

Abstract
Oncogenic extrachromosomal DNA elements (ecDNAs) play an important role in tumor evolution, but our understanding of ecDNA biology is limited. We determined the distribution of single-cell ecDNA copy number across patient tissues and cell line models and observed how cell-to-cell ecDNA frequency greatly varies. The exceptional intratumoral heterogeneity of ecDNA suggested ecDNA-specific replication and propagation mechanisms. To evaluate the transfer of ecDNA genetic material from parental to offspring cells during mitosis, we established the CRISPR-based ecTag method. EcTag leverages ecDNA-specific breakpoint sequences to tag ecDNA with fluorescent markers in living cells. Applying ecTag during mitosis revealed disjointed ecDNA inheritance patterns, enabling rapid ecDNA accumulation in individual cells. Post-mitosis, ecDNAs clustered into ecDNA hubs, and ecDNA hubs colocalized with RNA polymerase II, promoting transcription of cargo oncogenes. Our observations provide direct evidence for uneven segregation of ecDNA and shed new light on mechanisms through which ecDNAs contribute to oncogenesis.
Introduction
Tumor evolution drives intratumor heterogeneity which is a source of therapy failure and resistance (1,2). Genomic instability and chromosomal structural variations including oncogene amplification play a critical role in driving tumor evolution (3,4). Focal amplifications in cancer may occur on extrachromosomal DNA (ecDNA) elements, are observed in the majority of glioblastomas and at high frequencies in many cancer types (5–8), and contribute to an accelerated tumor growth and poor patient survival. EcDNAs are 50kb-5Mb genomic elements containing genes and/or regulatory sequences (9,10). Acentromeric and atelomeric features of ecDNAs suggest uneven ecDNA segregation during mitosis leading to discordant ecDNA inheritance and promoting rapid ecDNA accumulation in a subpopulation of cancer cells (11). Clonal variability of ecDNAs in neuroblastoma cells has been observed and provided evidence towards this hypothesis (12). There is limited knowledge of ecDNA behavior during DNA replication and our understanding of the ecDNA mobility in proliferating cancer cells. Fluorescence in situ hybridization (FISH) and 4′,6-diamidino-2-phenylindole (DAPI) staining of fixed cells have been used to demonstrate that oncogenes can reside extrachromosomally in cancer (5,6,8,9) . These static readouts of the number of ecDNA copies in single cells are unable to record behavioral patterns. Genome engineering technologies based on the CRISPR-associated RNA-guided inactive endonuclease Cas9 have been leveraged to visualize DNA in living cells. In recent studies, this technique allowed real-time tracing of the dynamic reorganization of genomic DNA during mitosis (13), programmable 3D genome interactions (14) and chromosome translocation induced by genome editing (15). Although originally designed for labeling of repetitive sequence (13), recent advances in CRISPR-based live-cell imaging techniques have additionally enabled visualization of non-repetitive chromosome loci (16,17). These techniques collectively help advance our ability to visualize genome organization during both, physiological and pathological cell states, and in response to CRISPR-guided perturbations. EcDNA sequences are indistinguishable from their parental chromosomal DNA, barring ecDNA-specific breakpoint sequences which provide an opportunity for live-cell ecDNA imaging. Here, we report a CRISPR-based DNA tracking system (‘ecTag’) that leverages DNA breakpoint junctions to label ecDNA elements with multiple fluorescent molecules. We applied this technology to understand ecDNA spatiotemporal dynamics and the mechanisms by which ecDNA contributes to intratumoral heterogeneity.
Results
EcDNA shows increased intratumoral copy number variability
While ecDNA has been nominated as a key factor contributing to intratumoral heterogeneity resulting from suspected unequal segregation (5,7,18), there is a paucity of direct, experimental evidence supporting this assumption. The proposed model of ecDNA inheritance, in contrast to canonical inheritance of linearly amplified DNA on chromosomes, can account for a high degree of intratumoral multiplicity of ecDNA copy number (Fig. 1A). We hypothesized that the number of ecDNA copies across single cells would be highly variable, whereas the copy number of genes amplified linearly on chromosomes is expected to be identical at the single-cell level. To evaluate the distribution of the number of ecDNA copies per cell, we performed interphase fluorescent in-situ hybridization (FISH) on four glioblastoma (GBM) tumor tissue samples (SM006, SM012, SM017 and SM018) and a pair of primary and recurrent GBM neurosphere lines, derived from the same patient (HF3016 and HF3177). We have previously found the GBM oncogene EGFR to be focally amplified in all four GBMs and both neurosphere lines (8,19). As a control, we included probes mapping to chromosome 7, which was broadly amplified at a low level in all six specimens. We evaluated the fit of EGFR-containing ecDNA copy number distributions to discrete probability distributions using the Akaike information criterion. We observed that a Gaussian distribution show the best fit in most cases, demonstrating that the number of ecDNA copies varied widely across cells and implicating uneven segregation of ecDNA (Fig. 1B-C and Supplementary Fig. 1A-D). These findings are a slight deviation from previous observations implying that uneven ecDNA segregation derive a binomial ecDNA distribution across cells (12). The copy numbers of chromosome 7 appeared more evenly distributed (Fig. 1B-C, lower panel) but not stable, possibly as a result of subclonal chromosome 7 tetraploidy which is common in GBM, cells residing in different stages of the cell cycle and noise levels of the assay. We calculated the median absolute deviation (MAD) of the distribution of copy numbers, which is a metric that represents variability independent of copy number level, and found the MAD of EGFR-ecDNA copies to be significantly higher than the MAD of chromosome 7 copies (average MAD 10.25 vs 1.61; Fligner-Killeen test, p-value < 1.5e-08 in all samples). Representative images of EGFR-containing ecDNA copies in cells containing identical numbers of chromosome 7 reflect the impact of ecDNA on intratumoral heterogeneity (Fig. 1B-C, upper panel).
Fig. 1 |. Unevenly segregated ecDNA drives intratumoral heterogeneity.
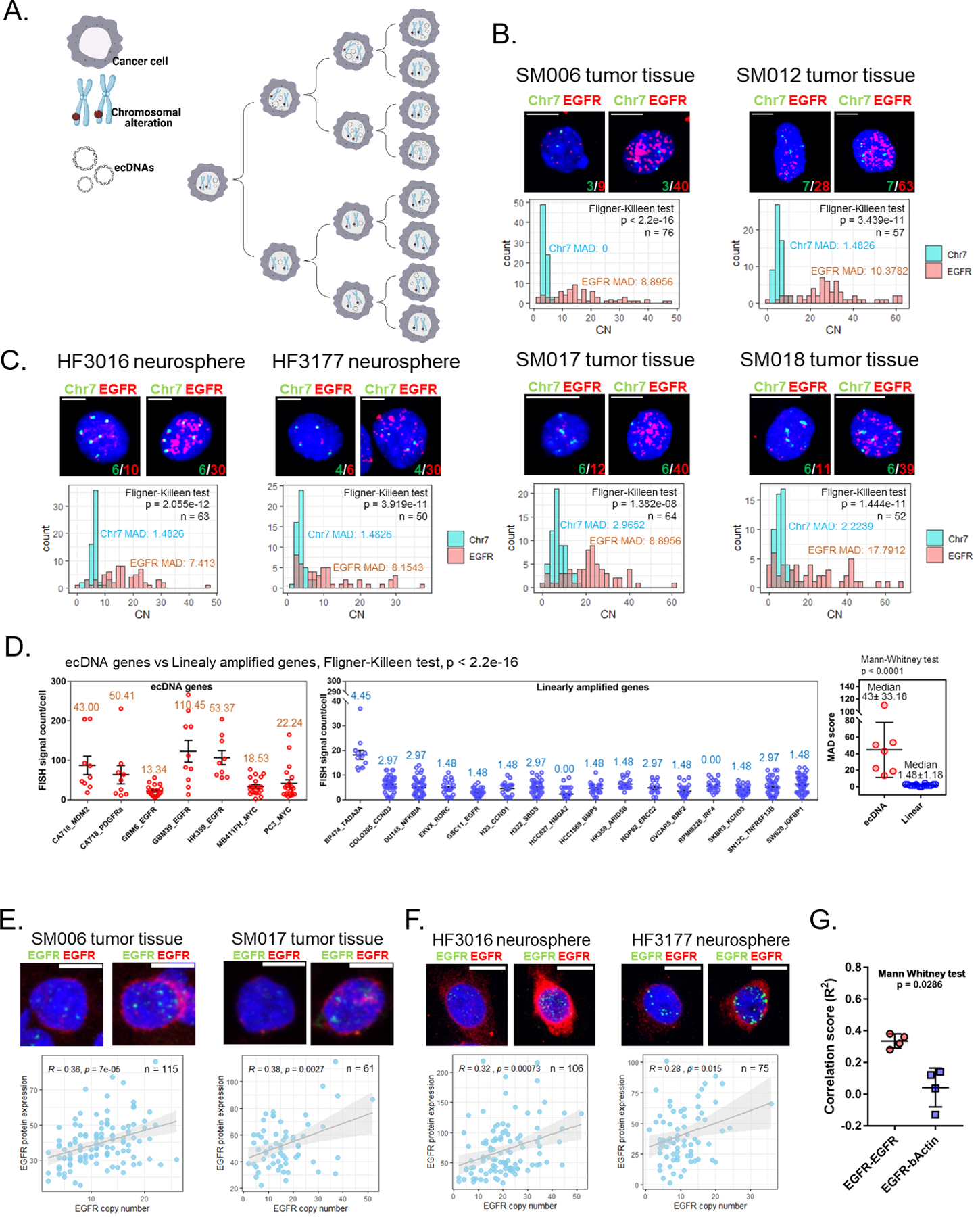
A. Cartoon representation of the pattern of inheritance of chromosomal alterations and ecDNAs. B-C. Representative EGFR/Chr7 FISH on four GBM tumor tissues (B, upper panel) and two neurosphere lines (C, upper panel). The MADs are indicated with the corresponding color in each image. Scale bar, 10 μm. Copy number count of each FISH probe per cell and p values indicating the homogeneity of variances between EGFR and Chr7 were determined by Fligner-Killeen test (lower panel). SM006 = Classical; SM012 = Proneural + Mesenchymal; SM017 = Mesenchymal + Classical; SM018 = Mesenchymal; HF3016 and HF3177 = Proneural. D. Copy number distribution of ecDNA genes (left panel) and linearly amplified genes (middle panel). The MADs indicated at the top of individual group. A p value indicating the homogeneity of variances between ecDNA genes and linearly amplified genes was determined by Fligner-Killeen test. The error bars represent standard error. The median MAD of ecDNA genes was significantly higher than the median MAD of linearly amplified genes. A p value indicating significant differences between two group was determined using a Mann-Whitney U test. The error bars represent standard deviation. E-F. ImmunoFISH experiment on two GBM tumor tissues (E) and two neurosphere lines (F). Scale bar, 10 μm. Green signal indicates EGFR FISH signal. Red signal indicates EGFR protein signal. Correlation between copy number of EGFR (number of EGFR DNA FISH signal foci) and EGFR protein expression (quantified based on signal intensity) per cell and p values were determined by Pearson’s correlation test (lower panel). EGFR protein signals that appears to be derived from the nucleus is in fact cytoplasmic and on the cell surface, but appears nuclear as two-dimensional images were obtained from a three-dimensional cell image. G. Comparison of Pearson’s correlation scores between EGFR-EGFR and EGFR-β-Actin.
To expand our observation, we assessed FISH images across a collection of different cancer cell lines and of genes that were recently shown to reside on either linear or circular amplicons by whole-genome sequencing (9). In total, we compared FISH signals from seven genes on ecDNA and 16 linearly amplified genes. While we observed considerable variability in single-cell copy number of both linear and ecDNA amplicons, ecDNA MADs (median MAD 43 +/- 33.18) were significantly higher than linear amplicon MADs (median MAD 1.48 +/- 1.18; Fig. 1D and Supplementary Fig. 2). The difference in copy number distribution between ecDNA and linear amplicons corroborates previous circumstantial evidence that ecDNA segregates unevenly (7,12).
Intratumoral heterogeneity, which impairs treatment response, is marked by genomic variability as well as intercellular diversity in gene and protein expression(20,21). To examine whether the aptitude of ecDNA for enhancing intratumoral diversity ultimately affects the heterogeneity of functional protein expression, we determined the association of EGFR copy number with EGFR protein expression at the single-cell level (Fig. 1E-F). In all samples, we observed a positive correlation between EGFR ecDNA copy number and EGFR protein expression (Fig. 1E-F, lower panel) but not with β-Actin protein expression (Supplementary Fig. 3A-B and Fig. 1G). The moderate correlation implies additional factors may determine expression levels.
CRISPR-based labeling enables live-cell ecDNA tracking
To understand how ecDNA heterogeneity is derived, we developed ecTag, a CRISPR-based DNA labeling method to study ecDNAs in live cells. EcDNAs are formed by DNA breakage followed by end-to-end ligation of DNA segments, resulting in one or more ecDNA breakpoint junctions (22–25) (Fig. 2A). The sequences covering the breakpoint sites are unique and cannot be detected in the parental linear chromosomes. EcDNA-specific breakpoint sequences provide an opportunity for the design of single guide-RNAs (sgRNA) to label or target ecDNA. ecTag was developed by employing Casilio (17,26), a hybrid technique that combines dead-Cas9 (dCas) labeling and Pumilio RNA-binding, to recruit multiple fluorescent protein molecules at a prespecified sgRNA target locus (Fig. 2A). SgRNAs designed with programmable Pumilio/FBF (PUF) RNA-binding sites (PUFBSs) achieve target DNA binding through a spacer sequence mapping the ecDNA breakpoint and also enable recruitment of fluorescent molecules conjugated with PUF (Fig. 2A). We evaluated ecTag to label ecDNA-specific breakpoints. To find targetable breakpoint sequences, we analyzed whole-genome sequencing (WGS) data of the HF3016 neurosphere line to reconstruct the structure of the ecDNA element (27) (Supplementary Fig. 4A-E). This identified four unique ecDNA structures, harboring an EGFR fragment (exon1), the full EGFR coding sequence, and the non-coding genes CCAT and CCDC26. We labeled these four lesions as ecEGFRx1, ecEGFR, ecCCAT1 and ecCCDC26 respectively. We designed four primer pairs (Supplementary Fig. 4D, yellow arrows) that allowed extraction of an ecDNA breakpoint fragment from each ecDNA on agarose gels (Supplementary Fig. 5A). We then performed Sanger sequencing to define the precise breakpoint sequence (Supplementary Fig. 5B)(28).
Fig. 2 |. CRISPR-based labeling enables live-cell ecDNA tracking.
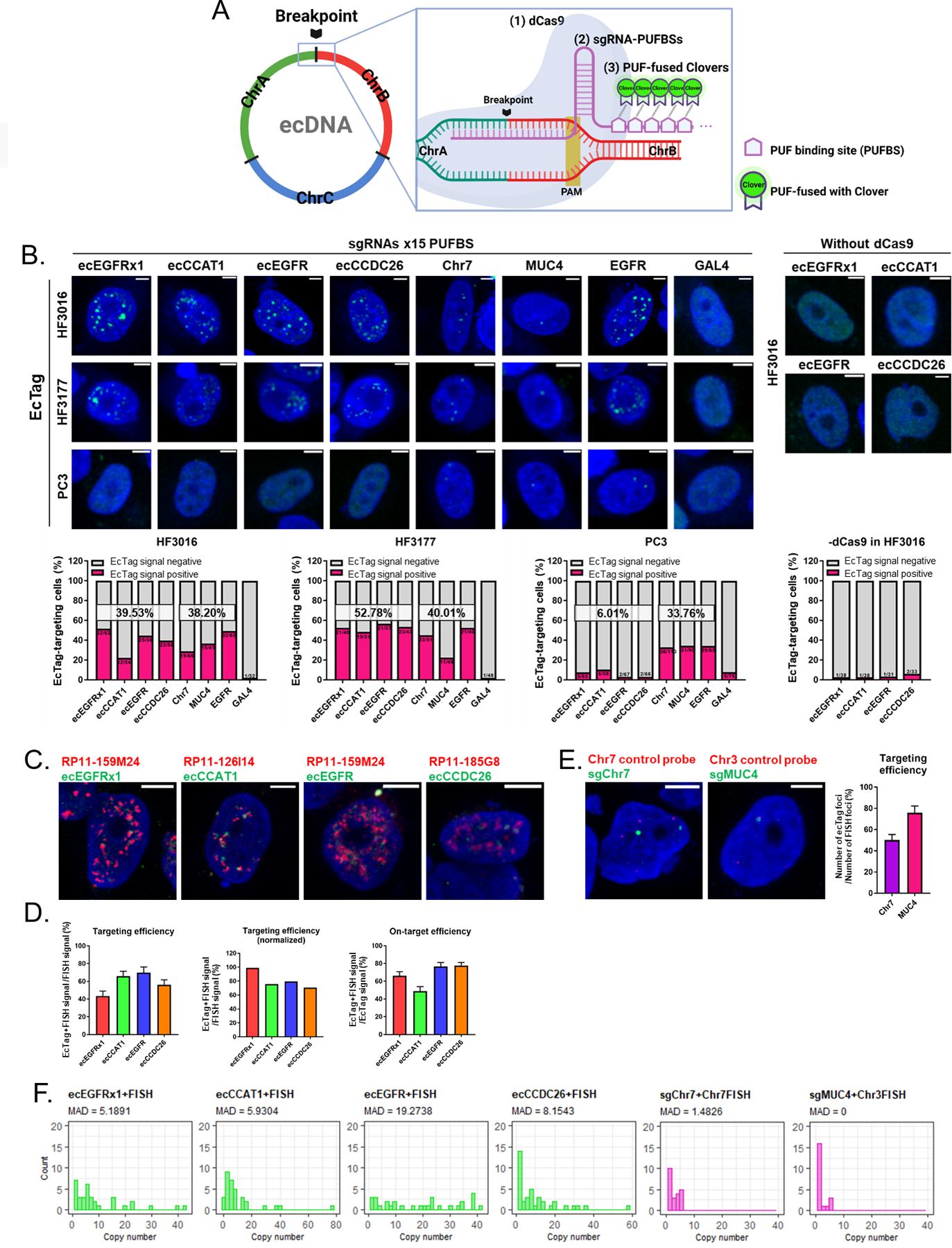
A. Schematic strategy of the ecTag ecDNA labeling system. B. Representative images of ecTag-transfected cells (upper panel). Scale bar, 10 μm. Proportion of ecTag-targeting cells out of the ecTag-transfected cells (bottom panel). The cells that have Clover signals spread out in the nucleus were counted as ecTag-transfected cells. Cells containing green spots in the nucleus were counted as ecTag-targeting cells. SgRNAs conjugated with 15 PUFBSs were used. (n = 31–110 cells per condition). C. Representative images of FISH validation (red BAC probe) performed on ecTag(green)-transfected HF3016 cells. Scale bar, 10 μm. SgRNAs conjugated with 25 PUFBSs were used. D. Proportion of ecTag and FISH double-positive signals relative to FISH (red) signals was calculated as targeting efficiency (left panel). The error bars represent S.E.M. Targeting efficiency normalized by the average proportion of Dual-FISH signals out of the red-color signals showed in Supplementary Figure 6D. Proportion of ecTag and FISH double-positive signals out of the ecTag (green) signals was calculated as on-target efficiency (right panel). n = 30–38 cells per condition. E. Representative images of FISH validation (red control chromosome probe) performed on control ecTag (Chr7 and MUC4, green)-transfected HF3016 cells. Scale bar, 10 μm. SgRNAs conjugated with 25 PUFBSs were used. Proportion of ecTag signals paired with FISH probe signals out of the FISH signals was calculated as targeting efficiency (right panel). The error bars represent S.E.M. n = 21–22 cells per condition. F. Copy-number distribution of ecTag/DNA FISH signals with a high MAD score in ecDNA-targeting group (green plots) confirmed that the ecTag system recapitulates the distribution pattern of ecDNA uneven segregation observed in oncogene-FISH and Dual-FISH. n = 21–38 cells per condition.
To validate that the target breakpoints were specific to the ecDNA and were extrachromosomal, we used Dual-FISH, in which two DNA BAC library FISH probes corresponding to DNA sequence on either side of the breakpoint were labeled with two different fluorescents (Methods, Supplementary Table 1. and Supplementary Fig. 6A). With this approach, a merged or adjacent fluorescent signal marked an extrachromosomal breakpoint (Supplementary Fig. 6B). We also included a second neurosphere line, HF3177, which was derived from the recurrent glioblastoma from the same patient from whom HF3016 was established, therefore both cell lines were likely to share the same ecDNA amplifications. We have previously found that the PC3 prostate cancer cell line contains ecDNAs (9) but very different in sequence from those in HF3016/HF3177, and therefore used PC3 as a negative control. The Dual-FISH analysis showed breakpoints co-labeled with two-color probes outside of chromosomes (Supplementary Fig. 6B). We counted breakpoint-positive metaphase cells and found that breakpoints were shared between primary (HF3016) and recurrent (HF3177) cell lines but in different ratios (Supplementary Fig. 6C, left and middle panel). We observed some signal in PC3 cells which is likely due to non-specific binding of the probes (Supplementary Fig. 6C, right panel). Only signals labeled with both color probes were counted as derived from ecDNA (Supplementary Fig. 6D). The distribution of the number of breakpoint signals per cell confirmed the random segregation pattern (Supplementary Fig. 6E). We observed that a subset of neurosphere cells presenting with HSRs stained with one of the two-color probes (Supplementary Fig. 6F). The variability of breakpoint quantities, which may reflect the evolution of the ecDNA structure (8), was confirmed by BP-PCR (Supplementary Fig. 7). These results showed that ecDNA-specific breakpoints can be leveraged to visualize ecDNA in single cells through fluorescence microscopy.
To engineer the ecTag live-cell ecDNA tracking system, we cloned breakpoint-specific sgRNAs with 15 or 25 PUFBS repeats. Co-transfection of sgRNAs, catalytically inactivated Cas9, and Clover-PUF fusion protein-expressing plasmids allows the enrichment of fluorescent signals at the targeted ecDNA breakpoint loci (Fig. 2A). To validate the targeting efficiency of breakpoint-specific sgRNAs, we performed an in vitro cleavage assay on HF3016 and HF3177 cells and confirmed on-target efficiency of sgRNA (Supplementary Fig. 8).
To verify the cell-type specificity of the four selected ecDNA breakpoints (ecEGFRx1, ecCCAT1, ecEGFR and ecCCDC26), the on-target HF3016, HF3177 cell lines and PC3 control cell line were co-transfected with three components: 1. breakpoint-specific sgRNA (15 PUFBS repeats), 2. dCas9 and 3. Clover-PUF fusion protein expressing plasmid. Each component was prepared as an individual plasmid. We included two positive control sgRNAs: 1) sgRNAs labeling an intronic region on chromosome 7 (Chr7) or the chr 3q29 gene MUC4, as representative linear DNA controls and 2) A non-repetitive EGFR-targeting sgRNA. As a negative control, we included an sgRNA targeting the yeast gene GAL4. Cells expressing the sgRNA target region showed an abundance of nuclear spot signals (Fig. 2B, representative images). The spot signals did not result from fluorescent molecules specifically aggregating in the nucleolus (Supplementary Fig. 9A). We confirmed that ecTag transfection did not affect the cell proliferation rate (Supplementary Fig. 9B) or result in nuclear atypia, which is a characteristic of HF3016 neurosphere cells (Supplementary Fig. 9C). The fraction of ecTag-targeting cells for ecDNA-sgRNAs in HF3016 and HF3177 was 39.5% and 52.8% respectively, compared to an off-target 6% in PC3, indicating that ecDNA-targeting sgRNAs were specific to HF3016 and HF3177 cells. The control-sgRNAs (non-repeat region of Chr7- and MUC4-, and EGFR-targeting sgRNA) showed comparable fractions of ecTag-targeting cells in HF3016, HF3177 and PC3 (38.2%, 40% and 33.8% respectively). The Gal4-targeting sgRNA used as negative control was detected in 1.9% in HF3016, 2.1% in HF3177, and 7.9% in PC3. The population of ecTag-targeting cells for ecDNA-sgRNAs without dCas9 in HF3016 (3.6%) demonstrated the minimal amount of non-specific signals generated by sgRNA-Clover coupling without dCas9. As the ecDNA-targeting sgRNAs map to a specific ecDNA breakpoint, not all ecDNA molecules are being tagged. Thus, the ecEGFRx1 or ecEGFR breakpoint regions may not be part of all EGFR-containing ecDNAs (Supplementary Fig. 4E), and these sgRNAs mark fewer signal foci compared to EGFR DNA FISH (Fig. 1C). The copy-number distribution pattern of ecTag signal foci per cell was comparable to those observed using FISH (Supplementary Fig. 10A-C). Two-color imaging of DNA-FISH and ecTag ecDNA labeling demonstrated that the spot signals derived from the ecTag accurately mapped the specific ecDNA breakpoint with 81.2% normalized targeting efficiency and 67.3% on-target efficiency, on average (Fig. 2C-D and Supplementary Fig. 11A-D). The targeting efficiency of the applying ecTag using guides designed to map chromosomal controls (Chr7 and MUC4) was determined by measuring signal foci paired with each control DNA probe out of the number of control DNA probe signal foci (Fig. 2E and Supplementary Fig. 11E-F). The ecTag targeting efficiency was also confirmed by performing EGFR DNA FISH on EGFR-targeting ecTag-transfected HF3016 cells (Supplementary Fig. 11G). We found that on average, EGFR-targeting ecTag was able to detect 42% of FISH-probe labeled EGFR. The lower specificity observed in this experiment can be explained by multiple ecDNAs containing a partial EGFR region which were unable to be captured by a single sgRNA mapping to a non-repetitive single region. The number of ecTag signals accompanying DNA-FISHs signal showed a broad distribution across cells but not in chromosomal controls (Chr7 and MUC4), demonstrating that ecTag is able to recapitulate the previously observed distribution pattern of ecDNA (Fig 2F).
EcDNA spatiotemporal tracking shows uneven segregation of ecDNA during mitosis
Centromeres provide attachment sites for spindle microtubules to enable chromosome segregation and the acentromeric character of ecDNA therefore implies unequal segregation in mitosis(5,29). Using our ecTag-based ecDNA tracing system, we sought to evaluate the distribution of ecDNA following cell division. We monitored mitosis in HF3016 neurosphere cells with fluorescent labels attached via ecTag to ecDNAs. We found that the fluorescent signal was diluted during cytoplasmic division, which may be explained by the level of DNA compaction during metaphase(30) or potential ecDNA clustering during mitosis (31), resulting in an inability of sgRNAs to bind target sequences (Fig. 3A). Once the telophase finished and the two daughter cells entered interphase, the fluorescent signals re-established again visualizing ecDNA molecules. We found that daughter cells often inherit different numbers of ecDNAs (Fig. 3A and Movie 1). We quantified fluorescent signals in offspring cells and observed that Chr7 and MUC4 derived signals showed uniform segregation (Supplementary Fig. 12A-B), reflected by a Pearson correlation of 1 or near 1, whereas the same analysis of ecDNA inheritance showed a marginal and non-significant correlation (Fig. 3B). To visualize the segregation of ecDNA over the full time window of a cell division, we established a ecTag-stable PC3 model through serial transduction and drug selection of each of the Casilio-based ecTag components (dCas9, Clover, and sgRNA) (Supplementary Fig 13A-C). Time-lapse images of the ecTag-stable PC3 model confirmed that ecTag signals are diluted as the cells enter into metaphase, thus, not only the ecDNA specific signal foci but also the entire Clover spread across nuclei was not visible during metaphase (Fig. 3C). Together, both the post-mitosis ecDNA distribution in the transient ecTag H3016 cells and the ecTag-stable PC3 cells demonstrate, unequivocally, that ecDNA segregates unevenly.
Fig. 3 |. Spatiotemporal tracking of ecDNA shows uneven segregation of ecDNA during mitosis.
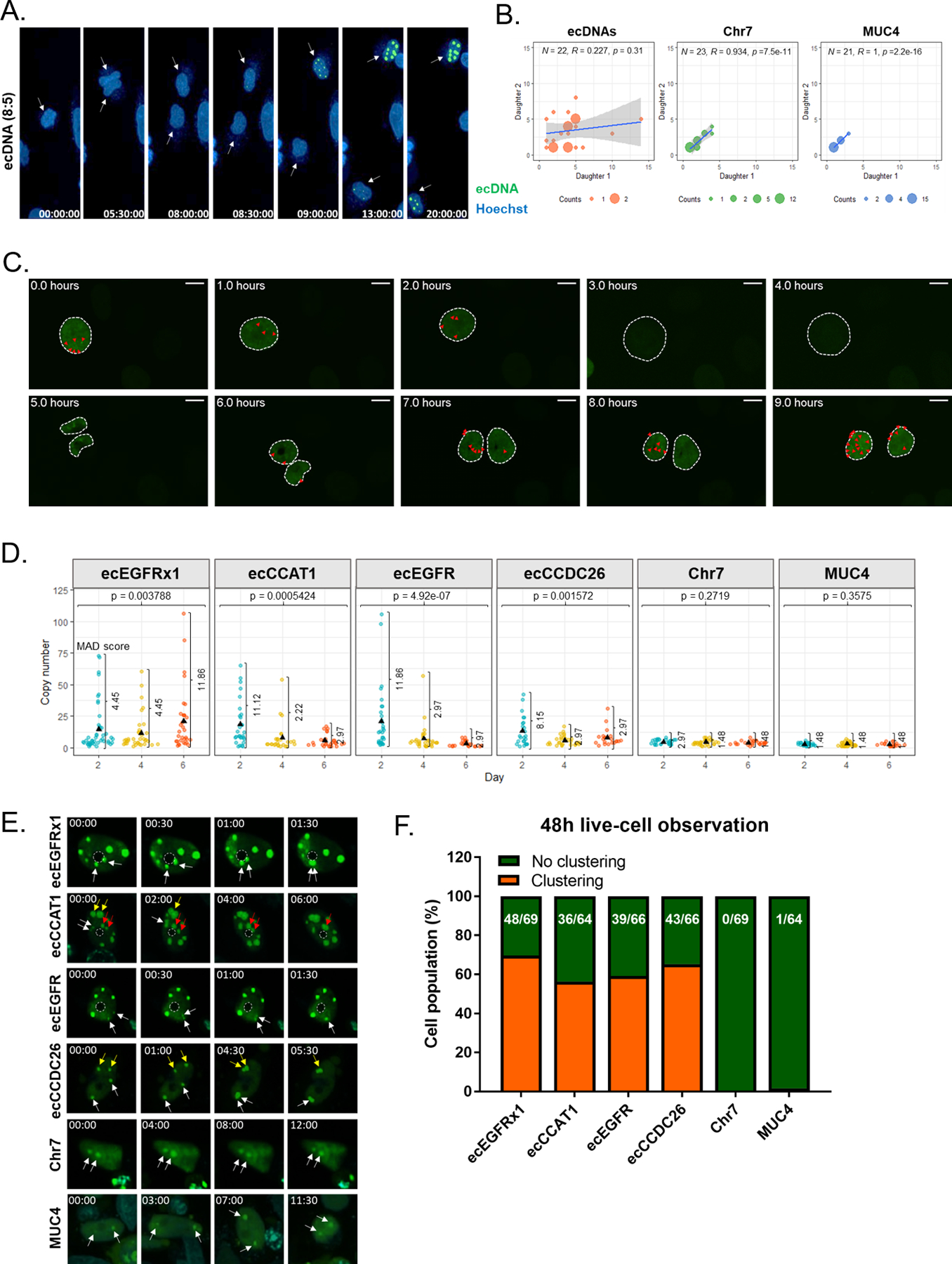
A. Captured time-lapse images of ecDNA segregation during mitosis. SgRNAs conjugated with 25 PUFBSs were used. B. Copy number of ecDNAs, Chr7, and MUC4 segregated into two daughter cells. (n > 20 dividing cells per each condition). We counted the number of ecDNAs when each daughter cell had the highest number of signal foci. Randomness of ecDNA segregation was determined by Pearson’s correlation test and the p value higher than 0.05 indicates the random distribution. C. Representative time-lapse images showing cell division process from interphase to interphase in PC3 cell model. Scale bar = 10 μm. Red triangles indicate the number of ecDNA signal spots determined based on adjusted green signal thresholding. D. Copy number distribution of ecDNAs, Chr7, and MUC4 in HF3016 neurosphere cells on three different days. Individual dots represent copy number counts of single-cells. The MADs are indicated. P values of the difference in copy number variance over time were determined using a Fligner-Killeen test. SgRNAs conjugated with 25 PUFBSs were used. The result is representative of a distribution of > 20 cells per sample. E. Captured time-lapse images of ecDNA hubs. The pair of arrows with the same color on each group showed the process of ecDNA hub formation. SgRNAs conjugated with 25 PUFBSs were used. The dashed circle indicates the nucleolus. (00:00 = Hour:Minute). F. The fraction of the cell population containing ecDNA hubs was counted across 48h and using live-cell imaging. The number of cells containing ecDNA hubs and the total number of observed cells are shown on each bar.
To obtain better insight into the discordant inheritance pattern of ecDNA, we determined the distribution of single-cell ecDNA and linear DNA copy numbers every two days, relative to the two days doubling time of HF3016 cells (Supplementary Fig. 14). EcDNA copy number continued to be highly variable over several cell doublings, in comparison to the Chr7 and MUC4 controls (Fig. 3D). In contrast, the average copy number of controls, Chr7 and MUC4 – linear DNA amplicons on canonical chromosomes - remained stable, indicating that the DNA replication of these regions is tightly regulated by cell cycle (7,18). To rule out the possibility that the distribution pattern of ecDNA copies was caused by cellular heterogeneity, we created three single-cell clones from the well-established PC3 cell line, which contains circularly amplified MYC. We performed MYC/Chr8 DNA FISH on three single-cell clones and the PC3 instance from which they were derived (Supplementary Fig. 15). We observed that MYC DNA copy numbers were highly variable (MAD = 7.413–14.826) in original PC3 (PC3-NCI) as well as in all isogenic clones (PC3-C4, PC3-C5, and PC3-C7), while Chr8 copy number were stable (MAD = 0), in support of uneven ecDNA segregation. Our results suggests that unlike linear DNA amplicons, ecDNA frequencies continuously fluctuate over time, and emphasizes the rapid mode of tumor evolution that ecDNA elements are able to direct in comparison to linear amplifications(8).
The live-cell ecDNA tracking experiments provided a spatiotemporally dynamic feature of ecDNA within a single cell, but also suggested that ecDNA showed a propensity to physically cluster together, not involving the nucleolus (Fig. 3E). We observed that ecDNA signals were significantly larger than controls (Supplementary Fig. 16A), implying a contribution by Clovers from multiple ecDNA molecules and indicating hubs of multiple ecDNAs. We performed stepwise photobleaching, a method to reveal the number of objectives within a molecule complex (32), on Chr7 and ecDNA foci. High laser intensity results in loss of fluorescent molecules and a longer time to signal intensity decrease indicates a higher number of fluorescent molecules. The resulting intensity drop of ecDNA signals is significantly less in comparison to Chr7, implicating that ecDNA signals consists of a higher number of Clover molecules than Chr7 (Supplementary Fig. 16B). To rule out the possibility that ecDNA clustering is an artefact explained by the high number of ecDNAs in the nucleus, we compared ecDNA interactions in hubs to interactions between telomeres, as there are similarly a large number of telomeres in each nucleus (Supplementary Fig. 17A-F). To achieve this, we used a sgRNA targeting repetitive telomere sequences. The distance between two interacting signal foci was measured from the core of one focus to another, every 30 minutes, for 7 hours (Supplementary Fig. 17A). We observed that ecDNA signal foci were stably contacting each other and over longer time periods, compared to the temporal and dynamic interactions taking place between telomeres (Supplementary Fig. 17A-E). To exclude the possibility that the circular nature of ecDNAs resulted in ecTag-induced adhesion, we evaluated ecTag in HF3016 transfected with a dsRED-expressing plasmid (Supplementary Fig. 17F). The interactions of dsRED-targeting ecTag signal showed a dynamic fluctuation pattern similar as to what we observe with respect to telomeres (Supplementary Fig. 17A), confirming previous similar observations that plasmids are not likely to form aggregates (33). Next, we employed a dual-color ecTag labeling system to determine whether larger ecTag signals were contributed by multiple individual ecDNA elements (Supplementary Fig. 18A). Two sgRNAs were designed mapping to the same breakpoint to visualize ecDNA aggregation using green and red fluorescent molecules. We observed complete merging of yellow signals or closely assembled two colors indicating ecDNA clustering (Supplementary Fig. 18B).
In live-cell imaging, ecDNA hubs were observed to take place in over 50% of cells within 48 hours of live cell imaging (Fig. 3F), expanding previous observations in anaphase cells(31) and suggesting functional relevance. We used multi-color ecTag labeling to tag two different ecDNAs and observed ecDNA containing different cargo genes both in close proximity as well as completely merged (Supplementary Fig. 19), suggesting that ecDNA hubs can form independent of ecDNA content. The similar ecDNA signal size distribution in HF3016 (primary) and HF3177 (recurrent) suggested that the ability for forming ecDNA hubs was preserved across time points (Supplementary Fig. 20A-B).
EcDNA hubs associate with RNA polymerase II activity
Recent studies have shown that ecDNA drives high levels of oncogene expression (9,34,35). We hypothesized that the generation of ecDNA hubs enhances transcriptional activity. First, we examined whether ecDNA hubs were associated with colocalization of nuclear bodies, including Cajal bodies and promyelocytic leukemia protein (PML) nuclear bodies (36,37). Cajal and PML bodies have been reported to contain hyperphosphorylated RNA polymerase II (RNAPII), nominating them as sites of active mRNA transcription (38,39). EcTag-transfected cells were stained using Cajal/PML marker protein antibodies and a secondary antibody conjugated with red fluorescent molecule was used to capture the primary marker protein antibody (Supplementary Fig. 21A and Supplementary Fig. 21B). We compared colocalization between nuclear bodies and ecDNA, using MUC4 and Chr7 colocalization as a control. Colocalization was defined as two different fluorescent signal foci partially or completely overlapping. The fraction of cells containing ecDNAs colocalizing with nuclear bodies was substantially higher than the fraction of cells containing MUC4 or Chr7 colocalizing with nuclear bodies (Cajal bodies: 45%, 6% and 0% for ecDNAs, MUC4, and Chr7, respectively; PML bodies, 67%, 10% and 17% for ecDNAs, MUC4, and Chr7, respectively)(Supplementary Fig. 21C and Supplementary Fig. 21D). Since the amount of colocalized signal is linearly correlated to the number of target copies and the amount of ecDNAs is much greater than the Chr7/MUC4 control, we normalized the number of colocalized signal loci per cell by the level of ecTag signal (0.07 to 0.24 in Cajal body and 0.09 to 0.16 in PML body, Mann-Whitney test, p-value < 3e-03 in all samples, 0.03 in Cajal body and 0.05 in PML body, ecDNAs vs Chr7, Mann-Whitney test, p-value = 0.5 in Cajal body and p-value = 0.72 in PML body, MUC4 vs Chr7, Mann-Whitney test,)(Supplementary Fig. 21E and Supplementary Fig. 21F). This showed that the interaction between ecDNA and nuclear bodies is not due to the abundance of ecDNAs. We additionally analyzed the size of colocalized area normalized by the total ecTag signal size. This analysis also showed that a significantly higher proportion of ecDNA signal is merged with nuclear bodies compared with Chr7 (0.02 to 0.06 in Cajal body and 0.03 to 0.06 in PML body, Mann-Whitney test, p-value < 2e-03 in all samples, ecDNAs vs Chr7) while the proportion of MUC4 area merged with nuclear bodies (0.01 in Cajal body and PML body, Mann-Whitney test, not significant, p-value = 0.24 in Cajal body and p-value = 0.64 in PML body, MUC4 vs Chr7), MUC4 vs Chr7) showed no significant differences (Supplementary Fig. 21G and Supplementary Fig. 21H). While we observed interactions between ecDNA and nuclear bodies at rates significantly higher than the controls (Supplementary Fig. 21A-H), there was no significant linear correlation between ecDNAs and the number of nuclear bodies (Supplementary Fig. 22A-B). This suggests that the relative increase of ecDNA with Cajal/PML body signal is due to the higher abundance of ecDNA and implies that ecDNA is not actively being trafficked towards the nuclear bodies. We measured the average greyscale intensity of nuclear bodies signals overlapping with ecTag signal foci over a 12 hour-window which demonstrated fluctuations indicating instable interaction between ecDNAs and nuclear bodies (Supplementary Fig. 23A-B), suggesting that ecDNAs generate their own clusters independently from those nuclear bodies.
To determine whether ecDNA signal regions are being actively transcribed, ecTag-transfected cells were stained with RNAPII antibody (Fig. 4Ai and Supplementary Fig. 24A). More than half of the cells (59.9 %) contained ecDNAs colocalizing with RNAPII (Fig. 4Aii). The number of colocalized loci per cell normalized by total ecTag signal showed that a significantly higher number of ecDNA loci colocalized with RNAPII compared with Chr7 (0.07 to 0.42, Mann-Whitney test, p-value < 8e-03 in all samples). A significantly higher number of ecDNA loci colocalized with RNAPII compared with MUC4 were observed in two ecDNA cases (Mann-Whitney test, p-value < 0.002, in ecEGFR and ecCCDC26). We also found significantly higher MUC4-RNAPII signal (0.14, Mann-Whitney test, p-value = 0.04, MUC4 vs Chr7) in comparison to Chr7, reflecting the active transcription of a linear chromosomal gene (Fig. 4Aiii). In addition to the increased number of overlapping loci, we detected a significantly higher total ecDNA area to be merged with RNAPII (0.01 to 0.08, Mann-Whitney test, p-value < 8e-03 in all samples, ecDNAs vs Chr7) compared with Chr7 (Supplementary Fig. 24B). The number of ecEGFRx1 and ecEGFR ecDNA breakpoints showed a linear correlation with RNAPII count, suggesting that the colocalization event of ecDNAs with RNPII is not random (Fig. 4B). EcDNA hubs colocalizing with RNAPII were found to be significantly larger than ecDNA without colocalization (Fig. 4C, Mann-Whitney test, p-value < 0.05 in all samples, ecDNAs vs Chr7). Chr7 and MUC4 signals did not show significant differences in size, suggesting that clustering and recruitment of functional transcriptional machinery is specific to ecDNA (Fig. 4C). As an alternative active transcription marker, we assessed correlation between ecDNA and 5-ethynyl uridine (EU) incorporation (Supplementary Fig. 25A). EcDNA signals colocalizing with EU incorporated region were significantly larger compared to ecDNA signals without EU colocalization. In contrast, the colocalization between ecDNA signals and Ki67, which is abundant in the nucleus without an association with transcriptional activity, was unaffected by ecDNA signal size (Supplementary Fig. 25B).
Fig. 4 |. EcDNA bodies enhances transcriptional activity by recruiting RNA polymerase II (RNAPII).
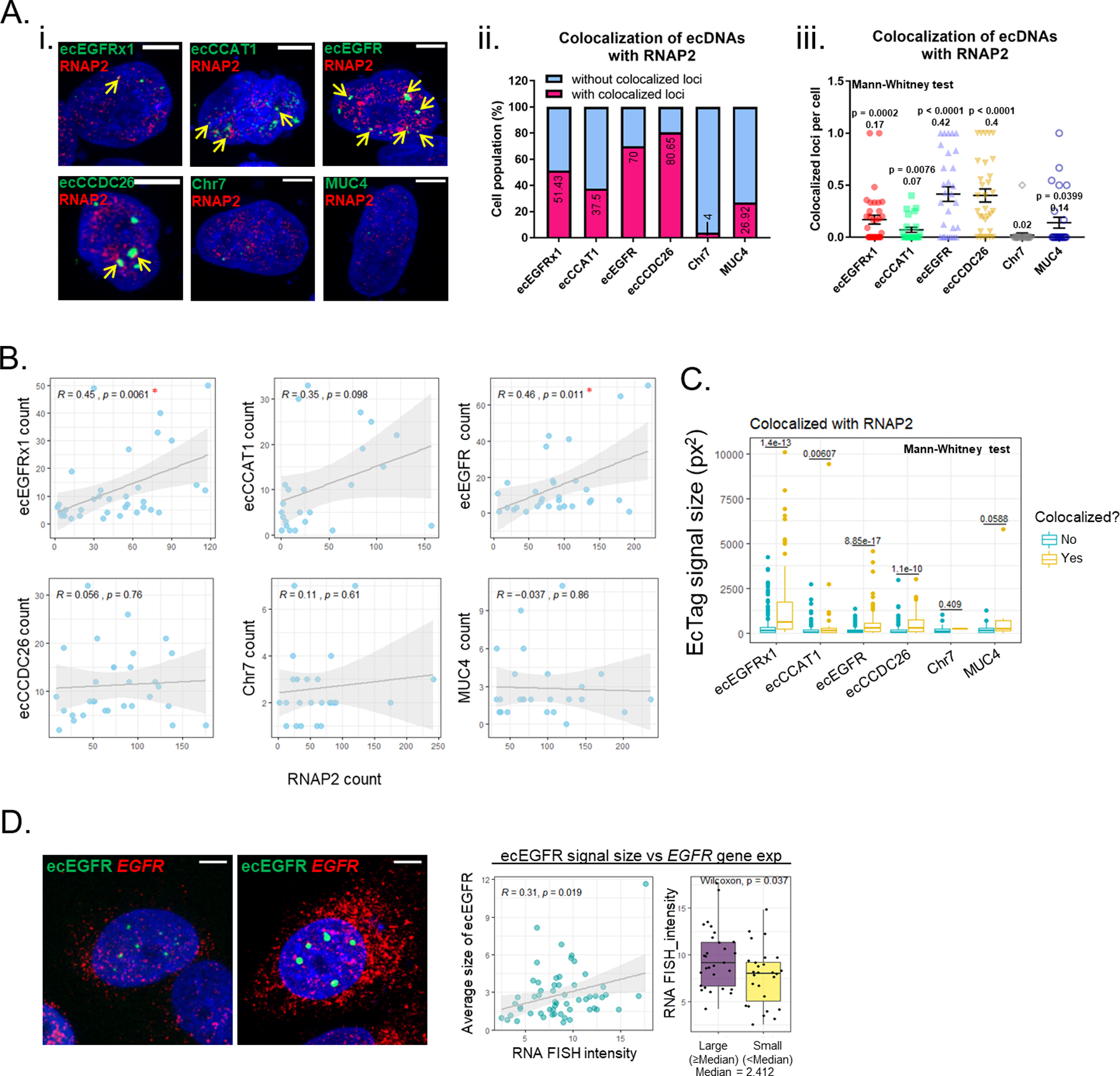
A. Representative images of RNAPII immunofluorescent staining. Scale bar, 10 μm (i). Colocalization was defined as two different fluorescent signal foci partially or completely overlapping. Proportion of cells with or without the loci colocalized with RNAPII (ii). Colocalized loci with RNAPII per cell (iii). All value was normalized by each ecTag signal. The values of ecDNAs and MUC4 were compared with Chr7. p values were determined by Mann-Whitney U test. Average values are indicated under each p value. At least 25 single-cell images per group were analyzed. The error bars represent SE. SgRNAs conjugated with 25 PUFBSs were used. B. Correlation between copy number of ecDNA and RNAPII count. Correlation score and p values were determined by Pearson’s correlation test. The positively correlated cases are marked with red star. At least 25 single-cell images per group were analyzed. C. Comparison of ecDNA signal size and colocalization with RNAP2. p values were determined by Mann-Whitney U test. The same images used in A were analyzed. D. Representative images of EGFR RNA FISH on ecTag-labeled cells with small ecDNA signals (left panel) and large ecDNA signals (right panel), Scale bar, 10 μm. SgRNAs conjugated with 25 PUFBSs were used. Correlation between ecDNA signal size and EGFR gene expression (right panel). EGFR gene expression was quantified based on signal intensity. The scatter plot and Pearson’s correlation score showed a positive correlation. The bar plots represent average EGFR gene expression in cells with large ecEGFR signal size and small ecEGFR signal size. The unit of signal size is μm. (median signal size = 2.412 μm, large size ≥ 2.412 μm, small size < 2.412 μm). 49 single-cell images were analyzed.
EGFR gene expression was additionally positively correlated with the size of ecEGFR signals, as cells with a comparable number but physically larger ecEGFR-ecTag foci expressed more EGFR mRNA (Fig. 4D). The number of ecEGFR signal foci did not correlate with EGFR mRNA abundance, potentially as a result of the ecTag tagging only the subset of EGFR-containing ecDNAs containing the target breakpoint (Supplementary Fig. 26A). We did not observe the same gene expression-ecTag signal size correlation for ecEGFRx1, which maps to ecDNAs containing only EGFR exon 1 (Supplementary Fig. 26B), and other ecDNA breakpoint tags (Supplementary Fig. 26C-D) or control loci (Supplementary Fig. 26E-F). In summary, these results support that ecDNA hubs can serve as site of active transcription. Clustering of ecDNA may drive increased transcriptional activity of its cargo gene, with a spatial interaction advantage provided by forming ecDNA hubs, thus exposing multiple ecDNA molecules to transcriptional machinery simultaneously.
Discussion
Here, we take advantage of a CRISPR dead-Cas9 technique that enables ecDNA-specific fluorescent tagging to interrogate undiscovered ecDNA biology, including inheritance pattern and dynamic behavior. By doing so, we extend previous in situ single time point observations, which are limited in the level of advance they are able to provide. The CRISPR-based genome visualization shown here expands beyond single time point observations and to live-cell tracking, to demonstrate the longitudinal development of extrachromosomal DNA dynamics.
Previous approaches have used three or more sgRNAs to map loci of interest (13,40) which is not feasible when tagging a unique breakpoint sequence. The Casilio system (26), which leverages the RNA binding domains of the PUF proteins fused with fluorescent molecule, enabled us to label a single non-repetitive target locus using a single sgRNA to create ecTag. Applying ecTag for tracing ecDNA in the process of cell mitosis visualized unequal ecDNA segregation during cell division despite of the technical challenges of live-cell imaging, such as limited time frames (~48 hours) as a result of laser-induced cellular stress. The limited accessibility of DNA during mitosis where all genomic components including ecDNA are condensed, restricted the ability to visualize DNA from metaphase to telophase. Future technological developments are needed to overcome this limitation. Our results show that ecDNA, through random segregation during mitosis, enhances intratumoral diversity at the genomic level, and thus allowing ecDNA accumulation over the course of just a few cell cycles. This observation was reflected by fluctuations in ecDNA copy number distribution over subsequent cell cycles, the rules of which are been further explored (41). The basis for ecTag is the use of a single sgRNA and its use is therefore not restricted to labeling of ecDNA, and includes labeling of other DNA as well. This includes tagging of small circular DNAs, also referred to as extrachromosomal circular DNA (eccDNA). EccDNAs are typically less than 1kb (42), creating challenges in the use of for example DAPI or FISH staining for visualization. EccDNAs derive through random ligation of genomic DNA fragments and ecTag could be used to target eccDNA-specific breakpoints, in combination with an available PAM sequence. EccDNAs have been implicated in the immune response marking them as interesting targets for further discovery (43).
We observed that ecDNAs tend to cluster into hubs leading to increased transcriptional activity. EcDNA hubs recruited RNAPII leading to active mRNA expression of cargo genes, highlighting an additional mechanism that explains the exceptionally high levels of ecDNA gene expression reported earlier (34,35). Our results compound recent discoveries of the wide-open chromatin accessibility of ecDNA (34), the topological advantage of ecDNA for communicating with regulators (35,44), ecDNA-driven oncogenic genome remodeling (45), and the function of ecDNAs as a mobile enhancer (46), and add to mounting evidence that the advantage of tumor cells for maintaining ecDNA extends beyond simple dosage effects on cargo gene transcription. The presence of ecDNA hubs has been independently and in parallel shown by others (47). Nuclear condensates resulting from phase separation have been associated with transcriptional processes, and future work may help to understand how ecDNA hubs relate to biomolecular condensation (48–50). The three-dimensional topological orientation of genomic loci on linear chromosomes enables physical interaction between distal regulatory elements and gene promoters (51). The circularization of oncogenes on ecDNAs increases enhancer interactions in ways restricted by insulators when on linear chromosomes (35). The ecDNA hubs described here creates additional interaction opportunities between ecDNA oncogene promoters and enhancers. Such ecDNA hubs can serve as transcriptional hotspots by sharing transcriptional machinery. Our results on ecDNA hubs reflect that the physical assembly of multiple ecDNA molecules may be required for optimal transcriptional activity. A comprehensive understanding of ecDNA clustering behavior will help explain the biological roles of ecDNA contributing to gene expression, cell proliferation, and cell motility in cancer. Taken together, our observations build upon genome engineering technologies to provide new insights into ecDNA biology, a factor contributing to intratumoral heterogeneity. Defining mechanisms of ecDNA replication and assembly will be needed to understand how ecDNA can be leveraged for cancer therapeutics.
Methods
Human tumor specimens
Human glioma resection specimens (SM006, SM012, SM017 and SM018) were obtained from St. Michael’s Hospital. All tissue donations were approved by the Institutional Review Board of the Jackson Laboratory and clinical institutions involved. This work was performed in accordance with the Declaration of Helskinki principles. The molecular subtypes of these glioma tissues were profiled in Johnson et al (19). SM006 = Classical; SM012 = Proneural + Mesenchymal; SM017 = Mesenchymal + Classical; SM018 = Mesenchymal.
Cell cultures and cell lines
Patient-derived glioblastoma spheroids (HF3016 and HF3177) were obtained with written informed consent from patients with protocol approved by the Henry Ford Hospital Institutional Review Board. These cells were cultured in neurophsere medium (NMGF): 500 ml DMEM/F12 medium (Invitrogen 11330) supplemented with N-2 (Gibco, 17502–048), 250 mg bovine serum albumin (BSA, Sigma, A4919), 12.5 mg gentamicin reagent (Gibco, 15710–064), 2.5 ml Antibiotic/Antimycotic (Invitrogen, 15240–062), 20 ng/ml EGF (Peprotech, 100–15), and 20 ng/ml bFGF (Peprotech, 100–18B). The molecular subtypes of two neurosphere lines were determined in Decarvalho et al (8). HF3016 and HF3177 = Proneural. Human prostate cancer cell line PC3 was a gift from Dr. Paul Mischel at University of California at San Diego, and cultured in F12-K (ATCC, 30–2004) with 10% fetal bovine serum (FBS, VWR, 97068–085). All cultured cells were tested for Mycoplasma contamination before use with MycoAlert Mycoplasma Detection Kit (Lonza).
FISH analysis
For patient tissues, the slightly thawed tissues were transferred to a positively charged glass slide by pressing against the surface of the specimen. The tissue slides were then immediately transferred into Carnoy’s fixative (3:1 methanol:glacial acetic acid, v/v), incubated at RT for 30 min and then air-dried. For interphase cell prep, neurospheres were dissociated into single cells and fixed in Carnoy’s fixative for 20 min, briefly washed with fixative and resuspended in fixative. Desired amounts of cells were then dropped onto the glass slide and air-dried. A hybridization buffer (Empire Genomics) mixed with EGFR-Chr7 probe (EGFR-CHR07–20-ORGR, Empire Genomics) was applied to the slides and the slides were denatured at 75°C for 5 min. The slides were then immediately transferred and incubated at 37°C overnight. The post-hybridization wash was with prewarmed 0.4x SSC at 75°C for 1 min followed by a second wash with 2x SSC/0.05% Tween20 for 2 min at RT. The slides were then briefly rinsed by water and air-dried. The VECTASHIELD mounting medium with DAPI (Vector Laboratories) was applied and the coverslip was mounted onto a glass slide. Tissue images were scanned under Leica STED 3X/DLS Confocal with oil-immersion objective (100x). As excitation laser, a 405 nm, a 488 nm, and a 561 nm were used. Z-stack acquired at 0.3–0.5 um step size was performed and all analysis conducted based on maximum intensity projection images of the 3D volume of the cells. Images were acquired and processed by LAS X software.
Evaluation of copy number distribution fitness
We evaluated fitness of EGFR-containing ecDNA copy number distribution to one or more discrete probability distributions. We first used descdist function in R package, fitdistrplus (v 1.1.3) to plot Skewness-kurtosis graph for the choice of distributions (Cullen and Frey 1999). Acknowledging limited data points (n < 100) for model fitness, results showed that our data was most likely sampled from a cell population following Gaussian, negative binomial distribution or Poisson distribution. Next, we estimated model fitness by fitting our data to each of these three distributions. Resulting relative likelihood of non-Gaussian models - based on Akaike information criterion (AIC) values - was significantly (>= 10-fold) lower for 8/12 fitted models for either negative binomial or Poisson distribution as compared to those from Gaussian distribution (Supplementary Fig. 1C-D).
Pan-cancer FISH analysis
We collected FISH images of four genes presented on ecDNA (MDM2, PDGFRA, EGFR, MYC) in six cell lines (CA718, GBM6, GBM39, HK359, MB411FH, and PC3) from GBM, medulloblastoma, and prostate cancer cell line (Supplementary Fig. 1, Red bar plots). We also collected FISH images of sixteen linearly amplified genes (TADA2A, CCND3, NFKBIA, RORC, EGFR, CCDN1, SBDS, HMGA2, BMP5, ARID5B, ERCC2, BRF2, IRF4, KCND3, TNFRSF13B, and IGFBP1) in sixteen cell lines (BP474, COLO205, DU145, EKVX, GSC11, H23, H322, HCC827, HCC1569, HK259, HOP62, OVCAR5, RPMI8226, SKBR3, SN12C, and SW620) from breast, colon, prostate, lung, GBM, ovary, hematopoietic, meduloblastoma, kidney cancer cell line (Supplementary Fig. 1, Blue bar plots). All images used here were obtained from https://figshare.com/s/6c3e2edc1ab299bb2fa0 and https://figshare.com/s/ab6a214738aa43833391.
ImmunoFISH analysis
Glioblastoma tumor tissue specimens were embedded in Tissue-Tek Optimal Cutting Temperature (OCT) compound and stored at −80°C until sectioning. Frozen 10- μM tissue sections were obtained on slides using a Cryostat (Leica, CM3050S) and slides were stored at −80°C until processing for ImmunoFISH analyses. For neurospheres, 1.2 x 105 cells were plated with 10% FBS-containing NMGF media into a glass-viewing area of confocal dish (VWR, 75856–740). The next day, cells were fixed with 4% PFA at RT for 10 min and briefly rinsed with 1X PBS. To allow permeabilization, fixed cells were incubated with 0.5% PBST for 20 min. Fixed cells were then dehydrated by incubating dishes in gradually increasing concentration (70%/80%/90%/100%) of EtOH and air dried. The dehydrated dishes stored at −20°C with foil.
FISH analysis was performed on the dehydrated tissue slides or dishes with cells as described above (FISH analysis section in Method). After the last washing step with water, the slides were then briefly rinsed with PBST and incubated with blocking buffer (5% BSA/0.3% Triton X-100/1X PBS) at RT for 1 hr. Without washing step, EGFR antibody (#4267, Cell signaling, 1:100) or beta-actin antibody (#4970, Cell signaling, 1:200) diluted in antibody diluent (1% BSA/0.3% Triton X-100/1X PBS) was applied to the slide and incubated at 4°C overnight (or RT for 3 hours). Then slides were washed with washing buffer (0.025% Tween-20/1X PBS) three times and the secondary antibody conjugated with Alexa 555 (ab150086, abcam, 1:1000) was applied. After 1 hr incubation, the slides were washed three times and counterstained with VECTASHIELD mounting medium with DAPI (Vector Laboratories). Images were scanned under Leica STED 3X/DLS Confocal with oil-immersion objective (100x). As excitation laser, a 405 nm, a 488 nm, and a 561 nm were used. Z-stack acquired at 0.3–0.5 um step size was performed and all analysis conducted based on maximum intensity projection images of the 3D volume of the cells. Images were acquired and processed by LAS X software.
Immunofluorescence staining
1.2 x 105 cells of HF3016 were plated with 10% FBS-containing NMGF media into a glass-viewing area of confocal dish (VWR, 75856–740). The next day, cells were transfected with ecTag plasmids (83.3 ng of dCas9, 83.3 ng of sgRNA and 83.3 ng of Clover) with Lipofectamine 3000 (Invitrogen, L3000015). After 24 h, the media was changed to fresh media. After 48 h post-transfection, the dishes were briefly rinsed with 1X PBS three times and fixed with 4% PFA at RT for 15 min. The fixed cells were then rinsed with 1X PBS and permeabilized by incubating with washing buffer at RT for 5 min. The dishes were then incubated with blocking buffer at RT for 1 hr and immediately the primary antibody (Coilin, ab87913 (1:200); PML, ab96051 (1:500); RNAPII (1:200), ab193468; Ki67, ab16667 (1:250); Abcam) diluted in antibody diluent was applied and incubated at RT for 1 hr (at 4°C overnight for Ki67). The dishes were then washed with washing buffer three times and the secondary antibody conjugated with Alexa 555 (ab150086, abcam, 1:1000) was applied. After 1 hr incubation, the slides were washed three times and counterstained with VECTASHIELD mounting medium with DAPI. Images were scanned under Leica STED 3X/DLS Confocal with oil-immersion objective (100x). As excitation laser, a 405 nm, a 488 nm, and a 561 nm were used. Z-stack acquired at 0.3–0.5 um step size was performed and all analysis conducted based on maximum intensity projection images of the 3D volume of the cells. Images were acquired and processed by LAS X software.
EU incorporation
ecTag-transfected cells were prepared as described above (Immunofluorescence staining section in Method). After 48 h of transfection, EU incorporation was performed was then performed following the manufacturer’s protocol (Click-iT™ RNA Alexa Fluor™ 594 lmaging Kit, C10330, Invitrogen).
Breakpoint-specific PCR
Genomic DNA was isolated from each cell line using the QIAamp DNA mini kit (Qiagen 51304) or Quick DNA Mini Prep Plus kit (Zymo Research, D4068). Breakpoint-specific PCR was performed in an automated thermal cycler (BioRad, C1000 Touch Thermal Cycler). Each reaction mixture was prepared with AccuPrime Taq DNA Polymerase system (Invitrogen, 12339016). The PCR protocol was: denaturation at 94 °C for 5 min followed by 30 cycles comprising denaturation at 94 °C for 30 sec, primer annealing at 61 °C for 30 sec and DNA elongation at 68 °C for 1 min, further extension at 72 °C for 5 min and rapid cooling to 4 °C. PCR products were analyzed by 1% agarose gel electrophoresis and visualized under UV illumination after SYBR-Safe staining (Invitrogen, S33102). For Sanger sequencing, the PCR amplicons were extracted using Nucleospin Gel and PCR Clean-Up kit (Macherey-Nager, 740609). The bidirectional Sanger sequencing was performed by EtonBio.
Metaphase Dual-FISH
Neurosphere cell cultures and PC3 were synchronized at metaphase by treating with 80 ng/ml Colcemid (Roche, 10–295-892–001) overnight. Cells were washed with PBS and incubated with 0.075 M KCl at 37 °C for 15 min. Samples were then fixed in Carnoy’s fixative (3:1 methanol:acetic acid, v/v) according to standard cytogenetic procedures. Metaphase cells were dropped onto glass slides and baked at 56 °C for 4–9 hours. A hybridization buffer (Empire Genomics) mixed with two probes (red probe:gold probe = 1:1.5, see Supplementary Table 1 for the list of BAC probes used for Dual-FISH) was applied to the slides and the slides were denatured at 75°C for 5 min. The slides were then immediately transferred and incubated at 37°C overnight. The post-hybridization wash was with prewarmed 0.4x SSC at 75°C for 1 min followed by a second wash with 2x SSC/0.05% Tween20 for 2 min at RT. The slides were then briefly rinsed by water and air-dried. The VECTASHIELD mounting medium with DAPI (Vector Laboratories) was applied and the coverslip was mounted onto a glass slide. Images were scanned under Leica STED 3X/DLS Confocal with oil-immersion objective (100x). As excitation laser, a 405 nm, a 488 nm, and a 561 nm were used. Z-stack acquired at 0.3–0.5 um step size was performed and all analysis conducted based on maximum intensity projection images of the 3D volume of the cells. Images were acquired and processed by LAS X software.
FISH validation for ecTag
1 x 105 cells of HF3016 were plated with 10% FBS-containing NMGF media into a glass-viewing area of confocal dish (VWR, 75856–740). The next day, cells were transfected with ecTag plasmids (100 ng of dCas9, 200 ng of sgRNA and 100 ng of Clover) with Lipofectamine 3000 (Invitrogen, L3000015). After 24 h, the media was changed to fresh media. The next day, cells were fixed with 4% PFA at RT for 10 min and briefly rinsed with 1X PBS. To allow permeabilization, fixed cells were incubated with 0.5% PBST for 20 min. Fixed cells were then dehydrated by incubating dishes in gradually increasing concentration (70%/80%/90%/100%) of EtOH and air dried. The dehydrated dishes stored at −20°C with foil. A hybridization buffer (Empire Genomics) mixed with the red probe from the set of Dual-FISH probes was applied to the slides and the slides were denatured at 68°C for 2 min. The slides were then immediately transferred and incubated at 37°C overnight. The post-hybridization wash was with prewarmed 0.4x SSC at 50°C for 1 min followed by a second wash with 2x SSC/0.05% Tween20 for 2 min at RT. The slides were then briefly rinsed by water and air-dried. The VECTASHIELD mounting medium with DAPI (Vector Laboratories) was applied and the coverslip was mounted onto a glass slide. Images were scanned under Leica STED 3X/DLS Confocal with oil-immersion objective (100x). As excitation laser, a 405 nm, a 488 nm, and a 561 nm were used. Z-stack acquired at 0.3–0.5 um step size was performed and all analysis conducted based on maximum intensity projection images of the 3D volume of the cells. Images were acquired and processed by LAS X software.
PC3-derived isogenic cell subcloning
Using PC3 parental cell line, cells were plated in serial dilutions in 96-well plates in such a way to obtain last two columns of the plates with 1 and 0.5 cell/well, respectively. Few hours after plating to allow attachment of cells, all wells in the last two columns of the plates were examined manually under microscope to confirm the presence of no more than single cell per well and wells with single cells were marked. Single cell clones were allowed to grow and used for analyses. The metaphase cells of these PC3-derived clones were prepared as described above (the metaphase Dual-FISH section in Method). The metaphase slides then used for MYC/Chr8 FISH analysis.
Cloning
The backbone vectors for cloning were gifted from Dr. Albert Cheng. The 5’-phosphorylated duplexed oligos for the guide sequences of each breakpoint were purchased from IDT. The sgRNA spacer sequences were digested via BbsI (NEB, R3539) and then were cloned into sgRNA-PUFBS expression vectors (single-color: pAC1373-pX-sgRNA-25xPUFBSa, dual-color: pCR8-sgRNA-15xPUFBSa and pCR8-sgRNA-15xPUFBSc). Guide sequences were then cloned into the digested vector using T4 ligase (Roche, 4898117001).
ecTag transfection for ecTag-targeting cell counting
1 x 105 cells of HF3016, HF3177 were plated with 10% FBS-containing NMGF media into a glass-viewing area of confocal dish (VWR, 75856–740). 1 x 105 cells of PC3 were plated with its regular media (F12K with 10% FBS) into a glass-viewing area of confocal dish (VWR, 75856–740). The next day, cells were transfected with ecTag plasmids (75 ng of dCas9, 150 ng of sgRNA and 75 ng of Clover) with Lipofectamine 3000 (Invitrogen, L3000015). After 24 h, the media was changed to fresh media. The next day, cells were fixed with 4% PFA at RT for 10 min and briefly rinsed with 1X PBS. The dishes then air dried, and the VECTASHIELD mounting medium with DAPI (Vector Laboratories) was applied and the coverslip was mounted onto a dish. Images were scanned under Leica STED 3X/DLS Confocal with oil-immersion objective (40x). As excitation laser, a 405 nm and a 488 nm were used. Z-stack acquired at 0.3–0.5 um step size was performed and all analysis conducted based on maximum intensity projection images of the 3D volume of the cells. Images were acquired and processed by LAS X software.
In vitro sgRNA test
The in vitro targeting efficiency of sgRNAs was tested using a Guide-it Complete sgRNA Screening System (Takara, 632636) according to the manufacturer’s instructions. Breakpoint-PCR amplicons were used as a targeting templates and incubated with appropriate sgRNA and recombinant Cas9 (rCas9) at 37°C for 1 hr. The cleaved fragments generated by sgRNA targeting were perceived on agarose gel.
Live-cell imaging
Neurospheres were plated into 100-mm tissue culture dishes at 1 million cells per dish with NMGF medium containing 10% FBS. The next day, cells were transfected with ecTag plasmids (1250 ng of dCas9, 1250 ng of sgRNA and 1250 ng Clover) with Lipofectamine 3000 (Invitrogen, L3000015). After 24 h, the cells were trypsinized and resuspended in DPBS including 10% FBS. To isolate ecTag-transfected cells, the Clover-positive cells were then sorted by FACSAria Fusion (BD Biosciences) with 130 μm nozzle. To recover the sorted cell fitness, the ecTag-transfected cells were then re-plated into 384-well plate at 5,000 cells per well with NMGF including 10% FBS and incubated at 37 °C overnight. The next day, cells were stained with CellMask Deep Red Plasma membrane stain (Invitrogen, C10046) and NucBlue Live ReadyProbe (Invitrogen, R37605). Live-cell imaging was performed on Opera Phenix High-Content Screening System (PerkinElmer) under 5% CO2 and 37 °C temperature. To avoid evaporation of medium, empty wells around the samples were filled with DPBS. All the images were acquired by taking 6 – 8 different focal planes, and shown as a maximum intensity projection. To track extrachromosomal DNA dynamics, images were acquired every 30 min for 24 h or 48 h with 20x magnification. Time-lapse movies and snapshot images were generated and analyzed by Harmony High-Content Imaging and Analysis Software (PerkinElmer).
Size threshold analysis of single ecTag signal
In the live-cell ecDNA tracking experiment (Figure 3D), we noticed saturated fluorescence for some cells. All plated cells were captured using the same laser settings throughout 48 hours, and the images were acquired with 20x magnification. Theses lead to larger single signal foci as shown in Chr7 and MUC4 images in Fig. 3D. EcTag signal dissection performed using sgRNAs tagged with fewer PUFBS repeats (x15), and imaged as fixed cells with a higher magnification (100x) detected the upper single signal focus size in four ecDNA groups compared to Chr7 and MUC4 (Chr7 = 0.551 μm2; MUC4 = 0.680 μm2) (Supplementary Fig. 18).
Stepwise photobleaching analysis
PC3 cells stably expressing ecTag system targeting Chr7 or ecDNA were fixed in 4% PFA. Microscopy imaging was performed on Dragonfly equipped with a Zyla camera (Andor) and 488 laser. Each dot was bleached using a 100% 488 laser, and images were acquired every 100 ms for 60 seconds. To measure spot intensity, each spot was cropped and the average intensity of the cropped region was calculated. Background spot intensity, obtained from an area outside spot loci, was subtracted.
Live-cell imaging of nuclear bodies
HF3016 cells were plated into 384-well plates at 4,000 cells per well with NMGF medium containing 10% FBS. The next day, cells were transfected with 10 ng of dCas9, 20 ng of sgRNA, 20 ng of mRuby and 25 ng of reporter plasmid (Cajal body, #36906; PML body, #118360; Addgene) with Lipofectamine 3000 (Invitrogen, L3000015). After 24 h, the media was changed to fresh media. The next day, cells were stained with NucBlue Live ReadyProbe (Invitrogen, R37605) at 48h post-transfection. Live-cell imaging was performed on Opera Phenix High-Content Screening System (PerkinElmer) under 5% CO2 and 37 °C temperature. To avoid evaporation of medium, empty wells around the samples were filled with DPBS. All the images were acquired by taking 6 different focal planes, and analyzed as a maximum intensity projection. Images were acquired every 30 min overnight with 40x magnification. Image analysis for measuring distance between two signal foci was performed in Fiji.
Live-cell imaging of telomeres and dsRED-plasmids
For the imaging of telomeres, 1.2 x 105 cells of HF3016 were plated with 10% FBS-containing NMGF media into one of the well of glass bottom dish (Greiner Bio-One, 627870). The next day, cells were transfected with ecTag plasmids (100 ng of dCas9, 200 ng of Telomere-sgRNA-PUFBSa and 100 ng of Clover-PUFa) with Lipofectamine 3000 (Invitrogen, L3000015). After 24 h, the media was changed to fresh media. The next day, cells were stained with 1.0 μg/ml Hoechst 33342 (Invitrogen, H3070) at 48h post-transfection. Live-cell imaging was acquired on Dragonfly Confocal (Andor) using an iXon EMCCD camera with Leica HC PL APO 63x/1.47NA OIL CORR TIRF objective mounted on a Leica DMi8 inverted microscope under humidified 5% CO2 and 37 °C temperature. Clover images were acquired with a 150 mW solid state 488 nm laser and 525/50 nm BP emission filter. Z-series covering the full nuclei was acquired with 0.3 μm step size every 30 minutes. For the imaging of dsRED-plasmids, 1 x 105 cells of HF3016 were plated with 10% FBS-containing NMGF media into one of the well of glass bottom dish (Greiner Bio-One, 627870). The next day, cells were transfected with dsRED-expressing plasmid (80 ng) and ecTag plasmids (80 ng of dCas9, 160 ng of dsRED-sgRNA and 80 ng of Clover) with Lipofectamine 3000 (Invitrogen, L3000015). After 24h, the media was changed to fresh media. The next day, cells were stained with NucBlue Live ReadyProbe (Invitrogen, R37605) at 48h post-transfection. Live-cell imaging was acquired on Dragonfly under 5% CO2 and 37 °C temperature. All the images were acquired by taking 61 different focal planes, and analyzed as a maximum intensity projection. All the images were acquired by taking 74 different focal planes, and analyzed as a maximum intensity projection. Images were acquired every 30 min overnight with 63x magnification. Image analysis for measuring distance between two signal foci was performed in Fiji.
Dual-color ecDNA labeling experiment
3 x 105 cells of HF3016 were plated with 10% FBS-containing NMGF media into a glass-viewing area of confocal dish (VWR, 75856–742). The next day, cells were transfected with ecTag plasmids (400ng of dCas9, 200 ng of sgRNA-PUFBSa, 200 ng of sgRNA-PUFBSc, 200 ng Clover-PUFa and 200 ng mRuby-PUFc) with Lipofectamine 3000 (Invitrogen, L3000015). After 24 h, the media was changed to fresh media. After 48 h post-transfection, the dishes were briefly rinsed with 1X PBS three times and fixed with 4% PFA at RT for 15 min. The fixed cells were then rinsed with 1X PBS and counterstained with VECTASHIELD mounting medium with DAPI. Images were scanned under Leica STED 3X/DLS Confocal with oil-immersion objective (100x). As excitation laser, a 405 nm, a 488 nm, and a 561 nm were used. Z-stack acquired at 0.3–0.5 um step size was performed and all analysis conducted based on maximum intensity projection images of the 3D volume of the cells. Images were acquired and processed by LAS X software.
Neurosphere doubling time test
HF3016 cells were plated in individual wells of a 96-well plate and viable cells were quantified using CellTiter-Glo 3D Cell Viability Assay (Promega) in triplicate wells as per manufacturer’s instructions. Luminescence readings, which represented viable cells, were taken on a Cytation 3 Cell Imaging Multi-Mode Reader (BioTek).
Test for the dynamic change of ecDNA copy number distribution
To image ecTag-transfected cells every two days (Day2, Day4, and Day6), three sets of cells were prepared. 1.2 x 105 cells of HF3016 were plated with 10% FBS-containing NMGF media into a glass-viewing area of confocal dish (VWR, 75856–740). The next day, cells were transfected with ecTag plasmids (83.3 ng of dCas9, 83.3 ng of sgRNA and 83.3 ng of Clover) with Lipofectamine 3000 (Invitrogen, L3000015). After 24 h, the media was changed to fresh media. After two days post-transfection, the dishes of the first set were briefly rinsed with 1X PBS three times and fixed with 4% PFA at RT for 15 min. The fixed cells were then rinsed with 1X PBS and counterstained with VECTASHIELD mounting medium with DAPI. Images were scanned under Leica STED 3X/DLS Confocal with oil-immersion objective (100x). As excitation laser, a 405 nm, and a 488 nm were used. Z-stack acquired at 0.3–0.5 um step size was performed and all analysis conducted based on maximum intensity projection images of the 3D volume of the cells. Images were acquired and processed by LAS X software. The second set of cells were processed after four days post-transfection and the third set of cells were processed after six days post-transfection.
EGFR RNA FISH
Fluorescence (Quasar 670 Dye)-conjugated EGFR DesignReady probe was purchased from Biosearch Technologies. RNA FISH was performed on HF3016 cells transfected with ecTag-labeling components using manufacturer’s protocol. Images were scanned under Leica STED 3X/DLS Confocal with oil-immersion objective (100x). As excitation laser, a 405 nm, a 488 nm, and a 647 nm were used. Z-stack acquired at 0.3–0.5 um step size was performed and all analysis conducted based on maximum intensity projection images of the 3D volume of the cells. Images were acquired and processed by LAS X software.
Image analysis and data availability
Macro scripting of FIJI (ImageJ 1.53c) was used for automated image analysis. Speckle inspector function in the BioVoxxel plugin was used for counting copy number of fluorescent signals. Colocalization analysis was done using the JACoP plugin (Pearson’s correlation test), the Image Calulator, and the Analyze Particle function of Fiji. Fiji Macro codes used for image processing and analysis are available on GitHub (https://github.com/yehyehyeh/EDTB_imageanalysis).
Statistics (GraphPad)
All sample sizes and statistical methods were indicated in the corresponding figure or figure legends. All data was tested for normality using the D’Agostino-Pearson omnibus test. According to the results of the normality test, all data in this study that was not normally distributed was then run through the Mann-Whitney U test (for two groups). The homogeneity of variances between groups was determined by Fligner-Kileen test. All statistical tests are two-sided. All plots are shown with median, upper and lower quartiles. All statistical tests were performed in GraphPad Prism 8 or R version 4.0.2.
Supplementary Material
Significance.
Extrachromosomal DNA elements (ecDNA) are vehicles for oncogene amplification. The circular nature of ecDNA affords unique properties, such as mobility, and ecDNA-specific replication and segregation behavior. We uncovered fundamental ecDNA properties by tracking ecDNAs in live cells, highlighting uneven and random segregation, and ecDNA hubs that drive cargo gene transcription.
Acknowledgements
We gratefully acknowledge the contribution of the Microscopy Service and the Single Cell Biology Laboratory at The Jackson Laboratory for expert assistance with the work described in this publication. This work was supported by the following NIH grants: R01 CA237208, R21 NS114873, R21 CA256575, and R33 CA236681 (R.G.W.V), Cancer Center Support Grant P30 CA034196 (R.G.W.V; A.W.C.); R01 HG009900 and CCF 1955712 (A.W.C); Department of Defense W81XWH1910246 (R.G.W.V); and grants from the Musella Foundation, the B*CURED Foundation and the Brain Tumour Charity (R.G.W.V). E.Y. is supported by a basic research fellowship from the American Brain Tumor Association (BRF1800014). K.C.J. is the recipient of an American Cancer Society Fellowship (130984-PF-17–141-01-DMC).
Footnotes
Disclosures
R.G.W.V. is a scientific co-founder of and has received research funding from Boundless Bio, Inc, and a consultant for Stellanova Therapeutics.
References
- 1.Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nature Reviews Cancer 2012;12(7):487–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andor N, Graham TA, Jansen M, Xia LC, Aktipis CA, Petritsch C, et al. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nature medicine 2016;22(1):105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amirouchene-Angelozzi N, Swanton C, Bardelli A. Tumor Evolution as a Therapeutic Target. Cancer discovery 2017;7:805–817 doi 10.1158/2159-8290.cd-17-0343. [DOI] [PubMed] [Google Scholar]
- 4.Li Y, Roberts ND, Wala JA, Shapira O, Schumacher SE, Kumar K, et al. Patterns of somatic structural variation in human cancer genomes. Nature 2020;578(7793):112–21 doi 10.1038/s41586-019-1913-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turner KM, Deshpande V, Beyter D, Koga T, Rusert J, Lee C, et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 2017;543(7643):122–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nathanson DA, Gini B, Mottahedeh J, Visnyei K, Koga T, Gomez G, et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science 2014;343(6166):72–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verhaak RGW, Bafna V, Mischel PS. Extrachromosomal oncogene amplification in tumour pathogenesis and evolution. Nature reviews Cancer 2019;19(5):283–8 doi 10.1038/s41568-019-0128-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Decarvalho AC, Kim H, Poisson LM, Winn ME, Mueller C, Cherba D, et al. Discordant inheritance of chromosomal and extrachromosomal DNA elements contributes to dynamic disease evolution in glioblastoma. Nature genetics 2018;50(5):708–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim H, Nguyen NP, Turner K, Wu S, Gujar AD, Luebeck J, et al. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat Genet 2020;52(9):891–7 doi 10.1038/s41588-020-0678-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wahl GM. The Importance of Circular DNA in Mammalian Gene Amplification. Cancer Research 1989;49(6):1333–40. [PubMed] [Google Scholar]
- 11.Shimizu N, Itoh N, Utiyama H, Wahl GM. Selective entrapment of extrachromosomally amplified DNA by nuclear budding and micronucleation during S phase. J Cell Biol 1998;140(6):1307–20 doi 10.1083/jcb.140.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lundberg G, Rosengren AH, Hakanson U, Stewenius H, Jin Y, Stewenius Y, et al. Binomial mitotic segregation of MYCN-carrying double minutes in neuroblastoma illustrates the role of randomness in oncogene amplification. PLoS One 2008;3(8):e3099 doi 10.1371/journal.pone.0003099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen B, Gilbert Luke A, Cimini Beth A, Schnitzbauer J, Zhang, Li G-W, et al. Dynamic Imaging of Genomic Loci in Living Human Cells by an Optimized CRISPR/Cas System. Cell 2013;155(7):1479–91 doi 10.1016/j.cell.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang H, Xu X, Nguyen CM, Liu Y, Gao Y, Lin X, et al. CRISPR-Mediated Programmable 3D Genome Positioning and Nuclear Organization. Cell 2018;175(5):1405–17.e14 doi 10.1016/j.cell.2018.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang H, Nakamura M, Abbott TR, Zhao D, Luo K, Yu C, et al. CRISPR-mediated live imaging of genome editing and transcription. Science 2019;365(6459):1301–5 doi 10.1126/science.aax7852. [DOI] [PubMed] [Google Scholar]
- 16.Qin P, Parlak M, Kuscu C, Bandaria J, Mir M, Szlachta K, et al. Live cell imaging of low- and non-repetitive chromosome loci using CRISPR-Cas9. Nature Communications 2017;8(1):14725 doi 10.1038/ncomms14725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clow PA, Jillette N, Zhu JJ, Cheng AW. CRISPR-mediated Multiplexed Live Cell Imaging of Nonrepetitive Genomic Loci. bioRxiv 2020:2020.03.03.974923 doi 10.1101/2020.03.03.974923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bailey C, Shoura MJ, Mischel PS, Swanton C. Extrachromosomal DNA—relieving heredity constraints, accelerating tumour evolution. Annals of Oncology 2020;31(7):884–93 doi 10.1016/j.annonc.2020.03.303. [DOI] [PubMed] [Google Scholar]
- 19.Johnson KC, Anderson KJ, Courtois ET, Gujar AD, Barthel FP, Varn FS, et al. Single-cell multimodal glioma analyses identify epigenetic regulators of cellular plasticity and environmental stress response. Nat Genet 2021;53(10):1456–68 doi 10.1038/s41588-021-00926-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014;344(6190):1396–401 doi 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh DK, Ku CJ, Wichaidit C, Steininger RJ III, Wu LF, Altschuler SJ. Patterns of basal signaling heterogeneity can distinguish cellular populations with different drug sensitivities. Molecular systems biology 2010;6(1):369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hahn PJ. Molecular biology of double‐minute chromosomes. Bioessays 1993;15(7):477–84. [DOI] [PubMed] [Google Scholar]
- 23.Singer MJ, Mesner LD, Friedman CL, Trask BJ, Hamlin JL. Amplification of the human dihydrofolate reductase gene via double minutes is initiated by chromosome breaks. Proceedings of the National Academy of Sciences 2000;97(14):7921–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cai M, Zhang H, Hou L, Gao W, Song Y, Cui X, et al. Inhibiting homologous recombination decreases extrachromosomal amplification but has no effect on intrachromosomal amplification in methotrexate‐resistant colon cancer cells. International journal of cancer 2019;144(5):1037–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shoshani O, Brunner SF, Yaeger R, Ly P, Nechemia-Arbely Y, Kim DH, et al. Chromothripsis drives the evolution of gene amplification in cancer. Nature 2020:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng AW, Jillette N, Lee P, Plaskon D, Fujiwara Y, Wang W, et al. Casilio: a versatile CRISPR-Cas9-Pumilio hybrid for gene regulation and genomic labeling. Cell Research 2016;26(2):254–7 doi 10.1038/cr.2016.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deshpande V, Luebeck J, Nguyen N-PD, Bakhtiari M, Turner KM, Schwab R, et al. Exploring the landscape of focal amplifications in cancer using AmpliconArchitect. Nature Communications 2019;10(1):392 doi 10.1038/s41467-018-08200-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Concordet J-P, Haeussler M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Research 2018;46(W1):W242–W5 doi 10.1093/nar/gky354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cleveland DW, Mao Y, Sullivan KF. Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell 2003;112(4):407–21 doi 10.1016/s0092-8674(03)00115-6. [DOI] [PubMed] [Google Scholar]
- 30.Hsiung CCS, Morrissey CS, Udugama M, Frank CL, Keller CA, Baek S, et al. Genome accessibility is widely preserved and locally modulated during mitosis. Genome Res 2015;25(2):213–25 doi 10.1101/gr.180646.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanda T, Sullivan KF, Wahl GM. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Current biology : CB 1998;8(7):377–85 doi 10.1016/s0960-9822(98)70156-3. [DOI] [PubMed] [Google Scholar]
- 32.Liesche C, Grussmayer KS, Ludwig M, Worz S, Rohr K, Herten DP, et al. Automated Analysis of Single-Molecule Photobleaching Data by Statistical Modeling of Spot Populations. Biophys J 2015;109(11):2352–62 doi 10.1016/j.bpj.2015.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mearini G, Nielsen PE, Fackelmayer FO. Localization and dynamics of small circular DNA in live mammalian nuclei. Nucleic acids research 2004;32(8):2642–51 doi 10.1093/nar/gkh587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu S, Turner KM, Nguyen N, Raviram R, Erb M, Santini J, et al. Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature 2019;575(7784):699–703 doi 10.1038/s41586-019-1763-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morton AR, Dogan-Artun N, Faber ZJ, MacLeod G, Bartels CF, Piazza MS, et al. Functional Enhancers Shape Extrachromosomal Oncogene Amplifications. Cell 2019;179(6):1330–41.e13 doi 10.1016/j.cell.2019.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Q, Sawyer IA, Sung M-H, Sturgill D, Shevtsov SP, Pegoraro G, et al. Cajal bodies are linked to genome conformation. Nature Communications 2016;7(1):10966 doi 10.1038/ncomms10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nature Reviews Molecular Cell Biology 2007;8(12):1006–16 doi 10.1038/nrm2277. [DOI] [PubMed] [Google Scholar]
- 38.Gall JG, Bellini M, Wu Za, Murphy C. Assembly of the Nuclear Transcription and Processing Machinery: Cajal Bodies (Coiled Bodies) and Transcriptosomes. Molecular Biology of the Cell 1999;10(12):4385–402 doi 10.1091/mbc.10.12.4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kiesslich A, von Mikecz A, Hemmerich P. Cell cycle-dependent association of PML bodies with sites of active transcription in nuclei of mammalian cells. Journal of structural biology 2002;140(1–3):167–79 doi 10.1016/s1047-8477(02)00571-3. [DOI] [PubMed] [Google Scholar]
- 40.Maass PG, Barutcu AR, Weiner CL, Rinn JL. Inter-chromosomal Contact Properties in Live-Cell Imaging and in Hi-C. Mol Cell 2018;69(6):1039–45 e3 doi 10.1016/j.molcel.2018.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lange JT, Chen CY, Pichugin Y, Xie L, Tang J, Hung KL, et al. Principles of ecDNA random inheritance drive rapid genome change and therapy resistance in human cancers. bioRxiv 2021:2021.06.11.447968 doi 10.1101/2021.06.11.447968. [DOI] [Google Scholar]
- 42.Shibata Y, Kumar P, Layer R, Willcox S, Gagan JR, Griffith JD, et al. Extrachromosomal microDNAs and chromosomal microdeletions in normal tissues. Science 2012;336(6077):82–6 doi 10.1126/science.1213307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y, Wang M, Djekidel MN, Chen H, Liu D, Alt FW, et al. eccDNAs are apoptotic products with high innate immunostimulatory activity. Nature 2021. doi 10.1038/s41586-021-04009-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Helmsauer K, Valieva ME, Ali S, Chamorro Gonzalez R, Schopflin R, Roefzaad C, et al. Enhancer hijacking determines extrachromosomal circular MYCN amplicon architecture in neuroblastoma. Nat Commun 2020;11(1):5823 doi 10.1038/s41467-020-19452-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koche RP, Rodriguez-Fos E, Helmsauer K, Burkert M, MacArthur IC, Maag J, et al. Extrachromosomal circular DNA drives oncogenic genome remodeling in neuroblastoma. Nature Genetics 2020;52(1):29–34 doi 10.1038/s41588-019-0547-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu Y, Gujar AD, Wong CH, Tjong H, Ngan CY, Gong L, et al. Oncogenic extrachromosomal DNA functions as mobile enhancers to globally amplify chromosomal transcription. Cancer Cell 2021;39(5):694–707 e7 doi 10.1016/j.ccell.2021.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hung KL, Yost KE, Xie L, Wu S, Lange JT, Duffy CV, et al. EcDNA hubs drive cooperative intermolecular oncogene expression. bioRxiv 2020:2020.11.19.390278 doi 10.1101/2020.11.19.390278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chong S, Dugast-Darzacq C, Liu Z, Dong P, Dailey GM, Cattoglio C, et al. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 2018;361(6400). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cho WK, Spille JH, Hecht M, Lee C, Li C, Grube V, et al. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 2018;361(6400):412–5 doi 10.1126/science.aar4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 2018;361(6400):eaar3958 doi 10.1126/science.aar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dekker J, Mirny L. The 3D Genome as Moderator of Chromosomal Communication. Cell 2016;164(6):1110–21 doi 10.1016/j.cell.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.