

Abstract
The ETS family of proteins consists of 28 transcription factors, many of which play critical roles in both normal tissue development and homeostasis and have been implicated in development and progression of a variety of cancers. In prostate cancer, gene fusion and overexpression of ETS factors ERG, FLI1, ETV1, ETV4 and ETV5 have been found in half of prostate cancer patients in Caucasian men and define the largest genetic subtype of prostate cancer. This review summarizes the data on the discovery, modeling, molecular taxonomy, lineage plasticity and therapeutic targeting of ETS family members in prostate cancer.
Keywords: Prostate Cancer, Transcription factors, Oncogenic translocations, Androgen receptor, ERG
1. Introduction
E26 transformation-specific (ETS) family proteins represent a large family of 28 human transcription factors that share a highly conserved DNA-binding domain with a winged helix–turn–helix (wHTH) topology that bind to 5’-GGA(A/T)-3’ motifs in DNA [1–3]. ETS factors have long been implicated in tumorigenesis. The founding member, ETS1, was first discovered in the E26 avian retrovirus v-ets, that cause erythroblastosis in chickens [4]. Subsequently, FLI1 and SPI1 were discovered to be the integration sites of Friend murine leukemia virus (F-MuLV) and Friend spleen focus-forming virus (SFFV)-induced erythroleukemias respectively [5, 6]. Definitive evidence of the causal role of ETS factors in human malignancy was first discovered by the finding of recurrent translocations between the EWSR1 gene on chr22 and the FLI1 gene on chr11 in Ewing’s Sarcoma [7]. “Ewing’s family sarcomas” are molecularly defined by a family of protein fusions with RNA-binding proteins, EWSR1 and FUS, comprising the amino-terminus and ETS factors, FLI1, ERG, FEV, ETV1 and ETV4, on the carboxyl-terminus [8]. Similar translocations were also discovered in a subset of leukemias [9].
In 2005, gene fusions between the androgen responsive gene TMPRSS2 and ETS factors ERG and ETV1 were found to occur in half of all prostate cancers [10]. The role of ETS in prostate cancer has been a novel subject of intense research and over 1,800 studies have been published on ETS factors and prostate cancer. Here, we review key findings on the mechanistic role of ETS factors in tumorigenesis from both modeling and biochemical studies, and provide an update on efforts targeting ETS fusions in prostate cancer.
Discovery of ETS family fusions in prostate cancer
The landmark discovery of the Philadelphia chromosome, a translocation between chr9 and chr22 in chronic myelogenous leukemia by Peter Nowell and David Hungerford in 1960, showed that human cancers are caused by genetic aberrations [11]. In the subsequent years, cytogenetics studies have found numerous recurrent translocations that defined molecular subtypes of leukemias, sarcomas, and lymphomas [12]. However, until the mid-2000’s, recurrent chromosomal translocations were not thought to occur in solid tumors. Next-generation sequencing technologies were not yet available, and the aneuploid genomes typical of solid tumors made cytogenetic studies difficult.
Chromosomal translocations and amplifications frequently cause outlier overexpression of the involved gene. In 2005, harnessing the newly available gene expression microarray datasets of clinical samples, Scott Tomlins, Arul Chinnaiyan and colleagues bioinformatically identified genes with outlier overexpression. In addition to finding already known amplifications and translocations, they discovered mutually exclusive outlier overexpression of ERG (21q22.3) in up to 50% and of ETV1 (7q21.2) in up to 10% of prostate cancers [10]. RACE (Rapid amplification of cDNA ends) found chimeric mRNA between the 5’ untranslated region of TMPRSS2 and 3’ end of ERG or ETV1 leading to the overexpression of slightly N-terminal truncated ETS proteins. The most common genomic event that causes ERG overexpression is a deletion of a ~3 Mb DNA fragment between TMPRSS2 and ERG on chr21. TMPRSS2 encodes a transmembrane serine protease that is highly expressed in prostate luminal epithelium as well as epithelial cells of other organs [13]. Since this initial discovery, several other ETS factors, including FLI1, ETV4, and ETV5 as well as multiple other fusion partners have been characterized (Figure 1) [10, 14–17]. After the landmark discovery of ETS fusions in prostate cancer, many other recurrent fusions have been discovered in solid tumors (e.g., ALK, ROS1, RET and NTRK) and have altered the molecular diagnosis and targeted therapy landscape in solid tumors [18, 19].
Figure 1.
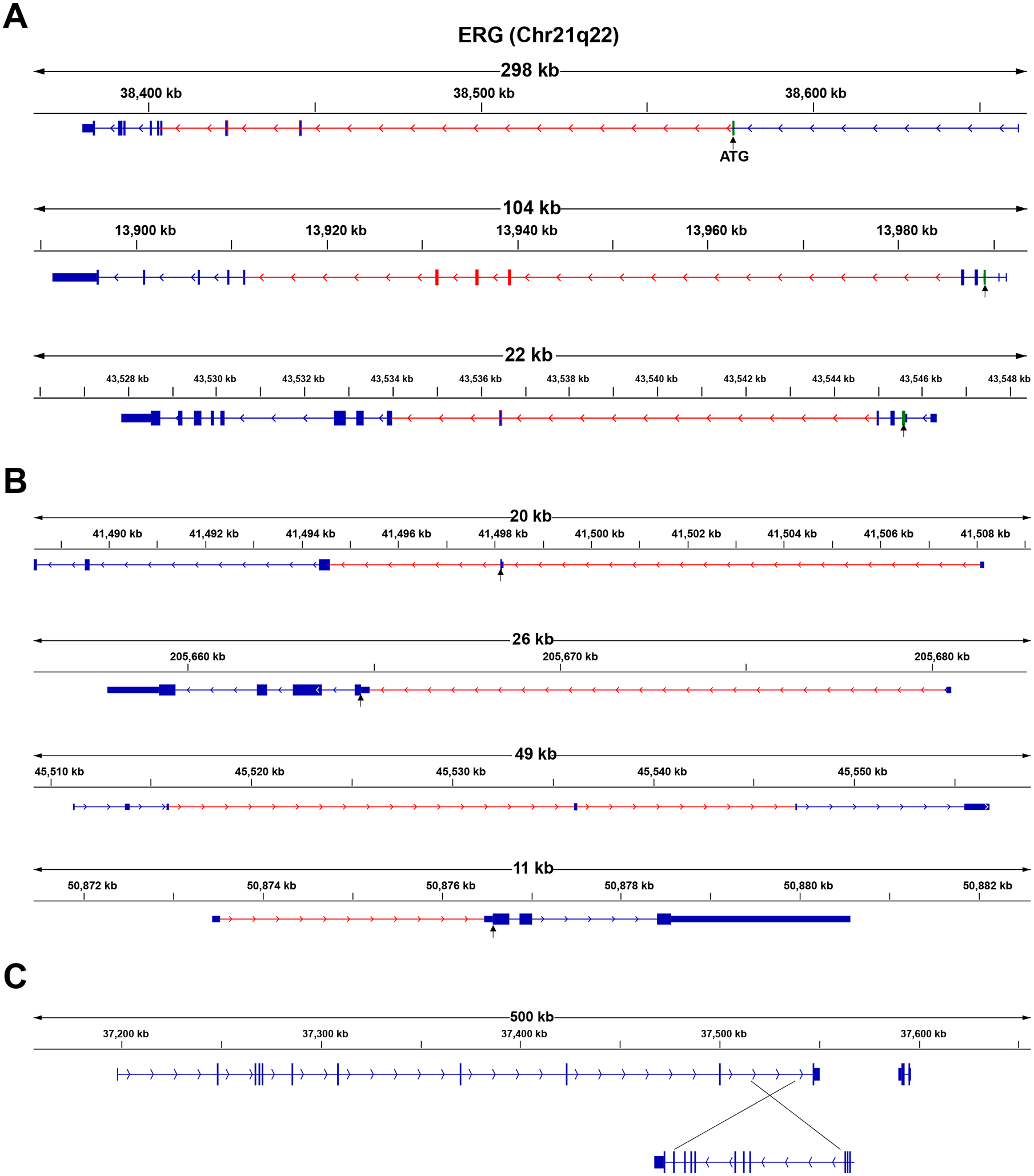
Structural variation that lead to overexpression of ETS proteins. A) Genomic view of ERG, ETV1, and ETV4, three most common ETS genes translocated in prostate cancer. The common introns with fusion breakpoints are in red. Breakpoints frequently occur after the first coding exon, before the ETS domain and usually in longer introns. B) Genomic view of common 5’ fusion partners, TMPRSS2, SLC45A3, C15orf21 and KLK2. The common introns with fusion breakpoints are in red. Most breakpoints occur before the first coding exon and some 5’ partners, such as SLC45A3, is a non-coding RNA. Fusions with coding components, such as TMPRSS2 intron 2 fusion, is limited by requirement to maintain correct coding frame. C) Schematic showing translocation of the entire ETV1 coding region into the MIPOL1-FOXA1 locus. This is a highly active transcriptional locus the prostate lineage and leads to aberrant overexpression of the full length ETV1 protein.
Genetic fusions can be classified as those that generate gain-of-function chimeric proteins (e.g., BCR-ABL that results in a constitutively activated ABL kinase) or promoter/enhancer hijacking that result in aberrant overexpression of a wild-type oncoprotein (e.g., IgG-MYC, IgG-BCL2 fusions in Burkitt’s and follicular lymphoma respectively) [12]. While EWS-FLI1 and EWS-ERG fusions in Ewing’s sarcoma generate a gain-of-function chimeric protein, prostate cancer ETS fusions rarely contain coding components of the fusion partner. Instead, ETS fusions lead to overexpression of either full length or slightly truncated proteins. For the ETS gene, the breakpoint tends to be in one of the larger introns at the beginning of the gene right after the first coding exon (Figure 1A). For the fusion partner, the breakpoint is at an early intron that may or may not include any coding components (Figure 1B). For example, TMPRSS2-ERG fusion, the most common breakpoints are intron 1 of TMPRSS2 and intron 3, intron 4 or intron 5 of ERG, leading to a transcript with the 5’ untranslated region (5’-UTR) of TMPRSS2 and a slightly amino-terminal truncated ERG protein that starts at exon 4, exon 5 or exon 6. Less frequently, intron 2 of TMPRSS2 can be involved leading to inclusion of short protein sequence of TMPRSS2. The multiple 5’ fusion partners share high prostate epithelial expression and including both coding as well as non-coding genes, such as the non-coding RNAs C15orf21, EST14, FLI35294 and the endogenous retroviral element HERV-K17.
The mechanistic basis that generate recurrent fusions in cancer is understood for only a few cases. For example, lymphomas and multiple myelomas harbor recurrent translocations involving the immunoglobin heavy, light chains or T-cell receptors (TCR) with oncogenes such as MYC, BLC2, BCL6 or FGFR3. These translocations arise from errors that occur during V(D)J recombination or class switch recombination that cause DNA breaks to generate antibody and TCR diversity [20]. In prostate cancer three studies have suggested that recurrent ETS rearrangements are not random events but a consequence of androgen receptor activity. In experimental systems, AR binding to TMPRSS2 and ERG introns can bring the two loci together and recruit enzymes, including topoisomerase II beta, cytidine deaminase, and LINE1ORF2, that can induce DNA double-stranded breaks and which are then ligated by DNA repair machinery to generate the fusion [21–23]. Recently, a study of human prostate specimens suggests that bacterial infection can lead to DNA breaks and ERG translocation [24]. Chronic inflammation and bacterial infection leading proliferative inflammatory atrophy (PIA) is a known precursor to invasive cancer [25]. Examining non-neoplastic areas of prostatectomy samples with bacterial prostatitis, the investigators identified TMPRSS2:ERG rearrangements.
In addition to gene-to-gene fusions that generate chimeric ETS-transcripts, other genomic structural variations have been characterized that also lead to aberrant ETS expression. For example, the entire ETV1 gene can be translocated to an actively transcribed locus that lead to overexpression of full length ETV1. The MIPOL1/FOXA1 locus on chr14q is a recurrent target for ETV1 translocation and this translocation is found in two prostate cancer cell lines, LNCaP and MDA-PCa2b (Figure 1C) [14, 26, 27]. In the TCGA cohort, while all cases ERG overexpression were due to gene fusion, a large fraction of ETV1, ETV4, and FLI1-positive tumors overexpressed the full-length transcript suggesting that “enhancer hijacking” where the entire normally transcriptionally silent locus is rearranged to an active locus leading to overexpression.
The importance of ETS overexpression and its activity in prostate cancer tumorigenesis is highlighted by the presence of additional alterations in the ETS pathway (Figure 2). The PEA3 subfamily of ETS factors, ETV1, ETV4, and ETV5 are degraded by the E3-ligase COP1, and COP1 loss causes dramatically increased protein levels of these ETS factors [28]. COP1 deletion is observed in a subset of prostate cancers and also the prostate cancer cell line PC3 [29]. In a genetically engineered mouse model (GEMM), Cop1 deletion in the prostate causes prostatic intraepithelial neoplasia (PIN) and cooperates with Pten deletion to cause invasive cancer [29]. ERF (Ets2 Repressor Factor) is an ETS repressive factor that is frequently mutated or deleted in prostate cancer, especially in patients of African American ancestry [30]. ERF binds to the same genomic sites as ERG but suppresses transcription. ERF loss phenocopies ERG overexpression and cooperates with Pten loss in tumorigenesis [31]. Capicua (CIC) is a transcriptional repressor of ETV1, ETV4, and ETV5 and a known tumor suppressor in oligodendroglioma and neuroblastoma [32, 33]. CIC is located adjacent to ERF on chromosome chr19q13.2 and co-deleted with ERF in prostate cancer [30].
Figure 2.
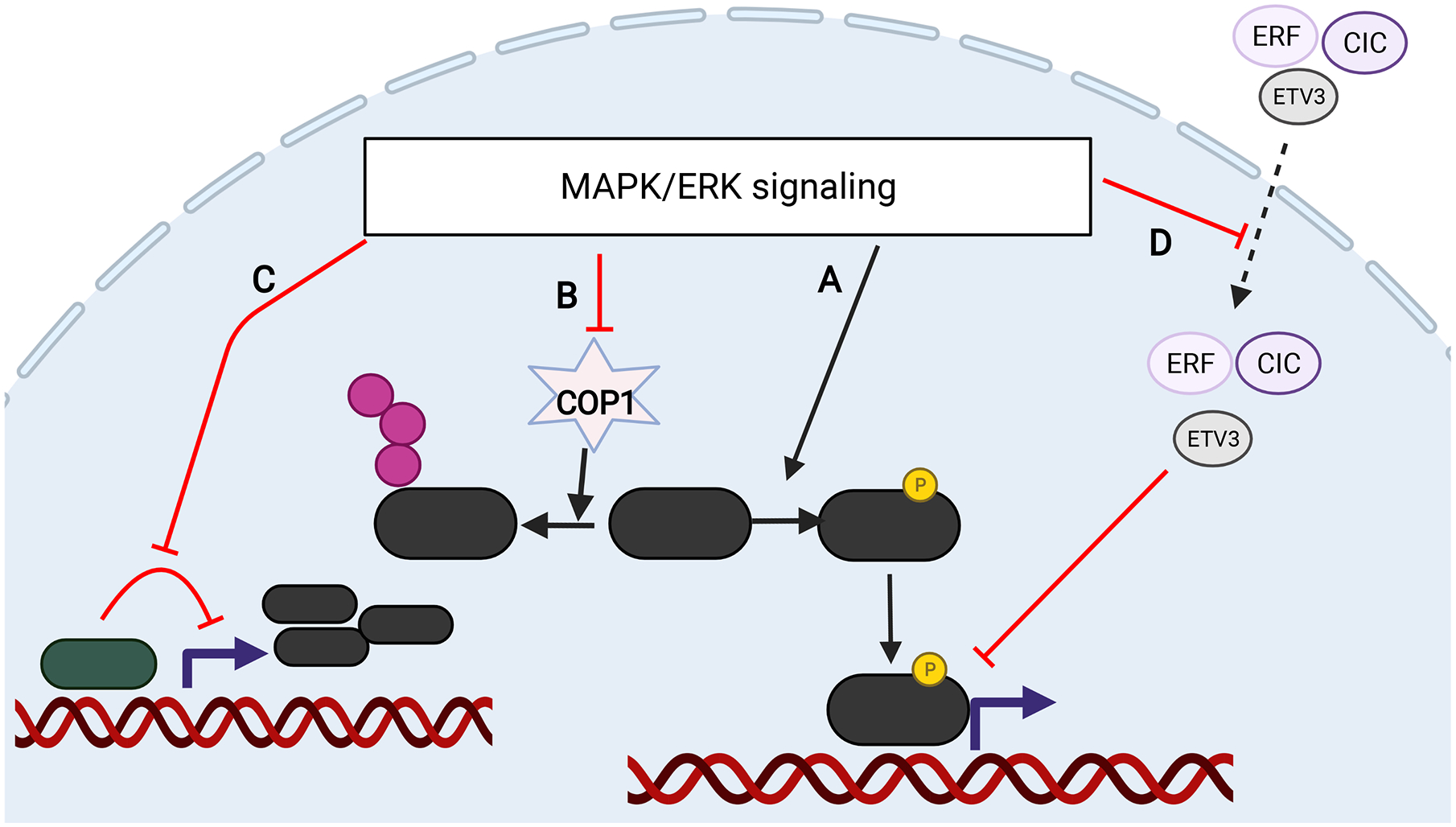
Schematic of ETS activation by MAPK and loss of tumor suppressors that activate ETS. A) ETS proteins ERG, ETV1, ETV4, and ETV5 can be phosphorylated by MAPK that activates transcriptional activity. B) PEA3 factors ETV1, ETV4, and ETV5 are ubiquitinated by the E3 ligase COP1 and this process is inhibited by MAPK signaling. COP1 is a tumor suppressor lost in prostate cancer that leads to elevated ETV1/4/5. In addition, most ETV1/4/5 fusions lose the N-terminal domain that contain the degron, leading to stabilized proteins. C) CIC is a transcriptional suppressor of PEA3 factors (especially ETV4) and its activity is inhibited by MAPK signaling. CIC, located next to ERF, is deleted in prostate cancer. D) ERF and ETV3 are ETS transcriptional suppressors that bind to ETS binding sites and inhibit transcription. When phosphorylated by MAPK, they are exported from the nucleus, facilitating positive ETS factors to bind and activate transcription. ERF is mutated and deleted in prostate cancer.
2. ETS fusions define a molecular subtype of prostate cancer
Strong evidence suggests that ETS fusions are an early genetic event that define a prostate cancer molecular subtype: 1) On IHC, ERG positivity can be detected in precursor high grade prostatic intraepithelial neoplasia (HGPIN) whereas PTEN loss, a common cooperating genetic event, is not detected [34]. 2) In ERG-positive prostate cancers, ERG IHC is uniformly positive within the clone (there does exist “collision tumors” were two or distinct primary cancer coexist). On the other hand, PTEN loss is frequently heterogeneous and subclonal [35]. 3) ERG fusion can be the only detectable copy number alteration and mutational event in primary prostate cancer [17, 35, 36]. 4) ERG-positive prostate cancers can be distinguished with a specific transcriptional program and epigenomic landscape defined by histone acetylation and DNA methylation [17, 37]. 6) Not only are different ETS fusions mutually exclusive with each other, they are also mutually exclusive with SPOP mutation, FOXA1 mutation, and IDH1 mutation form molecular distinct subgroups of prostate cancer [17].
Whether the ETS-fusion subtype of prostate cancer exhibits clinically distinct behavior is still controversial. Initial studies reported conflicting roles of ERG fusion in grade, stage, and recurrence rates [38–41]. However, larger cohorts suggest that ERG fusion status alone does not alter prognosis [42, 43]. In prostate cancer, ERG fusion is significantly associated with loss of several tumor suppressors, including PTEN, TP53 and a region of chr3p13 that includes FOXP1, SHQ1, RYBP. The concomitant loss of these tumor suppressors with ERG fusion does appear to have detrimental effect on clinical behaviors [36, 44–48].
Most studies of ETS-fusions have focused on ERG, given its high prevalence and the availability of IHC reagents. Whether different ETS fusions cause distinct transcriptional programs and definite separate subtypes or not are well known. ERG and FLI1 are close homologs in the ERG subfamily of ETS factors that are endogenously expressed in the hematopoietic and endothelial lineages. They are among the POINTED (PNT)-domains containing ETS factors [49, 50]. ETV1, ETV4, and ETV5 are in the PEA3 subfamily of ETS factors that are endogenously expressed in several lineages including neuronal and neural crest derivatives [51–53]. The TCGA showed that ETV1 and ETV4 overexpressing tumors were mostly clustered distinctly from ERG overexpressing ones. Several studies show that ERG and ETV1 may differentially regulate some downstream targets and ETV1 overexpressing tumors may have a worse clinical prognosis [27, 54].
Initial reports of the high prevalence ETS fusions focused on cohorts that were predominantly comprised of Caucasian men. Several studies have shown that the prevalence of ERG fusions in African-American and Asian men is less than half (~15–25% vs 40–60%) of that in Caucasian men [30, 55–58]. These studies also show that PTEN loss, which is significantly associated with ERG fusions, is also much less common in non-Caucasian populations. A recent study of Asian prostate cancer showed 41% of native patients had FOXA1 mutations [55]. These data indicate that genetic and/or environmental factors may affect mutagenic processes that drive prostate cancer.
3. Genetic Modeling of ETS factors
The fact that aberrant expression of wild-type ETS factors is oncogenic in the human prostate epithelium has led to a conundrum, that is, how does expression of a normal protein in other lineages, when aberrantly expressed in prostate cells, lead to tumorigenesis? For example, ERG staining by IHC in prostate cancer cells is comparable in intensity to adjacent endothelial cells that endogenously express ERG [59]; ETV1 expression in prostate cancer is no higher than normal interstitial cells of Cajal (ICC) or neural subsets that endogenously express the protein [51, 60]. The genomic binding (cistrome) and function of transcription factors depend strongly on the cellular lineage and epigenetic context. For example, endogenous ERG in endothelial cells is a master regulator of the lineage [61, 62] while endogenous ETV1 is a master regulator of the ICC lineage [51]. With ERG, several IHC studies of thousands of solid tumors have shown that ERG expression is highly specific for prostate cancer and vascular neoplasms where ERG is endogenously expressed [59]. Therefore, it is critical to study ETS expression in the prostate epithelial context.
Soon after ETS fusions were discovered, multiple laboratories generated genetic engineered mouse (GEM) models that overexpress ETS fusion in the prostate epithelium. Due to the ease rapidity to generate transgenic mice, the first generation GEM models involved transgenic overexpression of full-length ERG [63, 64], N-terminal truncated ERG that starts in exon 4 to recapitulate the fusion transcript ERG [65, 66], and N-terminal truncated ETV1 that starts in exon 4 [14, 67] all under the probasin promoter (ARR2Pb). These models have some limitations including the inability for temporal control to initiate gene expression. Moreover, probasin is a mouse specific gene that is expressed a subset of luminal cells and its expression highly suppressed in mouse prostate tumors whereas Tmprss2 is more broadly expressed in all luminal cells and its expression is unaltered by tumorigenesis. Several GEM models addressed some of these limitations. Our lab generated a knock-in of a conditional allele of truncated ERG in to the mouse constitutive Rosa26 locus [68] where expression can be initiated by selecting an appropriate Cre-driver, such as Pbsn-Cre or Tmrpss2-CreERT2 [68]. In this model, once recombined, the expression of ERG is not androgen dependent but constitutive under Rosa26. To mimic regulation of the ETS gene by TMPRSS2, Baena and colleagues knocked truncated ERG and truncated ETV1 into intron 2 of mouse Tmprss2 [69] while Casey and colleagues generated a human bacterial artificial chromosome (hBAC) transgenic with truncated ERG engineered into intron 2 of human TMPRSS2 [70].
While initial reports varied, the consensus is that in mice, aberrant ETS overexpression may cause hyperplasia and low-grade PIN with variable penetrance but does not cause invasive cancer [71, 72]. However, the most of these mice exhibit ETS expression levels that are much lower than that found in human prostate cancer, and for knock-in alleles Tmprss2 and Rosa26 expression in the mouse prostate epithelium are both lower than TMPRSS2 expression the human prostate. One transgenic strain with particularly high ERG expression developed prostate cancer over time, though there was still no early neoplastic phenotype despite high ERG expression [67, 73]. Of note, GEM models of recurrent SPOP mutations implicated to be an early genetic event that defines a separate subgroup of prostate cancer also only exhibit mild phenotype in mice [74].
As an alternative to GEM models, the Owen Witte laboratory used a renal capsule recombination model to study ETS function. They isolated prostate epithelial cells, transduced them with ETS-overexpressing lentiviral vectors, and recombined the cells with embryonic urogenital sinus mesenchymal (UGSM) cells in the renal capsule. When normal untransduced epithelial cells are used, each cell can generate a normal glandular structure containing both basal and luminal cells [75]. Using this system, they found that ERG and ETV1 overexpression alone caused hyperplasia but not invasive cancer, consistent with the GEM models. Notably, they showed that ETS-overexpressing cells generated glands with a skewed lineage composition and loss of CK5-positive basal cells [76]. One important advantage of this system is that it can be applied to human prostate epithelial cells to study human prostate cancer. Using this system, the Witte laboratory showed that overexpression of AKT and ERG in benign human basal cells, rather than luminal cells can induce HGPIN when grafted into SCID mice. The HGPIN strongly resembles human disease, where dysplastic pre-cancerous luminal cells sit on top of histologically normal basal cells. Additional overexpression of AR in this system led to invasive cancer with a luminal phenotype that recapitulates human prostate cancer [77].
4. Interaction between ETS expression, luminal fate, and AR
The prostate epithelium is comprised on luminal cells that secrete the prostatic fluid, basal cells and rare neuroendocrine cells. The cell or origin of prostate cancer has been extensively studied and continues to be a subject of ongoing debate [78]. Multiple recent studies have shown that both luminal and basal cells are self-sustaining and possess plasticity to generate both cell types, but basal cells are much more efficient stem cells in organoid and graft formation [79–81]. In prostate cancer, the bulk of cancerous cells exhibit luminal phenotype, expressing luminal cytokeratins, high AR, and secretory proteins such as PSA. The frequent TMPRSS2-ERG fusion suggests that the cancer cell of origin is a TMPRSS2-positive cell which is highest expressed in luminal cells but also expressed in basal cells. Independent studies of GEM models suggest that luminal cells can be easily transformed by Pten deletion whereas Pten-deleted basal cells seem to first transdifferentiate to luminal cells [82, 83]. As noted above, Witte and colleagues have shown that basal cells transduced with ERG, AR, and AKT will can a luminal prostate cancer that recapitulate the human disease when grafted while transduced luminal cells do not form grafts [77]. However, because basal cells are much hardier to survival ex-vivo transduction and subsequent grafting and because they can transdifferentiate into luminal cells, these data do not definitively implicate the cancer cell or origin in human prostate cancer.
Regardless of the cell of origin for prostate cancer, one hypothesis is that the transformation process leads to luminal linage commitment. Our lab used a complementary system of studying prostate organoids grown in 3D culture. In this system, single luminal cells and single basal cells are bi-potent and each can generate organoids with both outer layer of basal and inner layer of luminal cells [81, 84]. However, ERG overexpressing luminal cells generated organoids that are comprised of a single layer of luminal cells in both 3D organoid and when recombined with UGSM and grafted under the renal capsule [85]. A similar observation was made in human RWPE prostate epithelial cells, an immortalized “normal” line with basal cell features where ERG overexpression caused loss of basal markers and increased luminal specification [86]. These observations were independent seen in a GEM model of prostate-specific deletion of Pten and Trp53. These mice develop invasive prostate cancer with loss of luminal differentiation, decreased expression of AR and AR target genes and AR-independent growth. In this setting, transgenic ERG overexpression let to restored luminal differentiation and adenocarcinoma architecture and sensitivity to enzalutamide [87]. Mechanistically, ERG directly inhibits expression of TP63, a master regulator of the basal epithelial transcriptional program. ERG binds to a distal enhancer of TP63 and inhibits its 3D interaction with the TP63 promoter [85]. While normal epithelial cells exhibit differentiation potential into both basal and luminal cells, primary prostate cancer exhibits a luminal epithelial phenotype. Indeed, loss of basal cells is a pathological criterion to diagnose prostate cancer. Aberrant ETS expression, in addition to conveying a proliferative advantage, may serve to maintain a luminal lineage specification.
Another feature of untreated primary prostate cancer is dependence on AR signaling for growth. In organoids derived from mouse prostate epithelial cells, biochemical studies show that ERG directly binds to AR on DNA and increases AR affinity to DNA in vitro [88]. In mouse prostate epithelial cells, ERG reprograms the AR cistrome in vivo [68]. In addition, ERG also recruits AR co-activators such as NCOA3 to ERG/AR co-bound sites to increase expression of AR target genes [89]. Consistent with these observations, ERG overexpression cooperates with AR overexpression in tumorigenesis in the USGM recombination system [76]. However, other studies have shown opposite effects of ERG on AR-mediated transcription. In VCAP prostate cancer cells that harbor the ERG fusion, the cistromes of ERG and AR are highly overlapping at enhancers, similar to the mouse prostate epithelium. However, in VCAP cells, ERG knockdown results in increased expression of canonical AR-regulated genes such as KLK3 and FKBP5, suggesting that ERG is primarily a suppressor of AR signaling [90, 91]. One mechanism of this inhibition is that ERG recruits the arginine methyltransferase PRMT5 which methylates AR at arginine 761, suppressing AR transcriptional activity [92]. In two parallel GEM models of Tmprss2-ETS knock-in, ERG largely suppresses AR signaling while ETV1 activates AR signaling [69]. Taken together, these studies not only show robust AR and ETS interplay that may underlie the specificity of ETS fusions in prostate cancer, but also highlight the complexity of this interaction and the importance of cellular context. In reconciling the role of ETS in luminal specification and AR signaling, it is important to distinguish luminal state and terminal differentiation. In the normal prostate, AR signaling in luminal cells largely regulates the secretory program and is dispensable for luminal differentiation and cellular survival [93, 94], and ERG can enforce a luminal program in the absence of AR [85]. Neoplastic cells, on the other hand, are luminal and exhibit AR dependence and the AR cistrome is extensively reprogrammed and regulates growth and survival.
PTEN deletion is among the most common genetic alterations in primary prostate cancer, occurring approximately 15% of cases. It significantly co-occurs with ETS fusions, occurring in ~30% of ETS-positive and ~5% of ETS-negative cases in the TCGA cohort [17]. In GEM models, Pten deletion in the prostate epithelium causes HGPIN that may progress into invasive cancer over a variable timeframe [95, 96]. PTEN loss activates PI3K/AKT signaling and there is a well-characterized reciprocal feedback inhibition between the PI3K/AKT and AR signaling pathways [97]. Mouse prostate cancer from Pten-deleted epithelium exhibits not only loss of AR signaling, but also loss of AR dependence and expansion of both the basal and luminal compartments [96–100]. Multiple groups have shown that ERG and ETV1 overexpression cooperate with Pten deletion in prostate tumorigenesis, leading to more aggressive cancers than Pten loss alone [64, 65, 68, 69, 101]. Similarly, in the UGSM, ETS overexpression cooperates with Pten knockdown and with expression of activated AKT in tumorigenesis [76]. Compared to tumors with Pten-deletion alone, tumors with combined ERG expression and Pten-deletion exhibit partially restored AR signaling and a luminal phenotype [68, 85]. We recently uncovered another feedback loop between ERG and PI3K/AKT signaling as ERG overexpression leads to inhibition of PI-3K signaling. This may lead to the selection pressure to delete PTEN in ETS-positive prostate cancer [102]. These data provide a rational for the observed co-occurrence in human prostate cancer.
5. ETS factors are transcriptional mediators of MAPK signaling
In addition to their role in luminal specification, oncogenic ETS factors have a positive role in cellular proliferation and invasion. The mitogen activated protein kinase (MAPK) signaling pathway couples extracellular signals to a multitude of intracellular responses, including cellular proliferation. Multiple members of this pathway are mutated in human cancers, including BRAF, RAS, NF1, and upstream receptor tyrosine kinases. Activating MAPK pathway leads ultimately to downstream transcriptional response. ETS factors are well-known downstream transcriptional mediators of the RAS/MAPK signaling [103]. MAPK regulates ETS transcriptional output through multiple mechanisms: 1) First characterized in the ETS factor ELK1, ERK directly phosphorylates and activates several ETS factor subfamilies including PEA3 members ETV1, ETV4, and ETV5 (Figure 2) [104–109]. 2) PEA3 members are also activated by sumoylation and MAPK activation leads to their sumoylation. 3) PEA3 members are constitutively unstable and rapidly degraded by COP1 ubiquitin ligase and MAPK activity inhibits COP1 mediated degradation (Figure 2) [110]. 4) ERF and its homolog ETV3 are ETS transcriptional repressors. MAPK directly phosphorylates ETV3 and ERF which inhibits their DNA binding (Figure 2) [111, 112]. 5) CIC is a transcriptional repressor of PEA3 factors. MAPK activation leads to CIC phosphorylation and inactivation, resulting in transcriptional activation of PEA3 factors (Figure 2) [33, 113, 114].
In prostate cancer, mutations in the canonical MAPK signaling pathway is distinctly uncommon, with the exception of rare fusions involving RAF kinases that are mutually exclusive with ETS fusions [115]. Peter Hollenhorst and colleagues found that “oncogenic” ETS factors in the ERG and PEA3 families bind to composite ETS/AP1 sites that are typical of “RAS responsive elements” and activate a transcriptional program that mimic the MAPK program [116]. Since many ETS factors are activated by MAPK signaling pathway, in prostate cancer cells that do not have highly active MAPK signaling, it remains a question of whether overexpression alone is sufficient. For PEA3 members such as ETV1, their protein stability is highly MAPK dependent and in GIST and melanoma, high expression is married to constitutively active MAPK signaling to maintain high transcriptional output [51, 52, 117]. On the other hand, ERG is phosphorylated at serine 215 by ERK2 through a very high affinity interaction. This leads to ERG phosphorylation despite very low levels of MAPK activation [118]. Prostate cancer fusions of ETV1, ETV4, and ETV5 generate a truncated protein that lack the degron and are constitutively stable [28, 29, 110], bypassing the need to activate MAPK.
6. Therapeutic targeting of ETS factors
Transcription factors (TFs) are thought to be “undruggable” due to highly similar DNA binding interfaces within large transcription factor families and targeting of specific TFs remains a holy grail of cancer therapy. But the recent development and FDA approval of belzutifan that block dimerization of HIF-2α and HIF-1β, has led to renewed enthusiasm for TF targeting [119].
Targeting of ETS factors stays at the early stages, but there has been development of compounds that directly target ETS factors as well as drugs that modulate ETS activity. Liposomal delivery of ERG siRNA caused growth inhibition of ERG-positive prostate cancer xenograft in vivo, validating ERG as a therapeutic target [120]. ETS-mediated transcription requires PARP1, and PARP inhibitors inhibits the growth of ETS-positive prostate cancers models in vitro and in vivo [121]. However, in a clinical trial of the PARP inhibitor, veliparib, ETS status was not correlated with tumor response [122]. Using a high-content screen for compounds that lower ERG protein levels in prostate cancer VCAP cells but not in endothelial cells, Srivastava and colleagues discovered ERGi-USU which decreased ERG levels and inhibited growth of VCAP cells in vitro and when xenografted in vivo. ERGi-USU binds and inhibits the ribosomal biogenesis regulator atypical kinase RIOK2 which leads to decreased ERG levels [123]. Kittler and colleagues found that ERG is deubiquitinated by ubiquitin-specific peptidase 9, X-linked (USP9X). Treatment with the USP9X inhibitor WP1130 caused ERG degradation and WP1130 was active in ERG-positive xenografts in vivo [124].
Using phage display, Chinnaiyan and colleagues identified ERG inhibitory peptides that inhibit DNA binding. They then made cell permeable peptidomimetics that inhibited ERG-mediated transcription chromatin recruitment and tumor growth in vivo [125]. Using a small molecule microarray (SMM) to identify molecules that bind ETV1, Garraway and colleagues discovered BRD32048 which inhibits ETV1 transcriptional activity and ETV1 driven cellular invasion. BRD32048 further inhibited p300-mediated acetylation of ETV1, leading to its protein degradation.
YK-4-279 is a compound that was discovered through a binding screen to the EWS-FLI1 fusion protein. YK-4-279 inhibited EWS-FLI1 activity and specifically inhibited the growth of EWS-FLI1 driven Ewing’s sarcoma cell lines [126]. YK-4-279 was subsequently found to inhibit ERG and ETV1 mediated transcriptional activity in ETV1-positive LNCaP and ERG-positive VCaP cells [127]. Subsequently studies show that YK-4-279 exhibit in vivo activity against ERG and ETV1 positive xenografts [128, 129]. An analog of YK-4-279, TK216, is currently in a phase 1 clinical trial for patients with Ewing’s sarcoma (NCT02657005). BRD32048, identified by SMM screening, binds ETV1 directly and also modulates both ETV1-mediated transcriptional activity of ETV1-driven LNCaP and ERG-positive VCaP cells [130].
7. Conclusions and future perspectives
ETS fusions represent an early event in prostate tumorigenesis in half of all prostate cancers. Intense research has resulted in seminal discoveries on the role of ETS expression in prostate lineage specification, growth, and invasion. There has also been active research into therapeutic strategies to target ETS. Yet, key mechanistic questions remain to be answered and drug development remains challenging. Continued advances in biotechnology should lead to further discoveries that may be translated into diagnostic and therapeutic strategies in the coming years.
ETS gene fusions represent the most common genetic alteration in prostate cancer and lead to overexpression
ETS factors are downstream mediators of MAP kinase signaling and ETS fusion biologically mimic aspects of MAP kinase activation
ETS overexpression enforce luminal epithelial specification in prostate cancer
Acknowledgement
This work was supported by grants from the Prostate Cancer Foundation (PCF), STARR Cancer Consortium (YC, PC), Geoffrey Beene Cancer Research Center (YC, PC), Gerstner Family Foundation (YC) and the NCI (K08CA140946, R01CA208100, R01CA193837, P50CA092629, P30CA008748).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
Yu Chen owns stock and received royalties from ORIC pharmaceuticals.
References
- [1].Liang H, Mao X, Olejniczak ET, Nettesheim DG, Yu L, Meadows RP, Thompson CB, Fesik SW, Solution structure of the ets domain of Fli-1 when bound to DNA, Nat. Struct. Biol, 1 (1994) 871–875. [DOI] [PubMed] [Google Scholar]
- [2].Laudet V, Hanni C, Stehelin D, Duterque-Coquillaud M, Molecular phylogeny of the ETS gene family, Oncogene, 18 (1999) 1351–1359. [DOI] [PubMed] [Google Scholar]
- [3].Sharrocks AD, The ETS-domain transcription factor family, Nat. Rev. Mol. Cell Biol, 2 (2001) 827–837. [DOI] [PubMed] [Google Scholar]
- [4].Leprince D, Gegonne A, Coll J, de Taisne C, Schneeberger A, Lagrou C, Stehelin D, A putative second cell-derived oncogene of the avian leukaemia retrovirus E26, Nature, 306 (1983) 395–397. [DOI] [PubMed] [Google Scholar]
- [5].Ben-David Y, Giddens EB, Bernstein A, Identification and mapping of a common proviral integration site Fli-1 in erythroleukemia cells induced by Friend murine leukemia virus, Proc Natl Acad Sci U S A, 87 (1990) 1332–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Moreau-Gachelin F, Tavitian A, Tambourin P, Spi-1 is a putative oncogene in virally induced murine erythroleukaemias, Nature, 331 (1988) 277–280. [DOI] [PubMed] [Google Scholar]
- [7].Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, Kovar H, Joubert I, de Jong P, Rouleau G, et al. , Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours, Nature, 359 (1992) 162–165. [DOI] [PubMed] [Google Scholar]
- [8].Gamberi G, Cocchi S, Benini S, Magagnoli G, Morandi L, Kreshak J, Gambarotti M, Picci P, Zanella L, Alberghini M, Molecular diagnosis in Ewing family tumors: the Rizzoli experience--222 consecutive cases in four years, J Mol Diagn, 13 (2011) 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kong XT, Ida K, Ichikawa H, Shimizu K, Ohki M, Maseki N, Kaneko Y, Sako M, Kobayashi Y, Tojou A, Miura I, Kakuda H, Funabiki T, Horibe K, Hamaguchi H, Akiyama Y, Bessho F, Yanagisawa M, Hayashi Y, Consistent detection of TLS/FUS-ERG chimeric transcripts in acute myeloid leukemia with t(16;21)(p11;q22) and identification of a novel transcript, Blood, 90 (1997) 1192–1199. [PubMed] [Google Scholar]
- [10].Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, Lee C, Montie JE, Shah RB, Pienta KJ, Rubin MA, Chinnaiyan AM, Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer, Science, 310 (2005) 644–648. [DOI] [PubMed] [Google Scholar]
- [11].Nowell PC, Discovery of the Philadelphia chromosome: a personal perspective, J. Clin. Invest, 117 (2007) 2033–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rabbitts TH, Chromosomal translocations in human cancer, Nature, 372 (1994) 143–149. [DOI] [PubMed] [Google Scholar]
- [13].Li F, Han M, Dai P, Xu W, He J, Tao X, Wu Y, Tong X, Xia X, Guo W, Zhou Y, Li Y, Zhu Y, Zhang X, Liu Z, Aji R, Cai X, Li Y, Qu D, Chen Y, Jiang S, Wang Q, Ji H, Xie Y, Sun Y, Lu L, Gao D, Distinct mechanisms for TMPRSS2 expression explain organ-specific inhibition of SARS-CoV-2 infection by enzalutamide, Nat Commun, 12 (2021) 866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, Morris DS, Menon A, Jing X, Cao Q, Han B, Yu J, Wang L, Montie JE, Rubin MA, Pienta KJ, Roulston D, Shah RB, Varambally S, Mehra R, Chinnaiyan AM, Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer, Nature, 448 (2007) 595–599. [DOI] [PubMed] [Google Scholar]
- [15].Tomlins SA, Mehra R, Rhodes DR, Smith LR, Roulston D, Helgeson BE, Cao X, Wei JT, Rubin MA, Shah RB, Chinnaiyan AM, TMPRSS2:ETV4 gene fusions define a third molecular subtype of prostate cancer, Cancer Res, 66 (2006) 3396–3400. [DOI] [PubMed] [Google Scholar]
- [16].Helgeson BE, Tomlins SA, Shah N, Laxman B, Cao Q, Prensner JR, Cao X, Singla N, Montie JE, Varambally S, Mehra R, Chinnaiyan AM, Characterization of TMPRSS2:ETV5 and SLC45A3:ETV5 gene fusions in prostate cancer, Cancer Res, 68 (2008) 73–80. [DOI] [PubMed] [Google Scholar]
- [17].Cancer N Genome Atlas Research, The Molecular Taxonomy of Primary Prostate Cancer, Cell, 163 (2015) 1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, Asaka R, Hamanaka W, Ninomiya H, Uehara H, Lim Choi Y, Satoh Y, Okumura S, Nakagawa K, Mano H, Ishikawa Y, RET, ROS1 and ALK fusions in lung cancer, Nat. Med, 18 (2012) 378–381. [DOI] [PubMed] [Google Scholar]
- [19].Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, Nathenson M, Doebele RC, Farago AF, Pappo AS, Turpin B, Dowlati A, Brose MS, Mascarenhas L, Federman N, Berlin J, El-Deiry WS, Baik C, Deeken J, Boni V, Nagasubramanian R, Taylor M, Rudzinski ER, Meric-Bernstam F, Sohal DPS, Ma PC, Raez LE, Hechtman JF, Benayed R, Ladanyi M, Tuch BB, Ebata K, Cruickshank S, Ku NC, Cox MC, Hawkins DS, Hong DS, Hyman DM, Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children, N Engl J Med, 378 (2018) 731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lieber MR, Yu K, Raghavan SC, Roles of nonhomologous DNA end joining, V(D)J recombination, and class switch recombination in chromosomal translocations, DNA Repair (Amst), 5 (2006) 1234–1245. [DOI] [PubMed] [Google Scholar]
- [21].Lin C, Yang L, Tanasa B, Hutt K, Ju BG, Ohgi K, Zhang J, Rose DW, Fu XD, Glass CK, Rosenfeld MG, Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer, Cell, 139 (2009) 1069–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mani RS, Tomlins SA, Callahan K, Ghosh A, Nyati MK, Varambally S, Palanisamy N, Chinnaiyan AM, Induced chromosomal proximity and gene fusions in prostate cancer, Science, 326 (2009) 1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Haffner MC, Aryee MJ, Toubaji A, Esopi DM, Albadine R, Gurel B, Isaacs WB, Bova GS, Liu W, Xu J, Meeker AK, Netto G, De Marzo AM, Nelson WG, Yegnasubramanian S, Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements, Nat. Genet, 42 (2010) 668–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shrestha E, Coulter JB, Guzman W, Ozbek B, Hess MM, Mummert L, Ernst SE, Maynard JP, Meeker AK, Heaphy CM, Haffner MC, De Marzo AM, Sfanos KS, Oncogenic gene fusions in nonneoplastic precursors as evidence that bacterial infection can initiate prostate cancer, Proc Natl Acad Sci U S A, 118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].De Marzo AM, Marchi VL, Epstein JI, Nelson WG, Proliferative inflammatory atrophy of the prostate: implications for prostatic carcinogenesis, Am J Pathol, 155 (1999) 1985–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gasi D, van der Korput HA, Douben HC, de Klein A, de Ridder CM, van Weerden WM, Trapman J, Overexpression of full-length ETV1 transcripts in clinical prostate cancer due to gene translocation, PLoS One, 6 (2011) e16332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hermans KG, van der Korput HA, van Marion R, van de Wijngaart DJ, Ziel-van der Made A, Dits NF, Boormans JL, van der Kwast TH, van Dekken H, Bangma CH, Korsten H, Kraaij R, Jenster G, Trapman J, Truncated ETV1, fused to novel tissue-specific genes, and full-length ETV1 in prostate cancer, Cancer Res, 68 (2008) 7541–7549. [DOI] [PubMed] [Google Scholar]
- [28].Baert JL, Monte D, Verreman K, Degerny C, Coutte L, de Launoit Y, The E3 ubiquitin ligase complex component COP1 regulates PEA3 group member stability and transcriptional activity, Oncogene, 29 (2010) 1810–1820. [DOI] [PubMed] [Google Scholar]
- [29].Vitari AC, Leong KG, Newton K, Yee C, O’Rourke K, Liu J, Phu L, Vij R, Ferrando R, Couto SS, Mohan S, Pandita A, Hongo JA, Arnott D, Wertz IE, Gao WQ, French DM, Dixit VM, COP1 is a tumour suppressor that causes degradation of ETS transcription factors, Nature, 474 (2011) 403–406. [DOI] [PubMed] [Google Scholar]
- [30].Huang FW, Mosquera JM, Garofalo A, Oh C, Baco M, Amin-Mansour A, Rabasha B, Bahl S, Mullane SA, Robinson BD, Aldubayan S, Khani F, Karir B, Kim E, Chimene-Weiss J, Hofree M, Romanel A, Osborne JR, Kim JW, Azabdaftari G, Woloszynska-Read A, Sfanos K, De Marzo AM, Demichelis F, Gabriel S, Van Allen EM, Mesirov J, Tamayo P, Rubin MA, Powell IJ, Garraway LA, Exome Sequencing of African-American Prostate Cancer Reveals Loss-of-Function ERF Mutations, Cancer Discov, 7 (2017) 973–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bose R, Karthaus WR, Armenia J, Abida W, Iaquinta PJ, Zhang Z, Wongvipat J, Wasmuth EV, Shah N, Sullivan PS, Doran MG, Wang P, Patruno A, Zhao Y, S.U.C.P.C.F.P.C.D.T. International, Zheng D, Schultz N, Sawyers CL, ERF mutations reveal a balance of ETS factors controlling prostate oncogenesis, Nature, 546 (2017) 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Eleveld TF, Schild L, Koster J, Zwijnenburg DA, Alles LK, Ebus ME, Volckmann R, Tijtgat GA, van Sluis P, Versteeg R, Molenaar JJ, RAS-MAPK Pathway-Driven Tumor Progression Is Associated with Loss of CIC and Other Genomic Aberrations in Neuroblastoma, Cancer Res, 78 (2018) 6297–6307. [DOI] [PubMed] [Google Scholar]
- [33].Padul V, Epari S, Moiyadi A, Shetty P, Shirsat NV, ETV/Pea3 family transcription factor-encoding genes are overexpressed in CIC-mutant oligodendrogliomas, Genes Chromosomes Cancer, 54 (2015) 725–733. [DOI] [PubMed] [Google Scholar]
- [34].Morais CL, Guedes LB, Hicks J, Baras AS, De Marzo AM, Lotan TL, ERG and PTEN status of isolated high-grade PIN occurring in cystoprostatectomy specimens without invasive prostatic adenocarcinoma, Hum Pathol, 55 (2016) 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gumuskaya B, Gurel B, Fedor H, Tan HL, Weier CA, Hicks JL, Haffner MC, Lotan TL, De Marzo AM, Assessing the order of critical alterations in prostate cancer development and progression by IHC: further evidence that PTEN loss occurs subsequent to ERG gene fusion, Prostate Cancer Prostatic Dis, 16 (2013) 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, Socci ND, Lash AE, Heguy A, Eastham JA, Scher HI, Reuter VE, Scardino PT, Sander C, Sawyers CL, Gerald WL, Integrative genomic profiling of human prostate cancer, Cancer Cell, 18 (2010) 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kron KJ, Murison A, Zhou S, Huang V, Yamaguchi TN, Shiah YJ, Fraser M, van der Kwast T, Boutros PC, Bristow RG, Lupien M, TMPRSS2-ERG fusion co-opts master transcription factors and activates NOTCH signaling in primary prostate cancer, Nat. Genet, 49 (2017) 1336–1345. [DOI] [PubMed] [Google Scholar]
- [38].Perner S, Demichelis F, Beroukhim R, Schmidt FH, Mosquera JM, Setlur S, Tchinda J, Tomlins SA, Hofer MD, Pienta KG, Kuefer R, Vessella R, Sun XW, Meyerson M, Lee C, Sellers WR, Chinnaiyan AM, Rubin MA, TMPRSS2:ERG Fusion-Associated Deletions Provide Insight into the Heterogeneity of Prostate Cancer, Cancer Res, 66 (2006) 8337–8341. [DOI] [PubMed] [Google Scholar]
- [39].Wang J, Cai Y, Ren C, Ittmann M, Expression of Variant TMPRSS2/ERG Fusion Messenger RNAs Is Associated with Aggressive Prostate Cancer, Cancer Res, 66 (2006) 8347–8351. [DOI] [PubMed] [Google Scholar]
- [40].Saramaki OR, Harjula AE, Martikainen PM, Vessella RL, Tammela TL, Visakorpi T, TMPRSS2:ERG fusion identifies a subgroup of prostate cancers with a favorable prognosis, Clin. Cancer. Res, 14 (2008) 3395–3400. [DOI] [PubMed] [Google Scholar]
- [41].Nam RK, Sugar L, Wang Z, Yang W, Kitching R, Klotz LH, Venkateswaran V, Narod SA, Seth A, Expression of TMPRSS2 ERG Gene Fusion in Prostate Cancer Cells is an Important Prognostic Factor for Cancer Progression, Cancer Biol Ther, 6 (2007). [DOI] [PubMed] [Google Scholar]
- [42].Gopalan A, Leversha MA, Satagopan JM, Zhou Q, Al-Ahmadie HA, Fine SW, Eastham JA, Scardino PT, Scher HI, Tickoo SK, Reuter VE, Gerald WL, TMPRSS2-ERG gene fusion is not associated with outcome in patients treated by prostatectomy, Cancer Res, 69 (2009) 1400–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Esgueva R, Perner S, J.L. C, Scheble V, Stephan C, Lein M, Fritzsche FR, Dietel M, Kristiansen G, Rubin MA, Prevalence of TMPRSS2-ERG and SLC45A3-ERG gene fusions in a large prostatectomy cohort, Mod Pathol, 23 (2010) 539–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Krohn A, Diedler T, Burkhardt L, Mayer PS, De Silva C, Meyer-Kornblum M, Kotschau D, Tennstedt P, Huang J, Gerhauser C, Mader M, Kurtz S, Sirma H, Saad F, Steuber T, Graefen M, Plass C, Sauter G, Simon R, Minner S, Schlomm T, Genomic deletion of PTEN is associated with tumor progression and early PSA recurrence in ERG fusion-positive and fusion-negative prostate cancer, Am J Pathol, 181 (2012) 401–412. [DOI] [PubMed] [Google Scholar]
- [45].Krohn A, Seidel A, Burkhardt L, Bachmann F, Mader M, Grupp K, Eichenauer T, Becker A, Adam M, Graefen M, Huland H, Kurtz S, Steurer S, Tsourlakis MC, Minner S, Michl U, Schlomm T, Sauter G, Simon R, Sirma H, Recurrent deletion of 3p13 targets multiple tumour suppressor genes and defines a distinct subgroup of aggressive ERG fusion-positive prostate cancers, J. Pathol, 231 (2013) 130–141. [DOI] [PubMed] [Google Scholar]
- [46].Reid AH, Attard G, Ambroisine L, Fisher G, Kovacs G, Brewer D, Clark J, Flohr P, Edwards S, Berney DM, Foster CS, Fletcher A, Gerald WL, Moller H, Reuter VE, Scardino PT, Cuzick J, de Bono JS, Cooper CS, Molecular characterisation of ERG, ETV1 and PTEN gene loci identifies patients at low and high risk of death from prostate cancer, Br. J. Cancer, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hieronymus H, Iaquinta PJ, Wongvipat J, Gopalan A, Murali R, Mao N, Carver BS, Sawyers CL, Deletion of 3p13–14 locus spanning FOXP1 to SHQ1 cooperates with PTEN loss in prostate oncogenesis, Nat Commun, 8 (2017) 1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Leinonen KA, Saramaki OR, Furusato B, Kimura T, Takahashi H, Egawa S, Suzuki H, Keiger K, Ho Hahm S, Isaacs WB, Tolonen TT, Stenman UH, Tammela TL, Nykter M, Bova GS, Visakorpi T, Loss of PTEN is associated with aggressive behavior in ERG-positive prostate cancer, Cancer Epidemiol. Biomarkers Prev, 22 (2013) 2333–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Codrington R, Pannell R, Forster A, Drynan LF, Daser A, Lobato N, Metzler M, Rabbitts TH, The Ews-ERG fusion protein can initiate neoplasia from lineage-committed haematopoietic cells, PLoS Biol, 3 (2005) e242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zou J, Ichikawa H, Blackburn ML, Hu HM, Zielinska-Kwiatkowska A, Mei Q, Roth GJ, Chansky HA, Yang L, The oncogenic TLS-ERG fusion protein exerts different effects in hematopoietic cells and fibroblasts, Mol. Cell. Biol, 25 (2005) 6235–6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chi P, Chen Y, Zhang L, Guo X, Wongvipat J, Shamu T, Fletcher JA, Dewell S, Maki RG, Zheng D, Antonescu CR, Allis CD, Sawyers CL, ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours, Nature, 467 (2010) 849–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Jane-Valbuena J, Widlund HR, Perner S, Johnson LA, Dibner AC, Lin WM, Baker AC, Nazarian RM, Vijayendran KG, Sellers WR, Hahn WC, Duncan LM, Rubin MA, Fisher DE, Garraway LA, An oncogenic role for ETV1 in melanoma, Cancer Res, 70 (2010) 2075–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Arber S, Ladle DR, Lin JH, Frank E, Jessell TM, ETS gene Er81 controls the formation of functional connections between group Ia sensory afferents and motor neurons, Cell, 101 (2000) 485–498. [DOI] [PubMed] [Google Scholar]
- [54].Attard G, Clark J, Ambroisine L, Mills IG, Fisher G, Flohr P, Reid A, Edwards S, Kovacs G, Berney D, Foster C, Massie CE, Fletcher A, De Bono JS, Scardino P, Cuzick J, Cooper CS, Heterogeneity and clinical significance of ETV1 translocations in human prostate cancer, Br. J. Cancer, 99 (2008) 314–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Li J, Xu C, Lee HJ, Ren S, Zi X, Zhang Z, Wang H, Yu Y, Yang C, Gao X, Hou J, Wang L, Yang B, Yang Q, Ye H, Zhou T, Lu X, Wang Y, Qu M, Yang Q, Zhang W, Shah NM, Pehrsson EC, Wang S, Wang Z, Jiang J, Zhu Y, Chen R, Chen H, Zhu F, Lian B, Li X, Zhang Y, Wang C, Wang Y, Xiao G, Jiang J, Yang Y, Liang C, Hou J, Han C, Chen M, Jiang N, Zhang D, Wu S, Yang J, Wang T, Chen Y, Cai J, Yang W, Xu J, Wang S, Gao X, Wang T, Sun Y, A genomic and epigenomic atlas of prostate cancer in Asian populations, Nature, 580 (2020) 93–99. [DOI] [PubMed] [Google Scholar]
- [56].Mahal BA, Alshalalfa M, Kensler KH, Chowdhury-Paulino I, Kantoff P, Mucci LA, Schaeffer EM, Spratt D, Yamoah K, Nguyen PL, Rebbeck TR, Racial Differences in Genomic Profiling of Prostate Cancer, N Engl J Med, 383 (2020) 1083–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Magi-Galluzzi C, Tsusuki T, Elson P, Simmerman K, LaFargue C, Esgueva R, Klein E, Rubin MA, Zhou M, TMPRSS2-ERG gene fusion prevalence and class are significantly different in prostate cancer of Caucasian, African-American and Japanese patients, Prostate, 71 (2011) 489–497. [DOI] [PubMed] [Google Scholar]
- [58].Rosen P, Pfister D, Young D, Petrovics G, Chen Y, Cullen J, Bohm D, Perner S, Dobi A, McLeod DG, Sesterhenn IA, Srivastava S, Differences in frequency of ERG oncoprotein expression between index tumors of Caucasian and African American patients with prostate cancer, Urology, 80 (2012) 749–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Miettinen M, Wang ZF, Paetau A, Tan SH, Dobi A, Srivastava S, Sesterhenn I, ERG transcription factor as an immunohistochemical marker for vascular endothelial tumors and prostatic carcinoma, Am J Surg Pathol, 35 (2011) 432–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Yoneshima H, Yamasaki S, Voelker CC, Molnar Z, Christophe E, Audinat E, Takemoto M, Nishiwaki M, Tsuji S, Fujita I, Yamamoto N, Er81 is expressed in a subpopulation of layer 5 neurons in rodent and primate neocortices, Neuroscience, 137 (2006) 401–412. [DOI] [PubMed] [Google Scholar]
- [61].Sugimura R, Jha DK, Han A, Soria-Valles C, da Rocha EL, Lu YF, Goettel JA, Serrao E, Rowe RG, Malleshaiah M, Wong I, Sousa P, Zhu TN, Ditadi A, Keller G, Engelman AN, Snapper SB, Doulatov S, Daley GQ, Haematopoietic stem and progenitor cells from human pluripotent stem cells, Nature, 545 (2017) 432–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kalna V, Yang Y, Peghaire CR, Frudd K, Hannah R, Shah AV, Osuna Almagro L, Boyle JJ, Göttgens B, Ferrer J, Randi AM, Birdsey GM, The Transcription Factor ERG Regulates Super-Enhancers Associated With an Endothelial-Specific Gene Expression Program, Circ Res, 124 (2019) 1337–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Tomlins SA, Laxman B, Varambally S, Cao X, Yu J, Helgeson BE, Cao Q, Prensner JR, Rubin MA, Shah RB, Mehra R, Chinnaiyan AM, Role of the TMPRSS2-ERG gene fusion in prostate cancer, Neoplasia, 10 (2008) 177–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Carver BS, Tran J, Gopalan A, Chen Z, Shaikh S, Carracedo A, Alimonti A, Nardella C, Varmeh S, Scardino PT, Cordon-Cardo C, Gerald W, Pandolfi PP, Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate, Nat. Genet, 41 (2009) 619–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].King JC, Xu J, Wongvipat J, Hieronymus H, Carver BS, Leung DH, Taylor BS, Sander C, Cardiff RD, Couto SS, Gerald WL, Sawyers CL, Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis, Nat. Genet, 41 (2009) 524–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Klezovitch O, Risk M, Coleman I, Lucas JM, Null M, True LD, Nelson PS, Vasioukhin V, A causal role for ERG in neoplastic transformation of prostate epithelium, Proc Natl Acad Sci U S A, 105 (2008) 2105–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Shin S, Kim TD, Jin F, van Deursen JM, Dehm SM, Tindall DJ, Grande JP, Munz JM, Vasmatzis G, Janknecht R, Induction of prostatic intraepithelial neoplasia and modulation of androgen receptor by ETS variant 1/ETS-related protein 81, Cancer Res, 69 (2009) 8102–8110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Chen Y, Chi P, Rockowitz S, Iaquinta PJ, Shamu T, Shukla S, Gao D, Sirota I, Carver BS, Wongvipat J, Scher HI, Zheng D, Sawyers CL, ETS factors reprogram the androgen receptor cistrome and prime prostate tumorigenesis in response to PTEN loss, Nat. Med, 19 (2013) 1023–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Baena E, Shao Z, Linn DE, Glass K, Hamblen MJ, Fujiwara Y, Kim J, Nguyen M, Zhang X, Godinho FJ, Bronson RT, Mucci LA, Loda M, Yuan GC, Orkin SH, Li Z, ETV1 directs androgen metabolism and confers aggressive prostate cancer in targeted mice and patients, Genes Dev, 27 (2013) 683–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Casey OM, Fang L, Hynes PG, Abou-Kheir WG, Martin PL, Tillman HS, Petrovics G, Awwad HO, Ward Y, Lake R, Zhang L, Kelly K, TMPRSS2- Driven ERG Expression In Vivo Increases Self-Renewal and Maintains Expression in a Castration Resistant Subpopulation, PLoS One, 7 (2012) e41668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Ittmann M, Huang J, Radaelli E, Martin P, Signoretti S, Sullivan R, Simons BW, Ward JM, Robinson BD, Chu GC, Loda M, Thomas G, Borowsky A, Cardiff RD, Animal models of human prostate cancer: the consensus report of the New York meeting of the Mouse Models of Human Cancers Consortium Prostate Pathology Committee, Cancer Res, 73 (2013) 2718–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Carver BS, Tran J, Chen Z, Carracedo-Perez A, Alimonti A, Nardella C, Gopalan A, Scardino PT, Cordon-Cardo C, Gerald W, Pandolfi PP, ETS rearrangements and prostate cancer initiation, Nature, 457 (2009) E1; discussion E2–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Nguyen LT, Tretiakova MS, Silvis MR, Lucas J, Klezovitch O, Coleman I, Bolouri H, Kutyavin VI, Morrissey C, True LD, Nelson PS, Vasioukhin V, ERG Activates the YAP1 Transcriptional Program and Induces the Development of Age-Related Prostate Tumors, Cancer Cell, 27 (2015) 797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Blattner M, Liu D, Robinson BD, Huang D, Poliakov A, Gao D, Nataraj S, Deonarine LD, Augello MA, Sailer V, Ponnala L, Ittmann M, Chinnaiyan AM, Sboner A, Chen Y, Rubin MA, Barbieri CE, SPOP Mutation Drives Prostate Tumorigenesis In Vivo through Coordinate Regulation of PI3K/mTOR and AR Signaling, Cancer Cell, 31 (2017) 436–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Xin L, Ide H, Kim Y, Dubey P, Witte ON, In vivo regeneration of murine prostate from dissociated cell populations of postnatal epithelia and urogenital sinus mesenchyme, Proc Natl Acad Sci U S A, 100 Suppl 1 (2003) 11896–11903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Zong Y, Xin L, Goldstein AS, Lawson DA, Teitell MA, Witte ON, ETS family transcription factors collaborate with alternative signaling pathways to induce carcinoma from adult murine prostate cells, Proc Natl Acad Sci U S A, 106 (2009) 12465–12470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Goldstein AS, Huang J, Guo C, Garraway IP, Witte ON, Identification of a cell of origin for human prostate cancer, Science, 329 (2010) 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Lee SH, Shen MM, Cell types of origin for prostate cancer, Curr. Opin. Cell Biol, 37 (2015) 35–41. [DOI] [PubMed] [Google Scholar]
- [79].Chua CW, Shibata M, Lei M, Toivanen R, Barlow LJ, Bergren SK, Badani KK, McKiernan JM, Benson MC, Hibshoosh H, Shen MM, Single luminal epithelial progenitors can generate prostate organoids in culture, Nat. Cell Biol, 16 (2014) 951–961, 951–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Wang X, Kruithof-de Julio M, Economides KD, Walker D, Yu H, Halili MV, Hu YP, Price SM, Abate-Shen C, Shen MM, A luminal epithelial stem cell that is a cell of origin for prostate cancer, Nature, 461 (2009) 495–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Karthaus WR, Iaquinta PJ, Drost J, Gracanin A, van Boxtel R, Wongvipat J, Dowling CM, Gao D, Begthel H, Sachs N, Vries RG, Cuppen E, Chen Y, Sawyers CL, Clevers HC, Identification of multipotent luminal progenitor cells in human prostate organoid cultures, Cell, 159 (2014) 163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Wang ZA, Mitrofanova A, Bergren SK, Abate-Shen C, Cardiff RD, Califano A, Shen MM, Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity, Nat. Cell Biol, 15 (2013) 274–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Choi N, Zhang B, Zhang L, Ittmann M, Xin L, Adult murine prostate basal and luminal cells are self-sustained lineages that can both serve as targets for prostate cancer initiation, Cancer Cell, 21 (2012) 253–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, Dowling C, Wanjala JN, Undvall EA, Arora VK, Wongvipat J, Kossai M, Ramazanoglu S, Barboza LP, Di W, Cao Z, Zhang QF, Sirota I, Ran L, MacDonald TY, Beltran H, Mosquera JM, Touijer KA, Scardino PT, Laudone VP, Curtis KR, Rathkopf DE, Morris MJ, Danila DC, Slovin SF, Solomon SB, Eastham JA, Chi P, Carver B, Rubin MA, Scher HI, Clevers H, Sawyers CL, Chen Y, Organoid cultures derived from patients with advanced prostate cancer, Cell, 159 (2014) 176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Li F, Yuan Q, Di W, Xia X, Liu Z, Mao N, Li L, Li C, He J, Li Y, Guo W, Zhang X, Zhu Y, Aji R, Wang S, Tong X, Ji H, Chi P, Carver B, Wang Y, Chen Y, Gao D, ERG orchestrates chromatin interactions to drive prostate cell fate reprogramming, J. Clin. Invest, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Rickman DS, Soong TD, Moss B, Mosquera JM, Dlabal J, Terry S, MacDonald TY, Tripodi J, Bunting K, Najfeld V, Demichelis F, Melnick AM, Elemento O, Rubin MA, Oncogene-mediated alterations in chromatin conformation, Proc Natl Acad Sci U S A, 109 (2012) 9083–9088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Blee AM, He Y, Yang Y, Ye Z, Yan Y, Pan Y, Ma T, Dugdale J, Kuehn E, Kohli M, Jimenez R, Chen Y, Xu W, Wang L, Huang H, TMPRSS2-ERG Controls Luminal Epithelial Lineage and Antiandrogen Sensitivity in PTEN and TP53-Mutated Prostate Cancer, Clin. Cancer. Res, 24 (2018) 4551–4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Wasmuth EV, Hoover EA, Antar A, Klinge S, Chen Y, Sawyers CL, Modulation of androgen receptor DNA binding activity through direct interaction with the ETS transcription factor ERG, Proc Natl Acad Sci U S A, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Shah N, Kesten N, Font-Tello A, Chang MEK, Vadhi R, Lim K, Flory MR, Cejas P, Mohammed H, Long HW, Brown M, ERG-Mediated Coregulator Complex Formation Maintains Androgen Receptor Signaling in Prostate Cancer, Cancer Res, 80 (2020) 4612–4619. [DOI] [PubMed] [Google Scholar]
- [90].Chng KR, Chang CW, Tan SK, Yang C, Hong SZ, Sng NY, Cheung E, A transcriptional repressor co-regulatory network governing androgen response in prostate cancers, EMBO J, 31 (2012) 2810–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, Wang X, Wu L, Li J, Hu M, Gong Y, Cheng H, Laxman B, Vellaichamy A, Shankar S, Li Y, Dhanasekaran SM, Morey R, Barrette T, Lonigro RJ, Tomlins SA, Varambally S, Qin ZS, Chinnaiyan AM, An Integrated Network of Androgen Receptor, Polycomb, and TMPRSS2-ERG Gene Fusions in Prostate Cancer Progression, Cancer Cell, 17 (2010) 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Mounir Z, Korn JM, Westerling T, Lin F, Kirby CA, Schirle M, McAllister G, Hoffman G, Ramadan N, Hartung A, Feng Y, Kipp DR, Quinn C, Fodor M, Baird J, Schoumacher M, Meyer R, Deeds J, Buchwalter G, Stams T, Keen N, Sellers WR, Brown M, Pagliarini RA, ERG signaling in prostate cancer is driven through PRMT5-dependent methylation of the Androgen Receptor, Elife, 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].De Gendt K, Verhoeven G, Tissue- and cell-specific functions of the androgen receptor revealed through conditional knockout models in mice, Mol. Cell. Endocrinol, 352 (2012) 13–25. [DOI] [PubMed] [Google Scholar]
- [94].Shibata M, Epsi NJ, Xuan S, Mitrofanova A, Shen MM, Bipotent Progenitors Do Not Require Androgen Receptor for Luminal Specification during Prostate Organogenesis, Stem Cell Reports, 15 (2020) 1026–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, Khoo AS, Roy-Burman P, Greenberg NM, Van Dyke T, Cordon-Cardo C, Pandolfi PP, Pten dose dictates cancer progression in the prostate, PLoS Biol, 1 (2003) E59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy-Burman P, Nelson PS, Liu X, Wu H, Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer, Cancer Cell, 4 (2003) 209–221. [DOI] [PubMed] [Google Scholar]
- [97].Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, Arora VK, Le C, Koutcher J, Scher H, Scardino PT, Rosen N, Sawyers CL, Reciprocal Feedback Regulation of PI3K and Androgen Receptor Signaling in PTEN-Deficient Prostate Cancer, Cancer Cell, 19 (2011) 575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, Plaisier S, Garraway IP, Huang J, Graeber TG, Wu H, Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth, Cancer Cell, 19 (2011) 792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Wang S, Garcia AJ, Wu M, Lawson DA, Witte ON, Wu H, Pten deletion leads to the expansion of a prostatic stem/progenitor cell subpopulation and tumor initiation, Proc Natl Acad Sci U S A, 103 (2006) 1480–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Lu TL, Huang YF, You LR, Chao NC, Su FY, Chang JL, Chen CM, Conditionally ablated Pten in prostate basal cells promotes basal-to-luminal differentiation and causes invasive prostate cancer in mice, Am J Pathol, 182 (2013) 975–991. [DOI] [PubMed] [Google Scholar]
- [101].Higgins J, Brogley M, Palanisamy N, Mehra R, Ittmann MM, Li JZ, Tomlins SA, Robins DM, Interaction of the Androgen Receptor, ETV1, and PTEN Pathways in Mouse Prostate Varies with Pathological Stage and Predicts Cancer Progression, Horm Cancer, 6 (2015) 67–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Mao N, Gao D, Hu W, Gadal S, Hieronymus H, Wang S, Lee YS, Sullivan P, Zhang Z, Choi D, Rosen N, Sawyers CL, Gopalan A, Chen Y, Carver BS, Oncogenic ERG Represses PI3K Signaling through Downregulation of IRS2, Cancer Res, 80 (2020) 1428–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Yang SH, Sharrocks AD, Whitmarsh AJ, MAP kinase signalling cascades and transcriptional regulation, Gene, 513 (2013) 1–13. [DOI] [PubMed] [Google Scholar]
- [104].Marais R, Wynne J, Treisman R, The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain, Cell, 73 (1993) 381–393. [DOI] [PubMed] [Google Scholar]
- [105].Verger A, Buisine E, Carrere S, Wintjens R, Flourens A, Coll J, Stehelin D, Duterque-Coquillaud M, Identification of amino acid residues in the ETS transcription factor Erg that mediate Erg-Jun/Fos-DNA ternary complex formation, J. Biol. Chem, 276 (2001) 17181–17189. [DOI] [PubMed] [Google Scholar]
- [106].Gille H, Kortenjann M, Thomae O, Moomaw C, Slaughter C, Cobb MH, Shaw PE, ERK phosphorylation potentiates Elk-1-mediated ternary complex formation and transactivation, EMBO J, 14 (1995) 951–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Gille H, Sharrocks AD, Shaw PE, Phosphorylation of transcription factor p62TCF by MAP kinase stimulates ternary complex formation at c-fos promoter, Nature, 358 (1992) 414–417. [DOI] [PubMed] [Google Scholar]
- [108].Wasylyk C, Flores P, Gutman A, Wasylyk B, PEA3 is a nuclear target for transcription activation by non-nuclear oncogenes, EMBO J, 8 (1989) 3371–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].O’Hagan RC, Tozer RG, Symons M, McCormick F, Hassell JA, The activity of the Ets transcription factor PEA3 is regulated by two distinct MAPK cascades, Oncogene, 13 (1996) 1323–1333. [PubMed] [Google Scholar]
- [110].Xie Y, Cao Z, Wong EW, Guan Y, Ma W, Zhang JQ, Walczak EG, Murphy D, Ran L, Sirota I, Wang S, Shukla S, Gao D, Knott SR, Chang K, Leu J, Wongvipat J, Antonescu CR, Hannon G, Chi P, Chen Y, COP1/DET1/ETS axis regulates ERK transcriptome and sensitivity to MAPK inhibitors, J. Clin. Invest, 128 (2018) 1442–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Carlson SM, Chouinard CR, Labadorf A, Lam CJ, Schmelzle K, Fraenkel E, White FM, Large-scale discovery of ERK2 substrates identifies ERK-mediated transcriptional regulation by ETV3, Sci Signal, 4 (2011) rs11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Le Gallic L, Virgilio L, Cohen P, Biteau B, Mavrothalassitis G, ERF nuclear shuttling, a continuous monitor of Erk activity that links it to cell cycle progression, Mol. Cell. Biol, 24 (2004) 1206–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Wang B, Krall EB, Aguirre AJ, Kim M, Widlund HR, Doshi MB, Sicinska E, Sulahian R, Goodale A, Cowley GS, Piccioni F, Doench JG, Root DE, Hahn WC, ATXN1L, CIC, and ETS Transcription Factors Modulate Sensitivity to MAPK Pathway Inhibition, Cell Rep, 18 (2017) 1543–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Ren Y, Ouyang Z, Hou Z, Yan Y, Zhi Z, Shi M, Du M, Liu H, Wen Y, Shao Y, CIC Is a Mediator of the ERK1/2-DUSP6 Negative Feedback Loop, iScience, 23 (2020) 101635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Palanisamy N, Ateeq B, Kalyana-Sundaram S, Pflueger D, Ramnarayanan K, Shankar S, Han B, Cao Q, Cao X, Suleman K, Kumar-Sinha C, Dhanasekaran SM, Chen YB, Esgueva R, Banerjee S, LaFargue CJ, Siddiqui J, Demichelis F, Moeller P, Bismar TA, Kuefer R, Fullen DR, Johnson TM, Greenson JK, Giordano TJ, Tan P, Tomlins SA, Varambally S, Rubin MA, Maher CA, Chinnaiyan AM, Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma, Nat Med, 16 (2010) 793–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Hollenhorst PC, Ferris MW, Hull MA, Chae H, Kim S, Graves BJ, Oncogenic ETS proteins mimic activated RAS/MAPK signaling in prostate cells, Genes Dev, 25 (2011) 2147–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Mehra R, Dhanasekaran SM, Palanisamy N, Vats P, Cao X, Kim JH, Kim DS, Johnson T, Fullen DR, Chinnaiyan AM, Comprehensive Analysis of ETS Family Members in Melanoma by Fluorescence In Situ Hybridization Reveals Recurrent ETV1 Amplification, Transl Oncol, 6 (2013) 405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Selvaraj N, Kedage V, Hollenhorst PC, Comparison of MAPK specificity across the ETS transcription factor family identifies a high-affinity ERK interaction required for ERG function in prostate cells, Cell Commun Signal, 13 (2015) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Choueiri TK, Bauer TM, Papadopoulos KP, Plimack ER, Merchan JR, McDermott DF, Michaelson MD, Appleman LJ, Thamake S, Perini RF, Zojwalla NJ, Jonasch E, Inhibition of hypoxia-inducible factor-2alpha in renal cell carcinoma with belzutifan: a phase 1 trial and biomarker analysis, Nat. Med, 27 (2021) 802–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Shao L, Tekedereli I, Wang J, Yuca E, Tsang S, Sood A, Lopez-Berestein G, Ozpolat B, Ittmann M, Highly specific targeting of the TMPRSS2/ERG fusion gene using liposomal nanovectors, Clin. Cancer. Res, 18 (2012) 6648–6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Brenner JC, Ateeq B, Li Y, Yocum AK, Cao Q, Asangani IA, Patel S, Wang X, Liang H, Yu J, Palanisamy N, Siddiqui J, Yan W, Cao X, Mehra R, Sabolch A, Basrur V, Lonigro RJ, Yang J, Tomlins SA, Maher CA, Elenitoba-Johnson KS, Hussain M, Navone NM, Pienta KJ, Varambally S, Feng FY, Chinnaiyan AM, Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer, Cancer Cell, 19 (2011) 664–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Hussain M, Daignault-Newton S, Twardowski PW, Albany C, Stein MN, Kunju LP, Siddiqui J, Wu YM, Robinson D, Lonigro RJ, Cao X, Tomlins SA, Mehra R, Cooney KA, Montgomery B, Antonarakis ES, Shevrin DH, Corn PG, Whang YE, Smith DC, Caram MV, Knudsen KE, Stadler WM, Feng FY, Chinnaiyan AM, Targeting Androgen Receptor and DNA Repair in Metastatic Castration-Resistant Prostate Cancer: Results From NCI 9012, J Clin Oncol, 36 (2018) 991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Mohamed AA, Xavier CP, Sukumar G, Tan SH, Ravindranath L, Seraj N, Kumar V, Sreenath T, McLeod DG, Petrovics G, Rosner IL, Srivastava M, Strovel J, Malhotra SV, LaRonde NA, Dobi A, Dalgard CL, Srivastava S, Identification of a Small Molecule That Selectively Inhibits ERG-Positive Cancer Cell Growth, Cancer Res, 78 (2018) 3659–3671. [DOI] [PubMed] [Google Scholar]
- [124].Wang S, Kollipara RK, Srivastava N, Li R, Ravindranathan P, Hernandez E, Freeman E, Humphries CG, Kapur P, Lotan Y, Fazli L, Gleave ME, Plymate SR, Raj GV, Hsieh JT, Kittler R, Ablation of the oncogenic transcription factor ERG by deubiquitinase inhibition in prostate cancer, Proc Natl Acad Sci U S A, 111 (2014) 4251–4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Wang X, Qiao Y, Asangani IA, Ateeq B, Poliakov A, Cieslik M, Pitchiaya S, Chakravarthi B, Cao X, Jing X, Wang CX, Apel IJ, Wang R, Tien JC, Juckette KM, Yan W, Jiang H, Wang S, Varambally S, Chinnaiyan AM, Development of Peptidomimetic Inhibitors of the ERG Gene Fusion Product in Prostate Cancer, Cancer Cell, 31 (2017) 844–847. [DOI] [PubMed] [Google Scholar]
- [126].Erkizan HV, Kong Y, Merchant M, Schlottmann S, Barber-Rotenberg JS, Yuan L, Abaan OD, Chou TH, Dakshanamurthy S, Brown ML, Uren A, Toretsky JA, A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma, Nat. Med, 15 (2009) 750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Rahim S, Beauchamp EM, Kong Y, Brown ML, Toretsky JA, Üren A, YK-4-279 inhibits ERG and ETV1 mediated prostate cancer cell invasion, PLoS One, 6 (2011) e19343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Winters B, Brown L, Coleman I, Nguyen H, Minas TZ, Kollath L, Vasioukhin V, Nelson P, Corey E, Üren A, Morrissey C, Inhibition of ERG Activity in Patient-derived Prostate Cancer Xenografts by YK-4-279, Anticancer Res, 37 (2017) 3385–3396. [DOI] [PubMed] [Google Scholar]
- [129].Rahim S, Minas T, Hong SH, Justvig S, Çelik H, Kont YS, Han J, Kallarakal AT, Kong Y, Rudek MA, Brown ML, Kallakury B, Toretsky JA, Üren A, A small molecule inhibitor of ETV1, YK-4-279, prevents prostate cancer growth and metastasis in a mouse xenograft model, PLoS One, 9 (2014) e114260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Pop MS, Stransky N, Garvie CW, Theurillat JP, Hartman EC, Lewis TA, Zhong C, Culyba EK, Lin F, Daniels DS, Pagliarini R, Ronco L, Koehler AN, Garraway LA, A small molecule that binds and inhibits the ETV1 transcription factor oncoprotein, Mol Cancer Ther, 13 (2014) 1492–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]