

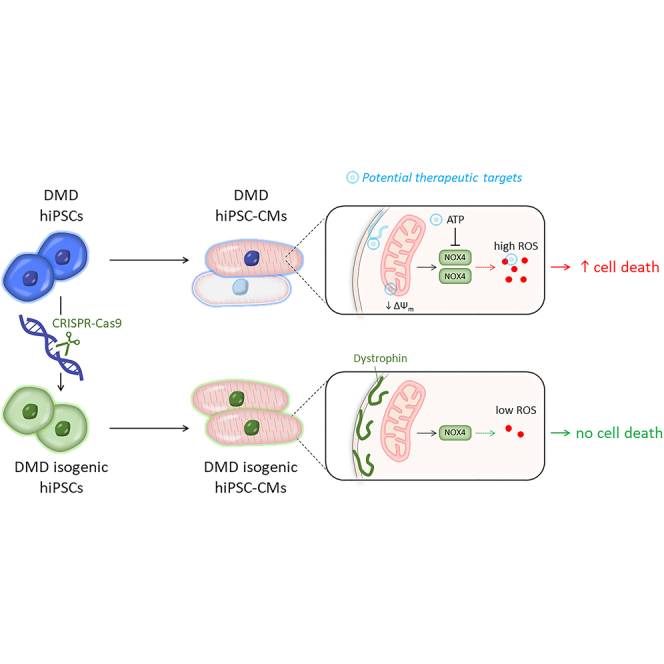
Summary
Duchenne muscular dystrophy (DMD) is a progressive muscle disorder caused by mutations in the Dystrophin gene. Cardiomyopathy is a major cause of early death. We used DMD-patient-specific human induced pluripotent stem cells (hiPSCs) to model cardiomyopathic features and unravel novel pathologic insights. Cardiomyocytes (CMs) differentiated from DMD hiPSCs showed enhanced premature cell death due to significantly elevated intracellular reactive oxygen species (ROS) resulting from depolarized mitochondria and increased NADPH oxidase 4 (NOX4). CRISPR-Cas9 correction of Dystrophin restored normal ROS levels. ROS reduction by N-acetyl-L-cysteine (NAC), ataluren (PTC124), and idebenone improved hiPSC-CM survival. We show that oxidative stress in DMD hiPSC-CMs was counteracted by stimulating adenosine triphosphate (ATP) production. ATP can bind to NOX4 and partially inhibit the ROS production. Considering the complexity and the early cellular stress responses in DMD cardiomyopathy, we propose targeting ROS production and preventing detrimental effects of NOX4 on DMD CMs as promising therapeutic strategy.
Keywords: Duchenne muscular dystrophy, cardiomyopathy, hiPSC modeling, CRISPR-Cas9, NADPH oxidase NOX4
Graphical abstract
Highlights
-
•
Human iPSC-based in vitro model for studying Duchenne cardiomyopathy
-
•
Premature cell death of untreated DMD hiPSC-CMs
-
•
DMD hiPSC-CMs show increased oxidative stress levels and NOX4
-
•
Inhibition of ROS-producing NOX4 by idebenone is beneficial for disease phenotype
In this article, Duelen and colleagues show that a hiPSC-based in vitro model reveals ROS-producing NOX4 as an important contributor of oxidative stress in Duchenne cardiomyopathy. They provide evidence that ROS reduction by a NAC scavenger, partial Dystrophin re-expression by ataluren (PTC124), and enhancing mitochondrial electron transport chain function by idebenone improved cell survival of DMD hiPSC-CMs.
Introduction
The shortage of human cardiac cell sources has challenged cardiovascular disease modeling and drug development. The generation of functional cardiomyocytes (CMs) differentiated from human induced pluripotent stem cells (hiPSCs) overcomes current limitations and offers an extraordinary platform to develop hiPSC-based models to study the genetic disease phenotype of cardiomyopathic pathologies in vitro (Mummery et al., 2012; Takahashi and Yamanaka, 2006).
Mutations in the Dystrophin gene cause the X-linked disorder Duchenne muscular dystrophy (DMD), the most common and severe phenotype among the muscular dystrophies (Mercuri et al., 2019). Most DMD patients develop adverse myocardial remodeling and chronic cardiomyopathy, a major cause of morbidity and early mortality (Emery, 2002). With the current standards of care, the median life expectancy at birth in individuals with DMD has improved during the last decades and ranges between 21.0 and 39.6 years (Landfeldt et al., 2020).
The Dystrophin protein has a crucial role during muscle contraction and stretch. Loss of function or absence lead to myocyte sarcolemma instability during contraction-relaxation cycles, making myocytes more susceptible to stretch-induced damage and necrosis (Davies and Nowak, 2006). The signaling-mediated roles of Dystrophin and the associated dystrophin glycoprotein complex are not yet fully understood (Allen et al., 2016). The pathophysiological role of Dystrophin in the heart is poorly defined, and multiple pathways are involved including dysregulation of calcium (Ca2+) homeostasis, oxidative stress, inflammation, and functional ischemia.
Oxidative stress is involved in the pathogenesis of heart failure. However, clinical trials using antioxidants have shown limited success (Sesso et al., 2008). Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) family enzymes generate reactive oxygen species (ROS) in a highly regulated manner, modulating several physiological aspects such as host defense, posttranslational processing of proteins, cellular signaling, regulation of gene expression, and cell differentiation (Vermot et al., 2021). However, NOX family enzymes also contribute to a wide range of pathological processes, including, in particular, cardiovascular diseases. The NOX4 isoform is predominantly expressed in CMs, although the precise location remains controversial. It is constitutively active at low levels, inducing cardioprotective effects under chronic stress. The exact role of NOX4 in CMs is still not clear, even though high levels of NOX4 could have severe detrimental effects (Ago et al., 2010; Varga et al., 2017; Zhang et al., 2013) including excessive ROS production (Spurney et al., 2008). Thus, targeting NOX isoforms may be a useful therapeutic strategy.
Several innovative therapeutic approaches focus on targeting the primary defect such as restoring the function or expression of Dystrophin through exon skipping (Wu et al., 2008), ribosomal readthrough technology (Welch et al., 2007), or gene (Moretti et al., 2020) and cell therapy (Bajek et al., 2015). Recent technological breakthroughs in genome editing successfully enabled the correction of the genetic mutation (Calos, 2016). In addition, compounds targeting downstream pathophysiology are under investigation in clinical trials (Verhaart and Aartsma-Rus, 2019). 2,3-dimethoxy-5-methyl-6-(10-hydroxy)decyl-1,4-benzoquinone (idebenone), a synthetic analogue of coenzyme Q10, has a dual mode of action. First, it detoxifies ROS by donating electrons to produce non-toxic reaction products. Second, it donates electrons directly to complex III of the mitochondrial electron transport chain (ETC), which restores electron flow, proton pumping activity of complexes III and IV, and adenosine triphosphate (ATP) production by complex V. Phase 2/3 randomized, placebo-controlled trials have demonstrated a beneficial role of idebenone in DMD patients (Buyse et al., 2015).
In this study, we used DMD-patient-specific hiPSC-derived CMs (hiPSC-CMs) to model cardiomyopathic features to explore pathological mechanisms. We observed mitochondrial dysfunction and high intracellular ROS concentrations in DMD hiPSC-CMs due to significantly increased NOX4. These features were not present in CRISPR-Cas9 genetically corrected DMD isogenic hiPSC-CMs. Additionally, by administration of the ROS scavenger N-acetyl-L-cysteine (NAC), the readthrough chemical drug ataluren (PTC124), or the synthetic benzoquinone idebenone, we observed beneficial outcomes regarding the survival and function of differentiated DMD hiPSC-CMs.
In conclusion, using DMD-patient-derived hiPSCs, we established an in vitro model to recapitulate DMD heart disease phenotypes and to study novel disease mechanisms that might become interesting therapeutic targets for cardiomyopathy in DMD patients.
Results
Generation of integration-free DMD hiPSCs
To obtain an unlimited cell source of CMs, recapitulating aspects of a single-gene disease phenotype, hiPSC lines were generated from human dermal fibroblasts (hFs) and human peripheral blood mononuclear cells (hPBMCs) obtained from DMD patients with known Dystrophin mutations (Table S1). Somatic cells were reprogrammed toward a pluripotent state using integration-free Sendai virus (SeV) vectors (Figures S1A–S1C), which expressed the OSKM (OCT3/4, SOX2, KLF4, and c-MYC) pluripotency markers. Subcutaneously injected DMD hiPSC lines into immunodeficient mice displayed teratoma formation, showing the differentiation capacity into all three developmental germ layers (ectoderm, mesoderm, and endoderm; Figures S1D and S1E). Furthermore, a detailed pluripotency analysis for related genes and proteins is given in the Supplemental information (Figures S1F and S1G). Three control hiPSC lines were used, which were generated from healthy donors with no neuromuscular disorders (Table S1).
CRISPR-Cas9-mediated correction of nonsense mutation in Dystrophin gene
Additionally, we created an isogenic control line to exclude genetic background variability. The isogenic control line was generated from DMD patient hiPSCs using CRISPR-Cas9 technology that were characterized by a genetic point mutation in exon 35 (c.4,996C>T; (p.Arg1,666X)) of the Dystrophin gene, resulting in a premature stop codon and, consequently, in the complete absence of a functional Dystrophin protein (Figure 1A). To restore the full-length expression of the Dystrophin gene, two 20 nt single-guide RNAs (sgRNAs) were designed to induce Cas9-mediated double-stranded breaks (DSBs) in the genomic DNA of the Dystrophin-deficient hiPSCs (Figure 1B). sgRNA specificity and CRISPR-Cas9 DSB cutting were evaluated in HEK293T cells by the appearance of non-homologous end joining (NHEJ) events after transfection of the sgRNA-Cas9 plasmids (Figures S2A and S2B). Cas9-mediated genome editing was performed via homology-directed repair (HDR), using a plasmid-based donor repair template with homology arm regions for the Dystrophin gene exon of interest, in order to substitute the premature stop codon into the original amino acid codon for arginine (Figure 1B). Sequencing analysis of exon 35 of the Dystrophin gene confirmed CRISPR-Cas9 correction of the DMD hiPSC line, further indicated as DMD isogenic control (Figure 1C). CRISPR-Cas9 off-target events were analyzed based on the sequence homology of sgRNAs (Figure S2C), and detailed comparative genomic hybridization (CGH) molecular karyotyping did not show additional chromosomal abnormalities due to unwanted Cas9-mediated DSB cuts (Figure S2D). To demonstrate that gene editing did not influence the pluripotency state of the DMD isogenic control line, pluripotency genes (c-MYC, GDF-3, KLF4, NANOG, OCT4, REX1, SOX2, and hTERT) and proteins (OCT4, NANOG, SSEA4, SOX2, TRA-1-60, and LIN28) were analyzed in several undifferentiated human pluripotent stem cell (hPSC) lines (Figures S1F and S1G). Furthermore, immunofluorescent staining showed the expression of the Dystrophin protein (green) in differentiated DMD hiPSC-CMs (cardiac troponin T [cTnT], red and Hoechst, blue) after CRISPR-Cas9 correction (Figure 1D).
Figure 1.

DMD hiPSC CRISPR-Cas9 gene editing of a nonsense mutation in exon 35 (c.4,996C>T; (p.Arg1,666X)) of the Dystrophin gene
(A) Schematic representation of the human Dystrophin gene sequence (top, transcript variant Dp427m) and the encoded Dystrophin protein (bottom, isoform Dp427m). The genetic point mutation is located in exon 35 of the Dystrophin gene, resulting in a premature stop codon.
(B) The 20 nt sgRNA (ATTTAACCACTCTTCTGCTC) to induce the Cas9-mediated DSB (indicated as black triangles). The donor repair template containing the genetic correction of the nonsense mutation in the Dystrophin gene is also shown.
(C) DNA sequencing of the mutated region of interest of Dystrophin before (DMD diseased) and after (DMD isogenic) CRISPR-Cas9 gene editing.
(D) Immunofluorescent staining showing the expression of Dystrophin protein levels (green) in differentiated DMD hiPSC-CMs (cTnT, red and Hoechst, blue) after CRISPR-Cas9-mediated correction. Scale bar: 50 μm. White boxes with corresponding insets are at a higher magnification. Scale bar: 10 μm.
See also Figures S1F, S1G, and S2A–S2D.
hiPSC-CMs to model diseased heart phenotype in DMD
Burridge et al. developed a fully chemically defined and small-molecule-based cardiac differentiation protocol that is effective for several hiPSC lines and has a high yield of mainly ventricular-like CMs (Burridge et al., 2014). Here, we differentiated control and DMD hiPSC lines to CMs, according to this monolayer-based cardiac differentiation strategy (Burridge et al., 2014), with additional 3D maturation in fibrin-based engineered heart tissue (EHT) constructs (Figures 2A and 2B) (Breckwoldt et al., 2017). During the early phases of cardiac differentiation, hiPSCs were treated with chemical Wnt signaling mediators (CHIR99021 and IWR-1) to obtain high CM yields (Figure 2A). Additional 3D maturation of hiPSC-CMs could significantly increase the expression of the cardiac-specific maturation isoforms MYL2 and TNNI3 (Figure 2C). Immunostaining of cTnT-positive hiPSC-CMs additionally matured in 3D EHTs showed a structural, aligned orientation due to the mechanical loading of the flexible microposts compared with those matured in classical 2D monolayer-based differentiation systems (Figure 2D). Importantly, differentiated hiPSC-CMs from DMD patients manifested pathologic features of cardiac involvement. They exhibited a significant reduction of the L-type Ca2+ current, indicating abnormal Ca2+ homeostasis (Figure 2E), and representative action potential (AP) recordings from DMD hiPSC-CMs displayed arrhythmogenic firing patterns including delayed afterdepolarizations (DADs) and oscillatory prepotentials (OPPs; Figure 2F), as reported in literature (Eisen et al., 2019; Lin et al., 2015; Pioner et al., 2020; Sato et al., 2019). Furthermore, patch-clamp recordings at day 24 of differentiation showed significantly longer mean AP durations at 90% repolarization (APD90) in DMD compared with in control hiPSC-CMs (Figure 2G). Other electrophysiological parameters including AP amplitude, resting membrane potential (RMP), cell capacitance, and beating frequency did not show significant differences (Figure 2H).
Figure 2.

Characterization of the hiPSC-CM differentiation protocol
(A) Schematic representation of the cardiac differentiation protocol. hiPSCs were differentiated to CMs in a monolayer cardiac differentiation protocol using chemical Wnt signaling mediators (CHIR99021 and IWR-1) and, eventually, further matured into 3D EHT constructs based on fibrinogen and thrombin polymerization.
(B) Representative example of 2D monolayer-based cardiac differentiation (left panel) and 3D mini-EHT construct between two flexible microposts positioned 7 mm from each other (right panel).
(C) Normalized gene expression ratios for isoforms of Myosin Heavy Chain (MYH7/MYH6), Myosin Light Chain (MYL2/MYL7), and Cardiac Troponin I (TNNI3/TNNI1) after 15 and 30 days of differentiation. Data are representative of three independent experiments (n = 3), and values are expressed as mean ± SEM. Significance of the difference is indicated as follows: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
(D) Immunostaining of cardiac troponin T (cTnT) -positive CMs (cTnT, red and Hoechst, blue) in monolayer-based cardiac differentiation (2D) or EHT constructs (3D). White dotted lines indicate the borders of the 3D EHT constructs. Scale bar: 50 μm.
(E) Voltage-current relation curve of the L-type Ca2+ current (pA/pF) assessed after whole-cell patch-clamp configuration. Each data point indicated biological replicates (DMD: N = 13, DMD isogenic: N = 7, healthy: N = 11), and values are expressed as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001 (DMD versus DMD isogenic control) or $p < 0.05, $$p < 0.01, $$$p < 0.001, and $$$$p < 0.0001 (DMD versus healthy control).
(F) Representative AP recordings. DMD hiPSC-CMs displayed arrhythmogenic firing patterns including DADs and OPPs.
(G) Patch-clamp recordings at day 24 of differentiation for mean APD90 (ms). Additional measurements were performed with di-4-ANEPPS (gray dots).
(H) Patch-clamp recordings for AP amplitude (mV), RMP (mV), cell capacitance (pF), and beating frequency (Hz). Each data point indicates biological replicates (DMD: N = 19, DMD isogenic: N = 7, healthy: N = 8). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
Enhanced cell death and excessive intracellular ROS levels in DMD hiPSC-CM cultures
The absence of the Dystrophin protein in differentiated hiPSC-CMs from DMD patients results in the progressive loss of CMs (Davies and Nowak, 2006; Lin et al., 2015). Here, we wanted to identify novel pathological cues that caused decreased cell survival of DMD hiPSC-CMs. We mainly used DMD hiPSCs that were characterized by the nonsense mutation in exon 35 (c.4,996C>T; (p.Arg1,666X)) of the Dystrophin gene (DMD #2 in Table S1). This DMD line represents a subgroup of DMD patients (approximately 13%) that is responsive to the readthrough chemical drug ataluren (PTC124; Figures 3A–3C). Cell death was examined by flow cytometric analyses using annexin V and 7-amino-actinomycin D (7AAD). DMD hiPSC-CMs underwent accelerated cell death compared with corresponding DMD isogenic and healthy controls (Figures 4A and S3A, left panels). A remarkable percentage of DMD hiPSC-CMs had high intracellular ROS concentrations compared with those found in controls (Figures 4B and S3A, middle panels). Moreover, the intracellular ROS content (mean fluorescence intensity [MFI]) in DMD hiPSC-CMs was significantly higher (Figures 4C and S3A, right panels). Upon treatment with NAC, PTC124 (alone or in combination), or idebenone, DMD hiPSC-CMs showed increased cell survival (Figures 4A and S3A, left panels) and reduced intracellular ROS levels (Figures 4B and S3A, middle panels) compared with those in untreated DMD hiPSC-CMs. The specificity of the drug effect on CM death and intracellular ROS levels of the experimental groups is shown in the Supplemental information (Figures S4A–S4D). Taken together, these results show increased intracellular ROS levels in DMD hiPSC-CMs. Interestingly, NAC, PTC124, and idebenone had beneficial effects on the cell survival, although idebenone exhibited superior effects in DMD cultures.
Figure 3.

Dystrophin re-expression in DMD hiPSC-CMs after PTC124 treatment
(A) Dystrophin gene expression profiles in DMD hiPSC-CMs, characterized by a genetic point mutation in exon 35 (c.4,996C>T; (p.Arg1,666X)) of the Dystrophin gene, upon NAC, PTC124, and idebenone addition. Each data point is represented as ΔCt and normalized for the housekeeping genes (GAPDH and RPL13a). Data are representative of five or more independent experiments (n ≥ 5), and values are expressed as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001 versus subjects within the treatment condition.
(B) Immunostaining at day 24 of differentiation demonstrating Dystrophin protein re-expression (green) upon PTC124 treatment in cTnT-positive DMD and control hiPSC-CMs (cTnT, red and Hoechst, blue). Scale bar: 100 μm.
(C) Western blot analysis quantifying Dystrophin proteins in ACTN2-positive DMD and control hiPSC-CMs, normalized to the loading protein ACTB.
Figure 4.

Characterization of the cardiomyopathic phenotype in vitro of DMD hiPSC-CMs, showing premature cell death, depolarized mitochondria, and increased intracellular ROS levels, which were counteracted by NAC, ataluren (PTC124), and idebenone
(A–C) Flow cytometric quantification at day 15 of cardiac differentiation showing the percentage of cell death of signal-regulatory protein alpha (SIRPA)-positive hiPSC-CMs (A), the percentage of CMs with high intracellular ROS levels (B), and the MFI of intracellular ROS in CMs (C) in conditions with (NAC, PTC124, and idebenone) or without (untreated) treatments. Data are representative of four independent experiments (n = 4), and values are reported as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
(D) Immunostaining of the fluorescent voltage-sensitive dye JC-1 was used to determine ΔΨm and mitochondrial health in 15-day-old differentiated hiPSC-CMs. Untreated DMD hiPSC-CMs were characterized by mitochondrial depolarization, as indicated by the decrease in mitochondrial aggregates (JC-1 red, top panels) and the increase in mitochondrial monomers (JC-1 green, middle panels) with respect to treated DMD hiPSC-CMs and controls. Corresponding histograms (bottom panels) showed the JC-1 fluorescence intensity ratios (aggregates/monomers). Scale bar: 5 μm.
(E) Representative flow cytometric analyses at day 15 of differentiation for JC-1 aggregates (phycoerythrin [PE]) and JC-1 monomers (fluorescein isothiocyanate [FITC]) in DMD hiPSC-CMs upon treatment. Data are representative of four independent experiments (n = 4).
(F) Flow cytometric analyses at day 15 of differentiation showing the mitochondrial superoxide production (MitoSOX; PE) in depolarized DMD mitochondria compared with in DMD isogenic and healthy controls. Data are representative of four independent experiments (n = 4). Flow cytometry data are reported as mean ± SEM.
See also Figures S3A–S3E and S4A–S4H.
Dystrophin-deficient hiPSC-CMs are characterized by depolarized mitochondria
DMD pathology is accompanied by abnormal intracellular Ca2+ handling and the accumulation of dysfunctional mitochondria with defective structures (Timpani et al., 2015). A distinctive feature of early phase cell death is the loss of the membrane potential of active mitochondria (ΔΨm) (Zorova et al., 2018). The carbocyanine compound JC-1, a fluorescent voltage-sensitive dye with membrane-permeant fluorescent lipophilic cationic properties (Mathur et al., 2000), was used to determine ΔΨm and mitochondrial health. Consistently with the previously observed accelerated death of untreated DMD hiPSC-CMs, these cultures were characterized by mitochondrial depolarization, indicated by the decrease in the red (aggregates)/green (monomers) JC-1 fluorescence intensity ratio (Figures 4D, 4E, and S3B). Interestingly, the combinatorial treatment of NAC and PTC124, as well idebenone treatment, displayed significantly beneficial effects on ΔΨm with respect to untreated DMD hiPSC-CMs. Furthermore, flow cytometric analyses confirmed a significant increased superoxide production in depolarized mitochondria compared with in controls (Figures 4F and S3C). No significant differences were observed for mitochondrial content upon the different treatments (Figures S3D and S3E). The specificity of the drug effect on ΔΨm and on the mitochondrial superoxide concentrations of the experimental groups is shown in the Supplemental information (Figures S4E–S4H). Taken together, these results indicate dysfunctional depolarized mitochondria in DMD hiPSC-CMs, which could lead to excessive ROS leakage. The combined treatment of NAC and PTC124, as well idebenone treatment, could rescue this condition.
NOX4 is overexpressed in DMD hiPSC-CMs
Several independent studies have reported increased NOX4 expression and activity in chronic heart failure, supporting the clinical relevance, although the role of NOX4 in CMs is still unclear (Ago et al., 2010; Spurney et al., 2008; Varga et al., 2017; Zhang et al., 2013). Here, NOX2 and NOX4, the predominantly expressed isoforms of the ROS-producing NOX family enzymes in the heart, were investigated. Gene expression profiles did not reveal a differential expression for NOX2 and accessory regulatory subunits (p47phox, p67phox, RAC2, and RAC3; Figure 5A). Interestingly, NOX4 and its regulatory subunit p22phox were significantly upregulated in DMD hiPSC-CMs. Moreover, DMD hiPSC-CMs treated with PTC124 alone or in combination with NAC exhibited decreased NOX4 and p22phox gene levels. In contrast, upon idebenone treatment, no reduction was observed in the expression of both genes. Flow cytometric analyses demonstrated a significant increased percentage of NOX4-positive DMD hiPSC-CMs compared with DMD isogenic and healthy controls (Figures 5B and 5C). The percentage of NOX4-positive DMD hiPSC-CMs was reduced upon idebenone treatment. The specificity of the drug on the expression of NOX4 among the experimental groups is shown in the Supplemental information (Figures S4I and S4J). Western blot analysis confirmed the increased protein levels of NOX4 in DMD hiPSC-CMs (Figure 5D). Upon idebenone addition, DMD hiPSC-CMs showed a downregulation of NOX4, as also observed in controls. These data demonstrate a significantly increased NOX4 expression in DMD hiPSC-CMs that upon treatment with idebenone could be reverted to basal levels.
Figure 5.

Increased expression levels of the ROS-producing NOX family enzyme NOX4 and its accessory regulatory subunit p22phox in Dystrophin-deficient hiPSC-CM cultures
(A) Gene expression profiles at day 24 of cardiac differentiation of NOX2 and NOX4, and the regulatory subunits (p22phox, p47phox, p67phox, RAC1, RAC2, and RAC3) in DMD, DMD isogenic, and healthy control hiPSC-CMs upon treatment with NAC, PTC124, and idebenone. Each data point is represented as ΔCt and is normalized for the housekeeping genes (GAPDH and RPL13a). Data are representative of five or more independent experiments (n ≥ 5), and values are expressed as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001 versus subjects within the treatment condition or $p < 0.05, $$p < 0.01, $$$p < 0.001, and $$$$p < 0.0001 versus treatment conditions within the subject group.
(B) Representative flow cytometric analyses at day 15 of differentiation showing the percentage of NOX4 (APC) protein expression in SIRPA (PE)-positive DMD hiPSC-CMs upon treatment. Data are representative of three independent experiments (n = 3). Flow cytometry data are reported as mean ± SEM.
(C) Flow cytometric quantification at day 15 of differentiation of the percentage of SIRPA-positive hiPSC-CMs expressing NOX4 upon treatment. Data are representative of three independent experiments (n = 3), and values are expressed as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
(D) Western blot analysis quantifying the protein expression levels of NOX4 in 15-day-old differentiated DMD and control hiPSC-CMs, normalized to the loading protein ACTB. See also Figures S4I and S4J.
Additionally, we demonstrated that the NOX4 upregulation in DMD hiPSC-CMs (DMD #2 in Table S1) was not a common downstream pathway of cell death. Therefore, we preincubated hiPSC-CMs with 1 μM staurosporine (STS), a potent cell death inducer, for 6 h (Belmokhtar et al., 2001), and we did not observe any increase in the NOX4 expression (Figures S5A and S5B). Interestingly, by analyzing ΔΨm and the mitochondrial superoxide production in various DMD-patient-specific hiPSC-CM lines (DMD #2, DMD #5, and DMD #6 in Table S1), we could observe an association between the levels of mitochondrial depolarization and ROS production with the gene and protein levels of NOX4, suggesting a crucial role of NOX4 (Figures S6A–S6G).
Idebenone stimulates ATP production in depolarized mitochondria, ameliorating NOX4-mediated ROS overproduction
Overall, oxidative stress, in synergy with intracellular Ca2+ overload, results in the progressive worsening of DMD cardiomyopathy (Allen et al., 2016). We hypothesized that Dystrophin gene mutations elicit excessive ROS generation via the mitochondrial ETC of depolarized mitochondria and via a NOX4-based NADPH-dependent process. To assess whether increased NOX4 could contribute to elevated intracellular ROS concentrations, NOX4 mRNA levels were transiently degraded by the addition of Antisense LNA GapmeRs to the DMD hiPSC-CM cultures (Figure 6A, left panel). Antisense LNA GapmeRs targeting MALAT1 mRNA were used as positive controls (Figure 6A, right panel). Interestingly, transient GapmeR-induced NOX4 mRNA degradation significantly reduced NOX4 activity, as monitored through changes in NADPH absorption (Figure 6B) (Shanmugasundaram et al., 2017). DMD hiPSC-CMs exhibited significantly elevated NOX4 activity compared with controls (Figure 6C). However, when idebenone was added to DMD hiPSC-CM cultures, the NOX4 NADPH-dependent ROS production was significantly reduced in isolated mitochondria (Figure 6C) and in the total CM fraction (Figure S7A). Moreover, idebenone restored ATP levels due to its electron donating property for mitochondrial ETC stimulation (Figures 6D and S7B).
Figure 6.

Idebenone could counteract the oxidative stress in DMD hiPSC-CMs through ATP stimulation of the mitochondrial ETC, which, in turn, reduced ROS-producing NOX4 activity
(A) Quantitative RT-PCR of NOX4 gene expression levels after the addition of NOX4-targeted Antisense LNA GapmeRs to the DMD hiPSC-CM cultures (left panel). As a positive control for the efficiency of the Antisense LNA GapmeRs, MALAT1 levels were determined after the addition of MALAT1-targeted Antisense LNA GapmeRs (right panel). Each data point is represented as ΔCt and is normalized for the housekeeping genes (GAPDH and RPL13a). Data are representative of three independent experiments (n = 3), and values are expressed as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
(B) Quantification of the NOX4 ROS production, measured via the NADPH-dependent ROS generation, in the isolated mitochondrial fraction of DMD hiPSC-CMs after a 6-day preincubation with GapmeRs. Each data point is represented as a percentage (%) and is normalized to the mitochondrial fraction of the untreated DMD hiPSC-CMs (vehicle).
(C) Quantification of the NADPH-dependent ROS production of NOX4 in the mitochondrial fraction of DMD hiPSC-CMs with or without idebenone treatment compared with in DMD isogenic and healthy controls.
(D) ATP luminescence detection showing the effect of idebenone treatment on the mitochondrial ATP levels in DMD hiPSC-CMs.
(E) Quantification of the ROS-producing NOX4 activity after 2.5 mM ATP addition in DMD hiPSC-CM and control cultures. Each data point is represented as a percentage (%) and is normalized to the mitochondrial fraction of the untreated DMD hiPSC-CMs.
(F) Quantification of the NADPH-dependent ROS production of NOX4 in the mitochondrial fraction of DMD hiPSC-CMs upon 2.5 mM ATP addition with or without idebenone treatment. Each data point is represented as a percentage (%) and is normalized to the mitochondrial fraction of the idebenone-treated DMD hiPSC-CM cultures. Data are representative of four or six independent experiments (n = 4 or n = 6), and values are expressed as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001. Colored rectangles represent the independent experiments.
See also Figures S7A–S7H.
Recent studies have identified an ATP-binding motif within NOX4 through which ATP, upon binding, could regulate NOX4 activity (Shanmugasundaram et al., 2017). Adding dose-dependent ATP concentrations to DMD hiPSC-CM cultures demonstrated that 2.5 mM ATP had a beneficial effect and significantly reduced ROS production of the NOX4 activity with respect to no ATP addition (Figures 6E and S7C). Interestingly, idebenone alone or in combination with a 2.5 mM ATP addition did ameliorate the activity of NOX4 in a similar manner, resulting in a significantly decreased NADPH-dependent ROS production compared with untreated DMD hiPSC-CMs (Figures 6F and S7D). The specificity of idebenone on the NOX4 ROS-producing activity and on the ATP levels of the experimental groups is shown in the Supplemental information (Figures S7E–S7H). These findings reveal an increased mitochondrial ROS-producing NOX4 activity in DMD hiPSC-CMs, which was counteracted by idebenone through ATP.
DMD EHTs show improved contractile function after idebenone administration
In order to assess the amplitude of contraction of 3D EHT constructs, we monitored the micropost deflection movements of the EHT devices, which were the result of a spontaneous contraction of the EHTs attached to the flexible microposts (Figure 2B). At physiological 1.8 mM Ca2+ concentrations, the contractile function of untreated DMD hiPSC-CM EHTs was significantly lower than of untreated EHTs generated from isogenic or healthy hiPSC-CMs (Figure 7A), confirming the validity of the 3D EHT model system for DMD. However, DMD EHTs treated with idebenone exhibited a significantly increased contraction, whereas the combined treatment of idebenone and PTC124 improved the contractile function even further. By incubating DMD EHTs with Ca2+ concentrations of 2.5 mM, we wanted to analyze the amplitude of contraction of DMD EHTs, mimicking the detrimental increased Ca2+ environment, as reported in hearts from DMD patients (Figure 7B) (Kyrychenko et al., 2017; Sato et al., 2019). At higher Ca2+ concentrations, treatment with idebenone alone no longer improved the contractile function of DMD EHTs. However, the contractile function remained significantly improved with the combinatorial treatment of idebenone and PTC124. These data point out the beneficial effect of a combinatorial treatment of idebenone and PTC124, highlighting the importance of targeting simultaneously different aspects of DMD cardiomyopathy in terms of heart functionality.
Figure 7.

Improved contraction of 3D EHT constructs after administration of idebenone alone or in combination with PTC124 under physiological Ca2+ levels
(A) Spontaneous contraction and relaxation cycles of EHTs were monitored under temperature-controlled conditions (37°C) at 1.8 mM physiological Ca2+ concentrations and measured by the deflection movements of the microposts (in μm). The effect of idebenone and PTC124 on the contractility of EHTs derived from DMD hiPSC-CMs (EHT diameter: 1,041.9 ± 74.1 μm) was compared with that from DMD isogenic (diameter: 938.0 ± 86.6 μm) and healthy control EHTs (diameter: 849.9 ± 80.5 μm). Data are representative of three or four independent experiments (n ≥ 3), and values are expressed as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
(B) EHTs derived from DMD hiPSC-CMs were incubated with Ca2+ concentrations of 1.8 and 2.5 mM to assess the amplitude of contraction. Data are representative of three or four independent experiments (n ≥ 3), and values are expressed as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001 (untreated versus idebenone + PTC124), $p < 0.05, $$p < 0.01, $$$p < 0.001, and $$$$p < 0.0001 (idebenone versus idebenone + PTC124), or #p < 0.05, ##p < 0.01, ###p < 0.001, and ####p < 0.0001 (untreated versus idebenone).
Discussion
hiPSCs have the potential to differentiate in functional cell types that can be used as an unlimited cell source of inaccessible tissues to study genetic disorders and, consequently, to gain novel insights in signaling pathways involved in the disease pathology.
In this study, we generated hiPSC-based cardiac disease models from three DMD patients to study the early stages of cardiomyopathy in DMD. hiPSCs were differentiated toward CMs according to the protocol of Burridge et al. (Burridge et al., 2014) and Breckwoldt et al. (Breckwoldt et al., 2017). First, hiPSCs were differentiated in a monolayer-based method using a fully chemically defined medium consisting of the basal medium RPMI 1640, rice-derived recombinant human albumin, and L-ascorbic acid 2-phosphate along with a small-molecule-based induction of differentiation (Burridge et al., 2014). L-ascorbic acid 2-phosphate has been shown to enhance cardiac differentiation and maturation through increased collagen production by promoting cardiac progenitor cell proliferation via the MEK-ERK1/2 pathway. Furthermore, L-ascorbic acid 2-phosphate-induced CMs exhibited better sarcomeric organization and enhanced responses of APs and Ca2+ transients to β-adrenergic and muscarinic stimulations (Cao et al., 2012). Second, hiPSC-CMs were further differentiated in 3D fibrin-based EHT constructs for contractility measurements (Breckwoldt et al., 2017). In several cancer-related studies, the effect of ascorbic acid on ROS production has been reported (Fukumura et al., 2012; Wei et al., 2017). In these studies, a ROS-scavenger effect was observed after the addition of 1 mM or higher concentrations of ascorbic acid. We used a lower final concentration, suggesting no significant antioxidative effect on ROS levels. Interestingly, Bartsch et al. (Bartsch et al., 2011) demonstrated an ascorbic-acid-enhanced cardiac differentiation accompanied by an upregulation of the NADPH oxidase isoforms NOX2 and NOX4 at basal expression levels with intracellular physiological ROS concentrations, indicating the suitability of the applied cardiac differentiation methods.
hiPSC-CMs obtained from DMD patients represent hallmarks of DMD-associated heart complications in in vitro cultures. Published studies showed that the lack of Dystrophin in DMD hiPSC-CMs resulted in enhanced cell death (Lin et al., 2015), Ca2+-handling abnormalities, and reduced contractile function (Kyrychenko et al., 2017; Sato et al., 2019). We observed premature cell death of DMD hiPSC-CMs due to significantly elevated intracellular oxidative stress levels. Furthermore, a detailed characterization demonstrated mitochondrial depolarization and significantly increased NOX4 expression. Whether the abnormal upregulation of NOX4 and its increased basal rate of ROS production are a direct or indirect consequence of the absence of Dystrophin is currently unknown. Increased Nox4 proteins have been found in left ventricular CMs of mdx mice and are associated with fibrosis and altered functional parameters in the heart (Spurney et al., 2008). Deep RNA sequencing of the cardiac transcriptome on explanted human heart samples, obtained from patients suffering from heart failure, indicated extensive alternative splicing of the NOX4 gene, which is associated with upregulation of the full-length NOX4 protein (Varga et al., 2017). Consistent with these results, we found significantly increased expression and activity of the cardiac-specific ROS-producing NOX4 isoform in DMD hiPSC-CMs. Dystrophin-deficient CMs are more vulnerable to mechanical stress due to their increased membrane fragility and stretch-induced Ca2+ influx, which results in cell death (Kyrychenko et al., 2017; Lin et al., 2015; Sato et al., 2019). The complexity of the DMD pathology results from signal amplification systems with bidirectional crosstalk and positive feedback loops. ROS generation in response to mechanical forces may originate from diverse sources including mitochondria and NOX isoforms (Ago et al., 2010; Zhang et al., 2013) or even other oxidase systems (Kerr et al., 2015; Khairallah et al., 2012; Prosser et al., 2011).
To ameliorate the DMD disease phenotype, we applied several therapeutic approaches. We investigated whether NAC, ataluren (PTC124), and idebenone could have beneficial effects on the dystrophic features observed in DMD hiPSC-CM cultures. PTC124 drug efficacy analyses were performed only on DMD hiPSCs with the nonsense mutation in exon 35 (c.4,996C>T; (p.Arg1,666X)) of the Dystrophin gene. This line represents a subgroup of DMD patients (approximately 13%) that is responsive to the readthrough chemical drug PTC124, which allowed us to investigate the effects of PTC124 on DMD cardiomyopathy in an in vitro hiPSC-based disease model. PTC124 is one of the gene-based therapeutic approaches for DMD, although it is applicable for only a small subgroup of DMD patients with a nonsense mutation (Welch et al., 2007). We demonstrated re-expression of Dystrophin after PTC124 addition in a fraction of differentiating DMD hiPSC-CMs. Recently, a phase 3 randomized, placebo-controlled trial evaluating an improvement in the 6-min walking test after 48 weeks has been completed (Campbell et al., 2020), and a clinical trial to study Dystrophin expression levels in a small cohort of PTC124-treated patients with DMD is currently ongoing. These clinical studies aim at targeting the primary cause of DMD progression.
Nowadays, several innovative therapeutic approaches focus on secondary pathology. In the last decade, researchers have shown growing interest in idebenone as potential treatment for DMD. The precise mechanism by which idebenone exerts its protective effect is still unknown. Yet, idebenone has been reported to protect mitochondria from oxidative damage and to boost their impaired function, delaying the disease progression of DMD (Buyse et al., 2015). Interestingly, given the dual mode of action of idebenone (ROS-scavenger function and stimulation of the mitochondrial ETC), we showed that idebenone exhibited a superior beneficial outcome on DMD hiPSC-CMs through increased ATP production that, in turn, decreased NOX4 activity. The exact mechanism of ATP-mediated inhibition of NOX4 activity is still unclear.
Recently, an ATP-binding motif within the NOX4 isoform has been identified, suggesting a potential novel mechanism through which NOX4 can be allosterically regulated. During normal respiration, OXPHOS-driven ATP production in the mitochondria binds NOX4 through the ATP-binding domain, keeping the NOX4-produced ROS levels low (Shanmugasundaram et al., 2017). The ATP-binding motif (AXXXXGKT) (Walker et al., 1982) that resides within the amino acids 534–541 of the C terminus, is unique to NOX4 (it is not found in other NOX isoforms) and is conserved in Homo sapiens, Rattus norvegicus, and Mus musculus (Shanmugasundaram et al., 2017). In line with these results, we demonstrated that the addition of idebenone to DMD hiPSC-CM cultures increased the intracellular and, more specifically, the mitochondrial ATP concentrations through idebenone-induced ETC stimulation. Moreover, idebenone could significantly reduce the ROS-producing NOX4 activity, assuming the allosterically regulation of NOX4 through ATP. Interestingly, the addition of external ATP to DMD hiPSC-CM cultures resulted in a similar reduction of the NADPH-dependent ROS production of NOX4.
Elevated ATP concentrations can be used by skeletal and cardiac myosin to increase cross-bridge binding and cycling, leading to stronger and faster contraction and relaxation (Moussavi-Harami et al., 2015). Cardiac-specific overexpression of the enzyme ribonucleotide reductase that converts adenosine diphosphate (ADP) to deoxy-ADP (dADP), which, in turn, is rapidly converted to deoxy-ATP (dATP) in cells, facilitated CM contraction and cardiac performance in normal rodent hearts and in rodent and pig infarcted hearts (Kolwicz et al., 2016). We showed improved contractile properties of EHTs derived from DMD hiPSC-CMs upon idebenone administration at physiological Ca2+ concentrations. Preincubation of idebenone with PTC124 further enhanced the contractility, probably due to the PTC124-induced re-expression of Dystrophin proteins. In line with these results, the Olson’s group performed CRISPR-Cas9-mediated exon skipping (“myoediting”) for DMD mutation corrections in order to rescue the contractile dysfunction of DMD hiPSC-CMs that were differentiated in 3D EHTs (Atmanli et al., 2021; Kyrychenko et al., 2017).
In conclusion, by using DMD-patient-derived hiPSC-CMs, we provided the first evidence that NOX4 expression and activity were significantly upregulated, contributing to increased intracellular ROS and cell death. Furthermore, we compared the effects of NAC, PTC124, and idebenone in an in vitro setting of cardiomyopathic DMD. Finally, we gained novel mechanistic insights into the mode of action of idebenone on the hyperactive state of NOX4 ROS production. Idebenone-mediated stimulation of ATP production by the ETC of mitochondria could increase the affinity of ATP to bind with NOX4, reducing its ROS production. Considering the early cellular stress responses present in CMs from DMD patients, interfering with any of these early cellular events that lead to excessive ROS signals would positively affect the mitochondrial activity, resulting in an improved contractile function.
Experimental procedures
hiPSC culture
Control and DMD-diseased hiPSC lines (Table S1) were cultured feeder-free on Geltrex LDEV-Free hESC-Qualified Reduced Growth Factor Basement Membrane Matrix and maintained in Essential 8 Flex Basal Medium supplemented with Essential 8 Flex Supplement (50×) and 0.1% penicillin-streptomycin (Pen/Strep) (all from Thermo Fisher Scientific) at 37°C under normoxic conditions (21% O2 and 5% CO2). Colonies were routinely passaged non-enzymatically with 0.5 mM EDTA in phosphate-buffered saline (PBS; both from Thermo Fisher Scientific). Mycoplasma contamination was assessed on a periodic basis for all cell cultures. No contaminated cells were used in the described experiments of this study.
Monolayer-based cardiac differentiation of hiPSCs
hiPSCs were differentiated into functional CMs according to a monolayer-based cardiac differentiation protocol, as previously described (Burridge et al., 2014). Briefly, prior to differentiation, control and DMD hiPSC lines were split into small colonies and subsequently cultured on a thin Matrigel Growth Factor Reduced (GFR) Basement Membrane Matrix layer (Corning) in complete Essential 8 flex medium at 37°C under hypoxic conditions (5% O2 and 5% CO2), in order to obtain the optimal confluency of 85%, 3 days after splitting. Mesoderm differentiation (day 0) was induced using 6 μM CHIR99021 (Axon Medchem) for 48 h in a chemically defined medium consisting of RPMI 1640 (Thermo Fisher Scientific), 500 μg/mL rice-derived recombinant human albumin, and 213 μg/mL L-ascorbic acid 2-phosphate (both from Merck). After 24 h of CHIR99021 stimulation, hiPSCs were transferred from hypoxia to normoxia. At day 2 of differentiation, hiPSC-derived mesodermal cells were fed with basal medium supplemented with 4 μM IWR-1 (Merck) for 48 h to induce cardiac progenitor cell differentiation. From day 4 onwards, the medium was changed every other day with CM maintenance medium (RPMI 1640, rice-derived recombinant human albumin, and L-ascorbic acid 2-phosphate). Contracting CMs appeared at day 8 or 9 of cardiac differentiation. DMD hiPSC-CMs were treated with 3 mM NAC and 0.5 μM idebenone from day 8 onwards, and 20 μg/mL ataluren (PTC124) was supplemented to the cardiac differentiation medium from day 4 onwards. In NOX4 knockdown experiments, 250 nM of single-stranded antisense oligonucleotides for silencing NOX4 mRNA, called Antisense LNA GapmeRs (Qiagen), were added to the cell cultures at day 8 of differentiation.
Generation of 3D EHT constructs
3D EHT constructs were generated from 8- to 10-day-old hiPSC-CMs, as previously described (Breckwoldt et al., 2017). CMs were dissociated with collagenase A (1 U/mL; Merck) for 20 min at 37°C and transferred to custom-made 2% agarose (UltraPure; Thermo Fisher Scientific) casting molds in 24-well plate formats. The single-cell suspension was maintained in DMEM low glucose medium containing 10% fetal bovine serum (FBS), 1% heat-inactivated horse serum (HS), 1% Pen/Strep (all from Thermo Fisher Scientific), and 0.1% Rho-associated protein kinase (ROCK) inhibitor (Y-27632; VWR). Each EHT construct consisted of 1.0 × 106 cells supplemented with GFR Matrigel, 5.06% fibrinogen (human plasma; Merck), 3U/mL thrombin (Stago BNL), and 1.44% aprotinin (Merck). The casting was performed around two flexible polydimethylsiloxane (PDMS) microposts within the agarose molds. After 2 h of incubation, polymerization formed a fibrin block around the microposts, embedding the single-cell suspension. The fibrin block was removed from the casting molds and transferred to 24-well plates containing an EHT medium composed of DMEM low glucose, 10% heat-inactivated HS, 1% Pen/Strep, 0.1% aprotinin, and 0.1% human insulin solution (Merck). Medium was changed every other day with EHT medium.
Author contributions
R.D. participated in conception and design, collection and assembly of data, data analysis and interpretation, and manuscript writing. D.C. participated in western blot collection and assembly of data, data analysis and interpretation, and reviewed the manuscript. G.G. participated in patch-clamp electrophysiology and Ca2+ recordings, data analysis and interpretation, and reviewed the manuscript. L.D.W. provided DMD patient study samples, participated in conception and design, data analysis and interpretation, and reviewed the manuscript. N.G. provided DMD patient study samples and reviewed the manuscript. K.D. participated in data analysis and interpretation and reviewed the manuscript. C.M.V. participated in conception and design, data analysis and interpretation, and reviewed the manuscript. K.R.S. participated in conception and design, data analysis and interpretation, and reviewed the manuscript. G.M.B. and M.S. participated in conception and design, data analysis and interpretation, reviewed the manuscript, and gave final approval for manuscript submission.
Conflict of interests
G.M.B. was an Investigator for clinical trials in DMD sponsored by Santhera Pharmaceuticals. G.M.B. is a co-inventor of relevant patent applications. The investigators and authors had sole discretion over study design, collection, analysis and interpretation of data, writing of the report, and the decision to submit the manuscript for publication. All other authors declare no competing interests.
Acknowledgments
The authors thank Santhera Pharmaceuticals (Pratteln, Switzerland) for providing idebenone. The authors gratefully acknowledge Hanne Grosemans and Sylvia Sauvage for technical assistance; Ewa Berlińska, Filippo Conti, Vittoria Marini, and Ilaria Tortorella for assistance during their internship period at the KU Leuven; and Christina Vochten and Vicky Raets for the administrative assistance. R.D. is supported by KU Leuven Rondoufonds voor Duchenne Onderzoek (EQQ-FODUCH-O2010) and an internal KU Leuven grant (C24/18/103). This work has been supported with the contributions of INTERREG – Euregio Meuse-Rhine (GYM – Generate your muscle 2020-EMR116), FWO (#G066821N) and KU Leuven C1-3DMuSyC (C14/17/111) grants. G.M.B. is Senior Clinical Investigator of the Research Foundation Flanders (FWO Vlaanderen, Belgium).
Published: January 27, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.stemcr.2021.12.019.
Supplemental information
References
- Ago T., Kuroda J., Pain J., Fu C., Li H., Sadoshima J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ. Res. 2010;106:1253–1264. doi: 10.1161/CIRCRESAHA.109.213116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen D.G., Whitehead N.P., Froehner S.C. Absence of dystrophin disrupts skeletal muscle signaling: roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol. Rev. 2016;96:253–305. doi: 10.1152/physrev.00007.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atmanli A., Chai A.C., Cui M., Wang Z., Nishiyama T., Bassel-Duby R., Olson E.N. Cardiac myoediting attenuates cardiac abnormalities in human and mouse models of duchenne muscular dystrophy. Circ. Res. 2021 doi: 10.1161/CIRCRESAHA.121.319579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajek A., Porowinska D., Kloskowski T., Brzoska E., Ciemerych M.A., Drewa T. Cell therapy in Duchenne muscular dystrophy treatment: clinical trials overview. Crit. Rev. Eukaryot. Gene Expr. 2015;25:1–11. doi: 10.1615/critreveukaryotgeneexpr.2015011074. [DOI] [PubMed] [Google Scholar]
- Bartsch C., Bekhite M.M., Wolheim A., Richter M., Ruhe C., Wissuwa B., Marciniak A., Muller J., Heller R., Figulla H.R., et al. NADPH oxidase and eNOS control cardiomyogenesis in mouse embryonic stem cells on ascorbic acid treatment. Free Radic. Biol. Med. 2011;51:432–443. doi: 10.1016/j.freeradbiomed.2011.04.029. [DOI] [PubMed] [Google Scholar]
- Belmokhtar C.A., Hillion J., Segal-Bendirdjian E. Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene. 2001;20:3354–3362. doi: 10.1038/sj.onc.1204436. [DOI] [PubMed] [Google Scholar]
- Breckwoldt K., Letuffe-Breniere D., Mannhardt I., Schulze T., Ulmer B., Werner T., Benzin A., Klampe B., Reinsch M.C., Laufer S., et al. Differentiation of cardiomyocytes and generation of human engineered heart tissue. Nat. Protoc. 2017;12:1177–1197. doi: 10.1038/nprot.2017.033. [DOI] [PubMed] [Google Scholar]
- Burridge P.W., Matsa E., Shukla P., Lin Z.C., Churko J.M., Ebert A.D., Lan F., Diecke S., Huber B., Mordwinkin N.M., et al. Chemically defined generation of human cardiomyocytes. Nat. Methods. 2014;11:855–860. doi: 10.1038/nmeth.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buyse G.M., Voit T., Schara U., Straathof C.S.M., D'Angelo M.G., Bernert G., Cuisset J.M., Finkel R.S., Goemans N., McDonald C.M., et al. Efficacy of idebenone on respiratory function in patients with Duchenne muscular dystrophy not using glucocorticoids (DELOS): a double-blind randomised placebo-controlled phase 3 trial. Lancet. 2015;385:1748–1757. doi: 10.1016/S0140-6736(15)60025-3. [DOI] [PubMed] [Google Scholar]
- Calos M.P. The CRISPR way to think about Duchenne's. N. Engl. J. Med. 2016;374:1684–1686. doi: 10.1056/NEJMcibr1601383. [DOI] [PubMed] [Google Scholar]
- Campbell C., Barohn R.J., Bertini E., Chabrol B., Comi G.P., Darras B.T., Finkel R.S., Flanigan K.M., Goemans N., Iannaccone S.T., et al. Meta-analyses of ataluren randomized controlled trials in nonsense mutation Duchenne muscular dystrophy. J. Comp. Eff. Res. 2020 doi: 10.2217/cer-2020-0095. [DOI] [PubMed] [Google Scholar]
- Cao N., Liu Z., Chen Z., Wang J., Chen T., Zhao X., Ma Y., Qin L., Kang J., Wei B., et al. Ascorbic acid enhances the cardiac differentiation of induced pluripotent stem cells through promoting the proliferation of cardiac progenitor cells. Cell Res. 2012;22:219–236. doi: 10.1038/cr.2011.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies K.E., Nowak K.J. Molecular mechanisms of muscular dystrophies: old and new players. Nat. Rev. Mol. Cell Biol. 2006;7:762–773. doi: 10.1038/nrm2024. [DOI] [PubMed] [Google Scholar]
- Eisen B., Ben Jehuda R., Cuttitta A.J., Mekies L.N., Shemer Y., Baskin P., Reiter I., Willi L., Freimark D., Gherghiceanu M., et al. Electrophysiological abnormalities in induced pluripotent stem cell-derived cardiomyocytes generated from Duchenne muscular dystrophy patients. J. Cell Mol. Med. 2019;23:2125–2135. doi: 10.1111/jcmm.14124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery A.E. The muscular dystrophies. Lancet. 2002;359:687–695. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- Fukumura H., Sato M., Kezuka K., Sato I., Feng X., Okumura S., Fujita T., Yokoyama U., Eguchi H., Ishikawa Y., Saito T. Effect of ascorbic acid on reactive oxygen species production in chemotherapy and hyperthermia in prostate cancer cells. J. Physiol. Sci. 2012;62:251–257. doi: 10.1007/s12576-012-0204-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr J.P., Robison P., Shi G., Bogush A.I., Kempema A.M., Hexum J.K., Becerra N., Harki D.A., Martin S.S., Raiteri R., et al. Detyrosinated microtubules modulate mechanotransduction in heart and skeletal muscle. Nat. Commun. 2015;6:8526. doi: 10.1038/ncomms9526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khairallah R.J., Shi G., Sbrana F., Prosser B.L., Borroto C., Mazaitis M.J., Hoffman E.P., Mahurkar A., Sachs F., Sun Y., et al. Microtubules underlie dysfunction in duchenne muscular dystrophy. Sci. Signal. 2012;5:ra56. doi: 10.1126/scisignal.2002829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolwicz S.C., Jr., Odom G.L., Nowakowski S.G., Moussavi-Harami F., Chen X., Reinecke H., Hauschka S.D., Murry C.E., Mahairas G.G., Regnier M. AAV6-mediated cardiac-specific overexpression of ribonucleotide reductase enhances myocardial contractility. Mol. Ther. 2016;24:240–250. doi: 10.1038/mt.2015.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyrychenko V., Kyrychenko S., Tiburcy M., Shelton J.M., Long C., Schneider J.W., Zimmermann W.H., Bassel-Duby R., Olson E.N. Functional correction of dystrophin actin binding domain mutations by genome editing. JCI Insight. 2017;2 doi: 10.1172/jci.insight.95918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landfeldt E., Thompson R., Sejersen T., McMillan H.J., Kirschner J., Lochmuller H. Life expectancy at birth in Duchenne muscular dystrophy: a systematic review and meta-analysis. Eur. J. Epidemiol. 2020;35:643–653. doi: 10.1007/s10654-020-00613-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin B., Li Y., Han L., Kaplan A.D., Ao Y., Kalra S., Bett G.C., Rasmusson R.L., Denning C., Yang L. Modeling and study of the mechanism of dilated cardiomyopathy using induced pluripotent stem cells derived from individuals with Duchenne muscular dystrophy. Dis. Model. Mech. 2015;8:457–466. doi: 10.1242/dmm.019505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur A., Hong Y., Kemp B.K., Barrientos A.A., Erusalimsky J.D. Evaluation of fluorescent dyes for the detection of mitochondrial membrane potential changes in cultured cardiomyocytes. Cardiovasc. Res. 2000;46:126–138. doi: 10.1016/s0008-6363(00)00002-x. [DOI] [PubMed] [Google Scholar]
- Mercuri E., Bonnemann C.G., Muntoni F. Muscular dystrophies. Lancet. 2019;394:2025–2038. doi: 10.1016/S0140-6736(19)32910-1. [DOI] [PubMed] [Google Scholar]
- Moretti A., Fonteyne L., Giesert F., Hoppmann P., Meier A.B., Bozoglu T., Baehr A., Schneider C.M., Sinnecker D., Klett K., et al. Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nat. Med. 2020;26:207–214. doi: 10.1038/s41591-019-0738-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussavi-Harami F., Razumova M.V., Racca A.W., Cheng Y., Stempien-Otero A., Regnier M. 2-Deoxy adenosine triphosphate improves contraction in human end-stage heart failure. J. Mol. Cell Cardiol. 2015;79:256–263. doi: 10.1016/j.yjmcc.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mummery C.L., Zhang J., Ng E.S., Elliott D.A., Elefanty A.G., Kamp T.J. Differentiation of human embryonic stem cells and induced pluripotent stem cells to cardiomyocytes: a methods overview. Circ. Res. 2012;111:344–358. doi: 10.1161/CIRCRESAHA.110.227512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pioner J.M., Guan X., Klaiman J.M., Racca A.W., Pabon L., Muskheli V., Macadangdang J., Ferrantini C., Hoopmann M.R., Moritz R.L., et al. Absence of full-length dystrophin impairs normal maturation and contraction of cardiomyocytes derived from human-induced pluripotent stem cells. Cardiovasc. Res. 2020;116:368–382. doi: 10.1093/cvr/cvz109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser B.L., Ward C.W., Lederer W.J. X-ROS signaling: rapid mechano-chemo transduction in heart. Science. 2011;333:1440–1445. doi: 10.1126/science.1202768. [DOI] [PubMed] [Google Scholar]
- Sato M., Shiba N., Miyazaki D., Shiba Y., Echigoya Y., Yokota T., Takizawa H., Aoki Y., Takeda S., Nakamura A. Amelioration of intracellular Ca(2+) regulation by exon-45 skipping in Duchenne muscular dystrophy-induced pluripotent stem cell-derived cardiomyocytes. Biochem. Biophys. Res. Commun. 2019;520:179–185. doi: 10.1016/j.bbrc.2019.09.095. [DOI] [PubMed] [Google Scholar]
- Sesso H.D., Buring J.E., Christen W.G., Kurth T., Belanger C., MacFadyen J., Bubes V., Manson J.E., Glynn R.J., Gaziano J.M. Vitamins E and C in the prevention of cardiovascular disease in men: the Physicians' Health Study II randomized controlled trial. JAMA. 2008;300:2123–2133. doi: 10.1001/jama.2008.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanmugasundaram K., Nayak B.K., Friedrichs W.E., Kaushik D., Rodriguez R., Block K. NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance. Nat. Commun. 2017;8:997. doi: 10.1038/s41467-017-01106-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurney C.F., Knoblach S., Pistilli E.E., Nagaraju K., Martin G.R., Hoffman E.P. Dystrophin-deficient cardiomyopathy in mouse: expression of Nox4 and Lox are associated with fibrosis and altered functional parameters in the heart. Neuromuscul. Disord. 2008;18:371–381. doi: 10.1016/j.nmd.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Timpani C.A., Hayes A., Rybalka E. Revisiting the dystrophin-ATP connection: how half a century of research still implicates mitochondrial dysfunction in Duchenne muscular dystrophy aetiology. Med. Hypotheses. 2015;85:1021–1033. doi: 10.1016/j.mehy.2015.08.015. [DOI] [PubMed] [Google Scholar]
- Varga Z.V., Pipicz M., Baan J.A., Baranyai T., Koncsos G., Leszek P., Kusmierczyk M., Sanchez-Cabo F., Garcia-Pavia P., Brenner G.J., et al. Alternative splicing of NOX4 in the failing human heart. Front. Physiol. 2017;8:935. doi: 10.3389/fphys.2017.00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaart I.E.C., Aartsma-Rus A. Therapeutic developments for Duchenne muscular dystrophy. Nat. Rev. Neurol. 2019;15:373–386. doi: 10.1038/s41582-019-0203-3. [DOI] [PubMed] [Google Scholar]
- Vermot A., Petit-Hartlein I., Smith S.M.E., Fieschi F. NADPH oxidases (NOX): an overview from discovery, molecular mechanisms to physiology and pathology. Antioxidants. 2021;10 doi: 10.3390/antiox10060890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker J.E., Saraste M., Runswick M.J., Gay N.J. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982;1:945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X., Xu Y., Xu F.F., Chaiswing L., Schnell D., Noel T., Wang C., Chen J., St Clair D.K., St Clair W.H. RelB expression determines the differential effects of ascorbic acid in normal and cancer cells. Cancer Res. 2017;77:1345–1356. doi: 10.1158/0008-5472.CAN-16-0785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch E.M., Barton E.R., Zhuo J., Tomizawa Y., Friesen W.J., Trifillis P., Paushkin S., Patel M., Trotta C.R., Hwang S., et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447:87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- Wu B., Moulton H.M., Iversen P.L., Jiang J., Li J., Li J., Spurney C.F., Sali A., Guerron A.D., Nagaraju K., et al. Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc. Natl. Acad. Sci. U S A. 2008;105:14814–14819. doi: 10.1073/pnas.0805676105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M., Perino A., Ghigo A., Hirsch E., Shah A.M. NADPH oxidases in heart failure: poachers or gamekeepers? Antioxid. Redox Signal. 2013;18:1024–1041. doi: 10.1089/ars.2012.4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorova L.D., Popkov V.A., Plotnikov E.Y., Silachev D.N., Pevzner I.B., Jankauskas S.S., Babenko V.A., Zorov S.D., Balakireva A.V., Juhaszova M., et al. Mitochondrial membrane potential. Anal. Biochem. 2018;552:50–59. doi: 10.1016/j.ab.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.