

Abstract
Insulin resistance is the pivotal pathogenic component of many metabolic diseases, including type 2 diabetes mellitus, and is defined as a state of reduced responsiveness of insulin-targeting tissues to physiological levels of insulin. Although the underlying mechanism of insulin resistance is not fully understood, several credible theories have been proposed. In this review, we summarize the functions of insulin in glucose metabolism in typical metabolic tissues and describe the mechanisms proposed to underlie insulin resistance, that is, ectopic lipid accumulation in liver and skeletal muscle, endoplasmic reticulum stress, and inflammation. In addition, we suggest potential therapeutic strategies for addressing insulin resistance.
Keywords: Diabetes mellitus, type 2; Insulin resistance; Metabolic syndrome; Therapeutics
INTRODUCTION
Evolution has equipped animals with highly efficient means of overcoming nutrient scarcity, which involves the synthesis of complex molecules and storage of redeemable energy sources when nutrients are plentiful. Insulin is a pivotal regulator of the transition from nutrient production to storage under such conditions. After nutrient intake, plasma glucose levels reach a threshold level that stimulates insulin secretion by pancreatic β-cells. Under normal circumstances, this insulin promotes carbohydrate uptake at key storage and consumption sites, such as in adipose tissue and skeletal muscle, in which carbohydrates and proteins are stored as lipids. However, over-nourished, sedentary modern lifestyles disrupt this system and can cause serious medical problems, which include metabolic syndrome, obesity, type 2 diabetes mellitus (T2DM), and cardiovascular disease.
Insulin resistance is defined physiologically as a state of reduced responsiveness in insulin-targeting tissues to high physiological insulin levels and is considered the pathogenic driver of many modern diseases, including metabolic syndrome, nonalcoholic fatty liver disease (NAFLD), atherosclerosis, and T2DM [1]. Insulin resistance precedes non-physiologic elevated plasma glucose levels, which is the primary clinical symptom of T2DM. In the prediabetic condition, insulin levels increase to meet normal insulin requirements leading to chronic hyperinsulinemia, hyperglycemia-induced β-cell failure, and eventually to T2DM [2].
Although the mechanism of insulin resistance has not been fully established, several theories are generally considered reasonable. This review summarizes the function of insulin in glucose metabolism in metabolic tissues, such as liver, skeletal muscle, and adipose tissue, and describes several putative mechanisms of insulin resistance, including the ectopic accumulation of lipids in liver and skeletal muscle. In addition, we suggest potential therapeutic strategies for fat-induced insulin resistance that target ectopic fat accumulation in liver and promote energy consumption by skeletal muscle.
INSULIN SIGNALING AND INSULIN RESISTANCE
Insulin signaling
In the fasted state, the liver secretes glucose into blood to maintain euglycemia and provide fuel for glucose-consuming tissues. This process is called hepatic glucose production (HGP), and involves the breakdown of hepatic glycogen (glycogenolysis) and de novo synthesis of glucose (gluconeogenesis) using fatty acids and glycerol derived from adipose tissues [3]. After food intake, insulin secreted by pancreatic β-cells promotes anabolism and suppresses catabolic programs. During glucose metabolism, insulin stimulates several glucose-consuming tissues, such as skeletal muscle and adipose tissues, to uptake glucose and then promotes the syntheses of glycogen and lipid in liver, skeletal muscle, and adipose tissue [4]. In addition, insulin suppresses HGP by inhibiting the expressions of gluconeogenic genes and lipolysis in adipose tissue [5]. Insulin also suppresses glucagon secretion from pancreatic α-cells [6,7] and reduces appetite via the central nervous system. In this review, we focus on the role of insulin in glucose metabolism in skeletal muscle, liver, and adipose tissue.
The intracellular functions of insulin are mediated through insulin receptor tyrosine kinase (IRTK) (Fig. 1). When insulin binds to the extracellular domain of IRTK, it induces a conformational change that results in the autophosphorylation of IRTK tyrosine residues and the subsequent activation of phosphotyrosine-binding proteins such as insulin receptor substrate (IRS), growth factor receptor-bound protein-2 (GRB-2), GRB-10, SHC-transforming protein (SHC), and SH2B adapter protein-2 (SH2B-2) [8]. The effects of insulin on glucose and lipid metabolism are mainly mediated by the IRTK-induced phosphorylation of IRS, which then recruits phosphatidylinositol-3-OH kinase (PI3K) and catalyzes the production of phosphatidylinositol-3,4,5-trisphosphate (PIP3) from phosphatidylinositol-4,5-bisphosphate (PIP2). After being recruited to the plasma membrane by PIP3, Akt is activated (phosphorylated) by 3-phosphoinositide-dependent kinase-1 (PDK1) and mechanistic target of rapamycin complex 2 (mTORC2) [9] and then phosphorylates various downstream substrates in metabolic tissues, including skeletal muscle, liver, and adipose tissue, which elicit insulin-induced nutrient reservation in these tissues.
Fig. 1.
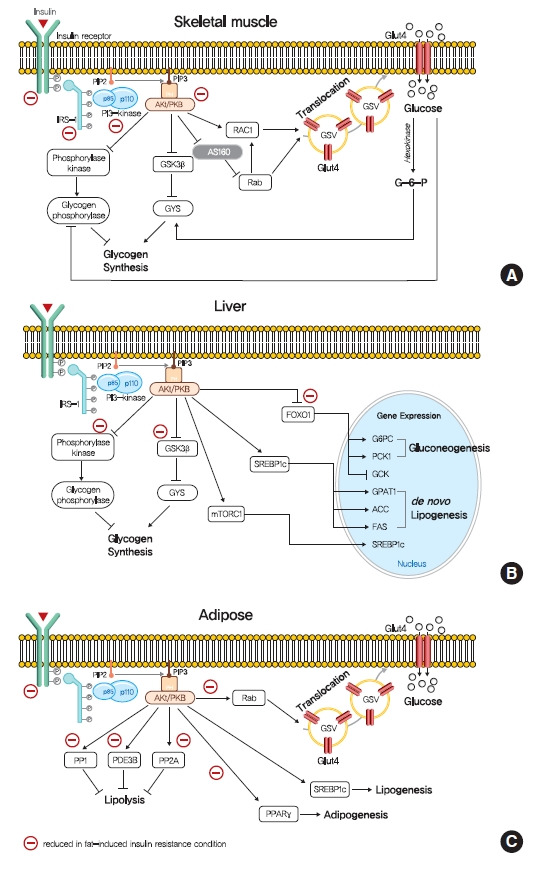
Role of insulin signaling in liver, skeletal muscle, and adipose tissue. (A) Insulin binds to insulin receptor tyrosine kinase (IRTK), and activate insulin receptor substrate-1 (IRS-1), which recruits phosphatidylinositol-3-OH kinase (PI3K) and activate Akt. In skeletal muscle, Akt promotes glucose uptake via the translocation of glucose transporter type 4 (GLUT4) storage vesicles (GSVs) to the plasma membrane, which is mediated by inactivation of GTPase-activating protein (GAP) AKT substrate of 160 kDa (AS160) and promotion of GTP-bound form of Ras-related C3 botulinum toxin substrate 1 (RAC1). Insulin stimulate glycogen synthesis via glycogen synthase kinase 3 (GSK3)-inhibition mediated glycogen synthease (GYS) activation and glycogen phosphorylase inactivation via the dephosphorylation of phosphorylase kinase. (B) In liver, Akt decreases gluconeogenesis by suppressing of forkhead box O1 (FOXO1)-mediated gluconeogenic gene expressions. In addition, insulin increases hepatic glycogen synthesis by regulating GYS2 and glycogen phosphorylase through GSK3 and protein phosphatase 1 (PP1). Also, insulin increases hepatic de novo lipogenesis by upregulating sterol regulatory element-binding protein 1c (SREBP-1c). (C) In white adipocyte, insulin suppress lipolysis, which in turn suppresses hepatic glucose production by reducing gluconeogenic substrates, which is believed to be mediated by phosphodiesterase 3B (PDE3B), PP1, and protein phosphatase-2A (PP2A). Insulin also promotes glucose transport, lipogenesis, and adipogenesis. PIP2, phosphatidylinositol-4,5-bisphosphate; PIP3, phosphatidylinositol-3,4,5-trisphosphate; PKB, protein kinase B; mTORC1, mechanistic target of rapamycin complex 1; G6PC, glucose-6-phosphatase; PCK1, phosphoenolpyruvate carboxykinase 1 (PEPCK); GCK, glucokinase; GPAT1, glycerol-3-phosphate acyltransferase 1; ACC, acetyl-CoA carboxylase; FAS, fatty acid synthase.
In skeletal muscle, insulin signaling promotes glucose uptake and net glycogen synthesis (Fig. 1A). Insulin increases glucose transport activity via the highly coordinated translocation and fusion of glucose transporter type 4 (GLUT4) storage vesicles (GSVs) to the plasma membrane in skeletal muscle [10]. After being activated by insulin signaling, Akt inactivates AS160 (GTPase-activating protein [GAP] AKT substrate of 160 kDa, also known as TBC1D4), which activates small Rab GTPase protein switches that control vesicle trafficking [10]. Insulin-induced Akt also promotes the guanosine triphosphate (GTP)-bound form of Ras-related C3 botulinum toxin substrate 1 (RAC1), which promotes GLUT4 translocation by inducing cortical actin reorganization [11]. On the other hand, insulin also regulates net glycogen synthesis in skeletal muscle by suppressing glycogenolysis and promoting glycogen synthesis. Insulin signaling promotes the activity of glycogen synthase (GYS) via the phosphorylation of glycogen synthase kinase 3 (GSK3) by Akt [12] and the activation of protein phosphatase 1 (PP1) to promote the dephosphorylation of GYS [13]. In addition, insulin regulates glycogen phosphorylase activity via the dephosphorylation of phosphorylase kinase [14].
Insulin in liver activates IRTK, which phosphorylates IRS1 and IRS2, and ultimately activates Akt2 [15], which decreases HGP, promotes glycogen synthesis, and transcriptionally activates lipogenesis (Fig. 1B). The primary function of hepatic insulin signaling is to decrease HGP by repressing gluconeogenesis mediated by the Akt-induced phosphorylation of forkhead box O1 (FOXO1) [15], which excludes FOXO1 from the nucleus, and thus, prevents the transcriptional activations of gluconeogenic gene expressions, such as glucose-6-phosphatase (G6PC) and phosphoenolpyruvate carboxylase (PEPCK) [15,16]. In addition to inhibiting gluconeogenic gene expressions, insulin suppresses hepatic gluconeogenesis by inhibiting adipocyte lipolysis, which reduces levels of gluconeogenesis substrates in liver [17]. Furthermore, besides suppressing HGP, insulin increases hepatic glycogen synthesis by regulating GYS (especially GYS2 in liver) and glycogen phosphorylase through GSK3 and PP1, as occurs in skeletal muscle [18]. Also, insulin activates lipid anabolism by upregulating sterol regulatory element-binding protein 1c (SREBP-1c; a master transcriptional regulator of hepatic de novo lipogenesis) and subsequently, enhancing the transcriptions of several lipogenic genes, including acetyl-CoA carboxylase 1 (ACC1), fatty acid synthase (FAS), and glycerol-3-phosphate acyltransferase 1 (GPAT1) [19,20].
The most important physiological function of insulin in white adipocyte tissue is to suppress lipolysis, which in turn suppresses HGP by reducing gluconeogenic substrates (Fig. 1C) [21]. The mechanism responsible for insulin-induced lipolysis suppression is not fully understood, though it is believed to be mediated by phosphodiesterase 3B (PDE3B) through reduced cyclic adenosine monophosphate (cAMP)-dependent protein kinase A (PKA) activity [22]. In addition, PP1 and protein phosphatase-2A (PP2A) appear to mediate PI3K-dependent insulin-induced lipolysis suppression through the dephosphorylations of lipolytic regulatory proteins [23,24]. Although insulin promotes glucose transport by signaling the phosphorylation of targets involved in vesicle tethering, docking, and fusion, its contribution to whole-body glucose disposal is comparatively minor [25]. Insulin also promotes lipogenesis in white adipose tissue by activating SREBP-1c, signaling the translocations of glucose or fatty acid transport proteins (FATPs), promoting fatty acid esterification [26], and stimulating adipogenesis through the transcription factor peroxisome proliferator-activated receptor-γ (PPARγ) [27].
Insulin resistance
As mentioned above, insulin resistance is physiologically defined as an inability of some type of tissues to respond to normal insulin levels, and thus, higher than normal levels of insulin are required to maintain the normal functions of insulin. Notably, the glucose-regulating effects of insulin such as the suppressions of HGP and lipolysis, cellular uptake of plasma glucose, and net glycogen synthesis are not observed in insulin resistant tissues at normal plasma levels [5]. Since skeletal muscle is a quantitatively central tissue for insulin-stimulated glucose disposal, and liver and adipose tissue are qualitatively the critical sites for glucose-induced insulin signaling, these tissues are considered central to the understanding of the mechanisms responsible for insulin resistance.
Insulin resistance in skeletal muscle
Insulin-stimulated glucose consumption is largely conducted in skeletal muscle, and thus, muscular insulin resistance could affect whole-body metabolism [28]. Muscle-specific insulin resistance by the muscle-specific deletion of IRTK or GLUT4 increased the susceptibility to hepatic steatosis, and increase adiposity in animal models [29,30]. Many molecular studies have demonstrated that insulin-stimulated muscle glucose uptake is highly susceptible to insulin resistance attributed to impaired GLUT4 translocation. However, the translocation of GLUT4 to the plasma membrane and glucose transport in T2DM are stimulated by hypoxia or exercise via the AMP-activated protein kinase (AMPK)-mediated regulation of GSV translocation [31,32], which suggests that defects of glucose transport in insulin resistance are abnormalities in the insulin signaling pathway rather than defects in the transport system itself. Furthermore, insulin resistance in skeletal muscle may be caused by defects at the proximal level of insulin signaling, for example, in the activities of IRTK, IRS1, PI3K, and AKT (Fig. 1A). In addition, in obese mice and obese/diabetic humans, the tyrosine kinase activity of IRTK is diminished in skeletal muscle, which supports this suggestion [33]. In addition, IRS1 tyrosine phosphorylation and IRS1-associated PI3K activity were found to be diminished in insulin resistant skeletal muscle [34].
Insulin resistance in liver and adipose tissue
The liver critically controls postprandial carbohydrate levels by suppressing HGP and stimulating the deposition of glucose as glycogen and is the primary source of glucose production during fasting [35]. In T2DM patients, insulin cannot regulate hepatic glycogen synthesis or glucose production, and increased hepatic gluconeogenesis is the primary cause of fasting hyperglycemia in T2DM [36,37]. Defective suppression of hepatic gluconeogenesis in insulin resistance is largely associated with lipolysis defects in adipose tissue and the de-suppression of FOXO1 transcription factor in liver [35]. Meanwhile, insulin resistance is also associated with defects in the insulin-induced stimulation of glycogen synthesis, for example, T2DM patients have lower fasting and postprandial hepatic glycogen content [38]. Furthermore, hepatic insulin resistance reduces the amplitude of hepatic glycogen metabolism induced by fasting and feeding [36].
Selective insulin resistance
Intriguingly, not all insulin functions are less responsive in the presence of insulin resistance. Some insulin pathways in hyperinsulinemia are highly responsive to insulin, a phenomenon referred to as selective insulin resistance or pathway-selective insulin responsiveness [39]. In the case of hepatic insulin resistance, insulin fails to suppress HGP but stimulates lipogenesis, which results in hyperglycemia, hyperlipidemia, and hepatic steatosis [39]. The underlying mechanism of selective insulin resistance has not been established but several hypotheses have been suggested. One involves glucose disposal potential differences in the substrate specificities of AKT phosphorylation between gluconeogenesis and lipogenesis [40]. Akt Ser473 phosphorylation may activate some AKT substrates related to gluconeogenesis, such as FOXO, and these activations might be suppressed in a background of insulin resistance, while other Akt substrates, such as GSK3β and tuberous sclerosis complex 2 (TSC2), which requires AKT phosphorylation at Thr308, might not be disrupted [41,42]. Another possible mechanism of selective insulin resistance in liver involves the different intrinsic sensitivities of insulin-induced SERBP-1c activation and gluconeogenesis suppression, which suggests that these functions require specific insulin levels. Recent studies have shown that insulin-regulated hepatic lipogenesis, but not gluconeogenesis, is mediated by stabilizing pleckstrin homology domain leucine-rich repeat protein phosphatase-2 (PHLPP-2), which terminates insulin-stimulated Akt activity [43,44]. These reports show that a high-fat diet (HFD) decreases PHLPP-2 stability, which delayed Akt dephosphorylation and then induces prolonged Akt activation in late stage after insulin pulse [43,44]. Since elevated Akt activity in the early postprandial stage is required to reduce HGP and Akt activity in the late stage is sufficient to increase de novo lipogenesis but not HGP [40,43,44], unterminated prolonged Akt activity might enhance de novo lipogenesis but not suppress HGP. The other hypothesis posits that selective insulin resistance in liver is caused by insulin-independent lipogenesis. Lipogenesis induction by nutrients is also mediated by the carbohydrate response element-binding protein (ChREBP), PPARγ coactivator 1-β, and liver X receptor-mediated SREBP-1c [45-47], and these alternative pathways of lipogenesis have been shown to be activated by fructose and monosaccharides [48,49].
MECHANISMS OF INSULIN RESISTANCE
Many epidemiological studies have concluded that obesity is a leading risk factor of T2DM, and reportedly, obese people are up to 80 times more likely to develop T2DM [50]. Furthermore, it has been shown an inverse correlation exists between plasma fatty acid concentration and insulin sensitivity in age and body mass index matched controls [51]. These results and others show that excess lipid content caused by obesity is a primary cause of insulin resistance and T2DM. Many theories have been proposed for the mechanism responsible for the development of insulin resistance due to excess lipid availability.
The glucose-fatty acid cycle (the Randle cycle)
In 1963, Randle et al. [52] hypothesized that lipid-induced insulin resistance in skeletal muscle characterized by a defect in glucose uptake is attributed to limited insulin-stimulated glucose utilization caused by increased fatty acid oxidation, which is referred to as the glucose-fatty acid or Randle cycle. It was asserted that increased fatty acid oxidation in the obese impaired in the use of glucose as a source of fuel by inhibiting the activities of key glycolytic enzymes. According to Randle et al. [52], fatty acid oxidation might increase mitochondrial acetyl-CoA levels through β-oxidation and subsequently inactivate pyruvate dehydrogenase, which in turn, would increase intracellular citrate levels and inhibit phosphofructokinase (a key glycolytic enzyme), and lead to the accumulation of intramyocellular glucose-6-phosphate, which inhibits hexokinase activity and causes the accumulation of intramyocellular glucose and reduced glucose uptake (Fig. 2). Furthermore, some experimental results have consistently shown that infusions of fatty acids decrease myocellular glucose utilization and increase intramyocellular glucose-6-phosphate concentrations in rat heart and diaphragm muscles [53-55].
Fig. 2.
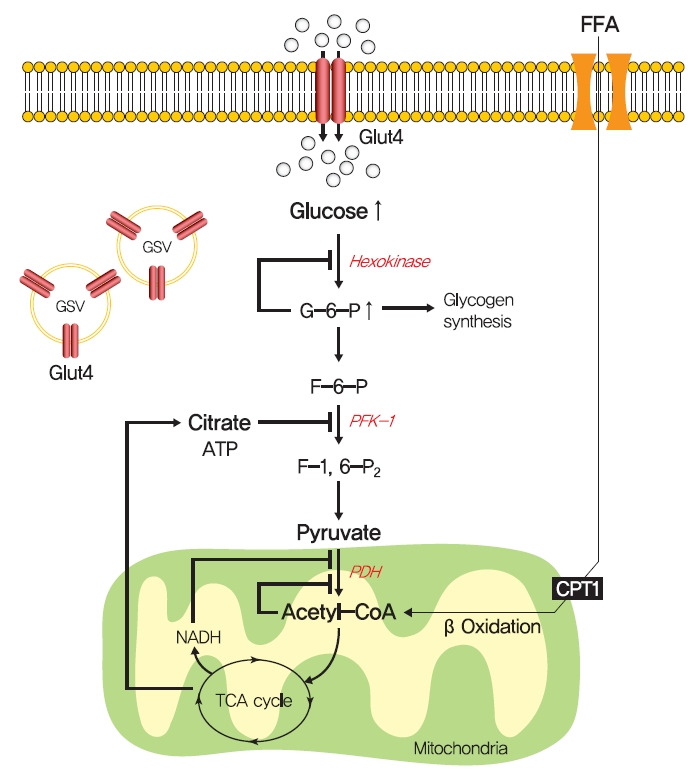
The glucose-fatty acid cycle hypothesis in insulin resistance. Randle et al. [52] proposed that lipid-induced insulin resistance in skeletal muscle is attributed to limited insulin-stimulated glucose utilization caused by increased fatty acid oxidation. Fatty acid oxidation might increase mitochondrial acetyl-CoA levels and subsequently inactivate pyruvate dehydrogenase (PDH), which in turn, would increase intracellular citrate levels and inhibit phosphofructokinase 1 (PFK-1), and lead to the accumulation of intramyocellular glucose-6-phosphate (G6-P), which inhibits hexokinase activity and causes the accumulation of intramyocellular glucose and reduced glucose uptake. FFA, free fatty acid; GLUT4, glucose transporter type 4; GSV, GLUT4 storage vesicle; ATP, adenosine triphosphate; CPT1, carnitine palmitoyltransferase 1; NADH, reduced nicotinamide adenine dinucleotide; TCA, trichloroacetic acid.
The glucose-fatty acid cycle shows that glucose-6-phosphate and glycogen synthesis levels should be increased by fatty acid and that glycolysis should be inhibited. However, exposure to high plasma fatty acid concentrations caused insulin resistance and reduced glucose-6-phosphate concentrations in the muscle of healthy individuals [56]. In addition, insulin-stimulated muscle glycogen synthesis and glucose oxidation were found to be suppressed in T2DM patients with chronic insulin resistance [57,58]. These studies demonstrate that the glucose-fatty acid cycle cannot fully explain all impairments of insulin resistance.
Hexosamine biosynthesis pathway
Although the glucose-fatty acid cycle does not satisfactorily explain lipid-induced insulin resistance, evidence indicates that muscular acetyl-CoA and citrate levels are elevated in line with increased fat oxidation, which suggests another pathway regulates glucose transport independently of glucose-6-phosphate levels in muscle (Fig. 3). The hexosamine biosynthesis pathway (HBP) offers another explanation for the development of insulin resistance. Fructose-6-phosphate is mainly produced from glucose-6-phosphate and is preferentially metabolized to fructose-1,6-bisphosphate during glycolysis, but approximately 5% of fructose-6-phosphate is converted to glucosamine-6-phosphate by glutamine:fructose-6-phosphate amidotransferase (GFAT), the rate-limiting enzyme of HBP [59]. Glucosamine-6-phosphate is converted to uridine 5’-diphosphate N-acetylglucosamine (UDP-GlcNAc), which serves as the donor sugar nucleotide for the glycosylation and O-GlcNAcylation of lipids and proteins [59], and these modifications, especially O-GlcNAcylation, could affect targeted proteins by regulating gene expressions or enzyme activities [60].
Fig. 3.

Hexosamine biosynthesis pathway (HBP) in insulin resistance. In HBP, fructose-6-phosphate (F-6-P) is converted to glucosamine-6-phosphate (Glucosamine-6-P) by glutamine:fructose-6-phosphate amidotransferase (GFAT), and glucosamine-6-P is converted to uridine 5’-diphosphate N-acetylglucosamine (UDP-GlcNAc), which serves as the donor sugar nucleotide for the O-GlcNAcylation of lipids and proteins. O-GlcNAcylation could affect target proteins by regulating gene expressions or enzyme activities. Insulin signaling pathway components, mammalian uncoordinated-18c (Munc18-c), and forkhead box O1 (FOXO1) are modified with O-GlcNAc. PIP2, phosphatidylinositol-4,5-bisphosphate; PIP3, phosphatidylinositol-3,4,5-trisphosphate; IRS-1, insulin receptor substrate-1; PKB, protein kinase B; GSK3, glycogen synthase kinase 3; GSV, GLUT4 storage vesicle; GLUT4, glucose transporter type 4; GYS, glycogen synthease; OGT, O-GlcNAc transferase; OGA, O-GlcNAcase; ATP, adenosine triphosphate; PFK-1, phosphofructokinase 1; PDH, pyruvate dehydrogenase; NADH, reduced nicotinamide adenine dinucleotide; TCA, trichloroacetic acid.
Thirty years ago, Marshall et al. [61] first suggested O-GlcNAcylation modulated insulin sensitivity and showed that the effects of glucose-induced insulin resistance could be blocked by inhibiting GFAT using amidotransferase inhibitors such as O-diazoacetyl-L-serine (azaserine) or 6-diazo-5-oxonorleucine (DON). Subsequently, it was established that glucosamine induces insulin resistance [62,63]. Furthermore, hyperglycemia and hyperinsulinemia reportedly increase UDP-GlcNAc levels in skeletal muscle [64,65], and O-GlcNAc levels have been reported to be elevated in the corneas, pancreases, and skeletal muscles of diabetic or insulin resistant rats [66,67]. Also, GFAT overexpression in adipose tissue or skeletal muscle developed whole-body insulin resistance characterized by a reduction in glucose disposal in mice [68]. Recently, skeletal muscle-specific O-GlcNAc transferase (OGT) knockout mice on a HFD were reported to have low plasma glucose levels and glucose tolerances [69]. Moreover, the overexpression of O-GlcNAcase (OGA), which removes O-GlcNAc from proteins, significantly improved whole-body glucose tolerance and insulin sensitivity in db/db mice, and O-(2-acetamido-2-deoxy-D-glucopyranosylidene) amino-N-phenylcarbamate (PUGNAc) (an OGA inhibitor) suppressed insulin-mediated glucose uptake in adipocytes [70,71]. Also, single-nucleotide polymorphisms in meningioma-expressed antigen 5 (MGEA5), a human OGA homolog, were associated with T2DM onset in Pima Indians and Mexican Americans [72,73].
The molecular mechanisms of HBP in insulin resistance have not been fully established, while O-GlcNAcylation provides the most acceptable mechanism of HBP in insulin resistance. O-GlcNAcylation of proteins has been suggested to compete with phosphorylation for sites, which could regulate the protein activity and signaling transduction [74]. In particular, insulin signaling pathway components, such as IRS-1/2, PI3K, PDK1, and Akt, are modified with O-GlcNAc [75,76]. In addition to these proximal components of insulin signaling, mammalian uncoordinated-18c (Munc18-c), a protein essential for insulin-stimulated GLUT4 translocation, was reported to be O-GlcNAc modified in a background of glucosamine-induced insulin resistance, and FOXO1, a master transcriptional factor for gluconeogenic genes in liver, was also O-GlcNacylated in diabetes [77,78].
Debate continues despite cumulating evidence regarding the role of HBP in insulin resistance. Robinson et al. [79] showed that adenovirus-induced OGA overexpression and OGT knockdown failed to restore impaired Akt activation by insulin stimulation and prevent glucose-induced insulin resistance in 3T3L1 adipocytes, and in another study, treatment with the OGA inhibitor 1,2-dideoxy-2’-propyl-α-ᴅ-glucopyranoso-[2,1, D]-Δ2’-thiazoline (NButGT) did not induce insulin resistance characterized by defective glucose uptake and Akt phosphorylation in 3T3-L1 adipocytes [80]. Taken together, discrepancies between the roles of HBP in vivo and in vitro suggest that more thorough systematic approaches using in vivo systems are needed to determine the role of HBP and the mechanism involved in insulin resistance.
Ectopic lipid accumulation
Obesity is an obvious and significant risk factor of T2DM, but numerous studies have shown that ectopic lipid accumulation in peripheral tissues, especially in liver and skeletal muscle, can lead to more severe insulin resistance, even in the absence of visceral adiposity [81]. Consistently, over 70% of obese individuals with T2DM are known to have NAFLD, and NAFLD patients almost universally have T2DM and hepatic insulin resistance [82]. In addition, interventions that reduce intrahepatic triglyceride content through modest weight loss or leptin treatment dramatically reverse hepatic insulin resistance in T2DM, NAFLD, and lipodystrophic patients [83]. Furthermore, intrahepatic and intramyocellular lipid contents are considered to be much better predictors of insulin resistance than visceral adipose tissue volume, which strongly indicates that increased lipid accumulation in liver and skeletal muscle could impair insulin signaling and induce insulin resistance [84,85].
Numerous animal experiments support the notion that insulin resistance is caused by ectopic lipid accumulation in liver or skeletal muscle. Several studies have shown that lipid accumulation in liver and skeletal muscle caused by short-term HFD feeding or lipid/heparin infusions induce insulin resistance in rats [86]. In addition, overexpression of lipoprotein lipase (LPL) in liver or muscle induced peripheral insulin resistance and the accumulation of lipid in respective tissues [87,88], and skeletal muscle-specific LPL deletion enhanced insulin signaling in HFD challenged muscle [89]. Furthermore, deleting fat transport proteins such as CD36 or FATP-1 increased insulin-mediated glucose uptake in skeletal muscle [90,91], and liver-specific knockdown of FATP2 or FATP5 significantly reduced HFD-induced hepatosteatosis and increased glucose tolerance [92,93].
Lipodystrophy provides the most well-established clinical evidence that sheds light on the role of ectopic lipid accumulation in the absence of any contribution from visceral adipose to insulin resistance, as this condition is characterized by a substantial reduction in fat cells in adipose tissue leading to hypertriglyceridemia, peripheral ectopic fat deposition, and severe insulin resistance. In lipodystrophy, postprandial fatty acid influx cannot be buffered by adipose tissue, and as a result fatty acids are delivered to other metabolic tissues, including liver and skeletal muscle, which impairs insulin signaling and causes severe insulin resistance [94]. Genetic lipodystrophic mice also exhibit insulin resistance induced by ectopic lipid accumulation in liver and skeletal muscle. Adipocyte-specific basic region-leucine zipper (B-ZIP) transcription factor knockout mice, which are called A-ZIP/F-1 fatless mice due to a lack of white fat tissue, are hyperinsulinemic and hyperglycemic, due to severe defects in IRS-1 and -2 associated PI3K activity in muscle and liver [95,96]. In addition, insulin functions in A-ZIP/F-1 fatless mice were dramatically restored by the transplantation of parametrial fat from littermates, suggesting that the distribution of triglyceride to skeletal muscle and liver might be a critical factor of insulin resistance [95]. The overexpression of preadipocyte factor-1 (Pref-1), a secreted protein that inhibits adipocyte differentiation, also induced the characteristics of lipodystrophic models, that is, reduced adipose tissue mass, dyslipidemia, and insulin resistance [97,98]. In addition, the inhibition of de novo sphingolipid biosynthesis by adipocyte-specific knockout of serine palmitoyltransferase 2 (Sptlc2), which catalyzes the first step of de novo sphingolipid synthesis, exhibited impaired adipose tissue development and a lipodystrophic phenotype, which progressed to systemic insulin resistance [99]. The ability to form unilocular lipid droplets in white adipocytes is required to maintain the ability of white adipocytes to store lipids. Knock out mice of fat-specific protein 27 (Fsp27) showed multilocular lipid droplets in adipocytes and increased lipolysis, resulting in hepatic steatosis and insulin resistance [100]. These studies demonstrate that excess lipid accumulation in liver and muscle can induce insulin resistance.
DAG
How excess lipid accumulation interferes with insulin signaling in liver and muscle has long been investigated, especially with respect to surface insulin receptor downregulation and impaired insulin signal transduction in typical obesity-associated insulin resistance [101]. The most plausible hypothesis of the mechanism whereby ectopic lipid accumulation induces insulin resistance is that several lipid metabolites, including diacylglycerol (DAG), lysophosphatidic acid (LPA), ceramides, and acylcarnitines, are involved in the pathogenesis of insulin resistance in liver and skeletal muscle.
Fatty acids are rapidly esterified in cells to fatty acyl-CoA, which is transferred to a glycerol backbone to form LPA, DAGs, and triacylglycerols (TAGs) through lipogenesis (Fig. 4). These lipid intermediates, especially DAG, might function as second messengers in key signaling pathways implicated in the pathogenesis of insulin resistance [102]. High-fat-induced and genetically obese rodents exhibited hepatic insulin resistance and elevated hepatic DAG contents [103,104], and hepatocytes treated with phorbol myristate acetate (PMA, a DAG analog) exhibited impaired IRTK tyrosine kinase activity and insulin-stimulated GYS activity [105].
Fig. 4.

Diacylglycerol (DAG)-protein kinase C (PKC) hypothesis in insulin resistance. Fatty acids are rapidly esterified in cells to fatty acyl-CoA, which form lysophosphatidic acid (LPA), DAG, and triacylglycerol (TAG) through lipogenesis. Increased hepatic DAG levels induced the translocation of nPKC (PKCε and PKCθ in liver and skeletal muscle, respectively) to the plasma membrane and inhibited insulin receptor tyrosine kinase (IRTK) tyrosine kinase activity by phosphorylating it at Thr1160, which inactivate insulin receptor substrate 2 (IRS-2), phosphatidylinositol-3-OH kinase (PI3K), and Akt2. PIP2, phosphatidylinositol-4,5-bisphosphate; PIP3, phosphatidylinositol-3,4,5-trisphosphate; PKB, protein kinase B; PLC, phospholipase C; FFA, free fatty acid; MGL, monoacylglycerol lipase; MAG, monoacylglycerol; HSL, hormone-sensitive lipase; ATGL, adipose triglyceride lipase; CGI-58, comparative gene identification-58; GPAT, glycerol-3-phosphate acyltransferase; AGPAT, acylglycerolphosphate acyltransferase; PA, phosphatidic acid; PAP, phosphatidic acid phosphatases; DGAT, diacylglycerol acyltransferase; ACS, acyl-CoA synthetases; FAS, fatty acid synthase; ACC, acetyl-CoA carboxylase; CPT1, carnitine palmitoyltransferase 1; TCA, trichloroacetic acid.
The DAG hypothesis of lipid-induced insulin resistance is that interference of insulin signaling by activated protein kinase C (PKC) results from the accumulation of DAG within insulin-sensitive tissues (Fig. 4). There are three groups of the PKC family (conventional, novel, and atypical), and novel PKC (nPKC), which has much greater affinity for DAG, is known to mediate the role of DAG in insulin resistance [106,107]. In liver, increased hepatic DAG levels induced the translocation of PKCε (the primary nPKC isoform in liver) to the plasma membrane and inhibited IRTK tyrosine kinase activity by phosphorylating it at Thr1160, which reduced insulin-stimulated phosphorylations (activations) of IRS2, PI3K, and Akt2 [108,109]. In the same vein, hepatic DAG contents and PKCε translocation to the plasma membrane were shown to be more potent predictors of hepatic insulin resistance than other factors such as ceramide, endoplasmic reticulum (ER) stress marker, and inflammatory cytokine levels in humans [106,110,111]. In a manner similar to that observed in liver, the accumulation of intramyocellular DAG impairs insulin signaling and muscle glucose uptake by activating PKCθ (muscle-type nPKC) [112,113], which elicits the phosphorylation of IRS-1 at Ser1101 and blocks the insulin-stimulated phosphorylation of IRS-1 [114,115]. Large-scale studies have also corroborated the DAG-PKC induced hypothesis of insulin resistance. High PKC levels have been reported in the membrane fractions of soleus muscles of diabetic rats and in the livers of obese rats [106,116]. Importantly, PKCε translocation to the plasma membrane was observed in rats with isolated hepatic steatosis and hepatic insulin resistance induced by HFD feeding [86,117]. Moreover, knockdown of PKCε using an antisense oligonucleotide (ASO) protected rats from fat-induced hepatic insulin resistance [109], and PKCε knockout mice (Prkce−/−) mice showed improved glucose tolerance than wild type controls after 7 days of high-fat feeding [118]. Apolipoprotein CIII (ApoC3) overexpression promoted diet-induced hepatic steatosis and hepatic insulin resistance, and resulted in higher DAG and membrane-localized PKCε [119]. Like PKCε, PKCθ modulated insulin signaling in myoblasts, and PKC-θ knockout prevented fat-induced defects in insulin signaling and glucose transport in mouse skeletal muscle [120]. Furthermore, PKCθ overexpression led to insulin resistance characterized by reduced insulin responsiveness in C2C12 myoblasts [121].
Notably, not all DAGs can stimulate PKC translocation to the plasma membrane and induce T2DM by inhibiting insulin signaling. The subcellular localizations of DAGs and DAG stereoisomer types were considered critical for activating PKC and inducing insulin resistance. Of the three DAG stereoisomers, sn-1,2-DAG, the product of the action of phospholipase C on PIP2, was reported to be the only stereoisomer that activate PKC [122]. Furthermore, the accumulation of DAG in lipid droplets, but not in plasma membrane, did not inhibit insulin signaling or induce hepatic insulin resistance, as shown in mice treated with adipose triglyceride lipase cofactor (comparative gene identification-58 [CGI-58]) ASO. CGI-58 ASO treated mice had increased hepatic DAG contents, but preserved hepatic insulin sensitivity [123]. Consequently, sn-1,2-DAG distribution in the plasma membrane disrupted insulin signaling and contributed to insulin resistance through PKC activation [123,124].
Ceramide
Ceramide is a sphingolipid and an essential bioactive lipid produced from an intracellular fatty acid and sphingosine and is believed to mediate lipid-induced insulin resistance [125]. Ceramide plays a crucial role in cell membrane stabilization and also regulates the distributions of signaling molecules. In obese humans, hepatic ceramides are known to be associated with homeostatic model assessment for insulin resistance (HOMA-IR) scores [126], and obese rats with insulin resistance have consistently been reported to exhibit elevated hepatic and muscle ceramide contents [103]. Inhibition of ceramide synthesis using myriocin, an inhibitor of serine palmitoyltransferase, prevented insulin resistance and attenuated ceramide contents in fat-fed mice [127,128]. In addition, mice heterozygous for dihydroceramide desaturase 1 (Des1), a gatekeeper of ceramide synthesis, had low fasting HOMA-IR scores and total ceramide levels in liver [127]. Also, liver-specific knock out of ceramide synthase 6 (CerS6) decreased hepatic ceramide levels (especially C16:0 species), protected against HFD-induced obesity, and improved glucose tolerance [129]. In addition, liver-specific overexpression of ceramidase protected HFD-fed mice against hepatic steatosis, enhanced hepatic insulin signaling, and reduced the levels of some ceramide species, including 16:0, 18:0, and 20:0 ceramides [130]. Furthermore, adipose- or liver-specific adiponectin receptor (AdipoR) overexpression reduced total ceramide contents and prevented HFD-induced insulin resistance [131]. The results of these studies suggest that ceramide levels influence lipid-induced insulin resistance.
The underlying molecular mechanism whereby ceramide induces insulin resistance has not been clearly demonstrated. However, some candidate mechanisms have been proposed, namely, the impairment of AKT translocation through the activation of atypical PKCζ, and the activation of PP2A [132,133]. Overexpression of ceramidase in liver and adipose tissue reduced PKCζ activity and prevented ceramide-induced insulin resistance and hepatic steatosis [130]. However, the ceramide analog, C2-ceramide, activated PP2A but not PKCζ and inactivated Akt activity in brown adipocytes [134], which suggested PP2A might mediate the role of ceramide in insulin resistance. However, LB1 (a PP2A inhibitor) increased hepatic insulin resistance as evidenced by elevated fasting plasma glucose and decreased the glucose infusion rate during hyperinsulinemic-euglycemic clamp testing in rats, despite strengthening hepatic insulin signaling, which indicated PP2A activation is not a major ceramide pathway to insulin resistance [135]. Furthermore, fibroblast growth factor 21 (FGF21), adiponectin, and NOD-like receptor pyrin domain-containing protein-3 (NLRP-3) inflammasome have been proposed to mediate or be associated with the function of ceramide [136-138]. Taken together, although growing evidence suggests ceramide can induce insulin resistance, further studies are needed to elucidate the molecular mechanism whereby ceramides impair insulin signaling or glucose transport translocation.
Other mechanisms: ER stress and inflammation
ER stress
Although ectopic lipid accumulation in muscle and liver plausibly explains the development of insulin resistance in obese or dyslipidemic subjects, several other hypotheses also have been proposed to explain the mechanism responsible for obesity-induced insulin resistance (Fig. 5). In particular, ER stress has been proposed to underlie insulin resistance in liver and pancreatic β-cells. ER stress is known to be enhanced by obesity [139]. When exposed to an over-nourished condition, the liver should produce an excess of enzymes to process nutrients and enhance unfolded protein response (UPR) by accumulating unfolded proteins in ER. During this process, the recruitment of glucose-regulated protein 78 (GPR78, also known as BiP) results in the activations of inositol requiring enzyme-1 (IRE1α), PKR-like ER kinase (PERK), and activating transcription factor 6 (ATF6), which inhibit protein translation, promote ER chaperones, and thus, reduce unfolded protein levels [140]. Experimentally, ER stress induced by tunicamycin suppresses insulin signaling via the serine phosphorylation of IRS-1 by c-Jun N-terminal kinase (JNK). In addition, the oral administration of chemical chaperones, such as 4-phenyl butyric acid or taurine-conjugated ursodeoxycholic acid, that alleviate ER stress, improved insulin signaling and restored insulin sensitivity in ob/ob mice [141]. As occurs in liver, increased demand for insulin secretion induces ER stress in pancreatic β-cells in chronic hyperglycemic diabetic humans and mice, and this contributes to the development of T2DM [142,143]. Pancreatic β-cell-specific knockout of X-box-binding protein-1 (XBP-1), a key transcription factor in ER stress, resulted in hyperglycemia and diet-induced insulin resistance resulting from β-cell dysfunction in mice [139,144]. Thus, it appears that, it is premature to conclude that ER stress directly induces insulin resistance, although some aspects of ER stress seem to regulate glucose and lipid metabolism, such as lipogenesis, lipid droplet formation, and lipid storage.
Fig. 5.

Other potential mechanisms for insulin resistance. Other hypotheses, such as endoplasmic reticulum (ER) stress, reactive oxygen species (ROS), and inflammation, have been proposed to explain the mechanism responsible for obesity-induced insulin resistance. PIP2, phosphatidylinositol-4,5-bisphosphate; PIP3, phosphatidylinositol-3,4,5-trisphosphate; PKB, protein kinase B; IKK, inhibitor of nuclear factor κ-B kinase; JNK, c-Jun N-terminal kinase; PKC, protein kinase C; LIPIN2, phosphatidic acid phosphatases; DAG, diacylglycerol; HFD, high-fat diet.
Inflammation
Systemic chronic inflammatory response characterized by altered cytokine production and the activations of inflammatory signaling pathways is being actively investigated to determine its role in obesity-related insulin resistance [145]. Inflammation-related cytokines, such as tumor necrosis factor-α (TNFα), have been reported to be excreted by macrophages recruited by adipocytes in obese and diabetic humans and animals [146,147]. When macrophages are recruited by adipose tissue due to the overexpression of monocyte chemoattractant protein 1 (MCP1, a chemokine ligand also known as CCL2), hepatic insulin resistance is observed with increased TNFα expression in adipose tissue, but without any change in body weight or adiposity [148]. In addition, knockout of MCP1 protected against HFD-induced insulin resistance [148]. Consistently, knockout of CCR2 (the receptor of MCP-1) and treatment with INCB3344 (a CCR2 antagonist), increased insulin sensitivity and reduced macrophage recruitment by adipose tissue [149]. The role of TNFα in insulin resistance is considered to be mediated by the JNK1, which phosphorylates serine 307 of IRS1 [150]. TNFα also activates inhibitor of nuclear factor κ-B kinase (IKK), and liver- or myeloid cell-specific deletion of IKK protected against hepatic insulin resistance, improved hepatic insulin signaling, and reduced inflammatory cytokine expression [151], which suggested IKK might mediate the role of TNFα in insulin resistance. These studies support the role of inflammation, at least the involvements of TNFα and MCP1, in insulin resistance.
However, liver-specific knockout of NF-κB essential modulator (NEMO) led to IKK activation, lowered fasting plasma glucose and fasting insulin, improved glucose tolerance, and increased inflammation in liver [152]. In addition, the overexpression of the NF-κB p65 subunit somewhat protected against diet-induced insulin resistance and improved hepatic and peripheral insulin sensitivities [153]. Furthermore, plasma levels of inflammatory cytokines, such as TNFα and interleukin-6, were unchanged in insulin resistant offspring of T2DM and were similar in insulin resistant elderly subjects compared with insulin-sensitive controls [154,155]. These studies demonstrate that chronic inflammation is not a primary causative factor of insulin resistance and that it is insufficient to disrupt systemic glucose metabolism. Thus, it seems that chronic inflammation indirectly exacerbates insulin resistance, and should not be considered a primary strategic target for insulin resistance and T2DM. Rigorously controlled additional studies are required to better determine the role of chronic inflammation in insulin resistance and glucose metabolism.
THERAPEUTIC STRATEGIES FOR INSULIN RESISTANCE
Pharmaceutical drugs for T2DM treatment
One hundred years ago, diabetes was considered a dreadful disease accompanied by continued weight loss, emaciation, ketoacidosis, and ultimately coma and death. After the use of insulin as an antidiabetic, diabetes-related mortality has dramatically reduced. However, sedentary lifestyles and the increased prevalence of obesity have increased the incidence of T2DM [156]. Although lifestyle modifications and weight loss are recommended to ameliorate T2DM, their effects are limited with low feasibility. Drugs currently prescribed for T2DM either stimulate insulin secretion or increase insulin sensitivity (Fig. 6). Since the currently available pharmaceutical drugs for T2DM treatment has been well described in many reviews, we briefly summarized the representative T2DM drugs in this review.
Fig. 6.
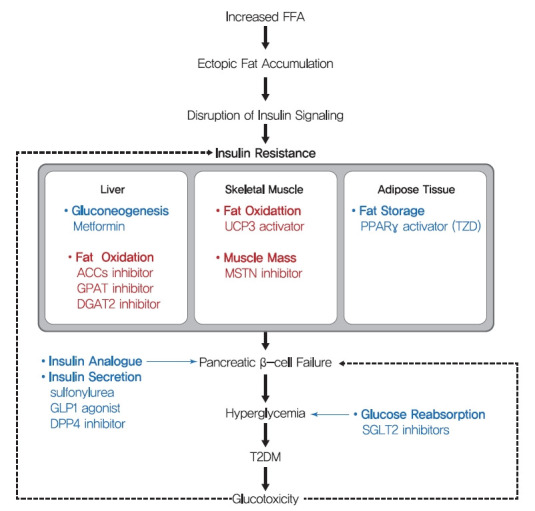
Schematic mechanism of type 2 diabetes mellitus (T2DM) and therapeutic strategies for insulin resistance. The main strategies of present (blue) treatment for T2DM and possible future (red) treatments for insulin resistance are summarized. Many T2DM drugs, such as sulfonylureas, glucagon-like peptide 1 (GLP-1) agonists, and dipeptidyl peptide-4 (DPP-4) inhibitors, target the ability of β-cells to secrete insulin. In addition, thiazolidinediones (TZDs) and metformin are insulin-sensitizing antidiabetic drugs, targeting fat storage capacity of adipose tissue and glucose production in liver, respectively. Key strategies of potential future treatment for insulin resistance suggested in this study are targeting enhancement of β oxidation in liver and skeletal muscle and stimulation of muscle quality. FFA, free fatty acid; ACC, acetyl-CoA carboxylase; GPAT, glycerol-3-phosphate acyltransferase; DGAT2, diacylglycerol acyltransferase 2; UCP3, uncoupling protein 3; MSTN, myostatin; PPARγ, peroxisome proliferator-activated receptor-γ; SGLT2, sodium-glucose cotransporter 2.
β-Cell dysfunction is a critical feature of the shift from prediabetes to T2DM [157], and many T2DM drugs target the ability of β-cells to secret insulin. Sulfonylureas stimulate β-cells to release insulin independently of glucose by blocking sulphonylurea receptor-1 (SUR-1), which is a regulatory subunit of ATP-sensitive K+ channel in β-cells [158]. In addition, glucagon-like peptide-1 (GLP-1), a hormone produced by L-cells in small intestine in response to food intake, stimulates the production and secretion of insulin by pancreatic β-cells and inhibits glucagon secretion by pancreatic α-cells. Since GLP-1 has poor stability, GLP-1 agonists with longer half-lives and dipeptidyl peptide-4 (DPP-4) inhibitors are used to treat T2DM. Although these drugs stimulate insulin secretion by β-cells to improve hyperglycemia and reduce the risks posed by glucotoxicity, they do not prevent disease progression.
Thiazolidinediones (TZDs) (such as pioglitazone and rosiglitazone) enhance insulin sensitivity in skeletal muscle, liver, and adipose tissue and promote fat redistribution from liver and skeletal muscle to adipocytes [159]. The effect of TZDs on insulin sensitivity is mediated by the activation of PPARγ, a nuclear receptor that regulates the transcriptions of several genes involved in glucose and lipid metabolism and energy balance and increases fat oxidation, adipocyte proliferation, lipogenesis, fat redistribution, and reduces plasma free fatty acid and pro-inflammatory cytokine levels [159]. Metformin, the most commonly prescribed drug for diabetes, may also increase insulin sensitivity in peripheral tissues during fasting by reducing HGP, which is considered to be mediated by the promotion of mitochondrial activity or the inhibition of glucagon signaling through AMPK activation [160,161].
In addition to drugs that increase insulin secretion and sensitivity, some T2DM drugs use other strategies. Alpha-glucosidase inhibitors (AGIs) inhibit gastrointestinal hydrolyzing enzymes, which delay the absorption of carbohydrates in small intestine and decrease postprandial blood glucose and insulin levels [162]. On the other hand, sodium-glucose cotransporter 2 (SGLT2) inhibitors lower blood glucose levels by blocking renal glucose reabsorption, which reduces glucotoxicity, improves β-cell function, and increases insulin sensitivity [163].
As indicated above, many drugs are prescribed to treat diabetes, and the enormous medical expenses associated with the development of diabetes treatments continue to burden pharmaceutical companies. Nevertheless, over recent years the prevalence and mortality of diabetes have increased rapidly. Considering the main causes of the development of T2DM, impaired insulin secretion and insulin resistance, the apparent inefficiency of current pharmaceutical strategies may be due to the undue focus place on alleviating symptoms, such as hyperglycemia by stimulating insulin secretion. Moreover, only two drugs targeting insulin resistance are available, while there are many drugs targeting insulin secretion [164]. Evidence shows insulin resistance is preceding and central to the development of T2DM and is mainly driven by ectopic fat accumulation in tissues. Moreover, insulin resistance is a common pathogenic component of the development of metabolic syndrome, which is strongly associated with obesity. Accordingly, we suggest different therapeutic strategies be adopted that target insulin resistance, and we believe this approach provides a more effective means of preventing and treating diabetes and metabolic syndrome.
Therapeutic strategy for insulin resistance
Inhibiting fat synthesis in liver
As mentioned above, the accumulation of lipid in hepatocytes induces hepatic insulin resistance via the activation of PKCε by hepatic DAG. Since liver function is central to glucose and lipid metabolism control, hepatic insulin resistance leads to systemic insulin resistance. Hence, the suppression of lipid synthesis through de novo lipogenesis and re-esterification are thought to constitute an effective means of reducing lipid accumulation in liver. This opinion is well supported by the results of animal studies. ACC1 and ACC2 are critical regulators of de novo lipogenesis and lipid oxidation, respectively. Inhibitions of ACC1 and ACC2 using ASO reduced liver DAG contents and PKCε translocation to the plasma membrane and protected mice from lipid-induced hepatic insulin resistance [165]. In addition, the adenovirus-induced overexpression of mitochondrial glycerol-3-phosphate acyltransferase (mGPAT, a lipogenesis rate-limiting enzyme) led to insulin resistance and increased DAG levels in hepatocytes [166]. In the same vein, mGPAT knockout mice had reduced hepatic DAG levels and were protected from HFD-induced hepatic insulin resistance [167], and shRNA knockdown of phosphatidic acid phosphatases (PAPs, also called Lipins) similarly reduced hepatic DAG levels and improved insulin sensitivity, while the overexpression of Lipin-2 using adenovirus perturbed glucose tolerance and insulin sensitivity [168,169]. Also, suppression of diacylglycerol acyltransferase 2 (DGAT2, an enzyme that catalyzes the final step of triglyceride synthesis from diacylglycerol) decreased hepatic DAG contents, reversed diet-induced hepatic steatosis and insulin resistance, and reduced PKCε levels in plasma membranes [170]. These studies strongly support the suggestion that therapies which reduce hepatic DAG and acetyl-CoA contents by suppressing hepatic de novo lipogenesis might effectively treat hepatic insulin resistance in T2DM. In this strategy, agents targeting ACC1, ACC2, and DGAT2 are in phase II clinical trials for the treatment of T2DM and nonalcoholic steatohepatitis (Table 1) [171].
Table 1.
Currently developing drugs targeting insulin resistance based on the strategy of inhibiting fat synthesis in the liver and stimulation of fat oxidation and muscle mass in the skeletal muscle
| Target | Drug development status | Indication | Sponsor |
|---|---|---|---|
| ACC1 | Phase II | Non-alcoholic steatohepatitis (NASH) | Gilead |
| Pfizer | |||
| Phase I | Type 2 diabetes | Pfizer | |
| ACC2 | Phase II | Non-alcoholic steatohepatitis (NASH) | Gilead |
| Pfizer | |||
| Shionogi | |||
| Phase I | Type 2 diabetes | Pfizer | |
| DGAT2 | Phase II | Non-alcoholic steatohepatitis (NASH) | Ionis Pharmaceuticals |
| Pfizer | |||
| Type 2 diabetes | Pfizer | ||
| Phase I | Obesity | Shionogi | |
| PPARδ | Registered (2021) | Type 2 diabetes | Chipscreen Biosciences |
| Phase III | Anti-diabetic drugs | Genfit | |
| Phase I | Fatty acid oxidation disorders | Reneo Pharmaceuticals | |
| Phase II | Type 2 diabetes | Pfizer | |
| CombinatoRx | |||
| GlaxoSmithKline | |||
| Phase I | Dyslipidemia | ABIONYX Pharma | |
| Sanofi | |||
| Type 2 diabetes | Kalypsys | ||
| Sanofi | |||
| Obesity | Kalypsis | ||
| MSTN | Phase I | Muscle loss (metabolic disorders) | Amgen |
| Cardiovascular disease | Pfizer |
Data from database Cortellis Drug Discovery Intelligence (Clarivate Analytics) [171].
ACC, acetyl-CoA carboxylase; DGAT2, diacylglycerol acyltransferase 2; PPAR, peroxisome proliferator-activated receptor; MSTN, myostatin.
Stimulation of fat oxidation and muscle mass
While the liver is qualitatively critical for glucose metabolism, skeletal muscle is quantitatively essential since skeletal muscle accounts for 35%–45% of body weight and up to 70%–80% of total glucose disposal after insulin stimulation. Therefore, increasing fat oxidation and/or skeletal muscle mass are considered potential strategies for reducing intramyocellular lipid accumulation, and eventually, improving insulin sensitivity. Skeletal muscle-specific overexpression of uncoupling protein 3 (UCP-3, an inner mitochondrial membrane transporter that dissipates mitochondrial proton gradients) protected against HFD-induced insulin resistance in skeletal muscle, enhanced energy expenditure, and reduced PKCθ activity and membrane-associated DAG levels [172]. The beneficial effect of fat oxidation in muscle on insulin resistance was also observed in ACC-2 (a key regulator of mitochondrial fat oxidation) knocked down mice [173]. In addition, skeletal muscle-specific deletion of autophagy-related gene-7 (ATG-7) protected mice from diet-induced obesity and insulin resistance and was accompanied by increased fat oxidation and adipocyte browning [174]. Also, skeletal muscle-specific overexpression of PPARδ improved glucose tolerance and increased number of slow-twitch muscle fibers (a mitochondria rich fiber type) in which mitochondrial oxidative metabolism contributes to energy production [175]. Furthermore, increased number of fast-twitch muscle fibers induced by myostatin (MSTN, also known as Gdf-8) ablation or the skeletal muscle-specific expression of a constitutively active form of Akt1 prevented dieti-nduced obesity, enhanced peripheral insulin sensitivity, and dramatically improved diet-induced insulin resistance [176], which were probably caused by increased glucose utilization and fat oxidation due to increased muscle mass. Despite the beneficial effects of increased muscle mass and quality improve glucose metabolism, few diabetic drugs under development target muscle. Monoclonal antibodies for MSTN are in phase I clinical trials for the treatment of metabolic disorders and cardiovascular disease (Table 1). As regards the developmental efforts to increase muscle quality, bezafibrate, which has been used to treat liver cirrhosis and targets PPARs, is the subject of a phase II clinical trial on T2DM, and chiglitazar sodium, a PPAR pan-agonist, recently approved in China for the treatment of T2DM (Table 1) [171].
CONCLUSIONS
According to the World Health Organization, the number of people with diabetes rose almost four-fold between 1980 and 2019 from 108 to 463 million, and this increase was accompanied by a steady increase in the prevalence of obesity. In addition, premature mortality attributed to diabetes increased by 5% between 2000 and 2016. In the general populations of most countries, the prevalence of hyperglycemia lies between 7% and 14% [156]. Furthermore, the global prevalence of diabetes has been forecast to increase to 700 million by 2045, and the rapidly increasing prevalence of metabolic disease is now viewed as a global health emergency [164].
As a primary and common cause of metabolic disease, insulin resistance should be considered a therapeutic target for metabolic diseases, including diabetes. However, no generally accepted theory explains the mechanism responsible for insulin resistance. Nevertheless, growing evidence demonstrates that ectopic lipid accumulation is more strongly related to diabetic physiology than other variables, such as ER stress and the plasma concentrations of inflammatory cytokine [110,111,126]. Numerous animal and epidemiological studies have shown that DAG accumulation in plasm membrane fractions caused by ectopic fat accumulation or reduced fat oxidation is a critical factor of insulin resistance development via the activation of nPKC in liver tissues and skeletal muscle. Based on this pathophysiological mechanism of insulin resistance, the suppression of lipid synthesis in liver and the stimulation of fat oxidation in skeletal muscle provide means of reducing ectopic lipid accumulation and potentially, improving insulin sensitivity, and eventually, preventing or delaying the onset of T2DM.
Like other metabolic diseases, diabetes is a multifaceted disease. Although ectopic lipid accumulation in peripheral tissue appears to be the primary cause of insulin resistance, increased ER stress, inflammation, and reactive oxygen species levels, dysregulations of adipokines contribute to the pathogenesis of insulin resistance in liver and skeletal muscle. Therefore, we suggest that treatments for insulin resistance should be based on multidisciplinary strategies that target physiological and metabolic impairments.
Acknowledgments
None
Footnotes
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
FUNDING
This research was supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government (MSIT) (No.2014M3A9D5A01073886) and by the Gachon University research fund of 2018 (GCU-2018-0683).
REFERENCES
- 1.Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001;414:782–7. doi: 10.1038/414782a. [DOI] [PubMed] [Google Scholar]
- 2.Kahn SE. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of type 2 diabetes. Diabetologia. 2003;46:3–19. doi: 10.1007/s00125-002-1009-0. [DOI] [PubMed] [Google Scholar]
- 3.Rizza RA. Pathogenesis of fasting and postprandial hyperglycemia in type 2 diabetes: implications for therapy. Diabetes. 2010;59:2697–707. doi: 10.2337/db10-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moore MC, Cherrington AD, Cline G, Pagliassotti MJ, Jones EM, Neal DW, et al. Sources of carbon for hepatic glycogen synthesis in the conscious dog. J Clin Invest. 1991;88:578–87. doi: 10.1172/JCI115342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev. 2018;98:2133–223. doi: 10.1152/physrev.00063.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asplin CM, Paquette TL, Palmer JP. In vivo inhibition of glucagon secretion by paracrine beta cell activity in man. J Clin Invest. 1981;68:314–8. doi: 10.1172/JCI110251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greenbaum CJ, Havel PJ, Taborsky GJ, Jr, Klaff LJ. Intra-islet insulin permits glucose to directly suppress pancreatic A cell function. J Clin Invest. 1991;88:767–73. doi: 10.1172/JCI115375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Youngren JF. Regulation of insulin receptor function. Cell Mol Life Sci. 2007;64:873–91. doi: 10.1007/s00018-007-6359-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khalid M, Alkaabi J, Khan MAB, Adem A. Insulin signal transduction perturbations in insulin resistance. Int J Mol Sci. 2021;22:8590. doi: 10.3390/ijms22168590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leto D, Saltiel AR. Regulation of glucose transport by insulin: traffic control of GLUT4. Nat Rev Mol Cell Biol. 2012;13:383–96. doi: 10.1038/nrm3351. [DOI] [PubMed] [Google Scholar]
- 11.Chiu TT, Jensen TE, Sylow L, Richter EA, Klip A. Rac1 signalling towards GLUT4/glucose uptake in skeletal muscle. Cell Signal. 2011;23:1546–54. doi: 10.1016/j.cellsig.2011.05.022. [DOI] [PubMed] [Google Scholar]
- 12.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–9. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 13.Newgard CB, Brady MJ, O’Doherty RM, Saltiel AR. Organizing glucose disposal: emerging roles of the glycogen targeting subunits of protein phosphatase-1. Diabetes. 2000;49:1967–77. doi: 10.2337/diabetes.49.12.1967. [DOI] [PubMed] [Google Scholar]
- 14.Agius L. Role of glycogen phosphorylase in liver glycogen metabolism. Mol Aspects Med. 2015;46:34–45. doi: 10.1016/j.mam.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 15.Dong XC, Copps KD, Guo S, Li Y, Kollipara R, DePinho RA, et al. Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 2008;8:65–76. doi: 10.1016/j.cmet.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tzivion G, Dobson M, Ramakrishnan G. FoxO transcription factors: regulation by AKT and 14-3-3 proteins. Biochim Biophys Acta. 2011;1813:1938–45. doi: 10.1016/j.bbamcr.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 17.Perry RJ, Camporez JG, Kursawe R, Titchenell PM, Zhang D, Perry CJ, et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell. 2015;160:745–58. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ros S, Garcia-Rocha M, Dominguez J, Ferrer JC, Guinovart JJ. Control of liver glycogen synthase activity and intracellular distribution by phosphorylation. J Biol Chem. 2009;284:6370–8. doi: 10.1074/jbc.M808576200. [DOI] [PubMed] [Google Scholar]
- 19.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–31. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krycer JR, Sharpe LJ, Luu W, Brown AJ. The Akt-SREBP nexus: cell signaling meets lipid metabolism. Trends Endocrinol Metab. 2010;21:268–76. doi: 10.1016/j.tem.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 21.Rebrin K, Steil GM, Mittelman SD, Bergman RN. Causal linkage between insulin suppression of lipolysis and suppression of liver glucose output in dogs. J Clin Invest. 1996;98:741–9. doi: 10.1172/JCI118846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi YH, Park S, Hockman S, Zmuda-Trzebiatowska E, Svennelid F, Haluzik M, et al. Alterations in regulation of energy homeostasis in cyclic nucleotide phosphodiesterase 3B-null mice. J Clin Invest. 2006;116:3240–51. doi: 10.1172/JCI24867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Begum N. Stimulation of protein phosphatase-1 activity by insulin in rat adipocytes. Evaluation of the role of mitogen-activated protein kinase pathway. J Biol Chem. 1995;270:709–14. doi: 10.1074/jbc.270.2.709. [DOI] [PubMed] [Google Scholar]
- 24.Resjo S, Goransson O, Harndahl L, Zolnierowicz S, Manganiello V, Degerman E. Protein phosphatase 2A is the main phosphatase involved in the regulation of protein kinase B in rat adipocytes. Cell Signal. 2002;14:231–8. doi: 10.1016/s0898-6568(01)00238-8. [DOI] [PubMed] [Google Scholar]
- 25.Virtanen KA, Lonnroth P, Parkkola R, Peltoniemi P, Asola M, Viljanen T, et al. Glucose uptake and perfusion in subcutaneous and visceral adipose tissue during insulin stimulation in nonobese and obese humans. J Clin Endocrinol Metab. 2002;87:3902–10. doi: 10.1210/jcem.87.8.8761. [DOI] [PubMed] [Google Scholar]
- 26.Kersten S. Mechanisms of nutritional and hormonal regulation of lipogenesis. EMBO Rep. 2001;2:282–6. doi: 10.1093/embo-reports/kve071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rieusset J, Andreelli F, Auboeuf D, Roques M, Vallier P, Riou JP, et al. Insulin acutely regulates the expression of the peroxisome proliferator-activated receptor-gamma in human adipocytes. Diabetes. 1999;48:699–705. doi: 10.2337/diabetes.48.4.699. [DOI] [PubMed] [Google Scholar]
- 28.DeFronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care. 2009;32 Suppl 2:S157–63. doi: 10.2337/dc09-S302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim JK, Michael MD, Previs SF, Peroni OD, Mauvais-Jarvis F, Neschen S, et al. Redistribution of substrates to adipose tissue promotes obesity in mice with selective insulin resistance in muscle. J Clin Invest. 2000;105:1791–7. doi: 10.1172/JCI8305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim JK, Zisman A, Fillmore JJ, Peroni OD, Kotani K, Perret P, et al. Glucose toxicity and the development of diabetes in mice with muscle-specific inactivation of GLUT4. J Clin Invest. 2001;108:153–60. doi: 10.1172/JCI10294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wojtaszewski JF, Higaki Y, Hirshman MF, Michael MD, Dufresne SD, Kahn CR, et al. Exercise modulates postreceptor insulin signaling and glucose transport in muscle-specific insulin receptor knockout mice. J Clin Invest. 1999;104:1257–64. doi: 10.1172/JCI7961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Neill HM, Maarbjerg SJ, Crane JD, Jeppesen J, Jorgensen SB, Schertzer JD, et al. AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci U S A. 2011;108:16092–7. doi: 10.1073/pnas.1105062108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Le Marchand-Brustel Y, Gremeaux T, Ballotti R, Van Obberghen E. Insulin receptor tyrosine kinase is defective in skeletal muscle of insulin-resistant obese mice. Nature. 1985;315:676–9. doi: 10.1038/315676a0. [DOI] [PubMed] [Google Scholar]
- 34.Frojdo S, Vidal H, Pirola L. Alterations of insulin signaling in type 2 diabetes: a review of the current evidence from humans. Biochim Biophys Acta. 2009;1792:83–92. doi: 10.1016/j.bbadis.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 35.Lewis GF, Carpentier AC, Pereira S, Hahn M, Giacca A. Direct and indirect control of hepatic glucose production by insulin. Cell Metab. 2021;33:709–20. doi: 10.1016/j.cmet.2021.03.007. [DOI] [PubMed] [Google Scholar]
- 36.Krssak M, Brehm A, Bernroider E, Anderwald C, Nowotny P, Dalla Man C, et al. Alterations in postprandial hepatic glycogen metabolism in type 2 diabetes. Diabetes. 2004;53:3048–56. doi: 10.2337/diabetes.53.12.3048. [DOI] [PubMed] [Google Scholar]
- 37.Basu R, Chandramouli V, Dicke B, Landau B, Rizza R. Obesity and type 2 diabetes impair insulin-induced suppression of glycogenolysis as well as gluconeogenesis. Diabetes. 2005;54:1942–8. doi: 10.2337/diabetes.54.7.1942. [DOI] [PubMed] [Google Scholar]
- 38.Magnusson I, Rothman DL, Katz LD, Shulman RG, Shulman GI. Increased rate of gluconeogenesis in type II diabetes mellitus. A 13C nuclear magnetic resonance study. J Clin Invest. 1992;90:1323–7. doi: 10.1172/JCI115997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu X, Chen K, Williams KJ. The role of pathway-selective insulin resistance and responsiveness in diabetic dyslipoproteinemia. Curr Opin Lipidol. 2012;23:334–44. doi: 10.1097/MOL.0b013e3283544424. [DOI] [PubMed] [Google Scholar]
- 40.Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A. 2010;107:3441–6. doi: 10.1073/pnas.0914798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, et al. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell. 2006;11:859–71. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 42.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–37. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 43.Kim K, Qiang L, Hayden MS, Sparling DP, Purcell NH, Pajvani UB. mTORC1-independent raptor prevents hepatic steatosis by stabilizing PHLPP2. Nat Commun. 2016;7:10255. doi: 10.1038/ncomms10255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim K, Ryu D, Dongiovanni P, Ozcan L, Nayak S, Ueberheide B, et al. Degradation of PHLPP2 by KCTD17, via a glucagon-dependent pathway, promotes hepatic steatosis. Gastroenterology. 2017;153:1568–80. doi: 10.1053/j.gastro.2017.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nagai Y, Yonemitsu S, Erion DM, Iwasaki T, Stark R, Weismann D, et al. The role of peroxisome proliferator-activated receptor gamma coactivator-1 beta in the pathogenesis of fructose-induced insulin resistance. Cell Metab. 2009;9:252–64. doi: 10.1016/j.cmet.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bindesboll C, Fan Q, Norgaard RC, MacPherson L, Ruan HB, Wu J, et al. Liver X receptor regulates hepatic nuclear O-GlcNAc signaling and carbohydrate responsive element-binding protein activity. J Lipid Res. 2015;56:771–85. doi: 10.1194/jlr.M049130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Postic C, Dentin R, Denechaud PD, Girard J. ChREBP, a transcriptional regulator of glucose and lipid metabolism. Annu Rev Nutr. 2007;27:179–92. doi: 10.1146/annurev.nutr.27.061406.093618. [DOI] [PubMed] [Google Scholar]
- 48.Erion DM, Popov V, Hsiao JJ, Vatner D, Mitchell K, Yonemitsu S, et al. The role of the carbohydrate response element-binding protein in male fructose-fed rats. Endocrinology. 2013;154:36–44. doi: 10.1210/en.2012-1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uyeda K, Repa JJ. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 2006;4:107–10. doi: 10.1016/j.cmet.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 50.Hauner H. Chichester: John Wiley & Sons; 2017. Obesity and diabetes. [Google Scholar]
- 51.Perseghin G, Ghosh S, Gerow K, Shulman GI. Metabolic defects in lean nondiabetic offspring of NIDDM parents: a cross-sectional study. Diabetes. 1997;46:1001–9. doi: 10.2337/diab.46.6.1001. [DOI] [PubMed] [Google Scholar]
- 52.Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1:785–9. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 53.Jucker BM, Rennings AJ, Cline GW, Shulman GI. 13C and 31P NMR studies on the effects of increased plasma free fatty acids on intramuscular glucose metabolism in the awake rat. J Biol Chem. 1997;272:10464–73. doi: 10.1074/jbc.272.16.10464. [DOI] [PubMed] [Google Scholar]
- 54.Schwartzman LI, Brown J. Glucose inhibition of fatty acid oxidation by rat diaphragm. Am J Physiol. 1960;199:235–7. doi: 10.1152/ajplegacy.1960.199.2.235. [DOI] [PubMed] [Google Scholar]
- 55.Christe ME, Rodgers RL. Cardiac glucose and fatty acid oxidation in the streptozotocin-induced diabetic spontaneously hypertensive rat. Hypertension. 1995;25:235–41. doi: 10.1161/01.hyp.25.2.235. [DOI] [PubMed] [Google Scholar]
- 56.Rothman DL, Shulman RG, Shulman GI. 31P nuclear magnetic resonance measurements of muscle glucose-6-phosphate. Evidence for reduced insulin-dependent muscle glucose transport or phosphorylation activity in non-insulin-dependent diabetes mellitus. J Clin Invest. 1992;89:1069–75. doi: 10.1172/JCI115686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cline GW, Petersen KF, Krssak M, Shen J, Hundal RS, Trajanoski Z, et al. Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in type 2 diabetes. N Engl J Med. 1999;341:240–6. doi: 10.1056/NEJM199907223410404. [DOI] [PubMed] [Google Scholar]
- 58.Shulman GI, Rothman DL, Jue T, Stein P, DeFronzo RA, Shulman RG. Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. N Engl J Med. 1990;322:223–8. doi: 10.1056/NEJM199001253220403. [DOI] [PubMed] [Google Scholar]
- 59.Marshall S, Bacote V, Traxinger RR. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J Biol Chem. 1991;266:4706–12. [PubMed] [Google Scholar]
- 60.Hawkins M, Barzilai N, Liu R, Hu M, Chen W, Rossetti L. Role of the glucosamine pathway in fat-induced insulin resistance. J Clin Invest. 1997;99:2173–82. doi: 10.1172/JCI119390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marshall S, Bacote V, Traxinger RR. Complete inhibition of glucose-induced desensitization of the glucose transport system by inhibitors of mRNA synthesis. Evidence for rapid turnover of glutamine:fructose-6-phosphate amidotransferase. J Biol Chem. 1991;266:10155–61. [PubMed] [Google Scholar]
- 62.McClain DA. Hexosamines as mediators of nutrient sensing and regulation in diabetes. J Diabetes Complications. 2002;16:72–80. doi: 10.1016/s1056-8727(01)00188-x. [DOI] [PubMed] [Google Scholar]
- 63.Rossetti L. Perspective: hexosamines and nutrient sensing. Endocrinology. 2000;141:1922–5. doi: 10.1210/endo.141.6.7566. [DOI] [PubMed] [Google Scholar]
- 64.Buse MG, Robinson KA, Gettys TW, McMahon EG, Gulve EA. Increased activity of the hexosamine synthesis pathway in muscles of insulin-resistant ob/ob mice. Am J Physiol. 1997;272(6 Pt 1):E1080–8. doi: 10.1152/ajpendo.1997.272.6.E1080. [DOI] [PubMed] [Google Scholar]
- 65.Robinson KA, Weinstein ML, Lindenmayer GE, Buse MG. Effects of diabetes and hyperglycemia on the hexosamine synthesis pathway in rat muscle and liver. Diabetes. 1995;44:1438–46. doi: 10.2337/diab.44.12.1438. [DOI] [PubMed] [Google Scholar]
- 66.Akimoto Y, Hart GW, Wells L, Vosseller K, Yamamoto K, Munetomo E, et al. Elevation of the post-translational modification of proteins by O-linked N-acetylglucosamine leads to deterioration of the glucose-stimulated insulin secretion in the pancreas of diabetic Goto-Kakizaki rats. Glycobiology. 2007;17:127–40. doi: 10.1093/glycob/cwl067. [DOI] [PubMed] [Google Scholar]
- 67.Akimoto Y, Kawakami H, Yamamoto K, Munetomo E, Hida T, Hirano H. Elevated expression of O-GlcNAc-modified proteins and O-GlcNAc transferase in corneas of diabetic Goto-Kakizaki rats. Invest Ophthalmol Vis Sci. 2003;44:3802–9. doi: 10.1167/iovs.03-0227. [DOI] [PubMed] [Google Scholar]
- 68.Hazel M, Cooksey RC, Jones D, Parker G, Neidigh JL, Witherbee B, et al. Activation of the hexosamine signaling pathway in adipose tissue results in decreased serum adiponectin and skeletal muscle insulin resistance. Endocrinology. 2004;145:2118–28. doi: 10.1210/en.2003-0812. [DOI] [PubMed] [Google Scholar]
- 69.Murata K, Morino K, Ida S, Ohashi N, Lemecha M, Park SY, et al. Lack of O-GlcNAcylation enhances exercise-dependent glucose utilization potentially through AMP-activated protein kinase activation in skeletal muscle. Biochem Biophys Res Commun. 2018;495:2098–104. doi: 10.1016/j.bbrc.2017.12.081. [DOI] [PubMed] [Google Scholar]
- 70.Vosseller K, Wells L, Lane MD, Hart GW. Elevated nucleocytoplasmic glycosylation by O-GlcNAc results in insulin resistance associated with defects in Akt activation in 3T3-L1 adipocytes. Proc Natl Acad Sci U S A. 2002;99:5313–8. doi: 10.1073/pnas.072072399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guinez C, Filhoulaud G, Rayah-Benhamed F, Marmier S, Dubuquoy C, Dentin R, et al. O-GlcNAcylation increases ChREBP protein content and transcriptional activity in the liver. Diabetes. 2011;60:1399–413. doi: 10.2337/db10-0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Farook VS, Bogardus C, Prochazka M. Analysis of MGEA5 on 10q24.1-q24.3 encoding the beta-O-linked N-acetylglucosaminidase as a candidate gene for type 2 diabetes mellitus in Pima Indians. Mol Genet Metab. 2002;77:189–93. doi: 10.1016/s1096-7192(02)00127-0. [DOI] [PubMed] [Google Scholar]
- 73.Lehman DM, Fu DJ, Freeman AB, Hunt KJ, Leach RJ, Johnson-Pais T, et al. A single nucleotide polymorphism in MGEA5 encoding O-GlcNAc-selective N-acetyl-beta-D glucosaminidase is associated with type 2 diabetes in Mexican Americans. Diabetes. 2005;54:1214–21. doi: 10.2337/diabetes.54.4.1214. [DOI] [PubMed] [Google Scholar]
- 74.Copeland RJ, Bullen JW, Hart GW. Cross-talk between GlcNAcylation and phosphorylation: roles in insulin resistance and glucose toxicity. Am J Physiol Endocrinol Metab. 2008;295:E17–28. doi: 10.1152/ajpendo.90281.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ball LE, Berkaw MN, Buse MG. Identification of the major site of O-linked beta-N-acetylglucosamine modification in the C terminus of insulin receptor substrate-1. Mol Cell Proteomics. 2006;5:313–23. doi: 10.1074/mcp.M500314-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kang ES, Han D, Park J, Kwak TK, Oh MA, Lee SA, et al. O-GlcNAc modulation at Akt1 Ser473 correlates with apoptosis of murine pancreatic beta cells. Exp Cell Res. 2008;314:2238–48. doi: 10.1016/j.yexcr.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 77.Housley MP, Rodgers JT, Udeshi ND, Kelly TJ, Shabanowitz J, Hunt DF, et al. O-GlcNAc regulates FoxO activation in response to glucose. J Biol Chem. 2008;283:16283–92. doi: 10.1074/jbc.M802240200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen G, Liu P, Thurmond DC, Elmendorf JS. Glucosamineinduced insulin resistance is coupled to O-linked glycosylation of Munc18c. FEBS Lett. 2003;534:54–60. doi: 10.1016/s0014-5793(02)03774-2. [DOI] [PubMed] [Google Scholar]
- 79.Robinson KA, Ball LE, Buse MG. Reduction of O-GlcNAc protein modification does not prevent insulin resistance in 3T3-L1 adipocytes. Am J Physiol Endocrinol Metab. 2007;292:E884–90. doi: 10.1152/ajpendo.00569.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Macauley MS, Bubb AK, Martinez-Fleites C, Davies GJ, Vocadlo DJ. Elevation of global O-GlcNAc levels in 3T3-L1 adipocytes by selective inhibition of O-GlcNAcase does not induce insulin resistance. J Biol Chem. 2008;283:34687–95. doi: 10.1074/jbc.M804525200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mann JP, Savage DB. What lipodystrophies teach us about the metabolic syndrome. J Clin Invest. 2019;129:4009–21. doi: 10.1172/JCI129190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Petersen KF, Dufour S, Feng J, Befroy D, Dziura J, Dalla Man C, et al. Increased prevalence of insulin resistance and nonalcoholic fatty liver disease in Asian-Indian men. Proc Natl Acad Sci U S A. 2006;103:18273–7. doi: 10.1073/pnas.0608537103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Petersen KF, Dufour S, Befroy D, Lehrke M, Hendler RE, Shulman GI. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes. 2005;54:603–8. doi: 10.2337/diabetes.54.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fabbrini E, Magkos F, Mohammed BS, Pietka T, Abumrad NA, Patterson BW, et al. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc Natl Acad Sci U S A. 2009;106:15430–5. doi: 10.1073/pnas.0904944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Krssak M, Falk Petersen K, Dresner A, DiPietro L, Vogel SM, Rothman DL, et al. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia. 1999;42:113–6. doi: 10.1007/s001250051123. [DOI] [PubMed] [Google Scholar]
- 86.Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, et al. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. 2004;279:32345–53. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- 87.Kim JK, Fillmore JJ, Chen Y, Yu C, Moore IK, Pypaert M, et al. Tissue-specific overexpression of lipoprotein lipase causes tissue-specific insulin resistance. Proc Natl Acad Sci U S A. 2001;98:7522–7. doi: 10.1073/pnas.121164498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Merkel M, Weinstock PH, Chajek-Shaul T, Radner H, Yin B, Breslow JL, et al. Lipoprotein lipase expression exclusively in liver. A mouse model for metabolism in the neonatal period and during cachexia. J Clin Invest. 1998;102:893–901. doi: 10.1172/JCI2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang H, Knaub LA, Jensen DR, Young Jung D, Hong EG, Ko HJ, et al. Skeletal muscle-specific deletion of lipoprotein lipase enhances insulin signaling in skeletal muscle but causes insulin resistance in liver and other tissues. Diabetes. 2009;58:116–24. doi: 10.2337/db07-1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Goudriaan JR, Dahlmans VE, Teusink B, Ouwens DM, Febbraio M, Maassen JA, et al. CD36 deficiency increases insulin sensitivity in muscle, but induces insulin resistance in the liver in mice. J Lipid Res. 2003;44:2270–7. doi: 10.1194/jlr.M300143-JLR200. [DOI] [PubMed] [Google Scholar]
- 91.Kim JK, Gimeno RE, Higashimori T, Kim HJ, Choi H, Punreddy S, et al. Inactivation of fatty acid transport protein 1 prevents fat-induced insulin resistance in skeletal muscle. J Clin Invest. 2004;113:756–63. doi: 10.1172/JCI18917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Doege H, Grimm D, Falcon A, Tsang B, Storm TA, Xu H, et al. Silencing of hepatic fatty acid transporter protein 5 in vivo reverses diet-induced non-alcoholic fatty liver disease and improves hyperglycemia. J Biol Chem. 2008;283:22186–92. doi: 10.1074/jbc.M803510200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Falcon A, Doege H, Fluitt A, Tsang B, Watson N, Kay MA, et al. FATP2 is a hepatic fatty acid transporter and peroxisomal very long-chain acyl-CoA synthetase. Am J Physiol Endocrinol Metab. 2010;299:E384–93. doi: 10.1152/ajpendo.00226.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Frayn KN. Adipose tissue as a buffer for daily lipid flux. Diabetologia. 2002;45:1201–10. doi: 10.1007/s00125-002-0873-y. [DOI] [PubMed] [Google Scholar]
- 95.Kim JK, Gavrilova O, Chen Y, Reitman ML, Shulman GI. Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. J Biol Chem. 2000;275:8456–60. doi: 10.1074/jbc.275.12.8456. [DOI] [PubMed] [Google Scholar]
- 96.Moitra J, Mason MM, Olive M, Krylov D, Gavrilova O, Marcus-Samuels B, et al. Life without white fat: a transgenic mouse. Genes Dev. 1998;12:3168–81. doi: 10.1101/gad.12.20.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lee K, Villena JA, Moon YS, Kim KH, Lee S, Kang C, et al. Inhibition of adipogenesis and development of glucose intolerance by soluble preadipocyte factor-1 (Pref-1) J Clin Invest. 2003;111:453–61. doi: 10.1172/JCI15924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Villena JA, Choi CS, Wang Y, Kim S, Hwang YJ, Kim YB, et al. Resistance to high-fat diet-induced obesity but exacerbated insulin resistance in mice overexpressing preadipocyte factor-1 (Pref-1): a new model of partial lipodystrophy. Diabetes. 2008;57:3258–66. doi: 10.2337/db07-1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lee SY, Lee HY, Song JH, Kim GT, Jeon S, Song YJ, et al. Adipocyte-specific deficiency of de novo sphingolipid biosynthesis leads to lipodystrophy and insulin resistance. Diabetes. 2017;66:2596–609. doi: 10.2337/db16-1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhou L, Park SY, Xu L, Xia X, Ye J, Su L, et al. Insulin resistance and white adipose tissue inflammation are uncoupled in energetically challenged Fsp27-deficient mice. Nat Commun. 2015;6:5949. doi: 10.1038/ncomms6949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Carpentier JL. Insulin receptor internalization: molecular mechanisms and physiopathological implications. Diabetologia. 1994;37 Suppl 2:S117–24. doi: 10.1007/BF00400835. [DOI] [PubMed] [Google Scholar]
- 102.Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148:852–71. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Turinsky J, O’Sullivan DM, Bayly BP. 1,2-Diacylglycerol and ceramide levels in insulin-resistant tissues of the rat in vivo. J Biol Chem. 1990;265:16880–5. [PubMed] [Google Scholar]
- 104.Turner N, Kowalski GM, Leslie SJ, Risis S, Yang C, Lee-Young RS, et al. Distinct patterns of tissue-specific lipid accumulation during the induction of insulin resistance in mice by high-fat feeding. Diabetologia. 2013;56:1638–48. doi: 10.1007/s00125-013-2913-1. [DOI] [PubMed] [Google Scholar]
- 105.Takayama S, White MF, Lauris V, Kahn CR. Phorbol esters modulate insulin receptor phosphorylation and insulin action in cultured hepatoma cells. Proc Natl Acad Sci U S A. 1984;81:7797–801. doi: 10.1073/pnas.81.24.7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Considine RV, Nyce MR, Allen LE, Morales LM, Triester S, Serrano J, et al. Protein kinase C is increased in the liver of humans and rats with non-insulin-dependent diabetes mellitus: an alteration not due to hyperglycemia. J Clin Invest. 1995;95:2938–44. doi: 10.1172/JCI118001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Karasik A, Rothenberg PL, Yamada K, White MF, Kahn CR. Increased protein kinase C activity is linked to reduced insulin receptor autophosphorylation in liver of starved rats. J Biol Chem. 1990;265:10226–31. [PubMed] [Google Scholar]
- 108.Petersen MC, Madiraju AK, Gassaway BM, Marcel M, Nasiri AR, Butrico G, et al. Insulin receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J Clin Invest. 2016;126:4361–71. doi: 10.1172/JCI86013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Samuel VT, Liu ZX, Wang A, Beddow SA, Geisler JG, Kahn M, et al. Inhibition of protein kinase C epsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J Clin Invest. 2007;117:739–45. doi: 10.1172/JCI30400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ter Horst KW, Gilijamse PW, Versteeg RI, Ackermans MT, Nederveen AJ, la Fleur SE, et al. Hepatic diacylglycerol-associated protein kinase Cε translocation links hepatic steatosis to hepatic insulin resistance in humans. Cell Rep. 2017;19:1997–2004. doi: 10.1016/j.celrep.2017.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Magkos F, Su X, Bradley D, Fabbrini E, Conte C, Eagon JC, et al. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology. 2012;142:1444–6. doi: 10.1053/j.gastro.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Schmitz-Peiffer C, Browne CL, Oakes ND, Watkinson A, Chisholm DJ, Kraegen EW, et al. Alterations in the expression and cellular localization of protein kinase C isozymes epsilon and theta are associated with insulin resistance in skeletal muscle of the high-fat-fed rat. Diabetes. 1997;46:169–78. doi: 10.2337/diab.46.2.169. [DOI] [PubMed] [Google Scholar]
- 113.Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. 2002;277:50230–6. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]
- 114.Li Y, Soos TJ, Li X, Wu J, Degennaro M, Sun X, et al. Protein kinase C Theta inhibits insulin signaling by phosphorylating IRS1 at Ser(1101) J Biol Chem. 2004;279:45304–7. doi: 10.1074/jbc.C400186200. [DOI] [PubMed] [Google Scholar]
- 115.Szendroedi J, Yoshimura T, Phielix E, Koliaki C, Marcucci M, Zhang D, et al. Role of diacylglycerol activation of PKCθ in lipid-induced muscle insulin resistance in humans. Proc Natl Acad Sci U S A. 2014;111:9597–602. doi: 10.1073/pnas.1409229111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Avignon A, Yamada K, Zhou X, Spencer B, Cardona O, Saba-Siddique S, et al. Chronic activation of protein kinase C in soleus muscles and other tissues of insulin-resistant type II diabetic Goto-Kakizaki (GK), obese/aged, and obese/Zucker rats. A mechanism for inhibiting glycogen synthesis. Diabetes. 1996;45:1396–404. doi: 10.2337/diab.45.10.1396. [DOI] [PubMed] [Google Scholar]
- 117.Qu X, Seale JP, Donnelly R. Tissue and isoform-selective activation of protein kinase C in insulin-resistant obese Zucker rats: effects of feeding. J Endocrinol. 1999;162:207–14. doi: 10.1677/joe.0.1620207. [DOI] [PubMed] [Google Scholar]
- 118.Raddatz K, Turner N, Frangioudakis G, Liao BM, Pedersen DJ, Cantley J, et al. Time-dependent effects of Prkce deletion on glucose homeostasis and hepatic lipid metabolism on dietary lipid oversupply in mice. Diabetologia. 2011;54:1447–56. doi: 10.1007/s00125-011-2073-0. [DOI] [PubMed] [Google Scholar]
- 119.Lee HY, Birkenfeld AL, Jornayvaz FR, Jurczak MJ, Kanda S, Popov V, et al. Apolipoprotein CIII overexpressing mice are predisposed to diet-induced hepatic steatosis and hepatic insulin resistance. Hepatology. 2011;54:1650–60. doi: 10.1002/hep.24571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kim JK, Fillmore JJ, Sunshine MJ, Albrecht B, Higashimori T, Kim DW, et al. PKC-theta knockout mice are protected from fat-induced insulin resistance. J Clin Invest. 2004;114:823–7. doi: 10.1172/JCI22230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Haasch D, Berg C, Clampit JE, Pederson T, Frost L, Kroeger P, et al. PKC theta is a key player in the development of insulin resistance. Biochem Biophys Res Commun. 2006;343:361–8. doi: 10.1016/j.bbrc.2006.02.177. [DOI] [PubMed] [Google Scholar]
- 122.Rando RR, Young N. The stereospecific activation of protein kinase C. Biochem Biophys Res Commun. 1984;122:818–23. doi: 10.1016/s0006-291x(84)80107-2. [DOI] [PubMed] [Google Scholar]
- 123.Cantley JL, Yoshimura T, Camporez JP, Zhang D, Jornayvaz FR, Kumashiro N, et al. CGI-58 knockdown sequesters diacylglycerols in lipid droplets/ER-preventing diacylglycerol-mediated hepatic insulin resistance. Proc Natl Acad Sci U S A. 2013;110:1869–74. doi: 10.1073/pnas.1219456110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Brown JM, Betters JL, Lord C, Ma Y, Han X, Yang K, et al. CGI-58 knockdown in mice causes hepatic steatosis but prevents diet-induced obesity and glucose intolerance. J Lipid Res. 2010;51:3306–15. doi: 10.1194/jlr.M010256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chavez JA, Summers SA. A ceramide-centric view of insulin resistance. Cell Metab. 2012;15:585–94. doi: 10.1016/j.cmet.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 126.Luukkonen PK, Zhou Y, Sadevirta S, Leivonen M, Arola J, Oresic M, et al. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J Hepatol. 2016;64:1167–75. doi: 10.1016/j.jhep.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 127.Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5:167–79. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 128.Ussher JR, Koves TR, Cadete VJ, Zhang L, Jaswal JS, Swyrd SJ, et al. Inhibition of de novo ceramide synthesis reverses dietinduced insulin resistance and enhances whole-body oxygen consumption. Diabetes. 2010;59:2453–64. doi: 10.2337/db09-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Turpin SM, Nicholls HT, Willmes DM, Mourier A, Brodesser S, Wunderlich CM, et al. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab. 2014;20:678–86. doi: 10.1016/j.cmet.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 130.Xia JY, Holland WL, Kusminski CM, Sun K, Sharma AX, Pearson MJ, et al. Targeted induction of ceramide degradation leads to improved systemic metabolism and reduced hepatic steatosis. Cell Metab. 2015;22:266–78. doi: 10.1016/j.cmet.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Holland WL, Xia JY, Johnson JA, Sun K, Pearson MJ, Sharma AX, et al. Inducible overexpression of adiponectin receptors highlight the roles of adiponectin-induced ceramidase signaling in lipid and glucose homeostasis. Mol Metab. 2017;6:267–75. doi: 10.1016/j.molmet.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Powell DJ, Hajduch E, Kular G, Hundal HS. Ceramide disables 3-phosphoinositide binding to the pleckstrin homology domain of protein kinase B (PKB)/Akt by a PKCzeta-dependent mechanism. Mol Cell Biol. 2003;23:7794–808. doi: 10.1128/MCB.23.21.7794-7808.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Stratford S, Hoehn KL, Liu F, Summers SA. Regulation of insulin action by ceramide: dual mechanisms linking ceramide accumulation to the inhibition of Akt/protein kinase B. J Biol Chem. 2004;279:36608–15. doi: 10.1074/jbc.M406499200. [DOI] [PubMed] [Google Scholar]
- 134.Teruel T, Hernandez R, Lorenzo M. Ceramide mediates insulin resistance by tumor necrosis factor-alpha in brown adipocytes by maintaining Akt in an inactive dephosphorylated state. Diabetes. 2001;50:2563–71. doi: 10.2337/diabetes.50.11.2563. [DOI] [PubMed] [Google Scholar]
- 135.Galbo T, Perry RJ, Nishimura E, Samuel VT, Quistorff B, Shulman GI. PP2A inhibition results in hepatic insulin resistance despite Akt2 activation. Aging (Albany NY) 2013;5:770–81. doi: 10.18632/aging.100611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Holland WL, Adams AC, Brozinick JT, Bui HH, Miyauchi Y, Kusminski CM, et al. An FGF21-adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell Metab. 2013;17:790–7. doi: 10.1016/j.cmet.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med. 2011;17:55–63. doi: 10.1038/nm.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–88. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–61. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 140.Okada T, Yoshida H, Akazawa R, Negishi M, Mori K. Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem J. 2002;366(Pt 2):585–94. doi: 10.1042/BJ20020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–40. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Laybutt DR, Preston AM, Akerfeldt MC, Kench JG, Busch AK, Biankin AV, et al. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia. 2007;50:752–63. doi: 10.1007/s00125-006-0590-z. [DOI] [PubMed] [Google Scholar]
- 143.Marchetti P, Bugliani M, Lupi R, Marselli L, Masini M, Boggi U, et al. The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia. 2007;50:2486–94. doi: 10.1007/s00125-007-0816-8. [DOI] [PubMed] [Google Scholar]
- 144.Lee AH, Heidtman K, Hotamisligil GS, Glimcher LH. Dual and opposing roles of the unfolded protein response regulated by IRE1alpha and XBP1 in proinsulin processing and insulin secretion. Proc Natl Acad Sci U S A. 2011;108:8885–90. doi: 10.1073/pnas.1105564108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115:1111–9. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 147.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116:1494–505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. 2006;116:115–24. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307) J Biol Chem. 2000;275:9047–54. doi: 10.1074/jbc.275.12.9047. [DOI] [PubMed] [Google Scholar]
- 151.Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–8. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- 152.Wunderlich FT, Luedde T, Singer S, Schmidt-Supprian M, Baumgartl J, Schirmacher P, et al. Hepatic NF-kappa B essential modulator deficiency prevents obesity-induced insulin resistance but synergizes with high-fat feeding in tumorigenesis. Proc Natl Acad Sci U S A. 2008;105:1297–302. doi: 10.1073/pnas.0707849104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tang T, Zhang J, Yin J, Staszkiewicz J, Gawronska-Kozak B, Jung DY, et al. Uncoupling of inflammation and insulin resistance by NF-kappaB in transgenic mice through elevated energy expenditure. J Biol Chem. 2010;285:4637–44. doi: 10.1074/jbc.M109.068007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–71. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–2. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.NCD Risk Factor Collaboration (NCD-RisC) Worldwide trends in diabetes since 1980: a pooled analysis of 751 population-based studies with 4.4 million participants. Lancet. 2016;387:1513–30. doi: 10.1016/S0140-6736(16)00618-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Boden G. Pathogenesis of type 2 diabetes. Insulin resistance. Endocrinol Metab Clin North Am. 2001;30:801–15. doi: 10.1016/s0889-8529(05)70216-4. [DOI] [PubMed] [Google Scholar]
- 158.Proks P, Reimann F, Green N, Gribble F, Ashcroft F. Sulfonylurea stimulation of insulin secretion. Diabetes. 2002;51 Suppl 3:S368–76. doi: 10.2337/diabetes.51.2007.s368. [DOI] [PubMed] [Google Scholar]
- 159.Hauner H. The mode of action of thiazolidinediones. Diabetes Metab Res Rev. 2002;18 Suppl 2:S10–5. doi: 10.1002/dmrr.249. [DOI] [PubMed] [Google Scholar]
- 160.Foretz M, Guigas B, Viollet B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat Rev Endocrinol. 2019;15:569–89. doi: 10.1038/s41574-019-0242-2. [DOI] [PubMed] [Google Scholar]
- 161.Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. 2017;60:1577–85. doi: 10.1007/s00125-017-4342-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kumar S, Narwal S, Kumar V, Prakash O. α-Glucosidase inhibitors from plants: a natural approach to treat diabetes. Pharmacogn Rev. 2011;5:19–29. doi: 10.4103/0973-7847.79096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Abdul-Ghani MA, Norton L, Defronzo RA. Role of sodium-glucose cotransporter 2 (SGLT 2) inhibitors in the treatment of type 2 diabetes. Endocr Rev. 2011;32:515–31. doi: 10.1210/er.2010-0029. [DOI] [PubMed] [Google Scholar]
- 164.Guerra J, Dias M, Brilhante A, Terra MF, Garcia-Arevalo M, Figueira A. Multifactorial basis and therapeutic strategies in metabolism-related diseases. Nutrients. 2021;13:2830. doi: 10.3390/nu13082830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Savage DB, Choi CS, Samuel VT, Liu ZX, Zhang D, Wang A, et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J Clin Invest. 2006;116:817–24. doi: 10.1172/JCI27300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Nagle CA, An J, Shiota M, Torres TP, Cline GW, Liu ZX, et al. Hepatic overexpression of glycerol-sn-3-phosphate acyltransferase 1 in rats causes insulin resistance. J Biol Chem. 2007;282:14807–15. doi: 10.1074/jbc.M611550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Neschen S, Morino K, Hammond LE, Zhang D, Liu ZX, Romanelli AJ, et al. Prevention of hepatic steatosis and hepatic insulin resistance in mitochondrial acyl-CoA:glycerol-sn3-phosphate acyltransferase 1 knockout mice. Cell Metab. 2005;2:55–65. doi: 10.1016/j.cmet.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 168.Ryu D, Seo WY, Yoon YS, Kim YN, Kim SS, Kim HJ, et al. Endoplasmic reticulum stress promotes LIPIN2-dependent hepatic insulin resistance. Diabetes. 2011;60:1072–81. doi: 10.2337/db10-1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ryu D, Oh KJ, Jo HY, Hedrick S, Kim YN, Hwang YJ, et al. TORC2 regulates hepatic insulin signaling via a mammalian phosphatidic acid phosphatase, LIPIN1. Cell Metab. 2009;9:240–51. doi: 10.1016/j.cmet.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 170.Choi CS, Savage DB, Kulkarni A, Yu XX, Liu ZX, Morino K, et al. Suppression of diacylglycerol acyltransferase-2 (DGAT2), but not DGAT1, with antisense oligonucleotides reverses diet-induced hepatic steatosis and insulin resistance. J Biol Chem. 2007;282:22678–88. doi: 10.1074/jbc.M704213200. [DOI] [PubMed] [Google Scholar]
- 171. Clarivate Analytics: Cortellis Drug Discovery Intelligence. Available from: http://www.cortellis.com/drugdiscovery (cited 2021 Dec 16)
- 172.Choi CS, Fillmore JJ, Kim JK, Liu ZX, Kim S, Collier EF, et al. Overexpression of uncoupling protein 3 in skeletal muscle protects against fat-induced insulin resistance. J Clin Invest. 2007;117:1995–2003. doi: 10.1172/JCI13579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Choi CS, Savage DB, Abu-Elheiga L, Liu ZX, Kim S, Kulkarni A, et al. Continuous fat oxidation in acetyl-CoA carboxylase 2 knockout mice increases total energy expenditure, reduces fat mass, and improves insulin sensitivity. Proc Natl Acad Sci U S A. 2007;104:16480–5. doi: 10.1073/pnas.0706794104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med. 2013;19:83–92. doi: 10.1038/nm.3014. [DOI] [PubMed] [Google Scholar]
- 175.Wang YX, Zhang CL, Yu RT, Cho HK, Nelson MC, Bayuga-Ocampo CR, et al. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol. 2004;2:e294. doi: 10.1371/journal.pbio.0020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wilkes JJ, Lloyd DJ, Gekakis N. Loss-of-function mutation in myostatin reduces tumor necrosis factor alpha production and protects liver against obesity-induced insulin resistance. Diabetes. 2009;58:1133–43. doi: 10.2337/db08-0245. [DOI] [PMC free article] [PubMed] [Google Scholar]