 10.1016/j.tim.2018.09.006. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y, Batra H, Dong J, Chen C, Rao VB, Tao P. 2019. Genetic engineering of bacteriophages against infectious diseases. Front Microbiol 10:954. 10.3389/fmicb.2019.00954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Augustin J, Götz F. 1990. Transformation of Staphylococcus epidermidis and other staphylococcal species with plasmid DNA by electroporation. FEMS Microbiol Lett 54:203–207. 10.1016/0378-1097(90)90283-v. [DOI] [PubMed] [Google Scholar]
- 10.Schenk S, Laddaga RA. 1992. Improved method for electroporation of Staphylococcus aureus. FEMS Microbiol Lett 73:133–138. 10.1016/0378-1097(92)90596-g. [DOI] [PubMed] [Google Scholar]
- 11.Löfblom J, Kronqvist N, Uhlén M, Ståhl S, Wernérus H. 2007. Optimization of electroporation-mediated transformation: Staphylococcus carnosus as model organism. J Appl Microbiol 102:736–747. 10.1111/j.1365-2672.2006.03127.x. [DOI] [PubMed] [Google Scholar]
- 12.Kraemer GR, Iandolo JJ. 1990. High-frequency transformation of Staphylococcus aureus by electroporation. Curr Microbiol 21:373–376. 10.1007/BF02199440. [DOI] [Google Scholar]
- 13.Monk IR, Shah IM, Xu M, Tan M-W, Foster TJ. 2012. Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. mBio 3:e00277-11. 10.1128/mBio.00277-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hisatsune J, Sato'o Y, Yu L, Kutsuno S, Hayakawa Y, Sugai M. 2016. Efficient transformation of Staphylococcus aureus using multi-pulse electroporation. J Microbiol Methods 130:69–72. 10.1016/j.mimet.2016.08.012. [DOI] [PubMed] [Google Scholar]
- 15.Grosser MR, Richardson AR. 2016. Method for preparation and electroporation of S. aureus and S. epidermidis. Methods Mol Biol 1373:51–57. 10.1007/7651_2014_183. [DOI] [PubMed] [Google Scholar]
- 16.Sjöström JE, Lindberg M, Philipson L. 1972. Transfection of Staphylococcus aureus with bacteriophage deoxyribonucleic acid. J Bacteriol 109:285–291. 10.1128/jb.109.1.285-291.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lindberg M, Sjöström JE, Johansson T. 1972. Transformation of chromosomal and plasmid characters in Staphylococcus aureus. J Bacteriol 109:844–847. 10.1128/jb.109.2.844-847.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sjöström JE, Lindberg M, Philipson L. 1973. Competence for transfection in Staphylococcus aureus. J Bacteriol 113:576–585. 10.1128/jb.113.2.576-585.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rudin L, Sjöström JE, Lindberg M, Philipson L. 1974. Factors affecting competence for transformation in Staphylococcus aureus. J Bacteriol 118:155–164. 10.1128/jb.118.1.155-164.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pattee PA, Neveln DS. 1975. Transformation analysis of three linkage groups in Staphylococcus aureus. J Bacteriol 124:201–211. 10.1128/jb.124.1.201-211.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Birmingham VA, Pattee PA. 1981. Genetic transformation in Staphylococcus aureus: isolation and characterization of a competence-conferring factor from bacteriophage 80 alpha lysates. J Bacteriol 148:301–307. 10.1128/jb.148.1.301-307.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riggs HG, Jr., Rosenblum ED. 1969. Transfection of lysostaphin-treated cells of Staphylococcus aureus. J Virol 3:33–37. 10.1128/JVI.3.1.33-37.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Götz F, Ahrné S, Lindberg M. 1981. Plasmid transfer and genetic recombination by protoplast fusion in staphylococci. J Bacteriol 145:74–81. 10.1128/jb.145.1.74-81.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stahl ML, Pattee PA. 1983. Confirmation of protoplast fusion-derived linkages in Staphylococcus aureus by transformation with protoplast DNA. J Bacteriol 154:406–412. 10.1128/jb.154.1.406-412.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stahl ML, Pattee PA. 1983. Computer-assisted chromosome mapping by protoplast fusion in Staphylococcus aureus. J Bacteriol 154:395–405. 10.1128/jb.154.1.395-405.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.GöTz F, Schumacher B. 1987. Improvements of protoplast transformation in Staphylococcus carnosus. FEMS Microbiol Lett 40:285–288. 10.1111/j.1574-6968.1987.tb02040.x. [DOI] [Google Scholar]
- 27.Nieuwlandt DT, Pattee PA. 1989. Transformation of a conditionally peptidoglycan-deficient mutant of Staphylococcus aureus with plasmid DNA. J Bacteriol 171:4906–4913. 10.1128/jb.171.9.4906-4913.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morikawa K, Inose Y, Okamura H, Maruyama A, Hayashi H, Takeyasu K, Ohta T. 2003. A new staphylococcal sigma factor in the conserved gene cassette: functional significance and implication for the evolutionary processes. Genes Cells 8:699–712. 10.1046/j.1365-2443.2003.00668.x. [DOI] [PubMed] [Google Scholar]
- 29.Morikawa K, Takemura AJ, Inose Y, Tsai M, Nguyen Thi LT, Ohta T, Msadek T. 2012. Expression of a cryptic secondary sigma factor gene unveils natural competence for DNA transformation in Staphylococcus aureus. PLoS Pathog 8:e1003003. 10.1371/journal.ppat.1003003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fagerlund A, Granum PE, Håvarstein LS. 2014. Staphylococcus aureus competence genes: mapping of the SigH, ComK1 and ComK2 regulons by transcriptome sequencing. Mol Microbiol 94:557–579. 10.1111/mmi.12767. [DOI] [PubMed] [Google Scholar]
- 31.Thi LTN, Romero VM, Morikawa K. 2016. Cell wall-affecting antibiotics modulate natural transformation in SigH-expressing Staphylococcus aureus. J Antibiot (Tokyo) 69:464–466. 10.1038/ja.2015.132. [DOI] [PubMed] [Google Scholar]
- 32.Cafini F, Thuy TL, Román F, Prieto J, Dubrac S, Msadek T, Morikawa K. 2017. Methodology for the study of horizontal gene transfer in Staphylococcus aureus. J Vis Exp 10:55087. 10.3791/55087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaiser AD, Hogness DS. 1960. The transformation of Escherichia coli with deoxyribonucleic acid isolated from bacteriophage lambda-dg. J Mol Biol 2:392–415. 10.1016/s0022-2836(60)80050-2. [DOI] [PubMed] [Google Scholar]
- 34.Ando H, Lemire S, Pires DP, Lu TK. 2015. Engineering modular viral scaffolds for targeted bacterial population editing. Cell Syst 1:187–196. 10.1016/j.cels.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith HO, Hutchison CA, Pfannkoch C, Venter JC. 2003. Generating a synthetic genome by whole genome assembly: phiX174 bacteriophage from synthetic oligonucleotides. Proc Natl Acad Sci USA 100:15440–15445. 10.1073/pnas.2237126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pulkkinen EM, Hinkley TC, Nugen SR. 2019. Utilizing in vitro DNA assembly to engineer a synthetic T7 Nanoluc reporter phage for Escherichia coli detection. Integr Biol 10.1093/intbio/zyz005. [DOI] [PubMed] [Google Scholar]
- 37.Marinelli LJ, Piuri M, Swigonova Z, Balachandran A, Oldfield LM, van Kessel JC, Hatfull GF. 2008. BRED: a simple and powerful tool for constructing mutant and recombinant bacteriophage genomes. PLoS One 3:e3957. 10.1371/journal.pone.0003957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wetzel KS, Guerrero-Bustamante CA, Dedrick RM, Ko C-C, Freeman KG, Aull HG, Divens AM, Rock JM, Zack KM, Hatfull GF. 2021. CRISPY-BRED and CRISPY-BRIP: efficient bacteriophage engineering. Sci Rep 11:6796. 10.1038/s41598-021-86112-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hug H, Hausmann R, Liebeschuetz J, Ritchie DA. 1986. In vitro packaging of foreign DNA into heads of bacteriophage T1. J Gen Virol 67:333–343. 10.1099/0022-1317-67-2-333. [DOI] [PubMed] [Google Scholar]
- 40.Lee CS, Guo P. 1995. In vitro assembly of infectious virions of double-stranded DNA phage phi 29 from cloned gene products and synthetic nucleic acids. J Virol 69:5018–5023. 10.1128/JVI.69.8.5018-5023.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shibata H, Fujisawa H, Minagawa T. 1987. Characterization of the bacteriophage T3 DNA packaging reaction in vitro in a defined system. J Mol Biol 196:845–851. 10.1016/0022-2836(87)90409-8. [DOI] [PubMed] [Google Scholar]
- 42.Rao VB, Thaker V, Black LW. 1992. A phage T4 in vitro packaging system for cloning long DNA molecules. Gene 113:25–33. 10.1016/0378-1119(92)90666-d. [DOI] [PubMed] [Google Scholar]
- 43.Rosenberg SM, Stahl MM, Kobayashi I, Stahl FW. 1985. Improved in vitro packaging of coliphage lambda DNA: a one-strain system free from endogenous phage. Gene 38:165–175. 10.1016/0378-1119(85)90215-x. [DOI] [PubMed] [Google Scholar]
- 44.Rustad M, Eastlund A, Jardine P, Noireaux V. 2018. Cell-free TXTL synthesis of infectious bacteriophage T4 in a single test tube reaction. Synth Biol (Oxf) 3:ysy002. 10.1093/synbio/ysy002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reilly BE, Spizizen J. 1965. Bacteriophage dexoyribonucleate infection of competent Bacillus subtilis. J Bacteriol 89:782–790. 10.1128/jb.89.3.782-790.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Epstein HT. 1968. Factors affecting bacterial competence for transfection and transfection enhancement. Bacteriol Rev 32:313–319. 10.1128/br.32.4_pt_1.313-319.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hamoen LW, Venema G, Kuipers OP. 2003. Controlling competence in Bacillus subtilis: shared use of regulators. Microbiology (Reading) 149:9–17. 10.1099/mic.0.26003-0. [DOI] [PubMed] [Google Scholar]
- 48.Dubnau D. 1991. Genetic competence in Bacillus subtilis. Microbiol Rev 55:395–424. 10.1128/mr.55.3.395-424.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen I, Christie PJ, Dubnau D. 2005. The ins and outs of DNA transfer in bacteria. Science 310:1456–1460. 10.1126/science.1114021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berka RM, Hahn J, Albano M, Draskovic I, Persuh M, Cui X, Sloma A, Widner W, Dubnau D. 2002. Microarray analysis of the Bacillus subtilis K-state: genome-wide expression changes dependent on ComK. Mol Microbiol 43:1331–1345. 10.1046/j.1365-2958.2002.02833.x. [DOI] [PubMed] [Google Scholar]
- 51.Rahmer R, Heravi KM, Altenbuchner J. 2015. Construction of a super-competent Bacillus subtilis 168 using the PmtlA-comKS inducible cassette. Front Microbiol 6:1431. 10.3389/fmicb.2015.01431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kilcher S, Studer P, Muessner C, Klumpp J, Loessner MJ. 2018. Cross-genus rebooting of custom-made, synthetic bacteriophage genomes in L-form bacteria. Proc Natl Acad Sci USA 115:567–572. 10.1073/pnas.1714658115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gumpert J, Taubeneck U. 1983. Characteristic properties and biological significance of stable protoplast type L-forms. Experientia Suppl 46:227–241. 10.1007/978-3-0348-6776-4_27. [DOI] [PubMed] [Google Scholar]
- 54.Kawai Y, Mickiewicz K, Errington J. 2018. Lysozyme counteracts β-Lactam antibiotics by promoting the emergence of L-form bacteria. Cell 172:1038–1049.e10. 10.1016/j.cell.2018.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klebe RJ, Harriss JV, Sharp ZD, Douglas MG. 1983. A general method for polyethylene-glycol-induced genetic transformation of bacteria and yeast. Gene 25:333–341. 10.1016/0378-1119(83)90238-x. [DOI] [PubMed] [Google Scholar]
- 56.Chen P, Liu H-H, Cui R, Zhang Z-L, Pang D-W, Xie Z-X, Zheng H-Z, Lu Z-X, Tong H. 2008. Visualized investigation of yeast transformation induced with Li and polyethylene glycol. Talanta 77:262–268. 10.1016/j.talanta.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 57.Sadykov MR. 2016. Restriction-modification systems as a barrier for genetic manipulation of Staphylococcus aureus. Methods Mol Biol 1373:9–23. 10.1007/7651_2014_180. [DOI] [PubMed] [Google Scholar]
- 58.Roberts GA, Houston PJ, White JH, Chen K, Stephanou AS, Cooper LP, Dryden DTF, Lindsay JA. 2013. Impact of target site distribution for Type I restriction enzymes on the evolution of methicillin-resistant Staphylococcus aureus (MRSA) populations. Nucleic Acids Res 41:7472–7484. 10.1093/nar/gkt535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, Bayles KW. 2013. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio 4:e00537–12–e00512. 10.1128/mBio.00537-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.El Haddad L, Ben Abdallah N, Plante P-L, Dumaresq J, Katsarava R, Labrie S, Corbeil J, St-Gelais D, Moineau S. 2014. Improving the safety of Staphylococcus aureus polyvalent phages by their production on a Staphylococcus xylosus strain. PLoS One 9:e102600. 10.1371/journal.pone.0102600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Duan Y, Llorente C, Lang S, Brandl K, Chu H, Jiang L, White RC, Clarke TH, Nguyen K, Torralba M, Shao Y, Liu J, Hernandez-Morales A, Lessor L, Rahman IR, Miyamoto Y, Ly M, Gao B, Sun W, Kiesel R, Hutmacher F, Lee S, Ventura-Cots M, Bosques-Padilla F, Verna EC, Abraldes JG, Brown RS, Jr., Vargas V, Altamirano J, Caballería J, Shawcross DL, Ho SB, Louvet A, Lucey MR, Mathurin P, Garcia-Tsao G, Bataller R, Tu XM, Eckmann L, van der Donk WA, Young R, Lawley TD, Stärkel P, Pride D, Fouts DE, Schnabl B. 2019. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 575:505–511. 10.1038/s41586-019-1742-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sheng Y, Mancino V, Birren B. 1995. Transformation of Escherichia coli with large DNA molecules by electroporation. Nucleic Acids Res 23:1990–1996. 10.1093/nar/23.11.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Briers Y, Staubli T, Schmid MC, Wagner M, Schuppler M, Loessner MJ. 2012. Intracellular vesicles as reproduction elements in cell wall-deficient L-form bacteria. PLoS One 7:e38514. 10.1371/journal.pone.0038514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Studer P, Borisova M, Schneider A, Ayala JA, Mayer C, Schuppler M, Loessner MJ, Briers Y. 2016. The absence of a mature cell wall sacculus in stable Listeria monocytogenes L-form cells is independent of peptidoglycan synthesis. PLoS One 11:e0154925. 10.1371/journal.pone.0154925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Studer P, Staubli T, Wieser N, Wolf P, Schuppler M, Loessner MJ. 2016. Proliferation of Listeria monocytogenes L-form cells by formation of internal and external vesicles. Nat Commun 7:13631. 10.1038/ncomms13631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Monk IR, Gahan CGM, Hill C. 2008. Tools for functional postgenomic analysis of Listeria monocytogenes. Appl Environ Microbiol 74:3921–3934. 10.1128/AEM.00314-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Monk IR, Tree JJ, Howden BP, Stinear TP, Foster TJ. 2015. Complete bypass of restriction systems for major Staphylococcus aureus lineages. mBio 6:e00308–15–e00315. 10.1128/mBio.00308-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mell JC, Redfield RJ. 2014. Natural competence and the evolution of DNA uptake specificity. J Bacteriol 196:1471–1483. 10.1128/JB.01293-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fouts DE. 2006. Phage_Finder: automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res 34:5839–5851. 10.1093/nar/gkl732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kreiswirth BN, Löfdahl S, Betley MJ, O'Reilly M, Schlievert PM, Bergdoll MS, Novick RP. 1983. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305:709–712. 10.1038/305709a0. [DOI] [PubMed] [Google Scholar]
- 71.Nair D, Memmi G, Hernandez D, Bard J, Beaume M, Gill S, Francois P, Cheung AL. 2011. Whole-genome sequencing of Staphylococcus aureus strain RN4220, a key laboratory strain used in virulence research, identifies mutations that affect not only virulence factors but also the fitness of the strain. J Bacteriol 193:2332–2335. 10.1128/JB.00027-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Waldron DE, Lindsay JA. 2006. Sau1: a novel lineage-specific type I restriction-modification system that blocks horizontal gene transfer into Staphylococcus aureus and between S. aureus isolates of different lineages. J Bacteriol 188:5578–5585. 10.1128/JB.00418-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Corvaglia AR, François P, Hernandez D, Perron K, Linder P, Schrenzel J. 2010. A type III-like restriction endonuclease functions as a major barrier to horizontal gene transfer in clinical Staphylococcus aureus strains. Proc Natl Acad Sci USA 107:11954–11958. 10.1073/pnas.1000489107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu S-Y, Corvaglia AR, Chan S-H, Zheng Y, Linder P. 2011. A type IV modification-dependent restriction enzyme SauUSI from Staphylococcus aureus subsp. aureus USA300. Nucleic Acids Res 39:5597–5610. 10.1093/nar/gkr098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Golding I, Coleman S, Nguyen VP, Yao T. 2021. Decision making by temperate phages, p 88–97. In Bamford DH, Zuckerman M (ed), Encyclopedia of Virology (Fourth Edition). Academic Press. [Google Scholar]
- 76.D’Souza R, White RC, Buzzeo R, Goglin K, Vashee S, Lee Y, Son B, Ryu S, Fouts DE. 2020. Complete genome sequence of Staphylococcus aureus phage SA75, isolated from goat feces. Microbiol Resour Announc 9:e00114-20. 10.1128/MRA.00114-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pang YY, Schwartz J, Thoendel M, Ackermann LW, Horswill AR, Nauseef WM. 2010. agr-dependent interactions of Staphylococcus aureus USA300 with human polymorphonuclear neutrophils. J Innate Immun 2:546–559. 10.1159/000319855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bryan EM, Bae T, Kleerebezem M, Dunny GM. 2000. Improved vectors for nisin-controlled expression in gram-positive bacteria. Plasmid 44:183–190. 10.1006/plas.2000.1484. [DOI] [PubMed] [Google Scholar]
- 79.Penewit K, Holmes EA, McLean K, Ren M, Waalkes A, Salipante SJ. 2018. Efficient and scalable precision genome editing in Staphylococcus aureus through conditional recombineering and CRISPR/Cas9-mediated counterselection. mBio 9:e00067-18. 10.1128/mBio.01839-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Babich K, Engle M, Skinner JS, Laddaga RA. 1991. Deletion mutant analysis of the Staphylococcus aureus plasmid pI258 mercury-resistance determinant. Can J Microbiol 37:624–631. 10.1139/m91-106. [DOI] [PubMed] [Google Scholar]
- 81.Larionov V, Kouprina N, Solomon G, Barrett JC, Resnick MA. 1997. Direct isolation of human BRCA2 gene by transformation-associated recombination in yeast. Proc Natl Acad Sci USA 94:7384–7387. 10.1073/pnas.94.14.7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ketner G, Spencer F, Tugendreich S, Connelly C, Hieter P. 1994. Efficient manipulation of the human adenovirus genome as an infectious yeast artificial chromosome clone. Proc Natl Acad Sci USA 91:6186–6190. 10.1073/pnas.91.13.6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Oldfield LM, Grzesik P, Voorhies AA, Alperovich N, MacMath D, Najera CD, Chandra DS, Prasad S, Noskov VN, Montague MG, Friedman RM, Desai PJ, Vashee S. 2017. Genome-wide engineering of an infectious clone of herpes simplex virus type 1 using synthetic genomics assembly methods. Proc Natl Acad Sci USA 114:E8885–E8894. 10.1073/pnas.1700534114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vashee S, Stockwell TB, Alperovich N, Denisova EA, Gibson DG, Cady KC, Miller K, Kannan K, Malouli D, Crawford LB, Voorhies AA, Bruening E, Caposio P, Früh K. 2017. Cloning, assembly, and modification of the primary Human Cytomegalovirus isolate Toledo by yeast-based transformation-associated recombination. mSphere 2:e00331-17. 10.1128/mSphereDirect.00331-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gros MF, Te Riele H, Ehrlich SD. 1987. Rolling circle replication of single-stranded DNA plasmid pC194. EMBO J 6:3863–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S3, Table S1. Download aem.01486-21-s0001.pdf, PDF file, 9.8 MB (10.1MB, pdf)
Data Availability Statement
Newly determined plasmid sequence data have been deposited in GenBank under accession number MN956986, and sequences for the synthetic yeast-assembled SA75 phage genome have been deposited in GenBank under accession number OL638402 as well as illustrated in Fig. S3.