

Abstract
Background
The comorbidity between polycystic ovary syndrome (PCOS) and obesity has long been observed in clinical settings, but their shared genetic basis remains unclear.
Methods
Leveraging summary statistics of large-scale GWAS(s) conducted in European-ancestry populations on body mass index (adult BMI, Nfemale=434,794; childhood BMI, N=39,620), waist-to-hip ratio (WHR, Nfemale=381,152), WHR adjusted for BMI (WHRadjBMI, Nfemale=379,501), and PCOS (Ncase=10,074, Ncontrol=103,164), we performed a large-scale genome-wide cross-trait analysis to quantify overall and local genetic correlation, to identify shared loci, and to infer causal relationship.
Results
We found positive genetic correlations between PCOS and adult BMI (rg=0.47, P=2.19×10−16), childhood BMI (rg=0.31, P=6.72×10−5), and WHR (rg=0.32, P=1.34×10−10), all withstanding Bonferroni correction. A suggestive significant genetic correlation was found between PCOS and WHRadjBMI (rg=0.09, P=0.04). Partitioning the whole genome into 1703 nearly independent regions, we observed a significant local genetic correlation for adult BMI and PCOS at chromosome 18: 57630483–59020751. We identified 16 shared loci underlying PCOS and obesity-related traits via cross-trait meta-analysis including 9 loci shared between BMI and PCOS (adult BMI and PCOS: 5 loci; childhood BMI and PCOS: 4 loci), 6 loci shared between WHR and PCOS, and 5 loci shared between WHRadjBMI and PCOS. Mendelian randomization (MR) supported the causal roles of both adult BMI (OR=2.92, 95% CI=2.33–3.67) and childhood BMI (OR=2.76, 95% CI=2.09–3.66) in PCOS, but not WHR (OR=1.19, 95% CI=0.93–1.52) or WHRadjBMI (OR=1.03, 95% CI=0.87–1.22). Genetic predisposition to PCOS did not seem to influence the risk of obesity-related traits.
Conclusions
Our cross-trait analysis suggests a shared genetic basis underlying obesity and PCOS and provides novel insights into the biological mechanisms underlying these complex traits. Our work informs public health intervention by confirming the important role of weight management in PCOS prevention.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12916-022-02238-y.
Keywords: Polycystic ovary syndrome, Obesity, Body mass index, Fat distribution, Genome-wide cross-trait analysis
Background
Polycystic ovary syndrome (PCOS) is the most common endocrine disorder affecting women of childbearing age, characterized by reproductive dysfunction including hyperandrogenism, menstrual and/or ovulatory irregularity together with subfertility, and metabolic dysfunction including hyperinsulinemia, insulin resistance, and type 2 diabetes [1, 2]. More than 50% of women with PCOS are either overweight or obese which further worsens all symptoms [3]. Indeed, epidemiological studies have observed a significant association between body mass index (BMI) and features of PCOS at all ages [4]. Clinically, even a modest weight loss (~ 5%) leads to meaningful improvements in the reproductive, hyperandrogenic, and metabolic features of PCOS [5], highlighting a biological link underlying obesity and PCOS.
The development of obesity and PCOS involves strong genetic components evidenced by recent discoveries from large-scale genome-wide associations studies (GWAS). These genetic data enable the utilization of a compiled analytical strategy—genome-wide cross-trait analysis—to determine shared and distinct genetic architecture which can provide better understandings and novel insights into disease mechanisms [6]. Such analysis features several analytic aspects: genetic correlation analysis to estimate overall and local genetic correlation, cross-trait meta-analysis to identify shared loci, and Mendelian randomizations (MR) to make causal inferences. Nevertheless, these advanced statistical genetics approaches have not been routinely applied to examine the genetic contribution to the epidemiologic associations between PCOS and its most common comorbidity, obesity [7, 8]. Despite three MR studies [7, 9, 10] have been conducted to explore the role of adult BMI in PCOS, these studies used a small number of index SNPs (< 100 instruments vs. ~ 300 female-specific BMI instruments currently identified by GWAS [11]); lacked sensitivity analyses to verify model assumptions; and lacked sex-specific analysis, i.e., using genetic data derived from a sex-mixed population instead of using female-specific data for a gynecological outcome PCOS.
In addition to the degree of adiposity, location and distribution of fat accumulation are informative predictors for obesity sequelae. For example, abdominal visceral fat, a known contributor to metabolic dysfunction including insulin resistance and abnormal adipokine and fatty acid release [12], is important for PCOS. Furthermore, early life weight pattern also influences obesity and metabolic alterations later on [13]. However, to the best of our knowledge, genetic analysis has rarely been conducted to examine the role of fat distribution or childhood BMI in PCOS [7].
Therefore, we aim to extend previous findings by providing a systematic evaluation of the relationship between obesity-related traits and PCOS, leveraging the hitherto largest GWAS summary statistics conducted for each trait. We examined the role of BMI (childhood (before age 10) (N=39,620) [14] and adult [11]), waist-to-hip ratio [15] (WHR, adult), and waist-to-hip ratio adjusted for BMI [15] (WHRadjBMI, adult) (all adult measures were restricted to female participants, NFemale=~ 400,000) in the development of PCOS (NPCOS=10,074; Ncontrol=103,164) [7], performing analyses to quantify overall and local genetic correlations, to identify shared loci and to infer causal relationships. A conceptual framework is shown in Fig. 1.
Fig. 1.

Overall study design. GWAS summary statistics on PCOS and 4 obesity-related traits (adult BMI, childhood BMI, WHR, and WHRadjBMI) were retrieved. First, we quantified the genome-wide and local genetic correlation between PCOS and obesity-related traits. Next, we identified shared loci contributing to PCOS and obesity-related traits, and further, conducted tissue enrichment analysis, and constructed 99% credible sets of causal variants for these loci. Finally, we conducted a bidirectional Mendelian randomization analysis to infer causality between obesity-related traits and PCOS
Methods
We carried out the current study by leveraging large-scale GWAS summary statistics and novel statistical genetics approaches. We included female-specific genetic data of adult obesity-related traits to best match with a female-specific outcome PCOS. However, sex-specific data for childhood BMI were unavailable. To reduce potential bias from population stratification, all genetic data were restricted to European population.
Obesity GWAS(s)
For adult obesity, the hitherto largest GWAS was conducted meta-analyzing data from UK Biobank and GIANT consortium totaling ~ 700,000 individuals of European ancestry, among which 434,794 female participants with information available for BMI, 381,152 for WHR, and 315,284 for WHRadjBMI [11, 15]. In each participating study, SNPs were imputed to the Haplotype Reference Consortium (HRC) reference panel and filtered by imputation quality score > 0.30, call rate > 0.95, minor allele frequency > 0.0001 and P value for Hardy-Weinberg equilibrium > 10−6. After quality control, genome-wide association testing was performed using a linear mixed model adjusting for age, recruitment center, genotyping batches, and principal components. A fixed-effect inverse-variance-weighted meta-analysis was conducted to combine effect sizes across studies. To identify independent top-associated SNPs, a PLINK clumping algorithm of P< 1×10− 9 and an LD window of ±5 Mb (r2 > 0.05) were first used to obtain LD-based clumps [16], followed by a proximal conditional and joint testing to identify primary and secondary signals within each of the clumping-based loci.
For childhood BMI, the hitherto largest GWAS was conducted at the latest time point between 2 and 10 years old among 61,111 children of European ancestry [14]. In the discovery stage, data from 26 studies (Ndiscovery = 39,620) imputed to the 1000 Genomes Project or the HRC were analyzed using a fixed-effect inverse variance-weighted meta-analysis. Top associated SNPs identified in the discovery stage at a P-threshold of 5×10−6 were taken forward for replication in 15 cohorts (Nreplication = 21,491) and results of the two stages were combined using meta-analysis. To identify independent top-associated SNPs, Genome-wide Complex Trait Analysis based on summary statistics with LD estimation from the Generation R Study as a reference sample was applied. Top associated SNPs in the combined meta-analysis reaching a stringent P-threshold of 5×10−8 were identified.
From these GWAS(s), we extracted the effect size and relevant information of instrumental variables (IVs) identified in women (281 BMI-associated index SNPs, 203 WHR-associated index SNPs, and 266 WHRadjBMI-associated index SNPs, Additional file 1: Tables S1-3) and children (25 childhood BMI-associated index SNPs, Additional file 1: Table S4). We also accessed and downloaded the full set GWAS summary statistics.
PCOS GWAS
The hitherto largest GWAS of PCOS was conducted based on international collaborations in 10,074 PCOS cases and 103,164 controls of European ancestry [7]. Data from 7 cohorts were imputed to the 1000 Genomes Project or HapMap2. Diagnosis of PCOS was based on the NIH (2540 cases/15,020 controls) or Rotterdam criteria (2669 cases/17,035 controls), or by self-reported diagnosis (5184 cases/82,759 controls, 23andMe). A fixed-effect inverse-variance weighted meta-GWAS was performed adjusting for age. To identify independent top-associated SNPs, a fixed-effect inverse-weighted-variance meta-analysis was applied to obtain the per-variant estimates from summary statistics of contributing studies. This GWAS identified 14 PCOS-associated variants reaching a P-threshold of 5×10−8. From PCOS GWAS, we extracted IVobesity-PCOS associations and relevant information. We also downloaded the full set GWAS summary statistics of PCOS in which data from 23andMe were excluded (due to data availability) (Additional file 1: Table. S5).
A table summarizing the information of all included GWASs is shown in Additional file 1: Table. S6 and a table summarizing all participating studies is shown Additional file 1: Table. S7. For all analyses, the human reference genome build 37 (or hg19) was used.
Statistical analysis
Genetic correlation analysis
Genome-wide genetic correlations (rg) quantifies the average sharing of genetic effect between two traits unaffected by environmental confounders. The estimate ranges from − 1 to 1, with − 1 indicating a perfect negative genetic correlation and 1 indicating a perfect positive genetic correlation. It can be quantified using GWAS summary data through an algorithm implemented in software linkage-disequilibrium score regression (LDSC) [17, 18] also described below:
where βj and γj are the effect sizes of SNP j on traits 1 and 2, rg is the genetic covariance, M is number of SNPs, N1 and N2 are the sample sizes for traits 1 and 2, Ns is the number of overlapping samples, r is the phenotypic correlation in overlapping samples, and lj is the linkage disequilibrium score. We used pre-computed LD scores obtained from ~ 1.2 million common SNPs in European ancestry represented in the HapMap3 reference panel, commonly recognized as of high imputation quality.
Genome-wide genetic correlations estimated by LDSC quantify the genome-wide contribution of genetic variation to the correlation between two traits. It is possible that even though two traits show negligible genome-wide genetic correlation, genetic variations localized at a specific genomic region contribute to the correlation between two traits. We, therefore, measured the pairwise local genetic correlations between each of the obesity-related traits and PCOS using ρ-HESS, an algorithm that partitions the genome-wide genetic sharing across 1703 nearly independent linkage disequilibrium (LD) regions of 1.5 Mb and precisely quantifies the genetic correlation between pairs of traits due to genetic variation restricted to these genomic regions.
Cross-trait meta-analysis
Cross-phenotype association analysis (CPASSOC) integrates GWAS summary statistics from multiple correlated traits to detect evidence for variants associated with multiple traits across studies while controlling population structure and cryptic relatedness [19]. CPASSOC provides two test statistics, SHom and SHet. SHom is based on the fixed-effect meta-analysis method and is the most powerful when genetic effect sizes are homogenous, which is unlikely to be true especially when multiple traits are analyzed. SHet is an extension of SHom with improved power that allows for heterogeneous effects of a trait from different study designs, environmental factors, or populations, as well as heterogeneous effects for different phenotypes, which is more common in practice. We used pairwise SHet to combine summary statistics for each of the obesity-related traits with PCOS. We applied PLINK [16] clumping function parameters: --clump-p1 5e-8 --clump-p2 1e-5 --clump-r2 0.2 --clump-kb 500) to obtain independent SNPs. Significant SNPs were defined as variants with Psingle trait< 1×10−3 (in each single trait) and PCPASSOC< 5×10−8 (in cross-traits). We used Ensembl Variant Effect Predictor (VEP) for detailed functional annotation for the variants identified by CPASSOC.
Fine-mapping credible set analysis
Index SNP does not necessarily represent causal variants. We further identified a 99% credible set of causal variants through a simplified Bayesian fine-mapping method named FM-summary (https://github.com/hailianghuang/FM-summary) [20]. Briefly, for each of the 16 shared loci identified as significant by the cross-trait meta-analysis, we extracted variants within 500 kb around the index SNP, which were used as input for FM-summary. FM-summary then set a flat prior and produced a posterior inclusion probability (PIP) of a true trait/disease association for each variant using the steepest descent approximation. A 99% credible set is equivalent to ranking SNPs from largest to smallest PIPs and taking the cumulative sum of PIPs until it is at least 99%. Details of the method were described elsewhere [21, 22].
Functional annotation and tissue enrichment analysis
To gain putative biological insights into the shared variants identified between obesity and PCOS, we performed GTEx tissue enrichment analysis including all genes in clumping regions for each trait identified by CPASSOC using software functional mapping and annotation (FUMA), the GENE2FUNC process with 54 tissue types from GTEx (version 8). FUMA obtained differentially expressed gene (DEG) sets for each tissue type by using the normalized expression (zero mean of log2(RPKM+1)) and conducting two-sided t-tests per gene per tissue. Genes were defined as DEG set in a specific tissue if Bonferroni corrected P-value<0.05 and had an absolute log-fold change ≥ 0.58 (background DEGs). Genes identified by CPASSOC were tested against those background DEG sets by hypergeometric tests to examine if they were overrepresented in DEG sets in specific tissue types, thus, identifying the most relevant tissue types.
To identify tissue and cell-type-specific enrichment of SNPs, we used LD score regression applied to specifically expressed genes (LDSC-SEG) [23] which tests for enrichment for per-SNP heritability. Pre-computed annotations constructed from epigenomics data from Roadmap Epigenomics Project [24] (DNase hypersensitivity, H3K27ac, H3K4me3, H3K4me1, H3K9ac, and/or H3K36me3 chromatin marks) were used in our analysis. The 396 cell-type annotations were further divided into 9 groups (adipose, central nervous system (CNS), digestive system, cardiovascular, musculoskeletal and connective tissue, immune and blood, liver, pancreas, and others). To correct for multiple comparison, a Bonferroni-corrected P-threshold (0.05/396) was used.
Mendelian randomization analysis
To test for the causal relationship between each of the obesity-related traits and PCOS, we conducted a two-sample MR. We applied an inverse-variance weighted (IVW) approach [25] as our primary MR analysis, an MR-Egger regression [26], and a weighted-median estimator approach [27] to examine the robustness of our findings under relaxed model assumptions.
We performed important sensitivity analyses to validify MR model assumptions. We excluded palindromic IVs with strand ambiguity (A/T or G/C SNPs with the same pair of letters on the forward and reverse strands, introducing ambiguity into strand identity) and pleiotropic SNPs (SNPs associated with potential confounding traits other than exposure and outcome of interest) according to GWAS catalog (https://www.ebi.ac.uk/gwas/, accessed on 03/18/2021). We conducted a leave-one-out analysis where we removed one IV each time and performed IVW using the remaining IVs to identify outlying instruments. We also examined a scenario through MR-Clust [28] where several distinct causal mechanisms may underlie the obesity-PCOS relationship (i.e., a risk factor influences outcome with different magnitudes and direction of causal effect). MR-Clust divides IVs into distinct clusters such that all variants in the cluster have similar causal estimates. To examine the causal effect of the genetic predisposition to PCOS on obesity, we finally performed a bidirectional MR analysis where instruments for outcomes were used to evaluate whether the “outcome” (here, PCOS) caused the “exposure” (here, obesity-related exposures). The 14 PCOS-associated independent loci with genome-wide significance were included as IVs in our reverse direction MR and their effects were extracted from the respective obesity GWAS(s).
Results
Genetic correlations between obesity-related traits and PCOS
We first estimated the overall genetic correlation between obesity-related traits and PCOS using cross-trait LDSC. After correcting for multiple testing (P< 0.05/4), we found a strong genetic correlation between BMI and PCOS (adult BMI: rg=0.47, P=2.19×10−16; childhood BMI: rg=0.31, P=6.72×10−5) (Table 1). A significant result was also observed for WHR and PCOS (rg=0.32, P=1.34×10−10). Given the complex interplay between BMI and WHR [29], we continued to investigate WHRadjBMI, a residual component of WHR in which the effect of BMI was removed. Perhaps not surprisingly, when the effect of BMI was removed from WHR (WHRadjBMI), the prior positive genetic correlation between PCOS and WHR was attenuated to null (WHRadjBMI: rg=0.09, P=0.04).
Table 1.
Genome-wide genetic correlation between PCOS and obesity-related traits
| Trait 1 | Trait 2 | rg | rg_SE | P value |
|---|---|---|---|---|
| PCOS | BMI | 0.4694 | 0.0572 | 2.19×10−16 |
| PCOS | WHR | 0.3198 | 0.0498 | 1.34×10−10 |
| PCOS | WHRadjBMI | 0.0931 | 0.0447 | 0.0371 |
| PCOS | CBMI | 0.3109 | 0.0078 | 6.72×10−5 |
rg, genetic correlation; SE, standard error; BMI, adult body mass index; PCOS, polycystic ovary syndrome; WHR; waist-to-hip ratio; WHRadjBMI, waist-to-hip ratio adjusted for body mass index; CBMI, childhood body mass index
Motivated by these findings, we further explored the local genetic correlation using ρ-HESS. As shown in Fig. 2 and Additional file 2: Fig. S1, after correcting for multiple testing (P< 0.05/1703), a significant local genetic correlation was only observed for adult BMI and PCOS at chr18: 57630483–59020751, a genetic region harboring MC4R, a locus previously reported to be associated with adult BMI, childhood BMI and obesity in PCOS [14, 30–33].
Fig. 2.

Local genetic correlation, genetic covariance, and SNP heritability between adult BMI and PCOS. Colored bars represent loci that have significant local genetic correlation and covariance after multiple testing adjustment. BMI, adult body mass index; PCOS, polycystic ovary syndrome
Cross-trait meta-analysis of obesity-related traits and PCOS
To identify individual SNPs affecting both obesity-related traits and PCOS, we next conducted a pairwise CPASSOC analysis. As shown in Table 2, in total we identified 9 independent loci shared between BMI and PCOS (adult: 5 loci, rs10938397, rs705696, rs1569979, rs10142183, and rs11672660; childhood: 4 loci, rs12641981, rs10987375, rs8050136, and rs7228430), 6 independent loci shared between WHR and PCOS (rs3767846, rs13032835, rs188488867, rs7774737, rs1421085, and rs310011) and 5 independent loci shared between WHRadjBMI and PCOS (rs3767846, rs13032835, rs7774737, rs11057429, and rs310011) (all these SNPs fulfilled Psingle trait< 1×10−3 and PCPASSOC< 5×10−8). Notably, none of these 16 SNPs identified by CPASSOC were previously reported to be associated with PCOS at genome-wide significance (0 out of 16 SNPs), while most of them were associated with obesity-related traits (14 out of 16 SNPs). For adult BMI and PCOS, the most significant shared SNP was rs10938397, (PCPASSOC=1.25×10−49) located at an intergenic region. The second most significant shared SNP (rs11672660, PCPASSOC=1.87×10−32) was located near GIPR, a gene associated with BMI and glucose tolerance [34, 35], and MIR642A, a gene involved in post-transcriptional regulation of gene expression. Notably, among the 5 adult BMI-PCOS shared SNPs, rs705696 was near ERBB3, a locus known to be associated with PCOS. For childhood BMI and PCOS, the most significant shared SNP (rs8050136, PCPASSOC=1.56×10−21) was near FTO, a locus known to affect BMI and the predisposition to childhood and adult obesity [36, 37]. Similarly, the most significant shared SNP for WHR and PCOS (rs1421085, PCPASSOC=1.97×10−51) was also near the FTO locus. Among the 6 variants associated with both WHR and PCOS, 4 were also found to be shared between WHRadjBMI and PCOS (rs3767846, rs13032835, rs7774737, and rs310011). For example, rs3767846 was near PROX1, a locus associated with fasting glucose-related traits [38]. The most significant variant shared by WHRadjBMI and PCOS (rs13032835, PCPASSOC=2.74×10−30) was located near genes SLC38A11 and RNA5SP111. SLC38A11 encodes a protein that belongs to the solute carrier family and has a role in sodium and amino acid transportation [39], and RNA5SP111 is a 5S ribosomal pseudogene [40]. Detailed annotations of each variant are shown in Additional file 1: Table. S8. Of note, some of the CPASSOC-identified significant SNPs were not mapped to any genes (7 out of 16).
Table 2.
Cross-trait meta-analysis between obesity-related traits and PCOS (P-CPASSOC < 5× 10−8, single trait P value < 1× 10−3)
| SNP | CHR | BP | Alleles (E/A) | BETA (SD) | P-trait | P-PCOS | P-CPASSOC | Genomic coordinates | Genes within clumping area | |
|---|---|---|---|---|---|---|---|---|---|---|
| trait | PCOS | |||||||||
| BMI and PCOS | ||||||||||
| rs10938397 | 4 | 45182527 | A/G | − 0.03 | − 0.10 | 1.66×10−45 | 8.90×10−04 | 1.25× 10−49 | 45068929–45186832 | |
| rs705696 | 12 | 56480648 | A/G | − 0.02 | 0.13 | 4.36×10−09 | 1.10× 10−04 | 6.39× 10−10 | 56364321–56564811 | CDK2, ERBB3, ESYT1, IKZF4, MYL6, MYL6B, PA2G4, RAB5B, RPL41, RPS26, SMARCC2, SUOX, ZC3H10 |
| rs1569979 | 14 | 29681294 | A/G | 0.02 | 0.12 | 1.56×10−17 | 9.80× 10−04 | 1.58× 10−18 | 29549249–29702590 | |
| rs10142183 | 14 | 29723729 | C/G | 0.02 | 0.17 | 1.34× 10−12 | 4.00× 10−05 | 8.00× 10−14 | 29662355–29856951 | |
| rs11672660 | 19 | 46180184 | T/C | − 0.03 | − 0.13 | 5.93×10−31 | 4.70× 10−04 | 1.87× 10−32 | 46172278–46214297 | FBXO46, GIPR, MIR642A, MIR642B, QPCTL, SNRPD2 |
| WHR and PCOS | ||||||||||
| rs3767846 | 1 | 214175126 | A/G | 0.02 | 0.13 | 5.69×10−11 | 5.50× 10−04 | 6.19× 10−12 | 214173840–214192133 | PROX1 |
| rs13032835 | 2 | 165748563 | A/T | − 0.02 | − 0.13 | 2.42× 10−18 | 5.00× 10−05 | 3.59× 10−20 | 165668018–165828042 | COBLL1, LOC101929633, SLC38A11 |
| rs188488867 | 2 | 219784256 | T/C | − 0.04 | 0.31 | 9.03×10−08 | 3.00× 10−04 | 1.35× 10−08 | 219755011–219849633 | CDK5R2, FEV, LINC00608, LOC101928537, WNT10A |
| rs7774737 | 6 | 81145901 | C/G | − 0.04 | − 0.38 | 3.22×10−10 | 5.10× 10−06 | 7.92× 10−11 | 80846547–81597411 | BCKDHB |
| rs1421085 | 16 | 53800954 | T/C | − 0.03 | − 0.13 | 6.44× 10−47 | 2.00× 10−05 | 1.97× 10−51 | 53797565–53848561 | FTO |
| rs310011 | 16 | 81471654 | A/G | 0.02 | 0.12 | 1.84×10−11 | 1.90× 10−04 | 2.30× 10−12 | 81449060–81471654 | |
| WHRadjBMI and PCOS | ||||||||||
| rs3767846 | 1 | 214175126 | A/G | 0.02 | 0.13 | 2.12×10−08 | 5.50× 10−04 | 9.99× 10−09 | 214173840–214189783 | PROX1 |
| rs13032835 | 2 | 165748563 | A/T | − 0.03 | − 0.13 | 5.10×10−27 | 5.00× 10−05 | 2.74× 10−30 | 165668018–165828042 | COBLL1, LOC101929633, SLC38A11 |
| rs7774737 | 6 | 81145901 | C/G | − 0.03 | − 0.38 | 4.02×10−08 | 5.10× 10−06 | 8.49× 10−09 | 81145901–81196080 | |
| rs11057429 | 12 | 124529194 | C/G | − 0.09 | 0.52 | 2.74× 10−09 | 4.20× 10−04 | 4.87× 10−10 | 124529194–124529194 | ZNF664-FAM101A |
| rs310011 | 16 | 81471654 | A/G | 0.02 | 0.12 | 1.20×10−14 | 1.90× 10−04 | 3.06× 10−16 | 81449060–81471654 | |
| CBMI and PCOS | ||||||||||
| rs12641981 | 4 | 45179883 | T/C | 0.04 | 0.10 | 4.19×10−08 | 8.90× 10−04 | 8.24× 10−10 | 45175691–45187622 | |
| rs10987375 | 9 | 129392365 | T/C | 0.04 | 0.14 | 2.37×10−06 | 3.60× 10−04 | 2.51× 10−08 | 129370576–129397014 | LMX1B |
| rs8050136 | 16 | 53816275 | A/C | 0.07 | 0.15 | 2.24×10−17 | 4.50× 10−06 | 1.56× 10−21 | 53797908–53848561 | FTO |
| rs7228430 | 18 | 57733925 | A/G | 0.05 | 0.12 | 5.02×10−09 | 9.50× 10−04 | 5.50× 10−11 | 57730096–57914679 | |
SNP, single nucleotide polymorphisms; CHR, chromosome; BP, physical position of SNP (base-pairs); E, effect allele; A, alternative allele; BETA, effect allele beta coefficient; P-trait, P value for each obesity-related trait; P-PCOS, P value for PCOS; P-CPASSOC, P value for cross-phenotype association; BMI, adult body mass index; PCOS, polycystic ovary syndrome; WHR, waist-to-hip ratio; WHRadjBMI, waist-to-hip ratio adjusted for body mass index; CBMI, childhood body mass index
Functional annotation and tissue enrichment analysis
We attempted to understand the underlying biological mechanisms by identifying relevant tissues using FUMA. As shown in Additional file 2: Fig. S2, for the expression of genes shared by adult BMI and PCOS, although failing to pass multiple correction, we observed enrichment in coronary artery tissue, digestive tract tissues including salivary gland, stomach, and colon, as well as both visceral omentum and subcutaneous adipose tissues. For WHR-PCOS and WHRadjBMI-PCOS shared genes, stomach showed the most significant enrichment although notwithstanding multiple correction. No significant tissue enrichment was identified for childhood BMI and PCOS. For LDSC-SEG enrichment analysis, as shown in Additional file 2: Fig. S3, despite significant enrichment of SNPs in adipose, brain, muscle tissues identified for obesity-related traits, none of the tissue/cell-type-specific enrichment withstood multiple correction (P< 0.05/396) for PCOS.
Fine-mapping credible set analysis
For each of the 16 shared loci identified as significant by the cross-trait meta-analysis, we identified a 99% credible set of causal variants. Lists of credible set SNPs in each shared locus for obesity-related traits and PCOS from fine mapping are shown in Additional file 1: Table. S9. We identified 39 SNPs in the 99% credible set for adult BMI and PCOS, 122 SNPs for childhood BMI and PCOS, 40 SNPs for WHR and PCOS, and 27 SNPs for WHRadjBMI and PCOS providing candidates for downstream experimental analysis.
Mendelian randomization analysis of obesity-related traits and PCOS
Finally, we conducted a bidirectional two-sample Mendelian randomization analysis to test for the causal relationship between obesity-related traits and PCOS. As shown in Figure 3, we observed an almost threefold increased risk of PCOS per-SD increment (4.8 kg/m2) in adult BMI (IVW OR=2.92, 95% CI=2.33–3.67) using 278 female-specific IVs. The effect did not alter using MR-Egger (OR=4.33, 95% CI=2.26–8.32) or weighted median (OR=3.06, 95% CI=2.14–4.36) approach. We did not observe any sign of horizontal pleiotropy (P for MR-Egger intercept=0.21). Sensitivity analysis removing palindromic SNPs or pleiotropic SNPs (Fig. 3, Supplementary Tables 1-4) revealed similar findings.
Fig. 3.

Estimates of causal effect sizes for genetically predicted obesity-related traits on PCOS using all genome-wide significant SNPs, excluding pleiotropic SNPs and excluding palindromic SNPs. Inverse-variance weighted approach was used as the primary analysis, MR-Egger and weighted median approaches were used as sensitivity analysis. BMI, adult body mass index; PCOS, polycystic ovary syndrome; WHR, waist-to-hip ratio; WHRadjBMI, waist-to-hip ratio adjusted for body mass index; CBMI, childhood body mass index
Consistent with results from the adult population, we observed a strong causal link with PCOS using 25 childhood BMI-associated IVs (IVW OR=2.76, 95% CI=2.09–3.66)—a less than 10% number of IVs compared to adult BMI. Unfortunately, and perhaps not surprisingly, we were underpowered to perform sensitivity analyses for this trait due to the limited number of IVs. Directional consistent results were observed in MR-Egger analysis although the significance was attenuated to null (MR-Egger OR=1.98, 95% CI=0.79–4.96) (Fig. 3).
On the contrary, we did not find any significant association between genetically predicted WHR and risk of PCOS (IVW OR=1.19, 95% CI=0.93–1.52; MR-Egger OR=1.04, 95% CI=0.57–1.90; weighted median OR=0.84, 95% CI=0.6–1.18). Our results imply a causal role of BMI but not WHR in the development of PCOS. We anticipate observing a null effect of WHRadjBMI with PCOS after eliminating the positive association of BMI from the null association of WHR. As expected, we did not identify any increased risk of PCOS with WHRadjBMI with all effect sizes close to 1.00 (IVW OR=1.00, 95% CI=0.87–1.22; MR-Egger OR=0.97, 95% CI=0.66–1.42; weighted median OR=0.96, 95% CI=0.71–1.28).
Our findings were corroborated by the results of two important sensitivity analyses. The leave-one-out analysis demonstrated the observed BMI-PCOS causal association (for both childhood and adult) was not driven by outlying variant. As shown in Additional file 2: Fig. S4, when iteratively removing one IV at a time and performing IVW using the remaining IVs, BMI-PCOS associations centered around an OR of 2.5–3.0, while WHR/WHRadjBMI-PCOS associations aggregated closely to 1.0. In the subsequent clustering analysis (Additional file 2: Fig. S5), for both childhood and adult BMI, we observed a clear linear trend in which BMI-increasing IVs also increased the risk of PCOS. On the contrary, no such pattern was observed for WHR or WHRadjBMI with PCOS, consistent with a null association.
Genetic predisposition to PCOS did not seem to affect any of the obesity-related traits in our reverse-directional MR (adult BMI beta = − 0.01, 95% CI = − 0.03–0.02; childhood BMI beta = 0.01, 95% CI = − 0.04–0.05; WHR beta = 0.01, 95% CI = − 0.02–0.04; WHRadjBMI beta = 0.02, 95% CI = − 0.01–0.04) (Fig. 4).
Fig. 4.
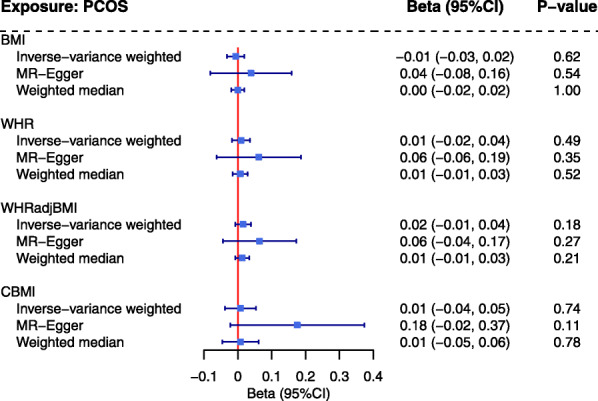
Estimates of causal effect sizes for genetical predisposition to PCOS on obesity-related traits using all genome-wide significant SNPs. Inverse-variance weighted approach was used as the primary analysis, MR-Egger and weighted median approaches were used as sensitivity analysis. BMI, adult body mass index; PCOS, polycystic ovary syndrome; WHR, waist-to-hip ratio; WHRadjBMI, waist-to-hip ratio adjusted for body mass index; CBMI, childhood body mass index
Discussion
To the best of our knowledge, this is the first large-scale genome-wide cross-trait analysis that investigates the shared genetic basis underlying obesity and PCOS. We found a positive overall genetic correlation between both adult and childhood BMI and PCOS. The significantly shared genetic basis between WHR and PCOS seems to be driven by BMI given the null finding of WHRadjBMI and PCOS. In local genetic correlation analysis, when the genome was partitioned into small regions, we identified one genomic region at chr18: 57630483–59020751 that showed a positive local genetic correlation between adult BMI and PCOS. Using cross-trait meta-analysis, we identified multiple shared loci between obesity and PCOS. Finally, MR analysis highlighted the causal role of adult BMI and childhood BMI in the development of PCOS.
Findings from our study are largely in line with those from the conventional epidemiological studies yet provide novel insights in several aspects. In line with the positive overall genetic correlation identified for adult BMI and PCOS (rg=0.34, P=8.21×10−18) by Day et al. [7], our results (adult BMI: rg=0.47, P=2.19×10−16) improved statistical power by using an enlarged sample size (434,794 female vs. 339,224 individuals) as well as reduced heterogeneity by using sex-specific adult BMI GWAS summary statistics, both of which previous studies did not have a chance for. Findings on the [41] overall and local genetic correlation between adult BMI and PCOS suggest a shared genetic basis underlying these two traits, which is either directly through variants affecting both traits (pleiotropy), or through the causal effect of one trait on the other. Our MR analyses exploring the causal relationships are in line with three existing MR(s) [7, 9, 10] conducted on BMI and PCOS while greatly extending those results. Firstly, we used the largest GWAS conducted in BMI with > 270 BMI-associated IVs—a more than three times augmented number of instruments (> 270 vs. 92) compared with previous analyses [7, 10]. Incorporating additional IVs substantially improves the strength of genetic instruments as well as both the accuracy and precision of our MR estimates. With the current sample size of outcome (N=113,238, 9% cases) and assuming phenotypic variance of the exposures explained by IVs to be around 4%, our study had 80% power to detect an association of 15% change for the risk of PCOS with BMI. Secondly, to ensure the validity of MR results, exposure and outcome samples should preferably be from the same underlying population. We conducted our analysis restricting to female participants—making it possible to utilize female-specific genetic instruments to study a female outcome PCOS, which previous analyses did not have the opportunity for. Finally, we conducted several sensitivity analyses to verify MR model assumptions. We selected the most significant SNPs (independent GWAS signals at a stringent P-threshold of 5×10−9) so all were robustly associated with exposure of interest, guaranteeing the “relevance” assumption. We excluded SNPs associated with potential confounders on the exposure-outcome relationship to satisfy the “exclusion restriction” assumption.
In addition to adult BMI, childhood BMI appears to influence the risk of PCOS later on [42, 43]. Findings from earlier observational studies including nearly 3000 participants from the Australian Childhood Determinants of Adult Health study (N=1516) and the biracial USA Babies substudy of the Bogalusa Heart Study (N=1247) have suggested an association between greater childhood BMI and PCOS among the white population (RR=4.05, 95% CI=1.10–14.83; RR=2.93, 95% CI=1.65–5.22) [43]. Our results of positive overall genetic, shared loci, and causal effect identified for childhood BMI with PCOS confirmed the observational association and provided genetic evidence for such association. For the null local genetic correlation, we note that the sample size of childhood BMI GWAS (N=39,620) is not sufficiently large for ρ-HESS recommendation (which 50,000 samples are preferably required). Future studies with larger sample sizes are warranted to replicate our findings.
Using WHR as a proxy to abdominal fat, we did not find any significant causal association for this trait with PCOS. Raw WHR is likely to be confounded by BMI as indicated by our previous genetic correlation analysis [29]. Dissecting the effect of BMI from WHR, the negative WHRadjBMI-PCOS causal association together with genetic correlation further confirmed the validity of our results. However, these results do not necessarily mean that abdominal visceral fat is not important in PCOS. While BMI is found to be highly correlated with excess fat mass (r=0.94) and abdominal visceral fat (r=0.71) [44], results for the association between fat distribution and PCOS from observational studies were contrasting. A study including 110 PCOS patients and 112 weight-matched controls assessed fat quantity and distribution using total-body dual x-ray absorptiometry concluded that women with PCOS had a higher quantity of central abdominal fat compared to weight-matched controls [45], however, recent imaging studies using a gold standard approach of magnetic resonance-based methodology demonstrated an equivalent visceral fat depot between women with PCOS and their BMI-/fat mass-matched controls [46–48]. Indeed, WHR poorly predicts the accumulation of visceral fat [49], which may explain our significant findings with BMI rather than with WHR. Moreover, studies have shown that waist circumference (WC) presents the highest correlation with magnetic resonance imaging measured abdominal visceral adiposity, the gold standard approach, as well as the best sensitivity and specificity in the receiver operating characteristic curve [50]—therefore is considered as a better indicator for abdominal obesity than WHR. Investigating WC and PCOS leveraging recently published female WC GWAS would be a future direction on this topic [51].
Genetic correlation provides genetic insights into the observational associations by estimating the degree of pleiotropy or causal overlaps shared by two traits while MR analysis infers causal relationships. We additionally performed a cross-trait meta-analysis to further dissect the complex genetic relationships between obesity and PCOS. Using cross-trait meta-analysis, we identified 16 SNPs shared between obesity-related traits and PCOS, indicating shared biological mechanisms underlying obesity and PCOS. Among these shared loci, we highlight the locus of ERBB3, FTO, PROX1, GIPR, and MC4R in relation to potential pathogenesis. ERBB3 encodes epidermal growth factor receptors (EGFRs) and has been reported to be associated with PCOS by a meta-GWAS [7]. Studies have shown that gonadotropins upregulate ERBB3 expression and EGFR signaling mediates LH-induced steroidogenesis, which plays an important role in follicular development [52]. EGFRs are also closely involved in obesity—experimental studies have shown that EGFRs are transactivated by leptin, a hormone of an elevated concentration in patients with obesity/metabolic syndrome [53]. For gene FTO, despite a large number of studies that have confirmed its contribution to adult obesity, childhood obesity, and obesity-related traits [36, 54–56], its role in PCOS remains controversial. Some studies have observed a positive association between FTO and PCOS whereas others have not [37, 57–59]. The association between FTO and PCOS may partially be mediated through obesity. Indeed, the most evident association between FTO and PCOS was observed in obese PCOS women, which was attenuated when adjusting for BMI [59]. Moreover, the impact of FTO on lipid oxidation in PCOS women might also contribute to the mechanism underlying the comorbidity of obesity and PCOS [60]. PROX1 encodes one of the proteins of the homeobox transcription factor family, which plays an essential role in organ development during embryogenesis [61]. Results from both large-scale population study and mice models have shown that PROX1 was associated with visceral fat accumulation [62, 63]. PROX1 has also been found to be differently methylated in adipose tissue in PCOS women and controls [64]. In addition to its role in obesity, PROX1 has also been linked to glycemic alteration (fasting glucose and type 2 diabetes) [38]—another important feature of PCOS. Similarly, GIPR influences glycemic traits including 2-h glucose level and insulin secretion [35]. GIPR encodes a G-protein coupled receptor for gastric inhibitory polypeptide (GIP), which has been demonstrated to stimulate insulin release in the presence of elevated glucose. Evidence has suggested that modulation of GIPR affects progesterone synthesis and expression of many progestogenic factors and enzymes that may involve in the subfertility feature of PCOS [65]. Notably, the shared region chr18: 57730096–57914679 (top SNP rs7228430) identified for childhood BMI and PCOS overlaps with the local genetic correlation identified for adult BMI and PCOS at chr18: 57630483–59020751, indicating shared biology underlying these traits. This region harbors MC4R, a gene previously found to be associated with higher adult BMI and childhood BMI. Using BMI-matched samples or BMI-adjusted statistical models, studies have found MC4R to be associated with an elevated BMI in PCOS [31, 66]. MC4R encodes melanocortin 4 receptor that plays an important role in central melanocortin neuronal pathways [67]. Furthermore, a study using PCOS rats models has also found an over-expression of the MC4R gene in the brain hypothalamus which may link to metabolic disorders [32].
Results from GTEx tissue enrichment analysis should be interpreted with caution due to the limited number of shared genes identified between obesity and PCOS. Similarly, the null findings from LDSC-SEG do not necessarily indicate a negligible shared biological mechanism given the limited sample size of PCOS GWAS and the positive findings identified by our other analyses.
We acknowledge several limitations. First, PCOS, as a complex disease, is classified into four phenotypes according to the presence or absence of ovulatory dysfunction, hyperandrogenism, and polycystic ovarian morphology [68]. Our study was not able to perform phenotype-specific analysis due to limited data availability. In addition, PCOS occurs in both obese/overweight and lean women, our findings of the role of obesity in PCOS may not be applicable to lean women with PCOS. A recent study using unsupervised clustering analysis suggested distinct genetic architecture underlying PCOS subtypes. Using biochemical and genotype data, PCOS can be classified into a “reproductive” subtype which presents higher luteinizing hormone (LH) and sex hormone-binding globulin (SHBG) levels with relatively low BMI, and a “metabolic” subtype which presents higher BMI, glucose, and insulin levels with lower SHBG and LH levels [69]. Our work was limited by the availability of subtype-specific PCOS GWAS, future work is needed to understand the role of BMI in lean PCOS. Second, the strong link between BMI and PCOS seems to be consistent across ethnicities, for example, using 78 BMI IVs discovered by a GWAS of Biobank Japan and 4,386 PCOS cases (8,017 controls) of East Asian ancestry, a twofold risk of PCOS was observed for heightened BMI (OR=2.21, 95% CI=1.54–3.17, P=1.8×10−5) [70], However, the generalizability of our findings of the shared genetic basis of obesity and PCOS is restricted to European population. Further genome-wide association studies on this topic leveraging data from other ethnicities are warranted. Third, despite our study being the (so far) largest in sample size, compared to adult obesity GWASs, PCOS has a much smaller sample size (4790 cases/20,405 controls vs. ~ 400,000 female), future studies with enlarged sample size are needed. Fourth, while our study identified genes relevant to obesity and PCOS, more data are needed to understand the underlying pathophysiological mechanisms.
Our work investigated the shared genetic basis underlying obesity and PCOS. Future work should be to perform large, prospective longitudinal clinical studies to define whether PCOS women carrying a specific genotype are at increased risk for developing, e.g., cardiovascular disease, type 2 diabetes, non-alcoholic fatty liver disease, and link to mortality. Of outmost importance, our study confirmed the causative role of BMI in PCOS prevention, future studies on whether medical weight reduction or bariatric surgery alleviates PCOS-related comorbidities are needed. Moreover, the use of gene-modified mice, e.g., knock-in/knock-out of obesity/PCOS risk genes is of importance to define the role of identified specific genes and rare genetic variants and would provide novel insights into the biological mechanisms underlying these complex traits.
Conclusions
In conclusion, leveraging the largest sex-specific GWAS summary statistics to date, the current study furthered our understanding of the observational association between obesity and PCOS by showing evidence of genetic correlation, revealing shared loci, and inferring causal relationships, all of which may provide insights into the biological pathways. Our work informs public health intervention by confirming the important role of weight management from childhood through adulthood in PCOS prevention.
Supplementary Information
Additional file 1: Table S1. Adult BMI IVs. Table S2. WHR IVs. Table S3. WHRadjBMI IVs. Table S4. childhood BMI IVs. Table S50. PCOS IVs. Table S6. Data sources, sample sizes, number of IVs and F-statistics. Table S7. Summary of contributing cohorts/consortia/studies in each GWAS. Table S8. Annotation of genome-wide significant SNPs from CPASSOC. Table S9. 99% credible set SNPs.
Additional file 2: Figure S1. Local genetic correlation between childhood BMI, WHR, WHRadjBMI and PCOS. Figure S2. GTEx tissue enrichment analysis. Figure S3. Cell-type specific enrichment analysis. Figure S4. Box plot of betas in leave-one-out analysis. Figure S5. MR-clust analysis.
Acknowledgements
Summary statistics for the GWAS(s) of adult BMI, childhood BMI, WHR, WHRadjBMI, and PCOS were obtained by Yengo L et al., Vogelezang S et al., Pulit SL et al. and Day FR et al. We thank all investigators and consortium for sharing the genome-wide summary statistics.
Abbreviations
- BMI
Body mass index
- CPASSOC
Cross-phenotype association analysis
- DEG
Differentially expressed gene
- EGFRs
Epidermal growth factor receptors
- FUMA
Functional mapping and annotation
- GWAS
Genome-wide associations studies
- HRC
Haplotype Reference Consortium
- IV
Instrumental variable
- IVW
Inverse-variance weighted
- LD
linkage disequilibrium
- LDSC
linkage-disequilibrium score regression
- LH
luteinizing hormone
- MR
Mendelian randomization
- PCOS
Polycystic ovary syndrome
- PIP
Posterior inclusion probability
- SHBG
Sex hormone-binding globulin
- VEP
Variant effect predictor
- WC
Waist circumference
- WHR
Waist-to-hip ratio
- WHRadjBMI
WHR adjusted for BMI
Authors’ contributions
QWL and XJ analyzed the data, interpreted the results, and wrote the manuscript. XJ and ESV designed the study. ZZZ provided statistical expertise and revised the manuscript. PK, QLD, and ESV revised the manuscript. All authors provided the corresponding author with permission to be named in the manuscript. XJ is the guarantor of this study. The authors read and approved the final version of the manuscript.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or non-profit sectors. Open Access funding was provided by Karolinska Institute.
Availability of data and materials
All data used in the present study were obtained from publicly available GWAS summary statistics.
Declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Naz MSG, Tehrani FR, Majd HA, Ahmadi F, Ozgoli G, Fakari FR, Ghasemi V. The prevalence of polycystic ovary syndrome in adolescents: a systematic review and meta-analysis. Int J Reprod Biomed. 2019;17(8):533–542. doi: 10.18502/ijrm.v17i8.4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.March WA, Moore VM, Willson KJ, Phillips DIW, Norman RJ, Davies MJ. The prevalence of polycystic ovary syndrome in a community sample assessed under contrasting diagnostic criteria. Hum Reprod Oxf Engl. 2010;25(2):544–551. doi: 10.1093/humrep/dep399. [DOI] [PubMed] [Google Scholar]
- 3.Lim SS, Davies MJ, Norman RJ, Moran LJ. Overweight, obesity and central obesity in women with polycystic ovary syndrome: a systematic review and meta-analysis. Hum Reprod Update. 2012;18(6):618–637. doi: 10.1093/humupd/dms030. [DOI] [PubMed] [Google Scholar]
- 4.Ollila M-ME, Piltonen T, Puukka K, Ruokonen A, Järvelin M-R, Tapanainen JS, Franks S, Morin-Papunen L. Weight gain and dyslipidemia in early adulthood associate with polycystic ovary syndrome: prospective cohort study. J Clin Endocrinol Metab. 2016;101(2):739–747. doi: 10.1210/jc.2015-3543. [DOI] [PubMed] [Google Scholar]
- 5.Barber TM, Hanson P, Weickert MO, Franks S. Obesity and polycystic ovary syndrome: implications for pathogenesis and novel management strategies. Clin Med Insights Reprod Health. 2019;13:117955811987404. doi: 10.1177/1179558119874042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu Z, Hasegawa K, Camargo CA, Liang L. Investigating asthma heterogeneity through shared and distinct genetics: insights from genome-wide cross-trait analysis. J Allergy Clin Immunol. 2021;147(3):796–807. doi: 10.1016/j.jaci.2020.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Day F, Karaderi T, Jones MR, Meun C, He C, Drong A, Kraft P, Lin N, Huang H, Broer L, Magi R, Saxena R, Laisk T, Urbanek M, Hayes MG, Thorleifsson G, Fernandez-Tajes J, Mahajan A, Mullin BH, Stuckey BGA, Spector TD, Wilson SG, Goodarzi MO, Davis L, Obermayer-Pietsch B, Uitterlinden AG, Anttila V, Neale BM, Jarvelin MR, Fauser B, Kowalska I, Visser JA, Andersen M, Ong K, Stener-Victorin E, Ehrmann D, Legro RS, Salumets A, McCarthy MI, Morin-Papunen L, Thorsteinsdottir U, Stefansson K, the 23andMe Research Team. Styrkarsdottir U, Perry JRB, Dunaif A, Laven J, Franks S, Lindgren CM, Welt CK. Large-scale genome-wide meta-analysis of polycystic ovary syndrome suggests shared genetic architecture for different diagnosis criteria. PLOS Genet. 2018;14(12):e1007813. doi: 10.1371/journal.pgen.1007813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joo YY, Actkins K, Pacheco JA, Basile AO, Carroll R, Crosslin DR, et al. A Polygenic and Phenotypic Risk Prediction for Polycystic Ovary Syndrome Evaluated by Phenome-Wide Association Studies. J Clin Endocrinol Metab. 2020;105(6):1918–1936. doi: 10.1210/clinem/dgz326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Day FR, Hinds DA, Tung JY, Stolk L, Styrkarsdottir U, Saxena R, Bjonnes A, Broer L, Dunger DB, Halldorsson BV, Lawlor DA, Laval G, Mathieson I, McCardle WL, Louwers Y, Meun C, Ring S, Scott RA, Sulem P, Uitterlinden AG, Wareham NJ, Thorsteinsdottir U, Welt C, Stefansson K, Laven JSE, Ong KK, Perry JRB. Causal mechanisms and balancing selection inferred from genetic associations with polycystic ovary syndrome. Nat Commun. 2015;6(1):8464. doi: 10.1038/ncomms9464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brower MA, Hai Y, Jones MR, Guo X, Chen Y-DI, Rotter JI, et al. Bidirectional Mendelian randomization to explore the causal relationships between body mass index and polycystic ovary syndrome. Hum Reprod Oxf Engl. 2019;34(1):127–136. doi: 10.1093/humrep/dey343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yengo L, Sidorenko J, Kemper KE, Zheng Z, Wood AR, Weedon MN, Frayling TM, Hirschhorn J, Yang J, Visscher PM, the GIANT Consortium Meta-analysis of genome-wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum Mol Genet. 2018;27(20):3641–3649. doi: 10.1093/hmg/ddy271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lebovitz HE, Banerji MA. Point: Visceral Adiposity Is Causally Related to Insulin Resistance. Diabetes Care. 2005;28(9):2322–2325. doi: 10.2337/diacare.28.9.2322. [DOI] [PubMed] [Google Scholar]
- 13.Simmonds M, Llewellyn A, Owen CG, Woolacott N. Predicting adult obesity from childhood obesity: a systematic review and meta-analysis. Obes Rev Off J Int Assoc Study Obes. 2016;17(2):95–107. doi: 10.1111/obr.12334. [DOI] [PubMed] [Google Scholar]
- 14.Vogelezang S, Bradfield JP, Ahluwalia TS, Curtin JA, Lakka TA, Grarup N, Scholz M, van der Most PJ, Monnereau C, Stergiakouli E, Heiskala A, Horikoshi M, Fedko IO, Vilor-Tejedor N, Cousminer DL, Standl M, Wang CA, Viikari J, Geller F, Íñiguez C, Pitkänen N, Chesi A, Bacelis J, Yengo L, Torrent M, Ntalla I, Helgeland Ø, Selzam S, Vonk JM, Zafarmand MH, Heude B, Farooqi IS, Alyass A, Beaumont RN, Have CT, Rzehak P, Bilbao JR, Schnurr TM, Barroso I, Bønnelykke K, Beilin LJ, Carstensen L, Charles MA, Chawes B, Clément K, Closa-Monasterolo R, Custovic A, Eriksson JG, Escribano J, Groen-Blokhuis M, Grote V, Gruszfeld D, Hakonarson H, Hansen T, Hattersley AT, Hollensted M, Hottenga JJ, Hyppönen E, Johansson S, Joro R, Kähönen M, Karhunen V, Kiess W, Knight BA, Koletzko B, Kühnapfel A, Landgraf K, Langhendries JP, Lehtimäki T, Leinonen JT, Li A, Lindi V, Lowry E, Bustamante M, Medina-Gomez C, Melbye M, Michaelsen KF, Morgen CS, Mori TA, Nielsen TRH, Niinikoski H, Oldehinkel AJ, Pahkala K, Panoutsopoulou K, Pedersen O, Pennell CE, Power C, Reijneveld SA, Rivadeneira F, Simpson A, Sly PD, Stokholm J, Teo KK, Thiering E, Timpson NJ, Uitterlinden AG, van Beijsterveldt CEM, van Schaik BDC, Vaudel M, Verduci E, Vinding RK, Vogel M, Zeggini E, Sebert S, Lind MV, Brown CD, Santa-Marina L, Reischl E, Frithioff-Bøjsøe C, Meyre D, Wheeler E, Ong K, Nohr EA, Vrijkotte TGM, Koppelman GH, Plomin R, Njølstad PR, Dedoussis GD, Froguel P, Sørensen TIA, Jacobsson B, Freathy RM, Zemel BS, Raitakari O, Vrijheid M, Feenstra B, Lyytikäinen LP, Snieder H, Kirsten H, Holt PG, Heinrich J, Widén E, Sunyer J, Boomsma DI, Järvelin MR, Körner A, Davey Smith G, Holm JC, Atalay M, Murray C, Bisgaard H, McCarthy MI, Early Growth Genetics Consortium. Jaddoe VWV, Grant SFA, Felix JF. Novel loci for childhood body mass index and shared heritability with adult cardiometabolic traits. PLOS Genet. 2020;16(10):e1008718. doi: 10.1371/journal.pgen.1008718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pulit SL, Stoneman C, Morris AP, Wood AR, Glastonbury CA, Tyrrell J, Yengo L, Ferreira T, Marouli E, Ji Y, Yang J, Jones S, Beaumont R, Croteau-Chonka DC, Winkler TW, GIANT Consortium. Hattersley AT, Loos RJF, Hirschhorn JN, Visscher PM, Frayling TM, Yaghootkar H, Lindgren CM. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum Mol Genet. 2019;28(1):166–174. doi: 10.1093/hmg/ddy327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, Sham PC. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bulik-Sullivan BK, Loh P-R, Finucane HK, Ripke S, Yang J, Patterson N, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47(3):291–295. doi: 10.1038/ng.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh P-R, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47(11):1236–1241. doi: 10.1038/ng.3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu X, Feng T, Tayo BO, Liang J, Young JH, Franceschini N, Smith JA, Yanek LR, Sun YV, Edwards TL, Chen W, Nalls M, Fox E, Sale M, Bottinger E, Rotimi C, COGENT BP Consortium. Liu Y, McKnight B, Liu K, Arnett DK, Chakravati A, Cooper RS, Redline S. Meta-analysis of correlated traits via summary statistics from GWASs with an application in hypertension. Am J Hum Genet. 2015;96(1):21–36. doi: 10.1016/j.ajhg.2014.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang H. hailianghuang/FM-summary. R. 2021. [Google Scholar]
- 21.Huang H, Fang M, Jostins L, Umićević Mirkov M, Boucher G, Anderson CA, et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature. 2017;547(7662):173–178. doi: 10.1038/nature22969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maller JB, McVean G, Byrnes J, Vukcevic D, Palin K, Su Z, et al. Bayesian refinement of association signals for 14 loci in 3 common diseases. Nat Genet. 2012;44(12):1294–1301. doi: 10.1038/ng.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Finucane HK, Reshef YA, Anttila V, Slowikowski K, Gusev A, Byrnes A, et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat Genet. 2018;50(4):621–629. doi: 10.1038/s41588-018-0081-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518(7539):317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burgess S, Scott RA, Timpson NJ, Davey Smith G, Thompson SG, EPIC- InterAct Consortium Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol. 2015;30(7):543–552. doi: 10.1007/s10654-015-0011-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–525. doi: 10.1093/ije/dyv080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet Epidemiol. 2016;40(4):304–314. doi: 10.1002/gepi.21965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Foley CN, Kirk PDW, Burgess S. MR-Clust: Clustering of genetic variants in Mendelian randomization with similar causal estimates. bioRxiv. 2019;:2019.12.18.881326. [DOI] [PMC free article] [PubMed]
- 29.Tang B, Shi H, Alfredsson L, Klareskog L, Padyukov L, Jiang X. Obesity-Related Traits and the Development of Rheumatoid Arthritis: Evidence From Genetic Data. Arthritis Rheumatol. 2021;73(2):203–211. doi: 10.1002/art.41517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bradnova O, Vejrazková D, Vaňková M, LUKÁŠOVÁ P, Včelák J, STANICKÁ S, et al. Metabolic and Hormonal Consequencies of the “Obesity Risk” MC4R Variant (rs12970134) in Czech Women. Physiol Res Acad Sci Bohemoslov. 2015;64:S187–S195. doi: 10.33549/physiolres.933119. [DOI] [PubMed] [Google Scholar]
- 31.Ewens KG, Jones MR, Ankener W, Stewart DR, Urbanek M, Dunaif A, Legro RS, Chua A, Azziz R, Spielman RS, Goodarzi MO, Strauss JF. FTO and MC4R Gene variants are associated with obesity in polycystic ovary syndrome. PLOS ONE. 2011;6(1):e16390. doi: 10.1371/journal.pone.0016390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nooranizadeh MH, Rahmanifar F, Ahmadloo S, Shaaban Z, Jafarzadeh Shirazi MR, Tamadon A. Enhancement of Melanocortin-4 Receptor (MC4R) and Constancy of Kiss1 mRNAs Expression in the Hypothalamic Arcuate Nucleus in a Model of Polycystic Ovary Syndrome Rat. Galen Med J. 2018;7:e1070. doi: 10.31661/gmj.v7i0.1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuan H, Zhu G, Wang F, Wang X, Guo H, Shen M. Interaction between common variants of FTO and MC4R is associated with risk of PCOS. Reprod Biol Endocrinol. 2015;13(1):55. doi: 10.1186/s12958-015-0050-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wen W, Cho Y-S, Zheng W, Dorajoo R, Kato N, Qi L, et al. Meta-analysis identifies common variants associated with body mass index in east Asians. Nat Genet. 2012;44(3):307–311. doi: 10.1038/ng.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saxena R, Hivert M-F, Langenberg C, Tanaka T, Pankow JS, Vollenweider P, Lyssenko V, Bouatia-Naji N, Dupuis J, Jackson AU, Kao WH, Li M, Glazer NL, Manning AK, Luan J, Stringham HM, Prokopenko I, Johnson T, Grarup N, Boesgaard TW, Lecoeur C, Shrader P, O'Connell J, Ingelsson E, Couper DJ, Rice K, Song K, Andreasen CH, Dina C, Köttgen A, le Bacquer O, Pattou F, Taneera J, Steinthorsdottir V, Rybin D, Ardlie K, Sampson M, Qi L, van Hoek M, Weedon MN, Aulchenko YS, Voight BF, Grallert H, Balkau B, Bergman RN, Bielinski SJ, Bonnefond A, Bonnycastle LL, Borch-Johnsen K, Böttcher Y, Brunner E, Buchanan TA, Bumpstead SJ, Cavalcanti-Proença C, Charpentier G, Chen YD, Chines PS, Collins FS, Cornelis M, J Crawford G, Delplanque J, Doney A, Egan JM, Erdos MR, Firmann M, Forouhi NG, Fox CS, Goodarzi MO, Graessler J, Hingorani A, Isomaa B, Jørgensen T, Kivimaki M, Kovacs P, Krohn K, Kumari M, Lauritzen T, Lévy-Marchal C, Mayor V, McAteer J, Meyre D, Mitchell BD, Mohlke KL, Morken MA, Narisu N, Palmer CN, Pakyz R, Pascoe L, Payne F, Pearson D, Rathmann W, Sandbaek A, Sayer AA, Scott LJ, Sharp SJ, Sijbrands E, Singleton A, Siscovick DS, Smith NL, Sparsø T, Swift AJ, Syddall H, Thorleifsson G, Tönjes A, Tuomi T, Tuomilehto J, Valle TT, Waeber G, Walley A, Waterworth DM, Zeggini E, Zhao JH, GIANT consortium. MAGIC investigators. Illig T, Wichmann HE, Wilson JF, van Duijn C, Hu FB, Morris AD, Frayling TM, Hattersley AT, Thorsteinsdottir U, Stefansson K, Nilsson P, Syvänen AC, Shuldiner AR, Walker M, Bornstein SR, Schwarz P, Williams GH, Nathan DM, Kuusisto J, Laakso M, Cooper C, Marmot M, Ferrucci L, Mooser V, Stumvoll M, Loos RJ, Altshuler D, Psaty BM, Rotter JI, Boerwinkle E, Hansen T, Pedersen O, Florez JC, McCarthy M, Boehnke M, Barroso I, Sladek R, Froguel P, Meigs JB, Groop L, Wareham NJ, Watanabe RM. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat Genet. 2010;42(2):142–148. doi: 10.1038/ng.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dina C, Meyre D, Gallina S, Durand E, Körner A, Jacobson P, Carlsson LMS, Kiess W, Vatin V, Lecoeur C, Delplanque J, Vaillant E, Pattou F, Ruiz J, Weill J, Levy-Marchal C, Horber F, Potoczna N, Hercberg S, le Stunff C, Bougnères P, Kovacs P, Marre M, Balkau B, Cauchi S, Chèvre JC, Froguel P. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet. 2007;39(6):724–726. doi: 10.1038/ng2048. [DOI] [PubMed] [Google Scholar]
- 37.Liu AL, Xie HJ, Xie HY, Liu J, Yin J, Hu JS, Peng CY. Association between fat mass and obesity associated (FTO) gene rs9939609 A/T polymorphism and polycystic ovary syndrome: a systematic review and meta-analysis. BMC Med Genet. 2017;18(1):89. doi: 10.1186/s12881-017-0452-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, Wheeler E, Glazer NL, Bouatia-Naji N, Gloyn AL, Lindgren CM, Mägi R, Morris AP, Randall J, Johnson T, Elliott P, Rybin D, Thorleifsson G, Steinthorsdottir V, Henneman P, Grallert H, Dehghan A, Hottenga JJ, Franklin CS, Navarro P, Song K, Goel A, Perry JR, Egan JM, Lajunen T, Grarup N, Sparsø T, Doney A, Voight BF, Stringham HM, Li M, Kanoni S, Shrader P, Cavalcanti-Proença C, Kumari M, Qi L, Timpson NJ, Gieger C, Zabena C, Rocheleau G, Ingelsson E, An P, O'Connell J, Luan J, Elliott A, McCarroll S, Payne F, Roccasecca RM, Pattou F, Sethupathy P, Ardlie K, Ariyurek Y, Balkau B, Barter P, Beilby JP, Ben-Shlomo Y, Benediktsson R, Bennett AJ, Bergmann S, Bochud M, Boerwinkle E, Bonnefond A, Bonnycastle LL, Borch-Johnsen K, Böttcher Y, Brunner E, Bumpstead SJ, Charpentier G, Chen YD, Chines P, Clarke R, Coin LJ, Cooper MN, Cornelis M, Crawford G, Crisponi L, Day IN, de Geus EJ, Delplanque J, Dina C, Erdos MR, Fedson AC, Fischer-Rosinsky A, Forouhi NG, Fox CS, Frants R, Franzosi MG, Galan P, Goodarzi MO, Graessler J, Groves CJ, Grundy S, Gwilliam R, Gyllensten U, Hadjadj S, Hallmans G, Hammond N, Han X, Hartikainen AL, Hassanali N, Hayward C, Heath SC, Hercberg S, Herder C, Hicks AA, Hillman DR, Hingorani AD, Hofman A, Hui J, Hung J, Isomaa B, Johnson PR, Jørgensen T, Jula A, Kaakinen M, Kaprio J, Kesaniemi YA, Kivimaki M, Knight B, Koskinen S, Kovacs P, Kyvik KO, Lathrop GM, Lawlor DA, le Bacquer O, Lecoeur C, Li Y, Lyssenko V, Mahley R, Mangino M, Manning AK, Martínez-Larrad MT, McAteer J, McCulloch L, McPherson R, Meisinger C, Melzer D, Meyre D, Mitchell BD, Morken MA, Mukherjee S, Naitza S, Narisu N, Neville MJ, Oostra BA, Orrù M, Pakyz R, Palmer CN, Paolisso G, Pattaro C, Pearson D, Peden JF, Pedersen NL, Perola M, Pfeiffer AF, Pichler I, Polasek O, Posthuma D, Potter SC, Pouta A, Province MA, Psaty BM, Rathmann W, Rayner NW, Rice K, Ripatti S, Rivadeneira F, Roden M, Rolandsson O, Sandbaek A, Sandhu M, Sanna S, Sayer AA, Scheet P, Scott LJ, Seedorf U, Sharp SJ, Shields B, Sigurethsson G, Sijbrands EJ, Silveira A, Simpson L, Singleton A, Smith NL, Sovio U, Swift A, Syddall H, Syvänen AC, Tanaka T, Thorand B, Tichet J, Tönjes A, Tuomi T, Uitterlinden AG, van Dijk K, van Hoek M, Varma D, Visvikis-Siest S, Vitart V, Vogelzangs N, Waeber G, Wagner PJ, Walley A, Walters GB, Ward KL, Watkins H, Weedon MN, Wild SH, Willemsen G, Witteman JC, Yarnell JW, Zeggini E, Zelenika D, Zethelius B, Zhai G, Zhao JH, Zillikens MC, DIAGRAM Consortium. GIANT Consortium. Global BPgen Consortium. Borecki IB, Loos RJ, Meneton P, Magnusson PK, Nathan DM, Williams GH, Hattersley AT, Silander K, Salomaa V, Smith GD, Bornstein SR, Schwarz P, Spranger J, Karpe F, Shuldiner AR, Cooper C, Dedoussis GV, Serrano-Ríos M, Morris AD, Lind L, Palmer LJ, Hu FB, Franks PW, Ebrahim S, Marmot M, Kao WH, Pankow JS, Sampson MJ, Kuusisto J, Laakso M, Hansen T, Pedersen O, Pramstaller PP, Wichmann HE, Illig T, Rudan I, Wright AF, Stumvoll M, Campbell H, Wilson JF, Anders Hamsten on behalf of Procardis Consortium. MAGIC investigators. Bergman RN, Buchanan TA, Collins FS, Mohlke KL, Tuomilehto J, Valle TT, Altshuler D, Rotter JI, Siscovick DS, Penninx BW, Boomsma DI, Deloukas P, Spector TD, Frayling TM, Ferrucci L, Kong A, Thorsteinsdottir U, Stefansson K, van Duijn C, Aulchenko YS, Cao A, Scuteri A, Schlessinger D, Uda M, Ruokonen A, Jarvelin MR, Waterworth DM, Vollenweider P, Peltonen L, Mooser V, Abecasis GR, Wareham NJ, Sladek R, Froguel P, Watanabe RM, Meigs JB, Groop L, Boehnke M, McCarthy M, Florez JC, Barroso I. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42(2):105–116. doi: 10.1038/ng.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Human Gene SLC38A11 (uc002ucw.2) Description and Page Index. https://genome-euro.ucsc.edu/cgi-bin/hgGene?hgg_gene=uc002ucw.2&hgg_prot=uc002ucw.2&hgg_chrom=chr2&hgg_start=165754708&hgg_end=165812035&hgg_type=knownGene&db=hg19&hgsid=268663950_ioYAdF36quunjngHgPqaO64jreTU#go. Accessed 24 Jun 2021.
- 40.NCBI RefSeq Other Annotations (not NM_*, NR_*, XM_*, XR_*, NP_* or YP_*) (RNA5SP111). https://genome-euro.ucsc.edu/cgi-bin/hgc?hgsid=268663950_ioYAdF36quunjngHgPqaO64jreTU&db=hg19&c=chr2&l=165752186&r=165752287&o=165752186&t=165752287&g=ncbiRefSeqOther&i=RNA5SP111.
- 41.Hoeger KM. Obesity and Lifestyle Management in Polycystic Ovary Syndrome. Clin Obstet Gynecol. 2007;50(1):277–294. doi: 10.1097/GRF.0b013e31802f54c8. [DOI] [PubMed] [Google Scholar]
- 42.Aarestrup J, Pedersen DC, Thomas PE, Glintborg D, Holm J-C, Bjerregaard LG, Baker JL. Birthweight, Childhood Body Mass Index, Height and Growth, and Risk of Polycystic Ovary Syndrome. Obes Facts. 2021;14(3):283–290. doi: 10.1159/000515294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He Y, Tian J, Blizzard L, Oddy WH, Dwyer T, Bazzano LA, Hickey M, Harville EW, Venn AJ. Associations of childhood adiposity with menstrual irregularity and polycystic ovary syndrome in adulthood: the Childhood Determinants of Adult Health Study and the Bogalusa Heart Study. Hum Reprod. 2020;35(5):1185–1198. doi: 10.1093/humrep/deaa069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bouchard C. BMI, fat mass, abdominal adiposity and visceral fat: where is the “beef”. Int J Obes. 2007;31:1552–1553. doi: 10.1038/sj.ijo.0803653. [DOI] [PubMed] [Google Scholar]
- 45.Carmina E, Bucchieri S, Esposito A, Del Puente A, Mansueto P, Orio F, et al. Abdominal Fat Quantity and Distribution in Women with Polycystic Ovary Syndrome and Extent of Its Relation to Insulin Resistance. J Clin Endocrinol Metab. 2007;92(7):2500–2505. doi: 10.1210/jc.2006-2725. [DOI] [PubMed] [Google Scholar]
- 46.Barber TM, Golding SJ, Alvey C, Wass JAH, Karpe F, Franks S, McCarthy MI. Global adiposity rather than abnormal regional fat distribution characterizes women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2008;93(3):999–1004. doi: 10.1210/jc.2007-2117. [DOI] [PubMed] [Google Scholar]
- 47.Dolfing JG, Stassen CM, van Haard PMM, Wolffenbuttel BHR, Schweitzer DH. Comparison of MRI-assessed body fat content between lean women with polycystic ovary syndrome (PCOS) and matched controls: less visceral fat with PCOS. Hum Reprod Oxf Engl. 2011;26(6):1495–1500. doi: 10.1093/humrep/der070. [DOI] [PubMed] [Google Scholar]
- 48.Mannerås-Holm L, Leonhardt H, Kullberg J, Jennische E, Odén A, Holm G, Hellström M, Lönn L, Olivecrona G, Stener-Victorin E, Lönn M. Adipose Tissue Has Aberrant Morphology and Function in PCOS: Enlarged Adipocytes and Low Serum Adiponectin, But Not Circulating Sex Steroids, Are Strongly Associated with Insulin Resistance. J Clin Endocrinol Metab. 2011;96(2):E304–E311. doi: 10.1210/jc.2010-1290. [DOI] [PubMed] [Google Scholar]
- 49.van der Kooy K, Leenen R, Seidell JC, Deurenberg P, Droop A, Bakker CJ. Waist-hip ratio is a poor predictor of changes in visceral fat. Am J Clin Nutr. 1993;57(3):327–333. doi: 10.1093/ajcn/57.3.327. [DOI] [PubMed] [Google Scholar]
- 50.Ahmad N, Adam SIM, Nawi AM, Hassan MR, Ghazi HF. Abdominal Obesity Indicators: Waist Circumference or Waist-to-hip Ratio in Malaysian Adults Population. Int J Prev Med. 2016;7(1):82. doi: 10.4103/2008-7802.183654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Christakoudi S, Evangelou E, Riboli E, Tsilidis KK. GWAS of allometric body-shape indices in UK Biobank identifies loci suggesting associations with morphogenesis, organogenesis, adrenal cell renewal and cancer. Sci Rep. 2021;11(1):10688. doi: 10.1038/s41598-021-89176-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Censin JC, Bovijn J, Holmes MV, Lindgren CM. Colocalization analysis of polycystic ovary syndrome to identify potential disease-mediating genes and proteins. Eur J Hum Genet. 2021;29:1–9. doi: 10.1038/s41431-021-00835-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jerzy B, Anna J-W. Transactivation of ErbB Receptors by Leptin in the Cardiovascular System: Mechanisms, Consequences and Target for Therapy. Curr Pharm Des. 2014;20(4):616–624. doi: 10.2174/138161282004140213155050. [DOI] [PubMed] [Google Scholar]
- 54.Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, Perry JRB, Elliott KS, Lango H, Rayner NW, Shields B, Harries LW, Barrett JC, Ellard S, Groves CJ, Knight B, Patch AM, Ness AR, Ebrahim S, Lawlor DA, Ring SM, Ben-Shlomo Y, Jarvelin MR, Sovio U, Bennett AJ, Melzer D, Ferrucci L, Loos RJF, Barroso IÌ, Wareham NJ, Karpe F, Owen KR, Cardon LR, Walker M, Hitman GA, Palmer CNA, Doney ASF, Morris AD, Smith GD, Hattersley AT, McCarthy MI. A Common Variant in the FTO Gene Is Associated with Body Mass Index and Predisposes to Childhood and Adult Obesity. Science. 2007;316(5826):889–894. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fischer J, Koch L, Emmerling C, Vierkotten J, Peters T, Brüning JC, Rüther U. Inactivation of the Fto gene protects from obesity. Nature. 2009;458(7240):894–898. doi: 10.1038/nature07848. [DOI] [PubMed] [Google Scholar]
- 56.Scuteri A, Sanna S, Chen W-M, Uda M, Albai G, Strait J, Najjar S, Nagaraja R, Orrú M, Usala G, Dei M, Lai S, Maschio A, Busonero F, Mulas A, Ehret GB, Fink AA, Weder AB, Cooper RS, Galan P, Chakravarti A, Schlessinger D, Cao A, Lakatta E, Abecasis GR. Genome-Wide Association Scan Shows Genetic Variants in the FTO Gene Are Associated with Obesity-Related Traits. PLOS Genet. 2007;3(7):e115. doi: 10.1371/journal.pgen.0030115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ramos RB, Spritzer PM. FTO gene variants are not associated with polycystic ovary syndrome in women from Southern Brazil. Gene. 2015;560(1):25–29. doi: 10.1016/j.gene.2015.01.012. [DOI] [PubMed] [Google Scholar]
- 58.Kim JJ, Choi YM, Cho YM, Hong MA, Chae SJ, Hwang KR, et al. Polycystic ovary syndrome is not associated with polymorphisms of the TCF7L2, CDKAL1, HHEX, KCNJ11, FTO and SLC30A8 genes. Clin Endocrinol (Oxf) 2012;77:439–445. doi: 10.1111/j.1365-2265.2012.04389.x. [DOI] [PubMed] [Google Scholar]
- 59.Barber TM, Bennett AJ, Groves CJ, Sovio U, Ruokonen A, Martikainen H, Pouta A, Hartikainen AL, Elliott P, Lindgren CM, Freathy RM, Koch K, Ouwehand WH, Karpe F, Conway GS, Wass JAH, Järvelin MR, Franks S, McCarthy MI. Association of variants in the fat mass and obesity associated (FTO) gene with polycystic ovary syndrome. Diabetologia. 2008;51(7):1153–1158. doi: 10.1007/s00125-008-1028-6. [DOI] [PubMed] [Google Scholar]
- 60.Kowalska I, Adamska A, Malecki MT, Karczewska-Kupczewska M, Nikolajuk A, Szopa M, et al. Impact of the FTO gene variation on fat oxidation and its potential influence on body weight in women with polycystic ovary syndrome. Clin Endocrinol (Oxf). 2012;77(1):120–125. doi: 10.1111/j.1365-2265.2012.04379.x. [DOI] [PubMed] [Google Scholar]
- 61.Elsir T, Smits A, Lindström MS, Nistér M. Transcription factor PROX1: its role in development and cancer. Cancer Metastasis Rev. 2012;31(3-4):793–805. doi: 10.1007/s10555-012-9390-8. [DOI] [PubMed] [Google Scholar]
- 62.Harvey NL, Srinivasan RS, Dillard ME, Johnson NC, Witte MH, Boyd K, Sleeman MW, Oliver G. Lymphatic vascular defects promoted by Prox1 haploinsufficiency cause adult-onset obesity. Nat Genet. 2005;37(10):1072–1081. doi: 10.1038/ng1642. [DOI] [PubMed] [Google Scholar]
- 63.Kretowski A, Adamska E, Maliszewska K, Wawrusiewicz-Kurylonek N, Citko A, Goscik J, Bauer W, Wilk J, Golonko A, Waszczeniuk M, Lipinska D, Hryniewicka J, Niemira M, Paczkowska M, Ciborowski M, Gorska M. The rs340874 PROX1 type 2 diabetes mellitus risk variant is associated with visceral fat accumulation and alterations in postprandial glucose and lipid metabolism. Genes Nutr. 2015;10(2):4. doi: 10.1007/s12263-015-0454-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kokosar M, Benrick A, Perfilyev A, Fornes R, Nilsson E, Maliqueo M, Behre CJ, Sazonova A, Ohlsson C, Ling C, Stener-Victorin E. Epigenetic and Transcriptional Alterations in Human Adipose Tissue of Polycystic Ovary Syndrome. Sci Rep. 2016;6(1):22883. doi: 10.1038/srep22883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nishiyama Y, Hasegawa T, Fujita S, Iwata N, Nagao S, Hosoya T, Inagaki K, Wada J, Otsuka F. Incretins modulate progesterone biosynthesis by regulating bone morphogenetic protein activity in rat granulosa cells. J Steroid Biochem Mol Biol. 2018;178:82–88. doi: 10.1016/j.jsbmb.2017.11.004. [DOI] [PubMed] [Google Scholar]
- 66.Louwers YV, Rayner NW, Herrera BM, Stolk L, Groves CJ, Barber TM, Uitterlinden AG, Franks S, Laven JSE, McCarthy MI. BMI-Associated Alleles Do Not Constitute Risk Alleles for Polycystic Ovary Syndrome Independently of BMI: A Case-Control Study. PLoS ONE. 2014;9(1):e87335. doi: 10.1371/journal.pone.0087335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, Christiansen LM, Edelstein E, Choi B, Boss O, Aschkenasi C, Zhang CY, Mountjoy K, Kishi T, Elmquist JK, Lowell BB. Divergence of Melanocortin Pathways in the Control of Food Intake and Energy Expenditure. Cell. 2005;123(3):493–505. doi: 10.1016/j.cell.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 68.Azziz R, Carmina E, Chen Z, Dunaif A, Laven JSE, Legro RS, Lizneva D, Natterson-Horowtiz B, Teede HJ, Yildiz BO. Polycystic ovary syndrome. Nat Rev Dis Primer. 2016;2(1):1–18. doi: 10.1038/nrdp.2016.57. [DOI] [PubMed] [Google Scholar]
- 69.Dapas M, Lin FTJ, Nadkarni GN, Sisk R, Legro RS, Urbanek M, Hayes MG, Dunaif A. Distinct subtypes of polycystic ovary syndrome with novel genetic associations: An unsupervised, phenotypic clustering analysis. PLOS Med. 2020;17(6):e1003132. doi: 10.1371/journal.pmed.1003132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhao Y, Xu Y, Wang X, Xu L, Chen J, Gao C, Wu C, Pan D, Zhang Q, Zhou J, Chen R, Wang Z, Zhao H, You L, Cao Y, Li Z, Shi Y. Body Mass Index and Polycystic Ovary Syndrome: A 2-Sample Bidirectional Mendelian Randomization Study. J Clin Endocrinol Metab. 2020;105(6):1778–1784. doi: 10.1210/clinem/dgaa125. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Adult BMI IVs. Table S2. WHR IVs. Table S3. WHRadjBMI IVs. Table S4. childhood BMI IVs. Table S50. PCOS IVs. Table S6. Data sources, sample sizes, number of IVs and F-statistics. Table S7. Summary of contributing cohorts/consortia/studies in each GWAS. Table S8. Annotation of genome-wide significant SNPs from CPASSOC. Table S9. 99% credible set SNPs.
Additional file 2: Figure S1. Local genetic correlation between childhood BMI, WHR, WHRadjBMI and PCOS. Figure S2. GTEx tissue enrichment analysis. Figure S3. Cell-type specific enrichment analysis. Figure S4. Box plot of betas in leave-one-out analysis. Figure S5. MR-clust analysis.
Data Availability Statement
All data used in the present study were obtained from publicly available GWAS summary statistics.