

Summary
Multi-omics data analysis is an important aspect of cancer molecular biology studies and has led to ground-breaking discoveries. Many efforts have been made to develop machine learning methods that automatically integrate omics data. Here, we review machine learning tools categorized as either general-purpose or task-specific, covering both supervised and unsupervised learning for integrative analysis of multi-omics data. We benchmark the performance of five machine learning approaches using data from the Cancer Cell Line Encyclopedia, reporting accuracy on cancer type classification and mean absolute error on drug response prediction, and evaluating runtime efficiency. This review provides recommendations to researchers regarding suitable machine learning method selection for their specific applications. It should also promote the development of novel machine learning methodologies for data integration, which will be essential for drug discovery, clinical trial design, and personalized treatments.
Subject area: machine learning, omics, systems biology
Graphical abstract
Highlights
-
•
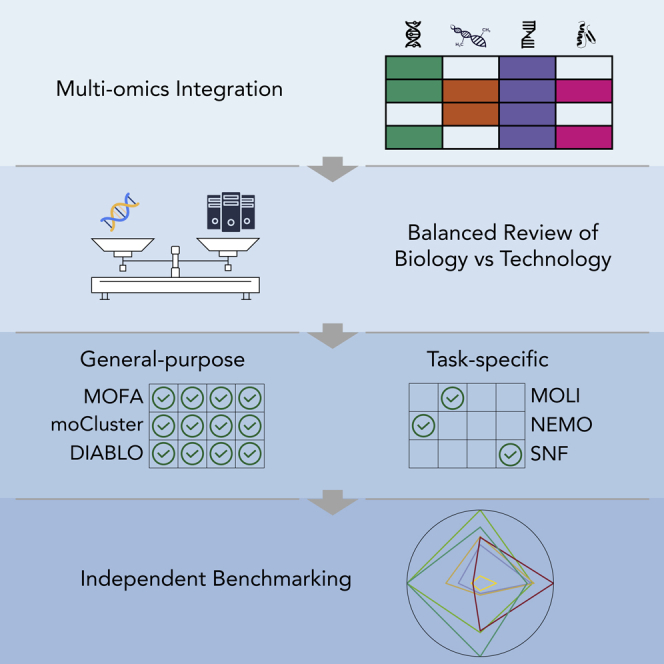
Featuring a balance of both biological and technical content
-
•
Categorizing the reviewed tools into general purpose and task-specific
-
•
Performing an independent benchmarking analysis using a publicly available dataset
Machine learning; Omics; Systems biology
Introduction
The discipline in molecular biology that aims for the collective characterization and quantification of the genome, transcriptome, and proteome, to influence the structure, function, and dynamics of a biological sample is termed omics (López de Maturana et al., 2019). Biotechnological advancements have enabled researchers to generate molecular datasets and perform individual or integrative analyses across various fields, such as genomics, transcriptomics, and proteomics (O’Donnell et al., 2020).
In human cancers, there are complex rearrangements at the genetic, transcriptional, and proteomic levels that drive oncogenesis. This process evolves through clonal selection and over time, contributing to resistance to treatment. Single-omics datasets such as those derived from the Human Genome Project (Lander et al., 2001) and initial genomic profiling from The Cancer Genome Atlas (TCGA) projects (Campbell et al., 2020), have failed to produce the revolution in cancer treatment that was expected for the vast majority of common cancer types (Tannock and Hickman, 2016). Next-generation sequencing of tumor genomes has been able to propose targeted treatments in only a small percentage of patients (Bohan et al., 2020), and no improvements in outcome have been found in randomized trials of targeted therapies (Le Tourneau et al., 2015). Consequently, developing a holistic view of cancer behavior and identification of new therapeutic vulnerabilities may only be possible through multi-omics analysis, which has become an area of increasing interest in cancer research over the last decades (Subramanian et al., 2020; Lee et al., 2020; Sathyanarayanan et al., 2020; Oh et al., 2021) (Figure 1). This has been exemplified by the addition of epigenomic, transcriptomic, proteomic, phosphoproteomic, and metabolomic data to the TCGA for many solid tumor subtypes in recent years (Clark et al., 2019; Wang et al., 2021). These large-scale integrative analyses on multi-omics data from various tumor cohorts have shed light on the complex systemic dysregulation associated with specific cancer phenotypes, producing essential insights that cannot be attained by examining only a single omics dataset. For example, a proteogenomic analysis of colon and rectal cancer showed moderate correlation between messenger RNA (mRNA) expression and protein abundance, and identified four cancer subtypes at the proteomic level to enable better prioritization of mutated (affecting DNA) or dysregulated (affecting RNA) cancer driver genes (Zhang et al., 2014). Another multi-omics study in an ethnically diverse lung adenocarcinoma cohort used machine learning to reveal four subgroups defined by mRNA transcripts, proteins, phosphoproteins, and acetylated proteins, with multiple potential therapeutic vulnerabilities to targeted therapy as well as immunotherapy resistance (Gillette et al., 2020).
Figure 1.

Growth of publications in omics
Line charts showing the number of articles published in each year from 1995 to 2020 in PubMed, colored by different omics. The y axis is plotted in log scale. Search terms used are “genomics,” “epigenomics,” “transcriptomics,” “proteomics,” and “multi-omics”.
Given the immense complexity of data integration across multiple omics, the computational algorithms required to tease out signals from noise become more complex. Therefore, strategies are required to systematically integrate heterogeneous multi-omics datasets to deliver actionable results that may advance biological sciences and eventually translate into clinical practice. There are three common strategies for multi-omics data integration: early, middle, and late integration (Rappoport and Shamir, 2018). Early integration, also known as early concatenation, is a simple concatenation of features from each omics layer into one single matrix. In late integration, modeling and analysis are performed at each omics layer separately, and the results are merged at the end. The difference between early, middle, and late integration is also summarized in Figure 2. Because both early and late integration do not involve additional statistical processing or modeling by machine learning, all methods reviewed in this article fall under middle integration, which focuses on using machine learning models to consolidate data without concatenating features or merging results.
Figure 2.

Illustration of early, middle, and late integration for merging data matrices generated by different omics
In early integration, features from different data matrices are concatenated. Middle integration uses machine learning models to consolidate data without concatenating features or merging results. In late integration, each omics layer is analyzed independently, and results are combined at the end.
The bedrock of multi-omics data analysis is machine learning, based upon which many tools have been developed (Argelaguet et al., 2020; Mo et al., 2018; Sharifi-Noghabi et al., 2019). Machine learning algorithms are trained to model complex patterns that cannot be accurately captured by traditional mathematical models in high dimensional data (Russell and Norvig, 2020). Publications of existing methods often emphasize the computational aspects of the proposed models, but lack a thorough introduction to the characteristics of individual omics. Recent reviews on multi-omics data integration focus on either biological applications or machine learning algorithms, rather than the combination of both (Nicora et al., 2020; Picard et al., 2021; Reel et al., 2021; Subramanian et al., 2020).
Published multi-omics data are usually stored in online portals for public access, serving as resources for both discovery and validation (Table 1). Among them is the TCGA project (Campbell et al., 2020) initiated by the National Cancer Institute in 2006, which generated multi-omics data for more than 20,000 tumors spanning 33 cancer types. The International Cancer Genome Consortium (ICGC) was initiated by multiple countries as a collaborative program, which incorporates some projects from TCGA and features a user-friendly online analysis interface (The International Cancer Genome Consortium, 2010). The Catalog of Somatic Mutations In Cancer (COSMIC) (Iorio et al., 2016; Tate et al., 2019) database is led by the Wellcome Sanger Institute and curates multi-omics data for both cancer cell lines and tumors. The Cancer Dependency Map (DepMap) (Broad, 2020) is a platform similar to COSMIC developed by the Broad Institute, which provides genome-wide CRISPR-Cas9 knockout screens with comprehensive multi-omics molecular characterization of cell lines and the corresponding drug screens.
Table 1.
Key portals for accessing publicly available multi-omics datasets
|
Name |
URL | Omic and other data types | Notes |
|---|---|---|---|
| TCGA (Campbell et al., 2020) | https://portal.gdc.cancer.gov/ |
|
|
| ICGC (Campbell et al., 2020) | https://dcc.icgc.org/ |
|
|
| CPTAC | https://cptac-data-portal.georgetown.edu/cptacPublic/ |
|
|
| COSMIC Cell Lines (Iorio et al., 2016) | https://cancer.sanger.ac.uk/cell_lines |
|
|
| DepMap (Broad, 2020) | https://depmap.org/portal/ |
|
|
| COSMIC (Tate et al., 2019) | https://cancer.sanger.ac.uk/cosmic |
|
|
The unique contribution of this review is three-fold (Figure 3). First, this review features a balance of both biological and technical content, so that readers from a range of backgrounds can benefit from the presented information and guidance when they seek multi-omics integration tools for cancer research. Other similar reviews primarily focus on only one of these aspects or lack comprehensiveness. Second, we propose a new classification that categorizes the reviewed tools into general-purpose and task-specific. This allows researchers to quickly determine which tools are the most applicable for their research questions. In addition, researchers who do not have a strong computational background may be not aware that general-purpose methods can also be applied to their research projects. Third, unlike most review articles, we perform an independent benchmarking analysis using a publicly available dataset called Cancer Cell Line Encyclopedia (CCLE) (Ghandi et al., 2019; Nusinow et al., 2020). The benchmarking exercise enables researchers to choose the most suitable tools for their research question and computational environment.
Figure 3.

Unique contribution of this review
First, we describe a balance of both biological and technical content covering topics from genomics to proteomics and from machine learning to multi-omics integration tools. Second, we propose a new classification that categorizes the reviewed tools into two categories, namely general-purpose and task-specific, and then review these tools for four types of applications in biomedical sciences. Third, we provide an independent benchmarking analysis to compare integration methods for cancer type classification and drug response prediction.
Omics overview
Understanding the biological underpinnings of the data in each omics layer and the data formats is crucial to method development and fully utilizing the available tools. For instance, genomic and epigenomic variants influence gene regulation and the quantities of transcribed mRNA (Haraksingh and Snyder, 2013). Splicing mechanisms and posttranslational modifications then impact the downstream measurements of the proteome. All of these mechanisms ultimately determine the cellular phenotype (Niklas et al., 2015). In this section, we review several common omics, describe data formats, and discuss corresponding analytical strategies.
Genomics
Genomics examines DNA sequences and seeks to understand the associations between diseases and genomic alterations (Stratton et al., 2009). Whole-exome sequencing (WES) and whole-genome sequencing (WGS) (Schwarze et al., 2018) are two popular technologies utilized in genomic studies. WES mostly examines the exonic (mRNA-coding) portion of the genome, whereas WGS aims to examine all nucleotides in the genome including the gene regulatory regions (Nakagawa and Fujita, 2018). WES usually involves a lower cost than WGS because it only covers the coding regions, although key regulatory and splice-site mutations that are not in coding regions could be missed by WES (Yang et al., 2013).
Genomic analysis focuses on single nucleotide variants (SNVs), insertions and deletions (INDELs), structural variation (SV), and copy number variation (CNV). SNVs are variants of only a single nucleotide that occurs at a specific genomic position. INDELs are small genetic variations with lengths usually shorter than 10,000 nucleotides. SV covers large variations in the chromosome, including deletions, duplications, insertions, inversions, and translocations of long nucleotide sequences. CNV is a particular form of SV and usually involves the amplification or deletion of a large region of a chromosome. The genome is the most fundamental layer of genetic information and is very well characterized because of the development of advanced sequencing technologies. Genomic analyses have revealed many significantly mutated genes in cancer, known as cancer driver genes, such as TP53 and KRAS (Stratton et al., 2009). Novel treatment strategies that have greatly improved outcome for subsets of cancers have been discovered by analyzing mutations at the genomic level (Behan et al., 2019). For example, EGFR tyrosine kinase inhibitors are used in the treatment of EGFR-mutant non-small cell lung cancer, whereas HER2-amplified breast cancers are treated with HER2 monoclonal antibodies (Cohen et al., 2021).
For computational analysis, the data matrices for SNVs, INDELs, and SVs can be summarized as binary values that indicate whether a gene is mutated or wild type. Genes are often filtered so that only those with mutations in sufficient numbers of samples are included to avoid a highly sparse matrix, and mutation frequencies are typically normalized against background mutation rates for that genomic locus (Lawrence et al., 2013). Filtering may also be required for specific mutation types, depending on the research question. For example, only missense mutations may be considered as mutants in certain situations. By contrast, CNV data are typically presented as a matrix of either counts or continuous values for each gene. The data matrix of CNV is sometimes in the format of log fold-change for cancer studies, reflecting changes of copy numbers compared with the normal ploidy. Outside of coding regions, genomic information can be used to understand elements of gene regulation and dysregulation in cancer (Andersson and Sandelin, 2020). Somatic, germline, and epigenetic variation affecting these regions can have profound effects on gene expression in cancer (Poulos and Wong, 2017). Although genomic technology is relatively more mature than other omics, discovering causal relationships in addition to associations remains as one of the biggest challenges (McGuire et al., 2020).
Epigenomics
The epigenome encompasses the set of indirect chemical modifications of nucleotides and proteins that regulate how genes are expressed, without changing the actual nucleotide sequence itself (Wang and Chang, 2018). The study of the epigenome is called epigenomics, which involves investigating DNA methylation (Jost and Saluz, 2013) and histone modification (Seligson et al., 2005), as well as understanding the three-dimensional structure of DNA, which is influenced by topologically-associating domains (Szabo et al., 2019). This three-dimensional structure is examined via sequencing technologies, such as ChiA-PET, 3C, 4C, 5C, and Hi-C (Van Berkum et al., 2010), whereas DNA methylation can be measured by a range of methods, such as bisulfite sequencing (Krueger et al., 2012; Wreczycka et al., 2017). Chromatin immunoprecipitation sequencing (ChIP-seq) (Valouev et al., 2008) experiments are often used for high-throughput measurement of histone modifications.
Methylomics is one of the best characterized aspects of epigenomics. It focuses primarily on the effects of promoter DNA methylation on silencing gene expression, but also commonly examines the effects of gene body methylation in cancer (Wong et al., 2014). Studies of DNA methylation play a pivotal role in biomedical research. For example, the promoter hypermethylation of MLH1 was found to result in hereditary nonpolyposis colon cancer (Cunningham et al., 1998). The processed methylation data matrix often contains continuous values ranging from 0 to 1, representing the proportion of cells in which the relevant nucleotide is found to be methylated. Methylation data usually require normalization and correction according to cancer types (Iorio et al., 2016). However, current high-throughput technologies for measuring methylomic data use probes, which may not properly cover the promoter regions of specific genes. This issue may be especially important if the research question is about one specific gene.
Transcriptomics
Genes are mostly transcribed into mRNA and introns are spliced out, leaving only exons in the mature mRNA, which consists of 5′ and 3' untranslated regions, and a protein-encoding open reading frame (Brouwer and Lenstra, 2019). The result is a large pool of cellular mRNA used by ribosomes for translation to proteins. This provides an indirect indicator of the protein expression in a cell, or the activity of the genome at a particular point in time. Transcriptomic analyses measure the abundance of the complete set of mRNA transcripts of each gene, which is also referred to as the gene expression level. Several methods are available to quantify the transcriptome, with the most popular approaches being microarrays and RNA-seq (Malone and Oliver, 2011). RNA-seq is now more commonly employed than microarrays, as it provides better performance and data consistency (Xu et al., 2013). Biomarkers found at the transcriptomic level can be used for identifying patient subtypes (Nielsen et al., 2010) and developing new cancer treatments. For example, the transcriptome can be more predictive of anticancer drug response than genomic mutations and DNA methylation data (Iorio et al., 2016).
RNA-seq data are usually normalized as transcripts per million bases, and many bioinformatic approaches have been proposed to identify differentially expressed genes (Law et al., 2014; Love et al., 2014; Ritchie et al., 2015; Robinson et al., 2010). RNA-seq data usually contain batch effects that significantly affect the downstream analysis, and many tools have been developed for batch correction (Risso et al., 2014; Zhang et al., 2020). When using RNA-seq data for multi-omics analysis, it is important to ensure that the data have been preprocessed to remove unwanted batch effects. The RNA expression matrix can contain either discrete raw counts or continuous values on a logarithmic scale. Genes with very low expression are often filtered out to highlight the most significant biological signals (Sha et al., 2015).
Proteomics
mRNAs are translated to produce proteins, which are sequences of amino acids (Kaeberlein and Kennedy, 2007). Analogous to the transcriptome, the proteome encompasses the entire set of expressed proteins in a cell or organism at a particular point in time. Proteins are the functional units that interact with other molecules like metabolites, lipids, or nucleotides (Spirin and Mirny, 2003). For this reason, along with the metabolome and lipidome, the proteome is more closely related to cellular phenotypes than the genome or transcriptome (Crick, 1970). Protein abundance often differs from gene expression levels because of a number of factors, including posttranslational modifications and protein stability or degradation (Hegde et al., 2003). For example, in clear cell renal cell carcinomas, genes related to oxidative phosphorylation-related metabolism, protein translation processes, and some phospho-signaling modules were found to be dysregulated only at the protein level (Clark et al., 2019).
Proteomics refers to the large-scale analysis of proteomes. Recent technological advances in mass spectrometry have enabled proteomics to become high-throughput and reproducible for large-scale cancer analyses (Poulos et al., 2020; Tully et al., 2019). However, the high prevalence of missing values of protein abundance in proteomic data presents unique analytical challenges (Poulos et al., 2020). The missing values need to be handled by applying domain-specific knowledge, statistical methods, or machine learning algorithms (Poulos et al., 2020; Välikangas et al., 2018). Normalized and imputed proteomic data matrices typically contain continuous values that are usually in the logarithmic scale.
Single-cell sequencing
Most omics studies are based on data generated from bulk samples. Therefore, the omics data are averaged measurements of multiple cells from samples. However, tumor samples exhibit a great degree of intertumoral and intratumoral heterogeneity, making analyses extremely challenging (Guo et al., 2018). The advancement of single-cell technologies has enabled researchers to investigate omics profiles at a single-cell level by tagging each cell from a sample (Nam et al., 2021), although single-cell analyses face a number of challenges that are distinct from analyses of bulk samples because of the difference of data resolution. Among the different omics data types, single-cell RNA sequencing (scRNA-seq) is relatively more mature and has yielded a number of discoveries (Argelaguet et al., 2019; Zhang et al., 2021). In the meantime, computational tools for integrating single-cell multi-omics data have been emerging. For example, Seurat 4.0 is able to integrate data from multiple single-cell technologies, including single-cell epigenomic, transcriptomic, and proteomic data (Hao et al., 2021). In this review, we focus on multi-omics data integration for bulk samples.
Machine learning for multi-omics integration
Having reviewed different omics, here we discuss core machine learning concepts that are involved in multi-omics data integration. Machine learning is the study of computational algorithms that make predictions and decisions based on experience and data, without having been explicitly programmed to do so (Koza et al., 1996; Mitchell, 1997). Machine learning models are generally divided into three categories: supervised, unsupervised, and reinforcement learning. Supervised learning methods use label information from samples for model training, whereas unsupervised learning infers patterns directly from unlabeled data. Reinforcement learning trains an agent to choose the best next action when the environment changes. Supervised and unsupervised learning are commonly applied to multi-omics data integration, but to the best of our knowledge, there have been no attempts to use reinforcement learning for this type of task.
As mentioned above, there are three approaches for multi-omics data integration: early, middle, and late. The most straightforward strategy is early concatenation. However, having a vast number of features while the number of available data points is low, known as the "curse of dimensionality" (Bellman, 1966), is a particular challenge for the use of early concatenation in multi-omics integration. For example, with the human genome containing more than 20,000 protein-coding genes, multi-omics datasets can easily comprise more than 50,000 features when the genome, transcriptome, and proteome are combined. By contrast, the number of available tumor samples in a dataset is often relatively small, with cancer cohorts typically comprising no more than a few hundred patients. Late integration only involves manual combination of the results from each omics layer, hence out of scope for this review. Therefore, we focus on middle integration that aims to overcome the challenge by using machine learning integration methods that are categorized either as general-purpose or task-specific (Figure 3). General-purpose methods couple dimensionality reduction with different downstream algorithms for a variety of applications, whereas task-specific methods are end-to-end models designed for one specific task.
In this section, we first provide a brief introduction to related machine learning concepts, followed by a more comprehensive review on existing general-purpose and task-specific methods for multi-omics data integration.
Basics of related machine learning concepts
Unsupervised learning
Unsupervised learning discovers patterns in multi-omics data without mapping the input data to output data. Most dimensionality reduction techniques are unsupervised methods. Principal component analysis (PCA) (Wold et al., 1987) projects each data point onto a lower-dimensional space by creating orthogonal principal components that are eigenvectors of the data's covariance matrix. Factor analysis (Singh and Gordon, 2008) can also be used to reduce dimensionality, assuming the existence of latent (unobserved) variables that are not limited to linear combinations of features. Factor analysis algorithms then seek such latent variables that can capture the common variance of the whole dataset. Joint latent variable models (Everett, 2013) extend factor analysis by allowing more assumptions and configurations on the statistical models. Canonical correlation analysis (CCA) (Hotelling, 1992) calculates how well different data matrices are correlated and derives a set of variables such that the correlations between data matrices are maximized. CCA can be applied to multi-omics data integration under the assumption that the correlation between different omics layers is to be maximized. Multiple kernel learning (MKL) (Lin et al., 2011) can be used with either supervised or unsupervised learning. Kernels allow the data to be transformed into a higher-dimensional space via kernel tricks (Aizerman, 1964). Multiple kernels are used in MKL so that data from different omics layers can be appropriately modeled.
Supervised learning
Supervised learning is usually used to make predictions. Given the input data and the output labels, supervised learning finds a mapping function that maps the input data to the label information. Label information in cancer research can be any phenotype of interest. For example, cancer types can be considered as label information. If the label information is discrete, then it is called classification. If the label is continuous, then it is regression. Linear regression and logistic regression are the two basic supervised learning models that only use linear predictor functions (Freedman, 2009). Elastic net (Zou and Hastie, 2005) adds both L1 (absolute-value norm) and L2 (Euclidean norm) regularization terms to the basic linear models. L1 regularization encourages non-informative features to have zero coefficients, and L2 regularization works well with correlated features by allocating roughly equivalent weights to strongly correlated features. Because of these characteristics, elastic net has been used widely in multi-omics analyses such as drug response studies (Iorio et al., 2016), where simple markers are preferred and interpretability is important. Random forest (Breiman, 2001) uses a set of decision trees (Rokach and Maimon, 2005) to make predictions based on votes over all the trees in the forest. Random forest is a nonlinear machine learning model that captures more complexity in the data than linear and logistic regression. The feature importance given by random forest represents how well each feature performs in terms of prediction, allowing researchers to prioritize the most important features for their studies. Neural networks are the root of deep learning algorithms that have attracted increasing attention recently and shown better predictive power than other traditional machine learning models in research areas such as natural language processing, computer vision, and biomedical sciences (Kim, 2016). Neural network models are versatile because they can be used for various purposes, including classification, regression, dimensionality reduction, and missing value imputation. Despite its superior predictive performance, deep learning is often criticized for its poor model interpretability. To overcome this limitation, deep learning algorithms that focus on model explanations have emerged in recent years (Lundberg and Lee, 2017; Ribeiro et al., 2016; Ullah et al., 2020).
Multi-omics data integration tools and their applications
In this section, we review machine learning tools that have been developed specifically for biomedical sciences and discuss how these tools are applied in three typical real-world applications, namely cancer type classification, drug response prediction, and patient stratification. Specifically, we used keywords “machine learning” and “multi-omics integration” to broadly search the methods using PubMed and Google Scholar. We then searched for existing reviews of similar topics. For each tool, we indicate whether it is general-purpose or task-specific, and whether it provides an easy-to-use interface for custom datasets (Table 2). Although no multi-omics dataset covers all omics layers, most integration tools are flexible enough to support the data analysis of any combination of available omics layers (Figure 3).
Table 2.
Machine learning tools for multi-omics data integration
Cancer type classification
Knowing the cancer subtype is crucial for disease classification, assessing prognosis, and planning treatment. For instance, breast carcinoma can be classified into five transcriptome-based subtypes that lead to different treatment responses and outcomes (Dai et al., 2015). Non-small cell lung cancer can also now be classified into a large number of subtypes depending on both histological appearance (squamous versus adenocarcinoma) and the presence/absence of particular driver mutations (Thomas et al., 2015). It would also be valuable to predict the cell of origin, and therefore the likely therapeutic sensitivity, for cancers of unknown primary (CUP) (Lu et al., 2021; Pavlidis and Pentheroudakis, 2012), as treatments differ significantly depending on the patient’s primary cancer type. Traditional methods for classifying cancer types mainly involve visual inspection by trained anatomical pathologists of cancer sections stained by H&E or by immunohistochemistry. Machine learning tools that integrate multi-omics data have provided more efficient diagnoses for patients with CUP, allowing the most effective treatment options to be identified (Bavafaye Haghighi et al., 2019). Relevant machine learning packages include the following.
mixOmics (general-purpose) (Rohart et al., 2017) is a well-implemented package enabling a set of supervised and unsupervised machine learning models based on linear discriminant analysis (LDA) (R.O. Duda et al., 2000), PCA, and CCA. mixOmics has been used extensively in studies to characterize cancer subtypes. It supports three modes: single omics analysis, data integration across different omics layers, and data integration across different samples. One specific multi-omics integration method proposed in the mixOmics package is DIABLO (Singh et al., 2019), which extends traditional CCA to a supervised mode. DIABLO has been compared with other methods in terms of classification and clustering using several cohorts, including 92 colon cancer samples, 122 kidney cancer samples, 213 glioblastoma samples, 106 lung cancer samples, and 989 breast cancer samples (Singh et al., 2019). PAM50 breast cancer subtype prediction was selected for the classification task, and colon cancer data from TCGA were used for clustering comparisons. The authors tested DIABLO via two model configurations. DIABLO_full represents correlations across all omics layers, which are modeled via CCA, whereas DIABLO_null does not consider correlations between omics layers by simply applying LDA on the dataset as early concatenation. Notably, DIABLO_null performed better than DIABLO_full in both classification and clustering tasks, suggesting that integration may not be effective because the connection of omics layers actually worsens the predictive accuracy. Apart from cancer type classification, mixOmics has been used for tasks such as analysing the association between gut microbial composition and the risk of asthma in childhood (Stokholm et al., 2018).
MOFA/MOFA2 (Multi-Omics Factor Analysis, general-purpose) (Argelaguet et al., 2018, 2020) seeks common factors that can explain the greatest variance of all data from different omics layers. MOFA (Argelaguet et al., 2018) uses factor analysis as the method of dimensionality reduction. It supports different data distributions and optimizes computational runtime for better performance in multi-omics integration. Various types of regularizations are supported in MOFA for better model interpretability. MOFA also handles missing values automatically and supports partial datasets. In their study, MOFA was able to classify four subtypes of chronic lymphocytic leukemia (Argelaguet et al., 2018). (Alcala et al., 2019) used MOFA on DNA methylation and gene expression data, and identified two latent factors that were associated with survival in large-cell neuroendocrine carcinomas. Apart from survival analysis, MOFA showed satisfactory results in other tasks, such as drug response prediction and patient stratification (Argelaguet et al., 2018). MOFA+/MOFA2 (Argelaguet et al., 2020) is a subsequent enhancement of MOFA that supports single-cell datasets and GPU-accelerated model inference, which is over twenty times faster than the original inference algorithm in MOFA. Although MOFA2 mostly highlighted its usage in analyzing single-cell datasets, bulk sample analyses also benefited from the faster inference of MOFA2 without any loss of functionalities (Argelaguet et al., 2020).
sCCA (sparse CCA, general-purpose) is a variation of CCA that imposes additional penalties in modeling so that the number of latent variables can be kept low for better interpretation (Rodosthenous et al., 2020). Several conventional CCA methods were compared with a customized sCCA variation that allowed data from more than two datasets to be used at the same time. The proposed sCCA variation was applied to TCGA gene expression, miRNA, and methylation data, and yielded the highest accuracy in a task of classifying three cancer types, namely breast, kidney, and lung cancer (Rodosthenous et al., 2020).
Drug response prediction
Using multi-omics profiles to predict drug responses allows researchers to discover new treatment opportunities and to provide recommendations on the design of early-phase clinical trials for either novel drugs or the repurposing of existing drugs for different cancers (Iorio et al., 2016). In this scenario, the problem can be either formulated as a regression task, where a model is trained to predict the half-maximal inhibitory concentration (IC50) value and area under the curve (AUC), or as a simplified classification task where the model predicts whether a given input is sensitive or resistant to a particular drug. Computational researchers have been focusing on designing machine learning models that are able to uncover explainable biomarkers with better prediction, facilitating personalized treatment.
MOLI (Multi-Omics Late Integration, task-specific) (Sharifi-Noghabi et al., 2019) is based on deep learning and predicts drug response as a classification task. MOLI uses a deep neural network (Rumelhart et al., 1986) as a feature extractor for each omics layer, and concatenates the last hidden layers with a triplet loss (Schroff et al., 2015) at the end to train the model. Triplet loss facilitates training by minimizing distances between similar samples and maximizing distances between different samples. Despite the “late integration” in its name, MOLI would be better classified as middle integration because MOLI integrates all omics layers using machine learning instead of merging results at the end. MOLI used gene mutation, expression, and copy number to predict cancer drug response by classifying patients as responders or nonresponders to cancer drugs. Various datasets were utilized, including GDSC (Iorio et al., 2016), PDX (Gao et al., 2015), and TCGA (Ding et al., 2016) drug response data. The number of samples included in each dataset for MOLI varied depending on the drugs, ranging from 16 to 856 samples. MOLI was only compared with early concatenation in its related publication (Schroff et al., 2015), without being thoroughly compared with similar integration tools.
CaDRReS (Cancer Drug Response prediction using a Recommender System, general-purpose) (Suphavilai et al., 2018) is a matrix-factorization-based model that treats drug response prediction as a regression task. CaDRReS was developed based on recommender systems, where matrix factorization has shown excellent performance. The fundamental intuition in the recommender system is that if a set of users rate a set of products similarly, they are also likely to give similar ratings on other products (Xue et al., 2017). This idea has been transformed into multi-omics data integration, where the molecular profile of a gene is considered to share similarities across different omics layers. CaDRReS was used to predict continuous drug responses IC50, using both GDSC (Iorio et al., 2016) and CCLE datasets (Barretina et al., 2012). CaDRReS has been benchmarked against two other matrix-factorization-based methods, as well as some basic models such as regularized linear regression (Suphavilai et al., 2018). Although CaDRReS is a general-purpose method with dimensionality reduction, the tool does not allow flexible usages for purposes other than drug response prediction.
HNMDRP (Heterogeneous Network-based Method for Drug Response Prediction, task-specific) (Zhang et al., 2018) focuses on constructing similarity networks among cell lines, drug structures, and drug target genes to predict drug responses. This method assumes that when a similar cell line is treated with a similar drug, then the drug response should be similar. Application of HNMDRP showed that protein-protein interactions and drug-target interactions were useful to improve prediction results, but drug chemical structure data were not widely accessible in many scenarios.
pairwiseMKL (multiple pairwise kernels for drug bioactivity prediction, general-purpose) (Cichonska et al., 2018) is an enhanced extension to MKL. It improves upon both runtime and memory efficiency of MKL and enables its application to drug response prediction, where the term “pairwise” refers to sample and drug pairs. The original study showed that pairwiseMKL ran approximately six times faster and consumed 95% less memory than KronRLS-MKL (Nascimento et al., 2016), when predicting continuous IC50 values of drugs in the GDSC (Iorio et al., 2016) dataset.
Patient stratification
Another key task for which researchers use multi-omics data is to cluster tumors and identify potential new cancer subtypes to facilitate better patient stratification (Li and Wong, 2019). These new subtypes could show different characteristics from existing known cancer subtypes, and new therapies may need to be developed for them. Computational methods for patient stratification are usually evaluated using simulated data, and methods that are able to clearly separate known phenotypes are generally considered high-performance methods (Argelaguet et al., 2018; Shen et al., 2009).
iCluster/iClusterPlus/iClusterBayes (general-purpose), are a family of machine learning methods based on joint latent variables. iCluster formulates the latent cancer subtypes as a joint variable, which results in a much smaller number of dimensions than early concatenation. The latent variables are modeled to capture common information from different omics layers. The iCluster study (Shen et al., 2009) shows that the best performance is achieved with less than ten dimensions in the latent variable. Based on iCluster, iClusterPlus (Mo et al., 2013) focuses on modeling different statistical distributions for discrete data types. iClusterBayes (Mo et al., 2018) is the latest version in the series and implements a fully Bayesian inference algorithm, which runs six times faster than iClusterPlus. Bass et al. used iCluster and discovered four subtypes of gastric cancer, enabling better patient stratification and treatment planning (Bass et al., 2014).
moCluster (general-purpose) (Meng et al., 2016) is also a joint latent variable-based machine learning model, and was benchmarked against iCluster and iClusterPlus. The main difference between moCluster and the iCluster series is the method of finding latent variables. Instead of using an expectation-maximization algorithm (Moon, 1996), moCluster uses consensus PCA (CPCA) (Westerhuis et al., 1998) to estimate the latent variables. CPCA is a variation of the typical PCA algorithm and allows modeling data from different groups, which can be naturally mapped to different omics layers. moCluster was shown to run 100 to 1,000 times faster than iCluster/iClusterPlus on a simulated dataset with better clustering performance (Meng et al., 2016). On a multi-omics dataset for the NCI-60 cancer cell lines (Gholami et al., 2013), moCluster was able to separate melanoma cell lines from the remaining cell lines, whereas iClusterPlus could not.
SNF (Similarity Network Fusion, task-specific) (Wang et al., 2014) is a network-based machine learning model developed for patient stratification as well as survival analysis. SNF focuses on patient similarity networks and uses a specific network fusion algorithm to iteratively update similarity networks for each omics layer, with information from other omics layers so that the similarity networks become more consistent. The fused network contains information from all omics layers, thus enabling multi-omics data integration. SNF is similar to HNMDRP, as both methods are based on similarity networks (Heckerman, 1990). However, SNF only uses molecular profile information, whereas HNMDRP requires drug chemical structure data and drug target information. Despite the similarity between the two methods, no comparison was made for these two methods. SNF revealed two clusters in pancreatic ductal adenocarcinoma using epigenomics and transcriptomics data, demonstrating potential personalized treatment opportunities (Raphael et al., 2017).
NEMO (NEighborhood based Multi-Omics clustering, task-specific) (Rappoport and Shamir, 2019) provides enhancements for other similarity-based methods, including SNF and HNMDRP. NEMO optimized the similarity inference algorithms to enable faster runtime and added support for partial datasets (i.e., datasets in which a sample may not have the same coverage across all the omics). In the study, gene expression, DNA methylation, and miRNA expression data from ten cancer types in the TCGA dataset (Ding et al., 2016) were included as the input. NEMO was able to identify patient subgroups that showed significant difference in terms of survival, and achieved superior performance compared with another nine clustering algorithms (Rappoport and Shamir, 2019). NEMO not only achieved comparable performance to several other multi-omics integration methods, but its interface is also more user-friendly. Evaluated on ten different cancer datasets, NEMO runs approximately 400 times faster than iClusterBayes and 20% faster than SNF (Wang et al., 2014).
Benchmarking
Previous sections about various omics data, fundamental machine learning concepts, and integration tools provide basic knowledge for researchers to perform their multi-omics data integration analysis. To facilitate the decision on which machine learning tools are suitable for specific applications, we performed a comparative analysis for two baseline methods (early concatenation and PCA) and five multi-omics data integration tools (MOFA2 v1.1, DIABLO v6.17.15, iClusterPlus v1.22.0, iClusterBayes v1.22.0, and moCluster v1.20.0) by using a common CCLE dataset. These five methods were selected from Table 2 because they satisfied the following criteria. First, the method provides a software package that allows users to apply the analysis to custom datasets. Second, the method has at least 10 citations. Third, the method was published within the past five years (Figure 4A). Because patient stratification aims at discovering new molecular subtypes, which are defined a priori, we only included cancer type classification and drug response prediction as the two specific applications (Figure 4B).
Figure 4.

Details of the benchmarking analysis
(A) The process of determining the scope of the benchmarking analysis.
(B) An overview of the steps included in the benchmarking analysis.
CCLE multi-omics data were retrieved from the DepMap portal (version 20Q2), which consists of information from four omics layers (Ghandi et al., 2019; Nusinow et al., 2020), including WES mutation, CNV, gene expression (RNA), and protein abundance. To compare the five general-purpose tools with each other on an equal footing and report both prediction accuracy and runtime efficiency, we selected random forest as the downstream algorithm, because general-purpose tools do not directly predict drug responses and cancer types (Figure 4B). Specifically, random forests with 500 trees were fitted using five-fold cross-validation, which was repeated 100 times by the Monte Carlo method to obtain a robust evaluation. Source code for this benchmarking analysis is provided in GitHub https://github.com/CMRI-ProCan/MODIBenchmark.
Cancer type classification
In the CCLE dataset, the number of features for WES mutation, CNV, gene expression (RNA), and protein abundance are 1,585, 18,466, 18,812, and 17,551, respectively (Figure 4B). The WES data were sequenced with 76-bp pair-end reads by Illumina HiSeq 2000 or Illumina GAIIX. The tool Picard was used to process the data, and mutation calling was done by MuTect. The copy number information was derived from WES data by ABSOLUTE. The methylation data were generated using reduced representation bisulfite sequencing and aligned by Bismark. For RNA-seq data, STAR was used for alignment and the read counts were normalized to transcripts per million bases. The protein data were generated by TMT10plex (Ghandi et al., 2019; Nusinow et al., 2020). No additional feature selection was performed to ensure unbiased downstream analysis. Only cell lines measured with all four omics layers were considered, because not all methods support partial datasets. A total of 217 cell lines from nine different cancer types were included in this benchmark comparison and CCLE metadata were used as the ground truth. Accuracy is chosen to be the evaluation metric, which is defined as the number of samples that are correctly predicted divided by the total number of samples.
DIABLO showed the highest accuracy in cancer type classification, with an average of 78.7% (Figure 5A). DIABLO is the only supervised integration tool, which uses tissue types as label information. Notably, iCluster series, moCluster, and MOFA2 did not perform better than PCA, which was not specifically developed for biomedical applications. MOFA2 and iClusterBayes had lower accuracy than early concatenation, suggesting no real improvement was gained from the statistical integration. In summary, our benchmarking study indicates that DIABLO is the most appropriate tool when label information such as tissue type is available, and general dimensionality reduction methods are most appropriate for cancer type prediction when unsupervised integration is intended.
Figure 5.

Benchmarking of machine learning-based integration tools using the CCLE multi-omics data
(A) Accuracy of each method for cancer type prediction, showing standard errors of the mean derived from 100 runs of five-fold cross-validation, totalling 500 experiments (∗ signifies p value < 0.05 and ∗∗∗ signifies p value < 0.001 by an unpaired two-tailed Student’s t test).
(B) MAE comparison for drug response prediction across 1,448 drugs, error bars representing standard errors of the mean (∗∗∗ signifies p value < 0.001 and n.s. stands for not significant by an unpaired two-tailed Student’s t test).
(C) Runtime comparison. PCA is omitted as the runtime was negligible compared with the five multi-omics integration methods.
(D) A summary of the benchmarking study, derived from the results of cancer type prediction, drug response prediction (MAE between the measured AUC and predicted AUC), runtime comparison, and the number of citations since publication. The number of citations for PCA was set to the maximum for better visualization and because of its widespread use. The inverse of the runtime and drug response prediction MAE values are plotted so that higher values indicate better performance in all dimensions, and all values are plotted in the range of 0 to 1 in the radar plot.
Drug response prediction
For drug response prediction, we used the PRISM repurposing secondary screening dataset as the target, covering 1,448 drugs across 248 cell lines (Corsello et al., 2020). The area under the curve (AUC) of plasma concentration of a drug versus time after dosage (Scheff et al., 2011) was used as the drug response measurement. Mean absolute error (MAE) between the measured AUC and predicted AUC was chosen as the evaluation metric, where MAE measures the arithmetic average of the absolute errors across all the samples.
The performance difference between models was relatively small for drug response prediction when compared with cancer type prediction (Figure 5B). Early concatenation yielded the best MAE of 0.1239, followed by moCluster with an MAE of 0.1252. This is similar to a previous study (Singh et al., 2019), where early concatenation also outperformed DIABLO. No evident difference was observed between DIABLO, PCA, MOFA2, and iClusterPlus. iClusterBayes produced the worst prediction, with an average MAE of 0.1309 and 0.1311, respectively. Therefore, for drug response prediction, early concatenation should be prioritized if computational resources suffice.
Runtime comparison
Finally, we reported runtime for modeling CCLE data with the two baseline methods and the five integration tools. Early concatenation was not included because it does not involve modeling. The computation was completed on a CPU of Intel® Core™ i9-9880H @ 2.30GH and with default settings.
We set five sample sizes (20%/40%/60%/80%/100% of the data) and compared the time consumed to run each of the methods (Figure 5C). PCA took less than one second even for the full dataset. Among the five multi-omics integration tools, DIABLO, and moCluster took the least runtime across all sizes with a linear runtime complexity. Although iClusterBayes also has a linear runtime complexity, it consistently ran slower than other tools for all sizes. MOFA2 has better scalability than iClusterBayes with a logarithmic runtime complexity. For sizes from 20% to 60%, iClusterPlus took less time than iClusterBayes and MOFA2, with a similar runtime compared to moCluster and DIABLO. However, the runtime of iClusterPlus surged dramatically from 10 minutes to 78 minutes when the number of samples was increased from 60% to 80%. Thus, iClusterPlus indicates a nonlinear growth, and is suboptimal for large datasets.
Recommendation
No single best method across all aspects can be found when analyzing the CCLE multi-omics dataset (Figure 5D). PCA was robust and scalable among unsupervised methods when compared with MOFA2, iCluster series, and moCluster. In terms of average ranking, moCluster showed satisfactory performance across all three types of comparisons. It was ranked as the fourth for cancer type prediction and the second for drug response prediction with a linear runtime complexity.
Conclusions
We comprehensively reviewed current machine learning tools for the integration of multi-omics data across various research tasks in the field of cancer research. Multi-omics data analysis is key to understanding the complex dysregulation that is associated with different cancer phenotypes. Despite the exponential growth of the number of multi-omics experiments and the amount of data available for analysis, limited efforts have been made to develop machine learning tools that automatically integrate these multi-omics datasets. Existing reviews on this topic have predominantly focused on computational approaches, leaving a gap in the literature for a review of multi-omics technologies and data formats. All tools reviewed in this article can only be used via a command-line interface, which is not user friendly for non-computational researchers. Therefore, future tools with a graphical user interface will facilitate wider adoption in cancer research. More importantly, here we only reviewed methods that support custom datasets, and we conducted an independent benchmarking with a range of evaluations, including cancer type prediction, drug response prediction, and runtime efficiency. We concluded that many multi-omics data integration tools did not show significantly enhanced performance than PCA on a common dataset.
One challenge for multi-omics data integration is to account for the inconsistency between data generated from multiple sites. A meta-analysis across 12 laboratories has shown that consistency for copy number and transcriptomic data is relatively high, whereas methylation and proteomic data only showed moderate to low consistency (Jaiswal et al., 2021). Other attempts at the genomics and proteomics levels have been made to partially mitigate this inconsistency (Collins et al., 2017; Zhong et al., 2018). We expect future machine learning approaches can resolve issues such as batch effects and normalization within the integration analysis.
Certain omics data types are not covered in detail in this review because significant challenges regarding data integration are yet to be addressed. For example, metabolomic data record the levels of small molecules that are involved in cell metabolism, and metabolomics has shown a significant impact on cancer research to date (Wang et al., 2021). However, metabolomic data are not stored in a gene-level format, making it difficult for machine learning to integrate metabolomic data with other omics data.
Another challenge for most machine learning tools is to incorporate biological knowledge into modeling approaches. Gene regulation is a fundamental biological mechanism that describes a hidden link between layers of multi-omics data, but this association is often inappropriately modeled. Most statistical methods focus on explaining the greatest amount of variation in a dataset by using a small number of surrogate variables. This approach can miss the detail of true biological relationships. We thus hypothesize that dynamical modeling of the regulation between genes and subsequent omics layers might be a promising direction for the future development of machine learning-based multi-omics data integration tools. Causal models are likely to be applied to modeling gene regulations.
Acknowledgments
ProCan® is supported by the Australian Cancer Research Foundation, Cancer Institute New South Wales (NSW) (2017/TPG001, REG171150), and NSW Ministry of Health (CMP-01), The University of Sydney, Cancer Council NSW (IG 18-01), Ian Potter Foundation, the Medical Research Futures Fund (MRFF-PD), National Health and Medical Research Council (NHMRC) of Australia European Union grant (GNT1170739, a companion grant to support the European Commission’s Horizon 2020 Program, H2020-SC1-DTH-2018-1, 'iPC - individualizedPaediatricCure' [ref. 826121]), and National Breast Cancer Foundation (IIRS-18-164). This work was done under the auspices of a Memorandum of Understanding between Children's Medical Research Institute and the U.S. National Cancer Institute’s International Cancer Proteogenome Consortium (ICPC). R.C.P. is supported by a National Health and Medical Research Council (NHMRC) of Australia Fellowship (GNT1138536).
Authors contributions
Conceptual development: Z.C. and Q.Z.; Drafting the manuscript: Z.C. and Q.Z.; Revision and editing: Z.C., R.P., J.L. and Q.Z.; Illustrating figures: Z.C.
Declaration of interests
JL has received grant funding from AstraZeneca for research unrelated to the current work.
References
- Aizerman M.A. Theoretical foundations of the potential function method in pattern recognition learning. Autom. Remote Control. 1964;25:821–837. [Google Scholar]
- Alcala N., Leblay N., Gabriel A.A.G., Mangiante L., Hervas D., Giffon T., Sertier A.S., Ferrari A., Derks J., Ghantous A., et al. Integrative and comparative genomic analyses identify clinically relevant pulmonary carcinoid groups and unveil the supra-carcinoids. Nat. Commun. 2019;10:3407. doi: 10.1038/s41467-019-11276-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson R., Sandelin A. Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet. 2020;21:71–87. doi: 10.1038/s41576-019-0173-8. [DOI] [PubMed] [Google Scholar]
- Andrew G., Arora R., Bilmes J., Livescu K. Deep canonical correlation analysis. Proc. 30th Int. Conf. Machine Learn. 2013;28:1247–1255. [Google Scholar]
- Argelaguet R., Velten B., Arnol D., Dietrich S., Zenz T., Marioni J.C., Buettner F., Huber W., Stegle O. Multi-omics factor analysis—a framework for unsupervised integration of multi-omics data sets. Mol. Syst. Biol. 2018;14:e8124. doi: 10.15252/msb.20178124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argelaguet R., Clark S.J., Mohammed H., Stapel L.C., Krueger C., Kapourani C.-A., Imaz-Rosshandler I., Lohoff T., Xiang Y., Hanna C.W., et al. Multi-omics profiling of mouse gastrulation at single-cell resolution. Nature. 2019;576:487–491. doi: 10.1038/s41586-019-1825-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argelaguet R., Arnol D., Bredikhin D., Deloro Y., Velten B., Marioni J.C., Stegle O. MOFA+: a statistical framework for comprehensive integration of multi-modal single-cell data. Genome Biol. 2020;21:111. doi: 10.1186/s13059-020-02015-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., Wilson C.J., Lehár J., Kryukov G.V., Sonkin D., et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass A.J., Thorsson V., Shmulevich I., Reynolds S.M., Miller M., Bernard B., Hinoue T., Laird P.W., Curtis C., Shen H., et al. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–209. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavafaye Haghighi E., Knudsen M., Elmedal Laursen B., Besenbacher S. Hierarchical classification of cancers of unknown primary using multi-omics data. Cancer Inform. 2019;18 doi: 10.1177/1176935119872163. 1176935119872163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behan F.M., Iorio F., Picco G., Gonçalves E., Beaver C.M., Migliardi G., Santos R., Rao Y., Sassi F., Pinnelli M., et al. Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature. 2019;568:511–516. doi: 10.1038/s41586-019-1103-9. [DOI] [PubMed] [Google Scholar]
- Bellman R. Dynamic programming. Science. 1966;153:34–37. doi: 10.1126/science.153.3731.34. [DOI] [PubMed] [Google Scholar]
- Bohan S.S., Sicklick J.K., Kato S., Okamura R., Miller V.A., Leyland-Jones B., Lippman S.M., Kurzrock R. Attrition of patients on a precision oncology trial: analysis of the I-PREDICT experience. Oncologist. 2020;25:e1803–e1806. doi: 10.1002/onco.13532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L. Random forests. Mach. Learn. 2001;45:5–32. [Google Scholar]
- Broad . 2020. DepMap. DepMap 20Q3 Public Figshare Dataset. https://figshare.com/articles/dataset/DepMap_21Q4_Public/16924132/1. [Google Scholar]
- Brouwer I., Lenstra T.L. Visualizing transcription: key to understanding gene expression dynamics. Curr. Opin. Chem. Biol. 2019;51:122–129. doi: 10.1016/j.cbpa.2019.05.031. [DOI] [PubMed] [Google Scholar]
- Campbell P.J., Getz G., Korbel J.O., Stuart J.M., Jennings J.L., Stein L.D., Perry M.D., Nahal-Bose H.K., Ouellette B.F.F., Li C.H., et al. Pan-cancer analysis of whole genomes. Nature. 2020;578:82–93. [Google Scholar]
- Cichonska A., Pahikkala T., Szedmak S., Julkunen H., Airola A., Heinonen M., Aittokallio T., Rousu J. Learning with multiple pairwise kernels for drug bioactivity prediction. Bioinformatics. 2018;34:i509–i518. doi: 10.1093/bioinformatics/bty277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark D.J., Dhanasekaran S.M., Petralia F., Pan J., Song X., Hu Y., da Veiga Leprevost F., Reva B., Lih T.-S.M., Chang H.-Y., et al. Integrated proteogenomic characterization of clear cell renal cell carcinoma. Cell. 2019;179:964–983.e31. doi: 10.1016/j.cell.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P., Cross D., Jänne P.A. Kinase drug discovery 20 years after imatinib: progress and future directions. Nat. Rev. Drug Discov. 2021;20:551–569. doi: 10.1038/s41573-021-00195-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins B.C., Hunter C.L., Liu Y., Schilling B., Rosenberger G., Bader S.L., Chan D.W., Gibson B.W., Gingras A.-C., Held J.M., et al. Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry. Nat. Commun. 2017;8:291. doi: 10.1038/s41467-017-00249-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsello S.M., Nagari R.T., Spangler R.D., Rossen J., Kocak M., Bryan J.G., Humeidi R., Peck D., Wu X., Tang A.A., et al. Discovering the anticancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer. 2020;1:235–248. doi: 10.1038/s43018-019-0018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F. Central dogma of molecular biology. Nature. 1970;227:561–563. doi: 10.1038/227561a0. [DOI] [PubMed] [Google Scholar]
- Cunningham J.M., Christensen E.R., Tester D.J., Kim C.-Y., Roche P.C., Burgart L.J., Thibodeau S.N. Hypermethylation of the hMLH1 promoter in colon cancer with microsatellite instability. Cancer Res. 1998;58:3455–3460. [PubMed] [Google Scholar]
- Dai X., Li T., Bai Z., Yang Y., Liu X., Zhan J., Shi B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015;5:2929. [PMC free article] [PubMed] [Google Scholar]
- Ding Z., Zu S., Gu J. Evaluating the molecule-based prediction of clinical drug responses in cancer. Bioinformatics. 2016;32:2891–2895. doi: 10.1093/bioinformatics/btw344. [DOI] [PubMed] [Google Scholar]
- Duda R.O., Hart P.E., Stork D. Wiley; 2000. Pattern Classification. [Google Scholar]
- Everett B. Springer Science & Business Media; 2013. An Introduction to Latent Variable Models. [Google Scholar]
- Freedman D.A. Cambridge University Press; 2009. Statistical Models: Theory and Practice. [Google Scholar]
- Gao H., Korn J.M., Ferretti S., Monahan J.E., Wang Y., Singh M., Zhang C., Schnell C., Yang G., Zhang Y., et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015;21:1318–1325. doi: 10.1038/nm.3954. [DOI] [PubMed] [Google Scholar]
- Ghandi M., Huang F.W., Jané-Valbuena J., Kryukov G.V., Lo C.C., McDonald E.R., Barretina J., Gelfand E.T., Bielski C.M., Li H., et al. Next-generation characterization of the cancer cell line encyclopedia. Nature. 2019;569:503–508. doi: 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gholami A.M., Hahne H., Wu Z., Auer F.J., Meng C., Wilhelm M., Kuster B. Global proteome analysis of the NCI-60 cell line panel. Cell Rep. 2013;4:609–620. doi: 10.1016/j.celrep.2013.07.018. [DOI] [PubMed] [Google Scholar]
- Gillette M.A., Satpathy S., Cao S., Dhanasekaran S.M., Vasaikar S.V., Krug K., Petralia F., Li Y., Liang W.-W., Reva B., et al. Proteogenomic characterization reveals therapeutic vulnerabilities in lung adenocarcinoma. Cell. 2020;182:200–225.e35. doi: 10.1016/j.cell.2020.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T., Li L., Zhong Q., Rupp N.J., Charmpi K., Wong C.E., Wagner U., Rueschoff J.H., Jochum W., Fankhauser C.D., et al. Multi-region proteome analysis quantifies spatial heterogeneity of prostate tissue biomarkers. Life Sci. Alliance. 2018;1:e201800042. doi: 10.26508/lsa.201800042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraksingh R.R., Snyder M.P. Impacts of variation in the human genome on gene regulation. J. Mol. Biol. 2013;425:3970–3977. doi: 10.1016/j.jmb.2013.07.015. [DOI] [PubMed] [Google Scholar]
- Heckerman D. Probabilistic similarity networks. Networks. 1990;20:607–636. [Google Scholar]
- Hegde P.S., White I.R., Debouck C. Interplay of transcriptomics and proteomics. Curr. Opin. Biotechnol. 2003;14:647–651. doi: 10.1016/j.copbio.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Hotelling H. Breakthroughs in Statistics. Springer; 1992. Relations between two sets of variates; pp. 162–190. [Google Scholar]
- Iorio F., Knijnenburg T.A., Vis D.J., Bignell G.R., Menden M.P., Schubert M., Aben N., Gonçalves E., Barthorpe S., Lightfoot H., et al. A landscape of pharmacogenomic interactions in cancer. Cell. 2016;166:740–754. doi: 10.1016/j.cell.2016.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal A., Gautam P., Pietilä E.A., Timonen S., Nordström N., Akimov Y., Sipari N., Tanoli Z., Fleischer T., Lehti K., et al. Multi-modal meta-analysis of cancer cell line omics profiles identifies ECHDC1 as a novel breast tumor suppressor. Mol. Syst. Biol. 2021;17:e9526. doi: 10.15252/msb.20209526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost J., Saluz H. Birkhäuser; 2013. DNA Methylation: Molecular Biology and Biological Significance. [Google Scholar]
- Kaeberlein M., Kennedy B.K. Protein translation, 2007. Aging Cell. 2007;6:731–734. doi: 10.1111/j.1474-9726.2007.00341.x. [DOI] [PubMed] [Google Scholar]
- Kim K.G. Book review: deep learning. Healthc. Inform. Res. 2016;22:351. [Google Scholar]
- Koza J.R., Bennett F.H., Andre D., Keane M.A. In: Artificial Intelligence in Design ’96. Gero J.S., Sudweeks F., editors. Springer Netherlands; 1996. Automated design of both the topology and sizing of analog electrical circuits using genetic programming; pp. 151–170. [Google Scholar]
- Krueger F., Kreck B., Franke A., Andrews S.R. DNA methylome analysis using short bisulfite sequencing data. Nat. Methods. 2012;9:145. doi: 10.1038/nmeth.1828. [DOI] [PubMed] [Google Scholar]
- Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Law C.W., Chen Y., Shi W., Smyth G.K. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M.S., Stojanov P., Polak P., Kryukov G.V., Cibulskis K., Sivachenko A., Carter S.L., Stewart C., Mermel C.H., Roberts S.A., et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Tourneau C., Delord J.-P., Gonçalves A., Gavoille C., Dubot C., Isambert N., Campone M., Trédan O., Massiani M.-A., Mauborgne C., et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015;16:1324–1334. doi: 10.1016/S1470-2045(15)00188-6. [DOI] [PubMed] [Google Scholar]
- Lee B., Zhang S., Poleksic A., Xie L. Heterogeneous multi-layered network model for omics data integration and analysis. Front. Genet. 2020;10:1381. doi: 10.3389/fgene.2019.01381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Wong K.-C. Evolutionary multiobjective clustering and its applications to patient stratification. IEEE Trans. Cybern. 2019;49:1680–1693. doi: 10.1109/TCYB.2018.2817480. [DOI] [PubMed] [Google Scholar]
- Lin Y., Liu T., Fuh C. Multiple kernel learning for dimensionality reduction. IEEE Trans. Pattern Anal. Mach. Intell. 2011;33:1147–1160. doi: 10.1109/TPAMI.2010.183. [DOI] [PubMed] [Google Scholar]
- Lock E.F., Hoadley K.A., Marron J.S., Nobel A.B. Joint and individual variation explained (JIVE) for integrated analysis of multiple data types. Ann. Appl. Stat. 2013;7:523–542. doi: 10.1214/12-AOAS597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López de Maturana E., Alonso L., Alarcón P., Martín-Antoniano I.A., Pineda S., Piorno L., Calle M.L., Malats N. Challenges in the integration of omics and non-omics data. Genes. 2019;10:238. doi: 10.3390/genes10030238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M.Y., Chen T.Y., Williamson D.F.K., Zhao M., Shady M., Lipkova J., Mahmood F. AI-based pathology predicts origins for cancers of unknown primary. Nature. 2021;594:106–110. doi: 10.1038/s41586-021-03512-4. [DOI] [PubMed] [Google Scholar]
- Lundberg S.M., Lee S.-I. In: Advances in Neural Information Processing Systems 30. Guyon I., Luxburg U.V., Bengio S., Wallach H., Fergus R., Vishwanathan S., Garnett R., editors. Curran Associates, Inc.; 2017. A unified approach to interpreting model predictions; pp. 4765–4774. [Google Scholar]
- Malone J.H., Oliver B. Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biol. 2011;9:1–9. doi: 10.1186/1741-7007-9-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire A.L., Gabriel S., Tishkoff S.A., Wonkam A., Chakravarti A., Furlong E.E.M., Treutlein B., Meissner A., Chang H.Y., López-Bigas N., et al. The road ahead in genetics and genomics. Nat. Rev. Genet. 2020;21:581–596. doi: 10.1038/s41576-020-0272-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng C., Helm D., Frejno M., Kuster B. moCluster: identifying joint patterns across multiple omics data sets. J. Proteome Res. 2016;15:755–765. doi: 10.1021/acs.jproteome.5b00824. [DOI] [PubMed] [Google Scholar]
- Mitchell T.M. McGraw-hill; 1997. Machine Learning. [Google Scholar]
- Mo Q., Wang S., Seshan V.E., Olshen A.B., Schultz N., Sander C., Powers R.S., Ladanyi M., Shen R. Pattern discovery and cancer gene identification in integrated cancer genomic data. Proc. Natl. Acad. Sci. U S A. 2013;110:4245–4250. doi: 10.1073/pnas.1208949110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo Q., Shen R., Guo C., Vannucci M., Chan K.S., Hilsenbeck S.G. A fully Bayesian latent variable model for integrative clustering analysis of multi-type omics data. Biostatistics. 2018;19:71–86. doi: 10.1093/biostatistics/kxx017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon T.K. The expectation-maximization algorithm. IEEE Signal. Process. Mag. 1996;13:47–60. [Google Scholar]
- Nakagawa H., Fujita M. Whole genome sequencing analysis for cancer genomics and precision medicine. Cancer Sci. 2018;109:513–522. doi: 10.1111/cas.13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam A.S., Chaligne R., Landau D.A. Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat. Rev. Genet. 2021;22:3–18. doi: 10.1038/s41576-020-0265-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimento A.C.A., Prudêncio R.B.C., Costa I.G. A multiple kernel learning algorithm for drug-target interaction prediction. BMC Bioinformatics. 2016;17:46. doi: 10.1186/s12859-016-0890-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicora G., Vitali F., Dagliati A., Geifman N., Bellazzi R. Integrated multi-omics analyses in oncology: a review of machine learning methods and tools. Front. Oncol. 2020;10:1030. doi: 10.3389/fonc.2020.01030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen T.O., Parker J.S., Leung S., Voduc D., Ebbert M., Vickery T., Davies S.R., Snider J., Stijleman I.J., Reed J., et al. A comparison of PAM50 intrinsic subtyping with immunohistochemistry and clinical prognostic factors in tamoxifen-treated estrogen receptor–positive breast cancer. Clin. Cancer Res. 2010;16:5222–5232. doi: 10.1158/1078-0432.CCR-10-1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niklas K.J., Bondos S.E., Dunker A.K., Newman S.A. Rethinking gene regulatory networks in light of alternative splicing, intrinsically disordered protein domains, and post-translational modifications. Front. Cell Dev. Biol. 2015;3:8. doi: 10.3389/fcell.2015.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusinow D.P., Szpyt J., Ghandi M., Rose C.M., McDonald E.R., Kalocsay M., Jané-Valbuena J., Gelfand E., Schweppe D.K., Jedrychowski M., et al. Quantitative proteomics of the cancer cell line encyclopedia. Cell. 2020;180:387–402.e16. doi: 10.1016/j.cell.2019.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell S.T., Ross R.P., Stanton C. The progress of multi-omics technologies: determining function in lactic acid bacteria using a systems level approach. Front. Microbiol. 2020;10:3084. doi: 10.3389/fmicb.2019.03084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh M., Park S., Kim S., Chae H. Machine learning-based analysis of multi-omics data on the cloud for investigating gene regulations. Brief. Bioinform. 2021;22:66–76. doi: 10.1093/bib/bbaa032. [DOI] [PubMed] [Google Scholar]
- Pavlidis N., Pentheroudakis G. Cancer of unknown primary site. Lancet. 2012;379:1428–1435. doi: 10.1016/S0140-6736(11)61178-1. [DOI] [PubMed] [Google Scholar]
- Picard M., Scott-Boyer M.-P., Bodein A., Périn O., Droit A. Integration strategies of multi-omics data for machine learning analysis. Comput. Struct. Biotechnol. J. 2021;19:3735–3746. doi: 10.1016/j.csbj.2021.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulos R.C., Wong J.W. ELS. American Cancer Society; 2017. cis-regulatory driver mutations in cancer genomes; pp. 1–10. [Google Scholar]
- Poulos R.C., Hains P.G., Shah R., Lucas N., Xavier D., Manda S.S., Anees A., Koh J.M.S., Mahboob S., Wittman M., et al. Strategies to enable large-scale proteomics for reproducible research. Nat. Commun. 2020;11:3793. doi: 10.1038/s41467-020-17641-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raphael B.J., Hruban R.H., Aguirre A.J., Moffitt R.A., Yeh J.J., Stewart C., Robertson A.G., Cherniack A.D., Gupta M., Getz G., et al. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell. 2017;32:185–203.e13. doi: 10.1016/j.ccell.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappoport N., Shamir R. Multi-omic and multi-view clustering algorithms: review and cancer benchmark. Nucleic Acids Res. 2018;46:10546–10562. doi: 10.1093/nar/gky889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappoport N., Shamir R. NEMO: cancer subtyping by integration of partial multi-omic data. Bioinformatics. 2019;35:3348–3356. doi: 10.1093/bioinformatics/btz058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reel P.S., Reel S., Pearson E., Trucco E., Jefferson E. Using machine learning approaches for multi-omics data analysis: a review. Biotechnol. Adv. 2021;49:107739. doi: 10.1016/j.biotechadv.2021.107739. [DOI] [PubMed] [Google Scholar]
- Ribeiro M.T., Singh S., Guestrin C. “Why should i trust you?”: explaining the predictions of any classifier. Proceedings of the 22nd ACM SIGKDD international conference on knowledge discovery and data mining. 2016:1135–1144. [Google Scholar]
- Risso D., Ngai J., Speed T.P., Dudoit S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014;32:896–902. doi: 10.1038/nbt.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodosthenous T., Shahrezaei V., Evangelou M. Integrating multi-OMICS data through sparse Canonical Correlation Analysis for the prediction of complex traits: a comparison study. Bioinformatics. 2020;36:4616–4625. doi: 10.1093/bioinformatics/btaa530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohart F., Gautier B., Singh A., Cao K.-A.L. mixOmics: an R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017;13:e1005752. doi: 10.1371/journal.pcbi.1005752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokach L., Maimon O. Data Mining and Knowledge Discovery Handbook. Springer; 2005. Decision trees; pp. 165–192. [Google Scholar]
- Rumelhart D.E., Hinton G.E., Williams R.J. Learning representations by back-propagating errors. Nature. 1986;323:533–536. [Google Scholar]
- Russell S., Norvig P. Pearson; 2020. Artificial Intelligence: A Modern Approach. [Google Scholar]
- Sathyanarayanan A., Gupta R., Thompson E.W., Nyholt D.R., Bauer D.C., Nagaraj S.H. A comparative study of multi-omics integration tools for cancer driver gene identification and tumour subtyping. Brief. Bioinform. 2020;21:1920–1936. doi: 10.1093/bib/bbz121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff J.D., Almon R.R., DuBois D.C., Jusko W.J., Androulakis I.P. Assessment of pharmacologic area under the curve when baselines are variable. Pharm. Res. 2011;28:1081–1089. doi: 10.1007/s11095-010-0363-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroff F., Kalenichenko D., Philbin J. Proceedings of the IEEE conference on computer vision and pattern recognition. 2015. Facenet: A Unified Embedding for Face Recognition and Clustering; pp. 815–823. [Google Scholar]
- Schwarze K., Buchanan J., Taylor J.C., Wordsworth S. Are whole-exome and whole-genome sequencing approaches cost-effective? A systematic review of the literature. Genet. Med. 2018;20:1122–1130. doi: 10.1038/gim.2017.247. [DOI] [PubMed] [Google Scholar]
- Seligson D.B., Horvath S., Shi T., Yu H., Tze S., Grunstein M., Kurdistani S.K. Global histone modification patterns predict risk of prostate cancer recurrence. Nature. 2005;435:1262–1266. doi: 10.1038/nature03672. [DOI] [PubMed] [Google Scholar]
- Sha Y., Phan J.H., Wang M.D. 2015 37th Annual International Conference of the IEEE Engineering in Medicine and Biology Society. EMBC; 2015. Effect of low-expression gene filtering on detection of differentially expressed genes in RNA-seq data; pp. 6461–6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharifi-Noghabi H., Zolotareva O., Collins C.C., Ester M. MOLI: multi-omics late integration with deep neural networks for drug response prediction. Bioinformatics. 2019;35:i501–i509. doi: 10.1093/bioinformatics/btz318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen R., Olshen A.B., Ladanyi M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics. 2009;25:2906–2912. doi: 10.1093/bioinformatics/btp543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A.P., Gordon G.J. In: Machine Learning and Knowledge Discovery in Databases. Daelemans W., Goethals B., Morik K., editors. Springer Berlin Heidelberg; 2008. A unified view of matrix factorization models; pp. 358–373. [Google Scholar]
- Singh A., Shannon C.P., Gautier B., Rohart F., Vacher M., Tebbutt S.J., Lê Cao K.-A. DIABLO: an integrative approach for identifying key molecular drivers from multi-omics assays. Bioinformatics. 2019;35:3055–3062. doi: 10.1093/bioinformatics/bty1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speicher N.K., Pfeifer N. Integrating different data types by regularized unsupervised multiple kernel learning with application to cancer subtype discovery. Bioinformatics. 2015;31:i268–i275. doi: 10.1093/bioinformatics/btv244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spirin V., Mirny L.A. Protein complexes and functional modules in molecular networks. Proc. Natl. Acad. Sci. U S A. 2003;100:12123–12128. doi: 10.1073/pnas.2032324100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanfield Z., Coşkun M., Koyutürk M. Drug response prediction as a link prediction problem. Sci. Rep. 2017;7:40321. doi: 10.1038/srep40321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokholm J., Blaser M.J., Thorsen J., Rasmussen M.A., Waage J., Vinding R.K., Schoos A.-M.M., Kunøe A., Fink N.R., Chawes B.L., et al. Maturation of the gut microbiome and risk of asthma in childhood. Nat. Commun. 2018;9:141. doi: 10.1038/s41467-017-02573-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton M.R., Campbell P.J., Futreal P.A. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian I., Verma S., Kumar S., Jere A., Anamika K. Multi-omics data integration, interpretation, and its application. Bioinforma. Biol. Insights. 2020;14 doi: 10.1177/1177932219899051. 1177932219899051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suphavilai C., Bertrand D., Nagarajan N. Predicting cancer drug response using a recommender system. Bioinforma. Oxf. Engl. 2018;34:3907–3914. doi: 10.1093/bioinformatics/bty452. [DOI] [PubMed] [Google Scholar]
- Szabo Q., Bantignies F., Cavalli G. Principles of genome folding into topologically associating domains. Sci. Adv. 2019;5:eaaw1668. doi: 10.1126/sciadv.aaw1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannock I.F., Hickman J.A. Limits to personalized cancer medicine. N. Engl. J. Med. 2016;375:1289–1294. doi: 10.1056/NEJMsb1607705. [DOI] [PubMed] [Google Scholar]
- Tate J.G., Bamford S., Jubb H.C., Sondka Z., Beare D.M., Bindal N., Boutselakis H., Cole C.G., Creatore C., Dawson E., et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47:D941–D947. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenenhaus A., Philippe C., Guillemot V., Le Cao K.-A., Grill J., Frouin V. Variable selection for generalized canonical correlation analysis. Biostat. Oxf. Engl. 2014;15:569–583. doi: 10.1093/biostatistics/kxu001. [DOI] [PubMed] [Google Scholar]
- The International Cancer Genome Consortium International network of cancer genome projects. Nature. 2010;464:993–998. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A., Liu S.V., Subramaniam D.S., Giaccone G. Refining the treatment of NSCLC according to histological and molecular subtypes. Nat. Rev. Clin. Oncol. 2015;12:511–526. doi: 10.1038/nrclinonc.2015.90. [DOI] [PubMed] [Google Scholar]
- Tully B., Balleine R.L., Hains P.G., Zhong Q., Reddel R.R., Robinson P.J. Addressing the challenges of high-throughput cancer tissue proteomics for clinical application: ProCan. PROTEOMICS. 2019;19:1900109. doi: 10.1002/pmic.201900109. [DOI] [PubMed] [Google Scholar]
- Ullah hsan, Rios A., Gala V., Mckeever S. Explaining deep learning models for structured data using layer-wise relevance propagation. Appl. Sci. 2020;12:136. [Google Scholar]
- Välikangas T., Suomi T., Elo L.L. A comprehensive evaluation of popular proteomics software workflows for label-free proteome quantification and imputation. Brief. Bioinform. 2018;19:1344–1355. doi: 10.1093/bib/bbx054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valouev A., Johnson D.S., Sundquist A., Medina C., Anton E., Batzoglou S., Myers R.M., Sidow A. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat. Methods. 2008;5:829–834. doi: 10.1038/nmeth.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Berkum N.L., Lieberman-Aiden E., Williams L., Imakaev M., Gnirke A., Mirny L.A., Dekker J., Lander E.S. Hi-C: a method to study the three-dimensional architecture of genomes. J. Vis. Exp. 2010;39:e1869. doi: 10.3791/1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K.C., Chang H.Y. Epigenomics: technologies and applications. Circ. Res. 2018;122:1191–1199. doi: 10.1161/CIRCRESAHA.118.310998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B., Mezlini A.M., Demir F., Fiume M., Tu Z., Brudno M., Haibe-Kains B., Goldenberg A. Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods. 2014;11:333–337. doi: 10.1038/nmeth.2810. [DOI] [PubMed] [Google Scholar]
- Wang L.-B., Karpova A., Gritsenko M.A., Kyle J.E., Cao S., Li Y., Rykunov D., Colaprico A., Rothstein J.H., Hong R., et al. Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell. 2021;39:509–528.e20. doi: 10.1016/j.ccell.2021.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerhuis J.A., Kourti T., MacGregor J.F. Analysis of multiblock and hierarchical PCA and PLS models. J. Chemom. 1998;12:301–321. [Google Scholar]
- Wold S., Esbensen K., Geladi P. Principal component analysis. Chemom. Intell. Lab. Syst. 1987;2:37–52. [Google Scholar]
- Wong C.C.Y., Meaburn E.L., Ronald A., Price T.S., Jeffries A.R., Schalkwyk L.C., Plomin R., Mill J. Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Mol. Psychiatry. 2014;19:495–503. doi: 10.1038/mp.2013.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wreczycka K., Gosdschan A., Yusuf D., Grüning B., Assenov Y., Akalin A. Strategies for analyzing bisulfite sequencing data. J. Biotechnol. 2017;261:105–115. doi: 10.1016/j.jbiotec.2017.08.007. [DOI] [PubMed] [Google Scholar]
- Xu X., Zhang Y., Williams J., Antoniou E., McCombie W.R., Wu S., Zhu W., Davidson N.O., Denoya P., Li E. Parallel comparison of Illumina RNA-Seq and Affymetrix microarray platforms on transcriptomic profiles generated from 5-aza-deoxy-cytidine treated HT-29 colon cancer cells and simulated datasets. BMC Bioinformatics. 2013;14:S1. doi: 10.1186/1471-2105-14-S9-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue H.-J., Dai X., Zhang J., Huang S., Chen J. Vol. 17. 2017. Deep Matrix Factorization Models for Recommender Systems; pp. 3203–3209. (IJCAI). [Google Scholar]
- Yang Y., Muzny D.M., Reid J.G., Bainbridge M.N., Willis A., Ward P.A., Braxton A., Beuten J., Xia F., Niu Z., et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 2013;369:1502–1511. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Wang J., Wang X., Zhu J., Liu Q., Shi Z., Chambers M.C., Zimmerman L.J., Shaddox K.F., Kim S., et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513:382–387. doi: 10.1038/nature13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F., Wang M., Xi J., Yang J., Li A. A novel heterogeneous network-based method for drug response prediction in cancer cell lines. Sci. Rep. 2018;8:3355. doi: 10.1038/s41598-018-21622-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Parmigiani G., Johnson W.E. ComBat-seq: batch effect adjustment for RNA-seq count data. NAR Genomics Bioinforma. 2020;2:lqaa078. doi: 10.1093/nargab/lqaa078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Wang D., Peng M., Tang L., Ouyang J., Xiong F., Guo C., Tang Y., Zhou Y., Liao Q., et al. Single-cell RNA sequencing in cancer research. J. Exp. Clin. Cancer Res. 2021;40:81. doi: 10.1186/s13046-021-01874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Q., Wagner U., Kurt H., Molinari F., Cathomas G., Komminoth P., Barman-Aksözen J., Schneider-Yin X., Rey J.-P., Vassella E., et al. Multi-laboratory proficiency testing of clinical cancer genomic profiling by next-generation sequencing. Pathol. - Res. Pract. 2018;214:957–963. doi: 10.1016/j.prp.2018.05.020. [DOI] [PubMed] [Google Scholar]
- Zou H., Hastie T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B Stat. Methodol. 2005;67:301–320. [Google Scholar]