

Abstract
The current conceptualization of Alzheimer disease (AD) is driven by the amyloid hypothesis, in which a deterministic chain of events leads from amyloid deposition and then tau deposition to neurodegeneration and progressive cognitive impairment. This model fits autosomal dominant AD but is less applicable to sporadic AD. Owing to emerging information regarding the complex biology of AD and the challenges of developing amyloid- targeting drugs, the amyloid hypothesis needs to be reconsidered. Here we propose a probabilistic model of AD in which three variants of AD (autosomal dominant AD, APOE ε4-related sporadic AD and APOE ε4-unrelated sporadic AD) feature decreasing penetrance and decreasing weight of the amyloid pathophysiological cascade, and increasing weight of stochastic factors (environmental exposures and lower-risk genes). Together, these variants account for a large share of the neuropathological and clinical variability observed in people with AD. The implementation of this model in research might lead to a better understanding of disease pathophysiology, a revision of the current clinical taxonomy and accelerated development of strategies to prevent and treat AD.
Alzheimer disease (AD) is the most common cause of dementia in elderly people and is becoming increasingly prevalent worldwide. The incidence of dementia doubles with every six years of age, from 3.9 per 1,000 person-years at the age of 60–64 years up to 104.8 per 1,000 person-years at the age of 90 years or older1. Thus, with an ever-ageing population, the number of people with dementia worldwide is predicted to climb to 131.5 million in 2050 (REF.1). In 2015, the global societal economic cost of dementia was estimated as US $818 billion, a sum similar in magnitude to the gross domestic products of countries such as the Netherlands, and this cost is forecast to steadily increase in the coming years (up to US $2 trillion by 2030)1.
According to its original pathological definition, AD is defined by extracellular amyloid plaques and intracellular neurofibrillary tangles2. The recent ‘ATN’ research framework of AD defines the disease on the basis of its underlying molecular pathology (plaques = amyloid (‘A’), neurofibrillary tangles = hyperphosphorylated tau (‘T’)) and the ensuing neurodegeneration (‘N’), irrespective of the clinical phenotype or underlying genetics3. Hence, the presence of both amyloid-β (Aβ) and tau deposits in the brain (A+T+) defines AD, whereas the presence of Aβ only (A+T−) defines a so-called Alzheimer pathological change3. Even though this framework is intended to be agnostic (that is, no direct assumptions regarding the causal role or order of biomarker abnormalities are made), it is strongly influenced by the amyloid hypothesis, which posits that Aβ is the earliest molecular driver of the disease4,5.
The amyloid hypothesis states that the peptide Aβ causes a cascade of downstream events that finally lead to cognitive impairment and dementia4,5. It has been the dominant model of AD pathogenesis for more than 30 years and the guiding influence for drug development, which to a large degree has aimed to produce compounds that either reduce Aβ production (secretase inhibitors) or increase Aβ clearance (immunotherapies). The hypothesis implicitly assumes a deterministic cause–effect model (that is, a chain of events that will invariably produce the same output from a given starting condition or state), in which the extracellular deposition of fibrillar Aβ (that is, amyloid) is the causative event and is followed by intracellular aggregation of hyperphosphorylated tau, synaptic dysfunction, neurodegeneration, cognitive dysfunction and, finally, loss of autonomy (that is, dementia)4,5. Some other diseases fit a deterministic model, such as certain cancers, in which a cancerogenic event always entails uncontrolled cell proliferation that is invariably followed by clinical cancer with final organ failure. For example, in chronic myeloid leukaemia, one single genetic event (that is, a fusion gene, BCR–ABL1, formed by a translocation between chromosomes 9 and 22) is necessary and sufficient (that is, conferring a 100% lifetime risk) to induce neoplastic proliferation. Interfering with the signalling pathway activated by BCR–ABL1 using tyrosine kinase inhibitors is an extremely effective therapeutic strategy at almost any disease stage6.
While the cornerstones of the amyloid hypothesis are supported by substantial evidence derived largely from autosomal dominant AD, Down syndrome, and cellular and animal models based on autosomal dominant mutations, its current formulation fails to account for a number of clinical and preclinical observations. The amyloid hypothesis has been heavily criticized7,8, as has the linear causal dynamics it implies9. Alternative models have been based largely on neurobiological arguments, and have failed to gain widespread acceptance in the community10. A revision of the amyloid hypothesis that fits the current clinical evidence more closely may help to redirect research and drug development towards more diverse pathways. In this Perspective, we consider supporting evidence for and inconsistencies in the current conceptualization of the amyloid hypothesis, and present an alternative model. We primarily leverage evidence from clinical studies and use preclinical findings as secondary evidence.
The amyloid hypothesis
Supporting evidence
The amyloid hypothesis of AD originated from evidence in Down syndrome11 and autosomal dominant AD, in which the proteins encoded by genes whose mutations are causative of familial AD — that is, mutations in PSEN1 (encoding presenilin 1 (PSEN1)), PSEN2 (encoding presenilin 2 (PSEN2)), or APP (encoding amyloid precursor protein (APP) — are unambiguously involved in the metabolism of brain Aβ2. These pathogenic mutations increase the production of Aβ42 — the form of Aβ most associated with AD — from APP or alter the Aβ42/Aβ40 ratios, both of which are thought to cause the deposition of Aβ42 into cortical plaques. PSEN1, PSEN2 and APP mutations have nearly 100% penetrance, and cognitive impairment almost invariably develops in people who carry these mutations12. Conversely, a protective mutation in APP (A673T, the Icelandic mutation) can reduce the risk of AD by decreasing the production of Aβ13.
The estimated prevalence of autosomal dominant AD is less than 1%14,15, with the vast majority of AD cases being sporadic; that is, determined by the effects of many genes (of which the apolipoprotein E (APOE) gene (APOE) is the most important16), environmental exposures and unknown factors. Nevertheless, it has been assumed that the amyloid hypothesis, as formulated on the basis of evidence from autosomal dominant AD and Down syndrome, is also applicable to sporadic AD. In the following sections we report some of the supporting evidence for the amyloid hypothesis.
The neuropathologies of autosomal dominant and sporadic AD are similar.
Autosomal dominant AD and sporadic AD cases with comparable disease duration are similar in terms of their Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) scores and their diffuse plaque scores17. Even Lewy body pathology, the pathognomonic lesion in Parkinson disease and a common pathological co- morbidity in AD, is similar between autosomal dominant AD and sporadic AD (being present in 27% of autosomal dominant cases and 31% of sporadic cases)17, even though the metabolism of α-synuclein (the major component of Lewy bodies) is not affected by PSEN1, PSEN2 or APP.
Evidence of a temporal sequence of events.
A large body of evidence supports the notion of a chronological ordering of the major pathophysiological events, starting with deposition of Aβ in plaques and followed by aggregation of hyperphosphorylated tau into tangles, leading to neurodegenerative changes and, finally, cognitive impairment18.
AD-related tau pathology spreads extensively from the medial temporal lobe to neocortical areas in the presence of Aβ, suggesting a facilitating role for Aβ in the development of tau pathology19. In animal models, evidence has been found that overexpression of APP accelerates the development20,21 and propagation22 of AD pathology. Human induced pluripotent stem cells develop tau pathology following the introduction of APP and PSEN1 mutations found in familial AD, suggesting that Aβ can lead to tau pathology in this model23. Conversely, Aβ immunotherapy reduces tau load in one mouse model24.
Individuals with detectable amyloid pathology but no detectable tau pathology (A+T− individuals), according to established cut-offs, are relatively frequent in the population (that is, they constitute 25% of cognitively unimpaired individuals and 28% of people with mild cognitive impairment (MCI)), whereas A−T+ individuals are exceedingly rare, accounting for 1% of cognitively unimpaired people and 3% of individuals with MCI25. This supports the hypothesis that extensive neocortical tau spread starts when Aβ has already been deposited.
There is evidence that tau pathology leads to neurodegeneration. The intensity and topography of tau deposition as revealed by positron emission tomography predicts future atrophy26. Tau accumulation in neurons leads to neurofibrillary tangle formation, and such neurofibrillary tangle- bearing neurons die in the course of the disease as indicated by ghost tangles27. Recently, it was shown that tau aggregates drive granulovacuolar degeneration, an AD- related pathological lesion associated with neuronal loss and expressing markers of the activated necrosome28,29. Necrosome activation is indicative of necroptosis, a programmed form of necrosis30,31.
The effect of tau on cognition seems to be mediated by neurodegeneration. Several studies have shown associations between tau pathology and cognition. In cognitively unimpaired elderly people, AD-like memory decline is associated with Aβ and tau deposition, and is predicted by hypometabolism in AD-specific cortical regions32. When neurodegeneration is taken into account in a formal mediation analyses, the association between tau and cognition disappears for some cognitive functions (semantic memory and visuospatial functions)33. This suggests that the effect of tau deposition is largely mediated by neurodegeneration, and supports the causal chain of tau deposition leading to neurodegeneration, which in turn leads to cognitive impairment33.
Evidence of anti-Aβ and anti-tau drug effects.
In people with sporadic AD, aducanumab (an anti-Aβ monoclonal antibody) strongly reduced brain amyloid plaque load34. Two twin phase III clinical trials, EMERGE and ENGAGE, were designed to evaluate the efficacy and safety of aducanumab in patients with either MCI owing to AD or mild AD dementia35,36. Interestingly, a decrease in the level of phosphorylated tau (p-tau) in the cerebrospinal fluid (CSF) and temporal tau tracer retention on positron emission tomography was observed in patients taking aducanumab37. Decreases in CSF p-tau and total-tau levels were also observed in patients taking the anti-Aβ monoclonal antibody gantenerumab38 or the anti- tau vaccine AADvac1 (REF.39). In addition, gantenerumab attenuated increases in the levels of neurofilament light polypeptide (a marker of neurodegeneration) in the CSF38, and AADvac1 reduced CSF levels of neurofilament light polypeptide and brain atrophy39. Even though full datasets for the trials involving these drugs have not been published yet, these results suggest that it is possible to reduce tau pathology and, potentially, neurodegeneration by targeting amyloid pathology, supporting a causal relationship between Aβ deposition and tau deposition.
For the first time in the history of phase III AD clinical trials of disease-modifying drugs, the EMERGE trial showed that patents taking the drug under study — a high dose of aducanumab for 78 weeks — exhibited a reduction in clinical decline (that is, −22% on the Clinical Dementia Rating Scale Sum of Boxes (CDR-SB)) compared with those taking placebo37. Even though the magnitude of the effect was minor (-0.39 CDR-SB points at 78 weeks37), it was achieved in a clinically symptomatic population in which individuals had probably been exposed to Aβ for an extended period40,41. The reduction of amyloid load and slowing of disease progression associated with an Aβ-clearing treatment were confirmed in phase II trials of the anti-Aβ monoclonal antibodies lecanemab and donanemab42,43. Clinical trials with similar drugs are ongoing in asymptomatic individuals with AD pathology, but the results will not be available for a few years44,45.
A+T+ individuals show relatively stereotypical features of disease
Decades of research have demonstrated a relatively consistent transition from preclinical to clinical stages in individuals with Aβ and tau pathology. Compared with A−T− individuals, A+T+ individuals exhibit a higher prevalence of APOE ε4 (REFS46–48; the strongest genetic risk factor for AD), older age46–49, a more frequent concurrent clinical diagnosis of MCI or prodromal AD46, poorer global cognition46,47,49,50, a predominantly amnestic cognitive profile46,49,50, increased posterior temporoparietal atrophy51 and elevated risk of future cognitive decline46–49 and dementia48,52.
Evidence of amyloid toxicity in other forms of amyloidosis.
Aβ is not the only amyloidogenic peptide or protein that causes a disease. In transthyretin (TTR) amyloidosis, TTR accumulates in cerebral blood vessels, in a similar way to Aβ, and in other organs, owing to a mutation in TTR that destabilizes the physiological TTR tetramer, leading to misfolded monomers that aggregate into amyloid fibrils. Treatment with protein stabilizers has been successful in patients with TTR amyloidosis. The TTR amyloid shows aggregation properties similar to those of Aβ and other amyloidogenic proteins. Thus, TTR amyloidosis is the proof- of- principle that amyloid formation can cause a clinical disorder that can be successfully treated by inhibiting the formation of amyloid aggregates53. A similar rationale could be translated to AD- related Aβ-amyloidosis.
Evidence against
Although there is evidence supporting the amyloid hypothesis, there is equally compelling evidence that does not support it, at least in the way that it is currently articulated. The amyloid hypothesis predicts that tau deposition will not happen in the absence of amyloid, A−T+ individuals should be, at most, rare, tau should colocalize with amyloid, A+T+ individuals should invariably develop neurodegenerative changes and cognitive impairment, and anti-Aβ and anti-tau treatments should halt or greatly slow the progression of neurodegeneration and cognitive impairment. The following sections provide evidence against these predictions and, in consequence, falsify the current articulation of the amyloid hypothesis.
Reports of tau positivity in the absence of amyloid.
As previously mentioned, the observed prevalence of A−T+ individuals is low if the established stringent cut- offs for tau positivity are applied. However, with use of more liberal cut-offs, earlier regions of interest (for example, tau deposition limited to the transentorhinal region or Braak stage I/II) or more sensitive tau positron emission tomography tracers, the presenceof A−T+ has been found in up to 45% of individuals without dementia50,54,55 This condition is referred to as ‘primary age- related tauopathy’56 and is sometimes considered a variant of AD57, undermining the initiating role of Aβ in the cascade.
At odds with the findings of imaging studies, which have generally been taken to indicate that Aβ deposition precedes tau deposition, neuropathology studies have supported the opposite sequence. Large cross- sectional autopsy studies have shown that tau pathology is found at young ages (that is, 21–40 years), when Aβ plaques are absent, in the brainstem and locus coeruleus; this pathology then expands into further subcortical nuclei and the entorhinal cortex. In these studies, individuals exhibiting only Aβ plaques in the absence of neurofibrillary tangle pathology were in a minority compared with individuals with tau pathology lacking Aβ deposition58,59. This finding argues against a deterministic model in which Aβ is the cause of tau pathology. Rather, this finding is consistent with the notion that pre-existing age-related tauopathy is a prerequisite for Aβ to act as driver for the spread of tau to the neocortex59. This may also explain why even people with autosomal dominant AD, in which the overproduction and deposition of Aβ are substantial, do not develop symptoms before 30–60 years of age. Indeed, this is the age when a considerable proportion of individuals exhibit age-related tau deposition mainly in the transentorhinal and entorhinal cortex of the medial temporal lobe58.
In mutant APP transgenic mice that overexpress Aβ, severe Aβ plaque pathology is not accompanied by significant tau or neurofibrillary tangle pathology60–62. Indeed to promote accelerated tau pathology by Aβ in transgenic mice, a minimum of pre- existing tau pathology, as present in double Aβ and tau transgenic animals, is required20. Recently, a molecular link between Aβ and tau was identified, namely the cellular prion protein (PrPC). In animal and cellular models, binding of Aβ and p- tau to PrPC is associated with accelerated spreading of tau pathology, which leads to toxic effects22,63. In humans, both Aβ and p- tau bind to PrPC22. As PrP knockdown in APP transgenic mice leads to improved performance of the animals in cognitive tests64, the critical involvement of proteins other than just Aβ argues against the pathogenesis of AD being driven solely by Aβ.
The ‘spatial paradox’ of the spread of amyloid and tau pathology.
If tau pathology. causally follows Aβ deposition, at least some degree of colocalization of tau deposition within amyloid- rich regions might be expected. The observations that Aβ and tau initially deposit in different brain regions (that is, Aβ in the neocortex and tau in the brainstem and (trans)entorhinal cortex) and that the topographical spreading patterns of Aβ and tau over time overlap only minimally65,66 are inconsistent with the amyloid hypothesis19,58. No comprehensive and persuasive explanation has been offered so far for this discrepancy, but a role for APOE4 in the toxicity of both Aβ and p-tau has been suggested67.
The rarity of A+T+ Braak stage V/VI cases without co-morbid pathology.
AD pathology is often contributory and less commonly the sole cause of dementia in autopsy series of patients with dementia. A recent pathology study of 375 brains showed that in individuals with dementia, the prevalence of isolated amyloidosis associated with tauopathy sufficiently extensive to justify the dementia (Braak stage V/VI) is below 20%68. Even when any severity of tauopathy is allowed (Braak stage I or higher) in association with plaques, the combination of plaques and tangles without other pathology accounts for barely 30% of dementia cases68. About two-thirds of patients with dementia show co-morbid molecular pathology in addition to plaques and tangles, namely Lewy bodies of α-synuclein aggregates, insoluble aggregates of TAR DNA-binding protein 43 (TDP43) or both68. Vascular pathology is also frequently co-morbid with neuropathological changes in AD69. Moreover, in people with AD, APOE ε4 carriers have 2.5 times greater odds of having quadruple brain pathologies (plaques, tangles, Lewy bodies and TDP43 aggregates) than non-carriers68.
The lifetime risk of cognitive deterioration in A+T+ cases.
The amyloid hypothesis implies that cognitive deterioration is the invariable end point of Aβ accumulation if people live long enough to reach the final steps of the cascade. However, the cumulative incidence of dementia (corrected for age, sex, education and APOE genotype) in cognitively unimpaired A+T+ individuals aged 74 years is less than 20% after 5 years of follow-up and less than 50% after 14 years of follow-up49. This observation indicates that being A+T+ is not a strong predictor of cognitive decline. Of course, it could be argued that the process of the Aβ cascade is slow, and that if follow-up were more extensive, the lifetime risk would approach 100%. However, data are currently not available to confirm or reject this hypothesis.
The extent of the clinical effects of anti-Aβ drugs is uncertain.
If AD fitted a deterministic model, treatment at the preclinical stage would interrupt the disease process and prevent clinical manifestations, whereas treatment at the symptomatic stage would be expected to at least halt clinical deterioration. This would hold true especially in the presence of good target engagement; that is, the clear reduction in Aβ production and the significant reduction in plaque load that follows treatment with β-site APP-cleaving enzyme 1 (BACE1; also known as β-secretase 1) inhibitors and anti-Aβ immunotherapies34,70. Halting clinical deterioration is indeed the effect of imatinib in chronic myeloid leukaemia, a drug that works as a specific inhibitor of tyrosine kinases, targeting oncogenic signalling cascades. Treatment with imatinib is necessary and sufficient to treat chronic myeloid leukaemia at any disease stage71.
By contrast, the many trials with anti-Aβ drugs conducted so far in AD have almost invariably failed to even slow, never mind halt, cognitive deterioration. In the DIAN-TU trial in autosomal dominant AD72, the monoclonal antibodies gantenerumab and solanezumab reduced amyloid load, indicating target engagement38,73, but they had no effect on cognitive decline74.
In sporadic AD, the 22% reduction in clinical decline associated with aducanumab treatment that was observed in the EMERGE trial was a barely significant effect, despite the agent’s drastic effect on amyloid load, consisting of almost normalization of amyloid load37. Moreover, the top-line results of the ENGAGE trial did not show any effect on clinical decline even in patients taking high-dose aducanumab (that is, +2% of CDR-SB, corresponding to +0.03 CDR-SB points, as compared with placebo after 78 weeks)37. In a phase II trial with the anti-Aβ monoclonal antibody lecanemab, cognitive decline was slowed by as much as 47%, as assessed on the cognitive scale ADAS-Cog14, although this was a secondary outcome of the study75. The anti-Aβ monoclonal antibody donanemab hit its primary outcome in a phase II trial by delaying clinical progression by 32%, although results on secondary outcomes were mixed43.
These observations suggest that, even assuming the best-case scenario for lecanemab in terms of its beneficial effect on cognitive decline, more than 50% of the clinical progression is independent of Aβ deposition, and sets the stage for alternative explanations and understanding the role of non-anti-Aβ therapies.
Topographically atypical cases.
Approximately 25% of AD cases deviate from typical AD in terms of the localization of neurodegeneration and the associated cognitive profile76. Atypical clinical presentations include posterior cortical atrophy (the ‘visual’ variant), logopenic primary progressive aphasia (the ‘language’ variant), the behavioural/dysexecutive variant (or the ‘frontal’ variant) and the corticobasal variant. These atypical presentations might be due to genetic77,78, neurodevelopmental79,80 or unknown factors affecting the cascade before or after Aβ deposition, supporting the notion that the outcome of the amyloid cascade can be heavily modulated. Among the genetic factors, APOE stands out as a powerful modifier of the amount and topography of Aβ and tau deposition, as well as of clinical presentation, as discussed later. The topography of pathology is even more atypical in autosomal dominant AD, in which deposition of Aβ in the basal ganglia happens as early as 10 years before expected symptom onset81. In people with sporadic AD, Aβ deposition in the striatum follows Aβ deposition in the cortex by many years82.
Cascade the other way around: head injury.
An alternative to Aβ aggregation causing neuronal damage might be that neuronal damage is upstream of Aβ deposition. Results from animal models of AD show that axonal defects can precede Aβ deposition and promote the amyloidogenic process83. Animal experimental studies on head injury, in which axonal damage is the main lesion, show accumulation of APP, BACE1 and PSEN1 in damaged axons, followed by an increase in Aβ aggregation and plaque formation84. Furthermore, autopsy and biopsy studies in patients with severe traumatic brain injury show extensive cortical Aβ deposition in the subacute phase after trauma, including in individuals as young as 35 years85,86.
The probabilistic model of AD
The current amyloid hypothesis considers Aβ deposition to be the causative agent of AD pathophysiology, being necessary and sufficient to initiate the cascade, and predicts that clinical symptoms invariably develop as the final stage. Here we propose an alternative model in which Aβ is still a key factor in AD pathophysiology, but that stipulates that the penetrance of the amyloid cascade is directly proportional to the penetrance of genetic risk factors (FIG. 1). This probabilistic model identifies three variants of the disease in which stochastic factors play an increasingly relevant role: autosomal dominant AD, APOE ε4-related sporadic AD and APOE ε4-unrelated sporadic AD. The model allows stochastic factors to account for the variability among variants of molecular pathology, onset and progression of neurodegeneration, age at clinical onset, biomarker and clinical features, and lifetime risk (FIGS 1,2). It should be acknowledged that although the proposed variants feature specific characteristics, a certain degree of overlap exists in their pathological and clinical features. The following sections outline the major arguments in favour of the probabilistic model.
Fig. 1 |. The probabilistic model of Alzheimer disease.
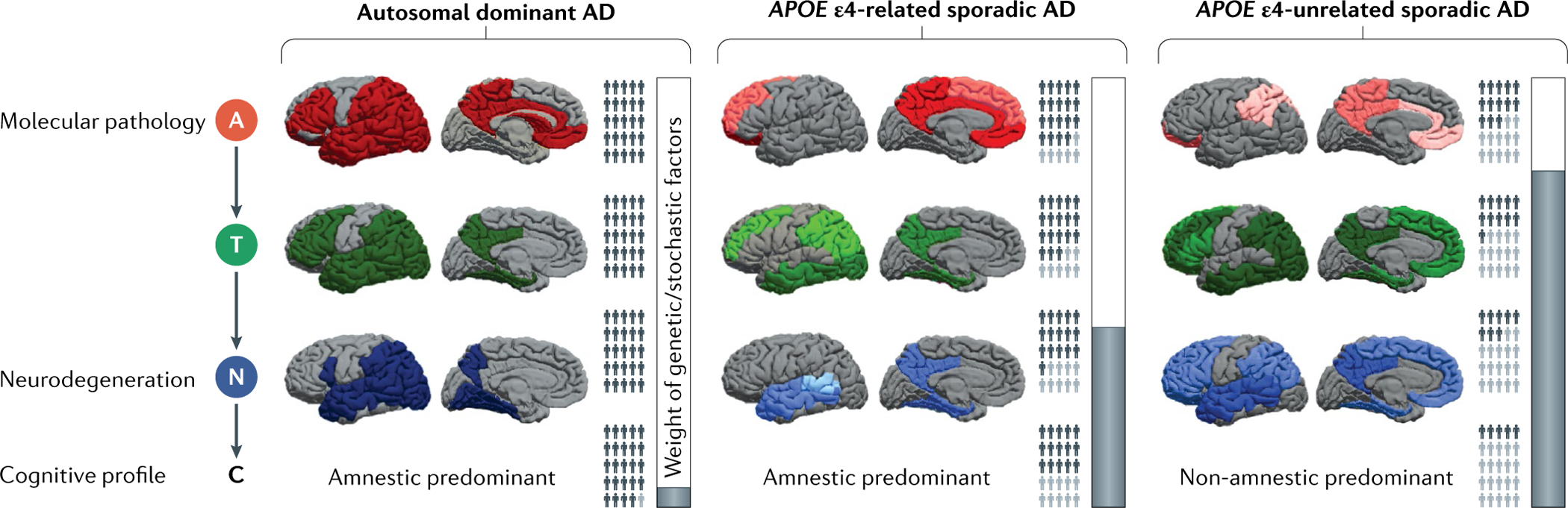
We propose three Alzheimer disease (AD) variants — autosomal dominant AD, APOE ε4-related sporadic AD and APOE ε4-unrelated sporadic AD — on the basis of genetic backgrounds and featuring differences in lifetime risk of dementia, the influence of stochastic factors, topography and the burden of amyloid pathology (A), tau pathology (T), neurodegeneration (N) and cognitive symptoms (C). A, T and N burdens in various brain regions are represented by the intensity of red, green and blue, respectively, with darker colours indicating greater burden. The topography of pathology and its global burden are reported in TABLE 1. The relative burden within and between variants for A is based on REFS113,117, for T on REFS124,126–128 and for N on REFS125–127,129. The proportions of dark and light grey people (affected and unaffected individuals, respectively) are approximate representations for the lifetime prevalence of A+, T+, N+ and cognitive impairment in the three AD variants. Autosomal dominant AD individuals almost invariably develop A, T, N and C. In sporadic AD, the interplay of APOE genotype with stochastic factors leads to a weaker cascade from A to T, N and C, resulting in fewer affected cases. The lifetime risk of dementia is very high (nearly 100%12) in autosomal dominant AD, intermediate (22–95%133,134) in APOE ε4-related AD and low (7–35%133,134) in APOE ε4-unrelated AD. The vertical bars graphically denote the weight of genetic and stochastic factors (white and grey, respectively).
Fig. 2 |. The lifetime dynamics of Aβ and tau in the three Alzheimer disease variants.
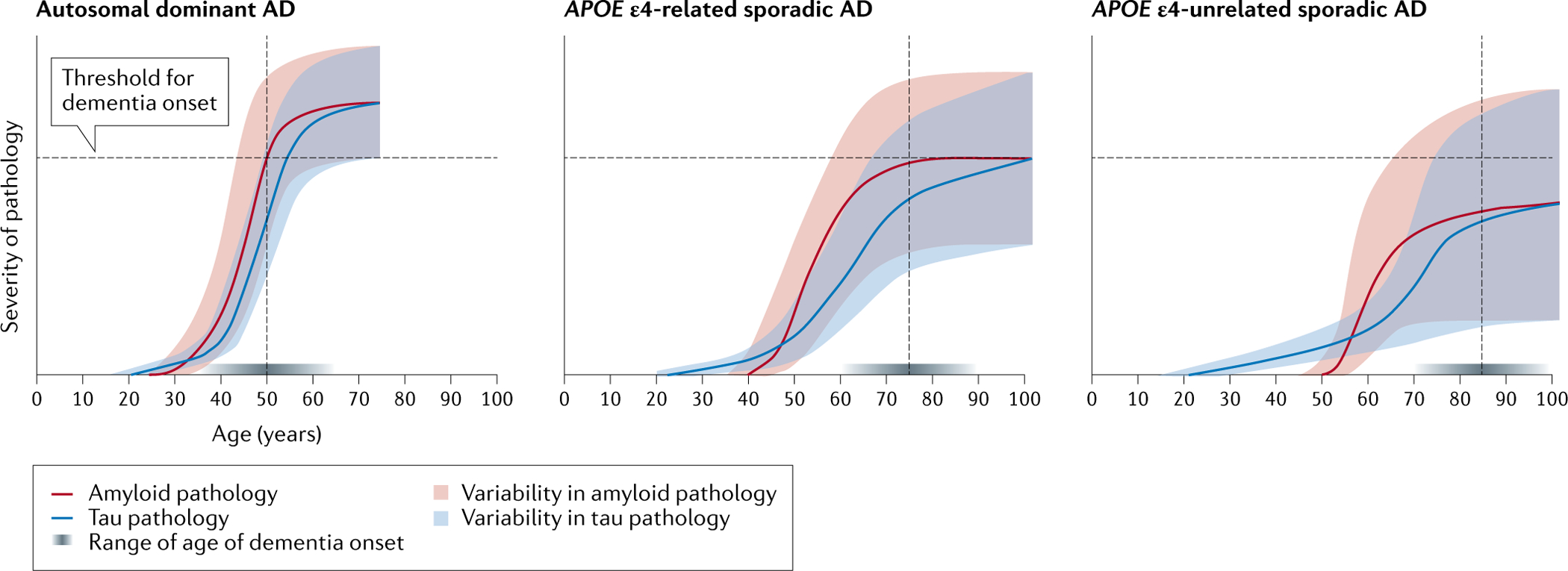
The curves represent the dynamics of Aβ (red) and tau (blue) deposition in the three Alzheimer disease (AD) variants. Age is shown on the x axis and the severity of the molecular pathology is shown on the y axis. The curves were generated by taking the dynamic biomarker model of Jack et al.18 as a template and warping it in accordance with evidence from the literature on the differences among autosomal dominant AD, APOE ε4-related sporadic AD and APOE ε4-unrelated sporadic AD regarding the following: the onset of Aβ and tau deposition (the intersections between the Aβ and tau curves with the x axis)58,59, the rates of change of patholog (curve slopes), the age of symptom onset (dotted vertical lines)87,106,107, the varoability of Aβ and tau pathology among individuals (shaded areas), the crossing of the clinical symptom threshold (dotted horizontal lines)12,133,134 and the range of age of dementia onset (horizontal shaded grey bar immediately above the x axis, where darker shades denote greater frequency density of the age of dementia onset)106–110. In the younger ages, the curve for tau lies above the Aβ curve to denote age-related tau deposition, which is thought to be necessary for Aβ to trigger further and extensive AD-related tau spread to neocortical areas58,59. The curves for Aβ and tau are steeper (denoting a more aggressive disease neurobiology) in autosomal dominant AD than in the APOE ε4-related and APOE ε4-unrelated sporadic variants. When the burdens of Aβ and tau pathologies reach a certain threshold, cognitive impairment becomes manifest. This occurs at around 50 years of age in autosomal dominant AD87, 75 years in APOE ε4-related sporadic AD106,107 and 85 years in APOE ε4-unrelated sporadic AD106,107. In all cases, in the final stages of the disease, the Aβ and tau curves reach a plateau. The intersection of the shaded areas with the threshold for dementia onset denotes the lifetime prevalence of dementia, with all individuals with autosomal dominant AD crossing the threshold at some point in their lifetime12, while a minority of non-APOE ε4 carriers133,134 and an intermediate proportion of APOE ε4 carriers133,134 do. Solid population-based data on the age of onset in APOE ε4 carriers and APOE ε4 non-carriers are lacking. The data in the literature were either estimated retrospectively on the basis of proxy reports in diagnostic cohorts not representative of the general population or estimated in well-conducted prospective population-based studies that had small sample sizes. Indeed, the two largest population-based studies (the Rotterdam Study and the Framingham Study) comprised only 134 and 43 cases, respectively106,107. However, it is widely accepted that carrying the APOE ε4 allele reduces the age of onset by about 12 years108–110. The curves for autosomal dominant AD are cut at around 60 years, assuming an average dementia duration of around 10 years88. The shaded areas denote the predicted variability of individual trajectories of Aβ and tau pathologies based on the probabilistic model that we are proposing. The variability of trajectories is inversely proportional to the penetrance of genetic factors and directly proportional to the impact of stochastic factors: smaller in autosomal dominant AD, intermediate in the APOE ε4-related variant and largest in the APOE ε4-unrelated variant.
Autosomal dominant AD
The deterministic view of the amyloid hypothesis fits reasonably well autosomal dominant AD, albeit imperfectly. Indeed, PSEN1, PSEN2 and APP mutations have nearly 100% penetrance as cognitive impairment develops almost invariably in individuals with these mutations12 (FIG. 1). The typical age of dementia onset is 35–55 years for PSEN1 mutations, 45–65 years for PSEN2 mutations and 45–60 years for APP mutations87; the duration of symptoms (before death) is around 10 years for all of these mutations88 (FIG. 2). Individuals with a PSEN1, PSEN2 or APP mutation feature the following characteristics: deposition of Aβ in the caudate nucleus81,89 and large parts of the frontal, parietal, temporal and occipital neocortex, sparing the superior frontal, medial temporal and medial occipital regions90; deposition of tau that largely overlaps with deposition of Aβ in the lateral frontal, temporal, parietal and occipital cortices but spares the mesial frontal and sensorimotor cortices and, rather, involves the medial temporal cortex91; neurodegeneration in the lateral temporal, parietal and occipital cortices, medial parietal and medial temporal cortices and medial occipitotemporal gyrus81,90,92; and amnesic-predominant cognitive impairment12,81 (FIG. 1; TABLE 1). Moreover, such individuals also feature variable TDP43 burden93,94 in the hippocampus and amygdala93, α-synuclein pathology94–96 in the amygdala96, cerebral amyloid angiopathy (CAA)97,98 with capillary involvement (CAA type 1) or without capillary involvement (CAA type 2)17, and neuroinflammation associated with tau and Aβ99 (TABLE 1).
Table 1 |.
Brain pathology associated with the three Alzheimer disease variants
| Pathology | Feature | Alzheimer disease variant | ||
|---|---|---|---|---|
| Autosomal dominant | APOE ε4 sporadic | Non-APOE ε4 sporadic | ||
| Aβ | Global burden | +++97,98 | ++117,118 | +117,118 |
| Topography | Precuneus; isthmus of cingulate gyrus, posterior cingulate and anterior cingulate gyri; rostral middle frontal gyrus, pars opercularis and pars triangularis of inferior frontal gyrus; orbital frontal gyrus; paracentral gyrus; postcentral gyrus; lateral temporal gyrus; lateral parietal gyrus; lateral occipital and pericalcarine gyri; and caudate nucleus81,89,90 | Precuneus; isthmus of cingulate gyrus, posterior cingulate and anterior cingulate gyri; medial and lateral orbitofrontal gyrus; rostral middle frontal and superior frontal gyri; and paracentral gyrus113,117 | Precuneus; isthmus of cingulate gyrus, posterior cingulate and anterior cingulate gyri; medial and lateral orbitofrontal gyri; paracentral gyrus; superior parietal lobe; and supramarginal gyrus113,117 | |
| Tau | Global burden | +++91 | ++126 | +++126 |
| Topography | Precuneus; isthmus of cingulate gyrus and posterior cingulate gyrus; middle and inferior frontal gyri; medial and lateral temporal gyri; lateral parietal lobe; lateral occipital lobe91 | Precuneus; isthmus of cingulate gyrus and posterior cingulate gyrus; middle frontal gyrus; medial, middle and inferior temporal gyri; lateral parietal lobe; lateral occipital lobe124,126–128 | Precuneus; isthmus of cingulate gyrus and posterior cingulate gyrus; superior, middle and inferior frontal gyri; medial, middle and inferior temporal gyri; lateral parietal lobe; lateral occipital lobe124,126–128 | |
| TDP43 | Global burden | Variable: −/+93,94 | ++130 | +130 |
| Topography | Hippocampus and amygdala93 | Amygdala, hippocampus, medial frontal gyrus130 | ||
| α-Synuclein | Global burden | Variable: −/+/++94–96 | ++131 | +131 |
| Topography | Amygdala96 | Not reported | Not reported | |
| Cerebral amyloid angiopathy | Global burden | +++97,98 | +++119 | +119 |
| Topography | Type 1 or type 2 (REF.17) | Type 1 (REFS120–123) | Type 2 (REFS120–123) | |
| Neuroinflammation | Topography | Colocalized with tau and Aβ pathology99 | Colocalized with tau and Aβ pathology99,167–170 | Weaker colocalization with tau and Aβ pathology99,167–170 |
| Neurodegeneration | Global burden | +++12 | +126 | ++126 |
| Topography | Precuneus; pars opercularis of inferior frontal gyrus; medial, superior and middle temporal gyri; lateral parietal lobe; lateral occipital lobe81,90,92 | Precuneus; isthmus of cingulate gyrus and posterior cingulate gyrus; medial, superior, and middle temporal gyri; temporoparietal junction125–127,129 | Precuneus; isthmus of cingulate gyrus and posterior cingulate gyrus; superior, middle and inferior frontal gyri; medial, superior and middle temporal gyri; temporoparietal junction; parietal lobe125–127,129 | |
The number of plus signs reflects the global burden of brain pathology based on studies comparing autosomal dominant Alzheimer disease (AD) with sporadic AD, and APOE ε4-related sporadic variants with APOE ε4-unrelated sporadic variants. Head-to-head comparisons among autosomal dominant AD, APOE ε4-related AD and APOE ε4-unrelated sporadic AD are not available. Global burden: +, low; ++, intermediate; +++, high. Regions listed under ‘topography’ are those mainly affected by pathology. Cerebral amyloid angiopathy type 1 is with capillary involvement, whereas type 2 is without capillary involvement. The topography of Aβ, tau and neurodegeneration and their burden are graphically represented in FIG. 1. Aβ, amyloid-β; TDP43, TAR DNA-binding protein 43.
Even among cases of autosomal dominant AD there is marked variability in the age of onset, neuropathology and clinical phenotype, suggesting that unknown stochastic factors are at play (FIG. 1). Indeed, in individuals with PSEN1 mutations, only 72% of the variance in dementia onset is explained by the mutations100, and around 16% of symptomatic PSEN1 mutation carriers have atypical presentations, including behavioural changes, language impairment, dyscalculia and dysexecutive syndrome100. Moreover, a recent description of a PSEN1-mutation carrier who did not develop cognitive impairment until she was in her 70s (nearly three decades after the typical age of onset) confirms the role of PSEN1-independent factors in influencing the course of disease even in autosomal dominant AD101. Imaging investigations in this patient showed a high Aβ burden but limited brain tau deposition and neurodegeneration as well as relatively preserved cognition. A protective effect has been proposed for the two copies of the APOE ε3 Christchurch (R136S) mutation that this patient was carrying101, but other genes are likely to be at play102. There is evidence that physical activity may delay symptom onset by 15 years even in autosomal dominant AD103.
Taken together, these observations indicate that despite the high penetrance, stochastic factors markedly affect the clinical phenotype in autosomal dominant AD (FIG. 1).
APOE ε4-related sporadic AD
The prevalence of the APOE ε4 allelic variant is 14% in cognitively unimpaired individuals and 38% in people with AD104, and increases to 64% in patients with amyloid- positive MCI and 66% in patients with AD dementia105. Different studies set the average age of dementia onset as 73–74 years in individuals with APOE ε4/ε4, 75–81 years in individuals with APOE ε3/ε4, and 76–82 years in individuals with APOE ε2/ε4 (REFS106,107) (FIG. 2). In general, it is widely accepted that carrying the APOE ε4 allele reduces the age of onset by about 12 years108–110. Although the APOE ε4 allele is strongly associated with a family history of dementia111, inheritance does not follow an autosomal dominant pattern, as is the case for APP, PSEN1 and PSEN2 mutations, in which the disease is transmitted from generation to generation with a probability of 50% in the offspring of mutation carriers. Since its identification, APOE has been regarded as a risk factor for sporadic forms of AD dementia112.
Several clinical and epidemiological observations suggest that APOE ε4 identifies a relatively distinct clinicopathological entity, as summarized next.
The burden and topography of pathology differs by APOE genotype.
In sporadic AD and irrespective of APOE genotype, Aβ deposition occurs first in the limbic cortex and extends posteriorly and dorsally into the precuneus and paracentral cortex and anteriorly and mesially to the orbitofrontal cortex113. Despite some inconsistent reports, possibly due to studies capturing different stages of the dynamic spread of Aβ deposition114,115, most evidence indicates that APOE ε4 carriers show Aβ deposition at an earlier age than APOE ε4 non- carriers116, and have a greater overall burden of Aβ pathology117,118 located mainly in the anterior and mesial frontal cortex117. They also feature more severe CAA119 with capillary involvement (CAA type 1)120–123. Conversely, APOE ε4 non-carriers feature preferential distribution of Aβ in lateral parietal regions117, and CAA lacking capillary involvement (CAA type 2)120–123 (FIG. 1; TABLE 1). Again irrespective of the APOE genotype, in people with AD, tau deposition is typically observed in large parts of the frontal, parietal, temporal and occipital neocortex, sparing the visual and sensorimotor regions124, with neurodegeneration largely overlapping with tau deposition125. When APOE ε4 status is taken into account, carriers feature a lower overall burden126 and less widespread126,127 tau pathology and neurodegeneration than non-carriers, preferentially affecting the medial temporal structures126–128. Conversely, APOE ε4 non-carriers show greater tau deposition and atrophy in frontal127,129 and parietal126,127 cortices (FIG. 1; TABLE 1). Finally, APOE ε4 is also associated with increased risk of TDP43 proteinopathy in elderly individuals130 and increased CSF α-synuclein levels131 (TABLE 1).
The profile of cognitive impairment differs by APOE genotype.
APOE ε4 carriers show a disproportionately more severe impairment in memory than APOE ε4 non-carriers, who are relatively more impaired in executive function, visuospatial abilities and language127 (FIG. 1; TABLE 1). In line with this, a recent study of patients with a pathologically confirmed diagnosis of AD found that individuals with an amnesic dementia phenotype were 2.5 times more likely to be APOE ε4 carriers than individuals with the primary progressive aphasia clinical phenotype132. These observations are consistent with the topography of tau pathology and neurodegeneration127.
The lifetime risk of dementia differs by APOE genotype
Independent studies have reported variable lifetime estimates of AD dementia risk, but the gap between APOE ε4 carriers and APOE ε4 non-carriers is consistently large (FIG. 1). According to Reiman and colleagues, the lifetime risk of developing AD dementia at 85 years is approximatively 95% for APOE ε4/ε4, 90% for APOE ε3/ε4, 35% for APOE ε2/ε3 and 20% for APOE ε2/ε2 (REF.133). According to Genin and colleagues, the lifetime risks associated with APOE genotypes are 51–68% for ε4/ε4, 22–35% for ε3/ε4, 7–12% for ε3/ε3 and 4–7% for ε2/ε2 and ε2/ε3 combined134.
Despite the high lifetime risk of the APOE ε3/ε4 and APOE ε4/ε4 genotypes, stochastic factors play a significant role. Indeed, although APOE ε4/ε4 carriers on average develop dementia about 10 years earlier than APOE ε2 carriers107, there is still significant variation in the age of onset for APOE ε4/ε4 carriers (standard deviation of 6 years)133, consistent with the notion of stochastic protective and risk factors (see later).
The largest genome-wide association study, by the European Alzheimer Disease DNA Biobank, identified 75 loci from almost as many genes implicated in AD135. Most of these variants are common, and although individually they have a relatively small effect on disease risk, the additive effect is relatively large. Numerous studies have found that these non-APOE AD genes may also affect the age of onset136–138. According to the law of Mendelian segregation in populations, these genes will be transmitted independently from APOE ε4 in the population. This leads to a large number of combinations of genetic variants that co-occur with APOE ε4 in the population, leading to stochastic variation in the age of onset in APOE ε4 carriers. Moreover, some people with the APOE ε4/ε4 genotype may even escape AD dementia. For example, the Dutch 100-plus Study reported a centenarian who stayed cognitively healthy despite having an APOE ε4/ε4 genotype139. Similarly to the interaction of PSEN1 with the APOE ε3 Christchurch mutation, the effect of APOE ε4/ε4 homozygosity may be modulated by both non-APOE genetic variants associated with AD and environmental exposures (see later). A similar phenomenon is known in the field of vascular diseases, in which individuals featuring supernormal vascular ageing (SUPERNOVA) have been described who do not show age-dependent increased arterial stiffness in spite of a heavy cardiovascular risk factor burden140,141. Epigenetic mechanisms have been invoked to explain the SUPERNOVA phenomenon142.
Amyloid-associated risk for dementia differs by APOE genotype.
Amyloid positivity and APOE ε4 carriage individually increase the risk of progression to MCI or dementia in a similar way (hazard ratio of 2.0 and 2.1, respectively, after 8 years)143. However, their interaction shows that A+ APOE ε4 carriers have the greatest risk of progression (hazard ratio 4.5), while the risk for amyloid-positive APOE ε4 non-carriers (hazard ratio 1.1) is similar to that for amyloid-negative APOE ε4 non-carriers143. This observation suggests the presence of a benign or a malign brain amyloidosis, according to its association with the APOE ε4 allele.
The influence of APOE ε4 on dementia risk declines after the age of 70 years.
The risk of dementia associated with the APOE ε4 allele is maximal between 55 and 70 years, and decreases afterwards144,145. This is consistent with the age of symptom onset of APOE ε4 carriers, and with the notion that the pathogenetic impact of non-AD co-morbidity increases at older ages146, thus diluting the effect of APOE ε4.
Potential mechanisms
A number of neurobiological mechanisms have been proposed that might account for the specific effect of the APOE ε4 variant on the neurodegenerative process of AD. First, it has been proposed that APOE plays a major role in Aβ clearance. APOE binds Aβ147 and transports it across the blood–brain barrier into the blood, facilitating its perivascular clearance148,149. Differences in the efficiency of this process among isoforms, the APOE4 isoform being the least efficient149, may explain Aβ accumulation in the brains of APOE ε4 carriers over time and the consequent greater risk of AD pathology. Genetic studies of Aβ levels in blood showed that APOE genotype is the most significant determinant of Aβ42 and the Aβ42/Aβ40 ratio, while mutations in BACE, APP and PSEN2 are significantly associated with higher Aβ40 levels150. Of note, an association with Aβ levels in blood is found for other non-APOE genes150.
Second, it has been proposed that APOE ε4 drives multiple AD-associated proteinopathies. In addition to its role as a regulator of Aβ deposition14, recent studies indicate that the APOE ε4 genotype is associated with the deposition of tau67,151, α-synuclein152,153 and TDP43 (REF.154), independently of Aβ deposition (FIG. 1; TABLE 1). When hybridized with tau transgenic mice, APOE ε4 mice develop more tangles than APOE ε3 mice67,151. Similarly, when crossed with A53T α-synuclein transgenic mice, APOE ε4 mice develop more α-synuclein pathology152. In both the tau model67 and the α-synuclein model153, neurodegeneration was accelerated by APOE ε4. In humans, APOE ε4 is associated with increased TDP43 burden, and with higher odds of hippocampal sclerosis and late-life cognitive impairment independent of Aβ pathology154. These data indicate that multiple proteinopathies might be an integral component of APOE-related neurodegeneration, and that, differently from PSEN1, PSEN2 and APP mutations, APOE ε4 has effects on pathologies other than AD.
Third, APOE ε4 may drive vascular deficits and blood–brain barrier dysfunction. APOE ε4 carriers exhibit an increase in blood–brain barrier breakdown in the hippocampus and medial temporal lobe, independent of tau and cortical Aβ burden155, and APOE4 impairs pericyte function155,156. APOE ε4 carriers have more severe CAA in the subiculum and entorhinal cortex and more frequent hippocampal microinfarcts than APOE ε4 non-carriers157. It is unknown to what extent these vascular changes contribute to cognitive impairment in APOE ε4 carriers.
Fourth, APOE ε4 is proposed to have adverse effects on brain structure and metabolism and cognition across the lifespan. There is consistent evidence that APOE ε4 adversely affects brain structure in infants158, and brain metabolism159 and cognition160 in midlife, well before the onset of AD- type pathology. A similar impact on brain metabolism has also been observed in younger adults161.
Last, APOE may have a role in the innate immune response in AD. APOE ε4 carriers show higher neuroinflammatory levels than non-carriers14,162,163. This observation might explain the proinflammatory state often reported in some patients with AD164–166, and the direct association of neuroinflammation with tau and Aβ pathology in APOE ε4 carriers99,167–170.
As is the case for APOE ε4, rare and relatively highly penetrant AD risk genes such as TREM2 could identify other AD variants with specific clinical and biological characteristics, but the low prevalence of carriers in clinical series has so far prevented any meaningful clinicopathological characterization or classification of AD subtypes driven by these genetic variants.
APOE ε4-unrelated sporadic AD
About 30–40% of individuals with amyloid-positive AD do not carry APOE ε4 (REF.105), and their average age of dementia onset ranges between 80 and 82 years in APOE ε3/ε3 cases to between 82 and 85 years in APOE ε2/ε2 and APOE ε2/ε3 cases106,107 (FIG. 2). In these patients, the amyloid cascade as described earlier herein does not differ significantly from that of autosomal dominant AD or APOE ε4-related sporadic AD, but a number of known and unknown modulating factors heavily influence the chain of events at all steps of the cascade (FIG. 1), making the disease process and clinical manifestations less predictable. Indeed, various lines of evidence from epidemiology, genetics, neuropsychology and biomarker studies support this view.
Non-APOE genes account for relatively few AD cases.
The genetic landscape of AD includes more than 60 genes other than APOE171. Twin studies have allowed estimation of the proportion of AD cases attributable to genetic factors as about 60–80%, while about 20–40% can be attributed to environmental factors172,173. APOE ε4 accounts for the largest share of the genetically attributable proportion, while non-APOE ε4 genes account for a minor share174. Indeed, a polygenic risk score, which takes into account a combination of risk loci, can discriminate individuals with AD and controls with an area under the curve of 75–84% (the remaining part being environmental risk factors or undiscovered genetics such as de novo mutations and/or rare variants), while APOE alone is able to discriminate them with an area under the curve of ~70%174.
It must be acknowledged, however, that knowledge of the effect of non-APOE genetic variants has increased just recently as a result of large multicohort studies. Indeed, a recent study of 13,959 patients with AD and 35,600 controls found that the dementia risk of individuals in the top decile of a full polygenic risk score (including APOE) is 57%, while the risk associated with APOE ε4 alone is 44%175. Interestingly, when APOE is removed from the polygenic risk score, the disease risk of individuals in the top decile drops to 36%175, a still substantial risk. Future studies of even larger cohorts may lead to a larger share of the attributable proportion of AD dementias being assigned to non-APOE genetic variants.
Genes other than APOE modulate APOE ε4-related risk.
Population-based studies have found that non-APOE genetic variants interact with APOE, for example, modifying the risk136–138 and age of onset of dementia136,137. In the Rotterdam Study and International Genomics of Alzheimer’s Project cohorts, non-APOE genetic variants account for 7–10 years of the variability of the age of onset in APOE ε4 homozygotes136. This suggests that non-APOE gene pathways may biologically interact with the APOE pathway.
Non-Aβ pathways are involved in AD pathophysiology.
The non-APOE genes described so far encompass multiple biochemical pathways, some of which are associated with neuroinflammation and cholesterol metabolism (for example, TREM2)176,177, ABCA7 (REF.178), PLCG2 (REF.179) and CLU180. Other described non-APOE genes, such as CR1 and CD33, are associated specifically with the innate immune system. Together, these data suggest that all the pathophysiological pathways mentioned earlier herein may, under certain conditions, contribute substantially to AD risk181.
A formal pathway analysis was conducted in a genome-wide association study of the International Genomics of Alzheimer’s Project for the common and rare variants separately (frequency 0.01 or greater and less than 0.01, respectively)182. Four function clusters were seen for the common variants, including APP metabolism and Aβ formation; tau protein binding; lipid metabolism (four pathways including protein–lipid complex assembly); and immune response. In line with the interaction of non-APOE genes with APOE, enrichment of the four clusters remained after removal of genes in the APOE region. Interestingly, when genes in the neighbourhood of APOE and other highly significant non-APOE genes were removed, tau protein binding, lipid metabolism and immune-related pathways remained significantly associated, suggesting that non-APOE genes are involved in these pathways182.
The more recent European Alzheimer’s Disease DNA BioBank consortium found very similar pathways (Aβ and hyperphosphorylated tau deposition, lipid metabolism and innate immunity, including macrophage and microglial cell activation)135. Its pathway analysis showed that genes in the Aβ pathways with the highest microglial expression show the strongest association with AD, suggesting a functional relationship between microglia and Aβ pathways135.
Protective genes.
While APOE ε4 confers an elevated risk of AD, APOE ε2 has a protective effect133,174. Indeed, when compared with the APOE ε3/ε3 genotype, APOE ε2/ε2 and APOE ε2/ε3 have significantly lower AD odds ratios (0.13 and 0.39, respectively)133. Such odds ratios are much smaller when the reference is APOE ε4/ε4 (0.004 for APOE ε2/ε2 and 0.012 for APOE ε2/ε3)133, consistent with high risk and high protection effects rather than an extreme effect of either of the genotypes. Other protective mutations and genes have been reported — for example, a mutation in APP (encoding A673T)13, PLCG2 (REFS183,184), BDNF185, KL (which encodes klotho)186, TMEM106B187 and POT1 (REF.188) — but the magnitude of the protection in each case has not yet been accurately estimated.
Lifestyles and vascular risk factors.
The Lancet Commission on Dementia Prevention, Intervention, and Care estimated that 40% of all cases of dementia are due to 12 modifiable risk factors. Of these, five are known to be general vascular disease risk factors (that is, hypertension and obesity in midlife, and smoking, physical inactivity and diabetes in later life) and seven are more specific to dementia (that is, lower education level in early life; hearing loss, traumatic brain injury and alcohol abuse in midlife; and depression, social isolation and air pollution in later life)173. As the APOE ε4 allele is the major genetic risk factor for sporadic AD, the influence of modifiable risk factors might be greater in the APOE ε4-unrelated variant than in the APOE ε4-related one, and this difference can be attributed to other modifiable or unknown genetic risk factors. Consistently, results from the population- based Rotterdam Study showed that favourable modifiable-risk profiles were associated with a lower risk of dementia only in APOE ε4 non-carriers, while no effect of modification was observed in APOE ε4 carriers189. Nevertheless, contrasting observational and intervention results have also been reported, indicating that lifestyle changes may be associated with decreased dementia risk also among people with a high baseline genetic risk190 and identifying better cognitive outcomes of a multidomain intervention (diet, exercise, cognitive training and vascular risk monitoring) in APOE ε4 carriers191.
Microbiota
Preliminary evidence suggests a role for the gut microbiota in AD pathogenesis192–194. Differences in the abundance of proinflammatory and antiinflammatory taxa have been described in amyloid- positive and amyloid- negative patients with cognitive impairment165, and they might be involved in the central and peripheral inflammatory state often reported in AD. It is not known whether there is an association between specific bacterial taxa and APOE genotype.
Resistance and resilience
Different combinations of the risk and protective factors described earlier herein can co-occur in the same individual, summing up to that person’s ultimate risk of developing neurodegeneration and dementia. The complex interplay of risk and protective factors has been conceptualized into the notions of resistance, brain resilience and cognitive resilience. ‘Resistance’ refers to the brain processes underlying the ability to prevent pathology195, despite the presence of risk factors. ‘Brain resilience’ refers to the neurobiological processes underlying the ability to better cope with pathology195. ‘Cognitive resilience’ refers to the functional process underlying the individual’s ability to sustain a better-than-expected cognitive performance in relation to the degree of pathology195. Brain resilience and cognitive resilience can modulate the effect of molecular pathology on neurodegeneration, and can delay the onset of symptoms. Resistance and resilience are at play in all variants of AD, and resilience appears to be largely independent of APOE genotype and clinical AD196, but their weight on the development of neurodegeneration and symptoms might be particularly relevant in the non-APOE ε4 sporadic AD variant. Unfortunately, resistance and resilience are theoretical constructs that have so far eluded direct observation. Future studies will need to operationalize them and test their effects in APOE ε4 and non-APOE ε4 AD variants.
Demographics.
Other variables play a role in the pathophysiology and clinical expression of AD. Indeed, increased age is one of the strongest risk factors for AD, and modulates the association between the APOE ε4 genotype and AD dementia, with the magnitude of the risk associated with APOE ε4 following an inverted U-shaped curve with a peak at 55–70 years of age144,145 How age modulates APOE-associated risk is far from fully understood. Stochastic theories hypothesize that biological ageing occurs randomly and persistently with time, through random error, free radicals and wear and tear197. Others suggest an effect of age-related decline of the immune system198 that is interwoven with telomere shortening, epigenetic alterations and insulin growth factor signalling. The effect of age comprises the joint effects of genetic and environmental factors discussed earlier herein, in line with studies in animal models that have shown that stochastic factors as well as genetic factors significantly contribute to ageing of nematodes199. Given that the late onset of AD dictates that patients have not died early in life, antagonistic pleiotropy has been suggested, implying that certain genes whose functions are beneficial during the reproductive age may exert adverse effects at a later age200.
Other demographic variables, such as sex and ethnicity, influence the effect of APOE ε4. Indeed, women carrying the APOE ε4 allele are at greater risk of developing AD than men carrying the APOE ε4 allele201,202, and they show a greater longitudinal reduction of hippocampal volume in the preclinical stage, denoting a stronger effect of APOE ε4 in women203. Moreover, APOE ε4 confers a greater risk of AD dementia in Japanese and white individuals than in African American and Hispanic individuals144. Consistently, African American APOE ε4 carriers show lower levels of CSF p-tau and total tau than white APOE ε4 individuals204, suggesting a differential effect of APOE ε4 as a function of ethnicity, which might be partially explained by environmental factors and non-APOE genetic variability.
Impact
The probabilistic amyloid hypothesis of AD has implications in the clinic and for research. Notably, APOE should be considered a major effect modifier in research and drug development. In all clinical and basic research studies, APOE should be considered a stratifying variable, not a mere covariate. When resistance to pathology and resilience to cognitive impairment are being investigated, APOE ε4-unrelated sporadic AD is the type for which effects are expected be most robust.
In drug development, APOE should be given more consideration as a drug target in AD104. According to the 2019 AD drug development pipeline, only one drug was targeting APOE-related mechanisms among the 132 under study in humans44, and only two more APOE-targeting drugs were mentioned in a more recent review205. The vast majority of drugs are still targeting Aβ, tau or other disease mechanisms. Research into drugs that target APOE and APOE-related mechanisms should be greatly expanded, and initiatives aiming to repurpose drugs with a potentially APOE- mediated mechanism should be encouraged. A recent report encouragingly showed that APOE immunotherapy reduces amyloid-related pathology while improving cerebrovascular function in mice206. As APOE ε4-unrelated sporadic AD pathophysiology is driven largely by non-APOE factors, and analogous to the treatment of risk factors for vascular diseases, therapeutic interventions in APOE ε4 non-carriers should prioritize combined preventive interventions (drugs acting on multiple molecular targets, multiple lifestyle interventions, or combined drug and lifestyle interventions). The major hurdles are the paucity of data on the specificity of response to treatment by APOE genotype and the need for combining drugs with individually proven efficacy on cognitive outcomes — which are currently unavailable.
The prevention of AD dementia should rely on reducing the risk rather than treating the disease. The amyloid hypothesis as a deterministic chain of events has understandably led to the unescapable conclusion that the clinical manifestations of AD (MCI and dementia) are but the last stage of a disease that starts much earlier (15–20 years) with Aβ deposition. The notion of preclinical diagnosis, in analogy to many malignant tumours, has been evoked, and criticized207–209. The probabilistic amyloid hypothesis does not necessarily imply disease starting before clinical manifestations. It views Aβ deposition and tau deposition as risk factors whence clinical manifestations do not necessarily follow, and ‘disease’ should be reserved for the clinical manifestations, in analogy to vascular diseases such as stroke and myocardial infarction. The clinical challenge is thus not accurate and early preclinical diagnosis, but accurate risk profiling. This will inform risk reduction interventions tailored on individual risk profiles210.
Research should estimate the risk associated with molecular and lifestyle risk factors by APOE ε4 carrier status. Accurate estimates of risk factors will allow stratification into strata of high, intermediate and low risk, and the devising of targeted interventions. Combined pharmacological (for example, anti-Aβ and anti-tau agents) and lifestyle interventions (for example, nutrition and physical activity) can be envisioned in specific patient populations to reduce both risk factors. Currently available risk estimates come from studies that have accurately investigated either modifiable lifestyle risk factors or molecular pathology, but seldom both, thus preventing accurate estimates of communality210. Future studies will need to estimate the risk of incident cognitive impairment and dementia in representative population samples with accurate assessment of both. Protocols for genetic counselling of people who carry one or two copies of the APOE ε4 allele and their relatives will need to be developed.
The molecular taxonomy of AD should stratify for APOE. The probabilistic amyloid hypothesis stresses the strong modulatory effect of APOE on amyloid-associated and tau-associated risk. The model implies that people should be classified as APOE ε4 carriers or non-carriers first, and then profiled according to the ATN framework. APOE ε4 carriers will be at greater risk than non-carriers at any ATN stage.
It may be argued that rare and relatively highly penetrant AD risk genes such as TREM2, PLCG2 or ABI3 could identify other relatively homogeneous high- risk groups with specific clinical and biological characteristics179. However, the low prevalence of carriers of the risk alleles in clinical series has so far prevented any meaningful clinicopathological characterization or classification of AD subtypes driven by these genetic variants136.
AD research should focus on pathways of resilience to AD pathology. Acknowledging the relevance of stochastic factors in AD opens a window of opportunity to modulate mechanisms that might slow the progression of the cascade from pathology to neurodegeneration and from neurodegeneration to cognitive impairment. Interventions that target vesicular trafficking211, neuroinflammation212, cell differentiation196, blood–brain barrier integrity155 and the microbiota194 are just a few of the potential strategies.
Developers of disease modifiers should prioritize people with no cognitive impairment. In the clinical trial space, the probabilistic model of AD means treating people with AD pathology when they are still in the preclinical phase. As of 2021, the overwhelming majority of new drugs were still being tested in people with cognitive impairment, with a few exceptions of trials in people without cognitive impairment at risk of AD dementia owing to AD pathology or genetic risk factors (GENERATION trial with the anti-Aβ vaccine CAD106; DIAN-TU-001 with the monoclonal antibodies gantenerumab and solanezumab; rrAD with amlodipine, losartan and atorvastatin; NCT02008357 with solanezumab; and NCT02719327 with omega-3 fatty acid icosapent ethyl)213. Individuals with cognitive impairment should be involved in trials only of symptomatic drugs, as supported by the recent success of pimavanserin in people with AD psychosis214.
Conclusions
Although AD is a multifactorial and heterogeneous disease215–217, much of the current drug development is driven by a deterministic model of the disease that concentrates on a single pathway. Progress in drug development is more likely to happen if a less rigid framework for AD pathophysiology is adopted, in which AD is driven by genetic factors of decreasing penetrance (autosomal dominant AD, APOE ε4-related sporadic AD and APOE ε4-unrelated sporadic AD) and stochastic factors whose weight is inversely related to penetrance. We acknowledge that a probabilistic model may gradually convert into a deterministic one when more knowledge is accrued, and predictions become increasingly accurate. However, the adoption of a deterministic model when knowledge is insufficient can lead to overly simplistic approaches. Adoption of a probabilistic model when knowledge is insufficient for a deterministic one is a more complex, but more informative and conceivably more successful, approach. The adoption of the probabilistic amyloid hypothesis will have implications for drug development, clinical and basic research, and clinical taxonomy. Future research embracing this model might make sense of the many conflicting findings that are currently slowing progression towards the effective prevention and treatment of AD and other neurodegenerative diseases.
Acknowledgements
This Perspective was the result of a workshop funded by the Swiss National Science Foundation entitled “How many roads lead to Rome? Insights in Alzheimer disease pathophysiology to lead future drug development” (grant number IZSEZ0_192840). G.B.F. received funding from the following sources: European Prevention of Alzheimer’s Dementia - EPAD (grant agreement number 115736) and Amyloid Imaging to Prevent Alzheimer’s Disease - AMYPED (grant agreement number 115952) funded by the EU–EFPIA Innovative Medicines Initiatives 2 Joint Undertaking; the Swiss National Science Foundation (“Brain connectivity and metacognition in persons with subjective cognitive decline (COSCODE): correlation with clinical features and in vivo neuropathology” (grant number 320030_182772)); Association Suisse pour la Recherche sur la Maladie d’Alzheimer, Geneva; Fondation Segré, Geneva; I. Pictet, Geneva; Fondazione Agusta, Lugano; Fondation Chmielewski, Geneva; and the VELUX Foundation. D.R.T. received funding from Fonds Wetenschappelijk Onderzoek Vlaanderen (FWO-G0F8516N Odysseus). R.v.d.K. was supported by an Alzheimer Nederland pilot grant (WE.03–2017-08) and a grant from the Selfridges Group Foundation (NR170059). K.B. is supported by the Swedish Research Council (2017–00915), the Swedish Alzheimer Foundation (AF-742881), Hjärnfonden, Sweden (FO2017–0243), and the Swedish state under an agreement between the Swedish government and the county councils, the ALF agreement (ALFGBG-715986). J.C. is supported by Keep Memory Alive, NIGMS grant P20GM109025, NINDS grant U01NS093334 and NIA grant R01AG053798.
Competing interests
G.B.F. has received grants from Avid Radiopharmaceuticals, Biogen, GE International, Guerbert, IXICO, Merz Pharma, Nestlé, Novartis, Eisai, Piramal, Roche, Siemens, Teva Pharmaceutical Industries and Vifor Pharma. He has received personal fees from AstraZeneca, Avid Radiopharmaceuticals, Biogen, Roche, Diadem, Neurodiem, Elan Pharmaceuticals, GE International, Lundbeck, Pfizer and TauRx Therapeutics. D.R.T. has received speaker honoraria from Novartis Pharma Basel (Switzerland) and Biogen (USA), has received travel reimbursement from GE Healthcare (UK), and UCB (Belgium) and has collaborated with GE Healthcare (UK), Novartis Pharma Basel (Switzerland), Probiodrug (Germany) and Janssen Pharmaceuticals (Belgium). K.B. has served as a consultant, on advisory boards or on data monitoring committees for Abcam, Axon, Biogen, Shimadzu, Julius Clinical, Lilly, MagQu, Novartis, Roche Diagnostics and Siemens Healthineers, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB, which is part of the GU Ventures incubator programme. J.C. has acted as a consultant for Acadia, Actinogen, Alkahest, Alzheon, Annovis, Avanir, Axsome, Biogen, Cassava, Cerecin, Cerevel, Cortexyme, Cytox, EIP Pharma, Eisai, Foresight, GemVax, Genentech, Green Valley, Grifols, Karuna, Merck, Novo Nordisk, Otsuka, Resverlogix, Roche, Samumed, Samus, Signant Health, Suven and United Neuroscience. J.C. also has stock options in ADAMAS, AnnovisBio, MedAvante and BiOasis, and owns the copyright of the Neuropsychiatric Inventory. P.S. has received consultancy fees (paid to Amsterdam UMC) from AC Immune, Brainstorm Cell, EIP, ImmunoBrain Checkpoint, Genentech, Novartis, and Novo Noridisk. He is a principal investigator on studies with AC Immune, FUJIFILM Toyama, UCB, and Vivoryon. He is a part-time employee of Life Sciences Partners Amsterdam. B.D. has received research funding (paid to the institution) from Merck-Avenir Foundation and Roche and consultancy fees from Biogen, Neurodiem, Green Valley, Cytox and Brainstorm. He is a principal investigator on clinical trials with Eisai, Genentech, Novartis, Biogen and Roche. D.A., F.R., R.v.d.K., R.O., C.v.D., P.M.N. and P.-Y.D. declare no competing interests.
Glossary
- Alzheimer disease
(AD). The co-occurrence of brain Aβ and tau pathology. AD dementia is the final stage of AD, in which cognitive impairment and loss of daily function are also present.
- Amyloid
In the brain, a 37–49-amino-acid polypeptide (amyloid-β (Aβ)) produced by the metabolism of the synaptic membrane protein amyloid precursor protein (APP). The amyloid fibrillar form is made mainly of the 42-amino-acid variant (Aβ42) and is the primary component of amyloid plaques found in the brains of individuals with Alzheimer disease. Soluble Aβ42 can be found in plasma and the cerebrospinal fluid and can give rise to soluble oligomers, thought to be the toxic form of Aβ.
- Braak stage
Braak stage denotes the degree of tau pathology in Alzheimer disease and assumes progressive spread of such pathology from the transentorhinal region of the brain. Braak stages I and II denote neurofibrillary tangle involvement confined mainly to the transentorhinal region, stages III and IV when there is also involvement of limbic regions such as the hippocampus, and stages V and VI when there is extensive neocortical involvement.
- Mild cognitive impairment
(MCI). A syndrome featuring cognitive impairment and no loss of daily function; Alzheimer disease is the underlying pathology in 60–80% of MCI cases. In these cases, the condition is also called prodromal Alzheimer disease or MCI due to Alzheimer disease.
- Neurodegeneration
Progressive loss of the structure or function of neurons, which may ultimately involve cell death. The earliest detectable event is thought to be synaptic loss, followed by neuronal loss. Neurodegeneration can be detected in vivo with volumetric MRI and positron emission tomography with 18F-labelled deoxyglucose.
- Tau
A protein whose primary role is in maintaining the stability of microtubules in axons. In the course of Alzheimer disease, tau becomes hyperphosphorylated, leading to axonal and synaptic dysfunction and aggregation of tau into intracellular neurofibrillary tangles.
Footnotes
Peer review information
Nature Reviews Neuroscience thanks G. Bu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
References
- 1.Prince M et al. World Alzheimer Report 2015. The Global Impact of Dementia - An Analysis of Prevalence, Incidence, Cost and Trends. https://www.alzint.org/u/WorldAlzheimerReport2015.pdf (2015).
- 2.Ballard C et al. Alzheimer’s disease. Lancet 377, 1019–1031 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Jack CR et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement 14, 535–562 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hardy JA & Higgins GA Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185 (1992). [DOI] [PubMed] [Google Scholar]
- 5.Selkoe DJ & Hardy J The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sorel I N, Cayssials É, Brizard F & Chomel JC Treatment and molecular monitoring update in chronic myeloid leukemia management. Ann. Biol. Clin. 75, 129–145 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Trojanowski JQ Tauists, baptists, syners, apostates, and new data. Ann. Neurol. 52, 263–265 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Makin S The amyloid hypothesis on trial. Nature 559, S4–S7 (2018). [DOI] [PubMed] [Google Scholar]
- 9.De Strooper B & Karran E The cellular phase of Alzheimer’s disease. Cell 164, 603–615 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Herrup K The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 18, 794–799 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Glenner GG & Wong CW Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 122, 1131–1135 (1984). [DOI] [PubMed] [Google Scholar]
- 12.Bateman RJ et al. Autosomal-dominant Alzheimer’s disease: a review and proposal for the prevention of Alzheimer’s disease. Alzheimer’s Res. Ther. 3, 1 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jonsson T et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 488, 96 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Liu CC, Kanekiyo T, Xu H & Bu G Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Cauwenberghe C, Van Broeckhoven C & Sleegers K The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet. Med. 18, 421–430 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sleegers K & Van Duijn CM Alzheimer’s disease: genes, pathogenesis and risk prediction. Community Genet. 4, 197–203 (2001). [DOI] [PubMed] [Google Scholar]
- 17.Ringman J et al. Neuropathology of autosomal dominant Alzheimer disease in the National Alzheimer Coordinating Center database. J. Neuropathol. Exp. Neurol. 75, 284–290 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jack CR et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 9, 119–128 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van der Kant R, Goldstein LSB &Ossenkoppele R Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat. Rev. Neurosci. 21,21–35 (2020). [DOI] [PubMed] [Google Scholar]
- 20.Götz J, Chen F, Van Dorpe J & Nitsch RM Formation of neurofibrillary tangles in P301L tau transgenic mice induced by Aβ42 fibrils. Science 293, 1491–1495(2001). [DOI] [PubMed] [Google Scholar]
- 21.Lewis J et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293, 1487–1491 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Gomes LA et al. Aβ-induced acceleration of Alzheimer-related τ-pathology spreading and its association with prion protein. Acta Neuropathol. 138,913–941 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Choi SH et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 51 5, 274–278 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oddo S, Billings L, Kesslak JP, Cribbs DH & LaFerla FM Aβ immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 43, 321–332 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Jack CR et al. The bivariate distribution of amyloid-β and tau: relationship with established neurocognitive clinical syndromes. Brain 142, 3230–3242 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.La Joie R et al. Prospective longitudinal atrophy in Alzheimer’s disease correlates with the intensity and topography of baseline tau-PET. Sci. Transl. Med. 12, eaau5732 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bancher C et al. Accumulation of abnormally phosphorylated τ precedes the formation of neurofibrillary tangles in Alzheimer’s disease. Brain Res. 477, 90–99 (1989). [DOI] [PubMed] [Google Scholar]
- 28.Koper MJ et al. Necrosome complex detected in granulovacuolar degeneration is associated with neuronal loss in Alzheimer’s disease. Acta Neuropathol. 1 39, 463–484 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Wiersma VI et al. Granulovacuolar degeneration bodies are neuron-selective lysosomal structures induced by intracellular tau pathology. Acta Neuropathol. 1 38, 943–970 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Galluzzi L et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 25, 486–541 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang D et al. The molecular machinery of regulated cell death. Cell Res. 29, 347–364 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanseeuw BJ et al. Fluorodeoxyglucose metabolism associated with tau-amyloid interaction predicts memory decline. Ann. Neurol. 81, 583–596 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bejanin A et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain 140, 3286–3300 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sevigny J et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537, 50–56 (2016). [DOI] [PubMed] [Google Scholar]
- 35.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02477800 (2021). [DOI] [PubMed]
- 36.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02484547 (2021). [DOI] [PubMed]
- 37.Haeberlein SB et al. EMERGE and ENGAGE topline results: two phase 3 studies to evaluate aducanumab in patients with early Alzheimer’s disease. https://investors.biogen.com/static-files/ddd45672-9c7e-4c99-8a06-3b557697c06f (2019). [Google Scholar]
- 38.ALZFORUM. Gantenerumab https://www.alzforum.org/therapeutics/gantenerumab (2021).
- 39.ALZFORUM. AADvac1 https://www.alzforum.org/therapeutics/aadvac1 (2021).
- 40.Villemagne VL et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 12, 357–367 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Jack CR et al. Brain β-amyloid load approaches a plateau. Neurology 80, 890–896 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swanson CJ et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res. Ther. 13, 80 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mintun MA et al. Donanemab in early Alzheimer’s disease. N. Engl. J. Med. 384, 1691–1704 (2021). [DOI] [PubMed] [Google Scholar]
- 44.Cummings J, Lee G, Ritter A, Sabbagh M & Zhong K Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement. Transl. Res. Clin. Interv. 5, 272–293 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.National Institute on Aging. NIA- funded active Alzheimer’s and related dementias clinical trials and studies. https://www.nia.nih.gov/research/ongoing-AD-trials#section2 (2021).
- 46.Altomare D et al. Applying the ATN scheme in a memory clinic population: the ABIDE project. Neurology 93, E1635–E1646 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Soldan A et al. ATN profiles among cognitively normal individuals and longitudinal cognitive outcomes. Neurology 92, E1567–E1579 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ebenau JL et al. ATN classification and clinical progression in subjective cognitive decline. Neurology 10.1212/wnl.0000000000009724 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vos SJB et al. Preclinical Alzheimer’s disease and its outcome: a longitudinal cohort study. Lancet Neurol. 12, 957–965 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weigand A et al. Is tau in the absence of amyloid on the Alzheimer’s continuum?: a study of discordant PET positivity. Brain Commun. 2, fcz046 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rabinovici GD et al. Distinct MRI atrophy patterns in autopsy- proven Alzheimer’s disease and frontotemporal lobar degeneration. Am. J. Alzheimers. Dis. Other Demen. 22, 474–488 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ekman U, Ferreira D & Westman E The A/T/N biomarker scheme and patterns of brain atrophy assessed in mild cognitive impairment. Sci. Rep. 8, 8431 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adams D, Koike H, Slama M & Coelho T Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat. Rev. Neurol. 15, 387–404 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Pascoal TA et al. 18F-MK-6240 PET for early and late detection of neurofibrillary tangles. Brain 10.1093/brain/awaa180 (2020). [DOI] [PubMed] [Google Scholar]
- 55.Leuzy A et al. Diagnostic performance of RO948 F 18 tau positron emission tomography in the differentiation of Alzheimer disease from other neurodegenerative disorders. JAMA Neurol. 77, 955–965 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Crary JF et al. Primary age- related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 128, 755–766 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Duyckaerts C et al. PART is part of Alzheimer disease. Acta Neuropathol. 129, 749–756 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Braak H, Thal DR, Ghebremedhin E & Del Tredici K Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 70, 960–969 (2011). [DOI] [PubMed] [Google Scholar]
- 59.Spires- Jones TL, Attems J & Thal DR Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol. 134, 187–205 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sturchler-Pierrat C et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl Acad. Sci. USA 94, 13287–13292 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Games D et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 373, 523–527 (1995). [DOI] [PubMed] [Google Scholar]
- 62.Hsiao K et al. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 274, 99–102 (1996). [DOI] [PubMed] [Google Scholar]
- 63.Corbett GT et al. PrP is a central player in toxicity mediated by soluble aggregates of neurodegeneration-causing proteins. Acta Neuropathol. 139, 503–526 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Salazar SV et al. Conditional deletion of Prnp rescues behavioral and synaptic deficits after disease onset in transgenic Alzheimer’s disease. J. Neurosci. 37, 9207–9221 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Braak H & Braak E Neuropathological stageing of Alzheimer-related changes. Acta Neuropathologica 82, 239–259 (1991). [DOI] [PubMed] [Google Scholar]
- 66.Thal DR, Rüb U, Orantes M & Braak H Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 58, 1791–1800 (2002). [DOI] [PubMed] [Google Scholar]
- 67.Shi Y et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Karanth S et al. Prevalence and clinical phenotype of quadruple misfolded proteins in older adults. JAMA Neurol. 10.1001/jamaneurol.2020.1741 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schneider JA, Arvanitakis Z, Bang W & Bennett DA Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 69, 2197–2204 (2007). [DOI] [PubMed] [Google Scholar]
- 70.Imbimbo BP & Watling M Investigational BACE inhibitors for the treatment of Alzheimer’s disease. Expert Opin. Investig. Drugs 28, 967–975 (2019). [DOI] [PubMed] [Google Scholar]
- 71.Hochhaus A et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 34, 966–984 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01760005 (2021). [DOI] [PubMed]
- 73.ALZFORUM. Solanezumab https://www.alzforum.org/therapeutics/solanezumab (2021).
- 74.Alzheimer’s Association. DIAN- TU phase 3 clinical trials, topline results–news https://www.alz.org/news/2020/dian-tu-phase-3-clinical-trials-topline-results (2020).
- 75.Eisai Co. Ltd. Eisai and Biogen announce presentation of additional data from the phase II clinical trial of BAN2401 in early Alzheimer’s disease and the 2018 Clinical Trials on Alzheimer’s Disease (CTAD) Conference https://www.eisai.com/news/2018/news201892.html (2018).
- 76.Murray ME et al. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 10, 785–796 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schott JM et al. Genetic risk factors for the posterior cortical atrophy variant of Alzheimer’s disease. Alzheimers Dement. 12, 862–871 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Carrasquillo MM et al. Late-onset Alzheimer disease genetic variants in posterior cortical atrophy and posterior AD. Neurology 82, 1455–1462 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Miller ZA et al. Prevalence of mathematical and visuospatial learning disabilities in patients with posterior cortical atrophy. JAMA Neurol. 75, 728–737 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Miller ZA et al. Handedness and language learning disability differentially distribute in progressive aphasia variants. Brain 136, 3461–3473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bateman RJ et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 367, 795–804 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hanseeuw BJ et al. PET staging of amyloidosis using striatum. Alzheimers Dement. 14, 1281–1292 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stokin GB et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s diseases. Science 307, 1282–1288 (2005). [DOI] [PubMed] [Google Scholar]
- 84.Johnson VE, Stewart W & Smith DH Traumatic brain injury and amyloid-β pathology: a link to alzheimer’s disease? Nat. Rev. Neurosci. 11, 361–370 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ikonomovic MD et al. Alzheimer’s pathology in human temporal cortex surgically excised after severe brain injury. Exp. Neurol. 190, 192–203 (2004). [DOI] [PubMed] [Google Scholar]
- 86.Roberts GW, Gentleman SM, Lynch A & Graham DI ßA4 amyloid protein deposition in brain after head trauma. Lancet 338, 1422–1423 (1991). [DOI] [PubMed] [Google Scholar]
- 87.Rossor MN, Fox NC, Mummery CJ, Schott JM & Warren JD The diagnosis of young- onset dementia. Lancet Neurol. 9, 793–806 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ryman DC et al. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta- analysis. Neurology 83, 253–260 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sanchez JS et al. Longitudinal amyloid and tau accumulation in autosomal dominant Alzheimer’s disease: findings from the Colombia- Boston (COLBOS) biomarker study. Alzheimers Res. Ther. 13, 27 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gordon BA et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: a longitudinal study. Lancet Neurol. 17, 241–250 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gordon BA et al. Tau PET in autosomal dominant Alzheimer’s disease: relationship with cognition, dementia and other biomarkers. Brain 142, 1063–1076 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cash DM et al. The pattern of atrophy in familial Alzheimer disease: volumetric MRI results from the DIAN study. Neurology 81, 1425–1433 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lloyd GM et al. Prominent amyloid plaque pathology and cerebral amyloid angiopathy in APP V717I (London) carrier - phenotypic variability in autosomal dominant Alzheimer’s disease. Acta Neuropathol. Commun. 8, 31 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sutovsky S et al. Neuropathology and biochemistry of early onset familial Alzheimer’s disease caused by 1 presenilin-1 missense mutation Thr 11 6Asn. J.Neural Transm. 125, 965–976 (2018). [DOI] [PubMed] [Google Scholar]
- 95.Gondim DD et al. Diffuse Lewy body disease and Alzheimer disease: neuropathologic phenotype associated with the PSEN1 p.A396T mutation. J. Neuropathol. Exp. Neurol. 78, 585–594 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lippa CF et al. Lewy bodies contain altered α- synuclein in brains of many familial Alzheimer’s disease patients with mutations in presenilin and amyloid precursor protein genes. Am. J. Pathol. 153, 1365–1370 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mann D et al. Predominant deposition of amyloid-beta 42(43) in plaques in cases of Alzheimer’s disease and hereditary cerebral hemorrhage associated with mutations in the amyloid precursor protein gene. Am. J. Pathol. 148, 1257–1266 (1996). [PMC free article] [PubMed] [Google Scholar]
- 98.Mann DMA et al. Amyloid β protein (Aβ) deposition in chromosome 14-linked Alzheimer’s disease: predominance of Aβ(42(43)). Ann. Neurol. 40, 149–156 (1996). [DOI] [PubMed] [Google Scholar]
- 99.Taipa R et al. Inflammatory pathology markers (activated microglia and reactive astrocytes) in early and late onset Alzheimer disease: a post mortem study. Neuropathol. Appl. Neurobiol. 44, 298–313 (2018). [DOI] [PubMed] [Google Scholar]
- 100.Ryan NS et al. Clinical phenotype and genetic associations in autosomal dominant familial Alzheimer’s disease: a case series. Lancet Neurol. 15, 1326–1335 (2016). [DOI] [PubMed] [Google Scholar]
- 101.Arboleda- Velasquez JF et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nat. Med. 25, 1680–1683 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yu CE, Chen S, Jayadev S & Bird T Lack of APOE Christchurch variant in five age of onset outliers with PSEN1, PSEN2 Alzheimer’s disease and MAPT frontotemporal dementia. J. Neurol. Sci. 418, 117143 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Müller S et al. Relationship between physical activity, cognition, and Alzheimer pathology in autosomal dominant Alzheimer’s disease. Alzheimers Dement. 14, 1427–1437 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yamazaki Y, Zhao N, Caulfield TR, Liu CC & Bu G Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat. Rev. Neurol. 15, 501–518 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mattsson N et al. Prevalence of the apolipoprotein E ε4 allele in amyloid β positive subjects across the spectrum of Alzheimer’s disease. Alzheimers Dement. 14, 913–924 (2018). [DOI] [PubMed] [Google Scholar]
- 106.Myers RH et al. Apolipoprotein E ε4 association with dementia in a population- based study: The Framingham Study. Neurology 46, 673–677 (1996). [DOI] [PubMed] [Google Scholar]
- 107.Slooter AJC et al. Risk estimates of dementia by apolipoprotein E genotypes from a population-based incidence study: The Rotterdam Study. Arch. Neurol. 55, 964–968 (1998). [DOI] [PubMed] [Google Scholar]
- 108.Belloy ME, Napolioni V & Greicius MD A quarter century of APOE and Alzheimer’s disease: progress to date and the path forward. Neuron 101, 820–838 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Corder EH et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923 (1993). [DOI] [PubMed] [Google Scholar]
- 110.Roses AD Apolipoprotein E alleles as risk factors in Alzheimer’s disease. Annu. Rev. Med. 47, 387–400 (1996). [DOI] [PubMed] [Google Scholar]
- 111.Jansen IE et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 51, 404–413 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Van Duijn CM et al. Apolipoprotein E4 allele in a population–based study of early–onset Alzheimer’s disease. Nat. Genet. 7, 74–78 (1994). [DOI] [PubMed] [Google Scholar]
- 113.Collij LE et al. Multitracer model for staging cortical amyloid deposition using PET imaging. Neurology 95, e1538–e1553 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ossenkoppele R et al. Differential effect of APOE genotype on amyloid load and glucose metabolism in AD dementia. Neurology 80, 359–365 (2013). [DOI] [PubMed] [Google Scholar]
- 115.Lehmann M et al. Greater medial temporal hypometabolism and lower cortical amyloid burden in ApoE4-positive AD patients. J. Neurol. Neurosurg. Psychiatry 85, 266–273 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Burnham SC et al. Impact of APOE-ε4 carriage on the onset and rates of neocortical Aβ-amyloid deposition. Neurobiol. Aging 95, 46–55 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Toledo JB et al. APOE effect on amyloid-β PET spatial distribution, deposition rate, and cut-points. J. Alzheimers Dis. 69, 783–793 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Schmechel DE et al. Increased amyloid β-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc. Natl Acad. Sci. USA 90, 9649–9653 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Olichney JM et al. Relationship between severe amyloid angiopathy, apolipoprotein E genotype, and vascular lesions in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 903, 138–143 (2000). [DOI] [PubMed] [Google Scholar]
- 120.Thal DR et al. Two types of sporadic cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 61, 282–293 (2002). [DOI] [PubMed] [Google Scholar]
- 121.Thal DR, Griffin WST, de Vos RAI & Ghebremedhin E Cerebral amyloid angiopathy and its relationship to Alzheimer’s disease. Acta Neuropathologica 115, 599–609 (2008). [DOI] [PubMed] [Google Scholar]
- 122.Thal DR et al. Capillary cerebral amyloid angiopathy identifies a distinct APOE ε4-associated subtype of sporadic Alzheimer’s disease. Acta Neuropathol. 120, 169–183 (2010). [DOI] [PubMed] [Google Scholar]
- 123.Greenberg SM et al. Cerebral amyloid angiopathy and Alzheimer disease — one peptide, two pathways. Nat. Rev. Neurol. 16, 30–42 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ossenkoppele R et al. Discriminative accuracy of [18F] flortaucipir positron emission tomography for Alzheimer disease vs other neurodegenerative disorders. JAMA 320, 1151–1162 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Frisoni GB et al. The topography of grey matter involvement in early and late onset Alzheimer’s disease. Brain 130, 720–730 (2007). [DOI] [PubMed] [Google Scholar]
- 126.Mattsson N et al. Greater tau load and reduced cortical thickness in APOE ε4-negative Alzheimer’s disease: a cohort study. Alzheimers Res. Ther. 10, 77 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Emrani S, Arain HA, DeMarshall C & Nuriel T APOE4 is associated with cognitive and pathological heterogeneity in patients with Alzheimer’s disease: a systematic review. Alzheimers Res. Ther. 12, 141 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Therriault J et al. Association of apolipoprotein E ε4 with medial temporal tau independent of amyloid-β. JAMA Neurol. 77, 470–479 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Geroldi C et al. APOE-ε4 is associated with less frontal and more medial temporal lobe atrophy in AD. Neurology 53, 1825–1832 (1999). [DOI] [PubMed] [Google Scholar]
- 130.Nelson PT et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 142, 1503–1527 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Twohig D et al. The relevance of cerebrospinal fluid a- synuclein levels to sporadic and familial Alzheimer’s disease. Acta Neuropathol. Commun. 6, 130 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Weintraub S et al. APOE is a correlate of phenotypic heterogeneity in Alzheimer disease in a national cohort. Neurology 94, e607–e612 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Reiman EM et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat. Commun. 11, 667 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Genin E et al. APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol. Psychiatry 16, 903–907 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bellenguez C et al. New insights on the genetic etiology of Alzheimer’s and related dementia. medRxiv 17, 10 (2020). [Google Scholar]
- 136.van der Lee SJ et al. The effect of APOE and other common genetic variants on the onset of Alzheimer’s disease and dementia: a community-based cohort study. Lancet Neurol. 17, 434–444 (2018). [DOI] [PubMed] [Google Scholar]
- 137.Desikan RS et al. Genetic assessment of age-associated Alzheimer disease risk: development and validation of a polygenic hazard score. PLoS Med. 14, e1002258 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chouraki V et al. Evaluation of a genetic risk score to improve risk prediction for Alzheimer’s disease. J. Alzheimers Dis. 53, 921–932 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Holstege H et al. The 100-plus Study of cognitively healthy centenarians: rationale, design and cohort description. Eur. J. Epidemiol. 33, 1229–1249 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Laurent S, Boutouyrie P, Cunha PG, Lacolley P & Nilsson PM Concept of extremes in vascular aging: from early vascular aging to supernormal vascular aging. Hypertension 74, 218–228 (2019). [DOI] [PubMed] [Google Scholar]
- 141.Bruno RM et al. Early and supernormal vascular aging. Hypertension 76, 1616–1624 (2020). [DOI] [PubMed] [Google Scholar]
- 142.Ding Y-N, Tang X, Chen H-Z & Liu D-P Epigenetic regulation of vascular aging and age- related vascular diseases. Adv. Exp. Med. Biol. 1086, 55–75 (2018). [DOI] [PubMed] [Google Scholar]
- 143.Dang C et al. Relationship between amyloid-β positivity and progression to mild cognitive impairment or dementia over 8 years in cognitively normal older adults. J. Alzheimers Dis. 65, 1313–1325 (2018). [DOI] [PubMed] [Google Scholar]
- 144.Farrer L et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. JAMA 278, 1349–1356 (1997). [PubMed] [Google Scholar]
- 145.Saddiki H et al. Age and the association between apolipoprotein E genotype and Alzheimer disease: a cerebrospinal fluid biomarker–based case–control study. PLoS Med. 17, e1003289 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Nelis SM et al. The impact of co-morbidity on the quality of life of people with dementia: findings from the IDEAL study. Age Ageing 48, 361–367 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Strittmatter WJ et al. Apolipoprotein E: High-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. U. S. A. 90, 1977–1981 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Thal DR et al. Occurrence and co-localization of amyloid β-protein and apolipoprotein E in perivascular drainage channels of wild-type and APP-transgenic mice. Neurobiol. Aging 28, 1221–1230 (2007). [DOI] [PubMed] [Google Scholar]
- 149.Deane R et al. apoE isoform-specific disruption of amyloid β peptide clearance from mouse brain. J. Clin. Invest. 118, 4002–4013 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Damotte V et al. Plasma amyloid β levels are driven by genetic variants near APOE, BACE1, APP, PSEN2: a genome-wide association study in over 12,000 non-demented participants. Alzheimers Dement. 10.1002/alz.12333 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wang C et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector article. Nat. Med. 24, 647–657 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Davis AA et al. APOE genotype regulates pathology and disease progression in synucleinopathy. Sci. Transl. Med. 12, eaay3069 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zhao N et al. APOE4 exacerbates α-synuclein pathology and related toxicity independent of amyloid. Sci. Transl. Med. 12, eaay1809 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Yang HS et al. Evaluation of TDP-43 proteinopathy and hippocampal sclerosis in relation to APOE ε4 haplotype status: a community-based cohort study. Lancet Neurol. 17, 773–781 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Montagne A et al. APOE4 leads to blood–brain barrier dysfunction predicting cognitive decline. Nature 581, 1–6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Blanchard JW et al. Reconstruction of the human blood–brain barrier in vitro reveals a pathogenic mechanism of APOE4 in pericytes. Nat. Med. 26, 1–12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hecht M, Krämer LM, von Arnim CAF, Otto M & Thal DR Capillary cerebral amyloid angiopathy in Alzheimer’s disease: association with allocortical/ hippocampal microinfarcts and cognitive decline. Acta Neuropathol. 135, 681–694 (2018). [DOI] [PubMed] [Google Scholar]
- 158.Dean DC et al. Brain differences in infants at differential genetic risk for late-onset Alzheimer disease: a cross-sectional imaging study. JAMA Neurol. 71, 11–22 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Reiman EM et al. Preclinical evidence of Alzheimer’s disease in persons homozygous for the ε4 allele for apolipoprotein E. N. Engl. J. Med. 334, 752–758 (1996). [DOI] [PubMed] [Google Scholar]
- 160.Evans SL et al. Mid age APOE ε4 carriers show memory-related functional differences and disrupted structure-function relationships in hippocampal regions. Sci. Rep. 10, 3110 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Reiman EM et al. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc. Natl Acad. Sci. USA 101, 284–289 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Reale,, M. et al. Relationship between inflammatory mediators, Aβ levels and ApoE genotype in Alzheimer disease. Curr. Alzheimer Res. 9, 447–457 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gorelick PB Role of inflammation in cognitive impairment: results of observational epidemiological studies and clinical trials. Ann. N. Y. Acad. Sci. 1207, 155–162 (2010). [DOI] [PubMed] [Google Scholar]
- 164.Morgan AR et al. Inflammatory biomarkers in Alzheimer’s disease plasma. Alzheimers Dement. 15, 776–787 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Cattaneo A et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 49, 60–68 (2017). [DOI] [PubMed] [Google Scholar]
- 166.Heneka MT et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Friedberg JS et al. Associations between brain inflammatory profiles and human neuropathology are altered based on apolipoprotein E ε4 genotype. Sci. Rep. 10, 29624 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sheng J, Mrak R & Griffin W Glial- neuronal interactions in Alzheimer disease: progressive association of IL-1alpha+ microglia and S100beta+ astrocytes with neurofibrillary tangle stages. J. Neuropathol. Exp. Neurol. 56, 285–290 (1997). [PubMed] [Google Scholar]
- 169.Griffin WST, Sheng JG, Roberts GW & Mrak RE Interleukin-1 expression in different plaque types in Alzheimer’s disease significance in plaque evolution. J. Neuropathol. Exp. Neurol. 54, 276–281 (1995). [DOI] [PubMed] [Google Scholar]
- 170.Akiyama H et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 21, 383–421 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Scheltens P et al. Alzheimer’s disease. Lancet 10.1016/S0140-6736(20)32205-4 (2021). [DOI] [Google Scholar]
- 172.Gatz M et al. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 63, 168–174 (2006). [DOI] [PubMed] [Google Scholar]
- 173.Livingston G et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 396, 413–446 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sims R, Hill M & Williams J The multiplex model of the genetics of Alzheimer’s disease. Nat. Neurosci. 23, 311–322 (2020). [DOI] [PubMed] [Google Scholar]
- 175.Zhang Q et al. Risk prediction of late- onset Alzheimer’s disease implies an oligogenic architecture. Nat. Commun. 11, 4799 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Andreone BJ et al. Alzheimer’s-associated PLCγ2 is a signaling node required for both TREM2 function and the inflammatory response in human microglia. Nat. Neurosci. 23, 927–938 (2020). [DOI] [PubMed] [Google Scholar]
- 177.Nugent AA et al. TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron 105, 837–854.e9 (2020). [DOI] [PubMed] [Google Scholar]
- 178.De Roeck A, Van Broeckhoven C & Sleegers K The role of ABCA7 in Alzheimer’s disease: evidence from genomics, transcriptomics and methylomics. Acta Neuropathol. 138, 201–220 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sims R et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet. 49, 1373–1384 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wollmer MA Cholesterol- related genes in Alzheimer’s disease. Biochim. Biophys. Acta 1801, 762–773 (2010). [DOI] [PubMed] [Google Scholar]
- 181.Cuyvers E & Sleegers K Genetic variations underlying Alzheimer’s disease: Evidence from genome- wide association studies and beyond. Lancet Neurol. 15, 857–868 (2016). [DOI] [PubMed] [Google Scholar]
- 182.Kunkle BW et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.van der Lee SJ et al. A nonsynonymous mutation in PLCG2 reduces the risk of Alzheimer’s disease, dementia with Lewy bodies and frontotemporal dementia, and increases the likelihood of longevity. Acta Neuropathol. 138, 237–250 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Magno L et al. Alzheimer’s disease phospholipase C-gamma-2 (PLCG2) protective variant is a functional hypermorph. Alzheimers Res. Ther. 11, 16 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Jiao SS et al. Brain- derived neurotrophic factor protects against tau- related neurodegeneration of Alzheimer’s disease. Transl. Psychiatry 6, e907 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Belloy ME, Napolioni V, Han SS, Le Guen Y & Greicius MD Association of Klotho-VS heterozygosity with risk of Alzheimer disease in individuals who carry APOE4. JAMA Neurol. 77, 849–862 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Satoh J-I et al. TMEM106B expression is reduced in Alzheimer’s disease brains. Alzheimers. Res. Ther. 6, 17 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Hohman TJ, Koran MEI & Thornton-Wells TA Genetic modification of the relationship between phosphorylated tau and neurodegeneration. Alzheimers Dement. 10, 637–645.e1 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Licher S et al. Genetic predisposition, modifiable- risk-factor profile and long-term dementia risk in the general population. Nat. Med. 25, 1364–1369 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lourida I et al. Association of lifestyle and genetic risk with incidence of dementia. JAMA 322, 430–437 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Solomon A et al. Effect of the apolipoprotein E genotype on cognitive change during a multidomain lifestyle intervention a subgroup analysis of a randomized clinical trial. JAMA Neurol. 75, 462–470 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Bostanciklioglu M The role of gut microbiota in pathogenesis of Alzheimer’s disease. J. Appl. Microbiol. 127, 954–967 (2019). [DOI] [PubMed] [Google Scholar]
- 193.Marizzoni M, Provasi S, Cattaneo A & Frisoni GB Microbiota and neurodegenerative diseases. Curr. Opin. Neurol. 30, 630–638 (2017). [DOI] [PubMed] [Google Scholar]
- 194.Cryan JF, O’Riordan KJ, Sandhu K, Peterson V & Dinan TG The gut microbiome in neurological disorders. Lancet Neurol. 19, 179–194 (2020). [DOI] [PubMed] [Google Scholar]
- 195.Arenaza- Urquijo EM & Vemuri P Resistance vs resilience to Alzheimer disease. Neurology 90, 695–703 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Dumitrescu L et al. Genetic variants and functional pathways associated with resilience to Alzheimer’s disease. Brain 143, 2561–2575 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Gladyshev VN Aging: progressive decline in fitness due to the rising deleteriome adjusted by genetic, environmental, and stochastic processes. Aging Cell 15, 594–602 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Partridge L, Deelen J & Slagboom PE Facing up to the global challenges of ageing. Nature 561, 45–56 (2018). [DOI] [PubMed] [Google Scholar]
- 199.Herndon LA et al. Stochastic and genetic factors influence tissue- specific decline in ageing C. Elegans. Nature 419, 808–814 (2002). [DOI] [PubMed] [Google Scholar]
- 200.Williams GC Pleiotropy, natural selection, and the evolution of senescence. Evolution 11, 398–411 (1957). [Google Scholar]
- 201.Altmann A, Tian L, Henderson VW & Greicius MD Sex modifies the APOE-related risk of developing Alzheimer disease. Ann. Neurol. 75, 563–573 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Fisher DW, Bennett DA & Dong H Sexual dimorphism in predisposition to Alzheimer’s disease. Neurobiol. Aging 70, 308–324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Shen S, Zhou W, Chen X & Zhang J Sex differences in the association of APOE ε4 genotype with longitudinal hippocampal atrophy in cognitively normal older people. Eur. J. Neurol. 26, 1362–1369 (2019). [DOI] [PubMed] [Google Scholar]
- 204.Morris JC et al. Assessment of racial disparities in biomarkers for Alzheimer disease. JAMA Neurol. 76, 264–273 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Williams T, Borchelt DR & Chakrabarty P Therapeutic approaches targeting Apolipoprotein e function in Alzheimer’s disease. Mol. Neurodegener. 15, 8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Xiong M et al. APOE immunotherapy reduces cerebral amyloid angiopathy and amyloid plaques while improving cerebrovascular function. Sci. Transl. Med. 13, eabd7522 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Langa KM & Burke JF Preclinical Alzheimer disease - early diagnosis or overdiagnosis? JAMA Intern. Med. 179, 1161–1162 (2019). [DOI] [PubMed] [Google Scholar]
- 208.Jicha GA & Rentz DM Cognitive and brain reserve and the diagnosis and treatment of preclinical Alzheimer disease. Neurology 80, 1180–1181 (2013). [DOI] [PubMed] [Google Scholar]
- 209.Frisoni GB et al. Re-aligning scientific and lay narratives of Alzheimer’s disease. Lancet Neurol. 18, 918–919 (2019). [DOI] [PubMed] [Google Scholar]
- 210.Frisoni GB et al. Precision prevention of Alzheimer’s and other dementias: anticipating future needs in the control of risk factors and implementation of disease-modifying therapies. Alzheimers Dement. 10.1002/alz.12132 (2020). [DOI] [PubMed] [Google Scholar]
- 211.Tavana JP et al. RAB10: an Alzheimer’s disease resilience locus and potential drug target. Clin. Interv. Aging 14, 73–79 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Barroeta- Espar I et al. Distinct cytokine profiles in human brains resilient to Alzheimer’s pathology. Neurobiol. Dis. 121, 327–337 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Cummings J, Lee G, Ritter A, Sabbagh M & Zhong K Alzheimer’s disease drug development pipeline: 2020. Alzheimers Dement. Transl. Res. Clin. Interv. 6, e12050 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Ballard C et al. Evaluation of the safety, tolerability, and efficacy of pimavanserin versus placebo in patients with Alzheimer’s disease psychosis: a phase 2, randomised, placebo-controlled, double- blind study. Lancet Neurol. 17, 213–222 (2018). [DOI] [PubMed] [Google Scholar]
- 215.Iqbal K & Grundke-Iqbal I Alzheimer neurofibrillary degeneration: significance, etiopathogenesis, therapeutics and prevention. J. Cell. Mol. Med. 12, 38–55 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Veitch DP et al. Understanding disease progression and improving Alzheimer’s disease clinical trials: recent highlights from the Alzheimer’s Disease Neuroimaging Initiative. Alzheimers Dement. 15, 106–152 (2019). [DOI] [PubMed] [Google Scholar]
- 217.Derry PJ et al. Revisiting the intersection of amyloid, pathologically modified tau and iron in Alzheimer’s disease from a ferroptosis perspective. Prog. Neurobiol. 184, 101716 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]