

Abstract
Sporadic cerebral small vessel disease (SVD) is a major contributor to vascular cognitive impairment and dementia in the aging human brain. On neuropathology, sporadic SVD is characterized by abnormalities to the small vessels of the brain predominantly in the form of cerebral amyloid angiopathy and arteriolosclerosis. These pathologies frequently coexist with Alzheimer’s disease changes, such as plaques and tangles, in a single brain. Conversely, during life MRI only captures the larger manifestations of SVD in the form of parenchymal brain abnormalities. There appears to be a major knowledge gap regarding the underlying neuropathology of individual MRI-detectable SVD abnormalities. Ex vivo MRI in post-mortem human brain tissue is a powerful tool to bridge this gap. This review summarizes current insights into the histopathological correlations of MRI-manifestations of SVD, their underlying etiology, presumed pathophysiology, and associated secondary tissue injury. Moreover, we discuss the advantages and limitations of ex vivo MRI-guided histopathological investigations and make recommendations for future studies.
Introduction
The concept of vascular contributions to cognitive impairment and dementia has arisen from a growing understanding of the role of vascular disease in cognitive impairment and overall brain health during aging(1). Sporadic cerebral small vessel disease (SVD) is considered the most common and significant driver of vascular cognitive impairment in older individuals. Neuropathologically, sporadic SVD constitutes of abnormalities to the small vessels of the brain including cerebral amyloid angiopathy (CAA) and arteriolosclerosis(2). Other forms of SVD not discussed in this review include hereditary variants such as Dutch-type CAA and cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. CAA and arteriolosclerosis frequently coexist with Alzheimer’s disease (AD) neuropathological changes (e.g. amyloid β plaques and tangles) at autopsy in older persons. A growing body of literature in large community- and hospital-based autopsy studies supports independent contributions of small vessel pathologies to ante-mortem cognition and dementia. During life, SVD is recognized by parenchymal abnormalities rather than direct visualization of small blood vessels on MRI. These larger manifestations of SVD have been well-characterized in terms of their MRI appearance and detection as well as associations with disease progression and cognitive trajectories(3). However, there appears to be a large gap between the MRI-manifestations of SVD at one end of the spectrum, and the microscopic abnormalities to individual small blood vessels detected at autopsy at the other end.
To address this gap, this qualitative review aims to provide an up-date on the neuropathological associations underlying individual MRI-manifestations of SVD. We discuss the use of ex vivo MRI-guided histopathological investigations as a powerful tool to bridge the gap between in vivo MRI findings and neuropathological observations in the context of sporadic SVD. We zoom in on the histopathological signature of MRI-detectable parenchymal SVD abnormalities and their presumed underlying etiologies, discuss novel insights regarding potential pathophysiological mechanisms, and evidence of peri-lesional tissue injury that may contribute to overall reduced brain health. Finally, we provide an overview of outstanding questions and knowledge gaps related to the neuropathology of SVD manifestations (Supplementary Box I).
Cerebral small vessel disease
Neuropathology of SVD
From a neuropathologic standpoint, arteriolosclerosis and CAA pathology have important similarities as well as distinctions. They are both microscopically identified SVD pathologies, common in aging and AD, occurring in small vessels, demonstrating often thickened, sometimes damaged vascular walls that may result in disruption of blood flow. Destruction of the vessel wall and flow may result in an assortment of neuropathologic sequelae including tissue ischemia (e.g. white matter degeneration), lacunar infarcts and microinfarcts, microbleeds and larger hemorrhages. Age is a prominent risk factor for both pathologies, yet the risk factors for arteriolosclerosis versus CAA are quite distinct. In addition to age, vascular risk such as diabetes and hypertension are central risk factors for arteriolosclerosis. By contrast, the most notable risk factor for sporadic CAA is AD and the Apolipoprotein E4 and E2 alleles.
By convention, arteriolosclerosis in the brain is histologically identified in the subcortical white matter and in the basal ganglia (Supplementary Figure I). The thalamus, basis pontis, and cerebellar white matter, also harbor penetrating vessels, and are also commonly affected by arteriolosclerosis. Brain arteriolosclerosis typically manifests with variable destruction of the intima and smooth muscle media with replacement by concentric fibrohyalinosis(4). In some instances, there may also be lipid deposition, historically named ‘lipohyalinosis’ and fibrinoid necrosis in severe cases. There are regional differences in severity of arteriolosclerosis with anterior and posterior white matter regions showing more severe vessel wall pathology(4). The spinal cord may also show evidence of arteriolosclerosis(5). Lacunar infarcts, microinfarcts, and microbleeds associated with arteriolosclerosis are typically located in subcortical regions of the brain. However, recent data suggests that in the presence of significant AD pathology (e.g. amyloid β plaques and tangles) arteriolosclerosis is associated with an increased odds of cortical microinfarcts(6).
Distinct from arteriolosclerosis, CAA preferentially affects the arterioles of the leptomeninges and the cerebral cortex with little to no involvement of the deeper gray or white matter structures (Supplementary Figure I). CAA is most notable across neocortical regions and tends to be somewhat more severe in the posterior parts of the brain. Indeed, larger hemorrhages associated with CAA tend to occur more often in the parietal-occipital regions of the brain(7). Capillaries are affected in a subset of cases and may have distinct roles in the disease(8). In CAA, the media is replaced with amyloid β 40, the lesser of the amyloidogenic species in AD. The vast majority of individuals with AD have some degree of CAA at autopsy, and about 40% have moderate to severe CAA(9). Conversely, there are few individuals with AD without any evidence of CAA, and those that meet the MRI-based Boston criteria for the diagnosis of CAA during life do not always have severe CAA on neuropathological examination of the brain(10). In the brain, vessels severely affected by CAA, may also show concentric splitting of the vessel wall or fibrinoid necrosis(8). In addition to ischemic and hemorrhagic manifestations there is a growing literature on the role of CAA in impairing perivascular drainage of amyloid β and other potentially toxic cellular metabolites(11). There are a few different published criteria for the staging of CAA for both sporadic cases with clinically significant hemorrhage(12) and in those with age-related CAA(8,9). In the former, there is emphasis of severe changes in the vessel walls, and in the latter an emphasis on overall burden of concentric vessel involvement. There are multiple potential pathways of tissue injury related to SVD. In addition to ischemia and hemorrhage, there may also be breakdown of the blood-brain barrier. Moreover, because small vessels and capillaries are central to perivascular clearance of amyloid β and other toxins, there has been increasing interest in the role of SVD in the export and abnormal retention of cellular waste.
MRI-manifestations of SVD
Because the individual small blood vessels are not resolved with current standard MRI techniques, SVD is often recognized on MRI by its more conspicuous manifestations in the brain in the form of parenchymal abnormalities. Conventionally, these manifestations include white matter hyperintensities (WMH), lacunar infarcts, microbleeds, microinfarcts, enlarged perivascular spaces, and cortical superficial siderosis (cSS) (Supplementary Figure I)(3). Other brain characteristics that have recently emerged in the context of SVD include diffusion-tensor imaging (DTI)-based microstructural changes and blood-brain barrier leakage / contrast enhancement(13,14). In this section we briefly summarize MRI techniques, diagnostic criteria, and clinical relevance related to each of these conventional and novel MRI-manifestations of SVD.
Conventional MRI-markers
White matter hyperintensities: are frequently observed white matter lesions in older individuals(3,15). WMH appear hyperintense on T2-weighted fluid-attenuated inversion recovery (FLAIR) MRI and are most commonly seen in the white matter near the lateral ventricles in community cohorts of older adults. The spatial pattern of WMH in the rest of the white matter is more variable and may reflect underlying small vessel pathologies. Multiple subcortical spots and posterior WMH have been linked to CAA, whereas WMH surrounding the basal ganglia appears to be indicative of arteriolosclerosis(16). Increasing WMH severity has consistently been found to predict cognitive decline, incident dementia, and stroke(17).
Lacunar infarcts: are relatively round or oval-shaped fluid-filled cavities with dimensions ranging from 3 to 15 mm, also termed ‘lacunes of presumed vascular origin’ and ‘recent small subcortical infarcts’ in the acute phase(3). Lacunar infarcts appear hyperintense on T2-weighted images, and hypointense on T1-weighted and FLAIR images. A hyperintense rim around the lacune is often visible on FLAIR. Small lacunar infarcts of the order of 3 to 5 mm can be confused with enlarged perivascular spaces. It has recently been suggested that the spatial pattern of lacunar infarcts may be indicative for the underlying small vessel pathologies, as lacunar infarcts in the white matter centrum semiovale were more commonly observed in patients with CAA-related intracerebral hemorrhage (ICH) whereas lacunar infarcts in deep areas of the brain were more frequently found in hypertensive ICH patients(18). Like WMH, lacunar infarcts have been associated with an increased risk of future stroke and cognitive decline(19).
Microbleeds: are visualized as relatively round regions of very low signal on T2*-weighted or susceptibility-weighted MRI(20). The size of the region with low signal is typically in the order of a few millimeters in diameter. Microbleeds are better visualized with higher magnetic field strengths due to the enhancement of magnetic susceptibility effects and higher signal to noise ratio. Microbleeds seen on conventional T2*-weighted or susceptibility-weighted MRI scans can be confused with calcifications, since in both cases the associated magnetic susceptibility effects lower the signal. Quantitative susceptibility mapping performed with multi-echo 3D gradient-echo sequences may resolve this problem(21). More so than WMH and lacunar infarcts, the topographical distribution of microbleeds has proven to be informative in the diagnosis of SVD subtype. The Boston criteria for the diagnosis of CAA relies on the detection of exclusively cortical microbleeds, whereas microbleeds in deep areas of the brain have been consistently associated with arteriolosclerosis(22,23). Although microbleeds have been associated with increased risk in future symptomatic ICH, the relationship with cognition appears to be modest(22).
Microinfarcts: are small ischemic lesions that can be detected as focal hyperintensities on diffusion-weighted MRI in the acute phase(24,25). Chronic microinfarcts appear hyperintense on T2-weighted and hypointense on T1-weighted MRI(24). Consensus criteria for the detection of chronic microinfarcts are currently limited to cortical areas of the brain, as microinfarcts in the white matter cannot easily be distinguished from WMH and enlarged perivascular spaces(24). Only large microinfarcts with sizes in the order of a millimeter remain visible over time, while the vast majority of microinfarcts escapes detection during life. Visualization of microinfarcts can obviously benefit from the use of high magnetic field strength(26). The presence of even a few microinfarcts on MRI has been associated with cognitive impairment, suggesting that despite their microscopic size, these lesions can have a profound impact on brain structure and function(24).
Perivascular spaces: are fluid-filled spaces surrounding blood vessels that facilitate transport between cerebrospinal and interstitial fluid(27). These spaces become enlarged during aging, which renders them visible on clinical MRI. They can be observed as linear structures following the course of vessels, varying in length from a few millimeters to a few centimeters. Enlarged perivascular spaces appear hyperintense on T2-weighted and hypointense on T1-weighted MRI and FLAIR images. Similar to microbleeds (and lacunar infarcts), the spatial pattern of enlarged perivascular spaces on MRI is indicative of the underlying small vessel pathologies. MRI-visible perivascular spaces in the centrum semiovale were more commonly observed in patients with CAA-related ICH, whereas perivascular spaces in deep areas of the brain were associated with hypertensive ICH(28).
Cortical superficial siderosis: appears as hypointense signals over the convexities of the brain and can be visualized using the same MRI sequences as those used for imaging microbleeds(29). cSS is believed to represent the chronic phase of convexity subarachnoid hemorrhage and is highly indicative of severe CAA. The presence of cSS on MRI dramatically increases the risk of both first-ever and recurrent ICH, and as such has emerged as one of the strongest and perhaps most clinically relevant marker in CAA(30).
See for a discussion on novel MRI-markers the Supplemental Material.
Overall, detection of several of the above MRI-manifestations of SVD can be enhanced with high spatial resolution and high signal to noise ratio imaging. Therefore, investigations of SVD may benefit from high magnetic field strengths (e.g. 7 Tesla), and the newest developments in MRI hardware (e.g. coils) and software (e.g. image reconstruction and image enhancement). Furthermore, most assessments of these manifestations are based on visual inspection and semiquantitative scales. SVD investigations will benefit from automated segmentation and fully quantitative assessment of SVD-related brain lesions. This has been achieved to a large degree for WMH(44), but a lot more work is needed for other lesions(27,45). An important next step that would move the field forward is to combine manually labeled datasets across centers to create a harmonized dataset that can be used for training appropriate deep learning algorithms. Moreover, an increased understanding of the neuropathology underlying these MRI-manifestations of SVD will greatly enhance our understanding of these clinically relevant lesions and metrics.
See for a discussion on post-mortem MRI the Supplemental Material.
Neuropathology of small vessel disease MRI-manifestations
Conventional MRI-markers
White matter hyperintensities: were the first SVD manifestation to be targeted in ex vivo MRI-histopathology studies. These early papers found that WMH have rather heterogenous neuropathological substrates(48). Areas with WMH on ex vivo MRI corresponded to white matter rarefaction on histopathology, including variable loss of myelin, axons, and oligodendrocytes (Figure 1). Moreover, these areas revealed reactive astrocytes, activated macrophages, and enlarged perivascular spaces, consistent with tissue injury. The etiology is often presumed ischemic but neuropathologic changes are not specific, and these changes may be related to other processes (e.g. Wallerian degeneration, trauma, or metabolic disease). Fibrohyalinosis and arteriolosclerosis were identified as the dominant small vessel changes, and were both observed in areas with incomplete as well as complete infarction(48). Later studies incorporated WMH location and pattern in their sampling strategy, as accumulating evidence from in vivo MRI studies suggested disparate clinical associations. Periventricular WMH, which is considered the most common form and perhaps the earliest manifestation of WMH during aging, corresponded to myelin loss around the ventricles, discontinuity and thinning of the ependyma, enlarged perivascular spaces, and tortuous vessels in the absence of arteriolosclerotic vessel changes(48). It has been hypothesized that this could be the result of aging-related cerebrospinal fluid leakage from the ventricle into the white matter. Conversely, punctate, early confluent, and confluent WMH in the deep white matter were found to reflect increasing severity of tissue changes, ranging from myelin loss and enlarged perivascular spaces, axonal loss and reactive astrocytes, to focally complete infarcts(48). The notion that severe WMH are primarily ischemic in origin has also been suggested by observations derived from serial in vivo MRI, where multiple acute microinfarcts appeared to precede WMH formation(64), although a more recent study could not confirm this hypothesis(65). Whether WMH are solely the result of ischemia or whether other mechanisms such as blood-brain barrier leakage or neuroinflammation may play a role in the pathogenesis of WMH(17,48,66,67) needs further clarification, and likely strongly depends on patient selection. Of particular interest in this regard is the contribution of cortical neurodegenerative pathology to WMH formation(68). A recent ex vivo MRI-histopathology study in a large (n=603) population-based sample revealed that WMH burden in the whole dataset was associated with both vascular (i.e. arteriolosclerosis, gross infarcts, and microinfarcts) and AD pathologies (i.e. increased amyloid β plaques)(46). Notably, in the subset of cases with mild or no cognitive impairment (n=332), WMH burden was solely related to gross infarcts and arteriolosclerosis, but not to amyloid β plaques(46). Interestingly, another ex vivo MRI-histopathology study investigating the nature of WMH in a small sample of AD cases (n=23) revealed that cortical hyperphosphorylated tau burden predicted the severity of WMH independent of both cortical amyloid β burden and SVD severity in the white matter(69). This intriguing association between cortical neurodegenerative pathology and WMH suggests a potential role for Wallerian degeneration in the pathogenesis of white matter lesions(70). Whether this mechanism may also contribute to the formation of WMH in CAA patients, where neurodegenerative pathologies and small vessel abnormalities frequently co-exist, remains to be determined(11). Future studies are needed to unravel the independent contributions of CAA, arteriolosclerosis, amyloid β plaques, and tau tangles to the formation of WMH. Other knowledge gaps include the histopathology underlying different topographical and morphological patterns of WMH, such as multi-spot and posterior dominant WMH, which have been described in the context of CAA(16).
Figure 1.
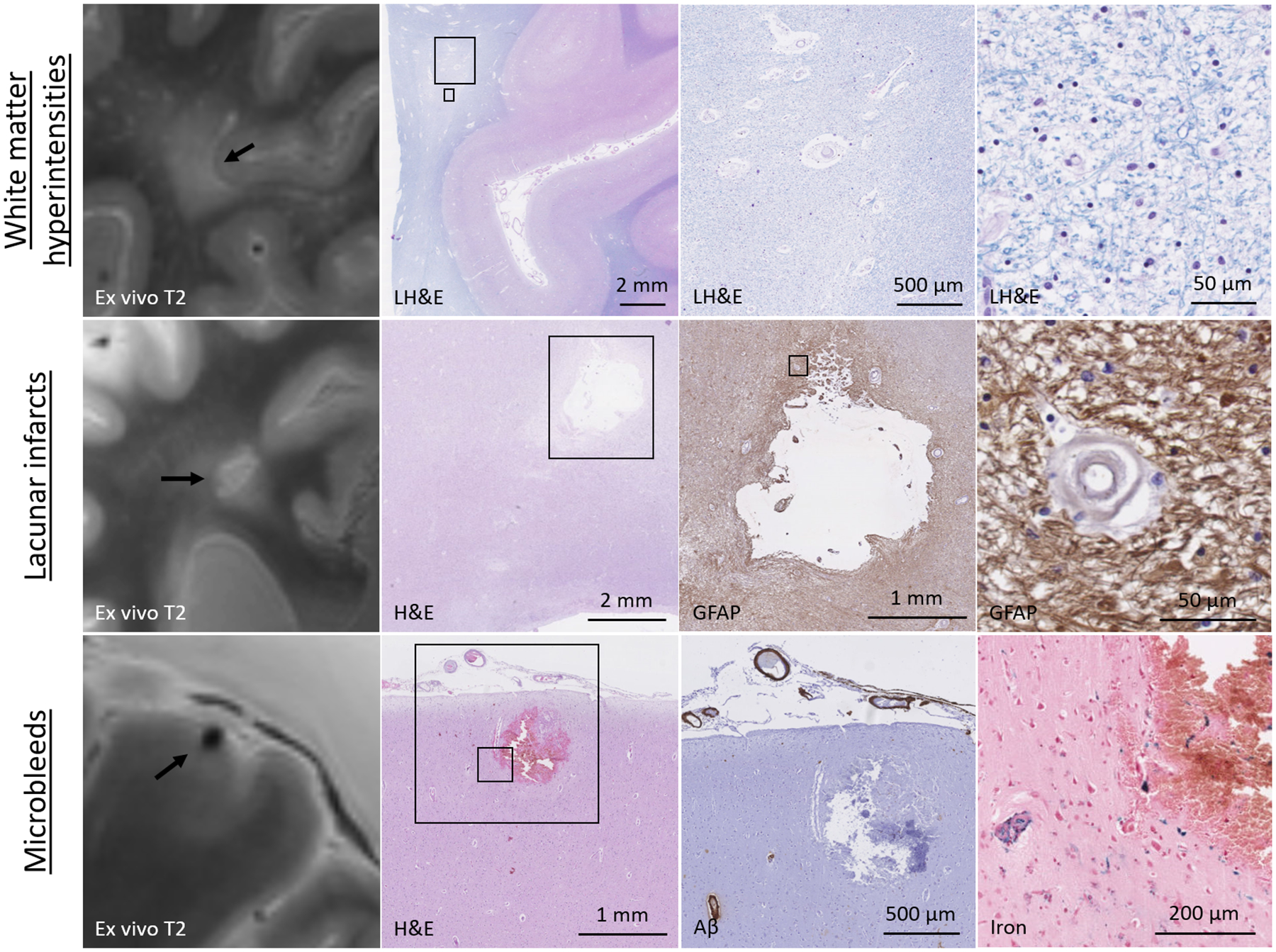
Neuropathology of white matter hyperintensities, lacunar infarcts, and microbleeds.
Tow row: a representative example of a white matter area with WMH on ex vivo MRI, which corresponded to white matter tissue rarefaction, enlarged perivascular spaces, and demyelination on histopathology. Middle row: a representative example of a lacunar infarct in the white matter on ex vivo MRI, which corresponded to a fluid-filled cavity with a rim of GFAP-positive astrocytes on histopathology. Note the concentric splitting of the wall of a nearby blood vessel. Bottom row: a representative example of a cortical microbleed on ex vivo MRI, which corresponded to a focal area of extravasated red blood cells and iron-positive blood-breakdown products, indicative of a subacute microbleed. Note the presence of vascular amyloid β in nearby blood vessels, but not the vessel responsible for the microhemorrhage.
Lacunar infarcts: only a handful of ex vivo MRI-histopathology studies have focused on lacunar infarcts, and confirmed their rather homogeneous neuropathological substrate as old cystic cavities in the white matter or basal ganglia with a rim of glial fibrillary acidic protein (GFAP)-positive reactive astrocytes (Figure 1)(48). They can be considered the larger equivalent of chronic cavitated microinfarcts, which are described in more detail below. Insights on the pathophysiology of lacunar infarcts are derived from the landmark papers by Dr. C.M. Fisher who utilized serial sectioning to find the blood vessel responsible for the infarct. These studies revealed occlusions in the upstream segment of the artery supplying the lesioned tissue(71). Besides prominent cell death and tissue loss at the core of lacunar infarcts, widespread tissue damage including loss of myelinated fibers have been observed in the peri-lesional areas (penumbra) as well(72). Therefore, lacunar infarcts can significantly affect brain structure and function, especially when occurring in strategic white matter tracts.
Microbleeds: ex vivo MRI-guided sampling has proven instrumental to study the histopathological nature of microbleeds, as they otherwise frequently escape detection on routine neuropathological examination at autopsy. Since the first description of microbleeds on clinical MRI, more than a dozen ex vivo MRI-histopathology studies have emerged, largely confirming the hemorrhagic nature of MRI-visible microbleeds in the context of small vessel abnormalities (Figure 1). In the cortex, MRI-visible microbleeds almost exclusively reflect small hemorrhages that originate from individual cortical blood vessels(7,47,73–75). As opposed to the high sensitivity and specificity for microbleeds in cortical areas, this appears to be lower in deep areas of the brain, such as the striatum(74). Although several studies did indeed confirm the hemorrhagic nature of microbleeds in deep areas, it remains unclear if the observed blood-breakdown products were the result of ruptured blood vessels or reflected hemorrhagic transformation after ischemic tissue injury(57,75–77). Besides being susceptible to ischemic tissue injury, these areas have also been associated with calcium accumulation, which is a common mimic of microbleeds on MRI(74). Future studies are needed to further assess the nature of deep microbleeds, using careful co-registration and serial sectioning approaches to retrieve the involved blood vessels. Based on their appearance on standard hematoxylin and eosin (H&E)-stained sections, microbleeds can be classified according to their presumed age. An acute microbleed can be identified by the presence of intact or lysed red blood cells in the parenchyma surrounding an affected blood vessel(75). In the subacute phase (>1 day after vessel rupture) extravasated red blood cells are broken down into biliverdin, hematoidin, and iron(57). This process triggers the recruitment of macrophages, which store these blood-breakdown products as hemosiderin. In the chronic stage, only a small focal cluster of hemosiderin-containing macrophages remain(75). Because these are positive for iron, even small clusters of hemosiderin-containing macrophages in the parenchyma can be seen as microbleeds on corresponding T2*-weighted MRI scans, an attribute known as ‘the blooming effect’(47,57). A combined in vivo MRI – ex vivo MRI – histopathology study in patients with sporadic CAA suggested that due to the blooming effect current in vivo research MRI protocols likely capture a large portion of the entire burden of microbleeds that are present in a single brain(47), although the smallest lesions still escape detection(78). Of note, increasing the spatial resolution of the ex vivo MRI scan to 75 μm3 isotropic in a small brain sample only resulted in increased detection of abnormal blood vessels (e.g. fibrinoid necrosis, microaneurysms), rather than frank hemorrhages(47). In terms of pathophysiology, microbleeds have been observed in close proximity to fibrohyalinotic and arteriolosclerotic vessels in deep areas(73,79) and to CAA in cortical areas of the brain(7,47,73,75). A recent study performed serial sectioning of tissue blocks in areas with multiple microbleeds on MRI, to study changes to the involved blood vessels at the site of rupture in patients with severe CAA(7). Bleeding was predominantly observed in cortical arterioles, at vessel segments with marked thickening of the walls, fragmentation, and reduced vascular amyloid β. Moreover, immunohistochemistry revealed drastic loss of vascular smooth muscle cells at the site of bleeding and cellular uptake of extravasated fibrin in surrounding immune cells(7). These recent observations suggest a cascade of events including blood-brain barrier leakage(80), extensive vascular remodeling, and amyloid β removal, prior to bleeding. Whether this process is mediated by neuroinflammation is a topic of ongoing investigations but has implications for the interpretation of microbleed formation in the context of anti-amyloid immunotherapy trials in patients with AD and concomitant CAA pathology(11,81). Whereas lacunar infarcts and microinfarcts can trigger extensive (peri-lesional) tissue loss, the impact of microbleeds on tissue integrity appears to be relatively modest(82), which may explain weak associations between microbleeds and cognition during life. Future studies are needed to address the histopathological nature and associated vessel abnormalities of deep microbleeds in patients with and without CAA, aided by serial sectioning to get at the site of rupture. It would be interesting to see if similar processes of vessel wall thickening, remodeling, and potentially immune-mediated vessel wall breakdown observed in the context of CAA also play a role in the formation of microhemorrhages in deep areas of the brain that are prone to hypertensive arteriopathy.
Microinfarcts: as opposed to microbleeds, microinfarcts are frequently observed on routine neuropathological examination and were first and foremost described in autopsy studies(83). Microinfarcts are considered the most widespread form of brain infarction and because of their high numbers have repeatedly been found to independently contribute to cognitive impairment and dementia(83). The clinical importance of these minute lesions hence fueled the search for their MRI signature, aided by high-resolution ex vivo MRI in cases with known microinfarcts(24,84,85). Like microbleeds, microinfarcts can be classified according to their presumed age based on their appearance on standard H&E-stained sections (which is also true for larger infarcts). An acute microinfarct can be identified by focal areas of tissue pallor and the appearance of eosinophilic or ‘red’ neurons in the cortex(26,86). The subacute phase is characterized by infiltration of macrophages and tissue loss. In the chronic phase only a few if any macrophages remain present, whereas the lesion boundaries are marked by an abundance of GFAP-positive reactive astrocytes (Figure 2)(26,86). Often, these old microinfarcts have a fluid-filled cavity at the core of the lesion, which likely aids their detection on MRI, but linear scarring within cortical regions is also a commonly observed phenotype neuropathologically. Even in the absence of a cavity, chronic microinfarcts can be seen on MRI as focal lesions in the cortex that are hypointense on T1 and hyperintense on T2(24,26,47,84,87). Their detection on MRI strongly depends on spatial resolution and field strength(47), and may be obscured by the presence of hemosiderin depositions when hemorrhagic transformation has occurred(86–88). With current research MRI scan protocols, only a small portion of the entire burden of microinfarcts can be captured during life. Moreover, the detection is limited to cortical areas of the brain, as microinfarcts in the white matter cannot be easily distinguished from WMH and enlarged perivascular spaces. A notable exception are acute microinfarcts, which are visible on diffusion-weighted scans for approximately two weeks and can be detected throughout the brain. Interestingly, a recent study found that some properties of acute microinfarcts are retained in formalin-fixed brains as they were still visible on ex vivo diffusion-weighted scans with high sensitivity and specificity(86). In terms of pathophysiology, cortical microinfarcts have been predominantly linked to CAA, whereas deep microinfarcts seem to be related to arteriolosclerosis (and atherosclerosis)(89,90). Only few studies have investigated small vessel abnormalities associated with individual microinfarcts and were predominantly performed in cases with CAA(91). Serial sectioning revealed severe CAA in penetrating cortical arterioles that supplied the lesioned tissue, as well as loss of vascular smooth muscle cells and fibrin deposition(7). The exact cascade of events through which these vessel changes result in ischemic injury in the surrounding tissue remains to be determined. Possible scenarios include extensive luminal narrowing through fibrin-amyloid β interactions, hypoperfusion in the context of reduced vascular reactivity, or trapped emboli originating from elsewhere(24). Several studies have suggested that microinfarcts can induce tissue changes far beyond the lesion boundaries that are visible on standard histology(72,92,93). By disrupting local and potentially even long-range projections(94), microinfarcts are capable of profoundly affecting brain structure and function, which could explain their strong link with cognition(24). Important knowledge gaps include the histopathological nature and associated small vessel abnormalities of microinfarcts in the white matter and deep areas of the brain. Future studies investigating deep microinfarcts may also aid the development of novel MRI criteria for the detection of microinfarcts in these areas. Another unknown concerns whether certain topographical vessel segments are more vulnerable than others, as has been suggested by some, but awaits replication in larger studies(95).
Figure 2.
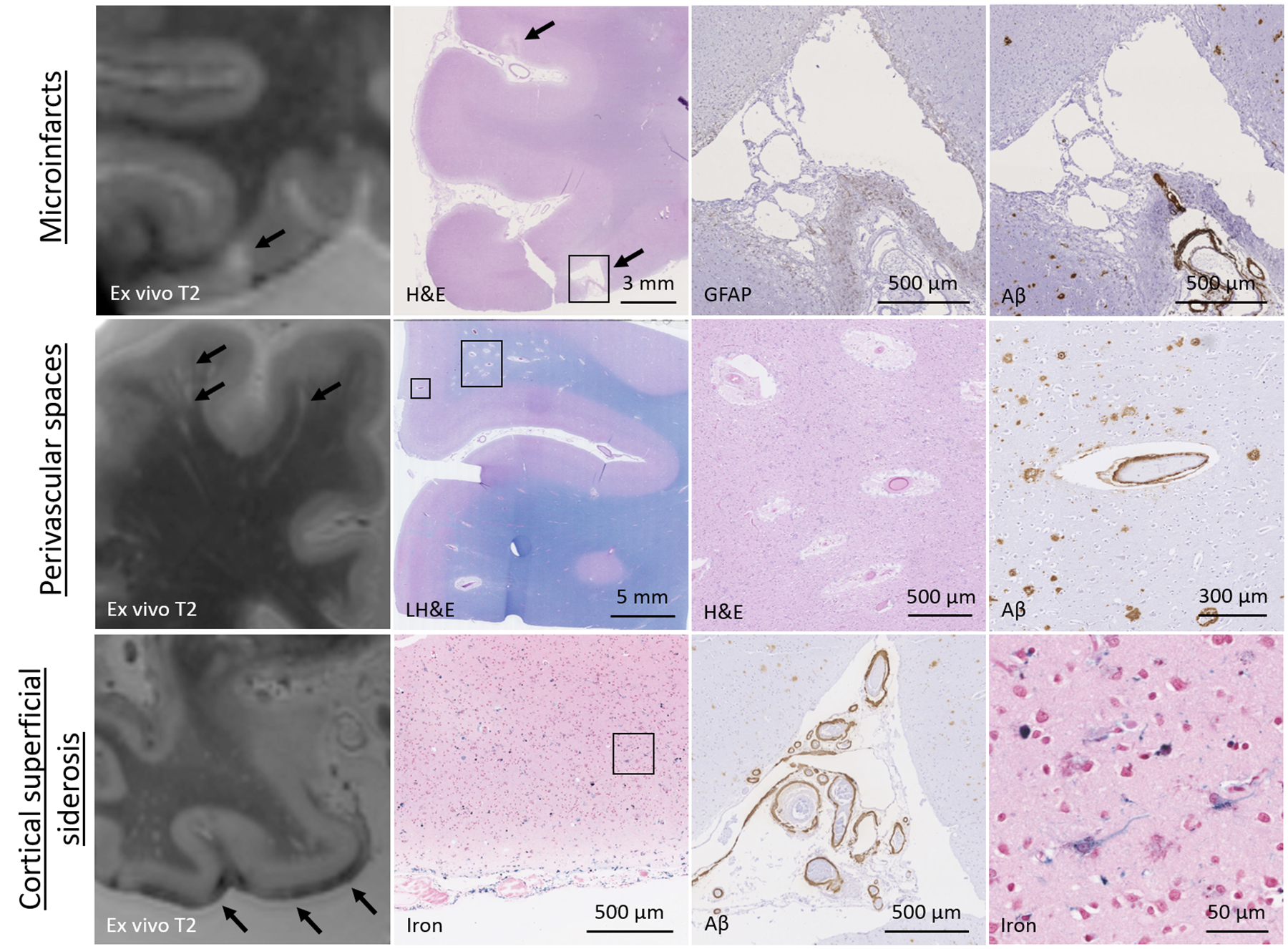
Neuropathology of microinfarcts, perivascular spaces, and cortical superficial siderosis.
Tow row: a representative example of a cortical microinfarct on ex vivo MRI, which corresponded to a focal area of cavitation with a rim of GFAP-positive astrocytes on histopathology, indicative of a chronic microinfarct. Note the presence of vascular amyloid β in nearby blood vessels and the additional microinfarct that was observed on neuropathological examination, but which escaped detection on ex vivo MRI. Middle row: a representative example of perivascular spaces on ex vivo MRI, which corresponded to enlarged fluid-filled spaces surrounding blood vessels on histopathology. Note the abnormal appearance of the blood vessels at the center of the enlarged perivascular spaces as well as amyloid β accumulation in the overlying cortex. Bottom row: a representative example of cSS on ex vivo MRI, which corresponded to iron-positive depositions in the subarachnoid space and superficial layers of the cortex, indicative of bleeding from the leptomeningeal vessels. Note the severe amyloid β accumulation and concentric vessel wall splitting in nearby leptomeningeal blood vessels as well as cellular uptake of iron in the cortex.
Perivascular spaces: a handful of ex vivo MRI studies that directly assessed the histopathological signature of MRI-visible perivascular spaces confirmed that they are abnormally enlarged spaces surrounding blood vessels (Figure 2)(26,56,96–98). One study in cases with neuropathological evidence of CAA found that the degree of juxtacortical perivascular space dilatation on ex vivo MRI was significantly correlated to CAA severity in the overlying cortex(97). This was in line with previous observations and suggests that dilation of perivascular spaces may be the result of a failure of drainage from the interstitial fluid caused by amyloid β deposition in the walls of cortical and leptomeningeal arterioles(99). Previous work has suggested that amyloid β drains along the basement membranes surrounding vascular smooth muscle cells towards the surface of the brain(100). When these compartments are altered in the context of aging, SVD, or Apolipoprotein E4, impaired perivascular drainage may result in accumulation of proteins (such as amyloid β in the form of CAA) surrounding smooth muscle cells. This lack of drainage and subsequent accumulation of protein may result in enlargement of the spaces between vascular basement membranes and the parenchyma in downstream portions of the vessels(100). Whether similar pathophysiological mechanisms play a role in enlarged perivascular spaces in the deep areas of the brain remains largely unknown. There are several other important knowledge gaps related to the neuropathology of enlarged perivascular spaces. It has been suggested that MRI-visible perivascular spaces primarily occur around arterioles(27), which warrants confirmation with high-resolution imaging techniques, coupled to detailed histopathological characterization of the connected vascular network. This has implications for the interpretation of the anatomy and potentially also directionality of clearance routes. In the context of CAA, amyloid β depositions are primarily observed in the walls of cortical arterioles, which suggests that peri-arteriolar (and not peri-venular) spaces are the main drainage pathways of amyloid β. Whether soluble amyloid β is primarily drained towards the surface of the brain or potentially along spaces into the white matter is currently not known. Other knowledge gaps include the pathophysiology of perivascular space dilatation and their contribution to tissue injury beyond the visible spaces. Notably, enlarged perivascular spaces have been described as one of the histopathological substrates of MRI-visible WMH(48). The mechanisms through which enlarged perivascular spaces contribute to white matter tissue injury and whether this might be mediated by blood-brain barrier leakage, neuroinflammation, or ischemia(56,101) remains to be determined.
Cortical superficial siderosis: since cSS has only recently emerged as a marker of SVD, only few studies have investigated the neuropathological correlates to date. On histopathology, MRI-visible cSS corresponds to hemosiderin-positive deposits in the subarachnoid space and the superficial layers of the cortex (Figure 2)(58,102,103). These observations are indicative of bleeding from the leptomeningeal vessels located over the convexities of the cortex and are consistent with neuropathological observations in the context of superficial siderosis of the central nervous system(104). Similar to microbleeds, the associated iron positivity of the hemosiderin-containing macrophages gives rise to enhanced visibility on blood-sensitive T2*-weighted MRI(58). cSS is very strongly (although not exclusively) associated with CAA, which confirms a growing body of literature using in vivo MRI(58,103). A recent case series that combined in vivo MRI with ex vivo MRI and histopathology found that cSS is strongly associated with advanced leptomeningeal CAA and especially concentric vessel wall splitting(58). Because cSS often extends over large areas of the pial surface, it is challenging to retrieve the individual vessel that has bled, even with serial sectioning. As such, it remains currently unclear whether similar mechanisms of vascular remodeling and Aβ removal that appear to play a role in the formation of microbleeds also occur at the level of the leptomeningeal vessels in the context of cSS. The severity of cSS was related to an increase in GFAP-positive reactive astrocytes in the cortex(58,102,105), as well as ischemic tissue changes(58,103). It has also been suggested that cSS may induce neurodegenerative changes in the form of neurofibrillary tangles and tau-positive deposits in astrocytes(105). These intriguing secondary tissue changes may be part of the pathophysiological mechanisms through which cSS increases the risk for future symptomatic bleeding in cases with CAA and therefore provide interesting targets to explore for future interventions. Other potential mechanisms that may play a role include neuroinflammation and blood-brain barrier leakage, which are topics of ongoing investigations.
See for a discussion on novel MRI-markers the Supplemental Material.
Conclusions
A growing body of literature utilizing ex vivo MRI in post-mortem human brain tissue has contributed to our increasing understanding of the neuropathological associations of MRI-visible manifestations of sporadic SVD during life. Future studies are needed to further unravel the sequence of events leading up to lesion formation, and to disentangle pathophysiological pathways in individuals with co-existing CAA, arteriolosclerosis, and AD pathologies.
Supplementary Material
Sources of funding
This work was supported by the NIH (R00AG059893 to SJvV, UH3NS100599 and P30AG010161 to KA and JAS, R01AG064233 and R01AG052200 to KA, and R01AG067482 to JAS), the American Heart Association (814728 to SJvV), and the Netherlands Organization for Scientific Research (Veni 016.196.021 to SJvV).
Non-standard Abbreviations and Acronyms
- AD
Alzheimer’s disease
- CAA
cerebral amyloid angiopathy
- DTI
diffusion-tensor imaging
- FLAIR
fluid-attenuated inversion recovery
- GFAP
glial fibrillary acidic protein
- H&E
hematoxylin and eosin
- ICH
intracerebral hemorrhage
- LH&E
luxol fast blue hematoxylin and eosin
- MRI
magnetic resonance imaging
- SVD
small vessel disease
- WMH
white matter hyperintensities
Footnotes
Conflicts of Interest / Disclosures
SJvV has served as a consultant on a CAA advisory board for Biogen. JAS serves as a consultant for Avid Radiopharmaceuticals, on the advisory panel of Alnylam Pharmaceuticals, and as an expert for the National Hockey League.
Supplemental Materials
References
- 1.Snyder HM, Corriveau RA, Craft S, Faber JE, Greenberg SM, Knopman D, Lamb BT, Montine TJ, Nedergaard M, Schaffer CB, et al. Vascular contributions to cognitive impairment and dementia including Alzheimer’s disease. Alzheimers Dement. 2015;11:710–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010;9:689–701. [DOI] [PubMed] [Google Scholar]
- 3.Wardlaw JM, Smith EE, Biessels GJ, Cordonnier C, Fazekas F, Frayne R, Lindley RI, O’Brien JT, Barkhof F, Benavente OR, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 2013;12:822–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blevins BL, Vinters HV, Love S, Wilcock DM, Grinberg LT, Schneider JA, Kalaria RN, Katsumata Y, Gold BT, Wang DJJ, et al. Brain arteriolosclerosis. Acta Neuropathol. 2021;141:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buchman AS, Leurgans SE, Nag S, VanderHorst VGJM, Kapasi A, Schneider JA, Bennett DA. Spinal Arteriolosclerosis Is Common in Older Adults and Associated With Parkinsonism. Stroke. 2017;48:2792–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kapasi A, Leurgans SE, Arvanitakis Z, Barnes LL, Bennett DA, Schneider JA. Aβ (Amyloid Beta) and Tau Tangle Pathology Modifies the Association Between Small Vessel Disease and Cortical Microinfarcts. Stroke. 2021;52:1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Veluw SJ, Scherlek AA, Freeze WM, Ter Telgte A, Van der Kouwe AJ, Bacskai BJ, Frosch MP, Greenberg SM. Different microvascular alterations underlie microbleeds and microinfarcts. Ann Neurol. 2019;86:279–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Love S, Chalmers K, Ince P, Esiri M, Attems J, Jellinger K, Yamada M, McCarron M, Minett T, Matthews F, et al. Development, appraisal, validation and implementation of a consensus protocol for the assessment of cerebral amyloid angiopathy in post-mortem brain tissue. Am J Neurodegener Dis. 2014;3:19–32. [PMC free article] [PubMed] [Google Scholar]
- 9.Boyle PA, Yu L, Nag S, Leurgans S, Wilson RS, Bennett DA, Schneider JA. Cerebral amyloid angiopathy and cognitive outcomes in community-based older persons. Neurology. 2015;85:1930–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinez-Ramirez S, Romero J-R, Shoamanesh A, McKee AC, Van Etten E, Pontes-Neto O, Macklin EA, Ayres A, Auriel E, Himali JJ, et al. Diagnostic value of lobar microbleeds in individuals without intracerebral hemorrhage. Alzheimers Dement. 2015;11:1480–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greenberg SM, Bacskai BJ, Hernandez-Guillamon M, Pruzin J, Sperling R, van Veluw SJ. Cerebral amyloid angiopathy and Alzheimer disease — one peptide, two pathways. Nat Rev Neurol. 2020;16:30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vonsattel JP, Myers RH, Hedley-Whyte ET, Ropper AH, Bird ED, Richardson EP. Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann Neurol. 1991;30:637–649. [DOI] [PubMed] [Google Scholar]
- 13.Freeze WM, Van der Thiel M, De Bresser J, Klijn CJM, Van Etten ES, Jansen JFA, Van der Weerd L, Jacobs HIL, Backes WH, Van Veluw SJ. CSF enhancement on post-contrast fluid-attenuated inversion recovery images; a systematic review. NeuroImage Clin. 2020;28:102456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Charidimou A, Boulouis G, Gurol ME, Ayata C, Bacskai BJ, Frosch MP, Viswanathan A, Greenberg SM. Emerging concepts in sporadic cerebral amyloid angiopathy. Brain. 2017;140:1829–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Debette S, Schilling S, Duperron M-G, Larsson SC, Markus HS. Clinical Significance of Magnetic Resonance Imaging Markers of Vascular Brain Injury: A Systematic Review and Meta-analysis. JAMA Neurol. 2019;76:81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Charidimou A, Boulouis G, Haley K, Auriel E, Van Etten ES, Fotiadis P, Reijmer Y, Ayres A, Vashkevich A, Dipucchio ZY, et al. White matter hyperintensity patterns in cerebral amyloid angiopathy and hypertensive arteriopathy. Neurology. 2016;86:505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alber J, Alladi S, Bae HJ, Barton DA, Beckett LA, Bell JM, Berman SE, Biessels GJ, Black SE, Bos I, et al. White matter hyperintensities in vascular contributions to cognitive impairment and dementia (VCID): Knowledge gaps and opportunities. Alzheimers Dement. 2019;5:107–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pasi M, Boulouis G, Fotiadis P, Auriel E, Charidimou A, Haley K, Ayres A, Schwab KM, Goldstein JN, Rosand J, et al. Distribution of lacunes in cerebral amyloid angiopathy and hypertensive small vessel disease. Neurology. 2017;88:2162–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ling Y, Chabriat H. Incident cerebral lacunes: A review. J Cereb Blood Flow Metab. 2020;40:909–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenberg SM, Vernooij MW, Cordonnier C, Viswanathan A, Al-Shahi Salman R, Warach S, Launer LJ, Van Buchem MA, Breteler MM, Microbleed Study Group. Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol. 2009;8:165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen W, Zhu W, Kovanlikaya I, Kovanlikaya A, Liu T, Wang S, Salustri C, Wang Y. Intracranial calcifications and hemorrhages: characterization with quantitative susceptibility mapping. Radiology. 2014;270:496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Puy L, Pasi M, Rodrigues M, Van Veluw SJ, Tsivgoulis G, Shoamanesh A, Cordonnier C. Cerebral microbleeds: from depiction to interpretation. 2021. [Online ahead of print]. [DOI] [PubMed]
- 23.Greenberg SM, Charidimou A. Diagnosis of Cerebral Amyloid Angiopathy: Evolution of the Boston Criteria. Stroke. 2018;49:491–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Veluw SJ, Shih AY, Smith EE, Chen C, Schneider JA, Wardlaw JM, Greenberg SM, Biessels GJ. Detection, risk factors, and functional consequences of cerebral microinfarcts. Lancet Neurol. 2017;16:730–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ter Telgte A, Wiegertjes K, Gesierich B, Baskaran BS, Marques JP, Kuijf HJ, Norris DG, Tuladhar AM, Duering M, De Leeuw F-El. Temporal Dynamics of Cortical Microinfarcts in Cerebral Small Vessel Disease. JAMA Neurol. 2020;77:643–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Veluw SJ, Zwanenburg JJM, Rozemuller AJM, Luijten PR, Spliet WGM, Biessels GJ. The spectrum of MR detectable cortical microinfarcts: A classification study with 7-tesla postmortem MRI and histopathology. J Cereb Blood Flow Metab. 2015;35:676–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wardlaw JM, Benveniste H, Nedergaard M, Zlokovic BV, Mestre H, Lee H, Doubal FN, Brown R, Ramirez J, MacIntosh BJ, et al. Perivascular spaces in the brain: anatomy, physiology and pathology. Nat Rev Neurol. 2020;16:137–153. [DOI] [PubMed] [Google Scholar]
- 28.Charidimou A, Boulouis G, Pasi M, Auriel E, Van Etten ES, Haley K, Ayres A, Schwab KM, Martinez-Ramirez S, Goldstein JN, et al. MRI-visible perivascular spaces in cerebral amyloid angiopathy and hypertensive arteriopathy. Neurology. 2017;88:1157–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Charidimou A, Linn J, Vernooij MW, Opherk C, Akoudad S, Baron JC, Greenberg SM, Jäger HR, Werring DJ. Cortical superficial siderosis: Detection and clinical significance in cerebral amyloid angiopathy and related conditions. Brain. 2015;138:2126–2139. [DOI] [PubMed] [Google Scholar]
- 30.Charidimou A, Boulouis G, Greenberg SM, Viswanathan A. Cortical superficial siderosis and bleeding risk in cerebral amyloid angiopathy: A meta-analysis. Neurology. 2019;93:E2192–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baykara E, Gesierich B, Adam R, Tuladhar AM, Biesbroek JM, Koek HL, Ropele S, Jouvent E, ADNI, Chabriat H, et al. A Novel Imaging Marker for Small Vessel Disease Based on Skeletonization of White Matter Tracts and Diffusion Histograms. Ann Neurol. 2016;80:581–592. [DOI] [PubMed] [Google Scholar]
- 32.McCreary CR, Beaudin AE, Subotic A, Zwiers AM, Alvarez A, Charlton A, Goodyear BG, Frayne R, Smith EE. Cross-sectional and longitudinal differences in peak skeletonized white matter mean diffusivity in cerebral amyloid angiopathy. NeuroImage Clin. 2020;27:102280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Low A, Mak E, Stefaniak JD, Malpetti M, Nicastro N, Savulich G, Chouliaras L, Markus HS, Rowe JB, O’Brien JT. Peak Width of Skeletonized Mean Diffusivity as a Marker of Diffuse Cerebrovascular Damage. Front Neurosci. 2020;14:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pasternak O, Sochen N, Gur Y, Intrator N, Assaf Y. Free water elimination and mapping from diffusion MRI. Magn Reson Med. 2009;62:717–730. [DOI] [PubMed] [Google Scholar]
- 35.Finsterwalder S, Vlegels N, Gesierich B, Araque Caballero MÁ, Weaver NA, Franzmeier N, Georgakis MK, Konieczny MJ, Koek HL, DIAN, et al. Small vessel disease more than Alzheimer’s disease determines diffusion MRI alterations in memory clinic patients. Alzheimers Dement. 2020;16:1504–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maillard P, Mitchell GF, Himali JJ, Beiser A, Fletcher E, Tsao CW, Pase MP, Satizabal CL, Vasan RS, Seshadri S, et al. Aortic Stiffness, Increased White Matter Free Water, and Altered Microstructural Integrity: A Continuum of Injury. Stroke. 2017;48:1567–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duering M, Finsterwalder S, Baykara E, Tuladhar AM, Gesierich B, Konieczny MJ, Malik R, Franzmeier N, Ewers M, Jouvent E, et al. Free water determines diffusion alterations and clinical status in cerebral small vessel disease. Alzheimers Dement. 2018;14:764–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ter Telgte A, Van Leijsen EMC, Wiegertjes K, Klijn CJM, Tuladhar AM, De Leeuw F-E. From a Focal To a Global Perspective. Nat Rev Neurol. 2018;14:387–98. [DOI] [PubMed] [Google Scholar]
- 39.Reijmer YD, Fotiadis P, Martinez-Ramirez S, Salat DH, Schultz A, Shoamanesh A, Ayres AA, Vashkevich A, Rosas D, Schwab K, et al. Structural network alterations and neurological dysfunction in cerebral amyloid angiopathy. Brain. 2015;138:179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farrall AJ, Wardlaw JM. Blood-brain barrier: ageing and microvascular disease--systematic review and meta-analysis. Neurobiol Aging. 2009;30:337–352. [DOI] [PubMed] [Google Scholar]
- 41.Thrippleton MJ, Backes WH, Sourbron S, Ingrisch M, Van Osch MJP, Dichgans M, Fazekas F, Ropele S, Frayne R, Van Oostenbrugge RJ, et al. Quantifying blood-brain barrier leakage in small vessel disease: Review and consensus recommendations. Alzheimers Dement. 2019;15:840–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tiwari YV, Lu J, Shen Q, Cerqueira B, Duong TQ. Magnetic resonance imaging of blood-brain barrier permeability in ischemic stroke using diffusion-weighted arterial spin labeling in rats. J Cereb Blood Flow Metab. 2017;37:2706–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shao X, Ma SJ, Casey M, D’Orazio L, Ringman JM, Wang DJJ. Mapping water exchange across the blood-brain barrier using 3D diffusion-prepared arterial spin labeled perfusion MRI. Magn Reson Med. 2019;81:3065–3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vanderbecq Q, Xu E, Ströer S, Couvy-Duchesne B, Diaz Melo M, Dormont D, Colliot O, ADNI. Comparison and validation of seven white matter hyperintensities segmentation software in elderly patients. NeuroImage Clin. 2020;27:102357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuijf HJ, de Bresser J, Geerlings MI, Conijn MMA, Viergever MA, Biessels GJ, Vincken KL. Efficient detection of cerebral microbleeds on 7.0 T MR images using the radial symmetry transform. Neuroimage. 2012;59:2266–2273. [DOI] [PubMed] [Google Scholar]
- 46.Arfanakis K, Evia AM, Leurgans SE, Cardoso LFC, Kulkarni A, Alqam N, Lopes LF, Vieira D, Bennett DA, Schneider JA. Neuropathologic Correlates of White Matter Hyperintensities in a Community-Based Cohort of Older Adults. J Alzheimers Dis. 2020;73:333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van Veluw SJ, Charidimou A, Van der Kouwe AJ, Lauer A, Reijmer YD, Costantino I, Gurol ME, Biessels GJ, Frosch MP, Viswanathan A, et al. Microbleed and microinfarct detection in amyloid angiopathy: A high-resolution MRI-histopathology study. Brain. 2016;139:3151–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gouw AA, Seewann A, van der Flier WM, Barkhof F, Rozemuller AM, Scheltens P, Geurts JJG. Heterogeneity of small vessel disease: a systematic review of MRI and histopathology correlations. J Neurol Neurosurg Psychiatry. 2011;82:126–135. [DOI] [PubMed] [Google Scholar]
- 49.Raman MR, Shu Y, Lesnick TG, Jack CR, Kantarci K. Regional T(1) relaxation time constants in Ex vivo human brain: Longitudinal effects of formalin exposure. Magn Reson Med. 2017;77:774–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dawe RJ, Bennett DA, Schneider JA, Vasireddi SK, Arfanakis K. Postmortem MRI of human brain hemispheres: T2 relaxation times during formaldehyde fixation. Magn Reson Med. 2009;61:810–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.D’Arceuil H, De Crespigny A. The effects of brain tissue decomposition on diffusion tensor imaging and tractography. Neuroimage. 2007;36:64–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Birkl C, Langkammer C, Haybaeck J, Ernst C, Stollberger R, Fazekas F, Ropele S. Temperature-induced changes of magnetic resonance relaxation times in the human brain: a postmortem study. Magn Reson Med. 2014;71:1575–1580. [DOI] [PubMed] [Google Scholar]
- 53.Grinberg LT, Amaro EJ, Teipel S, Dos Santos DD, Pasqualucci CA, Leite REP, Camargo CR, Gonçalves JA, Sanches AG, Santana M, et al. Assessment of factors that confound MRI and neuropathological correlation of human postmortem brain tissue. Cell Tissue Bank. 2008;9:195–203. [DOI] [PubMed] [Google Scholar]
- 54.Roseborough AD, Langdon KD, Hammond R, Cipriano LE, Pasternak SH, Whitehead SN, Khan AR. Post-mortem 7 Tesla MRI detection of white matter hyperintensities: A multidisciplinary voxel-wise comparison of imaging and histological correlates. NeuroImage Clin. 2020;27:102340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kotrotsou A, Bennett DA, Schneider JA, Dawe RJ, Golak T, Leurgans SE, Yu L, Arfanakis K. Ex vivo MR volumetry of human brain hemispheres. Magn Reson Med. 2014;71:364–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Javierre-Petit C, Schneider JA, Kapasi A, Makkinejad N, Tamhane AA, Leurgans SE, Mehta RI, Barnes LL, Bennett DA, Arfanakis K. Neuropathologic and Cognitive Correlates of Enlarged Perivascular Spaces in a Community-Based Cohort of Older Adults. Stroke. 2020;51:2825–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schrag M, McAuley G, Pomakian J, Jiffry A, Tung S, Mueller C, Vinters HV, Haacke EM, Holshouser B, Kido D, et al. Correlation of hypointensities in susceptibility-weighted images to tissue histology in dementia patients with cerebral amyloid angiopathy: a postmortem MRI study. Acta Neuropathol. 2010;119:291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Charidimou A, Perosa V, Frosch MP, Scherlek AA, Greenberg SM, Van Veluw SJ. Neuropathological correlates of cortical superficial siderosis in cerebral amyloid angiopathy. Brain. 2020;143:3343–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Evia AM, Kotrotsou A, Tamhane AA, Dawe RJ, Kapasi A, Leurgans SE, Schneider JA, Bennett DA, Arfanakis K. Ex-vivo quantitative susceptibility mapping of human brain hemispheres. PLoS One. 2017;12:e0188395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Takahashi E, Song JW, Folkerth RD, Grant PE, Schmahmann JD. Detection of postmortem human cerebellar cortex and white matter pathways using high angular resolution diffusion tractography: a feasibility study. Neuroimage. 2013;68:105–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Van Veluw SJ, Reijmer YD, Van Der Kouwe AJ, Charidimou A, Riley GA, Leemans A, Bacskai BJ, Frosch MP, Viswanathan A, Greenberg SM. Histopathology of diffusion imaging abnormalities in cerebral amyloid angiopathy. Neurology. 2019;92:E933–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McNab JA, Jbabdi S, Deoni SCL, Douaud G, Behrens TEJ, Miller KL. High resolution diffusion-weighted imaging in fixed human brain using diffusion-weighted steady state free precession. Neuroimage. 2009;46:775–785. [DOI] [PubMed] [Google Scholar]
- 63.Dawe RJ, Bennett DA, Schneider JA, Leurgans SE, Kotrotsou A, Boyle PA, Arfanakis K. Ex vivo T2 relaxation: Associations with age-related neuropathology and cognition. Neurobiol Aging. 2014;35:1549–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Conklin J, Silver FL, Mikulis DJ, Mell DM. Are acute infarcts the cause of leukoaraiosis? Brain mapping for 16 consecutive weeks. Ann Neurol. 2014;76:899–904. [DOI] [PubMed] [Google Scholar]
- 65.Ter Telgte A, Wiegertjes K, Gesierich B, Marques JP, Huebner M, De Klerk JJ, Schreuder FHBM, Caballero MAA, Kuijf HJ, Norris DG, et al. Contribution of acute infarcts to cerebral small vessel disease progression. Ann Neurol. 2019;86:582–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wardlaw JM, Smith C, Dichgans M. Small vessel disease: mechanisms and clinical implications. Lancet Neurol. 2019;18:684–696. [DOI] [PubMed] [Google Scholar]
- 67.Hainsworth AH, Minett T, Andoh J, Forster G, Bhide I, Barrick TR, Elderfield K, Jeevahan J, Markus HS, Bridges LR. Neuropathology of white matter lesions, blood-brain barrier dysfunction, and dementia. Stroke. 2017;48:2799–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tabaton M, Caponnetto C, Mancardi G, Loeb C. Amyloid beta protein deposition in brains from elderly subjects with leukoaraiosis. J Neurol Sci. 1991;106:123–127. [DOI] [PubMed] [Google Scholar]
- 69.McAleese KE, Firbank M, Dey M, Colloby SJ, Walker L, Johnson M, Beverley JR, Taylor JP, Thomas AJ, O’Brien JT, et al. Cortical tau load is associated with white matter hyperintensities. Acta Neuropathol Commun. 2015;3:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McAleese KE, Walker L, Graham S, Moya ELJ, Johnson M, Erskine D, Colloby SJ, Dey M, Martin-Ruiz C, Taylor J-P, et al. Parietal white matter lesions in Alzheimer’s disease are associated with cortical neurodegenerative pathology, but not with small vessel disease. Acta Neuropathol. 2017;134:459–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fisher CM. The arterial lesions underlying lacunes. Acta Neuropathol. 1969;12:1–15. [DOI] [PubMed] [Google Scholar]
- 72.Hinman JD, Lee MD, Tung S, Vinters HV, Carmichael ST. Molecular disorganization of axons adjacent to human lacunar infarcts. Brain. 2015;138:736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shoamanesh A, Kwok CS, Benavente O. Cerebral microbleeds: Histopathological correlation of neuroimaging. Cerebrovasc Dis. 2011;32:528–534. [DOI] [PubMed] [Google Scholar]
- 74.De Reuck J, Auger F, Cordonnier C, Deramecourt V, Durieux N, Pasquier F, Bordet R, Maurage CA, Leys D. Comparison of 7.0-T T₂*-magnetic resonance imaging of cerebral bleeds in post-mortem brain sections of Alzheimer patients with their neuropathological correlates. Cerebrovasc Dis. 2011;31:511–517. [DOI] [PubMed] [Google Scholar]
- 75.Van Veluw SJ, Biessels GJ, Klijn CJM, Rozemuller AJM. Heterogeneous histopathology of cortical microbleeds in cerebral amyloid angiopathy. Neurology. 2016;86:867–871. [DOI] [PubMed] [Google Scholar]
- 76.Fazekas F, Kleinert R, Roob G, Kleinert G, Kapeller P, Schmidt R, Hartung HP. Histopathologic analysis of foci of signal loss on gradient-echo T2*-weighted MR images in patients with spontaneous intracerebral hemorrhage: evidence of microangiopathy-related microbleeds. AJNR Am J Neuroradiol. 1999;20:637–642. [PMC free article] [PubMed] [Google Scholar]
- 77.Janaway BM, Simpson JE, Hoggard N, Highley JR, Forster G, Drew D, Gebril OH, Matthews FE, Brayne C, Wharton SB, et al. Brain haemosiderin in older people: pathological evidence for an ischaemic origin of magnetic resonance imaging (MRI) microbleeds. Neuropathol Appl Neurobiol. 2014;40:258–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Haller S, Scheffler M, Salomir R, Herrmann FR, Gold G, Montandon ML, Kövari E. MRI detection of cerebral microbleeds: size matters. Neuroradiology. 2019;61:1209–1213. [DOI] [PubMed] [Google Scholar]
- 79.Guidoux C, Hauw J-J, Klein IF, Labreuche J, Berr C, Duyckaerts C, Amarenco P. Amyloid Angiopathy in Brain Hemorrhage: A Postmortem Neuropathological-Magnetic Resonance Imaging Study. Cerebrovasc Dis. 2018;45:124–131. [DOI] [PubMed] [Google Scholar]
- 80.Freeze WM, Bacskai BJ, Frosch MP, Jacobs HIL, Backes WH, Greenberg SM, Van Veluw SJ. Blood-Brain Barrier Leakage and Microvascular Lesions in Cerebral Amyloid Angiopathy. Stroke. 2019;50:328–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sperling RA, Jack CR, Black SE, Frosch MP, Greenberg SM, Hyman BT, Scheltens P, Carrillo MC, Thies W, Bednar MM, et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: Recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement. 2011;7:367–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rosidi NL, Zhou J, Pattanaik S, Wang P, Jin W, Brophy M, Olbricht WL, Nishimura N, Schaffer CB. Cortical microhemorrhages cause local inflammation but do not trigger widespread dendrite degeneration. PLoS One. 2011;6:e26612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Smith EE, Schneider JA, Wardlaw JM, Greenberg SM. Cerebral microinfarcts: the invisible lesions. Lancet Neurol. 2012;11:272–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Van Veluw SJ, Zwanenburg JJM, Engelen-Lee J, Spliet WGM, Hendrikse J, Luijten PR, Biessels GJ. In vivo detection of cerebral cortical microinfarcts with high-resolution 7T MRI. J Cereb Blood Flow Metab. 2013;33:322–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jouvent E, Poupon C, Gray F, Paquet C, Mangin J-F, Le Bihan D, Chabriat H. Intracortical infarcts in small vessel disease: a combined 7-T postmortem MRI and neuropathological case study in cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Stroke. 2011;42:e27–30. [DOI] [PubMed] [Google Scholar]
- 86.Ter Telgte A, Scherlek AA, Reijmer YD, Van der Kouwe AJ, Van Harten T, Duering M, Bacskai BJ, De Leeuw F-E, Frosch MP, Greenberg SM, et al. Histopathology of diffusion-weighted imaging-positive lesions in cerebral amyloid angiopathy. Acta Neuropathol. 2020;139:799–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yilmazer-Hanke D, Mayer T, Müller HP, Neugebauer H, Abaei A, Scheuerle A, Weis J, Forsberg KME, Althaus K, Meier J, et al. Histological correlates of postmortem ultra-high-resolution single-section MRI in cortical cerebral microinfarcts. Acta Neuropathol Commun. 2020;8:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Van Veluw SJ, Zwanenburg JJ, Rozemuller AJ, Luijten PR, Spliet WG, Biessels GJ. The spectrum of MR detectable cortical microinfarcts: a classification study with 7-tesla postmortem MRI and histopathology. J Cereb Blood Flow Metab. 2015;35:676–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kövari E, Herrmann FR, Gold G, Hof PR, Charidimou A. Association of cortical microinfarcts and cerebral small vessel pathology in the ageing brain. Neuropathol Appl Neurobiol. 2017;43:505–513. [DOI] [PubMed] [Google Scholar]
- 90.Arvanitakis Z, Capuano AW, Leurgans SE, Buchman AS, Bennett DA, Schneider JA. The Relationship of Cerebral Vessel Pathology to Brain Microinfarcts. Brain Pathol. 2016;27:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Niwa A, Ii Y, Shindo A, Matsuo K, Ishikawa H, Taniguchi A, Takase S, Maeda M, Sakuma H, Akatsu H, et al. Comparative Analysis of Cortical Microinfarcts and Microbleeds using 3.0-Tesla Postmortem Magnetic Resonance Images and Histopathology. J Alzheimers Dis. 2017;59:951–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Coban H, Tung S, Yoo B, Vinters HV, Hinman JD. Molecular disorganization of axons adjacent to human cortical microinfarcts. Front Neurol. 2017;8:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Summers PM, Hartmann DA, Hui ES, Nie X, Deardorff RL, McKinnon ET, Helpern JA, Jensen JH, Shih AY. Functional deficits induced by cortical microinfarcts. J Cereb Blood Flow Metab. 2017;37:3599–3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lubart A, Benbenishty A, Har-Gil H, Laufer H, Gdalyahu A, Assaf Y, Blinder P. Single Cortical Microinfarcts Lead to Widespread Microglia/Macrophage Migration Along the White Matter. Cereb Cortex. 2021;31:248–266. [DOI] [PubMed] [Google Scholar]
- 95.De Reuck J, Deramecourt V, Auger F, Durieux N, Cordonnier C, Devos D, Defebvre L, Moreau C, Caparros-Lefebvre D, Bordet R, et al. Post-mortem 7.0-tesla magnetic resonance study of cortical microinfarcts in neurodegenerative diseases and vascular dementia with neuropathological correlates. J Neurol Sci. 2014;346:85–89. [DOI] [PubMed] [Google Scholar]
- 96.Song CJ, Kim JH, Kier EL, Bronen RA. MR imaging and histologic features of subinsular bright spots on T2-weighted MR images: Virchow-Robin spaces of the extreme capsule and insular cortex. Radiology. 2000;214:671–677. [DOI] [PubMed] [Google Scholar]
- 97.Van Veluw SJ, Biessels GJ, Bouvy WH, Spliet WG, Zwanenburg JJ, Luijten PR, Macklin EA, Rozemuller AJM, Gurol ME, Greenberg SM, et al. Cerebral amyloid angiopathy severity is linked to dilation of juxtacortical perivascular spaces. J Cereb Blood Flow Metab. 2016;36:576–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Van Veluw SJ, Wisse LEM, Kuijf HJ, Spliet WGM, Hendrikse J, Luijten PR, Geerlings MI, Biessels GJ. Hippocampal T2 hyperintensities on 7 Tesla MRI. NeuroImage Clin. 2013;3:196–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Roher AE, Kuo Y-M, Esh C, Knebel C, Weiss N, Kalback W, Luehrs DC, Childress JL, Beach TG, Weller RO, et al. Cortical and Leptomeningeal Cerebrovascular Amyloid and White Matter Pathology in Alzheimer’s Disease. 2003;9:112–122. [PMC free article] [PubMed] [Google Scholar]
- 100.Weller RO, Hawkes CA, Kalaria RN, Werring DJ, Carare RO. White matter changes in dementia: role of impaired drainage of interstitial fluid. Brain Pathol. 2015;25:63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Braffman BH, Zimmerman R a, Trojanowski JQ, Gonatas NK, Hickey WF, Schlaepfer WW. Brain MR: pathologic correlation with gross and histopathology. 2. Hyperintense white-matter foci in the elderly. AJR Am J Roentgenol. 1988;151:559–566. [DOI] [PubMed] [Google Scholar]
- 102.Torii Y, Iritani S, Fujishiro H, Sekiguchi H, Habuchi C, Umeda K, Matsunaga S, Mimuro M, Ozaki N, Yoshida M, et al. An autopsy case of cortical superficial siderosis with persistent abnormal behavior. Neuropathology. 2016;36:544–550. [DOI] [PubMed] [Google Scholar]
- 103.De Reuck J, Deramecourt V, Cordonnier C, Auger F, Durieux N, Pasquier F, Bordet R, Defebvre L, Caparros-Lefebvre D, Maurage CA, et al. Superficial Siderosis of the Central Nervous System : A Post-Mortem 7.0-Tesla Magnetic Resonance Imaging Study with Neuropathological Correlates. Cerebrovasc Dis. 2013;36:412–417. [DOI] [PubMed] [Google Scholar]
- 104.Koeppen AH, Michael SC, Li D, Chen Z, Cusack MJ, Gibson WM, Petrocine SV, Qian J. The pathology of superficial siderosis of the central nervous system. Acta Neuropathol. 2008;116:371–382. [DOI] [PubMed] [Google Scholar]
- 105.Takao M, Murayama S, Yoshida Y, Mihara B. Superficial siderosis associated with abundant τ and α-synuclein accumulation. BMJ Case Rep. 2011;1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kolasinski J, Stagg CJ, Chance SA, DeLuca GC, Esiri MM, Chang E-H, Palace JA, McNab JA, Jenkinson M, Miller KL, et al. A combined post-mortem magnetic resonance imaging and quantitative histological study of multiple sclerosis pathology. Brain. 2012;135:2938–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Absinta M, Vuolo L, Rao A, Nair G, Sati P, Cortese ICM, Ohayon J, Fenton K, Reyes-Mantilla M, Maric D, et al. Gadolinium-based MRI characterization of leptomeningeal inflammation in multiple sclerosis. Neurology. 2015;85:18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Van Nieuwenhuizen KM, Hendrikse J, Klijn CJM. New microbleed after blood-brain barrier leakage in intracerebral haemorrhage. BMJ Case Rep. 2017;1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zanon Zotin MC, Sveikata L, Viswanathan A, Yilmaz P. Cerebral Small Vessel Disease and Vascular Cognitive Impairment: from diagnosis to management. Curr Opin Neurol. 2021;34:246–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kozberg MG, Perosa V, Gurol ME, Van Veluw SJ. A practical approach to the management of cerebral amyloid angiopathy. Int J Stroke. 2020. [Online ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.