

Abstract
Background
Melioidosis is a potentially fatal infectious disease caused by Burkholderia pseudomallei and the disease is endemic in Southeast Asia and Northern Australia. It has been confirmed as endemic in Sri Lanka. Genomic epidemiology of B. pseudomallei in Sri Lanka is largely unexplored. This study aims to determine the biogeography and genetic diversity of clinical isolates of B. pseudomallei and the phylogenetic and evolutionary relationship of Sri Lankan sequence types (STs) to those found in other endemic regions of Southeast Asia and Oceania.
Methods
The distribution of variably present genetic markers [Burkholderia intracellular motility A (bimA) gene variants bimABP/bimABM, filamentous hemagglutinin 3 (fhaB3), Yersinia-like fimbrial (YLF) and B. thailandensis-like flagellum and chemotaxis (BTFC) gene clusters and lipopolysaccharide O-antigen type A (LPS type A)] was examined among 310 strains. Multilocus sequence typing (MLST) was done for 84 clinical isolates. The phylogenetic and evolutionary relationship of Sri Lankan STs within Sri Lanka and in relation to those found in other endemic regions of Southeast Asia and Oceania were studied using e BURST, PHYLOViZ and minimum evolutionary analysis.
Results
The Sri Lankan B. pseudomallei population contained a large proportion of the rare BTFC clade (14.5%) and bimABM allele variant (18.5%) with differential geographic distribution. Genotypes fhaB3 and LPSA were found in 80% and 86% respectively. This study reported 43 STs (including 22 novel). e-BURST analysis which include all Sri Lankan STs (71) resulted in four groups, with a large clonal group (group 1) having 46 STs, and 17 singletons. ST1137 was the commonest ST. Several STs were shared with India, Bangladesh and Cambodia.
Conclusion
This study demonstrates the usefulness of high-resolution molecular typing to locate isolates within the broad geographical boundaries of B. pseudomallei at a global level and reveals that Sri Lankan isolates are intermediate between Southeast Asia and Oceania.
Author summary
Burkholderia pseudomallei is an important cause of community acquired pneumonia, septicemia and abscesses in Sri Lanka. The risk of infection is increased after flooding following heavy rainfall. Risk groups include rice farmers and rural populations engaged in subsistence cultivation in home gardens. Nationwide surveillance has been carried out since 2006 and the state public health system offers free diagnostics and free antibiotic therapy. The incidence of melioidosis in Sri Lanka has increased in tandem with increased awareness among clinicians. This study reports the genetic diversity among Sri Lankan B. pseudomallei clinical isolates and shows that some variably present genes are regionally distributed. The population is intermediate between Southeast Asia and Oceania. This may reflect its past geological history.
Introduction
Burkholderia pseudomallei is a soil-dwelling, Gram-negative, saprophytic environmental bacterium that causes the potentially fatal infectious disease, melioidosis [1–3]. The disease is considered an emerging threat throughout tropical and subtropical regions globally and is endemic in Southeast Asia and Northern Australia [2,4]. The bacterium can be found in soil and fresh water bodies in endemic regions and infection is acquired via percutaneous inoculation, inhalation or ingestion [1,2,4]. Melioidosis has recently been confirmed as endemic in Sri Lanka [5] and the risk is increased during heavy rains [6]. Agricultural workers have been identified as vulnerable because of frequent exposure to soil and water [1,5]. Clinical manifestations of melioidosis are greatly diverse regardless of the route of infection, from localized abscesses to severe pneumonia or life-threatening sepsis, and occasionally neurological melioidosis [1,7,8]. The reported case fatality rates are up to 40% and relapse rates are ∼20% in certain settings [1,9,10]. Successful antibiotic treatment has proven to be crucial for favorable outcomes in humans. There are no vaccines currently available. Therefore, the organism was upgraded to Tier 1 Select Agent characterization by US Centers for Disease Control and Prevention in 2012 [11,12]. A request has been submitted to make it a notifiable disease in Sri Lanka.
A broad strain diversity has been reported from endemic regions such as northern Australia and Southeast Asia [2,9]. Multilocus sequence typing (MLST), which is based on sequence variation in seven housekeeping genes, has been successfully employed as an epidemiological tool to characterize the genetic diversity of B. pseudomallei within and across endemic regions [13,2]. e-BURST analysis (based upon sequence typing) has shown the presence of discrete clonal complexes which are specific to each endemic region indicating population groupings [3,14,15]. However, shared sequence types (STs) and single locus variants (SLVs) are widely distributed in India, Australia, Thailand and China [2,16,17].
A small number of Sri Lankan clinical strains of B. pseudomallei has been characterized so far using MLST and whole genome sequence analysis [5,15,18]. However, more work needs to be done to explore the biogeography and genetic diversity of B. pseudomallei in Sri Lanka. Therefore, the main aim of the present study was to determine genetic diversity among Sri Lankan clinical isolates and their regional distribution using MLST and real-time polymerase chain reaction (RT-PCR) based genotyping methods. The distribution of variably present epidemiological markers [Burkholderia intracellular motility A (bimA) gene variants bimABP/bimABM, filamentous hemagglutinin 3 (fhaB3), Yersinia-like fimbrial (YLF) gene cluster and B. thailandensis-like flagellum and chemotaxis (BTFC) gene cluster and lipopolysaccharide O-antigen type A (LPSA)] was examined among 310 clinical isolates while MLST was done for 84 and the STs of all isolates, including those previously reported from, Sri Lanka, were subjected to e-BURST analysis. The phylogenetic and evolutionary relationship of Sri Lankan STs within Sri Lanka and in relation to those found in other endemic regions of Southeast Asia and Oceania was studied.
Methods
Ethics statement
Ethics approval was obtained from the Ethics Review Committee, Faculty of Medicine, University of Colombo, Sri Lanka (EC-17-020). Written or verbal consent was not obtained in this study as all bacterial isolates and other information were used anonymously.
Clinical isolates
A total of 310 consecutive clinical isolates of B. pseudomallei collected from melioidosis cases (one isolate per case) across the country from 2006 to early 2018 were included in this study. All isolates were confirmed as B. pseudomallei by latex agglutination and lpxo RT-PCR assays [19,20]. Patient demographic and geographic data including age, sex, address, occupation, clinical presentation, underlying risk factors and outcome were recorded. MLST analysis was limited to 84 strains due to financial constraints (MLST for 109 isolates was already available in the B. pseudomallei MLST database).
Detection of variably present gene markers
Cryo-preserved bacterial cultures were grown overnight in blood agar medium at 35°C. Total genomic DNA was extracted from pure cultures using a commercially available bacterial genomic DNA extraction kit as per the manufacturer’s instructions (CEYGEN Biotech, Sri Lanka). The extracted DNA from each isolate was stored at -20°C until use. Optimized multiplex RT-PCR assays with gene specific oligonucleotides primers were performed to detect YLF and BTFC gene clusters and variably present genes fhab3, bimABp/bimABm and LPSA [21–23]. BRYT green qRT-PCR master mixture (Promega, USA) was used in this study. Known positive DNA extracted from B. pseudomallei clinical isolates previously confirmed by whole genome sequencing were used as positive controls [24].
MLST of 84 clinical isolates of B. pseudomallei
Amplification of housekeeping genes
Amplification of seven housekeeping genes (ace-gltB-gmhD-lepA-lipA-narK-ndh) was performed using established oligonucleotide primer sequences as reported previously [13], http://pubmlst.org/bpseudomallei.mlst.net/. Five microliters of PCR products were analyzed via 1.5% agarose gel electrophoresis to check for sufficient product, correct size and product purity.
Purification of amplicons and sequencing
PCR products were submitted for amplicon purification and SANGER sequencing to Macrogen Inc., Seoul, South Korea. Each DNA fragment was sequenced in forward and reverse directions using the same oligonucleotide primers that were used for the initial PCR amplification.
Sequence analysis and data submission to MLST database
For the sequence analysis, the forward and reverse sequences were aligned with a reference allele sequence obtained from the B. pseudomallei MLST website (http://pubmlst.org/bpseudomallei.mlst.net/) using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/). The sequences were edited by trimming to the appropriate size for each locus. The batch sequence (7 loci) from each isolate was queried in the MLST database to determine the allelic profile. The allelic profile of each isolate was queried for a match to the existing sequence types on the MLST database. The allele profile data for novel STs were submitted to the curator for confirmation and assignment of allelic numbers and STs. All isolate data and STs published in this study can be found at the B. pseudomallei MLST database (http://bpseudomallei.mlst.net/).
Multilocus sequence analysis (MLSA) was performed using e-BURST with SLV selected. A total of 193 isolate records with 71 STs reported from Sri Lanka (84 from this study and 109 submitted previously) was used for the e-BURST analysis and the criterion of six shared alleles out of seven alleles was used to delineate the clonal complexes [25]. Further, PHYLOVIZ 2.0 [26]. available at PubMLST (http://pubmlst.org/bpseudomallei.mlst.net/) was used to reveal the genetic relationship of STs within Sri Lanka and in relation to those found in Australia, India, Thailand, China, Cambodia, Malaysia and Vietnam.
Construction of minimum-evolutionary tree (MET)
A minimum -evolutionary tree was constructed from the concatenated sequences (3401 positions) in the order of loci ace-gltB-gmhD-lepA-lipA-narK-ndh for Sri Lankan STs and those STs that showed SLVs and double locus variants (DLVs) to Sri Lankan STs. The evolutionary history was inferred using the Maximum Likelihood method [27]. The bootstrap method was used to test the phylogeny. The evolutionary distances were computed using the Tamura and Nei Model. The MET was searched using the Close-Neighbor-Interchange (CNI) algorithm [28] at a search level of one. The Neighbor-joining algorithm was used to generate the initial tree [29]. Evolutionary analyses were conducted in MEGA X [30]. Measure of genetic differentiation (FST) was calculated for the Sri Lankan population using DNaSP Version: 6.12 [31].
Association of bimABM genotype with neurological melioidosis
Association of bimABM and neurological melioidosis was determined using the Fisher’s exact test in GraphPad Prism 8.4.3. Neurological syndromes such as meningitis, encephalitis and myelitis were considered as neurological melioidosis but cerebral abscesses were excluded.
Results
Melioidosis cases reported across Sri Lanka during 2006 to early 2018 (n = 310) comprised 215 (69%) males and 95 (31%) females, spanning an age range of 11 months to 98 years with a median of 51 years. Abscesses (with or without sepsis) were the most common clinical presentation with 48% (n = 150) of cases followed by pneumonia (29%, n = 89) and septic arthritis (15%, n = 47) (S1 Table). The case fatality rate was 24%, n = 75).
Eighty percent (n = 248) were from rural settings. Main occupational groups included housewives (16%, n = 50) and rice farmers (15%, n = 45). The commonest underlying risk factor was diabetes (64%, n = 199). Percentage distribution of STs (n = 193) is shown Fig 1. Culture positive melioidosis cases per 100,000 population for each of the nine provinces is shown in Table 1.
Fig 1. Percentage distribution of commonest STs.
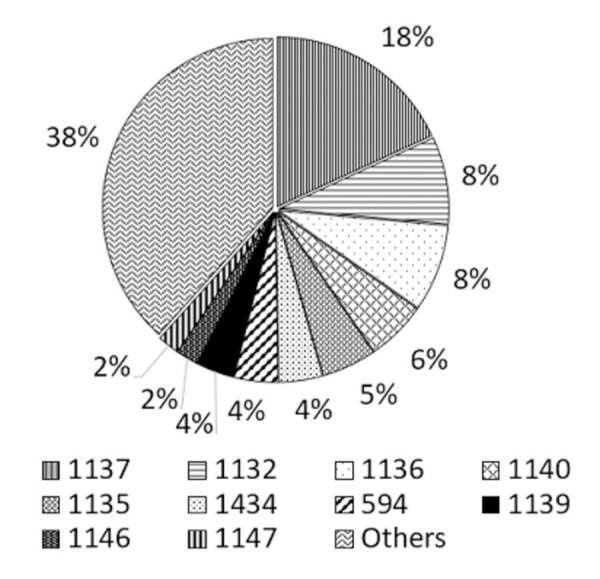
Table 1. Culture positive melioidosis cases per 100,000 population for each of the nine provinces.
| Province | No. of cases | Population (2012) | Cases/100,000 population |
|---|---|---|---|
| WP | 103 | 6,165,000 | 1.67 |
| NWP | 58 | 2,380,861 | 2.44 |
| EP | 47 | 1,555,510 | 3.02 |
| SP | 41 | 2,477,285 | 1.66 |
| CP | 16 | 2,571,557 | 0.62 |
| NCP | 9 | 1,266,663 | 0.71 |
| UVA | 8 | 1,266,463 | 0.63 |
| SG | 8 | 1,928,655 | 0.41 |
| NP | 12 | 1,061,315 | 1.13 |
WP-Western Province, SP-Southern Province
EP-Eastern Province, NP-Northern Province, NWP-North Western Province, SGP-Sabaragamuwa Province, CP-Central Province, NCP-North Central Province.
Genotyping and analyses were performed as shown in Fig 2.
Fig 2. Flowchart demonstrating genotyping and analyses of clinical isolates.

Distribution of epidemiology markers YLF/BTFC, bimA, fhaB3 and LPSA
Genotyping of clinical isolates (n = 310) showed that the majority of Sri Lankan isolates belong to the YLF group (85.5%, n = 265). However, about 14.5% (n = 45) of BTFC positive isolates was found. Further, this study examined the regional distribution of YLF/BTFC gene clusters within the country (Fig 3A and S2 Table). It was noted that the relative frequency of the BTFC group was found to vary among regional populations. Sri Lanka is administratively divided into 9 provinces and, while isolates with YLF gene cluster were seen in all provinces, isolates with BTFC gene cluster were predominantly present in the Eastern Province (36.17%, 17/47) (Fig 3A).
Fig 3.

A. Distribution of genotypes of Burkholderia pseudomallei in 9 provinces in Sri Lanka. A. Yersinia-like fimbrial (YLF) and B. thailandensis -like flagellum and chemotaxis (BTFC) gene clusters, B. Burkholderia intracellular motility factor A (bimABM bimABp), C. Filamentous hemagglutinin 3 (fhaB3). WP-Western Province, SP-Southern Province, Eastern Province, NP-Northern Province, NWP-North Western Province, SGP-Sabaragamuwa Province, CP-Central Province, NCP-North Central Province.
The rare allele variant bimABM was found in 18.5% (57/308) of the Sri Lankan B. pseudomallei population (S3 Table). Regional variation in its distribution was found with a predominance in the Northern, North Central and Eastern Provinces (Fig 3B). Among these, 19 isolates (33%) were in the BTFC clade.
As the literature has shown an association between neurological melioidosis and bimABM, we explored this phenomenon in our population. Five of eleven (45.45%) neurological melioidosis cases were caused by isolates with genotype bimABM although this genotype consisted of only 57 of 308 (18.5%) available genotypes. A Fisher’s exact Test of independence showed that there was a significant association between bimABM genotype and neurological melioidosis with (1, n = 308) = p = 0.0345).
The majority (80%, 238/298) of Sri Lankan clinical B. pseudomallei isolates possessed the fhaB3 variant. However, there was no regional preference in its distribution within the country (Fig 3C). Majority of Sri Lankan B. pseudomallei was found to carry the LPSA gene cluster (86%, 252/294). The four genotypes were randomly distributed among the strains showing no specific associations.
MLST analysis
MLST analysis of 84 clinical isolates resulted in 43 STs, of which 22 were novel: ST 1880–85, ST1887-95, ST1898, ST1900, ST1928-30, ST1933-34 (S4 Table). Two distinct alleles ace 55, and ndh 124 were found to be unique to Sri Lanka and are shown in bold numbers (Table 2). Considering the total Sri Lankan isolates in the database (a total of 193 records with 71 STs), the dominant alleles were ace-1, gltB-2, gmhD-6, lepA-2, lipA-1, narK-2 and ndh-3 (Table 2). Alleles gltB and ndh were the most variable with 7 variants each while ace had only 5 variants. ST1137 was found to be the most abundant ST (n = 35 isolates) followed by ST1136 (n = 16) and ST1132 (n = 16). When regional distribution is considered, ST1132 concentrated in the NWP (62.5%, 10/16) while ST1137 concentrated in the WP (60%, 21/35) (S5–S7 Tables).
Table 2. Prevalence of alleles in the Sri Lankan population (n = 193) of Burkholderia pseudomallei.
| Locus | Allele number and prevalence (%) |
|---|---|
| ace | 1 (76.5), 4 (18.4), 8 (3.6), 18 (1.0), 55 (0.5) |
| gltB | 2 (55.6), 4 (22.4), 12 (18.4), 1 (1.5), 5 (0.5), 6 (0.5), 16 (0.5) |
| gmhD | 6 (64.3), 3 (19.4), 14 (9.2), 10 (3.1), 13 (3.1), 124 (0.5) |
| lepA | 2 (74.5), 4 (21.9), 3 (1.0), 1 (1.0), 46 (0.5), 19 (0.5), |
| lipA | 1 (86.2), 5 (7.1), 20 (3.1), 3 (2.0), 25 (1.0), 6 (1.0) |
| narK | 2 (41.8), 1 (30.6), 8 (24.0), 42 (1.5), 60 (1.5), 21 (0.5) |
| ndh | 3 (55.1), 1 (23.0), 57 (15.8), 20 (2.6), 3 (1.5), 11 (1.0), 87 (0.5), |
Genetic relatedness among STs from Sri Lanka
e-BURST analysis revealed four groups with a large clonal group (Group1) having 46 STs distributed among eight sub-group founders (Fig 4). Group 2, 3 and 4 comprised four, two and two STs respectively. Seventeen (17) singletons (outliers) were also seen (Table 3). According to the current data, ST1132 was the predicted founder which has 16 clinical isolates reported so far (S4 Table). Single locus variants were ST293, ST501, ST1134, ST1146, ST1147, ST1880 and double locus variant ST194, ST912, ST1882, ST1898.
Fig 4. e-BURST tree showing the hypothetical pattern of descent for clinical isolates of Burkholderia pseudomallei in Sri Lanka.

Red shade depicts single locus variants (SLVs) while blue shade depicts double locus variants (DLVs).
Table 3. Description of the study isolates based on eBURST analysis.
| Group1 | ||||
| Sequence type | Frequency | Single locus variant | Double locus variant | Satellites (more distantly related isolates) |
| 13 | 2 | 2 | 1 | 42 |
| 194 | 1 | 3 | 7 | 35 |
| 293 | 1 | 4 | 15 | 26 |
| 474 | 1 | 1 | 2 | 42 |
| 501 | 1 | 5 | 10 | 30 |
| 590 | 2 | 1 | 4 | 40 |
| 594 | 8 | 4 | 8 | 33 |
| 598 | 1 | 2 | 2 | 41 |
| 615 | 1 | 1 | 4 | 40 |
| 655 | 1 | 5 | 14 | 26 |
| 733 | 1 | 1 | 4 | 40 |
| 912 | 2 | 1 | 4 | 40 |
| 1132* | 16 | 7 | 10 | 28 |
| 1134 | 1 | 3 | 8 | 34 |
| 1135 | 10 | 4 | 3 | 38 |
| 1136 | 16 | 3 | 5 | 37 |
| 1137 | 35 | 5 | 10 | 30 |
| 1138 | 1 | 2 | 6 | 37 |
| 1139 | 7 | 1 | 3 | 41 |
| 1140 | 12 | 4 | 5 | 36 |
| 1143 | 3 | 4 | 8 | 33 |
| 1145 | 1 | 5 | 12 | 28 |
| 1146 | 4 | 1 | 8 | 36 |
| 1147 | 4 | 6 | 9 | 30 |
| 1148 | 1 | 1 | 2 | 42 |
| 1152 | 2 | 2 | 7 | 36 |
| 1364 | 2 | 6 | 6 | 33 |
| 1413 | 1 | 2 | 5 | 38 |
| 1435 | 2 | 3 | 7 | 35 |
| 1437 | 1 | 2 | 10 | 33 |
| 1438 | 1 | 1 | 4 | 40 |
| 1439 | 2 | 1 | 3 | 41 |
| 1442 | 1 | 3 | 4 | 38 |
| 1692 | 1 | 5 | 9 | 31 |
| 1880 | 1 | 3 | 9 | 33 |
| 1882 | 1 | 2 | 7 | 36 |
| 1884 | 1 | 4 | 8 | 33 |
| 1885 | 1 | 2 | 6 | 37 |
| 1889 | 2 | 2 | 8 | 35 |
| 1891 | 1 | 2 | 2 | 41 |
| 1892 | 1 | 2 | 5 | 38 |
| 1894 | 1 | 1 | 7 | 37 |
| 1898 | 1 | 2 | 6 | 37 |
| 1928 | 1 | 1 | 1 | 43 |
| 1929 | 1 | 2 | 3 | 40 |
| 1933 | 1 | 2 | 7 | 36 |
| Group 2 | ||||
| 867 | 1 | 2 | 1 | |
| 1179 | 1 | 1 | 2 | |
| 1888 | 1 | 1 | 2 | |
| 1934 | 1 | 2 | 1 | |
| Group 3 | ||||
| 1434 | 8 | 1 | 0 | |
| 1436 | 1 | 1 | 0 | |
| Group 4 | ||||
| 1893 | 1 | 1 | 0 | |
| 1900 | 1 | 1 | 0 | |
| Singletons: 132, 202, 308, 338, 421, 944, 1133, 1141, 1142,1144, 1314,1881,1883, 1887,1890,1895,1930 | ||||
Shared STs observed were ST202, ST594 (Australia, Thailand), ST912 (Cambodia), ST501 (India, Thailand), ST912 (Cambodia and India), ST1692, ST293, ST1143, ST1152 (India), ST13, ST655, ST308 (Thailand), ST132 (Australia) and ST421 (Belgium). The shared STs ST501 and ST293 are SLVs of ST1132. The shared ST13 formed a subgroup founder with two SLVs ST1413 and ST1148 while shared ST655 was the subgroup founder of SLV ST1145 and was distantly related to ST1132 and the commonest ST1137. The shared ST1143 was found as a subgroup founder with shared ST1152 and the novel ST1891 (Fig 4).
Phylogenetic relatedness of Sri Lankan STs with a global collection
PHLOVIZ analysis showed that two distinct clades are formed by B. pseudomallei from Oceania and Southeast Asia. Sri Lankan STs (shaded in beige color) fall into four groups; A, B, C and D (Fig 5). Majority of Sri Lankan STs were clustered in Group A, B and C. Group A and D clustered with STs from Southeast Asia while group B and C clustered with STs from Oceania. Therefore, the phylogenetic relationship of Sri Lankan STs shows that they are intermediate to Australian and Southeast Asian B. pseudomallei populations.
Fig 5.

PHYLOVIZ analysis showing the genetic relationship among global collection of sequence types (STs) of Burkholderia pseudomallei reported from Thailand (ash), Malaysia (orange), China (light orange), Sri Lanka (dark green), India (light green), Cambodia (red) and Vietnam (pink). Sri Lankan STs (shaded in beige) clustered in four groups (A, B, C and D). Group A and D clustered with STs from Southeast Asia, group B and C clustered with STs from Oceania.
To ascertain the evolutionary relationship of Sri Lankan STs, Maximum Likelihood evolutionary tree (ME) was constructed using concatenated sequences of all seven housekeeping loci (Fig 6). B. pseudomallei MSR0688 was used as an outgroup. In ME analysis, the phylogenetic relatedness of all 17 singletons to the other Sri Lankan STs, which was not resolved by the e-BURST algorithm, was observed. For example, singletons ST1883 and ST1141 clustered together and singleton ST1887 clustered with Sri Lankan Group 4 ST1900. However, some singletons clustered with STs from other endemic regions such as singleton ST293 which clustered with the Indian ST1373. The singleton ST1133 was found to be phylogenetically close to Cambodian ST916. Further, singletons ST 308, ST1141, ST1881, ST1883 and Indian ST1555 formed a separate cluster in evolutionary analysis suggesting a common evolutionary linkage among those isolates. Furthermore, the shared ST202 (Sri Lanka and Thailand) was phylogenetically close to the shared ST1152 (Sri Lanka and India) and Indian ST1518 forming a single cluster. Several novel STs (ST1890, ST1881, ST1930) were found to have phylogenetic relatedness with STs from Australia and India (marked with parenthesis) and may represent recent importation. The measure of genetic differentiation (FST) for the Sri Lankan population was found to be 0.00139.
Fig 6. Minimum spanning tree showing the evolutionary relationship of 107 known STs including all 71 STs reported from Sri Lanka inferred using the Maximum likelihood method.
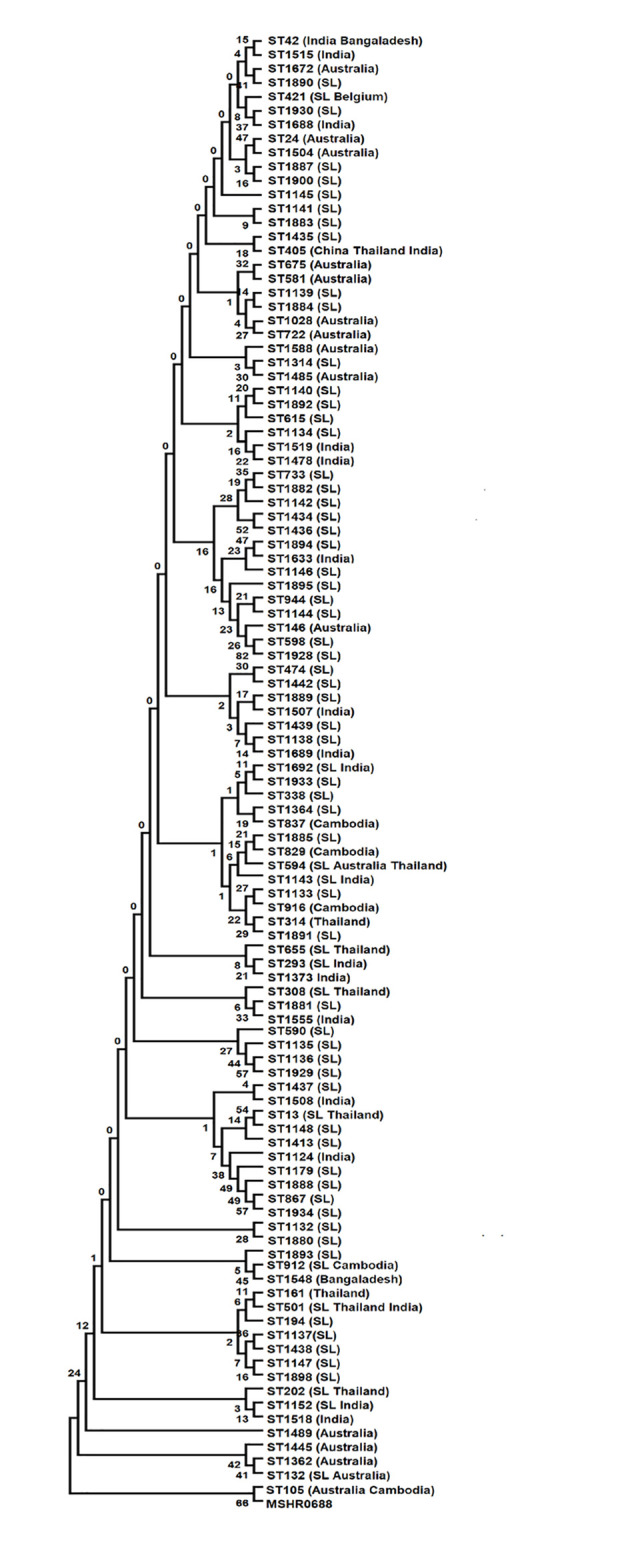
Tree constructed from the concatenated sequences of the seven MLST loci. The optimal tree with the sum of branch length = 0.0378947.
Discussion
B. pseudomallei is an important cause of community acquired pneumonia and septicemia in endemic countries. The risk of infection is increased after flooding and a case cluster was reported in eastern Sri Lanka in 2015 following heavy rainfall [24]. Risk groups in Sri Lanka include rice farmers and rural populations engaged in subsistence cultivation in home gardens. Nationwide surveillance has been carried out since 2006 and the state public health system offers free diagnostics and free antibiotic therapy. The incidence of melioidosis in Sri Lanka has increased in tandem with increased awareness among clinicians [5].
MLST is a widely used epidemiological tool to characterize the genetic diversity of B. pseudomallei within and across endemic regions [2,13,32]. The data generated using MLST can be further analyzed using phylogenetic tools such as e-BURST and PHYLOVIZ to elucidate the genetic relatedness of B. pseudomallei from distinct locations [26]. ME analysis based on concatenated nucleotide sequences of housekeeping loci may be used to further resolve the genetic relationship of B. pseudomallei strains. In addition, RT-PCR to detect variably present genetic markers such as YLF/BTFC gene clusters, bimABM/bimABP, fhaB3 and LPSA in B. pseudomallei have been used to elucidate strain origin, genetic relatedness and virulence [33,7]. Therefore, genotyping of these four selected genetic markers, along with MLST, may provide improved resolution of genetic diversity among B. pseudomallei across the country.
The YLF/BTFC gene clusters are mutually exclusive in the B. pseudomallei genome and is highly correlated with specific geographic regions with BTFC strains rarely found in Southeast Asian countries [21]. YLF gene cluster is known to be prevalent in Southeast Asia including Thailand (98%) and is uncommon in Australia (12%). Whereas BTFC is the most frequent genotype reported in Australia (79–88%) and is rarely found in Southeast Asia including Thailand (2%) [33]. Results of RT-PCR based genotyping showed that approximately 15% of Sri Lankan B. pseudomallei strains were BTFC. Therefore, it seems that the population in Sri Lanka comprises a mixed population of strains, intermediate between Laurasia and Oceania. BTFC clade is predominantly present in the Eastern Province (36.17% of local isolates). The reason for this regional distribution is poorly understood but it is interesting to note that the geological formation of this area (Eastern Province, 7.7853° N, 81.4279° E), known as the Vijayan Complex, is different to the rest of the country [34]. The Vijayan Complex is a distinct geological formation comprising Precambrian high-grade metamorphic rock (a composite unit of metamorphic rocks, migmatites and granites) that has undergone changes due to the climatic conditions of the dry zone which is reflected in its soil pattern and groundwater conditions.
Around 12% of the B. pseudomallei population in Australia contains B. mallei–like sequence variation of bimA, bimABM, but it is rarely found in other endemic regions in Southeast Asia. For example, prevalence of bimABM for isolates in Thailand is 0%. Interestingly, we found that 18.5% of the Sri Lankan B. pseudomallei population contains the bimABM allele variant similar to that observed in the Australia. This is further evidence of a population intermediate between Laurasia and Oceania. A strong association between isolates that carry bimABM variant and neurological melioidosis has been reported in northern Australia and India [7,8,35]. We also found a significant (P = 0.019) association between bimABM genotype and neurological melioidosis, although this genotype comprised only 57 isolates. In contrast to the findings of Webb et al. [33], in this collection of Sri Lankan isolates the proportion of the bimABM genotype among the LPSA positive strains was not different from that among LPSA negative strains (49/251, 20% and 4/42, 17% respectively. The majority of Sri Lankan clinical B. pseudomallei isolates possessed the fhaB3 variant (80.79%) and the LPSA gene cluster (86.57%,). The fhaB3 variant is found in 100% of Thai strains and 83% of Australian strains and LPSA is found in 97% of Thailand strains and 87% of Australian strains again suggesting a population intermediate between Laurasia and Oceania [22,33].
Considerable genetic diversity was observed among the 193 Sri Lankan clinical isolates of B. pseudomallei with multiple groups and sub-group founders within the clonal complex and singletons. Isolates with common STs were genetically diverse in terms of YLF/BTFC, bimA, fhaB3 and LPSA. For example, clinical isolate BPs 206 and BPs14 which carry the bimABM allele share ST1136 with BPs 211, 179, 173, 164, 158, 148 which carry bimABP. The majority of ST1136 isolates carried fhaB3, however BPs 14 and BPs179 did not possess this allele type. In contrast, genotyping of ST1132 (n = 16) ST1136 (n = 16) and ST1137 (n = 35) showed that all these isolates belonged to the YLF-group and contained bimABP (except two isolates of 1136), fhaB3 (except 9 isolates) and LPSA (except 6 isolates).
Several shared STs were observed with Australia, Thailand, Cambodia, India and Belgium. As observed previously, this could be due to homoplasy [34]. This demonstrates the limitation of the use of MLST for highly recombinogenic bacteria like Burkholderia species. Hence, analysis of whole genome data is important [36]. Migration of humans and animals might also have resulted in the dissemination of shared STs among these geographical locations [2].
PHYLOVIZ analysis has identified two distinct clades of B. pseudomallei in Oceania and Southeast Asia which could reflect geographical isolation over a long period of time [2,15,17]. However, some mixing of strains through exchange of flora and fauna between Australia and Southeast Asia may have occurred via transient land links [2]. ME analysis also supported the assumption that Sri Lankan B. pseudomallei genotypes are in between Oceania and Southeast Asia.
Phylogenetic analysis in this study revealed that Sri Lankan strains are intermediate to Australian and Southeast Asian B. pseudomallei populations. The relationship between Sri Lankan and Australian strains could date back to the period when they were linked in Gondwanaland [37]. However, the presence of several STs with phylogenetic relatedness with STs from India, Bangladesh and Cambodia may be partly attributable to multiple introductions of B. pseudomallei through anthropogenic sources, especially travel and trade routes [2,16].
Conclusion
This is the first study that describes the genetic diversity of the Sri Lankan B. pseudomallei population using MLST along with genotyping of 4 selected genetic markers and provides a better insight into the genetic diversity and biogeography of B. pseudomallei clinical strains. Considerable genetic diversity is observed among the B. pseudomallei population in Sri Lanka in terms of both STs and the other biomarkers. The B. pseudomallei population in Sri Lanka appears to be intermediate between Southeast Asia and Oceania in terms of the prevalence of YLF/BTFC, bimABP/bimABM and fhaB3. This may relate to its ancient position in Gondwanaland adjacent to Oceania. In contrast, several STs showed phylogenetic relatedness with STs from India, Bangladesh and Cambodia indicating the possibility of introduction of these STs on subsequent occasions.
In conclusion, this study demonstrates the usefulness of high-resolution molecular typing by including additional genetic markers along with the MLST to enhance the resolution of genetic diversity among clinical isolates to elucidate the true relationship of such isolates to each other. Further, genetic markers could help to locate the Sri Lankan isolates within the broad geographical boundaries of B. pseudomallei at a global level.
Supporting information
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
Acknowledgments
Authors would like to acknowledge technical staff in the Biomedical laboratory 1 at KDU and in the Department of Microbiology, University of Colombo for their valuable support.
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
Funding for this study was mainly through grant NRC 18079 to ADDS from National Research Council, Sri Lanka, and partial support was obtained from Fairmed organization, Sri Lanka (https://fairmedsrilanka.org/) to EMC. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Wiersinga WJ, Virk HS, Torres AG, Currie BJ, Peacock SJ, Dance DAB, et al. Melioidosis. Nat Rev Dis Primers. 2018; 4:17107. doi: 10.1038/nrdp.2017.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baker AL, Pearson T, Sahl JW, Hepp C, Price EP, Sarovich DS, et al. Burkholderia pseudomallei distribution in Australasia is linked to paleogeographic and anthropogenic history. PLoS One. 2018; 13(11): e0206845. doi: 10.1371/journal.pone.0206845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mukhopadhyay C, Shaw T, Varghese GM, Dance DAB. Melioidosis in South Asia (India, Nepal, Pakistan, Bhutan and Afghanistan). Trop Med Infect Dis. 2018; 3(2):51. doi: 10.3390/tropicalmed3020051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dance DAB. Ecology of Burkholderia pseudomallei and the interactions between environmental Burkholderia spp. and human-animal hosts. Acta Trop. 2000; 74:159–168. doi: 10.1016/s0001-706x(99)00066-2 [DOI] [PubMed] [Google Scholar]
- 5.Corea EM, de Silva AD, Thevanesam V. Melioidosis in Sri Lanka. Trop Med Infect. 2018; 21:3(1):22. doi: 10.3390/tropicalmed3010022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Corea EM, Merritt AJ, Ler YH, Thevanesam V, Inglis TJ. Sri Lankan national melioidosis surveillance program uncovers a nationwide distribution of invasive melioidosis. Am J Trop Med Hyg. 2016; 94(2):292–8. doi: 10.4269/ajtmh.15-0567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sarovich DS, Price EP, Webb JR, Ward LM, Voutsinos MY, Tuanyok A. Variable virulence factors in Burkholderia pseudomallei (melioidosis) associated with human disease. PLoS One. 2014; 9(3): e91682. doi: 10.1371/journal.pone.0091682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morris JL, Fane A, Sarovich DS, Price EP, Rush CM, Govan BL. Increased neurotropic threat from Burkholderia pseudomallei strains with a B. mallei-like Variation in the bimA Motility Gene, Australia. Emerg Infect Dis. 2017; 23(5):740–9. doi: 10.3201/eid2305.151417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Limmathurotsakul D, Wongratanacheewin S, Teerawattanasook N, Wongsuvan G, Chaisuksant S, Chetchotisakd P, et al. Increasing incidence of human melioidosis in Northeast Thailand. Am J Trop Med Hyg. 2010; 82(6):1113–7. doi: 10.4269/ajtmh.2010.10-0038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibney KB, Cheng AC. Reducing the melioidosis burden: public health, chronic disease prevention, or improved case management? Lancet Infect Dis. 2019; 19(8):800–802. doi: 10.1016/S1473-3099(19)30303-2 [DOI] [PubMed] [Google Scholar]
- 11.McRobb E, Kaestli M, Price EP, Sarovich DS, Mayo M, Warner J, et al. Distribution of Burkholderia pseudomallei in northern Australia, a land of diversity. Appl Environ Microbiol.2014; 80(11):3463–8. doi: 10.1128/AEM.00128-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hall CM, Jaramillo S, Jimenez R, Stone NE, Centner H, Busch JD. Burkholderia pseudomallei, the causative agent of melioidosis, is rare but ecologically established and widely dispersed in the environment in Puerto Rico. PLoS Negl Trop Dis. 2019;13(9): e0007727. doi: 10.1371/journal.pntd.0007727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Godoy D, Randle G, Simpson AJ, Aanensen DM, Pitt TL, Kinoshita R, et al. Multilocus sequence typing and evolutionary relationships among the causative agents of melioidosis and glanders, Burkholderia pseudomallei and Burkholderia mallei. J Clin Microbio. 2003; 41(5):2068–79. doi: 10.1128/JCM.41.5.2068-2079.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng AC, Ward L, Godoy D, Norton R, Mayo M, Gal D. Genetic diversity of Burkholderia pseudomallei isolates in Australia. J Clin Microbiol. 2008; 46(1):249–54. doi: 10.1128/JCM.01725-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sathkumara HD, Merritt AJ, Corea EM, Krishnananthasivam S, Natesan M, Inglis TJJ. et al. Clinical, Bacteriologic, and Geographic Stratification of Melioidosis Emerges from the Sri Lankan National Surveillance Program. Am J Trop Med Hyg. 2018; 98(2):607–615. doi: 10.4269/ajtmh.17-0441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mukhopadhyay C, Kaestli M, Vandana KE, Sushma K, Mayo M, Richardson L. et al. Molecular characterization of clinical Burkholderia pseudomallei isolates from India. Am. J. Trop. Med. Hyg. 2011; 85:121–123. doi: 10.4269/ajtmh.2011.11-0166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamthan A, Shaw T, Mukhopadhyay C, Kumar S. Molecular analysis of clinical Burkholderia pseudomallei isolates from southwestern coastal region of India, using multi-locus sequence typing. PLoS Negl Trop Dis. 2018;12(11):e0006915. doi: 10.1371/journal.pntd.0006915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jayasinghearachchi HS, Corea EM, Krishnananthasivam S, Sathkumara HD, Francis VR, Abeysekere TR, et al. Whole-Genome Sequences of Eight Clinical Isolates of Burkholderia pseudomallei from Melioidosis Patients in Eastern Sri Lanka. Microbiol Resour Announc. 2019; 8(33):e00645–19. doi: 10.1128/MRA.00645-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merritt A, Inglis TJJ, Chidlow G, Harnett G. PCR-based identification of Burkholderia pseudomallei. Rev. Inst. Med. Trop. Sao Paulo. 2006; 48:239–244. doi: 10.1590/s0036-46652006000500001 [DOI] [PubMed] [Google Scholar]
- 20.Hantrakun V, Thaipadungpanit J, Rongkard P, Srilohasin P, Amornchai P, Langla S, et al. Presence of B. thailandensis and B. thailandensis expressing B. pseudomallei-like capsular polysaccharide in Thailand, and their associations with serological response to B. pseudomallei. PLoS Negl Trop Dis. 2018;24;12(1):e0006193. doi: 10.1371/journal.pntd.0006193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tuanyok A, Auerbach RK, Brettin TS, Bruce DC, Munk AC, Detter JC, et al. A horizontal gene transfer event defines two distinct groups within Burkholderia pseudomallei that have dissimilar geographic distributions. J Bacteriol. 2007; 189:9044–9049. doi: 10.1128/JB.01264-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tuanyok A, Stone JK, Mayo M, Kaestli M, Gruendike J, Georgia S, et al. 2012) The genetic and molecular basis of O-antigenic diversity in Burkholderia pseudomallei lipopolysaccharide. PLoS Negl Trop Dis. 2012. 6(1):e1453. doi: 10.1371/journal.pntd.0001453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaestli M, Richardson LJ, Colman RE, Tuanyok A, Price EP, Bowers JR, et al. Comparison of TaqMan PCR assays for detection of the melioidosis agent Burkholderia pseudomallei in clinical specimens. J Clin Microbio. 2012; 50(6):2059–62. doi: 10.1128/JCM.06737-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jayasinghearachchi HS, Francis VR, Sathkumara HD, Krishnananthasivam S, Masakorala J, Muthugama T, et al. Nonclonal Burkholderia pseudomallei population in melioidosis case cluster, Sri Lanka. Emerg Infect Dis. 2021. Nov [date cited]. doi: 10.3201/eid2711.210219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol. 2004; 186(5):1518–1530. doi: 10.1128/JB.186.5.1518-1530.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nascimento M, Sousa A, Ramirez M, Francisco AP, Carrico JA, Vaz, C. PHYLOViZ 2.0: providing scalable data integration and visualization for multiple phylogenetic inference methods. Bioinformatics. 2017; 33:128–129. doi: 10.1093/bioinformatics/btw582 [DOI] [PubMed] [Google Scholar]
- 27.Tamura K. and Nei M. (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Molecular Biology and Evolution 10:512–526. doi: 10.1093/oxfordjournals.molbev.a040023 [DOI] [PubMed] [Google Scholar]
- 28.Nei M and Kumar S. Molecular evolution and phylogenetics. New York. Oxford University Press;2000. [Google Scholar]
- 29.Saitou N and Nei M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987; 4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454 [DOI] [PubMed] [Google Scholar]
- 30.Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol.2018; 1;35(6):1547–1549. doi: 10.1093/molbev/msy096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rozas J, Ferrer-Mata A, Sanchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsin SE, et al. DnaSP v6. DNA sequence polymorphism analysis of large data sets. Mol Biol Evol. 2017; 34: 3299–3302. doi: 10.1093/molbev/msx248 [DOI] [PubMed] [Google Scholar]
- 32.Tellapragada C, Kamthan A, Shaw T, Ke V, Kumar S, Bhat V, et al. Unravelling the Molecular Epidemiology and Genetic Diversity among Burkholderia pseudomallei Isolates from South India Using Multi-Locus Sequence Typing. PLoS One. 2016; 19;11(12):e0168331. doi: 10.1371/journal.pone.0168331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Webb JR, Rachlin A, Rigas V, Sarovich DS, Price EP, Kaestli M, et al. Tracing the environmental footprint of the Burkholderia pseudomallei lipopolysaccharide genotypes in the tropical "Top End" of the Northern Territory, Australia. PLoS Negl Trop Dis. 2019; 13(7):e0007369. doi: 10.1371/journal.pntd.0007369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cooray PG. The Precambrian of Sri Lanka: a historical review. Precambrian Res. 1994; 66: 3–18 [Google Scholar]
- 35.Shaw T, Tellapragada C, Kamath A, Kalwaje Eshwara V, Mukhopadhyay C. Implications of environmental and pathogen-specific determinants on clinical presentations and disease outcome in melioidosis patients. PLoS Negl Trop Dis. 2019. May 15;13(5):e0007312. doi: 10.1371/journal.pntd.0007312 ; PMCID: PMC6538188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Smet B, Sarovich DS, Price EP, Mayo M, Theobald V, Kham C, et al. Whole-genome sequencing confirms that Burkholderia pseudomallei multilocus sequence types common to both Cambodia and Australia are due to homoplasy. J Clin Microbiol. 2015; 53(1):323–6. doi: 10.1128/JCM.02574-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dissanayake CB, and Chandrajith R. Sri Lanka–Madagascar Gondwana Linkage: Evidence for a Pan-African Mineral Belt. The J Geol. 1999;107 (2):223–235. 10.1086/314342 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.