

ABSTRACT
Herpes simplex virus 1 (HSV-1) latency-associated transcript (LAT) plays a significant role in efficient establishment of latency and reactivation. LAT has antiapoptotic activity and downregulates expression of components of the type I interferon pathway. LAT also specifically activates expression of the herpesvirus entry mediator (HVEM), one of seven known receptors used by HSV-1 for cell entry that is crucial for latency and reactivation. However, the mechanism by which LAT regulates HVEM expression is not known. LAT has two small noncoding RNAs (sncRNAs) that are not microRNAs (miRNAs), within its 1.5-kb stable transcript, which also have antiapoptotic activity. These sncRNAs may encode short peptides, but experimental evidence is lacking. Here, we demonstrate that these two sncRNAs control HVEM expression by activating its promoter. Both sncRNAs are required for wild-type (WT) levels of activation of HVEM, and sncRNA1 is more important in HVEM activation than sncRNA2. Disruption of a putative start codon in sncRNA1 and sncRNA2 sequences reduced HVEM promoter activity, suggesting that sncRNAs encode a protein. However, we did not detect peptide binding using two chromatin immunoprecipitation (ChIP) approaches, and a web-based algorithm predicts low probability that the putative peptides bind to DNA. In addition, computational modeling predicts that sncRNA molecules bind with high affinity to the HVEM promoter, and deletion of these binding sites to sncRNA1, sncRNA2, or both reduced HVEM promoter activity. Together, our data suggest that sncRNAs exert their function as RNA molecules, not as proteins, and we provide a model for the predicted binding affinities and binding sites of sncRNA1 and sncRNA2 in the HVEM promoter.
IMPORTANCE HSV-1 causes recurrent ocular infections, which is the leading cause of corneal scarring and blindness. Corneal scarring is caused by the host immune response to repeated reactivation events. LAT functions by regulating latency and reactivation, in part by inhibiting apoptosis and activating HVEM expression. However, the mechanism used by LAT to control HVEM expression is unclear. Here, we demonstrate that two sncRNAs within the 1.5-kb LAT transcript activate HVEM expression by binding to two regions of its promoter. Interfering with these interactions may reduce latency and thereby eye disease associated with reactivation.
KEYWORDS: cornea, virus replication, transfection, infection, luciferase, HSV-1, HVEM, promoters, transfection systems
INTRODUCTION
Herpes simplex virus-1 (HSV-1) causes recurrent eye infections after primary infection that can produce corneal scarring and vision loss, commonly referred to as herpetic stromal keratitis, or HSK (1). HSK is the leading cause of corneal blindness in the United States, and approximately 70 to 90% of adults in the United States are latently infected with HSV-1 (2, 3). In ocular infections, HSV-1 first replicates in the eye, after which it establishes lifelong latency in the trigeminal ganglia (TG), where it can periodically reactivate. HSK is caused by host immune responses to recurrent HSV-1 infections (4); therefore, it is important to fully understand the mechanisms of HSV-1 latency and reactivation.
The HSV-1 latency-associated transcript (LAT) is not necessary for initial infection or virulence but does play a major role in enhancing latency reactivation, as viruses lacking LAT have reduced latency and reactivation (5–7). LAT also has antiapoptotic activity, which is important for its role in latency reactivation, because replacing LAT with other antiapoptotic genes restores latency and reactivation (8–13). Mice latently infected with LAT(−) virus have elevated expression of JAK-1 and JAK-2 as well as several interferon (IFN)-regulated genes (14), suggesting that LAT controls apoptosis by downregulating components of the type I IFN pathway. However, not all LAT functions can be attributed to LAT antiapoptotic activity. HSV-1 ocular infections result in CD8+ T cell exhaustion, which depends on LAT (4, 15, 16) and cannot be rescued by an unrelated antiapoptotic gene (14). There are two copies of LAT in the long terminal repeats of the HSV-1 genome. The 8.3-kb primary LAT transcript is spliced into a stable 2-kb LAT and an unstable 6.3-kb LAT (17–21). The 2-kb stable LAT is further spliced into a 1.5-kb LAT containing information needed for LAT antiapoptotic activity (8, 17, 18).
LAT is known to encode multiple microRNAs (miRNAs) and two small noncoding RNAs (sncRNAs). The two sncRNAs, sncRNA1 and sncRNA2, are 62 and 36 bp in length, respectively, located within the 1.5-kb stable LAT, and contribute to its antiapoptotic activity in vitro (22–26). Expression of sncRNA1 and -2 inhibits apoptosis and productive infection in vitro (27). Both contain an initiating ATG for open reading frames (ORFs), ORF3 and ORF8, encoding sncRNA1 and sncRNA2, respectively. Interestingly, mutation of ATG to TTG abolished the ability of the sncRNAs to inhibit apoptosis and virus replication, suggesting that either this mutation destabilizes the sncRNAs or these sncRNAs encode short peptides (27). However, this has not been experimentally tested.
Herpesvirus entry mediator (HVEM; TNFRSF14) is one of many host receptors used by HSV-1 for host cell entry (28). Other host receptors for HSV-1 include nectin-1, nectin-2, 3-O-sulfated heparan sulfate (3-OS-HS), paired immunoglobulin-like type 2 receptor α (PILR-α), nonmuscle myosin heavy chain IIA (NMHC-IIA), and myelin-associated glycoprotein (MAG) (28–32). HVEM is a member of the tumor necrosis factor (TNF) receptor superfamily (TNFRSF) and regulates immune cell activation or inactivation. When TNF-related ligand LIGHT or lymphotoxin-α (33) binds to HVEM, T cell activation occurs, whereas binding of CD160 or B and T lymphocyte attenuator to HVEM produces a T and B cell inhibitory signal (34–36). HVEM also has antiapoptotic activity by activating the NF-κB pathway.
HSV-1 LAT activates HVEM to establish latency (37). The viral glycoprotein D (gD) is the primary viral protein that binds to HVEM, facilitating viral entry via this receptor (38–40). Interestingly, LAT increases HVEM expression in latently infected TG (37). While HVEM is not necessary for virus replication during acute infection, latency and reactivation were reduced in HVEM knockout mice (37, 41, 42). This could be due to reduced apoptosis and impaired T cell activation in the absence of HVEM (37). We established that HVEM function in latency and reactivation is independent of its binding to gD (43). LAT activation of HVEM using LAT(+) and LAT(−) viruses could be due to one or both of the two sncRNAs encoded by LAT. However, the mechanism of sncRNAs regulating HVEM expression, and whether sncRNAs exert their function as peptides or as RNA molecules, is not known. Here, we demonstrate for the first time that (i) sncRNAs 1 and 2 activate HVEM promoter activity in vitro; (ii) these two sncRNAs likely function as RNA molecules; (iii) sncRNAs 1 and 2 are both required for wild-type (WT) levels of HVEM expression; (iv) ATG mutations within the two sncRNAs reduced HVEM promoter activation; and (v) we have identified predicted binding sites for sncRNAs in the HVEM promoter and show that deletion of these binding sites reduces promoter activity.
RESULTS
sncRNA1 and -2 peptide products do not bind to the HVEM promoter.
Since the original report of detection of latency-associated transcript (LAT) in trigeminal ganglia of latently infected host (17–19, 44), the possible presence of LAT products was extensively investigated without any convincing results (reviewed in Phelan et al. [45]). sncRNA1 and -2 contain a possible ATG start codon, which was shown to be necessary for their antiapoptotic activity (27). This raised the possibility that these sncRNAs can be translated into short peptides. We have shown that expression of LAT upregulated HVEM expression both in vitro and in vivo (37). To determine if sncRNA1 and -2 account for LAT control of HVEM expression and if they function as peptides by binding to its promoter, we performed chromatin immunoprecipitation (ChIP). 293T cells were transfected with plasmids expressing the mouse HVEM promoter and sncRNA1 and -2 sequences with an in-frame flag tag. Sheared chromatin from the transfected cells was probed with anti-Flag beads, and HVEM promoter-specific primers were used to amplify the pulled down chromatin. Lack of HVEM amplification suggested that the HVEM promoter was not pulled down with potential flag-tagged sncRNA-encoded peptides (Fig. 1). We repeated the assay by synthesizing flag-tagged sncRNA1 and sncRNA2 peptides (MLGSYCLGGGSADYKDDDDK and MFLFLSDYKDDDDK, respectively) and performed ChIP assay as described in Materials and Methods. Similar to the sncRNA expression plasmids, no HVEM amplification was seen with these sncRNA1 and -2 peptides (data not shown), suggesting that the sncRNAs are not translated into peptides but rather function as RNA molecules.
FIG 1.

Chromatin immunoprecipitation (ChIP) and PCR amplification of HVEM promoter. 293T cells were transfected with WT sncRNA1-FLAG or WT sncRNA2-FLAG as described in Materials and Methods. Chromatin from transfected cells was sheared and immunoprecipitated with anti-FLAG beads. Immunoprecipitated chromatin and input chromatin were amplified with primers corresponding to HVEM promoter sequences. Experiments were repeated twice.
Because we did not detect sncRNA binding to HVEM promoter as peptides, we used a computational tool to predict the likelihood that putative peptides encoded by sncRNA1 and sncRNA2 bind to HVEM DNA (http://biomine.cs.vcu.edu/servers/DRNApred/). This algorithm assigns DNA-binding propensity scores to each amino acid. All scores assigned to the sncRNA1 and sncRNA2 predicted amino acid sequences were below the threshold scores, suggesting that if these peptides were encoded by sncRNA1 and sncRNA2, they are not likely to bind to DNA (Table 1).
TABLE 1.
Prediction of putative DNA binding by peptides encoded by sncRNA1 and -2a
| Amino acid | Probability score | Binding (Y/N) |
|---|---|---|
| sncRNA1 | ||
| M | 0.1023 | N |
| L | 0.0774 | N |
| G | 0.0930 | N |
| S | 0.1380 | N |
| Y | 0.1571 | N |
| C | 0.0806 | N |
| L | 0.0798 | N |
| G | 0.1080 | N |
| G | 0.1015 | N |
| G | 0.1044 | N |
| S | 0.1519 | N |
| A | 0.0882 | N |
| M | 0.0848 | N |
| F | 0.0813 | N |
| L | 0.0773 | N |
| F | 0.0805 | N |
| L | 0.0780 | N |
| S | 0.0973 | N |
| sncRNA2 | ||
| M | 0.0848 | N |
| F | 0.0813 | N |
| L | 0.0773 | N |
| F | 0.0805 | N |
| L | 0.0780 | N |
| S | 0.0973 | N |
DNA binding probability of putative sncRNA1 and sncRNA2 amino acid sequences was predicted using the DRNApred (http://biomine.cs.vcu.edu/servers/DRNApred/) webserver. First column, amino acid sequence. Second column, putative DNA-binding propensity scores. Residues with a score of >0.4727 are annotated as predicted DNA-binding residues. The third column summarizes the predicted DNA binding of the residue (Y, likely to bind; N, not likely to bind).
WT sncRNAs 1 and 2 do not interfere with gB, ICP0, or ICP4 expression.
To determine if sncRNAs play a role in HSV-1 infections, we constructed plasmids expressing the stable 1.5-kb LAT sequence (Fig. 2A), WT sncRNA1 (Fig. 2B), and WT sncRNA2 (Fig. 2C). Rabbit skin (RS) cells were transfected with plasmids expressing WT sncRNA1, WT sncRNA2, or the stable 1.5-kb LAT control. Forty-eight hours after transfection, cells were infected with 1 PFU/cell of HSV-1 strain McKrae. Cells were lysed in TRIzol, and RNA was isolated at 2, 4, 8, and 12 h postinfection. Levels of ICP0, ICP4, and gB were measured by quantitative reverse transcription-PCR (qRT-PCR). There were no significant differences in levels of gB (Fig. 3A), ICP0 (Fig. 3B), or ICP4 (Fig. 3C) expression between cells transfected with sncRNA1, sncRNA2, or 1.5-kb LAT (P > 0.05). Thus, neither sncRNA1 nor sncRNA2 affected viral transcript levels in transfected and infected RS cells.
FIG 2.

sncRNA1 and sncRNA2 plasmids used in these studies. (A) Plasmid containing the 1.5-kb LAT (pGEM5317) and its promoter has been described previously (17, 18). Shaded boxes indicate sncRNA1 and sncRNA2 sequences within LAT. sncRNA1 (62 nt) (B) and sncRNA2 (36 nt) (C) were generated and inserted into the pSilencer and pcDNA3.1-FLAG plasmids. LAT sequences lacking ΔsncRNA1 sequence (D), ΔsncRNA2 sequence (E), or both sequences (E) and under the LAT promoter were generated and inserted into the pUC57 plasmid. sncRNA1 TTG (F) and sncRNA2 TTG (G) mutant constructs were established by inserting a 3,007-bp region of LAT corresponding to LAT 118641–121660 and containing a single base pair mutation of ATG to TTG in either sncRNA1 or sncRNA2 and inserted into pUC57. All constructs except those in panels B and C are under the LAT promoter.
FIG 3.
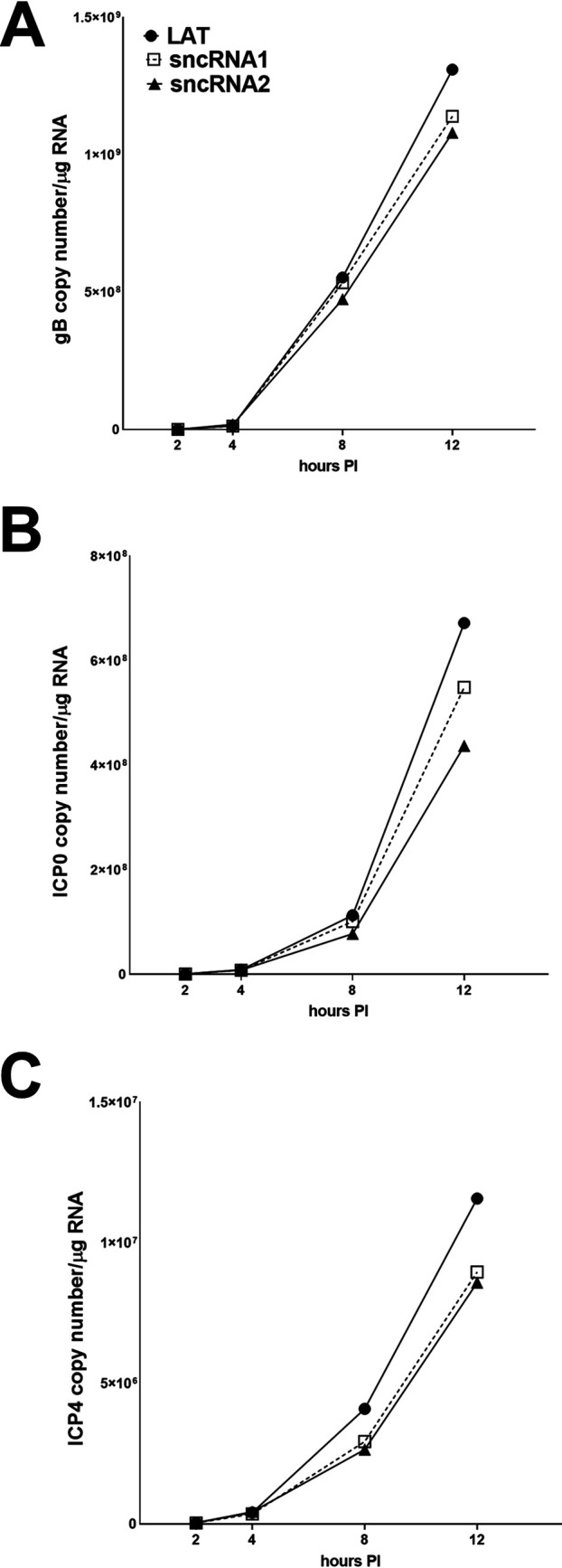
Expression of sncRNA1 or -2 does not affect expression of viral transcripts. RS cells were transfected with WT sncRNA1, sncRNA2, or LAT and infected with 1 PFU/cell of HSV-1 strain McKrae 48 h after transfection. Cells were collected at 2, 4, 8, and 12 h postinfection in TRIzol. RNA was extracted and amplified by qRT-PCR with gB (A), ICP0 (B), or ICP4 (C) primers. The copy number of any of the three sequences did not differ at any time point. Each point represents the means ± SEM for 3 experiments.
WT sncRNAs 1 and 2 enhance HVEM promoter activity.
We have previously shown that the presence of LAT activates HVEM expression in vitro and in vivo (37). LAT has multiple functions, including antiapoptotic activity (8). The antiapoptotic function of LAT resides within the stable 1.5-kb LAT transcript (6). The 1.5-kb LAT contains two small noncoding RNAs which have antiapoptotic effects (22–26, 47). To determine if these two sncRNAs play a role in LAT activation of HVEM and if one or both account for full LAT activation of HVEM expression, we measured the effects of LAT and sncRNA expression on HVEM promoter activity using a luciferase assay. 293T cells were cotransfected with sncRNA1-pSilencer (WT sncRNA1), sncRNA2-pSilencer (WT sncRNA2), or 1.5-kb stable LAT, together with the WT HVEM promoter. HVEM promoter activity was determined by measuring luciferase intensity at 8, 24, and 48 h after transfection as we described previously (46). At 8 h posttransfection, 1.5-kb LAT induced a nearly 6-fold increase in HVEM promoter activity (Fig. 4A) (P < 0.0001). Similarly, sncRNA1 and -2 increased HVEM promoter activity by approximately 4.5-fold (Fig. 4A) (P < 0.0001, sncRNA1) and 1.5-fold (Fig. 4A) (P < 0.0001, sncRNA2), respectively. At 24 h posttransfection, HVEM promoter activity was approximately 8-fold higher in LAT-transfected cells (Fig. 4A) (P < 0.0001) and 1.7-fold or 1.5-fold higher in sncRNA1- or sncRNA2-transfected cells, respectively (Fig. 4A) (P < 0.0001). At 48 h posttransfection, HVEM promoter activity in LAT-transfected cells was approximately 3.5-fold higher than that in empty vector-transfected cells (Fig. 4A) (P = 0.002) and approximately 1.8-fold higher in sncRNA1- and sncRNA2-transfected cells (Fig. 4A) (P < 0.0001). These results suggest that while sncRNA1 and -2 increase HVEM expression, it is not to the level of 1.5-kb LAT, suggesting that sncRNA1 and -2 both are necessary for full LAT function or that sncRNA1 and -2 are not as stable as 1.5-kb LAT after transfection. The results obtained with sncRNA1 and sncRNA2 are in agreement with our previous report showing increased HVEM expression in Neuro2A cells at 8 h posttransfection with sncRNA1 or sncRNA2 (37).
FIG 4.
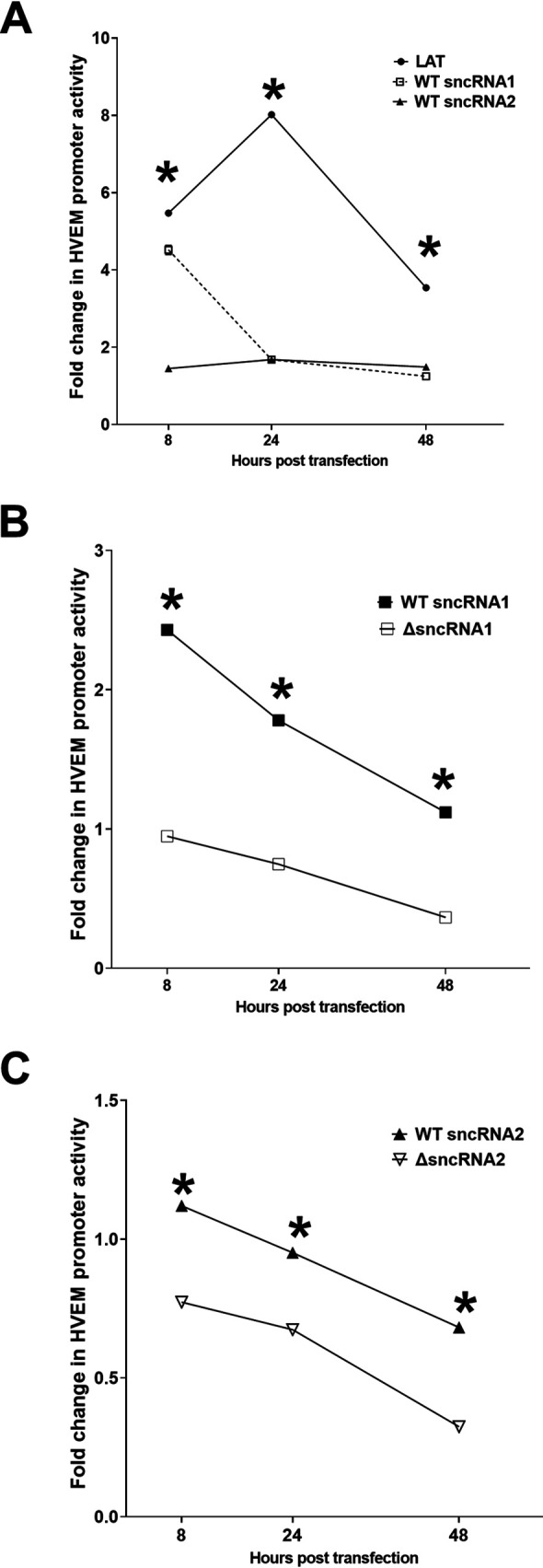
Expression of sncRNA1 and 2 increases HVEM promoter activity. 293T cells were transfected with pGL4-pHVEM and plasmids containing LAT, WT sncRNA1, WT sncRNA2, or empty vector (A), WT sncRNA1, sncRNA1 deletion plasmid (ΔsncRNA1), or empty vector (B), or WT sncRNA2, sncRNA2 deletion plasmid (ΔsncRNA2), or empty vector (C). HVEM promoter activity was measured 8, 24, and 48 h posttransfection. Expression relative to the empty vector is shown as fold change. Each point represents the mean ± SEM (n = 10 for A, n = 6 for B, C) from three separate experiments. *, P < 0.002.
To further demonstrate that sncRNA sequences play a role in increasing HVEM promoter activity, we constructed plasmids expressing LAT sequences lacking either sncRNA1 or sncRNA2 and compared luciferase activity in cells expressing WT sncRNA sequences or the deletion constructs (Fig. 2D and E, respectively). Luciferase activity was significantly lower at all time points in cells expressing the deletion mutants than in those expressing WT sncRNA (Fig. 4B and C) (P < 0.0001). Together, these results suggest that sncRNA1 increases HVEM promoter activity more than sncRNA2 at 8 h posttransfection and that both sncRNA sequences are important for LAT levels of HVEM expression. It also suggests that other regions of LAT do not contribute to HVEM activation.
Both sncRNA1 and -2 are required for WT level of HVEM promoter activity.
Results shown in Fig. 4 suggest reduced HVEM promoter activity in the absence of LAT plasmid lacking sncRNA1 or -2. However, the presence of each of the sncRNAs contributed to some HVEM promoter activity. Thus, to determine if both sncRNA sequences contribute to increases in HVEM promoter activity, we compared luciferase activity in cells expressing both sncRNA sequences (Fig. 2A, WT) with LAT plasmid lacking both the sncRNA1 and -2 sequences (Fig. 2F). Luciferase activity was significantly lower at all time points in cells expressing the double deletion mutants than in those expressing WT sncRNA (Fig. 5, P < 0.0001). Together, these results suggest that both sncRNA1 and -2 are contributing to increased HVEM promoter activity. It also further suggests that other regions of LAT do not contribute to HVEM activation.
FIG 5.

Deletion of both sncRNA1 and sncRNA2 sequences results in loss of HVEM promoter activity. 293T cells were transfected with pGL4-pHVEM and LAT or sncRNA1 and sncRNA2 deletion plasmid (ΔsncRNA1&2). The effects on HVEM promoter activity were determined 8, 24, and 48 h posttransfection. *, P < 0.0001.
ATGs within sncRNA1 and sncRNA2 are important for HVEM activation.
sncRNA1 and -2 both contain an ATG, and mutation of these ATGs is known to impair inhibition of apoptosis in vitro (27, 47), suggesting that these ATGs are important for either LAT stability or antiapoptotic activity. To determine if these two ATGs are also important for activating HVEM, we mutated the ATGs in sncRNA1 and sncRNA2 to TTG (Fig. 2G and H) and compared luciferase activity in cells expressing either the WT or TTG mutant version of sncRNA1 or sncRNA2. WT sncRNA1 (Fig. 6A) (P < 0.0001)- and WT sncRNA2 (Fig. 6B) (P < 0.0001)-transfected cells had significantly higher luciferase activity than their TTG mutant counterparts at all time points. Thus, similar to previous studies (27, 47), mutation of ATG to TTG in sncRNA1 and -2 significantly reduced HVEM promoter activity in vitro.
FIG 6.

sncRNA ATG is required to enhance HVEM promoter activity. 293T cells were transfected with pGL4-pHVEM and WT sncRNA1 or sncRNA1 TTG (A) or WT sncRNA2 or sncRNA2 TTG (B). The effects on HVEM promoter activity were determined 8, 24, and 48 h posttransfection. Each point represents the means ± SEM (n = 9) from three separate experiments. *, P < 0.0001.
TTG mutant sncRNAs have reduced ability to activate the HVEM promoter.
To determine the importance of the ATG element in HVEM activation, we compared the effects of expressing the sncRNA1 TTG with ΔsncRNA1 (LAT sequences lacking the sncRNA1 sequence) and sncRNA2 TTG with ΔsncRNA2 (LAT sequences missing the sncRNA2 sequence) (Fig. 2). 293T cells were cotransfected with plasmid sncRNA1 TTG (sncRNA1 TTG), plasmid ΔsncRNA1 (ΔsncRNA1), plasmid sncRNA2 TTG (sncRNA2 TTG), plasmid ΔsncRNA2 (ΔsncRNA2), and HVEM promoter. HVEM promoter activity was determined by measuring luciferase intensity at 8, 24, and 48 h after transfection as we described previously (46). No significant differences in HVEM promoter activity between ΔsncRNA1 and sncRNA1 TTG-transfected cells were seen at any time point (Fig. 7A) (P > 0.05). sncRNA2 TTG mutant showed higher HVEM promoter activity at 8 h after transfection than its deletion mutant counterpart (Fig. 7B) (P < 0.01). However, at 24 and 48 h after transfection, expression of TTG mutant sncRNA2 or LAT sequences lacking sncRNA2 sequence (ΔsncRNA2) had similar effects on HVEM promoter activity (Fig. 7B) (P > 0.05), suggesting that this mutation completely abolishes activation of the HVEM promoter by sncRNAs.
FIG 7.

HVEM promoter activation levels by TTG sncRNA and sncRNA deletion mutants are similar. 293T cells were transfected with pGL4-pHVEM and sncRNA1 TTG or sncRNA1 deletion plasmid (A) or sncRNA2 TTG or sncRNA2 deletion plasmid (B). Effect on HVEM promoter activity was determined as described for Fig. 3. Each point represents the mean ± SEM (n = 5) from two separate experiments. *, P < 0.01.
sncRNA binding sites in HVEM promoter are necessary for upregulation of mouse HVEM promoter activity.
We next asked whether the sncRNAs could bind to the HVEM promoter region as RNA molecules. We utilized an RNA hybrid algorithm (https://rna.urmc.rochester.edu/RNAstructureWeb/Servers/DuplexFold/DuplexFold.html), which predicted sncRNA1 and sncRNA2 binding to mouse HVEM promoter DNA at several positions, with the highest free energy of 50.6 kcal/mol at position −539/−478 upstream of the HVEM ATG start codon (Fig. 8A) for sncRNA1 and −19.9 kcal at position −484/−421 for sncRNA2 (Fig. 8B). Thus, to further verify the role of sncRNAs in regulating HVEM promoter activity, we synthesized HVEM promoter sequences lacking one or both of the predicted sncRNA binding sites (Fig. 8C to F) and measured promoter activity in cells 8 h after cotransfection with LAT. We chose the 8-h time point because we saw the highest HVEM promoter activity at 8 h posttransfection with various sncRNA constructs and decreased activity thereafter, and previously we showed that HVEM expression was highest in NeuroA2 cells 8 h posttransfection (37). HVEM promoter activity was significantly higher in cells transfected with WT HVEM promoter than cells transfected with HVEM promoter lacking the 61-bp binding site of sncRNA1, 63-bp binding site for sncRNA2, or both sncRNA1 and sncRNA2 binding sites (Fig. 9E) (P < 0.0001). Thus, the RNA hybrid algorithm program correctly identified the possible binding sites of sncRNA1 and sncRNA2 to the HVEM promoter. Although we observed a greater effect on promoter activity when expressing sncRNA1 than sncRNA2 at 8 h posttransfection (Fig. 4A), the absence of either binding site similarly reduced the HVEM promoter activity. This suggests that both sncRNA1 and sncRNA2 have similar effects on HVEM promoter activity. However, there is a 6-bp overlap between the two binding sites; therefore, disruption of one site could disrupt binding of both sncRNAs.
FIG 8.

Computational modeling predicts binding of sncRNA1 and sncRNA2 to mouse HVEM promoter. (A) Mouse HVEM promoter sequence corresponding to nucleotides 539/−478 upstream of the HVEM ATG start codon is predicted to bind to sncRNA1 with a free energy of binding of −50.6 kcal/mol. (B) mHVEM promoter sequence corresponding to −484/−421 upstream of the HVEM start codon is predicted to bind to sncRNA2 with a free energy of binding of −19.9 kcal/mol. Mfe refers to free energy of binding. Red boxes show the position of the ATGs in sncRNA sequences. Schematic diagram of (C) the wild-type mouse HVEM promoter construct, (D) the mouse HVEM promoter construct lacking the predicted sncRNA1 binding site, (E) the mouse HVEM promoter construct lacking the predicted sncRNA2 binding site, and (F) the mouse HVEM promoter construct lacking both the predicted sncRNA1 and sncRNA2 binding sites.
FIG 9.

sncRNA1 and -2 sequences are necessary for HVEM promoter activation. 293T cells were transfected with either LAT or empty vector and pGL4-pHVEM constructs lacking sncRNA1, sncRNA2, or both sncRNA1 and sncRNA2 binding sites. HVEM promoter activity was measured 8 h posttransfection. Expression relative to empty vector is shown as fold change. Each point represents the mean ± SEM (n = 10) from two separate experiments. *, P < 0.0001.
DISCUSSION
Both LAT and HVEM have been implicated in HSV-1 latency and reactivation, and both are known to have antiapoptotic activity (37, 48, 49). HVEM is one of several receptors HSV-1 uses for cell entry (28, 30). This binding is mediated by one of the HSV-1 glycoproteins, glycoprotein D (gD). We have shown that HVEM expression is upregulated in mice during latency in an LAT-dependent manner and that latency and reactivation were reduced in HVEM−/− mice (37, 41). The latency reactivation was rescued in mice where mouse HVEM was replaced with WT human HVEM or human or mouse HVEM where the gD binding site was disrupted, suggesting that human and mouse HVEM share similar functions (43). Additionally, it suggests that the latency reactivation defect is independent of HVEM binding to gD. In this study, we set out to determine the mechanism of LAT control of HVEM expression. Two sncRNAs encoded in the LAT region of the HSV-1 genome have also been shown to have antiapoptotic activity and are expressed during latency in TG of infected mice (24, 27). Because LAT RNA activates HVEM expression in vivo and in vitro (37), we sought to determine if activation of HVEM by LAT correlates with the presence of sncRNA1 and sncRNA2.
We first verified previous results by testing if expression of sncRNA1 or -2 affects expression of HSV-1 transcripts in vitro. Consistent with a previous study (27), we found that expression of ICP0, ICP4, and gB transcripts was similar in cells transfected with sncRNAs and then infected with HSV-1 strain McKrae. We previously reported that an RNA hybrid algorithm predicted sncRNA binding to the mouse HVEM promoter (37), and we found that LAT increased HVEM expression in vitro and in vivo (37). Here, we extended those results to show that LAT increased HVEM promoter activity, and this may be mediated by the two sncRNAs in LAT. In line with our previous results showing increased expression of HVEM transcript, we saw increased HVEM promoter activity in cells transfected with either sncRNA1 or sncRNA2, with sncRNA1 having a larger effect on HVEM promoter activity than sncRNA2. These results are also consistent with findings by the Jones group (27), who reported a greater antiapoptotic effect in cells transfected with sncRNA1 than with sncRNA2. These differences between the two sncRNAs are also in line with their predicted minimum free energy (MFE)-based secondary structures, as sncRNA1, but not sncRNA2, contains stem-loop structures (see Fig. S2A and C in the supplemental material), which are features often observed in regulatory RNAs (50, 51). Furthermore, while expression of both sncRNA1 and sncRNA2 increased HVEM promoter activity, neither sncRNA1 nor sncRNA2 increased it to levels seen with full-length LAT, suggesting that both are necessary to achieve full LAT activation of the HVEM promoter.
We further verified the role of sncRNAs in regulating HVEM promoter activity by coexpression studies with LAT and HVEM promoter plasmids lacking the sncRNA binding sites. We found that HVEM promoter sequences lacking the predicted sncRNA binding sites did not induce luciferase expression to the same level as the WT promoter. Although expression of sncRNA1 had a greater effect on HVEM promoter activity than sncRNA2, the difference between HVEM promoter lacking either sncRNA1 or sncRNA2 site was minor. This could be because the binding sites overlap by 6 bp (Fig. 9A); therefore, deletion of one binding site could affect binding of the other sncRNA. The remaining activity could be due to sncRNA binding to a different region with reduced affinity, as was predicted by an RNA-DNA hybrid algorithm (https://bibiserv.cebitec.uni-bielefeld.de/rnahybrid).
Interestingly, mutation of the sncRNA1 or -2 ATGs significantly reduced HVEM promoter activity. The importance of these ATGs was also reported by Shen et al. (27) in a study showing that while expression of WT sncRNA1 reduced virus production, a TTG mutant sncRNA1 did not. Additionally, Shen et al. reported that ICP4 protein was reduced in cells transfected with WT sncRNA2 but not in cells transfected with sncRNA2 TTG mutant. These results suggested that these ATGs are necessary for sncRNA function and/or stability. We found that TTG mutations within the sncRNAs reduced HVEM promoter activity to levels seen with sncRNA deletion mutants. The effect of mutating this ATG to TTG on virus replication, apoptosis, and HVEM promoter activity suggested that disruption of the ATG could alter folding of the sncRNAs, which may subsequently alter their function, stability, or binding to the HVEM promoter. The TTG mutation is predicted to cause only a minor change in the free energy of binding to HVEM promoter (Fig. S1) and to increase the free energy of the sncRNA1 TTG folded structure (Fig. S2A versus B). The increased stability of the unbound sncRNA1 could reduce the likelihood that the secondary structure is relaxed enough to allow binding to the HVEM promoter region. Alternatively, the TTG mutation disrupts one of the loop structures within sncRNA1 (Fig. S2A versus B), and recent findings have suggested the importance of stem-loop structures in activation of gene expression (51).
The possibility of LAT encoding a functional protein has been of interest in the field of HSV-1 latency for decades. Although some evidence suggests that the LAT locus encodes a protein (52–58), these proteins map outside the 1.5-kb region of LAT that is necessary and sufficient for antiapoptotic activity. Interestingly, eight putative open reading frames (ORFs) are located within the 1.5-kb LAT. Deletion studies concluded that out of these, neither ORF1 nor -2 is essential for latency reactivation (59–61) (reviewed in Phelan et al. [45]). A later study compared sequences of three HSV-1 strains (McKrae, KOS, and 17syn+) and showed that ORFs 5, 6, 7, and 8 are not conserved between the three strains and, thus, are unlikely to encode a functional protein (62). However, point mutations that disrupt ATGs in these ORFs reduce LAT antiapoptotic activity, suggesting these encode proteins (47). The effect of mutating these ORFs on antiapoptotic activity was graduated, with a more severe effect observed upon deletion of more than one ORF, suggesting that either multiple peptides cooperated or that LAT stability was affected by these mutations (47). In a subsequent study, Henderson et al. established peptide specific IgGs against all 8 ORFs and, using immunohistochemistry, detected a protein with only ORFs 2 and 8 (63). However, the results were not verified by Western blotting. Thus, despite intensive work done by various groups, no definitive evidence to support the presence of a peptide has been shown, and LAT is largely considered to exert its function in latency reactivation as a long noncoding RNA (61) (reviewed in Phelan et al. [45]).
Here, we tested the possibility that the sncRNAs within LAT encode peptides that bind the HVEM promoter using two approaches. However, neither chromatin immunoprecipitation (ChIP) with transfecting plasmids containing sncRNA sequences nor use of synthesized peptides detected binding to the HVEM promoter (Fig. 7 and data not shown). While our results do not irrefutably rule out the possibility of peptide, these results are consistent with the previous studies discussed above and the low probability of putative peptide binding to DNA predicted by computational algorithm (Table 1) (http://biomine.cs.vcu.edu/servers/DRNApred/). Furthermore, sncRNA1 is located within ORF3, and a previous study by Henderson et al. failed to detect a protein encoded by this ORF (63). The theoretical peptide encoded by sncRNA2 is within ORF8 but is predicted to migrate around 1.75 kDa, much smaller than the protein encoded within ORF8, which migrated around 13 kDa. Computational prediction analysis further suggests that LAT and the sncRNAs indeed function as RNA molecules. An RNA hybrid algorithm (https://bibiserv.cebitec.uni-bielefeld.de/rnahybrid) predicts both sncRNA1 and sncRNA2 binding to the HVEM promoter at several locations, with the highest free energy binding site located at positions −539/−478 and −484/−421 upstream of the HVEM ATG start codon (Fig. 8A and B, sncRNA1 and sncRNA2, respectively). We synthesized HVEM promoter constructs lacking one or both of these binding sites and showed that HVEM promoter activity is similarly reduced in the absence of one or both sites. The similar effect could be caused by possible disruption of both sites with deletion of one due to the 6-bp overlap between the binding sites, as we saw a greater effect on HVEM promoter activity when expressing sncRNA1 than sncRNA2 at 8 h posttransfection (Fig. 4A versus 9). These results confirm the binding sites predicted by the algorithm.
Currently, it is not known if sncRNAs activate HVEM via an intermediate host protein. Da Silva and Jones established a role for HSV-1 LAT sncRNA1 but not sncRNA2 in cooperating with RIG-I to increase IFN-β promoter activity (64). They showed sncRNA binding to RIG-I using a DNA probe corresponding to sncRNA1 sequence that recognized a 200-bp band that was pulled down with RIG-I. Further, sncRNA1 but not -2 stimulated NF-κB promoter activity in vitro (64), which is likely a conserved mechanism, as the BoHV-1 LR gene contains an miRNA with a similar function of interacting with RIG-I (65). It is currently not known if activation of HVEM by sncRNAs depends on recognition of RIG-I or whether sncRNAs function more directly. It is possible that, similar to IFN, sncRNA regulates HVEM promoter activity by binding to an unidentified protein. Of note, lncRNAs can act as scaffolds that recruit multiple protein complexes (66, 67).
In summary, our results reveal a novel role for HSV-1 sncRNAs within LAT in vitro in regulating HVEM promoter activity and that they likely function as RNA molecules. Our future studies will investigate a possible role for sncRNAs in vivo by constructing recombinant viruses lacking one or both of these sncRNAs and assessing their possible role in latency and reactivation. Overall, achieving a complete understanding of the role of sncRNAs in latency reactivation will lead to more effective management of HSV-1 infection and disease.
MATERIALS AND METHODS
Virus and cells.
Plaque-purified virulent HSV-1 strain McKrae was described previously (68). Rabbit skin (RS) cells were used to prepare virus stocks and determine growth kinetics. RS cells were grown in Eagle's minimal essential medium and supplemented with 5% fetal bovine serum (FBS). HVEM promoter activity was measured using HEK293T cells grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS as we described previously (46). These cells are referred to as 293T cells throughout the manuscript.
Plasmids.
WT LAT (pGEM-LAT5317) contains the LAT promoter and was described previously (17, 18). The 62-bp WT sncRNA1 and 36-bp sncRNA2 sequences were cloned into the pSilencer-GFP vector (GenScript, Piscataway, NJ). For ChIP assays, WT sncRNA1 and sncRNA2 sequences were synthesized and inserted into the pCDNA3.1-Flag backbone vector (GenScript). The sncRNA1 deletion construct was established by inserting a synthesized 2,945-bp DNA fragment corresponding to LAT 118641–121660, but lacking the 62-bp sncRNA1 sequence (GCCTGTGTTTTTGTGCCTGGCTCTCTATGCTTGGGTCTTACTGCCTGGGGGGGGGGAGTGCG), into pUC57 using EcoRI and HindIII restriction enzyme sites. The sncRNA2 deletion construct was established by inserting a synthesized 2,971-bp DNA fragment corresponding to LAT 118641–121660, but lacking the 36 bp sncRNA2 sequence (CATTCTTGTTTTCTAACTATGTTCCTGTTTCTGTCT), into pUC57 using EcoRI and HindIII restriction enzyme sites. The sncRNA1 TTG and sncRNA2 TTG mutant constructs were established by inserting a 3,007-bp region of LAT corresponding to LAT 118641–121660 and containing a single ATG-to-TTG mutation in both sncRNA1 (GCCTGTGTTTTTGTGCCTGGCTCTCTTTGCTTGGGTCTTACTGCCTGGGGGGGGGGAGTGCG) and sncRNA (CATTCTTGTTTTCTAACTTTGTTCCTGTTTCTGTCT) into pUC57 using EcoRI and HindIII restriction sites (GenScript, Piscataway, NJ). Boldface and underlining indicate the position of the ATG to TTG mutation. The sncRNA deletion constructs and TTG constructs contained the LAT promoter. WT HVEM promoter corresponding to (bp −1462 to −8, relative to ATG), promoter lacking the 61-bp sncRNA1 (corresponding to −539/−478), 63-bp sncRNA2 (corresponding to bp −484/−421), or both (118 bp, corresponding to bp −539/−421) upstream of the mouse TNFRSF14 gene were synthesized and inserted into the pGL4.17 vector using SacI and XhoI (GenScript, Piscataway, NJ).
Role of sncRNA1 sequence in HVEM promoter activity.
Either pGL4 empty vector or pGL4 vector containing the mouse HVEM promoter driving firefly luciferase expression (mHVEMp-pGL4) was cotransfected into 293T cells with WT sncRNA1, WT sncRNA2, ΔsncRNA1, ΔsncRNA2, sncRNA1 TTG, sncRNA2 TTG, or LAT-pGEM5317 using Xtremegene HP (Millipore-Sigma, St. Louis, MO). pRL-SV40 (Promega, Madison, WI), a renilla luciferase reporter plasmid, was added to all transfection mixtures as an internal control (46). Cells were harvested after 8, 24, or 48 h of culture, washed with PBS, and lysed in passive lysis buffer (Promega, Madison, WI) per the manufacturer’s instructions. Luciferase activity was measured with a luminometer (GloMax; Promega, Madison, WI) as we have described previously (46), and luciferase levels were normalized to the internal control and shown as fold change over cells transfected with empty vector alone. The means ± standard errors of means (SEM) were calculated from three separate experiments.
Effect of sncRNA on expression of gB, ICP0, and ICP4.
RS cells were transfected with sncRNA1, sncRNA2, LAT, or empty vector. Forty-eight hours posttransfection, cells were infected with HSV-1 McKrae at 1 PFU/cell for 2, 4, 8, and 12 h. At indicated times postinfection, cell lysates were collected, immersed in RNAlater RNA stabilizing reagent (Thermo Fisher Scientific, Waltham, MA), and stored at −80°C for processing. Total RNA was extracted as we have described previously (69, 70). Following RNA extraction, 500 ng of total RNA was reverse transcribed using random hexamer primers and murine leukemia virus reverse transcriptase using a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA) according to the manufacturer's instructions. Expression of gB, ICP0, and ICP4 was evaluated using the following custom primers and probes: gB-specific primers (forward, 5′-AACGCGACGCACATCAAG-3′; reverse, 5′-CTGGTACGCGATCAGAAAGC-3′, probe, 5′-6-carboxyfluorescein [FAM]-CAGCCGCAGTACTACC-3′), ICP0-specific primers (forward, 5′-CGGACACGGAACTGTTCGA-3′; reverse, 5′-CGCCCCCGCAACTG-3′; probe, 5′-FAM-CCCCATCCACGCCCTG-3′), and ICP4-specific primers (forward, 5′-GCGTCGTCGAGGTCGT-3′, reverse 5′-CGCGGAGACGGAGGAG-3′; probe, 5′-FAM-CACGACCCCGACCACC-3′) (amplicon length, 69 bp). As an internal control, glyceraldehyde-3-phosphate dehydrogenase primers (ASSAY I.D. m999999.15_G1; amplicon length, 107 bp; Applied Biosystems) were used.
Role of sncRNA1 and -2 sequences in HVEM promoter binding.
For in vivo analysis of putative sncRNA1 and -2 peptide binding to HVEM promoter, 293T cells were transfected with WT sncRNA1-pcDNA3.1-Flag, WT sncRNA2-pcDNA3.1-Flag, or the pcDNA3.1-Flag empty vector. Twenty-four hours after transfection, samples were processed for ChIP analysis using a ChIP-IT express kit (number 53008; Active Motif, Carlsbad, CA) as described by the manufacturer and as we have described previously (46). For in vitro peptide binding assay, 293T cell lysates were prepared and incubated with synthesized flag-tagged sncRNA1 and sncRNA2 peptides (MLGSYCLGGGSADYKDDDDK and MFLFLSDYKDDDDK, respectively; GenScript, Piscataway, NJ) for 4 h at 4°C and then processed for ChIP assay as described above. PCRs were prepared using the following primer sets: set 1 forward, 5′-CACAGATTCCTGTGGCTGGCCAC-3′; set 1 reverse, 5′-CTGCCCCTCCTGGTCCTGACTT-3′; set 2 forward, 5′-CAGGATGTGAGTGCACCAGG-3′; set 2 reverse, 5′-GTGGCCAGCCACAGGAATCTGTG-3′; set 3 forward, 5′-AAGTCAGGACCAGGAGGGGCAG-3′; set 3 reverse, 5′-AGGGAGACCTCCGGATGGAG-3′; set 4 forward, 5′-CTGTTCAGCAGAAGCTGAGATG-3′; set 4 reverse, 5′-AGCTCGGGAACCCAGCTAG-3′. Taq polymerase (New England Biolabs, Ipswich, MA) was used to amplify promoter regions.
Statistical analysis.
Student's t test, two-way analysis of variance (ANOVA) with Tukey’s multiple-comparison test, or one-way ANOVA with Dunnett’s multiple-comparison test were performed using the computer program Instat (GraphPad, San Diego, CA). Results were considered statistically significant when the P value was <0.05.
ACKNOWLEDGMENTS
This study was supported by Public Health Service NIH grants RO1 EY029160 and RO1 EY024649.
Footnotes
Supplemental material is available online only.
Contributor Information
Homayon Ghiasi, Email: ghiasih@CSHS.org.
Jae U. Jung, Lerner Research Institute, Cleveland Clinic
REFERENCES
- 1.Koujah L, Suryawanshi RK, Shukla D. 2019. Pathological processes activated by herpes simplex virus-1 (HSV-1) infection in the cornea. Cell Mol Life Sci 76:405–419. 10.1007/s00018-018-2938-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liesegang TJ, Melton LJ, III, Daly PJ, Ilstrup DM. 1989. Epidemiology of ocular herpes simplex. Incidence in Rochester, Minn, 1950 through 1982. Arch Ophthalmol 107:1155–1159. 10.1001/archopht.1989.01070020221029. [DOI] [PubMed] [Google Scholar]
- 3.Jaggi U, Varanasi SK, Bhela S, Rouse BT. 2018. On the role of retinoic acid in virus induced inflammatory response in cornea. Microbes Infect 20:337–345. 10.1016/j.micinf.2018.04.007. [DOI] [PubMed] [Google Scholar]
- 4.Mott KR, Bresee CJ, Allen SJ, BenMohamed L, Wechsler SL, Ghiasi H. 2009. Level of herpes simplex virus type 1 latency correlates with severity of corneal scarring and exhaustion of CD8+ T cells in trigeminal ganglia of latently infected mice. J Virol 83:2246–2254. 10.1128/JVI.02234-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perng GC, Dunkel EC, Geary PA, Slanina SM, Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. 1994. The latency-associated transcript gene of herpes simplex virus type 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J Virol 68:8045–8055. 10.1128/jvi.68.12.8045-8055.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perng GC, Ghiasi H, Slanina SM, Nesburn AB, Wechsler SL. 1996. The spontaneous reactivation function of the herpes simplex virus type 1 LAT gene resides completely within the first 1.5 kilobases of the 8.3- kilobase primary transcript. J Virol 70:976–984. 10.1128/JVI.70.2.976-984.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leib DA, Bogard CL, Kosz-Vnenchak M, Hicks KA, Coen DM, Knipe DM, Schaffer PA. 1989. A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J Virol 63:2893–2900. 10.1128/JVI.63.7.2893-2900.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perng GC, Jones C, Ciacci-Zanella J, Stone M, Henderson G, Yukht A, Slanina SM, Hofman FM, Ghiasi H, Nesburn AB, Wechsler SL. 2000. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science 287:1500–1503. 10.1126/science.287.5457.1500. [DOI] [PubMed] [Google Scholar]
- 9.Perng GC, Maguen B, Jin L, Mott KR, Osorio N, Slanina SM, Yukht A, Ghiasi H, Nesburn AB, Inman M, Henderson G, Jones C, Wechsler SL. 2002. A gene capable of blocking apoptosis can substitute for the herpes simplex virus type 1 latency-associated transcript gene and restore wild-type reactivation levels. J Virol 76:1224–1235. 10.1128/JVI.76.3.1224-1235.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henderson G, Peng W, Jin L, Perng GC, Nesburn AB, Wechsler SL, Jones C. 2002. Regulation of caspase 8- and caspase 9-induced apoptosis by the herpes simplex virus type 1 latency-associated transcript. J Neurovirol 8(Suppl 2):103–111. 10.1080/13550280290101085. [DOI] [PubMed] [Google Scholar]
- 11.Branco FJ, Fraser NW. 2005. Herpes simplex virus type 1 latency-associated transcript expression protects trigeminal ganglion neurons from apoptosis. J Virol 79:9019–9025. 10.1128/JVI.79.14.9019-9025.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin L, Perng GC, Mott KR, Osorio N, Naito J, Brick DJ, Carpenter D, Jones C, Wechsler SL. 2005. A herpes simplex virus type 1 mutant expressing a baculovirus inhibitor of apoptosis gene in place of latency-associated transcript has a wild-type reactivation phenotype in the mouse. J Virol 79:12286–12295. 10.1128/JVI.79.19.12286-12295.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin L, Carpenter D, Moerdyk-Schauwecker M, Vanarsdall AL, Osorio N, Hsiang C, Jones C, Wechsler SL. 2008. Cellular FLIP can substitute for the herpes simplex virus type 1 latency-associated transcript gene to support a wild-type virus reactivation phenotype in mice. J Neurovirol 14:389–400. 10.1080/13550280802216510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tormanen K, Allen S, Mott KR, Ghiasi H. 2019. The latency-associated transcript inhibits apoptosis via downregulation of components of the type I interferon pathway during latent herpes simplex virus 1 ocular infection. J Virol 93:e00103-19. 10.1128/JVI.00103-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allen SJ, Hamrah P, Gate DM, Mott KR, Mantopoulos D, Zheng L, Town T, Jones C, von Andrian UH, Freeman GJ, Sharpe AH, Benmohamed L, Ahmed R, Wechsler SL, Ghiasi H. 2011. The role of LAT in increased CD8+ T cell exhaustion in trigeminal ganglia of mice latently infected with herpes simplex virus type 1. J Virol 85:4184–4197. 10.1128/JVI.02290-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chentoufi AA, Kritzer E, Tran MV, Dasgupta G, Lim CH, Yu DC, Afifi RE, Jiang X, Carpenter D, Osorio N, Hsiang C, Nesburn AB, Wechsler SL, BenMohamed L. 2011. The herpes simplex virus 1 latency-associated transcript promotes functional exhaustion of virus-specific CD8+ T cells in latently infected trigeminal ganglia: a novel immune evasion mechanism. J Virol 85:9127–9138. 10.1128/JVI.00587-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wechsler SL, Nesburn AB, Watson R, Slanina S, Ghiasi H. 1988. Fine mapping of the major latency-related RNA of herpes simplex virus type 1 in humans. J Gen Virol 69:3101–3106. 10.1099/0022-1317-69-12-3101. [DOI] [PubMed] [Google Scholar]
- 18.Wechsler SL, Nesburn AB, Watson R, Slanina SM, Ghiasi H. 1988. Fine mapping of the latency-related gene of herpes simplex virus type 1: alternative splicing produces distinct latency-related RNAs containing open reading frames. J Virol 62:4051–4058. 10.1128/JVI.62.11.4051-4058.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rock DL, Nesburn AB, Ghiasi H, Ong J, Lewis TL, Lokensgard JR, Wechsler SL. 1987. Detection of latency-related viral RNAs in trigeminal ganglia of rabbits latently infected with herpes simplex virus type 1. J Virol 61:3820–3826. 10.1128/JVI.61.12.3820-3826.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zwaagstra JC, Ghiasi H, Slanina SM, Nesburn AB, Wheatley SC, Lillycrop K, Wood J, Latchman DS, Patel K, Wechsler SL. 1990. Activity of herpes simplex virus type 1 latency-associated transcript (LAT) promoter in neuron-derived cells: evidence for neuron specificity and for a large LAT transcript. J Virol 64:5019–5028. 10.1128/JVI.64.10.5019-5028.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deatly AM, Spivack JG, Lavi E, O'Boyle DR, Fraser NW. 1988. Latent herpes simplex virus type 1 transcripts in peripheral and central nervous system tissues of mice map to similar regions of the viral genome. J Virol 62:749–756. 10.1128/JVI.62.3.749-756.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jurak I, Kramer MF, Mellor JC, van Lint AL, Roth FP, Knipe DM, Coen DM. 2010. Numerous conserved and divergent microRNAs expressed by herpes simplex viruses 1 and 2. J Virol 84:4659–4672. 10.1128/JVI.02725-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. 2008. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 454:780–783. 10.1038/nature07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peng W, Vitvitskaia O, Carpenter D, Wechsler SL, Jones C. 2008. Identification of two small RNAs within the first 1.5-kb of the herpes simplex virus type 1-encoded latency-associated transcript. J Neurovirol 14:41–52. 10.1080/13550280701793957. [DOI] [PubMed] [Google Scholar]
- 25.Cui C, Griffiths A, Li G, Silva LM, Kramer MF, Gaasterland T, Wang XJ, Coen DM. 2006. Prediction and identification of herpes simplex virus 1-encoded microRNAs. J Virol 80:5499–5508. 10.1128/JVI.00200-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kramer MF, Jurak I, Pesola JM, Boissel S, Knipe DM, Coen DM. 2011. Herpes simplex virus 1 microRNAs expressed abundantly during latent infection are not essential for latency in mouse trigeminal ganglia. Virology 417:239–247. 10.1016/j.virol.2011.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen W, Silva M, Jaber T, Vitvitskaia O, Li S, Henderson G, Jones C. 2009. Two small RNAs encoded within the first 1.5 kilobases of the herpes simplex virus type 1 latency-associated transcript can inhibit productive infection and cooperate to inhibit apoptosis. J Virol 83:9131–9139. 10.1128/JVI.00871-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spear PG, Eisenberg RJ, Cohen GH. 2000. Three classes of cell surface receptors for alphaherpesvirus entry. Virology 275:1–8. 10.1006/viro.2000.0529. [DOI] [PubMed] [Google Scholar]
- 29.Satoh T, Arii J, Suenaga T, Wang J, Kogure A, Uehori J, Arase N, Shiratori I, Tanaka S, Kawaguchi Y, Spear PG, Lanier LL, Arase H. 2008. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 132:935–944. 10.1016/j.cell.2008.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taylor JM, Lin E, Susmarski N, Yoon M, Zago A, Ware CF, Pfeffer K, Miyoshi J, Takai Y, Spear PG. 2007. Alternative entry receptors for herpes simplex virus and their roles in disease. Cell Host Microbe 2:19–28. 10.1016/j.chom.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arii J, Goto H, Suenaga T, Oyama M, Kozuka-Hata H, Imai T, Minowa A, Akashi H, Arase H, Kawaoka Y, Kawaguchi Y. 2010. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 467:859–862. 10.1038/nature09420. [DOI] [PubMed] [Google Scholar]
- 32.Suenaga T, Satoh T, Somboonthum P, Kawaguchi Y, Mori Y, Arase H. 2010. Myelin-associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc Natl Acad Sci USA 107:866–871. 10.1073/pnas.0913351107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ware CF. 2011. The TNF receptor super family in immune regulation. Immunol Rev 244:5–8. 10.1111/j.1600-065X.2011.01065.x. [DOI] [PubMed] [Google Scholar]
- 34.Sedy JR, Gavrieli M, Potter KG, Hurchla MA, Lindsley RC, Hildner K, Scheu S, Pfeffer K, Ware CF, Murphy TL, Murphy KM. 2005. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat Immunol 6:90–98. 10.1038/ni1144. [DOI] [PubMed] [Google Scholar]
- 35.Cai G, Anumanthan A, Brown JA, Greenfield EA, Zhu B, Freeman GJ. 2008. CD160 inhibits activation of human CD4+ T cells through interaction with herpesvirus entry mediator. Nat Immunol 9:176–185. 10.1038/ni1554. [DOI] [PubMed] [Google Scholar]
- 36.Cheung TC, Oborne LM, Steinberg MW, Macauley MG, Fukuyama S, Sanjo H, D'Souza C, Norris PS, Pfeffer K, Murphy KM, Kronenberg M, Spear PG, Ware CF. 2009. T cell intrinsic heterodimeric complexes between HVEM and BTLA determine receptivity to the surrounding microenvironment. J Immunol 183:7286–7296. 10.4049/jimmunol.0902490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allen SJ, Rhode-Kurnow A, Mott KR, Jiang X, Carpenter D, Rodriguez-Barbosa JI, Jones C, Wechsler SL, Ware CF, Ghiasi H. 2014. Interactions between herpesvirus entry mediator (TNFRSF14) and latency-associated transcript during herpes simplex virus 1 latency. J Virol 88:1961–1971. 10.1128/JVI.02467-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoon M, Zago A, Shukla D, Spear PG. 2003. Mutations in the N termini of herpes simplex virus type 1 and 2 gDs alter functional interactions with the entry/fusion receptors HVEM, nectin-2, and 3-O-sulfated heparan sulfate but not with nectin-1. J Virol 77:9221–9231. 10.1128/jvi.77.17.9221-9231.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Montgomery RI, Warner MS, Lum BJ, Spear PG. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87:427–436. 10.1016/s0092-8674(00)81363-x. [DOI] [PubMed] [Google Scholar]
- 40.Connolly SA, Landsburg DJ, Carfi A, Wiley DC, Cohen GH, Eisenberg RJ. 2003. Structure-based mutagenesis of herpes simplex virus glycoprotein D defines three critical regions at the gD-HveA/HVEM binding interface. J Virol 77:8127–8140. 10.1128/jvi.77.14.8127-8140.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang S, Ljubimov AV, Jin L, Pfeffer K, Kronenberg M, Ghiasi H. 2018. Herpes simplex virus 1 latency and the kinetics of reactivation are regulated by a complex network of interactions between the herpesvirus entry mediator, its ligands (gD, BTLA, LIGHT, and CD160), and the latency-associated transcript. J Virol 92:e01451-18. 10.1128/JVI.01451-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang S, Hirose S, Ghiasi H. 2019. The absence of lymphotoxin-alpha, an HVEM ligand, affects HSV-1 infection in vivo differently than the absence of other HVEM cellular ligands. J Virol 93:e00707-19. 10.1128/JVI.00707-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tormanen K, Wang S, Jaggi U, Ghiasi H. 2020. Restoring herpesvirus entry mediator (HVEM) immune function in HVEM(-/-) mice rescues herpes simplex virus 1 latency and reactivation independently of binding to glycoprotein D. J Virol 94:e00700-20. 10.1128/JVI.00700-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stevens JG, Wagner EK, Devi-Rao GB, Cook ML, Feldman LT. 1987. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science 235:1056–1059. 10.1126/science.2434993. [DOI] [PubMed] [Google Scholar]
- 45.Phelan D, Barrozo ER, Bloom DC. 2017. HSV1 latent transcription and non-coding RNA: a critical retrospective. J Neuroimmunol 308:65–101. 10.1016/j.jneuroim.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 46.Matundan H, Ghiasi H. 2019. Herpes simplex virus 1 ICP22 suppresses CD80 expression by murine dendritic cells. J Virol 93:e01803-18. 10.1128/JVI.01803-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carpenter D, Henderson G, Hsiang C, Osorio N, BenMohamed L, Jones C, Wechsler SL. 2008. Introducing point mutations into the ATGs of the putative open reading frames of the HSV-1 gene encoding the latency associated transcript (LAT) reduces its anti-apoptosis activity. Microb Pathog 44:98–102. 10.1016/j.micpath.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Murphy KM, Nelson CA, Sedy JR. 2006. Balancing co-stimulation and inhibition with BTLA and HVEM. Nat Rev Immunol 6:671–681. 10.1038/nri1917. [DOI] [PubMed] [Google Scholar]
- 49.Ward-Kavanagh LK, Lin WW, Sedy JR, Ware CF. 2016. The TNF receptor superfamily in co-stimulating and co-inhibitory responses. Immunity 44:1005–1019. 10.1016/j.immuni.2016.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li R, Zhu H, Luo Y. 2016. Understanding the functions of long non-coding RNAs through their higher-order structures. Int J Mol Sci 17:702. 10.3390/ijms17050702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang F, Tanasa B, Micheletti R, Ohgi KA, Aggarwal AK, Rosenfeld MG. 2021. Shape of promoter antisense RNAs regulates ligand-induced transcription activation. Nature 595:444–449. 10.1038/s41586-021-03589-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lagunoff M, Roizman B. 1994. Expression of a herpes simplex virus 1 open reading frame antisense to the gamma(1)34.5 gene and transcribed by an RNA 3' coterminal with the unspliced latency-associated transcript. J Virol 68:6021–6028. 10.1128/JVI.68.9.6021-6028.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Naito J, Mukerjee R, Mott KR, Kang W, Osorio N, Fraser NW, Perng GC. 2005. Identification of a protein encoded in the herpes simplex virus type 1 latency associated transcript promoter region. Virus Res 108:101–110. 10.1016/j.virusres.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 54.Doerig C, Pizer LI, Wilcox CL. 1991. Detection of the latency-associated transcript in neuronal cultures during the latent infection with herpes simplex virus type 1. Virology 183:423–426. 10.1016/0042-6822(91)90159-9. [DOI] [PubMed] [Google Scholar]
- 55.Doerig C, Pizer LI, Wilcox CL. 1991. An antigen encoded by the latency-associated transcript in neuronal cell cultures latently infected with herpes simplex virus type 1. J Virol 65:2724–2727. 10.1128/JVI.65.5.2724-2727.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lock M, Miller C, Fraser NW. 2001. Analysis of protein expression from within the region encoding the 2.0-kilobase latency-associated transcript of herpes simplex virus type 1. J Virol 75:3413–3426. 10.1128/JVI.75.7.3413-3426.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Randall G, Lagunoff M, Roizman B. 1997. The product of ORF O located within the domain of herpes simplex virus 1 genome transcribed during latent infection binds to and inhibits in vitro binding of infected cell protein 4 to its cognate DNA site. Proc Natl Acad Sci USA 94:10379–10384. 10.1073/pnas.94.19.10379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thomas SK, Gough G, Latchman DS, Coffin RS. 1999. Herpes simplex virus latency-associated transcript encodes a protein which greatly enhances virus growth, can compensate for deficiencies in immediate-early gene expression, and is likely to function during reactivation from virus latency. J Virol 73:6618–6625. 10.1128/JVI.73.8.6618-6625.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fareed MU, Spivack JG. 1994. Two open reading frames (ORF1 and ORF2) within the 2.0-kilobase latency-associated transcript of herpes simplex virus type 1 are not essential for reactivation from latency. J Virol 68:8071–8081. 10.1128/JVI.68.12.8071-8081.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Farrell MJ, Hill JM, Margolis TP, Stevens JG, Wagner EK, Feldman LT. 1993. The herpes simplex virus type 1 reactivation function lies outside the latency-associated transcript open reading frame ORF-2. J Virol 67:3653–3655. 10.1128/JVI.67.6.3653-3655.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Perng GC, Jones C. 2010. Towards an understanding of the herpes simplex virus type 1 latency-reactivation cycle. Interdiscip Perspect Infect Dis 2010:262415. 10.1155/2010/262415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Drolet BS, Perng GC, Cohen J, Slanina SM, Yukht A, Nesburn AB, Wechsler SL. 1998. The region of the herpes simplex virus type 1 LAT gene involved in spontaneous reactivation does not encode a functional protein. Virology 242:221–232. 10.1006/viro.1997.9020. [DOI] [PubMed] [Google Scholar]
- 63.Henderson G, Jaber T, Carpenter D, Wechsler SL, Jones C. 2009. Identification of herpes simplex virus type 1 proteins encoded within the first 1.5 kb of the latency-associated transcript. J Neurovirol 15:439–448. 10.3109/13550280903296353. [DOI] [PubMed] [Google Scholar]
- 64.da Silva LF, Jones C. 2013. Small non-coding RNAs encoded within the herpes simplex virus type 1 latency associated transcript (LAT) cooperate with the retinoic acid inducible gene I (RIG-I) to induce beta-interferon promoter activity and promote cell survival. Virus Res 175:101–109. 10.1016/j.virusres.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Silva LF, Jones C. 2012. Two microRNAs encoded within the bovine herpesvirus 1 latency-related gene promote cell survival by interacting with RIG-I and stimulating NF-kappaB-dependent transcription and beta interferon signaling pathways. J Virol 86:1670–1682. 10.1128/JVI.06550-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chu C, Qu K, Zhong FL, Artandi SE, Chang HY. 2011. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell 44:667–678. 10.1016/j.molcel.2011.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Statello L, Guo CJ, Chen LL, Huarte M. 2021. Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol 22:96–118. 10.1038/s41580-020-00315-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ghiasi H, Kaiwar R, Slanina S, Nesburn AB, Wechsler SL. 1994. Expression and characterization of baculovirus expressed herpes simplex virus type 1 glycoprotein L. Arch Virol 138:199–212. 10.1007/BF01379126. [DOI] [PubMed] [Google Scholar]
- 69.Mott KR, Perng GC, Osorio Y, Kousoulas KG, Ghiasi H. 2007. A recombinant herpes simplex virus type 1 expressing two additional copies of gK is more pathogenic than wild-type virus in two different strains of mice. J Virol 81:12962–12972. 10.1128/JVI.01442-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mott KR, Osorio Y, Brown DJ, Morishige N, Wahlert A, Jester JV, Ghiasi H. 2007. The corneas of naive mice contain both CD4+ and CD8+ T cells. Mol Vis 13:1802–1812. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 and S2<br>. Download jvi.01985-21-s0001.pdf, PDF file, 0.1 MB (135.5KB, pdf)