

Abstract
Plant cells communicate information for the regulation of development and responses to external stresses. A key form of this communication is transcriptional regulation, accomplished via complex gene networks operating both locally and systemically. To fully understand how genes are regulated across plant tissues and organs, high resolution, multi-dimensional spatial transcriptional data must be acquired and placed within a cellular and organismal context. Spatial transcriptomics (ST) typically provides a two-dimensional spatial analysis of gene expression of tissue sections that can be stacked to render three-dimensional data. For example, X-ray and light-sheet microscopy provide sub-micron scale volumetric imaging of cellular morphology of tissues, organs, or potentially entire organisms. Linking these technologies could substantially advance transcriptomics in plant biology and other fields. Here, we review advances in ST and 3D microscopy approaches and describe how these technologies could be combined to provide high resolution, spatially organized plant tissue transcript mapping.
Combining spatial transcriptomics with 3D imaging technologies can demonstrate how genes are regulated across plant cells in a multi-dimensional space.
Introduction
Plants are multicellular organisms with complex organs and tissues that provide diverse functions including nutrient uptake, photosynthesis, and reproduction. These highly structured tissue domains are built of different cell types that are themselves heterogeneous as a result of their proximity and distance from other cell types and organs. The cells express and regulate diverse biological processes which can be represented as gene networks, with many networks active in individual cells at any time, and with a level of synchrony across adjacent cells within a tissue. Therefore, the multi-scale study and functional characterization of specialized tissue, cellular, and subcellular domains and their transcriptional changes is critical to understand their gene networks (Xia et al., 2019; Cho et al., 2021). That is, putting the data in a spatial context is important to interpreting these networks. Additionally, having cellular transcriptomics data in a spatial context, at a single-cell level, will surely result in the description of new cell types and resolve heterogeneity in data that currently confounds or over generalizes many studies of gene expression. The majority of gene expression studies in plants have been accomplished via “bulk” RNA-sequencing (RNA-seq)—that is, data are generated from intact organs (Stark et al., 2019)—leaving a number of molecular mechanisms undiscovered or poorly characterized, namely those that operate on a level of single or small numbers of cells.
A methodology known as “spatial transcriptomics (ST)” allows scientists to study gene expression from tissues and cells while retaining their spatial information (Crosetto et al., 2015; Marx, 2021). The field has advanced rapidly over the last few years, largely due to the development of many approaches to spatially resolve and capture RNA from tissue sections at or near a single-cell resolution and even subcellular mapping (Xia et al., 2019; Cho et al., 2021). ST can be divided into two main approaches, targeted and untargeted, based on the technology used. Targeted methods such as single molecule, fluorescent in situ hybridization (smFISH), yield precise and spatial gene expression data at a subcellular resolution (Chen et al., 2015b, 2018; Merritt et al., 2020). However, these methods are limited in the number of genes that can be analyzed per experiment. Untargeted methods, using array- or grid-based capture technologies, obtain transcripts in an unbiased manner from an entire tissue section (Ståhl et al., 2016; Rodriques et al., 2019; Vickovic et al., 2019; Stickels et al., 2021). However, the limitation of these capture methods is that their resolution is typically not at the single-cell level, and may be as low as one “spot” combining transcripts from a group of multiple cells. Nevertheless, the resolution is rapidly improving, and ST is now a powerful method with which to study gene expression in plants and mammals (Xia et al., 2019; Ali et al., 2021). Excitingly, there is also potential to generate these data in 3D, which requires reassembling the tissue from a serial array of 2D optical or physical tissue sections. With physical tissue sections (i.e., microtomy) this can be a computational challenge, as it requires the alignment of the microscopy-based consecutive individual 2D tissue sections produced by mechanical sectioning methods, which undergo non-linear distortions, compression, section tears, damage loss, etc., and then correlating with gene expression data from array/capture spots together with microscopy-based tissue images (Gurazada et al., 2021).
Understanding how different plant cell types are structured within 3D space is crucial to advanced analyses of tissue and organs, whether for interpreting cellular morphology (Huang et al., 2018), dynamic computational modeling (Shapiro et al., 2015), or in this case, integrating complex 3D transcriptome data. The application of optical techniques such as lightsheet fluorescence microscopy (LSFM) have shown tremendous potential for observing larger intact living 3D plant structures (Berthet and Maizel, 2016; Ovečka et al., 2018; Ovečka et al., 2021). In general, LSFMs use an objective lens to form a thin plane (sheet) of light into a specimen, illuminating only that plane. The orthogonal placement of an imaging objective lens and camera-based detector collects an image from the illuminated plane and by virtue of precise x–y–z positioning and rotation, full 3D volumes can be rapidly acquired in living and fixed specimens with less phototoxicity or photodamage. Of note, LSFM imaged samples in this form can be prepared for sectioning and array-based ST strategies. Furthermore, the combination of LSFM with expansion microscopy (ExM), a technique that anchors cell molecules to a nearby scaffold and expands many-fold while maintaining relative molecular relationships (Chen et al., 2015a; Asano et al., 2018; Bürgers et al., 2019), would allow in situ ST of bulk tissues. Alternatively, laboratory-based X-ray microscopy (XRM) is a specialized X-ray computed tomography (XCT) system that incorporates microscope objective lenses in the beam path, and it is an important addition to existing 3D imaging strategies (Jacobsen, 2020). Synchrotron-based XRM has distinct advantages in terms of faster imaging and the potential for elemental analysis of samples (Kopittke et al., 2018), but synchrotron beamline access is limited compared with commercial laboratory-based XRM instruments. In contrast to most laser- and electron-based systems, XRM allows high resolution, multiscale 3D imaging of relatively large and complex, yet intact samples, where X-rays can penetrate through the thick and highly scattering plant tissue. With commercial laboratory-based XRM instruments becoming more common in biology laboratories or core facilities, and with recent advancements in the methodology (Duncan et al., 2021), XRM can be a routine technology for obtaining 3D images over a wide range of plant systems.
These emerging technologies have the potential to further advance spatial genomics and transcriptomics studies in plant biology. Here, we first separately review some of the advances in ST in plants. We also discuss the potential opportunities to combine these approaches to generate 3D, spatially resolved transcriptome maps in plants, with a special emphasis on array-based approaches.
ST and single-cell RNA-seq: from mammals to plants
In recent years, many ST technologies have emerged, working toward the creation of a platform that produces high-resolution spatial data for many organisms or biological questions. While a comprehensive examination of all these approaches is beyond the scope of this review, here we will highlight several promising strategies for ST analysis. This includes recent advancements in the plant sciences, a field in which ST is in its infancy.
In 2016, a microarray-based system was described that allowed for the capture of spatial information of gene expression from tissue sections using mammalian specimens (Ståhl et al., 2016). This method integrated spotted microarrays with RNA-seq to generate barcoded libraries, indexed by the X/Y coordinate on the array from which the RNA was derived. Briefly, tissue sections were placed on microarrays; the arrays, unlike a conventional microarray of spotted, gene-specific sequences, were instead gridded with reverse transcription oligo(dT) nucleotides to capture poly(A) RNA, with each spot having a unique positional “barcode” (sequence) synthesized adjacent to the oligo(dT). The sample/tissue sections placed on the array were then fixed and imaged. Next, the mRNA was captured and reverse transcribed into cDNA while integrating the barcode into each captured molecule, before generating an RNA-seq library. Adapters for library construction are also added to the cDNA, in situ, and the cDNA was released from the array as a standard RNA-seq library for sequencing. Finally, the resulting gene expression data are mapped back to specific locations within the tissue section via the barcodes that were originally embedded during array construction. At the time, these microarrays consisted of 1,000 spots with a spot size of 100 µm, separated (i.e. the gap between spots) by 100 µm; this was an important seminal effort, however unable to achieve single-cell resolution. Subsequently, this technology was advanced and commercialized by 10× Genomics (as the Visium Spatial Gene Expression), increasing the spot number to 5,000 with a 55 µm resolution (Rao et al., 2020).
In order to improve the cellular resolution, other platforms sought to reduce the dimensions of the capture spots or “features.” A platform called Slide-seq used densely packed microbeads (Rodriques et al., 2019). Instead of synthesizing barcoded oligo(dT) in the form of capture probes, the probes were coated on 10 µm microbeads that were then randomly packed onto a “puck” and the spatial information of each bead (i.e. the sequence barcode at each location) was decoded by SOLiD sequencing. The imaging of the beads that was part of the SOLiD process determined the location on the puck of each barcode. Another technology that also used microbeads was known as high-definition ST (HDST). This approach utilized 2 µm beads and a sequential hybridization strategy to decode the sequences and thus positions of the spatial barcodes (Vickovic et al., 2019). The microbeads of Slide-seq and HDST achieve cellular and subcellular resolutions, respectively, and have been published only for applications to mammalian systems. However, one of the main drawbacks to these bead-based technologies was the requirement to decode the barcodes before each puck or array can be used, a highly complex process not easily performed by a typical molecular biology laboratory. Therefore, alternative methods continue to be explored to achieve spatially resolved data at a cellular resolution. A recently published method called XYZeq provides a workflow that encodes spatial data into single-cell RNA (scRNA)-seq libraries (Lee et al., 2021). In XYZeq, tissue sections were placed on an array of microwells that are spaced 500 µm center-to-center. These microwells have spatially barcoded oligo(dT), allowing for capture of cells from the tissue section while retaining their spatial information. After reverse transcription, the cells were collected, pooled, and individually assigned a second barcode via PCR before proceeding with scRNA-seq. This technology provides a platform to perform single-cell transcriptomic analysis while simultaneously assigning the spatial position within the tissue section. However, it has a lower spatial resolution and a more complex protocol compared with other ST technologies. Nevertheless, XYZeq presents an example of how single-cell and ST analysis can be combined to ultimately resolve gene expression in a tissue section.
While ST is also achievable in plants, its application has not advanced as rapidly as for mammalian systems. To date, the only published method and data for plant systems is the “first-generation” microarray-based approach from 2017 (Giacomello et al., 2017). It is likely that plant tissues could also be used on the Visium platform, which is, as mentioned above, a similar technology and protocol with higher resolution (Giacomello and Lundeberg, 2018; Rao et al., 2020). The paucity of publications of applications of the methods for plants is likely due to the plant cellular structures (i.e. cell wall, chloroplasts and vacuoles) that, with their presence in tissue sections, complicate transcriptional analysis (Cosgrove, 2005). These cellular structures will also present challenges to achieve ST at a subcellular resolution in the majority of plant systems. As mentioned above, a potential method that could be applied to reach this resolution is ExM, which involves embedding polymers to tissues and organs and expanding them 4–20 times their size, allowing for high-quality imaging at nanometer resolution (Chen et al., 2015a). Recently, a modified ExM protocol, containing a cocktail of cell wall enzymes, has shown promise for use in plant seed tissues for immunolocalization and shown to be compatible with fluorescent proteins (Kao and Nodine, 2019; Kao and Nodine, 2021). Additionally, published methods combined in situ sequencing (Ali et al., 2021) with ExM known as expansion sequencing (ExSeq), and multiplexed error-robust fluorescence in situ hybridization (MERFISH) with ExM (Xia et al., 2019) enabled large-scale, spatially resolved transcript identification and localization. However, these bulk sample/whole cell in situ approaches would require much further optimization to be applicable in plants for ST studies. Other subcellular or near subcellular platforms, such as HDST and Slide-seq, use randomly packed microbeads instead of capture probes, requiring more complex computational methods to deconvolute the data.
Currently, scRNA-seq is one of the more powerful approaches for quantifying and mapping gene expression at a cellular level in plants. scRNA-seq resolves the gene expression heterogeneity of tissue by extracting transcriptional data from individual cells. Within the last few years, a number of droplet-based scRNA-seq studies in Arabidopsis roots have been published (Jean-Baptiste et al., 2019; Ryu et al., 2019; Shulse et al., 2019; Shaw et al., 2021; Shahan et al., 2021) and also successfully applied in other plant species such as maize (Nelms and Walbot, 2019; Satterlee et al., 2020; Xu et al., 2021) and rice (Liu et al., 2021). While powerful, scRNA-seq is also still in its infancy for plant systems, as there are some challenges to overcome. scRNA-seq in plants typically requires removal of the cell wall, which can be achieved through protoplasting, something that is not yet routine in some plant species. In addition, plant cellular structure can influence the efficiency of protoplasting, leading to a bias in analyzing/detecting the cell types that are easier to isolate. Nevertheless, scRNA-seq is a promising technique to decipher heterogeneity between plant tissue types at a cellular level. Data from scRNA-seq potentially could be combined with data from ST to analyze thousands of genes from individual cells while retaining their spatial context in the plant tissue section, similar to the protocol of XYZeq (Lee et al., 2021).
While ST and scRNA-seq are powerful tools for high resolution studies of gene expression changes, these techniques heavily rely on computational algorithms and tools for data analysis—and thus the computational part of the workflow is as important as the laboratory-based work. Data analysis for ST begins with the data processing step that maps reads back to the spatial positions on the array and position within the section that was imaged on its surface. Paired-end sequencing data are typically generated, and the first read of the pair consists of the spatial barcode that uniquely identifies the position on the array, followed by the unique molecular identifier (UMI) portion of the oligo (from the array surface) used to define individual mRNA molecules (reducing PCR amplification bias at later stages), while the second read contains the cDNA from the gene transcript. The data processing workflow then typically utilizes the following steps to estimate transcript counts per spatial position: (1) removing sequencing adapters and low-quality reads, (2) grouping the reads based on the spatial barcode in the first read, (3) mapping the cDNA in the second read to reference gene transcripts and estimating gene expression by counting number of UMIs per cell/spot per gene, and finally, (4) normalizing expression by total reads per array feature. This workflow can be achieved using end-to-end computational pipelines from 10× Genomics such as the Cell Ranger pipeline and Space Ranger pipeline or using ST pipeline (Navarro et al., 2017) developed by the Swedish group behind the first published breakthrough approach of ST (Ståhl et al., 2016). Since each spatial position may contain from a partial to many cells, depending on the size of the array features and the positioning of the cells over each feature, it is also important to perform a high degree of replication; however, combining data across replicates presents new computational challenges that we discuss in further detail below.
3D optical and XRM in plants
In order to fully understand the impact of transcriptional variation on the structure and development of plant cells, tissues, and organs, they must be visualized within the context of the whole plant, ideally in 3D and at high resolution. Laser-based 3D imaging platforms (confocal, multiphoton, lightsheet, and super-resolution) have developed rapidly to image organisms in a mostly non-destructive manner, recording cellular and subcellular dynamics using a diverse range of fluorescent protein expressing systems (Komis et al., 2018; Ovečka et al., 2018, 2021). Lightsheet microscopy in particular, as highlighted in this review, has several advantages for 3D imaging of intact living or fixed and expanded volumes. Furthermore, volume electron microscopy (EM), another method with which subcellular 3D imaging at high resolution, has been achieved for important plant systems (Czymmek et al., 2020; Mursalimov et al., 2021), but generally requires encapsulation in a polymerized resin, which is problematic for array or in situ ST. Despite the unprecedented degree of cellular and subcellular visualization possible with these imaging platforms, they have several drawbacks including a necessarily smaller sample size, limited imaging depth, and in the case of volume EM, extensive sample preparation protocols. While their utility has limitations with respect to placing the imaging data in the context of the whole organ or plant, which 3D imaging strategy will serve as the ground truth is dependent upon the experimental question and tissue type. We are intrigued by the possibility that laboratory-based XRM can bridge this imaging gap and serve as a valuable complementary technology for high resolution, multiscale 3D visualization of relatively large and complex intact plant samples, ranging in sample size from 10 cm down to 1 mm with imaging resolution down to the sub-micrometer range (Prunet and Duncan, 2020).
A key challenge in imaging plant cells is the many refractive surfaces present in plant tissues, such as cuticular layers, cell walls, vacuoles, and air spaces, all of which substantially interfere with image formation and signal detection in the case of light-based platforms and with sample fixation and resin embedment in the case of volume EM. One solution to this problem was the development of XCT. Broadly speaking, XCT generates 3D image data by passing X-rays through a sample onto a detector, where variation in sample density is displayed as variation in grayscale in the resulting 2D radiograph. Hundreds or thousands of 2D radiographs are collected as the sample rotates, typically spanning 360 degrees (one complete sample rotation), and a 3D volume is computationally assembled. After it was first introduced commercially in the 1970s, XCT was quickly recognized as a useful means of addressing the challenges of imaging plants, by allowing deeper imaging into plant tissue for meaningful 3D visualization of many kinds of samples (Aylmore, 1993; Heeraman et al., 1997; Stuppy et al., 2003; Kaminuma et al., 2008; Leroux et al., 2009; Dhondt et al., 2010; van der Niet et al., 2010). However, most conventional XCT instruments rely solely on geometric magnification, and thus have functional resolution and sample size limits that are based on source–sample–detector geometry. Laboratory-based XRM systems insert a series of objective lenses into the beam path, which provide multiscale imaging of a single sample without the necessity of removing it from the instrument. Multiple high magnification scans can be overlaid onto a low magnification overview scan to set all the data within the context of the larger sample (Figure 1 and Supplemental Figure S1). This is distinct from synchrotron XRM which has recognized utility in plant biology (Kopittke et al., 2018; Gauthey et al., 2020; Peters et al., 2020), but has limited availability for routine imaging.
Figure 1.

X-ray microscope (XRM) and multiscale imaging of soybean (Glycine max) for floral anatomy. A, View inside the Zeiss XRM used to image soybean samples in B and C with the positions of the sample (S), X-ray source, detector, and objective lenses indicated. B, Axillary bud complex of soybean illustrates how an entire intact sample (box) can be prepared and imaged in high resolution 3D with XRM (C), followed by a region-of-interest scan at a higher magnification (orange), without removing the sample from the instrument. Reproductive organs such as ovaries (ov) and pollen-bearing anthers (an) are readily visualized.
In terms of X-ray energy, plant tissues are non-dense and provide little contrast with which an image can be generated. As a result, samples for high resolution (sub-micron) 3D XRM are fixed and contrast enhanced using familiar EM-based agents like iodine, phosphotungstic acid (PTA), or osmium tetroxide (Hayat, 1993). In a comprehensive analysis of plant sample preparation methods, Staedler et al. (2013) conducted an evaluation of conventional EM contrast agents as tools for examining plant samples with XRM. Using EM-based agents allows researchers to combine XRM with light and EM to study different biological questions. One example is a study that combined XRM with light and EM to study pollination morphology, with samples for XRM contrasted in PTA (Gamisch et al., 2013). Another study coupled XRM and light and EM, with metabolic analysis using GC–MS, using samples fixed in formaldehyde–acetic acid–alcohol (FAA) with 1% PTA for XRM imaging (Bellaire et al., 2014). Additionally, a variety of other fixation methods have been used for XRM, such as critical point drying (Jeiter et al., 2018, 2020). These studies demonstrate the utility of XRM for studying basic plant morphology by imaging, measuring, and comparing relatively large numbers of samples prepared in a variety of ways. Recent advances in XRM in plant biology (Duncan et al., 2021) have expanded the range of plant systems amenable to high resolution 3D imaging as well as the diversity of research questions that XRM can address. In particular, sample preparation methods were developed to allow XRM imaging of resin-embedded plant tissue, generating a high resolution 3D “road map” of the entire sample which was used to guide volume EM imaging of individual chloroplasts using the same sample block in a correlative workflow. Combining XRM and volume EM using a single sample preparation protocol for multiscale visualization of imaging data puts organelle-level imaging within the context of the larger plant tissue. Gold nanoparticles coated with affinity proteins are another tool we are testing to localize and visualize plant and microbial structures in situ with XRM. In addition, we are exploring the use of the ascorbate peroxidase (APEX, APEX2) fusion protein system (Ariotti et al., 2018) for localized expression of a compound that reacts with osmium when treated with 3,3-diaminobenzidine, depositing an electron-dense precipitate that is detectable with EM (Kong et al., 2020) and could potentially be visualized with XRM as well. Thus, multiscale correlative imaging would be a powerful tool if sample preparation methods could be similarly combined for XRM and ST (discussed in the next section).
Combining technologies to achieve 3D transcriptomics in plants
The studies described above demonstrate the utility of generating 3D image data of plant structures; many of those studies utilized complicated and delicate organs (Table 1). Applications of LSFM (Berthet and Maizel, 2016; Ovečka et al., 2018, 2021) and XRM have been greatly expanded to include intact imaging of inflorescence and meristematic structures, plant–microbe interactions, multiscale in situ imaging of root biology, and high resolution imaging of floral morphology and development. The unprecedented 3D image data they provide could complement ST approaches by serving as a model volumetric framework of individual plant cells within specific plant tissues and organs upon which expression data can be overlaid, to provide an authentic spatial context (Figure 2).
Table 1.
Important technologies reviewed
| Field | Technique | Description | Published plants study? |
|---|---|---|---|
| Transcriptomics | scRNA-seq | Extracting transcriptomic data from individual cells | Jean-Baptiste et al. (2019); Nelms and Walbot (2019); Ryu et al. (2019); Shulse et al. (2019); Satterlee et al. (2020); Shaw et al. (2021); Liu et al. (2021); Xu et al. (2021) |
| ST | First array-based ST platform; 100-µm spot capture resolution | Giacomello et al. (2017); Giacomello and Lundeberg (2018) | |
| Visium spatial gene expression (10× Genomics) | Upgraded resolution from ST; 55-µm spot capture resolution | No | |
| HDST | Uses microbeads and a sequential hybridization strategy for high resolution spatial capture; microbeads are 2 µm | No | |
| Slide-seq; Slide-seq V2 | Employs slides covered with pucks of 10-µm microbeads coated with poly-T capture probes | No | |
| XYZeq | Combines scRNA-seq and ST by using two rounds of split-pool indexing to capture the spatial information of each cell at a relatively low resolution (500 µm) followed by making indexed scRNA-seq libraries | No | |
| ExSeq | Combines ExM and in situ sequencing and hybridization to enable large-scale, spatially resolved transcript identification and localization | No | |
| 3D imaging | XRM | A specialized XCT system that incorporates microscope objective lenses in the beam path, allowing for high resolution, multiscale 3D imaging over a wide range of sample sizes | Duncan et al. (2021) |
| ExM | A technique that anchors cell molecules to a nearby scaffold and expands many-fold while maintaining relative molecular relationships | Kao and Nodine (2019, 2021) | |
| LSFM | A nondestructive technique that uses a thin plane of light to section and view tissues as low as subcellular resolution | Berthet and Maizel (2016); Ovečka et al. (2018, 2021) | |
| XCT | Generates 2D radiographs by passing X-rays through a sample onto a detector, and computationally reconstructing the images into a 3D volume | Aylmore (1993); Heeraman et al. (1997); Stuppy et al. (2003); Kaminuma et al. (2008); Leroux et al. (2009); Dhondt et al. (2010); and van der Niet et al. (2010) |
Figure 2.
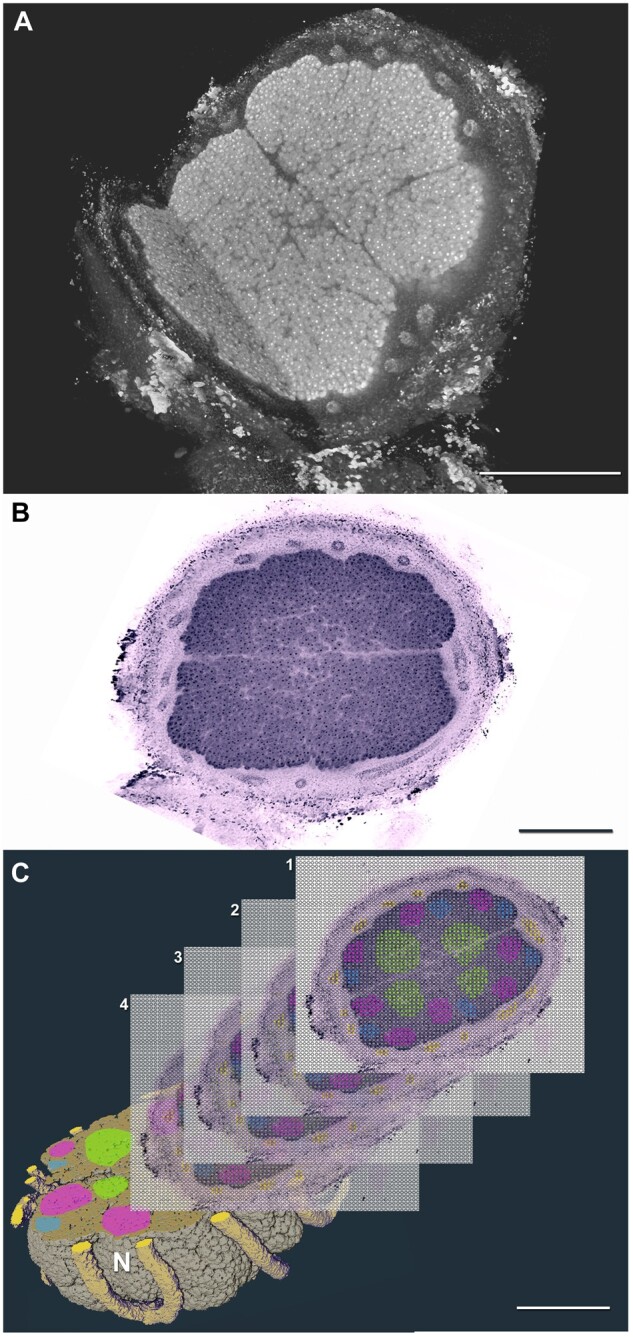
XRM and ST combined to illustrate localization of gene expression in 3D. A, Volume rendering from XRM scan of a soybean (G. max) nodule formed by nitrogen-fixing bacteria, with 3D cutaway view showing internal nodule structure. B, Physical microtome section collected from fixed and prepared nodule from A, sections are used for ST processing. C, Computationally segmented nodule (N) from A illustrates the 3D volume into which processed ST section data (1–4) are co-registered to identify where in the nodule specific genes are being expressed; the different colors illustrate localized expression of genes-of-interest on the ST sections. Scale bars = 500 μm.
Ideally, sample preparation methods can be developed so that the same sample that is imaged via fluorescence or scanned with XRM can then be sectioned and prepared for spatial transcriptome analysis. However, due to differences in sample preparation between the workflows of ST and XRM, tissue prepared for adequate contrast in XRM imaging may not be conducive to ST, and vice versa. Nevertheless, there are potential computational methods that would allow for expression data acquired through ST to be merged with the morphological data acquired through LSFM or XRM. This would involve using replicates, via both ST and respective imaging platforms, to generate a statistically guided and developmentally matched version of the plant structures being investigated, with the expression data mapped to the representation of where different cells are located (discussed in succeeding paragraphs). Understanding heterogeneities in the transcriptomes and cell-level patterning of tissues and organs will be essential to the Plant Cell Atlas endeavor (Cortijo and Locke, 2020), and the detailed 3D volume data provided by LSFM/XRM will serve as a useful framework upon which high-resolution expression data can be precisely understood.
Most current approaches in ST involve overlaying histological images of tissue sections with spatially resolved gene expression data from the ST arrays to create a comprehensive visual of spatial expression maps. Visualization in 3D space can then be achieved by stacking multiple serial sections to create a volume and integrating their molecular and imaging data into a reconstructed 3D framework using computational tools. ST spot detector (Wong et al., 2018), a computational tool developed specifically to analyze ST imaging data can overlay gene expression data defined by spatial coordinates in 2D, onto a high-quality tissue image. For 3D reconstruction, another recently developed computational package called ST utility (Bergenstråhle et al., 2020a) takes the counts files and images from 10× Genomics Visium (or other previous ST arrays) directly as input and creates visualizations of the combined data in 3D framework, giving a holistic view of the tissue and its spatially resolved expression landscape, although not at cellular resolution. ST utility begins with image processing by applying a series of transformation steps such as masking and auto-alignment of multiple tissue images to create a 3D visualization, and then provides downstream data analysis tools for identification of spatially correlated genes that can be visualized and explored within the 3D framework. However, with XRM providing volumetric 3D imaging of plant structures, the in silico re-constructed expression maps described above can be correlated and integrated with 3D reference data to establish representative ground truth spatial expression maps in 3D space (Figure 2). The integration of spatial gene expression data with 3D ground-truth volumes will require development of new computational strategies. One approach might be to expand the functionality of ST utility by replacing the turntable 3D model created by the software with the volumetric imaging data. Alternatively, a deep learning approach such as the one used in Xfuse (Bergenstråhle et al., 2020b) could be adopted to create a deep generative model of spatial expression data and fuse both XRM imaging data and spatial expression data as observable effects of a latent tissue state. The authors of Xfuse suggest that such a model can then be used to predict spatial gene expression from histological images that lack ST data, thereby creating a futuristic possibility for LSFM- or XRM-image-based in silico ST.
In terms of repeatability in ST, each tissue section is technically an independent sample. However, adjacent slices or layers of a tissue can be regarded as replicates in order to confirm the spatially correlated gene expression patterns and the distribution of cell types. Unlike bulk RNA-seq, aggregating gene expression across replicates in ST is nontrivial as we need to account for expression data across high-resolution spatial coordinates between the replicate sections with varying orientations. A computational method developed recently called PASTE (Probabilistic Alignment of ST Experiments) (Zeira et al., 2021) addresses this challenge by first formalizing and solving the problem of probabilistic pairwise alignment of adjacent ST layers based on both transcriptional and spatial similarity using Fused Gromov-Wasserstein Optimal Transport (FGW-OT) (Titouan et al., 2019). These pairwise alignments help construct a 3D representation of the tissue, similar to ST utility, however based on expression and spatial data in this case. The PASTE method then integrates multiple ST layers into a single layer and derives a consensus expression matrix for all replicates based on a “center” ST layer, by alternating between solving FGW-OT instances and solving a non-negative matrix factorization (NMF) (Lee and Seung, 2000) of a weighted expression matrix. As a further extension of this approach, we can similarly deal with replicates of complete 3D structures by iteratively applying the PASTE methodology to first reduce each 3D structure of adjacent slices into a single consensus layer for a replicate, and then derive a final consensus gene expression matrix across replicates using their respective consensus layers.
Conclusions
Characterizing the transcriptional activity of tissue sections at a cellular and subcellular level will be critical to understand how gene networks function across different plant compartments. The recent developments in genomics and transcriptomics are advancing the field in order to acquire high-resolution data from specialized plant organs to answer a variety of biological questions. As described in this review, combining ST with LSFM or XRM could potentially provide high-resolution, spatially structured data in a 3D context. Some possible applications combining these two emerging platforms together include studying how individual plant cells respond to a pathogen infection, or visualizing how individual cells divide, elongate, and related expression in plant roots. Additionally, these methods could be combined with spatio-temporal studies investigating how cellular and subcellular compartments are organized during the development of organs in different species; the floral meristem would be one well-studied example. This would likely require ST at single-cell resolution to do so, which has not yet been achieved in plants. However, using ST and 3D imaging could help further define the regulatory networks involved in spatio-temporal patterning during meristem development. While protocol development and optimization are needed, XRM and ST have the potential to accelerate plant biology and ultimately provide a more comprehensive understanding of how plant cells are organized and function with various environmental stimuli.
Supplemental data
Supplemental Figure S1. XCT and XRM.
Supplementary Material
Acknowledgments
We would like to thank August Thies for his assistance with developing Figure 2.
Funding
This work was supported by a Howard Hughes Medical Institute Hanna H. Gray Fellowship to K.L.C., and National Science Foundation Molecular and Cellular Biosciences award 1945854 to B.C.M. and K.J.C. Additional support for the work comes from the Donald Danforth Plant Science Center, including the Advanced Bioimaging Laboratory (RRID:SCR_018951). Financial support for the X-ray microscope came from a research collaboration between Valent BioSciences, Sumitomo Chemical Company, and the Donald Danforth Plant Science Center to C.N.T.
Conflict of interest statement. None declared.
ADVANCES.
ST and single cell technologies have rapidly advanced in recent years, including in the plant field.
XRM can be used to generate detailed 3D volume image data for a wide range of plant samples, particularly samples that cannot be effectively imaged with laser and electron tomography systems.
Improvements in 3D imaging platforms allow for cellular and subcellular imaging of plant organs in a non-destructive manner.
The combination of ST and 3D imaging technologies can better define transcript abundance patterns across complex plant tissues and organs, in a multi-dimensional context.
S.G.R.G., K.E.D., and K.J.C. designed the figures. K.L.C., S.G.R.G., K.E.D., K.J.C., C.N.T., and B.C.M. wrote the manuscript.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://academic.oup.com/plphys/pages/General-Instructions) is Blake C. Meyers (bmeyers@danforthcenter.org).
References
- Ali HR, Al Sa’d M, Alon S, Aparicio S, Battistoni G, Balasubramanian S, Becker R, Bodenmiller B, Boyden ES, Bressan D, et al. (2021) Expansion sequencing: Spatially precise in situ transcriptomics in intact biological systems. Science 371: eaax2656 (doi: 10.1126/science.aax2656) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariotti N, Rae J, Giles N, Martel N, Sierecki E, Gambin Y, Hall TE, Parton RG (2018) Ultrastructural localisation of protein interactions using conditionally stable nanobodies. PLoS Biol 16: e2005473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano SM, Gao R, Wassie AT, Tillberg PW, Chen F, Boyden ES (2018) Expansion microscopy: Protocols for imaging proteins and RNA in cells and tissues. Curr Protoc Cell Biol 80: e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylmore LAG (1993) Use of computer-assisted tomography in studying water movement around plant roots. InSparks DL, ed, Advances in Agronomy. Academic Press, Cambridge, pp 1–54 [Google Scholar]
- Bellaire A, Ischebeck T, Staedler Y, Weinhaeuser I, Mair A, Parameswaran S, Ito T, Schönenberger J, Weckwerth W (2014) Metabolism and development—integration of micro computed tomography data and metabolite profiling reveals metabolic reprogramming from floral initiation to silique development. New Phytol 202: 322–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergenstråhle J, Larsson L, Lundeberg J (2020a) Seamless integration of image and molecular analysis for spatial transcriptomics workflows. BMC Genomics 21: 482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergenstråhle L, He B, Bergenstråhle J, Andersson A (2020b) Super-resolved spatial transcriptomics by deep data fusion. bioRxiv [DOI] [PubMed]
- Berthet B, Maizel A (2016) Light sheet microscopy and live imaging of plants. J Microsc 263: 158–164 [DOI] [PubMed] [Google Scholar]
- Bürgers J, Pavlova I, Rodriguez-Gatica JE, Henneberger C, Oeller M, Ruland JA, Siebrasse JP, Kubitscheck U, Schwarz MK (2019) Light-sheet fluorescence expansion microscopy: Fast mapping of neural circuits at super resolution. Neurophotonics 6: 015005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Tillberg PW, Boyden ES (2015a) Optical imaging. Expansion microscopy. Science 347: 543–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X (2015b) RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348: aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Sun Y-C, Church GM, Lee JH, Zador AM (2018) Efficient in situ barcode sequencing using padlock probe-based BaristaSeq. Nucleic Acids Res 46: e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho C-S, Xi J, Si Y, Park S-R, Hsu J-E, Kim M, Jun G, Kang HM, Lee JH (2021) Microscopic examination of spatial transcriptome using Seq-Scope. Cell 184: 3559–3572.e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortijo S, Locke JCW (2020) Does gene expression noise play a functional role in plants? Trends Plant Sci 25: 1041–1051 [DOI] [PubMed] [Google Scholar]
- Cosgrove DJ (2005) Growth of the plant cell wall. Nat Rev Mol Cell Biol 6: 850–861 [DOI] [PubMed] [Google Scholar]
- Crosetto N, Bienko M, van Oudenaarden A (2015) Spatially resolved transcriptomics and beyond. Nat Rev Genet 16: 57–66 [DOI] [PubMed] [Google Scholar]
- Czymmek K, Sawant A, Goodman K, Pennington J, Pedersen P, Hoon M, Otegui MS (2020) Imaging plant cells by high-pressure freezing and serial block-face scanning electron microscopy. Methods Mol Biol 2177: 69–81 [DOI] [PubMed] [Google Scholar]
- Dhondt S, Vanhaeren H, Van Loo D, Cnudde V, Inzé D (2010) Plant structure visualization by high-resolution X-ray computed tomography. Trends Plant Sci 15: 419–422 [DOI] [PubMed] [Google Scholar]
- Duncan K. E., CzymmekKJ, , Jiang N,, Thies A C, Topp CN (2021) X-ray microscopy enables multiscale high-resolution 3D imaging of plant cells, tissues, and organs. Plant Physiol 34618094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamisch A, Staedler YM, Schönenberger J, Fischer GA, Comes HP (2013) Histological and micro-CT evidence of stigmatic rostellum receptivity promoting auto-pollination in the madagascan orchid Bulbophyllum bicoloratum. PLoS ONE 8: e72688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthey A, Peters JMR, Carins-Murphy MR, Rodriguez-Dominguez CM, Li X, Delzon S, King A, López R, Medlyn BE, Tissue DT, et al. (2020) Visual and hydraulic techniques produce similar estimates of cavitation resistance in woody species. New Phytol 228: 884–897 [DOI] [PubMed] [Google Scholar]
- Giacomello S, Lundeberg J (2018) Preparation of plant tissue to enable spatial transcriptomics profiling using barcoded microarrays. Nat Protoc 13: 2425–2446 [DOI] [PubMed] [Google Scholar]
- Giacomello S, Salmén F, Terebieniec BK, Vickovic S, Navarro JF, Alexeyenko A, Reimegård J, McKee LS, Mannapperuma C, Bulone V, et al. (2017) Spatially resolved transcriptome profiling in model plant species. Nat Plants 3: 17061. [DOI] [PubMed] [Google Scholar]
- Gurazada SGR, Cox KL, Czymmek KJ, Meyers BC (2021) Space: the final frontier—achieving single-cell, spatially resolved transcriptomics in plants. Emerg Top Life Sci 5: 179–188 [DOI] [PubMed] [Google Scholar]
- Hayat MA (1993) Stains and Cytochemical Methods. Plenum Press, New York [Google Scholar]
- Heeraman DA, Hopmans JW, Clausnitzer V (1997) Three dimensional imaging of plant roots in situ with X-ray computed tomography. Plant Soil 189: 167–179 [Google Scholar]
- Huang C, Wang Z, Quinn D, Suresh S, Hsia KJ (2018) Differential growth and shape formation in plant organs. Proc Natl Acad Sci USA 115: 12359–12364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen C (2020) X-ray Microscopy. Cambridge University Press, Cambridge [Google Scholar]
- Jean-Baptiste K, McFaline-Figueroa JL, Alexandre CM, Dorrity MW, Saunders L, Bubb KL, Trapnell C, Fields S, Queitsch C, Cuperus JT (2019) Dynamics of gene expression in single root cells of Arabidopsis thaliana. Plant Cell 31: 993–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeiter J, Langecker S, Weigend M (2020) Towards an integrative understanding of stamen–corolla tube modifications and floral architecture in Boraginaceae s.s. (Boraginales). Bot J Linn Soc 193: 100–124 [Google Scholar]
- Jeiter J, Staedler YM, Schönenberger J, Weigend M, Luebert F (2018) Gynoecium and fruit development in Heliotropium Sect. Heliothamnus (Heliotropiaceae). Int J Plant Sci 179: 275–286 [Google Scholar]
- Kaminuma E, Yoshizumi T, Wada T, Matsui M, Toyoda T (2008) Quantitative analysis of heterogeneous spatial distribution of Arabidopsis leaf trichomes using micro X-ray computed tomography. Plant J 56: 470–482 [DOI] [PubMed] [Google Scholar]
- Kao P, Nodine MD (2019) Transcriptional activation of Arabidopsis zygotes is required for initial cell divisions. Sci Rep 9: 17159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao P, Nodine MD (2021) Application of expansion microscopy on developing Arabidopsis seeds. Methods Cell Biol 161: 181–195 [DOI] [PubMed] [Google Scholar]
- Komis G, Novák D, Ovečka M, Šamajová O, Šamaj J (2018) Advances in imaging plant cell dynamics. Plant Physiol 176: 80–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong H, Zhang J, Li J, Wang J, Shin H-J, Tai R, Yan Q, Xia K, Hu J, Wang L, et al. (2020) Genetically encoded X-ray cellular imaging for nanoscale protein localization. Natl Sci Rev 7: 1218–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopittke PM, Punshon T, Paterson DJ, Tappero RV, Wang P, Blamey FPC, van der Ent A, Lombi E (2018) Synchrotron-based X-ray fluorescence microscopy as a technique for imaging of elements in plants. Plant Physiol 178: 507–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DD, Seung HS (2000) Algorithms for non-negative matrix factorization. InProceedings of the 13th International Conference on Neural Information Processing Systems. MIT Press, Cambridge, MA, USA, pp 535–541 [Google Scholar]
- Lee Y, Bogdanoff D, Wang Y, Hartoularos GC, Woo JM, Mowery CT, Nisonoff HM, Lee DS, Sun Y, Lee J, et al. (2021) XYZeq: Spatially resolved single-cell RNA sequencing reveals expression heterogeneity in the tumor microenvironment. Sci Adv. doi: 10.1126/sciadv.abg4755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroux O, Leroux F, Bellefroid E, Claeys M, Couvreur M, Borgonie G, Van Hoorebeke L, Masschaele B, Viane R (2009) A new preparation method to study fresh plant structures with X-ray computed tomography. J Microsc 233: 1–4 [DOI] [PubMed] [Google Scholar]
- Liu Q, Liang Z, Feng D, Jiang S, Wang Y, Du Z, Li R, Hu G, Zhang P, Ma Y, et al. (2021) Transcriptional landscape of rice roots at the single-cell resolution. Mol Plant 14: 384–394 [DOI] [PubMed] [Google Scholar]
- Marx V (2021) Method of the year: Spatially resolved transcriptomics. Nat Methods 18: 9–14 [DOI] [PubMed] [Google Scholar]
- Merritt CR, Ong GT, Church SE, Barker K, Danaher P, Geiss G, Hoang M, Jung J, Liang Y, McKay-Fleisch J, et al. (2020) Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat Biotechnol 38: 586–599 [DOI] [PubMed] [Google Scholar]
- Mursalimov S, Ohno N, Matsumoto M, Bayborodin S, Deineko E (2021) Serial block-face scanning electron microscopy reveals that intercellular nuclear migration occurs in most normal tobacco male meiocytes. Front Plant Sci 12: 672642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro JF, Sjöstrand J, Salmén F, Lundeberg J, Ståhl PL (2017) ST pipeline: An automated pipeline for spatial mapping of unique transcripts. Bioinformatics 33: 2591–2593 [DOI] [PubMed] [Google Scholar]
- Nelms B, Walbot V (2019) Defining the developmental program leading to meiosis in maize. Science 364: 52–56 [DOI] [PubMed] [Google Scholar]
- van der Niet T, Zollikofer CPE, de León MSP, Johnson SD, Linder HP (2010) Three-dimensional geometric morphometrics for studying floral shape variation. Trends Plant Sci 15: 423–426 [DOI] [PubMed] [Google Scholar]
- Ovečka M, Sojka J, Tichá M, Komis G, Basheer J, Marchetti C, Šamajová O, Kuběnová L, Šamaj J (2022) Imaging plant cells and organs with light-sheet and super-resolution microscopy. Plant Physiol 188: 683–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovečka M, von Wangenheim D, Tomančák P, Šamajová O, Komis G, Šamaj J (2018) Multiscale imaging of plant development by light-sheet fluorescence microscopy. Nat Plants 4: 639–650 [DOI] [PubMed] [Google Scholar]
- Peters JMR, Gauthey A, Lopez R, Carins-Murphy MR, Brodribb TJ, Choat B (2020) Non-invasive imaging reveals convergence in root and stem vulnerability to cavitation across five tree species. J Exp Bot 71: 6623–6637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prunet N, Duncan K (2020) Imaging flowers: A guide to current microscopy and tomography techniques to study flower development. J Exp Bot 71: 2898–2909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao N, Clark S, Habern O (2020) Bridging genomics and tissue pathology: 10x Genomics explores new frontiers with the visium spatial gene expression solution. Genet Eng Biotechnol News 40: 50–51 [Google Scholar]
- Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, Welch J, Chen LM, Chen F, Macosko EZ (2019) Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 363: 1463–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu KH, Huang L, Kang HM, Schiefelbein J (2019) Single-cell RNA sequencing resolves molecular relationships among individual plant cells. Plant Physiol 179: 1444–1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterlee JW, Strable J, Scanlon MJ (2020) Plant stem-cell organization and differentiation at single-cell resolution. Proc Natl Acad Sci USA 117: 33689–33699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahan R, Hsu C-W, Nolan TM, Cole BJ, Taylor IW, Vlot AHC, Benfey PN, Ohler U. (2021) A single cell Arabidopsis root atlas reveals developmental trajectories in wild type and cell identity mutants. bioRxiv (doi: 10.1101/2020.06.29.178863) [DOI] [PMC free article] [PubMed]
- Shapiro BE, Tobin C, Mjolsness E, Meyerowitz EM (2015) Analysis of cell division patterns in the Arabidopsis shoot apical meristem. Proc Natl Acad Sci USA 112: 4815–4820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw R, Tian X, Xu J (2021) Single-cell transcriptome analysis in plants: Advances and challenges. Mol Plant 14: 115–126 [DOI] [PubMed] [Google Scholar]
- Shulse CN, Cole BJ, Ciobanu D, Lin J, Yoshinaga Y, Gouran M, Turco GM, Zhu Y, O’Malley RC, Brady SM, et al. (2019) High-throughput single-cell transcriptome profiling of plant cell types. Cell Rep 27: 2241–2247.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staedler YM, Masson D, Schönenberger J (2013) Plant tissues in 3D via X-ray tomography: Simple contrasting methods allow high resolution imaging. PLoS ONE 8: e75295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, Giacomello S, Asp M, Westholm JO, Huss M, et al. (2016) Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353: 78–82 [DOI] [PubMed] [Google Scholar]
- Stark R, Grzelak M, Hadfield J (2019) RNA sequencing: The teenage years. Nat Rev Genet 20: 631–656 [DOI] [PubMed] [Google Scholar]
- Stickels RR, Murray E, Kumar P, Li J, Marshall JL, Di Bella DJ, Arlotta P, Macosko EZ, Chen F (2021) Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat Biotechnol 39: 313–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuppy WH, Maisano JA, Colbert MW, Rudall PJ, Rowe TB (2003) Three-dimensional analysis of plant structure using high-resolution X-ray computed tomography. Trends Plant Sci 8: 2–6 [DOI] [PubMed] [Google Scholar]
- Titouan V, Courty N, Tavenard R, Laetitia C, Flamary R (2019) Optimal transport for structured data with application on graphs. In K Chaudhuri, R Salakhutdinov, eds, Proceedings of the 36th International Conference on Machine Learning. PMLR, pp 6275–6284
- Vickovic S, Eraslan G, Salmén F, Klughammer J, Stenbeck L, Schapiro D, Äijö T, Bonneau R, Bergenstråhle L, Navarro JF, et al. (2019) High-definition spatial transcriptomics for in situ tissue profiling. Nat Methods 16: 987–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong K, Navarro JF, Bergenstråhle L, Ståhl PL, Lundeberg J (2018) ST spot detector: A web-based application for automatic spot and tissue detection for spatial transcriptomics image datasets. Bioinformatics 34: 1966–1968 [DOI] [PubMed] [Google Scholar]
- Xia C, Fan J, Emanuel G, Hao J, Zhuang X (2019) Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression. Proc Natl Acad Sci USA 116: 19490–19499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Crow M, Rice BR, Li F, Harris B, Liu L, Demesa-Arevalo E, Lu Z, Wang L, Fox N, et al. (2021) Single-cell RNA sequencing of developing maize ears facilitates functional analysis and trait candidate gene discovery. Dev Cell 56: 557–568.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeira R, Land M, Raphael B (2021) Alignment and integration of spatial transcriptomics data. bioRxiv https://www.biorxiv.org/content/10.1101/2021.03.16.435604v1 [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.