

Abstract
In cells, organs and bodies, nutrient sensing is key to maintaining homeostasis and adapting to a fluctuating environment1. In the digestive system of many animals, enteroendocrine cells harbour nutrient sensors; less is known about nutrient sensing in their cellular siblings – the absorptive enterocytes1. A genetic screen in Drosophila melanogaster identified Hodor: an enterocyte ionotropic receptor that sustains larval development particularly in nutrient-scarce conditions. Experiments in Xenopus oocytes and flies indicate that Hodor is a pH-sensitive zinc-gated chloride channel that mediates a previously unrecognised dietary preference for zinc. Hodor controls systemic growth from a subset of enterocytes (interstitial cells) by promoting food intake and insulin/IGF signalling. Although Hodor sustains gut luminal acidity and restrains microbial loads, its effect on systemic growth results from modulation of Tor signalling and lysosomal homeostasis within interstitial cells. Hodor-like genes are insect-specific, and may represent specific targets for disease vector control. Indeed, CRISPR/Cas9 genome editing revealed that the single Anopheles gambiae hodor orthologue is an essential gene. Our findings underscore the need to consider instructive contributions of metals and, more generally, micronutrients to energy homeostasis.
To investigate enterocyte nutrient sensing, we selected 111 putative nutrient sensors in Drosophila melanogaster based on their intestinal expression and predicted structure/function (Extended Data Fig. 1a, Source Data 1, Supplementary Information). Using two enterocyte-specific driver lines, we downregulated their expression in midgut enterocytes throughout development under two dietary conditions (nutrient-rich and nutrient-poor); we reasoned that dysregulation of nutrient-sensing mechanisms may increase or reduce the normal period of larval growth, and might do so in a diet-dependent fashion (Extended Data Fig. 1b–d). Enterocyte-specific knockdown of CG11340, also referred to as pHCl-22, resulted in developmental delay. This delay was exacerbated, with significantly reduced viability, under nutrient-poor conditions (Fig. 1a, Extended Data Figs. 1h, 2b): phenotypes that were confirmed using a second RNAi transgene and a new CG11340 mutant (Fig. 1b, c, Extended Data Fig. 1e–i and Source Data 1). In the tradition of naming Drosophila genes according to their loss-of-function phenotype, we named CG11340 “hodor”: acronym for “hold on, don’t rush”, describing the developmental delay.
Fig. 1.
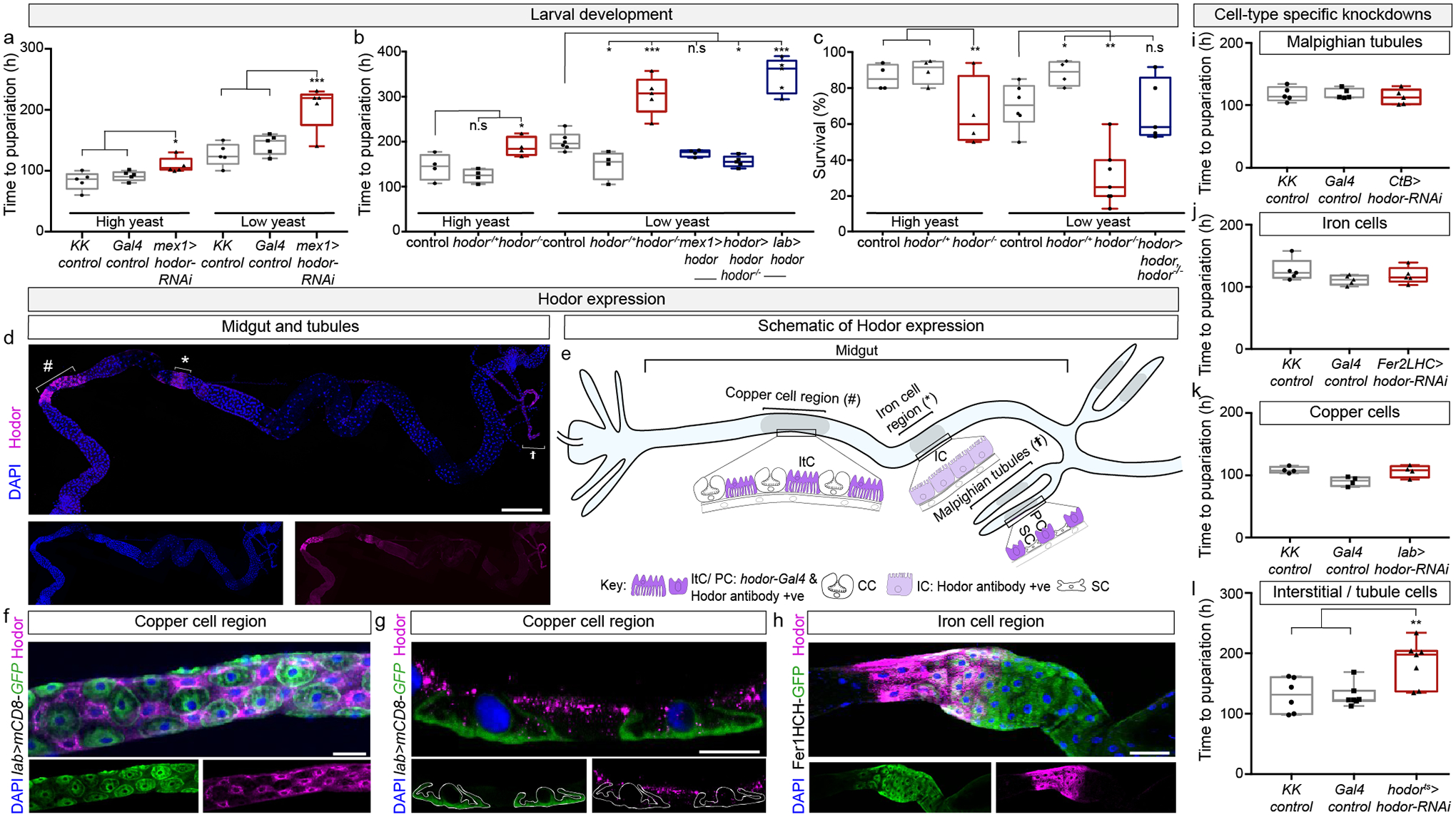
a, Intestinal Hodor sustains larval growth. Enterocyte-specific (mex1-Gal4 driven) hodor knockdown increases time to pupariation, particularly in nutrient-poor (low-yeast) conditions. b, Developmental delay of hodor mutants (increased time to pupariation) in both nutrient-rich (high yeast) and nutrient-poor (low-yeast) conditions, which can be fully rescued by overexpressing hodor in interstitial cells and Malpighian tubule principal cells (hodor-Gal4 driver), in migdut enterocytes (mex1-Gal4), but not in copper cells (labial (lab)-Gal4). c, The nutrient-dependent reduced viability of hodor mutants is rescued by hodor-Gal4-driven hodor re-expression. d, Hodor expression in copper (#) and iron cell (*) regions and Malpighian tubules (†) of a third-instar larval midgut. Expression in the large flat cell region flanked by the copper and iron cell regions was inconsistent. e, Hodor-expressing cell types: ItC – interstitial cells, IC – iron cells, CC – copper cells, PC – principal cells, SC – stellate cells. f, Hodor-positive interstitial cells are interspersed amongst copper cells (lab>mCD8-GFP-positive, Hodor-negative). g, Hodor is found on the apical (luminal, up) side of interstitial cells, flanked by lab>mCD8-GFP-expressing copper cells (outlined). h, Hodor in the anterior portion of the iron cell region (Fer1HCH-GFP-positive). i-k, Knockdown of hodor in principal cells (CtB-Gal4) (i), iron cells (Fer2LCH-Gal4) (j), or copper cells (lab-Gal4) (k) all fail to alter larval development. l, Post-embryonic hodor knockdown in interstitial and Malpighian tubule principal cells (by means of hodor-Gal4, tub-Ga80ts (hodorts in figure)-driven hodor RNAi) increases time to pupariation. See Supplementary information for sample sizes and full genotypes. One-way ANOVA with Tukey post-hoc tests were used for all graphs. Significance values: p< 0.05 *, p< 0.01 **, p< 0.001 ***. Box plots: line, median; box, 75th–25th percentiles; whiskers, minimum to maximum. Scale bars: d, 1mm; f, 40μm; g, 20μm; h, 100μm.
A transcriptional reporter revealed Hodor expression in the intestine3. A new antibody (Extended Data Fig. 2a, b) revealed that Hodor protein expression was confined to enterocytes in two midgut portions known to store metals: the copper and iron cell regions (Fig. 1d–h). Within the copper cell region, Hodor was only expressed in so-called interstitial cells (Fig. 1e, f, g). hodor-Gal4 was detected in the same cell types, apart from iron cells (Fig. 1e and Extended Data Fig. 2d, in contrast to published results3). Aside from the intestine, Hodor was only found in principal cells of the excretory Malpighian tubules2,3 (Fig. 1d, e). To identify the cells from which Hodor controls systemic growth, we conducted region- or cell-type specific downregulation/rescue experiments (Extended Data Fig. 1b, 2d–g). Only lines that downregulated hodor in interstitial cells slowed larval development (Fig. 1a, i–k, Extended Data Fig. 1j, 2c–h). This developmental delay persisted when hodor knockdown was induced post-embryonically during larval growth (Fig. 1l), and was rescued only by lines that re-instated hodor expression in cell types that included interstitial cells (Fig. 1b, c). The fat body (analogous to liver/adipose tissue) has long been known to couple nutrient availability with developmental rate4,5, but recent studies have revealed intestinal contributions, particularly in nutrient-poor conditions6,7. Our findings confirm a role for the intestine in coupling nutrient availability with larval growth, and further implicate a subpopulation of enterocytes – interstitial cells – as important mediators. Interstitial cells were described decades ago in blowfly8, but had remained relatively uncharacterised; their name only refers to their position9 – interspersed amongst the acid-secreting copper cells that control microbiota loads10–13.
How does Hodor control systemic growth from this intestinal cell subset? We established that hodor mutant/knockdown lethality was only apparent in the larval period (Extended Data Fig. 3a). hodor mutant development was slower throughout larval life; surviving mutants attained normal pupal and adult sizes (Extended Data Fig. 3b–d). Consistent with12, hodor mutation/knockdown reduced luminal acidity in the copper cell region (Extended Data Fig. 4a, b), suggesting a new role specifically for interstitial cells in this process. hodor mutants also had increased gut bacterial titres, consistent with the observed defects in copper cell region function13 (Extended Data Fig. 5a). Enlarged volumes of both the lumen of the copper cell region and the interstitial cells were also apparent after 1–3 days of (delayed) larval development (Extended Data Fig. 4e); ultrastructurally, this was apparent in interstitial cells as a reduction in the complexity of their characteristic basal infoldings14 (Extended Data Fig. 4d). We were, however, able to rule out all these defects as reasons for the developmental delay (Supplementary Information, Extended Data 4a–c, 4f–I, 5b–c). What then links Hodor function in interstitial cells with larval development?
We observed that hodor mutant larvae were more translucent than controls (Fig. 2a). This was suggestive of peripheral lipid depletion, which we confirmed by quantifying and staining for triacylglycerides (Fig. 2b, d, e). Reduced lipid stores did not result from disrupted enterocyte integrity: the intestinal barrier of mutants was intact, both anatomically and functionally (Extended Data Fig. 3g, h). We observed that hodor mutants had less food in their intestines (Fig. 2f) and accumulated insulin-like peptide Ilp2 in their brain (nutrient-dependent Ilp2 secretion promotes larval development; its accumulation in the brain is commonly interpreted as peptide retention in the absence of transcriptional changes5,15) (Fig. 2k, l). Consistent with reduced systemic insulin signalling, hodor mutant larval extracts had reduced phospho-Akt and phospho-S6 kinase (Fig. 2o and Extended Data Fig. 3e). As these are all indicators of starvation, we quantified food intake and observed reduced food intake in both hodor mutant larvae and in hodor knockdowns targeting interstitial cells (Fig. 2f, g, i, Extended Data Fig. 2c, 3f). Reduced food intake was apparent soon after hatching and persisted throughout larval development (Fig. 2f, g and Extended Data Fig. 3f). Ectopic expression of Ilp2 (which rescues developmental delay in larvae lacking insulin-like peptides15) in hodor mutants partially rescued their developmental delay, but not food intake (Fig. 2m, n). An “instructive” link between intestinal Hodor and food intake was further suggested by over-expression of hodor in otherwise wild-type enterocytes, which resulted in larvae that ate more, developed at a normal rate, but had increased lipid stores (Fig. 2c, h, j and Extended Data Fig. 3i). Thus, Hodor controls larval growth from a subset of enterocytes by promoting food intake and systemic insulin signalling. In its absence, larvae fail to eat sufficiently to proceed through development at the normal rate and are leaner. In excess, Hodor causes larvae to eat more and accumulate the energy surplus as fat.
Fig. 2. Intestinal Hodor/Tor signalling promotes food intake.

a, hodor mutants are more translucent than controls. b-c, Total triacylglycerides (TAG) normalised to weight in hodor (b) mutants and larvae overexpressing hodor (c). d, Lipid droplets within the fat body of hodor mutants and controls. e, Fat body lipid droplet (LD) size is reduced in hodor mutants. (L2 larvae were used in a-e). f, Reduced intestinal contents in L1 hodor mutants fed dye-laced food (45min). g-j, Food intake (g, h) or mouth hook contraction (i, j) quantifications for L1 hodor mutants (g, i) or L2 larvae overexpressing hodor in hodor-expressing cells (h,j). k-l, Ilp2 staining (quantification, k and representative images, l) of L2 brains of controls vs hodor mutants. m-n, Ectopic Ilp2 expression (hs-Ilp2) rescues the developmental delay of hodor mutants (m), but not their food intake (n). o, Reduced pAkt and pS6K in L2 hodor mutants compared to controls. p, The developmental delay of hodor knockdowns is exacerbated or rescued when the Tor pathway is simultaneously depleted (Tor-RNAi) or activated (S6KSTDETE), respectively, specifically in hodor-expressing cells. These manipulations did not affect the development of wild-type larvae (Extended Data Fig. 3j). q, The hodor mutant developmental delay is rescued by activation of the Tor pathway (S6KTE – weaker than S6KSTDETE – or UAS-Rheb) specifically in hodor-expressing cells. r, The reduced food intake of L2 hodor mutants is rescued by Tor pathway activation specifically in hodor-expressing cells (hodor>Rheb). See Supplementary information for sample sizes and full genotypes. Mann Whitney U tests or ordinary one-way ANOVA with Tukey post-hoc tests were used for two-group or more than two group comparisons, respectively. Significance values: p< 0.05 *, p< 0.01 **, p< 0.001 ***. Box plots: line, median; box, 75th–25th percentiles; whiskers, minimum to maximum. Scale bars: a, 500μm; b, 20μm; f, 100μm; k, 15μm.
In fly adipose tissue, amino acid availability activates Tor signalling to promote systemic growth4. Thus, we combined hodor knockout or knockdown with genetic manipulations to alter Tor signalling. Reduced or increased Tor signalling in hodor-expressing cells exacerbated or rescued the developmental delay of animals with reduced/absent Hodor function, respectively (Fig. 2p, q, Extended Data Fig. 3j). The reduced food intake of hodor mutants was also significantly rescued by activation of Tor signalling in hodor-expressing cells (Fig. 2r, Extended Data Fig. 3j, k). Genetic targeting of Rag GTPases or the Gator1 complex in these cells failed to affect the developmental delay of hodor mutants (Extended Data Fig. 3l), possibly suggesting non-canonical regulation of Tor signalling in Hodor-expressing cells. Thus, the systemic effects of Hodor on food intake and larval growth are modulated by Tor signalling within Hodor-expressing interstitial cells.
Hodor belongs to the (typically neuronal) Cys-loop subfamily of ligand-gated ion channels and is predicted to be a neurotransmitter-gated anion channel16 (Fig. 3a, Supplementary Information). It shows activity in response to alkaline pH in Xenopus oocytes2, but the acidic pH of the copper cell region prompted us to search for additional ligands. While we confirmed alkaline pH-induced Hodor activity in oocyte expression systems, Hodor did not respond to typical Cys-loop receptor ligands such as neurotransmitters or amino acids (Extended Data Table 1). Instead, our screen identified zinc as an unanticipated ligand, which elicited a strong, Hodor-dependent dose-dependent response (Fig. 3b, Extended Data Fig. 6e) with peak current amplitude values much greater than those observed in response to pH or other metals such as iron or copper (Extended Data Table 1). Force field-based structural stability and binding affinity calculations (Supplementary Information) identified the amino acid pair E255, E296 as a potential binding site for the divalent zinc ion. Mutating these residues did not abrogate zinc-elicited currents, but these had faster rise time and deactivation kinetics (Extended Data Fig. 6a–d), supporting the idea that zinc is a relevant Hodor ligand. Based on its sequence and conductance properties, Hodor has been proposed to transport chloride2,3, and the zinc-elicited currents we observed in oocytes had a reversal potential consistent with chloride selectivity. In flies in vivo, zinc supplementation of a low-yeast diet reduced chloride levels in interstitial cells, whereas hodor mutation increased them (Fig. 3c and Extended Data Fig. 6g, h). Thus, Hodor is a pH-modulated, zinc-gated chloride channel.
Fig. 3. Hodor is a zinc-gated chloride channel that controls dietary zinc preference and lysosomal functions.

a, Predicted pentameric complex; one monomer shown in blue. b, Left: only oocytes injected with Hodor respond to zinc. Middle graph: current-voltage (I-V) of zinc-activated currents. Right: zinc dose response (estimated EC50: 75.20μM, 95% confidence interval 58.63–94.65μM). c, Increased intracellular chloride (decreased 458nm/543nm ClopHensor ratio) in interstitial cells of L1 hodor mutants (20mM controls, 64mM in hodor mutants, calibration in Extended Data Fig. 6h). Representative 458nm images are shown. d, Zinc supplementation of a low-yeast diet increases food intake in controls, but not hodor mutants. e, Controls (but not hodor mutants) develop a preference (positive values) for a zinc-supplemented low-yeast diet, significant after 45h. ZnCl2 was used (ZnSO4 also elicited preference, not shown). f, Hodor is enriched on the apical (luminal) side of interstitial cells: on the brush border (arrow, phalloidin-positive) and intracellularly. g, h, A subpopulation of compartments positive for Lysotracker (g) and Lamp1-mCherry (h) co-express Hodor in interstitial cells (larvae were starved for 4h for improved lysosomal visualisation). i, A GFP-mCherry-Atg8a reporter reveals increased production of mCherry-positive autophagic punctae in interstitial cells; some are positive for GFP (normally quenched under acidic conditions). Single confocal slices for each channel are shown below. j, k, Knockdown of V-ATPase complex subunits from interstitial cells (hodor-Gal4) but not from other enterocytes (R2R4-Gal4) delays pupariation (j) and/or reduces food intake (k). See Supplementary information for sample sizes and full genotypes. Mann Whitney U tests or ordinary one-way ANOVA with Tukey post-hoc tests were used for two-group or more than two group comparisons, respectively. Significance values: p< 0.05 *, p< 0.01 **, p< 0.001 ***. Box plots: line, median; box, 75th–25th percentiles; whiskers, minimum to maximum. Some images were false-coloured for consistency. N: nucleus. Scale bars: e, 30μm; f, g and h, 10μm; i, j and k, 30μm: l, 50μm.
What is the significance of zinc binding to Hodor? We observed zinc enrichment in both the copper and iron cell regions of the larval gut (Extended Data Fig. 7a, b), revealing an unrecognised role for these Hodor-expressing regions in zinc handling. hodor mutation failed to affect this zinc accumulation, while dietary yeast levels did (Extended Data Fig. 5d, e and 7b, c), consistent with a role for Hodor in sensing rather than transporting zinc. (Notably, the white mutation – commonly used in the genetic background of Drosophila experiments – results in a small but significant reduction in both intestinal zinc accumulation and larval growth rate, although the status of the w gene neither exacerbated nor masked the more substantial, hodor-induced developmental delay (Extended Data Fig. 7b–e, Supplementary Information)). Furthermore, larvae fed a low-yeast diet ate significantly more when supplemented with zinc, which was abrogated in hodor mutants (Fig. 3d). And in a food choice experiment, control larvae developed a preference for zinc-supplemented food over time (Fig. 3e), suggesting that it develops post-ingestively. Consistent with this idea, zinc preference was specifically abrogated in hodor mutants (Fig. 3e; we confirmed their general ability to discriminate between other diets, Extended Data Fig. 6f). Thus, zinc sensing by Hodor is physiologically significant in vivo. Metals like zinc are primarily provided by yeasts in nature; Hodor may be one of several sensors used to direct larvae to nutrient-rich food sources.
What are the cellular roles of a zinc-gated chloride channel? The subcellular localisation of Hodor suggests that it may normally maintain low cytoplasmic chloride concentrations by transporting it out of the interstitial cells and/or into their lysosomes. Indeed, and consistent with its putative lysosomal localisation signals17, Hodor was specifically enriched in apical compartments positive for late endosome/lysosomal markers, as well as decorating the brush border of interstitial cells (Fig. 3f–h, Extended Data Fig. 8a–e). The presence of Hodor in a subpopulation of lysosomes caught our attention because chloride transport across lysosomal membranes often sustains the activity of the proton-pumping vacuolar-type ATPase (V-ATPase) that maintains lysosomal acidity and Tor activation on the lysosome18–20. To explore a role in enabling Tor signalling, we tested whether hodor absence induced autophagy: a hallmark of reduced Tor signalling21. We first confirmed induction of common autophagy markers in interstitial cells following knockdown of the V-ATPase complex, known to promote autophagy by reducing lysosomal acidity and Tor signalling20,22 (Extended Data Fig. 9a, b). Like V-ATPase knockdown, loss of hodor increased autophagy in interstitial cells (Extended Data Fig. 9a). Expression of the dual autophagosome/autolysosome reporter UAS-GFP-mCherry-Atg8a in intestinal cells of hodor mutants confirmed autophagy induction (Fig. 3i), and revealed two additional features. Firstly, the acidification of autophagic compartments was defective in hodor mutants (Fig. 3i, Extended Data Fig. 9c–e). Secondly, the increased autophagy and defective acidification of hodor mutants were particularly prominent in the two Hodor-expressing intestinal regions (copper and iron cell regions), consistent with cell-intrinsic roles for Hodor in these processes (Extended Data Fig. 9c, e). Also supporting roles for lysosomal function and Tor signalling in controlling whole-body growth from interstitial cells, most V-ATPase subunits were transcriptionally enriched in the copper cell region (“MidgutAtlas” RNA sequencing data12, confirmed with an endogenous protein reporter for the V-ATPase subunit Vha16–1, Extended Data Fig. 8f, g). Functionally, downregulation of V-ATPase subunits specifically in Hodor-expressing cells (but not in other subsets of enterocytes, such as those targeted by R2R4-Gal423, Extended Data Fig. 2h) led to developmental delay and reduced food intake comparable to those resulting from hodor downregulation (Fig. 3j, k). Hence, although the directionality of zinc sensing and chloride transport in interstitial cells remains to be established, our data are consistent with roles for brush border Hodor in transporting chloride out of interstitial cells, maintaining osmolarity and water balance, and for lysosomal Hodor in transporting chloride into the lysosome to sustain V-ATPase function, lysosomal acidification and TOR signalling, pointing to novel links between lysosomal homeostasis in specialised intestinal cells, food intake and systemic growth (Extended Data Fig. 11). Nutrients such as amino acids are important regulators of Tor signalling21,24,25. Our genetic data is consistent with novel metal/micronutrient input into Tor signalling. The nutrient-dependent zinc accumulation in lysosomal organelles recently described in mammalian cells and nematode worms26,27 suggest that links between zinc, lysosomes and Tor may be more broadly significant. Two attractive cell types in which to explore such links are the Paneth cells of the mammalian intestine, which accumulate zinc and regulate intestinal immunity and stem cell homeostasis28, and the “lysosome-rich enterocytes” recently described in fish and mice, with roles in protein absorption29.
An extensive reconstruction of the hodor family tree supported the presence of a single member of the family in the ancestor of insects (Extended Data Fig. 10, Supplementary Information). Since Hodor-like proteins are only present in insects, they may prove to be highly specific targets for chemical vector control, particularly given that mosquito genomes harbour a single gene rather than the three paralogues found in most flies. To test this idea, we used CRISPR/Cas9 genome editing to generate a mutant lacking the single hodor-like gene in the malaria vector Anopheles gambiae (AGAP009616), which is also expressed in the digestive tract (midgut and Malphighian tubules30) (Extended Data Fig. 10b, c and Supplementary Information). Three independent deletion alleles revealed that AGAP009616 function is essential for A. gambiae viability (Extended Data Fig. 10d). An intestinally expressed target like Hodor is particularly attractive for vector control as it may circumvent accessibility issues and could be directly targeted using ingestible drugs such as those applied to larval breeding sites.
Metals have received little attention in the contexts of development or whole-body physiology, and are commonly regarded as passive “building blocks”. By revealing roles for a metal sensor in food intake and growth control, our findings underscore the importance of investigating instructive contributions of metals and, more generally, micronutrients to energy homeostasis. These mechanisms may prove unexpectedly useful in insect vector control.
METHODS
Fly husbandry
Fly stocks were raised in incubators at 25°C, 65% humidity and on a 12h light/dark cycle, and were maintained on a standard cornmeal/agar diet (6.65% cornmeal, 7.1% dextrose, 5% yeast, 0.66% agar supplemented with 2.2% nipagin and 3.4% propionic acid) (“high yeast” food). For the “low yeast” food, all ingredients and quantity were the same as high yeast food, except for a lower yeast concentration (0.74%). All experiments were done at 25°C or 29°C. For experiments using Gal80ts, flies were initially raised at 18°C (permissive temperature), and were moved to 31°C (restrictive temperature) when Gal4 induction was required.
Fly stocks
The following fly stocks were used: hodor-Gal4 (3), lab-Gal4 (BDSC: 43652), CtB-Gal4 (31, gift from Barry Denholm), Fer2LCH-Gal4 (DGGR: 113517), mex1-Gal4 (32), Myo1A-Gal4 (DGGR: 112001), R2R4-Gal4 (23), tub-Gal80ts (BDSC: 7018), UAS-mCD8-GFP (BDSC: 5130), Fer1HCH-GFP (DGGR: 110620), UAS-Vha16–1-RNAi (GD17431, VDRC: v49291), UAS-Vha44-RNAi (GD10617, VDRC: v46563), UAS-Vha13-RNAi (GD10564, VDRC: v25985), UAS-nprl2-RNAi (KK101142, VDRC: v110579), UAS-iml-RNAi (KK101116, VDRC: v110386), UAS-hodor-RNAi (KK106835, VDRC: v108337), UAS-hodor-RNAi #2 (NIG: 11340R-3), UAS-hodor (this study, see below for details), hodor−/− (this study, see below for details), UAS-Stinger-GFP (BDSC: 65402), UAS-shiK44A (BDSC: 5811), KK control (VDRC: v60100), GD control (VDRC: v60000), UAS-Tor-RNAi (BDSC: 34639), UAS-Rheb (BDSC: 9688), UAS-S6KTE (BDSC: 6912), UAS-S6KSTDETE (BDSC: 6914), UAS-RagAT16N; UAS-RagAQ61L; UAS-RagCS54N; UAS-RagCQ99L (33,34, gift from Aurelio Teleman and Clive Wilson), UAS-p62-GFP (35), Lamp1-mCherry (36), hs-Ilp2 (15), Foxo-mCherry (37, gift from Elodie Prince), Vha16–1-GFP (DGGR: 110558), tub-Rab5-YFP, tub-Rab7-YFP and tub-Rab11-YFP (38, gifts from Clive Wilson), UAS-ClopHensor (39, gift from Aylin Rodan), UASp-GFP-mCherry-Atg8a (BDSC: 37749). Oregon R (OrR) and w1118 were used as control flies.
Developmental rate and viability assays
Enterocyte RNAi screen
UAS-RNAi lines for candidate genes were screened over three rounds to assess changes in developmental rate using two enterocyte Gal4 drivers (see Supplementary Information for a more detailed overview). Larvae were screened in batches of up to approximately 20 experimental crosses per diet per Gal4 driver, plus all appropriate controls. In the first two rounds, Gal4 and UAS parents were placed in experimental vials to seed them with test animals. Round two had a shorter laying period with more parent flies, compared to round one, and flies were mated prior to addition to the experimental vial. At the midpoint of the laying interval, animals were considered to be 0 days of age. In the third round, eggs were laid over 24h on egg collection plates, then 50 eggs were transferred to a vial using moist filter paper. At this collection, animals were considered to be 0 days of age. For all of these protocols, pupae were counted every 24h and the time to pupariation was calculated as an average (mean) for the vial.
Other experiments assessing developmental rate
Adult flies were allowed to lay eggs for 24h at 25°C on apple juice plates containing a small dollop of yeast paste. Embryos were collected and transferred to a new plate containing the appropriate diet, rinsing away yeast paste where necessary. After 4 hours of hatching, first-instar larvae were seeded into vials containing the appropriate diet (close to the food) at 15–20 (or, in some experiments, 25) per vial, or onto plates at 50 per plate. At the midpoint of the hatching interval, animals were considered to be 1 day of age. Pupae were counted once in the morning and once in the evening and time to pupariation was calculated as an average for the vial. For experiments requiring heat shock, control and experimental larvae were subjected to 37°C twice a day for 45min and then returned to either 25 or 29°C, as described in15). For experiments assessing larval transitions/survival, seeded larvae were checked every 24h. Developmental stage was assessed based on the size and maturity of their mouth hooks, and larvae were size-matched whenever appropriate.
Embryonic viability assays
Embryos on apple juice plates were collected after a 6hr egg laying window at 25°C. The number of hatched eggs and dead embryos were scored after 36hr. Embryonic viability was calculated as the percentage of first-instar larvae divided by the total number of embryos (hatched larvae plus dead embryos).
Quantifications of pupal size
Pupae from different experimental conditions were collected and placed onto a coverslip and imaged using a Leica 10450528 camera attached to a Leica M165FC stereomicroscope using a 0.5x c-mount. Dimensions (length and width) were measured with Fiji40. Pupal volume was calculated according to the following formula: V=4/3π(L/2)(w/2)241. Each data point represents one pupal case.
Immunohistochemistry
Larvae of the appropriate developmental stage were selected, dissected in PBS, transferred to a poly-lysine slide and fixed with 4% formaldehyde (16% formaldehyde (Thermo Fisher Scientific #28908) diluted in PBS) for 20–40min (depending on the specific antibody). Samples were washed with PBS, then PBT (PBS with 0.2% Triton X-100) and blocked with PBTN (PBT with 4% normal horse serum) for 1h. Primary antibody was diluted in PBTN and was incubated with samples overnight at 4°C, and washed with PBT the next day. For certain antibodies, guts were either cut or holes were made in the sample to improve antibody penetration into the tissue. Fluorescently-labeled secondary antibodies were then added for 1.5–5hr at room temperature or overnight at 4°C, and were washed away with PBT and PBS. For phalloidin staining, conjugated phalloidin in PBTN was added for 45min. Samples were then washed with PBS and mounted in Vectashield (with or without DAPI, Vector Labs #H-1200 or #H-1000 respectively). Staining of experimental and control samples was carried out on the same slide to allow direct comparisons.
To visualise lipid droplets, fat body tissue surrounding the male gonad of second-instar larvae was dissected in PBS, mounted on poly-lysine slides and fixed for 30min. Samples were then washed 3 times in PBT and Nile Red stain (Thermo Fisher Scientific #N1142) was applied 1:500 for 30min in the dark. Samples were then washed 3 times in PBS and mounted in Vectashield containing DAPI. Each data point corresponds to one dissected fat body from one larva (different data points correspond to different larvae).
For Lysotracker/Lysosensor stainings, guts were dissected in PBS and transferred to poly-lysine slides. Small punctures were made in the tissue using tungsten wire to allow entry of LysoTracker Red DND-99 (Thermo Fisher Scientific #L7528) or LysoSensor Green DND-189 (Thermo Fisher Scientific #L7535) which were applied at a 1:500 dilution in 4% paraformaldehyde for 15–30min (Fig. 3g) in dark or imaged immediately under live conditions (rest of panels). Samples were then washed with PBT and PBS, or blocked with PBTN, then immunostained. Samples were mounted in Vectashield.
For zinc staining, adult and larval guts were dissected in PBS, transferred to poly-lysine slides and fixed with 4% formaldehyde for 30min. They were then washed with ethanol, PBT and PBS. Guts were incubated with the zinc indicator FluoZin-3AM (1:3000 in PBS containing 0.02% Triton and 0.001% Tween) at 38°C for 45min in the dark. Guts were then washed with ethanol, PBT and PBS. Guts were mounted with Vectashield containing DAPI. To quantify zinc levels, integrated density was measured for the copper cell region using Fiji, ensuring that the area measured was the same between samples. Each data point corresponds to one gut.
For Ilp2 intensity measurements in the brain insulin-producing cells, staining were performed as usual. After imaging, the freehand selection tool was used to draw around the insulin-producing cells on both sides of the brain, and the mean grey scale was calculated after subtracting from background staining.
For autophagy/lysosomal acidity quantifications using the dual UAS-GFP-mCherry-Atg8a reporter, the total number of punctae in each channel (GFP and mCherry) was separately counted by importing raw data into Fiji and using the “find maxima” tool to highlight punctate structures. The same method was used to quantify Lysotracker, Lysosensor, p62 and Lamp-positive structures. For starvation experiments, larvae were placed in a moist clear dish for 7h or overnight.
The following antibodies were used: rabbit anti-Hodor (1:500, this study), mouse anti-α-Spectrin (1:10, DSHB #3A9), anti-Ilp2 (1:200, gift from Pierre Léopold), anti-mCherry (1:200, Thermo Fisher Scientific #PA534974), p70 S6K (Thr398) (1:1000, Cell signaling #9209S), pAKT (Ser505) (1:500, Cell Signaling #4054), Akt (1:500, Cell Signaling #9272S), tubulin (1:1000, DSHB #12G10). Conjugated fluorescent secondary antibodies (FITC-, Cy3- and Cy5) were obtained from Jackson Immunoresearch and used at 1:200. Phalloidin conjugated to AlexaFluor647 or AlexaFluor488 were obtained from Thermo Fisher Scientific (#A22287) and used at 1:100.
Hodor antibody generation
An antibody against Hodor was raised in rabbit by immunising with a short peptide sequence found in the extra-cytoplasmic region of the protein (PVVHNKDGEEVP; amino acids 91–102). Hodor antibody was purified from the serum. This entire procedure was outsourced to New England Peptide.
Assessments of midgut luminal acidity and diameter
Larvae were selected based on their developmental stage and placed on plates containing food supplemented with the pH-sensitive dye 0.04% bromophenol blue (which changes from yellow at pH 3.0 to blue at pH 4.6) for a minimum of 1h. Guts were dissected in unbuffered salt solution (80mM NaCl, 55mM KCl) and were immediately imaged using a Leica 10450528 attached to a Leica M165FC stereo microscope using a 0.5x c-mount. For gut diameter measurements, guts were acquired as stated above and the diameter of the copper cell region was calculated using Fiji40.
Food intake quantifications
Larvae from seeded plates were matched for developmental stage (using mouth hook anatomy) and size, and placed in plates containing 1% FCF-blue dye for 45min. For diets supplemented with zinc, ZnCl2 (Sigma #Z0152) was used; larvae were raised on either supplemented or un-supplemented food and developmental experiments were performed side by side. Larvae were gently washed in dH2O to remove excess dye remaining on their outer cuticle, and were either imaged using a Leica DFC420C camera to visualise blue food in the gut, or placed in 2ml Eppendorf tubes containing 45μl dH2O and a 5mm ball bearing. These larvae were then homogenised twice for 60s with a Qiagen TissueLyser II at 30Hz and then centrifuged for 60s at 13,000 RCF. The dye content of the supernatant was measured at 594nm either using a NanoDrop ND-1000 spectrophotometer or with a Fluostar Omega microplate reader.
For mouth hook contraction assays, larvae were placed on apple juice plates covered with a thin layer of yeast paste. Larvae were then given 5min to adjust to their new environment and mouth hook contractions were counted for 30s42,43. This value was multiplied by 2 to obtain counts per minute.
Food preference experiments
Developmentally matched first-instar larvae were raised on low or high yeast food and were starved for 3h. They were then placed onto a choice assay plate containing an agar separator with two sources of food on either side, as described in44, so that they had a choice between high- vs low-yeast, or zinc-supplemented vs non-zinc supplemented low-yeast food. ZnCl2 (Sigma #Z0152) was used to supplement low-yeast food to assess zinc preference. ZnSO4 supplementation also elicited preference (data not shown). The number of larvae on each side of the plate (and on the agar) was scored at the designated time points, and was used to calculate a preference index as follows based on45:
where Nx=number of larvae that preferred food x (x could be ZnCl2 or ZnSO4-supplemented low yeast diet, or a high yeast diet); Nl=number of larvae that preferred a low yeast diet and Na=number of larvae with no preference. Log2 was then applied to the PIx. If Log2(PIx) > 0, it means larvae prefer food x to low yeast; if Log2(PIx) < 0, it means larvae prefer low yeast to food x. Loess analysis was then performed to fit the data across the time points using ggplot2 geom_smooth() function with argument method=”loess” and se=”TRUE”.
Electron microscopy
First-instar larval guts were dissected and fixed in 2.5% glutaraldehyde in PB (0.1M phosphate buffer [pH 7.2]), followed by fixation in 1% osmium tetroxide + 1.5% potassium ferrocyanide for 60min at 4°C. After dehydration with ethanol, guts were infiltrated and embedded in Durcupan, and ultra-thin (70nm) sections were cut using a Leica Ultracut UCT. Ultra-thin sections were contrasted with 2% uranyl acetate for 10min and lead citrate for 5min. They were then analysed using a Morgagni 268 TEM (80kV) electron microscope (FEI Company) and imaged using a side-entry Morada CCD Camera (EMSIS).
Image acquisition and processing
All fluorescent images were acquired using a Leica SP5 II confocal microscope and Leica LAS AF software. The same confocal settings, including laser power, were applied to both experimental and control groups. Images were processed using Fiji40. All statistical analyses were performed on raw images. To visualise the whole gut, images were stitched together using the Pairwise Stitching plugin46. For cell volume measurements, images of midgut copper cell regions were imported into IMARIS 9.2.1 and cell volume was calculated by measuring green-labelled interstitial cells in 5μm intervals. For subcellular localization experiments (e.g. Hodor, Lysotracker Lamp1 or Rab5, 7, 11), the number of YFP and/or magenta-positive punctae were counted in a single plane of an interstitial cell (total 3 cells from per gut) and the percentage of YFP-positive punctae that were also positive for Hodor antibody signal was calculated. Some images were false-coloured for consistency with other images in the manuscript.
RT-qPCR
For each sample, RNA was extracted from 15 whole larvae (L2) using Trizol (Invitrogen), and cDNA was synthesized using iScript cDNA synthesis kit (Bio-Rad, #170–8890) from 500ng of total RNA. Quantitative PCR was performed by mixing cDNA sample (5ng) with iTaq Universal SYBR Green Supermix (Bio-Rad, #172–5124) and the optimised primer pairs (see below). Expression values were normalised to gapdh. For each gene at least three independent biological replicates were used, and two technical replicates were performed.
| Gene | Forward primer | Reverse primer |
|---|---|---|
| hodor | GAACACCACGGATGCTTTCAG | ATGGACTCTGCGTTTTTCAGC |
| gapdh | CATTGTGGGCTCCGGCAA | CGCCCACGATTTTCGCTATG |
Western analyses
For the pAkt Western blots, extracts of second-instar larvae were prepared by mechanical homogenisation and lysis in RIPA buffer (Thermo #89900) with complete protease inhibitor cocktail (Roche #11836170001) and phosphatase inhibitors (Sigma #4906837001). 60 larvae for each treatment group were pooled, and each experiment was repeated at least once. Lysates were cleared from debris and lipids by 10min centrifugation in a table top centrifuge at 4°C. Total protein concentrations were determined using the Pierce BCA Protein Assay kit (Thermo #23227) and concentrations of lysates were adjusted accordingly. For the pS6K Western blots, larvae were directly lysed in 1xLaemli containing protease and phosphatase inhibitors. Lysates were cleared from debris by 10min centrifugation at 4°C. Samples were boiled, resolved on SDS-PAGE, and transferred by standard protocols.
ClopHensor experiments
For ClopHensor experiments, first-instar larval intestines from the relevant genotypes were dissected in Drosophila saline, consisting of 117.5mM NaCl, 20mM KCl, 2mM CaCl2, 8.5mM MgCl2, 10.2mM NaHCO3, 4.3mM NaH2PO4, 15mM HEPES, and 20mM glucose, pH 7.0. They were mounted on poly-lysine slides and bathed in standard bathing medium consisting of a 1:1 mix of Drosophila saline and Schneider medium (ThermoFisher Scientific #21720024). Intestines were then imaged live using a Leica SP5 Inverted microscope with excitation set at 488nm (green emission), 458nm (cyan emission), and 543nm (red emission). Pixel intensity for the upper and lower limits of the whole copper cell region (avoiding the section where the gut lumen is visible) was measured, and the ratio between 488nm/458nm values was used to calculate pH, whilst the 458nm/543nm ratio was used for intracellular chloride measurements. For chloride calibration: larval intestines were dissected in Drosophila saline and then bathed in chloride calibration solution, consisting of NaCl2 (varying amounts), Na-gluconate (varying amounts), 50mM K-gluconate, 2mM Ca-gluconate, 8.5mM Mg-gluconate, 20mM glucose, 15mM HEPES pH 7.2, 10μM tributyltinchloride (Sigma), 5μM nigericin (Invitrogen), 5μM carbonyl cyanide 3-chlorophenylhydrazone (Sigma) and 5μM valinomycin (Sigma). Intestines were allowed to equilibrate for 1h in their respective solutions before imaging using a Leica SP5 Inverted microscope. The ratio of 458nm/543nm for each chloride concentration were interpolated as a sigmoidal curve using a logistic dose-response sigmoidal fit function in Prism.
Microbiome experiments
Bacterial strains and growth conditions
We used Acetobacter pomorumWJL 47 and Lactobacillus plantarumNC8 48. A. pomorum was grown in Mannitol Broth (Bacto peptone 3g/L, yeast extract 5g/L, D-mannitol 25g/L) for 24h at 30°C under 180rpm agitation. L. plantarum was grown in MRS Broth (Carl Roth) at 37°C overnight without agitation.
Germ-free flies
Flies were rendered germ-free (GF) following the protocol described in49. GF flies were maintained on fly medium supplemented with antibiotics: kanamycin 50μg/mL (Sigma #K1377), ampicillin50μg/mL (Sigma #A0166), tetracyclin 10μg/mL (Sigma #T7660), erythromycin 5μg/mL (Sigma #E5389). Axenicity was confirmed by crushing the flies and plating the lysate on LB Agar (Carl Roth) and MRS Agar (Carl Roth) plates.
Developmental timing
Larvae mono-associated with A. pomorum were reared on a medium composed of agar (7.14g/L), cornmeal (80g/L), yeast (50g/L or 7g/L for rich (high-yeast) and poor (low-yeast) medium, respectively), sucrose (45g/L), nipagin (0.7g/L, Sigma #85265) and propionic acid (0.1%, Sigma #P5561). GF larvae and larvae mono-associated with L. plantarum were reared on a medium composed of agar (7.1g/L), cornmeal (80g/L), yeast (50g/L or 7g/L for rich and poor medium, respectively), nipagin (5.2g/L) and propionic acid (0.4%). GF flies were allowed to lay eggs in sterile breeding cages overnight. GF embryos were collected and transferred in groups of 40 into fresh sterile tubes. Bacterial cultures were washed in PBS and inoculated on the eggs at the final concentration of ~107 CFUs per tube for A. pomorum and ~108 CFUs per tube for L. plantarum. Tubes were kept at 25°C and the number of newly emerged pupae was scored every day until the emergence of all pupae.
Bacterial loads
Larvae bi-associated with A. pomorum and L. plantarum were reared on a medium composed of agar (7.14 g/L), cornmeal (80g/L), yeast (50g/L), sucrose (45g/L), nipagin (0.7g/L, Sigma #85265) and propionic acid (0.1%, Sigma #P5561). GF flies were allowed to lay eggs in sterile breeding cages overnight. GF embryos were collected and transferred in groups of 40 into fresh sterile tubes. Bacterial cultures were washed in PBS and mixed together before inoculation on the eggs, yielding an initial concentration of 5×107 CFUs per tube for L. plantarum and ~5×106 CFUs per tube for A. pomorum. Size-matched third-instar larvae were collected, surface-sterilised in 70% ethanol and placed in microtubes containing 400μL PBS and 0.75–1mm glass microbeads (Carl Roth, A554.1). Larvae were then homogenised using a Precellys 24 Tissue Homogenizer (Bertin Technologies, Montigny-le-Bretonneux, France). Lysate dilutions were plated using an EasySpiral automatic plater (Intersciences, Saint Nom, France), on MRS Agar with selective antibiotics to select L. plantarum and A. pomorum; Kanamycin (50μg/mL) allowed selective growth of L. plantarum and Ampicilin (10g/L) allowed selective growth of A. pomorum. Plates were incubated at 30°C for 48h for A. pomorum and 37°C for 24h for L. plantarum, and colonies were counted using an automatic colony counter Scan1200 (Intersciences, Saint Nom, France).
Wing size measurements
The wings of 3–5 day old adult flies were dissected in isopropanol and mounted on a slide. The excess isopropanol was wiped off and several drops of Euparal (ALS - Anglian Lepidopterist Supplies #DS31) was added to the slide; a cover was slip placed on top. The slides were incubated at 60°C overnight and imaged using a Leica 10450528 attached to a Leica M165FC stereo microscope. To quantify wing size, a straight line was drawn from the distal tip of the L3 vein to the proximal tip of the L4 vein using Fiji.
Experimental design and statistical analyses
Sample sizes were not limiting and were chosen empirically based on the variability of each scored phenotype. Comparable sample sizes for each genotype/condition were used in every experiment. For sample size information (repeats, # of animals…) see Supplementary Information. All experiments were repeated at least three times yielding comparable outcomes. Further replicates were included if necessary, for example to account for variability resulting from incubator temperature fluctuations or food batch variation. Experimental and control flies were bred in identical conditions, and were randomised whenever possible (for example, with regard to housing, position in tray). Control and experimental samples were dissected and processed at the same time and on the same slides. The experimenter was typically not blind to the genotypes/conditions.
Data analysis was carried out in Prism 7. For comparisons involving two groups, a non-parametric Mann Whitney U test was used. Where more than two groups were compared, an ordinary one-way ANOVA test was performed with a Tukey post-hoc test. For Fig. 3d, each data point represents one set of day-matched experiments containing a minimum of 5 different biological replicates. A two-way ANOVA was used to test significance for this set of data. Significance values are denoted as follows: p< 0.05 *, p< 0.01 **, p< 0.001 ***. Data are displayed as boxplots with line, median; box, 75th–25th percentiles; and whiskers, minimum to maximum.
Smurf assays
These were conducted by adapting the adult protocol described in50. Briefly, experimental larvae were removed from seeded plates and washed in dH2O. They were then placed onto low-yeast food containing 1% FCF blue dye and allowed to feed overnight. The next day, larvae were washed and imaged using a Leica DFC420C camera attached to a Leica M165FC stereo microscope.
TAG quantifications
Triacylglycerides were quantified in whole second-instar larvae as previously described51. Briefly, five second-instar larvae were pooled per sample, weighed and then homogenised in PBS + 0.05% Tween. Samples were heated for 5min at 70°C to inactivate lipases, and were then centrifuged to remove debris. 20μL of supernatant for each sample was added to 200μL of Thermo Infinity Triglyceride Reagent (Thermo Scientific #TR22421) in duplicates, which was then incubated for 10min at 37°C. Absorbance was measured at 540nm using a plate reader, and TAG levels for each duplicate were averaged and corrected for weight.
hodor mutant generation
A hodor mutant line was generated as described in52. Homology arms 5’ and 3’ to the hodor gene were amplified from w1118 DNA using primers HA5 F/ R and HA3 F/ R (see below). The PCR products were cloned into pTV Cherry using NotI and Acc651 (NEB #R0189 and #R0599, respectively) or AscI and SpeI (NEB #R0558 and #R3133, respectively) restriction enzymes. The completed pTV Cherry vector with both homology arms was amplified, purified and injected into yw embryos with “Delta 2–3” Helper DNA (injections performed by Drosophila Transgenesis Service, Universidad Autonoma de Madrid) to randomly integrate into the genome by P-element insertion. Transformants were crossed to hs-FLP, hs-I-SceI flies and larval progeny were heat-shocked to induce homologous recombination. Mottled eyed adults were collected and crossed to ubiquitin-Gal4[3xP3-GFP]. Progeny from this cross were screened for the presence of red-eyed individuals, indicative of a successful recombination event. The ubiquitin-Gal4[3xP3-GFP] was later removed by selecting against the presence of GFP in the ocelli.
The following primers were used:
| Name | Forward primer | Reverse primer |
|---|---|---|
| HA3 F/ R | ACTAGTGTTCGTCAGGGAAAGAGAGCCATTC | GGCGCGCCTCCCATCATTGTTAACTCAAC |
| HA5 F/ R | GCGGCCGCAGACGCTTGCCAACGATTAAGTACC | GGTACCGAATCACGGGACTCAGTGGGTAAGTTTTCAGGAG |
Generation of UAS-hodor
To overexpress hodor, hodor complementary DNA (cDNA) was amplified from adult Oregon-R gut RNA using the primers Hodor F and Hodor R (see below). The PCR product was digested with NotI and EcoRI (Promega #R6435 and #R6017 respectively) and cloned into the pUASTattB vector53. hodor-containing pUASTattB was amplified, purified and then injected into ZH-attP-22A embryos53, which have an attP site on chromosome 2L (injections were carried out by Drosophila Transgenesis Service, Universidad Autonoma de Madrid). Injected flies were crossed to w1118 and progeny were screened for orange eyes, indicative of successful transgenesis.
| Name | Forward primer | Reverse primer |
|---|---|---|
| Hodor F/ R | CAACGACGTGCAAGACATGACTAAC | GCTCTAGGATCACAGAATGGCTCTC |
Modelling of Hodor structure and zinc binding
The 3D structure of Hodor was predicted using homology modelling by templating the sequence on to the 5vdi.pdb pentamer (https://www.rcsb.org/structure/5vdi). Potential zinc (Zn2+)-binding sites were predicted with the MIB: Metal Ion-Binding Site Prediction and Docking software using the fragment transformation method54. The residue pairs with the highest predicted binding score were E255,E296, C207,C221, and H94,D97. The top three binding sites were used to seed mutational binding affinity calculations. Models of all possible single (120) and double (2340) mutants of the three binding sites were prepared. Structure refinement was performed with 2000 steps of conjugate gradient and steepest descent energy minimization with a 2kcal/mol restraint on peptide backbone atoms, using the Amber ff14SB force-field55. The structural stability and zinc binding affinity were calculated using the molecular-mechanics Poisson-Boltzmann Surface Area (MMPBSA) method56.
Electrophysiology of Xenopus oocytes
cRNA synthesis
hodor cDNA was PCR-amplified from Canton S flies using the primers below, which introduced XbaI and NotI sites. The PCR product was digested with XbaI and NotI and ligated into pGH19 vector (a derivative of pGEMHE57. This vector was linearised using NotI-HF (NEB #R3189S) for 2h at 37°C. The linearised DNA was purified using a PCR purification Kit (Qiagen #28104) and eluted in 30μl RNAse-free water. RNA synthesis was performed with approximately 1μg DNA using mMessage mMachine T7 Transcription Kit including 15min of DNAse treatment (Ambion #AM1344). RNA was treated with a Zymo Clean & Concentrator Kit (Zymo # R1013) and aliquoted at a concentration of approximately 1μg/ul for injection.
The following primers were used:
| Name | Forward primer | Reverse primer |
|---|---|---|
| cRNA | GATCTCTAGACAAGACATGACTAACCACC | CTAGGCGGCCGCCTCAAAGGCAGTAGACCAGG |
Oocyte Preparation
Xenopus laevis ovaries (Nasco) were dissected and dissociated by incubating in Ca2+-free ND96 saline (96mM NaCl, 2mM KCl, 5mM HEPES, 3mM MgCl2, adjusted to pH 7.4 with NaOH) containing 50–60mg Type2 collagenase (lot dependent) (Worthington LS004176), 25mg BSA (Sigma, #A3311) and 12.5mg Trypsin inhibitor (from chicken egg white, Sigma #T9253) for 90–120min. Dissociated oocytes were then washed in Ca2+-free ND96 and manually selected into Barth’s medium (88mM NaCl, 1mM KCl, 0.33mM Ca(NO3)2, 0.41mM CaCl2, 0.82mM MgSO4, 2.4mM NaHCO3, 5mM Hepes, and 0.1mg/mL gentamycin, pH 7.6 with NaOH) for injection the following day. Oocytes were injected with 50ng RNA 24–36h prior to recording using Nanoject III (Drummond scientific) and kept in Barth’s medium at 17°C until recording.
Recordings
Two-electrode voltage recordings were carried out at room temperature with an Oocyte Clamp OC-725C amplifier (Warner Instruments) and digitised using a Digidata 1550B (Axon Instruments) interface and pClamp 11 software. Data were filter at 1kHz and sampled at 10 kHz. Recordings were performed using borosilicate glass pipettes with resistances of ~1 MΩ when filled with 3M KCl. ZnCl2 (Sigma #Z0152) was diluted into a standard ND96 extracellular solution (96mM NaCl, 2mM KCl, 5mM HEPES, 1mM MgCl2, 2 mM CaCl2 adjusted to pH 7.4 with NaOH). Current-voltage relationships were obtained using 200ms voltage ramps from −120mV to 120mV applied every 500ms with an inter-stimulus holding potential of −80mV. Dose-response relationships were calculated using peak currents measured at 100mV and normalised to maximal currents elicited in response to 1mM ZnCl2. Activation and deactivation kinetics were determined by fitting the rising and decaying phases of zinc-activated currents with single exponentials. Data were analysed in Clampfit 11 (Molecular Devices) and visualised with R (R version 3.5.1).
Phylogenetic analyses
hodor orthologue identification, alignment, and phylogenetic reconstruction
To retrace the evolutionary history of hodor, we first queried OrthoDB v9 to identify gene family members of hodor and its two paralogues in D. melanogaster (FBgn0029733, FBgn0036727). As our intention was to characterize the emergence of hodor rather than build a comprehensive tree that included evolutionarily distant orthologues, analysis was restricted to the Arthropoda (EOG090X08ZM), which in OrthoDB v9 principally covers insect species. This enabled careful manual curation, as detailed below and in the table below. We first retrieved coding sequences (CDS) corresponding to the proteins in EOG090X08ZM from the relevant source databases. Orthologue identification relies on single protein sequence per gene. As most metazoan genes have multiple splice isoforms, that single, often arbitrary sequence need not be the most suitable for comparison against a given focal sequence of interest (here hodor). To reduce alignment errors and provide maximum coverage of regions orthologous to the focal hodor protein sequence, we therefore systematically surveyed protein isoforms and swapped the CDS in EOG090X08ZM for a more suitable isoform if available as follows:
| Species | Gene | Action taken |
|---|---|---|
| Ceratitis capitata | CCAP005795 | Swapped in XM_004527025.1 |
| Drosophila grimshawi | Dgri\GH17038 | Swapped in Dgri\GH17038-PB |
| Drosophila grimshawi | Dgri\GH15188 | Swapped in Dgri\GH15188-PB |
| Bactrocera dorsalis | 1780586 | Swapped in 84262 (as provided by i5k) |
| Bactrocera dorsalis | 11780102 | Swapped in 244888 (as provided by i5k) |
| Drosophila yakuba | Dyak\GE19913 | The sequence in the source database (Flybase) is 1nt too long, an inserted C at position 109. This C was removed to make CDS length consistent with protein length. The resulting sequence has a predicted internal stop codon, which may be a sequencing error. |
| Megaselia scalaris | multiple | All proteins from this species were removed as they are partially unresolved. |
| Drosophila suzukii | multiple | All proteins from this species were removed. Annotated protein lengths are quite different from all other Drosophila spp., suggesting potential gene prediction issues. |
After manual curation, the surviving set of 109 proteins were aligned using mafft-linsi with default settings and alignments back-converted into CDS. The nucleotide-level alignment was then used to build a phylogenetic tree using RaxML v8.1.16 with the following parameters: -f a -x 12345 -p 12345 -# 1000 -m GTRGAMMA.
Subsequent tree exploration highlighted Ceratitis capitata as having only two paralogues in the EOG090X08ZM set where three would have been expected. A tblastn query of the annotated C. capitata transcriptome using hodor revealed 3 bona fide hits, one of which (LOC101460849) was missing from EOG090X08ZM. Alignment and tree building were therefore repeated after inclusion of a reconstructed CDS from this locus and the final tree was rooted in accordance with results from a prior phylogenomic analysis of insects58. There are 110 proteins in the final dataset.
Trees were rendered with FigTree v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/) and Adobe Illustrator, with silhouettes obtained from Phylopic (http://phylopic.org).
Testing for purifying, relaxed, and positive selection
Data on single nucleotide polymorphisms (SNPs) in hodor were retrieved from the PopFly genome browser (http://popfly.uab.cat/)59. Within-species diversity at non-synonymous (pN) and synonymous (pS) sites was then calculated globally (across all D. melanogaster populations) and for a defined high-diversity population (ZI, Zimbabwe) using the seqinr R package60. Pairwise rates of non-synonymous (dN) and synonymous (dS) divergence between hodor and its orthologues in other Drosophila species (as depicted in Extended Data Fig. 10 and Supplementary Fig. 2) were calculated using a relevant method61, implemented in the PopGenome R package62.
To test for positive, relaxed, and purifying selection in phylogenetic framework, we made use of a collection of likelihood ratio tests provided by the Datamonkey Adaptive Evolution Server (https://www.datamonkey.org/). In particular, we tested for relaxed selection using the RELAX statistical framework63, comparing rates of evolution in the hodor family clade (purple box in Extended Data Fig. 10a and Supplementary Fig. 2) with the two clades (grey boxes) containing hodor paralogues.
Mosquito strains and rearing
Mosquitoes were reared under standard conditions at 27°C and 80% relative humidity with access to fish food as larvae and 5% glucose solution as adults. The mosquito strain used in this project, the A. gambiae G3, is reasonably amenable to rearing and microinjection. We obtained this line from the MR4 (MRA-112) and was originally isolated from West Africa (MacCarthy Island, The Gambia) in 1975 (Malaria Research & Reference Reagent Resource Center). Cas9 mosquitoes were generated previously64 using human-codon optimised SpCas965 (https://www.addgene.org/42230/) under control of the vasa promoter within the pDSAY vector66 and inserted at the X locus (2L:10526503). The protocols and procedures used in this study were approved by the Animal Ethics Committee of Imperial College and are in compliance with United Kingdom Home Office regulations.
Identification of the A. gambiae hodor gene
AGAP009616 is the predicted one-to-many orthologue of Drosophila Hodor-family proteins67. To confirm this, the full-length protein sequence of D. melanogaster Hodor was used for tBLASTN searches of the A. gambiae genome transcript gene set (AgamP4.10) using VectorBase BLAST (https://www.vectorbase.org). Top ranking hits were manually searched and AGAP009616 was determined as the highest ranking candidate, with 48.4% overall identity. No sequence similarity was detected between the predicted coding part of exon 1 of the annotated AGAP009616 transcript and Drosophila hodor or its orthologues from more distantly related Aedes or Culex mosquito species. Although we found evidence for the existence of upstream exon 1 using RNAseq BAM alignment files from G3 adult females (Tony Nolan, unpublished) visualised by IGV software68, predictions regarding the structure of exon 1 differ between members of the Anopheles genus. For this reason, the conserved exon 2 was chosen as the target for Cas9 genome editing.
Protein sequence alignments
Protein sequence alignments were generated using Clustal Omega 1.2.3 using default parameters (www.ebi.ac.uk/Tools/msa/clustalo69) and were visually modified using ESPript 3.0 to highlight percentage equivalence between sequences (espript.ibcp.fr/ESPript/ESPript/70).
Generation of transgenic gRNA mosquito strains
To generate CRISPR gRNA germline transformation constructs, a single gRNA target site was identified within the second exon of AGAP009616 and assessed for potential off-targets using flyCRISPR (http://tools.flycrispr.molbio.wisc.edu/targetFinder/) and ZiFIT (http://zifit.partners.org/). Since predictions regarding the structure of exon 1 differed between the closely related A. gambiae, A. coluzzi and A. gambiae pimperena mosquito strains, we designed a single gRNA (GAGTGTCCCACGTTAGAAGGAGCGG) that targets coding exon two of the predicted AGAP009616 locus structure (Extended Data Fig. 10b), which codes for amino acids conserved between the majority of Hodor-family proteins. The gRNA spacer was cloned by BsaI-mediated Golden Gate Assembly using 9616gF (TGCTGTGTCCCACGCTAGAAGGAG) and 9616gR (AAACCTCCTTCTAGCGTGGGACAC) into a U6-expression vector, p125 (available from AddGene), to create p125–9616 containing the U6::gRNA cassette of p16564, a 3xP3::DsRed marker and piggyBac repeats for germline transformation. In order to generate transgenic mosquito lines, plasmid p125–9616 was injected into mosquito embryos at 200ng/μl using a Femtojet Express injector in a mixture containing 300ng/μl helper vector expressing piggyBack transposase to mediate genomic integration. Surviving G0 individuals were crossed to wild-type mosquitoes, and the progeny was screened under a fluorescent microscope for expression of DsRed to recover G1 transformants. Two independent gRNA-expressing strains were generated by random integration of which one line (g10) was used in subsequent crosses to generate mutant lines.
Generation, genotyping and phenotyping of A.gambiae hodor mutant strains
To generate AGAP009616 mutant strains, we crossed 10–20 GFP-positive females of vasa:hCas9 line with 10–20 RFP-positive males of guide RNA-bearing line g10. We selected 10–20 GPF- and RFP-positive male progeny of this cross, and crossed them en masse to wild-type females. To make sure that no source of Cas9 and guide RNA were present in the subsequent generations, we selected GFP- and RFP-negative male progeny of the second cross, and crossed each of these males separately to a batch of 5–10 wild-type females. After collecting the eggs from each single-male cross, males were sacrificed and genotyped to determine the presence of a possible mutation in the AGAP009616 gene. Among the different mutations we managed to recover three independent mutations that harboured 8bp, 16bp and 19bp deletions at the target site. To maintain these three mutant strains, potentially mutant females at each generation were crossed en masse to wild-type males. The pupal progeny was then genotyped by extracting the DNA of pupal exuviae using the QIAGEN DNeasy Blood & Tissue Kit with a final elution step in 50μL of buffer AE. For each sample, a PCR amplification was set up using the p9616 forward and reverse primers below, using the following thermocycling conditions: 30 cycles; Annealing 67°C, 30 seconds; Extension 72°C, 30 seconds. The PCR product was purified (QIAquick PCR Purification Kit, QIAGEN), and ca. 150ng of this template was exposed to the restriction enzyme BsrBI (NEB). This restriction enzyme was predicted to cut the wild-type amplicon once, but not the deletion alleles lacking the restriction site. The purified PCR product was further analysed with Sanger sequencing of amplicons using the p9616seq forward and reverse primers below. To genotype adults, genomic DNA was extracted from adult mosquitoes using the QIAGEN DNeasy Blood & Tissue Kit with a final elution step in 50uL of buffer AE. For each sample, two PCR amplifications were set up. We either used primers p9616 forward and reverse using the following Thermocycling conditions: 30 cycles; Annealing 67°C, 30 seconds; Extension 72°C, 30 seconds, or primers p9616 forward and pDEL1 (reverse primer designed to bind the wild-type allele) below using the following Thermocycling conditions: 30 cycles; Annealing 62°C, 30 seconds; Extension 72°C, 30 seconds. The PCR product was purified (QIAquick PCR Purification Kit, QIAGEN) and ~150ng were digested with ScaI (Thermo Fisher Scientific).
| Name | Forward primer | Reverse primer |
|---|---|---|
| p9616 F/ R | ACGCATTCATAACCAAGACGA | CGTTTGTACCGTTGATGGATTC |
| P9616seq F/R | GACTTAAATCGGCATAGCACTGTG | CGTTTGTACCGTTGATGGATTC |
| pDEL1 R | CCACGCTAGAAGGAGCG |
Viability assay for Anopheles mutant strains
Potentially heterozygous mosquitoes were separated at the pupal stage and were allowed to emerge as adults singly in cups. Exuviae were collected for each individual pupa and were genotyped as described in the previous section. All verified heterozygous individuals were used to set up a sibling cross in cages of 30 × 30 × 30cm size (BugDorm). Generally, 10 females and 10 males were crossed for each experiment. They were allowed to mate for at least 5 days, then fed with screened human blood provided by National Health Service (NHS) through Hemotek LTD apparatus. Two days later, an egg bowl containing rearing water (dH2O supplemented with 0.1% pure salt) was placed in the cage. One or two days after hatching, larvae were placed in trays containing rearing water, allowed to develop as adults and then sacrificed and genotyped. Control crosses with wild-type males and females were set up in parallel. The data collected from the control crosses (number of eggs laid, hatching rate, eclosion rate) were compared to the data obtained from the sibling heterozygous mutant crosses.
Extended Data
Extended Data Fig. 1. Enterocyte screen, hodor mutant validation and hodor knockdown phenotypes.

a, Design of enterocyte specific RNAi-screen and generation of hodor mutant. Distribution of the categories of genes targeted for intestinal knockdown and number of genes and lines tested in each round of the genetic screen. b, Larval gut expressing UAS-Stinger-GFP under the control of mex1-Gal4, showing expression in all enterocytes, including those in the copper cell region (#) and iron cell region (*). There is no expression in the Malpighian tubules (†). c, Flies carrying UAS-RNAi targeted against candidate genes were crossed to those carrying mex1-Gal4 to achieve enterocyte-specific knockdown in the resulting larval progeny, which were either placed on high or low yeast food and allowed to develop into pupae. d, Results from the first round of the RNAi screen using mex1-Gal4 with plots showing the average time to pupariation after egg laying (AEL). Blue stars represent four different control lines crossed to mex1-Gal4. Linear models for these control lines (analysed together) are displayed as dashed lines with a 90% prediction interval shown in dotted lines; knockdown of genes B (CG11340) and F (CG4797) frequently led to a delay to pupariation. See Source Data 1 for the lines/genes that specific letters correspond to, and Supplementary Information for details of – and reasons for – the percentage deviation data display. e, Strategy for generating hodor mutants using pTVcherry vector52 to direct homologous recombination. Candidate recombinants were recovered after several crosses, identified based on viability and eye colour. f, PCR verification of integration of pTVcherry construct at the hodor locus, no band is seen in w1118 controls (1,3), but a correctly-sized band of 3–4kbp (arrowheads) is seen in hodor +/− (2,4). g, Real-Time quantitative PCR of control and hodor mutant larvae relative to gapdh, showing absence of hodor transcripts in the mutant. h, Larval survival in low yeast conditions when hodor is knocked down in all enterocytes using mex1-Gal4. i, RNAi targeting a different segment of the hodor transcript also causes a developmental delay when expressed with mex1-Gal4. j, Limiting expression of hodor RNAi to interstitial cells and principal cells of the Malpighian tubules (using hodor-Gal4) causes a significant delay to development. See Supplementary information for sample sizes and full genotypes. Scale bar b: 1mm. Where more than two groups were compared, an ordinary one-way ANOVA test was performed with a Tukey post-hoc test. Significance values are denoted as follows: p< 0.05 *, p< 0.01 **, p< 0.001 ***. Box plots: line, median; box, 75th–25th percentiles; whiskers, minimum to maximum.
Extended Data Fig. 2. Gal4 driver lines used in this study.

a, Larval guts stained with anti-Hodor show immunoreactivity in the copper cell (#) and iron cell (*) regions of the gut and the Malpighian tubules (†) in control animals, whilst this staining pattern is absent in hodor mutants. b, RNAi-mediated hodor knockdown in enterocytes (using mex1-Gal4) substantially reduces Hodor protein levels. c, RNAi-mediated hodor knockdown using hodor-Gal4 reduces protein levels considerably in the copper cell region (#) but does not noticeably reduce levels in the iron cell region (*). d, Expression of UAS-Stinger-GFP in interstitial cells (#) and Malpighian tubules (†) using hodor-Gal4; note absence of GFP in the iron cell region (*). e, Staining of iron cells highlighted in green (Fer2LCH>mCD8-GFP) with Hodor antibody illustrating overlap between the two in the anterior portion. f, Expression of lab-Gal4 (visualised as lab>mCD8-GFP expression) is seen in the copper cells (but not the interstitial cells) of the copper cell region. The panel to the right shows a higher magnification image of the copper cell region. g, Expression of CtB-Gal4 (visualised as CtB>Stinger-GFP expression) is confined to the principal cells of Malpighian tubules. h, R2R4-Gal4 (visualised as R2R4>Stinger-GFP expression) is confined to a subset of enterocytes in the posterior midgut. Note its absence from the copper (#) and iron cell (*) regions as well as from Malpighian tubules (†). See Supplementary information for sample sizes and full genotypes. Scale bars: a, d, f and h: 1mm; e, b, 200μm; c, 300μm; g, 200μm; f inset, 50μm.
Extended Data Fig. 3. Hodor controls food intake and systemic growth.

a, Comparison of embryonic viability between control (w1118), heterozygous and homozygous hodor mutant larvae; there are no significant differences. b, Developmental progression of larvae lacking hodor compared to control animals (w1118). c, Pupal volume of hodor mutants compared to controls; each data point represents one pupa. d, Wing size measurements in control vs hodor mutant adults; no significant differences are apparent (see Methods for details of quantification, each data point represents one wing). e, Reduced pAkt relative to total protein in second-instar hodor mutants compared to controls, all raised on a low-yeast diet and repeated three times. pAkt in hodor mutants is comparable to that of wild-type larvae starved for 15h. f, Reduced food intake in hodor-Gal4-driven hodor knockdown when compared to control larvae. Experiments were performed using second-instar larvae raised on a low-yeast diet. g, Electron micrographs of the junctional region (arrow) between an interstitial cell and a copper cell, showing no obvious defects in first-instar hodor mutants. h, Smurf assay (see Methods) on second-instar control larvae and hodor mutants (examples are representative of at least 6 larvae per genotype). No leakage of blue dye from the intestine was seen in either group. i, Overexpression of hodor in interstitial cells using hodor-gal4 does not alter developmental rate in either high or low yeast conditions. j-k, Activation or inactivation of Tor signalling in hodor-expressing cells does not affect developmental rate (j) or food intake (k); none of the genetic manipulations are significantly different compared to their respective controls. l, Modulation of Rag and Gator1 complex components in the interstitial cells of hodor mutants (from hodor-Gal4) does not rescue/exacerbate their developmental delay. See Supplementary information for sample sizes and full genotypes. Scale bars: b, 0.5mm; d, 250μm; g, 500nm; h, 400μm. Where more than two groups were compared, an ordinary one-way ANOVA test was performed with a Tukey post-hoc test. Significance values are denoted as follows: p< 0.05 *, p< 0.01 **, p< 0.001 ***. Box plots: line, median; box, 75th–25th percentiles; whiskers, minimum to maximum.
Extended Data Fig. 4. Hodor sustains luminal acidity and luminal/cell volume.

a, The copper cell region (#) of Drosophila larvae is normally acidic (bromophenol blue dye appears yellow/orange, see Methods), but becomes less acidic (purple/blue) when using hodor RNAi in interstitial cells (hodor-gal4) or in hodor mutants. The latter phenotype can be rescued by re-expressing hodor in hodor-Gal4-expressing cells. Intestinal acidity is also lost by downregulating the gene coding for the Vha16–1 subunit of the V-ATPase proton pump in copper cells using lab-Gal4. b, Quantifications of intestinal acidity, depletion (by RNAi) or loss of hodor results in a reduction in the number of larvae with acidic middle midguts, as does depletion of the V-ATPase subunit Vha16–1 in copper cells using lab-gal4. c, Larval developmental rate is unaffected when acidity is lost due to reducing V-ATPase activity within copper cells (using lab-Gal4). d, Electron micrographs of interstitial cells of first-instar larvae, showing a reduction in their characteristic basal infoldings (arrows) in hodor mutants (* denotes basal lamina) relative to control cells. e, hodor-Gal4 driven mCD8-GFP expression in interstitial cells of control and hodor mutant larvae reveals an increase in luminal volume (*) and interstitial cell volume (insets with quantifications to the right) in first-instar mutant larvae when compared to controls (all raised on a low-yeast diet). See Methods for details of volume quantifications. f, Overexpression of the dominant-negative Shibire ShiK44A in hodor-expressing cells (using hodor-Gal4) reveals an increase in interstitial cell volume in hodor second-instar mutant larvae relative to controls (all raised on low-yeast diet). Lysotracker staining in green was used to reveal their cytoplasm. Quantifications are shown to the right. Second-instar larvae raised on a low-yeast diet were used for all experiments involving ShiK44A expression. g, This genetic manipulation also results in an increase in the width of the copper cell region (#) but does not affect the subcellular localisation of Hodor in interstitial cells (insets). h, Quantifications of copper cell region width in controls, hodor mutant larvae and larvae expressing ShiK44A from hodor-Gal4. i, Expression of Shikk44A in hodor-expressing cells (hodor> ShiK44A) does not alter developmental rate. See Supplementary information for sample sizes and full genotypes. Scale bars: a, 500μm; d, 500nm; e and f, 10μm; g: 250μm. For comparisons involving two groups, a non-parametric Mann Whitney U test was used. Where more than two groups were compared, an ordinary one-way ANOVA test was performed with a Tukey post-hoc test. Significance values are denoted as follows: p< 0.05 *, p< 0.01 **, p< 0.001 ***. Box plots: line, median; box, 75th–25th percentiles; whiskers, minimum to maximum.
Extended Data Fig. 5. The microbiota of hodor mutants.

a, Increased bacterial loads (CFU/larvae) in hodor mutants when compared to control larvae. Bacterial loads were assessed in third-instar larvae raised on a high-yeast diet. b-c, Developmental rate of control and hodor mutant larvae in germ-free conditions, or following re-colonisation with Acetobacter pomorum or Lactobacillus plantarum in either high (b) or low-yeast (c) conditions. hodor mutants remain developmentally delayed in germ-free conditions, particularly when reared on a low-yeast diet. Mono-association partially rescues the developmental delay of all larvae in low-yeast conditions, but the difference in developmental rate between control and hodor mutant larvae persists. d, Representative images of FluoZin-3AM stainings (a zinc dye) in the copper cell region of larvae reared in germ-free conditions or bi-associated with Acetobacter pomorum and Lactobacillus plantarum. More zinc is apparent in the copper cell region of high yeast-fed larvae relative to low yeast-fed larvae, but this is unaffected by the presence of microbiota. e, Quantifications of zinc staining in copper cell region. See Supplementary information for sample sizes and full genotypes. Scale bars: d, 30μm. For comparisons involving two groups, a non-parametric Mann Whitney U test was used. Where more than two groups were compared, an ordinary one-way ANOVA test was performed with a Tukey post-hoc test. Significance values are denoted as follows: p< 0.05 *, p< 0.01 **, p< 0.001 ***. Box plots: line, median; box, 75th–25th percentiles; whiskers, minimum to maximum.
Extended Data Fig. 6. Hodor gating, transport and effect on food intake.

a, Mutational free energy space, where each double mutant is plotted as zinc binding free energy and structural stability. The E255K-E296F mutant pair (black dot) was selected to increase the free energy of binding but keep the structural stability as low as possible to avoid refolding of the protein. b, Zinc-activated currents from oocytes expressing wild-type Hodor (top) or mutant Hodor-E255K-E296F (bottom) in response to the indicated concentrations (b). c, Activation (top) and deactivation (bottom) kinetics of currents elicited by 50μM ZnCl2 were significantly faster in Hodor-E255K-E296F (n=4–5, p<0.05 for ON, p<0.001 for OFF, Welch’s t-test). d, Concentration dependence of zinc-activated currents from oocytes expressing Hodor (sigmoidal fit from Figure 3B in gray) compared with that of Hodor-E255K-E296F (in red). The estimated EC50 for Hodor-E255K-E297F was comparable to wild-type Hodor (119.90μM, 95% confidence interval 104.70 to 137.10μM), with the only significant difference observed in response to 50μM ZnCl2 (p<0.05, two-way ANOVA with post hoc Bonferroni test, n = 5–9). Data represented as mean ± s.e.m., n denotes number of oocytes. e, Current-voltage (I-V) relationship of zinc-activated currents from uninjected oocytes in response to the indicated concentrations. f, Preference index plotted over time for larvae given a choice between high- and low-yeast diets. Both control and hodor mutant larvae develop a significant preference for a high-yeast diet (positive numbers) after 24h. g, hodor-Gal4-driven ClopHensor expression in live interstitial cells reveals a reduction in intracellular chloride levels (increased 458nm/543nm ratio) in first-instar larvae raised on a low-yeast diet supplemented with 0.4mM ZnSO4 compared to larvae raised on a low-yeast diet only. Chloride levels went from ca. 8.6mM in controls to ca. 5.7mM in larvae raised on a ZnSO4-supplemented diet, calculated based on calibration in Extended Data Fig. 6h. Representative 458nm fluorescence images are shown to the left. h, Calibration of the hodor-Gal4 driven ClopHensor in interstitial cells with eight different chloride concentrations (see Methods for details). The calibration graph to the left shows the sigmoidal curve interpolated from individual 458nm/543nm ratios obtained using the different chloride concentrations. This graph enables conversion of absorbance ratios to chloride concentration. Images to the right show representative 458nm signals for each concentration. See Supplementary information for sample sizes and full genotypes. Scale bars: g and h, 30μm. For comparisons involving two groups, a non-parametric Mann Whitney U test was used. Where more than two groups were compared, an ordinary one-way ANOVA test was performed with a Tukey post-hoc test. Significance values are denoted as follows: p< 0.05 *, p< 0.01 **, p< 0.001 ***. Boxplots show both minimum and maximum values. Box plots: line, median; box, 75th–25th percentiles; whiskers, minimum to maximum.
Extended Data Fig. 7. Intestinal zinc stainings.

a, Validation of the zinc-sensitive dye, FluoZin-3AM, in adult and larval Malpighian tubules. The tubules of w1118 adults have less zinc than those of wild-type (OrR) adults, which can be increased by supplementing their adult diet with 1mM ZnCl2 for 3 days (left panels). A more modest reduction in zinc levels is observed in larval tubules of second-instar w1118 larvae relative to wild-type OrR larvae (right panels). b, FluoZin-3AM staining in the middle midgut of second-instar wild-type larvae (OrR, which harbour a wild-type w gene), w mutant larvae (w1118), w mutant larvae with a mini-w transgene (UAS-Rheb/+) and hodor mutant larvae (which are mutant for w but carry mini-w transgenes). # denotes copper cell region, * denotes iron cell region. Panels to the right show higher magnification images of the copper cell region. Zinc levels are higher in the copper cell region of wild-type larvae relative to the other genotypes, which have comparable zinc. Bottom panel shows FluoZin-3AM staining of a wild-type (OrR) adult midgut. There is no apparent zinc enrichment in the copper cell region (#). c, Quantification of intestinal zinc intensity in the copper cell region. In both c and d, larvae were raised on a low-yeast diet. d, Wild-type OrR larvae are significantly faster to reach the pupal stage than w1118 in low yeast conditions, whilst hodor−/− still causes a significant developmental delay in either a genetic background with an intact w gene (w+; hodor−/−) or when backcrossed 8 times into a w mutant background lacking the w gene (w; hodor−/−). e, Heterozygous lines carrying mini-w are developmentally faster than w1118 larvae in low-yeast conditions. Scale bars; a: 50μm b: 500μm; insert 50μm. See Supplementary information for sample sizes and full genotypes. For comparisons involving two groups, a non-parametric Mann Whitney U test was used. Where more than two groups were compared, an ordinary one-way ANOVA test was performed with a Tukey post-hoc test. Significance values are denoted as follows: p< 0.05 *, p< 0.01 **, p< 0.001 ***. Boxplots show both minimum and maximum values. Box plots: line, median; box, 75th–25th percentiles; whiskers, minimum to maximum.
Extended Data Fig. 8. Subcellular localisation of Hodor.

a, Quantification of the fraction of Hodor-positive punctae that co-express Rab 5, 7, 11 (all of which are endogenously tagged with YFP), Lysotracker or Lamp1 (endogenously expressed Lamp1-mCherry). b-d, Co-expression analysis reveals limited overlap between Hodor immunoreactivity and the early endosome marker Rab5 (b) or the recycling endosome marker Rab11 (d), whilst more pronounced overlap is apparent with late endosome/lysosome marker Rab7 (c). e, The majority of Lamp1-positive structures co-expressed Hodor on the apical side of interstitial cells (* denotes the intestinal lumen). Larvae were briefly starved (4h) prior to dissection in order to visualise lysosomes as punctate structures. f, The endogenously expressed GFP-tagged Vha16–1 subunit of the V-ATPase complex is predominantly localised to the copper cell region (#) within the larval intestine. g, Expression of Vha16–1-GFP is apparent in both the copper cells and, to a lesser extent, the interstitial cells. See Supplementary information for sample sizes and full genotypes. Scale bars: b, c, d and e, 10μm; f, 200μm; g, 30μm. N: nucleus. Significance values are denoted as follows: p< 0.05 *, p< 0.01 **, p< 0.001 ***. Box plots: line, median; box, 75th–25th percentiles; whiskers, minimum to maximum.
Extended Data Fig. 9. Hodor regulates autophagy.

a, Representative expression of Lysosensor, Lysotracker, Lamp1-mCherry and hodor-Gal4-driven p62-GFP in the copper cell region of control larvae, larvae in which the V-ATPase subunit Vha44 has been downregulated in interstitial cells (using hodor-Gal4) or hodor mutant larvae. Vha44 knockdown and, to a lesser extent, hodor mutation result in an increase in the number of punctae positive for these markers. b, Quantifications of the number of punctae positive for the above mentioned markers in all three types of larvae shown in a. c, hodor mutants expressing the dual autophagosome/autolysosome marker UAS-GFP-mCherry-Atg8a in all enterocytes (using mex1-Gal4) show regional enrichment of autophagy in both the copper cell (#) and iron cell (*) regions when compared to an anterior portion of the gut (^). Note the appearance of GFP-positive punctae in the copper cell region (#), suggestive of defective autolysosomes unable to quench the GFP signal. d, hodor-Gal4-driven expression of GFP-mCherry-Atg8a in interstitial cells of starved hodor mutants. Large subcellular compartments positive for both GFP and mCherry are apparent. e, Quantification of GFP- and/or mCherry-positive Atg8a-expressing autophagosomes/autolysosomes in the copper cell region of fed or starved controls, and fed or starved hodor mutants (left graph, Atg8a reporter expressed from hodor-Gal4; right graph, Atg8a reporter expressed from mex1-Gal4 in fed hodor mutants). See Supplementary information for sample sizes and full genotypes. Scale bars: a, 30μm; c, 500μm; d, 45μm. Where more than two groups were compared, an ordinary one-way ANOVA test was performed with a Tukey post-hoc test. Significance values are denoted as follows: p< 0.05 *, p< 0.01 **, p< 0.001 ***. Box plots: line, median; box, 75th–25th percentiles; whiskers, minimum to maximum.
Extended Data Fig. 10. Hodor is an insect-specific gene, essential in A. gambiae.

a, Nucleotide-level maximum likelihood phylogeny of the hodor gene family highlighting successive duplication events at the base of the Schizophora (orange and red nodes, see Methods for details of phylogenetic reconstruction, and Extended Data Fig. 10 for a complete gene family tree). Bootstrap support is indicated along individual branches as a percentage of 1000 rapid bootstraps. b, gRNA target site within exon 2 of the Anopheles gambiae one-to-many orthologue AGAP009616 of fly hodor-like genes, the diagnostic primers used for genotyping and the three frameshift mutants recovered. PAM: protospacer adjacent motif. c, Recovering AGAP009616 mutants. d, Genotyping the progeny of crosses between verified heterozygote males and females revealed that AGAP009616 homozygous mutant adults are inviable. See Methods for details.
Extended Data Fig. 11. Current model of Hodor functions.

Hodor resides in the apical membrane and on the lysosomes of gut interstitial cells (highlighted in blue, adjacent to acid-secreting copper cells (#). Zinc sensing by Hodor promotes chloride transport and Tor signalling within interstitial cells. Hodor/Tor signalling in interstitial cells in turn promotes systemic growth through a neural relay, activating insulin-like signalling and thereby sustaining developmental rate, and 2) by promoting food intake via an as yet unknown mechanism independent of the brain insulin-producing cells. The reduced insulin signalling observed in hodor mutants may be secondary to their reduced food intake (hence the dashed arrow).
Extended Data Table 1. Compounds tested in Xenopus oocytes.
Specific compounds, concentrations and responses are listed.
| Compound | Concentration | Response |
|---|---|---|
| Acetylcholine | 10mM | No response |
| Alanine | 10mM | No response |
| Arginine | 10mM | No specific response |
| Asparagine | 10mM | No response |
| Aspartic acid | 10mM | No specific response |
| Cysteine | 10mM | No specific response |
| Cadmium | 500μM | Small response, approximately 233 ± 70 nA |
| Copper chloride | 500μM | Small response, approximately 20 nA |
| GABA | 10mM | No response |
| Glutamic acid | 10mM | No specific response |
| Glutamate | 10mM | No response |
| Glutamine | 10mM | No response |
| Glycine | 10mM | No response |
| Histidine | 10mM | No specific response |
| Histamine | 10mM | No response |
| Iron chloride | 500μM | Small response, approximately 20 nA |
| Isoleucine | 10mM | No response |
| Leucine | 10mM | No response |
| Lysine | 10mM | No specific response |
| Methionine | 10mM | No response |
| Phenylalanine | 10mM | No response |
| Proline | 10mM | No response |
| pH | pH 8, 9, 10 | Dose-dependent response |
| Serine | 10mM | No response |
| Serotonin | 10mM | No specific response |
| Threonine | 10mM | No response |
| Tryptophan | 10mM | No response |
| Tyrosine | 10mM | No response |
| Valine | 10mM | No response |
| Zinc chloride | 0.1, 1, 10, 50, 100, 250, 500, 1000μM | Dose-dependent response, see Fig. 3b |
| Zinc + picrotoxin | 100μM | Did not prevent zinc mediated activation |
| Zinc + pentylenetetrazol | 1mM | Did not prevent zinc mediated activation |
Supplementary Material
ACKNOWLEDGEMENTS
We thank Barry Denholm, Trent Perry, Elodie Prince, Aylin Rodan, Eric Rulifson, Aurelio Teleman, Alec Vincent and Clive Wilson for sharing reagents. Mahmoud Ardakani, Dirk Dormann, Cati Ware and Chad Whilding provided imaging advice. We are grateful to Marko Brankatschk, Gabor Juhasz, Fanis Missirlis, Lucia Prieto-Godino, Hui Gong and all members of the Miguel-Aliaga, Hirabayashi and Cochemé labs for helpful discussions and experimental advice and/or assistance. We thank Elisabeth Knust and Wieland Huttner for supporting the EM work. Tomotsune Ameku, Susumu Hirabayashi and Santiago Vernia provided comments on an earlier version of this manuscript. SR thanks Anna M L Coenen-Stass, Iman Al-Khatib and Siavoush Redhai for their advice and constant support. We thank the Bloomington Drosophila Stock Center, Vienna Drosophila Resource Center, the Kyoto Drosophila Genomics and Genetics Resource for flies, and the Developmental Studies Hybridoma Bank for antibodies. This work was funded by an ERC Advanced Grant to IM-A (ERCAdG 787470 “IntraGutSex”), an ERC Starting Grant to NW (ERCStG 335724 “VecSyn”), an Imperial Confidence in Concepts grant to IM-A, NW and SR, MRC intramural funding to IMA, a BBSRC grant to RAB (BB/L027690/1) and an Equipe FRM label to FL.
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
REPORTING SUMMARY
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
DATA AVAILABILITY
All raw data are available from the corresponding author on reasonable request.
References
- 1.Miguel-Aliaga I Nerveless and gutsy: intestinal nutrient sensing from invertebrates to humans. Semin Cell Dev Biol 23, 614–620, doi: 10.1016/j.semcdb.2012.01.002 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feingold D, Starc T, O’Donnell MJ, Nilson L & Dent JA The orphan pentameric ligand-gated ion channel pHCl-2 is gated by pH and regulates fluid secretion in Drosophila Malpighian tubules. J Exp Biol 219, 2629–2638, doi: 10.1242/jeb.141069 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Remnant EJ et al. Evolution, Expression, and Function of Nonneuronal Ligand-Gated Chloride Channels in Drosophila melanogaster. G3 (Bethesda) 6, 2003–2012, doi: 10.1534/g3.116.029546 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Colombani J et al. A nutrient sensor mechanism controls Drosophila growth. Cell 114, 739–749 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Geminard C, Rulifson EJ & Leopold P Remote control of insulin secretion by fat cells in Drosophila. Cell Metab 10, 199–207, doi: 10.1016/j.cmet.2009.08.002 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Rodenfels J et al. Production of systemically circulating Hedgehog by the intestine couples nutrition to growth and development. Genes Dev 28, 2636–2651, doi: 10.1101/gad.249763.114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Storelli G et al. Lactobacillus plantarum promotes Drosophila systemic growth by modulating hormonal signals through TOR-dependent nutrient sensing. Cell Metab 14, 403–414, doi: 10.1016/j.cmet.2011.07.012 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Waterhouse DF & Stay B Functional differentiation in the midgut epithelium of blowfly larvae as revealed by histochemical tests. Aust J Biol Sci, 253–277 (1955). [Google Scholar]
- 9.Poulson DF & Waterhouse DF Experimental studies on pole cells and midgut differentiation in Diptera. Aust J Biol Sci, 541–567 (1960). [Google Scholar]
- 10.Dubreuil RR et al. Mutations of alpha spectrin and labial block cuprophilic cell differentiation and acid secretion in the middle midgut of Drosophila larvae. Dev Biol 194, 1–11, doi: 10.1006/dbio.1997.8821 (1998). [DOI] [PubMed] [Google Scholar]
- 11.Li H, Qi Y & Jasper H Preventing Age-Related Decline of Gut Compartmentalization Limits Microbiota Dysbiosis and Extends Lifespan. Cell Host Microbe 19, 240–253, doi: 10.1016/j.chom.2016.01.008 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Overend G et al. Molecular mechanism and functional significance of acid generation in the Drosophila midgut. Sci Rep 6, 27242, doi: 10.1038/srep27242 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Storelli G et al. Drosophila Perpetuates Nutritional Mutualism by Promoting the Fitness of Its Intestinal Symbiont Lactobacillus plantarum. Cell Metab 27, 362–377 e368, doi: 10.1016/j.cmet.2017.11.011 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Filshie BK, Poulson DF & Waterhouse DF Ultrastructure of the copper-accumulating region of the Drosophila larval midgut. Tissue Cell 3, 77–102 (1971). [DOI] [PubMed] [Google Scholar]
- 15.Rulifson EJ, Kim SK & Nusse R Ablation of insulin-producing neurons in flies: growth and diabetic phenotypes. Science 296, 1118–1120, doi: 10.1126/science.1070058 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Dent JA Evidence for a diverse Cys-loop ligand-gated ion channel superfamily in early bilateria. J Mol Evol 62, 523–535, doi: 10.1007/s00239-005-0018-2 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Negi S, Pandey S, Srinivasan SM, Mohammed A & Guda C LocSigDB: a database of protein localization signals. Database (Oxford) 2015, doi: 10.1093/database/bav003 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sancak Y et al. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303, doi: 10.1016/j.cell.2010.02.024 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Senturk M et al. Ubiquilins regulate autophagic flux through mTOR signalling and lysosomal acidification. Nat Cell Biol 21, 384–396, doi: 10.1038/s41556-019-0281-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zoncu R et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 334, 678–683, doi: 10.1126/science.1207056 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valvezan AJ & Manning BD Molecular logic of mTORC1 signalling as a metabolic rheostat. Nature metabolism 1, 321–333 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Mauvezin C & Neufeld TP Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy 11, 1437–1438, doi: 10.1080/15548627.2015.1066957 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hudry B et al. Sex Differences in Intestinal Carbohydrate Metabolism Promote Food Intake and Sperm Maturation. Cell 178, 901–918 e916, doi: 10.1016/j.cell.2019.07.029 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gonzalez A & Hall MN Nutrient sensing and TOR signaling in yeast and mammals. EMBO J 36, 397–408, doi: 10.15252/embj.201696010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saxton RA & Sabatini DM mTOR Signaling in Growth, Metabolism, and Disease. Cell 168, 960–976, doi: 10.1016/j.cell.2017.02.004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blaby-Haas CE & Merchant SS Lysosome-related organelles as mediators of metal homeostasis. J Biol Chem 289, 28129–28136, doi: 10.1074/jbc.R114.592618 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roh HC, Collier S, Guthrie J, Robertson JD & Kornfeld K Lysosome-related organelles in intestinal cells are a zinc storage site in C. elegans. Cell Metab 15, 88–99, doi: 10.1016/j.cmet.2011.12.003 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holly MK & Smith JG Paneth Cells during Viral Infection and Pathogenesis. Viruses 10, doi: 10.3390/v10050225 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park J et al. Lysosome-Rich Enterocytes Mediate Protein Absorption in the Vertebrate Gut. Dev Cell 51, 7–20 e26, doi: 10.1016/j.devcel.2019.08.001 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baker DA et al. A comprehensive gene expression atlas of sex- and tissue-specificity in the malaria vector, Anopheles gambiae. BMC Genomics 12, 296, doi: 10.1186/1471-2164-12-296 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sudarsan V, Pasalodos-Sanchez S, Wan S, Gampel A & Skaer H A genetic hierarchy establishes mitogenic signalling and mitotic competence in the renal tubules of Drosophila. Development 129, 935–944 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Phillips MD & Thomas GH Brush border spectrin is required for early endosome recycling in Drosophila. J Cell Sci 119, 1361–1370, doi: 10.1242/jcs.02839 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Kim E, Goraksha-Hicks P, Li L, Neufeld TP & Guan KL Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol 10, 935–945, doi: 10.1038/ncb1753 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Romero-Pozuelo J, Demetriades C, Schroeder P & Teleman AA CycD/Cdk4 and Discontinuities in Dpp Signaling Activate TORC1 in the Drosophila Wing Disc. Dev Cell 42, 376–387 e375, doi: 10.1016/j.devcel.2017.07.019 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Pircs K et al. Advantages and limitations of different p62-based assays for estimating autophagic activity in Drosophila. PLoS One 7, e44214, doi: 10.1371/journal.pone.0044214 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hegedus K et al. The Ccz1-Mon1-Rab7 module and Rab5 control distinct steps of autophagy. Mol Biol Cell 27, 3132–3142, doi: 10.1091/mbc.E16-03-0205 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kakanj P et al. Insulin and TOR signal in parallel through FOXO and S6K to promote epithelial wound healing. Nat Commun 7, 12972, doi: 10.1038/ncomms12972 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marois E, Mahmoud A & Eaton S The endocytic pathway and formation of the Wingless morphogen gradient. Development 133, 307–317, doi: 10.1242/dev.02197 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Sun Q et al. Intracellular Chloride and Scaffold Protein Mo25 Cooperatively Regulate Transepithelial Ion Transport through WNK Signaling in the Malpighian Tubule. J Am Soc Nephrol 29, 1449–1461, doi: 10.1681/ASN.2017101091 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682, doi: 10.1038/nmeth.2019 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Layalle S, Arquier N & Leopold P The TOR pathway couples nutrition and developmental timing in Drosophila. Dev Cell 15, 568–577, doi: 10.1016/j.devcel.2008.08.003 (2008). [DOI] [PubMed] [Google Scholar]
- 42.Bhatt PK & Neckameyer WS Functional analysis of the larval feeding circuit in Drosophila. J Vis Exp, e51062, doi: 10.3791/51062 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shen P Analysis of feeding behavior of Drosophila larvae on solid food. Cold Spring Harb Protoc 2012, doi: 10.1101/pdb.prot069328 (2012). [DOI] [PubMed] [Google Scholar]
- 44.Almeida de Carvalho MJ & Mirth CK Food intake and food choice are altered by the developmental transition at critical weight in Drosophila melanogaster. Animal Behaviour 126, 195–208 (2017). [Google Scholar]
- 45.Toshima N & Tanimura T Taste preference for amino acids is dependent on internal nutritional state in Drosophila melanogaster. J Exp Biol 215, 2827–2832, doi: 10.1242/jeb.069146 (2012). [DOI] [PubMed] [Google Scholar]
- 46.Preibisch S, Saalfeld S & Tomancak P Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics 25, 1463–1465, doi: 10.1093/bioinformatics/btp184 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shin SC et al. Drosophila microbiome modulates host developmental and metabolic homeostasis via insulin signaling. Science 334, 670–674, doi: 10.1126/science.1212782 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Axelsson L et al. Genome sequence of the naturally plasmid-free Lactobacillus plantarum strain NC8 (CCUG 61730). J Bacteriol 194, 2391–2392, doi: 10.1128/JB.00141-12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Erkosar B et al. Drosophila microbiota modulates host metabolic gene expression via IMD/NF-kappaB signaling. PLoS One 9, e94729, doi: 10.1371/journal.pone.0094729 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rera M et al. Modulation of longevity and tissue homeostasis by the Drosophila PGC-1 homolog. Cell Metab 14, 623–634, doi: 10.1016/j.cmet.2011.09.013 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Musselman LP et al. A high-sugar diet produces obesity and insulin resistance in wild-type Drosophila. Dis Model Mech 4, 842–849, doi: 10.1242/dmm.007948 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baena-Lopez LA, Alexandre C, Mitchell A, Pasakarnis L & Vincent JP Accelerated homologous recombination and subsequent genome modification in Drosophila. Development 140, 4818–4825, doi: 10.1242/dev.100933 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bischof J, Maeda RK, Hediger M, Karch F & Basler K An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc Natl Acad Sci U S A 104, 3312–3317, doi: 10.1073/pnas.0611511104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin YF et al. MIB: Metal Ion-Binding Site Prediction and Docking Server. J Chem Inf Model 56, 2287–2291, doi: 10.1021/acs.jcim.6b00407 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Maier JA et al. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J Chem Theory Comput 11, 3696–3713, doi: 10.1021/acs.jctc.5b00255 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miller BR 3rd et al. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J Chem Theory Comput 8, 3314–3321, doi: 10.1021/ct300418h (2012). [DOI] [PubMed] [Google Scholar]
- 57.Liman ER, Tytgat J & Hess P Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron 9, 861–871 (1992). [DOI] [PubMed] [Google Scholar]
- 58.Misof B et al. Phylogenomics resolves the timing and pattern of insect evolution. Science 346, 763–767, doi: 10.1126/science.1257570 (2014). [DOI] [PubMed] [Google Scholar]
- 59.Hervas S, Sanz E, Casillas S, Pool JE & Barbadilla A PopFly: the Drosophila population genomics browser. Bioinformatics 33, 2779–2780, doi: 10.1093/bioinformatics/btx301 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Charif D & Lobry JR in Structural Approaches to Sequence Evolution 207–232 (Springer, 2007). [Google Scholar]
- 61.Li WH Unbiased estimation of the rates of synonymous and nonsynonymous substitution. J Mol Evol 36, 96–99 (1993). [DOI] [PubMed] [Google Scholar]
- 62.Pfeifer B, Wittelsburger U, Ramos-Onsins SE & Lercher MJ PopGenome: an efficient Swiss army knife for population genomic analyses in R. Mol Biol Evol 31, 1929–1936, doi: 10.1093/molbev/msu136 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wertheim JO, Murrell B, Smith MD, Kosakovsky Pond SL & Scheffler K RELAX: detecting relaxed selection in a phylogenetic framework. Mol Biol Evol 32, 820–832, doi: 10.1093/molbev/msu400 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hammond A et al. A CRISPR-Cas9 gene drive system targeting female reproduction in the malaria mosquito vector Anopheles gambiae. Nat Biotechnol 34, 78–83, doi: 10.1038/nbt.3439 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cong L et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823, doi: 10.1126/science.1231143 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Volohonsky G et al. Tools for Anopheles gambiae Transgenesis. G3 (Bethesda) 5, 1151–1163, doi: 10.1534/g3.115.016808 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kriventseva EV et al. OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res 47, D807–D811, doi: 10.1093/nar/gky1053 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Robinson JT et al. Integrative genomics viewer. Nat Biotechnol 29, 24–26, doi: 10.1038/nbt.1754 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Larkin MA et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948, doi: 10.1093/bioinformatics/btm404 (2007). [DOI] [PubMed] [Google Scholar]
- 70.Robert X & Gouet P Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42, W320–324, doi: 10.1093/nar/gku316 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data are available from the corresponding author on reasonable request.